

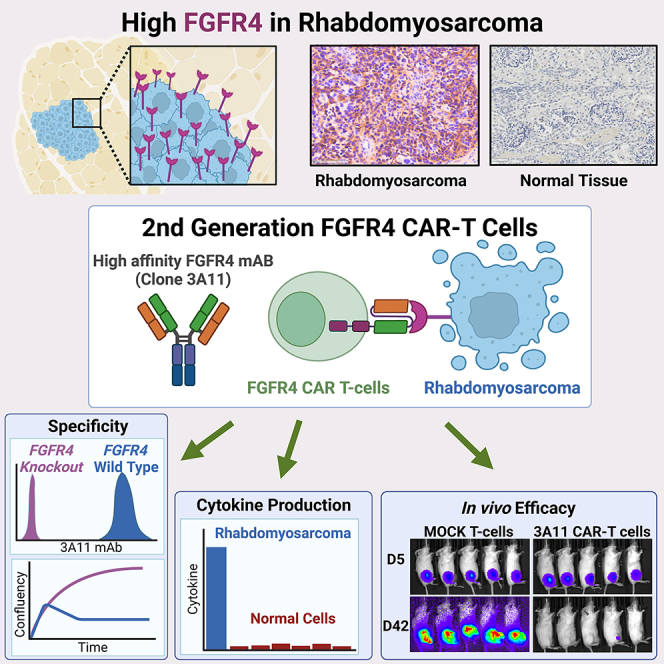
Summary
Pediatric patients with relapsed or refractory rhabdomyosarcoma (RMS) have dismal cure rates, and effective therapy is urgently needed. The oncogenic receptor tyrosine kinase fibroblast growth factor receptor 4 (FGFR4) is highly expressed in RMS and lowly expressed in healthy tissues. Here, we describe a second-generation FGFR4-targeting chimeric antigen receptor (CAR), based on an anti-human FGFR4-specific murine monoclonal antibody 3A11, as an adoptive T cell treatment for RMS. The 3A11 CAR T cells induced robust cytokine production and cytotoxicity against RMS cell lines in vitro. In contrast, a panel of healthy human primary cells failed to activate 3A11 CAR T cells, confirming the selectivity of 3A11 CAR T cells against tumors with high FGFR4 expression. Finally, we demonstrate that 3A11 CAR T cells are persistent in vivo and can effectively eliminate RMS tumors in two metastatic and two orthotopic models. Therefore, our study credentials CAR T cell therapy targeting FGFR4 to treat patients with RMS.
Keywords: rhabdomyosarcoma, FGFR4, CAR T cell therapy, specific cytotoxicity
Graphical abstract
Highlights
-
•
FGFR4 is highly expressed in rhabdomyosarcoma and lowly expressed in healthy human tissue
-
•
FGFR4 is a downstream target of the PAX3-FOXO1 fusion oncogene
-
•
FGFR4 CAR T cells demonstrate no cytokine release against primary human cells
-
•
FGFR4 CAR T cells eliminate metastatic and intramuscular solid RMS tumors in vivo
Tian et al. develop a potent clinical-grade CAR targeting FGFR4 that is highly expressed in rhabdomyosarcoma. FGFR4 CAR T cells show specific cytotoxicity against RMS cell lines and are non-reactive to healthy human primary cells. These CAR T cells eliminate tumors in metastatic and orthotopic mouse models of RMS.
Introduction
Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma in children, accounting for 5% of all childhood malignancies.1 Histologically, pediatric RMS is classified into two major subtypes, alveolar RMS (ARMS) and embryonal RMS (ERMS).1 However, RMS is more accurately defined by the expression of genes related to skeletal muscle differentiation.1,2,3 There are two major molecular subtypes of RMS based on the presence or absence of PAX3/7 gene translocations with FOXO1, which are referred to as fusion-positive RMS (FP-RMS) or fusion-negative RMS (FN-RMS), respectively.2,3 PAX3-FOXO1 is the primary oncogenic driver in FP-RMS, activating a myogenic transcription program,4 whereas FN-RMSs are primarily driven by mutations in the RAS pathway, including fibroblast growth factor receptor 4 (FGFR4).4,5,6 Despite clinical trials with multimodal therapies, including surgery, radiation, and aggressive chemotherapy, there has been no significant improvement in clinical outcomes for patients with RMS for over two decades.7 Because further dose escalation of chemotherapy has shown no benefit, alternative treatment strategies are necessary.
FGFR4 is a cell surface receptor tyrosine kinase that is involved in the regulation of cell proliferation, migration, cell survival, and angiogenesis in RMS.8,9 FGFR4 expression is lost after differentiation into mature skeletal muscle. And in healthy skeletal muscle tissue, FGFR4 is only transiently expressed in activated myoblasts in response to injury.10,11,12,13 We previously reported that FGFR4 is overexpressed in virtually all RMSs, and high expression is associated with poor outcome.9,10 We also have shown that PAX3-FOXO1 directly targets the FGFR4 gene locus, where it establishes a super-enhancer, driving its high expression in FP-RMS.6 Furthermore, approximately 7.5%–10% of FN-RMSs have activating mutations in FGFR4.2,14 Because of these important characteristics in RMS biology, we hypothesized that FGFR4 is an excellent antigen target for immunotherapy for this malignancy.
Chimeric antigen receptor (CAR) T cell therapy targeting cell surface tumor-associated antigens (TAAs) is an attractive treatment modality for tumors with a low mutational burden.15,16,17 Since its first report in the 1980s, multiple generations of CAR constructs have been developed. In general, second-generation CAR constructs contain an extracellular domain of single-chain variable fragment (scFv) that binds the target antigen, a CD8α or CD28 hinge and transmembrane domain, and an intracellular domain consisting of a costimulatory receptor (4-1BB and/or CD28) and a CD3ζ domain. When the scFv binds its target antigen, the intracellular costimulatory domain and the CD3ζ domain signal to activate the CAR T cell and induce cytolysis of the target cell.18 This strategy is rapidly becoming an important treatment option for children with cancer, with dramatic responses in leukemia and lymphoma, resulting in the approval of six CAR T cell therapies targeting CD19 or B cell maturation antigen (BCMA) by the FDA.19,20,21,22,23,24 In addition, CAR T cell therapies targeting ganglioside GD2 have recently shown promising clinical responses to neuroblastoma (NB)25,26 or H3K27M-mutated diffuse midline gliomas.27 In a phase 1 clinical trial,28,29 a patient with RMS went into long-term remission after receiving infusions of HER2 CAR T cells, which suggests that RMSs are susceptible to CAR T cell therapy. FGFR4 CAR T cells have been reported to show cytotoxicity against RMS cell lines in vitro,30 and recently, we reported a FGFR4-targeting CAR, which was only effective when combined with anti-myeloid polypharmacy (targeting CSF1R, IDO1, iNOS, transforming growth factor β [TGF-β], PDL1, MIF, and myeloid mis-differentiation).31 Here, we describe a potent engineered CAR targeting FGFR4 that is currently being developed for a phase 1 clinical trial at the National Cancer Institute (NCI).
Results
FGFR4 is highly expressed in RMS with low or no expression in human healthy tissue including vital organs
We previously reported that FGFR4 is highly expressed in all RMSs as a diagnostic and prognostic biomarker.9,10,32 Here, we confirm by RNA sequencing (RNA-seq) that FGFR4 is highly expressed in both FP-RMS and FN-RMS tumors and cell lines compared with other pediatric solid tumors and healthy human tissues (Figure 1A). To determine if FGFR4 is also expressed in other adult tumors, we compared its expression in RMS with all tumors of The Cancer Genome Atlas (TCGA). Notably, we found high expression of FGFR4 in liver hepatocellular carcinoma (LIHC) and cholangiocarcinoma (CHOL), suggesting that FGFR4-targeted therapy may also benefit individuals with these cancers (Figure 1B).
Figure 1.

FGFR4 is highly expressed in RMS and other cancers, with low expression in healthy tissue
(A) High expression of FGFR4 mRNA is found in both FP-RMS and FN-RMS tumors and cell lines compared with other pediatric cancers and healthy tissues. Expression levels measured as FPKM (fragments per kilobase of transcript per million mapped reads) for FGFR4 are summarized in violin plots with medians and quartiles. ASPS, alveolar soft part sarcoma; CCSK, clear cell sarcoma of the kidney; DSRCT, desmoplastic small round cell tumor; EWS, Ewing sarcoma; HBL, hepatoblastoma; ML, melanoma; NB, neuroblastoma; OS, osteosarcoma; SS, synovial sarcoma; UDS, undifferentiated sarcoma; WT, Wilms tumor; YST, yolk sac tumor).
(B) FGFR4 mRNA expression in TCGA data shows highest expression in liver hepatocellular carcinoma (LIHC), cholangiocarcinoma (CHOL), and individual tumors of other types. Abbreviations are as per TCGA (https://gdc.cancer.gov/resources-tcga-users/tcga-code-tables/tcga-study-abbreviations).
(C) Representative images of immunohistochemistry (IHC) for FGFR4 show high expression in RMS, with minimum or no expression in healthy organs. H score displayed in bottom right corner, and scale bars, 200 μm.
(D) Summary of membrane-staining H score of FGFR4 IHC of RMS and healthy tissues. Values represent mean ± SEM (error bars).
(E) Representative flow cytometry plots show differential levels of FGFR4 expression on FP-RMS or FN-RMS cell lines. Mean fluorescence intensity (MFI) of FGFR4 on indicated RMS cells is listed in the right table, stained with phycoerythrin (PE)-conjugated anti-human FGFR4 antibody or mouse IgG1 isotype control.
To examine FGFR4 expression at the protein level, we first measured the total FGFR4 level in the RMS cell line RH30 and in healthy tissues using an electrochemiluminescence assay. We found that the RH30 cell line has at least 3-fold higher FGFR4 protein expression (1,348 pg/mg) compared with healthy tissues (2–436 pg/mg) (Figure S1A). To validate these results, we performed western blotting using a commercially available FGFR4-specific monoclonal antibody on RMS and healthy tissues. FGFR4 is expressed in most RMS cell lines, and FP-RMS cell lines consistently expressed higher levels of FGFR4 (Figure S1B). FGFR4 was not detectable in any of the healthy tissues by western analysis (Figure S1C). Next, we performed immunohistochemistry (IHC) to evaluate the expression of FGFR4 protein on tissue microarrays (TMAs) containing both RMS tumors (22 ARMS and 28 ERMS) and healthy tissues (43 different human organs from 1 to 8 different individuals). Strong positive cell membrane staining of FGFR4 was observed in RMS tumors (17/22 [77%] ARMS and 5/28 [18%] ERMS with an H score >100) but not in healthy tissues, including muscle, heart, kidney, liver, and lung (Figures 1C and 1D). Finally, flow cytometry analysis demonstrated high expression of FGFR4 in FP-RMS and in RMS559, an FN-RMS with a FGFR4 driver mutation (V550L)33 in keeping with western blotting results (Figure 1E). Therefore, our RNA and protein expression data show that FGFR4 is a highly expressed cell surface TAA in RMS cells and primary tumors, while its expression is low or absent in healthy human tissues, demonstrating it is an ideal candidate target for CAR T cell therapy.
FGFR4 is a downstream target of the PAX3-FOXO1 fusion oncogene
FGFR4 is differentially expressed in RMS, with higher expression in PAX3-FOXO1-driven FP-RMS. Chromatin immunoprecipitation (ChIP) sequencing for the H3K27ac activation mark revealed the presence of super-enhancers in both FP-RMS and FN-RMS tumors and cell lines at the FGFR4 locus (Figure 2A). Some of these super-enhancers are only seen in myoblasts and myotubes but not in mature skeletal muscle tissues during healthy muscle development (Figure 2A). In addition, we also observed that PAX3-FOXO1 directly binds to these enhancers in FP-RMS cell lines including RH4, RH5, and SCMC (Figure 2A, top). To test whether PAX3-FOXO1 directly upregulates expression of FGFR4, we transduced a previously characterized human fibroblast cell line (7250) lacking FGFR4 expression with a PAX3-FOXO1 lentiviral construct (18). In the PAX3-FOXO1-expressing fibroblasts, we observed that the onco-fusion protein opens up chromatin, recruits BRD4, and establishes super-enhancers, which are indicated by an increase in H3K27ac at the FGFR4 locus (Figure 2B). These epigenetic changes, as defined by increased chromatin accessibility and deposition of BRD4 and H3K27ac, which are induced by the binding of PAX3-FOXO1, resulted in a dramatically increased FGFR4 transcription in the fibroblasts (Figure 2B). These results demonstrated that FGFR4 is a direct downstream target of the fusion protein PAX3-FOXO1 in RMS. In addition, RMS showed the most dependency on FGFR4 among all human cancers in the DepMap (21Q4) dataset (Figure 2C). Therefore, the FGFR4 locus, normally transcriptionally active only in developing muscle, becomes activated specifically in RMS.
Figure 2.

PAX3-FOXO1 establishes a super-enhancer at the FGFR4 locus, and RMSs are dependent on FGFR4 for survival
(A) PAX3-FOXO1 (top) and H3K27ac (bottom) ChIP-seq at the FGFR4 locus in FP-RMS cell lines and tumors (orange), FN-RMS cell lines and tumors (blue), and human skeletal muscle cell lines and tissues (gold).
(B) Top: FGFR4 expression is induced in fibroblasts after introduction of PAX3-FOXO1. Bottom: ChIP-seq demonstrates that PAX3-FOXO1 protein opens chromatin and establishes a super-enhancer at the FGFR4 locus. Open chromatin was assayed by DNase hypersensitivity; binding of PAX3-FOXO1 and BRD4 as well as chromatin H3K27ac status were assayed by ChIP-seq in human fibroblasts with or without PAX3-FOXO1.
(C) Average dependency score of RMS for FGFR4 was found to be the lowest among all human cancers, suggesting the highest dependency of RMS on this receptor for survival.
Development and characterization of the FGFR4-specific antibody 3A11
To develop FGFR4-specific antibodies, we generated hybridoma lines from mice immunized with the human FGFR4 extracellular domain protein. Monoclonal antibody (mAb) 3A11 was the lead clone, demonstrating specific binding to FGFR4+ RMS cell lines RH30 and RMS559 but not to CRISPR FGFR4 knockout RH30 cells (RH30 FGFR4-KO) or the FGFR4− human fibroblast cell line, 7250 (Figures 3A and 3B). We then constructed a chimeric human mAb 3A11 by fusing the sequence encoding the scFv of 3A11 to the human immunoglobulin G1 (IgG1) Fc domain to generate 3A11 scFvFc antibody and confirmed its specific binding to RMS cell lines only (Figures 3C and 3D). The dissociation constant (KD) of 3A11 scFvFc was determined to be 4.17 nM using biolayer interferometry (BLI) (Figure 3E). We also confirmed that 3A11 bound only to human FGFR4 but not to other members of the human FGFR family (FGFR1–3) and that it did not show cross-reactivity to mouse FGFR4 by ELISA (Figure 3F). Notably, the 3A11 scFvFc also recognized FGFR4 on RMS cell lines with differing levels of expression (Figure 3G). These data demonstrate that 3A11 scFvFc retains high specificity and affinity for human FGFR4 protein.
Figure 3.

Development and characterization of a specific FGFR4 binder from the monoclonal murine antibody 3A11
(A) Structure of the mouse anti-FGFR4 mouse monoclonal antibody (mAb) 3A11.
(B) Representative flow cytometry plots show the specificity of 3A11 mAb by staining with FGFR4+ cell lines, FGFR4 KO RH30, or FGFR4− cell 7250. Mouse IgG (msIgG) is used as isotype control for this mouse antibody 3A11.
(C) Structure of anti-FGFR4 antibody 3A11 in scFvFc format fused to the human IgG1 Fc region.
(D) Representative flow cytometry plots show 3A11 scFvFc chimeric antibody specifically binds to FGFR4+ cell lines RH30, RH4, and RMS559 but not to the RH30 FGFR4-KO, the RH4 FGFR4-KO, or fibroblast 7250 lines. Human IgG (huIgG) as an isotype control for 3A11 scFvFc. MFI fold change shown as orange font calculated by formula .
(E) Binding avidity of FGFR4 scFvFc, using 2-to-1 binding model and global fitting analysis, demonstrates the dissociation constant (KD) of 3A11 scFvFc against FGFR4 ECD is 4.17 nM.
(F) ELISA shows 3A11 scFvFc only recognizes human FGFR4 but not human FGFR1–3 or mouse FGFR4.
(G) Flow cytometry using 3A11 scFvFc shows FGFR4 is expressed in several RMS cell lines at various levels with higher expression in FP-RMS compared with in FN-RMS. MFI of 3A11 scFvFc or isotype control huIgG staining on above cells is shown in the table on the right.
3A11 CAR T cells demonstrate specific cytotoxicity in vitro
We next aimed to develop a potent clinical-grade FGFR4-targeting CAR by cloning the scFv of 3A11 into a proprietary second-generation CAR vector with a lentiviral backbone (Lentigen Technology) consisting of a 3A11 CAR and a truncated EGFR (tEGFR) safety switch (Figure 4A). The CliniMACS Prodigy system was used to transduce T cells isolated from three healthy donors, and anti-EGFR antibody was used to determine the transduction efficiency, which ranged from 66.5% to 74.7% at day 7 (Figure S2). To assess the in vitro efficacy of the 3A11 CAR T cells, we measured their cytotoxicity against RMS cell lines (RH30, RH4, and RMS559), control FGFR4 KO cell lines (RH30 FGFR4-KO and RH4 FGFR4-KO), and the FGFR4− human fibroblast cell line (7250). CAR T cells caused rapid and potent cytotoxicity to RMS cells but not to the FGFR4-KO or the FGFR4− cells at different effector-to-target (E:T) ratios (Figures 4B, 4C, S3A, and S3B). Cytokine production by 3A11 CAR T cells, including interferon γ (IFN-γ), tumor necrosis factor α (TNF-α), and interleukin-2 (IL-2), also increased when the CAR T cells were incubated with FGFR4+ cell lines in a dose-dependent manner but not when they were incubated with target-negative cell lines (Figures 4D and S3C). These data demonstrated specific killing activity of 3A11 CAR T cells to FGFR4-expressing target cells.
Figure 4.

Clinical-grade 3A11 CAR T cells show specific cytotoxicity to FGFR4+ cells
(A) Schematic of 3A11 CAR construct targeting FGFR4. HTM, hinge and transmembrane domain; hu tEGFR, human truncated EGFR.
(B) Cytotoxicity assays of 3A11 CAR T cells show potent killing activity toward target RMS cells at an E:T ratio of 0.75:1 in a xCELLigence Real-Time Cell Analysis (RTCA). Vertical black arrows show the time point for adding CAR T cells into a plate seeded with target cells. Representative of n = 3 independent experiments with n = 3 individual donors for (B)–(D). Values represent mean ± SD (standard deviation, error bars).
(C) Cytotoxic assay shows 3A11 CAR does not cause cytolysis to FGFR4-KO or FGFR4− cells at an E:T ratio of 0.75:1. Values represent mean ± SD (error bars).
(D) Cytokine release assay shows 3A11 CAR T cells only release high level of IFN-γ when cocultured with FGFR4-expressing RMS cells (RH30, RH4, or RMS559) rather than the respective FGFR4-KO cell lines or 7250. Values represent mean ± SEM (error bars). Two-way ANOVA is used to compare secreted IFN-γ by mock T or 3A11 CAR T cells cocultured with the target cells by calculating the p value. ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗p ≤ 0.0001; ns: no significant difference.
Clinical-grade 3A11 CAR T cells show no cytotoxicity to primary human cells
Low levels of FGFR4 mRNA expression are present in some healthy tissues including liver, lung, kidney, and pancreas (Figure 1A). Because CAR T cells require a high density of surface antigen for activation and cytotoxicity,34,35 we hypothesized that low levels of FGFR4 expression would not initiate CAR T cell reactivity in healthy tissues. To investigate this, we first measured FGFR4 protein expression in multiple primary human cells isolated from healthy organs by flow cytometry using 3A11scFvFc antibody. Slight peak shifts were observed with 3A11 scFvFc against cardiomyocytes, renal epithelial cells including cortical and proximal tubule epithelial cells, HEK293 (embryonic kidney cells), cholangiocytes, and hepatocytes pooled from 10 individuals, suggesting low FGFR4 expression in these healthy cells that is comparable with a FGFR4− control cell line, human fibroblast 7250 (Figure 5A).
Figure 5.

Low or absence of cell surface FGFR4 in primary human cells does not induce cytokine release when cocultured with 3A11 CAR T cells
(A) Representative flow cytometry plots show low or absence of FGFR4 cell surface expression on human cardiomyocytes, renal epithelial cells, renal cortical epithelial cells, renal proximal epithelial cells, HEK293, cholangiocytes, and hepatocytes pooled from 10 individuals using 3A11 scFvFc. MFI of 3A11 scFvFc or huIgG1 isotype control staining on these primary cells is shown in the top right table. Relative FGFR4 expression levels on these primary cells compared with RH30 cells are calculated as .
(B) Log2 ratio of cytokine (IFN-γ, IL-2, and TNF-α) release in the supernatant, by 3A11 CAR T cells cocultured with primary cells, as indicated, in their respective media, compared with the RMS cell RH30. Cell line 7250 serves as a FGFR4− control. Values represent n = 3 independent experiments with n = 3 individual donors. Values represent mean ± SEM (error bars).
To determine the FGFR4 expression threshold for cytotoxicity and cytokine release, we quantified the number of FGFR4 molecules per cell on a panel of RMS cell lines and the healthy embryonal kidney cell line HEK293 and performed coculture studies with the 3A11 CAR T cells. RH30, RH4, and RD with 9,606, 7,447, and 2,341 FGFR4 molecules per cell, respectively, showed significant cytotoxicity and cytokine release (Figure S4), whereas the low expressor cell lines HR (366 FGFR4 molecules per cell) and HEK293 (155) failed to elicit this response (Figures S4C and S4D). Of note, CTR with 3,282 FGFR4 molecules per cell did not induce significant cytotoxicity or cytokine release, indicating that other tumor-intrinsic factors are important to determine sensitivity to CAR T cells (Figures S4C and S4D).
To further assess the potential for on-target off-tumor reactivity of the 3A11 CAR, we cocultured 3A11 CAR T cells with primary cells and measured cytokine production individually. Again, we did not observe significant production of IFN-γ, IL-2, or TNF-α in coculture experiments using these healthy cells, which was comparable to that of coincubation with 7250 cells lacking FGFR4 expression (Figures 5B and S5). Collectively, these data indicate that 3A11 CAR T cells have little or no on-target off-tumor reactivity to healthy cells expressing low levels of FGFR4.
Clinical-grade 3A11 CAR T cells effectively eliminate metastatic RMS in vivo
Next, we evaluated the anti-tumor efficacy of the clinical-grade 3A11 CAR T cells in two in vivo metastatic model using both RH30, an FP-RMS cell line, and RMS559, an aggressive FN-RMS cell line with a high level of FGFR4 expression (Figure 1E) and a FGFR4 V550L mutation in the intracellular tyrosine kinase domain.33 First, luciferase-expressing RH30 or RMS559 cells were intravenously injected into NSG mice, and then we treated these mice with 3 million 3A11 CAR T cells, mock T cells, or HBSS vehicle control, 3 days after tumor inoculation, and monitored tumor progression via bioluminescence imaging (Figures 6A and 6E). Mice treated with 3A11 CAR T cells had a significantly lower tumor burden when compared with tumor only (p = 0.0010 and p < 0.0001 for RH30 and RMS559, respectively) or mock T cells (p = 0.0001 and p = 0.0047 for RH30 and RMS559, respectively). CAR T cell-treated groups had higher survival probability when compared with HBSS and mock (untransduced) T cell controls (Figures 6B–6D and 6F–6H). The study was ended at day 64 for RH30 and at day 73 for RMS559 due to the onset of graft-versus-host toxicity (dry skin, weight loss, hunched posture, and fur loss). Thus, these data demonstrate that 3A11 CAR T cells effectively suppress and eliminate metastatic RMS tumors in vivo.
Figure 6.

3A11 CAR T cell shows potent antitumor effect in RH30 and RMS559 metastatic xenograft models
(A) Schematic of an in vivo model testing the activity of 3A11 CAR T cells against an RH30 metastatic xenograft model.
(B) Bioluminescent imaging of untreated RH30 xenografts or RH30 xenografts treated with 3E6 mock transduced T cells or 3A11 CAR T cells (n = 5 per group).
(C) Total bioluminescence flux over time of individual mouse treated with HBSS, mock T cells, and 3A11 CAR T cells. Mean (lines) and individual replicates are shown (n = 5 per group). Vertical red arrow indicates the day of T cell infusion, also in (G). Mixed-effects or two-way repeated measures (RM) ANOVA analysis was used to calculate the p value between two groups, respectively. ∗∗∗p ≤ 0.001.
(D) Kaplan-Meier survival analysis of mice are shown in (D) (n = 5 per group). Study was ended at day 64 due to the onset of graft-versus-host toxicity (dry skin, weight loss, hunched posture, and fur loss).
(E) Schema of luciferase-expressing RMS559 metastatic model infused with HBSS or 3E6 of mock or CAR T cells on day 3 after tumor inoculation.
(F and G) Bioluminescence images (F) and bioluminescence kinetics (G) of RMS559 cell growth in the metastatic xenograft model. Means and each replicate are shown, n = 7 or 8. Mixed-effects analysis is used to calculate the p values between each two groups individually. ∗∗p = 0.0047; ∗∗∗∗p < 0.0001.
(H) Kaplan-Meier survival analysis of mice bearing RMS559 (7 or 8 mice/group). Log-rank (Mantel-Cox) test is used to compare the survival curves. ∗∗p = 0.0028.
(I and J) The frequencies of CAR+ or CAR− T cells (I) and total cell counts of CAR+ T cells (J) in the splenocytes from above mice at 70 days post-mock or CAR T cell infusion are examined by flow cytometry (n = 2 for mock T group, n = 4 for 3A11 CAR T cell-treated group; in mean ± SEM [error bars]).
(K and L) The percentages (K, values represent mean ± SD [error bars]) and total cell counts (L, values represent mean ± SEM [error bars]) of CD4+ and CD8+ cells in CAR− (tEGFR−) or CAR+ (tEGFR+) T cells from spleen of 3A11 CAR T cells treated mice are shown. Two-way ANOVA is used to calculate the p values. ∗p < 0.05.
To characterize T cell persistence, we analyzed the spleens from 3A11 CAR- or mock T cell-treated mice at day 73 post-tumor RMS559 inoculation (day 70 post-CAR or mock T cell infusion). We observed that human CAR+ T cells were only present in the spleens of 3A11 CAR T cell-treated mice (Figures 6I and 6J), and ∼60% of CD45+ cells were CAR+ T cells (Figure 6I). Interestingly, while CAR T cells were primarily CD4+ before infusion (Figure S2, donor 3), CD8+ CAR T cells were the predominant phenotype at day 70 post-CAR T cell infusion (Figure 6K). This suggests that CD8+ CAR T cells, which participate in the direct killing of the tumor cells,36 expanded more robustly and persisted in the spleen compared with non-CAR T cell controls (Figure 6L).
3A11 CAR T cells successfully infiltrate into subcutaneous RMS tumor and effectively eradicate intramuscular RMS tumor
Infiltration of CAR T cells into solid tumors is thought to be one of the major barriers of therapeutic efficacy.37 We evaluated the infiltration ability of 3A11 CAR T cells in a subcutaneous solid RMS RH30 tumor model. RH30 cells were subcutaneously injected into NSG mice, followed by intravenous injection of 3E6 3A11 CAR T cells, mock T cell control, or HBSS vehicle control 3 days after tumor inoculation (Figure S6A). 3A11 CAR T cells significantly inhibited the growth of the RH30 subcutaneous tumor (Figure S6B) and extended the survival of mice compared with mock T cell-treated mice (p = 0.0002) (Figure S6C). At day 21 post-tumor injection, four mice from each group were euthanized for immunohistochemistry (IHC). IHC staining showed significant CD8+ and CD4+ T cell infiltration only in the 3A11 CAR T cell-treated tumors but absent in tumors from no treatment or mock T cell control groups (Figures S6D and S6E). These data demonstrate that 3A11 CAR T cells can effectively infiltrate and suppress growth of RMS solid tumors.
To further evaluate the therapeutic efficacy of 3A11 CAR T cells in an orthotopic intramuscular RMS mouse model, luciferase-expressing RH30 cells (9,606 FGFR4 molecules per cell) were injected into the right gastrocnemius muscle of mice. Mock or 3A11 CAR T cells were intravenously infused at day 7 after tumor implantation (Figure 7A). 3A11 CAR T cells significantly controlled the tumor growth compared with the mock T cell-treated group (Figure 7B). Bioluminescence image data also demonstrated that 3A11 CAR T cells rapidly shrank the tumor and eventually eradicated the RMS tumors in 4/5 of mice (Figures 7C and 7D). At the end of this study (day 42 post-tumor implantation), all mock T cell-treated mice were euthanized, due to reaching the endpoint for tumor size, while 3A11 CAR T cell-treated mice were tumor free and survived (p = 0.0027; Figure 7E). We then repeated this experiment in another RMS orthotopic model using RMS RH4, which has a moderate FGFR4-expressing level (7,447 molecules per cell) (Figure 7F). Again, 3A11 CAR T cells mediated complete responses in 3/4 RH4-bearing mice (n = 8 per group) and demonstrated significant survival benefit compared with mock T cell-treated mice (Figures 7G–7J). When analyzing circulating blood T cells using flow cytometry, we found that 28%–96% (mean = 72%) CD45+ CD3+ T cells expressed the 3A11 CAR detected by an anti-EGFR antibody in the CAR T cell-treated RH30-bearing mice (Figure 7K), and they retained an average of 7.7E5 circulating CAR+ T cells per 100 μL in the blood (Figure 7L). Similarly, there is an average of 74% CD45+ CD3+ T cells expressing the 3A11 CAR in CAR T cell-treated RH4-bearing mice (Figures S7A and S7B) and about 4.6E4 CAR+ T cells per 100 μL circulating in blood at day 42 post-CAR T cell infusion (Figure S7C). Therefore, these data demonstrated the persistence of 3A11 CAR T cells combined with the eradication of RH30 and RH4 orthotopic tumor xenografts for the CAR T cell-treated mice.
Figure 7.

3A11 CAR T cells effectively eradicated RMS orthotopic intramuscular xenografts in two models
(A) Schema of the RH30 intramuscular xenograft model infused with mock or 10E6 CAR T cells on day 7 post-tumor inoculation.
(B) Tumor size was monitored over 42 days by measurement of leg volume before and after receiving mock or CAR T cell treatment. Vertical red arrow indicates the day of T cell infusion, also in (D), (G), and (I). Each replicate per group is shown, n = 5. Two-way RM ANOVA analysis is used to calculate the p values between two groups. ∗∗∗∗p < 0.0001.
(C) Bioluminescent images of RH30 intramuscular xenografts growth before and after infusion with mock T cells or 3A11 CAR T cells. The bottom row shows the tumor xenografts dissected from mice legs at the end time point of study.
(D) Total bioluminescence flux (photons per second) over time of individual mouse are shown (n = 5 per group). Two-way RM ANOVA analysis was used to calculate p value between two groups, respectively. ∗∗∗∗p < 0.0001.
(E) Kaplan-Meier survival analysis of mice bearing orthotopic RH30 tumors. ∗∗p = 0.0027.
(F) Schema of the RH4 intramuscular xenograft model infused with 10E6 mock or CAR T cells on day 7 post-tumor inoculation.
(G) Tumor size was monitored by leg volume. Each replicate per group is shown, n = 7 or 8. Two-way RM ANOVA analysis is used to calculate the p values between two groups. ∗∗∗∗p < 0.0001.
(H and I) Bioluminescent images of RH4 intramuscular xenografts for individual mice (H) and total bioluminescence flux (I) over time (n = 7 or 8 per group). Mixed-effects analysis was used to calculate p value between two groups, respectively. ∗∗p = 0.0021.
(J) Kaplan-Meier survival analysis of mice bearing orthotopic RH4 tumors. ∗∗p = 0.0011.
(K and L) Percentage of CAR+ and CAR− in CD45+ CD3+ T cells from peripheral blood mononuclear cells (PBMCs) (K) and total counts of the indicated T cells in 100 μL blood from RH30 orthotopic model (L). Data are shown as each replicate and the mean ± SEM (error bars; n = 5).
Discussion
Current multimodal therapy has improved outcomes of patients with RMS with localized disease. However, for patients with metastatic or relapsed RMS, outcomes remain poor, and dose-escalation chemotherapy has not improved survival.38,39,40,41 RMS tumors, especially FP-RMS, have a low mutation burden, and few tumors have directly actionable driver mutations.2 Attempts to target mTOR (ClinicalTrials.gov: NCT02567435),42 HDACs (ClinicalTrials.gov: NCT04299113), the RAS/MAPK pathway,43,44 and oxidative stress14 are in various stages of preclinical and clinical testing for RMS. Since RMSs are not intrinsically immunogenic possibly due to their low mutation burden,2,32 targeting tumor-associated cell surface antigens such as FGFR4 with engineered T cells is an attractive treatment strategy for high-risk RMS. In this study, we report the preclinical development of a CAR T cell therapy targeting FGFR4 in RMS. We identified a high-affinity murine antibody (3A11) that binds FGFR4 without cross-reactivity to other FGF receptor family members or murine FGFR4. 3A11 CAR T cells demonstrated strong FGFR4-specific T cell cytotoxicity to RMS cells in vitro and in vivo without any significant reactivity to healthy human primary cells.
CAR T cell therapies have shown remarkable response rates against refractory and relapsed leukemia and lymphoma.37 However, CAR T cell therapy for solid tumors has been less successful, and this is thought to be due to lack of targetable surface antigens, poor infiltration of T cells into the tumor, T cell exhaustion, and various immunosuppressive factors in the tumor microenvironment.37 Currently, there are several trials and preclinical studies of CAR T cells treating solid tumors, including targeting of PSMA (prostate-specific membrane antigen) for prostate cancers45; GD2,46 ALK,47 and GPC248,49 for NB; mesothelin for lung cancers50; HER2 for breast cancer51 or RMS28; EGFRvIII for glioblastoma52; and B7-H3 for multiple pediatric and adult solid tumors.53,54 Our 3A11 CAR T cell therapy showed promising activities against multiple RMS xenograft models, which suggests that relative high levels of FGFR4 expression on the surface of RMS tumor cells and 3A11 CAR design have made CAR T cell therapy effective to treat RMSs. In addition to our study, there has been anecdotal evidence of activity of HER2 CAR T cell therapies in solid tumors, including metastatic RMS.28,55
Despite low levels of FGFR4 mRNA detected in healthy lung, kidney, and liver tissues, we found little or undetectable FGFR4 protein expression in these vital organs by IHC staining and western blotting. However, the Human Protein Atlas (HPA) reports that FGFR4 is expressed at moderate levels in pancreas, kidney, liver, and lung. This discrepancy may be due to the use of a polyclonal FGFR4 antibody by the HPA resulting in non-specific staining. We further confirmed FGFR4 discrepant mRNA and protein expression detection in a wide spectrum of healthy human primary cells by bulk RNA sequencing (data not shown) and flow cytometry. To test if FGFR4 CAR T cells could be activated by human healthy cells, we cocultured CAR T cells with these primary human healthy cells and confirmed that 3A11 CAR T cells do not elicit a cytokine response against these primary cells from vital organs. We also discovered that low FGFR4 density on cells such as HR or HEK293 was unable to trigger cytotoxicity or cytokine release of 3A11 CAR T cells, indicating a safe therapeutic index for FGFR4 CAR T cells. However, we recognize that despite the lack of cytokine release against low-FGFR4-expressing cells in these short-term cultures, it may not preclude toxicity over longer period of observation. Therefore, the ultimate test for FGFR4 CAR T therapy toxicity will be evaluated in our planned human clinical trials. Nevertheless, to mitigate and treat any unforeseen toxicities from the 3A11 CAR T cells, we have engineered a tEGFR tag in our CAR construct that provides a safety switch for rapid depletion of CAR T cells after administration of cetuximab if required.56
In conclusion, our study further credentials FGFR4 as an immunotherapy target in RMS, and we have developed a clinical-grade potent CAR specifically targeting human FGFR4. Given the promising preclinical data presented here, 3A11 CAR T cell therapy is currently being developed for a phase 1 clinical trial in patients with RMS at the NCI. In addition, FGFR4 is also highly expressed in other human cancers including hepatoblastoma, hepatocellular carcinoma, and CHOL. Thus, 3A11 CAR T cell therapy may have a broader applicability to benefit patients with cancers expressing high levels of FGFR4.
Limitations of the study
Our in vitro tests showed that 3A11 CAR T cells did not cause cytokine release against either a panel of human primary cells or RMS cells with low FGFR4 expression. However, because our 3A11 binder is specific to human FGFR4 and does not recognize the murine protein, we could not determine the CAR T cells’ impact on healthy tissues that may express low levels of FGFR4 in the mouse models we used. Furthermore, the use of immunodeficient mouse models that lack specific immune cells, like regulatory T and myeloid-derived suppressor cells, may have artificially augmented the efficacy of our CAR T cell therapy. Given these factors, the test for toxicity and efficacy will be determined in upcoming human clinical trials.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FGFR4 (D3B12) XP® Rabbit mAb | Cell Signaling Technology | Cat# CST8562; RRID:AB_10891199 |
| Phycoerythrin (PE) conjugated mouse anti-human FGFR4 antibody (Clone 4FR6D3) | BioLegend | Cat# 324306; RRID:AB_2231698 |
| Ultra-LEAF™ Purified Mouse IgG1, κ Isotype Ctrl Antibody | BioLegend | Cat# 401414 |
| Ultra-LEAF™ Purified Human IgG1 Isotype Control Recombinant Antibody | BioLegend | Cat# 403502 |
| 3A11 mouse IgG antibody | This paper | N/A |
| 3A11 scFvFc | This paper | N/A |
| Recombinant Anti-CD4 antibody [EPR6855] | Abcam | Cat# ab133616; RRID:AB_2750883 |
| Recombinant Anti-CD8 alpha antibody [SP16] | Abcam | Cat# ab101500; RRID:AB_10710024 |
| HRP-conjugated β-actin (C4) | Santa Cruz | Cat# sc-47778; RRID:AB_626632 |
| Anti-rabbit, HRP labeled secondary antibody | Cell Signaling Technology | Cat# 7074S |
| FITC anti-human CD45 antibody | BioLegend, clone HI30 | Cat# 304006; RRID:AB_314394 |
| PE anti-human EGFR antibody | BioLegend, clone AY13 | Cat# 352904; RRID:AB_10896794 |
| PE anti-human CD3 antibody | BioLegend, clone HIT3a | Cat# 300308; RRID:AB_314044 |
| PE-Dazzle594 anti-human CD4 antibody | BioLegend, clone A161A1 | Cat# 357412; RRID:AB_2565664 |
| APC anti-human CD8a antibody | BioLegend, clone RPA-T8 | Cat# 301049; RRID:AB_2562054 |
| Ghost Dye™ Violet 510 | Cytek BioScience | SKU 13-0870-T100 |
| Bacterial and virus strains | ||
| NEB® Stable Competent E. coli (High Efficiency) | New England Biolabs | C3040H |
| Biological samples | ||
| Human Alveolar rhabdomyosarcoma tissue microarrays (ARMS TMAs) | Children’s Oncology Group (COG) | 3000–30-P8967 |
| Human embryonal rhabdomyosarcoma tissue microarrays (ERMS TMAs) | Children’s Oncology Group | 3000–30-P8968 |
| COG Normal Pediatric TMA | Children’s Oncology Group | N/A |
| human normal tissue microarray | US Biomax, Inc. | FDA999 |
| Buffy Coats | NIH blood bank | N/A |
| human peripheral blood mononuclear cells (PBMCs) | NIH blood bank | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant human FGFR1 | Sino Biological | 10616-H08H |
| Recombinant human FGFR2 | Sino Biological | 10824-H08H |
| Recombinant human FGFR3 | Sino Biological | 16486-H08H |
| Recombinant human FGFR4 | Sino Biological | 10538-H08H |
| Recombinant mouse FGFR4 | Sino Biological | 50194-M08H |
| Human IL-2 | NIH pharmacy | N/A |
| D-Luciferin | PerkinElmer, Inc. | 122799 |
| Critical commercial assays | ||
| V-PLEX human cytokine assay | Meso Scale Discovery (MSD) | Cat. K151AOH-2 |
| Dynabeads Human T-Expander CD3/CD28 | Thermo Fischer Scientific, Gibco | 11141D |
| Lipofectamine 3000 Transfection Reagent | Thermo Fischer Scientific | L3000075 |
| BD QuantiBRITE PE Beads | BD Biosciences | 340495 |
| Deposited data | ||
| dbGAP phs001928 for pediatric cancers | Brohl et al.32 | https://www.ncbi.nlm.nih.gov/gap/ |
| dbGaP phs000720 for RMS | Shern et al.2 | https://www.ncbi.nlm.nih.gov/gap/ |
| dbGaP phs001052 for Omics study | Chang et al.57 | https://www.ncbi.nlm.nih.gov/gap/ |
| TCGA data | https://portal.gdc.cancer.gov/ | TCGA |
| Chromatin immunoprecipitation (ChIP) sequencing | Gryder et al.58 | GSE116344 |
| Experimental models: Cell lines | ||
| RH30 | Gryder et al.58 | N/A |
| RH5 | Gryder et al.58 | N/A |
| RH41 | Gryder et al.58 | N/A |
| RH4 | Gryder et al.58 | N/A |
| JR | Gryder et al.58 | N/A |
| RMS559 | Dr. Jonathan Fletcher at Brigham and Women’s Hospital, Boston, USA | N/A |
| BIRCH | Brohl et al.32 | N/A |
| CTR | Brohl et al.32 | N/A |
| RD | Brohl et al.32 | N/A |
| RH30 FGFR4-KO | Dr. Michele Bernasconi at University Children’s Hospital Zurich, Switzerland | N/A |
| RH4 FGFR4-KO | Dr. Michele Bernasconi at University Children’s Hospital Zurich, Switzerland | N/A |
| 7250 | Gryder et al.6 | N/A |
| RMS238 | Gryder et al.58 | N/A |
| RMS209 | Gryder et al.58 | N/A |
| RMS224 | Gryder et al.58 | N/A |
| SCMC | Gryder et al.58 | N/A |
| NCI0082 | Gryder et al.58 | N/A |
| RH3 | Gryder et al.58 | N/A |
| RMS216 | Gryder et al.58 | N/A |
| SMS-CTR | Gryder et al.58 | N/A |
| RMS206 | Gryder et al.58 | N/A |
| NCI0075 | Gryder et al.58 | N/A |
| RMS008 | Gryder et al.58 | N/A |
| JR1 | Gryder et al.58 | N/A |
| RH18 | Gryder et al.58 | N/A |
| Myoblasts | Gryder et al.58 | N/A |
| Myotubes | Gryder et al.58 | N/A |
| Psoas Muscle | Gryder et al.58 | N/A |
| Skel. Muscle129 | Gryder et al.58 | N/A |
| Skel. Muscle134 | Gryder et al.58 | N/A |
| Human cardiomyocytes | Cellprogen | Cat# 36044-15 |
| cholangiocytes | Cellprogen | Cat# 36755-12 |
| pancreatic epithelial primary cells | Cellprogen | Cat# 36099-25 |
| human coronary artery smooth muscle cells (CASMC) | Lonza | Cat# CC-2583 |
| small airway epithelial cells (SAEC) | Lonza | Cat# CC-2547 |
| bronchial/tracheal epithelial cells (NHBE) | Lonza | Cat# CC-2541 |
| human renal epithelial cells (HRE) | Lonza | Cat# CC-2556 |
| renal cortical epithelial cells (HREC) | Lonza | Cat# CC-2554 |
| renal proximal tubule epithelial cells (RPTEC) | Lonza | Cat# CC-2553 |
| hepatocytes | Lonza | Cat# HUCPG |
| 10 Donors pooled hepatocytes | Lonza | Cat# HUCS10P |
| HEK 293 | ATCC | Cat# CRL-1573 |
| Lenti-X 293T lentiviral packaging cell line | Clontech | 632180 |
| Experimental models: Organisms/strains | ||
| NOD-scid IL2rgnull (NSG) mice | NCI CCR Animal Resource Program/NCI Biological Testing Branch |
N/A |
| Recombinant DNA | ||
| 3A11-CD8HTM-BBz CAR | This paper | N/A |
| pMDLg/pRRE | Addgene | Plasmid #12251 |
| PMD2.G | Addgene | Plasmid #12259 |
| pRSV-Rev | Addgene | Plasmid #12253 |
| Software and algorithms | ||
| FlowJo v10.7.2 | FlowJo, LLC | N/A |
| GraphPad Prism 8 | GraphPad Software Inc. | N/A |
| Integrative Genomics Viewer (IGV) | Robinson et al.59 | N/A |
| SnapGene 5.2.4 | SnapGene | N/A |
| Image Lab | Bio-Rad | N/A |
| Aperio ImageScope | Leica Biosystem | N/A |
| RTCA software 2.0 | Aligent | N/A |
| Living Image version (IVIS imaging) | Perkin Elmer | N/A |
| Biorender | Biorender | N/A |
| Other | ||
| Zombie UV™ Fixable Viability Kit | BioLegend | 423108 |
| CountBright Absolute Counting beads | Thermo Fischer Scientific | C36950 |
Resource availability
Lead contact
Requests for further information and reagents should be directed to and will be fulfilled by the lead contact, Javed Khan (khanjav@mail.nih.gov).
Materials availability
Materials created in this study will be available for the scientific community by contacting the corresponding author and completion of a material transfer agreement.
Experimental model and study participant details
Cell lines
FGFR4 knock out cell lines (RH4 FGFR4-KO, RH30 FGFR4-KO) were gifts from Dr. Michele Bernasconi at University Children’s Hospital Zurich, Switzerland. RMS559 was a gift from Dr. Jonathan Fletcher at Brigham and Women’s Hospital, Boston, USA. HR, RD, CTR, RH5, 7250, RH4, RH4 FGFR4-KO, RH30, RH30 FGFR4-KO and RMS559 were all grown in Dulbecco Modified Eagle Medium (DMEM, Quality Biological) with 10% FBS (Life Technologies), 1% L-glutamine, and 1% penicillin–streptomycin. JR was cultured with RPMI 1640 supplemented with 10% FBS, 1% L-glutamine, and 1% penicillin–streptomycin at 37°C in a humidified atmosphere with 5% CO2. All cell lines used were confirmed to be mycoplasma free by the MycoAlert kit (Lonza). STR DNA fingerprinting was performed for all cell lines used and confirmed their identity.
Primary cell culture
Peripheral blood mononuclear cells (PBMCs) obtained from blood of healthy donors were approved by NIH and isolated using Histopaque-1.077gm/mL (Sigma, Cat# 10771) according to the manufacturer’s instructions.
Human cardiomyocytes (Cat# 36044-15), cholangiocytes (Cat# 36755-12), and pancreatic epithelial primary cells (Cat# 36099-25) were obtained from Cellprogen; human coronary artery smooth muscle cells (CASMC, Cat# CC-2583), small airway epithelial cells (SAEC, Cat# CC-2547), bronchial/tracheal epithelial cells (NHBE, Cat# CC-2541), human renal epithelial cells (HRE, Cat# CC-2556), renal cortical epithelial cells (HREC, Cat# CC-2554), renal proximal tubule epithelial cells (RPTEC, Cat# CC-2553), hepatocytes (Cat# HUCPG), and 10 Donors pooled hepatocytes (Cat# HUCS10P) were purchased from Lonza; HEK 293 cells (Cat# CRL-1573) was purchased from ATCC. All these cells were grown in special growth medium stated in their product information.
In vivo studies
Animal studies were conducted using 5∼8-week-old female NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice, obtained from NCI CCR Animal Resource Program/NCI Biological Testing Branch. They were housed and treated under the protocol (GB-011) approved by the NCI Bethesda Animal Care and Use Committee at the NIH.
Briefly, luciferase transduced RH30 or RMS559 cells were trypsinized and neutralized when they reached 80% confluency in a T175 flask. Cells were then washed two times with cold HBSS and injected into NSG mice. For intravenous (i.v.) injection, each mouse received 1E6 RMS cells in 0.2mL HBSS; for subcutaneous (s.c.) injection, each mouse received 1E6 RMS cells in 0.2mL HBSS containing 50% Matrigel (Corning). Three days later each mouse received i.v. 3E6 CAR T-cells, untransduced control (mock T), or HBSS vehicle control. For intramuscular (IM) injection, each mouse received 1E6 RH30 cells or 4E6 RH4 cells expressing luciferase, 10E6 CAR T-cells or mock T-cells were infused at day 7 post tumor implantation. Tumor engraftment was measured by a caliper or Xenogen IVIS Lumina imager (PerkinElmer). For Xenogen imaging, the mice were first injected with luciferin at 300 mg/kg and imaged 20 min later. Images were analyzed using Living Image, version 3.1 software (PerkinElmer) and the bioluminescent signal flux for each mouse was expressed as average radiance (photons/second/cm2/steradian).
Method details
RNA-seq data analysis
RNA-seq data was analyzed as previously described.32 Briefly, total RNAs was isolated from freshly frozen tumors and polyA-selected or Ribozero-selected RNA libraries were prepared for RNA sequencing on Illumina HiSeq2000, 2500, and NextSeq500 according to the manufactures protocol (Illumina, San Diego, CA). FastQC were used to assess for quality of paired end reads. And then fastq files were mapped to GRCH37 reference genome using the STAR/2.5.3a alignment algorithm and subsequently quantified by RSEM program based upon Ensembl GRCh37.75 gene annotation.
Chip-seq data analysis
Previously published ChIP-seq data of active histone mark H3K27ac was analyzed in RMS tumors and cell lines, both FP and FN-RMS, and compared to H3K27ac data in myoblasts, myotubes and muscle.58 All samples were mapped to human genome build hg19 using BWA, and indexed BAMs were converted to compressed TDF format at 25 bp bin resolution after extension of reads to the median fragment length (∼200 bp extended past each mapped single-end, 75 bp read), using IGV command line tool toTDF. Files were visualized in IGV (https://software.broadinstitute.org/software/igv/download).
CRISPR data across the panel of cell lines was downloaded from Project Achilles (https://depmap.org/portal/).
Immunohistochemistry of normal tissue microarray (TMA) and RMS TMA
Normal tissue microarray (TMA) slide was purchased from US Biomax (FDA999w). Pediatric normal tissue TMA, aRMS TMA (P8967) and eRMS TMA (P8968) were obtained from the Children’s Oncology Group. The TMA slides were deparaffinized with xylene and then dehydrated through a graded ethanol series. Antigen retrieval was performed for 20 min in heat-activated antigen retrieval buffer pH 6.0 (Dako, Carpinteria, CA) using a pressure cooker (Dako) followed by 10 min of blocking using 3% hydrogen peroxidase. The sections were incubated with rabbit monoclonal anti-FGFR4 antibodies (Clone D3B12; Cell Signaling; Denver, CA) at 1:150 for 1 h. Subsequently, the reaction of antigen-antibody reaction was detected by EnVision+ Dual Link System-HRP (Dako) and visualized with DAB+ (3, 3′-Diaminobenzidine; Dako). TMA sections were lightly counterstained in a Mayer’s hematoxylin bath and then examined by light microscopy. Negative control rabbit immunoglobulin G (IgG) was used in place of a primary antibody to evaluate nonspecific staining and the TMA included appropriate positive control specimens. The stained sections were scanned using the NanoZoomer 2.0 HT (Hamamatsu Photonics K.K., Japan) at 20× objective magnification. Immunohistochemistry staining results were reviewed by a board-certified pathologist and H score was determined based on the staining intensity and percentage of positive stained cells using the formula: H-score = [(0 × % negative cells) + (1 × %weakly positive cells) + (2 × %moderately positive cells) + (3 × %strongly positive cells)]. For multiple cores with the same type of sample from the sample individual, only the highest H score was used for data analysis.
Generation of 3A11 scFvFc
3A11 is a mouse IgG that was developed from mice immunized with human FGFR4 Fc (R&D). The variable heavy and light chains encoding the scFv, were cloned into a plasmid containing the constant fragment (Fc) of human IgG1, thus creating the scFvFc expression vector. This scFvFc plasmid was transiently expressed in Expi293 system (Life Technologies). Protein was then purified by Affinity chromatography (GE Biosciences). The purified scFvFc fusion protein was analyzed by nanodrop for size and impurities.
ELISA assay
Recombinant human FGFR1-4 (Sino Sino Biological) proteins were coated on Corning EIA/RIA high binding 96 well plate (Corning, Inc) at 50ng per well for overnight at 4°C and blocked with 3% BSA in PBS. 3A11 scFvFc, at a starting concentration of 1000 ng/μL was further serially diluted to 3 ng/μL, added, and incubated at room temperature for 2 h. The plates were washed with PBS containing 0.05% Tween 20. Bound 3A11 scFvFc was detected by a goat anti-human IgG (Fc specific)- peroxidase (Jackson Laboratory). The assay was developed at room temperature with ABTS substrate (Roche) and absorbance quantified at 405nm.
3A11 scFvFc binding affinity determined by octet analysis system
The binding kinetics of the 3A11 scFvFc was determined by the Octet RED96 system (FortéBio) using Bio-Layer Interferometry (BLI). 3A11scFvFc protein was resuspended in 0.1% BSA, 0.1% Tween 20 PBS, pH 7.4 buffer. FGFR4 ECD protein was immobilized onto biosensors, 3A11 scFvFc was injected and allowed to flow through the sensor. The relative association measurements for a time window of 600 s and 1,400 s, respectively was collected. Data analysis was performed using the analysis software provided with the instrument. The KD value of 3A11 scFvFc was determined by global fitting analysis and 2:1 binding model.
Electrochemiluminescence assay
Normal human tissues were cut in small size, and lysates were prepared using Fastprep 24 homogenizer and lyser. RMS cell lysates were prepared using cell lysis buffer, RIPA and protease inhibitors. Both the tissue and cell lysates, after lysis, were processed by incubation on ice for 30 min in the lysis buffer. The lysates were centrifuged at 13200rpm for 10 min at 4C. The supernatant was isolated and used to measure total protein using BCA assay. All lysates were further diluted to a final total protein concentration of 1 mg/mL 25μL of this concentrate was used in Duoset ELISA kit that measures total human FGFR4. The detection reagent was conjugated as described by MSD Gold Sulfo Tag NHS-Ester kit. Recombinant FGFR4 protein, also supplied by the ELISA kit, was used to generate a standard curve, to quantify the amount of FGFR4 in tissue and cell lysates.
Western blot analysis
Western blot analysis was performed as previously described. Briefly whole cell lysates were prepared using RIPA buffer supplemented with protease and phosphatase inhibitors. Cells were lysed by sonication, rotated for 30 min at 4°C, and then lysates were centrifuged for 10 min, supernatant removed, and protein concentration quantified using a Bradford assay. 20 μg of lysate protein was resolved on 4–12% Bis-Tris gels and transferred to PVDF membrane, blocked in 5% nonfat milk in Tris-buffered saline and Tween 20 (TBS-T). Membranes were incubated at 4°C overnight in primary antibody, washed x 3 in TBS-T, then incubated in 1:2,000 diluted anti-rabbit, HRP-labeled secondary antibody (Cell Signaling Technology, Cat# 7074S) at room temperature for 1 h, washed an additional × 3 with TBST, and then developed with chemiluminescent reagents. The following antibodies were used: HRP-conjugated β-actin (Santa Cruz, C4, sc-47778); FGFR4 XP Rabbit mAb (Cell Signaling Technology, clone D3B12, CST8562).
Cell surface protein determination assay
1E6 RMS cell were detached from culturing flasks by 0.05% trypsin and then washed twice with cold PBS and resuspended in 100μL of cold FACS buffer (1× PBS, 22% FBS, 2mM EDTA and 25mM HEPES). Cells were incubated with 5μg of anti-FGFR4 mAb (Clone 4FR6D3, mouse anti-FGFR4 PE, BioLegend, Cat # 324306) or 1μg of 3A11 scFvFc or their isotype control, for 30 min at 4°C. After incubation, cells were washed twice with FACS buffer and resuspended in secondary goat anti mouse or human antibody (Jackson Laboratory) at 1:200 dilution in FACS buffer. Following incubation, cells were washed three times with FACS buffer. Stained cells were analyzed on BD FACS Caliber or LSRFortessa. Data was analyzed and MFI was calculated for FGFR4 surface levels measurement by using Flow Jo software. FGFR4 surface molecules per cell were calculated post subtracting background signal emanating from a respective isotype control antibody (Clone QA16A12 for human IgG1 antibody, Biolegend) by the Quantibrite PE Quantitation Kit (BD Biosciences, Cat# 340495) according to the manufacturer’s protocol.
Generation of the 3A11 CAR construct and lentivirus production
The FGFR4 CAR contains the targeting scFv sequence derived from a mouse 3A11 monoclonal antibody. The scFv sequence was linked in frame to CD8 hinge and transmembrane domain, 4-1BB/CD137 co-stimulatory domain, and CD3ζ activation domain. Leader sequence from CD8 alpha subunit was included upstream of the CAR, to facilitate CAR expression at the cell surface membrane. CAR sequence was followed by the ribosomal skip element T2A and the truncated epidermal growth factor receptor sequence (tEGFR, NP_958441.1, AA 334–627), used as a safety tag. The CAR expression cassette was cloned into a third-generation lentiviral plasmid backbone (Lentigen Technology, Gaithersburg, MD) under the regulation of a human EF-1α promoter. Clinical grade lentiviral vector (LV)-containing supernatants were generated by transient transfection of HEK 293 T-cells60 was used for all experiments except for efficacy testing in the RH4 IM model in Figure 7, which used in-house manufactured process as described previously.61 Harvested lentivirus supernatants were stored at −80°C.
3A11 CAR T cell manufacturing
Clinical grade 3A11 CAR T-cells were manufactured on the automated closed-system CliniMACS Prodigy (Miltenyi Biotec). Cultures were initiated from thawed CD4/CD8 T-cells that were previously selected on the CliniMACS Prodigy from healthy donor apheresis products. On Day 0 of culture, approximately 100E6 thawed CD4/CD8 T-cells were initiated on the Prodigy in TexMACS media (Miltenyi Biotec) supplemented with 3% human AB serum (Valley Biomedical) and 200 IU/mL IL-2 (Clinigen Inc.) and stimulated with TransAct anti-CD3/CD28 polymeric nanomatrix (Miltenyi Biotec). On Day 1, cells were transduced with FGFR4 lentiviral vector at a multiplicity of infection (MOI) of 20. On Day 3, transduction was stopped and TransAct was washed out, and a series of media addition/exchanges were performed daily on Days 5–8. On Day 9, the final product was collected and cryopreserved. For efficacy testing in the RH4 IM model in Figure 7, we used in-house CAR T cell production as described previously.61
xCELLigence Real-Time Cell Analysis and cytokine release assay
5000 target cells were seeded in each well of 96 E-plate (Roche). Cells were monitored continuously by the xCELLigence system. Six hours later, CAR T-cells and its mock T (un-transduced) controls at different E:T ratio and cells were added and continuously monitored for further 18 h. At the endpoint, cells were spun down, and supernatant was collected. V-PLEX Custom Human Biomarkers Proinflammatory Panel1 (Human IFN-γ, Human IL-2 and Human TNF-α, Meso Scale Discovery) was used to measure the cytokine level in the supernatant as per manufactory’s instruction.
In vitro toxicity study
For each of the toxicity experiment, 7250 and human RMS cells RH30 was used as negative and positive controls respectively. Briefly, controls and cells to be tested were first seeded in each well of 96 well flat bottom plate for 6 h. Then all culture medium for controls was replaced with culture medium used for the primary cells (DMEM for HEK293), CAR T-cells in the primary cell culture medium was then added at an E:T ratio of 1:1. Eighteen hours later, all cells were spun down and supernatant were taken for cytokine release assay. Since the growth medium is optimized for the primary cells and therefore may not be optimized for CAR T-cells, for each culture condition, the cytokine released was normalized with the cytokine detected in the positive control by the following formula: log2 (primary/RH30).
CAR T-cells persistence and exhaustion analysis by flow cytometry
For CAR T cell persistence analysis, splenocytes were isolated from RMS559 i.v. xenografted mice at day 70 post CAR T cell infusion. Blood was collected from RH30 IM xenografted mice at day 35 or RH4 IM implanted mice at day 42 post CAR T-cells treatment and then red blood cell (RBC) was lysed by RBC Lysis Buffer (BioLegend, 10×, Cat# 420301) before flow cytometry antibodies staining. T-cells were detected using the following anti-human antibodies: CD45-FITC (BioLegend, clone HI30), CD3-PE (BioLegend, clone HIT3a), CD4-PE-Dazzle594 (BioLegend, clone A161A1), CD8-APC (BioLegend, clone RPA-T8), EGFR-PE (BioLegend, clone AY13). The absolute CAR T cell counts in the spleen from tumor-bearing NSG mice were measured by using CountBright absolute counting beads for flow cytometry (Invitrogen) according to the manufacturer’s protocol. All staining was performed in 0.1 mL FACS buffer (PBS +0.5% BSA + 2mM EDTA). Flow cytometry was performed using a FACS LSRFortessa (BD Biosciences) and data analyzed with FlowJo software (Tree Star).
H&E and immunohistochemistry (IHC) for tumor infiltrating T-cells
Formalin-fixed tumors from RH30 subcutaneous xenograft model were embedded in paraffin, and 5 μm FFPE thick sections were cut and mounted for staining. Sections of neoplastic masses occasionally contained adjacent normal tissues, including fibroadipose connective tissue and skeletal muscle.
IHC was performed using CD4 (1:250 dilution, Abcam, Cat# Ab133616), CD8 (1:100 dilution, Abcam, Cat# Ab101500), FGFR4 (1:100 dilution, Cell Signaling Technology, Cat# CST8562). Positive IHC controls included human tonsil and cell pellets known to be positive and negative for FGFR4. Negative controls included replacing the primary antibody with nonspecific antibody from the same species and of the same isotype.
Whole Slide Imaging (WSI) was performed with an Aperio ScanScope XT (Leica) at 200× in a single z-plane. Annotations were made to include tumor tissue and exclude tumor necrosis and section artifacts for IHC quantification. Digital pathology for biomarker quantification was performed following WSI. Thresholds for positivity was determined using known positive controls. Tumor necrosis was estimated by a pathologist as % area from H&E images. CD4 and CD8 positive cells are reported as number of positive cells per mm2.
Quantification and statistical analysis
Statistical analysis
GraphPad Prism software was used for data analysis. Log rank statistics was used for calculation of p value of survival analyses, two-way repeated measures (RM) ANOVA or mixed-effect analysis was used for calculating significant difference of tumor growth curves, and two-way ANOVA was used for p value calculation of in vitro cytokines production data. p < 0.05 was considered statistically significant, and p values are denoted with asterisks as follows: p > 0.05; not significant, ns; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; and ∗∗∗∗p < 0.0001.
Acknowledgments
This work was supported by the Intramural Research Program of NIH, NCI, Center for Cancer Research, which included funds from NIH Cooperative Research and Development Agreement (CRADA, grant no. 3026, titled “Development of Fibroblast Growth Receptor 4 (FGFR4)-targeting Chimeric Antigen Receptors (CARs) for Cancer Therapy”). This research was also supported in part by the Stand Up to Cancer - St. Baldrick's Pediatric Dream Team Translational Research Grant (SU2CAACR-DT1113). Stand Up To Cancer is a division of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the Scientific Partner of SU2C. The authors acknowledge Yanyu Wang of the Lymphokine Testing Section at the NCI Clinical Support Laboratory (CSL) for measuring cytokine production. We acknowledge the NCI Flow Core for providing instruments and services related to flow cytometry. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US government.
Author contributions
J. Khan, A.T.C., N.S., and R.J.O. conceived and designed the experiments. A.T.C. and M.T. performed most of the experiments. A.T.C. and M.T. collected, analyzed, and interpreted the data. N.S. performed the antibody characterization studies. S.L.H., L.M., and H.S. manufactured clinical-grade CAR T cells. D.S. and B.D. made the lentiviral-expressing 3A11 CAR. B.E.G., H.-C.C., and Y.K.S. performed ChIP-seq and analyzed the RNA-seq and CRISPR data. G.T.B. manually quantified FGFR4 H score in IHC-stained TMAs. J.T.W. performed western blot assay for examining FGFR4 expression on RMS and healthy tissues. P.A. performed ELISA for 3A11 scFvFc specificity to FGFR1-4. B.B. and C.M.L. from Duke performed FGFR4 IHC staining. D.M., R.J.O., and A.R. provided technical or material support. M.T. organized data and made all figures. A.T.C. and M.T. wrote the original draft. M.T., J.S.W., and J. Khan revised this manuscript. J. Khan., M.T., J.S.W., C.L.M., D.M., A.T.C., K.E.M., Y.Y.K., R.J.O., S.L.H., B.B., C.M.L., D.S., D.S.D., and J. Kumar reviewed and edited the manuscript.
Declaration of interests
J. Khan, R.J.O., D.S.D., and A.T.C. are inventors on international patent application no. PCT/US2016/052496. The 3A11 CAR sequence is in this patent application (see https://patents.justia.com/patent/11078286) filed on September 19, 2016, titled “Monoclonal antibodies specific for fibroblast growth factor receptor 4 (FGFR4) and methods of their use.”
Published: September 28, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101212.
Contributor Information
Adam T. Cheuk, Email: cheukt@gmail.com.
Javed Khan, Email: khanjav@mail.nih.gov.
Supplemental information
Data and code availability
-
•
The FGFR4 expression data for this study are available in dbGaP: phs000720, phs001928, phs001052, and https://portal.gdc.cancer.gov/for the TCGA data. ChIPSeq data have been deposited at Gene Expression Omnibus (GSE116344) and is publicly available.
-
•
This paper does not report any original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
References
- 1.Skapek S.X., Ferrari A., Gupta A.A., Lupo P.J., Butler E., Shipley J., Barr F.G., Hawkins D.S. Rhabdomyosarcoma. Nat. Rev. Dis. Prim. 2019;5:1. doi: 10.1038/s41572-018-0051-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shern J.F., Chen L., Chmielecki J., Wei J.S., Patidar R., Rosenberg M., Ambrogio L., Auclair D., Wang J., Song Y.K., et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014;4:216–231. doi: 10.1158/2159-8290.CD-13-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shern J.F., Yohe M.E., Khan J. Pediatric Rhabdomyosarcoma. Crit. Rev. Oncog. 2015;20:227–243. doi: 10.1615/critrevoncog.2015013800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khan J., Bittner M.L., Saal L.H., Teichmann U., Azorsa D.O., Gooden G.C., Pavan W.J., Trent J.M., Meltzer P.S. cDNA microarrays detect activation of a myogenic transcription program by the PAX3-FKHR fusion oncogene. Proc. Natl. Acad. Sci. USA. 1999;96:13264–13269. doi: 10.1073/pnas.96.23.13264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gryder B.E., Wachtel M., Chang K., El Demerdash O., Aboreden N.G., Mohammed W., Ewert W., Pomella S., Rota R., Wei J.S., et al. Miswired Enhancer Logic Drives a Cancer of the Muscle Lineage. iScience. 2020;23 doi: 10.1016/j.isci.2020.101103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gryder B.E., Yohe M.E., Chou H.C., Zhang X., Marques J., Wachtel M., Schaefer B., Sen N., Song Y., Gualtieri A., et al. PAX3-FOXO1 Establishes Myogenic Super Enhancers and Confers BET Bromodomain Vulnerability. Cancer Discov. 2017;7:884–899. doi: 10.1158/2159-8290.CD-16-1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weigel B.J., Lyden E., Anderson J.R., Meyer W.H., Parham D.M., Rodeberg D.A., Michalski J.M., Hawkins D.S., Arndt C.A.S. Intensive Multiagent Therapy, Including Dose-Compressed Cycles of Ifosfamide/Etoposide and Vincristine/Doxorubicin/Cyclophosphamide, Irinotecan, and Radiation, in Patients With High-Risk Rhabdomyosarcoma: A Report From the Children's Oncology Group. J. Clin. Oncol. 2016;34:117–122. doi: 10.1200/JCO.2015.63.4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crose L.E.S., Etheridge K.T., Chen C., Belyea B., Talbot L.J., Bentley R.C., Linardic C.M. FGFR4 blockade exerts distinct antitumorigenic effects in human embryonal versus alveolar rhabdomyosarcoma. Clin. Cancer Res. 2012;18:3780–3790. doi: 10.1158/1078-0432.CCR-10-3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taylor J.G., Cheuk A.T., Tsang P.S., Chung J.Y., Song Y.K., Desai K., Yu Y., Chen Q.R., Shah K., Youngblood V., et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J. Clin. Invest. 2009;119:3395–3407. doi: 10.1172/JCI39703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khan J., Wei J.S., Ringnér M., Saal L.H., Ladanyi M., Westermann F., Berthold F., Schwab M., Antonescu C.R., Peterson C., Meltzer P.S. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat. Med. 2001;7:673–679. doi: 10.1038/89044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao P., Hoffman E.P. Embryonic myogenesis pathways in muscle regeneration. Dev. Dynam. 2004;229:380–392. doi: 10.1002/dvdy.10457. [DOI] [PubMed] [Google Scholar]
- 12.Lagha M., Kormish J.D., Rocancourt D., Manceau M., Epstein J.A., Zaret K.S., Relaix F., Buckingham M.E. Pax3 regulation of FGF signaling affects the progression of embryonic progenitor cells into the myogenic program. Genes Dev. 2008;22:1828–1837. doi: 10.1101/gad.477908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao P., Caretti G., Mitchell S., McKeehan W.L., Boskey A.L., Pachman L.M., Sartorelli V., Hoffman E.P. Fgfr4 is required for effective muscle regeneration in vivo. Delineation of a MyoD-Tead2-Fgfr4 transcriptional pathway. J. Biol. Chem. 2006;281:429–438. doi: 10.1074/jbc.M507440200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen X., Stewart E., Shelat A.A., Qu C., Bahrami A., Hatley M., Wu G., Bradley C., McEvoy J., Pappo A., et al. Targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell. 2013;24:710–724. doi: 10.1016/j.ccr.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee D.W., Kochenderfer J.N., Stetler-Stevenson M., Cui Y.K., Delbrook C., Feldman S.A., Fry T.J., Orentas R., Sabatino M., Shah N.N., et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grupp S.A., Kalos M., Barrett D., Aplenc R., Porter D.L., Rheingold S.R., Teachey D.T., Chew A., Hauck B., Wright J.F., et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Majzner R.G., Heitzeneder S., Mackall C.L. Harnessing the Immunotherapy Revolution for the Treatment of Childhood Cancers. Cancer Cell. 2017;31:476–485. doi: 10.1016/j.ccell.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 18.Sadelain M., Brentjens R., Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3:388–398. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bouchkouj N., Kasamon Y.L., de Claro R.A., George B., Lin X., Lee S., Blumenthal G.M., Bryan W., McKee A.E., Pazdur R. FDA Approval Summary: Axicabtagene Ciloleucel for Relapsed or Refractory Large B-cell Lymphoma. Clin. Cancer Res. 2019;25:1702–1708. doi: 10.1158/1078-0432.CCR-18-2743. [DOI] [PubMed] [Google Scholar]
- 20.Bouchkouj N., Zimmerman M., Kasamon Y.L., Wang C., Dai T., Xu Z., Wang X., Theoret M., Purohit-Sheth T., George B. FDA Approval Summary: Axicabtagene Ciloleucel for Relapsed or Refractory Follicular Lymphoma. Oncol. 2022 doi: 10.1093/oncolo/oyac054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haso W., Lee D.W., Shah N.N., Stetler-Stevenson M., Yuan C.M., Pastan I.H., Dimitrov D.S., Morgan R.A., FitzGerald D.J., Barrett D.M., et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121:1165–1174. doi: 10.1182/blood-2012-06-438002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kasamon Y.L., Price L.S.L., Okusanya O.O., Richardson N.C., Li R.J., Ma L., Wu Y.T., Theoret M., Pazdur R., Gormley N.J. FDA Approval Summary: Selinexor for Relapsed or Refractory Diffuse Large B-Cell Lymphoma. Oncol. 2021;26:879–886. doi: 10.1002/onco.13859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O'Leary M.C., Lu X., Huang Y., Lin X., Mahmood I., Przepiorka D., Gavin D., Lee S., Liu K., George B., et al. FDA Approval Summary: Tisagenlecleucel for Treatment of Patients with Relapsed or Refractory B-cell Precursor Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2019;25:1142–1146. doi: 10.1158/1078-0432.CCR-18-2035. [DOI] [PubMed] [Google Scholar]
- 24.Sharma P., Kanapuru B., George B., Lin X., Xu Z., Bryan W.W., Pazdur R., Theoret M.R. FDA Approval Summary: Idecabtagene Vicleucel for Relapsed or Refractory Multiple Myeloma. Clin. Cancer Res. 2022;28:1759–1764. doi: 10.1158/1078-0432.CCR-21-3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heczey A., Louis C.U., Savoldo B., Dakhova O., Durett A., Grilley B., Liu H., Wu M.F., Mei Z., Gee A., et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol. Ther. 2017;25:2214–2224. doi: 10.1016/j.ymthe.2017.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Straathof K., Flutter B., Wallace R., Jain N., Loka T., Depani S., Wright G., Thomas S., Cheung G.W.K., Gileadi T., et al. Antitumor activity without on-target off-tumor toxicity of GD2-chimeric antigen receptor T cells in patients with neuroblastoma. Sci. Transl. Med. 2020;12 doi: 10.1126/scitranslmed.abd6169. [DOI] [PubMed] [Google Scholar]
- 27.Majzner R.G., Ramakrishna S., Yeom K.W., Patel S., Chinnasamy H., Schultz L.M., Richards R.M., Jiang L., Barsan V., Mancusi R., et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature. 2022;603:934–941. doi: 10.1038/s41586-022-04489-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hegde M., Joseph S.K., Pashankar F., DeRenzo C., Sanber K., Navai S., Byrd T.T., Hicks J., Xu M.L., Gerken C., et al. Tumor response and endogenous immune reactivity after administration of HER2 CAR T cells in a child with metastatic rhabdomyosarcoma. Nat. Commun. 2020;11:3549. doi: 10.1038/s41467-020-17175-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitra S., Sydow S., Magnusson L., Piccinelli P., Törnudd L., Øra I., Ljungman G., Sandgren J., Gisselsson D., Mertens F. Amplification of ERBB2 (HER2) in embryonal rhabdomyosarcoma: A potential treatment target in rare cases? Genes Chromosomes Cancer. 2022;61:5–9. doi: 10.1002/gcc.22996. [DOI] [PubMed] [Google Scholar]
- 30.Alijaj N., Moutel S., Gouveia Z.L., Gray M., Roveri M., Dzhumashev D., Weber F., Meier G., Luciani P., Rössler J.K., et al. Novel FGFR4-Targeting Single-Domain Antibodies for Multiple Targeted Therapies against Rhabdomyosarcoma. Cancers. 2020;12 doi: 10.3390/cancers12113313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sullivan P.M., Kumar R., Li W., Hoglund V., Wang L., Zhang Y., Shi M., Beak D., Cheuk A., Jensen M.C., et al. FGFR4-targeted chimeric antigen receptors (CARs) combined with anti-myeloid poly-pharmacy effectively treats orthotopic rhabdomyosarcoma. Mol. Cancer Therapeut. 2022;21:1608–1621. doi: 10.1158/1535-7163.MCT-22-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brohl A.S., Sindiri S., Wei J.S., Milewski D., Chou H.C., Song Y.K., Wen X., Kumar J., Reardon H.V., Mudunuri U.S., et al. Immuno-transcriptomic profiling of extracranial pediatric solid malignancies. Cell Rep. 2021;37 doi: 10.1016/j.celrep.2021.110047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shukla N., Ameur N., Yilmaz I., Nafa K., Lau C.Y., Marchetti A., Borsu L., Barr F.G., Ladanyi M. Oncogene mutation profiling of pediatric solid tumors reveals significant subsets of embryonal rhabdomyosarcoma and neuroblastoma with mutated genes in growth signaling pathways. Clin. Cancer Res. 2012;18:748–757. doi: 10.1158/1078-0432.CCR-11-2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heitzeneder S., Bosse K.R., Zhu Z., Zhelev D., Majzner R.G., Radosevich M.T., Dhingra S., Sotillo E., Buongervino S., Pascual-Pasto G., et al. GPC2-CAR T cells tuned for low antigen density mediate potent activity against neuroblastoma without toxicity. Cancer Cell. 2022;40:53–69.e9. doi: 10.1016/j.ccell.2021.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Majzner R.G., Rietberg S.P., Sotillo E., Dong R., Vachharajani V.T., Labanieh L., Myklebust J.H., Kadapakkam M., Weber E.W., Tousley A.M., et al. Tuning the Antigen Density Requirement for CAR T-cell Activity. Cancer Discov. 2020;10:702–723. doi: 10.1158/2159-8290.CD-19-0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boulch M., Cazaux M., Loe-Mie Y., Thibaut R., Corre B., Lemaître F., Grandjean C.L., Garcia Z., Bousso P. A cross-talk between CAR T cell subsets and the tumor microenvironment is essential for sustained cytotoxic activity. Sci. Immunol. 2021;6 doi: 10.1126/sciimmunol.abd4344. [DOI] [PubMed] [Google Scholar]
- 37.June C.H., O'Connor R.S., Kawalekar O.U., Ghassemi S., Milone M.C. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361–1365. doi: 10.1126/science.aar6711. [DOI] [PubMed] [Google Scholar]
- 38.Bisogno G., Ferrari A., Prete A., Messina C., Basso E., Cecchetto G., Indolfi P., Scarzello G., D'Angelo P., De Sio L., et al. Sequential high-dose chemotherapy for children with metastatic rhabdomyosarcoma. Eur. J. Cancer. 2009;45:3035–3041. doi: 10.1016/j.ejca.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 39.Oberlin O., Rey A., Lyden E., Bisogno G., Stevens M.C.G., Meyer W.H., Carli M., Anderson J.R. Prognostic factors in metastatic rhabdomyosarcomas: results of a pooled analysis from United States and European cooperative groups. J. Clin. Oncol. 2008;26:2384–2389. doi: 10.1200/JCO.2007.14.7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weigel B.J., Breitfeld P.P., Hawkins D., Crist W.M., Baker K.S. Role of high-dose chemotherapy with hematopoietic stem cell rescue in the treatment of metastatic or recurrent rhabdomyosarcoma. J. Pediatr. Hematol. Oncol. 2001;23:272–276. doi: 10.1097/00043426-200106000-00007. [DOI] [PubMed] [Google Scholar]
- 41.Walterhouse D.O., Hoover M.L., Marymont M.A., Kletzel M. High-dose chemotherapy followed by peripheral blood stem cell rescue for metastatic rhabdomyosarcoma: the experience at Chicago Children's Memorial Hospital. Med. Pediatr. Oncol. 1999;32:88–92. doi: 10.1002/(sici)1096-911x(199902)32:2<88::aid-mpo3>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 42.Mascarenhas L., Chi Y.Y., Hingorani P., Anderson J.R., Lyden E.R., Rodeberg D.A., Indelicato D.J., Kao S.C., Dasgupta R., Spunt S.L., et al. Randomized Phase II Trial of Bevacizumab or Temsirolimus in Combination With Chemotherapy for First Relapse Rhabdomyosarcoma: A Report From the Children's Oncology Group. J. Clin. Oncol. 2019;37:2866–2874. doi: 10.1200/JCO.19.00576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yohe M.E., Gryder B.E., Shern J.F., Song Y.K., Chou H.C., Sindiri S., Mendoza A., Patidar R., Zhang X., Guha R., et al. MEK inhibition induces MYOG and remodels super-enhancers in RAS-driven rhabdomyosarcoma. Sci. Transl. Med. 2018;10 doi: 10.1126/scitranslmed.aan4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Odeniyide P., Yohe M.E., Pollard K., Vaseva A.V., Calizo A., Zhang L., Rodriguez F.J., Gross J.M., Allen A.N., Wan X., et al. Correction: Targeting farnesylation as a novel therapeutic approach in HRAS-mutant rhabdomyosarcoma. Oncogene. 2022;41:3037. doi: 10.1038/s41388-022-02342-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Santoro S.P., Kim S., Motz G.T., Alatzoglou D., Li C., Irving M., Powell D.J., Jr., Coukos G. T cells bearing a chimeric antigen receptor against prostate-specific membrane antigen mediate vascular disruption and result in tumor regression. Cancer Immunol. Res. 2015;3:68–84. doi: 10.1158/2326-6066.CIR-14-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Louis C.U., Savoldo B., Dotti G., Pule M., Yvon E., Myers G.D., Rossig C., Russell H.V., Diouf O., Liu E., et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6056. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walker A.J., Majzner R.G., Zhang L., Wanhainen K., Long A.H., Nguyen S.M., Lopomo P., Vigny M., Fry T.J., Orentas R.J., Mackall C.L. Tumor Antigen and Receptor Densities Regulate Efficacy of a Chimeric Antigen Receptor Targeting Anaplastic Lymphoma Kinase. Mol. Ther. 2017;25:2189–2201. doi: 10.1016/j.ymthe.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bosse K.R., Raman P., Zhu Z., Lane M., Martinez D., Heitzeneder S., Rathi K.S., Kendsersky N.M., Randall M., Donovan L., et al. Identification of GPC2 as an Oncoprotein and Candidate Immunotherapeutic Target in High-Risk Neuroblastoma. Cancer Cell. 2017;32:295–309.e12. doi: 10.1016/j.ccell.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li N., Fu H., Hewitt S.M., Dimitrov D.S., Ho M. Therapeutically targeting glypican-2 via single-domain antibody-based chimeric antigen receptors and immunotoxins in neuroblastoma. Proc. Natl. Acad. Sci. USA. 2017;114:E6623–E6631. doi: 10.1073/pnas.1706055114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morello A., Sadelain M., Adusumilli P.S. Mesothelin-Targeted CARs: Driving T Cells to Solid Tumors. Cancer Discov. 2016;6:133–146. doi: 10.1158/2159-8290.CD-15-0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun M., Shi H., Liu C., Liu J., Liu X., Sun Y. Construction and evaluation of a novel humanized HER2-specific chimeric receptor. Breast Cancer Res. 2014;16:R61. doi: 10.1186/bcr3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johnson L.A., Scholler J., Ohkuri T., Kosaka A., Patel P.R., McGettigan S.E., Nace A.K., Dentchev T., Thekkat P., Loew A., et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci. Transl. Med. 2015;7:275ra22. doi: 10.1126/scitranslmed.aaa4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Du H., Hirabayashi K., Ahn S., Kren N.P., Montgomery S.A., Wang X., Tiruthani K., Mirlekar B., Michaud D., Greene K., et al. Antitumor Responses in the Absence of Toxicity in Solid Tumors by Targeting B7-H3 via Chimeric Antigen Receptor T Cells. Cancer Cell. 2019;35:221–237.e8. doi: 10.1016/j.ccell.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Majzner R.G., Theruvath J.L., Nellan A., Heitzeneder S., Cui Y., Mount C.W., Rietberg S.P., Linde M.H., Xu P., Rota C., et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019;25:2560–2574. doi: 10.1158/1078-0432.CCR-18-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Navai S.A., Derenzo C., Joseph S., Sanber K., Byrd T., Zhang H., Mata M., Gerken C., Shree A., Mathew P.R., et al. Proceedings of the 110th Annual Meeting of the American Association for Cancer Research. 2019. Administration of HER2-CAR T cells after lymphodepletion safely improves T cell expansion and induces clinical responses in patients with advanced sarcomas. Abstract nr LB-147/4. [Google Scholar]
- 56.Paszkiewicz P.J., Fräßle S.P., Srivastava S., Sommermeyer D., Hudecek M., Drexler I., Sadelain M., Liu L., Jensen M.C., Riddell S.R., Busch D.H. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J. Clin. Invest. 2016;126:4262–4272. doi: 10.1172/JCI84813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chang W., Brohl A.S., Patidar R., Sindiri S., Shern J.F., Wei J.S., Song Y.K., Yohe M.E., Gryder B., Zhang S., et al. MultiDimensional ClinOmics for Precision Therapy of Children and Adolescent Young Adults with Relapsed and Refractory Cancer: A Report from the Center for Cancer Research. Clin. Cancer Res. 2016;22:3810–3820. doi: 10.1158/1078-0432.CCR-15-2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gryder B.E., Pomella S., Sayers C., Wu X.S., Song Y., Chiarella A.M., Bagchi S., Chou H.C., Sinniah R.S., Walton A., et al. Histone hyperacetylation disrupts core gene regulatory architecture in rhabdomyosarcoma. Nat. Genet. 2019;51:1714–1722. doi: 10.1038/s41588-019-0534-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kuroda H., Kutner R.H., Bazan N.G., Reiser J. Simplified lentivirus vector production in protein-free media using polyethylenimine-mediated transfection. J. Virol. Methods. 2009;157:113–121. doi: 10.1016/j.jviromet.2008.11.021. [DOI] [PubMed] [Google Scholar]
- 61.Tian M., Cheuk A.T., Wei J.S., Abdelmaksoud A., Chou H.C., Milewski D., Kelly M.C., Song Y.K., Dower C.M., Li N., et al. An optimized bicistronic chimeric antigen receptor against GPC2 or CD276 overcomes heterogeneous expression in neuroblastoma. J. Clin. Invest. 2022;132 doi: 10.1172/JCI155621. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The FGFR4 expression data for this study are available in dbGaP: phs000720, phs001928, phs001052, and https://portal.gdc.cancer.gov/for the TCGA data. ChIPSeq data have been deposited at Gene Expression Omnibus (GSE116344) and is publicly available.
-
•
This paper does not report any original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.