

Abstract
Background:
Clonal hematopoiesis (CH), resulting from an array of nonmalignant driver gene mutations, can lead to altered immune cell function and chronic disease, and has been associated with worse outcomes in patients with heart failure (HF) with reduced ejection fraction (HFrEF). However, the role of CH in the prognosis of heart failure with preserved ejection fraction (HFpEF) has been understudied. Therefore, this study sought to characterize CH in patients with HFpEF and elucidate its causal role in a murine model.
Methods/Results:
Using a panel of 20 candidate CH driver genes and a variant allele frequency (VAF) cutoff of 0.5%, ultra-deep error-corrected sequencing identified CH in a cohort of 81 patients with HFpEF (Age: 71 ± 6 years old, EF: 63% ± 5%) and 36 control individuals without a diagnosis of HFpEF (Age: 74 years old ±7 years old, EF: 61.5% ± 8%). Compared to control individuals, there was an enrichment of TET2-mediated CH in the HFpEF patient cohort (12% vs. 0%, respectively, p=0.02). Within the HFpEF cohort, patients with CH exhibited exacerbated diastolic dysfunction in terms of E/e’ (14.9 vs. 11.7, respectively, p=0.0096) and E/A (1.69 vs. 0.89, respectively, p=0.0206) compared to those without CH. The association of CH with exacerbated diastolic dysfunction was corroborated in a validation cohort of 59 individuals with HFpEF. Accordingly, HFpEF patients with CH and an age ≥ 70 years old exhibited worse prognosis in terms of 5-year CV-related hospitalization rate (HR = 5.06, p= 0.042) compared to HFpEF patients without CH and an age ≥ 70 years old. To investigate the causal role of CH in HFpEF, non-conditioned mice underwent adoptive transfer with Tet2-wildtype or Tet2-deficient bone marrow and were subsequently subjected to a high fat diet/L-NAME combination treatment to induce features of HFpEF. This model of Tet2-CH exacerbated cardiac hypertrophy by heart weight to tibia length and cardiomyocyte size, diastolic dysfunction by E/e’ and LV EDP, and cardiac fibrosis compared to the Tet2-wildtype condition.
Conclusions:
CH is associated with worse heart function and prognosis in patients with HFpEF, and a murine experimental model of Tet2-mediated CH displays greater features of HFpEF.
Keywords: Clonal hematopoiesis, Heart failure, Prognosis, Biomarkers, Translational Studies
INTRODUCTION:
Heart failure (HF) is a clinical syndrome of breathlessness and/or fatigue caused by impaired cardiac function. Heart failure (HF) can be further categorized into heart failure with reduced ejection fraction (HFrEF) or heart failure with preserved ejection fraction (HFpEF), which are classically associated with a predominant systolic dysfunction and diastolic dysfunction, respectively.1 Despite a similar prevalence of HFpEF and HFrEF, the mortality rate of HFrEF has dropped significantly due to advancements in care while the mortality rate of HFpEF has remained largely uncurbed due to limited FDA-approved treatments for HFpEF.2–4 The etiology of the HFpEF is poorly understood compared to that of HFrEF, yet recent findings suggest a pronounced role of inflammation, caused by diabetes, hyperlipidemia and hypertension, in the development of HFpEF.5 However, these conditions alone are insufficient for the development of HFpEF, suggesting the presence of additional factors that contribute to the etiology. Thus, a better understanding of the mechanisms that contribute to HFpEF is required to address this prevalent yet underserved disease.
Clonal hematopoiesis (CH) is an emerging immunological phenomenon that has been implicated in different diseases.6,7 In this process, hematopoietic stem cells (HSCs) incur somatic mutations as a consequence of aging, smoking, or other environmental/biological stresses. In some cases, these mutations occur in “driver” genes, such as DNMT3A, TET2, TP53, ASXL1, and JAK2.8–13 When these driver genes are mutated, they provide a selective growth advantage to the HSCs. As a result, the mutant cell line outcompetes neighboring cells in the bone marrow niche and undergoes a clonal expansion. Furthermore, these mutations are maintained in the progeny cells of the HSC, and they can potentially affect the function of leukocytes and promote a chronic inflammatory state.9,11–13 Recent studies have associated CH with worsened prognoses in HFrEF and in patients with an ischemic etiology of heart failure.14–17 However, there is no current literature connecting CH to the prognosis of patients with HFpEF. In this study, we elucidated the significance of CH in populations of patients with HFpEF and control individuals without a diagnosis of HFpEF. Furthermore, we characterized the effects of Tet2-mediated CH in a murine model of HFpEF.
METHODS:
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Clinical Data (Alberta HEART)
All participant data were sourced from the Alberta Heart Failure Analysis Research Team (HEART) cohort and described by Ezekowitz et. al.18,19 Briefly, the cohort was recruited in Alberta, Canada, from 2010 to 2014 in an outpatient setting from a variety of clinics and the community at large. The cohort has been prospectively followed for clinical outcomes since inception in 2010 via annual administrative health data abstraction. Standard baseline demographics, laboratory, and other medical history were collected via medical record review and direct contact with the patient during study entry and following visits. Transthoracic echocardiography was performed with the subjects at rest in left lateral decubitus position using commercially available Phillips iE33 ultrasound imaging system (Philips Medical Systems) equipped with S5-phased or X5-phased array transducer. All images were digitally stored for offline analysis (Xcelera, Philips Medical System). Standard apical four- and two-chamber views were recorded with care taken to avoid foreshortening. LV volumes were measured from the apical four- and two-chamber views. Left ventricular end-systolic volume and end-diastolic volume were calculated using Simpson’s biplane method of discs. LVEF was subsequently derived and expressed as a percentage. Patients received standardized follow-up every three months, during which additional information was collected. Median follow-up was 1355 days (25th–75th percentile 854–1774). Patients were assigned to the control group or HFpEF group through an adjudication process conducted by team members with clinical experience and expertise. The adjudication process required two expert clinicians to review each case independently while blinded to each other’s adjudication. The control population lacked a diagnosis of heart failure at any time during the study, were not risk of developing heart failure, and did not exhibit any symptomatology of heart failure at time of study entry. Given the diverse and ever-changing guidelines and criteria for HFpEF,20 no strict criteria for HFpEF were employed. Instead, the two expert clinicians reviewed each case independently and assigned patients to HFpEF group based on medical history, echocardiography, other radiology testing, and laboratory information. The data for adjudication was sourced from past medical records and testing performed at study entry. Blood samples for ultra-deep, error-corrected sequencing were taken at time of study entry.
Data management and regulatory, ethical and administrative review for Alberta HEART was provided in-kind by the Canadian VIGOUR Centre (Edmonton, Alberta, Canada), and biologic samples were managed by the Canadian BioSample Repository (Edmonton, Alberta, Canada) at the University of Alberta, Canada. Written informed consent was obtained from all participants. The study is registered (clinicaltrials.gov NCT02052804). All analyses on laboratories and echocardiographic measurements were performed on baseline values, and patients with missing data for a given variable were excluded from analysis for that variable.
Validation Clinical Cohort (SCAN-MP)
Participant data was sourced from the ongoing clinical trial Screening for Cardiac Amyloidosis with Nuclear Imaging in Minority Populations (SCAN-MP) cohort as described by Ruberg et. al.21 Briefly, subjects were recruited from Columbia University Irving Medical Center, Harlem Hospital, Yale University, and Boston Medical Center. All participants self-identified as Black or Hispanic of Caribbean origin. Heart failure was diagnosed by either the modified criteria utilized by Rich et al.22 or a score ≥3 on the National Health and Nutrition Examination Survey (NHANES) for heart failure criteria. Modified criteria utilized by Rich et al. which include a history of acute pulmonary edema or the occurrence of at least two of the following that improved with diuretic therapy without another identifiable cause: dyspnea on exertion, paroxysmal nocturnal dyspnea, orthopnea, bilateral lower extremity edema or exertional fatigue. Additionally, patients with HFpEF had an ejection fraction >30%. Only HFpEF patients, who lacked a diagnosis of cardiac amyloidosis, were included in the present analysis. At time of enrollment, medical history, medication inventory, laboratories, and transthoracic echocardiography were obtained. At time of enrollment, blood samples were also collected for assessment of clonal hematopoiesis.
SCAN-MP was approved by the Western Institutional Review Board (IRB) in a single IRB model (IRB#: 20183425). Written informed consent was obtained from all participants. The study is registered (clinicaltrials.gov NCT03812172). All analyses on laboratories and echocardiographic measurements were performed on baseline values, and patients with missing data for a given variable were excluded from analysis for that variable.
Animal Studies/Mice
Wild-type mice (Tet2+/+), Tet2-deficient mice (Tet2−/−: B6(Cg)-Tet2tm1.2Rao/J), and Pep Boy mice (B6.SJL-Ptprca Pepcb/BoyJ) were sourced from Jackson Laboratory (Stock #: 000664, 023359, 002014, respectively). All strains had a C57BL/6J background. Tet2+/− mice were used for breeding, and Tet2−/− and Tet2+/+ male offspring were used for the test group and the control group, respectively. The animal protocols for these experiments were approved by the Institutional Animal Care and Use Committee of the University of Virginia.
Nonmyeloablative Bone Marrow Transplantation
Nonmyeloablative bone marrow transplantation was performed as previously published.23 Briefly, 8- to 12-week-old Pep Boy mice were transplanted with suspensions of bone marrow cells from either Tet2+/+ mice or Tet2−/− mice. Unfractionated bone marrow cells (5 × 106 cells/day) were injected via retro-orbital vein into non-irradiated recipients per day over 3 consecutive days (total: 1.5 × 107 cells).
HFpEF Model
After nonmyeloablative bone marrow transplantation, mice were randomized to receive either a control diet and water or high fat diet (S1850, Bio-serv) and N[w]-nitro-l-arginine methyl ester (L-NAME; 1.0 g/L, Sigma Aldrich, N5751) in the drinking water.24 The drinking water was changed once per week. Throughout the length of the experiment, mice were maintained on a 12-hour light/dark cycle and had unrestricted access to food and water.
Statistical Analyses
Data are presented as mean and standard deviation unless otherwise noted. For continuous data with one variable, Shapiro Wilk test was performed to evaluate the data distribution. Non-normally distributed data was analyzed for statistical significance by Mann-Whitney U test while normally-distributed data was analyzed with Welch’s t test and 1-way ANOVA with Dunnett multiple-comparison for data with one independent variable and 2 groups or >2 groups, respectively. For categorical variables, Fisher exact tests were performed to test statistical significance. For continuous data with two variables, a 2-way ANOVA with post hoc Sidak multiple comparison test was used to test for statistical significance. For multivariable analysis of continuous variables, a multivariable linear model was created adjusting for differences in backgrounds between groups. Data are reported as the coefficient ± standard error. For univariable and multivariable analysis of outcomes, a Cox proportional hazards model was employed. Data are reported as a hazard ratio and its corresponding 95% confidence interval.
RESULTS:
HFpEF patients with CH display worse cardiac diastolic function and outcomes
To investigate the significance of clonal hematopoiesis in HFpEF, peripheral blood samples were sourced from the Alberta HEART cohort.19 Ultra-deep, error-corrected sequencing with a panel of 20 candidate driver genes was then employed to identify and quantify CH (Table S1). CH mutations only included frameshift, splicing, stop-gain, or nonsense variants, which had the potential to influence protein function. The sequencing methodology was sufficient to resolve mutations with variant allelic frequencies (VAFs) as low as 0.005. As such, we used this VAF as our threshold for diagnosing CH in patients. The cohort was composed of 81 patients with HFpEF and 36 control individuals without a diagnosis of HFpEF. As expected, the clinical parameters of the patients with HFpEF differed significantly from that of the control individuals with respect to hypertension, diabetes, smoking history, and a number of other comorbidities (Table S2). In this cohort, ultra-deep, error-corrected sequencing identified mutations in 12 known CH driver genes (Figure 1A). DNMT3A and TET2 were the most commonly mutated genes. Of all the individuals with CH in the cohort, individuals with 1 mutation, 2 mutations, and 3+ mutations accounted for 77%, 19%, and 4%, respectively (Figure 1B). Consistent with previous reports,7,16 CH prevalence increased with age (Figure 1C). Individuals between the ages of 60 to 69 years-old displayed a CH mutation prevalence of 24%, while 64% of individuals between 80 to 89 years-old displayed CH mutations. As shown in Figure 1D, patients with HFpEF possessed, on average, 4-fold larger CH clones than control individuals (VAF of 0.015 vs. 0.061, p=0.0197). Interestingly, there were no TET2 mutations identified in control individuals, while 10 patients with HFpEF harbored a detectable TET2 mutation (Figure 1E).
Figure 1. Characterization of clonal hematopoiesis in the Alberta HEART patient cohort.
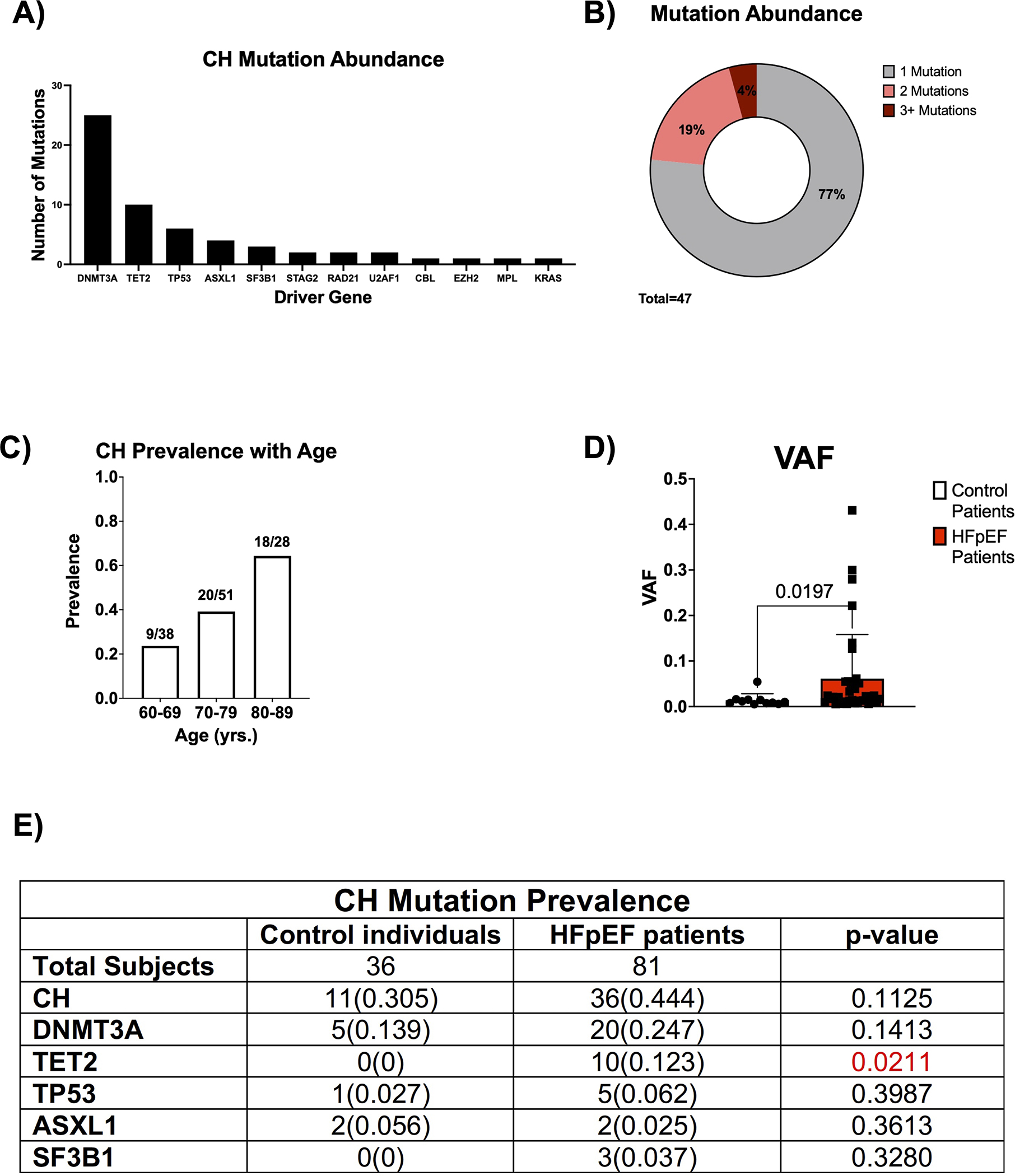
A. Abundance of the specified driver gene mutation in the patient cohort (n=58). B. Proportion of patients with the specified number of CH mutations (n=47). C. CH prevalence as a function of age in the patient cohort (n=117). D. VAF for the largest clone identified in patients with CH in control individuals and HFpEF patients. Statistical significance was determined by Mann-Whitney U test (n= 11 control individuals and 36 HFpEF patients). E. Prevalence of CH and driver gene mutations in control individuals and patients with HFpEF. Statistical significance was determined by Fisher’s Exact Test (n= 36 control individuals and 81 HFpEF patients).
Further analysis characterized the functional significance of CH within the HFpEF population. The backgrounds of HFpEF patients with CH and HFpEF patients without CH were similar (Table 1). However, HFpEF patients with CH tended to be older (77 vs. 72 years, p = 0.001) and have a lower BMI (28 vs. 32 kg/m2, p=0.008) and greater ACEi/ARB use (87% vs. 64%, p=0.01) than HFpEF patients without CH. HFpEF patients with CH had a worse baseline diastolic function compared to HFpEF patients without CH, as evidenced by an increase in both E/e’ and E/A and decreased deceleration time (Figure 2A–2C). HFpEF patients with CH had elevated levels of the BNP and NT-proBNP biomarkers of HFpEF severity compared to HFpEF patients without CH at baseline (Figure 2D and 2E). Multivariate analysis was performed to adjust for age, BMI, and ACEi/ARB use (Table S3). After adjustment, E/e’, E/A, and deceleration time remained statistically significant, and CH further became associated with increased left atrial volume index (LAVI). Additionally, many of these differences were maintained at different VAF cutoffs for identifying CH in both univariate and multivariate analyses, which corrected for corresponding differences in background characteristics (Tables S4–S5). Given the differences in heart function, we then evaluated whether this difference was maintained when only stratifying for the presence or absence of mutations in either of the two most common clonal hematopoiesis driver genes DNMT3A and TET2 as has been done previously. 15,16,25 HFpEF patients with a DNMT3A/TET2 mutation and HFpEF patients without DNMT3A/TET2 mutation had similar backgrounds (Table S6). However, HFpEF patients with a DNMT3A/TET2 mutation tended to be female (70% vs. 46%, p=0.0339), have a lower BMI (28 vs. 31kg/m2, p=0.0385), and have a lower prevalence of COPD (7% vs. 31%, p=0.0128). After adjusting for background differences, mutations in either DNMT3A or TET2 were associated with worse diastolic function by E/A and deceleration time (Table S7). Furthermore, if using 0.01 or 0.02 as VAF cutoffs for diagnosing CH, mutations in either DNMT3A or TET2 were associated with worse diastolic function by E/e’.
Table 1.
Clinical characteristics for the HFpEF patients with and without clonal hematopoiesis.
| HFpEF CH− vs HFpEF CH+ Patient Characteristics | |||
|---|---|---|---|
| Measurement | HFpEF CH− | HFpEF CH+ | p-value |
| Total | 45 | 36 | |
| Age (yrs) | 72(7) | 77(7) | 0.0012 |
| Female | 21(0.47) | 23(0.64) | 0.0929 |
| BMI (kg/m2) | 32(6) | 28(5) | 0.0083 |
| Hypertensive | 38(0.84) | 33(0.92) | 0.2635 |
| Diabetes | 19(0.42) | 11(0.31) | 0.1983 |
| Hyperlipidemia | 28(0.62) | 23(0.64) | 0.5316 |
| COPD | 14(0.31) | 5(0.14) | 0.0585 |
| CKD | 7(0.16) | 8(0.22) | 0.3144 |
| Prior history of cancer | 0(0) | 0(0) | 1.0000 |
| History of smoking | 24(0.53) | 17(0.47) | 0.3734 |
| Prior myocardial infarction | 11(0.24) | 6(0.17) | 0.2830 |
| Prior coronary revascularization | 0(0) | 0(0) | 1.0000 |
| Atrial fibrillation or flutter | 22(0.49) | 23(0.64) | 0.1302 |
| Cerebrovascular Disease | 4(0.09) | 8(0.22) | 0.0866 |
| Peripheral Vascular Disease | 1(0.02) | 2(0.06) | 0.4160 |
| ACE inhibitor or ARB | 39(0.87) | 23(0.64) | 0.0161 |
| Beta-blocker | 36(0.8) | 25(0.69) | 0.2015 |
| Diuretic | 36(0.8) | 31(0.86) | 0.3375 |
| CCB | 21(0.47) | 13(0.36) | 0.2331 |
| Digoxin | 3(0.07) | 4(0.11) | 0.3753 |
| Antiarrhythmic | 6(0.13) | 7(0.19) | 0.3283 |
| Anticoagulation | 21(0.47) | 21(0.59) | 0.2061 |
| Antiplatelet | 4(0.09) | 1(0.03) | 0.2570 |
Continuous variables are expressed as mean and standard deviation while categorical variables are described by absolute count and frequency. BMI: body mass index, COPD: chronic obstructive pulmonary disease, CKD: chronic kidney disease, ARB: angiotensin II receptor blocker, CCB: calcium channel blocker.
Figure 2. HFpEF patients with CH exhibit worse diastolic dysfunction and possess elevated levels of heart failure biomarkers at baseline.
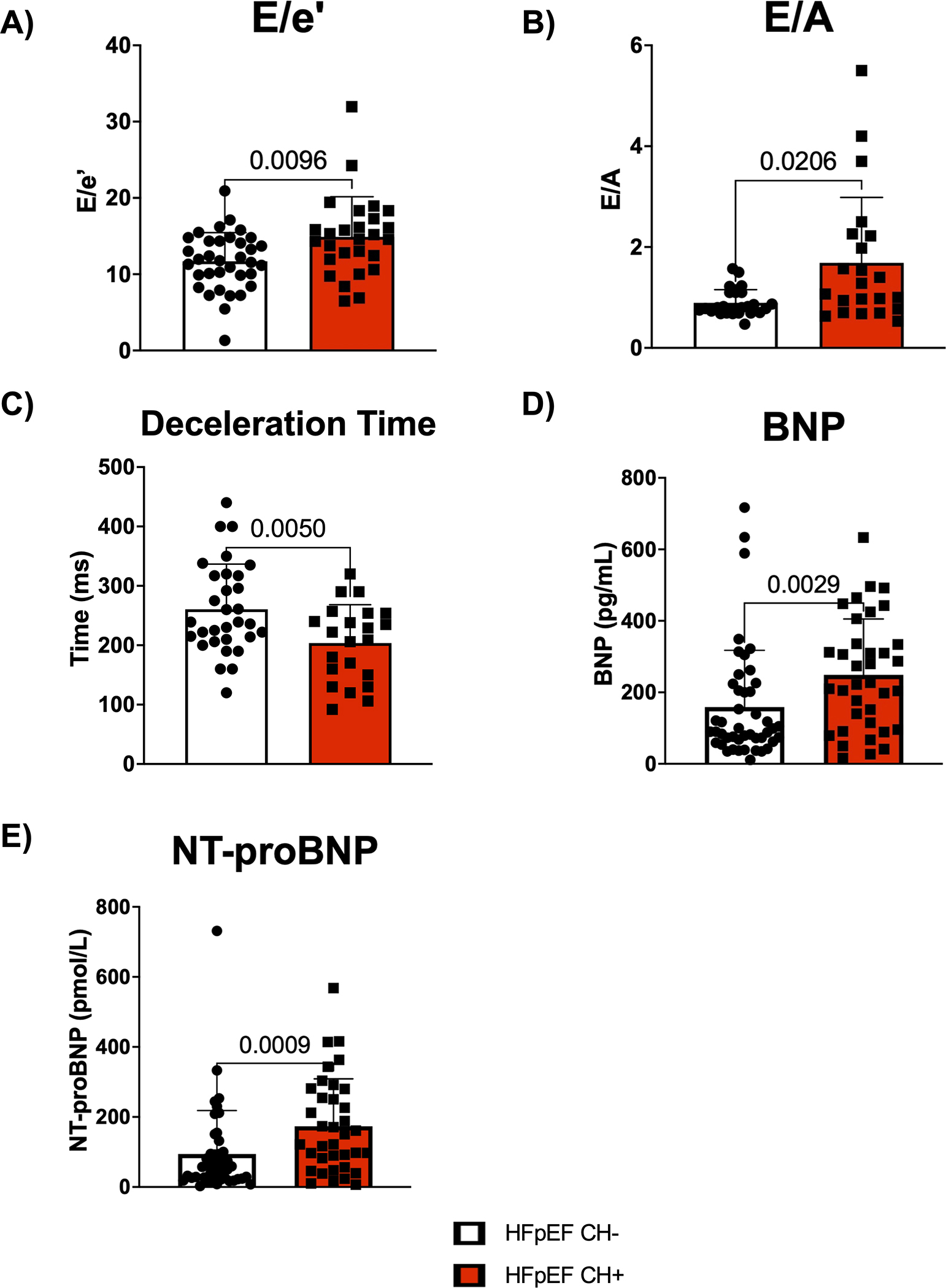
A. Echocardiographic analysis of E/e’ in HFpEF patients with and without CH. Statistical significance was determined by Mann-Whitney U test (n= 35 HFpEF CH− patients and 27 HFpEF CH+ patients). B. Echocardiographic analysis of E/A in HFpEF patients with and without CH. Statistical significance was determined by Mann-Whitney U test (n= 27 HFpEF CH− patients and 22 HFpEF CH+ patients). C. Echocardiographic analysis of deceleration time in HFpEF patients with and without CH. Statistical significance was determined by Welch’s t test (n= 31 HFpEF CH− patients and 22 HFpEF CH+ patients). D. Plasma brain natriuretic peptide (BNP) levels for HFpEF patients with and without CH. Statistical significance was determined by Mann-Whitney U test (n= 45 HFpEF CH− patients and 36 HFpEF CH+ patients). E. Plasma N-terminal pro b-type natriuretic peptide (NT-proBNP) levels for HFpEF patients with and without CH. Statistical significance was determined by Mann-Whitney U test (n= 45 HFpEF CH− patients and 36 HFpEF CH+ patients).
In a separate validation cohort, PBMCs were sourced from the ongoing SCAN-MP clinical trial,21 which comprised HFpEF patients from minority populations. Ultra-deep, error-corrected sequencing was again performed to identify and quantify CH. HFpEF patients with CH and HFpEF patients without CH displayed no differences in background characteristics (Table S8). However, HFpEF patients with CH exhibited worse diastolic function by both E/e’ (18.4 vs. 13.8, p=0.003) and e’ alone (5.0 vs. 6.2, p=0.03) compared to HFpEF patients without CH (Figure S1A,B). Furthermore, this association held if HFpEF patients were stratified for the presence or absence of mutations in either DNMT3A or TET2. Despite no difference in background characteristics (Table S9), HFpEF patients with a DNMT3A/TET2 mutation exhibited an increased E/e’ (18.7 vs. 14.3, p=0.005) compared to HFpEF patients without a DNMT3A/TET2 mutation (Figure S1C).
We further investigated the impact of CH on prognosis for patients with HFpEF, whose age ranged from 60 to 90 years old. HFpEF patients with CH and without CH displayed 5-year CV-related hospitalization rates of 43.5% and 26.9%, respectively (HR: 2.09, p=0.10, Figure S2A). We next examined prognosis by stratifying for an age ≥ 70 years old, at which many of these adverse events become more common. The backgrounds of HFpEF patients with CH and an age ≥ 70 years old and HFpEF patients without CH and an age ≥ 70 years old were similar (Table S10) except that HFpEF patients with CH tended to be older (80 vs. 77 years, p = 0.02) and have a lower BMI (28 vs. 31 kg/m2, p=0.008). HFpEF patients with CH and an age ≥ 70 years old displayed a 5-year CV-related hospitalization rate of 51% while HFpEF patients without CH and an age ≥ 70 years old only had a 5-year CV-related hospitalization rate of 10% (HR=6.83, p=0.0041, Figure 3A–B). To eliminate the contribution of age and BMI, a Cox proportional hazards model for HFpEF patient outcomes was performed (Figure 3C). After adjusting for both age and BMI, HFpEF patients with CH and an age ≥ 70 years old maintained a statistically significant increase in 5-year CV-related hospitalization rate compared to HFpEF patients without CH and an age ≥ 70 years old, suggesting worse prognosis (HR=5.06, p=0.042). Furthermore, the difference in CV-related hospitalization rate is maintained at VAF thresholds for CH of ≥0.005, ≥0.01, and ≥0.02 in in both univariate and multivariate analyses, which corrected for corresponding differences in background characteristics (Tables S11–S12).
Figure 3. HFpEF patients with mutations in CH driver genes exhibit worse long-term prognosis.
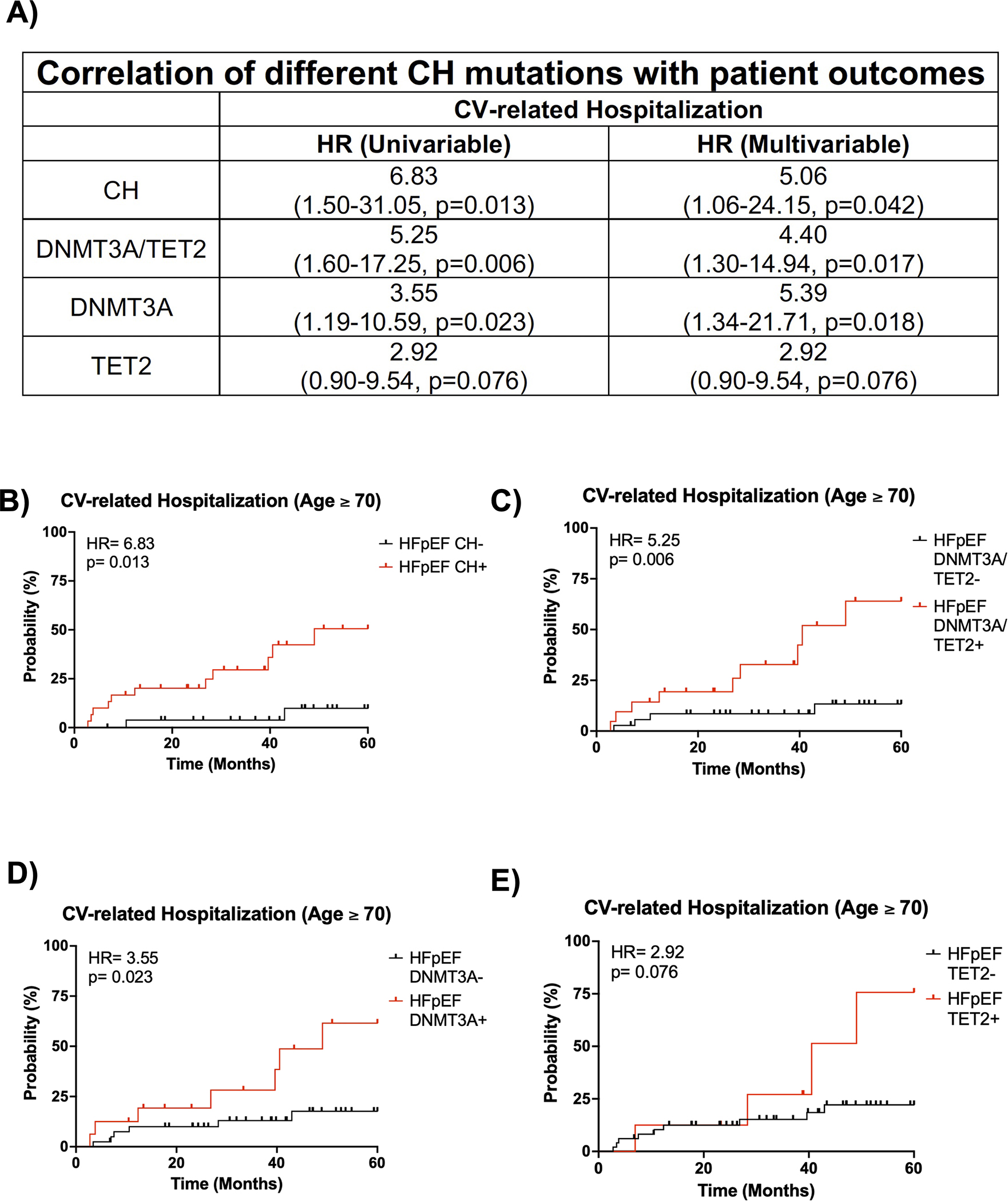
A. Univariate and multivariate analysis of CV-related hospitalization for HFpEF patients and an age ≥ 70 years old based on CH status, DNMT3A/TET2-driven CH status, DNMT3A-driven CH status, and TET2-driven CH status. Statistical significance and hazards ratio were determined by Cox proportional hazards model. B. 5-year CV-related hospitalization based on clonal hematopoiesis status for HFpEF patients with an age ≥ 70 years old. Statistical significance and hazards ratio were determined by Cox proportional hazards model (n= 27 HFpEF CH− patients and 30 HFpEF CH+ patients). C. 5-year CV-related hospitalization for HFpEF patients with and without DNMT3A/TET2-driven CH and an age ≥ 70 years old. Statistical significance and hazards ratio were determined by Cox proportional hazards model (n= 36 HFpEF DNMT3A/TET2- patients and 21 HFpEF DNMT3A/TET2+ patients). D. 5-year CV-related hospitalization for HFpEF patients with and without DNMT3A-driven CH and an age ≥ 70 years old. Statistical significance and hazards ratio were determined by Cox proportional hazards model (n= 41 HFpEF DNMT3A- patients and 16 HFpEF DNMT3A+ patients). E. 5-year CV-related hospitalization for HFpEF patients with and without TET2-driven CH and an age ≥ 70 years old. Statistical significance and hazards ratio were determined by Cox proportional hazards model (n= 49 HFpEF TET2- patients and 8 HFpEF TET2+ patients).
Given the difference in prognosis for patients with HFpEF and an age ≥ 70 years old, we then evaluated whether this difference was maintained at the age threshold when only stratifying for the presence or absence of mutations in either DNMT3A or TET2. Indeed, HFpEF patients with a DNMT3A/TET2 mutation and an age ≥ 70 years old had a 5-year CV-related hospitalization rate of 64% while HFpEF patients without a DNMT3A/TET2 mutation and an age ≥ 70 years old had a 5-year CV-related hospitalization rate of 13% (HR =5.25, p=0.006, Figure 3A,3C). HFpEF patients with a DNMT3A/TET2 mutation and an age ≥ 70 years old had a similar background to HFpEF patients without a DNMT3A/TET2 mutation and an age ≥ 70 years old; however, they tended to be female (71% vs. 44%, p=0.04, Table S13). After adjusting for sex by Cox proportional hazards model, this difference in CV-related hospitalizations was maintained (HR: 4.40, p=0.017, Figure 3A,3C). Furthermore, this difference in CV-related hospitalization rate was maintained at VAF cutoffs of ≥0.005, ≥0.01, and ≥0.02 in a univariate analysis and maintained at VAF cutoffs of ≥0.005 and ≥0.01 in a multivariate analysis, which corrected for corresponding differences in background characteristics (Table S14). To understand how individual mutations in either DNMT3A or TET2 modify prognosis, HFpEF patients were stratified for the presence or absence of either of these mutants. In multivariate analyses adjusting for differences in backgrounds of HFpEF patients with an age ≥ 70 years old (Table S15), DNMT3A-mediated CH was associated with a significant increase in CV-related hospitalization rate (HR: 5.39, p=0.018, Figure 3A,3D) while TET2-mediated CH was associated with a trending increase in CV-related hospitalization rate (HR; 2.92, p=0.076, Figure 3A,3E). In contrast to the findings with echocardiographic parameters and CV-related hospitalization data, analyses of all-cause mortality did not reveal associations with CH within the entire cohort (Figure S2B–C) or subgroups (Table S16), which may be due to the high degree of patient survival in the Alberta HEART cohort.
A murine model of HFpEF accelerates TET2-mediated hematopoietic cell expansion
Due to its enrichment in the HFpEF patient cohort, TET2 was chosen for further mechanistic studies in mice. To model a more physiologically relevant state of CH, a small number of CD45.2 Tet2-WT (Tet2+/+) or Tet2-deficient (Tet2−/−) bone marrow cells were adoptively transferred to CD45.1 Pep Boy mice and allowed to expand for one month. Mice were then either continued on control diet and water or placed on a high-fat diet (HFD) and L-NAME drinking water combination treatment to induce features of HFpEF.24 Serial measurements were performed at baseline, 5 weeks, and 12 weeks post-treatment induction (Figure 4A).
Figure 4. HFD/L-NAME treatment accelerates expansion of Tet2-deficient cells in peripheral blood.
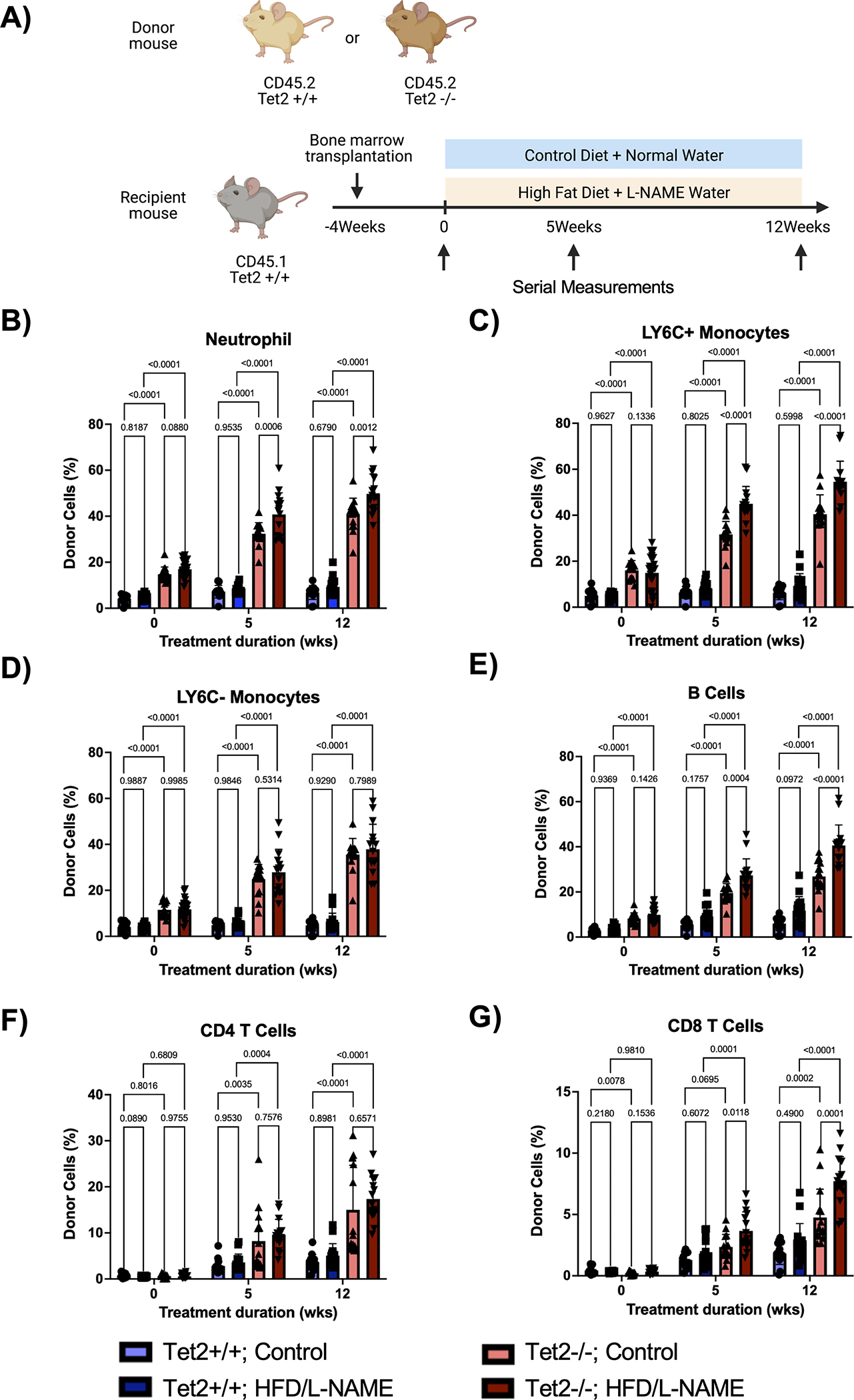
A. Schematic of the experimental design. CD45.2 Tet2-sufficient or Tet2-deficient bone marrow was adoptively transferred to CD45.1 Pep Boy mice. One month after bone marrow transplantation, mice were started on either HFD/L-NAME combination treatment or continued on control diet and water. Serial measurements were taken at baseline, 5 weeks, and 12 weeks. B-G. Flow cytometric quantification of donor cell chimerism for neutrophils, Ly6C+ monocytes, Ly6C− monocytes, B cells, CD4 T cells, and CD8 T cells in the peripheral blood at baseline and after 5 weeks and 12 weeks of treatment. Statistical significance was determined by 2-way ANOVA with post hoc Sidak multiple comparison test (n= 13–26 per group).
To evaluate clonal expansion in this model, flow cytometry of peripheral blood was performed. As previously published,23 donor-derived Tet2−/− cells expanded more rapidly throughout all white blood cell lineages compared to donor-derived Tet2+/+ cells, with a bias towards the myeloid and B cell populations (Figure 4B–4G). Interestingly, HFD/L-NAME enhanced Tet2-mediated cell expansion (Figure 4B, 4C, 4E, 4G). In accordance, flow cytometry of bone marrow revealed expansion of donor-derived Tet2−/− cells in the long-term and short-term hematopoietic stem cell pools (LT-HSC and ST-HSC, respectively) compared to donor-derived Tet2+/+cells (Figure 5A–5F). Further, in parallel with peripheral blood chimerism, donor chimerisms of hematopoietic stem cell pools were significantly higher for Tet2−/− cells subjected to the HFD/L-NAME regimen compared to Tet2−/− cells exposed to the control conditions. HFD/L-NAME induced a leukocytosis by 3 months with increases in the absolute number of neutrophils, monocytes, and leukocytes for the Tet2−/− group (Figure S3). There were no differences in platelet counts or hemoglobin concentration throughout the conditions.
Figure 5. HFD/L-NAME treatment promotes expansion of Tet2-deficient cells in the hematopoietic stem and progenitor cells.
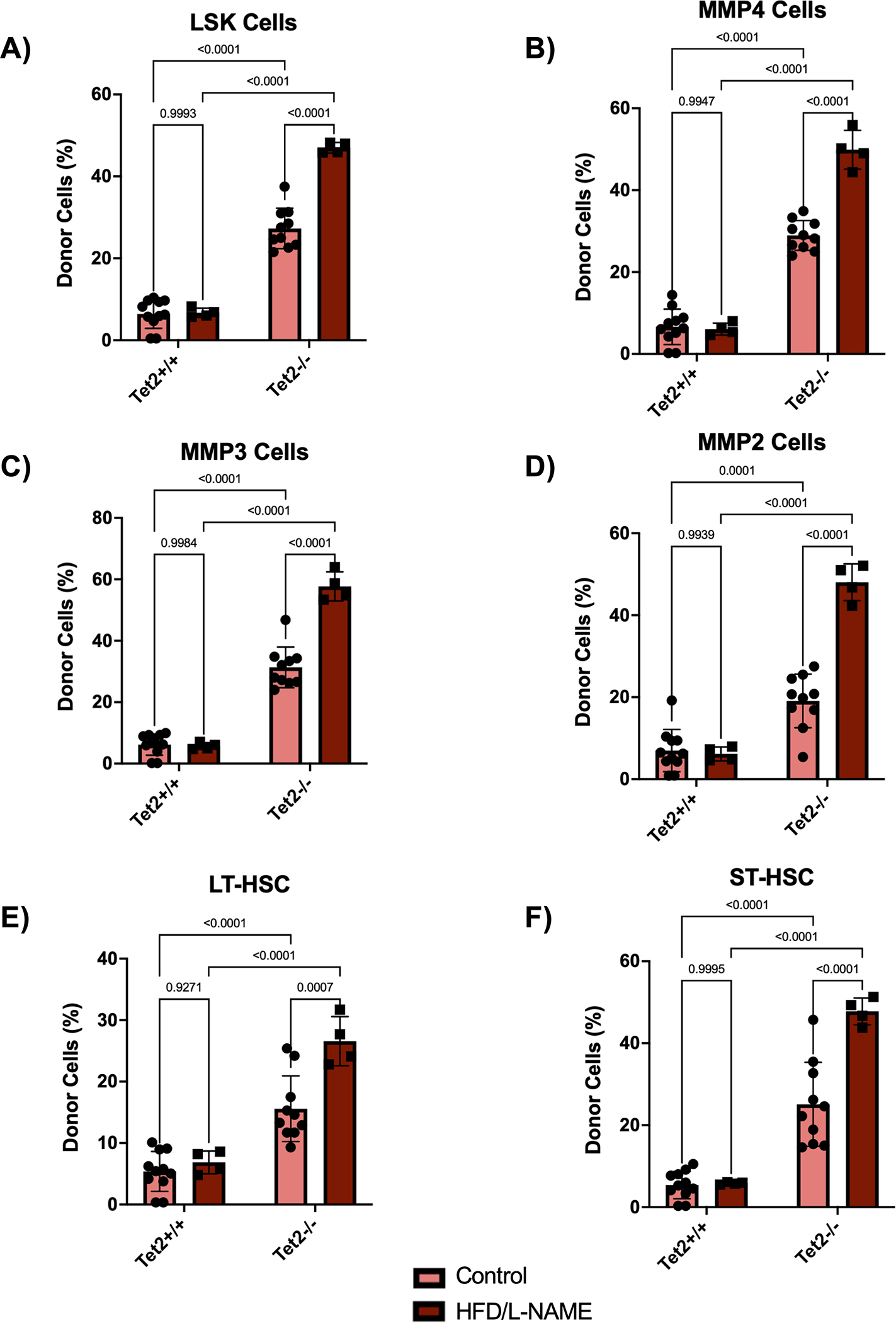
A-F. Flow cytometric quantification of donor cell chimerism in LSK cells, MMP4 cells, MMP3 cells, MMP2 cells, long-term hematopoietic stem cells (LT-HSC), and short-term hematopoietic stem cells (ST-HSC) of the bone marrow after 12 weeks of treatment. Statistical significance was determined by 2-way ANOVA with post hoc Sidak multiple comparison test (n=4–11 per group).
TET2-mediated CH exacerbates heart failure in a murine model of HFpEF
To induce HFpEF, a combination of an obesogenic diet and a hypertensive drug was employed. The HFD/L-NAME combination increased weight gain compared to respective mice on control diet and water (Figure 6A). Furthermore, Tet2−/− group exhibited increased weight gain compared to the Tet2+/+ group on the HFD/L-NAME treatment. Additionally, the HFD/L-NAME combination treatment led to increased systolic blood pressure compared to the respective control group (Figure 6B).
Figure 6. Tet2-mediated clonal hematopoiesis exacerbates cardiomyopathy in a model of HFpEF.
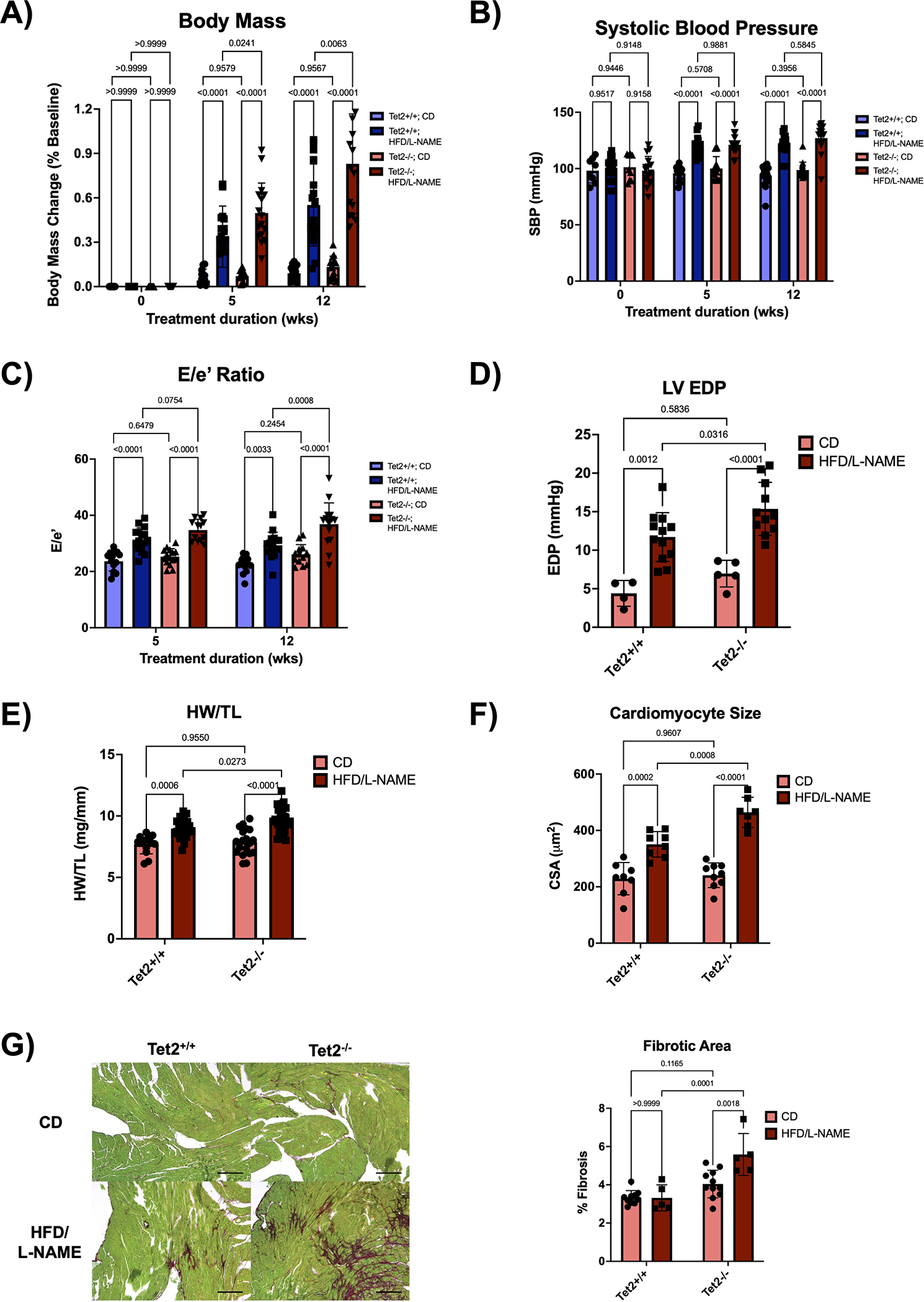
A. Normalized change in body mass relative to baseline body mass measured at the initiation of treatment. Measurements were performed at baseline and after 5 and 12 weeks of treatment. Statistical significance was determined by 2-way ANOVA with post hoc Sidak multiple comparison test (n=14–17 per group). B. Systolic blood pressure measured by tail-cuff plethysmography at baseline and after 5 and 12 weeks of treatment. Statistical significance was determined by 2-way ANOVA with post hoc Sidak multiple comparison test (n=8–17 per group). C. Serial echocardiographic analysis after 5 and 12 weeks of treatment. Statistical significance was determined by 2-way ANOVA with post hoc Sidak multiple comparison test (n= 13–16 per group). D. Left ventricular end diastolic pressure was obtained via LV catheterization after 12 weeks of treatment. Statistical significance was determined by 2-way ANOVA with post hoc Sidak multiple comparison test (n=4–12 per group). E. Heart weight normalized to tibia length after 12 weeks of treatment. Statistical significance was determined by 2-way ANOVA with post hoc Sidak multiple comparison test (n=13–28 per group). F. Quantification of cardiomyocyte cross-sectional area from heat germ agglutinin (WGA) stained hearts after 12 weeks of treatment. Statistical significance was determined by 2-way ANOVA with post hoc Sidak multiple comparison test (n=7–9 per group). G. Representative images and analysis of Picrosirius Red/Fast Green staining of the hearts after 12 weeks of treatment. Scale bars are included in the lower right corner and correspond to 200 μm. Statistical significance was determined by 2-way ANOVA with post hoc Sidak multiple comparison test (n=5–11 per group).
To test whether Tet2-mediated CH modifies cardiac function in this model of HFpEF, serial echocardiographic analysis was performed on these mice. The HFD/L-NAME combination induced an increase in left ventricular filling pressures as estimated by an increase in the Doppler-derived E/e’ ratio and measured directly via LV catheterization, collectively indicating worse diastolic heart function (Figure 6C–D). Moreover, the adoptive transplantation of Tet2−/− bone marrow exacerbated diastolic dysfunction compared to mice receiving Tet2+/+ bone marrow. The Tet2−/− group on the HFD/L-NAME treatment also displayed worse cardiac hypertrophy and fibrosis compared with the Tet2+/+ group of the same dietary treatment (Figure 6E–G).
DISCUSSION:
Clonal hematopoiesis is increasingly recognized as a key risk factor for cardiovascular disease. The accessibility of human peripheral blood samples and declining cost of ultra-deep error-corrected sequencing have streamlined the analysis of CH in clinical cohorts. In the last few years, numerous studies have been published on the associations between CH and CVD in patients7 and its additive effect with traditional CVD risk factors.26 Notably, CH is associated with increased risk of incident HF, and increased CVD morbidity and mortality in patients with HFrEF.14–17 Despite these advances, past studies were either not stratified for etiology or only constrained to patients with HFrEF.16 Thus, the present study has focused on the role of CH in HFpEF.
In this study, we investigated the significance of CH in control patients and HFpEF patients of the cardiometabolic phenogroup, as evidenced by the high BMI and diabetes prevalence. We found an enrichment of patients with TET2 mutations in the HFpEF cohort versus control individuals. In an analysis of all 20 CH driver genes assayed, there was a significant increase in variant clone size in the patients with HFpEF versus the control group. However, due to the differences between the patients with HFpEF and control individuals, it is uncertain whether these findings are being driven specifically by the disease state of HFpEF or by one or more of its comorbidities/covariates. Thus, focusing on patients with HFpEF, analyses revealed that the presence of CH was associated with worse echocardiographic metrics of heart function. Specifically, the increased E/e’ and E/A and decreased deceleration time collectively indicate worse diastolic heart function in patients with HFpEF and CH compared to those without CH. The increased left atrial volume index after multivariable adjustment suggests structural remodeling of the heart in HFpEF patients with CH compared to HFpEF patients without CH. Accordingly, these findings are associated with worse long-term prognosis as evidenced by increased 5-year CV-related hospitalization rate. Consistent with prior literature,15,16,25 we also stratified for the presence or absence of DNMT3A/TET2 mutations and found similar results. Notably, the association between CH and worse prognosis appears to become more pronounced with age. Patients with HFpEF and an age ≥ 70 years old had 6-fold greater odds of experiencing a CVD-related hospitalization if they possessed a DNMT3A or TET2 mutation. This age-dependent association of CH with CV-related hospitalization may be attributed to the greater prevalence of comorbidities and hospitalization in patients of advancing age. Furthermore, the significance of these findings is maintained when explicitly adjusting for covariates in a multivariable model (Table S17). In sum, the presence of CH appears to be an important determinant of the functional status and morbidity of HFpEF.
Our study extends upon previous clinical findings connecting CH and heart failure. Studies have reported that DNMT3A- and/or TET2-mediated clonal hematopoiesis is associated with worse prognosis in patients with chronic ischemic HF and following ST-segment elevation myocardial infarction.15,25 It has also been reported that patients with DNMT3A- and/or TET2-mediated clonal hematopoiesis exhibit adverse HF progression irrespective of ischemic or nonischemic etiology.16 Yu et al. found that CH, particularly mutations in the ASXL1, TET2 and JAK2 driver genes, was associated with incident heart failure in an analysis of 5 large biobanks.17 More recently, the study by Shi et al.27 focused specifically on the role of CH in incident HFpEF. This study reported that CH, using a VAF threshold of 2%, was associated with incident HFpEF in individuals younger than 65 years old, but this association was not found when the entire cohort was analyzed. Our data suggests that the association between CH and HFpEF can extend to patients older than 65 years old, and that VAFs as low as 0.5% may be predictive of this condition. The significance of this finding is bolstered by the fact that small clones become almost ubiquitous with advanced age.28 We found that lower VAF cutoffs tended to better stratify patients in terms of prognosis as evidence by the higher hazards ratio with lower VAF cutoffs (Table S13). This may be due to the fact that these small clones can expand over the course of follow-up and consequently, exert a greater biological effect with time. Thus, traditional DNA sequencing approaches, which are limited by their sequencing depth and therefore, VAF detection limit, are not able to detect these clones, which appear to harbor notable prognostic significance. As a consequence, though previous work has documented a pronounced effect of CH on prognosis,6,15,16,25,29,30 these data may be an underestimation of the true effect of CH.
To examine HFpEF in an experimental model, we utilized a combination of an obesogenic diet and the hypertensive drug L-NAME, as previously described.24 This model was chosen as it reproduces aspects of the metabolic syndrome and consequent features of HFpEF that can be observed in these patients and further mirrors the cardiometabolic phenogroup largely investigated in our clinical cohort. Furthermore, it does not exploit genetic perturbations that are generally not observed in patients. As such, it was deemed the more physiologically realistic and applicable model for our murine studies. Additionally, the non-irradiated adoptive transfer model of clonal hematopoiesis was employed to model the spontaneous development and expansion of Tet2-deficient clones. This model avoids dynamic changes induced in both the hematopoietic niche and cardiovascular system, and appetite loss as a consequence of irradiation.31–33 Together, we reasoned that this combined model more faithfully produces features of HFpEF and TET2-mediated CH for mechanistic studies.
As previously published in the adoptive transfer model,23 Tet2-deficient hematopoietic cells were found to expand more rapidly than Tet2-sufficient clones. However, the HFD/L-NAME treatment accelerated expansion of Tet2-deficient donor cells in both peripheral blood and hematopoietic stem cell lineages. Analogously, these data mirrored the increased VAF of CH clones observed in patients with HFpEF compared to control individuals. This accelerated expansion may be partially attributed to increased hematopoiesis. Compared to the Tet2−/− group on control conditions, the Tet2−/− group on HFD/L-NAME treatment exhibited leukocytosis with elevation of several WBC lineages. Furthermore, hypertension has been previously demonstrated to increase hematopoiesis in murine and human models.34 Thus, these differences may reconcile the lack of accelerated Tet2-deficient clonal expansion observed when mice are subjected to a high fat diet alone, atherogenic stimuli, or other models of heart failure.10,35,36 Moreover, under the conditions of experimental HFpEF, the increased chimerism may further exacerbate the sequelae of Tet2-mediated CH.
In this murine model, Tet2-mediated clonal hematopoiesis exacerbates several features of HFpEF. The Tet2−/− group manifested increased E/e’, LV EDP, HW/TL, and cardiac fibrosis compared to the Tet2+/+ group on the HFD/L-NAME treatment, suggesting worse diastolic function, cardiac hypertrophy, and cardiac fibrosis. This again parallels our clinical data, which showed that patients with HFpEF and CH have significantly worse diastolic function. In summary, our data suggests that Tet2-mediated clonal hematopoiesis directly exacerbates heart failure in a murine model of HFpEF and reproduces many findings observed in a human cohort of HFpEF.
This study extends upon previous work in elucidating causal connection between TET2-mediated CH and cardiovascular disease.7 Previously, our lab has shown that Tet2-deficiency in HSCs spontaneously leads to HFrEF in aging mice.23 Additionally, Tet2-deficiency in HSCs exacerbates heart failure in ischemic models of HFrEF such as myocardial infarction and non-ischemic models of HFrEF such as pressure overload and angiotensin II infusion.9,10 Collectively, Tet2-mediated CH exacerbates HFpEF and HFrEF in murine models of disease.
We acknowledge there are several limitations to the present study. First, for our clinical studies, the sample size is modest, and all patients were recruited from the same geographic location. However, in an analysis of a small cohort from the SCAN-MP study, we were able to corroborate that CH was associated with diastolic dysfunction. In the future, larger multicenter studies are required to further corroborate these findings and potentially reveal novel associations, whose effect sizes were too small to uncover in our analyses. Second, we acknowledge that HFpEF is a diverse disease state with myriad pheongroups.37,38 Our cohort was composed predominately of the cardiometabolic phenogroup, in which patients are typically obese and diabetic. As such, the role of CH in other HFpEF phenotypes remains outstanding. Third, it will be informative to discern whether CH has an impact on CV-related mortality through the analysis of larger cohorts or a longer follow-up. Regarding this point, cohorts of Canadian patients with HFpEF, such as the one examined herein, may exhibit reduced morbidity and mortality due to the predominance of Caucasians enrolled in the study.39,40 Fourth, the adoptive transfer of Tet2-deficient HSCs in experimental studies will generate larger homozygous clones rather than the heterozygous clones typically observed in humans. However, this is common for almost all experimental models, which employ exaggerated stimuli to model disease in a temporally and fiscally feasible manner. Additionally, it has been previously demonstrated that similar to Tet2 deficiency, Tet2 heterozygosity promotes clonal expansion and cardiac dysfunction, albeit to a lesser extent.10,23 Fifth, we acknowledge that we only employed one murine model of HFpEF. However, many other models of HFpEF don’t reliably reproduce many of the clinical phenotypes of HFpEF.41 Furthermore, models that resemble the clinical phenotype tend to utilize a combination of an obesogenic diet, hypertensive drug, and aging. Since the other robust models of HFpEF are largely redundant with our own, they weren’t examined in our study. Finally, male mice were used exclusively for this study since females are more resistant to HFD-induced metabolic disturbances and inflammation.42
In closing, TET2-mediated clonal hematopoiesis is enriched among patients with HFpEF, and HFpEF patients with CH display worse heart function and prognosis, particularly in individuals 70 years and older. Furthermore, experimental models of HFpEF and Tet2-mediated CH suggest this relationship to be causal and may serve as a platform to further elucidate the pathophysiology and possible treatments for HFpEF exacerbated by CH.
Supplementary Material
CLINICAL PERSPECTIVE.
1). What is new?
TET2-driven CH was enriched in a cohort of patients with HFpEF.
In patients with HFpEF, CH was associated with worse diastolic heart function and outcome.
In a murine model of HFpEF, Tet2-mediated CH led to higher echocardiographic E/e’, greater left ventricular end-diastolic pressure, and greater cardiac fibrosis.
2). What are the clinical implications?
TET2-driven CH may represent a novel pathophysiologic mechanism in HFpEF.
Our findings establish a rationale for measuring CH in patients with HFpEF to predict future outcomes.
Targeting TET2-mediated CH may be beneficial to prevent or treat HFpEF.
ACKNOWLEDGEMENTS
J. Cochran, Y. Yura, M. Thel, H. Doviak, A. Polizio, Yuka Arai, Yohei Arai, K. Horitani, E. Park, N. Chavkin, A. Kour, M. Evans, M. Huba, H. Sun, Y. Ban, N. Naya and S. Toldo conducted experiments and collected data. J. Dyck, J. Ezekowitz, F. Ruberg, and M. Maurer shared samples and corresponding patient data. T. Druley, N. Mahajan, M. Thel, A. Kour, and J. Cochran conducted and supervised ultra-deep error-correct sequencing J. Cochran analyzed data and generated figures. J. Cochran, Y. Yura, and K. Walsh designed the experiments. S. Sano and A. Abbate provided advice for the overall study. J. Cochran and K. Walsh wrote the manuscript. K. Walsh, J. Dyck, K. Hirschi, Y. Yura, and S. Sano acquired funding for this project. All affiliated authors revised and approved the manuscript.
Figure 4A was created with BioRender (BioRender.com).
SOURCES OF FUNDING
This work was supported by the National Institutes of Health (NIH) grants AG073249, AG072095, HL142650, and HL152174 and NASA grant 80NSSC21K0549 to K.W.; Grant-in-Aid for Young Scientists 22K16136, Kowa Life Science Foundation, MSD Life Science Foundation, Mitsubishi Foundation, Suzuken Memorial Foundation, THE HORI SCIENCE AND ARTS FOUNDATION, Mochida foundation, and JST FOREST Program (Grant Number JPMJFR2217, Japan) to Y.Y.; Grant-in-Aid for Research Activity Start-up 21K20879, Grant-in-Aid for Scientific Research C 22K08162, the Grant for Basic Research of the Japanese Circulation Society, the Japanese Heart Failure Society, the MSD Life Science Foundation, the Cardiovascular Research Fund, Kondou Kinen Medical Foundation to S.S.; In-kind contributions were received from Capital Health Regional Authority (now Alberta Health Services) and the Alberta HEART investigators. Funding for Alberta HEART was provided to JRBD by Alberta Innovates – Health Solutions (Grant # AHFMR ITG 200801018). SCAN-MP was supported by the National Institutes of Health (NIH) grants HL139671 to F.R. and M.M.. J.C. was supported by the University of Virginia Medical Scientist Training program (T32GM007267).
NON-STANDARD ABBREVIATIONS AND ACRONYMS
- CH
clonal hematopoiesis
- HSC
hematopoietic stem cells
- HfrEF
heart failure with reduced ejection fraction
- HfpEF
heart failure with preserved ejection fraction
- DNMT3A
DNA (cytosine-5)-methyltransferase 3A
- TET2
Tet Methylcytosine Dioxygenase 2
- TP53
Tumor protein P53
- ASXL1
ASXL transcriptional regulator 1
- SF3B1
Splicing factor 3b subunit 1
- STAG2
Stromal antigen 2
- RAD21
RAD21 cohesin complex component
- U2AF1
U2 small nuclear RNA auxiliary factor 1
- CBL
Cbl proto-oncogene
- EZH2
Enhancer of zeste 2 polycomb repressive complex 2 subunit
- MPL
MPL proto-oncogene, thrombopoietin receptor
- KRAS
KRAS proto-oncogene, GTPase
- IDH1
Calreticulin (CALR)Isocitrate dehydrogenase 1
- IDH2
Isocitrate dehydrogenase 2
- JAK2
Janus kinase 2
- NRAS
NRAS proto-oncogene, GTPase
- PTPN11
Protein tyrosine phosphatase non-receptor type 11
- RUNX1
RUNX family transcription factor 1
- ZRSR2
Zinc finger CCCH-type, RNA binding motif and serine/arginine rich 2
- Tet2+/+
wild-type mice
- Tet2−/−
Tet2-deficient mice
- HFD
high-fat diet
- L-NAME
N[w]-nitro-l-arginine methyl ester
- LT-HSC and ST-HSC
long-term and short-term hematopoietic stem cell pools
- CANTOS
Canakinumab Antiinflammatory Thrombosis Study
- ED
Emergency Department
- CV
Cardiovascular
- (MMP) cells
Multipotent progenitors
- (LSK) cells
Lin-Sca1+c-Kit+
- BMI
Body mass index
- COPD
Chronic obstructive pulmonary disease
- CKD
Chronic kidney disease
- ACEi
Angiotensin-converting enzyme inhibitors
- ARB
Angiotensin II receptor blocker
- CCB
calcium channel blocker
- LVEF
Left ventricular ejection fraction
- BSA
body surface area
- LAVI
left atrial volume index
- LVPWd
left ventricular posterior wall at end diastole
- LVMI
left ventricular mass index
- LVEDV
left ventricular end-diastolic volume
- LV EDP
Left ventricular end-diastolic pressure
- BNP
brain natriuretic peptide
- NT-proBNP
N-terminal pro b-type natriuretic peptide
Footnotes
DISCLOSURES
None.
REFERENCES:
- 1.Reddy YN, Borlaug BA. Heart Failure With Preserved Ejection Fraction. Curr Probl Cardiol. 2016;41:145–188. [DOI] [PubMed] [Google Scholar]
- 2.Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med. 2006;355:251–259. [DOI] [PubMed] [Google Scholar]
- 3.Pandey A, Omar W, Ayers C, LaMonte M, Klein L, Allen NB, Kuller LH, Greenland P, Eaton CB, Gottdiener JS, et al. Sex and Race Differences in Lifetime Risk of Heart Failure With Preserved Ejection Fraction and Heart Failure With Reduced Ejection Fraction. Circulation. 2018;137:1814–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoon S, Eom GH. Heart failure with preserved ejection fraction: present status and future directions. Exp Mol Med. 2019;51:1–9. doi: 10.1038/s12276-019-0323-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paulus WJ. Unfolding Discoveries in Heart Failure. N Engl J Med. 2020;382:679–682. [DOI] [PubMed] [Google Scholar]
- 6.Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med. 2017;377:111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evans MA, Walsh K. Clonal hematopoiesis, somatic mosaicism, and age-associated disease. Physiol Rev. 2023;103:649–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Min KD, Polizio AH, Kour A, Thel MC, Walsh K. Experimental ASXL1-Mediated Clonal Hematopoiesis Promotes Inflammation and Accelerates Heart Failure. J Am Heart Assoc. 2022;11:e026154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sano S, Oshima K, Wang Y, Katanasaka Y, Sano M, Walsh K. CRISPR-Mediated Gene Editing to Assess the Roles of Tet2 and Dnmt3a in Clonal Hematopoiesis and Cardiovascular Disease. Circ Res. 2018;123:335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sano S, Oshima K, Wang Y, MacLauchlan S, Katanasaka Y, Sano M, Zuriaga MA, Yoshiyama M, Goukassian D, Cooper MA, et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1beta/NLRP3 Inflammasome. J Am Coll Cardiol. 2018;71:875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sano S, Wang Y, Yura Y, Sano M, Oshima K, Yang Y, Katanasaka Y, Min KD, Matsuura S, Ravid K, et al. JAK2 (V617F) -Mediated Clonal Hematopoiesis Accelerates Pathological Remodeling in Murine Heart Failure. JACC Basic Transl Sci. 2019;4:684–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sano S, Wang Y, Ogawa H, Horitani K, Sano M, Polizio AH, Kour A, Yura Y, Doviak H, Walsh K. TP53-mediated therapy-related clonal hematopoiesis contributes to doxorubicin-induced cardiomyopathy by augmenting a neutrophil-mediated cytotoxic response. JCI Insight. 2021;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yura Y, Miura-Yura E, Katanasaka Y, Min KD, Chavkin N, Polizio AH, Ogawa H, Horitani K, Doviak H, Evans MA, et al. The Cancer Therapy-Related Clonal Hematopoiesis Driver Gene Ppm1d Promotes Inflammation and Non-Ischemic Heart Failure in Mice. Circ Res. 2021;129:684–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Assmus B, Cremer S, Kirschbaum K, Culmann D, Kiefer K, Dorsheimer L, Rasper T, Abou-El-Ardat K, Herrmann E, Berkowitsch A, et al. Clonal haematopoiesis in chronic ischaemic heart failure: prognostic role of clone size for DNMT3A- and TET2-driver gene mutations. Eur Heart J. 2021;42:257–265. [DOI] [PubMed] [Google Scholar]
- 15.Dorsheimer L, Assmus B, Rasper T, Ortmann CA, Ecke A, Abou-El-Ardat K, Schmid T, Brune B, Wagner S, Serve H, et al. Association of Mutations Contributing to Clonal Hematopoiesis With Prognosis in Chronic Ischemic Heart Failure. JAMA Cardiol. 2019;4:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pascual-Figal DA, Bayes-Genis A, Diez-Diez M, Hernandez-Vicente A, Vazquez-Andres D, de la Barrera J, Vazquez E, Quintas A, Zuriaga MA, Asensio-Lopez MC, et al. Clonal Hematopoiesis and Risk of Progression of Heart Failure With Reduced Left Ventricular Ejection Fraction. J Am Coll Cardiol. 2021;77:1747–1759. [DOI] [PubMed] [Google Scholar]
- 17.Yu B, Roberts MB, Raffield LM, Zekavat SM, Nguyen NQH, Biggs ML, Brown MR, Griffin G, Desai P, Correa A, et al. Supplemental Association of Clonal Hematopoiesis With Incident Heart Failure. J Am Coll Cardiol. 2021;78:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ezekowitz JA, Becher H, Belenkie I, Clark AM, Duff HJ, Friedrich MG, Haykowsky MJ, Howlett JG, Kassiri Z, Kaul P, et al. The Alberta Heart Failure Etiology and Analysis Research Team (HEART) study. BMC Cardiovasc Disord. 2014;14:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ezekowitz JA, McAlister FA, Howlett J, Alemayehu W, Paterson I, Belenkie I, Oudit GY, Kaul P, Dyck JR, Anderson T, et al. A prospective evaluation of the established criteria for heart failure with preserved ejection fraction using the Alberta HEART cohort. ESC Heart Fail. 2018;5:19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pfeffer MA, Shah AM, Borlaug BA. Heart Failure With Preserved Ejection Fraction In Perspective. Circ Res. 2019;124:1598–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruberg FL, Blaner WS, Chiuzan C, Connors LH, Einstein AJ, Fine D, Helmke S, Kurian D, Pandey S, Raiszadeh F, et al. Design and Rationale the SCAN-MP (Screening for Cardiac Amyloidosis With Nuclear Imaging in Minority Populations) Study. J Am Heart Assoc. 2023;12:e028534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rich MW, Beckham V, Wittenberg C, Leven CL, Freedland KE, Carney RM. A multidisciplinary intervention to prevent the readmission of elderly patients with congestive heart failure. N Engl J Med. 1995;333:1190–1195. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Sano S, Yura Y, Ke Z, Sano M, Oshima K, Ogawa H, Horitani K, Min KD, Miura-Yura E, et al. Tet2-mediated clonal hematopoiesis in nonconditioned mice accelerates age-associated cardiac dysfunction. JCI Insight. 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY, Luo X, Jiang N, May HI, Wang ZV, et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature. 2019;568:351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang S, Hu S, Luo X, Bao X, Li J, Liu M, Lv Y, Zhao C, Zeng M, Chen X, et al. Prevalence and prognostic significance of DNMT3A- and TET2- clonal haematopoiesis-driver mutations in patients presenting with ST-segment elevation myocardial infarction. EBioMedicine. 2022;78:103964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jaiswal S, Libby P. Clonal haematopoiesis: connecting ageing and inflammation in cardiovascular disease. Nat Rev Cardiol. 2020;17:137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi C, Aboumsallem JP, Suthahar N, de Graaf AO, Jansen JH, van Zeventer IA, Bracun V, de Wit S, Screever EM, van den Berg PF, et al. Clonal haematopoiesis of indeterminate potential: associations with heart failure incidence, clinical parameters and biomarkers. Eur J Heart Fail. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Young AL, Challen GA, Birmann BM, Druley TE. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun. 2016;7:12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Akhiyat N, Lasho T, Ganji M, Toya T, Shi CX, Chen X, Braggio E, Ahmad A, Corban MT, Stewart K, et al. Clonal Hematopoiesis of Indeterminate Potential Is Associated With Coronary Microvascular Dysfunction In Early Nonobstructive Coronary Artery Disease. Arterioscler Thromb Vasc Biol. 2023;43:774–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodriguez-Fraticelli AE, Wolock SL, Weinreb CS, Panero R, Patel SH, Jankovic M, Sun J, Calogero RA, Klein AM, Camargo FD. Clonal analysis of lineage fate in native haematopoiesis. Nature. 2018;553:212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sasi SP, Yan X, Zuriaga-Herrero M, Gee H, Lee J, Mehrzad R, Song J, Onufrak J, Morgan J, Enderling H, et al. Different Sequences of Fractionated Low-Dose Proton and Single Iron-Radiation-Induced Divergent Biological Responses in the Heart. Radiat Res. 2017;188:191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun J, Ramos A, Chapman B, Johnnidis JB, Le L, Ho YJ, Klein A, Hofmann O, Camargo FD. Clonal dynamics of native haematopoiesis. Nature. 2014;514:322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rohde D, Vandoorne K, Lee IH, Grune J, Zhang S, McAlpine CS, Schloss MJ, Nayar R, Courties G, Frodermann V, et al. Bone marrow endothelial dysfunction promotes myeloid cell expansion in cardiovascular disease. Nat Cardiovasc Res. 2022;1:28–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355:842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fuster JJ, Zuriaga MA, Zorita V, MacLauchlan S, Polackal MN, Viana-Huete V, Ferrer-Perez A, Matesanz N, Herrero-Cervera A, Sano S, et al. TET2-Loss-of-Function-Driven Clonal Hematopoiesis Exacerbates Experimental Insulin Resistance in Aging and Obesity. Cell Rep. 2020;33:108326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Samson R, Jaiswal A, Ennezat PV, Cassidy M, Le Jemtel TH. Clinical Phenotypes in Heart Failure With Preserved Ejection Fraction. J Am Heart Assoc. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shah SJ, Katz DH, Deo RC. Phenotypic spectrum of heart failure with preserved ejection fraction. Heart Fail Clin. 2014;10:407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lewis EF, Claggett B, Shah AM, Liu J, Shah SJ, Anand I, O’Meara E, Sweitzer NK, Rouleau JL, Fang JC, et al. Racial Differences in Characteristics and Outcomes of Patients With Heart Failure and Preserved Ejection Fraction in the Treatment of Preserved Cardiac Function Heart Failure Trial. Circ Heart Fail. 2018;11:e004457. [DOI] [PubMed] [Google Scholar]
- 40.Fuery MA, Chouairi F, Januzzi JL, Moe GW, Caraballo C, McCullough M, Miller PE, Reinhardt SW, Clark K, Oseran A, et al. Intercountry Differences in Guideline-Directed Medical Therapy and Outcomes Among Patients With Heart Failure. JACC Heart Fail. 2021;9:497–505. [DOI] [PubMed] [Google Scholar]
- 41.Withaar C, Lam CSP, Schiattarella GG, de Boer RA, Meems LMG. Heart failure with preserved ejection fraction in humans and mice: embracing clinical complexity in mouse models. Eur Heart J. 2021;42:4420–4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pettersson US, Walden TB, Carlsson PO, Jansson L, Phillipson M. Female mice are protected against high-fat diet induced metabolic syndrome and increase the regulatory T cell population in adipose tissue. PLoS One. 2012;7:e46057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brojakowska A, Kour A, Thel MC, Park E, Bisserier M, Garikipati VNS, Hadri L, Mills PJ, Walsh K, Goukassian DA. Retrospective analysis of somatic mutations and clonal hematopoiesis in astronauts. Commun Biol. 2022;5:828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wong WH, Bhatt S, Trinkaus K, Pusic I, Elliott K, Mahajan N, Wan F, Switzer GE, Confer DL, DiPersio J, et al. Engraftment of rare, pathogenic donor hematopoietic mutations in unrelated hematopoietic stem cell transplantation. Sci Transl Med. 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wong WH, Tong RS, Young AL, Druley TE. Rare Event Detection Using Error-corrected DNA and RNA Sequencing. J Vis Exp. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pich O, Reyes-Salazar I, Gonzalez-Perez A, Lopez-Bigas N. Discovering the drivers of clonal hematopoiesis. Nat Commun. 2022;13:4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.