

Abstract
Allylations are practical transformations that forge C–C bonds while introducing an alkene for further chemical manipulations. Here, we report a photoenzymatic allylation of α-chloroamides with allyl silanes using flavin-dependent ‘ene’-reductases (EREDs). An engineered ERED can catalyze annulative allylic alkylation to prepare 5, 6, and 7-membered lactams with high levels of enantioselectivity. Ultrafast transient absorption spectroscopy indicates that radical termination occurs via β-scission of the silyl group to afford a silyl radical, a distinct mechanism by comparison to traditional radical allylations involving allyl silanes. Moreover, this represents an alternative strategy for radical termination using EREDs. This mechanism was applied to intermolecular couplings involving allyl sulfones and silyl enol ethers. Overall, this method highlights the opportunity for EREDs to catalyze radical termination strategies beyond hydrogen atom transfer.
Keywords: biocatalysis, flavin, radical, allylation, photochemistry
Graphical Abstract
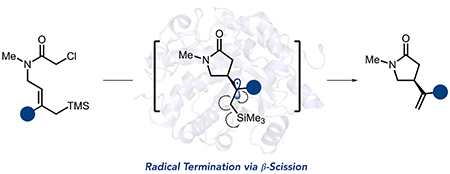
Asymmetric Csp3–Csp3 bond formation is indispensable for constructing societally essential molecules.1 Consequently, numerous catalytic methods have been developed to facilitate their construction.2 Among these, reactions involving open-shell radical intermediates are desirable because they have low activation barriers and can form sterically congested centers.3 However, strategies for rendering these transformations asymmetric remain underdeveloped compared to reactions involving other types of reactive intermediates.4
Enzymes are ideal catalysts for asymmetric synthesis because they can precisely orient reactive intermediates, and their activity can be optimized using directed evolution.5 However, biocatalysts are often restricted to their natural reaction mechanisms, limiting their ability to address selectivity challenges in chemical synthesis.6 An ongoing goal in the field has been to develop strategies to expand the synthetic capabilities of enzymes.7 We recently demonstrated that flavin-dependent ‘ene’-reductases (EREDs) could catalyze non-natural C–C bond-forming reactions involving radical intermediates.8 In nature, EREDs catalyze the reduction of activated alkenes via a hydride transfer mechanism.9 We found that these enzymes will template charge-transfer (CT) complexes between various alkyl halides and the reduced flavin hydroquinone (FMNhq) cofactor. Irradiation with visible light promotes an electron from the cofactor to the substrate. Upon mesolytic cleavage of the carbon–halogen bond, an alkyl radical is formed that can react with an alkene to forge a new C–C bond with high selectivity. Thus far, our studies have focused on radical termination via hydrogen atom transfer (HAT) from flavin semiquinone (FMNsq) (Figure 1a).10 To expand the synthetic utility of these catalysts, we sought to develop alternative radical termination mechanisms.
Figure 1.
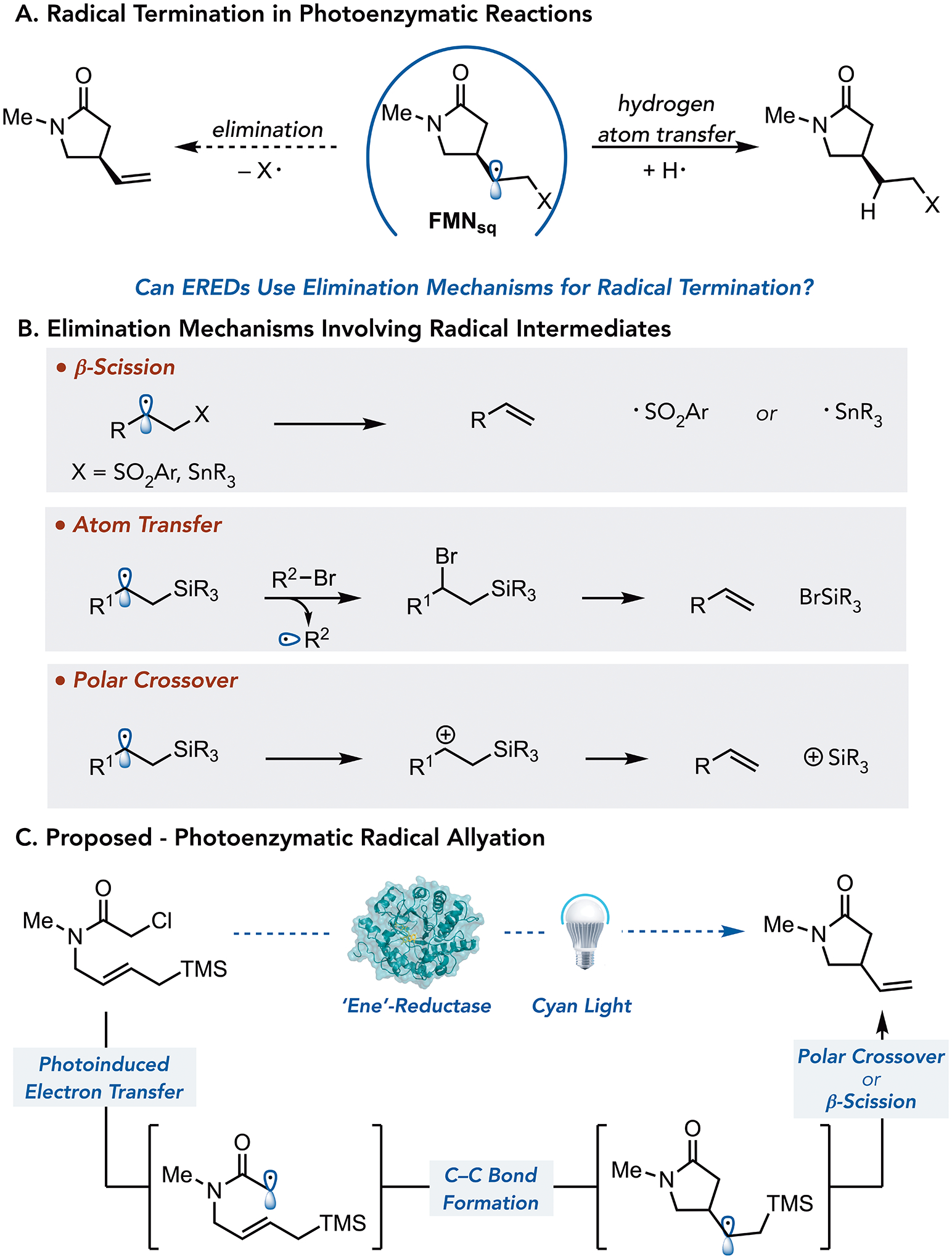
Mechanisms of Radical Allylation
Radical allylations are attractive reactions because they form a new C–C bond while also introducing a handle for subsequent functionalizations.11 The most common reagents for these reactions are allyl stannanes, silanes, and sulfones. In these reactions, the reagent largely dictates the mechanism of elimination. For allyl stannanes and sulfones, radical termination occurs via β-scission to produce stannyl or sulfinyl radicals (Figure 1b).12 As C–Si (76 kcal/mol) bonds are stronger than C–Sn (45 kcal/mol) or C–SO2Ar (65 kcal/mol), allyl silanes typically undergo different elimination mechanisms.13 In atom transfer reactions, the alkyl radical is trapped by a halide, followed by thermal elimination to form an alkene and halosilane (Figure 1b).14 Alternatively, the presence of an oxidant can enable a radical-polar crossover mechanism where the radical is oxidized to the β-silyl cation, which readily eliminates (Figure 1b).15 We hypothesized that a β-scission or polar crossover mechanism would be available to EREDs. However, it was unclear whether either of these mechanisms could be competitive with HAT (Figure 1c).
We tested the viability of the proposed reactivity in the cyclization of allyl silane 1 to afford γ-lactam 2. We found that GluER-T36A with an NADPH turnover system consisting of glucose as a terminal hydride source and glucose dehydrogenase (GDH) under visible light irradiation, afforded the desired product in 64% yield and >99:1 er, with < 5% yield of the reductive cyclization product 3 (Table 1, entry 1). A brief screening of GluER variants previously engineered in-house revealed that GluER-T36A-K317M-Y343F (GluER-G6) was the optimal enzyme (Table 1, entry 2).16 A control experiment confirmed that the cofactor turnover system is required to achieve high yields (Table 1, entry 3). Continuous light irradiation of GluER-G6 in buffer, without any turnover system present, is sufficient to generate both FMNhq and FMNsq in the protein active site (Supplemental Figure 9).17 The addition of cofactor turnover system favors formation of FMNhq, the oxidation state responsible for radical initiation. A control experiment confirms that radical initiation cannot occur from ground state FMNhq (Table 1, entry 4).18 Additionally, when GluER-G6 is photoreduced for 72 hours to generate a mixture of the FMNsq and FMNhq and then substrate is added after photoirradition was stopped, no product is formed (Table 1, entry 5). These results indicate that radical initiation does not occur from either the ground state FMNsq or FMNhq. While initial screens were run with six equivalents of glucose, we found that two equivalents provided comparable yields (Table 1, entry 6). Finally, we ran the reaction on a preparative scale using 0.75 mol % of GluER-G6 and isolated the desired lactam product in 46% yield with >99:1 er (Table 1, entry 7).
Table 1.
Reaction Optimization
| Entry | Deviation from “Initial Conditions” | Yield (%) | e.r. |
|---|---|---|---|
| 1 | none | 64 | 99:1 |
| 2 | GluER-G6 instead of GluER-T36A | 92 | 99:1 |
| 3 | GluER-G6 and no cofactor turnover system | 72 | 99:1 |
| 4 | GluER-G6 and no light | 0 | n.d. |
| 5 | GluER-G6 without cofactor turnover system and photoreduction of the enzyme prior to addition of the substrate | 0 | n.d |
| 6 | GluER-G6 and 2 equiv. of Glucose | 92 | 99:1 |
| 7 | 0.24 mmol scale at 43.6 mM using KRED P103 | 46 | 99:1 |
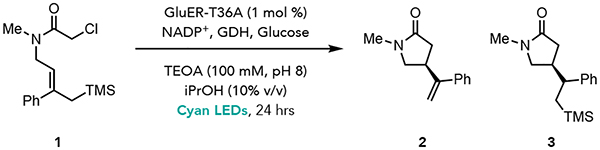
Reaction conditions: 1 (10 μmol, 3.1 mg), purified GluER enzyme (1 mol%, 100 nmol), 100 mM buffer (18 mM final substrate concentration), iPrOH (10 % v/v), glucose (60 μmol), GDH-105 (10 wt%, 0.3 mg/rxn), NADP+ (2 mol%), 24 hours, 25 °C. Yields determined by HPLC using a calibration curve for Entries 1–6. Reported yield for Entry 7 was for isolated and purified material. See Supplemental Information for detailed experimental procedure and additional optimization studies.
With the optimized conditions in hand, we explored the scope of the transformation (Figure 2). GluER-G6 accommodates substituents at the ortho-, meta-, and para-positions of the aromatic ring (Figure 2, 4–7). Electron-rich substrates are more reactive than electron-deficient ones. However, the enantioselectivity is high in all cases. Unsubstituted alkenes are also effective for both 5-exo-trig and 6-exo-trig cyclization, affording products in high yields but with modest levels of enantioselectivity (Figure 2, 8, 9, 10). We attribute the low enantioselectivity to the lack of substituents on the alkene moiety to help orient the substrate within the protein active site. This enzyme can also catalyze 7-exo-trig cyclizations in promising yields and enantioselectivities. Beyond aromatic substituents, aliphatic substituents are also tolerated. While the methyl-substituted substrates are only modestly selective (Figure 2, 12), larger i-propyl and c-hexyl substituents provide synthetically useful levels of enantioselectivity (Figure 2, 13, and 14). Furthermore, we found that heterocycles, such as furan, were also well tolerated (Figure 2, 15).
Figure 2.
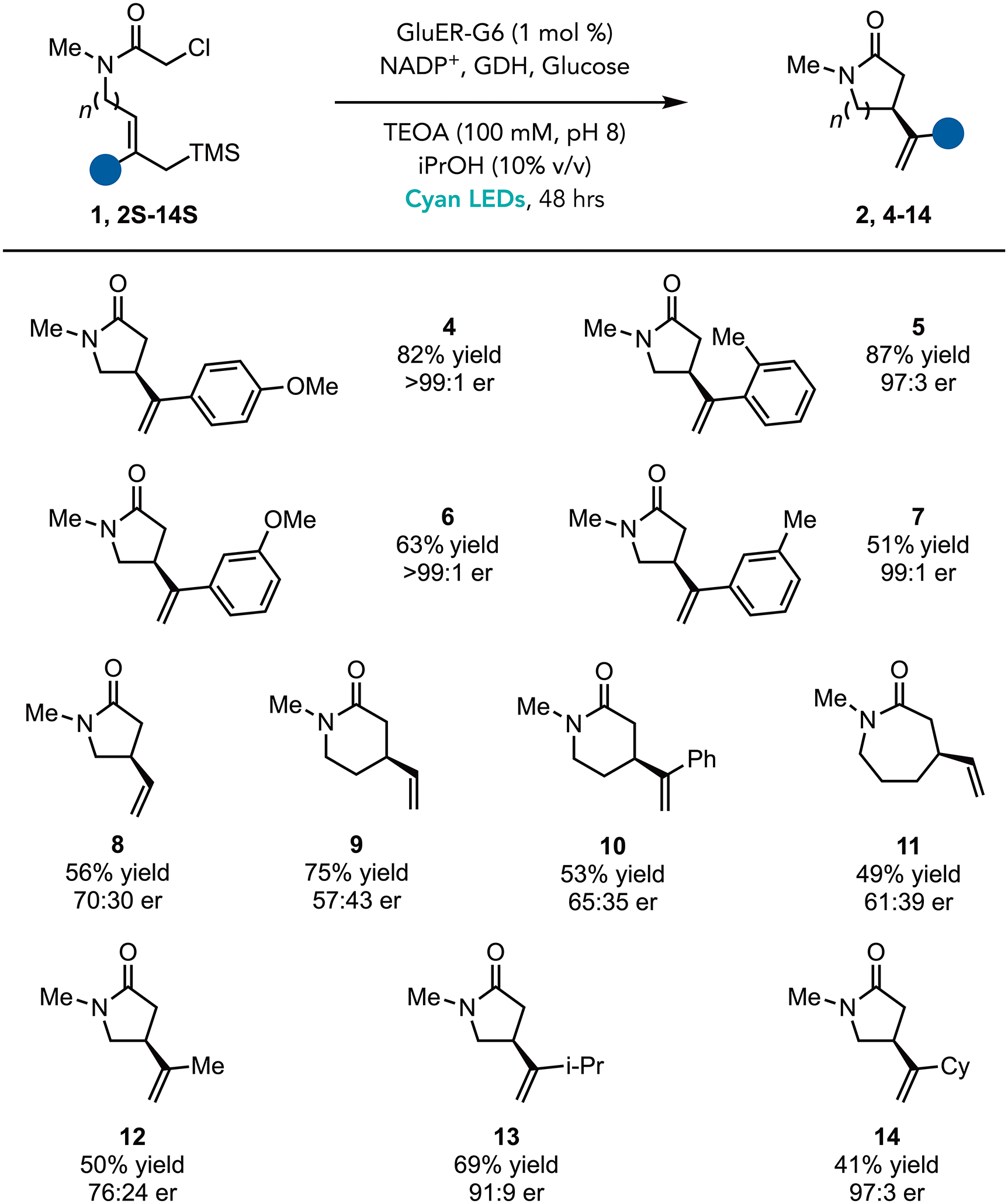
Substrate Scope
a. Reaction conditions: amide (20 μmol), purified GluER enzyme (1 mol%, 200 nmol), 100 mM buffer (18 mM final substrate concentration), iPrOH (10 % v/v), glucose (40 μmol), GDH-105 (10 wt%, 0.4–7.5 mg/rxn), NADP+ (2 mol%), 48 hours, 25 °C. Yields were determined by NMR analysis using trimethoxy benzene as an internal standard. See Supplemental Information for detailed experimental procedure and additional optimization studies.
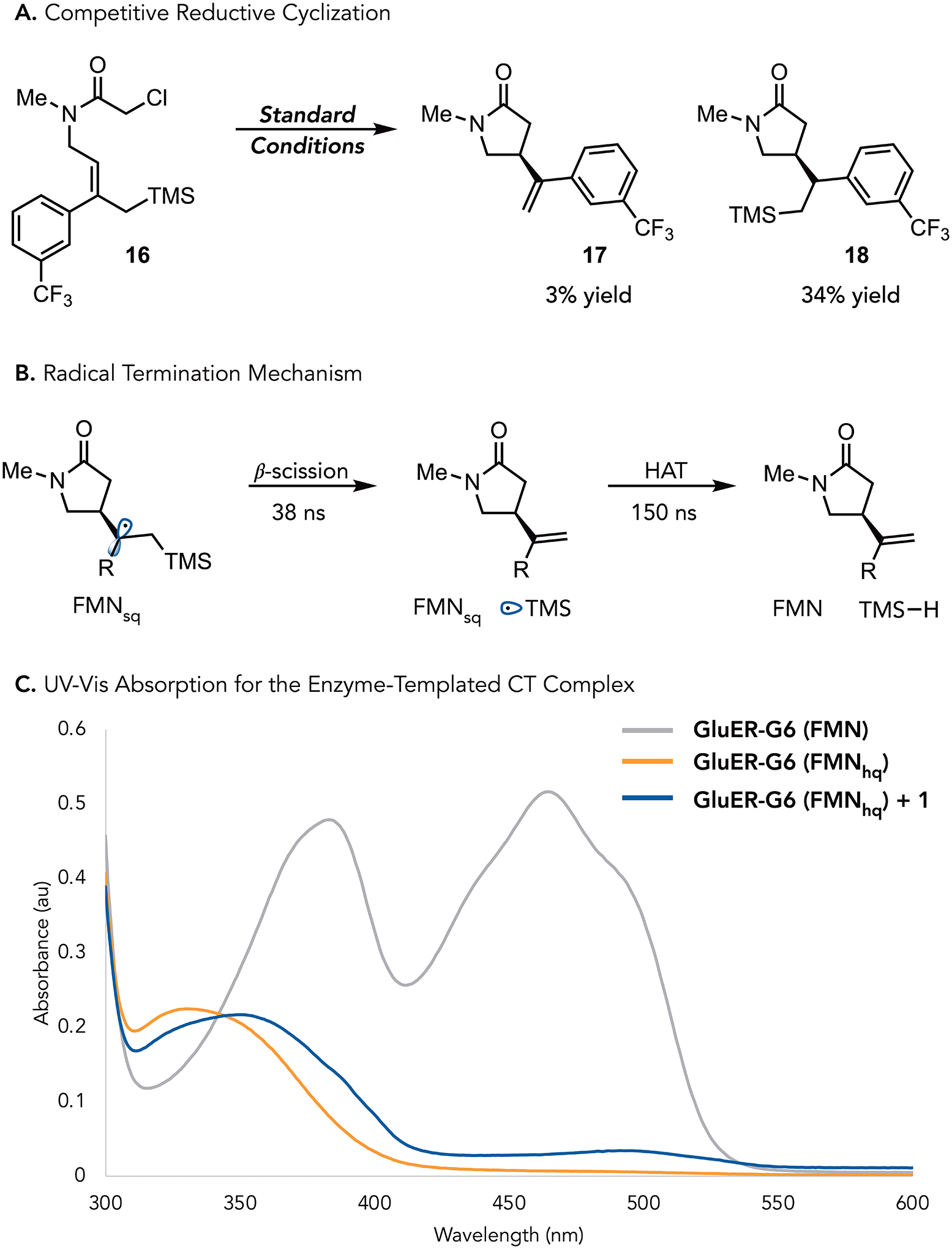
While evaluating the substrate scope, we found that aromatic substrates containing electron-withdrawing substituents provided lower yields of the desired product compared to those with electron-donating substituents. For example, meta-CF3 amide 16 afforded the allylated product 17 in only 3% yield. Upon further analysis, we found that the reductively cyclized product 18 is formed in 34% yield (Figure 3a). While performing a direct Hammett analysis might be difficult because substitution impacts substrate binding, this result suggests that more electrophilic radicals favor radical termination via hydrogen atom transfer rather than elimination.19
Figure 3.
Electronic Effects on Reaction Outcome
Next, we sought to interrogate the mechanism of radical termination. We envisioned two possibilities, i) a radical-polar crossover mechanism where the β-silyl radical is oxidized by FMNsq, forming FMNhq and a β-silyl cation that can eliminate to form the alkene and silanol, or ii) β-scission of the β-silyl radical producing the product and a silyl radical which can abstract a hydrogen atom from FMNsq to produce oxidized FMN. As the final flavin oxidation state differs between these two mechanisms, they can be distinguished using transient absorption spectroscopy.
Transient absorption spectroscopy (TAS) studies were conducted in a sealed quartz cuvette containing GluER-G6 reduced with sodium dithionite and chloroamide 1. The sample was excited with a 370 nm pulse and UV-Vis probe spectrum (400 – 750nm) was taken over various pump-probe delay times (maximum delay of 1000 ns). As these spectra contain multiple species, deconvolution was performed using global analysis and compared to the results of data obtained in our previous ERED catalyzed reductive cyclizations.10 The first-time component is mesolytic cleavage occurring with a lifetime of 10 ps, mirroring what was observed in the reductive cyclizations. This is followed by the growth a broad spectral feature that decays with a lifetime of 38 ns. As we know cyclization is fast (<700 ps) for structurally similar substrates used for reductive cyclizations, we attribute this feature to the TMS radical group undergoing β-scission.16 The extended lifetime of the radical intermediate is potentially due to stabilization of the radical by the electropositive β-silyl group.20 The spectrum formed after β-scission is consistent with the absorption profile of the neutral flavin semiquinone. This feature persists with a lifetime of 150 ns before decaying to the flavin quinone. This suggests that the silyl radical formed after β-scission abstracts a hydrogen atom from the neutral flavin semiquinone to form silane and flavin quinone (Figure 3b). In traditional radical chemistry, this mechanism is disfavored because of the strength of the C–Si bond. It is possible that this mechanism is available under biocatalytic conditions through the intermediacy of a silicon-ate complex.
Our previous studies indicate that this initiation event occurs via photoexcitation of an enzyme templated charge transfer complex that forms between the substrate and FMNhq. To confirm that this mechanism remains the case for these substrates, we prepared a sample with reduced GluER-G6 (containing FMNhq) and added the substrate. Consistent with our previous studies, we observed a new absorption band at 495 nm, suggesting the intermediacy of a CT complex (Figure 3c).
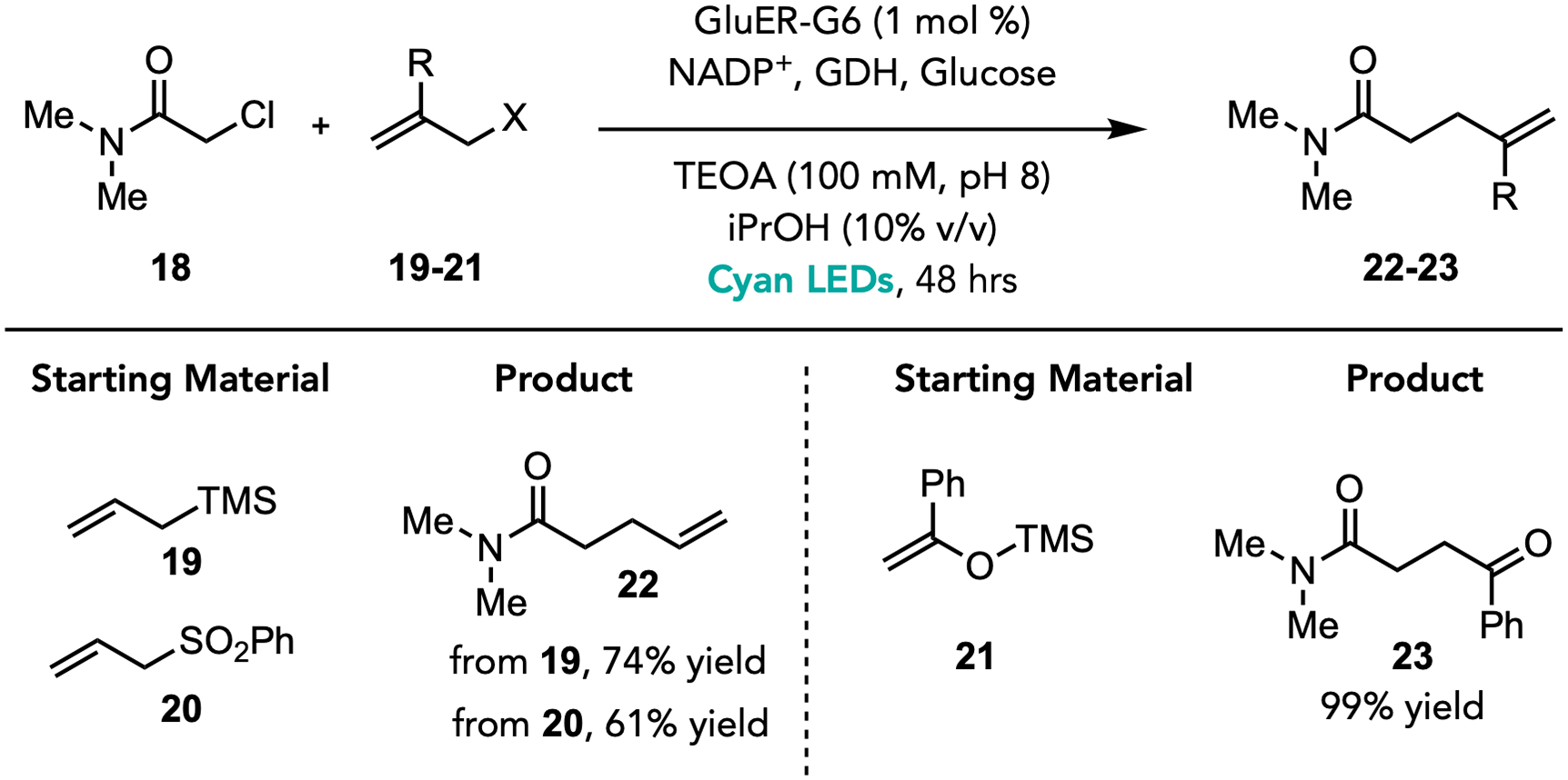
Having established that EREDs can catalyze intramolecular allylations, we explored whether they could facilitate intermolecular reactions. Using GluER-G6 under the standard reaction conditions, we found that chloroacetamide 19 could be coupled to trimethylallylsilane 20 to afford the γ,δ-unsaturated amide 23 in 61% yield (Figure 4). Beyond allyl silanes, silyl enol ether 22 is reactive and affords a 1,4-dicarbonyl product 24 in nearly quantitative yield. Finally, we hypothesized that allyl sulfones could be effective reagents for radical allylation because of their propensity to undergo β-scission elimination. When amide 19 is supplied with allylsulfone 21, the allylated product 23 is formed in 73% yield.21 Collectively, these examples suggest the generality of this radical termination mechanism.
Figure 4.
Intermolecular Allylation
a. Reaction conditions: 18 (20 μmol, 2.06μL), 19–21 (80 μmol) purified GluER enzyme (1 mol%, 200 nmol), 100 mM buffer (18 mM final substrate concentration), iPrOH (10 % v/v), glucose (40 μmol), GDH-105 (10 wt%, 0.4–7.5 mg/rxn), NADP+ (2 mol%), 48 hours, 25 °C. Yields were determined by HPLC using calibration curves. See Supplemental Information for detailed experimental procedure and additional optimization studies.
In conclusion, we have demonstrated that EREDs can catalyze asymmetric allylations using allyl silanes and allyl sulfones. Radical termination occurs via a β-scission mechanism that is competitive with hydrogen atom transfer from FMNsq. Beyond offering a new mechanism, this substitution pattern significantly expands the lifetime of radical intermediates within the protein active site. These observations offer new insights into non-natural chemistry with EREDs and unlock synthetic opportunities of this enzyme-catalyzed platform to enable novel, selective radical-based transformations as solutions to unaddressed selectivity challenges in radical chemistry.
Supplementary Material
ACKNOWLEDGMENT
Financial support for reaction development and evaluation of the scope was provided by the NIH (R01 GM127703). Mechanistic studies were supported by the Division of Chemical Science, Geosciences, and Biosciences, Office of Basic Energy Sciences of the U.S. Department of Energy through Grant DE-SC0019370. NL was supported by an NSF-GFRP. DGO was supported by the Postgraduate Scholarships Doctoral Program of NSERC. This work made use of the Cornell University NMR Facility, which is supported, in part, by the NSF through MRI Award CHE-1531632. The authors thank Ivan Keresztes for assistance in analyzing and collecting spectra.
Footnotes
The Supporting Information is available free of charge at https://pubs.acs.org/
Supplemental figures, detailed experimental procedures, spectroscopy information, characterization data (NMR spectra and HPLC traces) (PDF)
The authors declare no competing financial interest.
REFERENCES
- 1.(a) Williams K; Lee E Importance of Drug Enantiomers in Clinical Pharmacology. Drugs 1985, 30, 333–354. [DOI] [PubMed] [Google Scholar]; (b) Lovering F, Bikker J & Humblet C Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]
- 2.(a) Bar G; Parsons AF Stereoselective radical reactions. Chem. Soc. Rev 2003, 32, 251–263. [DOI] [PubMed] [Google Scholar]; (b) Choi J; Fu GC Transition metal-catalyzed alkyl-alkyl bond formation: another dimension in cross-coupling chemistry. Science 2017, 356, eaaf7230. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chen Z; Rong M, Nie J, Zhu X, Shi B Ma J Catalytic alkylation of unactivated C(sp3)–H bonds for C(sp3)–C(sp3) bond formation. Chem. Soc. Rev 2019, 48, 4921–4942. [DOI] [PubMed] [Google Scholar]; (d) Trost BM, Vranken DL Asymmetric Transition Metal-Catalyzed Allylic Alkylations. Chem Rev 1996, 96, 395–422. [DOI] [PubMed] [Google Scholar]; (e) Meggers E Asymmetric catalysis activated by visible light. Chem. Commun 2015, 51, 3290–3301. [DOI] [PubMed] [Google Scholar]
- 3.(a) Studer A; Curran DP Catalysis of Radical Reactions: A Radical Chemistry Perspective. Angew. Chem. Int. Ed 2016, 55, 58–102. [DOI] [PubMed] [Google Scholar]; (b) Prier CK; Ranik DA; MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Romero NA; Nicewicz DA Organic Photoredox Catalysis. Chem. Rev 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]; (d) Skubi KL; Blum TR; Yoon TP Dual catalysis strategies in photochemical synthesis. Chem. Rev 2016, 116, 10035–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Twilton J; Le C; Zhang P; Shaw MH; Evans RW; MacMillan DWC The merger of transition metal and photocatalysis. Nat. Chem. Rev 2017, 1, 0052. [Google Scholar]
- 4.(a) Sibi MP; Manyem S; Zimmerman J Enantioselective Radical Processes. Chem. Rev 2003, 103, 3263–3295. [DOI] [PubMed] [Google Scholar]; (b) Miyabe H; Kawashima A; Yoshioka, Eito; Kohtani, S. Progress in Enantioselective Radical Cyclizations. Chem. Eur. J 2017, 23, 6225–6236. [DOI] [PubMed] [Google Scholar]; (c) Nicewicz DA; MacMillan DWC Merging Photoredox Catalysis with Organocatalysis: the Direct Asymmetric Alkylation of Aldehydes. Science 2008, 322, 77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Du J; Skubi KL; Schultz DM; Yoon T A Dual-Catalysis Approach to Enantioselective [2 + 2] Photocycloadditions Using Visible Light. Science 2014, 344, 392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Huo H; Shen X; Wang C; Zhang L; Rose P; Chen L; Harms K;Marsch M; Hilt G; Meggers E Asymmetric photoredox transition-metal catalysis activated by visible light. Nature 2014, 515, 100–103. [DOI] [PubMed] [Google Scholar]; (f) Rono LJ; Yayla HG; Wang DY; Armstrong MF; Knowles RR Enantioselective Photoredox Catalysis Enabled by Proton-Coupled Electron Transfer: Development of an Asymmetric Aza-Pinacol Cyclization. J. Am. Chem. Soc 2013, 135, 17735–17738. [DOI] [PubMed] [Google Scholar]; (g) Blum TR; Miller ZD; Bates DM; Guzei IA; Yoon TP Enantioselective photochemistry through Lewis acid–catalyzed triplet energy transfer. Science 2016, 354, 1391–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Yi Dong; Bayer T; Badenhorst CPS; Wu S; Doerr M; Hohne M; Bornscheuer UT Recent trends in biocatalysis Chem. Soc. Rev 2021, 50, 8003–8049. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Devine PN, Howard RM, Kumar R; Thompson MP; Truppo MD; Turner NJ Extending the application of biocatalysis to meet the challenges of drug development. Nat. Rev. Chem 2018, 2, 409–421. [Google Scholar]; (c) Savile CK; Janey JM; Mundorff EC; Moore JC; Tam S; Jarvis WR; Colbeck JC; Krebber A; Fleitz FJ; Brands J; Devine PN; Huisman GW; Hughes GJ Biocatalytic Asymmetric Synthesis of Chiral Amines from Ketones Applied to Sitagliptin Manufacture. Science 2010, 329, 305–309. [DOI] [PubMed] [Google Scholar]; (d) Hall M Enzymatic strategies for asymmetric synthesis. RSC Chem. Biol 2021, 2, 958–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Sheldon RA; Brady D; Bode ML The Hitchhiker’s guide to biocatalysis: recent advances in the use of enzymes in organic synthesis. Chem. Sci 2020, 11, 2587–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schmidt NG; Eger E; Kroutil W Building Bridges: Biocatalytic C–C-Bond Formation toward Multifunctional Products. ACS Catal 2016, 6, 4286–4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Hammer SC; Knight AM; Arnold FH Design and evolution of enzymes for non-natural chemistry. Curr. Opin. Green Sustain. Chem 2017, 7, 23–30. [Google Scholar]; (b) Schwizer F; Okamoto Y; Heinisch T; Gu Y; Pellizzoni MM; Lebrun V; Reuter R; Köhler V; Lewis JC; Ward TR Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem Rev 2018, 118, 142–231. [DOI] [PubMed] [Google Scholar]; (c) Bornscheuer UT The fourth wave of biocatalysis is approaching. Phil. Trans. T. Soc. A 2017, 376.20170063. [DOI] [PubMed] [Google Scholar]
- 8.(a) Black MJ; Biegasiewicz KF; Meichan AJ; Oblinsky DG; Kudisch B; Scholes GD; Hyster TK Asymmetric redox-neutral radical cyclization catalysed by flavin-dependent ‘ene’-reductases. Nat. Chem 2020, 12, 71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Clayman PD; Hyster TK Photoenzymatic Generation of Unstabilized Alkyl Radicals: An Asymmetric Reductive Cyclization. J. Am. Chem. Soc 2020, 142, 15673–15677. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gao X; Turek-Herman JR; Choi YJ, Cohen RD; Hyster TK Photoenzymatic Synthesis of α-Tertiary Amines by Engineered Flavin-Dependent “Ene”-Reductases. J. Am. Chem. Soc 2021, 143, 19643–19647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roy TK; Sreedharan R; Ghosh P; Gandhi T; Maiti D Ene-Reductase: A Multifaceted Biocatalyst in Organic Synthesis Chem. Eur. J 2022. e202103949. [DOI] [PubMed] [Google Scholar]
- 10.(a) Biegasiewicz KF; Cooper SJ; Gao X; Oblinsky DG; Kim JH; Garfinkle SE; Joyce LA; Sandoval BA; Scholes GD; Hyster TK Photoexcitation of flavoenzymes enables a stereoselective radical cyclization Science 2019, 364, 1166–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Page C; Cooper SJ; DeHovitz JS; Oblinsky DG; Biegasiewicz KF; Antropow AH; Armbrust KW; Ellis JM; Hamann LG; Horn EJ; Oberg KM; Scholes G; Hyster TK Quaternary Charge-Transfer Complex Enables Photoenzymatic Intermolecular Hydroalkylation of Olefins. J. Am. Chem. Soc 2021, 143, 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Huang X; Wang B; Wang Y; Jiang G; Feng J; Zhao H Photoenzymatic enantioselective intermolecular radical hydroalkylation. Nature 2020, 584, 69–74. [DOI] [PubMed] [Google Scholar]
- 11.(a) Huang M; Bellotti P; Glorius F Transition metal-catalysed allylic functionalization reactions involving radicals. Chem. Soc. Rev 2020, 49, 6186–6197. [DOI] [PubMed] [Google Scholar]; (b) Tucker JW; Nguyen JD; Narayanam JMR; Krabbe SW; Stephenson CRJ Tin-free radical cyclization reactions initiated by visible light photoredox catalysis. Chem. Commun 2010, 46, 4985. [DOI] [PubMed] [Google Scholar]; (c) Mastrachhio A; Warkentin AA; Walji AM; MacMillan DWC Direct and enantioselective α-allylation of ketones via singly occupied molecular orbital (SOMO) catalysis. Proc. Natl. Acad. Sci. U.S.A 2010, 107, 20648–20651. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Pham PV; Ashton K; MacMillan DWC The intramolecular asymmetric allylation of aldehydes via organo-SOMO catalysis: A novel approach to ring construction. Chem. Sci 2011, 2, 1470–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Mizuta S; Engle KM; Verhoog S; Galicia-López O; O’Duill M; Médebielle, Wheelhouse K; Rassias G; Thompson AL; Gouverneur V Trifluoromethylation of Allylsilanes under Photoredox Catalysis. Org. Lett 2013, 15, 1250–1253. [DOI] [PubMed] [Google Scholar]
- 12.(a) Quiclet-Sire B; Zard SZ New Radical Allylation Reaction. J. Am. Chem. Soc 1996, 118, 1209–1210. [Google Scholar]; (b) Sibi MP; Ji J Acyclic Stereocontrol in Radical Reactions. Diastereoselective Radical Addition/Allylation of N-Propenoyloxazolidinone. J. Org. Chem 1996, 61, 6090–6091. [DOI] [PubMed] [Google Scholar]; (c) Curran DP; Chen MH; Spletzer E; Seong CM; Chang CT Atom transfer cyclization reactions of hex-5-ynyl iodides: synthetic and mechanistic studies. J. Am. Chem. Soc 1989, 111, 8872–8878. [Google Scholar]; (d) Structural and Chemical Properties of Silyl Radicals. Chatgilialoglu, C. Chem. Rev 1995, 95, 1229. [Google Scholar]
- 13.Luo Y-R; Cheng J-P in CRC Handbook of Chemistry & Physics 94th Edn (ed. Haynes WM) 9–65 (CRC, 2013). [Google Scholar]
- 14.Porter NA; Wu JH; Zhang G; Reed AD Enantioselective Free Radical Allyl Transfers from Allylsilanes Promoted by Chiral Lewis Acids. J. Org. Chem 1997, 62, 6702–6703. [Google Scholar]
- 15.(a) Pitzer L; Schwarz JL; Glorius F Reductive radical-polar crossover: traditional electrophiles in modern radical reactions. Chem Sci 2019, 10, 8285–8291. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sharma S; Singh J; Sharma A Visible Light Assisted Radical-Polar/Polar-Radical Crossover Reactions in Organic Synthesis. Adv. Synth. Catal 2021,363, 3146–3169. [Google Scholar]
- 16.Nicholls BT; Oblinsky DG; Kurtoic SI; Grosheva D; Ye Y; Scholes GD; Hyster TK Engineering a Non-Natural Photoenzyme for Improved Photon Efficiency. Angew. Chem. Int. Ed 2022, 61 e202113842. [DOI] [PubMed] [Google Scholar]
- 17.Massey V; Stankovich M; Hemmerich P Light-Mediated Reduction of Flavoproteins with Flavins as Catalysts. Biochemistry 1978, 17, 1–8. [DOI] [PubMed] [Google Scholar]
- 18.Fu H; Lam H; Emmanuel MA; Kim J; Sandoval B; Hyster TK Ground-State Electron Transfer as an Initiation Mechanism for Biocatalytic C–C Bond Forming Reactions. J. Am. Chem. Soc 2021, 143, 9622–9629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Vleeschouwer FD; Speybroeck VV; Waroquier M; Geerlings P; Proft FD Electrophilicity and Nucleophilicity Index for Radicals. Org Lett 2007, 9, 14, 2721–2724. [DOI] [PubMed] [Google Scholar]; (b) Fisher H; Radom L Factors Controlling the Addition of Carbon-Centered Radicals to Alkenes—An Experimental and Theoretical Perspective. Angew. Chem. Int. Ed 2001, 40, 1340–1371. [DOI] [PubMed] [Google Scholar]; (c) Héberger K; Lopata AJ Assessment of Nucleophilicity and Electrophilicity of Radicals, and of Polar and Enthalpy Effects on Radical Addition Reactions. Org. Chem 1998, 63, 8646–8653. [Google Scholar]
- 20.Parasee F; Senarathna MC; Kannangara PB; Alexander SN; Arche PDE; Weilin E Radical philicity and its role in selective organic transformations. Nat. Chem. Rev 2021, 5, 486–499. [DOI] [PubMed] [Google Scholar]
- 21. Replacing the TMS group with phenylsulfone for unsubstituted 5 exo substrate 8S-B affords product in 25% yield with 78:22 er.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.