

Abstract
Bacterial transposons are pervasive mobile genetic elements that use distinct DNA-binding proteins for horizontal transmission. For example, Escherichia coli Tn7homes to a specific attachment site using TnsD1, whereas CRISPR-associated transposons use type I or type V Cas effectors to insert downstream of target sites specified by guide RNAs2,3. Despite this targeting diversity, transposition invariably requires TnsB, a DDE-family transposase that catalyses DNA excision and insertion, and TnsC, a AAA+ ATPase that is thought to communicate between transposase and targeting proteins4. How TnsC mediates this communication and thereby regulates transposition fidelity has remained unclear. Here we use chromatin immunoprecipitation with sequencing to monitor in vivo formation of the type I-F RNA-guided transpososome, enabling us to resolve distinct protein recruitment events before integration. DNA targeting by the TniQ–Cascade complex is surprisingly promiscuous—hundreds of genomic off-target sites are sampled, but only a subset of those sites is licensed for TnsC and TnsB recruitment, revealing a crucial proofreading checkpoint. To advance the mechanistic understanding of interactions responsible for transpososome assembly, we determined structures of TnsC using cryogenic electron microscopy and found that ATP binding drives the formation of heptameric rings that thread DNA through the central pore, thereby positioning the substrate for downstream integration. Collectively, our results highlight the molecular specificity imparted by consecutive factor binding to genomic target sites during RNA-guided transposition, and provide a structural roadmap to guide future engineering efforts.
Transposable elements are pervasive across the tree of life and have fundamental roles in the biology and evolution of genomes5. Key to their dissemination are transposases, a ubiquitous class of enzymes that catalyse DNA excision and integration of class II DNA transposons6. Although most transposons are mobilized by single transposase polypeptides, which typically multimerize during the formation of synaptic complexes, bacterial Tn7-family transposons are unique in their reliance on heteromeric transposases, in which dedicated factors perform distinct functional tasks to properly position donor DNA and target DNA substrates for chemical transformation by the holo transpososome complex4. This architectural diversity has allowed Tn7-like transposons to adopt complex ‘lifestyles’ and targeting pathways, expanding the manner in which they evolve and diversify7.
E. coli Tn7 adopts a cut-and-paste transposition mechanism that involves multiple genes, tnsA, tnsB, tnsC, tnsD and tnsE (reviewed previously4). TnsA is structurally homologous to type II endonucleases, and TnsB is a member of the retroviral integrase superfamily, containing the characteristic DDE catalytic motif. TnsA and TnsB function as a heteromeric transposase to catalyse the excision and integration of transposon ends8. TnsC is an AAA+ (ATPase associated with various cellular activities) ATPase that was shown to mediate communication between the TnsAB transposase and one of two target-selecting proteins, TnsD or TnsE, which determines the specificity of transposition9–11. TnsD is a TniQ-family protein that directs transposon insertion into an attachment site downstream of the conserved glmS gene, whereas TnsE increases the frequency of non-specific integration into conjugative plasmids4. Biochemical studies suggest that TnsC is recruited to a local distortion in the target site, leading to TnsAB recruitment and DNA integration around 25 bp downstream of the TnsD binding site12. TnsC mutants with impaired ATP hydrolysis activity stimulate TnsAB transposition independently of targeting factors, implicating the ATPase activity of TnsC in toggling the TnsAB transposase between its active and inactive states11,13.
Tn7belongs to a phylogenetically diverse family of transposable elements that contain the tnsBC operon necessary for transposition, but possess diversified targeting factors. Recent comparative genomic analyses revealed a new class of elements that encode nuclease-deficient CRISPR–Cas systems14,15, including class I systems (types I-F3, I-B) that use Cascade2,16, and class 2 systems (type V-K) that use Cas12k3. We recently showed that the Vibrio cholerae (Vch) CRISPR-associated transposon (Tn6677) directs non-replicative, RNA-guided DNA integration with high efficiency and specificity by leveraging the DNA-targeting ability of the TniQ–Cascade complex2,17,18 (Fig. 1a). Although the genetic requirements for RNA-guided transposition have been determined, the hierarchy of interactions between TniQ–Cascade and TnsABC remains unclear, as well as how genome-wide specificity is achieved and influenced by the activity of TnsC. Sequence analysis unambiguously places it in the large family of AAA+ ATPases, which mediate diverse molecular processes ranging from protein folding to DNA translocation and transposition19. These ATPases share a common architectural framework built by ATP binding motifs, and additional accessory domains assist in sensing the presence of binding partners20. ATP-dependent oligomerization is a common theme, with ATP hydrolysis often acting as a switch mechanism to trigger disassembly or drive molecular movements that are propagated through the oligomer when a specific input is received20. It was proposed that the Tn7 post-transposition complex consists of around 25 copies of TnsC21; more recent structural studies of TnsC homologues have revealed a range of oligomeric states, including ring-like assemblies (E. coli TnsC)22 and large filaments that continuously wrap around double-stranded DNA (dsDNA) (Scytonema hofmannii TnsC)23,24. How the biochemical behaviour of TnsC from type I-F CRISPR-associated transposons (CAST systems) compares, and how TnsC recruitment affects the selection of genomic target sites for integration, has remained elusive.
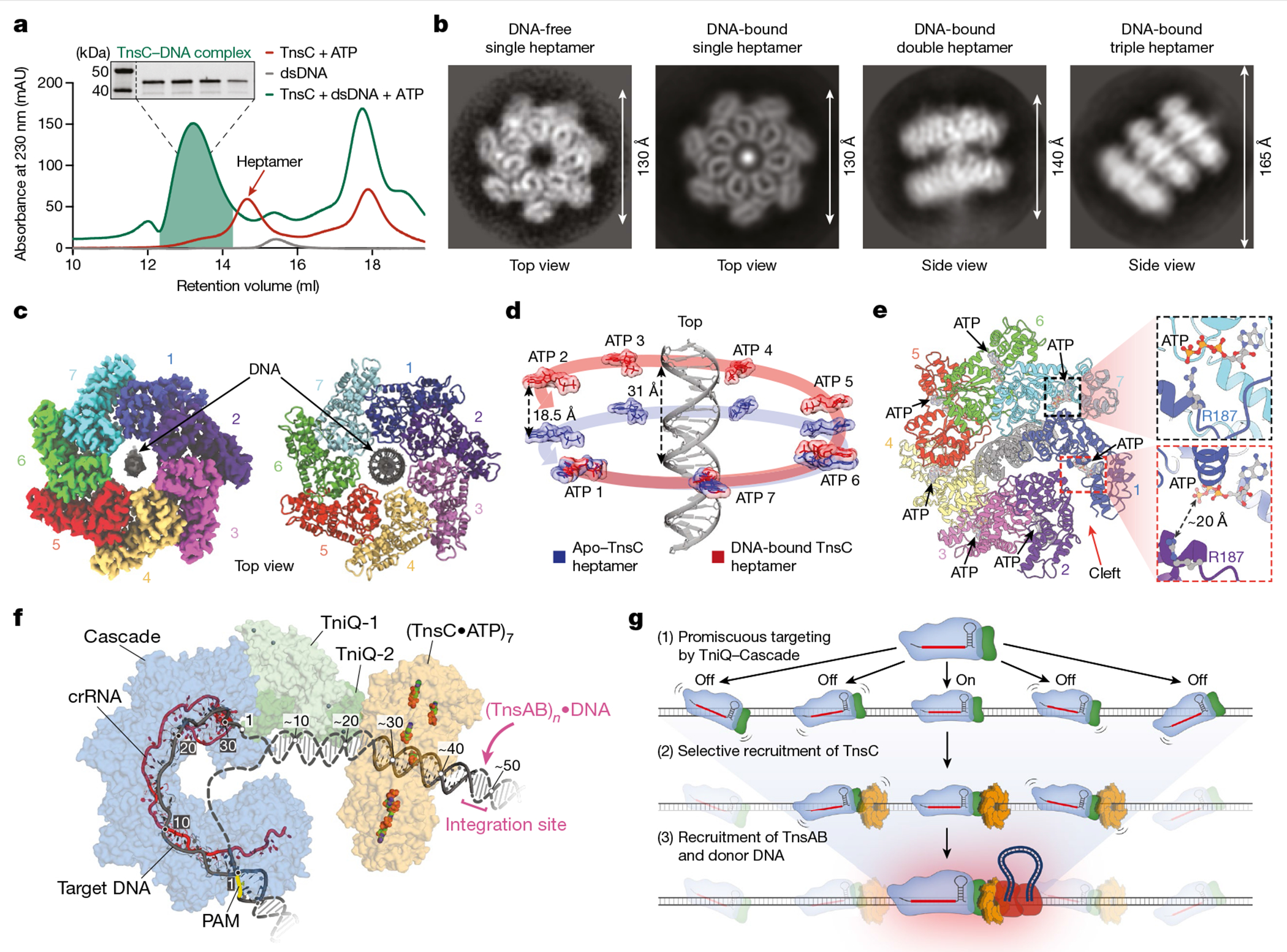
Fig. 1 |. In vivo assembly of the RNA-guided transpososome, monitored using ChIP–seq.
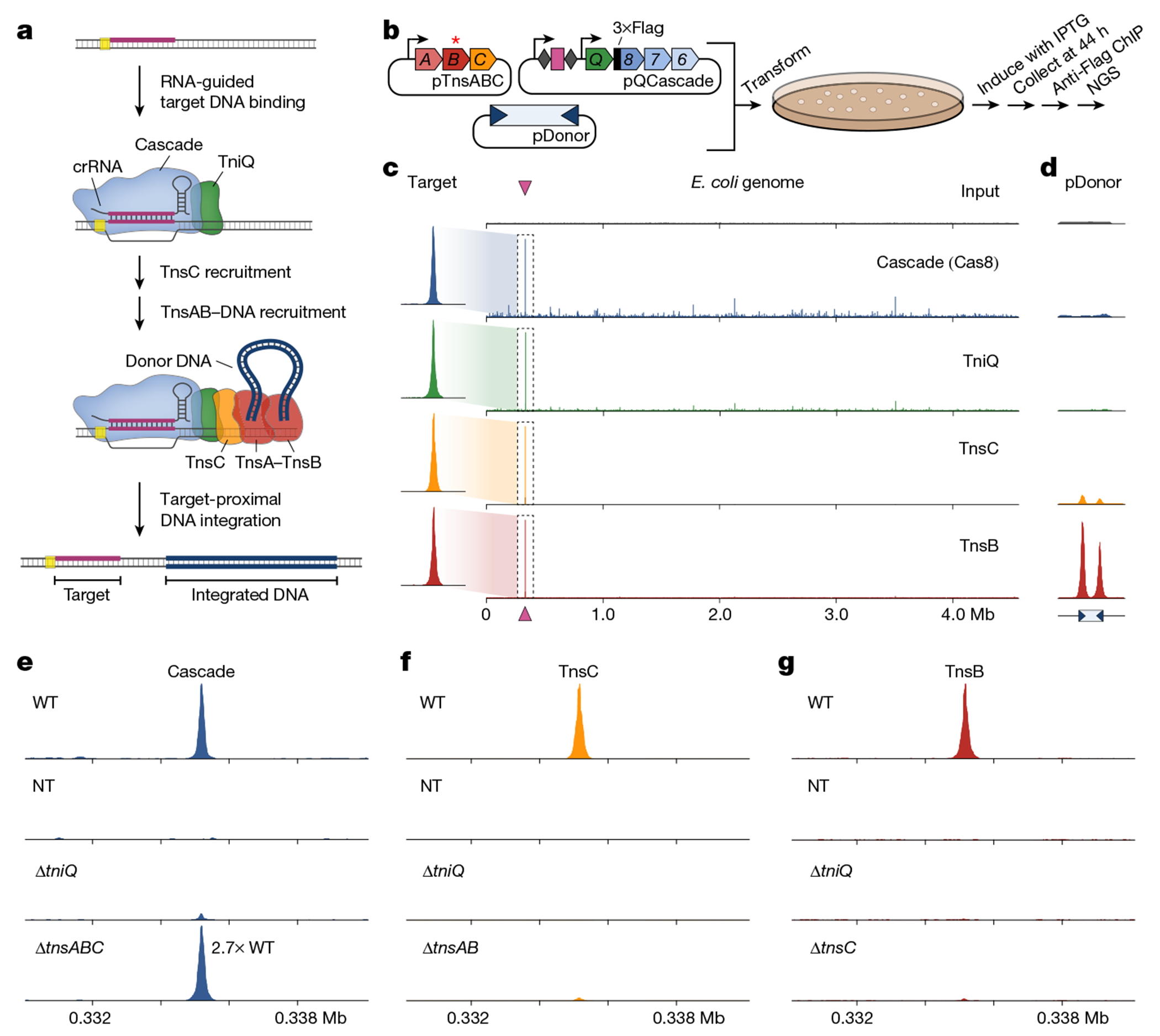
a, Schematic of RNA-guided DNA integration by transposons that encode type I-F CRISPR–Cas systems. R-loop formation by TniQ–Cascade is hypothesized to result in the sequential recruitment of TnsC and TnsA–TnsB bound to the transposon donor DNA, leading to integration about 50 bp downstream of the target site. Proteins are represented by a single shape for clarity, b, Schematic of the ChIP–seq workflow. E. coli cells were transformed with three plasmids encoding TnsABC, QCascade and the mini-transposon; one component was tagged with×Flag, and TnsB contains an inactivating point mutation indicated by the asterisk (*). After induction, cells were collected and lysed, protein–DNA cross-links were immunoprecipitated and NGS libraries were prepared. c, Representative ChIP–seq data for experiments with crRNA-4 reveal accurate recruitment of multiple distinct factors to the corresponding genomic target site (maroon triangle). Genome-wide graphs show read coverage (reads per kilobase per million mapped reads (RPKM)) normalized to the highest peak (Methods), and the insets show 3 kb of coverage centred at the target site. The Flag-tagged component in each experiment is indicated, and an input control (scaled proportionally to Cas8 ChIP–seq data) is shown at the top. d, pDonor-mapping reads are shown for the same samples as in c, normalized to the maximum peak height for TnsB. The inverted blue triangles represent transposon ends, which contain multiple TnsB binding sites. e–g, ChIP–seq control experiments provide evidence of a hierarchical transpososome assembly. Read coverage (RPKM) is shown for a 10-kb window centred on the target site for Cascade (e), TnsC (f) and TnsB (g) normalized to the WT peak in each panel (with the exception of Cascade ΔtnsABC, which is 2.7-fold the WT enrichment). Gene deletion or non-targeting (NT) crRNA control experiments are denoted.
We used cryo-electron microscopy to determine structures of VchTnsC•ATP complexes in the presence and absence of DNA, and chromatin immunoprecipitation with sequencing (ChIP–seq) to assay the genome-wide binding profiles of TnsC, TnsB and the TniQ–Cascade complex in vivo. These experiments offer insights into the way that potential target sites are identified, screened and licensed for RNA-guided DNA integration, and highlight the role of heteromeric factor recruitment in enhancing overall specificity. The structural outlook provides an understanding of how ATP binding drives oligomerization, how these oligomers engage DNA substrates and which regions of TnsC are likely to mediate critical interactions with TniQ–Cascade and TnsAB. Together, these findings will inform future efforts to develop CRISPR-associated transposases for the programmable delivery of cargo DNA to any locus of interest.
ChIP–seq captures transposase recruitment
Although our previous studies rigorously profiled the genome-wide specificity of RNA-guided transposition2,17,25, we wanted to further examine the reaction pathway by monitoring the specificity of protein recruitment steps upstream of DNA integration (Fig. 1a). Thus, we used ChIP–seq as an unbiased approach to capture in vivo genome-wide binding of several protein components, under conditions in which DNA excision and integration was prevented. We reconstituted the V. cholerae I-F system from Tn6677 (also referred to as VchCAST) using a three-plasmid expression vector design2, and included a point mutation in TnsB that prevents integration and a 3×Flag epitope tag on either Cas8 (Cascade large subunit), TniQ, TnsC, TnsA or TnsB (Fig. 1b and Extended Data Fig. 1a,b). After culturing cells under conditions that promote high-efficiency transposition activity (Extended Data Fig. 1c), cells were cross-linked to trap assembled transpososomes in a pre-integration complex state, ChIP was performed using standard methods and next-generation sequencing (NGS) libraries were generated (Methods).
The resulting ChIP–seq data revealed robust DNA enrichment directly at the CRISPR RNA (crRNA)-matching target site for parallel experiments in which distinct subunits were tagged, relative to a non-immunoprecipitated input control (Fig. 1c), with the exception of TnsA, which showed only weak target-specific enrichment (Extended Data Fig. 1d). Interestingly, whereas TnsC and TnsB both showed a paucity of visible off-target binding sites, both Cascade (3×Flag–Cas8) and TniQ exhibited dozens to hundreds of additional peaks above the background, which were reproducibly sampled in biological replicates (Fig. 1a and Extended Data Fig. 2a), suggesting a higher degree of promiscuity during the initial target search by the TniQ–Cascade complex (see below). ChIP–seq reads were also mapped to the three plasmids, revealing two additional insights into the biological pathway. First, both TnsA and TnsB exhibited strong enrichment at both transposon ends on pDonor, which contain several TnsB -binding sites2, providing a direct read-out of donor DNA recognition (Fig. 1d). TnsC also showed enrichment at the same sites, which we speculate reflects recruitment to the transposon DNA that probably occurs through protein–protein interactions with TnsA, as has been previously observed for E. coli Tn726. Second, we consistently observed enrichment of Cascade and downstream factors at the CRISPR array on pQCascade, which encodes the crRNA and therefore also represents a self-target (Extended Data Fig. 1f). These data agree with our prior observation of rare, self-targeting insertions and indicate that protospacer adjacent motif (PAM) recognition is relatively flexible, in contrast to the highly selective PAM requirements for other CRISPR–Cas effectors27. All of the above observations were consistent across experiments using a distinct crRNA sequence (Extended Data Fig. 1e,f).
We next sought to investigate anticipated protein dependencies during transpososome assembly through a series of carefully designed control experiments. Target DNA binding by Cascade was dependent on the crRNA sequence and independent of TnsABC expression, as expected, but exhibited some sensitivity to TniQ deletion (Fig. 1e). We concluded that this may result from expression and/or cross-linking efficiency differences, as the in vitro DNA binding behaviours of Cascade and TniQ–Cascade were similar, as were the in vivo binding activities as assessed by a fluorescence reporter assay (Extended Data Fig. 1g,h and Methods). TnsC recruitment was also strictly dependent on the presence of TniQ, which we previously speculated is a key docking platform when complexed with Cascade18. However, we were surprised to find that the removal of TnsA and TnsB significantly reduced target DNA enrichment, a result we verified using quantitative PCR (qPCR; Fig. 1f and Extended Data Fig. 1i). Although future experiments will be necessary, this result suggests a stabilizing function of TnsA, TnsB and/or the donor DNA complex during TnsC recruitment. Finally, TnsB recruitment was strictly dependent on the presence of both TniQ and TnsC (Fig. 1g), confirming a crucial role of these factors in mediating interactions that enable communication between the crRNA-guided targeting module and the terminal integration module. Interestingly, TnsB recruitment to the target site still proceeded independently of both TnsA and the donor DNA, albeit with reduced efficiency (Extended Data Fig. 1j), indicating that neither a paired-end complex nor TnsA/DNA-dependent heteromeric TnsB assembly are absolutely essential for its ability to engage other factors at genomic target sites.
Transpososome assembly enhances fidelity
Previous ChIP–seq studies provided insights into the molecular mechanisms governing target DNA recognition for both Streptococcus pyogenes Cas9 and E. coli Cascade28–32. We were therefore keen to further examine the pervasive VchCascade binding sites spread across the genome (Fig. 1c and Extended Data Fig. 1e). Using Cas8 ChIP–seq data from four independent datasets with distinct guides, we identified hundreds of off-target peaks using MACS333 and then used MEME34 to identify motifs that were common to each dataset. This analysis revealed that a majority of peaks shared a 5′-CN-3′ PAM and a highly conserved, crRNA-matching seed sequence ranging from 5–10 bp in length (Fig. 2a). These data are in excellent agreement with our PAM library integration experiments and earlier mechanistic studies of type I-E Cascade29,35. As further evidence that these motifs represented bona fide Cascade off-target sites, we observed low sequence discrimination at the +6 position, consistent with structural research demonstrating that every sixth nucleotide of the crRNA spacer is ‘flipped out’ and does not participate in target DNA base-pairing18.
Fig. 2 |. Analysis of off-target binding events by Cascade and downstream transposase factors.
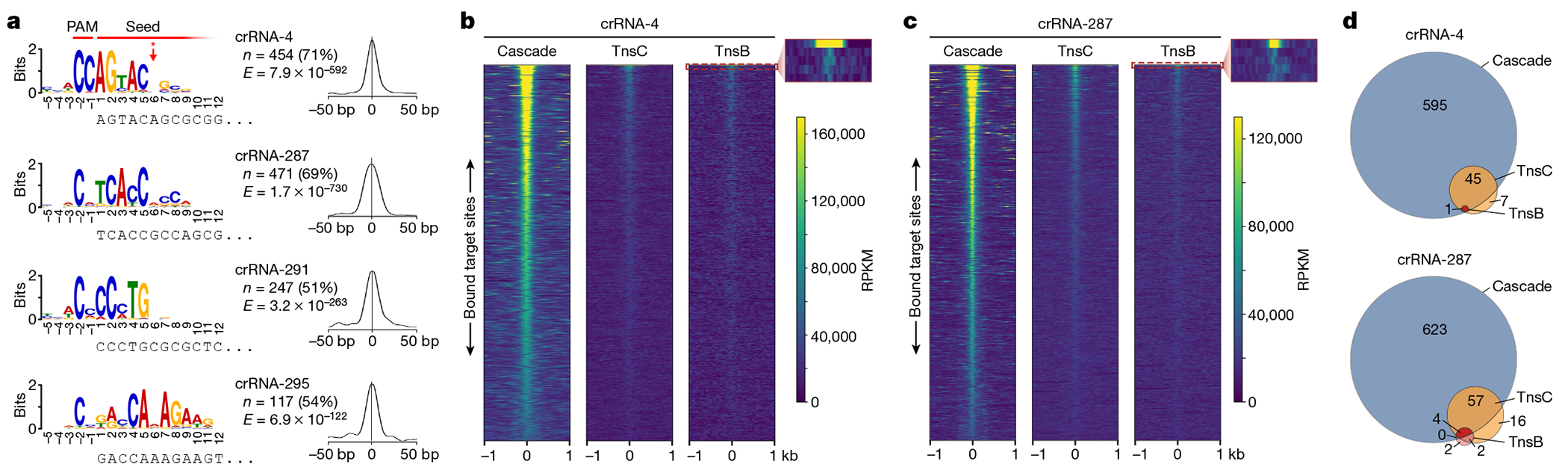
a, An analysis using MEME to identify motifs that are common to Cascade off-target peaks revealed conserved PAM and seed sequences. The motif visualized above the corresponding nucleotides at the 5′end of crRNA spacer (left) and a graph showing the probability of a motif match occurring at a given position in the input sequence (right) are shown for four datasets with distinct crRNAs (top to bottom). The PAM and seed regions are indicated at the top, as well as the lack of discrimination at position 6 (red asterisk/arrow). n indicates the number of peaks contributing to the motif (and their percentage of the total peaks called); E, E-value significance of the motif generated from the MEME analysis. b, Global visualization of ChIP–seq peaks with crRNA-4 for off-target sites using heat maps, plotted side by side for Cascade (left), TnsC (middle) and TnsB (right). A 2-kb window for 641 genomic loci (y axis) is plotted in order of decreasing peak enrichment for ChIP–seq peaks called for the Cascade dataset. TnsC and TnsB enrichments are shown for the same positions as the Cascade peaks and are ordered on the basis of Cascade enrichment. The inset for TnsB highlights the strong enrichment of the on-target site (row 1) and the immediate drop-off for the other Cascade off-target sites (rows 2–5). c, Global visualization of ChIP–seq peaks with crRNA-287, plotted as described in b. d, The overlap in off-target peaks called for Cascade, TnsC and TnsB. Peaks were called individually for each ChIP–seq dataset using MACS3, and overlaps were analysed based on the coordinates for each peak. Data are shown for the same two crRNAs as in b and c.
Although the observation of frequent off-target binding events was not unanticipated, given the extensive literature documenting similar trends for other CRISPR–Cas effectors, it nevertheless conflicted with our previous findings that DNA integration is highly accurate2,17. We therefore hypothesized that Cascade may licence the recruitment of downstream, transposon-associated factors at only a subset of off-target binding sites. To test this, we extracted the genomic coordinates of Cascade peaks for each crRNA and compared the levels of TnsC and TnsB enrichment at the same sites. The resulting heat maps revealed that the vast majority of off-target sites bound by Cascade failed to recruit either TnsC or TnsB, and that TnsB in particular showed robust enrichment at only a single genomic site: the on-target (Fig. 2b,c and Extended Data Fig. 2d,e). When we compared all peaks that were independently called for each ChIP–seq dataset, we consistently observed a decrease in the number of off-target sites bound by TnsB and TnsC, as compared to Cascade (Fig. 2d and Extended Data Fig. 2f). To support the conclusion that TnsC binding generally occurred at genomic sites at which Cascade was present, we found that enrichment values for overlapping peaks were positively correlated, and that the coordinates of their peak maxima were predominantly within 50 bp of each other (Extended Data Fig. 2b,c).
Collectively, these results demonstrate that initial target recognition by Cascade occurs in a promiscuous manner, but that most off-target sites fail to recruit the downstream factors necessary to form catalytically competent transpososome complexes. Our results suggest a funnel-like recruitment pathway in which Cascade identifies a large set of candidate integration sites, of which a subset is bound by TnsC and an even smaller subset is bound by TnsB—resulting in an overall gain in the fidelity of RNA-guided transposition. We speculate that the bound lifetime and/or conformational heterogeneity of DNA-bound Cascade complexes control the binding of TnsC, similar to how canonical type I Cascade complexes regulate Cas3 nuclease recruitment and priming versus interference decision making30,36.
One oddity that arose during our analyses was a class of off-target sites shared across multiple Cascade ChIP–seq datasets, which all exhibited the same PAM and seed-sequence motifs (Extended Data Fig. 2g). These off-target sites could be explained by pseudo-crRNAs that use a spacer-like sequence downstream of the CRISPR array and presumably assemble Cascade-like complexes that lack Cas6 but are nevertheless capable of RNA-guided DNA binding (Extended Data Fig. 2h). These results are consistent with previous studies of other type I systems showing that Cas6 is dispensable for Cascade assembly and target DNA recognition37. Although Cas6-deficient Cascade in our system would probably fail to direct downstream DNA integration2,18, pseudo-crRNAs accounted for 15–30% of off-target sites (Extended Data Fig. 2g) and could titrate protein components away from the primary (intended) crRNA. These results highlight the importance of carefully considering all design parameters when generating guide RNA expression cassettes.
Mismatch sensitivity of transposition
When we analysed Cascade ChIP–seq peaks for crRNA-291, we readily identified off-target (OT) sequences corresponding to the most highly enriched sites (Fig. 3a), including OT1 and OT2 that differed in only 3–4 positions at the PAM-distal end, where Cascade generally exhibits minimal discrimination30,38. These sites were nearly completely ignored during the TnsC recruitment step, as indicated by a comparative analysis of TnsC ChIP–seq data (Fig. 3a). We previously investigated crRNA sequence requirements during RNA-guided transposition and found that mismatches at positions 25–28, as well as in the well-known seed sequence, eliminated DNA integration, but we were unable to resolve where in the pathway mismatch discrimination was occurring2. Thus, the ChIP–seq data suggested that stable TnsC recruitment was being specifically inhibited, a hypothesis that we confirmed by performing quantitative TnsC ChIP–qPCR analysis at the on-target site with similarly mismatched crRNAs. TnsC barely enriched the target DNA locus above the background when positions 1–4 (seed) or 25–28 (PAM-distal) were mutated, whereas mismatches elsewhere led to variable levels of DNA enrichment (Fig. 3b).
Fig. 3 |. PAM-distal RNA–DNA mismatches affect TnsC recruitment and RNA-guided transposition.
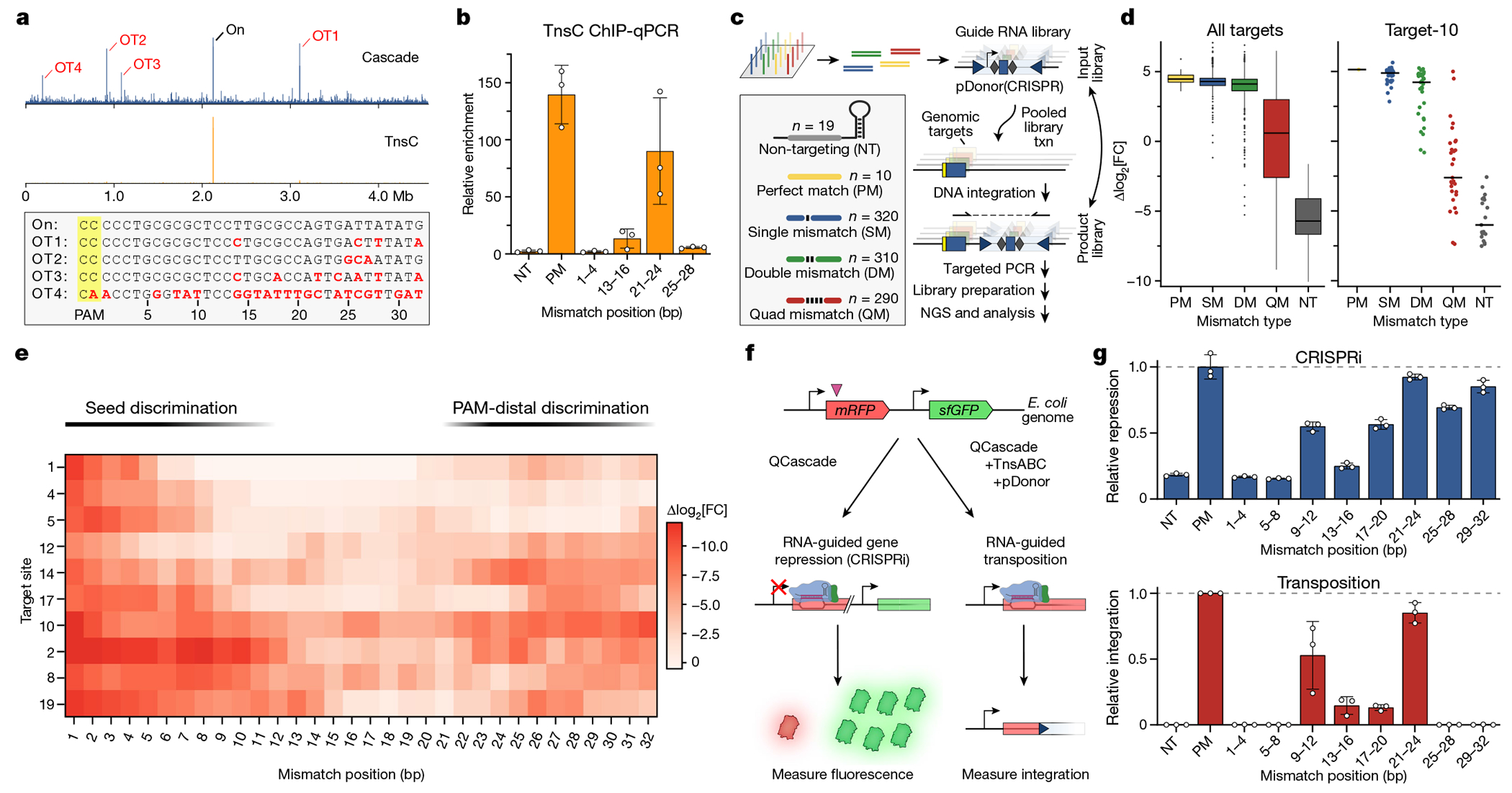
a, Genome-wide ChIP–seq profiles for Cascade and TnsC using crRNA-291, with the indicated on- and off-target sites (OT1–4). The corresponding DNA sequences are shown below, with PAM (yellow) and mismatches (red), b, TnsC ChIP–qPCR quantification of target DNA enrichment for crRNA-4 and mismatched variants. Enrichment values (ΔΔCq) are measured relative to a reference locus. NT, non-targeting; PM, perfect match. c, Schematic of the pooled crRNA library experiment to measure the effect of RNA–DNA mismatches at ten unique target sites. By encoding the crRNA within the mini-transposon on pDonor, amplicon deep sequencing captures the abundances of library members driving integration to each target site. Txn, transformation. d, Enrichment scores describing the crRNA abundance change in the integration product versus input library, plotted as log2-transformed fold change (FC) with box plots for all targets (left, n of mismatch library samples as shown in c) or individual data points for target-10 library members (right). The boxes show the median (centre line) and interquartile range (box limits), and the whiskers indicate the range or extend to 1.5× the interquartile range from the 25th and 75th percentile in the presence of outliers (dots). SM, single mismatch; DM, double mismatch; QM, quad mismatch. e, Position-specific enrichment scores for mismatched library members against ten target sites (y axis), plotted as the difference in log2-transformed fold change relative to the perfectly matching crRNA. f, Schematic of CRISPR interference (CRISPRi) experiments to monitor in vivo target binding by QCascade through transcriptional repression of mRFP (left). In parallel experiments, the inclusion of TnsABC and pDonor leads to RNA-guided transposition (right). g, CRISPRi (top) and transposition activity (bottom) for mismatched crRNAs, demonstrating that PAM-distal mismatches (positions 25–32) minimally perturb DNA binding but ablate DNA integration. CRISPRi repression was measured as optical density-normalized mRFP fluorescence; transposition was measured using junction qPCR. For b and g, data are shown as mean ± s.d. of n = 3 independent biological replicates.
To build a more comprehensive picture of how mismatches affect RNA-guided DNA integration by type I-F CAST systems, we developed a scalable, guide RNA library-based transposition assay (Fig. 3c and Methods). The majority of CRISPR libraries rely on inferring the activity of a given Cas9 sgRNA by perturbing cells (such as by gene editing), applying some type of selection and then deep sequencing the sgRNA expression cassette, without directly detecting the edit itself. In contrast, we reasoned that a programmable integration technology would uniquely enable the guide RNA library member to be sequenced together with the DNA insertion product in the same step. We redesigned our expression vectors so that the crRNA was encoded within the mini-transposon cargo, confirmed that high integration activity was preserved and then cloned a library of spacers that would introduce tiled mismatches of various length (single, dual or quad) across ten different target sites in the E. coli genome (Fig. 3c and Extended Data Fig. 3a,b). We amplified all of the integration products for each target site by PCR after culturing cells transformed with the pDonor(+CRISPR) library. Enrichment/depletion scores were then calculated for each library member by determining its fold change in the integration product library relative to the plasmid input library (Methods).
As expected, library members were more depleted with increasing numbers of RNA–DNA mismatches, indicating a reduction in transposition efficiency; this trend was readily apparent for multiple target sites (Fig. 3d and Extended Data Fig. 3c). Single mismatches incurred a minor penalty, if any at all, whereas quadruple mismatches resulted in a wide range of enrichment scores, which we suspected reflected position-specific effects on transposition efficiency. Indeed, when we plotted enrichment scores across the 32-bp target sequence as heat maps, some clear trends emerged from the data (Fig. 3e), despite heterogeneity among the tested target sites as has been observed previously39. Mismatches in the seed sequence consistently led to large penalties in transposition efficiency, as did mismatches in a PAM-distal region around positions 25–32 (Fig. 3e and Extended Data Fig. 3d,e). This pattern was particularly evident for target-19, for which only mismatches within the middle of the crRNA spacer (15–23) were well tolerated. We also included library members that contained additional mismatch combinations focused within the last 10 bp of the target site, and identified positions for which transposition was especially sensitive or insensitive to mutation (Extended Data Fig. 3f). These data corroborated our earlier results2 and provided critical evidence that the PAM-distal region has a generalizable role in regulating whether off-target sites are licensed for RNA-guided DNA integration.
Type I Cascade complexes engage potential DNA target sites by first recognizing the PAM, promoting DNA unwinding within the seed sequence, and directionally unzipping the DNA from the PAM-proximal to the PAM-distal end40. In canonical type I-E CRISPR–Cas systems, Cas3 nuclease recruitment is highly sensitive to the Cascade conformational state, which is affected by both PAM mutations and RNA–DNA mismatches30,38. Analogously, in our type I-F system, we hypothesized that PAM-distal mismatches were primarily inhibiting TnsC recruitment, as suggested by ChIP–qPCR data (Fig. 3b), by modulating the conformational state of Cascade and/or rendering the R-loop structure unfavourable for TnsC binding. Importantly, as with other studies of DNA binding by type I-E Cascade, we anticipated that PAM-distal mismatches would not entirely ablate DNA binding (Fig. 3a). To test this, we developed an in vivo DNA binding assay, in which targeting of genomically integrated mRFP leads to transcriptional repression through CRISPR interference (CRISPRi)41 (Fig. 3f). Consistent with previous dCas9 studies41, TniQ–Cascade binding resulted in strand-specific gene knockdown that could be switched between mRFP and GFP depending on the crRNA sequence (Extended Data Fig. 3g,h). When we introduced a panel of quadruple mismatches tiled throughout the crRNA spacer, transcriptional repression was eliminated with seed-sequence mutations, as expected. By contrast, repression was minimally affected by PAM-distal mutations at positions 21–32 (Fig. 3g (top)), demonstrating that Cascade retained the ability to bind to these mismatched targets stably enough to prevent RNA polymerase elongation. We next tested the exact same crRNAs for RNA-guided DNA integration with the full protein machinery and found that, while seed-sequence mutations again led to a complete loss of activity, mismatches at positions 25–32 similarly abrogated transposition (Fig. 3g (bottom)).
These results expand our understanding of how type I-F CRISPR-associated transposons achieve high degrees of accuracy during RNA-guided transposition. Although off-target sites exhibiting RNA–DNA mismatches within the seed sequence are rejected during initial targeting by TniQ–Cascade, exquisite discrimination of PAM-distal mismatches is achieved not at the target DNA binding step, but specifically during subsequent TnsC recruitment. This mechanism helps to explain the substantially different genome-wide ChIP–seq profiles observed for Cascade and TnsC under otherwise identical experimental conditions (Fig. 3a), and highlights the key role of TnsC in providing a selective proofreading checkpoint during RNA-guided transposition.
Structural analysis of TnsC using cryo-EM
To provide additional insights into the molecular function of TnsC, we pursued structural characterization using single-particle cryogenic electron microscopy (cryo-EM). Heterologous expression of VchTnsC in E. coli and initial purification attempts yielded a poorly soluble protein. In contrast to EcoTnsC22, purified VchTnsC (hereafter TnsC) was stable only in high-ionic-strength (1 M NaCl) aqueous buffers. In the presence of ATP (or a non-hydrolysable ATP analogue), however, TnsC remained stable in lower-ionic-strength buffers and assembled into a high-molecular-weight oligomer, as assessed by size-exclusion chromatography (SEC) (Fig. 4a). A comparison to a SEC calibration curve yielded an estimated molecular weight of about 280 kDa (Fig. 4b), compatible with a supramolecular assembly of seven TnsC monomers (predicted molecular weight of 265 kDa), and this heptameric architecture was confirmed using an orthogonal biophysical approach, interferometric scattering mass spectrometry (iSCAMS)42 (Fig. 4c). The agreement between distinct techniques affirmed the stability and relevance of the heptameric TnsC•ATP species.
Fig. 4 |. Biophysical characterization and overall architecture of the TnsC•ATP heptamer.
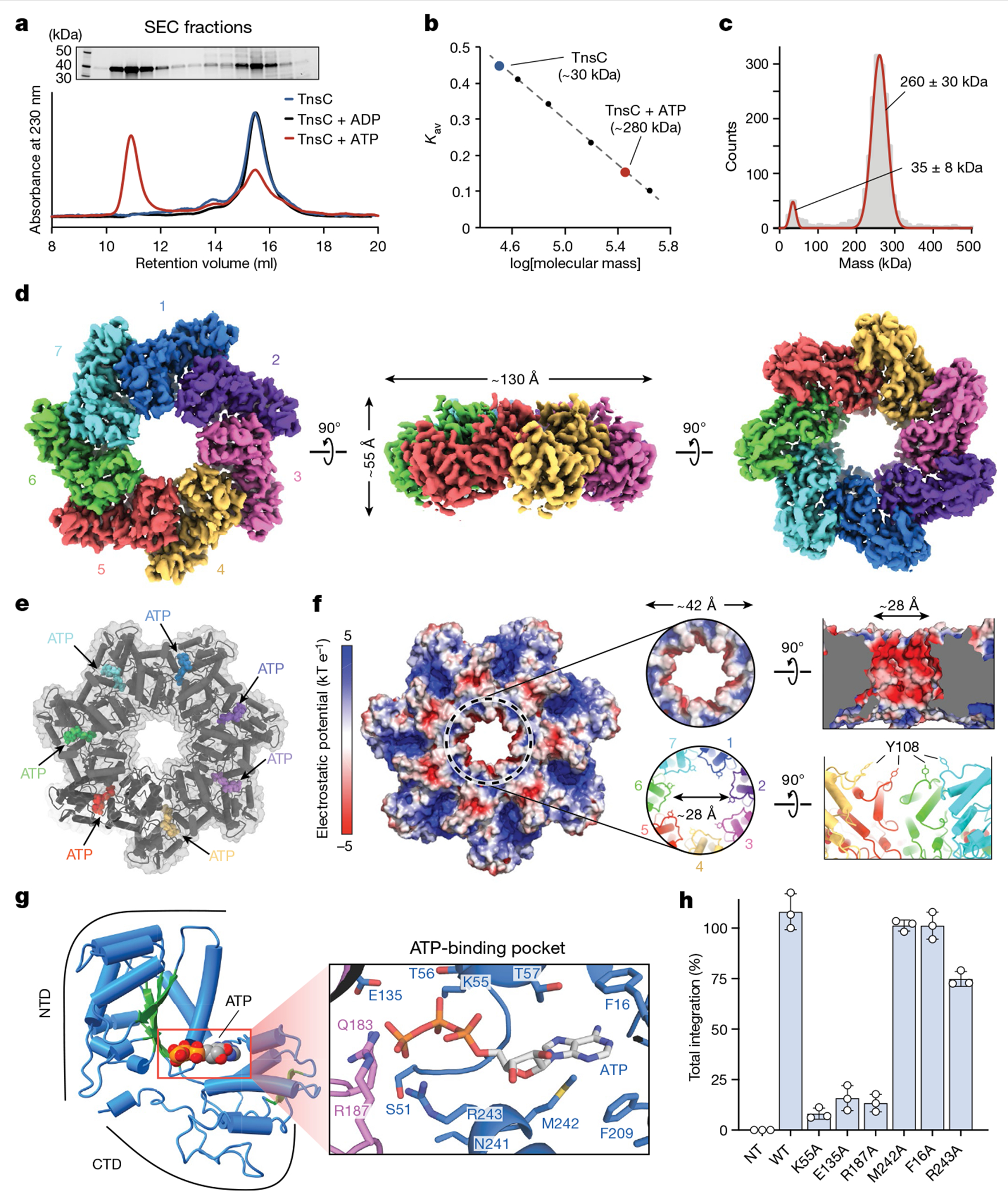
a, SEC profile of TnsC alone (blue) or in the presence of ADP (black) or ATP (red) (bottom). A high-molecular-weight oligomer is observed only in the presence of ATP. Top, SDS–PAGE analysis of SEC fractions. Gel source data are provided in Supplementary Fig. 1. b, SEC calibration curve with molecular weight standards. The estimated mass for the TnsC oligomer was about 280 kDa, which is compatible with a heptameric architecture. Kav, partition coefficient. c, iSCAMS analysis of a TnsC•ATP sample. d, Three orthogonal views of the final cryo-EM map for the TnsC•ATP heptamer. The overall dimensions are indicated in the side view (centre). e, Cartoon representation of TnsC heptamer from the refined model, configured as in d, with ATP molecules shown as coloured spheres. f, Electrostatic surface potential representation of TnsC heptamer, configured as in d. Right, a magnified view of the central pore is shown from a top and side view. The inner diameter (~28 Å) is defined in part by Tyr108 contributed by each monomer. g, Cartoon representation of a TnsC monomer from the refined model, with the NTD and CTD indicated. The central β-sheet characteristic of AAA+ domains is coloured in green, and the bound ATP molecule is shown in spheres representation. Right, a magnified view of the ATP-binding pocket, with residues labelled, h, RNA-guided transposition assays for WT TnsC and the indicated mutants within the ATP-binding pocket, as measured using qPCR. NT, non-targeting crRNA control. Data are shown as mean ± s.d. of n = 3 independent biological replicates.
Initial image processing and analysis of a large cryo-EM dataset revealed a symmetrical entity compatible with a heptameric architecture (Extended Data Fig. 4). Three-dimensional classifications in Relion343 identified a homogeneous group of particles that could be refined to high resolution (overall 3.5 Å, Extended Data Table 1), with local resolution values ranging from 5 Å (peripheral loops) to 3 Å (central region of larger TnsC domain) (Extended Data Fig. 5a–d). The post-processed map enabled de novo building of 310 amino acid residues out of the 333 residues present in the cloned construct, as well as 7 molecules of ATP situated within each TnsC monomer (Fig. 4d,e). The overall heptameric TnsC architecture can be described as a flat disk with a diameter of around 130 Å and a thickness of about 55 Å, with distinguishable apical surfaces projecting above and below the disk (Fig. 4d). The centre features a ~28-Å-diameter pore or circular aperture, which would allow binding of dsDNA (20 Å diameter for the B-form), with the minimal diameter defined by the Tyr108 side chain contributed by each of the 7 monomers (Fig. 4f). The TnsC monomer structure contains N-terminal (NTD) and C-terminal (CTD) domains, which enclose a kinked interface where ATP is bound (Fig. 4g and Extended Data Fig. 5e). The NTD exhibits a general fold that agrees well with other AAA+ family members, encompassing a central β-sheet flanked on both sides by α-helices; the CTD comprises three large α-helices, two smaller ones and a short β-strand at the distal edge of the disk. The ATP molecules have a scaffolding role by coordinating both intramolecular NTD–CTD interactions, as well as intermolecular monomer-to-monomer contacts within the heptamer (Fig. 4e,g). The side chains from multiple residues tightly approach the adenine heterocycle and phosphates of the ATP molecule, with putative density for a Mg2+ ion near the γ-phosphate, in close proximity to Walker A (Lys55), Walker B (Glu135) and arginine finger (Arg187) residues (Fig. 4g).
We examined the essentiality of ATP-coordinating residues by testing TnsC mutants for transposition activity in vivo, and found that residues involved in binding to the adenine nucleobase (Met242, Phe16) or stabilizing the triphosphate (Arg243) exhibited variable tolerance to alanine substitution (Fig. 4h). By contrast, mutation of residues belonging to the Walker A, Walker B and arginine finger motifs led to severely compromised activity, and the same mutants failed to form stable heptameric species in the presence of ATP in vitro (Fig. 4h and Extended Data Fig. 6a). Importantly, though, under these experimental conditions in which the wild-type (WT) system directs around 100% integration efficiency (Methods), all of the ATPase mutants had partial activity (>5% of the WT), suggesting that ATP hydrolysis is non-essential for transpososome assembly and excision/integration chemistry, in agreement with previous studies on other Tn7-family transposons1,13,23. Aside from the intermolecular interactions involved in ATP coordination, additional NTD–NTD and NTD–CTD contacts form a wide surface area (Extended Data Fig. 6b), and reciprocal mutagenesis of interacting loops between contiguous monomers completely abolished transposition in vivo and prevented heptamer formation in vitro (Extended Data Fig. 6c,d).
Within the context of the transpososome, VchTnsC acts as a mediator to bridge interactions between the RNA-guided DNA-targeting module (TniQ–Cascade) and the DNA excision and integration module (TnsAB), similar to the Tn7 system9,21,22. The EcoTnsC homologue encoded by Tn7 must similarly communicate between the DNA-targeting protein (EcoTnsD) and heteromeric transposase (EcoTnsAB), and previous work identified key interaction sites for both modules near the N and C termini, respectively4. Although these regions are not highly conserved between EcoTnsC and VchTnsC (see below), we hypothesize that the N-terminal (top) and C-terminal (bottom) faces of the VchTnsC heptameric disc are uniquely responsible for engaging TniQ and TnsAB, respectively (Extended Data Fig. 7a). C-terminal truncations had severe effects on transposition, even though these regions are not expected to disrupt the general heptameric architecture of the TnsC•ATP complex (Extended Data Fig. 7b). On the N-terminal side, two flexible loops protrude from the top of the disc that were within the NTD but distal from the AAA+ ATPase core. We suspected they would be important for interactions with TniQ (see below) and, indeed, deletion of either loop led to a complete loss of transposition activity (Extended Data Fig. 7b), but did not affect the ability of TnsC to form ATP-dependent heptamers (Extended Data Fig. 7c). Finally, we were curious to investigate the functional importance of the two conserved zinc-finger (ZnF) motifs in VchTniQ18, given recent data from a type V-K CRISPR-associated transposon (ShCAST) that places ShoTniQ and its conserved ZnF motifs in direct proximity to ShoTnsC23. Whereas mutations of all four coordinating residues of the ZnF-2 motif had almost no effect on transposition, mutations within the ZnF-1 motif, which is conserved between VchTniQ and EcoTnsD, led to a complete loss of activity (Extended Data Fig. 7b). These results highlight the critical role of TnsC oligomerization in the assembly of a catalytically competent transpososome.
TnsC threads DNA through its central pore
Given the dimensions of the central aperture, we investigated interactions between TnsC and dsDNA. Using a fluorescence polarization assay, we found that TnsC bound to DNA weakly, with no striking preference for dsDNA over single-stranded DNA (ssDNA) and only a mild dependence on ATP (Extended Data Fig. 8a). Complementary ATPase assays revealed that both ssDNA and dsDNA stimulated ATP hydrolysis, further supporting the ability of TnsC to functionally engage nucleic acid substrates (Extended Data Fig. 8b). We attempted to reconstitute and structurally characterize a stable TnsC–DNA co-complex and ultimately found that 3×Flag-tagged TnsC exhibited improved solubility and could be isolated in complex with 52-bp dsDNA as a high-molecular-weight species that ran distinct from the unbound (apo) TnsC heptamer (Fig. 5a).
Fig. 5 |. The architecture of the DNA-bound TnsC•ATP heptamer and an updated model for transposition mechanism.
a, SEC profiles of TnsC•ATP (red), dsDNA (grey) and TnsC•ATP•dsDNA (green). Inset: SDS–PAGE analysis of the fractions used for cryo-EM. Gel source data are provided in Supplementary Fig. 1. b, Representative 2D class averages from the sample in a reveal apo DNA-free TnsC heptamers (left) and single, double and triple DNA-bound heptamers. c, The final cryo-EM map (left) and ribbon representation (right) of single heptamer TnsC•ATP•DNA. Seven protomers are labelled; DNA is coloured grey, d, Structural alignment of apo and DNA-bound TnsC reveals that the heptamer is distorted after DNA binding, as visualized by tracking the ATP molecules (red or blue) bound to each subunit. Subunit 2 of the DNA-bound heptamer is displaced by 18.5 Å relative to the apo-TnsC ring. For comparison, the helical rise per turn of the bound DNA is around 31 Å. e, Distortion of the DNA-bound heptamer creates a cleft between subunits 1 and 2 (left) that disrupts the ATP binding pocket (bottom right) compared with an undisrupted ATP binding pocket (top right), f, The model for TnsC recruitment and assembly at DNA target sites bound by TniQ–Cascade for type I-F CRISPR-associated transposons (Methods). The PAM is highlighted (yellow), and the hypothesized path of DNA from target-bound TniQ–Cascade to TnsC is indicated by dotted lines. Numbered positions for the target DNA (grey) hybridized to the crRNA (red) are labelled, as are the hypothetical numbered positions from the 3′ edge of the target DNA to the integration site approximately 50 bp downstream. The donor DNA is probably recruited to the bottom (C-terminal) face of the TnsC oligomer through protein–protein interactions with the heteromeric TnsAB transposase. g, Genome-wide accuracy of RNA-guided DNA integration is increased through sequential assembly of the catalytically competent transpososome complex.
The TnsC–DNA co-complex was imaged under two conditions (cryo-grids without support or with amorphous carbon as support), and all of the images were combined into a single cryo-EM dataset. Initial image processing revealed a highly heterogeneous sample, with multiple species corresponding to the canonical TnsC heptamer previously described, alongside larger complexes comprising two or three stacked heptameric rings bound to dsDNA, in excellent agreement with iSCAMS measurements (Fig. 5b and Extended Data Figs. 8c and 9). We were able to identify and refine to a high resolution two of these species: a single heptamer with dsDNA in the central pore (overall resolution of 3.46 Å) and a double-heptamer species with dsDNA threading through the central pore of both heptamers (overall resolution of 3.92 Å) (Extended Data Fig. 8d–l and Supplementary Video 1). A third class corresponding to a multimer of three stacked TnsC heptamers was also present in the dataset, as revealed by clear 2D reference-free class averages (Fig. 5b (right) and Supplementary Video 2); however, owing to the highly flexible nature of this class, no 3D high-resolution information could be attained. Importantly, we did not observe any continuous filaments reminiscent of MuB44 or the ShoTnsC homologue from type V-K CRISPR-associated transposons (ShCAST)23,24, suggesting that VchTnsC may be restricted to forming only rings.
Density attributable to DNA was readily apparent in the central pore of TnsC rings of both single- and double-heptameric species, with clear features reminiscent of the major and minor grooves of dsDNA, particularly for the double-ring complexes (Fig. 5c, Extended Data Fig. 9a,d and Supplementary Video 1). The likely heterogeneity between protein–DNA contacts across averaged particles led to a noticeable shrinkage of the dsDNA density volume, an interpretation that is consistent with the absence of identifiable protein–DNA contacts; this included residues Arg143 and Arg146, which are near to the bound site of DNA and were crucial for transposition activity (Extended Data Fig. 7a,b). Each heptameric ring covers about 20 bp of dsDNA in a non-sequence-specific manner, and we did not detect obvious sites of molecular interaction between adjacent heptamers in the double-heptamer form, suggesting that they are unlikely to represent biologically relevant assemblies within the context of the eventual transpososome, as was recently deduced for similar, multi-ring architectural states of EcoTnsC22.
TnsC rings maintained their heptameric assembly state within each species, and the structure of individual TnsC monomers in single- and double-ring complexes was consistent with the monomers in apo-TnsC rings (root-mean-square deviation = 0.532 and 0.643 Å, respectively, Extended Data Fig. 10a). Despite the absence of large-scale conformational changes, we noticed that DNA-bound heptamers exhibited a notable rise of about 18 Å relative to the DNA helical axis, in contrast to the flat disc architecture of apo-TnsC rings (Fig. 5d and Supplementary Video 3). This rearrangement is insufficient to enable continuous filaments to form, but does enforce a modified interface that creates a cleft between TnsC subunits 1 and 2, preventing the arginine finger from one monomer to properly contact the ATP molecule bound in the adjacent monomer (Fig. 5e). Interestingly, a similar cleft was recently reported for the heptameric DNA-bound structure of EcoTnsC22, suggesting that this remodelled state is conserved within certain Tn7-like transposons. Ring opening (ring-breaker mechanisms) could enable the loading of the otherwise closed and locked apo-TnsC heptameric ring onto target DNA during transpososome assembly45. Consistent with this hypothesis, weaker density was observed on one side of the ring in all of the DNA-free and DNA-bound structures (Extended Data Figs. 5b and 8e,i), indicative of a dynamic state that may facilitate nucleic acid engagement. Further studies will be needed to investigate the individual molecular steps leading to TnsC recruitment and assembly at the target site and, in particular, how interactions with other protein components modulate the architecture and dynamics of TnsC oligomers.
Discussion
Using previous structures of the target DNA-bound VchTniQ–Cascade complex18,46, our structure of heptameric VchTnsC•ATP•DNA and a recently described structure of a ShoTnsC•TniQ•DNA co-complex from an evolutionarily related transposon23, we present an updated model for a transpososome intermediate in the RNA-guided DNA integration pathway of type I-F CRISPR-associated transposons (Fig. 5f and Extended Data Fig. 10b–h). This model predicts a critical interaction surface between the N-terminal loops of the VchTnsC ring and the ZnF domain of TniQ, consistent with the finding that both regions were essential for transposition (Extended Data Fig. 7a,b). We propose that the two DNA strands reanneal after exiting the R-loop formed by TniQ–Cascade and are stabilized through interactions with a basic patch of one TniQ monomer that is well positioned to thread dsDNA into the central pore of a TnsC heptamer (Extended Data Fig. 10i,j). The model, which features TniQ and a single heptameric TnsC ring, elegantly explains the strict ~50 bp spacing between the Cascade-bound target and the downstream integration site (Fig. 5f and Extended Data Fig. 10j), and furthermore predicts a severe kink in the DNA when assessing the angle between dsDNA that enters Cascade (upstream of the PAM) and exits the TnsC•ATP oligomer. Not surprisingly, previous studies of the E. coli Tn7 transpososome using atomic force microscopy also suggest that the target DNA is probably bent during transposition21 and, more generally, DNA distortion is a common theme that is exploited by diverse transposases and related enzymes47.
Nevertheless, our DNA-bound structures suggest that VchTnsC does not substantially remodel DNA after binding, similar to structures of EcoTnsC (from non-CRISPR-associated Tn7)22, but in contrast to ShoTnsC (from type V-K CRISPR-associated transposons)23,24 (Extended Data Fig. 10). In that case, ShoTnsC structurally distorts dsDNA to match its own helical symmetry, necessitating an increased helical pitch, and notably, forms stable continuous filaments23,24,44. By contrast, VchTnsC forms heptameric rings in the presence of ATP, whether unbound or bound to DNA substrates, but does not appear to form higher-order filaments after nucleic acid engagement (Figs. 4 and 5), similar to EcoTnsC22. The helical filaments observed with ShoTnsC are reminiscent of those also formed by MuB44—a related AAA+ ATPase that functions together with MuA during phage Mu transposition. As Mu uses only MuB (TnsC homologue) and MuA (TnsB homologue) while undergoing non-specific transposition, in which random genomic target sites are identified through the formation of ATP-dependent MuB filaments44,48, it is tempting to speculate that type V-K systems may exhibit untargeted (Cas12k/sgRNA-independent) transposition using similar TnsC filamentation properties; the same pathway is probably exploited by E. coli TnsC gain-of-function mutants11. The biochemical and structural propensity to form continuous DNA-bound filaments may be intrinsically linked to the ability of the composite heteromeric transposase to non-specifically transpose without using targeting factors. Although more experiments will be needed to test this hypothesis, the DNA-binding activity of VchTnsC and EcoTnsC may have evolved to suppress off-target insertions by relying more exclusively on target-selection proteins (for example, TniQ–Cascade or TnsD/TnsE) for active recruitment to potential sites of transposition. The weak DNA-binding affinity of VchTnsC observed in vitro (Extended Data Fig. 8a), together with the paucity of stable genomic binding sites observed using ChIP–seq in vivo (Fig. 1), are consistent with this hypothesis.
TnsC oligomerization is dependent on ATP and may be further modulated after protein interaction with TniQ–Cascade and TnsAB, suggesting that TnsC dynamics are important during the formation of the holo-transpososome complex. A region of notable local flexibility, a feature shared by all DNA-free and DNA-bound VchTnsC heptamers (Fig. 5e, Extended Data Figs. 5b and 8e,i) and also observed in DNA-bound EcoTnsC heptamers (Extended Data Fig. 10e), suggests a potential ring-breaker mechanism that may explain how TnsC could be loaded onto DNA45. Our findings that VchTnsC forms stable heptamers in the presence of ATP alone, and that monomeric VchTnsC exhibits poor solubility, suggest that the oligomeric assembly may be the predominant state in the cell before target recruitment. Structural superposition of TnsC homologues suggests that, despite considerable conservation of the N-terminal AAA+ core domain, the efficiency of subunit packing and the position of helix α1 may determine the hexameric or heptameric architecture (Extended Data Fig. 10e–g). Further experiments will be needed to investigate these questions more thoroughly, including the role of ATP hydrolysis in regulating rearrangements and disassembly of DNA-bound TnsC rings.
We envision that TnsC recruitment and oligomerization function as a dynamic and critical point of control during the identification and licensing of candidate target sites for successful DNA integration. The results from our genome-wide profiling experiments using ChIP–seq strongly agree with this conclusion. By tracking each major component of the overall transposition pathway (the RNA-guided Cascade complex, the catalytic TnsB transposase and the TnsC mediator protein), we provide insights into the degree of on- and off-target binding and reveal that—although DNA targeting by Cascade is relatively promiscuous—TnsC and TnsB are selectively recruited to only a small subset of those sites (Fig. 2). Off-target sites share conserved 5′-CN-3′ PAM and 5–10 bp seed-sequence motifs (Fig. 2a), as seen in ChIP–seq studies of functionally analogous Cascade and dCas9–RNA complexes28–30,32, but exhibit large variation in the number and positions of mismatches elsewhere in the target site (Fig. 2a). A combination of other experimental approaches, including CRISPR interference and transposon-encoded guide RNA libraries, shows that DNA targeting by Cascade readily tolerates PAM-distal mismatches, whereas these mismatches severely abrogate TnsC recruitment and downstream integration (Fig. 3). In light of numerous studies highlighting the critical role of conformational dynamics in controlling the fate of DNA sites targeted by CRISPR effector complexes49, we conclude that the target-bound TniQ–Cascade complex probably functions as a sensitive beacon for TnsC recruitment, and that TnsC integrates information about the TniQ–Cascade binding lifetime and/or conformational state to regulate whether candidate targets are certified for transposase–transposon DNA recruitment and integration (Fig. 5g). In this manner, assembly of the oligomeric TnsC complex may act as a proofreading checkpoint to ensure high-fidelity selection of genomic sites for transposition, although we acknowledge that ChIP–seq lacks the ability to detect transient, rare and/or stochastic binding events that are heterogeneously sampled across a cellular population50, and thus certain binding behaviours may remain invisible using this approach. An outstanding question is whether a similar proofreading mechanism operates in other Tn7-family transposons and, in particular, why type V-K CRISPR-Tn systems exhibit lower levels of RNA-guided DNA integration fidelity3,17.
RNA-guided transposases hold great promise for future biotechnological and human therapeutic applications, given their ability to insert kilobase-scale payloads without generating DNA double-stranded breaks or requiring homologous recombination machinery51. Our research provides important structural and mechanistic insights that will inform the design and delivery of CRISPR- and transposon-associated components for optimal transpososome assembly and integration specificity. Rational, structure-guided engineering of CRISPR–Cas9 to tune the energetics of DNA binding enabled development of the first high-fidelity genome editors, and we similarly envision leveraging our emerging understanding of TnsC recruitment and ATP-dependent oligomerization to facilitate future deployment of RNA-guided transposase technology.
Methods
Plasmid construction
All V. cholerae plasmid constructs were subcloned from pTnsABC, pQCascade and pDonor, as described previously2,17, which derive from the V. cholerae Tn6677 type I-F CRISPR-associated transposon (also referred to as VchCAST). In brief, variants of these plasmids were generated using a combination of inverse (around-the-horn) PCR, Gibson assembly, ligation of hybridized oligonucleotides and restriction digestion. All PCR steps were performed using Q5 DNA polymerase (NEB). Constructs were cloned in NEB Turbo E. coli cells, and plasmid DNA samples were isolated using the Miniprep kit (QIAGEN) and verified by Sanger sequencing (GENEWIZ). For ChIP experiments, 3×Flag tags were added using around-the-horn PCR, and the D223N mutation was introduced into the tnsB gene to prevent transposition. Gibson assembly was also used to generate a minimal pDonor to reduce plasmid reads from a lacZ gene fragment present on both the original pUC19 vector and the E. coli BL21(DE3) reference genome. Expression plasmids for protein purification were subcloned into a p1S vector (QB3 MacroLab) containing an N-terminal hexahistidine tag and a SUMO tag, followed by a TEV protease cleavage site. Sequences and descriptions of all plasmids used in this study are provided in Supplementary Table 1; sequences of purified protein constructs are provided in Supplementary Table 2; and a list of crRNA spacer sequences is provided in Supplementary Table 3.
E. coli culturing and general transposition assays
All E. coli transposition assays were generally performed as described previously17. In brief, a three-plasmid system consisting of pQCascade, pTnsABC and pDonor (or derivatives thereof) was used. Expression of the crRNA and the TniQ–Cascade proteins from pQCascade was driven by separate T7 promoters (Fig. 1b). Chemically competent E. coli BL21(DE3) cells containing a combination of two of the three plasmids were transformed with a third plasmid by heat shock and then recovered in LB at 37 °C. Transformed cells were initially cultured on LB agar plates with the appropriate antibiotics (200 μg ml−1 carbenicillin, 200 μg ml−1 spectinomycin, 100 μg ml−1 kanamycin) for 16 h at 37 °C. Colonies were subsequently scraped, replated on LB agar with the same antibiotics and supplemented with 0.1 mM IPTG, and cultured for 44 h at room temperature (~25 °C). Our previous study found that reduced temperature increases transposition efficiency for Tn667717. qPCR analysis of transposition was performed as described previously2.
ChIP–qPCR and ChIP–seq
For all ChIP experiments, an active site mutation (D223N) was introduced into the tnsB gene in pTnsABC to block transposition and trap transpososome complexes in a pre-integration state2 (Extended Data Fig. 1c). A single protein was 3×Flag-tagged in each ChIP experiment, while the other proteins remained untagged. Transformations using the three-plasmid system were performed according to the general transposition assay protocol. After incubation for 16 h at 37 °C on LB agar plates with antibiotics but no IPTG induction, colonies were scraped and resuspended in 1 ml of fresh LB. The optical density at 600 nm (OD600) was measured, and approximately 4.0 × 108 cells (equivalent to 1 ml with an OD600 of 0.25) were spread onto two LB agar plates containing antibiotics and supplemented with 0.1 mM IPTG. We chose to culture cells on solid LB agar to reduce any competition-induced effects of culturing in liquid LB medium. Plates were incubated at room temperature (~25 °C) for 44 h, during which time a bacterial lawn formed. All cell material from both plates was scraped and spread onto the wall of a 50 ml conical tube in a thin layer, to allow for efficient resuspension and cross-linking in the next step.
Cross-linking and immunoprecipitation were generally performed according to previously established protocols52. Formaldehyde (1 ml of 37% solution; Thermo Fisher Scientific) was added to a separate tube containing 40 ml of LB medium (~1% final concentration), and the solution was mixed immediately by inverting. This solution was added to the conical tube containing the scraped cells, and the cells were fully resuspended in the LB formaldehyde solution by vigorous vortexing at room temperature. Cross-linking was performed by gently shaking at room temperature for 20 min. To stop cross-linking, 4.6 ml of 2.5 M glycine (~0.25 M final concentration) were added, followed by 10 min incubation with gentle shaking. Cells were pelleted at 4 °C by centrifuging at 4,000g for 8 min. The following steps were performed on ice using buffers that had been sterile-filtered through a 0.22 μm filter. The supernatant was discarded and the pellets were fully resuspended in 40 ml TBS buffer (20 mM Tris-HCl pH 7.5, 0.15 M NaCl) by vortexing. After centrifuging again at 4,000g for 8 min at 4 °C, the supernatant was removed, and the pellet was again resuspended in 40 ml TBS buffer. Next, the OD600 was measured for a 1:1 mixture of the cell suspension and fresh TBS buffer, and a standardized volume equivalent to 40 ml of OD600 = 0.6 was aliquoted into new 50 ml conical tubes. A final 8 min centrifugation step at 4,000g and 4 °C was performed, cells were pelleted and the supernatant was discarded. Residual liquid was removed by briefly inverting the tube, and cell pellets were flash-frozen using liquid nitrogen and stored at −80 °C or kept on ice for the subsequent steps.
Bovine serum albumin (GoldBio) was dissolved in 1× PBS buffer (Gibco) and sterile-filtered to generate a 5 mg ml−1 BSA solution. For each sample, 25 μl of Dynabeads Protein G (Thermo Fisher Scientific) slurry (hereafter, beads or magnetic beads) were prepared for immunoprecipitation. Beads from up to 250 μl of the initial slurry were processed together in a single tube, and washes were performed at room temperature, as follows: the slurry was transferred to a 1.5 ml tube and placed onto a magnetic rack until the beads had fully settled. The supernatant was removed carefully, 1 ml BSA solution was added, and the beads were fully resuspended by vortexing, followed by rotating for 30 s. This was repeated for three more washes. Finally, the beads were resuspended in 25 μl (× n samples) of BSA solution, followed by addition of 4 μl (× n samples) of monoclonal anti-Flag M2 antibodies produced in mouse (Sigma-Aldrich). The suspension was moved to 4 °C and rotated for >3 h to conjugate antibodies to the magnetic beads. While conjugation was proceeding, cross-linked cell pellets were thawed on ice, resuspended in FA lysis buffer 150 (50 mM HEPES-KOH pH 7.5, 0.1% (w/v) sodium deoxycholate, 0.1% (w/v) SDS, 1 mM EDTA, 1% (v/v) Triton X-100, 150 mM NaCl) with protease inhibitor cocktail (Sigma-Aldrich) and transferred to a 1 ml milliTUBE AFA Fiber (Covaris). The samples were sonicated on a M220 Focused-ultrasonicator (Covaris) with the following SonoLab 7.2 settings: minimum temperature, 4 °C; set point, 6 °C; maximum temperature, 8 °C; peak power, 75.0; duty factor, 10; cycles/bursts, 200; 17.5 min sonication time. After sonication, samples were cleared of cell debris by centrifugation at 20,000g and 4 °C for 20 min. The pellet was discarded, and the supernatant (~1 ml) was transferred into a fresh tube and kept on ice for immunoprecipitation. For non-immunoprecipitated input control samples, 10 μl (~1%) of the sheared cleared lysate were transferred into a separate 1.5 ml tube, flash-frozen in liquid nitrogen and stored at −80 °C.
After >3 h, the conjugation mixture of magnetic beads and antibodies was washed four times as described above, but at 4 °C. Next, the beads were resuspended in 30 μl (× n samples) FA lysis buffer 150 with protease inhibitor, and 31 μl of resuspended antibody-conjugated beads were mixed with each sample of sheared cell lysate. The samples were rotated overnight for 12–16 h at 4 °C for immunoprecipitation of Flag-tagged proteins. The next day, tubes containing beads were placed on a magnetic rack, and the supernatant was discarded. Then, six bead washes were performed at room temperature, as follows, using 1 ml of each buffer followed by sample rotation for 1.5 min: (1) two washes with FA lysis buffer 150 (without protease inhibitor); (2) one wash with FA lysis buffer 500 (50 mM HEPES-KOH pH 7.5, 0.1% (w/v) sodium deoxycholate, 0.1% (w/v) SDS, 1 mM EDTA, 1% (v/v) Triton X-100, 500 mM NaCl); (3) one wash with ChIP wash buffer (10 mM Tris-HCl pH 8.0, 250 mM LiCl, 0.5% (w/v) sodium deoxycholate, 0.1% (w/v) SDS, 1 mM EDTA, 1% (v/v) Triton X-100, 500 mM NaCl); and (4) two washes with TE buffer 10/1 (10 mM Tris-HCl pH 8.0, 1 mM EDTA). The beads were then placed onto a magnetic rack, the supernatant was removed and the beads were resuspended in 200 μl of ChIP elution buffer (1% (w/v) SDS, 0.1 M NaHCO3) made fresh on the day of the washes. To release protein–DNA complexes from beads, the suspensions were incubated at 65 °C for 1.25 h with gentle vortexing every 15 min to resuspend settled beads. During this incubation, the non-immunoprecipitated input samples were thawed, and 190 μl of ChIP Elution Buffer was added, followed by the addition of 10 μl of 5 M NaCl. After the 1.25 h incubation of the immunoprecipitated samples was complete, the tubes were placed back onto a magnetic rack, and the supernatant containing eluted protein–DNA complexes was transferred to a new tube. Then, 9.75 μl of 5 M NaCl was added to ~195 μl of eluate, and the samples (both immunoprecipitated and non-immunoprecipitated controls) were incubated at 65 °C overnight (without shaking) to reverse-cross-link proteins and DNA. The next day, samples were mixed with 1 μl of 10 mg ml−1 RNase A (Thermo Fisher Scientific) and incubated for 1 h at 37 °C, followed by addition of 2.8 μl of 20 mg ml−1 proteinase K (Thermo Fisher Scientific) and 1 h incubation at 55 °C. After adding 1 ml of buffer PB (QIAGEN recipe), the samples were purified using QIAquick spin columns (QIAGEN) and eluted in 40 μl TE buffer 10/0.1 (10 mM Tris-HCl pH 8.0, 0.1 mM EDTA).
For ChIP–qPCR, 1 μl of each purified sample was diluted with 14 μl of TE buffer 10/0.1. Pairs of genomic qPCR primers (Supplementary Table 4) were designed for each target site, and previously established primers against a reference locus (rssA) were used2. For both samples (immunoprecipitated and input control), parallel qPCR reactions were performed using both the genomic target locus primer pair and the reference locus primer pair. The fold change (FC) at the target locus was calculated using the ΔΔCq method: A = Cq(immunoprecipitated sample at target locus) − Cq(immunoprecipitated sample at reference locus); B = Cq(input sample at target locus) − Cq(input sample at reference locus); ΔΔCq = 2−(A−B).
For ChIP–seq, Illumina libraries were generated for immunoprecipitated and input samples using the NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB). Sample concentrations for ChIP–seq were determined using the DeNovix dsDNA Ultra High Sensitivity Kit. Starting DNA amounts were standardized such that an approximately equal mass of all input and immunoprecipitated DNA was used for library preparation. After adapter ligation, a single PCR amplification was performed to add Illumina barcodes, and ~450 bp DNA fragments were selected using two-sided AMPure XP bead (Beckman Coulter) size selection, as follows: the volume of barcoded immunoprecipitated and input DNA was brought up to 50 μl with TE Buffer 10/0.1; in the first size-selection step, 0.55× AMPure beads (27.5 μl) were added to the DNA, the sample was placed onto a magnetic rack, and the supernatant was discarded and the AMPure beads were retained; in the second size-selection step, 0.35× AMPure beads (17.5 μl) were added to the DNA, the sample was placed onto a magnetic rack, and the AMPure beads were discarded and the supernatant was retained. The concentration of DNA was determined for pooling using the DeNovix dsDNA High Sensitivity Kit.
Illumina libraries were sequenced in either single- or paired-end mode on the Illumina MiniSeq and NextSeq platforms with automated demultiplexing and adapter trimming (Illumina). For each ChIP–seq sample, >1,800,000 raw reads (including genomic and plasmid-mapping reads) were obtained. ChIP–seq read and metadata information is provided in Supplementary Table 5.
ChIP–seq data analysis
ChIP–seq data processing and analysis was performed using standard methods. ChIP–seq single- and paired-end reads were trimmed and mapped to an E. coli BL21(DE3) reference genome (GenBank: CP001509.3) using fastp53 and Bowtie254 with the default parameters, respectively. Two genomic lacZ/lacI regions and the flanking regions, which are partially identical to plasmid-encoded sequences, were masked in all of the alignments (genomic coordinates: 335600–337101 and 748601–750390). Enrichment analysis on pDonor (Fig. 1d and Extended Data Fig. 1d,e) and pQCascade (Extended Data Fig. 1f) was performed by mapping reads (Bowtie254, using the default parameters) to the same E. coli BL21(DE3) reference genome concatenated to the respective pQCascade, pTnsABC and pDonor plasmid sequences used in each experiment. For those concatenated reference genome files, sequence elements were separated from one another by inserting 1,000 bp represented as Ns.
Mapped reads were sorted and indexed using the SAMtools software package55 (with the samtools sort and samtools index commands, respectively). For all datasets, the command samtools view was used to eliminate multi-mapping reads with a MAPQ score < 10 (-bq 10 option). Alignments were normalized by RPKM and converted to 1-bp-bin bigwig files using the deepTools256 command bamCoverage, with the following parameters: --normalizeUsing RPKM -bs 1. After normalization, genome- and plasmid-mapping reads were visualized in IGV57. The Bedtools58 software package (bedtools makewindows -w 1000 and bedtools map -o max) and IGV were used to generate all genome-wide views. Maximum read coverage values were plotted in 1-kb bins across the genome30.
Peaks were called on prenormalized alignments using MACS333, with the following parameters: -g 4500000 --nomodel --extsize 400 -q 0.05 -B. A non-immunoprecipitated control library of Cas8 was used as an input for standardizing all library peak calls (-n option). Heat maps of peak calls were first generated by computing a heat map matrix on the alignments using the deepTools function (computeMatrix reference-point) with the default parameters. Subsequently, the deepTools function plotHeatmap was used on the heat map matrices with the following parameters: --whatToShow ‘heatmap and colorbar’ --sortUsing max. Venn diagrams were generated using custom R code on MACS3 peak output files. Sequence motifs were identified using MEME34 (v.4.12.0) with the default parameters.
Off-target mismatch analysis of peak calls was performed using a custom Python script. In brief, a 200-bp sequence around each peak was examined, and the 10-bp sequence most closely matching a 5′-CN-3′ PAM and the first 8 nucleotides of the seed sequence, with the 6th bp set as N, was identified. The best-matching sequence was inferred to be the off-target binding site, and the full 34-bp PAM and target sequence was extracted for each peak.
crRNA library experiments
A ssDNA oligo pool comprising 1,629 crRNA library members (Supplementary Table 6) was synthesized (Twist Bioscience) and amplified in two parallel 15 μl PCR reactions, each containing 5 ng of oligo pool template, 400 μM dNTPs, 2 μM primers, 1× Q5 reaction buffer and 0.3 U of Q5 DNA polymerase (NEB). The PCR reactions were performed with 12 amplification cycles, and DNA was pooled and purified, digested with SalI and BamHI (NEB), and purified for ligation into pDonor. The pDonor parent vector was digested with SalI and BamHI (NEB) and gel-purified on a 1% agarose gel. Each 20 μl ligation reaction included 60 ng of digested vector, 20 ng of digested PCR products, and 400 U of T4 DNA ligase (NEB). Five parallel ligations were performed at room temperature for 30 min, and were subsequently pooled and purified using the MinElute Spin Column (QIAGEN); around 40% of the elution was used to transform electrocompetent 10-beta E. coli cells (NEB). Sufficient numbers of individual colonies across transformations were generated for >200× library coverage. Colonies were scraped and resuspended in liquid LB, and pDonor plasmid libraries were isolated using the Midiprep kit (QIAGEN).
Transposition assays were performed in a BL21(DE3) E. coli strain that contained a pEffector plasmid expressing Tn6677 (also referred to as VchCAST) protein components driven by an IPTG-inducible T7 promoter. Parallel transformations of cells with the pDonor plasmid library were performed to generate sufficient numbers of individual colonies for >200× library coverage. Transformed cells were plated on LB agar with antibiotic selection and 0.1 mM IPTG inducer, cultured at room temperature for 44 h, and then scraped and resuspended in liquid LB medium. Plasmids were extracted using Miniprep (QIAGEN), and genomic DNA (gDNA) was extracted using the Wizard Genomic DNA Purification Kit (Promega). The entire crRNA library cloning process, subsequent transposition assays and sequencing were performed in two independent biological replicates.
To sequence the input library, we amplified the CRISPR array from extracted plasmids by performing a PCR-1 step using primers flanking the CRISPR array within the mini-Tn. In a 25 μl PCR-1 reaction (16 cycles, 65 °C annealing temperature), 1 μl of 1:100 diluted plasmid (~1 ng of plasmid) was mixed with 200 μM dNTPs, 0.5 μM primers, 1× Q5 reaction buffer and 0.5 U of Q5 DNA polymerase (NEB). Then, 1 μl of the PCR-1 product was used as the template for a 10 μl PCR-2 step (10 cycles) to attach indexed p5/p7 Illumina adapters. To sequence the integration product library at each target site, we amplified the CRISPR array in PCR-1 using a transposon-specific primer downstream of the CRISPR array paired with a genome-specific primer, as shown in Fig. 3c (a list of all of the primers used in this experiment is provided in Supplementary Table 4). From the PCR-1 products, amplicons for groups of 2–4 distinct target sites were combined to generate PCR-2 products, with PCR-1 and PCR-2 reaction conditions as described above. All PCR-2 products were resolved using 2.5% agarose gel electrophoresis, and bands representing the expected single insertion products (~410–430 bp) were extracted using the Gel Extraction Kit (Qiagen). Notably, any PCR amplicons resulting from nested insertion events due to self-targeting of the pDonor-encoded CRISPR array were eliminated during this size-selective gel extraction step. NGS libraries were quantified using the NEBNext Library Quant Kit for Illumina (NEB) and sequenced on the Illumina NextSeq platform using a 150-cycle high-output kit.
After automated adapter trimming and demultiplexing, the sequence of the CRISPR array in each i7-end read was identified using known anchor sequences and extracted and matched to crRNA members with no mismatch allowance. All crRNA members were detected with input library sequencing. The FC value for each library member at each target was calculated as the ratio of abundance in the genomic integration product amplicons from that target, compared with its overall abundance in the starting pDonor library. log2[FC] was determined by taking the log2 of fold change values; for each member with no reads detected in the output sequencing and a fold change value of 0, log2[FC] was assigned a value equal to the most negative log2[FC] of all members for that target site. The Δlog2[FC] value for each crRNA library member was determined as the difference of the log2[FC] values for that member and for the corresponding perfectly matching crRNA member. log2[FC] and Δlog2[FC] values used in Fig. 3d,e and Extended Data Fig. 3c–f were averaged from two independent biological replicates. For visualization purposes, heat maps for individual targets in Fig. 3e were clustered with Seaborn and SciPy using the Nearest Point Algorithm (single linkage method). All raw read counts and analysed scores for crRNA library members are provided in Supplementary Table 6.
GFP and RFP repression experiments
An E. coli strain expressing mRFP and sfGFP from the chromosome41, a gift from L. S. Qi, was transformed with pQCascade or pCascade (ΔTniQ), in which the crRNA and protein components are constitutively expressed. Spacers were designed to target either mRFP or sfGFP proximal to the 5′ end of the coding region (Extended Data Fig. 3g), and targeted either the template or non-template strand. Transformed cells were plated on LB agar with antibiotic selection, and the resulting colonies were scraped before inoculation of an overnight liquid LB culture. For each sample, 1 μl of the overnight culture was used to inoculate 200 μl of LB medium on a 96-well optical-bottom plate. The OD600 was measured in parallel with fluorescence signals for sfGFP and mRFP using a Synergy Neo2 microplate reader (Biotek), while shaking at 37 °C for 16 h. The fluorescence intensities in each well at OD600 = 1.0 were used to determine the fold repression of each gene, and the data were normalized to the WT (perfectly matching) spacer.
For the transposition assays shown in Fig. 3g, the same E. coli strain was transformed with a pDonor plasmid encoding a mini-Tn, cells were made chemically competent, and subsequently transformed with a pEffector plasmid expressing the crRNA and all seven protein components from Tn6677, driven by a constitutive promoter. Transformed cells were plated on LB agar with antibiotic selection and incubated at 37 °C for 24 h. Colonies were then scraped and lysed, and transposition efficiencies were determined from cell lysates using qPCR, as described above.
TnsC purification
A plasmid encoding full-length V. cholerae TnsC (GenBank: WP_002024152.1) with an N-terminal His6–SUMO–TEV or His6–SUMO–TEV–3×Flag tag was used to transform E. coli BL21(DE3) competent cells. Cells were grown in 2xYT media supplemented with 50 μg ml−1 kanamycin at 37 °C until the OD600 reached 0.5–0.7. Cells were induced with 0.5 mM IPTG at 15 °C for 16–20 h and then collected by centrifugation at 4,000g for 10 min at 4 °C. Cell pellets were resuspended in lysis buffer (20 mM HEPES-KOH pH 7.5, 1 M NaCl, 5 mM magnesium acetate, 0.2 mM EDTA, 0.1% (v/v) Triton X-100, 5% (v/v) glycerol, 1 mM DTT) supplemented with 0.5× cOmplete protease inhibitor cocktail tablet (Roche), 1 mM PMSF and 0.1 mg ml−1 lysozyme, and were lysed by sonication. The soluble His6–SUMO–TEV–VchTnsC or His6–SUMO–TEV–3×Flag–VchTnsC proteins were isolated by centrifugation at 30,000g for 45 min at 4 °C, and the supernatant was incubated with Ni-NTA agarose resin that was pre-equilibrated with equilibration buffer (20 mM HEPES-KOH pH 7.5, 1 M NaCl, 0.2 mM EDTA, 5% (v/v) glycerol, 10 mM imidazole, 1 mM DTT). Protein-bound Ni-NTA resin was collected in a gravity column and washed with 10 column volumes (CV) equilibration buffer. To remove contaminating proteins, the resin was washed with 5 CV of wash buffer (20 mM HEPES-KOH pH 7.5, 1 M NaCl, 0.2 mM EDTA, 5% (v/v) glycerol, 30 mM imidazole, 1 mM DTT). Purified protein was eluted with 5 CV elution buffer (20 mM HEPES-KOH pH 7.5, 1 M NaCl, 0.2 mM EDTA, 5% (v/v) glycerol, 300 mM imidazole, 1 mM DTT). To remove the His6–SUMO tag, 5% (w/v) TEV protease was added to the eluted protein, and the sample was dialysed overnight in dialysis buffer (20 mM HEPES-KOH pH 7.5, 1 M NaCl, 0.2 mM EDTA, 1% (v/v) glycerol, 1 mM DTT, 10 mM imidazole). Any precipitated protein was removed by centrifugation at 20,000g for 20 min at 4 °C, and the sample was next incubated with Ni-NTA agarose resin for about 30 min at 4 °C. Cleaved VchTnsC was collected from the flow-through and processed for initial SEC on a Superdex 200 Increase gl 10/300 column equilibrated in high-salt buffer (20 mM HEPES-KOH pH 7.5, 1 M NaCl, 0.2 mM EDTA, 1% (v/v) glycerol, 1 mM DTT). Monomeric VchTnsC eluted at ~18.4 ml, as verified by SDS–PAGE analysis, and was stored at −80 °C after snap-freezing in liquid nitrogen. TEV-cleaved 3×Flag–VchTnsC was collected from the flow-through and processed for initial SEC on a HiLoad 16/600 Superdex 200 column equilibrated in high-salt buffer. Monomeric 3×Flag-VchTnsC eluted at ~81.4 ml, as verified by SDS–PAGE analysis, and was stored at −80 °C after snap-freezing in liquid nitrogen. All SDS–PAGE gel source data are provided in Supplementary Fig. 1.
To monitor formation of ATP-dependent heptamers, monomeric VchTnsC in 1 M NaCl was dialysed in medium salt dialysis buffer (20 mM HEPES-KOH pH 7.5, 300 mM NaCl, 0.6 mM ATP, 0.6 mM MgCl2, 1 mM DTT) overnight, and the sample was injected onto a Superdex 200 Increase gl 10/300 column equilibrated in medium-salt dialysis buffer. Heptameric VchTnsC eluted at ~15.6 ml. VchTnsC mutants were expressed and purified using the same protocol.
Cascade and TniQ–Cascade purification
Protein components for Cascade and TniQ–Cascade were expressed from a pET-derivative vector containing an N-terminal His10–MBP–TEV site fusion on Cas8 and TniQ, respectively. The crRNA for Cascade and TniQ–Cascade were expressed separately from a pACYC-derivative vector (Supplementary Table 1). The Cascade and TniQ–Cascade complexes were overexpressed and purified as described previously2,18.
In vitro TniQ–Cascade DNA binding assay
A 52-bp 6-FAM-labelled dsDNA substrate corresponding to target-4 was generated by annealing two oligonucleotides (IDT; Supplementary Table 4). In vitro DNA binding reactions (30 μl) were prepared using 5 nM 6-FAM-labelled 52-bp dsDNA and purified Cascade or TniQ–Cascade (containing crRNA-4) ranging in final concentration from 0–400 nM, in TniQ–Cascade DNA binding buffer (20 mM HEPES pH 7.5, 75 mM NaCl, 1 mM DTT). The reactions were incubated at 37 °C for 30 min and then transferred into a 384-well black flat-bottom plate. The fluorescence polarization (FP) signal was measured at 37 °C using a Synergy Neo2 microplate reader (Biotek) with the requested polarization set to 25 mP. The normalized FP signal of each well was calculated by subtracting the FP signal of a well containing TniQ–Cascade DNA Binding Buffer and 6-FAM labelled 52-bp DNA only.
In vitro TnsC DNA binding assay
FAM-labelled ssDNA or dsDNA substrates (10 nM) corresponding to target-4 (Supplementary Table 4) were titrated with varying concentrations of TnsC (0.1–10 μM) in TnsC DNA binding buffer (20 mM HEPES pH 7.5, 2 mM MgCl2, 100 mM NaCl, 10 μM ZnCl2, 1 mM DTT) in the presence or absence of 2 mM ATP, in a final volume of 30 μl. The samples were incubated for 30 min at 37 °C in a 384-well plate, and fluorescence polarization was measured using the Synergy Neo2 microplate reader (Biotek) plate reader. The instrument gain for each sample was adjusted to a well containing no TnsC, maintaining a requested polarization of 40 mP for all measurements. The error bars represent the s.d. measured across two experimental replicates (n = 2).
In vitro TnsC ATPase assay
Relative ATP hydrolysis for WT and mutant TnsC constructs were measured using the Malachite Green Phosphate Assay Kit (Sigma-Aldrich) according to the manufacturer’s protocol. TnsC (10 μM) was incubated with 5 μM ssDNA or dsDNA substrate (Supplementary Table 4) and 1 mM ATP for 30 min at 37 °C in TnsC ATPase reaction buffer (20 mM HEPES pH 7.5, 2 mM MgCl2, 180 mM NaCl, 10 μM ZnCl2, 1 mM DTT) in a final volume of 5 μl. Next, the reaction was diluted with water to a volume of 40 μl and incubated with 10 μl working reagent for 5 min. Absorbance at 620 nm was monitored on a plate reader. All ATPase activities were normalized to WT TnsC incubated with ssDNA, which liberated 127.6 μM phosphate. Error bars represent the s.d. measured across two technical replicates (n = 2).
TnsC–DNA complex formation for cryo-EM
A 52-bp dsDNA substrate corresponding to target-4 was generated by annealing two oligonucleotides (IDT; Supplementary Table 4). To isolate the DNA-bound 3×Flag–VchTnsC complex, 40 μM purified 3×Flag–VchTnsC (monomer concentration) was combined with 2 μM dsDNA in a sterile-filtered TnsC–DNA complex reaction buffer (20 mM HEPES pH 7.5, 1 M NaCl, 1% (v/v) glycerol, 1 mM ATP, 1 mM MgCl2, 1 mM DTT) in a final volume of 500 μl. The reaction was dialysed overnight at 4 °C against TnsC–DNA low-salt dialysis buffer (20 mM HEPES pH 7.5, 50 mM NaCl, 1 mM ATP, 1 mM MgCl2, 1 mM DTT) in a 10 kDa molecular weight cut-off Slide-A-Lyzer MINI Dialysis Device (Thermo Fisher Scientific). After centrifugation at 20,000g for 20 min at 4 °C, the cleared supernatant was injected onto a Superose 6 Increase 10/300 GL column equilibrated with TnsC–DNA low-salt dialysis buffer. DNA-bound VchTnsC complexes eluted at ~13.3 ml, whereas unbound dsDNA eluted at ~15.4 ml (Fig. 5a).
iSCAMS
Reconstituted heptameric VchTnsC and VchTnsC–DNA co-complex were analysed using iSCAMS42 with a mass photometer (Refeyn). The instrument was calibrated using NativeMark Unstained Protein Standard (Thermo Fisher Scientific). A total of 10 μl of VchTnsC buffer (20 mM HEPES-KOH pH 7.5, 300 mM NaCl, 0.6 mM ATP, 0.6 mM MgCl2, 1 mM DTT) or VchTnsC–DNA co-complex buffer (20 mM HEPES-KOH pH 7.5, 50 mM NaCl, 1 mM ATP, 1mM MgCl2, 1 mM DTT) was loaded onto a glass coverslip. Once the ratiometric reading stabilized, 1 μl of 1 μM heptameric VchTnsC or VchTnsC–DNA co-complex was diluted into the respective buffer, and mass measurements were recorded. Analysis was performed using the Discover MP software.
Cryo-EM
For cryo-EM, 3 μl of purified heptameric VchTnsC and 3×Flag–VchTnsC (prepared as described in the previous sections) was incubated for 15 s on glow-discharged (30 s plasma treatment in a Gatan-Solarus plasma cleaner with a gas mix 25/75% O2/Ar at 25 W) UltrAuFoil 0.6/1 gold grids59, before vitrification in a FEI Vitrobot Mark II at 4 °C and 100% humidity (blot force setting 3, drain time 0 s, blotting time between 3.0 and 4.5 s). The concentration of VchTnsC and 3×Flag–VchTnsC was quantified by Bradford assay and ranged between 1–6 μM for all of the cryo-EM experiments.
Vitrified grids were screened in a Glacios microscope (Thermo Fisher Scientific) equipped with a Falcon III direct detector. Microscope operation and data collection were carried out using the Leginon60 software. Initial screening revealed almost exclusively top or bottom views, with the sample exhibiting a strong preferential orientation in cryogenic conditions. Addition of the detergent lauryl maltose neopentyl glycol (LMNG; Anatrace; gift from A. I. Sobolevsky) to the buffer before vitrification ameliorated the tendency of VchTnsC to adopt exclusively top or bottom orientations. A systematic examination of LMNG concentration in the final buffer revealed that 0.0002–0.0004% (w/v) was ideal to induce 30–40% of side views without affecting the concentration or reproducibility of the cryo-grid-making process. An initial dataset of 200 images collected using the Glacios microscope with the Falcon-III detector in counting mode (pixel size, 0.55 Å per pixel; fluence, 0.5 e Å−2 s−1; total dose, 35 e Å−2) revealed clear 2D classes with all views needed for three-dimensional reconstruction. An initial image-processing workflow was established by manually picking ~2,000 particles, including top, bottom and side view classes, after 2D classification in Relion343. A promising subnanometre 3D reconstruction with C7 symmetry was obtained. The cryo-grid was recovered and saved for high-resolution data collection.
High-resolution data were collected at the New York Structural Biology Center using the Titan Krios equipped with a Cs corrector and BioQuantum-K3 imaging system. Around 13,000 images were collected from two grids in counting mode with a pixel size of 0.678 Å per pixel at a fluence of 10 e Å−2 s−1 and a total exposure time of 4 s. The defocus range was set between −0.6 μm and −2 μm.
Image processing was integrally performed in Relion343 with CTF estimation using a wrapper to CTFFIND461. After motion correction and CTF estimation, an autopicking job using the selected 2D class averages from the previous Glacios dataset as templates (picking parameter, 0.01; and interparticle distance limit of 100 Å) found 12 million particles in the entire dataset. Initial particle extraction was performed with a bin size of 8 (pixel size of 5.42 Å per pixel), and a 2D classification job (200 classes, tau fudge 4 and the subset option deactivated) was used to discard bad or poorly aligned particles. The selected particles were processed for a 3D refinement job using as an initial model a volume obtained by stochastic gradient descent (SGD) from the Glacios dataset. After 3D refinement, particles were re-extracted at bin 4 (pixel size of 2.712 Å per pixel) with the re-centre option activated. Next, a 3D classification job using a three-pixel extended soft mask and without alignment (20 classes, tau-fudge parameter set to 3) found three or four major populations (classes 3, 11 and 19; Extended Data Fig. 4c). Detailed analysis of these classes revealed very similar maps with variations in peripheral loops. Class 3 clearly showed features compatible with helices, and particles belonging to this class (~84,000) were selected, re-extracted to bin 2 (pixel size 1.356 Å per pixel) and re-refined at this pixel size. This reconstruction reached ~4 Å resolution, which was further improved by refinement of the CTF parameters and Bayesian polishing of the beam-induced particle movements62,63. The final subset of shiny particles was re-refined with and without imposing C7 symmetry. The symmetrical reconstruction reached 3.5 Å resolution after post-processing.
Map exploration revealed features compatible with the reported resolution, including clear side-chain density particularly for tyrosine, phenylalanine, leucine and tryptophan residues. Several loops facing the inner pore or the periphery of the heptameric disk showed weaker density. A re-refinement of the same set of shiny particles without symmetry imposition (C1) yielded a 3.6 Å reconstruction, with good overall agreement to the symmetrical reconstruction. However, one of the seven VchTnsC monomers exhibited weaker density, which could be interpreted as flexibility or lower occupancy for this monomer (Extended Data Fig. 4c (bottom)). To improve the density of the flexible loops in the symmetrical reconstruction, several approaches were explored. First, symmetry relaxation to C7 was activated in a 3D refinement job64. Moreover, local symmetry conditions (local symmetry operators) were manually defined and refined in Relion343 with the aim to find a set of conditions specific to the VchTnsC heptamer. Finally, symmetry expansion with local subtraction and classification was pursued for the monomer exhibiting weaker density. None of these approaches worked ideally, though some improvements were obtained, particularly with the symmetry relaxation strategy. A final approach consisting of a masked local refinement, including only the four contiguous monomers of the VchTnsC heptamer that showed the best density in the C1 reconstruction, revealed improved density for the disordered loops of the symmetrical reconstruction, which aided model building of these areas.
Both symmetrical and masked asymmetrical post-processed maps were used for de novo model building in Coot65. Clear density, especially for bulky residues, allowed proper tracing of the polypeptide chain and correction of the register, yielding a final model consisting of 310 amino acids (starting from Arg5 to His314) out of the 333 residues present in the cloned construct (following TEV protease cleavage). The final model was refined in real space with Phenix66 imposing secondary structure and Ramachandran restraints. This refined model was checked in Coot, corrected in problematic areas and re-refined in the reciprocal space with Refmac67, using secondary structure restraints computed in ProSmart68. The quality of the final model was monitored by Molprobity69. Pymol (The PyMOL Molecular Graphics System, v.2.0 Schrodinger), Chimera and ChimeraX were used for analysing the structure and for figure generation. Cryo-EM data collection, refinement and validation statistics can be found in Extended Data Table 1.
For the TnsC–DNA complex, we followed a similar approach as for the apo-TnsC sample, using similar conditions for grid optimization, data collection and image processing. However, the TnsC–DNA sample exhibited a strong preferential orientation which could not be overcome by adding a small amount of the detergent LMNG as with the apo-TnsC sample. To address this issue, we collected a second dataset for the TnsC–DNA sample in the presence of 2 nm amorphous carbon (Quantifoil R1.2/1.3 Au 300 mesh, 2 nm C). A joint dataset (combining images with and without support) was processed analogously to the apo-TnsC dataset with the only variation of increasing to 8 the number of classes in the SGD step. With this approach, 3–4 relevant classes of particles were identified by SGD. Of these, one class corresponded to the single heptamer with additional density at the central pore that was interpreted as dsDNA and a second class corresponded to a double heptamer class with strong density in the central cavity that clearly exhibited features compatible with dsDNA. These particle classes were further processed independently with no symmetrical impositions, yielding, after CTF parameter refinement, Bayesian particle polishing and postprocessing, maps with resolution estimations of ~3.5 Å for the single heptamer with DNA and ~3.9 Å for the double-heptamer class with DNA.
The refined apo-TnsC model was fitted monomer by monomer in the new maps and ideal B-form dsDNA was generated and fitted with Coot65. For the single-heptamer class with DNA, a real-space-refinement step in Phenix66 of the model against the map was performed followed by a reciprocal-space refinement step in Refmac67. For the double-heptamer class, the reciprocal-space refinement step in Refmac was omitted. The quality of the final model was monitored by Molprobity69.
TniQ–Cascade–TnsC–DNA model
A VchTnsC monomer (chain 4) of the DNA-bound VchTnsC single heptamer (this study) was aligned to chain H (ShoTnsC monomer) of the ShoTnsC–TniQ–DNA structure (Protein Data Bank (PDB): 7N6I)23, with a global root-mean-square deviation (RMSD) of 1.758 Å. This placed ShoTniQ-2 (chain B from PDB 7N6I) in direct proximity to chain 4 in the VchTnsC–DNA structure. Next, VchTniQ-2 (chain J) of a VchTniQ–Cascade structure (PDB: 6VBW)46 was aligned to ShoTniQ-2 (chain B from PDB 7N6I), with a global RMSD of 0.807 Å.
Statistics and reproducibility
The SDS–PAGE gels shown are representative of three independent experiments for TnsC•ATP (Fig. 4a) and TnsC•ATP•DNA (Fig. 5a), and representative of two independent experiments for TnsC mutants (Extended Data Figs. 6a,c and 7c). All experiments for generating cryo-EM micrographs were repeated at least twice independently with similar results (Extended Data Figs. 4a and 8d).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Design of Flag-tagged transpososome components and analysis of ChIP data.
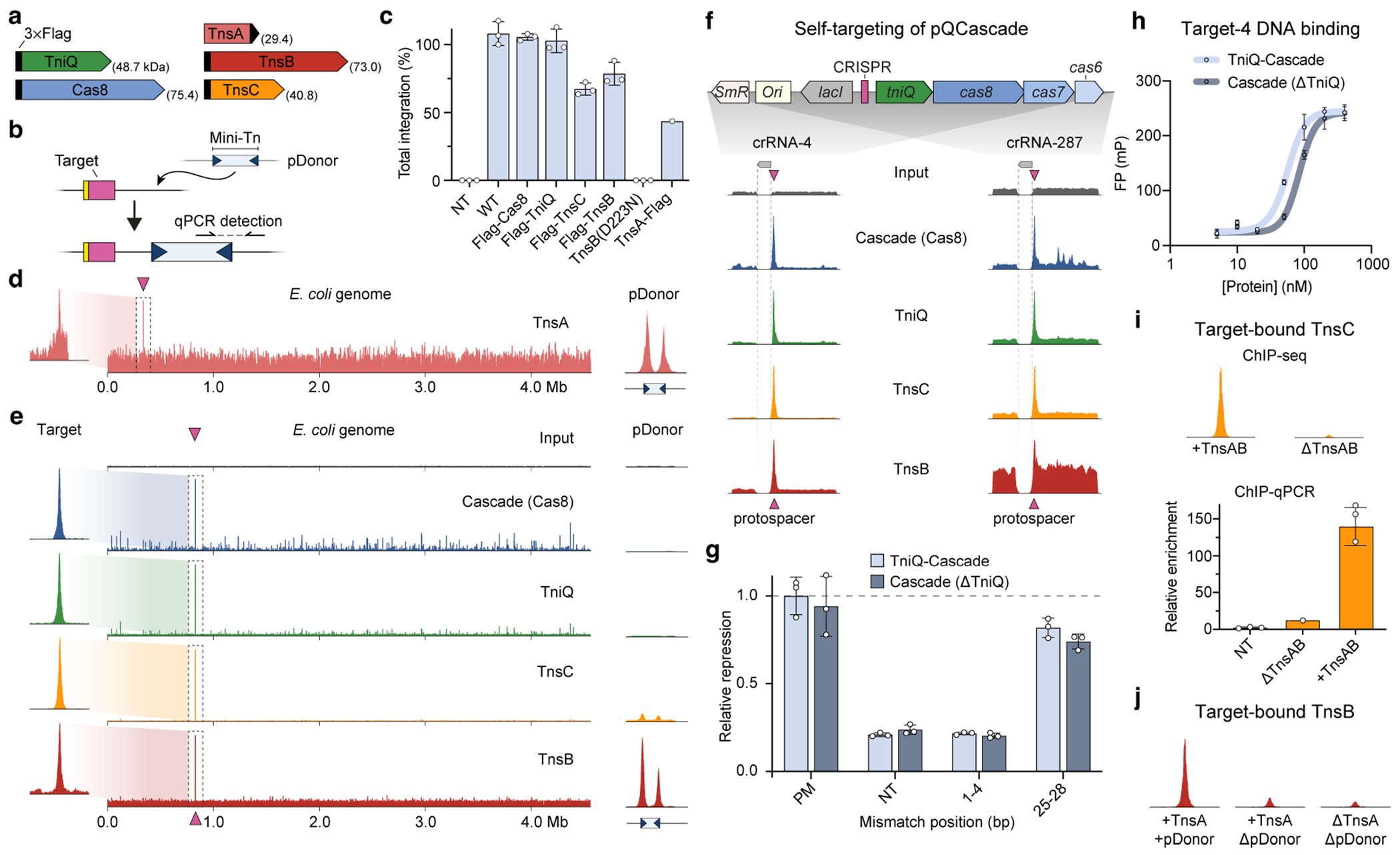
a, Schematic highlighting the 3×Flag-tag positions in each protein component that was used for ChIP–seq experiments. Molecular weights are indicated in parentheses. b, Simplified schematic of the in vivo transposition assay used to quantify RNA-guided DNA integration in E. coli. Mini-transposon (Mini-Tn) DNA substrates are mobilized from pDonor to genomic DNA, downstream of the target site complementary to crRNA, and integration is quantified by junction PCR. c, RNA-guided transposition assays for 3×Flag-tagged constructs demonstrate that near-wild-type activity is maintained. A single point mutation in the TnsB active site (D223N) completely ablates DNA integration. TnsA-Flag was not pursued further after weak enrichment at the target site was observed (see panel d), and hence was only tested once. NT, non-targeting crRNA control. d, ChIP–seq data for 3×Flag-tagged TnsA with crRNA-4, shown as in Fig. 1c, d, reveal inefficient DNA enrichment at the genomic target site (left). However, the prominent peaks at both transposon ends on pDonor (right) indicate that the protein retains in vivo function and likely exhibits inefficient crosslinking to the target DNA within the pre-integration transpososome complex. Read coverage (RPKM) is scaled to the highest peak (E. coli genome mapping reads) or TnsB ChIP-seq data (pDonor mapping reads). e, Representative ChIP–seq data for experiments with crRNA-287, shown as in Fig. 1c, d, reveal accurate recruitment of multiple distinct factors to the corresponding genomic target site. f, ChIP–seq data reveal robust self-targeting of the CRISPR array across multiple crRNA datasets. Reads mapping to pQCascade are shown for two distinct crRNAs for the indicated Flag-tagged components, and were normalized to the highest peak, except for input samples, which were normalized to the highest peak in their corresponding Cas8 sample. The vector map of pQCascade is shown (top), and the position of the CRISPR spacer is denoted with maroon triangles (bottom). Non-unique reads mapping to lacI were not analysed (dotted lines). g, CRISPR interference (CRISPRi)-based repression of genomically-encoded mRFP using Cascade in the presence (light blue) or absence (ΔTniQ, dark blue) of TniQ. Transcriptional repression is measured as OD-normalized mRFP fluorescence relative to the perfectly matching (PM) crRNA. NT, non-targeting; 1–4, crRNA mismatches at position 1–4; 25–28, crRNA mismatches at position 25–28. h, Target DNA binding by Cascade (crRNA-4) with and without TniQ, as measured by fluorescence polarization. Colouring is as shown in g. i, TnsC may be stabilized in the pre-integration transpososome complex by the presence of TnsAB. ChIP–seq reads mapping to a 3-kb window centred at the genomic target site are shown for 3×Flag-tagged TnsC in experiments that differed only in the presence or absence of TnsA and TnsB (top), with read coverage (RPKM) scaled the same in both graphs. Note that the same data are presented in Fig. 1f. Quantitative PCR was performed on the same ChIP samples alongside a non-targeting control, confirming the diminished target DNA enrichment levels when TnsA-TnsB were omitted (bottom). Relative enrichment is calculated as ΔΔCq between the target locus and reference gene. j, TnsB can be recruited to the RNA-guided target site in the absence of TnsA and donor DNA, albeit with reduced efficiency. ChIP–seq reads mapping to a 3-kb window centred at the genomic target site are shown for 3×Flag-tagged TnsB in experiments that differed only in the presence or absence of TnsA and pDonor, with read coverage (RPKM) scaled the same in all three graphs. Data in c, g, h and i are shown as mean ± s.d. of n = 3 independent biological replicates, except for TnsA-Flag (c, n = 1) and ΔTnsAB (i, n = 1).
Extended Data Fig. 2 |. Additional analyses of off-target DNA binding from ChIP–seq data.
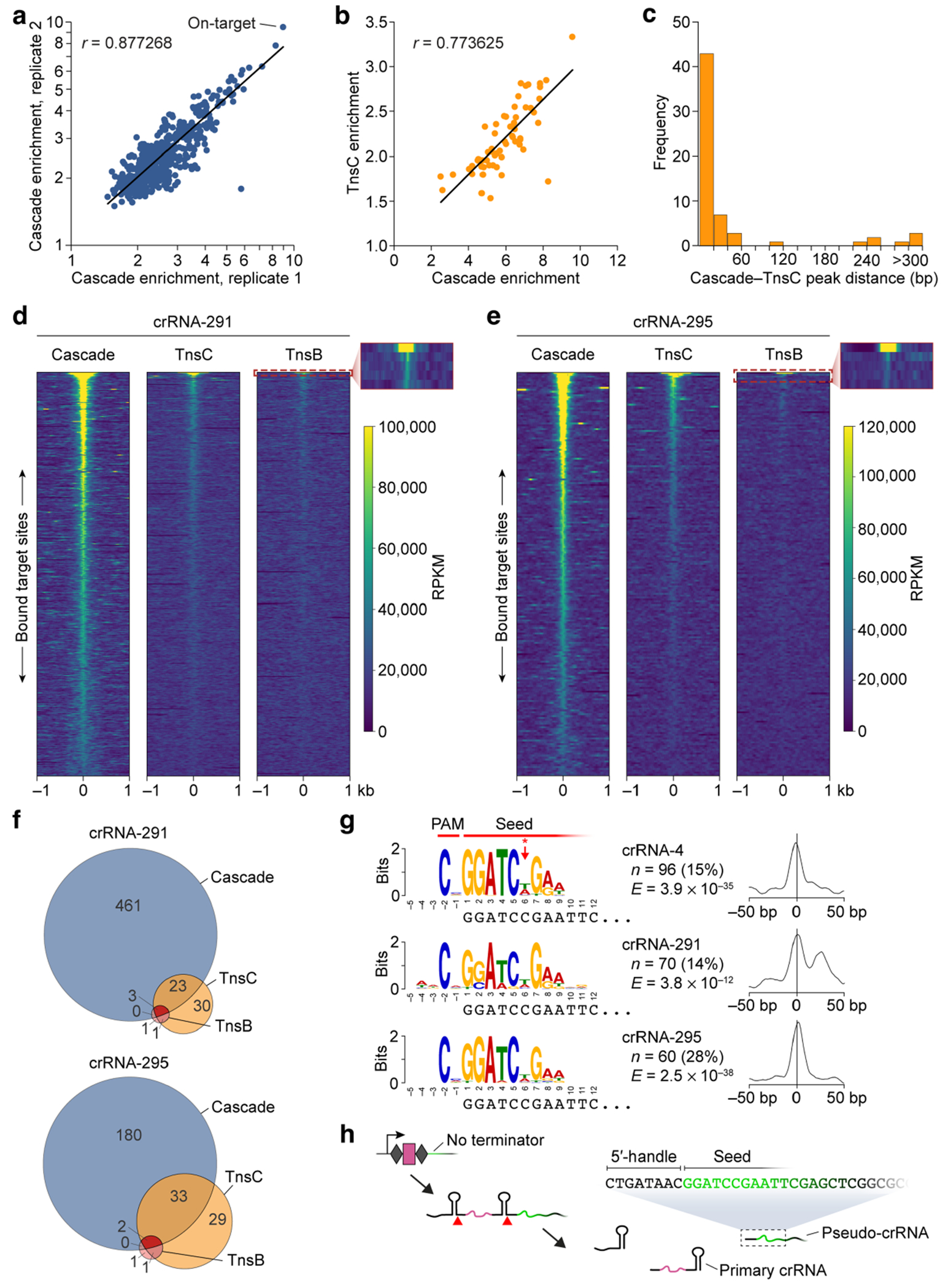
a, Scatter plot showing correlation between two biological replicates of ChIP–seq experiments with 3×Flag-tagged Cas8 and crRNA-4. ChIP–seq enrichment values are plotted for n = 424 peaks called by MACS3 that were present in both datasets, as identified by overlapping peak start and end coordinates. A linear regression fit and Pearson linear correlation coefficient (r) are shown; the on-target site is indicated. b, Scatter plot showing correlation between off-target peaks observed for Cas8 and TnsC ChIP–seq experiments with crRNA-287. ChIP–seq enrichment values are plotted for n = 60 peaks called by MACS3 that were present in both datasets, as identified by overlapping peak start and end coordinates. A linear regression fit and Pearson linear correlation coefficient (r) are shown; the on-target site is off-scale for TnsC and was not included. c, Histogram of the distance between the genomic coordinates of Cas8 and TnsC peak summits for the overlapping peaks in b. Most peaks are within 20 bp of each other. d, Global visualization of ChIP–seq peaks with crRNA-291 for off-target sites using heat maps, plotted side-by-side for Cascade (left), TnsC (middle), and TnsB (right); data are shown as in Fig. 2b, c. A 2-kb window for 487 genomic loci (y axis) is plotted in order of decreasing peak enrichment for ChIP–seq peaks called for the Cascade dataset, with the RPKM scale bar shown. The inset for TnsB highlights the strong enrichment of the on-target site (row 1) and immediate drop-off for other Cascade off-targets (rows 2–5). e, Global visualization of ChIP–seq peaks with crRNA-295, plotted as in d. f, Venn diagrams showing the overlap in off-target peaks called for Cascade, TnsC, and TnsB. Peaks were called individually for each ChIP–seq dataset using MACS3, and overlaps were analysed based on the coordinates for each peak. Data are shown for the same two crRNAs as in d and e. g, Multiple Cascade ChIP–seq datasets from experiments with distinct crRNAs exhibit common off-target peaks that share a common motif, as assessed by MEME analysis. Shown for each dataset (top to bottom) are the motif visualized above the corresponding nucleotides at the 5’ end of pseudo-crRNA spacer (left), and the motif probability graph showing the probability of a motif match occurring at a given position in the input sequence (right). The PAM and seed regions are indicated at the top, as well as the lack of discrimination at position 6 (red asterisk/arrow). n, the number of peaks contributing to the motif (and their percentage of total peaks called); E, the E-value significance of the motif resulting from MEME analysis. h, The motifs for common off-target peaks in g can be ascribed to pseudo-crRNAs that use the spacer-like sequence downstream of the terminal repeat within the CRISPR array. The schematic shows the architecture of the CRISPR array encoded by pQCascade and the presumed mechanism of crRNA biogenesis. A pseudo-crRNA relies on a 5′-handle derived from the second repeat and a pseudo-spacer derived from the downstream sequence, which is common to all pQCascade vectors used in this study. Notably, the pseudo-crRNA will lack the repeat-derived 3′-handle (stem-loop bound by Cas6), is unlikely to have a single defined length, and is predicted to form a minimal Cascade complex lacking both Cas6 and TniQ.
Extended Data Fig. 3 |. Additional analyses of crRNA library and CRISPRi data.
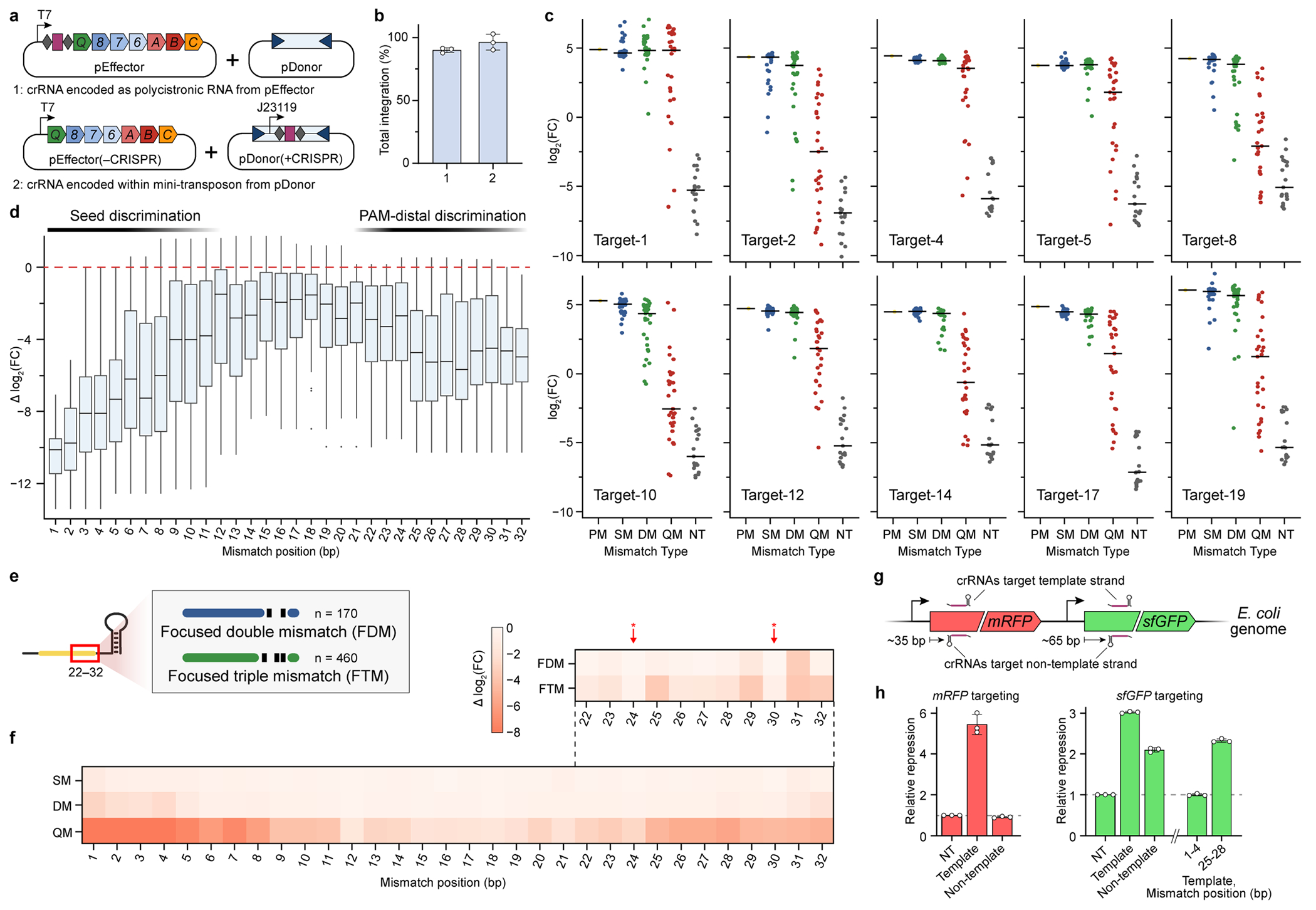
a, Two-plasmid system used for in vivo transposition assays with crRNA libraries. We relocated the CRISPR array from pEffector to inside the mini-transposon within pDonor itself (bottom), relative to pEffector and pDonor plasmids we described previously17 (top), thereby generating pEffector(−CRISPR) and pDonor(+CRISPR). b, RNA-guided transposition assays for the two sets of plasmid constructs shown in a, as measured by qPCR. Transposon-encoded crRNAs direct indistinguishable integration efficiencies. c, Enrichment scores for crRNA library members associated with their corresponding target sites, plotted as in Fig. 3d. d, Position-specific enrichment scores are plotted as the change in log2(fold change) relative to the perfectly matching crRNA, for crRNAs containing quadruple mismatches across all 10 target sites. Data are shown as box plots with single-bp resolution along the crRNA; boxes show the median and interquartile range, whiskers indicate the range or extend 1.5× the interquartile range from the 25th and 75th percentile in the presence of outliers (dots). The data reveal off-target discrimination in both the PAM-proximal seed sequence and a PAM-distal region (indicated at the top). e, Schematic of mismatched crRNA library members focused on positions 22–32 in the PAM-distal region. Members were designed to test the effects of non-adjacent mismatches on transposition. f, Position-specific enrichment scores were averaged across all 10 target sites and plotted for each mismatch type (SM, DM, QM) as the change in log2(fold change), similarly to Fig. 3e. Data from focused mismatches within positions 22–32 (schematised in e) are shown at the top, revealing a lack of discrimination at positions 24 and 30 (red arrows), where crRNA nucleotides are flipped out. g, Schematic showing the location of crRNAs targeting genomically encoded mRFP and sfGFP genes. Guides were designed to target both the template and non-template strands, and were positioned ~35 bp and ~65 bp downstream of the gene start codon, respectively. h, Transcriptional repression was measured for each of the targeting crRNAs alongside a non-targeting (NT) control; the crRNA targeting the template strand shows strongest repression. Mismatches at positions 1–4 and 25–28 for an sfGFP-targeting crRNA show similar effects as with the mRFP data shown in Fig. 3g. Data in b and h are shown as mean ± s.d. of n = 3 independent biological replicates. The absolute sample numbers shown in d are n = 10, 20, 30, 40, 30, 20, and 10 for mismatch position 1, 2, 3, 4–29, 30, 31, and 32, respectively.
Extended Data Fig. 4 |. Raw electron micrographs and image processing workflow for TnsC•ATP heptamer.

a, Representative cryo-micrographs for the TnsC•ATP heptamer at various defoci; the corresponding CTF is shown (top right), b, Representative reference-free 2D class averages with four types of views identified: top and bottom views, a lateral side view and intermediate views. The size of circular masks used during the classification run is indicated in the first class average for each type. c, Image processing workflow employed for the identification and high-resolution refinement of a homogenous class of particles for the TnsC•ATP heptamer. One of the seven TnsC•ATP monomers exhibited weaker density (orange asterisk/arrow).
Extended Data Fig. 5 |. Fourier Shell Correlation curves, local resolution, representative densities, and secondary structure diagram for TnsC.
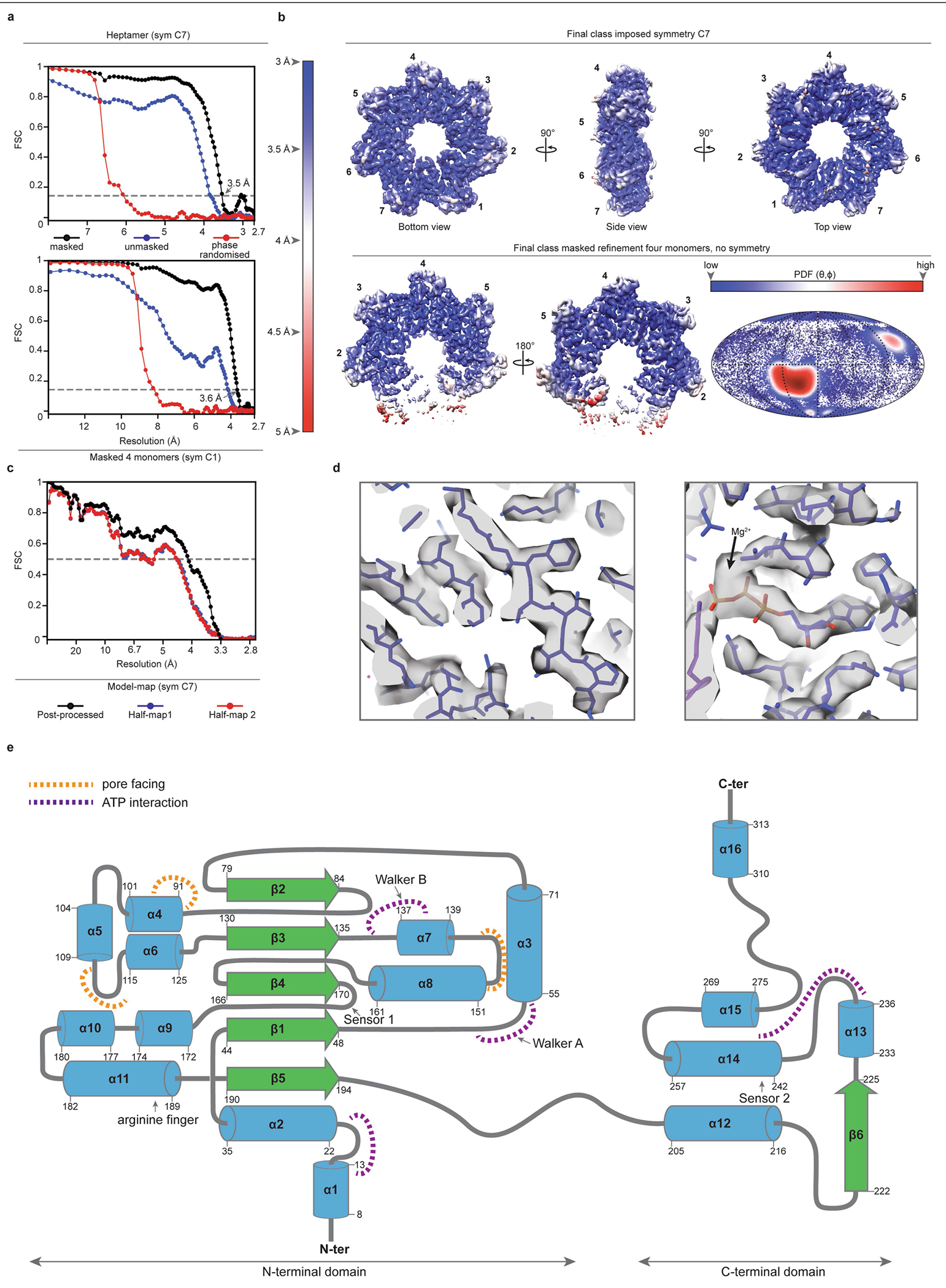
a, Fourier Shell Correlation (FSC) curves computed for two independently refined half-maps of the final sub-class of particles before masking (blue), after masking (black), and after phase randomization (red). Curves are shown for the refinement with a C7 symmetry condition imposed (top) and for a masked refinement of the four best TnsC monomers without the imposition of any symmetrical constraint (bottom). The final resolution by the FSC-0.143 criteria is 3.5 Å for the symmetrical reconstruction and 3.6 Å for the masked refinement without symmetry impositions. b, Unsharpened maps coloured according to local resolution calculations, showing the symmetrical map in orthogonal orientations (top) and the masked map without symmetrical impositions (bottom). A Mollweide diagram with the Euler angle distribution for the masked map refined in C1 is shown (bottom right), coloured according to the density distribution. c, Model-map FSC curve (black line) and model overfitting test performed by randomly displacing 0.5 Å of the atomic coordinates of the final model and re-refining the displaced coordinates against half-map 1. The coincidence of the blue curve (FSC against half-map 1) with the red curve (FSC against half-map 2, not included in the refinement) guarantees the absence of overfitting in the original model. d, Representative final cryo-EM densities from the post-processed symmetrical map, with the refined model represented as sticks. The putative density for a Mg2+ ion is indicated by a black arrow (right). e, Secondary structure diagram of VchTnsC derived from the structural data. α-helices are depicted as blue cylinders, β-strands as green arrows and loops as grey traces. ATP binding regions, pore facing regions and hallmark sequences from the AAA+ ATPase family are indicated.
Extended Data Fig. 6 |. Inter-monomer interactions, biochemical characterization, and TnsC heptamer formation.
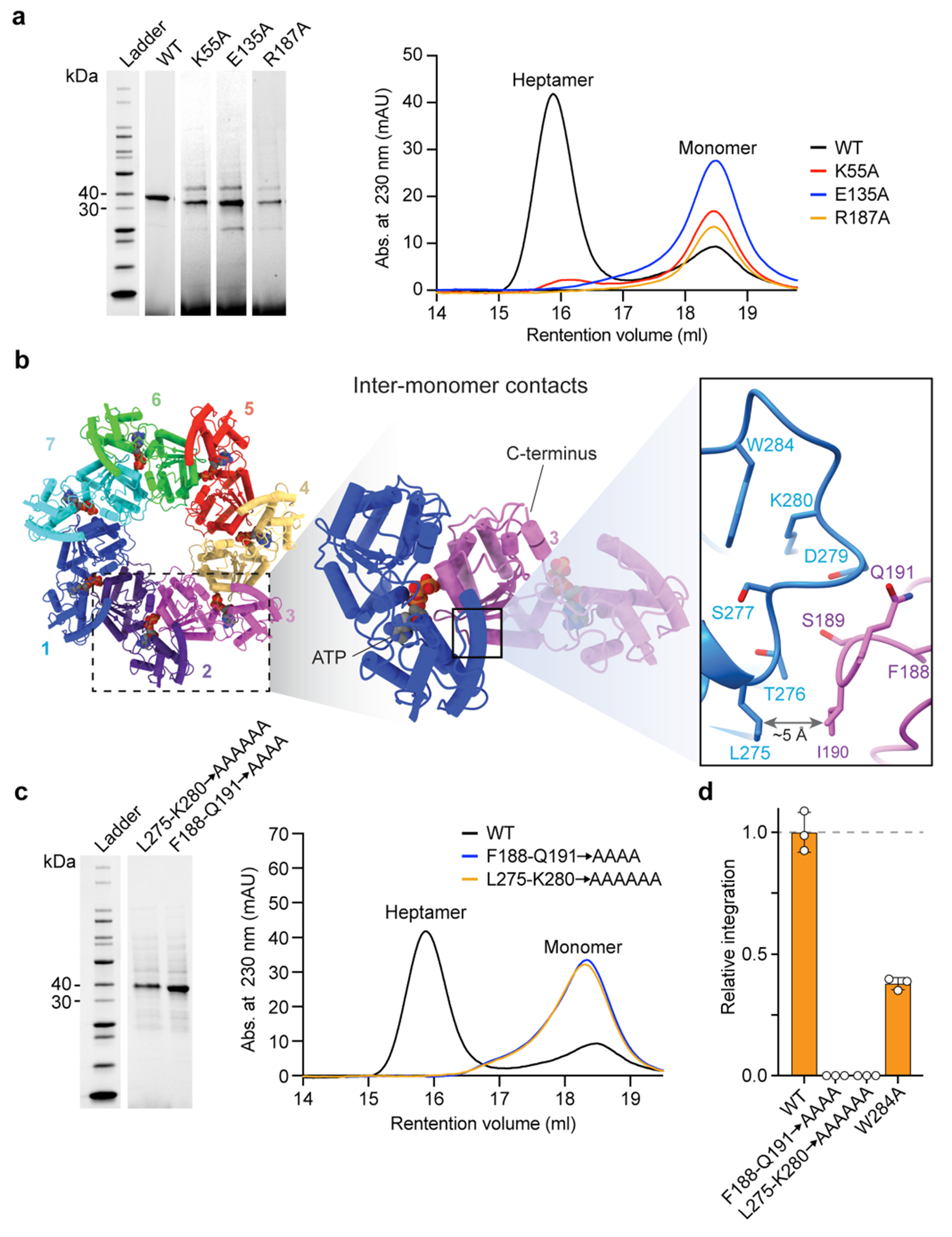
a, Purification and heptamer assembly experiments with ATPase mutants of TnsC. SDS–PAGE analysis (left) and SEC chromatograms (right) are shown for the indicated mutants, with TnsC monomer and heptamer peaks indicated. Mutations in the Walker A (K55A), Walker B (E135A), and arginine finger (R187A) motifs prevent formation of stable heptamers. b, Adjacent TnsC monomers interact via two proximal loops, in which residues 275–280 from one monomer engage residues 188–191 from the neighbouring monomer (inset on the right) c, Purification and heptamer assembly experiments with interface and pore loop mutants of TnsC, shown as in a. Mutations at the monomer-monomer interface are unable to form stable heptamers in the presence of ATP. For gel source data, see Supplementary Fig. 1. d, CRISPR RNA-guided transposition assays for the indicated loop mutants, as measured by qPCR. Data are normalized relative to WT. Data in d are shown as mean ± s.d. for n = 3 independent biological replicates.
Extended Data Fig. 7 |. Characterization of flexible loops in TnsC.
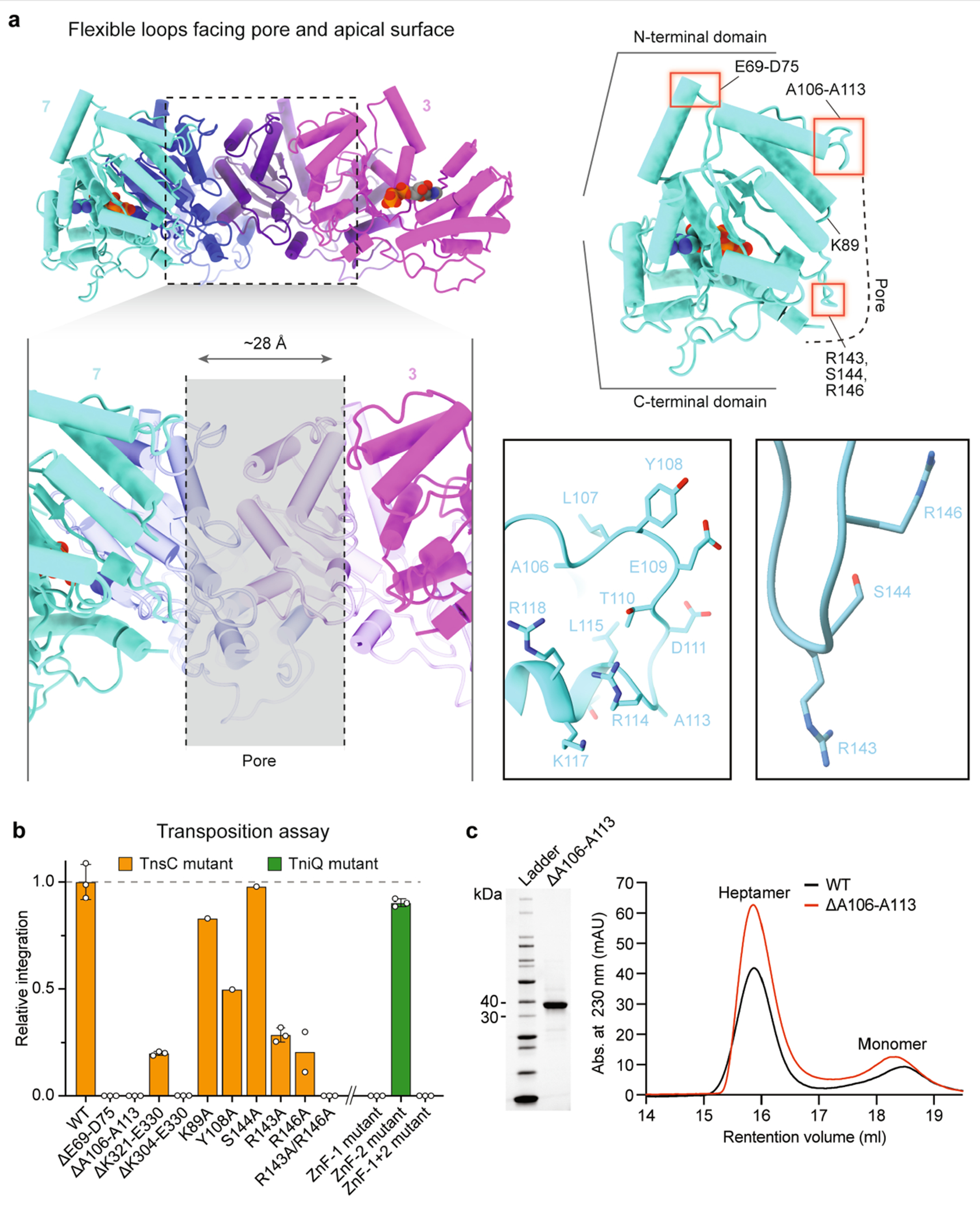
a, Multiple flexible loops within the NTD protrude from the top of the heptameric TnsC disc (top left), which are potential sites of engagement with the TniQ–Cascade complex. Apical loops E69-D75 and A106–A113 are highlighted by red boxes, as are putative DNA-interacting residues including R143, S144 and R146 (top right). A long flexible tail at the CTD folds back towards the centre of the pore, and is likely to engage TnsAB, based on functional similarity to EcoTnsC. Magnified views of A106–A113 and the putative DNA binding region are shown in ribbon representation (centre right), b, CRISPR RNA-guided transposition assays for the indicated TnsC loop mutants, CTD deletions and pore-facing residues (left; coloured in orange), and TniQ ZnF mutations (right; coloured in green), as measured by qPCR. Data are normalized relative to WT. Mutation of R143 and R146 together abrogates transposition. Data are shown as mean ± s.d. of n = 3 independent biological replicates, with the exception of R146A (n = 2) and K89A, Y108A and S144A (n = 1). c, Purification and heptamer assembly experiments with the TnsC A106–A113 loop deletion mutant. SDS-PAGE analysis (left), and SEC chromatograms (right) are shown, with TnsC monomer and TnsC•ATP heptamer peaks indicated. Despite abrogating transposition, this loop deletion mutant is still able to form ATP-dependent heptamers. For TnsC(ΔA106–A113), the baseline was set to 0 at the 5 ml retention volume mark, for clarity. For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 8 |. DNA binding, representative micrographs, Fourier Shell Correlation curves, local resolution, and representative densities for DNA-bound TnsC.
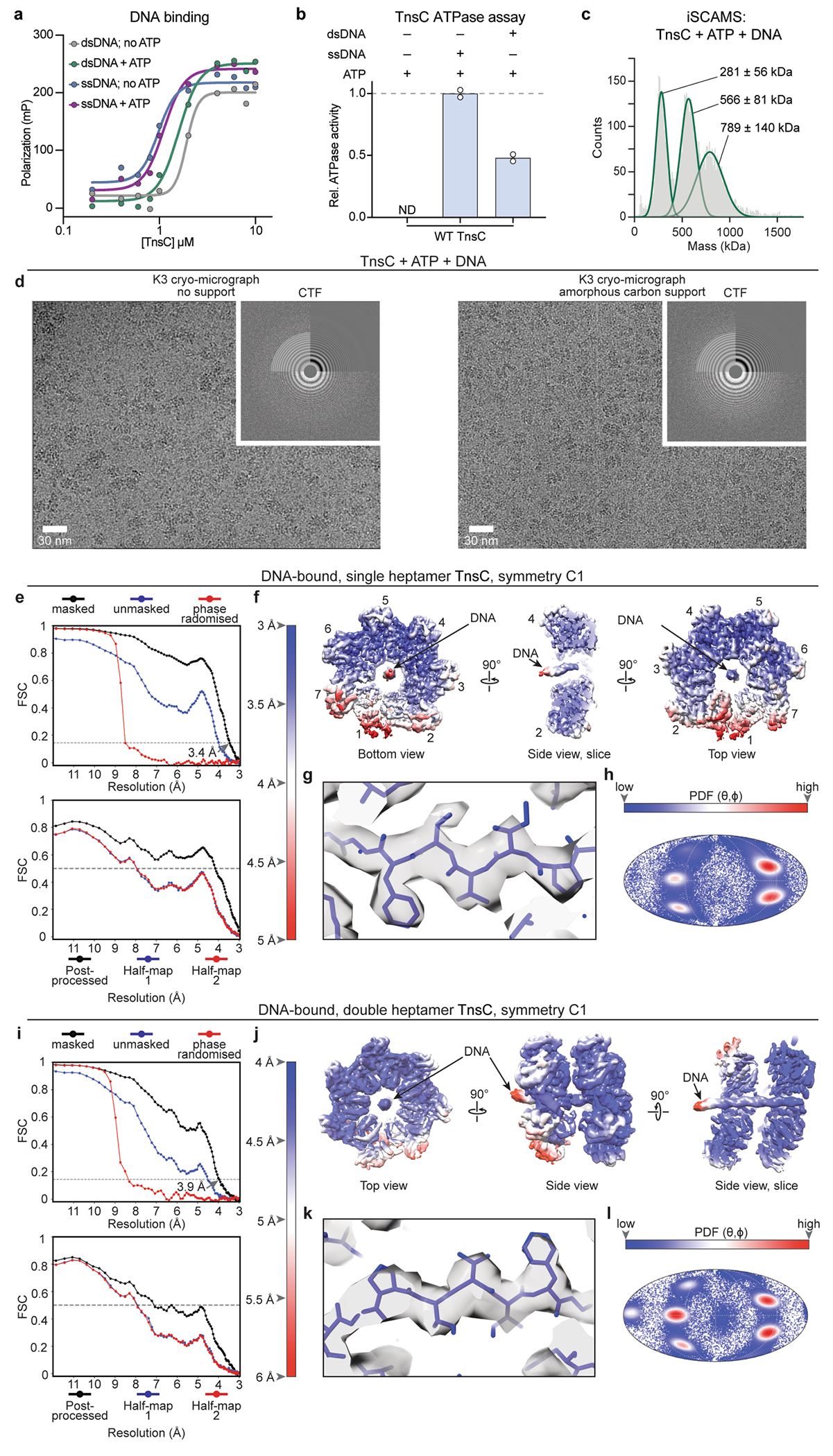
a, DNA binding by TnsC as measured by fluorescence polarization, tested with ssDNA and dsDNA substrates +/− ATP. Data are shown as mean ± s.d. of n = 2 experimental replicates. b, ATPase activity of TnsC in the absence or presence of ssDNA and dsDNA; ND, not detected. Data are shown as mean ± s.d. of n = 2 technical replicates. c, iSCAMS analysis of a TnsC sample after incubation with ATP and dsDNA reveals three distinct high-molecular-weight species, in good agreement with the DNA-bound single, double, and triple heptameric rings observed by cryo-EM. d, Representative cryo-micrographs for the DNA-bound TnsC sample at various defoci; the corresponding CTF is shown at the top right for each panel. e–h, DNA-bound single heptamer complex. i–l, DNA-bound double heptamer complex. e,i, Fourier Shell Correlation (FSC) curves (top) computed for the final independently refined half-maps before masking (blue), after masking (black), and after phase randomization (red). The final resolution by the FSC-0.143 criteria is 3.46 Å (single heptamer) and 3.9 Å (double heptamer) for the refinement without symmetry impositions. The bottom graph shows a model-map FSC curve (black line) and model overfitting test performed by randomly displacing 0.5 Å of the atomic coordinates of the final model and re-refining the displaced coordinates against half-map 1. The coincidence of the blue curve (FSC against half-map 1) with the red curve (FSC against half-map 2, not included in the refinement) guarantees the absence of overfitting in the original model. f,j, Unsharpened maps coloured according to local resolution calculations shown in orthogonal orientations. g,k, Representative final cryoEM densities from the post-processed maps, with the refined model represented as sticks. h,l, Mollweide diagrams showing the Euler angle distribution for the final refined maps, coloured according to the density distribution.
Extended Data Fig. 9 |. Structural diversity of DNA-bound TnsC heptamers.
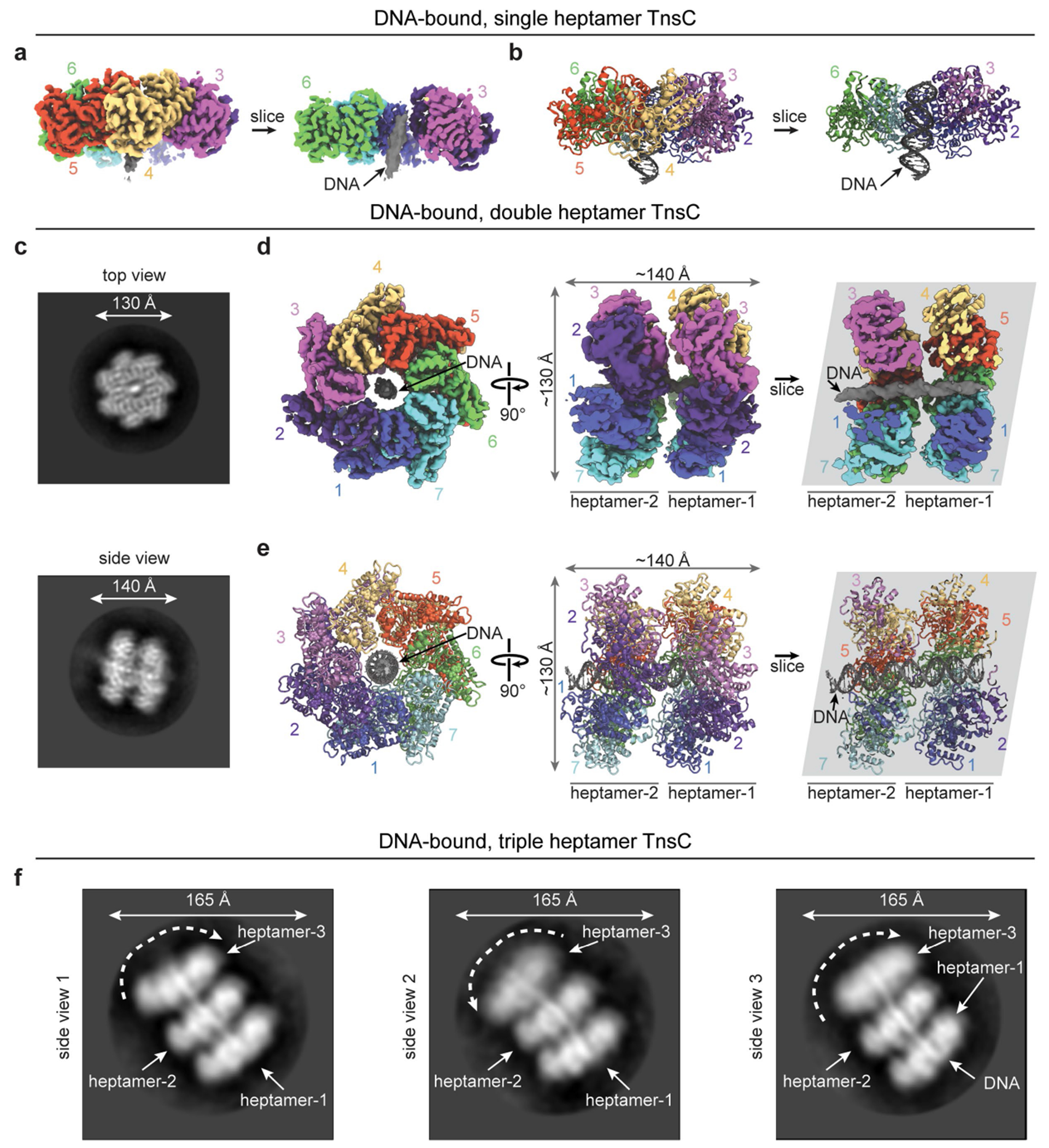
a, Final cryo-EM density of a single DNA-bound TnsC heptamer showing unsliced (left) and sliced (right) side views to highlight DNA that is threaded through the central pore. Individual subunits are labelled. b, Ribbon representation of the views shown in a. c, Representative 2D class averages for the DNA-bound TnsC double heptamer. d, Two orthogonal views of the final cryo-EM map for the DNA-bound TnsC double heptamer. Each of the seven protomers is labelled, and DNA density is shown in grey. Both heptameric rings bind DNA in the same orientation. e, Ribbon representation of the DNA-bound TnsC double heptamer shown as in b, with ATP molecules present in all protomers shown as spheres. f, Three representative 2D class averages of the DNA-bound TnsC triple heptamer. Dynamics of individual heptamers are highlighted by dotted arrows, and TnsC heptamers located at the end of the bound DNA exhibit flexibility relative to the adjacent heptamers (see Supplementary Video 2).
Extended Data Fig. 10 |. Comparison of VchTnsC and TnsC homologues from evolutionarily diverse transposons.
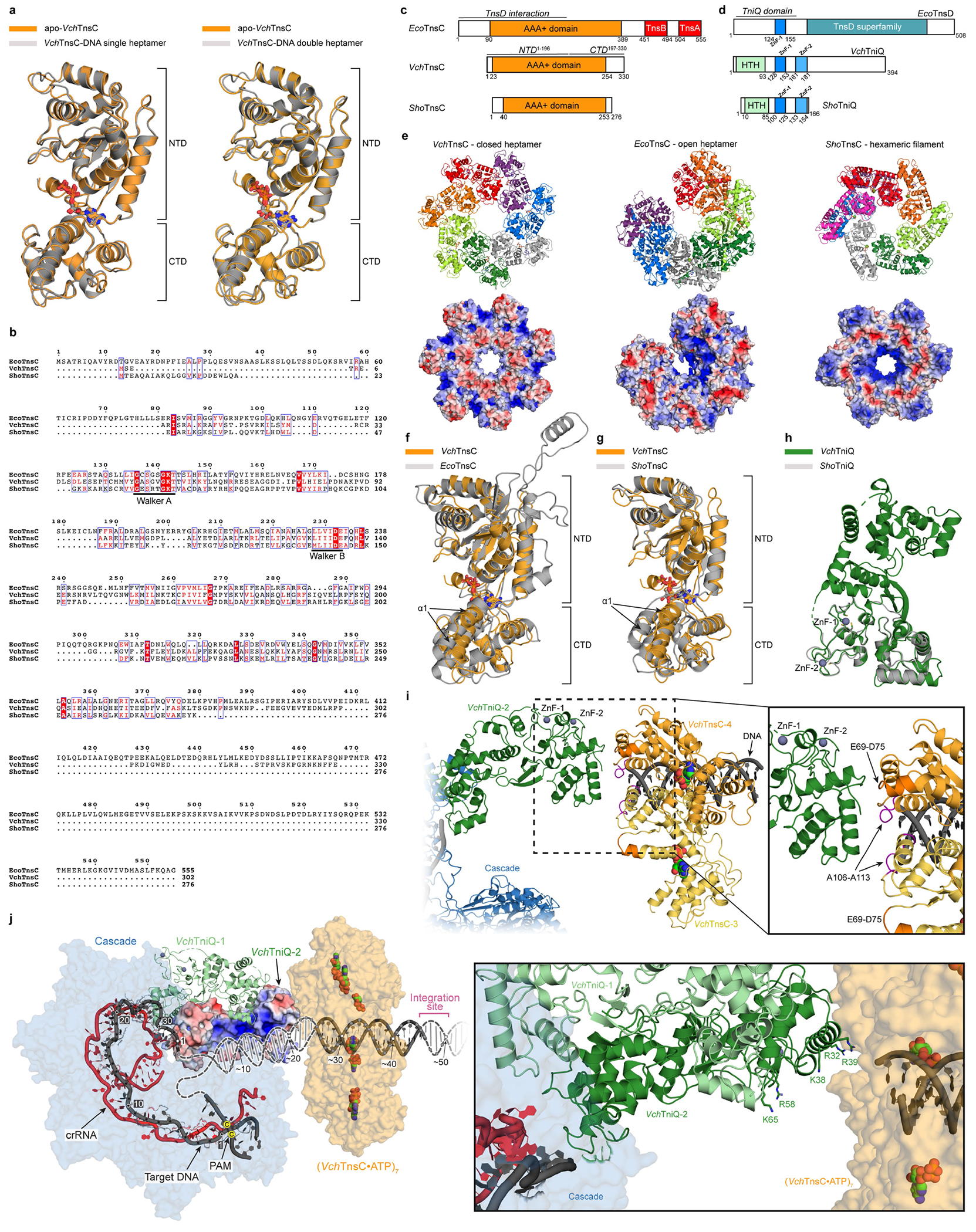
a, The structure of TnsC is highly similar between apo and DNA-bound states. Structural superposition of a monomer from the apo-TnsC and DNA-bound TnsC single heptamer structure yields a global RMSD of 0.53 Å (left); a superposition with the double heptamer yields a global RMSD of 0.64 Å (right). ATP molecules are shown as sticks, and the NTD and CTD are labelled. b, Multiple sequence alignment of EcoTnsC22 from Tn7 (non-CRISPR associated), VchTnsC from this study (type I-F CRISPR-associated), and ShoTnsC from ShCAST3,23,24 (type V-K CRISPR-associated). Residues critical for ATPase activity are indicated. Sequence identities are as follows: VchTnsC and EcoTnsC, 18.84%; VchTnsC and ShoTnsC, 22.18%; EcoTnsC and ShoTnsC, 20.83%. c, Comparison of domain organization between three TnsC homologues, with EcoTnsC regions that interact with TnsA, TnsB, and TnsD indicated4. Note the significantly shorter N- and C-termini for VchTnsC and ShoTnsC. d, Comparison of domain organization between three TniQ-family protein homologues; note that the E. coli Tn7 protein is referred to as TnsD. Zinc-finger (ZnF) motifs are highlighted. e, Comparison of overall oligomeric architecture (top) and APBS electrostatic potential70 (bottom) for DNA-bound VchTnsC•ATP (left; this study), DNA-bound EcoTnsC(A225V)•AMPPNP (centre; PDB ID: 7MCS)22 and DNA-bound ShoTnsC•ATPɣS (right; PDB ID: 7M99)23, with the C-terminal face of the pore pointing up in each oligomer. DNA was omitted for clarity and for all electrostatics calculations. Heptamer formation of VchTnsC and EcoTnsC may be favoured by more efficient protomer-protomer packing, which appears less apparent in the hexameric ShoTnsC complex. f, Structural superposition of DNA-bound VchTnsC•ATP (this study) and EcoTnsC (PDB ID: 7MCS)22 monomers, by alignment of NTD residues 16–196 (VchTnsC) and residues 70–90 and 128–295 (EcoTnsC), yields an RMSD of 2.42 Å. The conformation of α1 (labelled) may create a more open protomer architecture enabling efficient packing of adjacent subunits, and thus, heptamer formation. ATP and AMPPNP are shown as sticks. g, Structural superposition of DNA-bound VchTnsC•ATP (this study) and ShoTnsC (PDB ID: 7M99)23 monomers, by alignment of NTD residues 16–196 (VchTnsC) and 34–198 (ShoTnsC), yields an RMSD of 1.27 Å. In ShoTnsC, the conformation of α1 may cause a more closed protomer architecture that obstructs efficient packing of adjacent subunits and promotes hexamer formation. ATP and ATPɣS are shown as sticks. h, Structural superposition of VchTniQ (PDB ID: 6V9P)46 and ShoTniQ monomers (PDB ID: 7N6I)23, which align with a global RMSD of 1.01 Å. ZnF motifs are indicated. i, Model of VchTniQ•TnsC interaction and its positioning relative to dsDNA, based on structural superpositions with the TniQ-bound TnsC complex from S. hofmannii (PDB ID: 7N6I; Methods)23, as shown in Fig. 5f. Protomers 3 and 4 of VchTnsC are shown in yellow and orange, respectively. Loops protruding from the N-terminal face of VchTnsC, which we found were essential for RNA-guided transposition (Extended Data Fig. 7b), are near the putative VchTniQ contact point. E69-D75 and A106–A113 are coloured dark orange and pink, respectively. j, An electrostatic rendering of VchTniQ-2 from the model shown in i suggests that DNA exiting the R-loop (dark grey) is well positioned to interact with a positively charged (blue) surface (left); TniQ-1 is shown in ribbon representation. DNA strand re-annealing may occur on this TniQ surface that is rich in lysine and arginine residues (right), guiding DNA from Cascade into the central pore of the VchTnsC heptamer. The 5′-CC-3′ PAM nucleotides are indicated by yellow dots.
Extended Data Table 1 |.
Cryo-EM data collection, refinement and validation statistics
| TnsC (single heptamer) (EMDB-24783) (PDB 7RZY) |
TnsC-DNA (single heptamer) (EMDB-26476) (PDB 7UFI) |
TnsC-DNA (double heptamer) (EMDB-26477) (PDB 7UFM) |
|
|---|---|---|---|
| Data collection and processing | |||
| Magnification | 105,000 | 130,000 | |
| Voltage (kV) | 300 | 300 | |
| Electron exposure (e–/Å2) | 53.55 | 73.94 | |
| Defocus range (μm) | −0.5/−2 | −0.5/−2 | |
| Pixel size (Å) | 0.678 | 0.6485 | |
| Symmetry imposed | C7 | C1 | C1 |
| Initial particle images (no.) | 2.4M | 4.2M | 4.2M |
| Final particle images (no.) | 84,309 | 125,371 | 125,833 |
| Map resolution (Å) | 3.54 | 3.46 | 3.92 |
| FSC threshold | 0.143 | 0.143 | 0.143 |
| Map resolution range (Å) | 3-5 | 3.5-6 | 4-8 |
| Refinement | |||
| Initial model used (PDB code) | Manual built | Manual built | Manual built |
| Model resolution (Å) | 3.9 | 4.2 | 5.3 |
| FSC threshold | 0.5 | 0.5 | 0.5 |
| Model resolution range (Å) | 3-5 | 3.5-5 | 4-8 |
| Map sharpening B factor (Å2) | −111 | −85.23 | −112.5 |
| Model composition | |||
| Non-hydrogen atoms | 17,458 | 18,319 | 36,392 |
| Protein residues | 2,111 | 2,177 | 4,354 |
| Ligands | ATP (7 molecules) | ATP (7 molecules) | ATP (14 molecules) |
| DNA nucleotides | - | 21 | 44 |
| B factors (Å2) | |||
| Protein | 84.23 | 74.49 | 129.21 |
| Ligand | 65.70 | 53.53 | 108.9 |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.014 | 0.014 | 0.0087 |
| Bond angles (°) | 1.73 | 1.81 | 1.82 |
| Validation | |||
| MolProbity score | 1.54 | 2.41 | 3.05 |
| Clashscore | 1.86 | 2.75 | 7.84 |
| Poor rotamers (%) | 0.74 | 1.22 | 1.92 |
| Ramachandran plot | |||
| Favored (%) | 88.40 | 90.38 | 89.00 |
| Allowed (%) | 98.57 | 99.49 | 11.00 |
| Disallowed (%) | 1.43 | 0.51 | 0 |
Supplementary Material
Acknowledgements
We thank N. Jaber and S. R. Pesari for laboratory support; D. J. Villano, X. Chen and C. Lu for discussions about ChIP–seq data analysis; D. Chen for statistics consultation; C. Lu for Covaris sonicator access; N. E. Sanjana for helpful discussions about crRNA library experiments; F. Vallese and O. B. Clarke for assistance with iSCAMS experiments and Refeyn instrument access; L. F. Landweber for qPCR instrument access; and the staff at the JP Sulzberger Columbia Genome Center for NGS support. Cryo-grids were generated and screening data were collected at the Columbia University Cryo-Electron Microscopy Center. Some cryo-EM data collection was performed at the Simons Electron Microscopy Center and the National Resource for Automated Molecular Microscopy, located at the New York Structural Biology Center, supported by grants from the Simons Foundation (SF349247) and NIH National Institute of General Medical Sciences (GM103310), with additional support from NYSTAR and the New York State Assembly Majority. J.T.G. was supported by the International Human Frontier Science Program Organization Postdoctoral Fellowship LT001117/2021-C. This research was supported by NIH grant DP2HG011650-01, NIH grant R01EB031935-01, a Pew Biomedical Scholarship and Sloan Research Fellowship, and a start-up package from the Columbia University Irving Medical Center Dean’s Office and the Vagelos Precision Medicine Fund (to S.H.S.).
Footnotes
Code availability
Custom scripts used for the described ChIP–seq data analyses are available at GitHub (https://github.com/sternberglab/Hoffmann_Kim_Beh_etal_2022).
Competing interests Columbia University has filed patent applications related to CRISPR-transposon systems, for which S.H.S. and P.L.H.V. are listed as inventors. S.H.S. is a co-founder and scientific advisor to Dahlia Biosciences, a scientific advisor to CrisprBits and Prime Medicine, and an equity holder in Dahlia Biosciences and CrisprBits.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information, details of author contributions and competing interests, and statements of data and code availability are available at https://doi.org/10.1038/s41586-022-05059-4.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-022-05059-4.
Data availability
Next-generation sequencing data are available at the National Center for Biotechnology Information (NCBI) Sequence Read Archive (Bio-Project accession: PRJNA759215) and the Gene Expression Omnibus (GSE183114). The published genome used for analyses was obtained from the NCBI (GenBank: CP001509.3). Maps and models for the apo-TnsC heptamer, DNA-bound single-ring complex and DNA-bound double-ring complex have been deposited at the Electron Microscopy Data Bank under accession codes EMDB-24783, EMDB-26476 and EMDB-26477, and the PDB under accession codes 7RZY, 7UFI and 7UFM, respectively. Other atomic models used in this study are available at the PDB under accession codes 7N6I (TniQ-bound ShoTnsC), 7M99 (ShoTnsC), 6VBW (VchTniQ–Cascade), 6V9P (VchTniQ) and 7MCS (EcoTnsC). Datasets generated and analysed in the current study are available from the corresponding authors on reasonable request.
References
- 1.Bainton RJ, Kubo KM, Feng J & Craig NL Tn7 transposition: target DNA recognition is mediated by multiple Tn7-encoded proteins in a purified in vitro system. Cell 72, 931–943 (1993). [DOI] [PubMed] [Google Scholar]
- 2.Klompe SE, Vo PLH, Halpin-Healy TS & Sternberg SH Transposon-encoded CRISPR–Cas systems direct RNA-guided DNA integration. Nature 571, 219–225 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Strecker J et al. RNA-guided DNA insertion with CRISPR-associated transposases. Science 365, 48–53 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peters JE Tn7. Microbiol. Spectr 2, MDNA3-0010-2014 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Koonin EV Viruses and mobile elements as drivers of evolutionary transitions. Philos. Trans. R. Soc. B 371, 20150442 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hickman AB & Dyda F Mechanisms of DNA Transposition. Microbiol. Spectr 3, MDNA3-0034-2014 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peters JE Targeted transposition with Tn7 elements: safe sites, mobile plasmids, CRISPR/Cas and beyond. Mol. Microbiol 112, 1635–1644 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sarnovsky RJ, May EW & Craig NL The Tn7 transposase is a heteromeric complex in which DNA breakage and joining activities are distributed between different gene products. EMBO J. 15, 6348–6361 (1996). [PMC free article] [PubMed] [Google Scholar]
- 9.Choi KY, Spencer JM & Craig NL The Tn7 transposition regulator TnsC interacts with the transposase subunit TnsB and target selector TnsD. Proc. Natl Acad. Sci. USA 111, E2858–E2865 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peters JE & Craig NL Tn7 recognizes transposition target structures associated with DNA replication using the DNA-binding protein TnsE. Gene Dev. 15, 737–747 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stellwagen AE & Craig NL Gain-of-function mutations in TnsC, an ATP-dependent transposition protein that activates the bacterial transposon Tn7. Genetics 145, 573–585 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuduvalli PN, Rao JE & Craig NL Target DNA structure plays a critical role in Tn7 transposition. EMBO J. 20, 924–932 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stellwagen AE & Craig NL Analysis of gain-of-function mutants of an ATP-dependent regulator of Tn7 transposition. J. Mol. Biol 305, 633–642 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Peters JE, Makarova KS, Shmakov S & Koonin EV Recruitment of CRISPR-Cas systems by Tn7-like transposons. Proc. Natl Acad. Sci. USA 114, E7358–E7366 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Faure G et al. CRISPR–Cas in mobile genetic elements: counter-defence and beyond. Nat. Rev. Microbiol 17, 513–525 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saito M et al. Dual modes of CRISPR-associated transposon homing. Cell 184, 2441–2453 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vo PLH et al. CRISPR RNA-guided integrases for high-efficiency, multiplexed bacterial genome engineering. Nat. Biotechnol 39, 480–489 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Halpin-Healy TS, Klompe SE, Sternberg SH & Fernández IS Structural basis of DNA targeting by a transposon-encoded CRISPR–Cas system. Nature 577, 271–274 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Snider J, Thibault G & Houry WA The AAA+ superfamily of functionally diverse proteins. Genome Biol. 9, 216 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Puchades C, Sandate CR & Lander GC The molecular principles governing the activity and functional diversity of AAA+ proteins. Nat. Rev. Mol. Cell Biol 21, 43–58 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holder JW & Craig NL Architecture of the Tn7 posttransposition complex: an elaborate nucleoprotein structure. J. Mol. Biol 401, 167–181 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen Y et al. Structural basis for DNA targeting by the Tn7 transposon. Nat. Struct. Mol. Biol 29, 143–151 (2022). [DOI] [PubMed] [Google Scholar]
- 23.Park J-U et al. Structural basis for target site selection in RNA-guided DNA transposition systems. Science 373, 768–774 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Querques I, Schmitz M, Oberli S, Chanez C & Jinek M Target site selection and remodelling by type V CRISPR-transposon systems. Nature 599, 497–502 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vo PLH, Acree C, Smith ML & Sternberg SH Unbiased profiling of CRISPR RNA-guided transposition products by long-read sequencing. Mob. DNA 12, 13 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ronning DR et al. The carboxy-terminal portion of TnsC activates the Tn7 transposase through a specific interaction with TnsA. EMBO J. 23, 2972–2981 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leenay RT & Beisel CL Deciphering, communicating, and engineering the CRISPR PAM. J. Mol. Biol 429, 177–191 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuscu C, Arslan S, Singh R, Thorpe J & Adli M Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol 32, 677–683 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Wu X et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol 32, 670–676 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cooper LA, Stringer AM & Wade JT Determining the specificity of cascade binding, interference, and primed adaptation in vivo in the Escherichia coli type I-E CRISPR-Cas system. mBio 9, e02100–17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thakore PI et al. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 12, 1143–1149 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Geen H, Henry IM, Bhakta MS, Meckler JF & Segal DJ A genome-wide analysis of Cas9 binding specificity using ChIP-seq and targeted sequence capture. Nucleic Acids Res. 43, 3389–3404 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bailey TL et al. MEME suite: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klompe SE et al. Evolutionary and mechanistic diversity of type I-F CRISPR-associated transposons. Mol. Cell 82, 616–628 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiao Y et al. Structure basis for directional R-loop formation and substrate handover mechanisms in type I CRISPR-Cas system. Cell 170, 48–60 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Semenova E et al. The Cas6e ribonuclease is not required for interference and adaptation by the E. coli type I-E CRISPR-Cas system. Nucleic Acids Res. 43, 6049–6061 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jung C et al. Massively parallel biophysical analysis of CRISPR-Cas complexes on next generation sequencing chips. Cell 170, 35–47 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen C-H et al. Improved design and analysis of CRISPR knockout screens. Bioinformatics 34, 4095–4101 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rutkauskas M et al. Directional R-loop formation by the CRISPR-Cas surveillance complex cascade provides efficient off-target site rejection. Cell Rep. 10, 1534–1543 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Qi LS et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Young G et al. Quantitative mass imaging of single biological macromolecules. Science 360, 423–427 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zivanov J et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mizuno N et al. MuB is an AAA+ ATPase that forms helical filaments to control target selection for DNA transposition. Proc. Natl Acad. Sci. USA 110, E2441–E2450 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davey MJ & O’Donnell M Replicative helicase loaders: ring breakers and ring makers. Curr. Biol 13, R594–R596 (2003). [DOI] [PubMed] [Google Scholar]
- 46.Jia N, Xie W, de la Cruz MJ, Eng ET & Patel DJ Structure–function insights into the initial step of DNA integration by a CRISPR–Cas–transposon complex. Cell Res. 30, 182–184 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arinkin V, Smyshlyaev G & Barabas O Jump ahead with a twist: DNA acrobatics drive transposition forward. Curr. Opin. Struct. Biol 59, 168–177 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walker DM, Freddolino PL & Harshey RM A well-mixed E. coli Genome: widespread contacts revealed by tracking Mu transposition. Cell 180, 703–716 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jackson RN, van Erp PB, Sternberg SH & Wiedenheft B Conformational regulation of CRISPR-associated nucleases. Curr. Opin. Microbiol 37, 110–119 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmiedeberg L, Skene P, Deaton A & Bird A A temporal threshold for formaldehyde crosslinking and fixation. PLoS ONE 4, e4636 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anzalone AV, Koblan LW & Liu DR Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol 38, 824–844 (2020). [DOI] [PubMed] [Google Scholar]
- 52.Bonocora RP & Wade JT Bacterial transcriptional control, methods and protocols. Methods Mol. Biol 1276, 327–340 (2015). [DOI] [PubMed] [Google Scholar]
- 53.Chen S, Zhou Y, Chen Y & Gu J fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramírez F et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Robinson JT et al. Integrative genomics viewer. Nat. Biotechnol 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Russo CJ & Passmore LA Ultrastable gold substrates for electron cryomicroscopy. Science 346, 1377–1380 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Suloway C et al. Automated molecular microscopy: the new Leginon system. J. Struct. Biol 151, 41–60 (2005). [DOI] [PubMed] [Google Scholar]
- 61.Rohou A & Grigorieff N CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zivanov J, Nakane T & Scheres SHW Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION-3.1. IUCrJ 7, 253–267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zivanov J, Nakane T & Scheres SHW A Bayesian approach to beam-induced motion correction in cryo-EM single-particle analysis. IUCrJ 6, 5–17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Abrishami V et al. Localized reconstruction in Scipion expedites the analysis of symmetry mismatches in cryo-EM data. Prog. Biophys. Mol. Biol 160, 43–52 (2021). [DOI] [PubMed] [Google Scholar]
- 65.Casañal A, Lohkamp B & Emsley P Current developments in Coot for macromolecular model building of electron Cryo-microscopy and crystallographic data. Protein Sci. 29, 1055–1064 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Afonine PV et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D 74, 531–544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Murshudov GN, Vagin AA & Dodson EJ Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D 53, 240–255 (1997). [DOI] [PubMed] [Google Scholar]
- 68.Brown A et al. Tools for macromolecular model building and refinement into electron cryo-microscopy reconstructions. Acta Crystallogr. D 71, 136–153 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Prisant MG, Williams CJ, Chen VB, Richardson JS & Richardson DC New tools in MolProbity validation: CaBLAM for CryoEM backbone, UnDowser to rethink “waters,” and NGL Viewer to recapture online 3D graphics. Protein Sci. 29, 315–329 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Baker NA, Sept D, Joseph S, Holst MJ & McCammon JA Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl Acad. Sci. USA 98, 10037–10041 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Next-generation sequencing data are available at the National Center for Biotechnology Information (NCBI) Sequence Read Archive (Bio-Project accession: PRJNA759215) and the Gene Expression Omnibus (GSE183114). The published genome used for analyses was obtained from the NCBI (GenBank: CP001509.3). Maps and models for the apo-TnsC heptamer, DNA-bound single-ring complex and DNA-bound double-ring complex have been deposited at the Electron Microscopy Data Bank under accession codes EMDB-24783, EMDB-26476 and EMDB-26477, and the PDB under accession codes 7RZY, 7UFI and 7UFM, respectively. Other atomic models used in this study are available at the PDB under accession codes 7N6I (TniQ-bound ShoTnsC), 7M99 (ShoTnsC), 6VBW (VchTniQ–Cascade), 6V9P (VchTniQ) and 7MCS (EcoTnsC). Datasets generated and analysed in the current study are available from the corresponding authors on reasonable request.