

Abstract
Dietary fibres can exert beneficial anti-inflammatory effects through microbially fermented short-chain fatty acid metabolites1,2, although the immunoregulatory roles of most fibre diets and their microbiota-derived metabolites remain poorly defined. Here, using microbial sequencing and untargeted metabolomics, we show that a diet of inulin fibre alters the composition of the mouse microbiota and the levels of microbiota-derived metabolites, notably bile acids. This metabolomic shift is associated with type 2 inflammation in the intestine and lungs, characterized by IL-33 production, activation of group 2 innate lymphoid cells and eosinophilia. Delivery of cholic acid mimics inulin-induced type 2 inflammation, whereas deletion of the bile acid receptor farnesoid X receptor diminishes the effects of inulin. The effects of inulin are microbiota dependent and were reproduced in mice colonized with human-derived microbiota. Furthermore, genetic deletion of a bile-acid-metabolizing enzyme in one bacterial species abolishes the ability of inulin to trigger type 2 inflammation. Finally, we demonstrate that inulin enhances allergen- and helminth-induced type 2 inflammation. Taken together, these data reveal that dietary inulin fibre triggers microbiota-derived cholic acid and type 2 inflammation at barrier surfaces with implications for understanding the pathophysiology of allergic inflammation, tissue protection and host defence.
The beneficial microbes that constitute the intestinal microbiota influence multiple physiological processes, including development3, metabolism4, immune responses1,5,6 and cognitive function7,8. Metabolites produced by the microbiota—including aromatic amino acid derivatives, branched-chain amino acids, short-chain fatty acids (SCFAs) and bile acid derivatives—cross the intestinal epithelium, enter the circulation and regulate multiple aspects of systemic host physiology6,9,10. Among these metabolites, SCFAs derived from the microbial degradation of fermentable dietary fibre are best studied and have been shown to influence multiple cell types, including epithelial cells, dendritic cells and regulatory T (Treg) cells to promote immunoregulatory effects in multiple tissues2,11–14. However, not all fibre diets are the same, their effects on the microbiota and host physiology are not generalizable, and the systemwide influence of fibre diets on microbial metabolites and the mammalian immune system remain incompletely defined. Here we used an unbiased metabolomic approach to experimentally define the effects of a fermentable inulin fibre diet15 on the composition of the microbiota, on the profile of the microbiota-derived metabolome and on the subsequent immune responses.
Inulin alters the systemic metabolome
To test whether dietary-fibre-induced changes in the microbiota are associated with changes in microbial metabolites, conventionally raised specific-pathogen free (SPF) wild-type (WT) C57BL/6 mice were exposed to a control or inulin-based high-fibre diet for 2 weeks (Fig. 1a (left) and Extended Data Table 1). After exposure to inulin, sequencing of 16S ribosomal RNA (rRNA) genes from the faeces revealed an increase in Bacteroidetes and decrease in Firmicutes compared with the controls (Extended Data Fig. 1). This inulin-fibre-induced shift in the abundance of Bacteroidetes is consistent with a range of clinical studies in humans16,17. Moreover, several of the differentially abundant members of the microbiota, including the major phyla members, could be mapped to operational taxonomic units (OTUs) in the Human Microbiome Project (HMP) database (Fig. 1a (right)), specifically Erysipelotrichaceae bacterium 6_1_45 (a major Firmicute) and Bacteroides sp. 3_1_19 (a major Bacteroidetes).
Fig. 1 |. Inulin fibre diet upregulates systemic bile acids and tissue eosinophils.
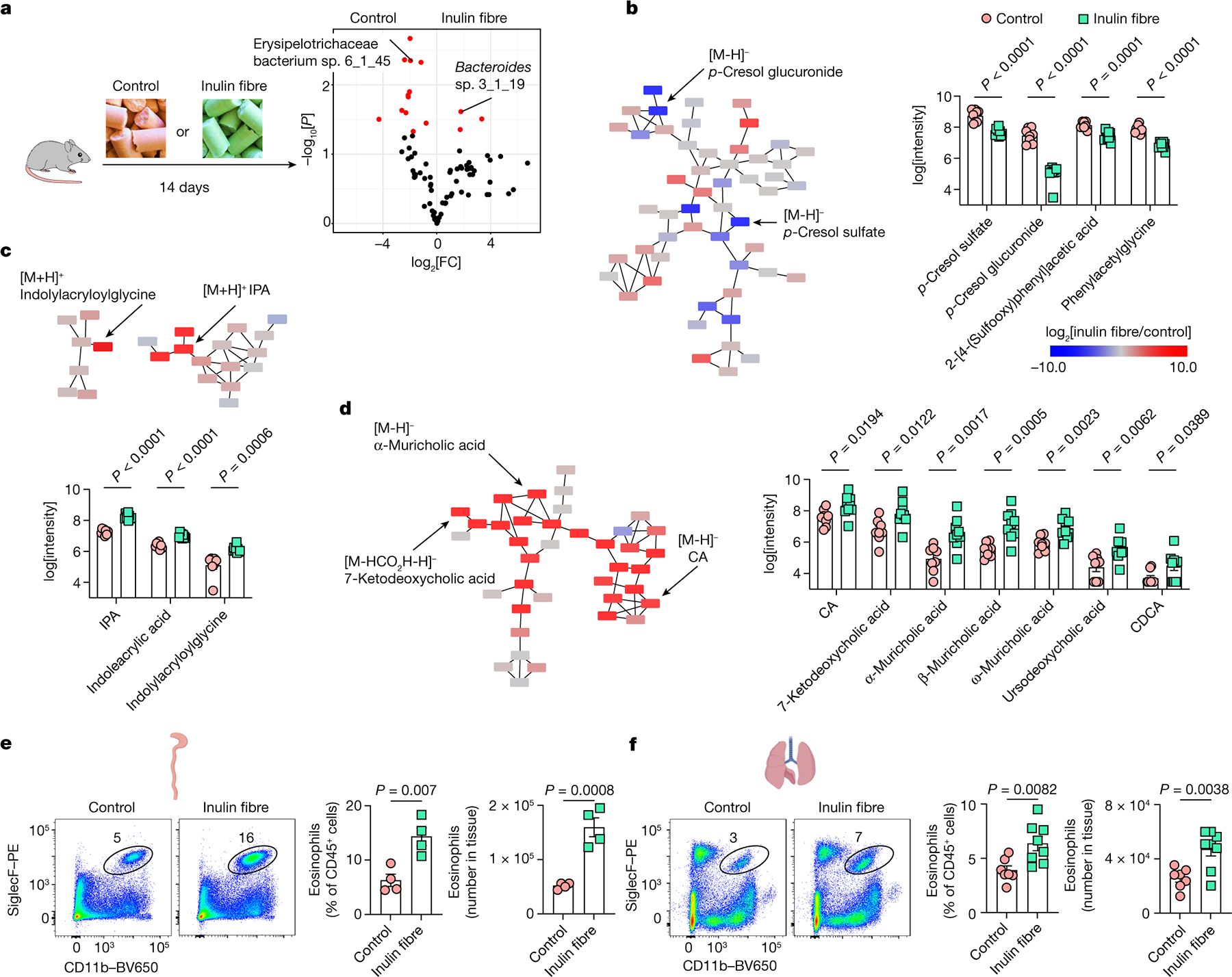
a, Schematic of the experimental design (left). Right, the differential abundance of intestinal microbiota in control versus inulin fibre diet groups mapped to the HMP database. n = 5 mice. The red dots show differentially abundant taxa (P < 0.05, determined using DESeq2 Wald test). FC, fold change. b–d, Partial representation of the MS2 network for mouse serum, highlighting three clusters for phenolics (b), indoles (c) and bile acids (d). n = 8 (control) and n = 9 (inulin fibre) mice. Each node represents a unique feature, and the connections between nodes indicate the similarity between MS2 spectra. Blue represents downregulated and red represents upregulated features in the inulin fibre group compared with the control group. The arrows indicate some of the differentially abundant compounds. The bar graphs show the relative abundance of metabolites that were significantly different between control and inulin fibre groups. Peak integration data from HPLC–MS analysis were log-transformed48 before statistical analysis. e,f, Representative flow cytometry plots, the frequency of CD11b+SiglecF+ eosinophils in the CD45+ population and the total number in the colonic lamina propria (n = 4 mice) (e) and the lungs (n = 7 (control) and n = 8 (inulin fibre) mice) (f). Data are representative of three independent experiments (a) or pooled from two independent experiments (b–d and f). For b–f, data are mean ± s.e.m. Statistical analysis was performed using unpaired two-tailed t-tests (b–f). The diagrams in a, e and f were created using BioRender.
Untargeted comparative metabolomic analyses of serum samples isolated from control or inulin-administered WT mice were performed using high-resolution liquid chromatography coupled with mass spectrometry (LC–MS). Analysis of LC–MS and tandem MS (MS/MS) data revealed several hundred features of which the abundances were increased or decreased by inulin (Extended Data Fig. 2a and Supplementary Figs. 1 and 2), demonstrating that the effect of dietary fibre on the host metabolome is much broader than the known changes in SCFAs17,18 (Extended Data Fig. 2b). Molecular networking analyses of the MS/MS data identified several major families of structurally related metabolites with differential abundance. Among these families, phenolics were significantly downregulated, and indoles were significantly upregulated by inulin (Fig. 1b,c and Extended Data Figs. 2a and 3a–c). However, the largest and most abundant family of differentially abundant metabolites was represented by bile acids (Supplementary Figs. 1 and 2). Serum levels of all major primary unconjugated bile acids and one secondary unconjugated bile acid were significantly upregulated by inulin, including the most abundant primary unconjugated bile acid in mouse serum—cholic acid (CA)19,20 (Fig. 1d and Extended Data Figs. 2a,c and 3a,b,d). We also performed metabolomic analyses of faecal and caecal contents and observed that the faecal levels of some bile acids, but not the highly abundant CA and β-muricholic acid, were significantly reduced in the inulin-fibre-diet group compared with the control-diet group (Extended Data Fig. 3e–g), suggesting that their reabsorption was promoted by the presence of high levels of inulin in the intestine. Together, comparative metabolomic analyses indicate that crosstalk between the microbiota and host after this dietary perturbation could be driven by a diverse network of metabolites.
Defined fibre diets trigger eosinophilia
Previous studies reported that dietary-fibre-derived SCFAs can limit inflammation in multiple tissues2,12–14. Given the impact of inulin fibre diet on the composition of the intestinal microbiota and the production of metabolites (Extended Data Fig. 1 and Supplementary Figs. 1 and 2), we hypothesized that diet-induced changes in other microbial metabolites could have important immunoregulatory effects. To directly test this, we profiled antigen-presenting dendritic cells and macrophages, T cell subsets, neutrophils and eosinophils within the intestinal lamina propria (Extended Data Fig. 4a–c). After exposure to inulin, a modest increase in Treg cells was observed (Extended Data Fig. 5a), as reported in previous studies13, but no significant changes in most other immune cell types were observed (Extended Data Fig. 5b). However, mice that were exposed to inulin exhibited significantly higher levels of eosinophils in the colon compared with mice fed a control diet (Fig. 1e). Heightened eosinophilia was observed within one week and remained elevated in mice beyond 2 months of inulin exposure (Extended Data Fig. 5c). A significant accumulation of eosinophils was also observed in the lungs (Fig. 1f and Extended Data Fig. 5d), indicating that a diet of inulin fibre has a systemic effect on type 2 inflammation.
In addition to in-house bred mice (Fig. 1d–f), we tested the effects of inulin on mice purchased from a commercial vendor ( Jackson Laboratory) that were kept isolated in the animal facility throughout the experiment. We observed similar metabolic and immunological effects (Extended Data Fig. 5e,f), indicating that these effects are not restricted to the source of the mice. We also tested fibre-rich diets based on psyllium (a fermentable fibre structurally distinct from inulin) or cellulose (a non-fermentable fibre). Notably, whereas the cellulose fibre diet had no significant effects compared to the control diet, the psyllium fibre diet induced elevated levels of bile acids (Extended Data Fig. 5e) and eosinophilia in the colon and lungs, comparable to those induced by the inulin fibre diet (Extended Data Fig. 5f). These results indicate that diverse types of fermentable dietary fibre can produce similar metabolomic and immunological changes.
Inulin-induced eosinophilia requires ILCs and IL-33
Next, we tested the role of innate and adaptive lymphoid cells—two major regulators of type 2 inflammation21,22. After administration of inulin, eosinophilia was observed in the colons of Rag2−/− mice (deficient in adaptive B cells and T cells) but not in Rag2−/−Il2rg−/− mice (deficient in B cells, T cells and ILCs) (Fig. 2a), indicating that ILCs are essential in promoting inulin-induced eosinophilia.
Fig. 2 |. Inulin fibre diet-induced eosinophilia requires ILC2s and IL-33.
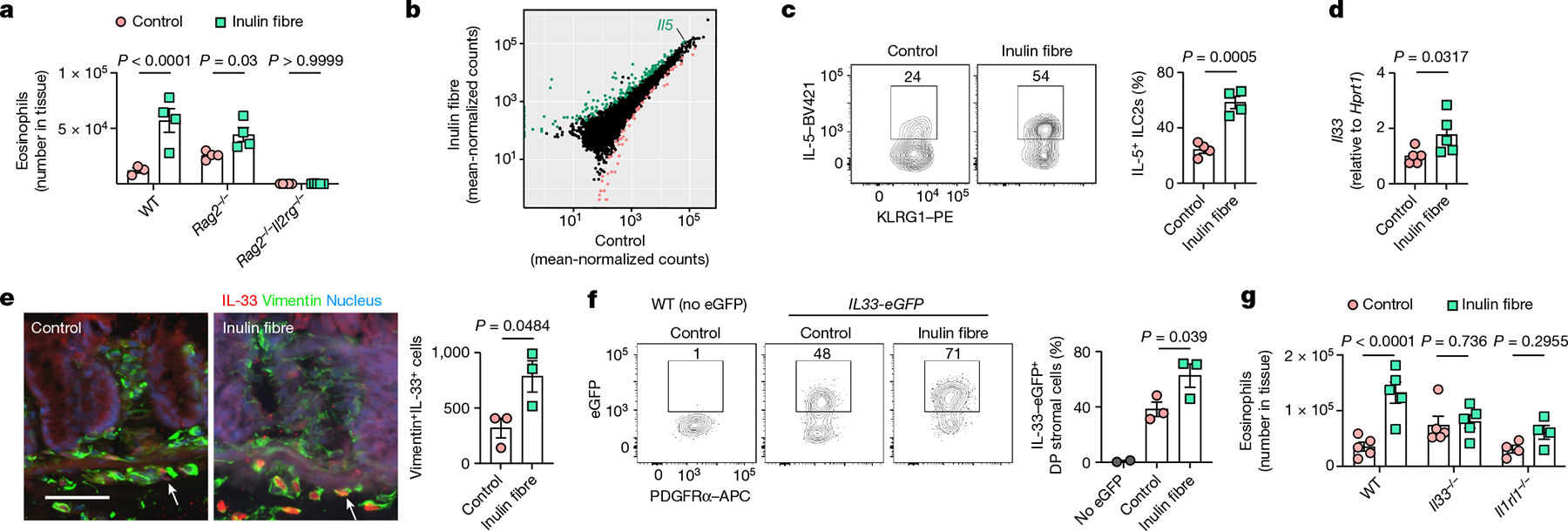
a, The number of eosinophils in the colons of WT, Rag2−/− and Rag2−/−Il2rg−/− mice. n = 3 (WT control), n = 4 (WT inulin fibre or Rag2−/−) and n = 5 (Rag2−/−Il2rg−/−) mice. b, The differential gene expression of colonic ILC2s in mice fed the control or inulin fibre diet. Genes upregulated by inulin fibre diet are shown in green and genes downregulated by inulin fibre diet are shown in light red (DESeq2 Wald test, false-discovery rate (FDR) < 0.1). c, The frequency of IL-5-expressing KLRG1+ ILC2s in the colonic lamina propria, determined by intracellular cytokine staining. n = 4 mice. Flow cytometry plots were gated from CD45+Lin−CD90.2+CD127+ cells. d, Expression of Il33 in the distal colon determined by quantitative PCR with reverse transcription (RT–qPCR) analysis. n = 5 mice. e, Representative immunofluorescence staining showing IL-33 expression in vimentin+ stromal cells in the colons (n = 3 mice). The arrows indicate IL-33+ cells. Scale bar, 50 μm. The bar graph shows the quantification of IL-33+vimentin+ cells per cross-section of whole colon ‘Swiss rolls’. f, Flow cytometry plots and bar graphs showing IL-33-eGFP expression in PDGFRα+ Sca-1+ double-positive (DP) mesenchymal stromal cells in the colon. n = 2 (no eGFP) and n = 3 (control or inulin fibre) mice. g, The number of eosinophils in the colons of WT, Il33−/− and Il1rl1−/− mice fed the control or inulin fibre diet. n = 4 (Il1rl1−/−) and n = 5 mice (WT or Il33−/−) mice. Data are representative of two independent experiments (a and c–g). For a and c–g, data are mean ± s.e.m. Statistical analysis was performed using two-way (a and g) or one-way (f) analysis of variance (ANOVA) with Holm–Šidák’s multiple-comparisons test or unpaired two-tailed t-tests (c and e) or Mann–Whitney U-tests (d).
Group 2 innate lymphoid cells (ILC2s) produce type 2 cytokines and other bioactive molecules that can promote eosinophilia and regulate inflammation, metabolic homeostasis and tissue protection21,23–25. To test the effects of inulin on the activation and effector functions of ILC2s, control or inulin fibre diet was administered to WT mice, and RNA-sequencing (RNA-seq) analysis of FACS-sorted ILC2s from mouse colons was performed. Gene Ontology (GO) enrichment analyses revealed substantial reprogramming of the transcriptome of ILC2s after exposure to inulin, with enrichment of pathways that are associated with regulation of inflammation and cytokine production (Extended Data Fig. 6a). Notably, a significant upregulation of Il5—a key cytokine in the maintenance of tissue-resident eosinophils26—was observed (Fig. 2b). Although CD4+ T helper type 2 (TH2) cells and ILC2s are two major sources of IL-5, inulin induced an increased frequency of IL-5-expressing ILC2s but not CD4+ T cells (Fig. 2c and Extended Data Fig. 6b,c), indicating that inulin preferentially activates ILC2-derived IL-5 production and eosinophilia.
IL-33 is an important activator of ILC2s and has an important role in promoting pathological or protective type 2 inflammation after exposure to allergens or helminth infection23,27,28. After exposure to inulin, expression of Il33 was significantly upregulated in the colon (Fig. 2d). Immunofluorescence microscopy of colon tissue confirmed fibre-mediated upregulation of IL-33 in stromal cells—a major source of IL-33 (refs. 29–31) (Fig. 2e). Using Il33-eGFP reporter mice, we identified that inulin upregulated IL-33 primarily in PDGFRα+Sca-1+ double-positive mesenchymal stromal cells in the colon (Fig. 2f and Extended Data Fig. 6d,e). By contrast, inulin induced IL-33 in epithelial cells but not in stromal cells in the lungs (Extended Data Fig. 6f). Next, to test whether the IL-33–IL-33R pathway is required for inulin-induced eosinophilia, control versus inulin fibre diet was administered to WT mice, IL-33-deficient (Il33−/−) mice or IL-33-receptor-deficient (T1/ST2) (Il1rl1−/−) mice. Whereas inulin promoted significant eosinophilia in WT mice, there was a significant defect in the eosinophil response in mice deficient in IL-33 or IL-33 receptor (Fig. 2g), indicating that the IL-33–IL-33R pathway promotes inulin-induced type 2 inflammation.
CA promotes eosinophilia
To test whether the microbiota is required for the effects of inulin on colonic eosinophilia, SPF and germ-free (GF) WT mice were exposed to control or inulin fibre diets. In contrast to SPF mice, inulin did not promote eosinophilia in GF mice (Fig. 3a), indicating that inulin-induced eosinophilia requires the presence of the microbiota. Next, to test the role of microbiota-derived metabolites in the regulation of colonic eosinophilia, we analysed indolepropionic acid (IPA) (the most abundant indole), acetate, propionate and butyrate (three major SCFAs), and CA (the most abundant bile acid in the serum of mice19) (Fig. 1c,d). β-Muricholic acid was not selected for testing in this experiment given that this bile acid is not present in humans32. After oral administration of SCFAs, an increase in the frequency of RORγt+ Treg cells was observed (Extended Data Fig. 7a), as previously reported12–14, but there were no changes in eosinophilia (Fig. 3b). By contrast, a combination of CA and IPA triggered eosinophilia to a level similar to inulin (Fig. 3b). When applied individually, only CA triggered eosinophilia (Fig. 3b and Extended Data Fig. 7b,c), indicating that inulin promotes eosinophil responses at least in part through CA. Chenodeoxycholic acid (CDCA)—another inulin-fibre-induced bile acid that is the most abundant primary bile acid in humans and has a stronger receptor affinity19,32—also triggered eosinophilia (Fig. 3c and Extended Data Fig. 7d).
Fig. 3 |. Microbiota-derived CA and host FXR mediate inulin fibre diet-triggered type 2 inflammation.
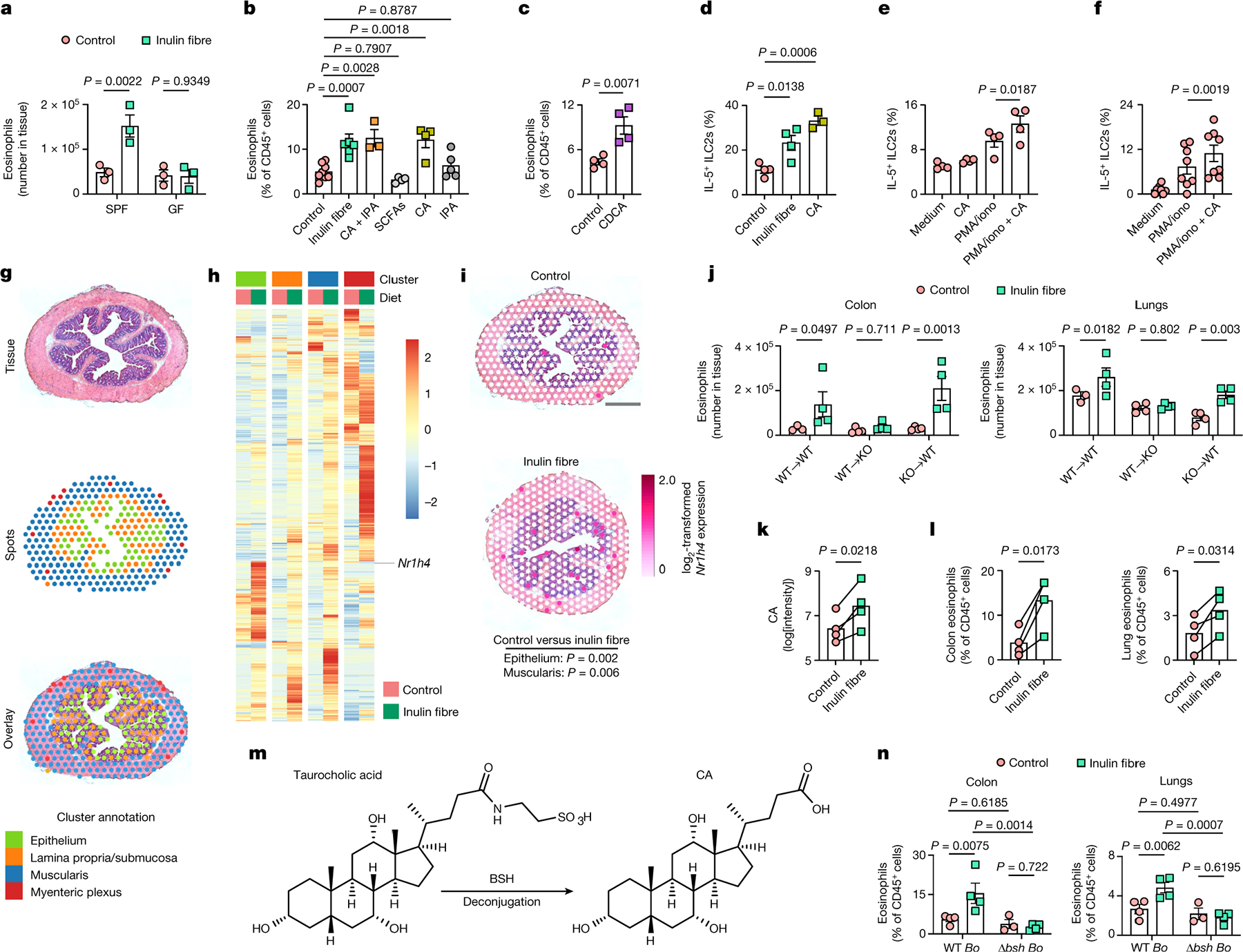
a, The number of eosinophils in the colon. n = 3 mice. b, The frequency of colon eosinophils after administration of the diets (n = 8 (control) and n = 6 (inulin fibre) mice) or metabolites (n = 3 (CA + IPA), n = 4 (SCFAs or CA) and n = 5 (IPA) mice). c, The frequency of colon eosinophils. n = 4 mice. d, The frequency of IL-5-expressing ILC2s in the lungs. n = 4 (control or inulin fibre) and n = 3 (CA) mice. e, The frequency of IL-5-expressing ILC2s in naive mouse lung cell preparations after incubation in vitro under the indicated conditions. n = 4 mice. f, The frequency of IL-5-expressing ILC2s in PBMCs from human donors (n = 8 donors) co-cultured with the human colonic stromal cell line Ccd-18Co. g–i, Spatial transcriptomic analyses of distal colon tissues. g, A representative H&E-stained colon section, which was overlaid by ‘spots’ containing transcriptional information. h, Heat map showing the genes in each layer of the colon sections with uncorrected P < 0.01 (Wilcoxon rank-sum test). i, Spatial representation of the relative expression of FXR-encoding Nr1h4 in representative colon tissues. Scale bar, 500 μm. j, The frequency of colon and lung eosinophils in the bone marrow chimeric mice with indicated donor to recipient conditions. WT→WT: n = 3 (control mice) and n = 4 (inulin fibre) mice; irradiated Nr1h4−/− mice with WT bone marrow (WT→Nr1h4−/− (KO)): n = 4 mice; and irradiated WT mice with Nr1h4−/− bone marrow (KO→WT): n = 4 mice. k,l, Serum CA (k) and tissue eosinophil (l) levels in gnotobiotic mice with human microbiota. Each pair of dots represents the mean levels of all of the mice receiving microbiota from a single human donor. n = 4 donors. m, Schematic of the bile acid deconjugation pathway. n, The frequency of colon and lung eosinophils in GF mice monocolonized with WT or bsh-deleted B. ovatus (WT Bo or Δbsh Bo, respectively). n = 3 (Δbsh Bo control diet group) and n = 4 (other groups). Data are representative of (a, c–e, j and n) or pooled from (b and f) two independent experiments. Data are mean (k and l) or mean ± s.e.m. (a–f, j and n). Statistical analysis was performed using two-way ANOVA with uncorrected Fisher’s LSD test (a, j and n); one-way ANOVA with Dunnett’s multiple-comparisons test (b and d); unpaired (c) or paired (e, f, k and l) two-tailed t-tests; or pairwise Wilcoxon test with Bonferroni correction (i).
CA also upregulated the frequency of IL-5-expressing ILC2s (Fig. 3d). To test the influence of bile acids on IL-5 expression ex vivo, single-cell preparations from colon or lung tissues were stimulated with or without CA. Exposure to CA significantly increased the frequency of ILC2s expressing IL-5 (Fig. 3e and Extended Data Fig. 7e). Furthermore, after CA stimulation of peripheral blood mononuclear cells (PBMCs) isolated from healthy humans, there was a significant increase in the frequency of IL-5+ ILC2s (Fig. 3f and Extended Data Fig. 7f). Together, these data demonstrate that the ability of CA to mimic inulin-fibre-diet-induced type 2 immune responses is conserved in mice and humans.
Inulin-induced type 2 inflammation requires FXR
Primary and secondary bile acids can activate immune cells, including macrophages, dendritic cells, natural killer T cells, TH17 cells and Treg cells, through multiple cell-surface or nuclear bile acid receptors1,33–35. However, the alarmin cytokine IL-33, which is required for inulin-fibre-diet-mediated activation of type 2 inflammatory responses (Fig. 2g), is primarily secreted from non-immune cell types, such as epithelial and stromal cells29,30, suggesting that CA may also directly act on these cell types to upregulate the production of IL-33 and promote ILC2-dependent type 2 inflammation. Inulin-induced changes in gene expression were determined using spatial transcriptomic analyses, enabling the detection of differentially expressed genes simultaneously in diverse cell types, together with information about their anatomical location in the tissue36. Unbiased clustering divided the colon into four distinct zones, which corresponded to the epithelium, lamina propria and submucosa, muscularis and myenteric plexus, as visualized by haematoxylin and eosin (H&E) staining (Fig. 3g). Exposure to an inulin fibre diet reprogrammed the transcriptome in each of these regions (Fig. 3h). The genes that were significantly upregulated by inulin at distinct tissue layers include those with known relevant functions, such as carbohydrate binding and antioxidant activity (Extended Data Fig. 7g). Notably, after exposure to an inulin fibre diet, Nr1h4—which encodes a nuclear receptor for bile acids (farnesoid X receptor (FXR))20,32—was upregulated in epithelial and muscularis regions of the colon (Fig. 3h,i). Deletion of FXR abrogated inulin-induced Il33 expression and eosinophilia in the colon and lungs (Extended Data Fig. 7h,i), indicating that inulin promotes eosinophilia through this specific bile acid nuclear receptor. Consistent with this, CA-induced Il33 expression in stromal cells was abrogated in the presence of DY268, a potent FXR antagonist (Extended Data Fig. 7j). As FXR is expressed by diverse cell types in both the haematopoietic and non-haematopoietic compartments32, bone marrow chimeras were used to test the cell-type-specific requirement of FXR in vivo. The inulin triggered IL-5 production and eosinophilia in WT mice reconstituted with either WT or Nr1h4−/− bone marrow, but the enhanced IL-5 and eosinophilia were absent in Nr1h4−/− mice reconstituted with WT bone marrow (Fig. 3j and Extended Data Fig. 7k), indicating that expression of the FXR receptor by non-haematopoietic cell types including epithelial and stromal cells is required for inulin-induced type 2 inflammation.
Human microbiota-dependent effects of inulin
To determine the relevance of the inulin-fibre-diet-mediated metabolic and immunoregulatory effects in the context of the human microbiota, we performed human faecal microbiota transfer (FMT) from healthy individuals into GF mice (Supplementary Table 1). Groups of GF mice received human FMT and were then divided into control-diet-versus inulin-fibre-diet-fed cohorts (Extended Data Fig. 8a). After administration of inulin, GF mice that received human FMT exhibited increases in the abundance of Bacteroidetes (Extended Data Fig. 8b). Inulin also upregulated serum bile acids including CA in all of the groups of mice that were colonized by human microbiota (Fig. 3k and Extended Data Fig. 8c,e,f), and triggered type 2 inflammation, including upregulation of Il33, IL-5 production and eosinophilia (Fig. 3l and Extended Data Fig. 8d,g,h). Collectively, these data indicate that the microbiota-dependent immunoregulatory effects of inulin are conserved in mice colonized with mouse or human microbiotas.
A bacterial enzyme regulates the effects of inulin
Primary bile acids produced in the liver are conjugated to taurine or glycine before they are released into the gastrointestinal tract. After reaching the large intestine, conjugated bile acids are hydrolysed into unconjugated bile acids by intestinal bacterial bile salt hydrolases (BSH) (Fig. 3m) and then partly reabsorbed into the systemic circulation20,32,37. After exposure to inulin fibre diet, faecal levels of several bile acids were reduced (Extended Data Fig. 3e), indicating that increased reabsorption could contribute to their elevated systemic levels38. However, faecal levels of CA, the most abundant bile acid in control mice, were not significantly reduced (Extended Data Fig. 3e). We therefore sought to determine whether bacterial hydrolysis of conjugated bile acids contributes to the effects of inulin on systemic bile acid concentrations. GF mice were monocolonized with WT Bacteroides ovatus, a prominent Bacteroidetes member of the human and murine intestinal microbiota39, or a mutant strain of B. ovatus in which we deleted the BSH-encoding gene BO_02350 (Δbsh B. ovatus)40 (Supplementary Table 2), rendering this mutant unable to deconjugate bile acids (Extended Data Fig. 8i,j). Monocolonization of GF mice with WT B. ovatus was sufficient to restore the ability of inulin to upregulate levels of serum CA (Extended Data Fig. 8k–m) and trigger type 2 inflammation (Fig. 3n and Extended Data Fig. 8n,o). In these monocolonized mice, inulin did not alter bacterial colonization but significantly upregulated the expression of bsh (Extended Data Fig. 8p,q). By contrast, whereas BSH deficiency did not affect bacterial colonization (Extended Data Fig. 8p), GF mice colonized with Δbsh B. ovatus exhibited a significantly lower levels of CA in the serum compared with those colonized with WT B. ovatus (Extended Data Fig. 8k), and a significant defect in inulin-induced type 2 inflammation (Fig. 3n and Extended Data Fig. 8n,o). Together, these data demonstrate that expression of BSH in a single bacterial species and subsequent bile acid metabolism are sufficient for inulin-induced type 2 inflammation.
Inulin promotes type 2 barrier inflammation
Eosinophilia is a hallmark of type 2 inflammation after exposure to allergens or helminth infection. Furthermore, increased levels of bile acids, including CA, have been associated with chronic obstructive pulmonary diseases in patients41,42. To determine the effects of inulin-induced eosinophilia in the lungs, mice were exposed to papain—a proteolytic enzyme commonly used in the food industry that is known to cause type 2 inflammation and lung damage43 (Fig. 4a). After sensitization with papain, mice receiving inulin exhibited more severe inflammation in the lungs, characterized by increased inflammatory cell infiltration in the perivascular and peribronchial regions (Fig. 4b). Eosinophil responses were significantly elevated in both the lungs and the bronchoalveolar lavage fluid (BALF) in inulin-exposed mice (Fig. 4c,d). WT mice exposed to control or inulin fibre diet were also challenged with house dust mite (HDM), which triggers both protease-induced type 2 inflammation as well as endotoxin-induced type 3 inflammation44 (Extended Data Fig. 9a). After exposure to low-endotoxin HDM, mice exposed to inulin exhibited significantly elevated eosinophil levels in the lungs and BALF (Extended Data Fig. 9b,c) and increased airway resistance (Extended Data Fig. 9d), supporting a role for inulin in exacerbating protease allergen-induced type 2 inflammation.
Fig. 4 |. Inulin-fibre-diet-induced eosinophils promote allergic inflammation and anti-helminth defence.
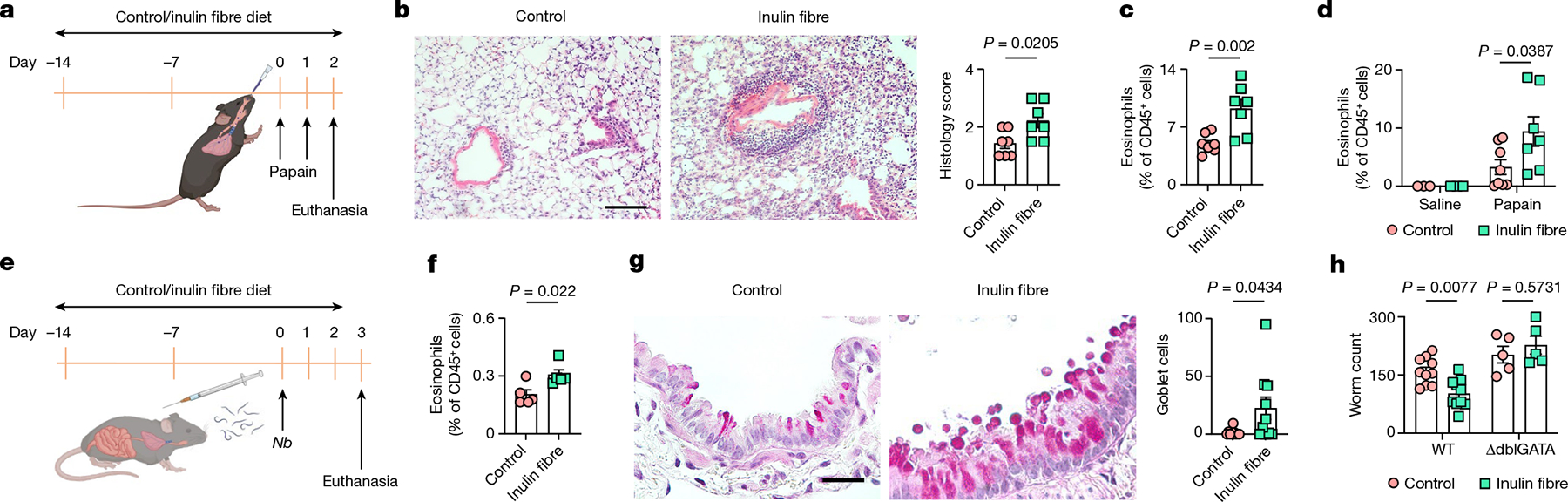
a–d, Papain-induced lung inflammation. a, Schematic of sensitization with papain. b, Representative H&E-stained lung sections and perivascular/peribronchial inflammation as scored by histology. n = 7 mice. Scale bar, 100 μm. c,d, The frequency of eosinophils in the lungs (c) or BALF (d) 24 h after the second dose of papain. n = 8 (papain with control diet), n = 7 (papain with inulin fibre diet) and n = 3 (saline) mice. e–h, N. brasiliensis (Nb) model of helminth infection. e, Schematic of the infection model. f, The frequency of eosinophils in mesenteric lymph nodes. n = 5 mice. g, Representative histology of the lung airways stained with periodic acid Schiff (PAS). Scale bar, 50 μm. Each data point in the graph represents the average number of goblet cells per bronchiole in a section of the left lung from one animal. n = 10 mice. h, Worm count in the small intestine of WT (n = 10 mice) or ΔdblGATA (n = 5 mice) mice. Data are representative of (f) or pooled from (b–d, g and h) two independent experiments. Data are mean ± s.e.m. Statistical analysis was performed using unpaired two-tailed t-tests (b, c, f and g) or two-way ANOVA with Šidák’s multiple-comparisons test (d and h). The schematics in a and e were created using BioRender.
To determine the effects of the inulin on protective type 2 immune responses after helminth infection, mice were infected with Nippostrongylus brasiliensis, a nematode parasite that migrates from the skin through the lung to the small intestine45 (Fig. 4e). Inulin-exposed WT mice exhibited elevated type 2 inflammation marked by heightened eosinophilia, exaggerated goblet cell responses and accelerated expulsion of parasites (Fig. 4f–h). By contrast, eosinophil-deficient (ΔdblGATA) mice exposed to inulin did not exhibit accelerated expulsion of parasites compared with the control mice (Fig. 4h), demonstrating that an inulin fibre diet promotes protective type 2 immunity after helminth infection in an eosinophil-dependent manner.
Discussion
Here we demonstrate that exposure to an inulin fibre diet regulates the composition of the microbiota and alters diverse classes of microbiota-derived metabolites in the peripheral circulation that results in exaggerated pathological and protective type 2 inflammation at barrier surfaces. Mechanistically, inulin alters the microbial metabolism leading to increases in the systemic levels of bile acids, specifically CA. Bile acids trigger increases in stromal-cell-derived IL-33, subsequently activating ILC2s to produce IL-5, resulting in tissue eosinophilia and type 2 inflammation (Extended Data Fig. 9e). Inulin triggered type 2 inflammation in the context of both the mouse and human microbiota, whereas CA mimicked the effects of inulin on mouse and human immune cells, indicating that the effects of inulin on the microbiota and type 2 inflammation are conserved across species. Several bile acids have recently been linked to the generation of intestinal Treg cells and TH17 cells1,33–35. Furthermore, dysregulation of bile acid metabolism by multiple types of dietary fibre has been reported to contribute to hepatic diseases such as non-alcoholic fatty liver diseases and cholestatic cancer, which have been linked to neutrophilic inflammation38,46. Here we report on the ability of a specific dietary fibre or bile acid to promote type 2 inflammation at multiple barrier tissues. Spatial transcriptomic analyses accompanied by genetic deletion studies identified FXR as a bile acid receptor that mediates signals from inulin to promote type 2 inflammatory responses. Moreover, using genetically engineered B. ovatus strains, we show that microbiota-intrinsic bile acid metabolism is essential to mediate inulin-induced type 2 inflammatory responses.
Several of the inulin-induced microbiota species that we identified in mice are also present in the human microbiota and express genes encoding bile acid deconjugation enzymes (for example, choloylglycine hydrolase in Bacteroides sp. 3_1_19; EnsemblBacteria: EFI07923). Notably, we and others have previously shown that SCFAs derived from dietary fibre exert anti-inflammatory effects through diverse mechanisms, including activation of G-protein-coupled receptors and inhibition of histone deacetylases (HDAC)11–14. Related to this, intervention with inulin has been shown to reduce the expression of HDAC9 in patients with asthma as well as reduced airway eosinophils and an improvement in asthma control47. Bile acids may also be upregulated in the inulin-supplemented patients with asthma in these studies; however, the beneficial effects of SCFAs could supersede the negative effects of bile acids, resulting in an overall improved clinical outcome. We therefore propose that the immunological outcome of consumption of inulin fibre diet depends on the interplay between various microbiota-derived metabolites and distinct immunoregulatory pathways.
Taken together, our findings delineate a pathway through which dietary intake of inulin fibre directly influences the composition of the microbiota and microbiota-derived metabolites to promote type 2 inflammation. The finding that an inulin fibre diet selectively influences major subsets of microbiota-derived metabolites, including CA, highlights the complex and multidimensional effects of diet on physiological and immunoregulatory processes. Furthermore, the ability of microbiota-dependent bile acids to reprogram tissue microenvironments towards ILC2-mediated type 2 inflammatory responses could provide therapeutic insights into the regulation of barrier immunity during infection, allergic inflammation and tissue protection.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-022-05380-y.
Methods
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized, and the investigators were not blinded to allocation when conducting experiments and assessing outcomes. No humans or animals were excluded from the analysis unless clearly indicated.
Mice and diets
C57BL/6 (Jax, 000664), Il33 fl/fl-eGFP (Jax, 030619)49, Nr1h4−/− (Jax, 007214)50 and ΔdblGATA (Jax, 005653)51 mice were originally purchased from The Jackson Laboratories. Rag2−/− (RAGN12) and Rag2−/−Il2rg−/− (4111) mice were purchased from Taconic Farms. These strains, as well as Il33−/− (ref. 52) (provided by M. Colonna) and Il1rl1−/− (ref. 53) (provided by A. N. J. McKenzie) mice on a C57BL/6 background were bred at Weill Cornell Medicine (WCM). For some of the experiments, mice purchased from commercial vendors were kept isolated from other in-house mice in the animal facility throughout the experiment. GF C57BL/6J mice and gnotobiotic mice were bred and maintained in flexible PVC isolators (Park Bioservices) at WCM. All of the other mice were maintained under SPF conditions. Both female and male mice were included for all of the strains used in this study. All of the mice used were between 6 and 12 weeks old, and age- and sex-matched for each experiment. All of the mice were maintained in facilities under a 12 h–12 h light–dark cycle, an average ambient temperature of 21 °C and an average humidity of 48%, and were provided food and water ad libitum. When studying the effects of dietary fibre, mice were given an inulin fibre diet (D16052309, Research Diets) supplemented with 30% fibre (26% inulin and 4% cellulose) or a calorie-matched control diet (D12450J-1.5, Research Diets) containing 4.7% cellulose (Extended Data Table 1), which is comparable to the crude fibre content of standard chow. For psyllium (D20052607) or cellulose (D20052606) fibre diets, inulin was substituted with psyllium or cellulose, respectively. The duration of dietary intervention was 2 weeks unless otherwise stated. Double-irradiated sterile diets were used for all GF, FMT and monocolonized mice. All of the mouse experiments were approved by, and performed in accordance with, the Institutional Animal Care and Use Committee guidelines at WCM.
Isolation of cells from various tissues
Mouse colons were removed, cleaned and washed in ice-cold PBS (Sigma-Aldrich). They were then opened longitudinally and washed again in ice-cold PBS. Dissociation of epithelial cells was performed by shaking at 37 °C in HBSS (Sigma-Aldrich) containing 10 mM HEPES and 5 mM EDTA (Thermo Fisher Scientific) twice for 15 min. After each step, the samples were vortexed, and the supernatant containing the epithelial fraction was collected and centrifuged at 300g for 3 min, and dissociated with TrypLE express (Invitrogen) for 1 min at 37 °C. The single-cell suspension was then passed through a 100 μm filter, washed with PBS and resuspended in RPMI with 5% FBS. The remaining tissue was chopped into small pieces, and enzymatic digestion was performed using collagenase II or III (1 mg ml−1; Worthington), dispase (0.4 U ml−1; Thermo Fisher Scientific), DNase I (20 μg ml−1; Sigma-Aldrich) and 4% FBS for 40 min in a shaker at 37 °C. The single-cell suspension was then filtered through a 70 μm cell strainer and centrifuged through a Percoll (Sigma-Aldrich) gradient and washed. Mesenteric lymph nodes were chopped and incubated in RPMI 1640 medium (Sigma-Aldrich) supplemented with 1% FBS (Sigma-Aldrich), collagenase II (1 mg ml−1; Sigma-Aldrich) and DNase I (20 μg ml−1) for 20 min in a shaker at 37 °C. Cells were then dissociated using a Pasteur pipette and filtered through a 70 μm cell strainer. Lungs were minced and incubated in RPMI medium supplemented with collagenase D or Liberase (50 μg ml−1; Roche) and DNase I (20 μg ml−1) for 30–40 min at 37 °C. The remaining tissues were mashed with a syringe plunger and single-cell suspensions were filtered through a 70 μm cell strainer. Red blood cells were lysed with ACK lysing buffer (Lonza). BALF was collected by three sequential lavages through the punctured trachea, each using 1 ml of 0.1 mM EDTA in PBS, delivered using a 3 ml syringe with a 19G blunt cannula that was secured onto the trachea with a surgical suture.
Flow cytometry and cell sorting
Mouse single-cell suspensions were pretreated with anti-CD16/32 antibodies and then incubated on ice with conjugated antibodies in PBS. Dead cells were routinely excluded using Fixable Aqua Dead Cell Stain (Thermo Fisher Scientific). Lineage (Lin) markers used were as follows: Lin1: CD3ε (145–2C11), CD5 (53–7.3) and CD8a (53–6.7); Lin2: CD19 (1D3) and CD11c (N418). FcεRI (MAR-1) was included in either Lin1 or Lin2. NK1.1 (PK136) was included in Lin2 for intracellular cytokine staining. CD45 (30-F11), CD4 (GK1.5), CD127 (A7R34), CD90.2 (30-H12), KLRG1 (2F1), CD11b (M1/70), SiglecF (E50–2440), CD64 (X54–5/7.1), F4/80 (BM8), Ly6G (1A8), Ly6C (HK1.4), NK1.1 (PK136), KIT (CD117) (2B8), Ter119 (TER119), EPCAM (G8.8), CD31 (390), Sca-1 (D7) and PDGFRα (CD140a) (APA5) were used for surface staining. CD45.1 (30-F11) and CD45.2 (30-F11) were used to detect congenic markers. Transcription factors were stained using FoxP3 (FJK-16s), GATA3 (TWAJ) and RORγt (B2D). For human samples, the Lin markers used were as follows: Lin1: CD3, CD5, CD19 and FcεRIα; Lin2: CD14, CD16 and CD11b. Surface markers used were CD45, Nkp44, CD56 and CD127. For the detection of intracellular cytokines, isolated cells were stimulated with 100 ng ml−1 phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich) and 1 μg ml−1 ionomycin (Sigma-Aldrich) for 4 h in the presence of 10 μg ml−1 brefeldin A (Sigma-Aldrich) and/or golgistop (BD) in complete RPMI1640 medium (containing 10% FBS, 50 mM 2-mercaptoethanol, 1 mM l-glutamine, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin) to evaluate their cytokine production. IL-5+ cells were detected using anti-IL-5 (TRFK5) antibodies. All antibodies used for flow cytometry were purchased from Thermo Fisher Scientific, BioLegend or BD. All antibodies above were used at dilutions of 1:200, except for CD127-BV421, IL-5-BV421 and FoxP3-AF488, which were used at 1:100 dilutions. The BD Cytofix/Cytoperm fixation and permeabilization solution (BD Bioscience) and the eBioscience Foxp3/transcription factor staining buffer set (Thermo Fisher Scientific) were used for cytokine staining and intranuclear staining, respectively. Stained cells were analysed on a 5-laser, 18-colour custom-configuration BD LSRFortessa (BD) or sorted on a 5-laser, 18-colour custom-configuration FACSAria III. Data were collected using BD FACSDiva v.8.0.1 or v.9.0 and analysed using FlowJo (v.10.6.1 or v.10.7.1, Tree Star).
RT–qPCR
Total RNA was isolated from tissues or sorted cells using the RNeasy Plus mini kit (Qiagen), and cDNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit with Multiscribe Reverse Transcriptase (Thermo Fisher Scientific), according to the protocol provided by the manufacturer. When the number of sorted cells was low, 10× lysis buffer (Takara) or TRIzol reagent (Thermo Fisher Scientific) was used for RNA isolation and Superscript II Reverse Transcriptase or the SuperScript VILO cDNA Synthesis Kit (Thermo Fisher Scientific) was used for cDNA synthesis. qPCR reactions were set up using the Power SYBR Green PCR Master Mix (Thermo Fisher Scientific) and run on a QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems) using QuantStudio Real-Time PCR software (v.1.0). The following primers were used: Il5 (QT00099715, Qiagen), Il13 (QT02423449, Qiagen), Areg (QT00112217, Qiagen), Il33 (QT00135170, Qiagen), Hprt1 (forward, 5′-CTTGCTGGTGAAAAGGACCTCTC-3′; reverse, 5′-GAAGTACT CATTATAGTCAAGGGCA-3′), bsh (BO_02350; forward, 5′-CAGCTCA CGTCATTCACTCC-3′; reverse, 5′-TACAACAGCCAGACCAACCA-3′), universal 16S rRNA gene (forward, 5′-ACTCCTACGGGAGGCAGCAGT-3′; reverse, 5′-ATTACCGCGGCTGCTGGC-3′). Mouse tissue samples were normalized to Hprt1 and faecal bacterial samples were normalized to the 16S gene and displayed as the fold change compared with the controls.
Immunofluorescence microscopy
Colons were coiled into a Swiss roll and fixed in 4% paraformaldehyde (BioWorld) in phosphate-buffered saline (PBS) for 4 h at 4 °C. Tissues were then placed in 30% sucrose dissolved in PBS at 4 °C overnight. Tissues were then embedded in OCT medium (Tissue-Tek, Sakura) and stored at −80 °C until sectioning. Frozen tissues were sectioned at 10 μm using a cryotome (Leica Instruments), immobilized on Superfrost Plus slides (VWR) and stored at −20 °C until immunostaining. For immunostaining, the slides were allowed to come to room temperature and any excess OCT medium was washed off using PBS. Tissue sections were then blocked in PBS with 5% normal goat serum (Jackson Immunoresearch), 5% normal donkey serum (Jackson Immunoresearch) and 0.2% Triton X-100 (Sigma-Aldrich) for 30 min. Tissue sections were then stained with the following primary antibodies diluted in blocking buffer overnight at 4 °C: anti-IL-33 1:50 (R&D, AF3626), anti-Vimentin 1:200 (BioLegend, 699301). The sections were washed three times with PBS and then incubated with secondary antibodies (anti-rat IgG-A488 and anti-goat IgG-A555, Invitrogen) diluted 1:500 in blocking buffer for 1 h at room temperature. Tissue sections were then washed three times in PBS and mounted with Prolong Diamond antifade reagent with DAPI (Invitrogen). Stained sections were imaged on an inverted Nikon Eclipse Ti microscope (Nikon) and analysed using Fiji (v.1.53g).
Administration of metabolites
CA or CDCA (sodium salt form, VWR) was administered in drinking water at a 6 mM concentration. IPA (VWR) was dissolved in 0.1 M NaOH, diluted with ultrapure water, and its pH was adjusted to 7.2. A total of 20 mg kg−1 body weight IPA or the vehicle (NaOH diluted and pH-adjusted) was delivered intraperitoneally daily in a volume of 100 μl10,54,55. SCFAs (acetate, propionate and butyrate, sodium salt form, all from VWR) were supplied in drinking water containing 50 mM concentration of each SCFA14. All metabolites were administered for a duration of two weeks.
Ex vivo stimulation of human cells
PBMCs were isolated from buffy coats (New York Blood Center) with a Ficoll-Paque (GE Healthcare) gradient and added on a monolayer of the human colonic stromal cell line CCD-18Co (ATCC)56. This coculture of human PBMCs and stromal cells was then subjected to PMA/ionomycin stimulation with or without CA for 4 h.
Generation of bone marrow chimeras
Mice were irradiated with 11 Gy from an X-ray irradiator split in two doses of 5.5 Gy with a 4 h break between the two cycles. CD45.1 WT mice were reconstituted with bone marrow from CD45.2 Nr1h4−/− mice. In parallel, CD45.2 WT or Nr1h4−/− mice were reconstituted with bone marrow from CD45.1 WT mice. Antibiotics (sulfamethoxazol and trimethoprim) were delivered for 2 weeks after bone marrow transplantation and mice were reconstituted for at least 8 weeks before experimentation.
FMT
For faecal microbiota transplantation (FMT) studies, donor faecal samples were collected from healthy individuals and resuspended in PBS with 10% glycerol in an anaerobic chamber57. The samples were either retrieved from the JRI IBD Live Cell Bank or collected fresh at Weill Cornell Medicine. Faecal suspensions from individual donors were administered to recipient GF mice by oral gavage (100 μl per mouse). Transplanted animals were maintained in sterile isocages for 2 to 4 weeks before administration of diets. Animals were evaluated for successful transplantation by comparing 16S sequencing between human donors and recipient mice. The study protocol was approved by the Institutional Review Board at Weill Cornell Medicine and informed consent was obtained from all Live Cell bank study participants.
Monocolonization of GF mice
WT or mutant B. ovatus strains were grown in a TYGB/Mega medium at 37 °C overnight inside an anaerobic chamber. GF mice were monocolonized by oral gavage of the bacterial culture (200 μl; ~1 × 107 colony-forming units (CFU)) and maintained in Sentry sealed positive pressure cages (SPP, Allentown) for the duration of the experiments. The level of colonization was determined by quantifying the bacterial load in mouse faecal pellets (CFU per gram of faeces). In brief, approximately 5 mg of faecal material was resuspended in 200 μl of prereduced Gibco PBS buffer (pH 7.4). Then a tenfold serial dilution (to 10−4) was made in the same buffer on a 96-well plate, and 50 μl from each 10−4-diluted well was plated onto anaerobically prereduced TSAB agar at 37 °C. After 24 h, colonies appeared and the CFUs for faecal samples collected from WT and mutant colonized GF mice were calculated after normalizing to faecal weight. Monocolonized mice were maintained in sterile isocages for at least 2 weeks before administration of diets.
Models of allergic airway inflammation
Mice were sensitized to allergic responses by intranasal administration of papain, an allergenic protease or HDM extract (Der p 1, Greer Laboratories)28. In brief, mice were anaesthetized with isoflurane (Isothesia, Henry Schein Animal Health) and 10 μg of papain (5125, Calbiochem) in 30 μl of PBS was administered for 2 consecutive days (day 0 and 1). For HDM, mice were sensitized by 25 μg (protein content) in 25 μl of PBS for 3 days (days 0, 1 and 2), and challenged after two weeks with 5 μg (protein content) in 25 μl of PBS for 3 days (days 14, 15 and 16)28. The mechanical properties of the respiratory system were measured using a rodent lung function testing system (FlexiVent, SCIREQ). In brief, the mice were anaesthetized by intraperitoneal injection of sodium pentobarbital (100 mg kg−1, Esconarkon, Streuli Pharma), then tracheotomized and mechanically ventilated followed by intraperitoneal administration of pancuronium (0.08 mg kg−1, Hospita). Airway constriction was induced by administering increasing aerosolized doses (0.3125 kg−1, 12.5 kg−1 and 50 mg kg−1) of methacholine chloride (Sigma-Aldrich). Airway resistance was calculated from forced-oscillation measurements of changes in pressure, volume and flow.
Helminth infection
For N. brasiliensis infection, third-stage larvae (L3) were purified with a Baermann apparatus. After washing three times in PBS, living worms were counted, and 500 purified worms in 250 μl PBS were injected subcutaneously. Tissues were collected on day 3 after infection. Collection and enumeration of worms in the small intestine was performed as previously described45.
Histological analysis
The left lung lobe was inflated and fixed with 4% paraformaldehyde, followed by embedment in paraffin. Sections (5–6 μm) were used for staining with H&E or periodic acid Schiff (PAS) by IDEXX BioResearch. Histological scoring of H&E-stained lung tissues was performed on the basis of cellular infiltration in the perivascular and peribronchial regions as previously described58. In brief, a score of 1 represents a tissue section with occasional veins cuffed by inflammatory cells; a score of 2 indicates that most veins in the section are surrounded by a thin layer (1–5 cells thick) of inflammatory cells; a score of 1.5 indicates a level of infiltration between scores 1 and 2; a score of 3 indicates that most veins are surrounded by a thick layer (more than 5 cells thick) of inflammatory cells. For quantification of bronchoepithelial goblet cells, the number of PAS+ goblet cells in each bronchiole was counted, then the average number of goblet cells per bronchiole in each mouse tissue section was used for statistical analysis.
16S amplicon sequencing of intestinal microbiota
Library preparation, sequencing and OTU table generation were performed by the Microbiome Core Lab at Weill Cornell Medicine. Each faecal pellet was deposited into a Qiagen PowerBead tube with 0.1 mm glass beads (13118–50). Using a Promega Maxwell RSC PureFood GMO and Authentication Kit (AS1600), 1 ml of CTAB buffer and 20 μl of RNase A solution were added to the PowerBead tube that contained the sample. Then the sample/buffer was mixed for 10 s on a Vortex Genie2 and then incubated at 95 °C for 5 min on an Eppendorf ThermoMixer F2.0, with shaking at 1,500 rpm. The tube was removed and clipped to a horizontal microtube attachment on a Vortex Genie2 and vortexed at high-speed for 20 min. The sample was then removed from the Vortex and centrifuged at 40 °C and 12,700 rpm for 10 min. After completion, the sample was centrifuged again for an additional 10 min to eliminate foam. The sample tube cap was removed, and the sample was checked for foam and particulates. If foam or particulates were found in the sample, they were carefully removed using a P1000 pipette. The opened tube was then added to a Promega MaxPrep Liquid Handler tube rack. The Liquid Handler instrument was loaded with proteinase K tubes, lysis buffer, elution buffer, 1000 ml tips, 50 ml tips, 96-sample deep-well plate and Promega Maxwell RSC 48 plunger tips. The Promega MaxPrep Liquid Handler instrument was programmed to use 300 μl of the sample and transfer all of the sample lysate into a Promega Maxwell RSC 48 extraction cartridge for DNA extraction. After completion, the extraction cartridge was loaded into a Promega Maxwell RSC 48 for DNA extraction and elution. DNA was eluted in 100 μl and transferred to a standard 96-well plate. DNA was quantified using a Quant-iT dsDNA High Sensitivity Assay Kit using a Promega GloMax plate reader on a microplate.
Libraries were prepared according to the protocol of the Earth Microbiome Project (https://earthmicrobiome.org/protocols-and-standards/16s/). Amplicon libraries were washed using Beckman Coulter AMPure XP magnetic beads. Library quality and size verification was performed using a PerkinElmer LabChip GXII instrument and a DNA 1K Reagent Kit (CLS760673). Library concentrations were quantified using the Quant-iT dsDNA High Sensitivity Assay Kit and the Promega GloMax plate reader on a microplate. Library molarity was calculated on the basis of library peak size and concentration. Libraries were normalized to 2 nM using the PerkinElmer Zephyr G3 NGS Workstation and pooled together using the same volume across all normalized libraries into a 1.5 ml Eppendorf DNA tube.
Pooled libraries were sequenced on the Illumina MiSeq instrument at a loading concentration of 8 pM with 10% PhiX, using MiSeq Reagent Kit v2, 500-cycles (MS-102–2003), producing paired 250 bp reads. Raw reads were processed and clustered into OTUs using USEARCH v.11.0.667. Specifically, reads were demultiplexed and read pairs were merged, with a maximum of five mismatching bases in the overlap region, as well as a minimum sequence agreement of 80%. PhiX contaminant sequences were removed, and the merged sequences were filtered according to FASTQ quality scores using a maximum expected nominal error number of 1.0 per read. Filtered sequences were clustered into OTUs at a 97% identity threshold using the USEARCH cluster_otus command with the default settings. Unfiltered merged reads were mapped to the OTU representative sequences, generating an OTU table. Taxonomic classification of OTU representative sequences was performed with the USEARCH SINTAX command with a confidence score of 0.8, using version 16 of the RDP 16S training set59. A phylogenetic tree was generated from the OTU representative sequences using the USEARCH -cluster_aggd command with the default settings. Diversity estimation and principal coordinates analysis ordination were performed using the phyloseq R package (v.1.30.0)60 after subsampling the OTU table to even depth. For mapping 16S data to the HMP database, the raw 16S reads were checked for quality and then mapped to a DNA library containing the full-length 16S rRNA sequences of 395 intestinal microbes using Bowtie 2 (high sensitivity)61. The mapped sequences with high sequence identity (number of nucleotide sequences with quality) were normalized to the total reads of the 16S rRNA samples to evaluate the relative abundance of that specific microbial species within the whole intestinal microbiome community. Differential abundance was assessed using Student’s t-tests, performed only for strains for which at least one sample had a minimum of 50 nucleotide sequences with quality, with significance determined by a 10% FDR.
RNA-seq analysis of ILC2s
RNA-seq libraries were prepared from 1,000 sorted colonic lamina propria ILC2s (CD45+Lin−CD90.2+CD127+KLRG1+) by the Epigenomics Core at WCM using the Clontech SMARTer Ultra Low Input RNA Kit V4 (Clontech Laboratories). Libraries were sequenced on the Illumina HiSeq 2500 system, generating 50 bp single-end reads. Two samples from two control-diet-fed WT SPF mice and two samples from two inulin-fibre-diet-fed WT SPF mice were used. Reads were demultiplexed using Illumina’s CASAVA (v.1.8.2), adapter-trimmed using FLEXBAR (v.2.4)62 and aligned to the mouse genome (NCBI GRCm38/mm10) using STAR aligner (v.2.3.0)63 with the default parameters. Read counts per gene (RefSeq annotation) were determined using the Rsubread R package (v.3.10 I)64. Normalization and differential expression analysis were performed using DESeq2 (v.1.26.0)65. Before differential expression testing, genes were prefiltered to include only genes that had a minimum of 50 raw reads in at least two samples. Tests were corrected for multiple comparisons, using a FDR of 10% to determine significance. GO enrichment analysis was performed using the enrichGO function of the clusterProfiler R package (v.3.14.3)66, using the biological process ontology. Enrichment P values were corrected for multiple comparisons using the Benjamini and Hochberg method67.
Spatial transcriptomic analysis of the colon
Distal colon samples (four samples from two control-diet-fed mice and four samples from two inulin-fibre-diet-fed mice) were processed using the Visium Spatial Gene Expression Slide & Reagent Kit (PN-1000184, 10x Genomics) according to the manufacturer’s instructions. In brief, snips from the distal colon were snap frozen in OCT and stored at −20 °C until they were sectioned. Cross-sections (10 μm) were cryosectioned and placed on the designated areas of the Visium Spatial Gene Expression Slide. The slide was then thawed, fixed with methanol at −20 °C for 30 min, stained with H&E (ab245880, Abcam) and imaged at ×10 magnification using the Nikon Ti Eclipse microscope and the large image feature (based on tiling and stitching) of the NIS Elements Viewer software (v.4.30.02). The slide was then permeabilized for RNA extraction according to the manufacturer’s protocol. The permeabilization time for colon tissue sections was determined to be 20 min. This was established using the instructions provided by the manufacturers of the Visium Spatial Tissue Optimization Slide Kit (PN-1000191) and the Visium Spatial Tissue Optimization Reagent Kit (PN-1000192) prior to the actual experiment. Libraries were constructed using the Visium Library Construction Kit (PN-1000190) according to the manufacturer’s instructions and sequenced on the Illumina NovaSeq 6000 system. Sequencing data were aligned on H&E-stained images using Space Ranger v.1.1.0 (10x Genomics).
Spatial transcriptomics data were analysed using the Seurat R package68 (v.3.2.1). Space Ranger output was imported into R using the Seurat Load10X_Spatial function. Spots that did not align with tissue slices were removed before analysis. Each tissue sample was separately normalized using the Seurat SCTransform function. For the purpose of clustering spots across all of the samples, one of the samples (Ctrl1) was selected as a reference dataset. The k-means clusters (with k = 4) generated for the reference sample by Space Ranger were then used to predict cluster identities for the other samples using the FindTransferAnchors and TransferData functions of Seurat. All of the samples (2 control and 2 inulin fibre) were then merged into a single Seurat object, which was used in all further analysis. Cluster marker genes, as well as genes differentially expressed between control and inulin fibre diets within the clusters, were determined using the Seurat FindMarkers function, implementing the Wilcoxon test. Genes were considered to be significant if they had a Bonferroni-adjusted P value < 0.05 and an absolute log-transformed (base 2) fold change of >0.25. GO enrichment analysis of differentially expressed genes was performed using the web service STRING69. Spatial images were obtained using Loupe Browser v.4.1.0.
Metabolomic analysis of serum, faeces and caecal contents
Metabolite extraction.
Metabolites were extracted from serum, faeces and caecal samples with methanol. For serum, a total of 800 μl of methanol was added to 200 μl of serum in 1.7 ml Eppendorf tubes. The tubes were then vortexed and sonicated for 1 min. For faeces or caecal samples, 30 mg of dried faecal pellet or caecal content was vortexed and sonicated in 500 μl of 100% methanol for 1 min. Extracts from serum, faecal and caecal samples were pelleted at 5,000g for 10 min at 4 °C, and the supernatants were transferred to 2 ml HPLC vials. The samples were then dried in a SpeedVac (Thermo Fisher Scientific) vacuum concentrator. The dried materials were resuspended in 1 ml of methanol and vortexed for 1 min. The samples were pelleted at 5,000g for 5 min at 22 °C, and the supernatants were transferred to HPLC vials and dried in a SpeedVac vacuum concentrator. The samples were then resuspended in 100 μl of methanol and centrifuged at 5,000g for 10 min at 22 °C. Clarified extracts were transferred to fresh HPLC vials and stored at −20 °C until analysis.
MS analysis.
LC–MS was performed on the Thermo Fisher Scientific Vanquish UHPLC system coupled with a Thermo Q Exactive HF hybrid quadrupole-orbitrap high-resolution mass spectrometer equipped with a HESI ion source. Metabolites were separated using a water−acetonitrile gradient on an Agilent Zorbax Eclipse XDB-C18 column (150 mm × 2.1 mm, particle size 1.8 μm) maintained at 40 °C (solvent A: 0.1% formic acid in water; solvent B: 0.1% formic acid in acetonitrile). The A/B gradient started at 1% B for 3 min after injection and increased linearly to 100% B at 20 min, then 100% B for 5 min, and down to 1% B for 3 min at a flow rate of 0.5 ml min−1. MS parameters were as follows: spray voltage, 3.5 kV; capillary temperature, 380 °C; probe heater temperature, 400 °C; 60 sheath flow rate, 20 auxiliary flow rate, and one spare gas; S-lens RF level, 50; resolution, 240,000; AGC target, 3 × 106. The instrument was calibrated weekly with positive and negative ion calibration solutions (Thermo Fisher Scientific). Each sample was analysed in negative and positive ionization modes using an m/z range of 100 to 800. Data were collected using Thermo Fisher Scientific Xcalibur software (v.4.1.31.9) and quantified by integration in Excalibur Quan Browser (v.4.1.31.9).
Feature detection and characterization.
LC–MS RAW files from biospecimens from control and inulin fibre diet-fed mice were converted to mzXML format (centroid mode) using MSconvert v.3.0.20315–7da487568 (ProteoWizard), followed by analysis using the XCMS analysis feature in Metaboseek v.0.9.7 (https://metaboseek.com)70 based on the centWave XCMS algorithm to extract features71. Peak detection values were set as follows: 4 ppm, 3 to 20 peakwidth, 3 snthresh, 3 and 100 prefilter, FALSE fitgauss, 1 integrate, TRUE firstBaselineCheck, 0 noise, wMean mzCenterFun and −0.005 mzdiff. XCMS feature grouping values were set as follows: 0.2 minfrac, 2 bw, 0.002 mzwid, 500 max, 1 minsamp and FALSE usegroup. Metaboseek peak filling values were set as follows: 5 ppm_m, 5 rtw, and TRUE rtrange. Resulting tables of all detected features were then processed with the Metaboseek data explorer. To select differential features, we applied a filter that retained only entries with peak area ratios of less than 0.25 (down in inulin-fibre-diet mice) or greater than 4 (up in inulin-fibre-diet mice), with a retention time window of 1 to 20 min and >0.95 peak quality as calculated using Metaboseek. We manually curated the resulting list to remove false-positive entries (that is, features that, after manual inspection of raw data, were not differential). For verified differential features, we examined elution profiles, isotope patterns and MS1 spectra to find molecular ions and remove adducts, fragments and isotope peaks. The remaining masses were put on the inclusion list for MS/MS (ddMS2) characterization. Positive and negative ionization mode data were processed separately. To acquire MS2 spectra, we ran a top-10 data-dependent MS2 method on the Thermo Q Exactive HF mass spectrometer with an MS1 resolution of 60,000; AGC target, 1 × 106; maximum injection time (IT), 50 ms; MS2 resolution, 45,000; AGC target, 5 × 105; maximum IT, 80 ms; isolation window, 1.0 m/z; stepped normalized collision energy (NCE), 10 and 30 for positive ion mode and 40 and 60 for negative ion mode; dynamic exclusion, 3 s.
Compound identification.
The identities of bile acids, IPA, indoleacrylic acid, phenylacetylglycine and p-cresol sulfate were confirmed by coinjection with authentic standards (Schymanski annotation level one, according to previously published standards72). CA, β-muricholic acid, ω-muricholic acid, CDCA, ursodeoxycholic acid, 7-ketodeoxycholic acid, IPA, indoleacrylic acid and phenylacetylglycine were purchased from Sigma-Aldrich. α-Muricholic acid was purchased from Cambridge Isotope Laboratories. p-Cresol sulfate was purchased from Cayman Chemical. The structures of indolylacryloylglycine73, p-cresol glucuronide74 and 2-[4-(sulfooxy)phenyl]acetic acid75 were proposed based on MS2 fragmentation (Schymanski annotation level three72).
Quantification of bile acids.
A methanol stock solution of standard bile acids was prepared and diluted at five different concentrations for the construction of calibration curves. The resulting mixtures and sample extracts were analysed using the LC–MS conditions described above. Quantification was performed by integration in Xcalibur Quan Browser (Thermo Fisher Scientific). For the quantification of bile acids in faecal samples, the amounts were normalized on the basis of the weight of the lyophilized samples before extraction.
MS2-based molecular networking.
An MS2 molecular network76 was created using Metaboseek70 (v.0.9.6) and visualized in Cytoscape77. Features were matched with their respective MS2 scan within an m/z window of 5 ppm and a retention time window of 15 s, using the MS2scans function. To construct a molecular network, tolerance of the fragment peaks was set to an m/z of 0.002 or 5 ppm, the minimum number of peaks was set to 3 and the noise level was set to 2%. Once the network was constructed, a cosine value of 0.7 was used, the number of possible connections was restricted to 4 for negative ion mode and 5 for positive ion mode, and the maximum cluster size was restricted to 200 for positive ion mode.
Deletion of the bsh gene in B. ovatus
Vector assembly.
We conducted gene deletion of the annotated candidate bsh gene BO_02350 in the B. ovatus genome. The conventional double-crossover gene deletion method was used to generate the bsh mutant78,79. First, we amplified two fragments with a length of around 600–1,000 bp around the target gene and integrated them into a single unit by fusion PCR. The new fragment was then assembled with a PCR-amplified pExchange79 vector using Gibson assembly and transformed into Escherichia coli S17 directly to form a suicide vector (after diagnostic PCR and sequencing verification) that was introduced into Bacteroides by conjugation as described below. A list of all of the primers used in cloning is provided in Supplementary Table 2.
Introducing vectors by conjugation into B. ovatus.
We introduced the suicide vectors into B. ovatus (ATCC-8483) based on previously used methods78. First, a single colony of B. ovatus Δtdk (a strain deleted for the BO_02336 gene, which encodes a thymidine kinase) was inoculated in 3 ml TYGB/Mega broth culture in an anaerobic chamber at 37 °C under an atmosphere consisting of 10% CO2, 5% H2, 85% N2 for 12 h. E. coli S17 containing a suicide vector was grown in 3 ml LB broth supplemented with carbenicillin (100 μg ml−1) at 37 °C with shaking at 225 rpm. After around 10–12 h, when the optical density at 600 nm of E. coli S17 reached 0.8–1.0, 3 ml of E. coli S17 culture was centrifuged at 1,500g for 3 min. The supernatant was discarded, and the cell pellet was washed once with 1.5 ml PBS buffer (pH 7.4). The washed E. coli S17 cell pellet was resuspended in 3 ml overnight culture of the Bacteroides strain. The mixture was filtered through a 0.2 μm filter. The filtered liquid was discarded and the filter membrane with the mixture of donor and recipient cells was placed onto the surface of a prereduced TSAB plate, and the plate was incubated aerobically in a 37 °C incubator.
After aerobic incubation at 37 °C for 24 h, the filter membrane was soaked in 2 ml of prereduced TYGB medium, and the cells on the filter were resuspended into the medium by gentle vortexing. The mixture was then transferred into an anaerobic chamber and 100 μl was plated onto a prereduced TSAB plate with 200 μg ml−1 gentamycin and 25 μg ml−1 erythromycin. Colonies with plasmid inserted into the genome typically appeared after 36–48 h. Eight colonies were picked and restreaked on prereduced TSAB plates with 200 μg ml−1 gentamycin and 25 μg ml−1 erythromycin to isolate single colonies. After around 1–2 days, a single colony of each single recombinant was picked into TYGB medium without antibiotics and grown overnight for 16–24 h. Equal amounts of each of the TYGB-grown single recombinants were then combined into one pooled stock. Finally, 100 μl of 2–3 serial tenfold dilutions (that is, 1×, 0.1× and so on) of the pooled single recombinants were streaked onto TSAB plates containing FUdR (200 μg ml−1) and grown anaerobically at 37 °C for 2–3 days. When colonies grew, 10 FUdR-resistant colonies were restreaked on TSAB plates with FUdR (200 μg ml−1). Finally, the genome of these colonies was extracted using a liquid culture and a diagnostic PCR was performed to identify the successful gene-deleted constructs. Those mutants were then ready for the in vitro testing for their ability to transform TCA into CA. On the basis of our in vitro MS results, the ability to transform TCA into CA was almost fully abolished in the mutant (Δbsh B. ovatus) in which the BO_02350 gene was deleted (Extended Data Fig. 8i,j).
MS analysis of bacterial cultures and faecal SCFA
Quantification of bile acids.
For in vitro quantification, bacterial broth was mixed with an equal volume of methanol and centrifuged at 21,000g for 20 min. The mixture was then centrifuged at 21,000g for 15 min. The supernatant of the bacterial broth was analysed using the Agilent 1290 LC system coupled to an Agilent 6530 quadrupole time-of-flight (QTOF) mass spectrometer with a 2.1 μm, 2.1 × 50 mm Zorbax Eclipse Plus C18 column (Agilent). Water with 0.2% formic acid (A) and acetone with 0.2% formic acid (B) was used as the mobile phase at a flow rate of 0.40 ml min−1 over an 11 min gradient: 0–1 min, 0% B; 1–3 min, 0–40% B; 3–10 min, 40–100% B; 10–11 min, 100–0% B. Reagents were MS grade and were purchased from Thermo Fisher Scientific. All data were collected in negative-ion mode.
Quantification of SCFAs.
To quantify SCFAs in mouse biological samples, ~10 mg of faecal samples was resuspended in 50 μl of 50% methanol (in H2O) and vortexed for 10 min (some beads were added to disperse the faecal material). The mixture was then centrifuged and 10 μl of supernatant was mixed with 190 μl SCFA derivatization solution (freshly prepared acetonitrile solution containing 1 mM 2,2′-dipyridyl disulfide, 1 mM triphenylphosphine and 1 mM 2-hydrazinoquinoline80). The resulting mixture was vortexed and incubated at 60 °C for 1 h, centrifuged at 21,000g for 20 min, and its supernatant was analysed by LC–MS. We used the following solvent system for detecting SCFA derivatives: A: H2O with 0.1% formic acid; B: methanol with 0.1% formic acid. A total of 1 μl of each sample was injected at a flow rate of 0.35 ml min−1 and a column temperature of 40 °C. The gradient for HPLC–MS analysis was: 0–6.0 min, 99.5% A to 70.0% A; 6.0–9.0 min, 70.0% A to 2.0% A; 9.0–9.4 min, 2.0% A to 2.0% A; 9.4–9.6 min, 2.0% A to 99.5% A. Peaks were assigned by comparison with authentic standards and absolute analyte concentrations were quantified by comparing their peak areas with those of gradient standards and then further normalized by sample weight.
Statistical analysis
Statistical tests were performed with GraphPad Prism v.9.4.1 or R v.3.6.3. P values of datasets were determined by unpaired or paired (where applicable) two-tailed t-tests with a 95% confidence interval. Where appropriate, Mann–Whitney U-tests or one or two-way ANOVA followed by post hoc tests were performed. P values for RNA-seq datasets were determined by DESeq2 Wald test or Wilcoxon test. P < 0.05 was considered to be significant. The entity n represents biologically independent samples and not technical replicates unless specified otherwise. Error bars show the s.e.m.
Extended Data
Extended Data Fig. 1 |. Changes in gut microbiota by inulin fibre diet.
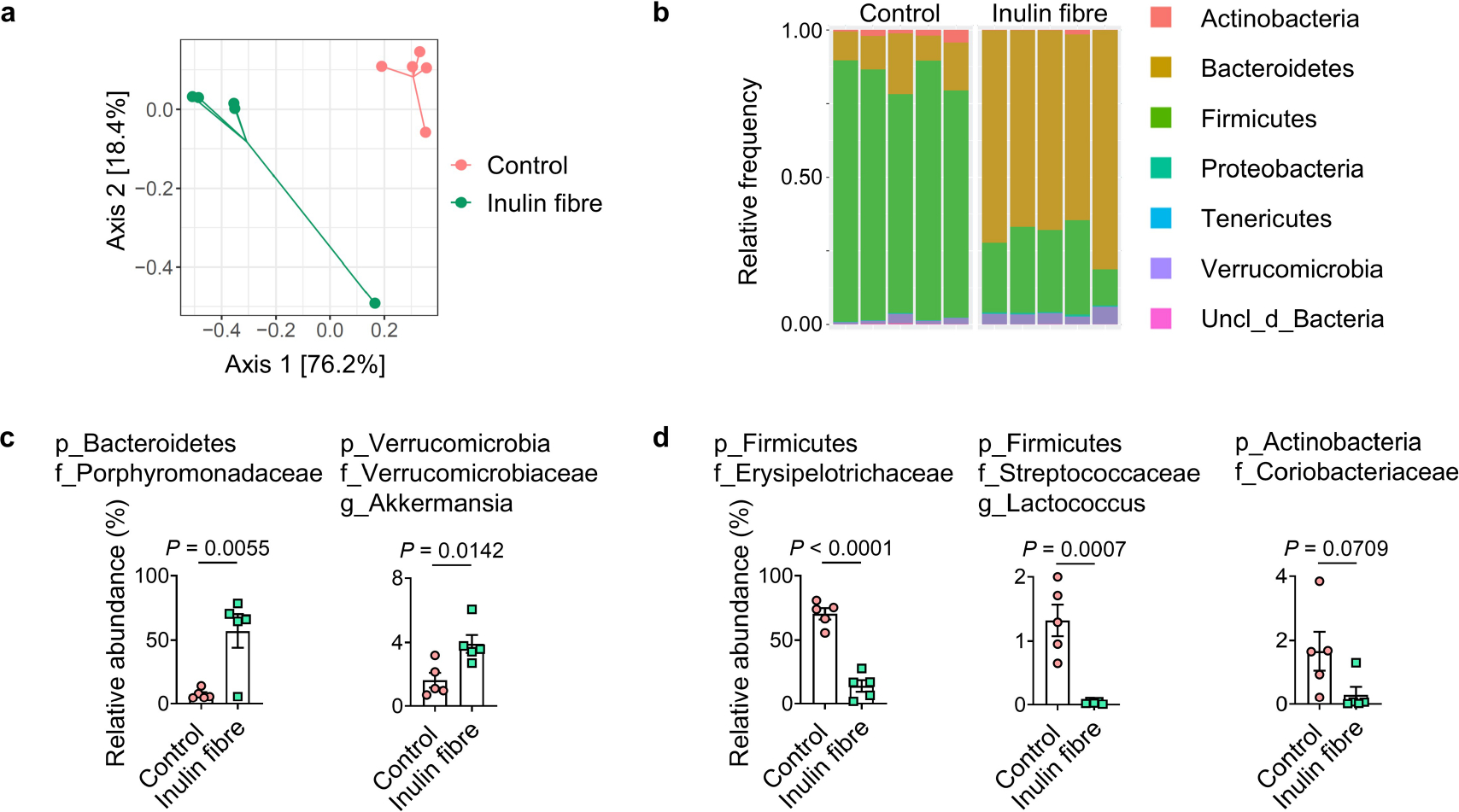
a–d, Mice were fed the control or inulin fibre diets for two weeks (n = 5 mice). Weighted UniFrac PCoA (a) and taxonomic classification (b) of 16S rRNA in stool pellets were shown. For PCoA plot PERMANOVA: F = 12.6, Df = 1, P = 0.008. ‘uncl_d_’ stands for ‘unclassified_domain_’. ‘uncl_d_Bacteria’ matches exactly to mitochondria or chloroplasts, probably from the food. Relative abundance of families or genera increased (c) or decreased (d) by the inulin fibre diet was shown. Data in a–d are representative of three independent experiments. Data are means ± s.e.m. Statistics were calculated by unpaired two-tailed t-test (c,d).
Extended Data Fig. 2 |. Changes in systemic metabolome by inulin fibre diet.
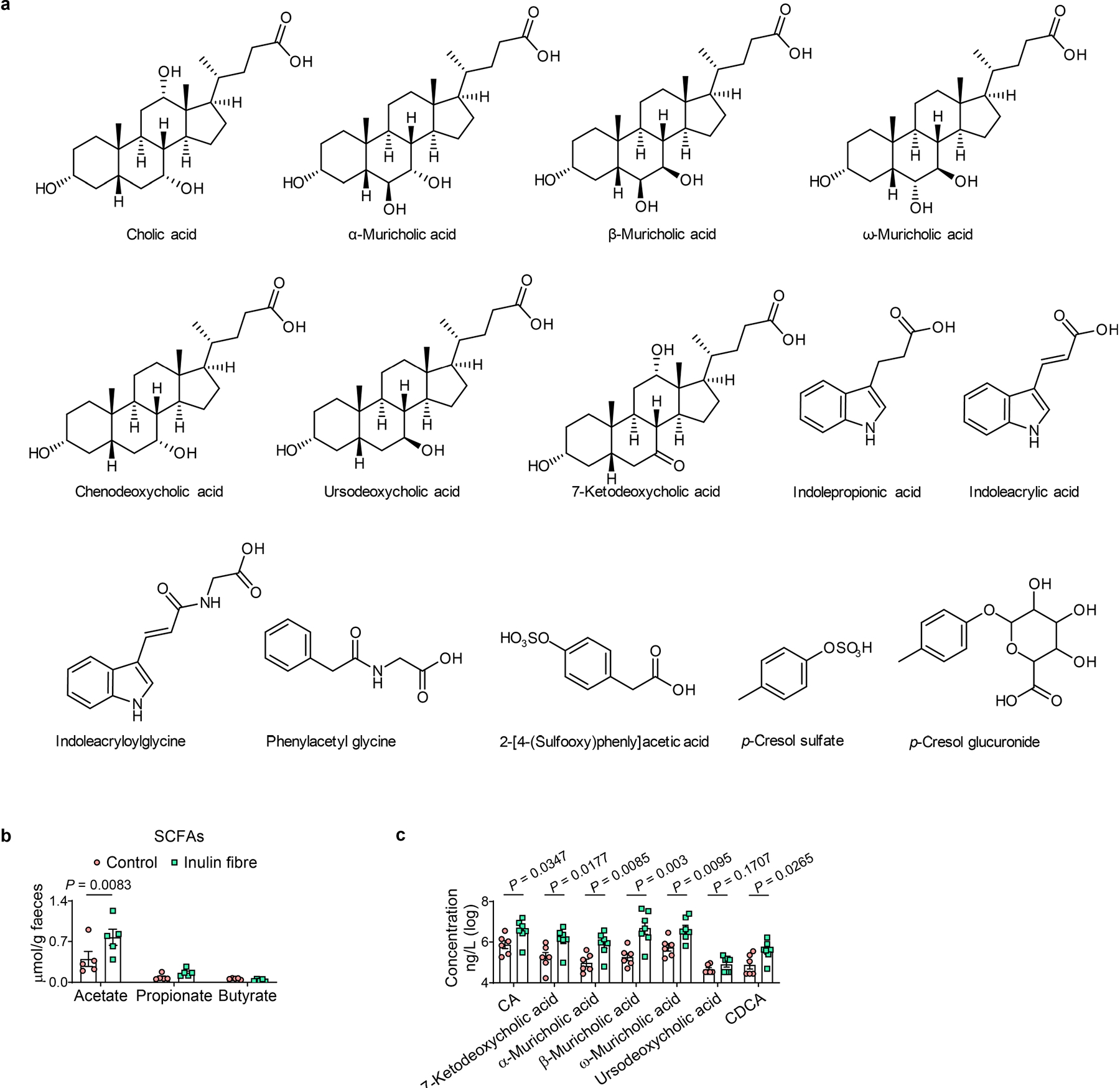
a, Chemical structures of major differentially abundant compounds identified in this study. b, Faecal concentration of SCFAs in mice from control and inulin fibre diet groups (n = 5 mice). c, Integration of serum bile acids shown in Fig. 1d (n = 6 mice for control or 7 mice for inulin fibre). Data in b,c are representative of at least two independent experiments. Data are means ± s.e.m. Statistics were calculated by two-way ANOVA with Šidák’s multiple-comparisons test (b) or unpaired two-tailed t-test (c).
Extended Data Fig. 3 |. MS analyses of various samples.
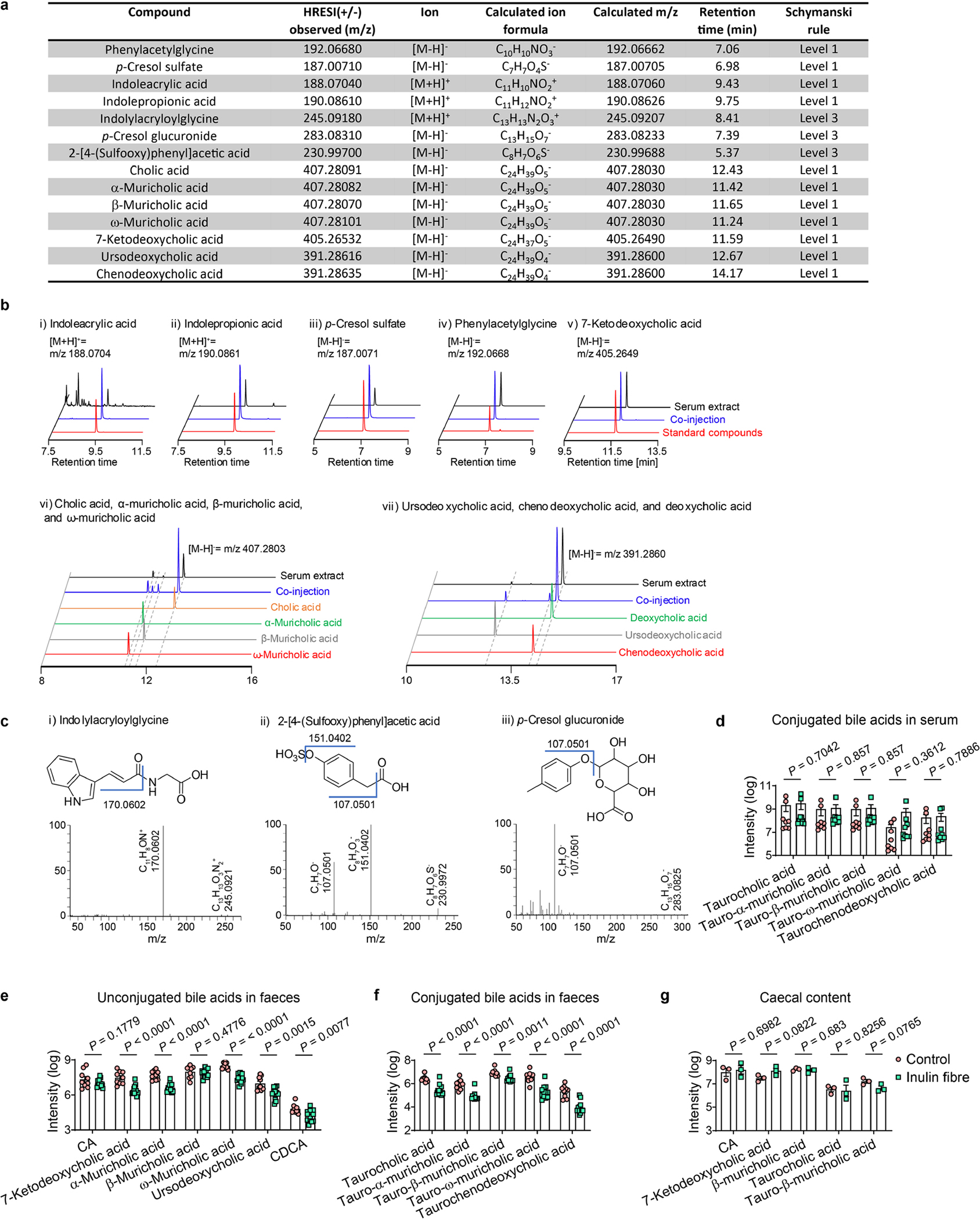
a, Table depicting detection level and other parameters for the metabolites analysed. b–c, Coinjection plots (b) or MS/MS spectra (c) for the metabolites analysed. d–g, Relative abundance of conjugated bile acids in serum (n = 8 mice for control or 9 mice for inulin fibre) (d), faecal unconjugated (e) and conjugated bile acids (f) (n = 9 mice for control or 12 mice for inulin fibre), and conjugated and unconjugated bile acids in caecal content (n = 3 mice) (g). Data are representative of (g) or combined (d–f) from two independent experiments. Data are means ± sem. Statistics were calculated by unpaired two-tailed t-test (d–g).
Extended Data Fig. 4 |. Gating strategies shown using samples from colon.
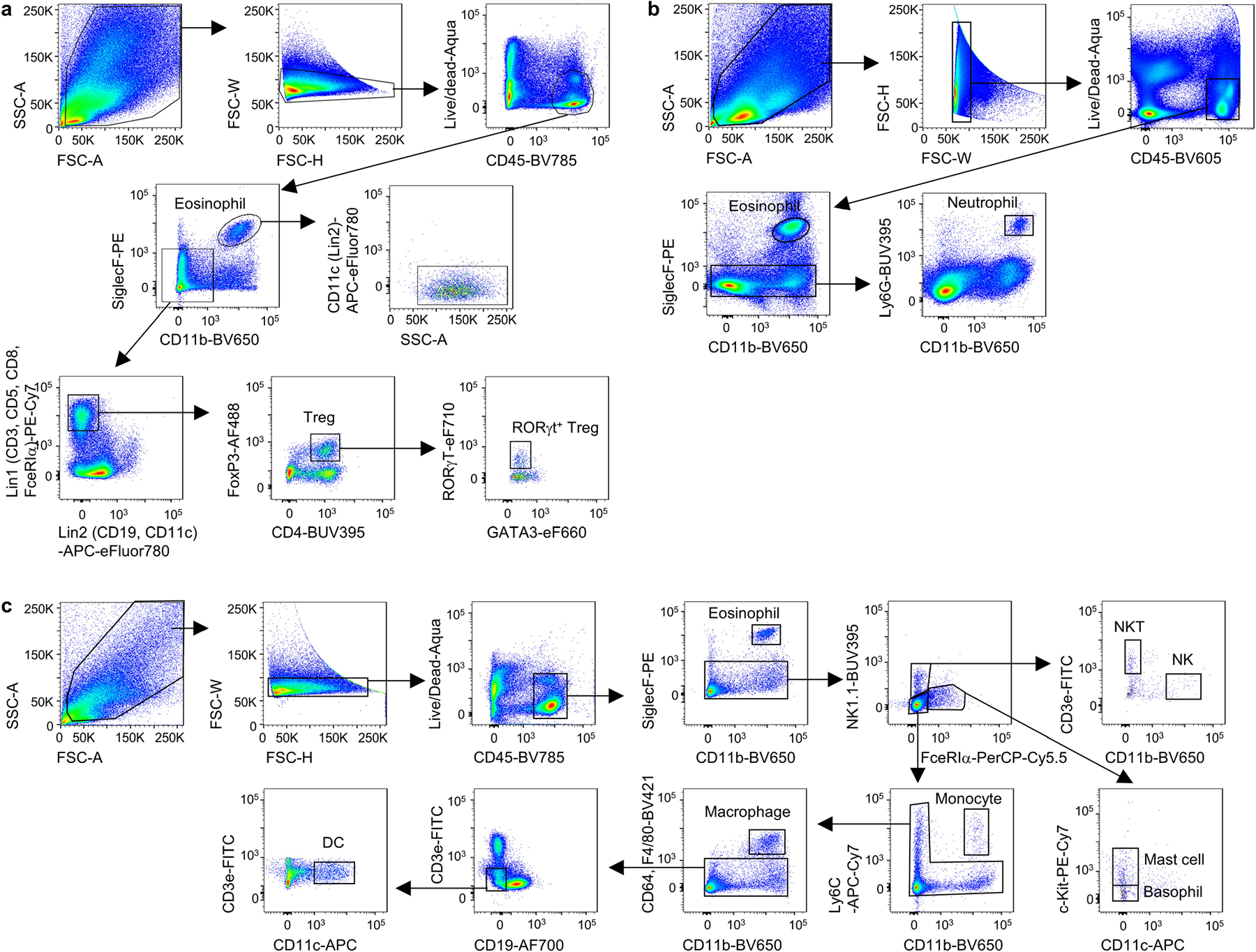
a, Gating strategy for eosinophils in bar graphs including Fig. 1e and Tregs in Extended Data Figs. 5a and 7a. CD45+CD11b+SiglecF+ cells were further verified as SSChiCD11c−. b–c, Gating strategies for neutrophils (b) and various other immune cells (c) in Extended Data Fig. 5b.
Extended Data Fig. 5 |. Effects of inulin fibre diet on immune cells in various tissues.
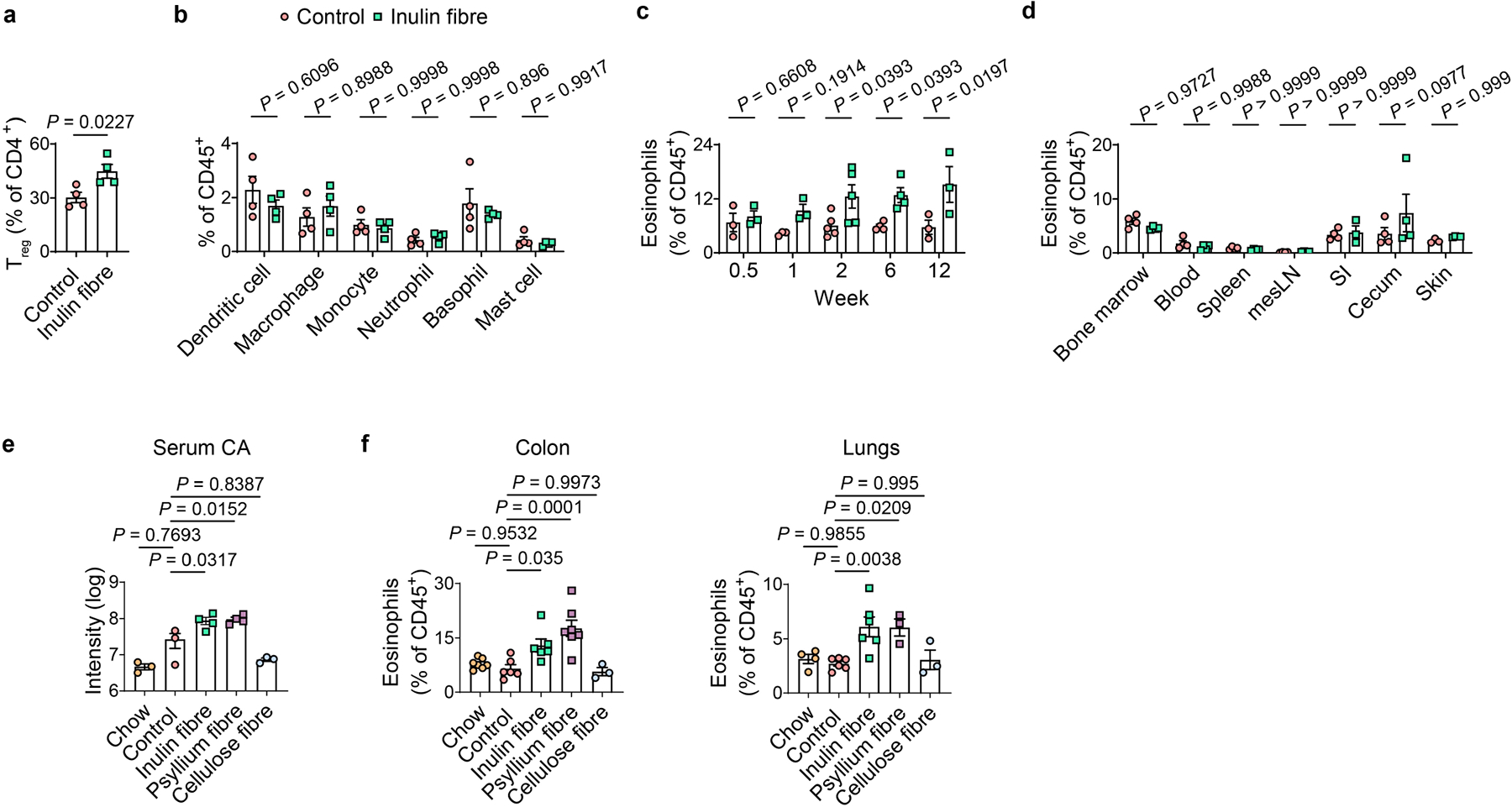
a,b, Mice were fed control or inulin fibre diet for two weeks (n = 4 mice). Bar graph shows frequencies of FoxP3+ Treg cells (a) and various immunocytes (b) in the colonic lamina propria of control or inulin fibre diet-fed mice. c, Frequency of colonic eosinophils in mice fed control or inulin fibre diet for the indicated periods of time (n = 3 mice for 0.5-, 1-, or 12-week timepoints, n = 5 mice for 2-week timepoint, and n = 4 mice for 6-week timepoint). d, Percentage of eosinophils in various tissue sites from mice administered control or inulin fibre diet for two weeks. mesLN, mesenteric lymph nodes; SI, small intestine. n = 4 for bone marrow, blood, spleen, mesLN, caecum, SI control or skin inulin fibre, n = 3 for SI inulin fibre or skin control. e,f, Mice were fed chow, control diet, or various high fibre diets, and serum levels of CA (e) were measured after two weeks (n = 3 mice for chow, control, or cellulose fibre, and n = 4 mice for inulin or psyllium fibre). Frequency of eosinophils in the colon and lung (f) of mice fed the indicated diets for two weeks (n = 7 mice (colon) or 4 mice (lung) for chow, n = 6 mice for control or inulin fibre, n = 3 mice for cellulose fibre, and n = 7 mice (colon) or 3 mice (lung) for psyllium fibre). Data are representative of (a–d) or combined (e,f) from two independent experiments. Data are means ± s.e.m. Statistics were calculated by unpaired two-tailed t-test (a), two-way ANOVA with Šidák’s (b,d) or Holm–Šidák’s (c) multiple-comparisons test, or one-way ANOVA with Šidák’s multiple-comparisons test (e,f).
Extended Data Fig. 6 |. Inulin fibre diet-induced gene and protein expression in various immune and non-immune cells.
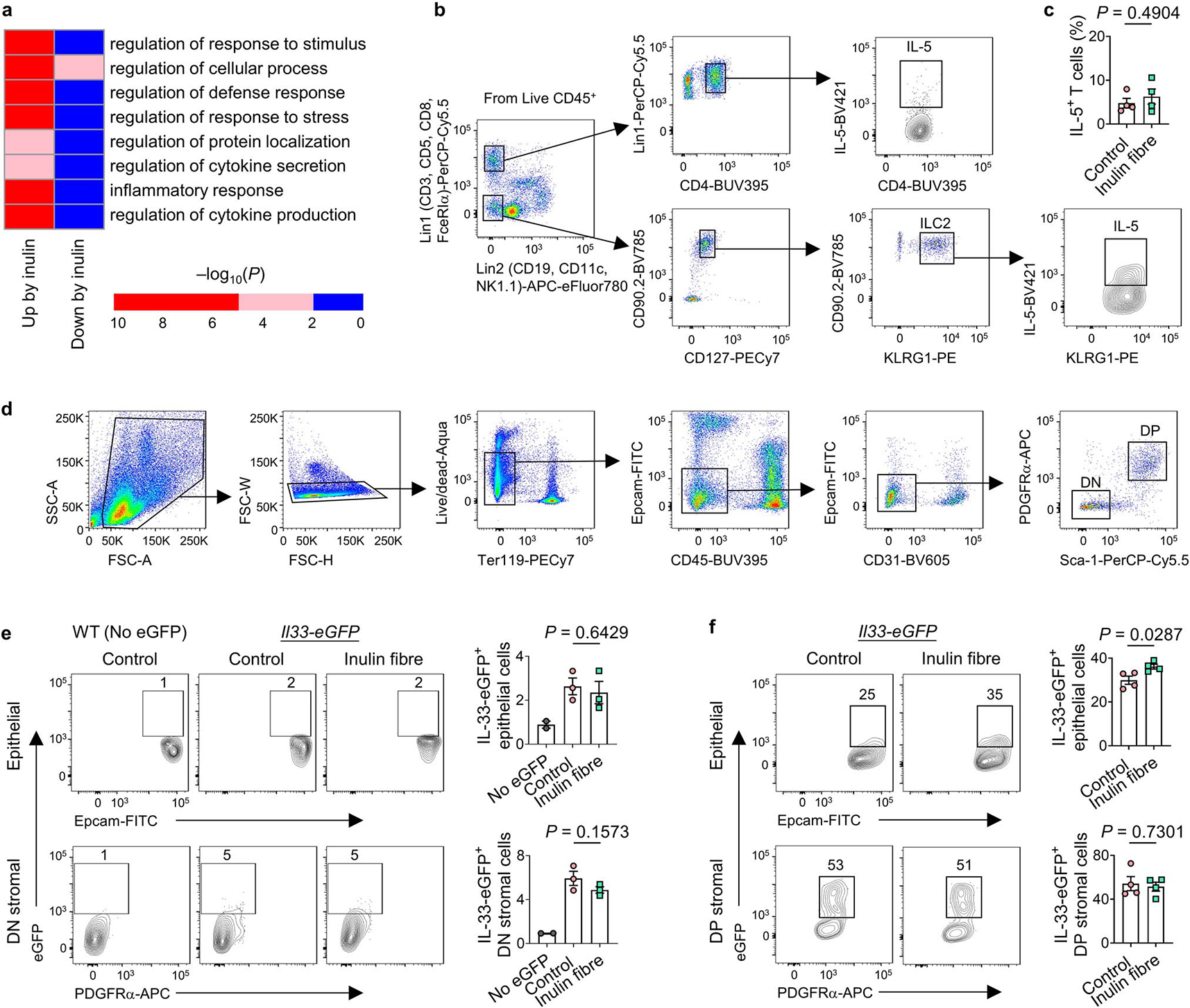
a, Mice were fed control or inulin fibre diet for two weeks and ILC2s (CD45+Lin−CD90.2+CD127+ KLRG1+) were sorted from colonic lamina propria cells. Heatmap showing level of significance of GO enrichment tests in the colonic ILC2 RNASeq data, as measured by −log10(Pcorrected). Blue, not significant (Pcorrected > 0.01), Red, significant (Pcorrected < 0.01). b,c Gating strategy for bar graphs for IL-5+ cells (including Fig. 2c) (b) and frequency of IL-5-expressing CD4+ T cells (c) in the colonic lamina propria of mice (n = 4 mice). d, Gating strategy for detecting PDGFRα+Sca-1+ (double positive, DP) mesenchymal stromal cells (for bar graphs including Fig. 2f) and double negative (DN) stromal cells in colon and lung tissues. e,f Flow cytometry plots and bar graphs showing IL-33-eGFP expression in epithelial and stromal cell subsets in the colon (e) and lung (f). n = 2 mice (No eGFP) or 3 mice (colon control or inulin fibre) or 4 mice (lung control or inulin fibre). Data in c,e,f, are representative of two independent experiments. Data are means ± s.e.m. Statistics were calculated by unpaired two-tailed t-test (c, f) or one-way ANOVA with Holm–Šidák’s multiple-comparisons test (e).
Extended Data Fig. 7 |. Effects of bile acid metabolites on immune and non-immune cells.
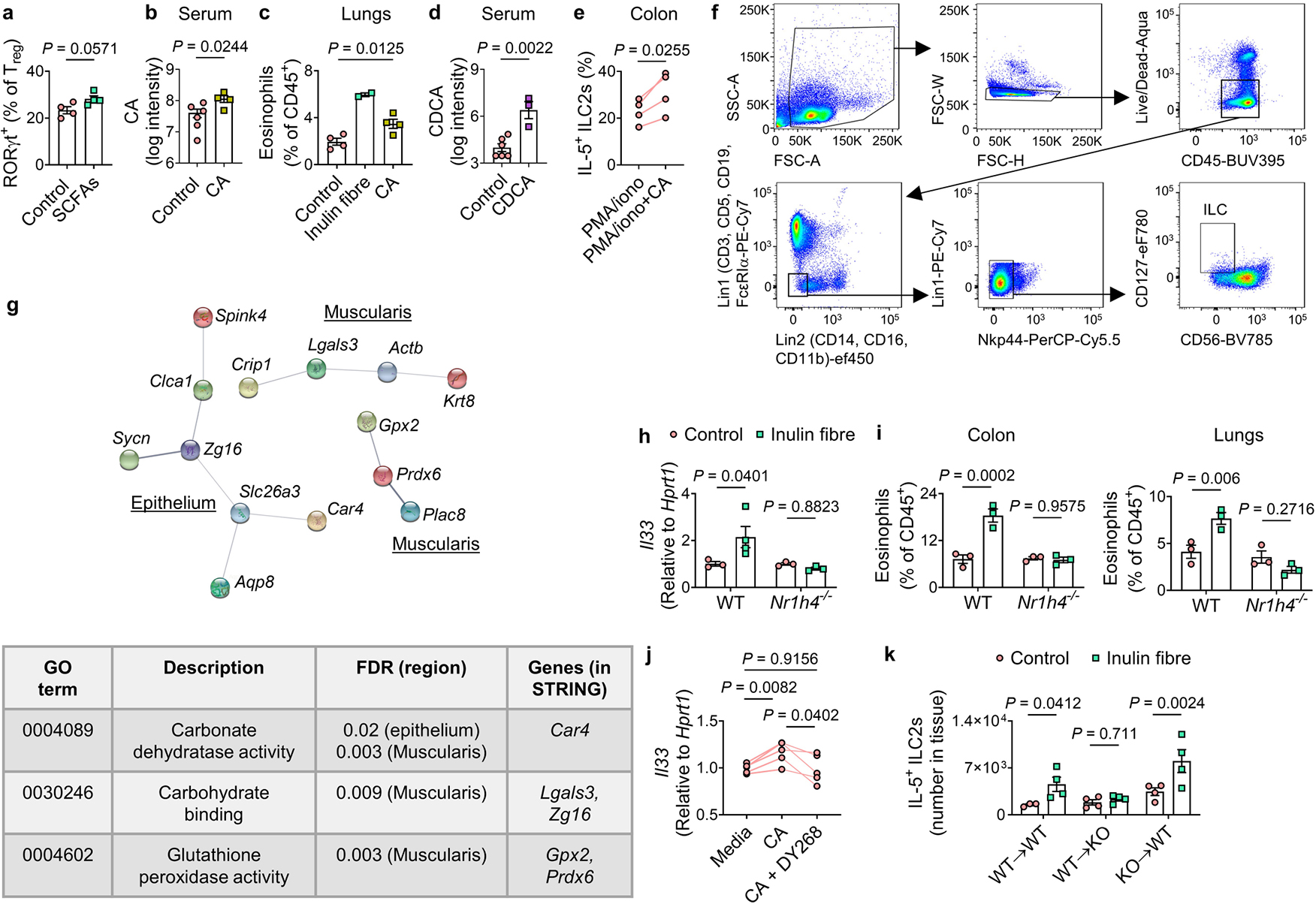
a, Frequency of RORγt+ subset of Tregs in the colons of mice administered SCFAs in drinking water, or water alone (control) (n = 4 mice). b, Serum levels of CA in mice provided with regular or CA-supplemented drinking water (control n = 6 mice, CA n = 4 mice). c, Frequency of eosinophils in the lungs of indicated groups (control n = 4 mice, inulin fibre n = 2 mice, CA n = 4 mice). d, Serum levels of CDCA in mice administered 6 mM CDCA in drinking water for two weeks (control n = 6 mice, CDCA n = 3 mice). e, Percentage of IL-5+ ILC2s in single cells from the colons of naïve WT mice in vitro stimulation with or without 50 μM CA (n = 4 mice). f, Gating strategy for detection of human ILCs (for bar graphs including Fig. 3f). g, Top, STRING network visualization of the genes upregulated by the inulin fibre diet (FDR < 10%) in the indicated layers of mouse colon determined by spatial transcriptomic analyses as shown in Fig. 3h. Lines represent protein–protein associations. Connected clusters of less than 3 nodes (genes), as well as all disconnected nodes were excluded. Bottom, a table shows selected significantly enriched KEGG pathways for the genes shown in the STRING network. h, Il33 expression in WT or Nr1h4−/− mice fed control or inulin fibre diet for two weeks (n = 4 mice for WT inulin fibre group and 3 mice for other groups). i, Frequency of eosinophils in the colons and lungs of WT and Nr1h4−/− mice on control or inulin fibre diet (n = 3 mice). j, Il33 levels determined by qRT-PCR in sorted stromal cells cultured for 7 days followed by stimulation for 24 h with media control or 50 μM CA with or without 10 μM DY268 (n = 5 mice). k, Number of IL-5-expressing ILC2s in the colon of bone marrow chimeric mice on indicated diets (For WT→WT, n = 3 mice for control or 4 mice for inulin fibre, For WT→Nr1h4−/−(KO) or KO→WT, n = 4 mice). Data in a–e, h–k are representative of two independent experiments. Data are means ± s.e.m. Statistics were calculated by Mann–Whitney U-test (a), unpaired (b,d) or paired (e, j) two-tailed t-test or one-way (c) or two-way (h,i,k) ANOVA with Fisher’s LSD test.
Extended Data Fig. 8 |. Metabolic and immunological parameters in gnotobiotic mice on inulin fibre diet.
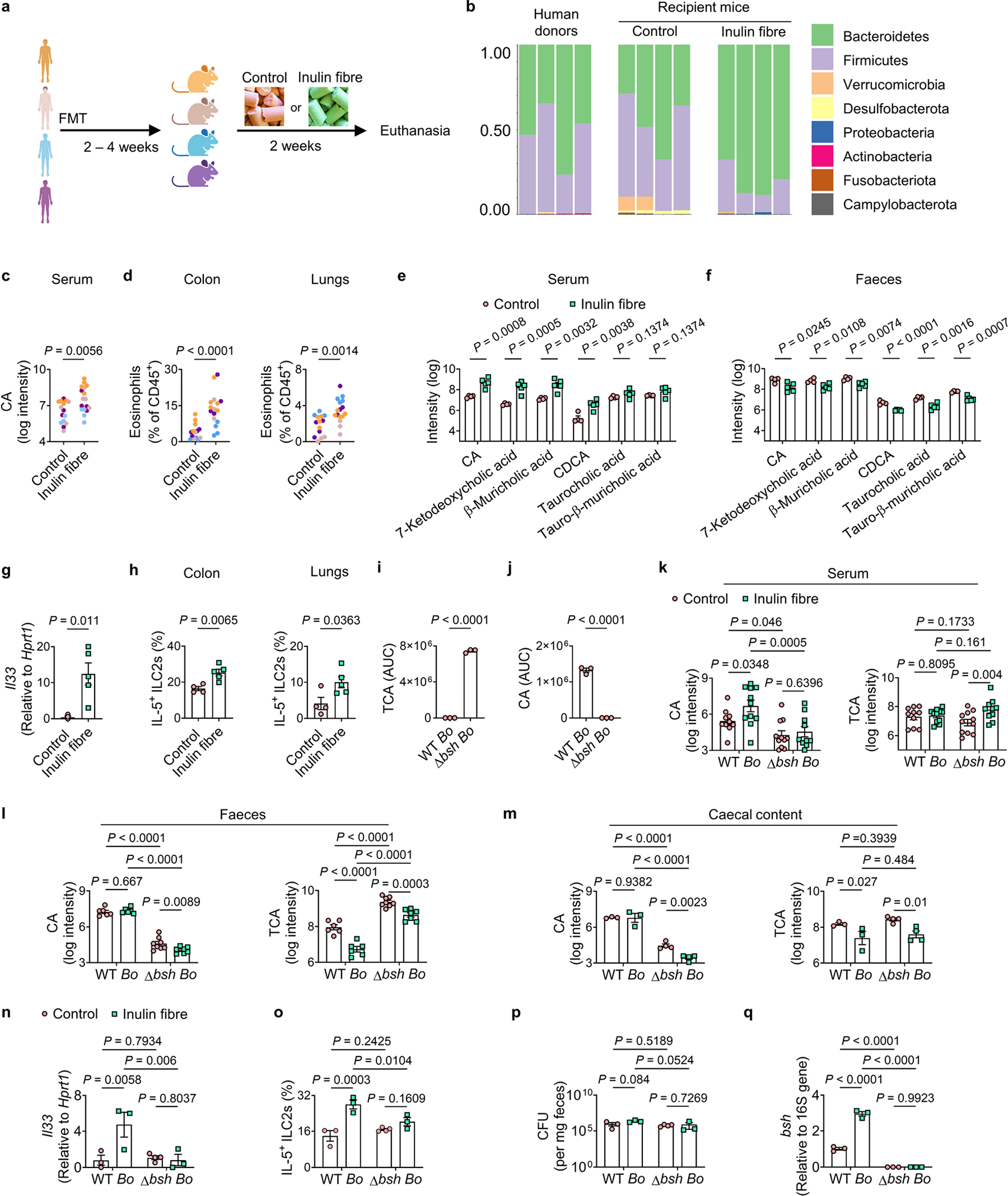
a, Schematic diagram for the FMT experiment with human microbiota. b, Taxonomic classification of 16S rRNA genes in faecal suspension from individual human donors or stool pellets collected from representative recipient mice with corresponding human microbiota on control or inulin fibre diet (n = 4 human donors or recipient mice). c–d, Serum CA (c) and tissue eosinophil (d) levels in the diet-fed recipient mice. Each dot represents one animal, and each colour represents one donor. For serum CA levels, n = 15 control mice or 17 inulin fibre mice. For tissue eosinophil levels, n = 14 control mice or 16 inulin fibre mice. e–h, Representative metabolic and immunological parameters of mice colonized with faecal content from one donor two weeks post initiation of the indicated diets (n = 4 mice for control or 5 mice for inulin fibre). Levels of various unconjugated and conjugated bile acid were measured in serum (e) and faeces (f). Il33 expression was measured in the colon (g) and IL-5+ ILC2s in colon and lung (h) two weeks post initiation of the indicated diets. i–j, Single colonies of WT or Δbsh B. ovatus (Bo) strains were cultured in Mega medium with 100 μM taurocholic acid (TCA) for 72 h and then TCA (i) and CA (j) in 100 μl culture supernatants were quantified, n = 3 independent bacterial colonies. k–q, GF mice were monocolonized with WT or Δbsh Bo and fed control or inulin fibre diet. CA and TCA were measured. For serum, n = 10 mice (WT Bo control or Δbsh Bo inulin fibre) or n = 11 mice (WT Bo inulin fibre or Δbsh Bo control). For faeces, n = 6 mice (WT Bo) or n = 8 mice (Δbsh Bo control) or n = 7 mice (Δbsh Bo inulin fibre), and for caecal content, n = 3 mice (WT Bo) or 4 mice (Δbsh Bo) (k–m). Il33 expression in the colon (n), IL-5+ ILC2s in the lung (o), and faecal CFUs (p) were measured (n = 3 mice for WT Bo, n = 4 mice for Δbsh Bo control, n = 3 mice for Δbsh Bo inulin fibre). Expression levels of bsh gene BO_02350 in faecal samples were also quantified (n = 3 mice) (q). Data are representative of (b–j,m–q) or combined (c,d,k,l) from 2–4 independent experiments. Data are means ± s.e.m. Statistics were calculated by unpaired two-tailed t-test (c–j) or two-way ANOVA with Fisher’s LSD test (k–q). The diagram in a was created using BioRender.
Extended Data Fig. 9 |. The effects of inulin fibre diet in mice challenged with HDM.
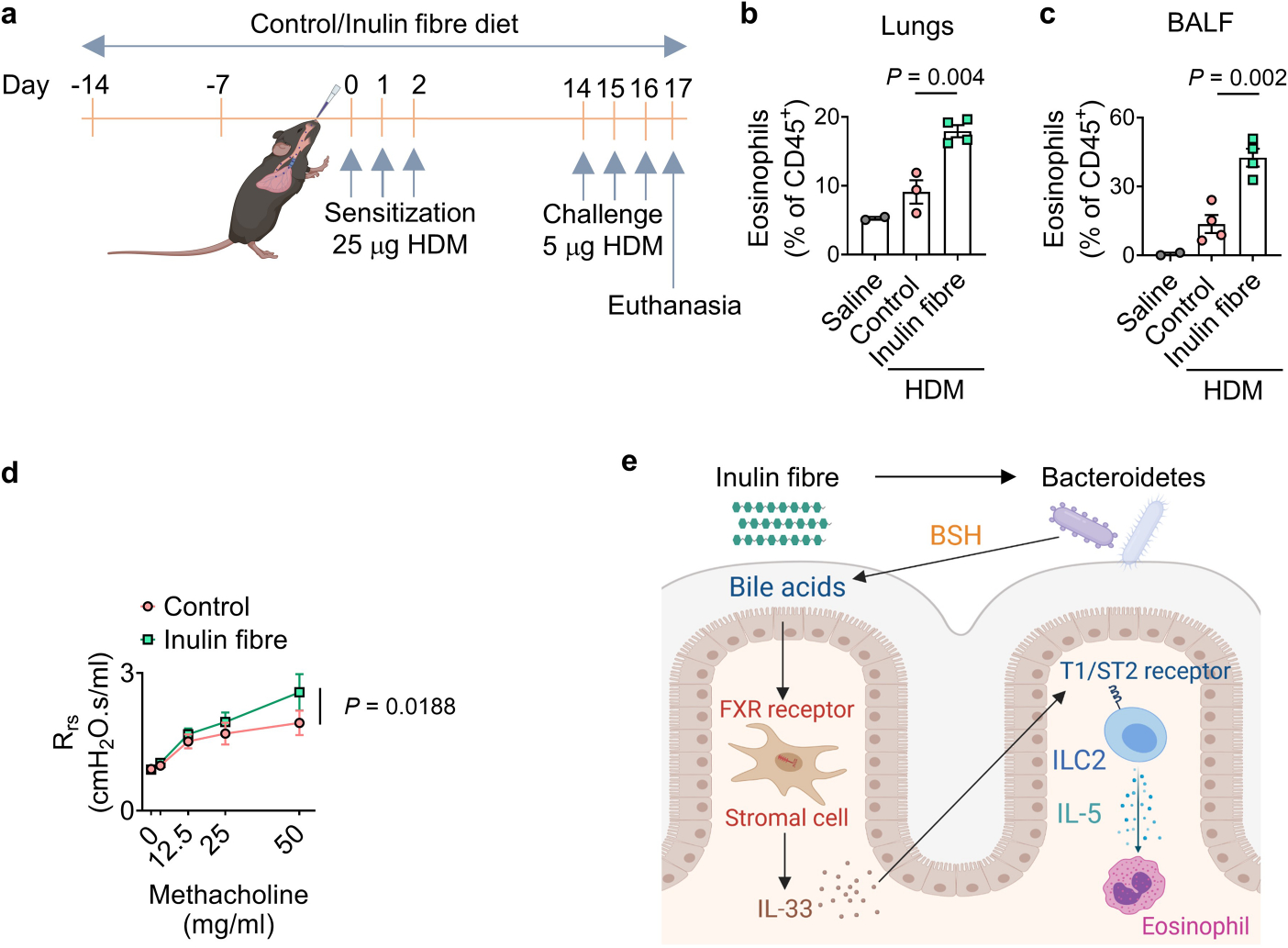
a, Schematic diagram for HDM model of allergic airway inflammation. b–d, Frequency of eosinophils in lung (b) and BALF (c) in naïve (saline) or HDM-challenged mice fed the control or inulin fibre diet (saline n = 2 mice, HDM control diet n = 3 mice for lung or 4 mice for BALF, HDM inulin fibre n = 4 mice). d, Airway hyperresponsiveness in HDM-challenged mice measured as resistance of the respiratory system (Rrs) to increasing doses of methacholine (n = 6 mice). Data are representative of (b–c) or combined (d) from 2 independent experiments. Data are means ± s.e.m. Statistics were calculated by unpaired two-tailed t-test (b–c) or two-way ANOVA with Fisher’s LSD test (d). e,Proposed model. The diagrams in a and e were created using BioRender.
Extended Data Table 1.
| Composition of diets used in this study
| Product # | Control diet D12450J-1.5V | Inulin fibre diet D16052309 | Cellulose fibre diet D20052606 | Psyllium fibre diet D20052607 | ||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| gm% | kcal% | gm% | kcal% | gm% | kcal% | gm% | kcal% | |
|
| ||||||||
| Protein | 19 | 20 | 16 | 20 | 14 | 20 | 14 | 20 |
| Carbohydrate | 67 | 70 | 47 | 70 | 49 | 70 | 48 | 70 |
| Fat | 4 | 10 | 4 | 10 | 3 | 10 | 3 | 10 |
|
| ||||||||
| kcal/gm | 100 | 100 | 100 | 100 | ||||
|
| ||||||||
| Ingredient | gm | kcal | gm | kcal | gm | kcal | gm | kcal |
|
| ||||||||
| Casein | 200 | 800 | 200 | 800 | 200 | 800 | 200 | 800 |
| L-Cystine | 3 | 12 | 3 | 12 | 3 | 12 | 3 | 12 |
| Com starch | 506.2 | 2025 | 383.25 | 1533 | 506.2 | 2025 | 506.2 | 2025 |
| Maltodextrin 10 | 125 | 500 | 125 | 500 | 125 | 500 | 125 | 500 |
| Sucrose | 63.8 | 255 | 63.8 | 255 | 63.8 | 255 | 63.8 | 255 |
| Cellulose BW200 | 50 | 0 | 50 | 0 | 430.7 | 0 | 50 | 0 |
| Inulin | 0 | 0 | 328 | 492 | 0 | 0 | 0 | 0 |
| Psyllium | 0 | 0 | 0 | 0 | 0 | 0 | 432 | 0 |
| Soybean oil | 25 | 225 | 25 | 225 | 25 | 225 | 25 | 225 |
| Lard | 20 | 180 | 20 | 180 | 20 | 180 | 20 | 180 |
| Mineral mix, SI0026 | 10 | 0 | 10 | 0 | 10 | 0 | 10 | 0 |
| Dicalcium phosphate | 13 | 0 | 13 | 0 | 13 | 0 | 13 | 0 |
| Calcium carbonate | 5.5 | 0 | 5.5 | 0 | 5.5 | 0 | 5.5 | 0 |
| Potassium citrate, 1 H2O | 16.5 | 0 | 16.5 | 0 | 16.5 | 0 | 16.5 | 0 |
| Vitamin mix, VI0001 | 15 | 60 | 15 | 60 | 15 | 60 | 15 | 60 |
| Choline bitartrate | 2 | 0 | 2 | 0 | 2 | 0 | 2 | 0 |
| Dye | 0.05 | 0 | 0.05 | 0 | 0.05 | 0 | 0.05 | 0 |
|
| ||||||||
| Total | 1055.05 | 4057 | 1260.1 | 4057 | 1435.75 | 4057 | 1487.05 | 4057 |
|
| ||||||||
| Total fibre (%) | 4.7 | 30 | 30 | 30 | ||||
Supplementary Material
Acknowledgements
We thank the members of the Artis laboratory for discussions and reading the manuscript; A. Alonso and other members of the Epigenomics Core and the Microbiome Core of Weill Cornell Medicine for spatial transcriptomic analyses and RNA-seq; all of the contributing members of the JRI IBD Live Cell Bank consortium, which is supported by the JRI, Jill Roberts Center for IBD, Cure for IBD, the Rosanne H. Silbermann Foundation, the Sanders Family and the Weill Cornell Medicine Division of Pediatric Gastroenterology and Nutrition. The cartoons and illustrations were created using BioRender. The chemical structures were created with ChemDraw. This work was supported by the Crohn’s & Colitis Foundation (851136 to M.A., 901000 to W.Z., 937437 to H.Y.); the Sackler Brain and Spine Institute Research (to C.C.); Thomas C. King Pulmonary Fellowship and Weill Cornell Medicine Fund for the Future (to C.N.P.); the WCM Department of Pediatrics Junior Faculty Pilot Award and The Jill Roberts Center Pilot Award for Research in IBD (to A.M.T.); AGA Research Foundation, WCM-RAPP Initiative, the W. M. Keck Foundation (to C.-J.G.); the Howard Hughes Medical Institute (to F.C.S.); the LEO foundation, CURE for IBD, the Jill Roberts Institute for Research in IBD, the Sanders Family Foundation and Rosanne H. Silbermann Foundation (to D.A.); and the National Institutes of Health (5T32HL134629 to C.N.P., DP2 HD101401–01 to C.-J.G., AI140724 to S.W., KL2 TR002385 to A.F.H., R35 GM131877 to F.C.S., and DK126871, AI151599, AI095466, AI095608, AR070116, AI172027 and DK132244 to D.A.).
Footnotes
Competing interests D.A. has contributed to scientific advisory boards at Pfizer, Takeda, FARE and the KRF. F.C.S. is a cofounder of Ascribe Bioscience and Holoclara. The other authors declare no competing interests.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-022-05380-y.
Peer review information Nature thanks Pieter Dorrestein and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Reprints and permissions information is available at http://www.nature.com/reprints.
JRI IBD Live Cell Bank Consortium
David Artis1,2,3, Randy Longman1,2,3, Gregory Sonnenberg1,2,3, Ellen Scherl1, Robbyn Sockolow6, Dana Lukin1, Robert Battat1, Thomas Ciecierega6, Aliza Solomon6, Elaine Barfield6, Kimberley Chien6, Johanna Ferreira6, Jasmin Williams1, Shaira Khan1, Peik Sean Chong1, Samah Mozumder1,2,3, Lance Chou1,2,3, Wenqing Zhou1,2,3, Anees Ahmed1,2,3, Connie Zhong1,2,3, Ann Joseph1,2,3, Sanchita Kashyap1,2,3, Joseph Gladstone1,2,3 & Samantha Jensen1,2,3
Data availability
All data necessary to understand and evaluate the conclusions of this paper are provided in the Article and its Source Data and Supplementary Information. The 16S rRNA-seq data are available at the NCBI Sequence Read Archive under accession number BioProject PRJNA761331. The RNA-seq and spatial transcriptomic data are available at Gene Expression Omnibus (GEO) under accession numbers GSE183443 and GSE183696, respectively. The mouse genome data (NCBI GRCm38/mm10) used for the alignment of RNA-seq data are available under accession number BioProject PRJNA20689. The MS1 and MS2 data for mouse samples analysed in this study are available at the GNPS website (https://massive.ucsd.edu) under MassIVE ID number MSV000086890. Source data are provided with this paper.
References
- 1.Brestoff JR & Artis D Commensal bacteria at the interface of host metabolism and the immune system. Nat. Immunol. 14, 676–684 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rooks MG & Garrett WS Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 16, 341–352 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robertson RC, Manges AR, Finlay BB & Prendergast AJ The human microbiome and child growth—first 1000 days and beyond. Trends Microbiol. 27, 131–147 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Fan Y & Pedersen O Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 19, 55–71 (2021). [DOI] [PubMed] [Google Scholar]
- 5.Belkaid Y & Hand TW Role of the microbiota in immunity and inflammation. Cell 157, 121–141 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blander JM, Longman RS, Iliev ID, Sonnenberg GF & Artis D Regulation of inflammation by microbiota interactions with the host. Nat. Immunol. 18, 851–860 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morais LH, Schreiber HLT & Mazmanian SK The gut microbiota–brain axis in behaviour and brain disorders. Nat. Rev. Microbiol. 19, 241–255 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Chu C et al. The microbiota regulate neuronal function and fear extinction learning. Nature 574, 543–548 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quinn RA et al. Global chemical effects of the microbiome include new bile-acid conjugations. Nature 579, 123–129 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wikoff WR et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl Acad. Sci. USA 106, 3698–3703 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maslowski KM et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461, 1282–1286 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arpaia N et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504, 451–455 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Furusawa Y et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Smith PM et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vinelli V et al. Effects of dietary fibers on short-chain fatty acids and gut microbiota composition in healthy adults: a systematic review. Nutrients 14, 2559 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chambers ES et al. Dietary supplementation with inulin-propionate ester or inulin improves insulin sensitivity in adults with overweight and obesity with distinct effects on the gut microbiota, plasma metabolome and systemic inflammatory responses: a randomised cross-over trial. Gut 68, 1430–1438 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Filippo C et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl Acad. Sci. USA 107, 14691–14696 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schnorr SL et al. Gut microbiome of the Hadza hunter-gatherers. Nat. Commun. 5, 3654 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thakare R, Alamoudi JA, Gautam N, Rodrigues AD & Alnouti Y Species differences in bile acids I. Plasma and urine bile acid composition. J. Appl. Toxicol. 38, 1323–1335 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Wahlstrom A, Sayin SI, Marschall HU & Backhed F Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 24, 41–50 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Klose CS & Artis D Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat. Immunol. 17, 765–774 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Saenz SA et al. IL25 elicits a multipotent progenitor cell population that promotes TH2 cytokine responses. Nature 464, 1362–1366 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klose CSN & Artis D Innate lymphoid cells control signaling circuits to regulate tissue-specific immunity. Cell Res. 30, 475–491 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klose CSN et al. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature 549, 282–286 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brestoff JR et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature 519, 242–246 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nussbaum JC et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature 502, 245–248 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flamar AL et al. Interleukin-33 induces the enzyme tryptophan hydroxylase 1 to promote inflammatory group 2 innate lymphoid cell-mediated immunity. Immunity 52, 606–619 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Halim TY et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 40, 425–435 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dahlgren MW et al. Adventitial stromal cells define group 2 innate lymphoid cell tissue niches. Immunity 50, 707–722 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahapatro M et al. Programming of intestinal epithelial differentiation by IL-33 derived from pericryptal fibroblasts in response to systemic infection. Cell Rep. 15, 1743–1756 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Mahlakoiv T et al. Stromal cells maintain immune cell homeostasis in adipose tissue via production of interleukin-33. Sci. Immunol. 4, eaax0416 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thomas C, Pellicciari R, Pruzanski M, Auwerx J & Schoonjans K Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Discov. 7, 678–693 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Campbell C et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 581, 475–479 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hang S et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature 576, 143–148 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song X et al. Microbial bile acid metabolites modulate gut RORγ+ regulatory T cell homeostasis. Nature 577, 410–415 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stahl PL et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Ridlon JM, Kang DJ & Hylemon PB Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 47, 241–259 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Singh V et al. Dysregulated microbial fermentation of soluble fiber induces cholestatic liver cancer. Cell 175, 679–694 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qin J et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yao L et al. A selective gut bacterial bile salt hydrolase alters host metabolism. eLife 7, e37182 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cruickshank-Quinn CI et al. Metabolomics and transcriptomics pathway approach reveals outcome-specific perturbations in COPD. Sci. Rep. 8, 17132 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim DJ et al. Metabolic fingerprinting uncovers the distinction between the phenotypes of tuberculosis associated COPD and smoking-induced COPD. Front. Med. 8, 619077 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Novey HS, Marchioli LE, Sokol WN & Wells ID Papain-induced asthma–physiological and immunological features. J. Allergy Clin. Immunol. 63, 98–103 (1979). [DOI] [PubMed] [Google Scholar]
- 44.Lajoie S et al. Complement-mediated regulation of the IL-17A axis is a central genetic determinant of the severity of experimental allergic asthma. Nat. Immunol. 11, 928–935 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Camberis M, Le Gros G & Urban J Jr. Animal model of Nippostrongylus brasiliensis and Heligmosomoides polygyrus. Curr. Protoc. Immunol. 55, 19.12.1–19.12.27 (2003). [DOI] [PubMed] [Google Scholar]
- 46.Janssen AWF et al. Modulation of the gut microbiota impacts nonalcoholic fatty liver disease: a potential role for bile acids. J. Lipid Res. 58, 1399–1416 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McLoughlin R et al. Soluble fibre supplementation with and without a probiotic in adults with asthma: a 7-day randomised, double blind, three way cross-over trial. EBioMedicine 46, 473–485 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karpievitch YV, Dabney AR & Smith RD Normalization and missing value imputation for label-free LC-MS analysis. BMC Bioinform. 13, S5 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Han H et al. IL-33 promotes gastrointestinal allergy in a TSLP-independent manner. Mucosal Immunol. 11, 394–403 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sinal CJ et al. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 102, 731–744 (2000). [DOI] [PubMed] [Google Scholar]
- 51.Yu C et al. Targeted deletion of a high-affinity GATA-binding site in the GATA-1 promoter leads to selective loss of the eosinophil lineage in vivo. J. Exp. Med. 195, 1387–1395 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ameri AH et al. IL-33/regulatory T cell axis triggers the development of a tumor-promoting immune environment in chronic inflammation. Proc. Natl Acad. Sci. USA 116, 2646–2651 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hsu CL, Neilsen CV & Bryce PJ IL-33 is produced by mast cells and regulates IgE-dependent inflammation. PLoS ONE 5, e11944 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Poeggeler B et al. Indole-3-propionate: a potent hydroxyl radical scavenger in rat brain. Brain Res. 815, 382–388 (1999). [DOI] [PubMed] [Google Scholar]
- 55.Rothhammer V et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 22, 586–597 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Friedrich M et al. IL-1-driven stromal-neutrophil interactions define a subset of patients with inflammatory bowel disease that does not respond to therapies. Nat. Med. 27, 1970–1981 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goc J et al. Dysregulation of ILC3s unleashes progression and immunotherapy resistance in colon cancer. Cell 184, 5015–5030 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Curtis JL, Byrd PK, Warnock ML & Kaltreider HB Requirement of CD4-positive T cells for cellular recruitment to the lungs of mice in response to a particulate intratracheal antigen. J. Clin. Invest. 88, 1244–1254 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cole JR et al. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42, D633–D642 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McMurdie PJ & Holmes S phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dodt M, Roehr JT, Ahmed R & Dieterich C FLEXBAR—flexible barcode and adapter processing for next-generation sequencing platforms. Biology 1, 895–905 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liao Y, Smyth GK & Shi W The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 47, e47 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yu G, Wang LG, Han Y & He QY clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Benjamini Y & Hochberg Y Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300 (1995). [Google Scholar]
- 68.Stuart T et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Szklarczyk D et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Helf MJ, Fox BW, Artyukhin AB, Zhang YK & Schroeder FC Comparative metabolomics with Metaboseek reveals functions of a conserved fat metabolism pathway in C. elegans. Nat. Commun. 13, 782 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tautenhahn R, Bottcher C & Neumann S Highly sensitive feature detection for high resolution LC/MS. BMC Bioinform. 9, 504 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schymanski EL et al. Identifying small molecules via high resolution mass spectrometry: communicating confidence. Environ. Sci. Technol. 48, 2097–2098 (2014). [DOI] [PubMed] [Google Scholar]
- 73.Carr K, Whiteley P & Shattock P Development and reproducibility of a novel high-performance liquid-chromatography monolithic column method for the detection and quantification of trans-indolyl-3-acryloylglycine in human urine. Biomed. Chromatogr. 23, 1108–1115 (2009). [DOI] [PubMed] [Google Scholar]
- 74.Letertre MPM et al. A targeted ultra performance liquid chromatography—tandem mass spectrometric assay for tyrosine and metabolites in urine and plasma: application to the effects of antibiotics on mice. J. Chromatogr. B 1164, 122511 (2021). [DOI] [PubMed] [Google Scholar]
- 75.Correia MSP, Lin W, Aria AJ, Jain A & Globisch D Rapid preparation of a large sulfated metabolite library for structure validation in human samples. Metabolites 10, 415 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang M et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 34, 828–837 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shannon P et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Salyers AA, Shoemaker N, Cooper A, D’Elia J & Shipman JS 8 genetic methods for Bacteroides species. Methods Microbiol. 29, 229–249 (1999). [Google Scholar]
- 79.Koropatkin NM, Martens EC, Gordon JI & Smith TJ Starch catabolism by a prominent human gut symbiont is directed by the recognition of amylose helices. Structure 16, 1105–1115 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lu Y, Yao D & Chen C 2-Hydrazinoquinoline as a derivatization agent for LC-MS-based metabolomic investigation of diabetic ketoacidosis. Metabolites 3, 993–1010 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data necessary to understand and evaluate the conclusions of this paper are provided in the Article and its Source Data and Supplementary Information. The 16S rRNA-seq data are available at the NCBI Sequence Read Archive under accession number BioProject PRJNA761331. The RNA-seq and spatial transcriptomic data are available at Gene Expression Omnibus (GEO) under accession numbers GSE183443 and GSE183696, respectively. The mouse genome data (NCBI GRCm38/mm10) used for the alignment of RNA-seq data are available under accession number BioProject PRJNA20689. The MS1 and MS2 data for mouse samples analysed in this study are available at the GNPS website (https://massive.ucsd.edu) under MassIVE ID number MSV000086890. Source data are provided with this paper.