

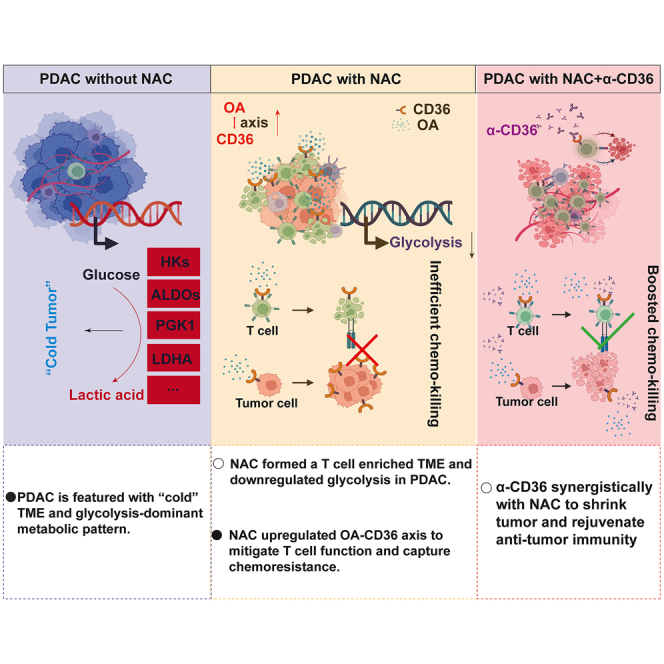
Summary
The molecular dynamics of pancreatic ductal adenocarcinoma (PDAC) under chemotherapy remain incompletely understood. The widespread use of neoadjuvant chemotherapy (NAC) provides a unique opportunity to investigate PDAC samples post-chemotherapy. Leveraging a cohort from Fudan University Shanghai Cancer Center, encompassing PDAC samples with and without exposure to neoadjuvant albumin-bound paclitaxel and gemcitabine (AG), we have compiled data from single-cell and spatial transcriptomes, proteomes, bulk transcriptomes, and metabolomes, deepening our comprehension of the molecular changes in PDACs in response to chemotherapy. Metabolic flux analysis reveals that NAC induces a reprogramming of PDAC metabolic patterns and enhances immunogenicity. Notably, NAC leads to the downregulation of glycolysis and the upregulation of CD36. Tissue microarray analysis demonstrates that high CD36 expression is linked to poorer survival in patients receiving postoperative AG. Targeting CD36 synergistically improves the PDAC response to AG both in vitro and in vivo, including patient-derived preclinical models.
Keywords: pancreatic cancer, immune microenvironment, tumor metabolism, neoadjuvant chemotherapy
Graphical abstract
Highlights
-
•
The proteo-transcriptomic differences in chemo-treated and untreated PDAC are shown
-
•
Analyzing immune and metabolic changes tied to chemotherapy in PDAC
-
•
Boosting the potency of chemotherapy via CD36 targeting, with translational potential
Tang et al. conduct a comprehensive multi-modal analysis, uncovering that neoadjuvant chemotherapy has a profound impact on the immune microenvironment and metabolic patterns in pancreatic ductal adenocarcinoma. Their findings suggest that targeting CD36 could potentially enhance the effectiveness of chemotherapy and bolster the anti-tumor immune response.
Introduction
Almost 80% of patients with pancreatic ductal adenocarcinoma (PDAC) miss the opportunity for upfront resection (UR) and must undergo chemotherapy.1 Even for resectable PDACs, regular postoperative adjuvant chemotherapy is highly recommended by current guidelines, which makes chemotherapy a cornerstone for PDAC clinical management. Nonetheless, PDAC is still one of the most lethal diseases, with a 5-year overall survival (OS) of less than 10%,2 which means that additional treatment is warranted to improve the efficacy of chemotherapy for patients with PDAC.
As of now, an understanding of how PDAC responds to chemotherapy remains limited. The adoption of neoadjuvant chemotherapy (NAC), administering chemotherapy before surgery, enables the acquisition of treated PDAC samples for exploring novel targets associated with enhancing chemotherapeutic efficacy. NAC presents a valuable opportunity to investigate strategies for improving therapeutic outcomes in PDAC patients.3,4,5
Recent studies have provided evidence suggesting that some types of chemotherapy-induced tumor cell death can elicit immunogenic cell death, thereby activating antitumor immunity within the tumor microenvironment,6 suggesting that chemotherapy may facilitate antitumor immunity and synergistically promote immunotherapy. However, chemotherapy-induced damage to bone marrow homeostasis also leads to systemic immune inhibition, which seems to add difficulties for fueling sustainable antitumor immunity. Notably, a recent study alerted that NAC may dampen interferon (IFN)-α and IFN-γ responses in cancer, which could support immune evasion.7 As an immunologically cold tumor, most PDAC cases do not show a response to any common immunotherapy, such as immune checkpoint inhibitors.8 Whether NAC can heat PDAC and create novel immunoregulatory targets deserves to be investigated.
Here, we performed multiple complementary approaches to explore the differences in molecular characteristics between PDAC treated with preoperative chemotherapy versus UR tumor samples. We observed that NAC significantly reprogrammed the metabolic pattern of PDAC and reshaped the tumor microenvironment. Furthermore, systematic upregulation of CD36, a metabolic receptor, was found to be a side effect at the molecular level, attributed to chemotherapy based on our data, whose blockade synergistically improved the efficacy of albumin-bound paclitaxel and gemcitabine (AG) on PDAC in vivo.
Results
NAC triggered broad alterations in the transcriptome and proteome of PDAC samples to achieve survival benefits
To study PDAC’s molecular response to NAC, we analyzed transcriptome and proteome in resected human tumor tissues from patients treated with or without NAC. Patient characteristics are summarized in Table S1. Prior trials have reported NAC’s survival benefits by enabling timely surgical resection after chemotherapy-induced tumor size reduction,9,10 visible in decreased Ki67-positive malignant cells (Figure S1A). Our analysis revealed 4473 differentially expressed genes (DEGs) between PDAC samples treated with NAC and UR samples at the transcriptome level based on DESEQ2 algorithms (Figures 1A and S1B). Among these DEGs, 3,603 genes were upregulated and 870 genes were downregulated in the NAC samples (Table S2). Hence, we wondered whether NAC conferred a survival benefit by affecting gene expression. To explore this hypothesis, we assessed the effect of identified DEGs on patient OS using three large PDAC cohorts (Figures 1A and 1B). Among the downregulated genes, 69 genes were significantly associated with patient OS, and 68 (98.6%) of these genes were related to poor OS in The Cancer Genome Atlas cohort. In contrast, among the upregulated genes after NAC, 120 genes were significantly associated with patient OS, and 109 (90.8%) of these were associated with prolonged OS.
Figure 1.

NAC rebuilt a survival-benefitted transcriptome and proteome landscape in PDAC
(A) Heatmap showing that more DEGs related to good prognosis were overexpressed in NAC samples. The hazard ratios (HR) were calculated by batched univariate Cox regression. Z score referred to relative gene expression level.
(B) Kaplan-Meier curve indicating that NAC-upregulated genes were associated with a prolonged survival period and downregulated genes were associated with poor prognoses in E-MTAB-6134, The Cancer Genome Atlas (TCGA), and GSE71729 cohorts. The statistical significance of survival difference was detected by log rank test.
(C) The genes that were downregulated in NAC samples were mostly associated with the proliferative phenotype in pancreatic cell lines (DepMap data). The statistical significance of percentages difference was detected by chi-square test.
(D) Higher percentages of metabolic quiescent and less immune-cold and fibrotic PDAC subtypes were found in the NAC groups. The statistical significance of percentages difference was detected by chi-square test. RR, relative risk.
(E) A complex heatmap showing the DEPs and the distribution of clinical parameters between NAC and UR groups.
(F) A high proportion of DEPs were immunity or metabolism associated.
In the GSE71729 cohort, 92.7% of downregulated genes associated with prognosis correlated with worse OS, while 57.0% of upregulated genes associated with prognosis correlated with better OS. Similar trends were observed in the E-MTAB-6134 cohort, where 84.2% of downregulated genes associated with prognosis correlated with worse OS, and 65.8% of upregulated genes associated with prognosis correlated with better OS. Using the single-sample gene set enrichment analysis (ssGSEA) algorithm, we identified enriched gene sets (NeoHsig and NeoLsig) in the NAC group and examined their relationship with patient OS. NeoHsig was associated with prolonged OS, while NeoLsig predicted poorer survival across the three independent PDAC cohorts (Figure 1B). Additionally, the Wilcoxon test, another statistical method, was applied to identify DEGs and evaluate their association with patient prognoses. The results were consistent with the aforementioned findings (Figures S1C and S1D).
Furthermore, a CRISPR-based screening method was used to analyze the effects of upregulated and downregulated genes in pancreatic cancer cell lines. The results indicated that most downregulated genes after NAC potentially contributed to the proliferative phenotype of pancreatic cancer cells, and vice versa (Figure 1C). Previous studies have divided PDAC into many molecular subtypes based on their different molecular characteristics, including Moffitt subtypes,11 Collisson subtypes,12 microenvironment subtypes,13 and metabolic subtypes.14 To explore whether NAC promoted the transition of molecular subtypes in PDACs, we analyzed the percentage of each subtype between PDAC samples with and without NAC using chi-square test. We found that the Moffitt subtype percentages were similar between the two groups, while fewer immune-cold and -fibrotic and more metabolic-quiescent subtypes were observed in NAC samples, suggesting that stringent immune and metabolic reprogramming occurred within PDAC during NAC (Figure 1D). In this study, data-independent acquisition proteome analysis identified a total of 478 differentially expressed proteins (DEPs) (Table S3). Among them, approximately 29.7% of these DEPs belonged to immune or metabolic protein categories. This suggests that significant immune and metabolic rewiring occurred in the PDAC microenvironment after NAC treatment (Figures 1E and 1F). Then, we performed functional annotations and enrichment analyses (Figures S2A–S2G and Tables S4–S9). The results showed a significant enrichment of DEPs and DEGs in various metabolic pathways. Notably, proteins associated with cholesterol metabolism and fatty acid absorption were enriched among the upregulated proteins in NAC samples (Figure S2C). In contrast, genes related to central carbon metabolism, such as glycolysis, were enriched among the downregulated genes in NAC samples (Figure S2F). GSEA analysis further revealed the universal attenuation of mitotic cytokinesis, basal transcription factor activity, nucleotide salvage, and nucleosome positioning activity in NAC samples, potentially triggering downstream reactions (Figure S2I). Overall, the integrated analysis demonstrated an intermediate correlation between gene expression in the transcriptome and proteome (Figure S2J).
NAC shaped an immunogenic antitumor microenvironment in PDAC
Single cell RNA-sequencing (sc-RNA-seq) was performed on fresh PDAC samples to delineate the molecular alterations associated with chemotherapy in the microenvironment. The basic clinical information for each sample is presented in Table S10. After appropriate quality control, a total of 46,993 cells, including 37,626 cells originating from NAC samples and 9,367 cells from UR tumor tissues, were analyzed (Figures S3A and S3B). Cells were further clustered into eight subgroups based on their unique molecular markers (Figures 2A–2C). Then, we refined T cell subclusters and found that the percentage of CD8+ T cells, an essential antitumor component in the tumor microenvironment, was significantly increased in PDACs that received NAC (Figures 2D, 2E, and S3C). In addition, we conducted T cell receptor (TCR) sequencing analysis, revealing that the clonotype of TCRs was increasingly expanded in PDAC samples with NAC, which indicated enhanced reactivity of intratumoral immune components to eliminate tumor cells after chemotherapy (Figures 2F, 2G, and S3D).
Figure 2.

NAC reprogrammed the immune microenvironment in PDAC
(A) UMAP plot revealed cell clusters in PDACs with or without NAC. NK, natural killer.
(B) The distribution of marker genes among different cell clusters. Red dots refer to enriched gene expression.
(C) The percentage of different cell clusters in PDAC samples with and without NAC.
(D) UMAP plot revealing subclusters for NK and T cells.
(E) More CD8+ T cells were enriched in PDACs with AG treatment, which was showed by chi-square test.
(F) PDACs that received NAC showed higher TCR clonotype expansion. n, means the number of expanded clonotypes.
(G) The Gini index for TCR clonotype was increased in PDACs with NAC (mean with standard deviation).
(H) Correlation network for immune cells in either the NAC or UR group. The size of circles reflected the infiltration level for cells.
(I) Comparison of immune cell constitution between the UR and NAC groups.
(J) IHC analysis revealed increased CD8+ T cells in PDACs treated with NAC (n = 54). The upper panel shows the representative graph for positive staining of CD8+ T cells.
(K) The spatial transcriptome showed that more CD8+ T cells were enriched in the tissue slices of PDAC samples with NAC.
(L) mIF showed that an increase in GZMB+ CD8+ T cells was enriched in PDACs treated with NAC (n = 20) (mean with standard deviation). The Left) panel showed the representative graph for GZMB+ CD8+ T cells in PDACs.
(M) More patients who had received NAC were predicted to be responders to immunotherapy, which analyzed using ImmuneCellAI algorithm. The statistical significance shown in this figure was detected using t test.
To evaluate the global alteration of immune infiltrates, we calculated the relative abundance of immune cells within the PDAC microenvironment using two bioinformatic algorithms, ssGSEA and CIBERSORT, which were based on the bulk transcriptome. As expected, we found that the level of infiltrating CD8+ T cells was increased in the NAC group compared with the UR group (Figures 2H, 2I, and S3E). We also generated a spatial transcriptomic source for PDACs with and without NAC (Figure S3F). The spatial transcriptome, three additional transcriptome-based algorithms (QUANTISEQ, TIMER, and XCELL), and immunohistochemistry (IHC) results also supported the enrichment of CD8+ T cells in tumors after NAC (Figures 2J, 2K, and S4A). Subsequently, we conducted a comparison of the activity of 49 knowledge-based immune signatures between the two cohorts. Our analysis revealed that both CD8+ T cells and the cytolytic activity signature exhibited a trend toward enrichment in NAC samples (Figure S4B). Granzyme was the main substance released by cytotoxic T cells mediating the killing of tumor cells through apoptotic or immunogenic cell death, as we previously summarized.15 The proteome data generated in the present study also revealed an upregulation trend for granzymes A, B, and K in tumor tissues with NAC, exhibiting marginal significance (Figure S4C). Furthermore, we found that the infiltration level of granzyme B+ CD8+ T cells within the tumor microenvironment was elevated in pretreated PDACs (Figure 2L). Then, we investigated whether the levels of infiltrating regulatory T cells (Tregs) in the PDAC microenvironment after NAC were different from those in UR samples, given that Tregs played a notorious immunosuppressive function in tumors. To our surprise, no distinct difference in either Treg infiltration or FOXP3+ cell density was found between the NAC and UR groups (Figure S4D) (p = 0.42 and 0.87, respectively), while another immunosuppressive cell,16,17,18 the infiltrated CD163+ macrophages, potentially decreased in PDACs treated with NAC (Figure S4E). TIMER and EPIC algorithms respectively indicated that NAC increased the abundance of B cells and CD4+ T cells based on the t-test, which are important components of tertiary lymphoid structures (TLS) (Figures S4F and S4G). Recent studies have shown that TLSs could be a sensitive biomarker for predicting immunotherapy response and patient prognosis. Notably, our results also demonstrated that TLS density was significantly increased in the NAC group (p=0.003) (Figures S4H and S4I). In addition, we calculated the immunophenoscore for each PDAC sample to evaluate the comprehensive immunogenic status of the tumor. The results showed that samples collected from patients who underwent NAC had a higher immunophenoscore (Figure S4J, p=0.008). All these results reflected enhanced immune activity and the potential benefit from immunotherapy after NAC in samples of PDAC. Using the ImmuCellAI algorithm, a method used to predict the response to immunotherapy, we found that only the NAC group included potential responders to immunotherapy (response rate 12.8% in the NAC group vs. 0.0% in the UR group), which indicated that NAC could be an effective method to boost the efficacy of subsequent immunotherapy (Figure 2M). Interestingly, the expression levels of programmed cell death 1, programmed cell death 1 ligand (PDL1), and CTLA4 were comparable between NAC and UR samples (Figure S4K), suggesting that traditional immune checkpoints may not be the optimal target of immunotherapy for PDAC patients who receive chemotherapy, and novel targets associated with chemotherapeutic exposure for immunotherapy may be required.
Chemotherapy-induced metabolic heterogeneity in PDAC is linked to immune microenvironment remodeling
Clustering analysis classified PDAC cells into two major clusters (Figure S5A), which is consistent with a previous report.19 Further analysis annotated 11 subclusters for PDAC cells, of which a FXYD3+TFF1– subtype was significantly decreased in NAC samples (Figures 3A and S5B). Metabolic reprogramming in cancer cells has been regarded as the hallmark of cancers and plays an important role in immune evasion. Through single-cell-level analysis of metabolic enzyme expression, we found that lipid and glutamine metabolism were significantly upregulated in malignant cells that survived chemotherapy. In contrast, as the core metabolic approach of tumor cells to generate energy, glycolysis was apparently lower in PDACs treated with NAC (Figure 3B). Fructose metabolism, which shared many metabolites and enzymes with glycolytic flux, also showed a decrease in the NAC samples (Figure 3B). Then, we showed a broad downregulation of glycolytic enzymes by analyzing the differential expression of metabolic enzymes along the glycolytic axis (Figure 3C). In addition, through gene list-based annotation, we found that the percentage of glycolytic malignant cells was significantly decreased in NAC samples (Figure 3D), while metabolic pathways such as cholesterol synthesis, fatty acid metabolism, sialic acid metabolism, and tryptophan metabolism were significantly upregulated (Figure S5C).
Figure 3.

Single-cell and spatial transcriptome analyses revealed that metabolic enzyme alterations may contribute to immune infiltration in a treatment-associated background
(A) UMAP analysis showed subclusters for ductal cells in PDACs with or without NAC treatment. The lower right panel presented the percentage of c10 ductal cluster was lower in PDAC samples treated with NAC.
(B) Single-cell metabolism analysis showed differentially activated metabolic pathways between NAC and UR groups.
(C) Transcriptome analysis at the single-cell level revealed differentially expressed glycolytic enzymes in pancreatic ductal cells.
(D) The percentage of glycolytic ductal cells was lower in PDACs with NAC treatment. The statistical significance of percentage difference was detected by chi-square test.
(E) Consensus clustering distinguished four types of ductal cells based on the single-cell level activity of metabolic pathways.
(F) Heatmap showing the distribution of metabolic pathway activity in distinct ductal subtypes and treatment cohorts.
(G) C4 ductal cells were more enriched in the NAC group, while C1 ductal cells were more enriched in the UR group. The statistical significance of percentage difference was detected by chi-square test.
(H) Spatial transcriptome visualized the distribution of subtype-associated signatures in PDACs with or without NAC treatment. MA, malignant area; NA, normal pancreas area; SA, stroma area.
(I) Most genes downregulated in treated PDACs were negatively correlated with the high level of cytotoxic signature and CD8+ T cell infiltration, and vice versa.
(J) CellChat algorithm showed PDACs with or without NAC were featured with different communication patterns.
(K) CellChat algorithm deregulated communication signaling from ductal cells to T cells in PDACs with or without NAC treatment.
Furthermore, we classified PDAC cells into four subtypes using an unsupervised clustering algorithm based on the activities of metabolic pathways, which are referred to as C1–C4 (Figures 3E and S5D). We next compared the distribution of each ductal subtype between PDAC samples with and without NAC and found that C4, characterized by high cholesterol biosynthesis, was significantly enriched in PDACs with NAC, while another subtype C1 was enriched in samples without chemotherapeutic exposure (Figures 3F and 3G). Here, we also generated a subtype-specific gene signature for each ductal cell subtype based on the transcriptome at the single-cell level and projected it to the slices coded by sequenced spatial transcriptome data. The results validated that C1 was the dominant ductal cell subtype in PDAC without NAC; in contrast, the distribution of C4 ductal cells was more enriched in treated PDACs (Figures 3H and S5E). All these results suggested that PDAC cells under chemotherapeutic pressure experienced stringent metabolic reprogramming compared with treatment-naïve cells, which may influence other cells in the tumor microenvironment. In this context, we explored the correlation between upregulated/downregulated metabolic genes and cytotoxic signatures/CD8+ T cell infiltration at the bulk level. Notably, all the genes upregulated in the NAC group were positively associated with CD8+ T cell infiltration and/or cytotoxic signatures, while almost all the genes that were downregulated in the NAC samples showed an inverse trend (Figures 3I and Table S11). Similarly, based on sc-RNA-seq, 71% of the downregulated genes in the NAC group were negatively associated with CD8+ T cell infiltration, which was calculated by integrating nine algorithms for microenvironment estimation (Figure S5F).
Then, we compared the differences in cell-cell interactions between NAC and UR samples. Interestingly, both the number and strength of the ductal-T cell interaction were significantly increased in PDAC samples after NAC (Figure 3J). In detail, interactions including HLA-CD8s, SPP1-CD44 and SPP1-(ITGA4+ITGB1) were more frequently seen in the microenvironment after chemotherapeutic exposure, while interactions such as LAMB1-CD44 were reduced in the NAC group (Figure 3K).
PDACs with chemotherapeutic exposure captured chemoresistance potentially by promoting the accumulation of oleic acid
Compared with single-cell analysis, bulk-level metabolic analysis via transcriptome, proteome, and metabolome had advantages in reflecting the global metabolic landscape of PDAC. The expression of metabolic enzymes either at the protein or transcription level experienced significant alterations after NAC, which is the same as in the single-cell analysis. All the core enzymes along the glycolytic pathway were downregulated at the protein and/or transcriptional level, such as HKs, ALDOs, PGK1, LDHA, and the transporter of lactic acid SLC16A1 (Figure 4A). In contrast, many enzymes in other metabolic pathways, including lipid metabolism, glutamine utilization, and oxidative phosphorylation, were universally upregulated after NAC, potentially for energy compensation (Figure 4A). As an essential enzyme for lactic acid production, LDHA was significantly decreased after NAC at the mRNA level. In addition, proteome data showed a downregulation trend of LDHA with marginal significance, which was further validated by IHC (Figures S5G and S5H). Furthermore, the expression pattern of LDHA and CD8 showed an inverse trend in samples with and without NAC (Figure S5I). Through analysis of bulk transcriptomic data, we found that the negative correlation between LDHA and CD8 was more significant in PDAC samples with NAC than in treatment-naïve samples (Figure S5J). In addition, we found no correlation between LDHA and CD8 in treatment-naïve samples (Figure S5K). To better monitor glycolytic activity before and after NAC, we generated PDX models and administered chemotherapy or placebo to PDX mice with comparable xenograft volumes. Next, we established PDXOs using dissociated xenografts with or without chemotherapeutic exposure and cultivated them in vitro (Figures 4B and 4C). To explore whether glycolytic addiction varied in surviving PDAC cells after NAC, we performed glucose metabolism flux analysis with uniformly labeled C13-glucose by using PDXOs derived from treated or naive mice. The results revealed a significant increase in C13-glucose-derived tricarboxylic acid cycle metabolites (e.g., m+2 citrate) in PDXOs with previous chemotherapy, while the C13-labeled glycolytic metabolites (e.g., m+3 lactate) were dramatically decreased (Figure 4D). Although most the tricarboxylic acid (TCA) carbon in pancreatic cancer was noted as being derived from glutamine,20 the decreases in C13-labeled glycolytic metabolites could still indicate that PDAC cells that survived NAC exhibited reduced dependency on glycolysis; nonetheless, how metabolic compensation works remained unclear. To systematically investigate the perturbance of metabolic flux in PDACs during chemotherapeutic exposure, we estimated the metabolic flux in both NAC and UR samples and revealed that the flux to acetyl-coenzyme A (CoA) and succinyl-CoA, the products of fatty acid beta-oxidation, was significantly increased in chemotherapy-treated samples, which could either enter the TCA cycle or generate ketone bodies for energy preservation. Meanwhile, PDACs with NAC upregulated cholesterol use, while treatment-naïve tumors featured more active nucleic acid flux (Figure 4E). Furthermore, we performed untargeted metabolome analysis to screen NAC-specific metabolites. A total of 270 differential metabolites (DMs) were detected between the NAC and UR groups (Figure S6A and Table S12). Bray-Curtis NMDS, PCoA and PCA confirmed the capability of these DMs in distinguishing the NAC and UR samples (Figures S6B–S6D). Notably, lactic acid abundance was dramatically decreased in NAC samples, while many metabolites involved in fatty acid metabolism, oxidative phosphorylation, and glutamine metabolism were significantly increased (Figure S6E), which supported the metabolic enzyme-based findings of chemotherapy-associated metabolic reprogramming. We further conducted energy metabolism-targeted metabolome for samples with NAC and UR. The results validated that the glycolytic products lactic acid and 3-P-glycerate were decreased in samples treated with NAC, while alpha-ketoglutarate was increased (Figure 4F). In addition, we conducted spatial metabolome analysis for a slice of resected PDAC with NAC. Overall, regional differences in metabolite distribution were observed and could be classified into several clusters (Figure S6F). Some metabolites were specifically abundant in CD8+ T cell-enriched regions; for example, PG_38:5 was significantly upregulated in cluster_7 regions (Figures S6G and S6H).
Figure 4.

Multiomics analysis supported that NAC decreased glycolysis but developed compensatory approaches
(A) Transcriptome and proteome analysis revealed alterations in metabolic enzymes associated with AG treatment. The log fold-change value of transcriptome alteration is shown in yellow, while proteome alteration is shown in black.
(B) Construct PDXO models from PDX mice with and without AG treatment.
(C) A representative graph for PDXO in bright fields of microscope.
(D) Metabolic flux experiments validated that PDXO separated from PDX mice treated with AG showed less glycolytic activity. The upper panel showsfocused isoforms of metabolites in glycolysis and TCA cycle. The heatmap reflects the relative abundance of isoforms shown in the ideographs. “m” referred to the number of C13 in the metabolite structure (n = 4).
(E) Comparison of metabolic flux between the AG and control groups based on RNA-seq data (scFEA algorithm).
(F) Targeted metabolomics analysis showed the differences of lactic acids, 3-PD, phosphoenolpyruvate, and alpha-ketoglutarate between PDAC samples with and without NAC, which implied NAC is associated with downregulated glycolytic activity in PDAC (n= 44) (mean with standard deviation).
(G) Cell-Counting-Kit-8 (CCK-8) results showed oleic acid (50 μM) promoted the proliferation of panc-1 cells and could be blocked by targeting CD36, on the contrary (the upper panel), palmitic acid (50 μM) had no effect on the proliferation of panc-1 cells (the lower panel) (n = 5).
(H and I) Oleic acid, as opposed to palmitic acid, was found to enhance the growth of PDOs according to the viability assay. The upper section of the figure displaysRepresentatives of PDOs cultured under the indicated conditions for 7 days. The lower section features a bar plot illustrating the relative viability of different groups, measured using the CellTiter-Glo 3D Cell Viability Assay (n = 5).
(J) EdU assay showed oleic acid may fuel the drug resistance to AG but could be blocked by targeting CD36 (n = 4). The statistical significance shown in this figure was detected using t test.
To explore the impact of metabolites that were upregulated in NAC tissues, we stimulated human T cells isolated from PBMCs with the top ten available upregulated metabolites or their synthetic substitutions (Figures S6I and S6J). Then, we measured the secretion of IL-2 and IFN-γ by T cells after metabolite treatment, which showed that most of these metabolites are capable of promoting the secretion of cytokines, while only oleic acid showed an inverse trend, suggesting that oleic acid might be a chemotherapy-associated unfavorable factor for T cell function (Figure S6K). Oleic acid treatment significantly enhanced the growth of pancreatic cells, as demonstrated by the Cell-Counting-Kit-8 assay. Instead, palmitic acid, which has also been reported to enter tumor cells via CD36, did not show effect on the growth of pancreatic cancer cells. Furthermore, our investigation using the 3D cell viability assay revealed that PDO exhibited accelerated growth in a medium supplemented with oleic acid, compared with the normal control. These effects could be further rescued by blocking oleic acid absorption using an anti-CD36 neutralizing antibody (Figures 4G–4I). Through EdU experiments, we further revealed that panc-1 cells with oleic acid treatment showed higher proliferative rate compared with the control group, which could be mitigated by blocking oleic acid absorption (Figures 4J and S6L), indicating that increased use of oleic acid may render PDAC cells resistant to AG.
Additionally, we performed CD36 knockdown in Panc-1 cells to investigate its impact on cell proliferation and chemotherapeutic resistance (Figure S6M). The results demonstrated that, under normal conditions, CD36 knockdown did not affect cell proliferation. However, it blocked the stimulatory effects of oleic acid on cell proliferation (Figure S6N). Similarly, when we treated panc-1 cells with AG in vitro, we observed that oleic acid significantly enhanced chemoresistance in CD36-intact cells, but not in CD36-knockdown cells (Figure S6O). These findings further confirm that oleic acid may exert pro-tumoral effects through its interaction with CD36.
NAC triggered systematic upregulation of CD36 in both the PDAC microenvironment and circulation
Notably, CD36, a transporter of oxidized lipids and free fatty acids such as oleic acid, was also significantly overexpressed in PDACs after NAC, as shown by both proteome sequencing and IHC validation (Figures 5A and S7A). Among all metabolism-related molecules upregulated in the NAC group, CD36 was the only protein distributed on the membrane, which made it easier to target (Figure S7B). To ensure the upregulated oleic acid-CD36 axis after NAC, we compared the oleic acid uptake ability between PDXOs derived from PDX mice with and without AG treatment. Based on tracing 13C-labeled oleic acid, we found that the abundance of 13C-labeled oleic acid was significantly increased in PDXOs derived from AG-treated mice, confirming the enhanced capability of PDAC cells to absorb oleic acid after chemotherapy (Figure 5B). As shown in the last section, targeting CD36 overcomes oleic acid-mediated tumor growth and chemoresistance in vitro. Given that chemotherapy is a systematic and unselected treatment, we further investigated whether CD36 was only induced in tumor cells. To our surprise, through analysis of resected PDAC tissues, we found that CD36 was also overexpressed on tumor-infiltrating CD8+ T cells and widely distributed in TLSs (Figures 5C and 5D). Then, we performed a large-scale correlational analysis between CD36 expression and 112 cell-specific signatures. As expected, CD36 was highly correlated with signatures of endothelial cells because of its constitutive expression on many types of vascular structures. However, CD36 also showed a high correlation with many signatures of immune cells, especially CD8+ T cells, only in the NAC group, which meant that CD36 could be induced in immune cells when encountered with chemotherapeutic exposure (Figure 5E). Beyond tumor-infiltrating immune cells, we detected CD36 expression on circulatory immune cells in patients with and without NAC. Through an analysis of immune cells with myeloid linkage, we found that, although the percentage of immunosuppressive CD11b+ CD45+ cells was decreased in NAC samples, the distribution of CD36 was significantly expanded in CD11b+ CD45+ cells, especially for CD11b+ CD14+ cells, which are monocyte-like myeloid-derived suppressor cells (Figure S7C). Overall, increased CD36 expression was found on CD45+ cells with NAC, of which the trend for CD45+ CD8+ T cells was the most significant (Figure 5F). To further explore the impact of the upregulated oleic-CD36 axis on the function of CD8+ T cells, we measured the IFN-γ secretion with the intended treatment. Flow cytometry results revealed that oleic acid accumulation may dampen the secreting ability of CD8+ T cells, and this effect could be rescued by blocking CD36 (Figure 5G).
Figure 5.

CD36 was systematically upregulated in tumor cells and resident and circulating immune cells
(A) Immunohistochemical staining indicated that CD36 expression is increased in PDACs treated with NAC compared with UR samples (n= 54) (mean with standard deviation). Representative graphs are shown on the left.
(B) Metabolic flux experiments showed that PDXOs derived from PDXs treated with AG had increased capability to uptake oleic acid (n = 3).
(C) The percentages of CD36+CD8+ T cells and CD36+GZMB+CD8+ T cells were significantly upregulated in PDACs treated with NAC (n = 10) (mean ± SD). Representative graphs are shown on the left.
(D) Representative graph by mIF showed co-localization of CD36 and TLS in PDAC.
(E) Heatmap showing the correlation between CD36 expression and infiltration of immune cells, which indicated that CD36 expression was highly correlated with CD8+ T cell abundance only in PDACs treated with NAC.
(F) Flow cytometry for PBMCs from PDAC patients showed CD36 was upregulated in circulating CD8+ T cells from patients treated with NAC. Left panel displays t-Distributed stochastic neighbor embedding (TSNE) analysis for labeled cell clusters (mean with standard deviation). Right panel displays the higher percentage of both CD36+ CD8+ T cells and CD36+ CD45+ immune cells in samples with NAC.
(G) Flow cytometry showed that oleic acid decreased the percentage of IFN-γ+ CD8+ T cells and could be rescued by CD36 blockage (n = 3).
(H) T cells treated with lysates from different PDAC samples manifested distinct tumor-killing ability, which is showed by LDH-releasing experiments (n= 5).
(I) T cells treated with lysates from CD36-low NAC samples showed significantly higher IFN-γ secretion compared with UR samples.
(J) Caspase3/7 detection indicated that targeting CD36 synergistically enhanced the killing effect of AG based on a PDAC organoid/PBMC coculture system. Left panel shows the representative graph of caspase3/7-positive organoids at the time points 0 and 48 h. Right panel shows the percentage of apoptotic organoids in each group, which was evaluated through flow cytometry.
(K) mIF showed CD36 blockage enhanced the killing effect of AG, which was showed by detecting Ki67+ organoids via mIF technology.
(L) Successful construction of ovalbumin (OVA+) murine KPC organoids.
(M) The OVA+organoid/OT-1-cell coculture system validated the synergistic effect of CD36 blockade on AG-mediated tumor killing (n= 3). Left panel shows the representative graph of caspase3/7-positive organoids. Right panel shows the percentage of apoptotic organoids in each group, which was evaluated through flow cytometry. The statistical significance shown in this figure was detected using t test.
While CD8+ T cells were more highly enriched in the PDAC microenvironment with NAC, the percentage of CD36+ CD8+ T cells also increased, which may mitigate the immunogenic effect of preoperative chemotherapy. In this context, we treated CD8+ T cells with PDAC tissue lysates with different levels of CD36 expression and further detected the cytotoxicity and IFN-γ secretion of the treated cells. Overall, both the cytotoxicity and IFN-γ release of CD8+ T cells were increased in the group treated with the tissue lysates from NAC samples; notably, that increase was more obvious in the cases from CD36-low NAC samples (Figures 5H, 5I, and S7D–S7F). In samples treated with NAC, CD36 expression in lysate was significantly negatively correlated with the LDH-releasing level (Figure S7G). In this context, we generated an oleic acid-containing patient-derived organoid-peripheral blood mononuclear cell (PDO-PBMC) coculture system with the indicated treatment and detected the percentage of apoptotic organoids. We observed a synergistic effect of CD36 blockade and AG treatment on PBMC-mediated tumor killing, suggesting the potential clinical value of combining CD36 blockade with AG agents in NAC or adjuvant treatment for PDAC patients (Figures 5J and 5K). Moreover, we constructed ovalbumin-expressing KPC organoids to achieve antigen-specific immune responses mediated by OT-1 cells (Figure 5L). Based on this experimental design, we observed similar results to those reflected in the PDO-PBMC coculture system. These findings further confirmed the synergistic effect of combining CD36 targeting with AG treatment (Figure 5M).
Targeting CD36 synergistically promoted AG efficacy for PDAC in preclinical models
Next, we investigated whether anti-CD36 therapy could improve the efficacy of AG in vivo based on several preclinical animal models. Tumor xenografts that received AG combined with the neutralizing antibody of CD36 grew significantly slower than those treated with either single AG or anti-CD36 therapy, suggesting that additional anti-CD36 treatment with AG may optimize NAC efficacy (Figures 6A–6D). Further analyses validated that AG exposure increased both the intratumoral infiltration of CD8+ T cells and the percentage of granzyme B+ CD8+ T cells (Figures 6E and 6F). Likewise, we found that AG treatment increased CD36 expression on CD8+ T cells but not CD4+ T cells, which was consistent with what we found in the previous sections (Figure 6F). Interestingly, AG treatment also increased the percentages of CD36+ myeloid cells in PDAC (Figures S7H and S7I). The combination of anti-CD36 therapy with AG effectively increased the expression of cytotoxic signatures and the levels of IFN-γ and tumor necrosis factor α in tumor xenografts, further revealing that anti-CD36 was a targetable immunoregulatory molecule during AG treatment (Figures 6G and S7J). Subsequently, we generated orthotopic murine models of pancreatic cancer and grouped them into different treatment arms (Figure 6H). The group treated with AG and anti-CD36 antibody had the longest survival interval (combination vs. AG alone, p=0.042; combination vs. anti-CD36 antibody alone, p < 0.0001; log rank test) (Figure 6I). Moreover, we generated a huPBMC-PDX model to test whether the combination of anti-CD36 and AG under an immunocompetent background had a synergistic effect. During follow-up, we found that humanized PDX mice treated with the combination of CD36 blockade and AG significantly slowed tumor growth compared with AG alone, while a single use of CD36 blockade did not achieve satisfactory efficacy (Figure 6J). The DepMap database integrated information on drug resistance and data on gene-dependent tumor growth. By analyzing gemcitabine sensitivity and CD36 dependency in pancreatic cancer cell lines, we found that gemcitabine-insensitive pancreatic cancer cell lines are more dependent on CD36, supporting the importance of CD36 in gemcitabine-based chemoresistance for PDAC (Figure S7K).
Figure 6.

Targeting CD36 synergistically promoted AG-mediated killing of PDAC in preclinical models
(A) Visual presentation of subcutaneous xenograft murine PDAC tumor models (C57 mice) for each group.
(B) Measurement of tumor volumes showed CD36 blockage synergistically promoted AG-mediated killing of PDAC in subcutaneous xenograft murine PDAC tumor models.
(C) Measurement of tumor weights showed CD36 blockage synergistically promoted AG-mediated killing of PDAC in subcutaneous xenograft murine PDAC tumor models (n = 5).
(D) Representative IHC staining showed Ki67 expression in subcutaneous xenografts treated with different regimens.
(E) t-Distributed stochastic neighbor embedding (TSNE) analyses showed the clustering for CD36+ CD8+ T cells and GZMB+ CD8+ T cells.
(F) Flow cytometry revealed that more CD8+ T cells infiltrated PDAC with NAC, while the percentage of CD36+ CD8+ T cells also increased (n = 5) (mean with standard deviation).
(G) ELISA results showed the combination of AG and CD36 blockade significantly improved IFN-γ and tumor necrosis factor α (TNF-α) levels intratumorally (n = 5).
(H) Representative image of orthotopic murine models of PDAC.
(I) Kaplan-Meier curve revealed the combination of CD36 blockade and AG significantly prolonged the survival interval of mice that received orthotopic PDAC cell transplantation (n = 10). Circle or square referred to a happened event (death or censored). Censored event means the mice is still alive at the time point that we ended follow-up.
(J) CD36 blockade synergistically with AG regimens optimally narrowed the PDAC tumor size in a humanized PDX model (n = 10).
(K) Representative IHC staining image of CD36-high and -low PDAC.
(L) Kaplan-Meier curve showed increased CD36 expression predicted worse prognosis of PDAC patients with adjuvant AG chemotherapy. The statistical significance shown in this figure was detected using t test.
Next, we detected CD36 expression for PDAC samples in a tissue microarray (TMA) cohort (Table S13). After screening participants who underwent adjuvant AG treatment after surgery, we discovered a significant association between upregulated CD36 expression and poorer prognosis in this specific population (Figures 6K and 6L), suggesting that anti-CD36 plus AG could be a promising strategy not only for the clinical management of neoadjuvant PDAC, but also extended to adjuvant chemotherapy for postoperative patients.
Discussion
The wide acceptance and use of NAC marks a new treatment era for PDAC. Beyond the strengths of NAC observed in clinical practice, the idea of chemotherapy before surgery has made it possible to investigate human resected PDAC tissues under chemotherapeutic exposure. Although almost all patients diagnosed with PDAC should accept chemotherapy, little is known about how the molecular landscape of tumors varies under chemotherapeutic pressure.
A previous study showed that PDAC treated with FOLFIRINOX regimens exhibited the densest CD8+ cell infiltration, but less M2 macrophage infiltration compared with a treatment-naïve group.21 Hwang et al.22 recently applied single-nucleus RNA-seq to analyze frozen samples treated with NAC or UR, which also showed CD8+ T cells increased in samples with NAC. In contrast, Farren et al.23 compared the differences in immune infiltrates and immune-related markers between PDAC patients treated with UR or neoadjuvant FOLFIRINOX. Their results indicated that FOLFIRINOX did not alter the infiltration of CD8+ T cells in either the tumor-enriched area or the immune cell-enriched area, while the combination of neoadjuvant stereotactic radiotherapy with FOLFIRINOX even decreased the number of CD8+ T cells within these regions.23 The regimen for NAC studied in the present study is AG. Compared with FOLFIRINOX, which is already composed of four chemotherapeutic agents, AG not only had fewer adverse reactions that could be tolerated by more patients, but also potentially allowed for additional treatment, such as immune checkpoint inhibitors or anti-CD36 treatment we proposed here. Moreover, our previous study showed that the 4- and 5-year survival probabilities of the two strategies were comparable.24 Here, based on bulk and single-cell level sequencing techniques and IHC validation, we showed that PDAC samples with neoadjuvant AG were enriched with more CD8+ T cells and TLSs, but fewer M2-polarized macrophages, indicating that AG could be an immunogenic regimen to transform cold PDAC to an immune cell-enriched state.
Unfortunately, the rate of objective response to AG treatment is only 23%.25 According to another clinical trial, the surgical conversion rate of NAC was only approximately 35.9% (95% confidence interval, 24.3–48.9).10 Hence, overcoming resistance to AG is still a huge but significant challenge for improving the prognosis of patients with PDAC. Our study revealed that broad metabolic reprogramming occurred in PDAC samples treated with NAC by metabolome and metabolic flux assays. We showed systemic oleic acid accumulation and the upregulation of its receptor CD36 in NAC samples. As a targetable metabolic protein, CD36 was broadly upregulated on both tumor and immune cells after NAC. We proposed that tumor cells could increase the absorption of oleic acid by CD36 to survive when they are in chemotherapeutic attack. In contrast, oleic acid exposure and increased CD36 expression on CD8+ T cells significantly damaged their antitumor capability. Interestingly, we also observed increased CD36 expression on myeloid cells in NAC samples. According to a previous study, myeloid cells can take up tumor-derived long-chain fatty acids to fuel themselves and trigger their tumor-promoting activities, which may also be a side effect of NAC.26 Overall, CD36 upregulation potentially undermined the efficacy of NAC, which was supported by multiple preclinical models, suggesting the synergistic value of CD36 blockade with AG treatment.
In clinical practice, not every pancreatic cancer patient is sensitive to NAC. For instance, some patients only receive two cycles of NAC, during which their carbohydrate antigen 19-9 (CA 19-9) levels increase and/or their tumors enlarge. Nevertheless, some of these patients may ultimately undergo radical resection, as their tumors remain anatomically resectable. Because of the abundant stroma, radiographic findings often do not exhibit clear shrinkage after NAC in pancreatic cancer patients, even when the tumor responds to NAC. This implies that the majority of pancreatic cancer patients have stable disease according to RECIST 1.1 after NAC. The NCCN guideline for pancreatic cancer (version 2. 2021) states that "radiographic findings may appear stable despite dramatic falls in CA 19-9" and "exploration for resection should be considered if there is a >50% decrease in CA 19-9 level and clinical improvement indicating response to therapy." Consequently, in the inclusion criteria for this study, we considered both radiographic and CA 19-9 changes after NAC when designing the study.
Notably, a highlight of the present study is that CD36 was a pan-upregulated target in both tumor cells and T effector cells; meanwhile, the blockage of CD36 not only restrain tumor chemoresistance but also recover anti-tumor immunity, which endowed with fewer off-target effects and a greater potential for clinical translation based on CD36 targeting.
In summary, multimodal analysis of PDAC specimens with and without NAC revealed broad immune and metabolic reprogramming, the findings of which might be extended to advanced PDACs treated with adjuvant chemotherapy. The combination of CD36 blockade and AG is expected to increase the surgical conversion rate in the NAC context and the response rate in the background of adjuvant chemotherapy.
Limitations of the study
The present study has some limitations. On the one hand, although we performed many metabolic analyses in vitro and in silico, we did not explore the metabolome disturbance at the single-cell level. This limitation could not be well addressed because relevant techniques are still not defined clearly. In this context, we tried to use single-cell and bulk-level variations in metabolic enzymes, bulk-level metabolome detection, and isotope-labeled metabolic flux evaluation by a patient-derived PDAC model to explore chemotherapy-associated metabolic changes and support our findings. On the other hand, the PADC samples with NAC and UR analyzed in the present study were unpaired. The primary reason for this limitation is the clinical practice for pancreatic cancer patients. When a patient is diagnosed with unresectable disease based on radiographic evidence, they are typically recommended to undergo chemotherapy. As a result, it becomes challenging to obtain matched samples because of the treatment decisions made in the management of pancreatic cancer.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-CD45 | BD | Cat# 564225; RRID:AB_2716861 |
| anti-CD36 | BD | Cat# 565933; RRID:AB_2739397 |
| anti-CD4 | BD | Cat# 550954; RRID:AB_393977 |
| anti-CD8a | BD | Cat# 557654; RRID:AB_396769 |
| anti-Gr-1 | Biogend | Cat# 108412; RRID:AB_313377 |
| anti-CD11B | Invitrogen | Cat# 25-0112-82; RRID:AB_469588 |
| anti-CD25 | BD | Cat# 564370; RRID:AB_2738772 |
| anti-FOXP3 | BD | Cat# 12-4771-82; RRID:AB_529580 |
| anti-CD45 | BD | Cat# 557833; RRID:AB_396891 |
| anti-CD3 | BD | Cat# 555339; RRID:AB_395745 |
| anti-CD4 | BD | Cat# 560650; RRID:AB_1727476 |
| anti-CD8 | BD | Cat# 557746; RRID:AB_396852 |
| anti-CD25 | BD | Cat# 555432; RRID:AB_395826 |
| anti-FOXP3 | EB | Cat# MA518160; RRID:AB_2539534 |
| anti-CD36 | BD | Cat# 744766; RRID:AB_2871599 |
| anti-CD11B | BD | Cat# 557396; RRID:AB_396679 |
| anti-CD68 | BD | Cat# 565595; RRID:AB_2739298 |
| anti-CD33 | BD | Cat# 555450; RRID:AB_395843 |
| anti-CD14 | BD | Cat# 555399; RRID:AB_398596 |
| anti-CD15 | BD | Cat# 560828; RRID:AB_10563612 |
| CD36 neutralizing antibody (Clone JC63.1) (Low Endotoxin) | Cayman | Cat# 10009893; RRID:AB_10613953 |
| anti-IgA, kappa [S107] | abcam | Cat# ab37322; RRID:AB_2927403 |
| anti-CD36 | Proteintech | Cat# 18836-1-AP; RRID:AB_10597244 |
| anti-LDHA | Abcam | Cat# ab52488; RRID:AB_2134961 |
| anti-GLS | Abcam | Cat# ab156876; RRID:AB_2721038 |
| anti-Ki67 | Abcam | Cat# ab15580; RRID:AB_443209 |
| anti-CD20 | Abcam | Cat# ab64088; RRID:AB_1139386 |
| anti-CD163 | Proteintech | Cat# 16646-1-AP; RRID:AB_2756528 |
| anti-CD4 | Abcam | Cat# ab133616; RRID:AB_2750883 |
| anti-FOXP3 | Abcam | Cat# ab215206; RRID:AB_2860568 |
| anti-CD8 | Abcam | Cat# ab189926 |
| anti-CD36 | Abcam | Cat# ab133625; RRID:AB_2716564 |
| Granzyme B | Abcam | Cat# ab255598; RRID:AB_2860567 |
| Pan-CK | Abcam | Cat# ab7753; RRID:AB_306047 |
| Biological samples | ||
| Resected PDAC tissues with or without NAC treatment | This study | FUSCCNAC |
| Chemicals, peptides, and recombinant proteins | ||
| Oleic-13 C18 | Merck | 490431 |
| D-glucose-C13 | Merck | 389374 |
| Ovalbumin | Merck | S7951 |
| Diphenylamine hydrochloride | MCE | HY-N7133 |
| Pyridoxine | MCE | HY-B1328 |
| Isocitric acid trisodium salt | MCE | HY-W009362 |
| Isovaleric acid | MCE | HY-W012980 |
| Palmitic acid | MCE | HY-N0830 |
| Cadaverine | Sigma-Aldrich | 462-94-2 |
| N-Acetylneuraminic acid | MCE | 131-48-6 |
| L-Proline | MCE | HY-Y0252 |
| Oleic acid | Selleck | 112-80-1 |
| Deoxycholic acid | MCE | HY-N0593 |
| L−2−Hydroxyglutaric acid | MCE | HY-113039 |
| Gemcitabine | MCE | LY 188011 |
| Albumin-bound paclitaxel | Abraxane | Abraxane |
| T cell Activation/Expansion Kit | Miltenyi Biotec | 130-091-441 |
| IL-2(human) | Absin | Abs04045 |
| IL-2(mice) | Absin | Abs00970 |
| Critical commercial assays | ||
| Free Fatty Acid Uptake Assay Kit | Abcam | ab176768 |
| Tumor dissociation kit | MACS | 130-096-730 |
| Stain buffer | BD | 554656 |
| Fc blocker | BD | 564219 |
| Transcription factor buffer set | BD | 562574 |
| Green-fluorescent caspase 3/7 probe reagent | Invitrogen | R37111 |
| Matrigel | Corning | 3533-010-02 |
| LDH Cytotoxicity Assay Kit | Cayman | 601170 |
| CellTiter-Glo 3D Cell viability assay | Promega | G9683 |
| Human lymphocyte isolation solution | Dayou | 7111011 |
| RBC lysis buffer | Biolegend | 420302 |
| CCK-8 kit | Selleck | B34302 |
| EdU Kit | Beyotime | C0071L |
| Deposited data | ||
| Bulk and single-cell RNA sequencing data (FUSCCNAC) | This study | National Omics Data Encyclopedia (OEP003152 and OER330659). |
| Proteome and metabolome data (FUSCCNAC) | This study | National Omics Data Encyclopedia (OEP003152 and OER330659). |
| TCGA pancreatic cancer cohort | Cancer Genome Atlas Network, 2012 | http://www.cbioportal.org/ |
| Moffitt pancreatic cancer cohort | Moffitt et al.11 | GSE71729 |
| Puleo pancreatic cancer cohort | Puleo et al.27 | E-MTAB-6134 |
| TCPA pancreatic cancer cohort | Li et al.28 | http://bioinformatics.mdanderson.org/main/TCPA:Overview |
| Experimental models: Cell lines | ||
| Panc 02 cell-line | the National Intrastructure of Cell Line Resource | 1101MOU-PUMC000446 |
| KPC-0116 cells | This study | N/A |
| Ovalbumin (OVA)-expressing KPC cells | This study | N/A |
| Panc-1 cells | ATCC | CRL-1469 |
| Experimental models: Organisms/strains | ||
| Female C57BL/6N mice (6-7 week-old) | Shanghai Jihui Laboratory Animal Care Co.,Ltd | N/A |
| Female NPSG mice (6-7 week-old) | N/A | |
| OT-1 mice | Cyagen | N/A |
| Oligonucleotides | ||
| 5′ GAAGTTACATATTAGGCCAT 3′ (shCD36#1) | Ribo Bio | N/A |
| 5′ CCGACGTTAATCTGAAAGGA 3′ (shCD36#2) | Ribo Bio | N/A |
| Software and algorithms | ||
| GraphPad Prism software version 9.0.0 | GraphPad Software | https://www.graphpad.com |
| R version 3.5.2 | R Foundation for Statistical Computing | https://cran.r-project.org |
| FlowJo version 10.7.1 | FlowJo, LLC | https://www.flowjo.com/ |
| FeatureCounts v1.5.0-p3 | Liao et al.29 | http://subread.sourceforge.net |
| Proteome Discoverer 2.2 | search engines | Thermo |
| DESeq2 | Love et al.30 | http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Gene Set Enrichment Analysis (GSEA) version 3.0 | Liberzon et al.31 | http://www.gsea-msigdb.org/gsea/index.jsp |
| Kyoto Encyclopedia of Genes and Genomes (KEGG) | Ogata et al.32 | http://www.genome.ad.jp/kegg/ |
| Gene set variation analysis (GSVA) | Hanzelmman et al.33 | http://www.bioconductor.org/packages/release/bioc/html/GSVA.html |
| Pheatmap 1.0.12 | Kolde et al. | https://cran.r-project.org/web/packages/pheatmap/index.html |
| Cibersort | Alizadeh Lab and Newman Lab | https://cibersortx.stanford.edu/ |
| TIMER | Liu XS Lab | http://timer.cistrome.org/ |
| Immunedeconv 2.0.4 | Gregor and Sturm et al. | https://icbi-lab.github.io/immunedeconv |
| Immunophenoscore | TCIA | https://tcia.at/home |
| ImmuneCellAI | Guo Lab | http://bioinfo.life.hust.edu.cn/ImmuCellAI#!/ |
| ConsensusClusterPlus | Wilkerson et al.34 | https://doi.org/10.1093/bioinformatics/btq170. |
| ScFEA | Alghamdi et al.35 | https://doi.org/10.1101/gr.271205.120 |
| Seurat | satijalab | https://satijalab.org/seurat/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be facilitated by the lead contact, Si Shi (shisi@fudanpci.org).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Sample collection for multimodal analyses
PDAC tissues with neoadjuvant albumin-bound paclitaxel plus gemcitabine and upfront resection were obtained from January 1, 2015, to December 31, 2020, were collected for at least one of transcriptome, proteome and metabolome. Proteome sequencing was conducted on pancreatic ductal adenocarcinoma (PDAC) samples from 93 patients (56 NAC and 37UR). Of these, 70 samples underwent transcriptome sequencing (47 NAC and 23UR). To make sure the metabolite landscape we detected could mostly recapture the effect of chemotherapy on PDAC samples, we only detected samples well-conserved within six months. In preparation for the untargeted metabolome analysis, we proactively collected 42 NAC PDAC samples and 31 UR PDAC samples. Additionally, we performed targeted analyses of energy metabolism on 20 NAC PDAC samples and 24 UR PDAC samples. In addition, 11 fresh PDAC samples (8 for NAC and 3 for UR) were collected for single-cell transcriptome sequencing (scRNA-seq) and TCR sequencing, and 3 of them (2 for NAC and 1 for UR) were further processed for spatial transcriptome sequencing. We used a tissue microarray (TMA) obtained from 278 PDAC patients who were diagnosed with PDAC using histopathological tests at the FUSCC from 2015 to 2020. The postoperative adjuvant chemotherapy regimens for the patients were followed-up. Human peripheral blood mononuclear cells (PBMCs) were isolated from the peripheral blood of patients with NAC or UR preoperatively by Ficoll density gradient centrifugation. For flow cytometry analysis, samples were preserved in liquid nitrogen with cryoprotectant (90% FBS and 10% DMSO) until use. Only patients with partial response and/or a CA19-9 decrease ≥50% after NAC were enrolled in the cohort. All samples enrolled in the present study were obtained after approval of the study by the Fudan University Shanghai Cancer Center (FUSCC) Ethics Committee (serial number: 2109243-16), and each patient provided written informed consent.
PDAC cell lines
The murine pancreatic cancer cells used in the present study included KPC-0116 and Panc 02 cells. KPC-0116 cells were derived from KrasLSL-G12D/+; Trp53LSL-R172H/+; Pdx1-Cre (KPC) mice and cultivated in our laboratory in vitro. Panc-02 cells were purchased from the National Intrastructure of Cell Line Resource. Ovalbumin (OVA)-expressing KPC cells (KPC-OVA) were generated as target cells with lentiviral vector-OVA, and empty LV was transfected as a control. The human pancreatic cancer cell line Panc-1 was purchased from the American Type Culture Collection. All these cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum. CD36-KD panc-1 cell-line was constructed using lentivirus plasmid for stable RNA interference. HEK-293 T cells were transfected with target/control vectors, psPAX2 packaging plasmid and pMD2.G envelope plasmid with Lipofectamine 3000. After 48h, lentivirus was harvested and transduced into Panc-1 cells, which were then selected with 3 μg/mL puromycin (Sangon Biotech, Shanghai, China) for 14 days.
Patient- and murine-derived PDAC organoids and in vitro analysis
Human PDAC tissue and spontaneous KPC tumors were collected and digested into single-cell suspensions using PDAC dissociation reagent. The components of the reagent and procedures for digestion as well as culture media preparation were described in a previous publication.36 OVA-expressing KPC organoids were constructed using dissociated OVA-expressing KPC cells.
Organoids and PBMCs were cocultured roughly as described previously.37 In brief, PBMCs were stimulated with PDAC organoid culture media, a T cell Activation/Expansion Kit (Miltenyi Biotec, 130-091-441) and 30 U recombinant IL-2 in 96-well plates for 24 h before starting coculture. Activated PBMCs and PDAC organoids were directly cocultured at a 5:1 ratio on Matrigel (Corning)-coated 96-well plates and treated with the indicated drugs. For in vitro treatment with gemcitabine, paclitaxel and CD36 neutralizing antibody, the final concentrations for these regimens were 100 nM, 10 nM and 8 μg/mL. At the beginning of coculture, a green-fluorescent caspase 3/7 probe reagent (R37111, Invitrogen) was added to visualize apoptotic organoids according to the manual. OVA-specific TCR transgenic (OT-1 cells) were isolated from the spleen of OT-1 mice, and the OVA–derived peptide SIINFEKL was used to generate cytotoxic T lymphocytes. The coculture of OT-1 cells and OVA-expressing KPC organoids was performed according to the mentioned procedures.
The LDH release assay was performed using an LDH Cytotoxicity Assay Kit (Cayman, 601170) according to the manufacturer’s instructions. When determining the cytotoxicity of PBMCs or OT-1 cells in the organoid coculture system, we performed flow cytometry analysis to determine the percentage of apoptotic CD45-negative cells. In the present study, given that we used a green-fluorescent caspase 3/7 probe reagent to label apoptotic cells, the signal of the FITC channel was set to evaluate the apoptotic situation. In addition, we performed immunofluorescent staining to detect proliferative organoid cells, where the following antibodies were used in the experiment: Ki67 (1:1000, Abcam, ab15580) and Panc-CK (1:1000, Abcam ab7753). The ratio of Ki67-positive nuclei to DAPI could be used to reflect the proliferative activity of organoids. Beyond the coculture system, the viability of organoid cells was evaluated by a CellTiter-Glo 3D Cell viability assay (Promega, G9683) based on the manufacturer’s instructions. Organoids were digested into single cells before experiments started, so the numbers of organoids could be accurately counted and seeded in the same preliminary density for each group. Under this circumstance, the viability of organoids at the timepoint of measurement would reflect the growth speed of organoid with distinct treatment.
Animal experiments
C57BL/6N mice (female, 6–8 weeks of age, 18–20 g) were housed in ventilated caging units in the Shanghai Cancer Center Specific Pathogen Free (SPF) facility with standard housing and husbandry and free access to food and water. For subcutaneous transplantation, the left flank of the mice was transplanted with 0.5×106 cells. Tumor volume was measured every four days using calipers and calculated using the following formula: length × width × height. Euthanasia was performed using CO2 inhalation prior to tumor stripping. For orthotopic murine models, we performed a left flank incision and step-by-step dissection until the spleen could be pushed out from the ventral cavity. The pancreas is hidden behind the spleen but can be turned over by rolling using a sterile cotton swab. After sufficient exposure of the pancreas, 50 μL volumes of 0.5×106 cells were injected along the long axis of the pancreas from the end near the spleen. A puffy transparent bubble without leakage was regarded as a successful case.
To acquire an immunologically humanized PDX model, we began by constructing a PDX model according to the procedures described in our previous study.38 Briefly, surgically resected PDAC samples were cut into five equal blocks of approximately 10 mm3 for subcutaneous transplantation into the flanks of female NPSG mice (6–8 weeks). Then, 2x10ˆ7 isolated human PBMCs were resuspended in 500 μL PBS and intravenously injected into PDX mice seven days after transplantation. For immune reconstruction evaluation, the peripheral blood of PDX mice was collected for flow cytometry analysis. PDX mice that had over 25% hCD45+ CD3+ cells in the peripheral blood were considered qualified cases.
Gemcitabine powder (MedChemExpress (MCE), LY 188011) was resuspended in sterile normal saline at 10 mg/mL. The drug was administered at 50 mg/kg by intraperitoneal injection every four days. nab-paclitaxel was resuspended in sterile normal saline and intravenously administered at 30 mg/kg at the same time as gemcitabine treatment. Mice in the corresponding groups were treated with 20 μg CD36 monoclonal antibody (Clone JC63.1) (Low Endotoxin) (Cayman, 10009893) and were injected intraperitoneally every other day. The control group was treated with corresponding IgA (mouse IgA, kappa [S107], Abcam, ab37322).
Method details
High-throughput sequencing
Proteome sequencing
Ninety-three frozen tissue samples (56 NAC and 37 UR) were taken from an −80°C refrigerator. The samples were ground into powder at a low temperature and quickly transferred to centrifuge tubes pre-cooled with liquid nitrogen. An appropriate amount of PASP protein lysis solution (100 mM ammonium bicarbonate, 8 M urea, pH = 8) was added, and the mixture was shaken and mixed thoroughly. The samples were then lysed in an ice water bath. Subsequently, the lysates were further processed to construct the library for DIA (data-independent acquisition) protein identification. Other regents used for preparation of proteome sequencing are listed as follows: Mass spectrometry-grade trypsin (purchased from Promega/V5280), iRT kit (purchased from Biognosys), Bradford protein quantification kit (purchased from Beyotime), Dithiothreitol (DTT, purchased from Sigma/D9163-25G), Iodoacetamide (IAM, purchased from Sigma/I6125-25G), Sodium dodecyl sulfate (SDS, purchased from Guoyao), Urea (purchased from Guoyao/10023218), Ammonium bicarbonate (purchased from Sigma/5330050050), LC-MS grade ultrapure water (purchased from Thermo Fisher Chemical/W6-4), Triethylammonium bicarbonate buffer (TEAB, purchased from Sigma/T7408-500ML), LC-MS grade acetonitrile (purchased from Thermo Fisher Chemical/A955-4), LC-MS grade formic acid (purchased from Thermo Fisher Scientific/A117-50), Acetone (purchased from Beijing Chemical Factory/11241203810051), Ammonium hydroxide (purchased from Sigma/221228-500ML-A), ProteoMiner low-abundance protein enrichment kit (purchased from Bio-Rad/1633007), Trifluoroacetic acid (TFA, purchased from Sigma/T6508-100ML). The whole scanning range of mass spectrometry could be divided into several windows according to the mass charge ratio (m/z) and collect fragment ion information of all parent ions for protein characterization and quantification.39 The separated peptides were analyzed by Q ExactiveTM HF-X mass spectrometer (Thermo Fisher), with ion source of Nanospray Flex(ESI), spray voltage of 2.1 kV and ion transport capillary temperature of 320°C. For DIA acquisition, the m/z range covered from 350 to 1500. MS1 resolution was set to 60000 (at m/z 200), full scan AGC target value was 5 × 105, the maximum ion injection time was 20 ms. Peptides were fragmented by HCD in MS2, in which resolution was set to 30000 (at 200 m/z), AGC target value was 1 × 106, a normalized collision energy of 27%. The resulting spectra from DDA scan were searched separately against the “homo_sapiens_uniprot_2020_7_2.fasta (192320 sequences) database” by the search engine Proteome Discoverer 2.2 (PD 2.2, Thermo). The search parameters were set as follows: mass tolerance for precursor ion was 10 ppm and mass tolerance for product ion was 0.02 Da. Carbamidomethyl was specified as a fixed modification, oxidation of methionine (M) was specified as a dynamic modification, and acetylation was specified as an N-terminal modification in PD 2.2. A maximum of 2 missed cleavage sites were allowed. The quality controlled DDA data is imported into a software tool called Spectronaut, where a DDA library is constructed. The DIA scan data, in the form of raw files, is then analyzed using Spectronaut. The proteins whose quantitation significantly different between experimental and control groups, (p < 0.05 and |log2FoldChange| > 1), were defined as differentially expressed proteins (DEP).
Transcriptome sequencing
70 frozen tissue samples (47 NAC and 23UR) were taken from an −80°C refrigerator. A total amount of 1 μg RNA per sample was used as input material for the RNA sample preparations. RNA integrity was assessed using the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, CA, USA). To preferentially select cDNA fragments 370–420 bp in length, the library fragments were purified with an AMPure XP system (Beckman Coulter, Beverly, USA). The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumina) according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina Novaseq platform, and 150 bp paired-end reads were generated.
FeatureCounts v1.5.0-p3 was used to count the read numbers mapped to each gene. Then, the FPKM of each gene was calculated based on the length of the gene and read count mapped to this gene. FPKM, the expected number of fragments per kilobase of transcript sequence per million base pairs sequenced, considers the effect of sequencing depth and gene length on the read count at the same time and is currently the most commonly used method for estimating gene expression levels. Differential expression analysis for transcriptomic profiling was performed using DESeq2 R package. The genes whose quantitation significantly different between experimental and control groups, (fdr <0.05 and |log2FoldChange| > 0.5), were defined as differentially expressed genes (DEG). In addition, we also performed wilcox test for DEG selection to validate the findings.
Metabolome analysis
42 NAC PDAC samples and 31 UR PDAC samples were accurately weighed and processed for untargeted metabolomics analysis. Chromatographic separation was performed with an ACQUITY UPLC HSS T3 column (150 × 2.1 mm, 1.8 μm, Waters) maintained at 40°C. The ESI-MSn experiments were used with spray voltages of 3.5 kV and −2.5 kV in positive and negative modes, respectively. Data-dependent acquisition (DDA) MS/MS experiments were performed with an HCD scan. The normalized collision energy was 30 eV. Dynamic exclusion was implemented to remove some unnecessary information in the MS/MS spectra. The main parameters set in peak identification, filtration and alignment were bw = 2, ppm = 15, peakwidth = c(5,30), mzwid = 0.015, mzdiff = 0.01, and method = centWave. Then, matrixes including information on the mass-to-charge ratio, retention time and intensity were generated. Databases including HumanMetabolome, Metlin, massbank, LipidMaps, mzclound and the self-built database of BioNovoGene were used to match the information of metabolites. Metabolites with VIP >1 and false discovery rate (fdr) < 0.05 were considered significant.
Targeted metabolome for 44 PDAC samples, of which 20 PDAC samples treated with neoadjuvant AG and 24 PDAC samples that underwent UR, was performed using Selective Reaction Monitoring/Multiple Reaction Monitoring (SRM/MRM) techniques, by referring to standard products, can specifically and selectively detect and analyze specific metabolite groups. SRM/MRM techniques selectively collect mass spectrometry signals based on known or assumed reaction ion information, record signals for ions that meet the rules, and eliminate interference from signals of ions that do not. During the quantitative analysis process, this technique first screens for the parent ions specific to the target metabolites. It collects mass spectrometry signals only for the selected MS/MS2 ions, thus achieving more specific, sensitive, and accurate analysis of the target metabolite molecules. Chromatographic conditions: The samples were separated using the Agilent 1290 Infinity LC ultra-high performance liquid chromatography system. The samples were placed in an auto-sampler at 4°C, the column temperature was 35°C, the mobile phase A was a 50mM aqueous solution of ammonium acetate +1.2% ammonium hydroxide, and the mobile phase B was a 1% acetonitrile solution of acetylacetone. The flow rate was 300 μL/min, and the injection volume was 2 μL. The related liquid phase gradient was as follows: From 0 to 1min, 70% of phase B; from 1 to 10min, phase B linearly decreased from 70% to 60%; from 10 to 12 min, phase B linearly decreased from 60% to 30%; from 12.1–15min, phase B remained at 30%; from 15 to 15.5min, phase B linearly changed from 30% to 70%; from 15.1–22min, phase B remained at 70%.
In the sample queue, a QC sample was set after a certain number of experimental samples to detect and evaluate the stability and repeatability of the system. The target substance standard mixture of the sample queue was used for the correction of the chromatographic retention time. Mass spectrometry conditions: Mass spectrometry analysis was performed using a 5500 QTRAP mass spectrometer (SCIEX) in negative ion mode. The 5500 QTRAP ESI source conditions were as follows: source temperature 450°C, Ion Source Gas1 (Gas1): 45, Ion Source Gas2 (Gas2): 45, Curtain gas (CUR): 30, Ion Spray Voltage Floating (ISVF) −4500 V.
Single-cell RNA-seq (scRNA-seq)
scRNA-seq data were generated by NovelBio Bio-Pharm Technology Co., Ltd. A single-cell suspension was prepared as follows: Fresh PDAC tumor samples were obtained immediately after tumor resection and transferred to a 50 mL centrifuge tube filled with tissue storage solution (Miltenyi, Germany). Specimens were then washed three times with cold 1× PBS and digested with a Miltenyi Tumor Dissociation Kit and GentleMACS (Miltenyi, Germany) following the manufacturer’s instructions. When the digestion finished, dissociated cells were subsequently passed through a 70 μm cell-strainer (Miltenyi, Germany) to remove undigested tissue. After centrifugation (300 × g for 5 min), red blood cells were removed with RBC lysis buffer (Beyotime, China). Then, the cell pellet was washed three times with MACS buffer (PBS containing 1% FBS, 0.5% EDTA, and 0.05% gentamycin) and resuspended in sorting buffer (PBS supplemented with 1% FBS). Single-cell RNA sequencing was performed when the single-cell suspensions had viability >70%. The cell suspension was loaded onto the Chromium Single Cell Controller (10x Genomics) to generate single-cell gel beads in the emulsion according to the manufacturer’s protocol using a Single-Cell 5′ Library and Gel Bead Kit (10x Genomics, 1000006) and Chromium Single Cell A Chip Kit (10x Genomics, 120236).
Cell Ranger software was obtained from the 10x Genomics website (https://support.10xgenomics.com). Alignment, filtering, barcode counting, and UMI counting were performed with the Cell Ranger count module to generate a feature-barcode matrix and determine clusters. Cells containing over 200 expressed genes and mitochondrial UMI rates below 30% passed the cell quality filtering, and mitochondrial genes were removed from the expression table.
Spatial transcriptome sequencing (stRNA-seq)
stRNA-seq data were generated by NovelBio Bio-Pharm Technology Co., Ltd.
Tissue section preparation
The tissues were surgically removed and embedded in optimal cutting temperature (OCT) compound (SAKURA). The tissues with OCT compound were quick-frozen on dry ice immediately and stored at −80°C until cryosectioning. The cryosectioning was placed in a cryostat (Leica, CM1950) to cryosect the OCT embedded tissue blocks into appropriately sized sections for Visium Spatial slides while keeping the samples frozen. Tissue sections were 10 μm thick each. Tissue sections were placed within the frames of capture areas on Visium Spatial slides (10X Genomics).
Fixation, staining and imaging
Tissue section slides were incubated for 1 min at 37°C and then fixed in methanol at −20°C for 30 min. For staining, the slides were incubated in hematoxylin for 7 min and in Bluing Buffer for 2 min. Then, eosin was added to the slides and incubated for 1 min. After each staining step, the slides were washed with DNase and RNase-free water. Stained tissue sections were imaged by a microscope (Pannoramic MIDI, 3DHISTECH).
Tissue prepermeabilization
Prepermeabilization was performed to optimize the suitable permeabilization time. Visium Spatial Tissue Optimization Slides & Reagent Kits (10X Genomics) were used for prepermeabilization. The tissues were permeabilized in permeabilization enzyme for varying amounts of time, and then Fluorescent RT Master Mix was added to the tissue sections. For tissue removal, the tissue sections were incubated in Tissue Removal Mix for 60 min at 56°C. The best permeabilization time was selected through a fluorescence microscope (Pannoramic MIDI, 3DHISTECH).
Tissue permeabilization and spatial transcriptomic sequencing
Tissue permeabilization and spatial transcriptomic sequencing were performed using Visium Spatial Gene Expression Slides & Reagent Kits. The stained slides were incubated in RT Master Mix for 45 min at 53°C for reverse transcription after permeabilization for the appropriate time. Next, Second Strand Mix was added to the tissue sections on the slide and incubated for 15 min at 65°C to initiate second strand synthesis. After transfer of cDNA from the slides, barcoded cDNA was purified and amplified. The amplified barcoded cDNA was fragmented, A-tailed, ligated with adaptors and index PCR amplified. The final libraries were quantified using the Qubit High Sensitivity DNA assay (Thermo Fisher Scientific), and the size distribution of the libraries was determined using a High Sensitivity DNA chip on a Bioanalyzer 2200 (Agilent). All libraries were sequenced by an Illumina sequencer (Illumina, San Diego, CA) on a 150 bp paired-end run.
Spatial metabolome
Spatial metabolome data were generated by PANOMIX Co., Ltd. To prepare frozen sections, we utilized a freezing microtome obtained from Fisher Scientific (Loughborough, UK). The process began by activating the Thermo CRYOSTAR NX50 cryostat and lowering the freezer temperature to −20°C. Tissue samples, initially stored at −80°C, were placed in the freezer for 3 h to equilibrate. The sample was then affixed to the positionable sample head using an embedding solution, and the orientation was adjusted for optimal slicing. Sections intended for mass spectrometry imaging were cut to a thickness of 20 μm, while those for HE stains were sliced to 10 μm. Using a pre-cooled brush, the sections were transferred to a pre-cooled ITO conductive glass slide and allowed to dry for 20 min.
If immediate testing was not required, the ITO conductive glass slides could be stored at −80°C. For matrix spray, an automatic matrix spraying instrument from Sun Chrome (SunCollect, Germany) was used. The matrix solution was sprayed onto the tissue section surface using this device, with the nozzle temperature set to room temperature and a moving velocity of 1000 mm/min. Twelve matrix passes were deposited at a gradient flow rate between 15 and 60 μL/min. The matrix α- Cyano-4-neneneba hydroxycinnamic acid (5 mg/ml) was used for the spatial metabolome analysis.
Mass spectrometry imaging was conducted using Thermo Q Exactive Plus connected AP/MALDI (MassTech, Columbia, MD). An AP/MALDI (ng) UHR ion source (MassTech, Columbia, USA) was coupled with a Thermo QE Plus mass spectrometer (Thermo Fisher Scientific, USA) for all data acquisition. The laser energy used ranged from 10 to 30%. The mass spectrometer was operated in full MS mode with positive polarity, a capillary temperature set to 320°C, and an S-lens RF level of 50%. Full scans utilized a mass range of m/z 120–1300 with a resolution of 35,000. An automatic gain control (AGC T) target of 1e6 with a 100 ms maximum injection time was used, and an isolation window of m/z 2 was set.
Bioinformatic analysis, software and computational algorithms
Immune cell infiltration estimation
RNA-seq-based algorithms can predict the relative percentages of various infiltrating immune cells. The R package “immunedeconv (2.0.4)” was applied to compute quanTIseq,40 CIBERSORT,41 TIMER,42 MCPcounter,43 EPIC44 and xCell45 data. In addition, single-sample gene set enrichment analysis (ssGSEA) was performed to estimate immune cell infiltration and other immune signatures.46 The immunophenoscore was calculated according to the guidance of The Cancer Immune Atlas (TCIA; https://tcia.at/home). ImmuneCellAI was used to deduce the therapeutic response of PDAC to immunotherapy.
CRISPR-based gene and drug dependency analysis
To analyze the impact of a specific gene on tumor cell proliferation, the DepMap database (DepMap 21Q4 Public+Score, Chronos) was used for screening. To analyze the correlation between drug sensitivity and gene dependency in pancreatic cell lines, the data in PRISM 19Q4 were also analyzed (https://depmap.org/portal/download/).
Metabolism-related gene list
The metabolism-related gene list consisted of two sections: 1661 genes were originated from the KEGG human metabolic pathway gene list and 870 genes collected from five metabolism-related Gene Set Enrichment Analysis (GSEA) hallmark gene sets. The two sections were merged into a single gene list comprising 2041 metabolic genes after duplicates were removed.
Estimation of metabolic activity
The single-cell level metabolic activity of tumor cells was estimated by scMetabolism and AUCell algorithms, which are publicly available R packages. Glycolytic ductal cells were defined as the top 25% of cells ranked by AUC score for the glycolysis pathway. The gene list for performing AUCell analysis was downloaded from the Molecular Signature Database – Reactome (https://www.gsea-msigdb.org/gsea/msigdb/index.jsp). Metabolic flux estimation was performed using Python according to the instruction of published paper.47
Cell communication analysis
To enable a systematic analysis of cell–cell communication molecules, we applied cell communication analysis based on the Cellchat package, a public repository of ligands, receptors and their interactions. Membrane, secreted and peripheral proteins of the cluster of different time points were annotated. Significant mean and cell communication significance (p value < 0.05) were calculated based on the interaction and the normalized cell matrix achieved by Seurat normalization.
Consensus clustering
The clustering of ductal cells was performed via the R package “ConsensusClusterPlus” with the parameters (clusterAlg = "km" and distance = "euclidean"). The optimal number for clustering was determined by the relative change in the area under the CDF curve.
Preparation of pancreatic lysate
In brief, PDAC tissues were accurately weighed, thoroughly washed, and homogenized in 1 mL of cold sterile PBS. The homogenization solution was centrifuged at 15,000 × g for 10 min at 4 °C, followed by passing through a 0.22-μm filter. The supernatant was collected as the pancreatic lysate as described in a previous study.48 The remaining tumor tissues were homogenized using RIPA buffer supplemented with proteinase and phosphatase inhibitors, and the protein concentration was determined with BCA reagent (Beyotime, China). Then, Western blot was conducted according to the procedures as shown in supplementary methods.
Enzyme-linked immunosorbent assay (ELISA)
For the analysis of cytotoxic mediators, we performed ELISA for tumor homogenate or cell supernatant according to the manufacturers’ instructions of each ELISA kit. The ELISA kits used in the present study were as follows: Human IFN-gamma ELISA Kit (absin, abs510007); Human IL-2 ELISA Kit (Boster, EK0397); Mouse TNF alpha ELISA Kit (absin, abs520010) and Mouse IFN- gamma ELISA Kit (absin, abs520007).
Knowledge-based immune signatures
We integrated three sets of immune signatures into the knowledge-based immune signatures and analyzed their activities for each sample using RNA-seq data. Signature I, including 29 genes, was widely reported in published studies49; Signature II, including M1-polarization, M2-polarization and MDSC was summarized from classical reviews and Xiao et al. work.50,51,52 Signature III consisted of 17 immune signaling pathways from ImmPort database (https://www.immport.org/home).
Flow cytometry
Single-cell suspensions from PDAC or mouse xenograft tissues and PBMCs were obtained as described above. For staining of cell surface markers, the cells were incubated with fixable viability dye (BD) and Fc block (BD) for 15 min before staining with fluorochrome-conjugated antibodies for 45 min at 4°C. For intracellular staining, the cells were fixed and permeabilized with a BD Cytofix/CytopermTM Fixation/Permeabilization Kit according to the manufacturer’s protocol and incubated with the indicated antibody for 45 min at 4°C. Flow cytometry analysis was performed with a Beckman CytoFlex S or Celesta flow cytometer. The fluorophore-conjugated antibodies and other agents used in the present study are listed as follows:
For flow cytometry targeting mouse tumors, the following antibodies were used: anti-CD45 (BD, 564225); anti-CD36 (BD, 565933); anti-CD4 (BD, 550954); anti-CD8a (BD, 557654); anti-Gr-1 (BioLegend, 108412); anti-CD11B (Invitrogen, 25-0112-82); anti-CD25 (BD, 564370); and anti-FOXP3 (Invitrogen, 12-4771-82). A tissue digestion kit was purchased from MACS (Miltenyi, 130-096-730). Fixable viability stain 510 (BD, 564406) and FC blocker (BD, 553141) were also used. For flow cytometry targeting human peripheral blood mononuclear cells (PBMCs), the following antibodies were used: anti-CD45 (BD, 557833); anti-CD3 (BD, 555339); anti-CD4 (BD, 560650); anti-CD8 (BD, 557746); anti-CD25 (BD, 555432); anti-FOXP3 (EB, MA518160); anti-CD36 (BD, 744766); anti-CD11B (BD, 557396); anti-CD68 (BD, 565595); anti-CD33 (BD, 555450); anti-CD14 (BD, 555399); anti-CD15 (BD, 560828); fixable viability stain 700 (BD, 564997); and an FC blocker (BD, 564219). To detect IFN-y+ cells using flow cytometry, the culture system was supplemented with the following reagents for 4 h prior to detection: 40 ng/mL PMA, 1 μg/mL lonomycin, and 10 μg/mL BFA.
Western blot
Equal amounts of protein were electrophoresed on 10% SDS‒PAGE gels and were then transferred onto PVDF membranes (Millipore), which were soaked for 2 h in 5% skim milk. Subsequently, the membranes were probed with the indicated primary antibodies: CD36 (1:1000, ab133625, Abcam) and β-actin (1:10000, 66,009–1-lg, Proteintech). Then, the PVDF membranes were incubated with corresponding secondary antibodies (1:1000, CST) for 60 min. After washing three times (15 min each time), the targeted proteins were visualized using enhanced chemiluminescence reagent (Millipore, MA, USA).
T cell extraction, culture, and in vitro analysis
PDAC patient peripheral blood was collected preoperatively under the approval of the Clinical Research Ethics Committee of FUSCC. For the isolation of T cells, PBMCs were depleted of B cells, monocytes, natural killer cells, dendritic cells, early erythroid cells, platelets and basophils via magnetic cell sorting (Miltenyi Biotec, 130-096-535). For isolation human CD8+ T cells, we performed positive selection from PBMCs (Miltenyi Biotec, 130-045-201). Activation of T cells was based on a T cell Activation/Expansion Kit (Miltenyi Biotec, 130-091-441). T cells were cultured in RPMI 1640 (Gibco) supplemented with 20 IU/mL IL-2.
The metabolites used in the present study were as follows: diphenylamine hydrochloride (MCE, HY-N7133); pyridoxine (MCE, HY-B1328); DL-isocitric acid trisodium salt (MCE, HY-W009362); isovaleric acid (MCE, HY-W012980); cadaverine (Sigma‒Aldrich, 462-94-2); N-acetylneuraminic acid (MCE, 131-48-6); L-proline (MCE, HY-Y0252); oleic acid (Selleck, 112-80-1); deoxycholic acid (MCE, HY-N0593); and L−2−hydroxyglutaric acid (MCE, HY-113039). Twenty-four hours after activation, specific metabolites were added to the T cell medium for stimulation.
Metabolic flux assay
13C-oleic acid and 13C-D-glucose were purchased from Merck Company (490431 and 389374, respectively). 13C-oleic acid was added to FBS- and glucose-free DMEM at 2 mM for the culture of the indicated PDXOs for 12 h, and 10 μM Y27632 was added to the culture medium.13C-D-glucose was supplemented to complete organoid culture medium at 2 mg/mL for the indicated PDXOs for 6 h. Then, the culture medium was removed, and the PDXOs were washed with cold PBS three times. Liquid nitrogen was added to the dish to quickly terminate the cell viability. Next, cold methyl alcohol was added to the dish when liquid nitrogen was evaporated. The UHPLC-HRMS platform was used for subsequent metabolic flux analysis. Chromatographic separation was performed on a ThermoFisher Ultimate 3000 UHPLC system. The eluents were analyzed on a ThermoFisher Q Exactive Hybrid Quadrupole-Orbitrap mass spectrometer (QE) in heated electrospray ionization negative (HESI-) mode. Natural isotope correction analysis was performed according to a previous study.53
Immunohistochemistry and multispectral fluorescent immunohistochemistry
Immunohistochemistry (IHC) was performed based on formalin-fixed paraffin-embedded (FFPE) slices from 22 NAC and 32 UR PDAC samples for LDHA, CD36, Ki67, CD20, CD8 and CD163 staining. Sections (5-μm thick) were stained using a two-step method as we previously described.54 Protein expression levels were calculated by multiplying the positivity (0, <5% of the total cells; 1, 5–25% of the total cells; 2, 25–50% of the total cells; 3, 50–75% of the total cells; and 4, 75%–100% of the total cells) and intensity scores (0, no color; 1, pale yellow; 2, yellow; and 3, clay bank). Evaluation of the Ki67 level in human PDAC tissue slices and animal models was based on the fraction of Ki67-positive cells. All procedures were performed after obtaining approval from the Clinical Research Ethics Committee of FUSCC, and informed consent was obtained from each patient prior to the analyses. The cut-off criteria to distinguish CD36-low and CD36-high samples was IHC score ≤ 4. Two independent pathologists conducted the strict pathological diagnoses and postoperative follow-ups.
The antibodies used for immunohistochemistry (IHC) analysis in the present study are as follows: anti-CD36 (Proteintech, 18836-1-AP; dilution 1:300); anti-LDHA (Abcam, ab52488; dilution 1:1500); anti-Ki67 (Abcam, ab15580; dilution 1:1000); anti-CD20 (Abcam, ab64088; dilution 1:800); and anti-CD163 (Proteintech, 16646-1-AP; dilution 1:1000).
FFPE slides from 10 NAC and 10 UR specimens were subjected to multispectral immunohistochemistry (mIHC) staining using an Opal color kit (PerkinElmer, Hopkinton, Massachusetts, USA). Various antibodies were used to identify the presence of immune cells in tumor tissues, including anti-CD4 (Abcam, ab133616; dilution 1:1000), anti-FOXP3 (Abcam, ab215206; dilution 1:200), anti-CD8 (Abcam, ab189926; dilution 1:800), anti-CD36 (Abcam, ab133625; dilution 1:300), and anti-granzyme B (Abcam, ab255598; dilution 1:2500), Ki67 (Abcam, ab15580; dilution 1:1000) and Panc-CK (Abcam ab7753; dilution 1:1000). Cell nuclei were stained with DAPI.
Two independent observers (RT and QM) evaluated the stained sections, and a third observer (SS) verified the numbers of tumor-infiltrating immune cells in cases of disagreement between the two observers. The details for multispectral fluorescent immunohistochemistry are listed in the supplementary methods.
Quantification and statistical analysis
Unpaired Student’s t test, one-way analysis of variance, the Kruskal–Wallis test and the Mann–Whitney–Wilcoxon test were applied to compare the differences in continuous or ordered categorical variables if required, while the comparison of unordered categorical variables was based on chi-square test or Fisher’s exact test. Kaplan–Meier curves were generated to show survival differences, and the log rank test was performed to test the statistical significance of these differences. Statistical tests were two-sided, and statistical significance was indicated with a p value <0.05. The error bars in the figures represent the standard deviation (SD).
Acknowledgments
This manuscript was written on behalf of the Chinese Study Group for Pancreatic Cancer. We are grateful to the patients and their families who contributed to this study.
This study was jointly supported by the National Natural Science Foundation of China (U21A20374), Shanghai Municipal Science and Technology Major Project (21JC1401500), Scientific Innovation Project of Shanghai Education Committee (2019-01-07-00-07-E00057), Clinical Research Plan of Shanghai Hospital Development Center (SHDC2020CR1006A), Shanghai Rising-Star Program (20QA1402100), and Xuhui District Artificial Intelligence Medical Hospital Cooperation Project (2021-011).
Author contributions
X.-J.Y. and S.S. participated in the conception and design of the study; R.T., J.X., and W.W. determined the workflow and developed the methodology; R.T., Q.-C.M., C.L., Z.-F.Z., and J.H. conducted the experiments and bioinformatic analysis; B.Z., H.Z., Y.-Y.Z., Y. Lei, Y Liu, X.-J.S., Q.D., and D.W. provided technical guidance; R.T. and S.S. wrote, reviewed and revised the manuscript; R.T., J.X., and W.W. contributed equally to this work.
Declaration of interests
The authors declare no competing interests.
Published: October 17, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101234.
Contributor Information
Xianjun Yu, Email: yuxianjun@fudan.edu.cn.
Si Shi, Email: shisi@fudanpci.org.
Supplemental information
Data and code availability
-
•
Bulk transcriptome, metabolome, proteome and single-cell RNA sequencing data have been deposited in National Omics Data Encyclopedia (http://www.biosino.org/node):OEP003152 and OER330659.
-
•
This study did not generate original codes. All software and algorithms used in this study are publicly available and listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Strobel O., Neoptolemos J., Jäger D., Büchler M.W. Optimizing the outcomes of pancreatic cancer surgery. Nat. Rev. Clin. Oncol. 2019;16:11–26. doi: 10.1038/s41571-018-0112-1. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R.L., Miller K.D., Wagle N.S., Jemal A. Cancer statistics, 2023. CA A Cancer J. Clin. 2023;73:17–48. doi: 10.3322/caac.21763. [DOI] [PubMed] [Google Scholar]
- 3.Gillen S., Schuster T., Meyer Zum Büschenfelde C., Friess H., Kleeff J. Preoperative/neoadjuvant therapy in pancreatic cancer: a systematic review and meta-analysis of response and resection percentages. PLoS Med. 2010;7:e1000267. doi: 10.1371/journal.pmed.1000267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Versteijne E., Vogel J.A., Besselink M.G., Busch O.R.C., Wilmink J.W., Daams J.G., van Eijck C.H.J., Groot Koerkamp B., Rasch C.R.N., van Tienhoven G., Dutch Pancreatic Cancer Group Meta-analysis comparing upfront surgery with neoadjuvant treatment in patients with resectable or borderline resectable pancreatic cancer. Br. J. Surg. 2018;105:946–958. doi: 10.1002/bjs.10870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jang J.Y., Han Y., Lee H., Kim S.W., Kwon W., Lee K.H., Oh D.Y., Chie E.K., Lee J.M., Heo J.S., et al. Oncological Benefits of Neoadjuvant Chemoradiation With Gemcitabine Versus Upfront Surgery in Patients With Borderline Resectable Pancreatic Cancer: A Prospective, Randomized, Open-label, Multicenter Phase 2/3 Trial. Ann. Surg. 2018;268:215–222. doi: 10.1097/sla.0000000000002705. [DOI] [PubMed] [Google Scholar]
- 6.Galluzzi L., Humeau J., Buqué A., Zitvogel L., Kroemer G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2020;17:725–741. doi: 10.1038/s41571-020-0413-z. [DOI] [PubMed] [Google Scholar]
- 7.Zhou H., Li G., Yin J., Wang T., Hu H., Li T., Zhou Q., Hu J., Wang L., Dong S., et al. Neoadjuvant chemotherapy alters peripheral and tumour-infiltrating immune cells in breast cancer revealed by single-cell RNA sequencing. Clin. Transl. Med. 2021;11:e621. doi: 10.1002/ctm2.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bear A.S., Vonderheide R.H., O'Hara M.H. Challenges and Opportunities for Pancreatic Cancer Immunotherapy. Cancer Cell. 2020;38:788–802. doi: 10.1016/j.ccell.2020.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghaneh P., Palmer D., Cicconi S., Jackson R., Halloran C.M., Rawcliffe C., Sripadam R., Mukherjee S., Soonawalla Z., Wadsley J., et al. Immediate surgery compared with short-course neoadjuvant gemcitabine plus capecitabine, FOLFIRINOX, or chemoradiotherapy in patients with borderline resectable pancreatic cancer (ESPAC5): a four-arm, multicentre, randomised, phase 2 trial. The lancet. Lancet. Gastroenterol. Hepatol. 2023;8:157–168. doi: 10.1016/s2468-1253(22)00348-x. [DOI] [PubMed] [Google Scholar]
- 10.Kunzmann V., Siveke J.T., Algül H., Goekkurt E., Siegler G., Martens U., Waldschmidt D., Pelzer U., Fuchs M., Kullmann F., et al. Nab-paclitaxel plus gemcitabine versus nab-paclitaxel plus gemcitabine followed by FOLFIRINOX induction chemotherapy in locally advanced pancreatic cancer (NEOLAP-AIO-PAK-0113): a multicentre, randomised, phase 2 trial. The lancet. Lancet. Gastroenterol. Hepatol. 2021;6:128–138. doi: 10.1016/s2468-1253(20)30330-7. [DOI] [PubMed] [Google Scholar]
- 11.Moffitt R.A., Marayati R., Flate E.L., Volmar K.E., Loeza S.G.H., Hoadley K.A., Rashid N.U., Williams L.A., Eaton S.C., Chung A.H., et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015;47:1168–1178. doi: 10.1038/ng.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collisson E.A., Bailey P., Chang D.K., Biankin A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019;16:207–220. doi: 10.1038/s41575-019-0109-y. [DOI] [PubMed] [Google Scholar]
- 13.Bagaev A., Kotlov N., Nomie K., Svekolkin V., Gafurov A., Isaeva O., Osokin N., Kozlov I., Frenkel F., Gancharova O., et al. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell. 2021;39:845–865.e7. doi: 10.1016/j.ccell.2021.04.014. [DOI] [PubMed] [Google Scholar]
- 14.Karasinska J.M., Topham J.T., Kalloger S.E., Jang G.H., Denroche R.E., Culibrk L., Williamson L.M., Wong H.L., Lee M.K.C., O'Kane G.M., et al. Altered Gene Expression along the Glycolysis-Cholesterol Synthesis Axis Is Associated with Outcome in Pancreatic Cancer. Clin. Cancer Res. 2020;26:135–146. doi: 10.1158/1078-0432.Ccr-19-1543. [DOI] [PubMed] [Google Scholar]
- 15.Tang R., Xu J., Zhang B., Liu J., Liang C., Hua J., Meng Q., Yu X., Shi S. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J. Hematol. Oncol. 2020;13:110. doi: 10.1186/s13045-020-00946-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dai E., Han L., Liu J., Xie Y., Kroemer G., Klionsky D.J., Zeh H.J., Kang R., Wang J., Tang D. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy. 2020;16:2069–2083. doi: 10.1080/15548627.2020.1714209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu S.W., Pan H.C., Hsu Y.H., Chang K.C., Wu L.W., Chen W.Y., Chang M.S. IL-20 antagonist suppresses PD-L1 expression and prolongs survival in pancreatic cancer models. Nat. Commun. 2020;11:4611. doi: 10.1038/s41467-020-18244-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X., Luo G., Zhang K., Cao J., Huang C., Jiang T., Liu B., Su L., Qiu Z. Hypoxic Tumor-Derived Exosomal miR-301a Mediates M2 Macrophage Polarization via PTEN/PI3Kγ to Promote Pancreatic Cancer Metastasis. Cancer Res. 2018;78:4586–4598. doi: 10.1158/0008-5472.Can-17-3841. [DOI] [PubMed] [Google Scholar]
- 19.Peng J., Sun B.F., Chen C.Y., Zhou J.Y., Chen Y.S., Chen H., Liu L., Huang D., Jiang J., Cui G.S., et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019;29:725–738. doi: 10.1038/s41422-019-0195-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Son J., Lyssiotis C.A., Ying H., Wang X., Hua S., Ligorio M., Perera R.M., Ferrone C.R., Mullarky E., Shyh-Chang N., et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–105. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Michelakos T., Cai L., Villani V., Sabbatino F., Kontos F., Fernández-Del Castillo C., Yamada T., Neyaz A., Taylor M.S., Deshpande V., et al. Tumor Microenvironment Immune Response in Pancreatic Ductal Adenocarcinoma Patients Treated With Neoadjuvant Therapy. J. Natl. Cancer Inst. 2021;113:182–191. doi: 10.1093/jnci/djaa073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hwang W.L., Jagadeesh K.A., Guo J.A., Hoffman H.I., Yadollahpour P., Reeves J.W., Mohan R., Drokhlyansky E., Van Wittenberghe N., Ashenberg O., et al. Single-nucleus and spatial transcriptome profiling of pancreatic cancer identifies multicellular dynamics associated with neoadjuvant treatment. Nat. Genet. 2022;54:1178–1191. doi: 10.1038/s41588-022-01134-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farren M.R., Sayegh L., Ware M.B., Chen H.R., Gong J., Liang Y., Krasinskas A., Maithel S.K., Zaidi M., Sarmiento J.M., et al. Immunologic alterations in the pancreatic cancer microenvironment of patients treated with neoadjuvant chemotherapy and radiotherapy. JCI insight. 2020;5:e130362. doi: 10.1172/jci.insight.130362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang R., Meng Q., Wang W., Liang C., Hua J., Xu J., Yu X., Shi S. Head-to-head comparison between FOLFIRINOX and gemcitabine plus nab-paclitaxel in the neoadjuvant chemotherapy of localized pancreatic cancer: a systematic review and meta-analysis. Gland Surg. 2021;10:1564–1575. doi: 10.21037/gs-21-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Von Hoff D.D., Ervin T., Arena F.P., Chiorean E.G., Infante J., Moore M., Seay T., Tjulandin S.A., Ma W.W., Saleh M.N., et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang P., Qin H., Li Y., Xiao A., Zheng E., Zeng H., Su C., Luo X., Lu Q., Liao M., et al. CD36-mediated metabolic crosstalk between tumor cells and macrophages affects liver metastasis. Nat. Commun. 2022;13:5782. doi: 10.1038/s41467-022-33349-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puleo F., Nicolle R., Blum Y., Cros J., Marisa L., Demetter P., Quertinmont E., Svrcek M., Elarouci N., Iovanna J., et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology. 2018;155:1999–2013. doi: 10.1053/j.gastro.2018.08.033. [DOI] [PubMed] [Google Scholar]
- 28.Li J., Lu Y., Akbani R., Ju Z., Roebuck P.L., Liu W., Yang J.Y., Broom B.M., Verhaak R.G., Kane D.W., et al. TCPA: a resource for cancer functional proteomics data. Nat Methods. 2013;10:1046–1047. doi: 10.1038/nmeth.2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 30.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ogata H., Goto S., Sato K., Fujibuchi W., Bono H., Kanehisa M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hänzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilkerson M.D., Hayes D.N. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26:1572–1573. doi: 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alghamdi N., Chang W., Dang P., Lu X., Wan C., Gampala S., Huang Z., Wang J., Ma Q., Zang Y., et al. A graph neural network model to estimate cell-wise metabolic flux using single-cell RNA-seq data. Genome Res. 2021;31:1867–1884. doi: 10.1101/gr.271205.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Driehuis E., Kretzschmar K., Clevers H. Establishment of patient-derived cancer organoids for drug-screening applications. Nat. Protoc. 2020;15:3380–3409. doi: 10.1038/s41596-020-0379-4. [DOI] [PubMed] [Google Scholar]
- 37.Dijkstra K.K., Cattaneo C.M., Weeber F., Chalabi M., van de Haar J., Fanchi L.F., Slagter M., van der Velden D.L., Kaing S., Kelderman S., et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell. 2018;174:1586–1598.e12. doi: 10.1016/j.cell.2018.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liang C., Shi S., Qin Y., Meng Q., Hua J., Hu Q., Ji S., Zhang B., Xu J., Yu X.J. Localisation of PGK1 determines metabolic phenotype to balance metastasis and proliferation in patients with SMAD4-negative pancreatic cancer. Gut. 2020;69:888–900. doi: 10.1136/gutjnl-2018-317163. [DOI] [PubMed] [Google Scholar]
- 39.Zhang F., Ge W., Ruan G., Cai X., Guo T. Data-Independent Acquisition Mass Spectrometry-Based Proteomics and Software Tools: A Glimpse in 2020. Proteomics. 2020;20:e1900276. doi: 10.1002/pmic.201900276. [DOI] [PubMed] [Google Scholar]
- 40.Finotello F., Mayer C., Plattner C., Laschober G., Rieder D., Hackl H., Krogsdam A., Loncova Z., Posch W., Wilflingseder D., et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 2019;11:34. doi: 10.1186/s13073-019-0638-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Newman A.M., Liu C.L., Green M.R., Gentles A.J., Feng W., Xu Y., Hoang C.D., Diehn M., Alizadeh A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li B., Severson E., Pignon J.C., Zhao H., Li T., Novak J., Jiang P., Shen H., Aster J.C., Rodig S., et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol. 2016;17:174. doi: 10.1186/s13059-016-1028-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Becht E., Giraldo N.A., Lacroix L., Buttard B., Elarouci N., Petitprez F., Selves J., Laurent-Puig P., Sautès-Fridman C., Fridman W.H., de Reyniès A. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016;17:218. doi: 10.1186/s13059-016-1070-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Racle J., de Jonge K., Baumgaertner P., Speiser D.E., Gfeller D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife. 2017;6:e26476. doi: 10.7554/eLife.26476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aran D., Hu Z., Butte A.J. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18:220. doi: 10.1186/s13059-017-1349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barbie D.A., Tamayo P., Boehm J.S., Kim S.Y., Moody S.E., Dunn I.F., Schinzel A.C., Sandy P., Meylan E., Scholl C., et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alghamdi N., Chang W., Dang P., Lu X., Wan C., Gampala S., Huang Z., Wang J., Ma Q., Zang Y., et al. A graph neural network model to estimate cell-wise metabolic flux using single-cell RNA-seq data. Genome Res. 2021;31:1867–1884. doi: 10.1101/gr.271205.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lv J., Liu Y., Mo S., Zhou Y., Chen F., Cheng F., Li C., Saimi D., Liu M., Zhang H., et al. Gasdermin E mediates resistance of pancreatic adenocarcinoma to enzymatic digestion through a YBX1-mucin pathway. Nat. Cell Biol. 2022;24:364–372. doi: 10.1038/s41556-022-00857-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.He Y., Jiang Z., Chen C., Wang X. Classification of triple-negative breast cancers based on Immunogenomic profiling. J. Exp. Clin. Cancer Res. 2018;37:327. doi: 10.1186/s13046-018-1002-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiao Y., Ma D., Zhao S., Suo C., Shi J., Xue M.Z., Ruan M., Wang H., Zhao J., Li Q., et al. Multi-Omics Profiling Reveals Distinct Microenvironment Characterization and Suggests Immune Escape Mechanisms of Triple-Negative Breast Cancer. Clin. Cancer Res. 2019;25:5002–5014. doi: 10.1158/1078-0432.Ccr-18-3524. [DOI] [PubMed] [Google Scholar]
- 51.Mantovani A., Sozzani S., Locati M., Allavena P., Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–555. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 52.Locati M., Curtale G., Mantovani A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. 2020;15:123–147. doi: 10.1146/annurev-pathmechdis-012418-012718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Millard P., Delépine B., Guionnet M., Heuillet M., Bellvert F., Létisse F. IsoCor: isotope correction for high-resolution MS labeling experiments. Bioinformatics. 2019;35:4484–4487. doi: 10.1093/bioinformatics/btz209. [DOI] [PubMed] [Google Scholar]
- 54.Zhang W.H., Wang W.Q., Gao H.L., Xu S.S., Li S., Li T.J., Han X., Xu H.X., Li H., Jiang W., et al. Tumor-Infiltrating Neutrophils Predict Poor Survival of Non-Functional Pancreatic Neuroendocrine Tumor. The Journal of clinical endocrinology and metabolism. 2020;105 doi: 10.1210/clinem/dgaa196. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Bulk transcriptome, metabolome, proteome and single-cell RNA sequencing data have been deposited in National Omics Data Encyclopedia (http://www.biosino.org/node):OEP003152 and OER330659.
-
•
This study did not generate original codes. All software and algorithms used in this study are publicly available and listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.