

Abstract
Unscheduled increases in ploidy underlie defects in tissue function, premature aging, and malignancy. A concomitant event to polyploidization is the amplification of centrosomes, the main microtubule organization centers in animal cells. Supernumerary centrosomes are frequent in tumors, correlating with higher aggressiveness and poor prognosis. However, extra centrosomes initially also exert an onco‐protective effect by activating p53‐induced cell cycle arrest. If additional signaling events initiated by centrosomes help prevent pathology is unknown. Here, we report that extra centrosomes, arising during unscheduled polyploidization or aberrant centriole biogenesis, induce activation of NF‐κB signaling and sterile inflammation. This signaling requires the NEMO‐PIDDosome, a multi‐protein complex composed of PIDD1, RIPK1, and NEMO/IKKγ. Remarkably, the presence of supernumerary centrosomes suffices to induce a paracrine chemokine and cytokine profile, able to polarize macrophages into a pro‐inflammatory phenotype. Furthermore, extra centrosomes increase the immunogenicity of cancer cells and render them more susceptible to NK‐cell attack. Hence, the PIDDosome acts as a dual effector, able to engage not only the p53 network for cell cycle control but also NF‐κB signaling to instruct innate immunity.
Keywords: centrosome, immunosurveillance, NF‐κB, PIDDosome, sterile inflammation
Subject Categories: Cancer, Cell Cycle, Immunology
Centrosome amplification downstream of polyploidization can drive sterile inflammation and NK‐cell attack via NEMO‐PIDDosome‐dependent NF‐kB signaling.
Introduction
Polyploidization, the acquisition of multiple copies of the genome, is developmentally programmed in certain cell types found in highly differentiated tissues such as the liver, bone marrow, or heart. However, ploidy increases are also a critical part of the cellular response to both external and internal insults. Physiologic and pathologic conditions such as hypoxia, viral infection, or aging, can induce polyploidization by different processes such as cell fusion or endomitosis. (Rosenstiel et al, 2008; Lee et al, 2009; Lopez‐Sanchez et al, 2014, 2017). Polyploidy is also a characteristic alteration in premalignant lesions that can drive genetic instability and aneuploidy in tumors. According to different pan‐cancer analyses, up to 37% of human cancers undergo whole‐genome duplication (WGD) during the transformation process (Zack et al, 2013; Bielski et al, 2018). To preserve genomic integrity during genome duplication, different cell cycle checkpoints protect it (Gordon et al, 2012), but chromosomal content is also subjected to indirect control by the immune system. Different innate and adaptive immune effectors contribute to recognizing and eliminating hyperploid cancer cells (Aranda et al, 2018). Among them, natural killer (NK) cells play a critical part in the early stages of tumor prevention, as evidenced by the correlation of higher cancer susceptibility with defects in NK cell function (Guerra et al, 2008; Lopez‐Soto et al, 2017b). NK cell activity against tumors is independent of HLA recognition and enforced by inflammatory cues produced by innate immune cells such as dendritic cells and macrophages (Maghazachi et al, 1994; Mantovani et al, 2022). Different events associated with aneuploidy, such as ER stress, DNA damage, or cytosolic DNA detection by the cGAS/STING axis, have been implicated in the immunogenicity of cancer cells after unscheduled polyploidization (Ishikawa & Barber, 2008; Senovilla et al, 2012; Santaguida et al, 2017). However, it is presently unclear whether integral aspects of polyploidization itself could be responsible for the increased immune recognition of these cells.
A hallmark of polyploidization is the concomitant increase of centrosome number. In mammalian cells, centrosomes act as microtubule organizing centers (MTOCs) that orchestrate mitotic spindle formation, ciliogenesis, or immune synapse formation in effector cells (Tanos et al, 2013; Yi et al, 2013; Ritter et al, 2015; Dieckmann et al, 2016; Nigg & Holland, 2018). Centrosomes comprise two centrioles, defined as mature and immature, assembled in a cloud of pericentriolar matrix proteins that allows microtubule nucleation. Centrosome amplification is a common feature of many solid and hematological tumors that correlates with their aggressiveness (D'Assoro et al, 2002; Nigg, 2006; Chan, 2011). Centrosome amplification can lead to chromosome missegregation and promote chromosomal instability (CIN) and aneuploidy (D'Assoro et al, 2002; Ganem et al, 2009; Silkworth et al, 2009). Moreover, extra centrosomes have been proposed to increase the metastatic potential of tumor cells, facilitating paracrine invasion (Godinho et al, 2014; Arnandis et al, 2018; Ganier et al, 2018; LoMastro & Holland, 2019). In mice and flies, centrosome amplification by overexpression of Polo‐like kinase 4 (PLK4), a key‐regulator of centriole biogenesis, suffices to induce spontaneous tumors with complex karyotypes that resemble those observed in humans (Basto et al, 2008; Coelho et al, 2015; Levine et al, 2017). These reports suggest that extra centrosomes can be active drivers of tumorigenesis and not just a consequence of the transformation process.
Nonetheless, supernumerary centrosomes can also initially exert anti‐oncogenic effects. During early transformation, (pre‐)malignant cells are subjected to intrinsic onco‐suppressive mechanisms leading to the limitation or removal of genomic abnormalities by either permanent cell cycle arrest (senescence) or programmed cell death. Limited experimental evidence has been provided connecting aberrant centrosome number and the observed block in proliferation (Hachet et al, 2007), but a prominent role of these structures triggering p53 pathway activation has emerged in the last decade (Holland et al, 2012; Bazzi & Anderson, 2014; Lambrus et al, 2016). We recently identified the PIDDosome multi‐protein complex as the missing link between extra centrosomes and the p53 network. The core protein of this complex, p53‐induced death‐domain protein 1 (PIDD1), interacts with the mature centriole distal appendage protein ANKRD26 and patrols their numbers (Burigotto et al, 2021; Evans et al, 2021). The presence of extra centrosomes suffices to trigger PIDDosome pathway activation, resulting in caspase‐2‐dependent MDM2 cleavage, p53 stabilization, and p21‐mediated cell cycle arrest (Fava et al, 2017).
PIDDosome formation was originally described under genotoxic stress conditions, and in this scenario, two PIDD1‐containing complexes with potential antagonistic functions were characterized: the Caspase‐2‐ and the NEMO‐PIDDosome (Janssens & Tinel, 2012). The ability of PIDD1 to ensemble these different complexes relies on its constitutive intein‐like autoproteolysis that sequentially originates different active fragments. A first cleavage generates two halves: PIDD1‐N, with currently no clear function assigned, and PIDD1‐C, containing a C‐terminal death domain (DD). Additional autocleavage of the PIDD1‐C fragment can yield a shorter C‐terminal fragment, dubbed PIDD1‐CC (Tinel & Tschopp, 2004; Sladky et al, 2017). Although PIDD1‐CC recruits the dual adaptor RAIDD, needed for caspase‐2 binding and activation, PIDD1‐C was proposed to interact with receptor‐interacting protein kinase 1 (RIPK1) and NF‐κB essential modulator (NEMO/IKKγ) to promote NF‐κB activation and facilitate DNA repair and cell survival (Tinel et al, 2007, 2011). Further studies reported that PIDD1 could act as a molecular switch between cell survival and death, which may be flipped by ATM phosphorylation (Ando et al, 2012). However, despite these initial findings, the capacity of PIDD1 to activate the NF‐κB network and promote cell survival could not yet be placed into a firm physiological or pathological context (Bock et al, 2013; Weiler et al, 2022).
Here, we combined RNA‐sequencing, live‐cell imaging, and genome editing to establish that PIDD1 activates NF‐κB in response to supernumerary centrosomes. Moreover, we show that this activation requires interaction with RIPK1 and NEMO and confirm the formation of this complex in cells that fail cytokinesis. Our results provide the first evidence that PIDD1 mediates sterile inflammation in cells with extra centrosomes. This signaling can induce macrophage polarization into a pro‐inflammatory phenotype and increases cancer cell–intrinsic immunogenicity, rendering them more susceptible to NK cell–driven elimination. We conclude that extra centrosomes act as signaling hubs that elicit not only cell cycle arrest but also auto‐ and paracrine inflammatory responses, placing the NEMO‐PIDDosome next to other inflammasomes in its ability to instruct innate immunity.
Results
Polyploidization elicits an NF‐κB‐driven transcriptional program and sterile inflammation
To gain insight into the potential consequences of unscheduled polyploidization, we analyzed the transcriptional profile of cells undergoing cytokinesis failure. For this aim, primary mouse embryonic fibroblasts (MEF) were treated with the Aurora kinase inhibitor ZM447439 (referred to as ZM hereafter) and subjected to 3′ mRNA sequencing, comparing ZM vs. DMSO treatments. Aurora kinase inhibition induced a significant number of differentially expressed genes (DEGs), most of which presented recognizable binding motifs for RELA and/or p53 in their promoter region (Fig 1A and B). Further prediction analysis performed using the TRRUST database (transcriptional regulatory relationships unraveled by sentence‐based text mining) also suggested that in addition to TP53, RELA, and NFκB1, other transcription factors involved in cell cycle regulation, such as E2F1, E2F3, E2F4, as well as STAT1 and STAT2, the latter two tightly related to innate immune responses, were likely regulating the DEGs in ZM treated MEF (Fig EV1A).
Figure 1. Aurora kinase inhibition elicits an inflammatory and immunoregulatory response.
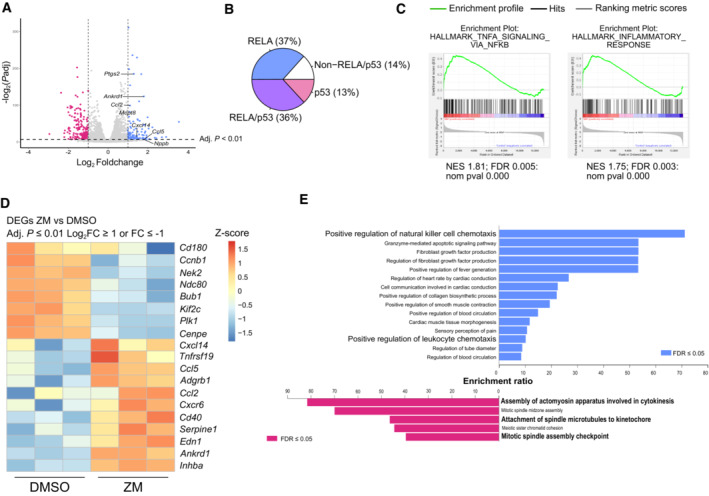
- Volcano plot representation of differentially expressed genes (DEGs) between ZM and DMSO treatment. Adjusted P value ≤ 0.01 and log2 FC ≥ 1 or log2 FC ≤ −1.
- Pie chart of DEGs presenting consensus binding sequences for p53 and/or RELA transcription factors, in their promotor regions. Adjusted P value ≤ 0.05.
- Gene set enrichment analysis (GSEA) analysis of transcripts differentially regulated between ZM and DMSO treatment.
- Curated heatmap of significant DEGs involved in inflammatory signaling or cell cycle control. Adjusted P value ≤ 0.01 and log2 FC ≥ 1 or log2 FC ≤ −1.
- Gene Ontology (GO) term analysis for significant DEGs for ZM vs DMSO stimulated cells. FDR ≤ 0.05, adjusted P ≤ 0.01, and cutoff values log2 FC ≥ 1 or log2 FC ≤ −1.
Figure EV1. Aurora kinase inhibition regulates transcriptional pathways that promote inflammation and immune recognition.
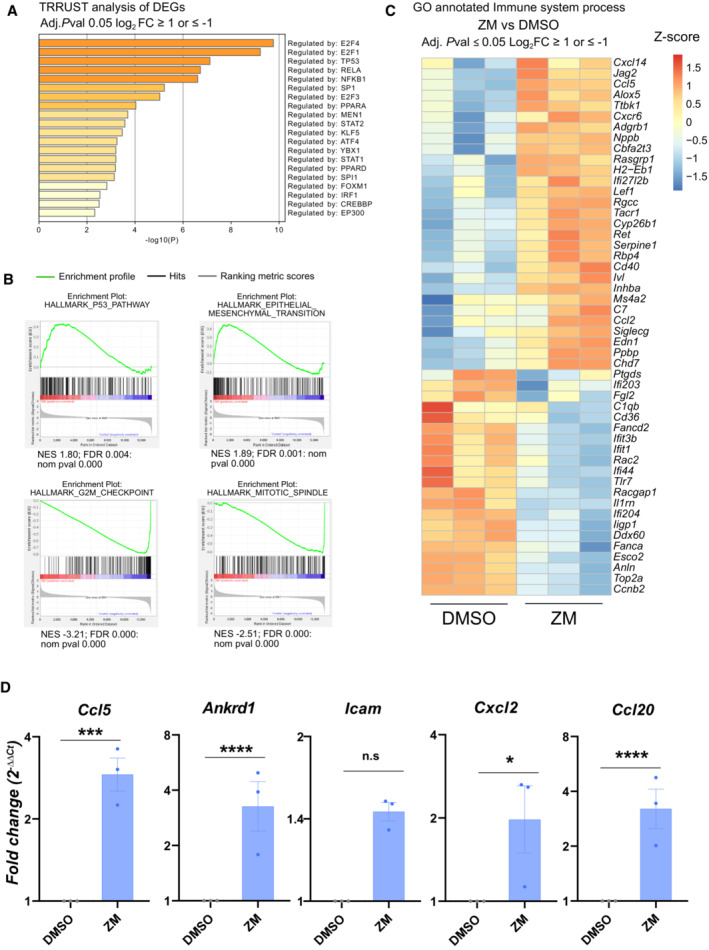
- TRRUST analysis of significant DEGs in ZM vs. DMSO treated cells displaying transcription factors predicted to participate in their regulation. Adjusted P ≤ 0.05 and cut‐off values log2 FC ≥ 1 or log2 FC ≤ −1.
- Gene set enrichment analysis (GSEA) analysis of transcripts differentially regulated between ZM and DMSO treatment.
- Curated heatmap for significant DEGs involved in the GO‐term “immune system process” (GO: 0002376) that are enriched in ZM vs. DMSO treated MEF. Adjusted P ≤ 0.05 and cut‐off values log2 FC ≥ 1 or log2 FC ≤ −1.
- qRT‐PCR analysis of selected pro‐inflammatory cytokines and chemokines induced in ZM‐treated MEF, plotted as fold‐change expression relative to untreated cells. Hprt1, β‐actin and Gapdh were used as house‐keeping gene expression controls. n = 3 biological replicates; data represent mean values ± SEM. The statistical significance was determined using two‐tailed t‐test; P ≤ 0. 1 (*), P ≤ 0.01 (**), P ≤ 0.001 (***), P ≤ 0.0001 (****).
We explored the magnitude of transcriptional changes by performing gene set enrichment analysis (GSEA) and observed that genes related to NF‐κB, p53, and inflammatory responses were consistently upregulated upon ZM treatment. In contrast, genes associated with G2M checkpoint and mitotic spindle regulation were decreased, being congruent with previous work (Andreassen et al, 2001; Ganem & Pellman, 2007; Figs 1C and EV1B). In support of an inflammatory and immunoregulatory response, we next compared the significant DEGs to a selected list of genes associated with inflammation and the cell cycle (Fig 1D) and generated a curated heatmap of immune system–associated genes derived from the GO term “immune system process” enriched in our RNA‐seq dataset (Fig EV1C). In addition, gene ontology (GO) analysis of significant DEGs pointed out that terms related to immune cell attraction were enriched in ZM‐treated cells (Fig 1E). Finally, a set of genes related to leukocyte chemotaxis and inflammatory response was subsequently also validated by RT‐qPCR (Fig EV1D). Taken together, these analyses suggest that acute polyploidization induced by Aurora kinase inhibition, next to mounting a recognizable anti‐proliferative response, also upregulates the NF‐κB pathway and induces signs of sterile inflammation.
PIDD1 promotes NF‐κB signaling upon cytokinesis failure
NF‐κB regulates multiple aspects of innate and adaptive immunity and plays an essential role in inflammation (Karin et al, 2002; Muthuswamy et al, 2012; Park & Hong, 2016; Taniguchi & Karin, 2018). To substantiate our RNA‐seq findings, we generated stable A549 lung adenocarcinoma cells containing a NF‐κB reporter (A549κB) where expression of destabilized EGFP is driven by a minimal CMV promoter including four NF‐κB binding elements. Aiming to exclude a possible off‐target effect of Aurora inhibition unrelated to polyploidization, cytokinesis was also perturbed by interfering with actomyosin ring contraction using dihydrocytochalasin‐B (DHCB). Induction of polyploidization led to a significant increase of NF‐κB reporter activity in A549κB cells, tracked individually by time‐lapse live‐cell imaging (Fig 2A; Movies [Link], [Link]). These findings prompted us to further investigate NF‐κB activation using alternative readouts in unmodified cells.
Figure 2. Cytokinesis failure induces NF‐κB activation via PIDD1.
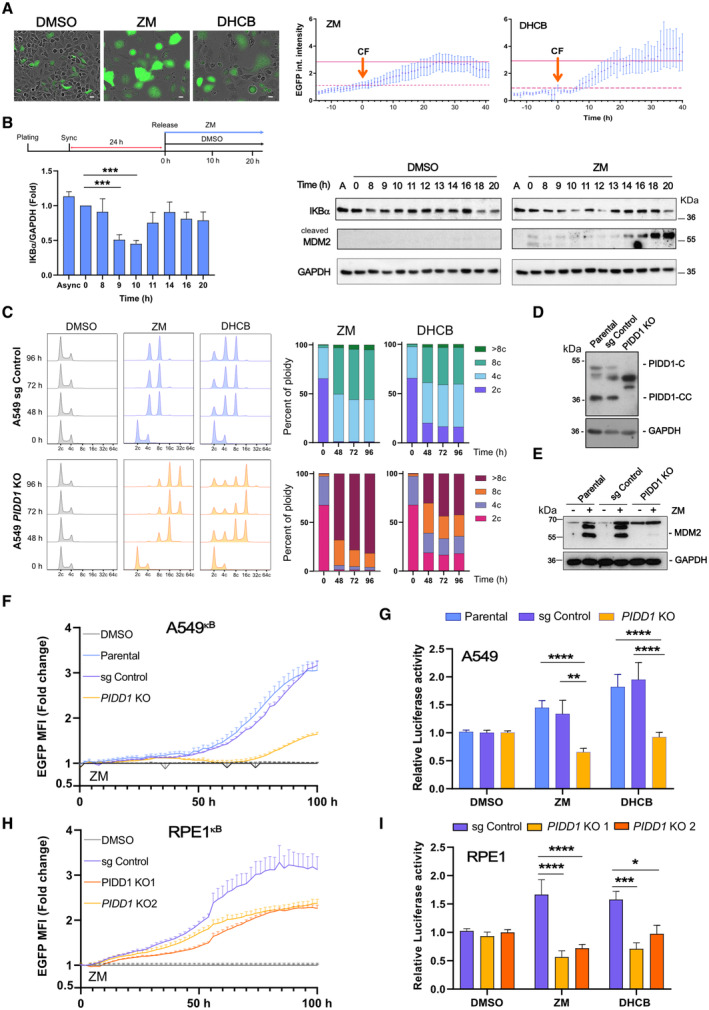
-
ATime‐lapse analysis of NF‐κB‐driven EGFP reporter activity in A549kB cells where cytokinesis failure was induced by treatment with ZM (2 μM) or DHCB (4 μM). Single cells were imaged every 20 min using IncuCyte life cell imaging technology. Left, representative images of A549kB cells 48 h after starting the treatment. Scale bars: 10 μm. See also Movies [Link], [Link]. Right, EGFP integrated fluorescence intensity of individually tracked cells was measured over time. Time zero was determined as the moment of cytokinesis failure (CF) of each individual cell analyzed. Dashed lines indicate the integrated EGFP intensity of the cells at time zero that was used to calculate the fold‐change over time. Solid lines indicate the mean EGFP integrated fluorescence intensity of the cells 24 h after failing cytokinesis. An n ≥ 10 cells per condition were followed individually in a single experiment, data points represent means ± SEM.
-
BTop, experimental design. Left, densitometric analysis of IKBα protein levels in synchronized A549 cells detected over time after release. Bars represent ZM values normalized with respect to DMSO. n = 3 independent experiments, data represent mean values ± SEM. Right, time course immunoblot analysis of synchronized A549 cells, released in DMSO or ZM (2 μM).
-
CFlow cytometric analysis of DNA content in A549 cells after induction of cytokinesis failure with ZM (2 μM) or DHCB (4 μM) over time. Two independent experiments were performed.
-
D, E(D) Western blot analysis confirming lack of PIDD1 expression in A549 cells and (E) MDM2 processing after induction of cytokinesis failure using ZM (2 μM) for 48 h.
-
FLive‐cell imaging analysis of NF‐κB EGFP‐reporter activity in CRISPR‐edited polyclonal A549κB cells. NF‐κB activity of ZM‐treated cells is represented as fold‐change with respect to their untreated counterparts (DMSO‐treated cells, gray lines). n = 5 independent experiments. Error bars represent ± SEM.
-
GLuciferase‐based reporter assay of NF‐κB activity in CRISPR‐edited polyclonal PIDD1‐competent or –deficient A549 cells. Cells were transfected with a firefly 2x(κB)‐luciferase reporter and treated with ZM (2 μM) or DHCB (4 μM) for 48 h to induce cytokinesis failure. Luciferase activity was normalized respective a co‐transfected renilla control. n ≥ 3 experiments, bars represent means ± SEM.
-
HLive‐cell imaging analysis of NF‐κB EGFP‐reporter activity in CRISPR‐edited polyclonal RPE1kB cells analyzed as in as in F.
-
IAnalysis of CRISPR‐edited polyclonal PIDD1‐competent or deficient RPE1 cells, analyzed for luciferase reporter activity as in G. n ≥ 5 experiments; bars represent mean values ± SEM.
Data information: The statistical significance was determined using two‐way ANOVA; P ≤ 0. 1 (*), P ≤ 0.001 (***), P ≤ 0.0001 (****).
Source data are available online for this figure.
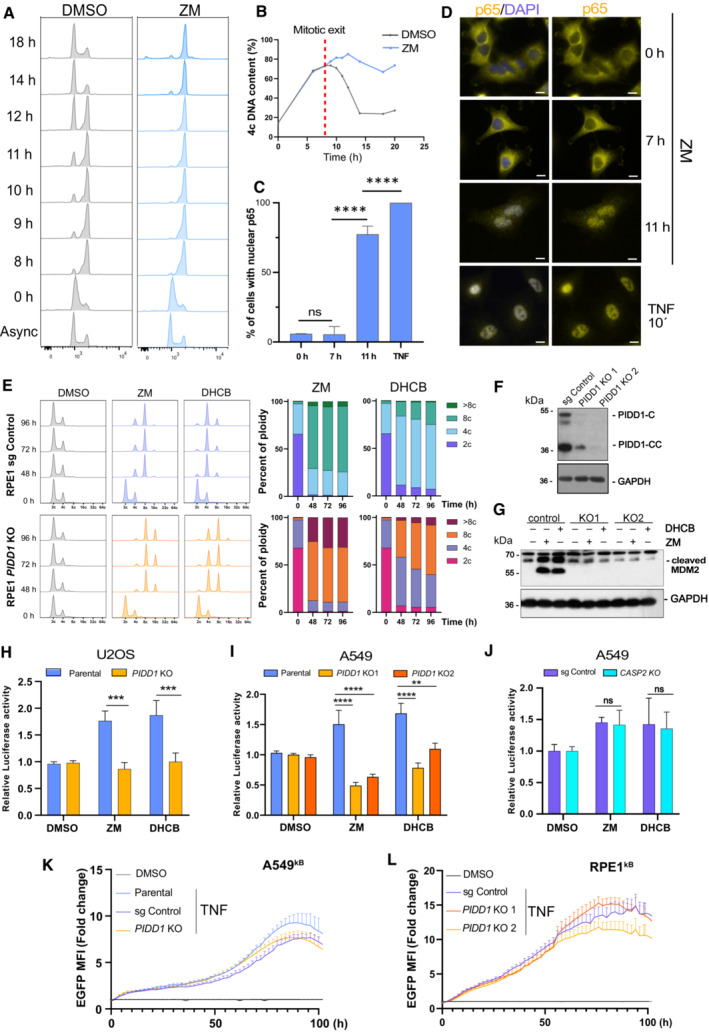
We reasoned that cytokinesis failure would define the cue driving NF‐κB activation. Hence, to obtain a more readily detectable signal, A549 cells were synchronized at the G1/S phase boundary prior to their release into DMSO or Aurora kinase inhibitor (Fig 2B). Polyploid cells first appeared between 9 and 11 h after ZM treatment, as revealed by flow cytometric DNA content analysis, when comparing ZM‐ with DMSO treatment (Fig EV2A and B). Of note, degradation of IKBα protein was found to be most pronounced between 9 and 10 h after release into ZM, supporting the idea that NF‐κB is activated after cytokinesis failure (Fig 2B). Likewise, a sharp increase in p65‐positive nuclei was also observed 11 h after release into ZM, but not at earlier times (Fig EV2C and D). We previously reported that failure in cytokinesis induces a cell cycle arrest response driven by the PIDDosome, enabling caspase‐2‐mediated MDM2 processing and p53 stabilization. Analysis of MDM2 protein levels showed a detectable accumulation of its cleavage product starting around 13 h after release into ZM (Fig 2B), in agreement with previous observations (Fava et al, 2017). Altogether, our findings suggest an orchestrated and potentially sequential activation of NF‐κB and p53 signaling pathways in cells failing cytokinesis that motivated us to investigate the role PIDD1 in this process.
Figure EV2. Cytokinesis failure induces p65 nuclear translocation and NF‐κB activation via PIDD1.
-
AFlow cytometric DNA content analysis of single‐thymidine synchronized A549 cells, released in DMSO or ZM.
-
BQuantification of the percentage of polyploid cells of samples shown in (A). Red dashed line indicates the beginning of mitotic exit in DMSO control cells.
-
CNuclear translocation of p65/RELA was quantified over time and is represented as percentage of cells displaying nuclear p65 signal (n ≥ 50 cells per condition). Bars represent means ± SEM of two technical replicates.
-
DRepresentative immunofluorescence images of synchronized A549 cells released in ZM and analyzed at different time points by IF. Cells were incubated with TNF (50 ng/ml) for 10 min, as a positive control. Scale bars: 10 μm.
-
EFlow cytometric analysis of DNA content in RPE1 cells of the indicated genotypes after induction of cytokinesis failure with ZM (2 μM) or DHCB (4 μM) compared to DMSO. Two independent experiments were performed.
-
F, G(F) Western blot analysis confirming lack of PIDD1 expression and (G) impaired MDM2 processing in RPE1 cells lacking PIDD1 after induction of cytokinesis failure using ZM (2 μM) for 48 h.
-
H–J(H) Luciferase reporter assay of NF‐κB activity in U2OS single cell clone lacking PIDD1 or (I) A549 single cell clones generated with two different guide RNAs targeting PIDD1 or (J) lacking caspase‐2. n ≥ 3 independent biological experiments, mean ± SEM.
-
K, L(K) Live‐cell imaging analysis of NF‐κB EGFP‐reporter activity in CRIPSR‐edited polyclonal PIDD1‐competent or –deficient A549κB cells or (L) RPE1 cells treated with TNF (20 ng/ml). n ≥ 3 independent experiments; bars represent mean values ± SEM.
Data information: The statistical significance was determined using two‐way ANOVA; P ≤ 0.0 1 (**), P ≤ 0.001 (***), P ≤ 0.0001 (****).
Cell treatment with either ZM or DHCB led to a substantial ploidy increase that, as expected, was enhanced in PIDD1‐deficient A549 or U2OS cells (Fava et al, 2017), which failed to cleave MDM2 and reached a DNA content of 32C or more (Fig 2C–E; Appendix Fig S1). Induction of cytokinesis failure revealed a steady increase in NF‐κB‐reporter activity over time, which was indistinguishable between both parental and CRISPR control A549κB cells but clearly reduced in PIDD1 knockout derivatives (Fig 2F). Similar findings were observed for ZM and DHCB treatments using a luciferase‐based NF‐κB reporter assay (Fig 2G). Importantly, PIDD1‐dependent NF‐κB activation was not restricted to A549 cells but also detected in retinal pigment epithelial (RPE1) cells and U2OS osteosarcoma cells (Figs 2H and I, and EV2E–H). Two different sgRNAs targeting PIDD1 led to the same results upon forced polyploidization, but the lack of PIDD1 did not affect canonical NF‐κB activation by TNF (Fig EV2I, K and L). We also noted that loss of caspase‐2 did not affect the induction of NF‐κB after cytokinesis failure (Fig EV2J), suggesting the involvement of alternative PIDD1 interactors. Collectively, these data indicate that unscheduled increases in ploidy induce NF‐κB activation via PIDD1‐dependent signaling. Nonetheless, the initiating cue of this response connecting PIDD1 to NF‐κB activation remained undefined at this stage.
PIDD1 mediates NF‐kB activation in response to supernumerary centrosomes
An inherent feature of polyploidiztion is the accumulation of supernumerary centrosomes (Holland & Cleveland, 2009), reported by us to activate the PIDDosome (Fava et al, 2017). Thus, we wondered whether extra centrosomes could initiate the observed NF‐κB signaling. To address this question, we subjected A549κB cells to centrinone treatment, a PLK4 inhibitor able to block centriole duplication (Wong et al, 2015), and subsequently induced cytokinesis failure. As shown by γ‐tubulin staining, centrinone treatment caused centriole depletion in about half of cells within 24 h, without blocking cellular ploidy increases after ZM treatment (Figs 3A and EV3A). Live‐cell imaging analysis revealed that pretreatment with centrinone prevented a significant NF‐κB activation upon cytokinesis failure, but had no impact on TNF‐induced reporter activity (Fig 3B). These results show that blocking centrosome duplication blunts NF‐κB‐reporter activity, implying that the presence of supernumerary centrosomes, and not the increase in ploidy, triggers NF‐κB activation in cells that fail cytokinesis.
Figure 3. PIDD1 mediates NF‐κB activation in response to supernumerary centrosomes § .
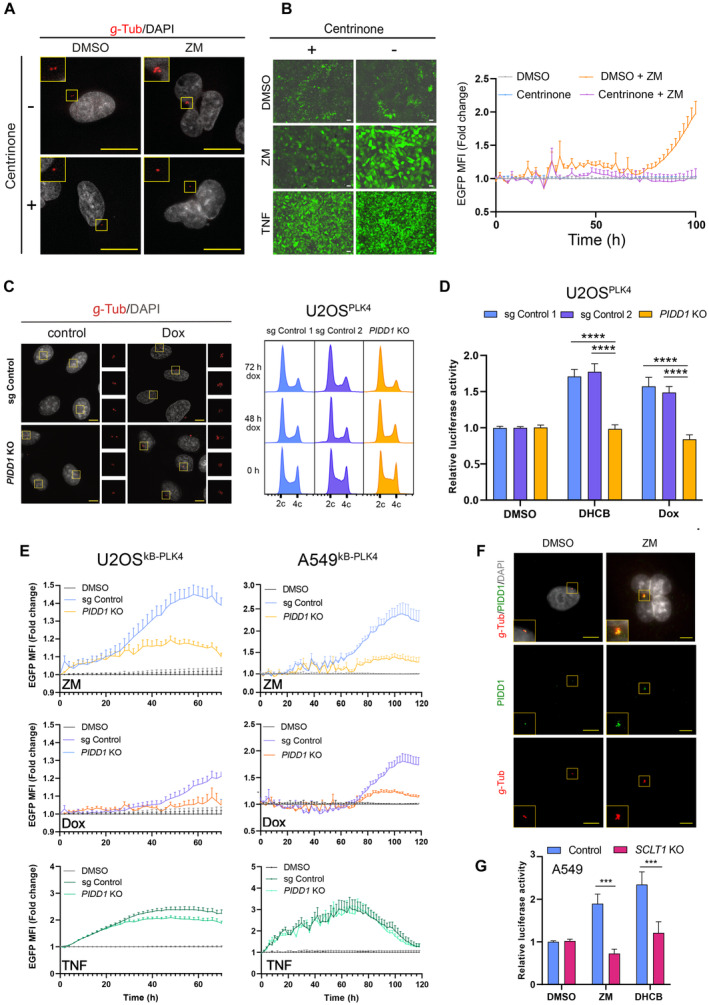
- Representative immunofluorescence images of γ‐tubulin staining to mark and enumerate centrosomes (Fig EV3A) in A549κB reporter cells. Cells were pre‐treated for 24 h with centrinone (125 nM) or solvent control (DMSO) and cultured further in medium either containing DMSO or ZM to promote cytokinesis failure. Size bar: 10 μM.
- Cells treated as indicated in A were analyzed for NF‐κB‐driven EGFP‐reporter activation by IncuCyte live‐cell imaging. Left, representative pictures of live‐cell imaging NF‐κB activation after treatment with DMSO, ZM (2 μM, 48 h) or TNF (20 ng/ml, 24 h). Size bar: 20 μm. Right, EGFP MFI quantification of ZM‐treated cells was plotted as fold‐change expression relative to DMSO‐treated controls. Data points represent means of n = 4 biological replicates ± SEM.
- Left, representative immunofluorescence images of supernumerary centrosomes originated from Dox‐inducible PLK4 overexpression in CRISPR‐edited polyclonal U2OSPLK4 cells. Size scale bar: 10 μm. Right, representative cell cycle profiles of U2OSPLK4 control and PIDD1‐deficient cells.
- Luciferase reporter assay of NF‐κB activity in CRISPR‐edited polyclonal PIDD1‐competent or –deficient U2OSPLK4 cells treated with DHCB (4 μM, 48 h) or Dox (1 μg/ml, 72 h). n ≥ 6 independent experiments; mean values ± SEM.
- Live‐cell imaging analysis of NF‐κB EGFP‐reporter activity in CRISPR‐edited polyclonal PIDD1‐competent or ‐deficient U2OSκB‐PLK4 and A549κB‐PLK4 cells. NF‐κB activity of ZM, Dox or TNF‐treated cells is represented as fold‐change with respect to their untreated counterparts. Bars represent means of n = 5 independent experiments ± SEM.
- Representative immunofluorescence images of A549 cells co‐stained with the indicated antibodies and documenting PIDD1 localization at centrosomes. Scale bars: 10 μm.
- Luciferase reporter analysis of NF‐κB activity in CRISPR‐edited SCLT1‐ competent or –deficient A549 cells. Luciferase activity was normalized to renilla control. n = 3 independent experiments; mean values ± SEM.
Data information: The statistical significance was determined using two‐way ANOVA; P ≤ 0.001 (***), P ≤ 0.0001 (****).
Source data are available online for this figure.
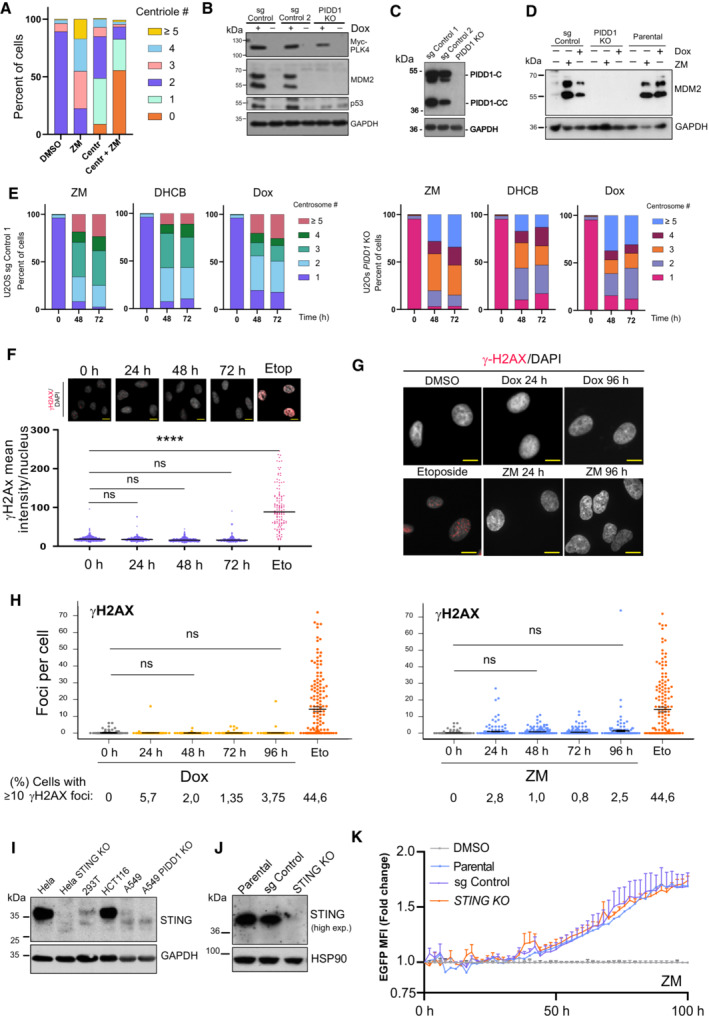
Figure EV3. Assessment of impact of DNA‐damage and cGAS/STING signaling on NF‐kB activation induced by extra centrosomes.
-
AEnumeration of centriole numbers in A549 cells after centrinone pretreatment (24 h) followed by induction of cytokinesis failure using ZM (2 μM) for additional 48 h by immunofluorescence imaging and γ‐Tubulin staining. A minimum of 50 cells per condition were analyzed in two independent experiments. Data are presented as mean values.
-
B, C(B) Immunoblot analysis of PIDD1 pathway competence, monitored by MDM2 processing and assessed in the indicated (C) CRISPR‐edited polyclonal U2OSPLK4 cells after induction of MYC‐PLK4 expression using Dox (1 μg/ml, 48 h).
-
DA549PLK4 reporter cells lacking or expressing PIDD1 were treated for the indicated times with ZM or Dox and analyzed for MDM2 processing by western blot analysis.
-
ECentrosome number was quantified based on γ‐Tubulin staining in the indicated genotypes of U2OS cells treated as indicated. A minimum of 50 cells per condition were analyzed in two independent experiments. Data are presented as mean values.
-
FDNA damage levels were assessed in U2OSPLK4 cells exposed to Dox treatment (1 μg/ml) for the indicated times. Nuclear γH2AX levels were determined by immunofluorescence and plotted as mean intensity per nucleus. Etoposide treatment (10 μg/ml, 24 h) was used as positive control. Top, representative immunofluorescence images. n ≥ 200 cells per condition were analyzed. Data represented as mean values ± SEM. The statistical significance was determined using one‐way ANOVA; P ≤ 0.0001 (****). Scale bar: 10 μm.
-
GRepresentative immunofluorescence images of nuclear γH2AX foci in A549PLK4 cells forming upon the indicated treatments. Scale bar: 10 μm.
-
HQuantification of DNA damage levels in A549PLK4 cells determined by the number of γH2AX nuclear foci per cell upon Dox or ZM treatment. n ≥ 200 cells per condition, n = 3 independent experiments; mean ± SEM. The statistical significance was determined using one‐way ANOVA.
-
IImmunoblot analysis of STING endogenous protein levels in different cancer cell lines.
-
JSTING levels were assessed by immunoblot in parental and CRISPR‐edited polyclonal STING‐deficient A549 derivatives.
-
KLife cell imaging analysis of NF‐κB EGFP‐reporter activity in polyclonal CRISPR‐edited STING‐competent or ‐deficient A549κB cells after ZM treatment. n = 5 independent experiments. Data represent mean values ± SEM.
To further explore whether centrosome amplification suffices to promote NF‐κB signaling in absence of ploidy increases, we uncoupled these two events using a Doxycycline (Dox)‐inducible PLK4 expression system. Overexpression of PLK4 drove centrosome amplification without altering cellular ploidy (Fig 3C) and caused PIDDosome‐dependent p53 stabilization in both U2OS and A549 cells (Fig EV3B and C; Appendix Fig S2). Analysis by immunofluorescence staining revealed that either cytokinesis failure or PLK4 overexpression increased centrosome numbers independently of PIDD1 expression status (Fig EV3E; Appendix Fig S2). Interestingly, supernumerary centrosomes significantly increased NF‐κB activation in U2OSPLK4 control cells but had no such effect on PIDD1‐deficient derivatives (Fig 3D). These results were comparable to those observed upon induction of cytokinesis failure with DHCB (Fig 3D). To corroborate this finding, we additionally tested PLK4 overexpression in combination with the NF‐κB EGFP reporter in our PIDD1‐proficient or ‐deficient cell lines. Analysis of U2OSκB‐PLK4 and A549κB‐PLK4 showed that although extra centrosomes wired cells toward NF‐κB activation, this effect was repressed in PIDD1 KO cell lines (Fig 3E). Of note, TNF‐induced stimulation did not depend on PIDD1, indicating a similar ability to activate NF‐κB in its absence (Fig 3E). Altogether, our data strongly indicate that the appearance of extra centrosomes suffices to trigger a PIDD1‐mediated signal that activates the NF‐κB network.
We and others have previously reported that Caspase‐2‐PIDDosome activation depends on PIDD1 recruitment to mother centrioles via the peripheral distal appendage protein ANKRD26 (Fava et al, 2017; Burigotto et al, 2021; Evans et al, 2021). Previous work establishing a hierarchy for centriolar distal appendage protein assembly determined that ANKRD26 recruitment occurs downstream of SCLT1 (Tanos et al, 2013; Bowler et al, 2019). Hence, we corroborated the presence of PIDD1 at centrosomes in either diploid or induced polyploid cells (Fig 3F) and investigated whether SCLT1 deficiency would compromise NF‐κB signaling. Taking advantage of previously characterized SCLT1‐deficient A549 cells (Burigotto et al, 2021), we observed that SCLT1 depletion abrogated NF‐κB transcriptional activity in response to perturbing cytokinesis using ZM or DHCB (Fig 3G). These data indicate that PIDD1 recruitment to distal appendages is necessary to promote NF‐κB activity upon centrosome accumulation, similar to its need for mounting a p53 response.
Finally, we wondered whether alternative effects induced by our treatments could be contributing to the NF‐κB activity observed. With this aim, we turned our attention to DNA damage, a secondary event linked to centrosome amplification (Ganem et al, 2009; Silkworth et al, 2009). We first examined the levels of genotoxic stress in U2OSPLK4 cells using an early marker of DNA damage, γH2AX. In sharp contrast to etoposide treatment, PLK4 overexpression did not significantly increase levels of γH2AX as compared to untreated cells (Fig EV3F). These results were also corroborated in A549PLK4 cells by analyzing the number of nuclear γH2AX foci in Dox‐treated cells (Fig EV3G and H). Noteworthy, induction of cytokinesis failure by ZM treatment caused no significant accumulation of double‐strand breaks over time, as observed by either γ‐H2AX immunofluorescence or western blotting analysis (Fig EV3H; Appendix Fig S3). As we could not exclude micronuclei formation during the course of Aurora kinase inhibition, we also explored a possible contribution of STING. Potential breakdown of micronuclei exposes self‐DNA to the cytosol, activating the cGAS/STING axis, which can promote NF‐κB signaling (Mackenzie et al, 2017). Initial analysis of STING protein levels displayed differences among cell lines and were comparatively low for A549 cells. However, expression levels did not differ between PIDD1 competent or deficient cells (Fig EV3I). Remarkably, upon ZM treatment, STING KO cells presented a robust induction of NF‐κB reporter activity, which was indistinguishable from the response of parental or CRISPR control cells (Fig EV3J and K). We conclude that neither secondary DNA damage nor that the cGAS/STING axis exerts a significant contribution to the NF‐κB activation detected in response to PLK4 overexpression or cytokinesis failure in our assays. Altogether, our findings not only document that extra centrosomes are sufficient to induce NF‐κB activation but also document that this signaling is transmitted by PIDD1 and initiated from extra centrosomes.
NF‐κB activation by supernumerary centrosomes requires the NEMO‐PIDDosome
Based on findings by Janssen et al (Janssens et al, 2005), PIDD1 is able to interact with the NF‐κB pathway components, RIPK1 and NEMO/IKKγ under genotoxic stress conditions forming the alternative NEMO‐PIDDosome (Fig 4A). Hence, we first investigated whether the lack of these proteins would affect NF‐κB activity after cytokinesis failure. In response to ZM or DHCB treatment, loss of RIPK1 and NEMO significantly abrogated NF‐κB activation, showing the requirement of the NEMO‐PIDDosome in this setting (Fig 4B).
Figure 4. Supernumerary centrosomes activate NF‐κB through the NEMO‐PIDDosome formation.
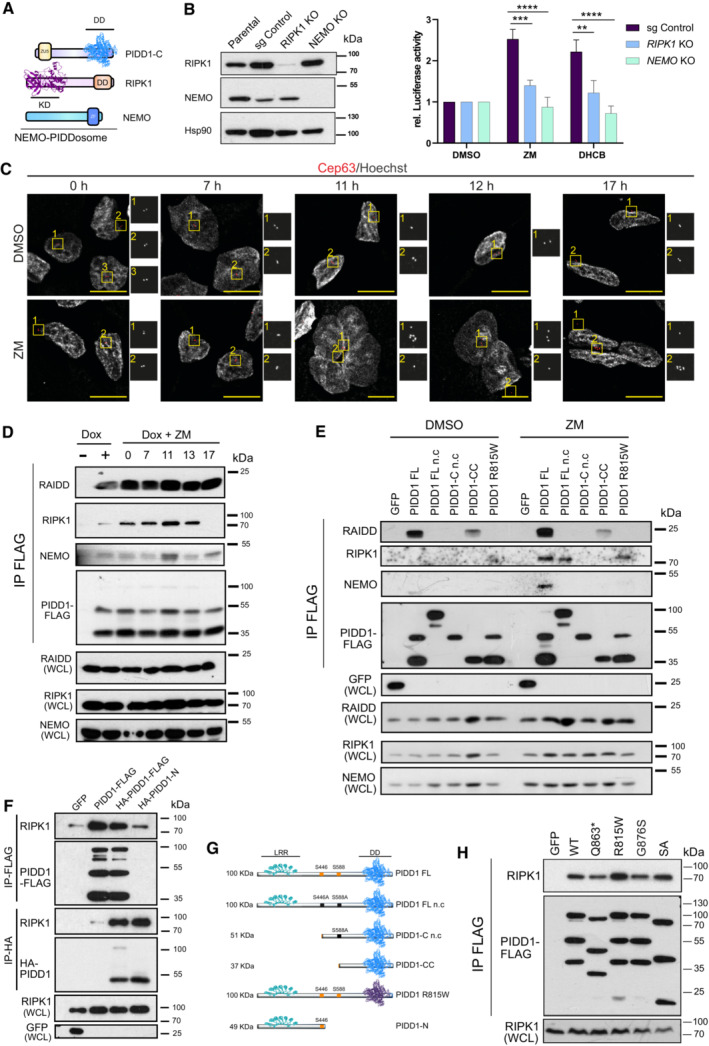
- Graphical representation of NEMO‐PIDDosome components.
- Left, immunoblot of CRISPR‐edited polyclonal RIPK1‐ or NEMO‐deficient A549κB derivatives. Right, luciferase assay of A549 cells of indicated genotypes, treated for 48 h with either ZM (2 μM) or DHCB (4 μM). n = 4 independent experiments. Data are represented as the mean values ± SEM. The statistical significance was determined using two‐way ANOVA; P < 0.01 (**); P < 0.001 (***); P < 0.0001 (****).
- Representative confocal fluorescence images documenting centrosome accumulation, as identified by CEP63 staining in 293TRex‐Flp‐In cells. Time course analysis was performed in cells synchronized in G1/S after a 24 h single thymidine block and released in either DMSO or ZM (2 μM). Cells were fixed at the indicated times for IF analyses. Scale bars: 10 μm.
- Different time point immunoprecipitations (IP) after release in ZM (2 μM) of endogenous RAIDD, RIPK1 and NEMO proteins in PIDD1‐deficient 293‐TRex‐Flp‐In cells re‐expressing a FLAG‐tagged PIDD1‐FL protein. One representative experiment out of three is shown.
- Analysis of NEMO‐PIDDosome formation in PIDD1‐deficient 293 TRex‐Flp‐In cells re‐expressing different PIDD1 mutants depicted in (G). Cells were treated as in (C) and processed for FLAG‐IP 11 h after release into DMSO or ZM (2 μM). One out of two independent experiments is shown.
- 293T cells were transiently transfected with constructs allowing expression of GFP, a full length (FL) single (PIDD1‐FL_FLAG) or double‐tagged (HA_PIDD1‐FL_FLAG) version of PIDD1, or an HA‐tagged N‐terminal fragment of PIDD1 (HA_PIDD1‐N). Co‐immunoprecipitation was performed to probe for interaction with endogenous RIPK1 after either anti‐FLAG or anti‐HA pull‐down. One out of two independent experiments is shown.
- Scheme of C‐terminally FLAG‐tagged PIDD1 mutants used in (E) or (F) for IP analyses. Mutation of S446 and/or S588 give rise to a non‐cleavable (n.c.) version of full‐length PIDD1 or PIDD1‐C that cannot be processed further into PIDD1‐CC, while the R815W mutation impairs the death domain in PIDD1. PIDD1‐N comprises aa 1–446.
- IP of endogenous RIPK1 protein in 293T‐Trex‐Flip‐in cells transiently transfected with different pathogenic PIDD1 variants with a compromised DD. One out of n = 3 independent experiments is shown. Abbreviations used: Death domain (DD), Kinase Domain (KD), Zinc Finger (ZF), Leucine Rich Repeats (LRR).
Source data are available online for this figure.
Next, we assessed whether PIDD1, RIPK1, and NEMO would physically interact in response to supernumerary centrosomes. To address this question, we exploited inducible 293 TRex‐Flp‐In cells lacking PIDD1 that were reconstituted with PIDD1 carrying a C‐terminal FLAG epitope tag, that allowed us to control protein expression and perform immunoprecipitation studies. As before, the timing of the apparence of extra centrosomes was narrowed down by performing confocal imaging analysis on cells released into ZM after a single thymidine block. Staining for γ‐tubulin revealed that 11 h after cell cycle reentry, cells that were released into solvent exhibited normal centrosome numbers, while most of those forced to fail cytokinesis became polynucleated and concomitantly acquired extra centrosomes (Fig 4C). These results were in line with the observation of a peak in phospho‐Histone H3+ cells at 10–11 h, followed by a pronounced reduction, indicative of mitotic exit of the majority of cells (Appendix Fig S3). PIDD1 complex formation was assessed over time by performing co‐immunoprecipitation assays of FLAG‐tagged PIDD1 (Fig 4D) with endogenous protein interactors. Using this strategy, we observed that RAIDD robustly interacted with PIDD1 as soon as it was re‐expressed (Fig 4D), suggesting that increases in PIDD1 protein levels above a certain threshold are likely sufficient to allow Caspase‐2‐PIDDosome formation. Remarkably, the interaction with RIPK1 gradually increased over time, reaching a peak after 11 h that eventually vanished. Most importantly, NEMO co‐elution with PIDD1 was best detectable at 11 h post release, supporting the idea of a transient NEMO‐PIDDosome complex forming in response to the appearance of supernumerary centrosomes (Fig 4D).
PIDD1 is constitutively cleaved at S446 and S588 giving rise to two C‐terminal fragments, PIDD1‐C and PIDD1‐CC (Janssens et al, 2005; Tinel et al, 2007). Upon DNA damage, PIDD1‐C interaction with RIPK1, predictably via their respective death domains (DD), was proposed to induce NEMO recruitment and activation of NF‐κB. However, this was never formally tested. Therefore, we set out to investigate whether PIDD1 autoprocessing or homotypic DD interactions were indeed critical in a scenario with supernumerary centrosomes. For this purpose, we re‐expressed diverse FLAG‐tagged PIDD1 variants in PIDD1 −/− 293 TRex‐Flp‐In cells and performed co‐immunoprecipitation assays at the time point of the appearance of extra centrosomes (Fig 4E and G). Confirming our previous observations, 11 h after the release of synchronized cells, PIDD1‐FLAG in its full‐length version (FL) robustly interacted with RAIDD, independently of the presence (ZM) or absence (DMSO) of supernumerary centrosomes (Fig 4E). This interaction was abolished when auto‐processing into PIDD1‐CC was compromised by mutating S446 (PIDD1 FL n.c. and PIDD1‐C n.c.). Similarly, a DD mutation at position R815W, which is known to abrogate MDM2 cleavage (Sheikh et al, 2021), also abolished RAIDD binding (Fig 4E). Although RAIDD interaction was constitutive, RIPK1 binding clearly depended on the presence of extra centrosomes (ZM treatment). Notably, preventing PIDD1 autoprocessing did not block RIPK1 recruitment, but in sharp contrast, the absence of the PIDD1 N‐terminal fragment impaired RIPK1 recruitment (Fig 4E), pointing toward a role of the N‐terminus of PIDD1 in the formation of the NEMO‐PIDDosome complex. Turning our attention to NEMO, we noted a selective interaction only when autoprocessing‐competent PIDD1 FL was expressed in the presence of supernumerary centrosomes, indicating that the formation of the complex is tightly regulated.
To explore the relevance of PIDD1 N‐terminus in the interaction with RIPK1, we generated an HA‐tagged PIDD‐N fragment (HA‐PIDD1‐N) and a double‐tagged version of PIDD1‐FL (HA‐PIDD1‐FL‐FLAG) to compare with the previously used PIDD1‐FL‐FLAG construct. Co‐immunoprecipitation using anti‐FLAG beads identified a similar RIPK1 interaction to either of the PIDD1 full‐length versions expressed (Fig 4F). To our surprise, HA‐pull down of the N‐terminus fragment revealed that RIPK1 specifically co‐eluted with both PIDD1‐FL and PIDD1‐N versions, suggesting that the PIDD1‐C fragment would be dispensable for RIPK1 binding. Furthermore, overexpression of different pathogenic PIDD1 mutants with either a functionally compromised DD (R815W, G876S), a truncated DD (Q863*), or a splice‐acceptor mutant (SA), documented that association with endogenous RIPK1 was not impaired (Fig 4H). These findings challenge prior hypotheses (Janssens et al, 2005; Tinel et al, 2007) and reinforce the notion that PIDD1‐RIPK1 DD‐DD interaction is not required or rate‐limiting, but PIDD1‐N likely assists other PIDDosome components in NEMO‐PIDDosome formation. Taken together, we conclude that the NEMO‐PIDDosome complex is selectively forming in polyploid cells with an increase in centrosome number and is essential for the activation of NF‐κB.
Supernumerary centrosomes trigger sterile inflammation and polarization of macrophages
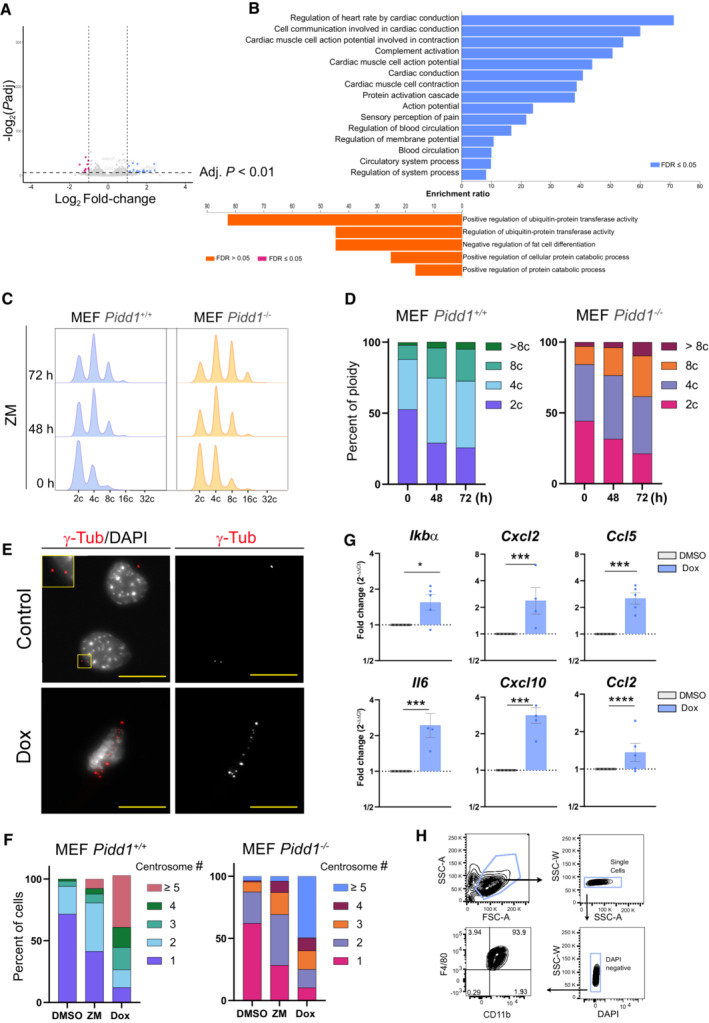
In light of the relevance of PIDD1 for NF‐κB activation upon cytokinesis failure, we also analyzed by mRNA‐sequencing the gene expression signature of Pidd1‐deficient primary MEF treated with ZM. In contrast to Pidd1‐proficient MEF (Fig 1), the same analysis carried out in Pidd1 −/− cells yielded a considerably smaller number of DEGs (Fig EV4A). GO term analysis did not reveal any immune or inflammatory response processes, and terms related to cell cycle progression were not downregulated in these cells (Fig EV4B). Notably, flow cytometric analysis confirmed a higher level of ploidy in primary PIDD1‐mutant MEF when compared to wild‐type controls (Fig EV4C and D), but a comparable increase in centrosome number between genotypes (Fig EV4F). Comparison of a set of genes related to inflammation and immune cell chemotaxis (Ccl2, Ccl7, Ccl11, CxCl14) revealed important differences between PIDD1‐proficient and deficient MEF (Fig 5A). Some of these changes were validated by qRT‐PCR, presenting significant differences in Il6 and Ccl2 transcriptional levels and an overall reduction of inflammatory gene expression in Pidd1 −/− MEF failing cytokinesis (Fig 5B). Together, these results suggested that PIDD1 is necessary to regulate sterile inflammatory responses upon unscheduled polyploidization induced by Aurora kinase inhibition.
Figure EV4. Extra centrosomes induce sterile inflammation in a PIDD1‐dependent manner.
-
AVolcano plot of DEGs in Pidd1 −/− primary MEF induced by ZM vs. DMSO treatment for 72 h. Adjusted P value ≤ 0.01 and log2 FC ≥ 1 or log2 FC ≤ −1.
-
BGO‐term analysis for significant DEGs for ZM vs. DMSO stimulated primary Pidd1 −/− MEF. FDR ≤ 0.05, adjusted P ≤ 0.01 and cut‐off values log2 FC ≥ 1 or log2 FC ≤ −1.
-
C, D(C) Representative histograms of DNA content analysis in primary MEF after induction of cytokinesis failure with ZM (2 μM) for the indicated times, quantified in (D). Two independent experiments were performed.
-
E–G(E) Primary MEF derived from TET‐PLK4 mice were treated with Dox for 48 h and processed for immunofluorescence detection of extra centrosomes using γ‐tubulin, quantified in (F), or RNA extraction for qRT‐PCR analyses of NF‐κB target genes (G). Scale bars: 10 μm. β‐actin was used as housekeeping control. n = 5 biological replicates; mean ± SEM. The statistical significance was determined using two‐way ANOVA; P ≤ 0. 1 (*), P ≤ 0.001 (***), P ≤ 0.0001 (****).
-
HFlow cytometric analysis of F4/80 and CD11b in BMDM on day 7 after isolation.
Figure 5. Supernumerary centrosomes induce sterile inflammation and macrophage polarization.
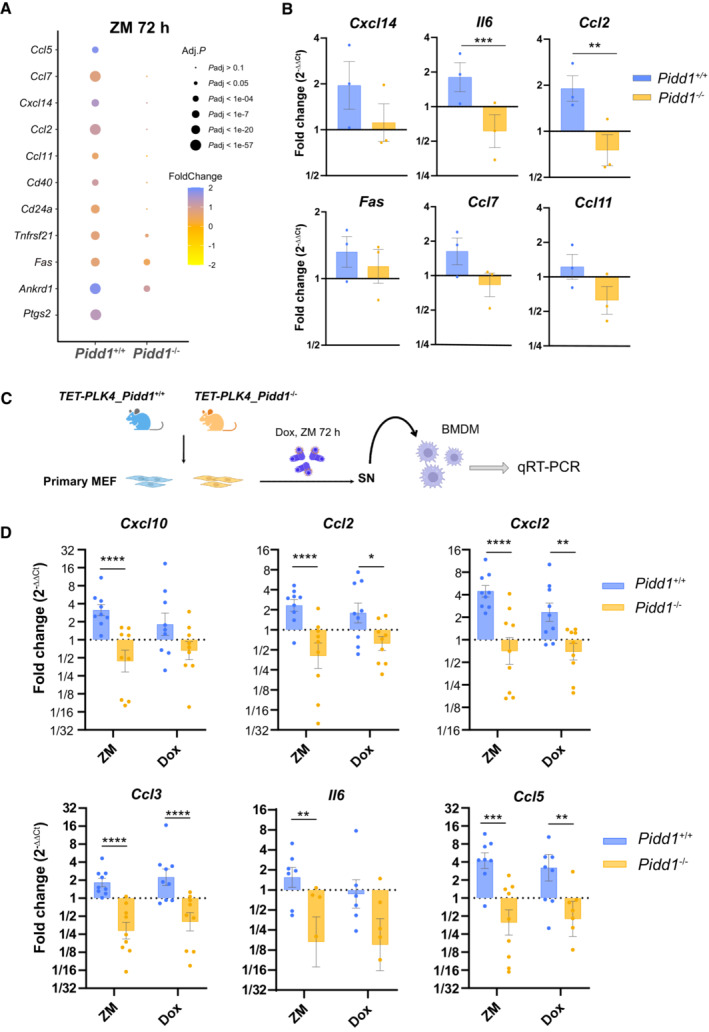
- Dot plot depicting the expression of the indicated genes related to inflammatory and immunoregulatory processes in Pidd1 +/+ and Pidd1 −/− MEF.
- qRT‐PCR analysis to confirm upregulation of different pro‐inflammatory genes in Pidd1 +/+ but not Pidd1 −/− MEF. Hprt1, β‐actin and Gapdh were used as housekeeping gene expression controls. n = 3 independent samples; data represent mean values ± SEM. The statistical significance was determined using two‐way ANOVA test; P ≤ 0.01 (**), P ≤ 0.001 (***).
- Schematic representation of the experimental work flow to study macrophage polarization. Bone marrow–derived macrophages (BMDM) were treated for 24 h with conditioned medium derived from Pidd1 +/+ and Pidd1 −/− MEFPLK4 cells where centrosome amplification was induced prior to RNA isolation by induction of cytokinesis failure (ZM) or PLK4 overexpression (Dox).
- qRT‐PCR analysis of macrophage activation markers indicative of a pro‐inflammatory state. Relative expression values are plotted as the fold‐change expression relative to BMDM stimulated with conditioned medium from untreated MEFPLK4. β‐actin was used as housekeeping gene for normalization. Bars represent mean ± SEM (n = 9) biological replicates. The statistical significance was determined using two‐way ANOVA; P ≤ 0.1 (*), P ≤ 0.01 (**), P ≤ 0.001 (***), P ≤ 0.0001 (****).
We next investigated whether the mere presence of extra centrosomes would induce similar transcriptional changes. For this, we employed primary MEF from TET‐Plk4 transgenic mice (Levine et al, 2017) and induced centrosome amplification by doxycycline treatment for 48 h. Alongside with extra centrosomes (Fig EV4E and F), increased mRNA levels of the NF‐κB target Ikba, as well as Il6, Cxcl2, and other pro‐inflammatory genes, were observed (Fig EV4G). These data pointed out that supernumerary centrosomes can elicit an inflammatory and immunoregulatory transcriptional response.
To characterize possible paracrine effects engaging the innate immune system, we studied the phenotype of primary bone marrow–derived macrophages (BMDM; Fig EV4H) exposed to the supernatant from pretreated Pidd1 competent or deficient TET‐PLK4 MEF (Fig 5C). Conditioned media from cells that failed cytokinesis after ZM treatment promoted macrophage polarization toward an “activated” M1‐like phenotype (Fig 5D) with increased pro‐inflammatory Il6, Cxcl2 or Ccl3, among other chemokines, but undetectable Ym1 mRNA expression, a hallmark of M2‐like macrophages. Consistent with this, BMDM incubated with media from Dox‐treated cells that experience centrosome amplification also displayed an increase in the transcript levels of these pro‐inflammatory genes (Fig 5D). These effects required PIDD1, since media transferred from ZM or Dox‐treated MEF lacking Pidd1 failed to activate macrophages. These data show that the presence of supernumerary centrosomes exerts a PIDD1‐dependent paracrine effect that can promote an inflammatory macrophage activation phenotype.
PIDD1 orchestrates immune clearance of polyploid cells after cytokinesis failure
The transcriptional and paracrine immunoregulatory response observed raised the possibility that supernumerary centrosomes played a role in immunosurveillance. Inspired by the GO‐term analysis of our initial RNA‐seq results revealing “positive regulation of NK cell chemotaxis” as the top hit (Fig 1E), we addressed whether polyploid cells resulting from cytokinesis failure were targeted by natural killer (NK) cells. To test our hypothesis, we took advantage of the well‐established human NK cell line, NK‐92 (Gong et al, 1994). NK cell killing was quantified in EGFP‐expressing A549 target cells (A549EGFP) where acute polyploidization was induced by ZM or, as an alternative measure, DHCB treatment. Mimicking the conditions of our RNA‐seq experiment, we removed the drug after 72 h and established a co‐culture system to monitor the interactions between polyploid tumor cells and NK effector cells (Fig EV5A). Of note, analysis of centrosome numbers in A549EGFP cells confirmed an increase upon polyploidization that was maintained over the entire observation period of our assay, without any signs of adaptation or counter‐selection (Fig EV5B). Time‐lapse microscopy revealed that NK cells were effectively attracted to polyploid cells and eliminated them in a period of around 18 h. This killing was accompanied by loss of target cell integrity, pyknotic nuclei, EGFP release, and a concomitant reduction of their fluorescence and confluence (Fig 6A; Movie EV4). In this setup, polyploid A549EGFP cells presented a significant NK cell–mediated reduction of their confluence of around 50% when compared to euploid cells derived from DMSO treatment (Fig 6B). These data indicated that aberrant polyploid cells are killed by NK‐92 cells more efficiently than their euploid counterparts.
Figure EV5. PIDD1 is necessary to activate NK cells targeting polyploid cells.
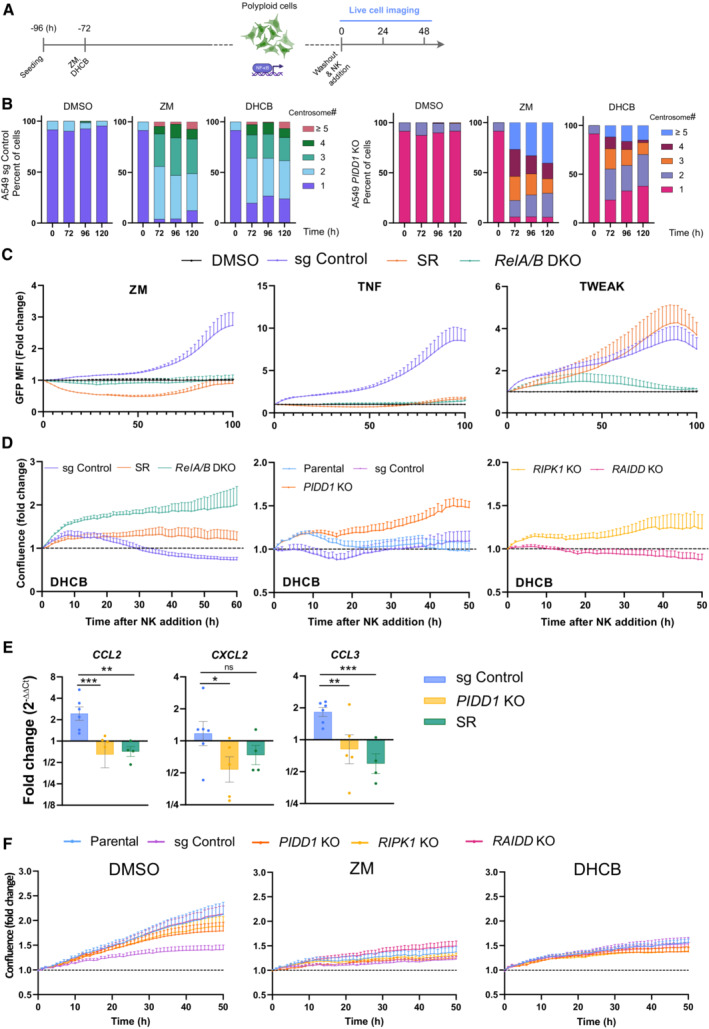
- Experimental design of NK cell killing assays.
- Evaluation of centriole counts based on IF analysis and γ‐tubulin staining. More than 50 cells were evaluated per condition in two independent experiments. Data are presented as means values.
- Evaluation of reporter activity in A549 cells of the indicated genotypes proficient or deficient in NF‐κB activation, due to loss of RELA/B or overexpression of the IkBα super‐repressor (SR) after treatment with ZM, the canonical NF‐κB pathway agonist, TNF or the non‐canonical NF‐κB pathway agonist TWEAK. n = 3 independent experiments; mean ± SEM.
- Confluence curves from A549EGFP derivatives of the indicated genotypes, pretreated with DHCB (4 μM) to induce cytokinesis defects and co‐cultured with NK‐92 cells. n ≥ 3 independent experiments; mean ± SEM.
- Conditioned medium from ZM pretreated A549 cells of the indicated genotypes was applied to human monocyte–derived macrophages for 24 h followed by RNA isolation and qRT‐qPCR analysis of macrophage activation markers. Expression values are plotted as the fold‐change relative to macrophages stimulated with conditioned medium from untreated A549 cells. HPRT1 expression was used for normalization. Bars represent mean ± SEM of n ≥ 4 independent experiments. The statistical significance was determined using two‐way ANOVA; P ≤ 0. 1 (*), P ≤ 0.01 (**), P ≤ 0.001 (***).
- Proliferation curves of different A549EGFP derivatives, pretreated with ZM (2 μM) or DHCB (4 μM), for 72 h in the absence of NK cells. Time zero is defined as the moment of the drug wash out. Live‐cell imaging of target cells without NK cells was performed in parallel to killing experiments using the same conditions as described in the methods section. n = 5 independent experiments; mean ± SEM.
Figure 6. PIDD1 induces NK cell–mediated clearance of polyploid cells.
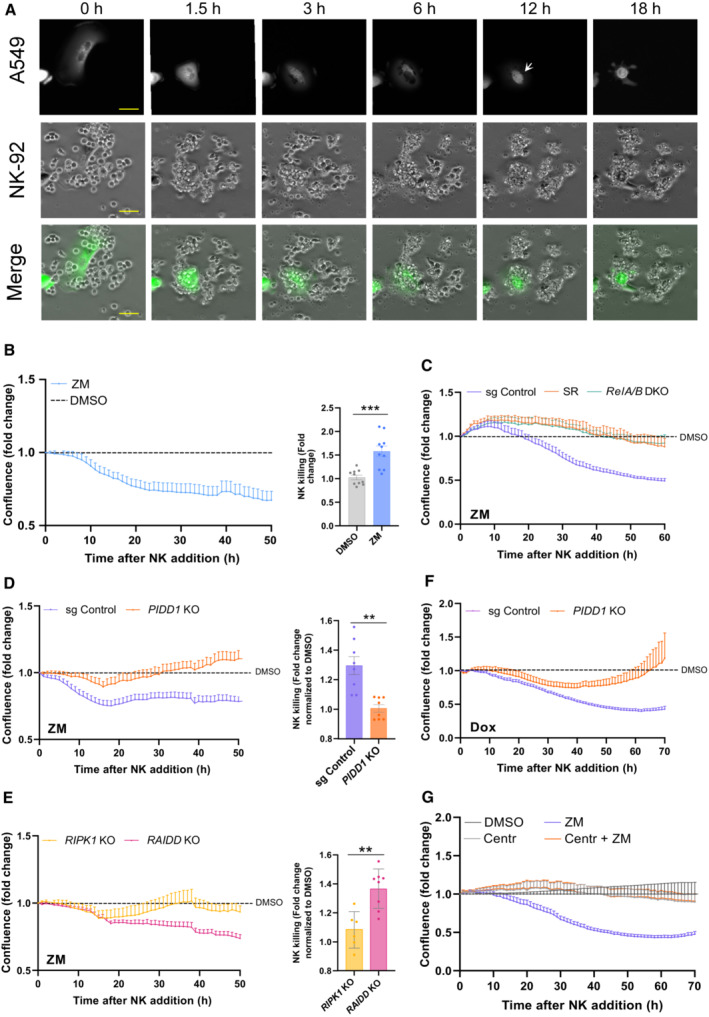
- Representative images of target A549EGFP cells pretreated for 72 h with ZM (2 μM) and co‐cultured subsequently with NK‐92 cells in a 1:10 target to effector ratio. Interactions were recorded by live‐cell imaging for 18 h at a 1 min interval. White arrow indicate polyploid cell undergoing cell death. EGFP release coincides with loss of cell membrane integrity. All images were acquired at the same exposure time and light intensity. Scale bar: 10 μm. View also Movie EV4.
- Left, measurement of A549EGFP cell elimination by NK cells monitored for a period of 50 h. Cell density was followed over time by live‐cell imaging using the integrated EGFP fluorescence area as a readout of A549EGFP growth or depletion. Confluence was normalized with respect to time zero (NK cell addition) and compared over time between ZM treated (polyploid) and DMSO treated (euploid) cells, respectively. Middle, NK cell‐mediated killing interpolated from cell confluence graphs at 50 h. n = 5 independent experiments, mean ± SEM.
- Confluence analysis of ZM pre‐treated A549EGFP cells, impaired in NF‐κB activation, was measured over time in the presence of NK cells and normalized with respect to their DMSO treated controls. n = 4 independent experiments, mean ± SEM.
- Confluence of ZM pre‐treated A549EGFP cells expressing or lacking PIDD1 was measured over time in presence of NK cells and normalized with respect to their DMSO treated counterparts. Middle, NK cell–mediated killing interpolated from cell confluence graphs at 50 h. n ≥ 4 independent experiments performed in duplicates, mean ± SEM.
- Confluence measurement over time of A549EGFP derivatives impaired in either NEMO‐PIDDosome (RIPK1 KO) or Caspase‐2‐PIDDosome formation (RAIDD KO). Middle, NK cell‐mediated killing interpolated from cell confluence graphs at 50 h. n ≥ 3 independent experiments; mean ± SEM; unpaired t‐test.
- Confluence analysis of Dox pre‐treated A549EGFP cells overexpressing PLK4 was performed over time in the presence of NK cells and normalized with respect to their DMSO treated controls. n = 5 independent experiments; mean ± SEM.
- Confluence analysis of centriole‐depleted and ZM pretreated A549EGFP cells cultured in presence of NK cells. Values are normalized with respect to their DMSO ± Centrinone (125 nM) treated controls. n = 4 independent experiments; mean ± SEM.
Data information: The statistical significance was determined using two‐tailed t‐test; P < 0.001 (**); P < 0.001 (***).
Source data are available online for this figure.
To investigate whether this immune clearance was indeed dependent on NF‐κB activity, we generated RELA/B double mutant A549EGFP cells or blocked the canonical NF‐κB pathway by expressing a dominant negative form of IκBα (Super‐Repressor; SR) (Feltham et al, 2018). In A549SR cells, ZM as well as TNF‐induced reporter activity was fully blunted, validating our system and excluding a role for non‐canonical NF‐κB signaling (Fig EV5C). Compared to A549EGFP CRISPR control cells, RELA/B depletion, as well as SR overexpression, protected ZM‐ or DHCB‐induced polyploid cells against NK killing (Figs 6C and EV5D). Moreover, conditioned medium from ZM‐treated A549 cells increased CCL2, CXCL2 and CCL3 mRNA expression in human monocyte–derived macrophages, while supernatants from A549SR or PIDD1 KO cells had no such effect (Fig EV5E). These data strongly suggest that increased cellular immunogenicity upon cytokinesis failure leans on canonical NF‐κB signaling.
Next, we assessed the role of the PIDDosome in the acquired immunogenic potential of abnormally polyploid cells. Polyploid or euploid CRISPR control and PIDD1 KO A549EGFP cells were co‐cultured with NK‐92 cells, and their confluence was assessed by live‐cell imaging. In this setup, PIDD1 KO cells presented decreased immunogenicity and were not targeted as effectively as CRISPR controls by NK‐92 cells when rendered polyploid by ZM or DHCB treatment (Figs 6D and EV5D). Of note, we did not find significant differences among the diverse sgRNA cells in their basal proliferation rates after drug washout in the absence of NK‐92 cells (Fig EV5F). Afterward, we also addressed the contribution of the NEMO‐ vs. Caspase‐2‐PIDDosome to immune recognition after cytokinesis failure. While aberrant polyploid RIPK1 KO cells behaved similar to PIDD1 KO A549EGFP cells, the impairment of Caspase‐2‐PIDDosome complex formation due to loss of RAIDD did not impact A549EGFP cells elimination as effectively (Fig 6E). Similar results were observed in A549EGFP cells forced to fail cytokinesis by DHCB treatment (Fig EV5D). Finally, we wondered if manipulation of centrosome number would suffice to stimulate NK killing in the absence of ploidy changes. Hence, we either overexpressed PLK4 or inhibited it with centrinone to alter their numbers. These experiments revealed that indeed extra centrosomes suffice to promote NK‐cell attack while loss of centrioles rendered cells inert to NK killing (Fig 6F and G).
Altogether, our findings indicate that unscheduled polyploidization after cytokinesis failure induces a PIDD1‐dependent immunosurveillance response, elicited from extra centrosomes, which promotes NK‐mediated clearance of polyploid cancer cells in vitro. This cell immunogenic behavior is independent of PIDD1's ability to promote a p53 response but relies on the NEMO‐PIDDosome for activation of the NF‐κB signaling network.
Discussion
Polyploidization is considered an early event in transformation that can drive aneuploidy and oncogenesis (Fujiwara et al, 2005; Davoli & de Lange, 2011). Hyperploidy of (pre) malignant cells is subjected to intrinsic and extrinsic control mechanisms, most notably by cellular components of the innate and adaptive immune system (Acebes‐Huerta et al, 2016; Santaguida et al, 2017; Lopez‐Soto et al, 2017a). The results included in this article strongly support the concept that supernumerary centrosomes are one of the key triggers of this cellular immunogenicity (Figs 1, EV1, 2, EV2, 3).
Centrosome amplification has been linked to chromosomal instability and DNA damage, one of the common triggers of NF‐κB activity, also associated with senescence (Rodier et al, 2009; Kang et al, 2015). PIDDosome formation was initially described in response to genotoxic damage, and more recent studies have reported its nucleolar assembly together with NPM1 in response to DNA damage (Janssens et al, 2005; Ando et al, 2017). Furthermore, it was recently described that after unscheduled whole‐genome duplication, high rates of DNA damage were observed in the immediate following S phase. This was proposed to be caused by the lack of enzymatic components needed for effective DNA replication after tetraploidization (Gemble et al, 2022). Altogether, a contribution of DNA damage to PIDDosome activation in our experiments seemed plausible but could be excluded experimentally (Fig EV3). Moreover, the phenomenon described by Gemble et al seems to be limited to the first S phase of tetraploid cells, since similar levels of genotoxic stress were found between G1 tetraploid and diploid cells. Moreover, the grade of DNA damage was considerably reduced in the subsequent S phases of those tetraploid cells that escaped cell cycle arrest. In our experiments, NF‐κB activity was assessed either before or long after the first S phase after polyploidization (Figs 2 and 3). Moreover, in agreement with previous publications (Ganem et al, 2014; Levine et al, 2017), we did not observe significant increases in DNA damage upon PLK4‐induced centrosome amplification or accumulation after cytokinesis failure, suggesting that DNA damage is not a confounding factor in our analyses (Fig EV3).
It is worth mentioning that across many of our experiments the effects induced by PLK4 overexpression were weaker in U2OS cells, presenting the mildest increase in centrosome numbers in our hands, which could potentially explain the differences in the amplitude of NF‐κB reporter activity noted (Figs 3 and EV3; Appendix Fig S2). In general, ZM treatment appeared more potent in inducing NF‐κB activation, but this did not lead to enhanced NK cell killing, when compared to PLK4 overexpression or DHCB treatment (Fig 6). Yet, we cannot fully exclude that additional effects triggered by Aurora kinase inhibition may add to the reporter activity observed.
As mentioned above, under DNA damaging conditions, exogenous PIDD1 was proposed to interact with RIPK1, which in turn facilitated NEMO recruitment for NF‐κB activation (Janssens et al, 2005). Even though it was assumed that PIDD1‐RIPK1 association occurred through their respective death domains, experimental proof of it, as well as definitive evidence confirming direct interaction of PIDD1, RIPK1, and NEMO, was not presented in these studies (Janssens et al, 2005; Tinel et al, 2007). To our surprise, we noted that the DD of PIDD1 is actually dispensable for RIPK1 recruitment (Fig 4), as different PIDD1 mutants compromising the functionality of its DD were still capable to bind RIPK1 (Sheikh et al, 2021). This finding prompted us to speculate which domains in PIDD1 might be required for interaction. We noticed that the inability of PIDD1 to be autoprocessed perturbed NEMO but not RIPK1 recruitment (Fig 4). These results are in agreement with the original experiments of Tinel et al (2007) where, based on luciferase reporter analysis, lack of PIDD1‐N hampered NF‐κB activation. Interestingly, the same experiments also suggested a negative effect of the PIDD1 LRR domain in NF‐κB activation, but the mechanism of this inhibitory effect remains unclear. According to our findings, it appears plausible that the ZU5 domain in PIDD1‐N could facilitate RIPK1 recruitment, as the N‐terminal portion of exogenous PIDD1 allowed pull‐down of endogenous RIPK1 (Fig 4). Whether the LRR domain exerts some kind of steric hindrance due to its bulkiness, or if its possible inhibitory effect is relieved by ligand binding or posttranslational modification, like seen in inflammasome activation (Lopez‐Castejon, 2020), needs to be defined. It will be interesting to dissect what defines formation of the NEMO‐ vs. Caspase‐2‐PIDDosome under these conditions, which, as suggested by our western analysis (Fig 1), seems to be sequential. In the context of DNA damage, ATM/ATR kinases have been implicated in regulating Caspase‐2‐PIDDosome function (Ando et al, 2012; Shah et al, 2021). As we did not find evidence for DNA damage in response to cytokinesis failure or PLK4 overexpression, a decisive contribution of these kinases in regulating the switch between NEMO‐ and Caspase‐2‐PIDDosome assembly in response to centrosome amplification seems rather unlikely.
A new role of centrosomes to tune innate immunity toward infection is currently emerging (Moller et al, 2022; Weier et al, 2022). Recent studies have also expanded our view identifying the centrosome as a signaling hub able to facilitate the formation of NLRP3‐ and Pyrin‐inflammasome (Magupalli et al, 2020; Yang et al, 2020). Here, we propose that supernumerary centrosomes act as an activation platform for the formation of the NEMO‐PIDDosome and NF‐κB activation. Our RNA‐seq data and transcriptional analysis in MEF that undergo cytokinesis failure or centrosome amplification indicate an increase of proinflammatory cytokines that support a sterile inflammatory response triggered by extra centrosomes (Figs 1 and EV4). The impact of extra centrosomes is not limited to the cells carrying them, but also exerts a secretory response with paracrine effects (Fig 5). Despite that this secretory pathway has been mainly linked to non‐cell‐autonomous invasion (Arnandis et al, 2018; Adams et al, 2021), we observed that supernumerary centrosomes can also mount a transcriptional pro‐inflammatory response, in which paracrine effects increase cellular immunogenicity (Fig 5). This begs the question of how naturally polyploid cells, such as hepatocytes or cardiomyocytes, experience centrosome amplification during development and avoid sterile inflammation. At least for the liver, we documented that Pidd1 levels are repressed by inhibitory E2F7/8 in mature hepatocytes (Sladky et al, 2020a), while postnatal rat cardiomyocytes were reported to lose their centrosome integrity (Zebrowski et al, 2015), which, extrapolating this observation to mice, would also disable PIDD1. Among the different transcripts upregulated after cytokinesis failure (Fig 1), we found Ccl2, Ccl7, and Cxcl14, well‐known chemokines that act as positive attractors of NK cells and other innate immune cell types, such as macrophages (Morrison et al, 2003; Hara & Tanegashima, 2012; Sturmlechner et al, 2021). Curiously, although transcripts of pro‐inflammatory cytokines, such as Il6 or Cxcl2, were upregulated upon treatment, we could not detect a transcriptional increase in TNF. These findings resemble those of Vertii et al (2016), which described that centrosome maturation is necessary for macrophages during inflammatory processes for the effective secretion of several cytokines and chemokines, such as IL‐6, IL‐10, and CCL2, but not TNF.
Different studies have focused on the molecular mechanisms underlying the cellular immunogenic potential of polyploid or aneuploid cells. A connection between tetraploidy and immunosurveillance was established in cancer cells, based on the cell membrane exposure of calreticulin, which facilitates cell elimination by macrophages (Gardai et al, 2005; Chao et al, 2010; Senovilla et al, 2012). While endoplasmic reticulum (ER) stress, aneuploidy, and cGAS/STING pathway activation are proposed as important molecular players of cell immunogenicity, we conclude that supernumerary centrosomes add to this and play a critical role by engaging different effector arms that have cell intrinsic as well as extrinsic effects. NK immune surveillance could be potentiated by macrophages in tissue (Maghazachi et al, 1994), since we found that macrophages polarized by conditioned media from cells with extra centrosomes exhibited transcriptional upregulation of Ccl2, Ccl5, and Ccl3 chemokines (Figs 5 and EV4). Notably, we found that PIDD1 facilitates macrophage polarization, as well as NK cell–induced elimination of aberrant polyploid cells (Figs 5 and 6). Hence, it appears surprising that Pidd1 has been shown to exert oncogenic effects, as its loss delayed the onset of MYC‐driven B cell lymphomas (Manzl et al, 2012) or DEN‐induced hepatocellular carcinoma in mice (Sladky et al, 2020b). While in the latter case an increase in hepatocyte ploidy, paralleled by an increase in tumor suppressor genes, may account for the delay of tumor onset (Lin et al, 2020) and dominate over impaired immune clearance, the situation in MYC‐driven lymphomas is less clear and awaits clarification. Despite these open questions, our evidence presented here suggests that activation of NF‐κB signaling in cells at risk to develop aneuploidies can increase their immunological visibility and prime them for NK cell recognition. Similar findings have been made in drug‐induced hyperploid cells and investigating transcriptional profiles of model cell lines forced to become aneuploid upon spindle assembly checkpoint override (Acebes‐Huerta et al, 2016; Wang et al, 2021).
Effective cancer immunotherapies exploiting NK cell cytotoxicity, for example, in the form of cytokine priming or CAR‐NK cells, have received increased attention in recent years. However, the efficacy of these therapies has been limited so far, potentially due to impairment of NK cell activity by the tumor microenvironment (Gotthardt & Sexl, 2016; Kuhne et al, 2017; Liao et al, 2018). A better understanding of the molecular mechanisms underlying the intrinsic immunogenicity of cancer cells and how NF‐κB pathway signaling is modulated during tumor development, evolution, and drug treatment will be fundamental steps to improve cancer treatment. Here, we conclude that extra centrosomes act as signaling hubs that elicit cell cycle arrest as well as inflammatory responses, placing the NEMO‐PIDDosome next to other inflammasomes in its ability to instruct innate immunity that may be harnessed for future cancer immunotherapies.
Materials and Methods
Cell culture and cell lines
A549 (ATCC CCL‐185), U2OS (ATCC HTB‐96), and RPE1 (ATCC CRL‐2302) cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Sigma‐Aldrich, D5671), supplemented with 10% fetal calf serum (FCS, Invitrogen, 10270106), 1% L‐glutamine (Sigma‐Aldrich, G7513), 100 U/ml penicillin and 100 μg/ml streptomycin (P/S) (Sigma‐Aldrich, P0781). NK‐92 cells were obtained from DSMZ (ACC 488) and cultured in alpha‐MEM with ribo‐ and deoxyribonucleosides (Gibco, 12571063), including 12.5% h.i. FCS (Invitrogen, 10270106), 12.5% h.i. horse serum (Gibco, 16050122), 2 mM L‐glutamine (Sigma‐Aldrich, G7513) and 5 ng/ml IL‐2 (Preprotech, 200‐02). All cells were grown at 37°C with 5% CO2 in a humidified atmosphere. Primary mouse embryonic fibroblasts (MEF) were obtained from 13.5 embryos by tryptic digestion. MEF was cultured in DMEM containing 10% FCS, 1% L‐glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (P/S). All experiments were performed with at least three independent bulk cultures derived from individual embryos.
Bone marrow–derived macrophages (BMDM) and human monocyte–derived macrophages
Bone marrow–derived macrophages were prepared from bone marrow isolated from femora of C57BL/6N mice. Briefly, bones were flushed with PBS and cells plated at a density of 1 × 106 cells/ml. Progenitor cells were differentiated into macrophages by culturing them for 6 days in DMEM GlutaMAX medium (ThermoFisher, 31966047) containing 10% FCS, 100 U/ml penicillin, and 100 μg/ml streptomycin (P/S) and supplemented with 15% L929 cell‐conditioned medium (CM). Medium was replaced after 3 days. Human monocytes were isolated as reported before (Ploner et al, 2021) and resuspended in RPMI medium (PAN Biotech, Germany) containing 10% FCS, 2 mM l‐glutamine, and MCSF (50 ng/ml). Cells were differentiated for 7 days. After this time, cells were washed and cultivated for another 24 h without MCSF prior to adding conditioned medium harvested from different ZM‐treated A549 variants.
Drug treatment, synchronization procedure, and centriole depletion
Drugs were used at the following final concentrations: ZM447439 (2 μM; Selleck Chemicals, S1103), DHCB (4 μM; Sigma‐Aldrich, D1641), doxycycline (1 μg/ml; sigma‐aldrich, D9891), human TNF (20 ng/ml; Peprotech, 300‐01A), human TWEAK (100 ng/ml; Peprotech, 310‐06), etoposide (10 μg/ml). For synchronization purposes, cells were subjected to a single pulse of thymidine (2 mM, Sigma‐Aldrich, T1895) for 24 h. Cells were washed twice with PBS prior to release in ZM‐ (2 μM) or DMSO‐containing medium. For centriole depletion, cells were pretreated for 24 h with centrinone (125 nM; MedChemExpress, HY‐18682), or DMSO. Afterward, fresh medium containing ZM ± centrinone was added to the samples. Live cell images were taken using IncuCyte technology in 2 h intervals.
Lentiviral production and transduction
To generate lentiviral particles, 293T cells were plated at 6.5 × 104 cells/cm2 and transfected with packaging plasmids psPAX2 and pCMV‐VSV‐G (Addgene, #8454 and #12260, respectively) and the corresponding lentiviral plasmids in a ratio 1:1:2. Briefly, 2 μg of lentiviral vector, 1 μg of psPAX, and 1 μg of pCMV‐VSV‐G were diluted in a total volume of 80 μl of OptiMEM (Gibco, 31985070) and incubated at room temperature for 5 min. Separately, 6 μl of polyethylenimine (PEI, 1 μg/μl; Polysciences, Hirschberg, Germany, 23966‐1) was diluted in 80 μl of OptiMEM. After incubation, the DNA and PEI mixtures were combined and incubated at room temperature for 20 min. The transfection mixture was added dropwise to the 293T cells to a final volume of 1.5 ml. Cell culture medium was changed the next day and after 48 h, viral supernatants were collected and filtered through a 0.45 μM PVDF syringe filter. Filtered supernatants were combined with 10 μg/ml of polybrene (Sigma‐Aldrich) and added to the cells.
Generation of knockout cell lines
CRISPR/Cas9‐mediated loss‐of‐function cell lines were generated using pLentiCRISPR‐v2 puromycin backbone (Addgene, #52961) containing a small guide RNA (sgRNA), targeting the gene of interest (Appendix Table S1). These sgRNAs were designed using CRISPR tool (Concordet & Haeussler, 2018), and all plasmids were verified by Sanger sequencing. The following sgRNA were used: PIDD1_1 (5′ gccgatagcggatggtgatg); PIDD1_2 (5′ ggcccggcgctgccgtgaag); CASP 2 (5′ tggtgagcaacatatcctcc); RAIDD (5′ ccagctccaggcgaagtgag); RIPK1 (5′ tagtctgacggataaacacc); NEMO (5´acgccgccgtccgagagacg); SCLT1 (5′ gggcctcagtcatatgttcc); RELA (5′ actacgacctgaatgctgtg); RELB (5′ gaaagcacaggatccatc). As control for possible CRISPR off‐target effects, sgRNA targeting mouse CD8 (5´ggctgggtgagtcgattatcc) or a none‐targeting sgRNA (5´tatctaatcgcggagtcgta) from the Gecko human none‐targeting primer list (https://crispr.mit.edu) were used. Transduced cells were selected in 1 μg/ml puromycin (Sigma‐Aldrich, P8833) for at least 7 days. Unless otherwise specified in the figure legends, all cell lines were used as polyclonal bulks. Gene disruption was validated by immunoblot analysis of the targeted protein and functional assays. For the generation of single cell clones, puromycin selected cells were seeded at a density of 0.1 cells per well, in 96‐well plates, and incubated for up to 3 weeks.
Generation of stable cell lines
To generate A549PLK4 cells, the human PLK4 ORF was cloned into the lentiviral pInducer21 plasmid (Addgene #46948) using the GATEWAY system. The resulting backbone was digested with NdeI (NEB) and SphI (NEB) restriction enzymes to replace the EGFP cassette for hygromycin and make it compatible with our NF‐κB‐EGFP reporter system. Hygromycin resistance and IRES coding sequences were amplified including primer overhangs (Appendix Table S1) and the PCR products, as well as the digested backbone, were purified from agarose gels and fused using Gibson assembly (Gibson et al, 2009). A549 cells were transduced with the resulting pPLK4‐Hygr lentiviral plasmid and selected with 400 μg/ml of hygromycin for 10 days. Cells stably expressing a NF‐κB EGFP reporter were generated after lentiviral transduction with pTRH1 mCMV NF‐κB dscGFP plasmid (Vince et al, 2008). The vector contains a destabilized codon optimized GFP fluorescent protein, similar to EGFP but brighter, with a C‐terminal destabilizing peptide, which shortens the half‐life time of the mature protein to 1 h in absence of additional transcription. Transduced cells were expanded and stimulated with TNF (20 ng/ml) for 16 h prior sorting. To obtain a more homogeneous response, cells with similar levels of EGFP fluorescence were sorted in an FACS Aria III (BD Bioscience).
To create reconstituted cells stably expressing different PIDD1 constructs, 293Trex Flp‐In cells were previously transduced with pLentiCRISPR‐v2 containing sgRNA against PIDD1 and single cell clones were generated as described above. PIDD1 non‐cleavable mutants were generated by introduction of S446A and/or S588A mutations using site‐directed mutagenesis (Quick‐change Lightning site‐directed mutagenesis kit; Agilent Technologies, Santa Clara, CA). PIDD1‐C non‐cleavable and PIDD1‐CC constructs were cloned by PCR using primers encoding HindIII and NotI restriction enzymes. The R815W point mutation was introduced by site‐directed mutagenesis using Quick‐change. Silent mutations were additionally introduced to disrupt the PAM sequence and neutralize the sgRNA (5′ccccatcaccatccgctatcggc) used to target the PIDD1 locus. All constructs were cloned into pcDNA5‐FRT‐TO expression plasmid. 293 Flp‐In cells were transfected with the different cDNAs encoding FLAG‐tagged PIDD1 and the pOG44 Flp‐Recombinase expression vector in a 1:5 ratio. Cells were selected using 100 μg/ml Hygromycin (Roth, CP12.2) and 15 μg/ml Blasticidin.
Protein sample preparation and immunoblotting
Cells were washed with room‐temperature PBS and subjected to direct lysis in the well using Tris–HCl 50 mM, EDTA 1 mM, NaCl 150 mM, pH 7.4 buffer, supplemented with 1% triton X‐100, NaF 50 mM, Na3VO4 1 mM, PMSF 1 mM, and protease inhibitor cocktail (Roche, 11873580001). Cell lysates were incubated in ice for 30 min prior to centrifugation. Protein concentration was measured by Bradford analysis (Bio‐Rad, 500‐0006) using a TECAN plate reader. For immunoblotting analysis, equal amounts of protein samples were separated by SDS‐PAGE, transferred using a wet transfer system (Bio‐Rad) onto nitrocellulose membranes (GE Healthcare), and blocked with 5% w/v non‐fat milk or bovine serum albumin (BSA) in TBS‐tween 0.1% v/v for 1 h. Membranes were incubated overnight at 4°C with the following primary antibodies, diluted in blocking buffer: GAPDH (rabbit, Cell signaling, 2118, 1:5,000), Hsp90 (F‐8) (mouse, Santa Cruz, 13119; 1:1,000), MDM2 (IF2) (mouse, Invitrogen, MA1‐113, 1:500), IKBα (rabbit, Cell Signaling, 9242, 1:1,000), p53 (1C12) (mouse, Cell Signaling, 2524, 1:1,000), MYC‐tag (9E10) (mouse, Invitrogen, MA1‐980, 1: 1000), STING (D2P2F) (rabbit, Cell signaling, 13647; 1:1,000), RIPK1 (D94C12) (rabbit, Cell signaling, 3463; 1:1,000), NEMO (DA10‐12) (mouse, Cell signaling, 2695; 1:100), RAIDD (4B12) (LSBio, LS‐C179788; 1:1,000), FLAG‐M2 (mouse, Sigma, F1804; 1:1,000). Blots were washed with TBS‐Tween 0.1% and then incubated with HRP‐conjugated antibodies (Dako, P0161; 1:10,000) for 1 h at room temperature.
Co‐immunoprecipitation analyses
HEK 293Trex Flp‐In cells stably expressing different inducible PIDD1‐FLAG mutants were seeded in 15 cm dishes and treated with doxycycline (1 μg/ml) for 24 h prior to synchronization, released into ZM (2 μM) or DMSO, and collected at different time points. For transient expression assays, PIDD1‐mutant 293Trex Flp‐In cells were transiently transfected with polyethylenimine (PEI; Polysciences, Hirschberg, Germany, 23966‐1) in a 1:5 w/w ratio (DNA:PEI). For co‐immunoprecipitation, cells were lysed in co‐IP lysis buffer (50 mM Tris pH 8.0, 0.5% NP‐40, 150 mM NaCl, 50 mM NaF, 1 mM Na3VO4, 1 mM PMSF, and EDTA‐free protease inhibitor mix) and incubated on ice for 30 min. Soluble extracts were prepared, and supernatants were incubated with anti‐DYKDDDDK magnetic agarose beads (Pierce, A36797). Beads were washed three times in lysis buffer, and immunopurified protein was analyzed by western blot.
Immunofluorescence
Treated cells grown on 10 mm glass coverslips were washed in PBS and fixed either using 4% PFA for 10 min at room temperature, or absolute methanol for 10 min at −20°C. Cells fixed in PFA required an additional step of permeabilization with 0.2% Triton X‐100 for 10 min at room temperature. Cells were blocked in 5% BSA + 0.2% Tween in PBS for 1 h followed by 1 h of incubation with antibodies diluted in the blocking solution. DNA was detected using DAPI (Sigma, D9542), and samples were mounted in Mowiol. Staining was performed with the following primary antibodies: p65 (D14E12) (rabbit, Santa Cruz, SC‐109; 1:1,000), γ‐tubulin (rabbit, DQ19, Sigma‐Aldrich, T3195; 1:2,000 or mouse, Santa Cruz, sc‐51715; 1:500), PIDD1 (mouse, Enzo, ALX‐804‐837‐C100; 1:500), γH2AX (Ser139) (rabbit, Cell Signaling, 9718; 1:500). Secondary goat antibodies coupled to Alexa Fluor 488 or Alexa Fluor 546 (Invitrogen) were used at 1:750 dilution.
High‐resolution images were captured with an LSM 980 confocal laser scanning microscope (ZEISS) using a 63× Olympus oil objective. Immunofluorescence images were collected at room temperature using a Zeiss Axiovert 200M microscope with an oil immersion objective (Ph3 Plan‐Neofluar 100×/1.3 oil, Zeiss, Oberkochen, Germany). Images were obtained with the acquisition software VisiView 4.1.0.3 (Visitron Systems, Puchheim, Germany). Images were deconvolved with Huygens Professional software (Scientific Volume Imaging, Hilversum, The Netherlands).
Time‐lapse video microscopy
A549 cells stably expressing EGFP were seeded into Ibidi 35 mm μ‐Dishes (Ibidi, 81156) and treated with ZM (2 μM) for 72 h to render them polyploid. Two hours before imaging, cells were washed, counted, and switched to NK cell growth medium. NK‐92 cells were co‐cultured with target cells at a 1:10 target:effector ratio. Images of the cell co‐culture were taken every minute for a time period of for 18 h using a Leica DMi8 microscope with a ×40 objective and a Hamamatsu OrcaFlash 4.0 camera (Hamamatsu Photonics, Hamamatsu, Japan) and an environmental chamber set to 37°C.
NK cell killing assay
A549EGFP cells were seeded in 96‐well plates and incubated for 72 h in the presence of DMSO, ZM (2 μM), DHCB (4 μM), or doxycycline (1 μg/ml). Cells were washed with PBS, counted, and switched to NK cell growth medium. NK‐92 cells were co‐cultured with target cells at a 1:10 target:effector ratio right before the beginning of the filming. Interactions were recorded by Incucyte live‐cell imaging for 50 h at 1 h interval. Confluence data were normalized respect the moment of the addition of NK‐92 cells and plotted referred to the confluence of DMSO‐pretreated (euploid) cells also co‐cultured with NK‐92 cells.
Live cell imaging NF‐κB reporter assays
Cells stably expressing pTRH1 mCMV NF‐κB dscoGFP plasmid were plated in 96‐well plates in technical duplicates. Cells were treated with the indicated compounds and imaged every 2 h in an IncuCyte S3 microscope (Sartorius) using the 10× objective. Four positions per well were imaged. Data were analyzed in the IncuCyte software and normalized to image EGFP MFI values at time zero to determine the fold change in MFI. When different cell lines were compared, each one was normalized to their respective DMSO control.
Luciferase reporter assays
For NF‐κB reporter luciferase assays, cells were seeded in 24 well plates and transiently co‐transfected with plasmids pRL‐TK renilla luc and pGL3‐2x(κB)‐luc (Reintjes et al, 2016) in a ratio 1:10 using Metafectene (Biontex, Munich, Germany, T020‐1.0). The next day after transfection, medium was changed, and cells were subjected to the indicated treatments. Cells were collected after treatment and analyzed for luciferase activity using Dual Stop and Glo Luciferase Kit (Promega). NF‐κB activity was determined by measuring the luciferase activity present in cell extracts. Luciferase relative light unit (RLU) values were normalized for differences in transient transfection efficiency on the basis of renilla activity in the same extracts and expressed as fold change values relative to unstimulated cells.
Flow cytometry
To determine the effect of cytokinesis failure in DNA content, A549 cells were seeded in duplicates for each time point and synchronized once with thymidine (2 mM) for 24 h. After washout and release, one well was treated with ZM (2 μM) and the other with DMSO. Cell suspensions were washed with PBS and fixed overnight in 1 ml ice‐cold 70% ethanol at −20°C. After fixation, cells were collected by centrifugation (2000 r.p.m., 2 min), washed twice with PBS to remove ethanol, and incubated during 45 min with 40 μg/ml propidium iodide (Sigma‐Aldrich, P4170) at 37°C in the presence of RNase A (Sigma‐Aldrich, R4875). After this, cells were transferred to microtiter tubes and acquired on LOW mode.
For intracellular phospho‐Histone H3 staining, 1 × 106 cells were fixed in 1 ml ice‐cold 70% ethanol in FACS tubes overnight at −20°C. Cells were washed twice in PBS (2000 r.p.m., 2 min), transferred to 96‐well plates, and permeabilized with 200 μl of 0.25% Triton X‐100 (Sigma T8787) in PBS for 20 min. Cells were washed with PBS and incubated for 60 min with phospho‐Histone 3 Ser‐10 antibody (1:200; Cell Signalling, 9701) in 50 μl of staining buffer (PBS with 3% FCS). Cells were washed twice with PBS with 0.25% Triton X‐100 and incubated 30 min with Alexa Fluor 488 goat α‐rabbit (1:1,000; Invitrogen, A11034) in 50 μl of staining buffer. Cells were washed twice with PBS, resuspended in 100 μl of PBS containing 250 μg/ml RNase A, and incubated for 30 min at 37°C. Prior to acquisition, 50 μl of 10 μg/ml DAPI (Sigma, D9542) in PBS was added.
For cell surface marker expression analysis, BMDM was harvested after 6 days of differentiation using 250 μl of Accutase for 30 min at 37°C. Accutase activity was neutralized with 500 μl of DMEM GlutaMax medium, and BMDM was collected by centrifugation. Cells were washed with 0.5% FCS in PBS and transferred to 96‐well plates. To prevent unspecific antibody‐binding, cells were pre‐incubated with 1 μg/ml of Fc‐block (BioLegend, 101310) in 30 μl staining buffer containing 2 mM EDTA (Sigma, ED4S) for 15 min. After this, cells were collected (2,000 r.p.m., 2 min), washed, and stained with F4/80‐PE (1:200; BioLegend, 123110) and CD11b‐PerCP (1:200; BioLegend, 101228) antibodies, alone or in combination, in 50 μl of staining buffer for 30 min. Cells were washed twice and resuspended in 200 μl of staining buffer. Cell viability was assessed adding DAPI (1:10,000; Sigma‐Aldrich, D‐9542) 10 min prior flow cytometric analysis.
During all preparations and staining steps, cells were kept at 4°C. Data were acquired on a LSRII cytometer (BD Bioscience) in “low” acquisition mode and analyzed by FlowJo software (Tree star, Ashland, OR, USA). Non‐singlet events were typically excluded from analysis based on characteristics of FSC‐H/FSC‐W and SSC‐H/SSC‐W.
Bone marrow–derived macrophage polarization assays
Primary MEFPLK4 derived from Pidd1 +/+ and Pidd1 −/− mice was seeded in 10 cm plates and incubated for 72 h in the presence of DMSO, ZM (2 μM) or doxycycline (1 μg/ml) until they reached ~ 80% confluence. After treatment, cells were washed three times with PBS to remove the drugs, and their medium was replaced for DMEM without serum for 16 h. The conditioned medium (CM) was collected, centrifuged at 1,500 rpm for 10 min, and filtered through a 0.2 μm pore filter. CM was added to BMDM previously seeded in 6 cm plates and differentiated as described above.
RNA isolation and qRT‐PCR
Total RNA from untreated or stimulated cells was extracted using RNAeasy Mini Kit (Quiagen) including an optional DNA on‐column digestion using the RNAse‐Free DNAse Set (Quiagen, 79254). First‐strand cDNA was generated from 100 ng of total RNA with a 260/280 ratio of > 1.8 using the iScript cDNA Synthesis Kit (Bio‐Rad, Hercules, CA, USA, 170‐8891), following manufacturer's instructions. Samples were assayed using the AceQ qPCR SYBR Green Master Mix (Vazyme Q111‐02) on a StepOnePlus Real‐time PCR system (Applied Biosystems, Foster City, CA, USA). Gene expression was normalized to the geometrical mean of the house‐keeping genes and displayed as the fold change referred to unstimulated control samples, as previously described (Livak & Schmittgen, 2001). Where different groups (genotype, medium conditions) are compared side‐by‐side, gene expression of one group is displayed as the fold‐change, compared to an unstimulated control sample of that group, to ensure that intrinsic differences in untreated conditions are accounted for. Melting curve analysis was performed for every run to verify the presence of a single PCR product. Raw data were analyzed with LinReg PCR software (Ruijter et al, 2009) and N0 fluorescence values were calculated using the same program. A no‐template blank served as negative control. The following primers were used at a final concentration of 100 nM: Ccl5 fw: GCTGCCCTCACCATCATCC, Ccl5 rv: TGATGTATTCTTGAACCCACTTCTTC; Ankrd1 fw:CGGAAAAGCGAGAAACTGCGA, Ankrd1 rv:TCAGAAACCTCGGCACATCCA; Icam fw: TGGAACTGCACGTGCTGTATG, Icam rv: CCGTCTGCAGGTCATCTTAGG; Cxcl2 fw: GATACTGAACAAAGGCAAGGCTAAC, Cxcl2 rv:GGCACATCAGGTACGATCCAG; Ccl20 fw: CGTCTGCTCTTCCTTGCTTTG, Ccl20 rv:GCCATCTGTCTTGTGAAACCC; Hprt fw: GTCATGCCGACCCGCAGTC, Hprt rv: GTCCTTCCATAATAGTCCATGAGGAATAAAC; Cxcl14 fw: GAAGATGGTTATCGTCACCACC, Cxcl14 rv: CGTTCCAGGCATTGTACCACT; IL6 fw: GCTACCAAACTGGATATAATCAGGA, IL6 rv: CCAGGTAGCTATGGTACTCCAGAA; β‐actin fw: TGGCACCACACCTTCTACAATG, β‐actin rv:GACCAGAGGCATACAGGGACAG; Ccl2 fw: ATTGGGATCATCTTGCTGGT, Ccl2rv: CCTGGTGTTCACAGTTGCC; Fas fw: ACTGCGATTCTCCTGGCTGT, Fas rv: GCACGGCTCAAGGGTTCCAT; Ccl7 fw: CCTGGGAAGCTGTTATCTTCAAG, Ccl7 rv:CCTCCTCGACCCACTTCTGA; Ikba fw: TCCTGCAGGCCACCAACTAC, Ikba rv: GCTGTCCGGCCATTGC; Ccl3 fw: CACCAGAAGGATACAAGCAGCAG, Ccl3 rv:TAGGAGAAGCAGCAGGCAGTC; Cxcl10 fw: GCCCACGTGTTGAGATCATTG, Cxcl10 rv:AGGCTCTCTGCTGTCCATCC.
RNAseq analysis
Total RNA from cultured primary MEF was extracted using RNAeasy Mini Kit (Quiagen) as per the manufacturer's instructions. DNA was removed by on‐column digestion using the RNAse‐Free DNAse Set (Quiagen, 79254), and RNA concentration was determined in a NanoDrop device. High‐quality RNA was used for downstream application. mRNA‐sequencing libraries were prepared using 200 ng of total RNA using NEBNext Ultra II Directional RNA library prep kit with UMI Adaptors. The libraries were pooled equimolarly and sequenced on 2 lanes of Hiseq 3000/4000 with the following configuration: 51 + 19 + 8 (single read). Single‐end RNA files quality was checked using Fastqc (Wingett & Andrews, 2018; v 0.11.8), and adapters were trimmed using trim_galore (v 0.6.4_dev). Alignment was done with STAR (Dobin et al, 2013; v 2.6.1) to mouse genome (mm10). Aligned files were processed using samtools (Li et al, 2009; v 1.9) and bedtools (Quinlan & Hall, 2010; v 2.25.0), and only reads with mapq > 30 were considered for further analysis. Counts were quantified to exons using Rsubread (Liao et al, 2019a; v 2.0.1) and Mus_musculus.GRCm38.99.gtf from Ensembl. Differential expression (DE) analysis was performed in those genes with at least 10 counts per sample, −globally more than 60 as the comparison was 3 × 3‐ with Deseq2 (Love et al, 2014; v 1.26.1), and gene annotation was performed with biomaRt (Durinck et al, 2009; v 2.42.1) in R (v 3.6.1). The analysis was performed using the likelihood‐ratio test, and sex differences were removed with the reduced argument. Volcano and bubble plots were performed with ggplot2 (v 3.3.3). The gene ontology and gene set enrichment analysis were performed with WebGestalt (Liao et al, 2019b) and with GSEA (Mootha et al, 2003; Subramanian et al, 2005; v 4.2.3) with the filters mentioned in each figure, respectively. The genes for the GO term Immune System Process were obtained from AmiGO2 (Carbon et al, 2009). For heatmaps, Pheatmap (v1.1.12) was used in R (v4.3.1). For the pie charts, we selected those genes with Padj < 0.05 and considering the strand we chose 4,000 bp upstream and 1,000 bp downstream of the transcription start site (TSS) as the promoter region. In this set, Fimo (Grant et al, 2011; v 5.1.0) was tested for TP53 and RELA motif presence using the transcription factor binding profiles from Jaspar2022 (Castro‐Mondragon et al, 2022; MA0106.3 and MA0107.1 respectively).
Quantification and statistical analysis
Statistical analysis was performed using GraphPad (San Diego, CA, USA) Prism software. Differences between samples were tested as stated in the respective figure legends. Statistical comparisons of different genotypic groups receiving the same treatment were performed using one‐way or two‐way ANOVA analysis, followed by Tukey's post‐hoc correction for multiple comparisons. Replicates from experiments with immortalized cell lines were acquired on different days and with cells in different passages. Each data point from graphs of primary MEF or BMDM represents an independent biological replicate. In all analyses, significance was annotated following the nomenclature: ns (P > 0.05), *(P ≤ 0.05), **(P ≤ 0.01), ***(P ≤ 0.001), and **** (P ≤ 0.0001).
Author contributions
Irmina García‐Carpio: Conceptualization; data curation; investigation; methodology; writing – original draft. Vincent Z Braun: Investigation; methodology. Elias S Weiler: Data curation; investigation. Marina Leone: Investigation; methodology. Sergio Niñerola: Data curation. Angel Barco: Data curation; supervision. Luca L Fava: Resources. Andreas Villunger: Conceptualization; supervision; funding acquisition; investigation; writing – original draft; project administration.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix S1
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We are grateful to I. Gaggl, J. Heppke, and C. Soratroi for excellent technical assistance. We thank, A. Holland, T. Valovka, E. Nigg, P. Meier, J. Silke, F. Eichin, W. Posch, for cells or reagents, as well as S. Geley, for help with microscopy and fruitful discussion. This work was supported by AIRC under MFAG 2019 ‐ ID. 23560 project ‐ awarded to LLF, Spanish Ministry of Science and Innovation (MICINN) awarded to SN, PID2020‐118169RB‐100 and AEI/10.13039/ 501100011033 awarded to AB, and the ERC‐AdG “POLICE” (#787171) awarded to AV.
The EMBO Journal (2023) 42: e113510
See also: AN Brown-Suedel & L Bouchier-Hayes (October 2023)
Footnotes
Correction added on 11 September 2023 after first online publication: Panel E of Figure 3 has been replaced.
Contributor Information
Irmina Garcia‐Carpio, Email: irmina.garcia@i-med.ac.at.
Andreas Villunger, Email: andreas.villunger@i-med.ac.at.
Data availability
The RNAseq data have been deposited in Gene Expression Omnibus with the accession number GSE235120. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE235120.
References
- Acebes‐Huerta A, Lorenzo‐Herrero S, Folgueras AR, Huergo‐Zapico L, Lopez‐Larrea C, Lopez‐Soto A, Gonzalez S (2016) Drug‐induced hyperploidy stimulates an antitumor NK cell response mediated by NKG2D and DNAM‐1 receptors. Onco Targets Ther 5: e1074378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams SD, Csere J, D'Angelo G, Carter EP, Romao M, Arnandis T, Dodel M, Kocher HM, Grose R, Raposo G et al (2021) Centrosome amplification mediates small extracellular vesicle secretion via lysosome disruption. Curr Biol 31: 1403–1416.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando K, Kernan JL, Liu PH, Sanda T, Logette E, Tschopp J, Look AT, Wang J, Bouchier‐Hayes L, Sidi S (2012) PIDD death‐domain phosphorylation by ATM controls prodeath versus prosurvival PIDDosome signaling. Mol Cell 47: 681–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando K, Parsons MJ, Shah RB, Charendoff CI, Paris SL, Liu PH, Fassio SR, Rohrman BA, Thompson R, Oberst A et al (2017) NPM1 directs PIDDosome‐dependent caspase‐2 activation in the nucleolus. J Cell Biol 216: 1795–1810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreassen PR, Lohez OD, Lacroix FB, Margolis RL (2001) Tetraploid state induces p53‐dependent arrest of nontransformed mammalian cells in G1. Mol Biol Cell 12: 1315–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda F, Chaba K, Bloy N, Garcia P, Bordenave C, Martins I, Stoll G, Tesniere A, Kroemer G, Senovilla L (2018) Immune effectors responsible for the elimination of hyperploid cancer cells. Onco Targets Ther 7: e1463947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnandis T, Monteiro P, Adams SD, Bridgeman VL, Rajeeve V, Gadaleta E, Marzec J, Chelala C, Malanchi I, Cutillas PR et al (2018) Oxidative stress in cells with extra centrosomes drives non‐cell‐autonomous invasion. Dev Cell 47: 409–424.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basto R, Brunk K, Vinadogrova T, Peel N, Franz A, Khodjakov A, Raff JW (2008) Centrosome amplification can initiate tumorigenesis in flies. Cell 133: 1032–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzi H, Anderson KV (2014) Acentriolar mitosis activates a p53‐dependent apoptosis pathway in the mouse embryo. Proc Natl Acad Sci USA 111: E1491–E1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielski CM, Zehir A, Penson AV, Donoghue MTA, Chatila W, Armenia J, Chang MT, Schram AM, Jonsson P, Bandlamudi C et al (2018) Genome doubling shapes the evolution and prognosis of advanced cancers. Nat Genet 50: 1189–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock FJ, Krumschnabel G, Manzl C, Peintner L, Tanzer MC, Hermann‐Kleiter N, Baier G, Llacuna L, Yelamos J, Villunger A (2013) Loss of PIDD limits NF‐kappaB activation and cytokine production but not cell survival or transformation after DNA damage. Cell Death Differ 20: 546–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowler M, Kong D, Sun S, Nanjundappa R, Evans L, Farmer V, Holland A, Mahjoub MR, Sui H, Loncarek J (2019) High‐resolution characterization of centriole distal appendage morphology and dynamics by correlative STORM and electron microscopy. Nat Commun 10: 993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burigotto M, Mattivi A, Migliorati D, Magnani G, Valentini C, Roccuzzo M, Offterdinger M, Pizzato M, Schmidt A, Villunger A et al (2021) Centriolar distal appendages activate the centrosome‐PIDDosome‐p53 signalling axis via ANKRD26. EMBO J 40: e104844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B, Lewis S, Ami GOH, Web Presence Working Group (2009) AmiGO: online access to ontology and annotation data. Bioinformatics 25: 288–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro‐Mondragon JA, Riudavets‐Puig R, Rauluseviciute I, Lemma RB, Turchi L, Blanc‐Mathieu R, Lucas J, Boddie P, Khan A, Manosalva Perez N et al (2022) JASPAR 2022: the 9th release of the open‐access database of transcription factor binding profiles. Nucleic Acids Res 50: D165–D173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JY (2011) A clinical overview of centrosome amplification in human cancers. Int J Biol Sci 7: 1122–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao MP, Jaiswal S, Weissman‐Tsukamoto R, Alizadeh AA, Gentles AJ, Volkmer J, Weiskopf K, Willingham SB, Raveh T, Park CY et al (2010) Calreticulin is the dominant pro‐phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci Transl Med 2: 63ra94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho PA, Bury L, Shahbazi MN, Liakath‐Ali K, Tate PH, Wormald S, Hindley CJ, Huch M, Archer J, Skarnes WC et al (2015) Over‐expression of Plk4 induces centrosome amplification, loss of primary cilia and associated tissue hyperplasia in the mouse. Open Biol 5: 150209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concordet JP, Haeussler M (2018) CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res 46: W242–W245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Assoro AB, Barrett SL, Folk C, Negron VC, Boeneman K, Busby R, Whitehead C, Stivala F, Lingle WL, Salisbury JL (2002) Amplified centrosomes in breast cancer: a potential indicator of tumor aggressiveness. Breast Cancer Res Treat 75: 25–34 [DOI] [PubMed] [Google Scholar]
- Davoli T, de Lange T (2011) The causes and consequences of polyploidy in normal development and cancer. Annu Rev Cell Dev Biol 27: 585–610 [DOI] [PubMed] [Google Scholar]
- Dieckmann NM, Frazer GL, Asano Y, Stinchcombe JC, Griffiths GM (2016) The cytotoxic T lymphocyte immune synapse at a glance. J Cell Sci 129: 2881–2886 [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Spellman PT, Birney E, Huber W (2009) Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc 4: 1184–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans LT, Anglen T, Scott P, Lukasik K, Loncarek J, Holland AJ (2021) ANKRD26 recruits PIDD1 to centriolar distal appendages to activate the PIDDosome following centrosome amplification. EMBO J 40: e105106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fava LL, Schuler F, Sladky V, Haschka MD, Soratroi C, Eiterer L, Demetz E, Weiss G, Geley S, Nigg EA et al (2017) The PIDDosome activates p53 in response to supernumerary centrosomes. Genes Dev 31: 34–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltham R, Jamal K, Tenev T, Liccardi G, Jaco I, Domingues CM, Morris O, John SW, Annibaldi A, Widya M et al (2018) Mind bomb regulates cell death during TNF signaling by suppressing RIPK1's cytotoxic potential. Cell Rep 23: 470–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D (2005) Cytokinesis failure generating tetraploids promotes tumorigenesis in p53‐null cells. Nature 437: 1043–1047 [DOI] [PubMed] [Google Scholar]
- Ganem NJ, Pellman D (2007) Limiting the proliferation of polyploid cells. Cell 131: 437–440 [DOI] [PubMed] [Google Scholar]
- Ganem NJ, Godinho SA, Pellman D (2009) A mechanism linking extra centrosomes to chromosomal instability. Nature 460: 278–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Cornils H, Chiu SY, O'Rourke KP, Arnaud J, Yimlamai D, Thery M, Camargo FD, Pellman D (2014) Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell 158: 833–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganier O, Schnerch D, Oertle P, Lim RY, Plodinec M, Nigg EA (2018) Structural centrosome aberrations promote non‐cell‐autonomous invasiveness. EMBO J 37: e98576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy‐Ullrich JE, Bratton DL, Oldenborg PA, Michalak M, Henson PM (2005) Cell‐surface calreticulin initiates clearance of viable or apoptotic cells through trans‐activation of LRP on the phagocyte. Cell 123: 321–334 [DOI] [PubMed] [Google Scholar]
- Gemble S, Wardenaar R, Keuper K, Srivastava N, Nano M, Mace AS, Tijhuis AE, Bernhard SV, Spierings DCJ, Simon A et al (2022) Genetic instability from a single S phase after whole‐genome duplication. Nature 604: 146–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, Smith HO (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6: 343–345 [DOI] [PubMed] [Google Scholar]
- Godinho SA, Picone R, Burute M, Dagher R, Su Y, Leung CT, Polyak K, Brugge JS, Thery M, Pellman D (2014) Oncogene‐like induction of cellular invasion from centrosome amplification. Nature 510: 167–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong JH, Maki G, Klingemann HG (1994) Characterization of a human cell line (NK‐92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 8: 652–658 [PubMed] [Google Scholar]
- Gordon DJ, Resio B, Pellman D (2012) Causes and consequences of aneuploidy in cancer. Nat Rev Genet 13: 189–203 [DOI] [PubMed] [Google Scholar]
- Gotthardt D, Sexl V (2016) STATs in NK‐cells: the good, the bad, and the ugly. Front Immunol 7: 694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, Noble WS (2011) FIMO: scanning for occurrences of a given motif. Bioinformatics 27: 1017–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, Knoblaugh S, Cado D, Greenberg NM, Raulet DH (2008) NKG2D‐deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity 28: 571–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hachet V, Canard C, Gonczy P (2007) Centrosomes promote timely mitotic entry in C. elegans embryos. Dev Cell 12: 531–541 [DOI] [PubMed] [Google Scholar]
- Hara T, Tanegashima K (2012) Pleiotropic functions of the CXC‐type chemokine CXCL14 in mammals. J Biochem 151: 469–476 [DOI] [PubMed] [Google Scholar]
- Holland AJ, Cleveland DW (2009) Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol 10: 478–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland AJ, Fachinetti D, Zhu Q, Bauer M, Verma IM, Nigg EA, Cleveland DW (2012) The autoregulated instability of Polo‐like kinase 4 limits centrosome duplication to once per cell cycle. Genes Dev 26: 2684–2689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Barber GN (2008) STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455: 674–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens S, Tinel A (2012) The PIDDosome, DNA‐damage‐induced apoptosis and beyond. Cell Death Differ 19: 13–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens S, Tinel A, Lippens S, Tschopp J (2005) PIDD mediates NF‐kappaB activation in response to DNA damage. Cell 123: 1079–1092 [DOI] [PubMed] [Google Scholar]
- Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, Lu T, Yankner BA, Campisi J, Elledge SJ (2015) The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349: aaa5612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Cao Y, Greten FR, Li ZW (2002) NF‐kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer 2: 301–310 [DOI] [PubMed] [Google Scholar]
- Kuhne L, Jung B, Poth H, Schuster A, Wurm S, Ruemmele P, Banas B, Bergler T (2017) Renal allograft rejection, lymphocyte infiltration, and de novo donor‐specific antibodies in a novel model of non‐adherence to immunosuppressive therapy. BMC Immunol 18: 52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrus BG, Daggubati V, Uetake Y, Scott PM, Clutario KM, Sluder G, Holland AJ (2016) A USP28‐53BP1‐p53‐p21 signaling axis arrests growth after centrosome loss or prolonged mitosis. J Cell Biol 214: 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HO, Davidson JM, Duronio RJ (2009) Endoreplication: polyploidy with purpose. Genes Dev 23: 2461–2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine MS, Bakker B, Boeckx B, Moyett J, Lu J, Vitre B, Spierings DC, Lansdorp PM, Cleveland DW, Lambrechts D et al (2017) Centrosome amplification is sufficient to promote spontaneous tumorigenesis in mammals. Dev Cell 40: 313–322.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao L, Guo Y, Zhuang X, Li W, Zou J, Su Q, Zhao J, Liu Y, Liao X, Du Z et al (2018) Immunosuppressive effect of ticagrelor on dendritic cell function: a new therapeutic target of antiplatelet agents in cardiovascular disease. J Biomed Nanotechnol 14: 1665–1673 [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W (2019a) The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res 47: e47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B (2019b) WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res 47: W199–W205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YH, Zhang S, Zhu M, Lu T, Chen K, Wen Z, Wang S, Xiao G, Luo D, Jia Y et al (2020) Mice with increased numbers of polyploid hepatocytes maintain regenerative capacity but develop fewer hepatocellular carcinomas following chronic liver injury. Gastroenterology 158: 1698–1712.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
- LoMastro GM, Holland AJ (2019) The emerging link between centrosome aberrations and metastasis. Dev Cell 49: 325–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Castejon G (2020) Control of the inflammasome by the ubiquitin system. FEBS J 287: 11–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Sanchez LM, Jimenez C, Valverde A, Hernandez V, Penarando J, Martinez A, Lopez‐Pedrera C, Munoz‐Castaneda JR, De la Haba‐Rodriguez JR, Aranda E et al (2014) CoCl2, a mimic of hypoxia, induces formation of polyploid giant cells with stem characteristics in colon cancer. PloS One 9: e99143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Sanchez N, Fontan‐Lozano A, Palle A, Gonzalez‐Alvarez V, Rabano A, Trejo JL, Frade JM (2017) Neuronal tetraploidization in the cerebral cortex correlates with reduced cognition in mice and precedes and recapitulates Alzheimer's‐associated neuropathology. Neurobiol Aging 56: 50–66 [DOI] [PubMed] [Google Scholar]
- Lopez‐Soto A, Gonzalez S, Lopez‐Larrea C, Kroemer G (2017a) Immunosurveillance of malignant cells with complex karyotypes. Trends Cell Biol 27: 880–884 [DOI] [PubMed] [Google Scholar]
- Lopez‐Soto A, Gonzalez S, Smyth MJ, Galluzzi L (2017b) Control of metastasis by NK cells. Cancer Cell 32: 135–154 [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A et al (2017) cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548: 461–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maghazachi AA, al‐Aoukaty A, Schall TJ (1994) C‐C chemokines induce the chemotaxis of NK and IL‐2‐activated NK cells. Role for G proteins. J Immunol 153: 4969–4977 [PubMed] [Google Scholar]
- Magupalli VG, Negro R, Tian Y, Hauenstein AV, Di Caprio G, Skillern W, Deng Q, Orning P, Alam HB, Maliga Z et al (2020) HDAC6 mediates an aggresome‐like mechanism for NLRP3 and pyrin inflammasome activation. Science 369: eaas8995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, Marchesi F, Garlanda C (2022) Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discov 21: 799–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzl C, Peintner L, Krumschnabel G, Bock F, Labi V, Drach M, Newbold A, Johnstone R, Villunger A (2012) PIDDosome‐independent tumor suppression by Caspase‐2. Cell Death Differ 19: 1722–1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller K, Brambach M, Villani A, Gallo E, Gilmour D, Peri F (2022) A role for the centrosome in regulating the rate of neuronal efferocytosis by microglia in vivo . Elife 11: e82094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E et al (2003) PGC‐1alpha‐responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34: 267–273 [DOI] [PubMed] [Google Scholar]
- Morrison BE, Park SJ, Mooney JM, Mehrad B (2003) Chemokine‐mediated recruitment of NK cells is a critical host defense mechanism in invasive aspergillosis. J Clin Invest 112: 1862–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthuswamy R, Berk E, Junecko BF, Zeh HJ, Zureikat AH, Normolle D, Luong TM, Reinhart TA, Bartlett DL, Kalinski P (2012) NF‐kappaB hyperactivation in tumor tissues allows tumor‐selective reprogramming of the chemokine microenvironment to enhance the recruitment of cytolytic T effector cells. Cancer Res 72: 3735–3743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg EA (2006) Origins and consequences of centrosome aberrations in human cancers. Int J Cancer 119: 2717–2723 [DOI] [PubMed] [Google Scholar]
- Nigg EA, Holland AJ (2018) Once and only once: mechanisms of centriole duplication and their deregulation in disease. Nat Rev Mol Cell Biol 19: 297–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MH, Hong JT (2016) Roles of NF‐kappaB in cancer and inflammatory diseases and their therapeutic approaches. Cell 5: 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploner C, Rauchenwald T, Connolly CE, Joehrer K, Rainer J, Seifarth C, Hermann M, Nagl M, Lobenwein S, Wilflingseder D et al (2021) Oxidant therapy improves adipogenic differentiation of adipose‐derived stem cells in human wound healing. Stem Cell Res Ther 12: 280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reintjes A, Fuchs JE, Kremser L, Lindner HH, Liedl KR, Huber LA, Valovka T (2016) Asymmetric arginine dimethylation of RelA provides a repressive mark to modulate TNFalpha/NF‐kappaB response. Proc Natl Acad Sci USA 113: 4326–4331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter AT, Asano Y, Stinchcombe JC, Dieckmann NM, Chen BC, Gawden‐Bone C, van Engelenburg S, Legant W, Gao L, Davidson MW et al (2015) Actin depletion initiates events leading to granule secretion at the immunological synapse. Immunity 42: 864–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J (2009) Persistent DNA damage signalling triggers senescence‐associated inflammatory cytokine secretion. Nat Cell Biol 11: 973–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenstiel PE, Gruosso T, Letourneau AM, Chan JJ, LeBlanc A, Husain M, Najfeld V, Planelles V, D'Agati VD, Klotman ME et al (2008) HIV‐1 Vpr inhibits cytokinesis in human proximal tubule cells. Kidney Int 74: 1049–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruijter JM, Ramakers C, Hoogaars WM, Karlen Y, Bakker O, van den Hoff MJ, Moorman AF (2009) Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res 37: e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Richardson A, Iyer DR, M'Saad O, Zasadil L, Knouse KA, Wong YL, Rhind N, Desai A, Amon A (2017) Chromosome mis‐segregation generates cell‐cycle‐arrested cells with complex karyotypes that are eliminated by the immune system. Dev Cell 41: 638–651.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senovilla L, Vitale I, Martins I, Tailler M, Pailleret C, Michaud M, Galluzzi L, Adjemian S, Kepp O, Niso‐Santano M et al (2012) An immunosurveillance mechanism controls cancer cell ploidy. Science 337: 1678–1684 [DOI] [PubMed] [Google Scholar]
- Shah RB, Kernan JL, van Hoogstraten A, Ando K, Li Y, Belcher AL, Mininger I, Bussenault AM, Raman R, Ramanagoudr‐Bhojappa R et al (2021) FANCI functions as a repair/apoptosis switch in response to DNA crosslinks. Dev Cell 56: 2207–2222.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh TI, Vasli N, Pastore S, Kharizi K, Harripaul R, Fattahi Z, Pande S, Naeem F, Hussain A, Mir A et al (2021) Biallelic mutations in the death domain of PIDD1 impair caspase‐2 activation and are associated with intellectual disability. Transl Psychiatry 11: 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silkworth WT, Nardi IK, Scholl LM, Cimini D (2009) Multipolar spindle pole coalescence is a major source of kinetochore mis‐attachment and chromosome mis‐segregation in cancer cells. PLoS One 4: e6564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladky V, Schuler F, Fava LL, Villunger A (2017) The resurrection of the PIDDosome – emerging roles in the DNA‐damage response and centrosome surveillance. J Cell Sci 130: 3779–3787 [DOI] [PubMed] [Google Scholar]
- Sladky VC, Knapp K, Soratroi C, Heppke J, Eichin F, Rocamora‐Reverte L, Szabo TG, Bongiovanni L, Westendorp B, Moreno E et al (2020a) E2F‐family members engage the PIDDosome to limit hepatocyte ploidy in liver development and regeneration. Dev Cell 52: 335–349.e7 [DOI] [PubMed] [Google Scholar]
- Sladky VC, Knapp K, Szabo TG, Braun VZ, Bongiovanni L, van den Bos H, Spierings DC, Westendorp B, Curinha A, Stojakovic T et al (2020b) PIDDosome‐induced p53‐dependent ploidy restriction facilitates hepatocarcinogenesis. EMBO Rep 21: e50893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturmlechner I, Zhang C, Sine CC, van Deursen EJ, Jeganathan KB, Hamada N, Grasic J, Friedman D, Stutchman JT, Can I et al (2021) p21 produces a bioactive secretome that places stressed cells under immunosurveillance. Science 374: eabb3420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi K, Karin M (2018) NF‐kappaB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol 18: 309–324 [DOI] [PubMed] [Google Scholar]
- Tanos BE, Yang HJ, Soni R, Wang WJ, Macaluso FP, Asara JM, Tsou MF (2013) Centriole distal appendages promote membrane docking, leading to cilia initiation. Genes Dev 27: 163–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinel A, Tschopp J (2004) The PIDDosome, a protein complex implicated in activation of caspase‐2 in response to genotoxic stress. Science 304: 843–846 [DOI] [PubMed] [Google Scholar]
- Tinel A, Janssens S, Lippens S, Cuenin S, Logette E, Jaccard B, Quadroni M, Tschopp J (2007) Autoproteolysis of PIDD marks the bifurcation between pro‐death caspase‐2 and pro‐survival NF‐kappaB pathway. EMBO J 26: 197–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinel A, Eckert MJ, Logette E, Lippens S, Janssens S, Jaccard B, Quadroni M, Tschopp J (2011) Regulation of PIDD auto‐proteolysis and activity by the molecular chaperone Hsp90. Cell Death Differ 18: 506–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertii A, Ivshina M, Zimmerman W, Hehnly H, Kant S, Doxsey S (2016) The centrosome undergoes Plk1‐independent interphase maturation during inflammation and mediates cytokine release. Dev Cell 37: 377–386 [DOI] [PubMed] [Google Scholar]
- Vince JE, Chau D, Callus B, Wong WW, Hawkins CJ, Schneider P, McKinlay M, Benetatos CA, Condon SM, Chunduru SK et al (2008) TWEAK‐FN14 signaling induces lysosomal degradation of a cIAP1‐TRAF2 complex to sensitize tumor cells to TNFalpha. J Cell Biol 182: 171–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RW, Vigano S, Ben‐David U, Amon A, Santaguida S (2021) Aneuploid senescent cells activate NF‐kappaB to promote their immune clearance by NK cells. EMBO Rep 22: e52032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weier AK, Homrich M, Ebbinghaus S, Juda P, Mikova E, Hauschild R, Zhang L, Quast T, Mass E, Schlitzer A et al (2022) Multiple centrosomes enhance migration and immune cell effector functions of mature dendritic cells. J Cell Biol 221: e202107134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler ES, Szabo TG, Garcia‐Carpio I, Villunger A (2022) PIDD1 in cell cycle control, sterile inflammation and cell death. Biochem Soc Trans 50: 813–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingett SW, Andrews S (2018) FastQ Screen: a tool for multi‐genome mapping and quality control. F1000Res 7: 1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YL, Anzola JV, Davis RL, Yoon M, Motamedi A, Kroll A, Seo CP, Hsia JE, Kim SK, Mitchell JW et al (2015) Cell biology. Reversible centriole depletion with an inhibitor of Polo‐like kinase 4. Science 348: 1155–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XD, Li W, Zhang S, Wu D, Jiang X, Tan R, Niu X, Wang Q, Wu X, Liu Z et al (2020) PLK4 deubiquitination by Spata2‐CYLD suppresses NEK7‐mediated NLRP3 inflammasome activation at the centrosome. EMBO J 39: e102201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi J, Wu X, Chung AH, Chen JK, Kapoor TM, Hammer JA (2013) Centrosome repositioning in T cells is biphasic and driven by microtubule end‐on capture‐shrinkage. J Cell Biol 202: 779–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhsng CZ, Wala J, Mermel CH et al (2013) Pan‐cancer patterns of somatic copy number alteration. Nat Genet 45: 1134–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zebrowski DC, Vergarajauregui S, Wu CC, Piatkowski T, Becker R, Leone M, Hirth S, Ricciardi F, Falk N, Giessl A et al (2015) Developmental alterations in centrosome integrity contribute to the post‐mitotic state of mammalian cardiomyocytes. Elife 4: e05563 [DOI] [PMC free article] [PubMed] [Google Scholar]