

Abstract

Unactivated olefins are converted to alkyl azides with bench-stable NaN3 in the presence of FeCl3·6H2O under blue-light irradiation. The products are obtained with anti-Markovnikov selectivity, and the reaction can be performed under mild ambient conditions in the presence of air and moisture. The transformation displays broad functional group tolerance, which renders it suitable for functionalization of complex molecules. Mechanistic investigations are conducted to provide insight into the hydroazidation reaction and reveal the role of water from the iron hydrate as the H atom source.
Organic azides are an integral part of an array of drug molecules, energetic materials, and chemical probes.1 They also are valuable building blocks in the synthesis of natural products, pharmaceuticals, and agrochemicals.2 As synthetic handles, azides have found widespread application in classic methods such as Staudinger reduction and ligation,3 Huisgen cycloaddition4 and click chemistry,5 as well as Schmidt6 and aza-Wittig reactions.7 More recently, azides have been utilized as nitrene precursors in transition-metal-catalyzed C–H bond aminations.8 Especially in the context of multistep synthesis, R–N3 can serve as a protected amine.9 Herein, we report the first iron- and light-mediated anti-Markovnikov hydroazidation of unactivated olefins (Figure 1). The transformation employs NaN3 as a bench-stable10 azide source, tolerates air and moisture, and proceeds under mild conditions allowing for a wide functional group compatibility.
Figure 1.

Iron-mediated photochemical anti-Markovnikov hydroazidation of unactivated olefins.
The widespread application of organic azides in synthetic chemistry and biology necessitates methodologies to access them directly from readily available starting materials. Traditionally, organic azides have been synthesized via nucleophilic substitution as well as diazo- and azido-transfer reactions.11 In a complementary approach, direct transformations of olefins to alkyl azides have been explored. Early studies by Hassner and Kropp focused on the addition of HN3 to alkenes, affording the corresponding Markovnikov products (Figure 2A).12
Figure 2.

Approaches toward (A) Markovnikov and (B) anti-Markovnikov hydroazidation of unactivated olefins and (C) desired transformation.
Milder and more broadly applicable conditions for the Markovnikov hydroazidation of unactivated olefins were developed in our group which employ a cobalt catalyst, silane, and TsN3.13 Boger later disclosed a Markovnikov hydroazidation that is presumed to proceed via an iron hydride species.14 To obtain anti-Markovnikov addition products, multistep sequences were required. Only recently has the direct anti-Markovnikov azidation of double bonds been reported (Figure 2B). Chiba and Gagosz have investigated a hypervalent iodine reagent (azidobenziodoxolone, ABX) for the hydroazidation of homoallylic benzyl ethers with the latter serving as intramolecular H atom donor in the reaction.15 Yu and co-workers have documented a hydroazidation reaction of unsaturated aryl amides using an Ir photocatalyst and TMSN3.16 Most recently, Xu and Liu independently generated the active hypervalent iodine reagent ABX in situ from TMSN3 and a benziodoxolone to achieve anti-Markovnikov hydroazidation.17 Despite these important advances, convenient procedures using NaN3 as an off-the-shelf, inexpensive azide source are lacking.
Our interest in metal-mediated olefin hydroazidation reactions and, more recently, alkene functionalization and photochemical methods has led us to examine approaches to alkyl azides.13,18 Applications of transition-metal salts (e.g., CuII, TiIV, and FeIII) under visible-light irradiation caught our attention.19 The excitation of these salts with visible light induces ligand-to-metal charge transfer (LMCT), resulting in, for example, dichlorination, diazidation, or Giese reaction.19c−19g,20 We focused on the most abundant transition metal and hypothesized that the use of radicals generated from an iron complex under blue-light irradiation may be suitable to effect hydroazidation in a mild and selective manner. To this end, 4-phenylbutene (1a) was subjected to NaN3 (3.0 equiv) and Fe(NO3)3·9H2O (1.0 equiv) in dichloromethane (0.2 M) and irradiated (λmax = 446 nm, 350 W blue LED photoreactor; for technical details see the Supporting Information) for 16 h at room temperature. In this experiment, primary azide 2a was observed in 36% yield (Table 1, entry 1). Further studies revealed that FeCl3·6H2O in CH2Cl2 afforded the highest yield among all investigated iron salts (for details see the Supporting Information). As azides are known to displace the chlorides in CH2Cl2, subsequently generating highly explosive intermediates,21 solvent alternatives were investigated. The use of polar solvents such as acetone, ethyl acetate, and MeCN did not yield any hydroazidation, instead giving the diazide in a range of yields (for details see the Supporting Information).19c,19d In contrast, employing less polar solvents such as haloarenes yielded monoazide in a good yield. Of the alternatives investigated, α,α,α-trifluorotoluene (PhCF3) performed best. Under optimized conditions (1.5 equiv of FeCl3·6H2O, 3.0 equiv of NaN3, PhCF3 (0.2 M), 0 °C for 16 h, blue LEDs, entry 2), 2a was formed in 83% yield.
Table 1. Optimization of the Reaction Conditions.

| entry | deviation from standard conditions | 2aa (%) |
|---|---|---|
| 1 | Fe(NO3)3·9H2O (1.0 equiv), CH2Cl2, rt | 36 |
| 2 | none | 83 |
| 3 | TMSN3 instead of NaN3 | 17 |
| 4 | no iron salt | 0 |
| 5 | no light | 0 |
| 6 | FeCl3 instead of FeCl3·6H2O | <5 |
| 7 | 40 W blue LED Kessil light, rt | 80 |
Yield obtained by 1H NMR with mesitylene as internal standard.
Control experiments were performed to gain further insights into the transformation (for details including oxygen sensitivity, see the Supporting Information). When the reaction was conducted with TMSN3 under otherwise identical conditions, 2a was produced in 17% yield (entry 3). The reaction did not provide the product in the absence of iron salt or light, and 1a was fully recovered (entries 4 and 5). If anhydrous FeCl3 was used in oven-dried glassware under otherwise identical conditions, merely traces of the product were formed, indicating the necessity of water (entry 6). When using a 40 W blue LED Kessil light at 25% intensity, the best results were obtained at room temperature (entry 7; for details see the Supporting Information).
With the optimized conditions in hand, we set out to investigate the functional group tolerance of the hydroazidation reaction (Figure 3). To this end, a variety of alkyl azides were accessed in moderate to high yields from the corresponding alkenes.22 When vinyl silane 1b was subjected to the reaction conditions, β-silyl azide 2b was obtained in 80% yield. Alkyl azides 2c and 2d were isolated in yields of 73 and 80% from primary haloalkenes 1c and 1d. Notably, no substitution of either chloride or bromide was observed under the reaction conditions. 1-Dodecene yielded 2e in 86% yield. A variety of electron-poor and -rich arenes were tolerated, furnishing 2f–2h in 81–90% yield. Substrates containing protic groups, such as alcohol 1i, tosyl amide 1j, carbamate 1k, and N-tosyl imide 1l, were converted to the corresponding azides in 65–82% yield. Acid-labile t-BuMe2Si-protected alcohol 1m afforded 2m in 61% yield. Methyl carbonate 1n and thioether 1o were competent under reaction conditions, resulting in 2n and 2o in 85 and 43% yield, respectively. Azides 2p–2s were accessed in 62–75% yield. Markedly, alkenes bearing a heterocycle, such as thiophene, furan, phthalimide, oxetane, and pyridine, were also well tolerated and gave rise to products 2t–2x in 44–86% yield. The practical aspects of the method were demonstrated by the synthesis of 2a on a larger scale (2.0 mmol) in 75% yield. All terminal olefins underwent anti-Markovnikov hydroazidation in excellent selectivity (rr =12:1 to >20:1; for details see the Supporting Information).
Figure 3.
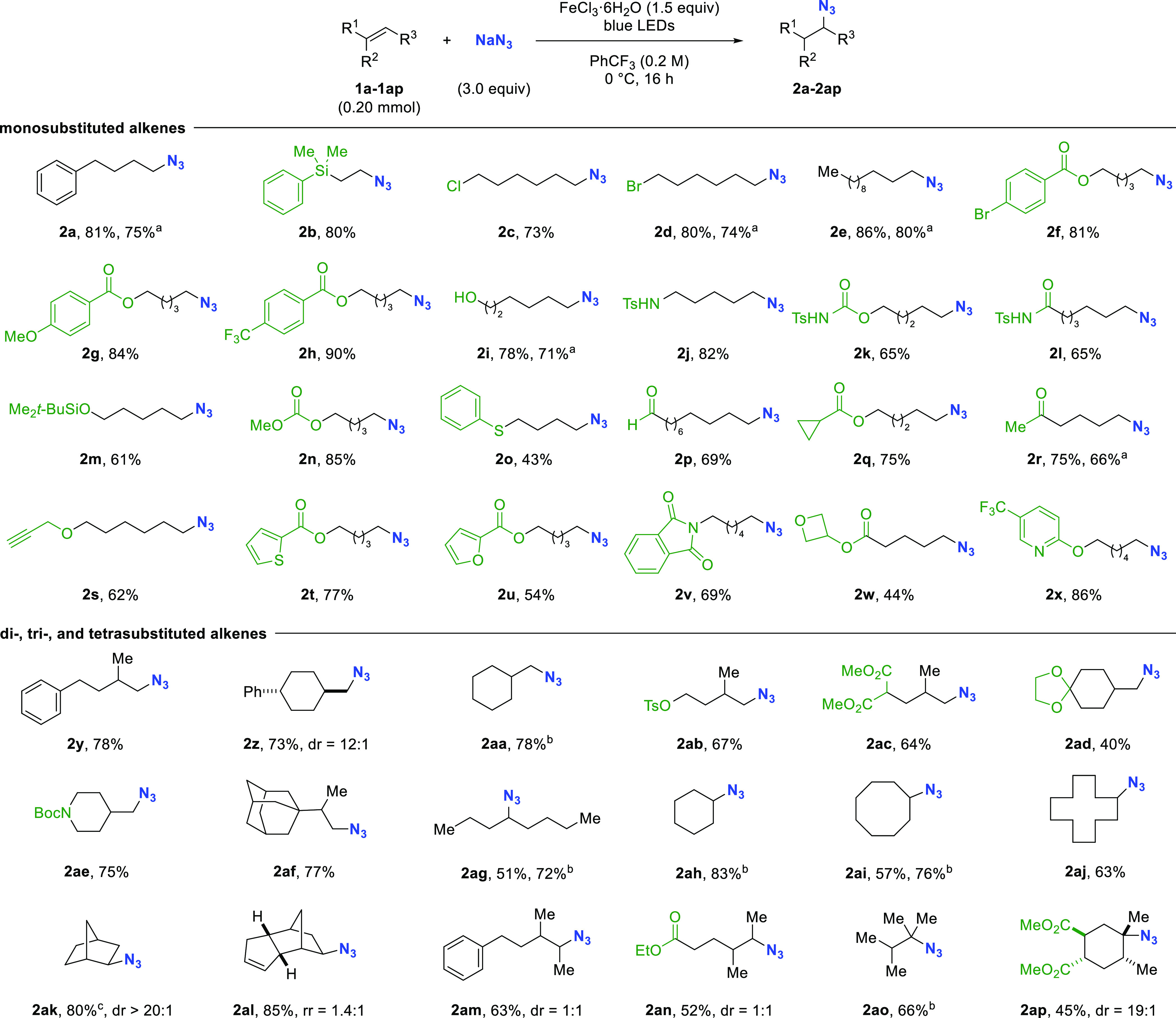
Substrate scope. Reaction conditions: olefin (0.20 mmol), NaN3 (0.60 mmol), FeCl3·6H2O (0.30 mmol) in PhCF3 (1.0 mL), irradiation in 350 W photoreactor at 0 °C for 16 h. dr determined by 1H NMR of the unpurified reaction mixture. aCarried out on 2.0 mmol scale with a 40 W blue LED Kessil light at rt. bYield obtained by 1H NMR with mesitylene as internal standard. c8 h reaction time.
Next, the effects of the alkene substitution patterns were examined. The reaction was amenable to di-, tri-, and tetrasubstituted olefins. 1,1-Disubstituted olefins 1y to 1aa afforded primary azides in 73–78% yield. Substrates bearing a tosylate, malonate, acetal, or carbamate were successfully converted to products 2ab–2ae in 40–75% yield. β-Adamantyl azide 2af was accessed in 77% yield. Acyclic and cyclic 1,2-disubstituted alkenes were subjected to the reaction conditions, furnishing the corresponding azides 2ag–2aj in 51–83% yield. Strained olefins, in particular norbornene and dicyclopentadiene, were hydroazidated in high yield (80 and 85%, respectively).23 Trisubstituted olefins were well tolerated, giving rise to secondary azides with 2am and 2an isolated in 63 and 52% yield. All di- and trisubstituted olefins employed underwent anti-Markovnikov azidation in good regioselectivity (for details see the Supporting Information). Finally, tetrasubstituted alkenes 1ao and 1ap yielded 2ao and 2ap in 66 and 45% yield, respectively.
Encouraged by the broad functional group tolerance of the hydroazidation reaction, we set out to explore the generality of the protocol in a more complex setting. An array of alkenes derived from active pharmaceutical ingredients and natural products was subjected to the reaction conditions (Figure 4). To our delight, these substrates led to the formation of desired azides 4a–4u in good yields (40–86%). In particular, molecules featuring diversely substituted arenes and heterocycles, such as oxazoles, indoles, thiazoles, β-lactams, and diazoles were well tolerated, indicating the potential of this method for late-stage application.
Figure 4.
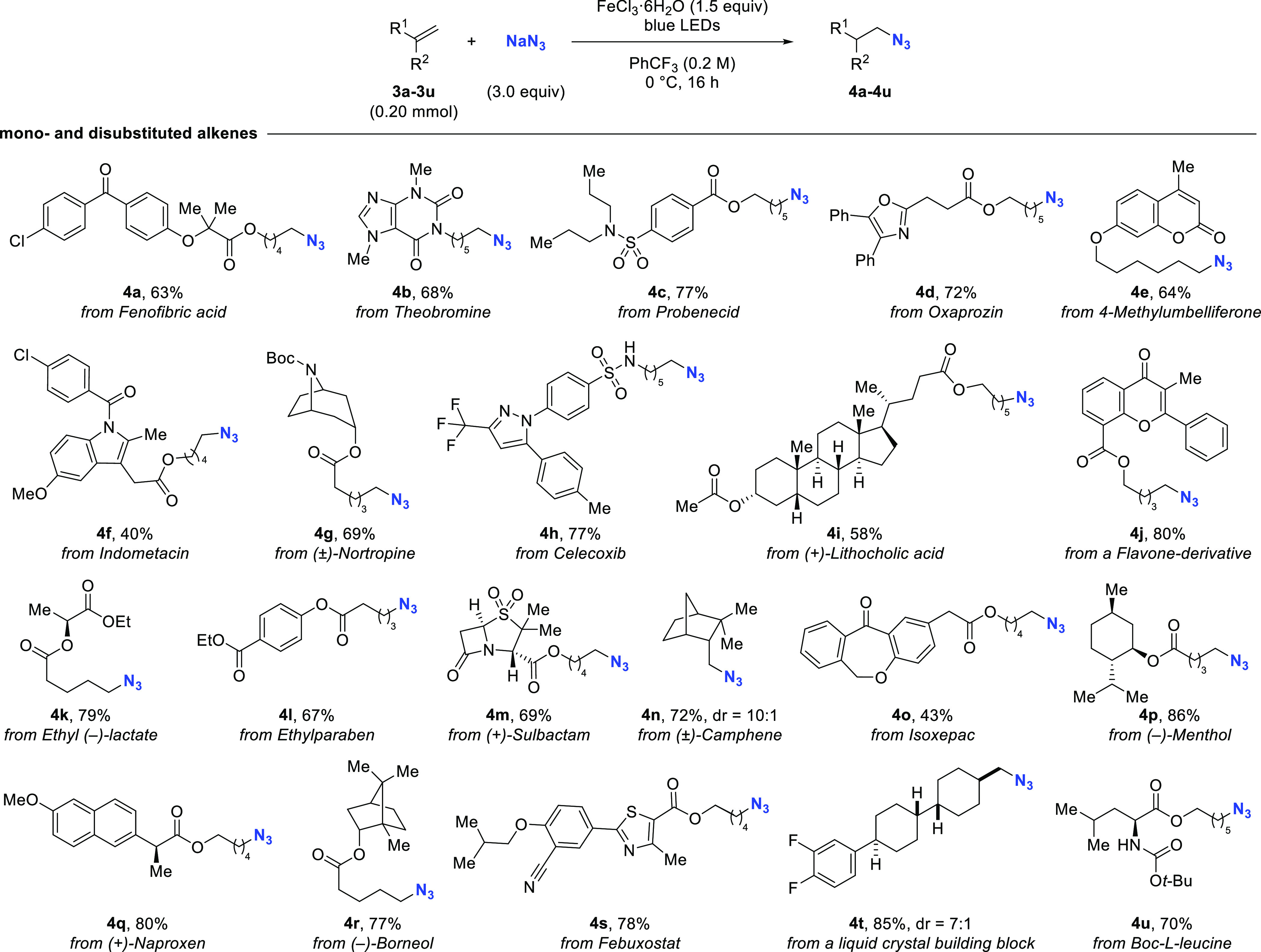
Substrate scope for olefins derived from natural products and drugs. dr determined by 1H NMR of the unpurified reaction mixture.
In order to deal with the volatility and inherent risk of low-molecular-weight azides, we examined several transformations subsequent to hydroazidation. One-pot procedures would avoid work-up, solvent evaporation, purification, isolation, and handling of the azide intermediates (Scheme 1). First, attempts to perform a Cu(I)-catalyzed azide–alkyne click reaction in situ were unsuccessful. We observed that the addition of NEt3 was crucial for the formation of triazole 7a in 76% yield. When cyclooctyne was employed as a reaction partner, triazole 7b was isolated in 54% yield from volatile azide 2ao. Primary amine 7c was accessed in 72% yield through Pd-catalyzed hydrogenation, and Staudinger reduction of cyclohexyl azide 2ah with subsequent Boc-protection furnished the corresponding carbamate 7d in 62% yield.
Scheme 1. Sequential One-Pot Transformations.

To gain mechanistic insights into the hydroazidation, a series of experiments was conducted (Figure 5). Initial investigations focused on determining whether radical species are involved in the reaction. The addition of 2.0 equiv TEMPO as radical scavenger24 under standard conditions suppressed the reaction, and no hydroazidation product could be detected by either 1H NMR or HRMS. Instead, alkene starting material was recovered.
Figure 5.

Mechanistic investigations and the proposal of a plausible mechanistic construct. dr determined by 1H NMR of the unpurified reaction mixture. aYield obtained by 1H NMR with mesitylene as internal standard.
In the context of our examination of the substrate scope, when aldehyde 8 was subjected to the standard reaction conditions, we observed 1-azido-4-methylpentane (9) as the sole product (Figure 5B). This is consistent with the formation of a secondary carbon-centered radical from the olefin followed by a 1,5-radical hydrogen atom abstraction from the aldehyde. Decarbonylation affords the more stable tertiary radical which is quenched.25
This result prompted us to examine substrates 10a and 10b which could undergo 5-exo-trig cyclization reactions (Figure 5C). N-Tosyl diallyl amine furnished pyrrolidine 11a in 55% yield, and diethyl diallylmalonate delivered the corresponding cyclopentyl product 11b in 79% yield. For both substrates, only cyclization products were isolated, suggesting that quenching is slower than cyclization for 10a and 10b.26
It has been proposed in the literature that under visible-light irradiation Fe(III) azides generate Fe(II) salts along with azidyl radicals.19c,19d Based on these reports, in the system we describe the azidyl radical can then add (anti-Markovnikov) to the olefin to provide a reactive carbon-centered radical.27 This intermediate is ultimately quenched by an H atom donor. Accordingly, we set out to identify the origin of the hydrogen atom involved in the quenching.
It has previously been observed that hydrates of boron (Me3B·OH2) and titanium (Cp2ClTi·OH2) serve as H atom donors in radical reactions.28 Consequently, we hypothesized that the iron-bound water in FeCl3·6H2O might be involved as an H atom source. In an experiment in oven-dried glassware under a nitrogen atmosphere, D2O was added to anhydrous FeCl3 followed by addition of PhCF3/NaN3/4-phenylbutene (Figure 5D). After being stirred for 16 h under blue-light irradiation, azidodeuterated product 2a-D1 was isolated in 18% yield. In parallel experiments using H2O under otherwise identical conditions, product 2a was obtained in 77% yield. The difference in yield between the two experiments suggests a strong primary kinetic isotope effect (kH/kD ≫ 1; for details see the Supporting Information). These data support water as the terminal H atom source and indicate that the H atom transfer to the secondary carbon-centered radical likely is the rate-limiting step of the transformation.17a,29 Although free water is not known to be an H atom source (HBDE(HO–H) = 118 kcal/mol), its coordination to iron dramatically decreases the bond-dissociation energy (HBDE(FeII(H2O)5(HO–H)) = 77 kcal/mol).30,31 Our observation of solvent effects described in the optimization reactions suggests that the nature of the solvent affects speciation of Fe(III) complexes in the presence of azide and chloride counterions as well as water. Further mechanistic studies to understand the nature of Fe complexes formed, including μ2-bridged dimers, are clearly necessary as they may provide additional avenues for the development of new transformations.32
In conclusion, we have developed a photochemical anti-Markovnikov hydroazidation of unactivated alkenes with FeCl3·6H2O. The transformation shows broad functional group tolerance and was amenable to terminal as well as highly substituted olefins. Salient features of the reaction are its tolerance to air and moisture and the successful use of NaN3 as a bench-stable, low-cost, and easy-to-handle azide source. We demonstrated that the simplicity and generality of this method make it ideally positioned for late-stage applications, allowing for the efficient and versatile synthesis of diverse organic azides featuring biologically active motifs.
Acknowledgments
This work was funded by the European Research Council (OLECAT, Grant ID 833540). We are grateful to Dr. M.-O. Ebert, R. Frankenstein, S. Burkhardt, and R. Arnold for support with NMR experiments, Dr. N. Trapp and M. Solar for X-ray crystallographic analysis, and C. A. Bärtschi, C. Marro, and H. Benz for the maintenance and construction of the photoreactor (all ETH Zürich). W.M.A. is grateful for partial support with funding from the SSCI (Scholarship Fund of the Swiss Chemical Industry).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c09122.
Experimental procedures and characterization data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Fischl M. A.; Richman D. D.; Grieco M. H.; Gottlieb M. S.; Volberding P. A.; Laskin O. L.; Leedom J. M.; Groopman J. E.; Mildvan D.; Schooley R. T.; et al. The Efficacy of Azidothymidine (AZT) in the Treatment of Patients with AIDS and AIDS-Related Complex. N. Engl. J. Med. 1987, 317, 185–191. 10.1056/NEJM198707233170401. [DOI] [PubMed] [Google Scholar]; b Ek S.; Goede P.; Latypov N.; Wang L. Y.; Gou-Ying Y.. New chemical compound suitable for use as a plasticiser in explosive and propellant compositions. Patent WO2009072955A1, 2009.; c Sarott R. C.; Westphal M. V.; Pfaff P.; Korn C.; Sykes D. A.; Gazzi T.; Brennecke B.; Atz K.; Weise M.; Mostinski Y.; et al. Development of High-Specificity Fluorescent Probes to Enable Cannabinoid Type 2 Receptor Studies in Living Cells. J. Am. Chem. Soc. 2020, 142, 16953–16964. 10.1021/jacs.0c05587. [DOI] [PubMed] [Google Scholar]; d Badgujar D. M.; Talawar M. B.; Asthana S. N.; Mahulikar P. P. Advances in science and technology of modern energetic materials: An overview. J. Hazard. Mater. 2008, 151, 289–305. 10.1016/j.jhazmat.2007.10.039. [DOI] [PubMed] [Google Scholar]; e Jiang S.; Iliopoulos-Tsoutsouvas C.; Tong F.; Brust C. A.; Keenan C. M.; Raghav J. G.; Hua T.; Wu S.; Ho J.-H.; Wu Y.; et al. Novel Functionalized Cannabinoid Receptor Probes: Development of Exceptionally Potent Agonists. J. Med. Chem. 2021, 64, 3870–3884. 10.1021/acs.jmedchem.0c02053. [DOI] [PubMed] [Google Scholar]; f Kumar R.; Wiebe L. I.; Knaus E. E. Synthesis and antiviral activity of novel 5-(1-azido-2-haloethyl) and 5-(1-azido-, amino-, or methoxyethyl) analogs of 2’-deoxyuridine. J. Med. Chem. 1993, 36, 2470–2474. 10.1021/jm00069a004. [DOI] [PubMed] [Google Scholar]; g Clark S. C.; Jasinski D. R.; Pevnick J. S.; Griffith J. D. Azidomorphine: Subjective effects and suppression of morphine abstinence. Clin. Pharmacol. Ther. 1976, 19, 295–299. 10.1002/cpt1976193295. [DOI] [PubMed] [Google Scholar]; h Raeburn J. A.; Devine J. D. Pharmacological Findings during Azidocillin Treatment of Chest Infections. Scand. J. Infect. Dis. 1973, 5, 135–139. 10.3109/inf.1973.5.issue-2.08. [DOI] [PubMed] [Google Scholar]; i Bhuta P.; Chung H. L.; Hwang J.-S.; Zemlicka J. Analogs of chloramphenicol: circular dichroism spectra, inhibition of ribosomal peptidyltransferase, and possible mechanism of action. J. Med. Chem. 1980, 23, 1299–1305. 10.1021/jm00186a004. [DOI] [PubMed] [Google Scholar]; j Wang G.; Ge Z.; Luo Y. Synthesis and Characterization of Poly(3-azidomethyl-3-methyl oxetane) by the Azidation of Poly(3-mesyloxymethyl-3-methyl oxetane). Propellants Explos. Pyrotech. 2015, 40, 920–926. 10.1002/prep.201500064. [DOI] [Google Scholar]; k Mellor B.A Preliminary Technical Review of DMAZ: A Low-Toxicity Hypergolic Fuel. In ESA Special Publication; Wilson A., Ed.; 2004, Vol. 557, p 22.1. [Google Scholar]
- a Hein C. D.; Liu X.-M.; Wang D. Click Chemistry, A Powerful Tool for Pharmaceutical Sciences. Pharm. Res. 2008, 25, 2216–2230. 10.1007/s11095-008-9616-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sivaguru P.; Ning Y.; Bi X. New Strategies for the Synthesis of Aliphatic Azides. Chem. Rev. 2021, 121, 4253–4307. 10.1021/acs.chemrev.0c01124. [DOI] [PubMed] [Google Scholar]; c Shee M.; Singh N. D. P. Chemical versatility of azide radical: journey from a transient species to synthetic accessibility in organic transformations. Chem. Soc. Rev. 2022, 51, 2255–2312. 10.1039/D1CS00494H. [DOI] [PubMed] [Google Scholar]; d Schock M.; Bräse S. Reactive & Efficient: Organic Azides as Cross-Linkers in Material Sciences. Molecules 2020, 25, 1009. 10.3390/molecules25041009. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Nicolaou K. C.; Bunnage M. E.; Koide K. Total Synthesis of Balanol. J. Am. Chem. Soc. 1994, 116, 8402–8403. 10.1021/ja00097a072. [DOI] [Google Scholar]; f Diethelm S.; Schindler C. S.; Carreira E. M. Synthesis of Microcin SF608 through Nucleophilic Opening of an Oxabicyclo[2.2.1]heptane. Org. Lett. 2010, 12, 3950–3953. 10.1021/ol1017189. [DOI] [PubMed] [Google Scholar]; g Xu Z.; Wang Q.; Zhu J. Enantioselective Total Syntheses of Leuconolam-Leuconoxine-Mersicarpine Group Monoterpene Indole Alkaloids. J. Am. Chem. Soc. 2013, 135, 19127–19130. 10.1021/ja4115192. [DOI] [PubMed] [Google Scholar]; h Yang Y.; Bai Y.; Sun S.; Dai M. Biosynthetically Inspired Divergent Approach to Monoterpene Indole Alkaloids: Total Synthesis of Mersicarpine, Leuconodines B and D, Leuconoxine, Melodinine E, Leuconolam, and Rhazinilam. Org. Lett. 2014, 16, 6216–6219. 10.1021/ol503150c. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Leng L.; Zhou X.; Liao Q.; Wang F.; Song H.; Zhang D.; Liu X.-Y.; Qin Y. Asymmetric Total Syntheses of Kopsia Indole Alkaloids. Angew. Chem., Int. Ed. 2017, 56, 3703–3707. 10.1002/anie.201700831. [DOI] [PubMed] [Google Scholar]
- a Bednarek C.; Wehl I.; Jung N.; Schepers U.; Bräse S. The Staudinger Ligation. Chem. Rev. 2020, 120, 4301–4354. 10.1021/acs.chemrev.9b00665. [DOI] [PubMed] [Google Scholar]; b Staudinger H.; Meyer J. Über neue organische Phosphorverbindungen III. Phosphinmethylenderivate und Phosphinimine. Helv. Chim. Acta 1919, 2, 635–646. 10.1002/hlca.19190020164. [DOI] [Google Scholar]; c Schilling C. I.; Jung N.; Biskup M.; Schepers U.; Bräse S. Bioconjugation viaazide-Staudinger ligation: an overview. Chem. Soc. Rev. 2011, 40, 4840–4871. 10.1039/c0cs00123f. [DOI] [PubMed] [Google Scholar]
- a Huisgen R.Proc. Chem. Soc. London 1961, 357–396. [Google Scholar]; b Huisgen R. 1,3-Dipolar Cycloadditions. Past and Future. Angew. Chem., Int. Ed. 1963, 2, 565–598. 10.1002/anie.196305651. [DOI] [Google Scholar]; c Breugst M.; Reissig H.-U. The Huisgen Reaction: Milestones of the 1,3-Dipolar Cycloaddition. Angew. Chem., Int. Ed. 2020, 59, 12293–12307. 10.1002/anie.202003115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kolb H. C.; Finn M. G.; Sharpless K. B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. . [DOI] [PubMed] [Google Scholar]; b Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. . [DOI] [PubMed] [Google Scholar]; c Tornøe C. W.; Christensen C.; Meldal M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002, 67, 3057–3064. 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]; d Baskin J. M.; Prescher J. A.; Laughlin S. T.; Agard N. J.; Chang P. V.; Miller I. A.; Lo A.; Codelli J. A.; Bertozzi C. R. Copper-free click chemistry for dynamic in vivo imaging. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 16793–16797. 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Aube J.; Milligan G. L. Intramolecular Schmidt reaction of alkyl azides. J. Am. Chem. Soc. 1991, 113, 8965–8966. 10.1021/ja00023a065. [DOI] [Google Scholar]; b Nyfeler E.; Renaud P. Intramolecular Schmidt Reaction: Applications in Natural Product Synthesis. Chimia 2006, 60, 276. 10.2533/000942906777674714. [DOI] [Google Scholar]; c Desai P.; Schildknegt K.; Agrios K. A.; Mossman C.; Milligan G. L.; Aubé J. Reactions of Alkyl Azides and Ketones as Mediated by Lewis Acids: Schmidt and Mannich Reactions Using Azide Precursors. J. Am. Chem. Soc. 2000, 122, 7226–7232. 10.1021/ja000490v. [DOI] [Google Scholar]
- a Shah S.; Protasiewicz J. D. ‘Phospha-variations’ on the themes of Staudinger and Wittig: phosphorus analogs of Wittig reagents. Coord. Chem. Rev. 2000, 210, 181–201. 10.1016/S0010-8545(00)00311-8. [DOI] [Google Scholar]; b Palacios F.; Alonso C.; Aparicio D.; Rubiales G.; de los Santos J. M. The aza-Wittig reaction: an efficient tool for the construction of carbon-nitrogen double bonds. Tetrahedron 2007, 63, 523–575. 10.1016/j.tet.2006.09.048. [DOI] [Google Scholar]; c Lao Z.; Toy P. H. Catalytic Wittig and aza-Wittig reactions. Beilstein J. Org. Chem. 2016, 12, 2577–2587. 10.3762/bjoc.12.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Shin K.; Kim H.; Chang S. Transition-Metal-Catalyzed C-N Bond Forming Reactions Using Organic Azides as the Nitrogen Source: A Journey for the Mild and Versatile C-H Amination. Acc. Chem. Res. 2015, 48, 1040–1052. 10.1021/acs.accounts.5b00020. [DOI] [PubMed] [Google Scholar]; b Uchida T.; Katsuki T. Asymmetric Nitrene Transfer Reactions: Sulfimidation, Aziridination and C-H Amination Using Azide Compounds as Nitrene Precursors. Chem. Rec. 2014, 14, 117–129. 10.1002/tcr.201300027. [DOI] [PubMed] [Google Scholar]
- a Wuts P. G. M.; Greene T. W.. Protection for the Amino Group. In Greene’s Protective Groups in Organic Synthesis; John Wiley & Sons: 2006; pp 696–926. [Google Scholar]; b Titz A.; Radic Z.; Schwardt O.; Ernst B. A safe and convenient method for the preparation of triflyl azide, and its use in diazo transfer reactions to primary amines. Tetrahedron Lett. 2006, 47, 2383–2385. 10.1016/j.tetlet.2006.01.157. [DOI] [Google Scholar]; c Breder A.; Chinigo G. M.; Waltman A. W.; Carreira E. M. Towards the Synthesis of Massadine: A Unified Strategy for the Stereoselective Synthesis of the Carbocyclic C,D-Ring Subunit. Chem.—Eur. J. 2011, 17, 12405–12416. 10.1002/chem.201101862. [DOI] [PubMed] [Google Scholar]
- Bräse S.; Mende M.; Jobelius H. H.; Scharf H.-D.. Hydrazoic Acid and Azides. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: 2015; pp 1–11. [Google Scholar]
- a Boyer J. H.; Canter F. C. Alkyl and Aryl Azides. Chem. Rev. 1954, 54, 1–57. 10.1021/cr60167a001. [DOI] [Google Scholar]; b Scriven E. F. V.; Turnbull K. Azides: their preparation and synthetic uses. Chem. Rev. 1988, 88, 297–368. 10.1021/cr00084a001. [DOI] [Google Scholar]; c Bräse S.; Gil C.; Knepper K.; Zimmermann V. Organic Azides: An Exploding Diversity of a Unique Class of Compounds. Angew. Chem., Int. Ed. 2005, 44, 5188–5240. 10.1002/anie.200400657. [DOI] [PubMed] [Google Scholar]; d Goswami M.; de Bruin B. Metal-Catalysed Azidation of Organic Molecules. Eur. J. Org. Chem. 2017, 2017, 1152–1176. 10.1002/ejoc.201601390. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Sharma A.; Hartwig J. F. Metal-catalysed azidation of tertiary C-H bonds suitable for late-stage functionalization. Nature 2015, 517, 600–604. 10.1038/nature14127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hassner A.; Fibiger R.; Andisik D. Synthetic methods. 19. Lewis Acid Catalyzed Conversion of Alkenes and Alcohols to Azides. J. Org. Chem. 1984, 49, 4237–4244. 10.1021/jo00196a025. [DOI] [Google Scholar]; b Breton G. W.; Daus K. A.; Kropp P. J. Surface-mediated reactions. 2. Addition of hydrazoic acid to alkenes. J. Org. Chem. 1992, 57, 6646–6649. 10.1021/jo00050a053. [DOI] [Google Scholar]
- a Waser J.; Gaspar B.; Nambu H.; Carreira E. M. Hydrazines and Azides via the Metal-Catalyzed Hydrohydrazination and Hydroazidation of Olefins. J. Am. Chem. Soc. 2006, 128, 11693–11712. 10.1021/ja062355+. [DOI] [PubMed] [Google Scholar]; b Waser J.; Nambu H.; Carreira E. M. Cobalt-Catalyzed Hydroazidation of Olefins: Convenient Access to Alkyl Azides. J. Am. Chem. Soc. 2005, 127, 8294–8295. 10.1021/ja052164r. [DOI] [PubMed] [Google Scholar]
- Leggans E. K.; Barker T. J.; Duncan K. K.; Boger D. L. Iron(III)/NaBH4-Mediated Additions to Unactivated Alkenes: Synthesis of Novel 20′-Vinblastine Analogues. Org. Lett. 2012, 14, 1428–1431. 10.1021/ol300173v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonca G. H.; Ong D. Y.; Tran T. M. H.; Tejo C.; Chiba S.; Gagosz F. Anti-Markovnikov Hydrofunctionalization of Alkenes: Use of a Benzyl Group as a Traceless Redox-Active Hydrogen Donor. Angew. Chem., Int. Ed. 2017, 56, 11440–11444. 10.1002/anie.201705368. [DOI] [PubMed] [Google Scholar]
- a Wang J.-J.; Yu W. Anti-Markovnikov Hydroazidation of Alkenes by Visible-Light Photoredox Catalysis. Chem.—Eur. J. 2019, 25, 3510–3514. 10.1002/chem.201806371. [DOI] [PubMed] [Google Scholar]; (b) Yu and co-workers reported 42 examples for the hydroazidation of aryl amides; however, five examples for the hydroazidation of phenyl ketones and phenyl esters under metal photoredox catalysis are also shown.
- a Li H.; Shen S.-J.; Zhu C.-L.; Xu H. Direct Intermolecular Anti-Markovnikov Hydroazidation of Unactivated Olefins. J. Am. Chem. Soc. 2019, 141, 9415–9421. 10.1021/jacs.9b04381. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Li X.; Chen P.; Liu G. Iodine(III) reagent (ABX—N3)-induced intermolecular anti-Markovnikov hydroazidation of unactivated alkenes. Sci. China Chem. 2019, 62, 1537–1541. 10.1007/s11426-019-9628-9. [DOI] [Google Scholar]
- a Müller N.; Schreib B. S.; Leutenegger S. U.; Carreira E. M. Picolinamides and Iodoalkynes Enable Palladium-Catalyzed syn-Aminoalkynylation of Di- and Trisubstituted Alkenes to Give Pyrrolidines. Angew. Chem., Int. Ed. 2022, 61, e202204535. 10.1002/anie.202204535. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Schreib B. S.; Carreira E. M. Palladium-Catalyzed Regioselective C-H Iodination of Unactivated Alkenes. J. Am. Chem. Soc. 2019, 141, 8758–8763. 10.1021/jacs.9b03998. [DOI] [PubMed] [Google Scholar]; c Fischer D. M.; Freis M.; Amberg W. M.; Lindner H.; Carreira E. M. Organophotocatalytic carbo-heterofunctionalization of unactivated olefins with pendant nucleophiles. Chem. Sci. 2023, 14, 7256–7261. 10.1039/D3SC02250A. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Fischer D. M.; Lindner H.; Amberg W. M.; Carreira E. M. Intermolecular Organophotocatalytic Cyclopropanation of Unactivated Olefins. J. Am. Chem. Soc. 2023, 145, 774–780. 10.1021/jacs.2c11680. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Fischer D. M.; Balkenhohl M.; Carreira E. M. Cobalt-Catalyzed Cyclization of Unsaturated N-Acyl Sulfonamides: a Diverted Mukaiyama Hydration Reaction. JACS Au 2022, 2, 1071–1077. 10.1021/jacsau.2c00186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kochi J. K. Photolyses of Metal Compounds: Cupric Chloride in Organic Media. J. Am. Chem. Soc. 1962, 84, 2121–2127. 10.1021/ja00870a025. [DOI] [Google Scholar]; b Hossain A.; Vidyasagar A.; Eichinger C.; Lankes C.; Phan J.; Rehbein J.; Reiser O. Visible-Light-Accelerated Copper(II)-Catalyzed Regio- and Chemoselective Oxo-Azidation of Vinyl Arenes. Angew. Chem., Int. Ed. 2018, 57, 8288–8292. 10.1002/anie.201801678. [DOI] [PubMed] [Google Scholar]; c Bian K.-J.; Kao S.-C.; Nemoto D.; Chen X.-W.; West J. G. Photochemical diazidation of alkenes enabled by ligand-to-metal charge transfer and radical ligand transfer. Nat. Commun. 2022, 13, 7881. 10.1038/s41467-022-35560-3. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Zhang M.; Zhang J.; Li Q.; Shi Y. Iron-mediated ligand-to-metal charge transfer enables 1,2-diazidation of alkenes. Nat. Commun. 2022, 13, 7880. 10.1038/s41467-022-35344-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Lian P.; Long W.; Li J.; Zheng Y.; Wan X. Visible-Light-Induced Vicinal Dichlorination of Alkenes through LMCT Excitation of CuCl2. Angew. Chem., Int. Ed. 2020, 59, 23603–23608. 10.1002/anie.202010801. [DOI] [PubMed] [Google Scholar]; f Juliá F. Ligand-to-Metal Charge Transfer (LMCT) Photochemistry at 3d-Metal Complexes: An Emerging Tool for Sustainable Organic Synthesis. ChemCatChem 2022, 14, e202200916. 10.1002/cctc.202200916. [DOI] [Google Scholar]; g Yamane M.; Kanzaki Y.; Mitsunuma H.; Kanai M. Titanium(IV) Chloride-Catalyzed Photoalkylation via C(sp3)-H Bond Activation of Alkanes. Org. Lett. 2022, 24, 1486–1490. 10.1021/acs.orglett.2c00138. [DOI] [PubMed] [Google Scholar]
- For early work on the use of Fe salts in olefin diazidations as well as chloro- and alkoxyazidations, see:; a Minisci F.; Galli R.; Pallini U. Reazioni radicaliche in soluzione. Reattività di radicali liberi al carbonio provenienti da butadiene in presenza di sistemi ossido-riduttivi. Gazz. Chim. Ital. 1961, 90, 1023–1029. [Google Scholar]; b Minisci F.; Galli R.; Cecere M. Nuovi processi radicalici in presenza di sistemi ossido-riduttivi. Indroduzione diretta di gruppi azidici. Gazz. Chim. Ital. 1964, 94, 67–90. [Google Scholar]
- a Conrow R. E.; Dean W. D. Diazidomethane Explosion. Org. Process Res. Dev. 2008, 12, 1285–1286. 10.1021/op8000977. [DOI] [Google Scholar]; b Hassner A.; Stern M.; Gottlieb H. E.; Frolow F. Synthetic methods. 33. Utility of a Polymeric Azide Reagent in the Formation of Di- and Triazidomethane. Their NMR Spectra and the X-ray Structure of Derived Triazoles. J. Org. Chem. 1990, 55, 2304–2306. 10.1021/jo00295a014. [DOI] [Google Scholar]
- For an overview of unsuccessful substrates see the Supporting Information.
- The obtained exo selectivity for both regioisomers of azide 2al was determined via X-ray crystallographic analysis after one-pot diversification to the corresponding triazole (for details see the Supporting Information, compound SI-7).
- Zhang B.; Studer A. Stereoselective Radical Azidooxygenation of Alkenes. Org. Lett. 2013, 15, 4548–4551. 10.1021/ol402106x. [DOI] [PubMed] [Google Scholar]
- a Chatgilialoglu C.; Crich D.; Komatsu M.; Ryu I. Chemistry of Acyl Radicals. Chem. Rev. 1999, 99, 1991–2070. 10.1021/cr9601425. [DOI] [PubMed] [Google Scholar]; b Fischer H.; Paul H. Rate constants for some prototype radical reactions in liquids by kinetic electron spin resonance. Acc. Chem. Res. 1987, 20, 200–206. 10.1021/ar00137a007. [DOI] [Google Scholar]; c Gorin E. Photolysis of Aldehydes and Ketones in the Presence of Iodine Vapor. J. Chem. Phys. 1939, 7, 256–264. 10.1063/1.1750427. [DOI] [Google Scholar]
- Newcomb M. Competition Methods and Scales for Alkyl Radical Reaction Kinetics. Tetrahedron 1993, 49, 1151–1176. 10.1016/S0040-4020(01)85808-7. [DOI] [Google Scholar]
- Workentin M. S.; Wagner B. D.; Lusztyk J.; Wayner D. D. M. Azidyl Radical Reactivity. N6•- as a Kinetic Probe for the Addition Reactions of Azidyl Radicals with Olefins. J. Am. Chem. Soc. 1995, 117, 119–126. 10.1021/ja00106a015. [DOI] [Google Scholar]
- a Boekell N. G.; Flowers R. A. II. Coordination-Induced Bond Weakening. Chem. Rev. 2022, 122, 13447–13477. 10.1021/acs.chemrev.2c00254. [DOI] [PubMed] [Google Scholar]; b Gansäuer A.; Shi L.; Otte M.; Huth I.; Rosales A.; Sancho-Sanz I.; Padial N. M.; Oltra J. E.. Hydrogen Atom Donors: Recent Developments. In Radicals in Synthesis III; Heinrich M., Gansäuer A., Eds.; Springer: Berlin, 2011; pp 93–120. [DOI] [PubMed] [Google Scholar]; c Spiegel D. A.; Wiberg K. B.; Schacherer L. N.; Medeiros M. R.; Wood J. L. Deoxygenation of Alcohols Employing Water as the Hydrogen Atom Source. J. Am. Chem. Soc. 2005, 127, 12513–12515. 10.1021/ja052185l. [DOI] [PubMed] [Google Scholar]; d Barrero A. F.; Oltra J. E.; Cuerva J. M.; Rosales A. Effects of Solvents and Water in Ti(III)-Mediated Radical Cyclizations of Epoxygermacrolides. Straightforward Synthesis and Absolute Stereochemistry of (+)-3α-Hydroxyreynosin and Related Eudesmanolides. J. Org. Chem. 2002, 67, 2566–2571. 10.1021/jo016277e. [DOI] [PubMed] [Google Scholar]
- a Simmons E. M.; Hartwig J. F. On the Interpretation of Deuterium Kinetic Isotope Effects in C-H Bond Functionalizations by Transition-Metal Complexes. Angew. Chem., Int. Ed. 2012, 51, 3066–3072. 10.1002/anie.201107334. [DOI] [PubMed] [Google Scholar]; b Wiberg K. B. The Deuterium Isotope Effect. Chem. Rev. 1955, 55, 713–743. 10.1021/cr50004a004. [DOI] [Google Scholar]
- a Goldsmith C. R.; Jonas R. T.; Stack T. D. P. C-H Bond Activation by a Ferric Methoxide Complex: Modeling the Rate-Determining Step in the Mechanism of Lipoxygenase. J. Am. Chem. Soc. 2002, 124, 83–96. 10.1021/ja016451g. [DOI] [PubMed] [Google Scholar]; b Ruscic B.; Wagner A. F.; Harding L. B.; Asher R. L.; Feller D.; Dixon D. A.; Peterson K. A.; Song Y.; Qian X.; Ng C.-Y.; et al. On the Enthalpy of Formation of Hydroxyl Radical and Gas-Phase Bond Dissociation Energies of Water and Hydroxyl. J. Phys. Chem. A 2002, 106, 2727–2747. 10.1021/jp013909s. [DOI] [Google Scholar]
- (a) We cannot rule out that H-atom transfer occurs from in situ generated hydrazoic acid (HBDE(N3–H) = 85 kcal/mol).; b Chiang M.; Wheeler R. H—N3 and CH3—N3 bond dissociation energies. Can. J. Chem. 1968, 46, 3785–3788. 10.1139/v68-629. [DOI] [Google Scholar]
- (a) Indeed, it has been noted that characteristic features of iron hydrates in noncoordinating solvents are its complexity and sensitivity to conditions.; b Cotton F. A.; Wilkinson G.; Murillo C. A.; Bochmann M.. Advanced Inorganic Chemistry, 6th ed.; Wiley-Interscience: 1999; p 787. [Google Scholar]; c Albright H.; Riehl P. S.; McAtee C. C.; Reid J. P.; Ludwig J. R.; Karp L. A.; Zimmerman P. M.; Sigman M. S.; Schindler C. S. Catalytic Carbonyl-Olefin Metathesis of Aliphatic Ketones: Iron(III) Homo-Dimers as Lewis Acidic Superelectrophiles. J. Am. Chem. Soc. 2019, 141, 1690–1700. 10.1021/jacs.8b11840. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Jones T. K.; Denmark S. E. Silicon-Directed Nazarov Reactions III. Stereochemical and Mechanistic Considerations. Helv. Chim. Acta 1983, 66, 2397–2411. 10.1002/hlca.19830660803. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.