

ABSTRACT
Staphylococcus aureus is a facultative intracellular pathogen in many host cell types, facilitating its persistence in chronic infections. The genes contributing to intracellular pathogenesis have not yet been fully enumerated. Here, we cataloged genes influencing S. aureus invasion and survival within human THP-1 derived macrophages using two laboratory strains (ATCC2913 and JE2). We developed an in vitro transposition method to produce highly saturated transposon mutant libraries in S. aureus and performed transposon insertion sequencing (Tn-Seq) to identify candidate genes with significantly altered abundance following macrophage invasion. While some significant genes were strain-specific, 108 were identified as common across both S. aureus strains, with most (n = 106) being required for optimal macrophage infection. We used CRISPR interference (CRISPRi) to functionally validate phenotypic contributions for a subset of genes. Of the 20 genes passing validation, seven had previously identified roles in S. aureus virulence, and 13 were newly implicated. Validated genes frequently evidenced strain-specific effects, yielding opposing phenotypes when knocked down in the alternative strain. Genomic analysis of de novo mutations occurring in groups (n = 237) of clonally related S. aureus isolates from the airways of chronically infected individuals with cystic fibrosis (CF) revealed significantly greater in vivo purifying selection in conditionally essential candidate genes than those not associated with macrophage invasion. This study implicates a core set of genes necessary to support macrophage invasion by S. aureus, highlights strain-specific differences in phenotypic effects of effector genes, and provides evidence for selection of candidate genes identified by Tn-Seq analyses during chronic airway infection in CF patients in vivo.
KEYWORDS: Staphylococcus aureus, facultatively intracellular pathogens, transposons, Tn-Seq, genomics, cystic fibrosis, chronic infection, macrophages, persistence, cell invasion
INTRODUCTION
Staphylococcus aureus is a prevalent and well-studied bacterium capable of causing a range of diseases in humans and animals alike but has only recently been recognized as a facultative intracellular pathogen (1, 2). Multiple studies conducted both in vitro and in vivo have demonstrated that S. aureus is able to enter, replicate within, and persist inside various host cell types, including professional and non-professional phagocytes (1 – 4). Intracellular pathogenesis is believed to perpetuate chronic infections by allowing S. aureus to evade both the human immune system and the action of extracellular antibiotics (5). Infected host cells can consequently serve as reservoirs for quiescent S. aureus that later maintain persistent infection and facilitate dissemination of bacteria (2, 6). Indeed, small colony variants (SCVs) of S. aureus, slow-growing auxotrophic mutants that arise frequently in chronic infection, are notable for their elevated capacity for intracellular pathogenesis (7 – 11).
The fate of intracellular S. aureus depends on both host and strain-encoded factors. Host cell type is one such determinant (4). During invasion of non-professional phagocytes, S. aureus utilizes adhesins to trigger uptake by host cells. After internalization, S. aureus can escape the endosome into the cytosol and replicate. In contrast, professional phagocytes actively engulf bacteria into phagosomes, which ultimately fuse with lysosomes to mature as bactericidal phagolysosomes. S. aureus can persist and replicate in that compartment and encode molecular pathways providing protection against lysozyme, antimicrobial peptides, reactive oxygen species, and low pH environments (4). Prior work has separately demonstrated that various S. aureus lineages differ in their inherent capacity for pathogenesis within various host cell types, likely due to differences in the complement or expression of relevant virulence factors (12).
Though substantial work has identified genes critical for S. aureus pathogenesis in host cells, knowledge of the pathways and factors involved remains incomplete (1, 3, 4). Moreover, the extent to which strain-specific accessory genes or genetic backgrounds impact intracellular pathogenesis has not yet been extensively explored (4). To address these questions, here we conducted studies to more comprehensively catalog factors contributing to the invasion and early survival of two phylogenomically distinct S. aureus laboratory strains (ATCC2913 and JE2) within human macrophages. We developed methods to generate high saturation Tn5-based transposon mutant libraries for each strain and performed transposon insertion sequencing (Tn-Seq) (13) of the population remaining viable after entry into THP-1 cell-line derived human macrophages (14, 15). The contributions of select candidate genes were subsequently verified using isogenic knockdowns generated by CRISPR interference (CRISPRi). The potential role of the complete set of implicated genes in chronic human infection was ascertained using mutational analysis of strains isolated from the airways of individuals with cystic fibrosis (CF).
MATERIALS AND METHODS
Strains and growth conditions
S. aureus ATCC29213 was obtained from the American Type Culture Collection (ATCC, Manassas, Virginia), and JE2 was obtained from the Biodefense and Emerging Infections Research Resources Repository. S. aureus transposon mutants were grown using LB (ThermoFisher, Waltham, Massachusetts) supplemented with 5 µg/mL thymidine, 1 µg/mL hemin, and 1 µg/mL menadione to support auxotrophic mutants (16) and containing 10 µg/mL chloramphenicol (SupLB-CAM). Escherichia coli DH5-alpha was from NEB (Ipswich, MA) and grown in LB containing 100 µg/mL ampicillin to maintain plasmids. All strains were cultured at 37°C. THP-1 cells were obtained from ATCC and cultured at 37°C in a humidified 5% (vol/vol) CO2 air atmosphere in RPMI 1640 medium (ThermoFisher) supplemented with 0.1 mg/mL l-glutamine, 0.1 mg/mL streptomycin, 100 U/mL penicillin, and 20% (vol/vol) Nu-Serum Serum Replacement (Corning, Corning, NY).
Transposon vector and transposon mutant libraries
Oligonucleotides and synthetic gene sequences (gBlocks) were synthesized by IDT (Coralville, IA) (Table S1).
The transposon vector (pAureus-TnCAM) was generated by joining two gBlocks (Transposon_CAM_part I and Transposon_CAM_part II) into pUC19 vector. Transposomes were generated using this vector as previously (17), with some modifications. Briefly, phosphorylated primers (transposon_mosaic_F and transposon_mosaic_R) were used to amplify the transposon cassette by PCR. The PCR product was purified using Monarch PCR & DNA clean up kit (NEB), eluting in TE buffer. 1 µL transposon DNA at 400 ng/µL, 2 uL EZ-TN transposase (1 U/µL, Lucigen, Middleton, WI), and 1 µL 100% glycerol were combined, incubated at room temperature for 45 min and then at 4°C overnight, prior to long-term storage at −20°C.
Transposon mutant libraries were generated as elsewhere (17), with some modifications. Electrocompetent S. aureus were prepared as elsewhere (18). Per S. aureus transformation (~108 cells), 0.5 µl transposome complex was electroporated with 1 µl TypeOne restriction inhibitor (Lucigen) as elsewhere (18), except excluding the use of pellet paint and using a Bio-Rad MicroPulser set to 2.3 kV and 2.5 ms time constant. Transformants were incubated in 950 µL recovery medium (18) for 2 h. Nineteen transformations were performed per strain, and transformants were pooled after recovery. The transformant pool was cryopreserved using 75 µL DMSO per milliliter culture, and aliquots were stored at −80°C.
Transformants were then expanded by culture on solid media to generate the initial transposon mutant pools. Frozen transformants were thawed on ice for 30 min then at room temperature for 15 min. One aliquot was initially plated onto SupLB-CAM to determine the titer of viable transposon insertion mutants. Bacteria were then plated on a series of 74 150 mm SupLB-CAM agar plates and incubated overnight. Colonies were harvested from each dish by applying 3 mL SupLB-CAM and resuspending colonies using a sterile cell spreader. Harvested colonies were pooled on ice and cryopreserved as above, yielding the initial transposon mutant library pools.
Macrophage invasion
All studies were conducted in quadruplicate. Forty-eight hours prior to bacterial infection, ~1 million THP-1 cells per infection were differentiated into macrophages using PMA (Sigma-Aldrich, St. Louis, MO) solubilized in DMSO at a concentration of 10 µg/mL (19 – 21). The PMA-containing medium was removed 24 h after treatment, and cells were washed with RPMI 1640 and incubated for 24 h. To maximize the number of transposon mutants that could be analyzed, the initial transposon mutant library pools were applied at a multiplicity of infection (MOI) of 100 in serum-free minimal essential medium (MEM) for 1 h. This condition resulted in successful internalization of 0.6% of the JE2 and 0.04% ATCC29213 bacterial inoculum while retaining 72% (JE2) and 50% (ATCC29213) THP-1 macrophage viability at the conclusion of the experiment. Following incubation, the medium was replaced with MEM containing 50 µg/mL lysostaphin (Sigma-Aldrich) for 3 h to kill extracellular bacteria (22). Host cells were washed with DPBS and lysed with 0.025% Triton X-100 in water (Sigma-Aldrich). Lysate was plated onto SupLB-CAM for overnight expansion of viable bacteria, then pooled and harvested as above. As an outgrowth control, smaller quantities of the initial library pools (105 bacteria) were inoculated into cell culture media in the absence of macrophages and lysostaphin and plated directly after 4 h incubation.
Quantitative measures of invasion were conducted as above, but serial dilutions of inoculum and cell lysate were plated onto LB to evaluate the count of viable bacteria, and 10 µg/mL chloramphenicol was included in all culture media.
Tn-Seq
Tn-Seq library preparation followed existing protocols (17, 23). Bacterial DNA was extracted using Qiagen DNeasy UltraClean Microbial Kit. Using Covaris E220 (peak power 140 w, duty factor 10%, 200 cycles/burst, time 80 s), 1.5 µg DNA was sheared to ~300 bp. End repair was performed in a 40 µL reaction containing 5 × Quick Ligation Buffer (NEB), 1.675 mM each dNTP (NEB), 3 µL E. coli DNA Polymerase I (NEB), 0.5 µL T4 PNK (NEB), incubated at 37°C for 30 min and 72°C for 20 min. DNA was purified using Monarch PCR & DNA Cleanup Kit (NEB), using two sequential elutions of 10 µL EB buffer each. C-tailing was as described previously (17) except that DNA was eluted in 7.5 µL EB buffer, followed by rounds 1 and 2 of PCR (17). Multiplexing of specimens was accomplished by incorporating sample-specific indexed primers (TnSeq barcode primers 1–12) during round 2 of PCR (17). Size selected 100–500 bp fragments were purified using Monarch DNA Gel Extraction Kit (NEB). Sequencing utilized a Nextseq500 (Illumina, San Diego, CA) with “Mid-Output” flow cells and a 75 bp single-end read using a custom read 1 sequencing primer (T26_SEQ-6). Demultiplexing was performed using bcl2fastq v2.20.0.422 (Illumina).
Tn-Seq data analysis
Analysis was performed with TRANSIT (version 3.2.7) (24) using the appropriate reference genome (GenBank accession CP000255.1 for JE2 or the ATCC29213 reference genome supplied by ATCC), for all protein coding features. Preprocessing used the TPP tool. Transposon mutant library sizes were estimated for each strain by combining sequencing data from all four replicates of the initial transposon mutant pool and tallying the number of unique coding insertions using the Tn5Gaps method. ANOVA with default parameters was used to perform pairwise comparisons across conditions, with Benjamini-Hochberg adjusted P < 0.05 considered significant. Pathway enrichment analysis of significant genes was performed using TRANSIT. REVIGO (25) was used to summarize and visualize pathway enrichment analyses. Gene clustering to identify homologous genes between the S. aureus lineages was performed as previously (26).
CRISPRi knockdown
CRISPRi knockdown of candidate genes was performed using vector pCRISRPi, as described elsewhere (D. R. Long, E. A. Holmes, H.-Y. Lo, K. Penewit, J. Almazan, T. Hodgson, N. F. Berger, Z. H. Bishop, D. J. Wolter, J. D. Lewis, A. Waalkes, and S. J. Salipante, submitted for publication). sgRNA were designed using CRISPOR (27) (Table S1). Gene knockdown was assessed using gene-targeted real-time PCR of cDNA prepared from mid-log phase growth cells, normalizing expression to that of gyrA. Gene expression levels in individual CRISPRi mutants were compared to relative gene expression for a pCRISRPi vector targeted to a neutral gene sequence (GFP) using the ΔΔCt method.
Analysis of gene selection in vivo
Whole-genome sequencing data from 1,382 S. aureus isolates longitudinally collected from 246 children with CF (26) were utilized for analysis of in vivo strain adaptation. The isolates comprise 237 clonal groups related by descent (26). Gene clustering was repeated as above for reference genomes in this study against pre-existing clustering models from clonal groups (26) to identify homologous genes. The presence of disruptive (stop-gain and frameshift) and synonymous (silent) de novo mutations within each clonal group was tallied for candidate genes from Tn-Seq. As a control, for each strain we identified genes within the upper quartile of P-value (i.e., least significant) by Tn-Seq ANOVA and having average insertion counts among replicates of the initial transposon mutant pool ≥1, identified homologs shared between the two strains (n = 183), and similarly analyzed those genes. For each gene, Fisher’s exact test was used to assess the proportion of clonal groups with one or more disruptive or synonymous de novo mutations occurring across clonal groups. The distribution of -log10-transformed P-values, corresponding to evidence of selection, was compared between candidate genes and control genes using the Wilcoxon rank-sum test.
RESULTS
Development of a facile Tn-Seq strategy for S. aureus
Based on prior work (28), we developed a novel vector, pAureus-TnCAM, enabling transposome-mediated saturation mutagenesis in S. aureus (Fig. 1). The transposon cassette is flanked by mosaic end sequences recognized by Tn5 transposase and contains a cat194 chloramphenicol resistance gene driven by the constitutive sarA promoter and sodB ribosome binding site (29, 30). A bidirectional blaZ transcriptional terminator (31) downstream of the resistance cassette limits polar effects from the read-through transcription of adjacent genes (32) and interference of resistance gene expression from opposing transcripts in the bacterial genome. Transposome complexes derived from PCR-amplified transposon were generated in vitro ( 28) and subsequently electroporated into S. aureus ( 18) with purified phage ocr protein (33), facilitating bypass of the S. aureus type I restriction system. Nineteen transformations were combined for each S. aureus strain to generate transposon mutant libraries. Sequence analysis identified 391,070 unique insertions in ATCC29213 and 316,248 in JE2 for the initial transposon mutant library pools, about 20- and 16-fold greater, respectively, than those considered saturated in prior work (34). Based on genome size, this averaged an insertion every 7 bp and 9 bp, respectively. Library pools encompassed an average of 149 (ATCC21913) and 124 (JE2) insertions per genome feature (Tables S2 and S3). These estimated library pool sizes are expected to be conservative as they do not include insertions occurring in intergenic regions. However, the bacterial transformation rate (1 in 4,859 for ATCC29213 and 1 in 6,007 for JE2) remains low enough that the risk of more than one transposon inserted into the genome of any given bacterium is exceedingly improbable.
Fig 1.

Transposon mutant generation in S. aureus. (A) The transposon vector is built into a pUC19 backbone and integrates a constitutively expressed chloramphenicol antibiotic resistance marker (cat194) with a bidirectionally active transcriptional terminator. (B) Transposon is amplified using phosphorylated primers and (C) Combined with Tn5 transposase in vitro to generate stable transposome complexes. (D) Transposomes are electroporated into S. aureus with an inhibitor of the type one restriction system (Ocr), and transformants are selected for antibiotic resistance on solid media (E) to yield transposon mutant libraries.
TnSeq identifies S. aureus genes relevant to macrophage pathogenesis
We used Tn-Seq to identify genes influencing invasion and early survival of S. aureus in macrophages (Fig. 2). We infected human macrophages derived from cultured THP-1 cells, which have previously been established as a model for S. aureus macrophage invasion in vivo (14), with sufficient quantities of transposon mutant libraries to ensure ~100-fold redundancy of each individual mutant. Following invasion, extracellular bacteria were killed (22), and intracellular organisms were harvested and expanded by overnight growth on solid media. Primary analyses compared the composition of the initial transposon mutant library pools to those following invasion, but as additional controls for outgrowth, library aliquots were also inoculated into cell culture media without macrophages and lysostaphin and plated after the incubation period. All experiments were performed in quadruplicate, followed by Tn-Seq analysis. Sequencing was performed to an average depth of 9.3 million reads per specimen and ultimately yielded an average of 4.6 million uniquely mapped reads per specimen.
Fig 2.

Experimental design. (A) Initial transposon mutant libraries are incubated with THP-1 derived macrophages to allow invasion (B) to occur. (C) Extracellular bacteria are killed by the application of lysostaphin. (D) Macrophages are lysed, and surviving bacteria are expanded by growth on solid media (E). (F) To provide a control for outgrowth, initial transposon mutant libraries are incubated in cell culture media and then plated directly on solid media (G). (H) Tn-Seq is performed on the initial library population, the library after macrophage invasion, and the outgrowth control. (I) Tn-Seq output is compared across conditions to identify genes for which insertional inactivation is significantly overrepresented (gene C), underrepresented (gene A), or unchanged (gene B) in mutants successfully invading macrophages relative to counts in initial and/or outgrowth control libraries.
ANOVA identified genes (Table 1) exhibiting statistically significant variability (Benjamini-Hochberg adjusted P < 0.05) in transposon insertion counts when comparing the initial transposon mutant library pools to the mutant pools having successfully invaded host cells (Tables S4 and S5). This analysis identifies populations of genes whose corresponding transposon mutants exhibit significant increases or decreases in fold change relative to the initial library state, corresponding to genes whose disruption enhances or diminishes macrophage invasion, respectively. The number and identity of genes significantly affecting macrophage invasion varied substantially between strains. Comparing the initial library to the mutant pool recovered after invasion identified 321 significant genes for ATCC29213 and 215 for JE2. For both strains, the majority of significant genes showed decreases in fold change abundance (83% for ATCC29213 and 92% for JE2), suggesting they were “conditionally essential” for the phenotype. The substantially smaller proportion of genes having increases in fold change likely reflects the minority of factors promoting increased fitness of corresponding mutants when disrupted.
TABLE 1.
Genes significant to macrophage pathogenesis identified by Tn-Seq
| S. aureus ATCC29213 | S. aureus JE2 | |
|---|---|---|
| Positive fold change abundance | 54 | 17 |
| Conditionally essential | 267 | 198 |
| Total | 321 | 215 |
Conserved and strain-specific pathways are relevant to intracellular pathogenesis in S. aureus
Pathway analysis ascertained enrichment of significant genes sharing functionally related roles, determined by comparing initial transposon mutant library pools to mutants surviving after invasion (Fig. 3). Transposon mutagenesis may produce opposite phenotypic effects within the same pathway, for example, knocking out an effector gene compared to disrupting its repressor. Analysis was, therefore, performed per strain on all implicated genes, regardless of the direction of fold change after invasion. This identified several consistently enriched pathways across strains: DNA replication, lysine biosynthetic process via diaminopimelate, tricarboxylic acid cycle, tetrahydrofolate biosynthetic process, tRNA pseudouridine synthesis, and enterobacterial common antigen biosynthetic process. Genes contributing to several other functional pathways were similarly enriched but were highly strain-specific.
Fig 3.

Pathway enrichment of genes contributing to macrophage invasion in S. aureus. Genes significant by Tn-Seq after macrophage invasion at adjusted P-value < 0.05 are represented, as determined by comparing initial transposon mutant library pools to mutants surviving following invasion. The color of circles corresponds to the P-value for each Gene Ontology (GO) category, and the size is proportional to the Log10 size of each GO term. The thickness of connecting lines represents semantic similarity between categories, and the spatial arrangement of discs approximately reflects the grouping of categories by semantic similarity. Results are separately displayed for strains ATCC29213 (A) and JE2 (B), with pathways common to both strains labeled in red.
To more directly compare specific implicated genes from the two strains, we identified gene homologs on the basis of sequence identity. Three hundred of the genes identified from ATCC29213 (93%) and 204 genes from JE2 (94%) had homologs present in the opposing strain. One hundred eight homologs were concordantly identified as significant in both strains (Table S6). Moreover, 106 of those 108 genes had negative fold changes after invasion in both strains, consistent with the majority of shared gene content being conditionally essential for macrophage invasion.
These results indicate that genes affecting macrophage pathogenesis comprise several core functions that are relevant to both strains and necessary to support the virulence phenotype but that a significant number of genes are strain-specific.
Assessment of candidate invasion genes by isogenic CRISPRi knockdown
We next tested the contributions of candidate macrophage invasion genes by generating gene-specific, isogenic CRISPRi knockdowns. Given the number of genes implicated, we prioritized a limited set for functional validation in each strain. We required that candidate genes achieved statistical significance by ANOVA when compared against both the initial library (Tables S4 and S5) and the outgrowth control (Tables S7 and S8) and that the direction of their fold change was consistent across both comparisons. These criteria identified 31 unique genes for functional testing (Table 2): 13 mutants with positive fold change in abundance and 11 conditionally essential mutants from ATCC29213 and one mutant with positive fold change in abundance and seven conditionally essential mutants from JE2, with a single candidate gene, liaR, shared between strains.
TABLE 2.
Prioritized S. aureus candidate genes involved in macrophage pathogenesis
| Strain of gene orign | Tn-Seq result | Pan-genome gene identifier | ATCC locus tag | JE2 locus tag | Common gene name | Gene function |
|---|---|---|---|---|---|---|
| ATCC21913 | Positive fold change abundance | SAUPAN004469000 | LNEJMEBC_00229 | SAUSA300_1721 | putative staphylococcal protein | |
| ATCC21913 | Positive fold change abundance | SAUPAN004146000 | LNEJMEBC_00424 | SAUSA300_1525 | glyQS | glycyl-tRNA synthetase |
| ATCC21913 | Positive fold change abundance | SAUPAN003559000 | LNEJMEBC_00728 | SAUSA300_1152 | frr | ribosome recycling factor |
| ATCC21913 | Positive fold change abundance | SAUPAN002319000 | LNEJMEBC_01408 | SAUSA300_0532 | fusA | translation elongation factor G |
| ATCC21913 | Positive fold change abundance | SAUPAN002146000 | LNEJMEBC_01525 | SAUSA300_0420 | membrane protein | |
| ATCC21913 | Positive fold change abundance | SAUPAN001028000 | LNEJMEBC_01759 | SAUSA300_0188 | brnQ_3 | branched-chain amino acid transport system |
| ATCC21913 | Positive fold change abundance | SAUPAN000035000 | LNEJMEBC_01860 | SAUSA300_0023 | yycI | two-component system WalR/WalK regulatory protein |
| ATCC21913 | Positive fold change abundance | SAUPAN000017000 | LNEJMEBC_01874 | SAUSA300_0009 | serS | serine—tRNAligase |
| ATCC21913 | Positive fold change abundance | SAUPAN006410000 | LNEJMEBC_01929 | SAUSA300_2599 | icaR | ica operon repressor |
| ATCC21913 | Positive fold change abundance | SAUPAN006269000 | LNEJMEBC_02007 | SAUSA300_2526 | pyrD | Dihydroorotate dehydrogenase(quinone) |
| ATCC21913 | Positive fold change abundance | SAUPAN006235000 | LNEJMEBC_02026 | SAUSA300_2506 | isaA | putative trans glycosylase |
| ATCC21913 | Positive fold change abundance | SAUPAN005904000 | LNEJMEBC_02216 | SAUSA300_2326 | NA | hypothetical protein |
| ATCC21913 | Positive fold change abundance | SAUPAN005440000 | LNEJMEBC_02446 | SAUSA300_2096 | gmuF | putative mannose-6-phosphate isomerase |
| ATCC21913 | Conditionally essential | SAUPAN003893000 | LNEJMEBC_00550 | SAUSA300_1331 | ald1 | alanine dehydrogenase |
| ATCC21913 | Conditionally essential | SAUPAN003264000 | LNEJMEBC_00932 | SAUSA300_0958 | lytR_1 | cell envelope-related transcriptional attenuator |
| ATCC21913 | Conditionally essential | LNEJMEBC_00949 | hypothetical protein | |||
| ATCC21913 | Conditionally essential | SAUPAN003193000 | LNEJMEBC_00971 | SAUSA300_0917 | ltaA | major facilitator superfamily transporter |
| ATCC21913 | Conditionally essential | SAUPAN005415000 | LNEJMEBC_02466 | SAUSA300_2077 | qsrR | transcriptional regulator |
| ATCC21913 | Conditionally essential | SAUPAN004592000 | LNEJMEBC_00188 | SAUSA300_1769 | lukEv | leucotoxin |
| ATCC21913 | Conditionally essential | LNEJMEBC_00223 | hypothetical protein | |||
| ATCC21913 | Conditionally essential | SAUPAN003833000 | LNEJMEBC_00574 | SAUSA300_1307 | arlS | Signal transduction histidine-protein kinase |
| ATCC21913 | Conditionally essential | SAUPAN003749000 | LNEJMEBC_00627 | SAUSA300_1255 | mprF | Phosphatidyl glycerol lysyl transferase |
| ATCC21913 | Conditionally essential | SAUPAN003673000 | LNEJMEBC_00670 | SAUSA300_1213 | hypothetical protein | |
| JE2 | Positive fold change abundance | SAUPAN002234000 | LNEJMEBC_01475 | SAUSA300_0473 | purR | pur operon repressor |
| JE2 | Conditionally essential | SAUPAN002709000 | LNEJMEBC_01182 | SAUSA300_0759 | gpmI | 2,3-bisphosphoglycerate-independent phosphoglycerate mutase |
| JE2 | Conditionally essential | SAUPAN003031000 | LNEJMEBC_01054 | SAUSA300_0835 | dltA | D-alanine--poly(phosphoribitol) ligase subunit 1 |
| JE2 | Conditionally essential | SAUPAN003810000 | LNEJMEBC_00591 | SAUSA300_1290 | dapD | 2,3,4,5-tetrahydropyridine-2,6-dicarboxylate N-acetyltransferase |
| JE2 | Conditionally essential | SAUPAN004782000 | LNEJMEBC_00149 | SAUSA300_1799 | airS | sensor histidine kinase |
| JE2 | Conditionally essential | SAUPAN005279000 | SAUSA300_1991 | agrC | accessory gene regulator protein C | |
| JE2 | Conditionally essential | SAUPAN006159000 | LNEJMEBC_02081 | SAUSA300_2455 | putative fructose-1,6-bisphosphatase | |
| JE2 and ATCC29213 | Conditionally essential | SAUPAN004781000 | LNEJMEBC_00150 | SAUSA300_1798 | liaR | DNA-binding response regulator |
Despite multiple attempts, we were unable to achieve significant knockdown for eight of the candidate genes (LNEJMEBC_00229, LNEJMEBC_00424, LNEJMEBC_01874, LNEJMEBC_02026, LNEJMEBC_00949, LNEJMEBC_02466, LNEJMEBC_00627, and LNEJMEBC_00670). Successful isogenic knockdowns were generated for liaR as well as 15 additional genes relevant for S. aureus ATCC29213 and seven for JE2 (Table S9). CRISPRi knockdown mutants were assessed for their capability to invade THP-1 derived macrophages relative to a strain-matched control bearing a CRISPRi vector targeting an irrelevant biological target (Fig. 4).
Fig 4.

Intracellular invasion phenotypes of isogenic CRISPRi knockdowns. Results are shown for the knockdown of candidate genes from Tn-Seq analysis of post-macrophage invasion mutant pools that were identified as significant and showing consistent effects relative to both the initial transposon mutant library pool and the outgrowth control. Panels indicate genes identified by Tn-Seq as: (A) ATCC29213 genes with positive fold change in abundance, (B) conditionally essential ATCC29213 genes, (C) JE2 genes with positive fold change in abundance, and (D) conditionally essential JE2 genes. Y-axes indicate the fold difference in THP-1 derived macrophage invasion for isogenic CRISPRi knockdowns in both S. aureus strains relative to invasion activity of the matched parental strain carrying a silencing vector targeted to an irrelevant biological target (GFP), represented by a dashed line (red, at 0). Error bars indicate SEM. Measured values that are significantly different (by 2-tailed t test) are indicated by asterisks: *, P < 0.05; **, P < 0.01; ***, P < 0.001, either against the parental strain (displayed above each bar plot) or for the indicated comparisons between strains (black lines). NS, not significant. Common gene names, where applicable, are indicated in parentheses below each gene ID. liaR, relevant to both strains, is in bold.
Four of the nine knockdowns of ATCC29213-relevant genes that showed positive fold change in abundance after selection (Fig. 4A) recapitulated anticipated gains in invasive capacity (frr, fusA, yycl, and LNEJMEBC_02216), two had no significant impact (LNEJMEBC_01525 and brnQ3), and three paradoxically decreased invasive capacity (icaR, pyrD, and gmuF). Unexpectedly, all seven conditionally essential candidate genes from the ATCC29213 library produced statistically significant increases in invasiveness following their knockdown in ATCC29213 (Fig. 4B).
Only one JE2 gene in the validation set showed positive fold change in abundance after invasion (purR), and its knockdown accordingly resulted in a significant increase in invasiveness (Fig. 4C). Knockdown of the seven conditionally essential candidate genes in the transposon mutant pool (Fig. 4D) resulted in significantly decreased invasion for three of the genes (gpmI, dltA, and dapD) as anticipated, did not significantly impact invasion for two genes (liaR and agrC), and produced significantly increased invasion capacity for two genes (airS and SAUSA300_2455).
We conclude that most candidate genes prioritized for functional validation measurably affect intracellular invasion, however, enhancement or reduction of the invasion phenotype does not always match predictions based on Tn-Seq analysis.
Strain-specific effects of effector genes contributing to S. aureus cell invasion
Given the paucity of observed overlap in prioritized candidate genes between the two S. aureus strains, we next evaluated whether those genes could impact intracellular pathogenesis in a strain-dependent fashion. We, therefore, transformed CRISPRi vectors targeting candidate genes identified from one strain into the opposing S. aureus strain and again assessed the invasion capacity of transformants relative to a strain-matched neutral CRISPRi control. The effects of individual gene knockdowns were then compared between the two strains (Fig. 4).
Knockdown of seven candidate genes with positive fold change in abundance identified from ATCC29213 resulted in significantly decreased invasiveness for JE2, while one had no significant impact and one significantly increased invasive capacity (Fig. 4A). For only two genes (pyrD and gmuF) were these results consistent between the two strains. Similarly, while knockdown of all seven conditionally essential ATCC29213 candidate genes resulted in enhanced invasiveness in that background, only three genes showed concordant effects in JE2, while two significantly decreased invasiveness in that strain and the remaining two showed no effect (Fig. 4B). Inversions of phenotypic effect were also seen for JE2 candidate genes knocked down in ATCC29213. While knockdown of the single candidate gene with positive fold change in abundance from JE2 (Fig. 4C) augmented the invasion phenotypes of both strains, five of the seven conditionally essential candidate genes (Fig. 4D) showed discordant phenotypes between JE2 and ATCC29213.
These findings indicate that multiple genes contributing to macrophage invasion exert disparate functional effects, depending on the S. aureus strain background being evaluated.
Selection of candidate intracellular invasion genes occurs in chronic cystic fibrosis airway infections in vivo
We performed analyses to ascertain the relevance of candidate genes identified from Tn-Seq during chronic S. aureus infection in vivo. We used respiratory infection in CF as a model, as S. aureus is postulated to undergo selection for increased macrophage pathogenesis in that environment (10, 35). We examined 237 groups of clonally related S. aureus isolates longitudinally collected from children with CF over a period of several years (26). Studies from our group using the isolate collection have accordingly shown that the capacity of strains for macrophage invasion increases during the course of chronic infection in vivo (D. R. Long, E. A. Holmes, H.-Y. Lo, K. Penewit, J. Almazan, T. Hodgson, N. F. Berger, Z. H. Bishop, D. J. Wolter, J. D. Lewis, A. Waalkes, and S. J. Salipante, submitted for publication). We therefore hypothesized that, in chronically infecting S. aureus populations, natural selection would be stronger in genes involved in macrophage invasion than in genes not contributing to that phenotype.
Individuals with CF become infected with a variety of S. aureus lineages (26). We consequently considered genes identified by Tn-Seq as having significant effects in both ATCC29213 and JE2 (Table S6), which are most likely to affect consistent phenotypes among a diverse population of S. aureus strains. As comparators, we included 183 control genes least associated with the macrophage invasion phenotype. Tn-Seq identifies gene disruptions that are conditionally essential for macrophage invasion as those showing negative fold changes in abundance after invasion, which are expected to be under purifying selection in vivo. Conversely, genes whose inactivation enhances macrophage invasion are identified as those having positive fold changes in abundance and should be subject to positive selection. This set contained a paucity of genes expected to be under positive selection (n = 2), with nearly all expected to be subjected to purifying selection (n = 106). For this reason, and because missense mutations may produce either gain-of-function or loss-of-function effects, which are difficult to predict, analysis was based on de novo mutations which unequivocally disrupted gene function (i.e., frameshift and stop-gain mutations, comparable to insertional inactivation), and the two candidate genes predicted to be under positive selection were not included.
We assessed the frequencies of de novo disruptive versus silent mutations in candidate and control genes occurring within clonally related S. aureus lineages from patient specimens (Table S10). For each gene, we ascertained whether there was a statistically significant deficit of disruptive relative to synonymous mutations observed across clonal groups, which would be indicative of purifying selection in vivo (36). The distribution of resultant P-values, showing the strength of evidence for selection in each gene, was then compared between candidate genes and control genes. This analysis showed significantly lower frequencies of disruptive mutation in candidate invasion genes relative to control genes (Fig. 5, P = 0.002, Wilcoxon rank-sum test, Table S10), consistent with predictions from Tn-Seq analysis.
Fig 5.
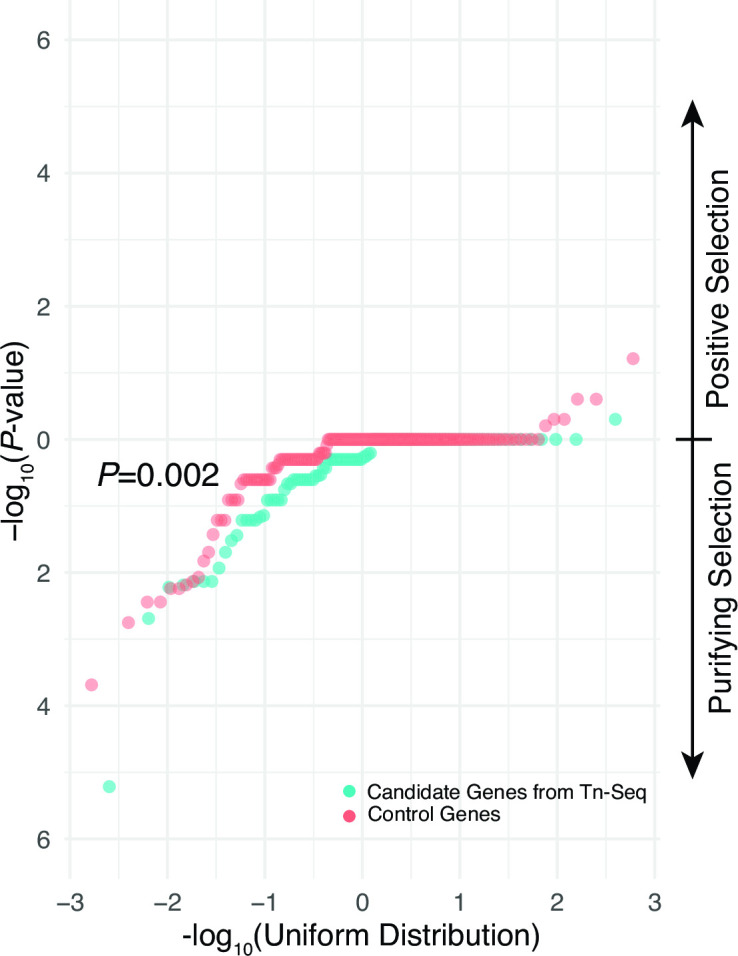
Selection of candidate macrophage invasion genes in chronic CF respiratory infections in vivo. QQ-plots of -log10 transformed P-values testing the proportion of disruptive versus synonymous mutations in candidate genes Tn-Seq identified as having significant effects on macrophage invasion for both ATCC29213 and JE2 and corresponding control genes not implicated in that phenotype. P-values in ranges consistent with positive (more disruptive than synonymous mutations) and purifying selection (more synonymous than disruptive mutations) are indicated by the arrows at right. Significance of difference between the distributions of candidate and control genes by Wilcoxon rank-sum test is shown.
These studies provide evidence that conditionally essential macrophage invasion genes implicated by Tn-Seq were collectively under greater purifying selection during chronic infection in CF patient airways in vivo than genes not associated with that phenotype, highlighting their relevance in human disease.
DISCUSSION
We sought to comprehensively identify genes relevant to S. aureus invasion of human macrophages using Tn-Seq. To this end, we developed a novel system for generating saturation-level transposon mutant libraries in S. aureus through electroporation of transposon-transposase complexes (transposomes, Fig. 1).
Our transposition system offers several advantages over prior approaches used in S. aureus. The earliest methods involved co-transformation of two temperature sensitive plasmids that separately encode mariner-based bursa aurealis transposon and the mariner transposase gene, resulting in random transposition into the S. aureus genome when combined (34, 37 – 42). After transposition, high temperature plasmid-curing steps are required to remove the vectors, however, plasmid curing may be incomplete (43), resulting in ongoing or unstable transposition events, and heat stress can inadvertently select for temperature sensitive mutants or otherwise bias the mutant pool (44). Alternatively, Φ11 bacteriophage has been used to transduce transposon cassette-bearing plasmids with a conditional replication origin into transgenic S. aureus recipient strains expressing transposase (44 – 46). This eliminates the need for temperature-dependent plasmid curing, and the high efficiency of phage transduction allows ultra-high density mutant libraries (44 – 46). However, not all S. aureus isolates are susceptible to Φ11 transduction (47). Labor intensive manipulations are also required to remove Φ11 family prophages from recipient strains to prevent non-transposase-catalyzed insertions mediated by phage-encoded integrases or homologous recombination (44). Moreover, recipient strains in this system retain the transposase expression vector, preventing them from becoming fully isogenic.
In contrast, electroporation of transposon-transposase complexes (transposomes) is a simple and efficient way to generate transposon mutant libraries (28). Such methods have been applied to multiple bacterial species (48 – 51), but until this work, have not been adapted for use in S. aureus. Our transposon encodes a robust selectable marker and a bidirectional transcriptional terminator intended to limit polar effects and is electroporated into target strains in the presence of type I restriction enzyme inhibitor to bypass S. aureus DNA restriction systems (18). Because transposition is mediated by complexes prepared in vitro, there is no dependence on host-encoded factors and no need to remove transgenic elements, and transposition events are stable after genomic integration. The approach can be applied to any S. aureus strain for which electrocompetent cells can be prepared, which is readily achievable following published protocols (18). We rapidly generated highly saturated S. aureus transposon mutant libraries in two strains, comprising ~300,000 to 400,000 unique insertions each.
We used Tn-Seq to compare initial transposon mutant library pools to populations having successfully invaded host macrophages, thereby identifying genes whose disruption positively or negatively impacts intracellular pathogenesis (Table 1). A substantial proportion of S. aureus total gene complement was implicated by Tn-Seq, corresponding to ~12% and 8% of coding sequences in ATCC29213 and JE2, respectively. This observation supports prior studies indicating that complex epistatic interactions govern many virulence traits in S. aureus (52). In both strains, the majority of genes identified as significant by Tn-seq showed negative fold change in abundance following invasion relative to the initial transposon library pool, consistent with most relevant factors being required (or conditionally essential) for effective invasion and early survival in macrophages. Virtually all (98%) candidate genes which had identifiable homologs between the two strains consistently evidenced negative fold change in abundance from Tn-Seq experiments of both strains, identifying a core set of 106 conditionally essential genes that are necessary to support macrophage invasion in S. aureus (Table S6). A smaller proportion of knockouts showed positive fold change in abundance from the initial transposon mutant pools after macrophage invasion, indicating that spontaneous chromosomal mutations in S. aureus can also enhance this virulence phenotype (53) and may facilitate adaptation in chronic infection. Collectively, physiological and metabolic functions consistently contributing to macrophage invasion in both strains (Fig. 3) may reflect the association between intracellular pathogenesis and the SCV phenotype (7 – 11), as disruption of many such pathways has been identified in SCVs (54 – 57).
Comparing initial and selected transposon mutant libraries is used in most Tn-Seq studies of complex bacterial phenotypes (34, 41, 42, 58, 59) and is generally considered robust. However, this approach does not account for population dynamics during the selection period and may introduce bias from differences in the fitness or growth of mutants during that time (28). Given these uncertainties, we prioritized genes for functional validation in each strain as those which remained significant after comparing to a secondary population controlling for outgrowth. It should be noted that this outgrowth control is also expected to be biased, only differently so. As a case in point, in both strains, disruptions of purine biosynthesis pathway genes showed significant increases in fold change after invasion relative to the outgrowth control (Tables S7 and S8) but not in comparison to the initial library (Tables S4 and S5). Previous studies have found that purine biosynthesis supports intracellular pathogenesis in S. aureus (53, 60), making the outgrowth control result consistent with a growth-related artifact. Nevertheless, we reasoned that genes having the strongest contributions to intracellular pathogenesis would be independently identified by comparison against both the initial and outgrowth control populations.
These conservative criteria identified 31 unique candidate genes for functional validation, 24 in ATCC29213, and eight in JE2, with liaR shared in common (Table 2). 23 genes could be empirically tested as isogenic CRISPRi knockdown mutants. Twenty of these significantly impacted macrophage invasion when knocked down by CRISPRi in their strain of origin and were considered validated, although we note that all 23 genes significantly affected invasion in at least one strain, after testing was conducted in both ATCC29213 and JE2 (Fig. 4). Unexpectedly, 12 of the 20 functionally validated mutants produced phenotypic effects opposing expectations based on the direction of fold change of genes observed by Tn-Seq in their corresponding strain of origin. Phenotypic discordance between knockdown and knockout mutants of specific genes has been well documented for eukaryotic organisms (61, 62), with similar discrepancies reported in studies of Salmonella (63) and could reflect activation of compensatory networks buffering against deleterious mutations (61), insufficient gene knockdown, or unintended off-target silencing effects, although gRNA design algorithms limit that possibility (27). Moreover, we found that many validated genes identified from one strain (Fig. 4) produced contradicting phenotypic effects when knocked down in the other. These well-controlled studies, where the same knockdown vector transferred to two different S. aureus strains resulted in opposing consequences to macrophage invasion, provide strong evidence that effects of such factors are strain-specific.
Five of the 20 validated genes [purR (53, 60), icaR (64), yycI (65), arlS (66), and ltaA(67)] have previously described roles in S. aureus virulence, while two others [lukEv(68) and airS(69)] specifically impact survival against professional phagocytes, offering encouraging external validation of our findings. The remaining 13 validated genes are pathogenesis factors newly implicated by this study. Two such genes (LNEJMEBC_02216 and LNEJMEBC_00223) are functionally uncharacterized; however, the described roles of other factors highlight several key pathways that mimic the higher-level functional roles enriched within the total set of genes identified by Tn-Seq (Fig. 2) and provide greater insight into specific contributory roles.
First, the largest functional group of validated genes participate in metabolism and biosynthesis. Ribosome recycling factor frr (70) and elongation factor-encoding fusA (71) have functions in protein synthesis. Genes involved in the biosynthesis [dapD (72)] or catabolism of amino acids [ald1 (73)] also impacted macrophage invasion, as did three genes involved in glycolysis [gpmI (74), gmuF (75), and putative fructose-1,6-bisphosphatase SAUSA300_2455] and one relevant to pyrimidine biosynthesis [pyrD (76)]. Of note, several of such genes have known roles in S. aureus persistence [frr (77) and dapD (72)] or SCV [fusA(71)] phenotypes or adaptation during respiratory infection [ald1(73)], phenotypes which are consistent with enhanced intracellular pathogenicity and which lend greater credence to the findings of the present study. Second are genes relevant to survival within the toxic intracellular environment presented by macrophages. airS comprises half of the two-component airSR regulatory system, which controls resistance to reactive oxygen species (69), while gpmI, primarily identified as a metabolic gene, also contributes to S. aureus fitness during nitric oxide exposure (78). Third are regulatory effectors. Previously described genes purR (53, 60), yycI (65), and arlS (66) are thought to influence virulence by this mechanism. Like members of other two-component regulatory systems, liaR (79) and lytR, (80, 81) serve to translate external sensory signals to appropriate cytoplasmic responses within S. aureus. The responses elicited by such systems are frequently pleiotropic and may encode multiple phenotypes relevant to invasion and survival within macrophages. The fourth category encompasses bacterial cell wall regulation. Flippase ltaA has been shown to adaptively modify S. aureus cell wall teichoic acids under acidic conditions (82), and previous Tn-Seq studies have identified its contributions to metastatic S. aureus bloodstream infections (67). dltA has a similar role in teichoic acid alteration (83). Implication of these genes in macrophage pathogenesis is concordant with findings from other groups identifying S. aureus cell wall modification as important for bacterial survival during infection in vivo (84, 85), with some studies further suggesting that these factors promote adherence of bacteria to host cells (86, 87). The two remaining genes, icaR and lukEv, provide unique functions within this set, likely impacting macrophage intracellular pathogenesis by host cell adhesion (64) and attenuation of cytotoxicity (68), respectively.
Excitingly, genomic studies of isolates collected from individuals with CF provide evidence for the importance of genes identified by Tn-Seq during chronic S. aureus infection of human hosts in vivo. Studies of S. aureus obtained from individuals with CF have indicated that there is a selection for mutants having enhanced capacity for intracellular pathogenesis of host cells, especially airway macrophages (10, 35). In a large, longitudinally banked collection of isolates obtained from individuals with CF (26), we found genes implicated by Tn-Seq as conditionally essential for macrophage intracellular pathogenesis in vitro were collectively under significantly greater purifying selection in vivo than control genes least significantly associated with that phenotype (Fig. 5). This is particularly striking given the diversity of adaptive phenotypes that selected S. aureus during chronic respiratory infections in CF, including antibiotic resistance or tolerance, increased formation of biofilms or biofilm-like aggregates, and metabolic adaptations (10, 26, 88 – 90). It is likely that at least some control genes used for this comparison are themselves under selection for other phenotypes relevant to chronic infection, but even so, genes associated with intracellular pathogenesis are under significantly greater selective pressures. For example, the candidate gene with the strongest signal of purifying selection in vivo (adsA) plays a critical role in S. aureus escape from phagocytic clearance and survival in human blood (91). Collectively, these findings offer evidence that genes identified by this study are relevant to chronic human infection in vivo and support the importance of macrophage pathogenesis in contributing to S. aureus persistence in CF (10, 35).
Although illuminating, our study is subject to several limitations. Cell lines cultured in vitro are not physically or biochemically identical to analogous cells or tissues found in vivo (12), making our Tn-Seq assay necessarily contrived. However, our finding that clinical isolates evidence selection in many candidate genes identified using the THP-1 model employed here and by others (11, 14, 15) offers encouraging support that it reasonably approximates in vivo conditions. We considered two distinct laboratory strains from different phylogenomic backgrounds, but it is likely that multiple strain-specific macrophage invasion genes have yet to be identified in additional lineages. Tn-Seq monitors count of insertions that disrupt gene function; therefore, it will only identify genes that impact a phenotype by loss of function. Genes able to contribute to a process of interest through overexpression or gain-of-function mutations may exist but will not be identified by Tn-Seq analysis. Due to the large number of genes identified by this study, we were unable to subject all to empiric validation, leaving their biological contributions unverified. Finally, use of CRISPRi as an orthologous validation method did not allow interrogation of all targets of interest, and valid effector genes that were insufficiently knocked down might not have imparted measurable phenotypic effects.
These data provide a detailed catalog of the factors influencing S. aureus invasion and initial survival within a host cell type important to chronic infection. Our analyses identify conserved and strain-specific genes and pathways used by S. aureus when invading macrophages and reveal that individual genes may have opposing effects when disrupted in different S. aureus lineages. Future work will seek to better understand the mechanisms by which implicated genes influence intracellular pathogenesis, to further explore the diversity of S. aureus genetic factors that facilitate human cell invasion, to explore the basis of strain-specific gene effects, and to identify genes necessary for the maintenance of chronic infection within macrophages following initial host cell invasion. Genes and pathways implicated here may present novel therapeutic targets that could be leveraged to disrupt persistent S. aureus infection in vivo.
ACKNOWLEDGMENTS
This work was supported by grants from Vertex Pharmaceuticals (to S.J.S.), the Cystic Fibrosis Foundation (SINGH19R0 to S.J.S.), and NIH (P30 DK089507 to S.J.S., K23 AR080209 to D.R.L.).
Contributor Information
Stephen J. Salipante, Email: stevesal@uw.edu.
Kimberly A. Kline, Universite de Geneve, Geneva, Switzerland
DATA AVAILABILITY
Sequence data from this study are available from the NCBI Sequence Read Archive under accession PRJNA942332.
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/iai.00228-23.
Oligonucleotide sequences.
Composition of initial transposon mutant pool for ATCC29213.
Composition of initial transposon pool JE2.
Tn-Seq results for ATCC29213, initial vs invasion library.
Tn-Seq results for JE2, initial vs invasion library.
Gene homologs identified as significant for invasion in both S. aureus strains.
Tn-Seq results for ATCC29213, outgrowth control vs invasion library.
Tn-Seq results for JE2, outgrowth control vs invasion library.
CRISPRi gene knockdown results.
Mutational characteristics of candidate macrophage invasion genes in vivo.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Fraunholz M, Sinha B. 2012. Intracellular Staphylococcus aureus: live-in and let die. Front Cell Infect Microbiol 2:43. doi: 10.3389/fcimb.2012.00043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Horn J, Stelzner K, Rudel T, Fraunholz M. 2018. Inside job: Staphylococcus aureus host-pathogen interactions. Int J Med Microbiol 308:607–624. doi: 10.1016/j.ijmm.2017.11.009 [DOI] [PubMed] [Google Scholar]
- 3. Moldovan A, Fraunholz MJ. 2019. In or out: phagosomal escape of Staphylococcus aureus. Cell Microbiol 21:e12997. doi: 10.1111/cmi.12997 [DOI] [PubMed] [Google Scholar]
- 4. Hommes JW, Surewaard BGJ. 2022. Intracellular habitation of Staphylococcus aureus: molecular mechanisms and prospects for antimicrobial therapy. Biomedicines 10:1804. doi: 10.3390/biomedicines10081804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Garzoni C, Kelley WL. 2009. Staphylococcus aureus: new evidence for intracellular persistence. Trends Microbiol 17:59–65. doi: 10.1016/j.tim.2008.11.005 [DOI] [PubMed] [Google Scholar]
- 6. Garzoni C, Kelley WL. 2011. Return of the Trojan horse: intracellular phenotype switching and immune evasion by Staphylococcus aureus. EMBO Mol Med 3:115–117. doi: 10.1002/emmm.201100123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. von Eiff C, Peters G, Becker K. 2006. The small colony variant (SCV) concept -- the role of staphylococcal SCVs in persistent infections. Injury 37:S26–S33. doi: 10.1016/j.injury.2006.04.006 [DOI] [PubMed] [Google Scholar]
- 8. Atalla H, Gyles C, Mallard B. 2011. Staphylococcus aureus small colony variants (SCVs) and their role in disease. Anim Health Res Rev 12:33–45. doi: 10.1017/S1466252311000065 [DOI] [PubMed] [Google Scholar]
- 9. Loss G, Simões PM, Valour F, Cortês MF, Gonzaga L, Bergot M, Trouillet-Assant S, Josse J, Diot A, Ricci E, Vasconcelos AT, Laurent F. 2019. Staphylococcus aureus small colony variants (SCVs): news from a chronic prosthetic joint infection. Front Cell Infect Microbiol 9:363. doi: 10.3389/fcimb.2019.00363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tan X, Coureuil M, Ramond E, Euphrasie D, Dupuis M, Tros F, Meyer J, Nemazanyy I, Chhuon C, Guerrera IC, Ferroni A, Sermet-Gaudelus I, Nassif X, Charbit A, Jamet A. 2019. Chronic Staphylococcus aureus lung infection correlates with proteogenomic and metabolic adaptations leading to an increased intracellular persistence. Clin Infect Dis 69:1937–1945. doi: 10.1093/cid/ciz106 [DOI] [PubMed] [Google Scholar]
- 11. Gabryszewski SJ, Wong Fok Lung T, Annavajhala MK, Tomlinson KL, Riquelme SA, Khan IN, Noguera LP, Wickersham M, Zhao A, Mulenos AM, Peaper D, Koff JL, Uhlemann A-C, Prince A. 2019. Metabolic adaptation in methicillin-resistant Staphylococcus aureus pneumonia. Am J Respir Cell Mol Biol 61:185–197. doi: 10.1165/rcmb.2018-0389OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Strobel M, Pförtner H, Tuchscherr L, Völker U, Schmidt F, Kramko N, Schnittler H-J, Fraunholz MJ, Löffler B, Peters G, Niemann S. 2016. Post-invasion events after infection with Staphylococcus aureus are strongly dependent on both the host cell type and the infecting S. aureus strain. Clin Microbiol Infect 22:799–809. doi: 10.1016/j.cmi.2016.06.020 [DOI] [PubMed] [Google Scholar]
- 13. van Opijnen T, Bodi KL, Camilli A. 2009. Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat Methods 6:767–772. doi: 10.1038/nmeth.1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nguyen HA, Denis O, Vergison A, Tulkens PM, Struelens MJ, Van Bambeke F. 2009. Intracellular activity of antibiotics in a model of human THP-1 macrophages infected by a Staphylococcus aureus small-colony variant strain isolated from a cystic fibrosis patient: pharmacodynamic evaluation and comparison with Isogenic normal-phenotype and revertant strains. Antimicrob Agents Chemother 53:1443–1449. doi: 10.1128/AAC.01146-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chanput W, Mes JJ, Wichers HJ. 2014. THP-1 cell line: an in vitro cell model for immune modulation approach. Int Immunopharmacol 23:37–45. doi: 10.1016/j.intimp.2014.08.002 [DOI] [PubMed] [Google Scholar]
- 16. Precit MR, Wolter DJ, Griffith A, Emerson J, Burns JL, Hoffman LR. 2016. Optimized in vitro antibiotic susceptibility testing method for small-colony variant Staphylococcus aureus. Antimicrob Agents Chemother 60:1725–1735. doi: 10.1128/AAC.02330-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gallagher LA. 2019. Methods for Tn-Seq analysis in Acinetobacter baumannii. Edited by Biswas I. and Rather P. N.. Methods Mol Biol 1946:115–134. doi: 10.1007/978-1-4939-9118-1_12 [DOI] [PubMed] [Google Scholar]
- 18. Monk IR, Shah IM, Xu M, Tan M-W, Foster TJ. 2012. Transforming the untransformable: application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. mBio 3:e00277-11. doi: 10.1128/mBio.00277-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tsuchiya S, Kobayashi Y, Goto Y, Okumura H, Nakae S, Konno T, Tada K. 1982. Induction of maturation in cultured human monocytic leukemia cells by a phorbol diester. Cancer Res 42:1530–1536. [PubMed] [Google Scholar]
- 20. Maeß MB, Wittig B, Cignarella A, Lorkowski S. 2014. Reduced PMA enhances the responsiveness of transfected THP-1 macrophages to polarizing stimuli. J Immunol Methods 402:76–81. doi: 10.1016/j.jim.2013.11.006 [DOI] [PubMed] [Google Scholar]
- 21. Aldo PB, Craveiro V, Guller S, Mor G. 2013. Effect of culture conditions on the phenotype of THP-1 monocyte cell line. Am J Reprod Immunol 70:80–86. doi: 10.1111/aji.12129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim J-H, Chaurasia AK, Batool N, Ko KS, Kim KK, Torres VJ. 2019. Alternative enzyme protection assay to overcome the drawbacks of the gentamicin protection assay for measuring entry and intracellular survival of staphylococci. Infect Immun 87:e00119–19. doi: 10.1128/IAI.00119-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Salipante SJ, Sengupta DJ, Cummings LA, Robinson A, Kurosawa K, Hoogestraat DR, Cookson BT. 2014. Whole genome sequencing indicates Corynebacterium jeikeium comprises 4 separate genomospecies and identifies a dominant genomospecies among clinical isolates. Int J Med Microbiol 304:1001–1010. doi: 10.1016/j.ijmm.2014.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. DeJesus MA, Ambadipudi C, Baker R, Sassetti C, Ioerger TR. 2015. TRANSIT - a software tool for Himar1 Tnseq analysis. PLOS Comput Biol 11:e1004401. doi: 10.1371/journal.pcbi.1004401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Supek F, Bošnjak M, Škunca N, Šmuc T. 2011. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 6:e21800. doi: 10.1371/journal.pone.0021800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Long DR, Wolter DJ, Lee M, Precit M, McLean K, Holmes E, Penewit K, Waalkes A, Hoffman LR, Salipante SJ. 2021. Polyclonality, shared strains, and convergent evolution in chronic cystic fibrosis Staphylococcus aureus airway infection. Am J Respir Crit Care Med 203:1127–1137. doi: 10.1164/rccm.202003-0735OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Concordet J-P, Haeussler M. 2018. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res 46:W242–W245. doi: 10.1093/nar/gky354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gallagher LA. 1946. Methods for Tn-Seq analysis in Acinetobacter baumannii. Methods Mol Biol Clifton NJ:115–134. doi: 10.1007/978-1-4939-9118-1 [DOI] [PubMed] [Google Scholar]
- 29. Kato F, Nakamura M, Sugai M. 2017. The development of fluorescent protein tracing vectors for multicolor imaging of clinically isolated Staphylococcus aureus. Sci Rep 7:2865. doi: 10.1038/s41598-017-02930-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. de Jong NWM, van der Horst T, van Strijp JAG, Nijland R. 2017. Fluorescent reporters for markerless genomic integration in Staphylococcus aureus. Sci Rep 7:43889. doi: 10.1038/srep43889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Charpentier E, Anton AI, Barry P, Alfonso B, Fang Y, Novick RP. 2004. Novel cassette-based shuttle vector system for gram-positive bacteria. Appl Environ Microbiol 70:6076–6085. doi: 10.1128/AEM.70.10.6076-6085.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hutchison CA 3rd, Merryman C, Sun L, Assad-Garcia N, Richter RA, Smith HO, Glass JI. 2019. Polar effects of transposon insertion into a minimal bacterial genome. J Bacteriol 201:e00185-19. doi: 10.1128/JB.00185-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Walkinshaw MD, Taylor P, Sturrock SS, Atanasiu C, Berge T, Henderson RM, Edwardson JM, Dryden DTF. 2002. Structure of OCR from bacteriophage T7, a protein that mimics B-form DNA. Mol Cell 9:187–194. doi: 10.1016/s1097-2765(02)00435-5 [DOI] [PubMed] [Google Scholar]
- 34. Bae T, Banger AK, Wallace A, Glass EM, Aslund F, Schneewind O, Missiakas DM. 2004. Staphylococcus aureus virulence genes identified by bursa aurealis mutagenesis and nematode killing. Proc Natl Acad Sci U S A 101:12312–12317. doi: 10.1073/pnas.0404728101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li C, Wu Y, Riehle A, Ma J, Kamler M, Gulbins E, Grassmé H. 2017. Staphylococcus aureus survives in cystic fibrosis macrophages, forming a reservoir for chronic pneumonia. Infect Immun 85:e00883-16. doi: 10.1128/IAI.00883-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jeffares DC, Tomiczek B, Sojo V, Reis M. 2015. A beginners guide to estimating the non-synonymous to synonymous rate ratio of all protein-coding genes in a genome. Methods Mol Biol Clifton NJ 1201:65–90. doi: 10.1007/978-1-4939-1438-8 [DOI] [PubMed] [Google Scholar]
- 37. Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, Bayles KW. 2013. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. mBio 4:e00537-12. doi: 10.1128/mBio.00537-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Valentino MD, Foulston L, Sadaka A, Kos VN, Villet RA, Santa Maria J Jr, Lazinski DW, Camilli A, Walker S, Hooper DC, Gilmore MS. 2014. Genes contributing to Staphylococcus aureus fitness in abscess- and infection-related ecologies. mBio 5:e01729-14. doi: 10.1128/mBio.01729-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bae T, Glass EM, Schneewind O, Missiakas D. 2008. Generating a collection of insertion mutations in the Staphylococcus aureus genome using Bursa aurealis , p 103–116. In Osterman AL, Gerdes SY (ed), Microbial gene essentiality: protocols and bioinformatics. Humana Press. doi: 10.1007/978-1-59745-321-9 [DOI] [PubMed] [Google Scholar]
- 40. Rajagopal M, Martin MJ, Santiago M, Lee W, Kos VN, Meredith T, Gilmore MS, Walker S. 2016. Multidrug intrinsic resistance factors in Staphylococcus aureus identified by profiling fitness within high-diversity transposon libraries. mBio 7:e00950-16. doi: 10.1128/mBio.00950-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Benton BM, Zhang JP, Bond S, Pope C, Christian T, Lee L, Winterberg KM, Schmid MB, Buysse JM. 2004. Large-scale identification of genes required for full virulence of Staphylococcus aureus. J Bacteriol 186:8478–8489. doi: 10.1128/JB.186.24.8478-8489.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mei JM, Nourbakhsh F, Ford CW, Holden DW. 1997. Identification of Staphylococcus aureus virulence genes in a murine model of bacteraemia using signature‐tagged mutagenesis. Mol Microbiol 26:399–407. doi: 10.1046/j.1365-2958.1997.5911966.x [DOI] [PubMed] [Google Scholar]
- 43. Chen J, Ram G, Yoong P, Penadés JR, Shopsin B, Novick RP. 2015. An rpsL-based allelic exchange vector for Staphylococcus aureus. Plasmid 79:8–14. doi: 10.1016/j.plasmid.2015.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Santiago M, Matano LM, Moussa SH, Gilmore MS, Walker S, Meredith TC. 2015. A new platform for ultra-high density Staphylococcus aureus transposon libraries. BMC Genomics 16:252. doi: 10.1186/s12864-015-1361-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Coe KA, Lee W, Stone MC, Komazin-Meredith G, Meredith TC, Grad YH, Walker S. 2019. Multi-strain Tn-seq reveals common daptomycin resistance determinants in Staphylococcus aureus. PLOS Pathog 15:e1007862. doi: 10.1371/journal.ppat.1007862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang H, Claveau D, Vaillancourt JP, Roemer T, Meredith TC. 2011. High-frequency transposition for determining antibacterial mode of action. Nat Chem Biol 7:720–729. doi: 10.1038/nchembio.643 [DOI] [PubMed] [Google Scholar]
- 47. Winstel V, Liang C, Sanchez-Carballo P, Steglich M, Munar M, Bröker BM, Penadés JR, Nübel U, Holst O, Dandekar T, Peschel A, Xia G. 2013. Wall teichoic acid structure governs horizontal gene transfer between major bacterial pathogens. Nat Commun 4:2345. doi: 10.1038/ncomms3345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Goryshin IY, Jendrisak J, Hoffman LM, Meis R, Reznikoff WS. 2000. Insertional transposon mutagenesis by electroporation of released Tn5 transposition complexes. Nat Biotechnol 18:97–100. doi: 10.1038/72017 [DOI] [PubMed] [Google Scholar]
- 49. Osorio H, Jara C, Fuenzalida K, Rey-Jurado E, Vásquez M. 2019. High-efficiency nuclear transformation of the microalgae Nannochloropsis oceanica using Tn5 transposome for the generation of altered lipid accumulation phenotypes. Biotechnol Biofuels 12:134. doi: 10.1186/s13068-019-1475-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chimukuche NM, Williams MJ. 2021. Genetic manipulation of non-tuberculosis mycobacteria. Front Microbiol 12:633510. doi: 10.3389/fmicb.2021.633510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vernyik V, Karcagi I, Tímár E, Nagy I, Györkei Á, Papp B, Györfy Z, Pósfai G. 2020. Exploring the fitness benefits of genome reduction in Escherichia coli by a selection-driven approach. Sci Rep 10:7345. doi: 10.1038/s41598-020-64074-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Laabei M, Recker M, Rudkin JK, Aldeljawi M, Gulay Z, Sloan TJ, Williams P, Endres JL, Bayles KW, Fey PD, Yajjala VK, Widhelm T, Hawkins E, Lewis K, Parfett S, Scowen L, Peacock SJ, Holden M, Wilson D, Read TD, van den Elsen J, Priest NK, Feil EJ, Hurst LD, Josefsson E, Massey RC. 2014. Predicting the virulence of MRSA from its genome sequence. Genome Res 24:839–849. doi: 10.1101/gr.165415.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. McLean K, Holmes EA, Penewit K, Lee DK, Hardy SR, Ren M, Krist MP, Huang K, Waalkes A, Salipante SJ. 2019. Artificial selection for pathogenicity mutations in Staphylococcus aureus identifies novel factors relevant to chronic infection. Infect Immun 87:e00884-18. doi: 10.1128/IAI.00884-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jin Q, Xie X, Zhai Y, Zhang H. 2023. Mechanisms of folate metabolism-related substances affecting Staphylococcus aureus infection. Int J Med Microbiol 313:151577. doi: 10.1016/j.ijmm.2023.151577 [DOI] [PubMed] [Google Scholar]
- 55. Alreshidi Mousa M, Dunstan RH, Gottfries J, Macdonald MM, Crompton MJ, Ang C-S, Williamson NA, Roberts TK. 2016. Changes in the cytoplasmic composition of amino acids and proteins observed in Staphylococcus aureus during growth under variable growth conditions representative of the human wound site. PLOS ONE 11:e0159662. doi: 10.1371/journal.pone.0159662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Alreshidi M. M., Dunstan RH, Macdonald MM, Gottfries J, Roberts TK. 2020. The uptake and release of amino acids by Staphylococcus aureus at mid-exponential and stationary phases and their corresponding responses to changes in temperature, pH and osmolality. Front. Microbiol 10:3059. doi: 10.3389/fmicb.2019.03059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Somerville GA, Chaussee MS, Morgan CI, Fitzgerald JR, Dorward DW, Reitzer LJ, Musser JM. 2002. Staphylococcus aureus aconitase inactivation unexpectedly inhibits post-exponential-phase growth and enhances stationary-phase survival. Infect Immun 70:6373–6382. doi: 10.1128/IAI.70.11.6373-6382.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ramsey KM, Ledvina HE, Tresko TM, Wandzilak JM, Tower CA, Tallo T, Schramm CE, Peterson SB, Skerrett SJ, Mougous JD, Dove SL. 2020. Tn-seq reveals hidden complexity in the utilization of host-derived glutathione in Francisella tularensis. PLOS Pathog 16:e1008566. doi: 10.1371/journal.ppat.1008566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Coulter SN, Schwan WR, Ng EY, Langhorne MH, Ritchie HD, Westbrock-Wadman S, Hufnagle WO, Folger KR, Bayer AS, Stover CK. 1998. Staphylococcus aureus genetic loci impacting growth and survival in multiple infection environments. Mol Microbiol 30:393–404. doi: 10.1046/j.1365-2958.1998.01075.x [DOI] [PubMed] [Google Scholar]
- 60. Goncheva MI, Flannagan RS, Heinrichs DE. 2020. De novo purine biosynthesis is required for intracellular growth of Staphylococcus aureus and for the hypervirulence phenotype of a purR mutant. Infect Immun 88:e00104-20. doi: 10.1128/IAI.00104-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rossi A, Kontarakis Z, Gerri C, Nolte H, Hölper S, Krüger M, Stainier DYR. 2015. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 524:230–233. doi: 10.1038/nature14580 [DOI] [PubMed] [Google Scholar]
- 62. De Souza AT, Dai X, Spencer AG, Reppen T, Menzie A, Roesch PL, He Y, Caguyong MJ, Bloomer S, Herweijer H, Wolff JA, Hagstrom JE, Lewis DL, Linsley PS, Ulrich RG. 2006. Transcriptional and phenotypic comparisons of Ppara knockout and siRNA knockdown mice. Nucleic Acids Res 34:4486–4494. doi: 10.1093/nar/gkl609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Santiviago CA, Reynolds MM, Porwollik S, Choi S-H, Long F, Andrews-Polymenis HL, McClelland M. 2009. Analysis of pools of targeted Salmonella deletion mutants identifies novel genes affecting fitness during competitive infection in mice. PLoS Pathog 5:e1000477. doi: 10.1371/journal.ppat.1000477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cramton SE, Gerke C, Schnell NF, Nichols WW, Götz F. 1999. The intercellular adhesion (ica) locus is present in Staphylococcus aureus and is required for biofilm formation. Infect Immun 67:5427–5433. doi: 10.1128/IAI.67.10.5427-5433.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gajdiss M, Monk IR, Bertsche U, Kienemund J, Funk T, Dietrich A, Hort M, Sib E, Stinear TP, Bierbaum G. 2020. YycH and YycI regulate expression of Staphylococcus aureus autolysins by activation of WalRK phosphorylation. Microorganisms 8:870. doi: 10.3390/microorganisms8060870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fournier B, Klier A, Rapoport G. 2001. The two-component system ArlS-ArlR is a regulator of virulence gene expression in Staphylococcus aureus: virulence regulation in Staphylococcus aureus. Mol Microbiol 41:247–261. doi: 10.1046/j.1365-2958.2001.02515.x [DOI] [PubMed] [Google Scholar]
- 67. Groma M, Horst SA, Das S, Huettel B, Klepsch M, Rudel T, Medina E, Fraunholz M, Fey PD, Gilmore MS. 2020. Identification of a novel LysR-type transcriptional regulator in Staphylococcus aureus that is crucial for secondary tissue colonization during metastatic bloodstream infection. mBio 11:e01646–20. doi: 10.1128/mBio.01646-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Alonzo F 3rd, Benson MA, Chen J, Novick RP, Shopsin B, Torres VJ. 2012. Staphylococcus aureus leucocidin ED contributes to systemic infection by targeting neutrophils and promoting bacterial growth in vivo. Mol Microbiol 83:423–435. doi: 10.1111/j.1365-2958.2011.07942.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hall JW, Yang J, Guo H, Ji Y, Fang FC. 2017. The Staphylococcus aureus AirSR two-component system mediates reactive oxygen species resistance via transcriptional regulation of staphyloxanthin production. Infect Immun 85:e00838–16. doi: 10.1128/IAI.00838-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Basu A, Shields KE, Yap M-N. 2020. The hibernating 100S complex is a target of ribosome-recycling factor and elongation factor G in Staphylococcus aureus. J Biol Chem 295:6053–6063. doi: 10.1074/jbc.RA119.012307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lannergård J, Cao S, Norström T, Delgado A, Gustafson JE, Hughes D. 2011. Genetic complexity of fusidic acid-resistant small colony variants (SCV) in Staphylococcus aureus. PLoS ONE 6:e28366. doi: 10.1371/journal.pone.0028366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Howden BP, Smith DJ, Mansell A, Johnson PDR, Ward PB, Stinear TP, Davies JK. 2008. Different bacterial gene expression patterns and attenuated host immune responses are associated with the evolution of low-level vancomycin resistance during persistent methicillin-resistant Staphylococcus aureus bacteraemia. BMC Microbiol 8:39. doi: 10.1186/1471-2180-8-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chaffin DO, Taylor D, Skerrett SJ, Rubens CE. 2012. Changes in the Staphylococcus aureus transcriptome during early adaptation to the lung. PLoS ONE 7:e41329. doi: 10.1371/journal.pone.0041329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Radin JN, Kelliher JL, Solórzano PKP, Grim KP, Ramezanifard R, Slauch JM, Kehl-Fie TE. 2019. Metal-independent variants of phosphoglycerate mutase promote resistance to nutritional immunity and retention of glycolysis during infection. PLOS Pathog 15:e1007971. doi: 10.1371/journal.ppat.1007971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. San-Martin-Galindo P, Rosqvist E, Tolvanen S, Miettinen I, Savijoki K, Nyman TA, Fallarero A, Peltonen J. 2021. Modulation of virulence factors of Staphylococcus aureus by nanostructured surfaces. Mater Des 208:109879. doi: 10.1016/j.matdes.2021.109879 [DOI] [Google Scholar]
- 76. Buvelot H, Roth M, Jaquet V, Lozkhin A, Renzoni A, Bonetti E-J, Gaia N, Laumay F, Mollin M, Stasia M-J, Schrenzel J, François P, Krause K-H. 2021. Hydrogen peroxide affects growth of S. aureus through downregulation of genes involved in pyrimidine biosynthesis. Front Immunol 12:673985. doi: 10.3389/fimmu.2021.673985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Peyrusson F, Varet H, Nguyen TK, Legendre R, Sismeiro O, Coppée J-Y, Wolz C, Tenson T, Van Bambeke F. 2020. Intracellular Staphylococcus aureus persisters upon antibiotic exposure. Nat Commun 11:2200. doi: 10.1038/s41467-020-15966-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Grosser MR, Paluscio E, Thurlow LR, Dillon MM, Cooper VS, Kawula TH, Richardson AR. 2018. Genetic requirements for Staphylococcus aureus nitric oxide resistance and virulence. PLOS Pathog 14:e1006907. doi: 10.1371/journal.ppat.1006907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jani S, Sterzenbach K, Adatrao V, Tajbakhsh G, Mascher T, Golemi-Kotra D. 2020. Low phosphatase activity of LiaS and strong liar-DNA affinity explain the unusual LiaS to liar in vivo stoichiometry. BMC Microbiol 20:104. doi: 10.1186/s12866-020-01796-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sharma-Kuinkel BK, Mann EE, Ahn J-S, Kuechenmeister LJ, Dunman PM, Bayles KW. 2009. The Staphylococcus aureus LytSR two-component regulatory system affects biofilm formation. J Bacteriol 191:4767–4775. doi: 10.1128/JB.00348-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Yang S-J, Xiong YQ, Yeaman MR, Bayles KW, Abdelhady W, Bayer AS. 2013. Role of the LytSR two-component regulatory system in adaptation to cationic antimicrobial peptides in Staphylococcus aureus. Antimicrob Agents Chemother 57:3875–3882. doi: 10.1128/AAC.00412-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhang B, Liu X, Lambert E, Mas G, Hiller S, Veening J-W, Perez C. 2020. Structure of a proton-dependent lipid transporter involved in lipoteichoic acids biosynthesis. Nat Struct Mol Biol 27:561–569. doi: 10.1038/s41594-020-0425-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wood BM, Santa Maria JP, Matano LM, Vickery CR, Walker S. 2018. A partial reconstitution implicates DltD in catalyzing lipoteichoic acid d-alanylation. J Biol Chem 293:17985–17996. doi: 10.1074/jbc.RA118.004561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Weidenmaier C, Peschel A. 2008. Teichoic acids and related cell-wall glycopolymers in gram-positive physiology and host interactions. Nat Rev Microbiol 6:276–287. doi: 10.1038/nrmicro1861 [DOI] [PubMed] [Google Scholar]
- 85. van Dalen R, Peschel A, van Sorge NM. 2020. Wall teichoic acid in Staphylococcus aureus host interaction. Trends Microbiol 28:985–998. doi: 10.1016/j.tim.2020.05.017 [DOI] [PubMed] [Google Scholar]
- 86. Wobser D, Ali L, Grohmann E, Huebner J, Sakinc T, Lemos JA. 2014. A novel role for d-alanylation of lipoteichoic acid of Enterococcus faecalis in urinary tract infection. PLoS ONE 9:e107827. doi: 10.1371/journal.pone.0107827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Cooper VS, Honsa E, Rowe H, Deitrick C, Iverson AR, Whittall JJ, Neville SL, McDevitt CA, Kietzman C, Rosch JW, Shade A. 2020. Experimental evolution in vivo to identify selective pressures during pneumococcal colonization . mSystems 5:e00352–20. doi: 10.1128/mSystems.00352-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kahl BC, Becker K, Löffler B. 2016. Clinical significance and pathogenesis of staphylococcal small colony variants in persistent infections. Clin Microbiol Rev 29:401–427. doi: 10.1128/CMR.00069-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hirschhausen N, Block D, Bianconi I, Bragonzi A, Birtel J, Lee JC, Dübbers A, Küster P, Kahl J, Peters G, Kahl BC. 2013. Extended Staphylococcus aureus persistence in cystic fibrosis is associated with bacterial adaptation. Int J Med Microbiol 303:685–692. doi: 10.1016/j.ijmm.2013.09.012 [DOI] [PubMed] [Google Scholar]
- 90. Schwartbeck B, Birtel J, Treffon J, Langhanki L, Mellmann A, Kale D, Kahl J, Hirschhausen N, Neumann C, Lee JC, Götz F, Rohde H, Henke H, Küster P, Peters G, Kahl BC. 2016. Dynamic in vivo mutations within the ica operon during persistence of Staphylococcus aureus in the airways of cystic fibrosis patients. PLOS Pathog 12:e1006024. doi: 10.1371/journal.ppat.1006024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Thammavongsa V, Kern JW, Missiakas DM, Schneewind O. 2009. Staphylococcus aureus synthesizes adenosine to escape host immune responses. J Exp Med 206:2417–2427. doi: 10.1084/jem.20090097 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Oligonucleotide sequences.
Composition of initial transposon mutant pool for ATCC29213.
Composition of initial transposon pool JE2.
Tn-Seq results for ATCC29213, initial vs invasion library.
Tn-Seq results for JE2, initial vs invasion library.
Gene homologs identified as significant for invasion in both S. aureus strains.
Tn-Seq results for ATCC29213, outgrowth control vs invasion library.
Tn-Seq results for JE2, outgrowth control vs invasion library.
CRISPRi gene knockdown results.
Mutational characteristics of candidate macrophage invasion genes in vivo.
Data Availability Statement
Sequence data from this study are available from the NCBI Sequence Read Archive under accession PRJNA942332.