

Abstract
Proteolysis Targeting Chimeras (PROTACs) are a promising therapeutic strategy to selectively promote the degradation of protein targets by exploiting the ubiquitin-proteasome system. Among the limited number of E3 ligase ligands discovered for the PROTAC technology, ligands of cereblon (CRBN) E3 ligase such as pomalidomide, thalidomide, or lenalidomide are the most frequently used for the development of PROTACs. Our group previously reported that a phenyl group could be tolerated on the C4-position of lenalidomide as the ligand of CRBN to develop PROTACs. Herein, we report a modular chemistry platform for the efficient attachment of various ortho, meta, and para substituted phenyls to the C4-position of the lenalidomide via Suzuki cross-coupling reaction, which allows the systematic investigation of the linker effect for the development of PROTACs against any target. We examined the substrate scope by preparing twelve lenalidomide-derived CRBN E3 ligase ligands with different linkers.
Keywords: Suzuki cross-coupling, lenalidomide, partial PROTAC, phthalimide, Cereblon ligands
Graphical Abstract
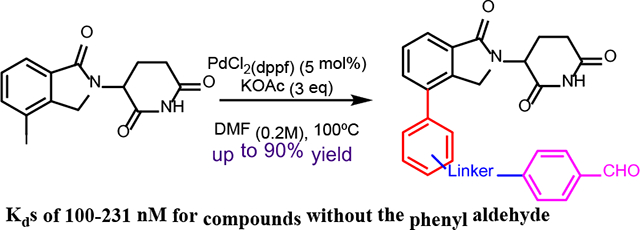
Here we described a modular platform for the synthesis of ortho-, meta-, and para- substituted phenyl-connected lenalidomide-derived CRBN E3 ligase ligands and a partial PROTAC library under Suzuki cross-coupling conditions. This method allowed us to systematically introduce linkers with subtle differences in length and orientation to examine their effect for the activity of PROTACs against any potential targets.
Introduction
Proteolysis targeting chimeras (PROTACs) are heterobifunctional small molecules that can degrade a protein of interest (POI) by employing the ubiquitin (Ub)-Proteasome System.1 These molecules consist of a linker bound by two ligands. One of the ligands binds to the POI and the other one binds to an E3 ubiquitin ligase.2 The simultaneous binding forms a ternary complex which initiates an E1-E2-E3 ubiquitin ligase cascade to enable targeted protein’s polyubiquitination followed by proteasome degradation of the POI.2 Since the PROTAC molecule is used catalytically, the PROTAC molecule recycles and promotes the process continuously.
Although the PROTAC technology has been applied to a variety of disease-associated proteins, only a handful of E3 ligase ligands have been successfully discovered. Among them, CRBN and von Hippel-Lindau (VHL) are the two most widely used E3 ligase ligands for the development of PROTACs.3 Specifically, CRBN E3 ligase ligands have been more commonly used in PROTACs for the degradation of protein targets related to many diseases, such as cancer, neurodegenerative diseases like Alzheimer’s disease, and immune disorders.4,5 Although there are extensive patent literature on various CRBN ligands,6 no detailed structure activity relationship were disclosed in these patents. Most of the frequently used CRBN ligands still hold limitations7 such as low potency, permeability, or poor selectivity and efforts on improving them are actively pursued.8,9 Moreover, the chemistry developed for the assembly of CRBN ligands with various linkers bearing diverse properties is also limited.1,3,10,11 This is particularly important for the development of PROTACs, as the types of linkers and their attachment points are essential for the pharmacological properties of PROTACs, such as potency, selectivity, solubility, and metabolic stability.12
To date, most of the CRBN E3 ligase ligands in PROTACs rely on pomalidomide, 4-hydroxythalidomide, alkyl-based thalidomide, and lenalidomide derivatives (Figure 1).5 Because lenalidomide lacks a carbonyl group in the phthalimide ring, which contributes to the better chemical and metabolic stability over pomalidomide or thalidomide,13 lenalidomide-based ligands are found more often over pomalidomide-derived ligands in the development of PROTAC type of degraders, when better stability is more important than other parameters.1,5
Figure 1.
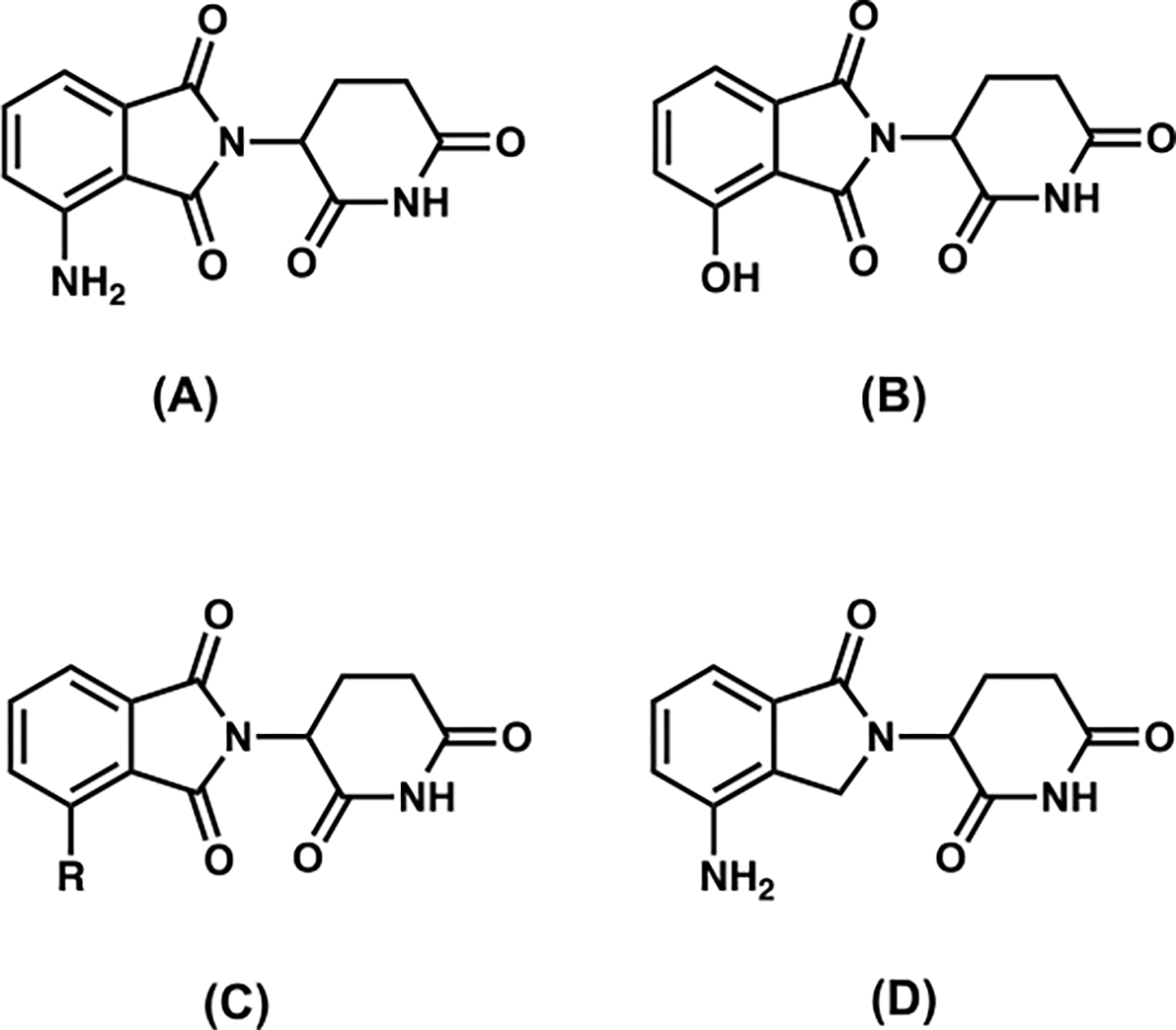
Structures of pomalidomide (A), 4-hydroxythalidomide (B), alkyl-based thalidomide (C), and lenalidomide (D).
A recent study involving the development of a cell-based target engagement assay by our group suggests that lenalidomide-based analogs with a phenyl substituent displayed high affinity and better selectivity to CRBN over thalidomide (Figure 2).14 These compounds containing the phenyl substituent had an activity comparable to pomalidomide, yet they do not degrade IKZFs neo-substrates like pomalidomide or thalidomide.14 Moreover, ortho, meta, or para substitutions of the phenyl ring did not decrease the activity significantly14 indicating the possibility of the introduction of new phenyl-connected linkers at any of these positions for the development of PROTACs, which will allow the systematic evaluation of the effect of linker to the activity of PROTACs.
Figure 2.
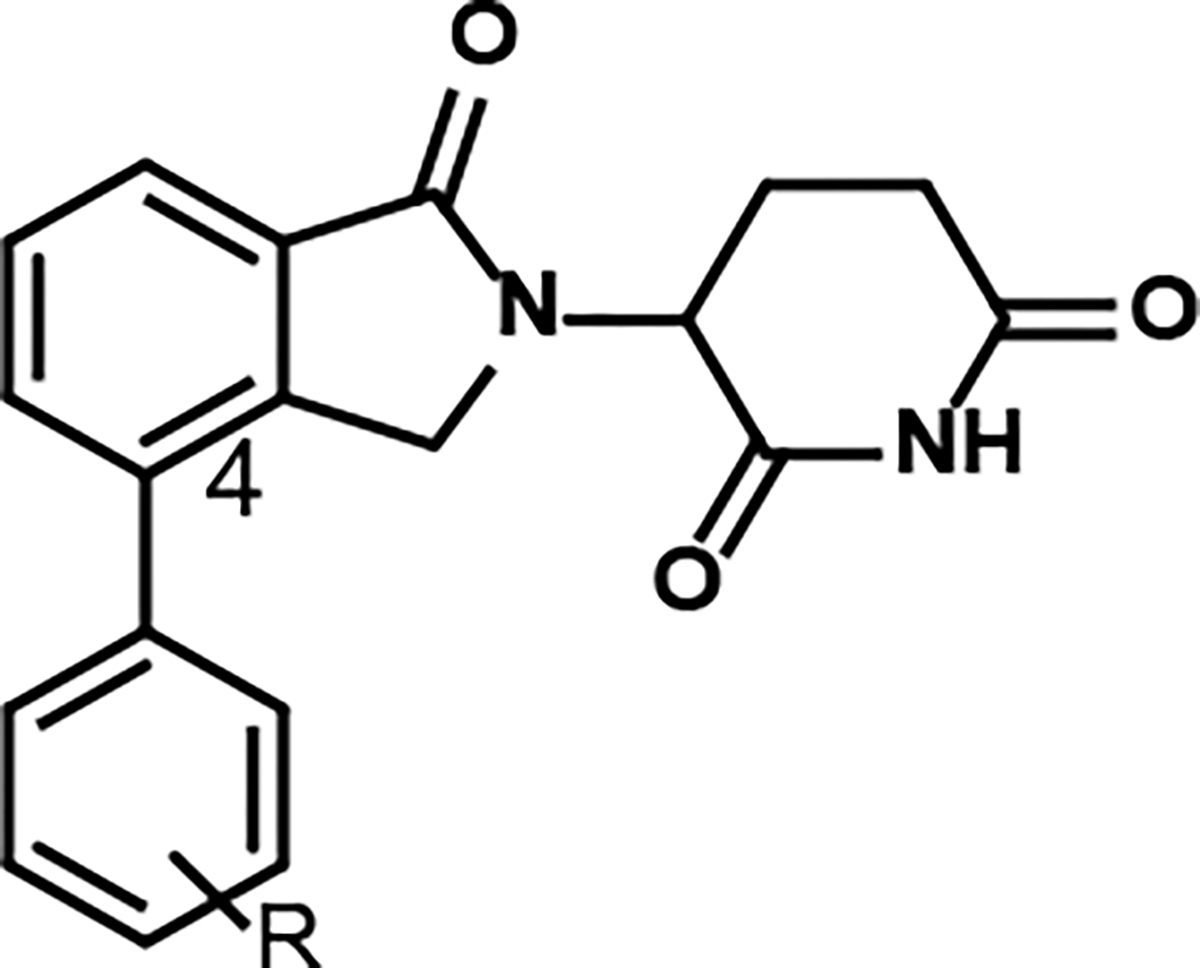
Structure of C4-lenalidomide-derived substituted phenyl ring.
Optimizing commonly used synthetic conditions for PROTACs could benefit their applications as chemical probes and therapeutics. Most current lenalidomide-based E3 ligase ligands have linkers attached to the C4- or C5- position of the phthalimide ring.10 However, compared to pomalidomide-based ligands, the synthesis of lenalidomide-based PROTACs can be limited since the decrease of the phthalimide ring’s electrophilicity made some methods such as SNAr not accessible.10 In addition, some cross-coupling reactions, which are widely used in PROTAC synthesis to attach linkers to the bicyclic ring, can lead to the hydrolysis of the cyclic imides.
Existing alkyl linkers in lenalidomide-based PROTACs have been focused on introducing alkynes followed by reduction.5 Recently, a palladium-catalyzed Buchwald-Hartwig amination protocol was reported for the synthesis of lenalidomide-based PROTACs from aryl bromides (Scheme 1A).10 A similar Suzuki cross-coupling strategy was reported by using an isoindolinone boronic acid intermediate to introduce an ester to the phthalimide ring of lenalidomide-based analogs, which can be used for the attachment of amine linkers after hydrolysis (Scheme 1B).15
Scheme 1.
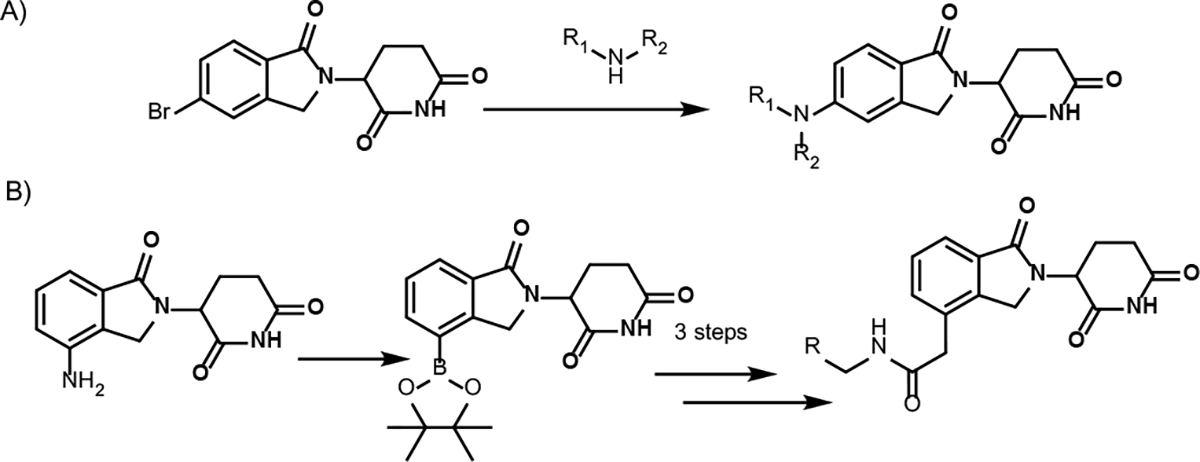
Current methods for the synthesis of lenalidomide-derived CRBN E3 ligase ligands with an attachment.
We herein describe the development of a modular chemistry platform for the direct attachment of phenyl groups to the phthalimide moiety of lenalidomide-based ligands by Suzuki cross-coupling. This strategy provides an opportunity to introduce ortho, meta, and para substituted phenyl groups to lenalidomide, which allows systematic evaluation of the effect of the attachment positions to the property of PROTACs against various targets. Finally, we applied some of the lenalidomide-based ligands with phenyl substitutions to the creation of a pre-assembled partial PROTAC library composed of twelve compounds that can later be used in the synthesis of full PROTAC libraries for any protein of interest.
Results and Discussion
We first prepared the iodolenalidomide derivative 2, a key intermediate for the Suzuki cross-coupling reaction. The synthesis of the iodo-lenalidomide intermediate 2 was achieved in three steps as previously reported14,16 from commercially available starting material 1 (Scheme 2). Because more diverse aryl iodides are available than aryl boronic acids, we decided to replace the iodide in 2 by a boronic ester functional group so that we can couple it with various functionalized aryl iodides if necessary. We adapted the cross-coupling reaction condition reported in the literature for this transformation17 (Scheme 3). Essentially, a palladium-catalyzed coupling reaction using a pinacol ester diboron and aryl halides can afford various arylboronic esters.17
Scheme 2.
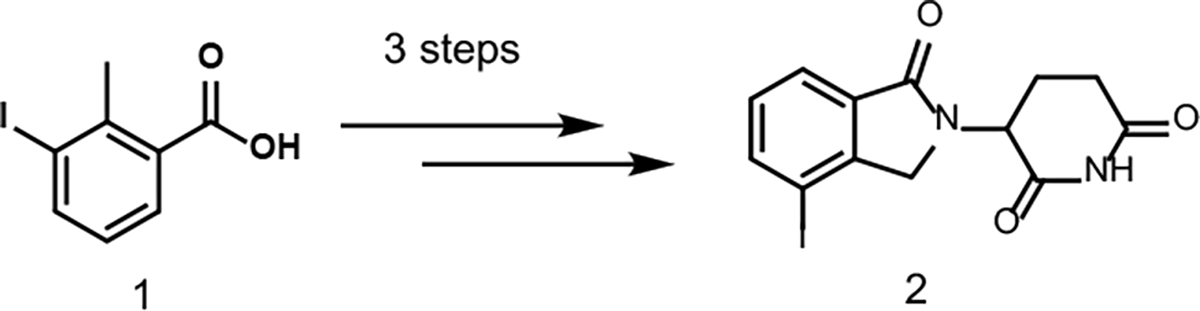
Synthesis of key iodide-lenalidomide starting material 2.
Scheme 3.
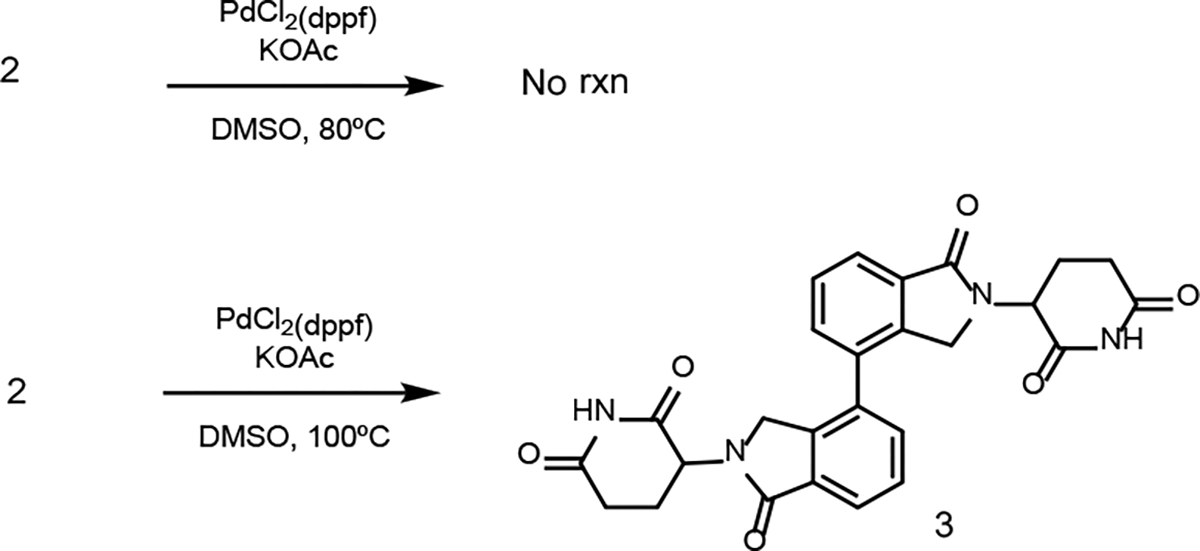
Optimization of the Suzuki cross-coupling reaction using 2 (1 eq), bis(pinacolato) diboron (2 eq), PdCl2(dppf) (5 mol %), KOAC (3 eq), and DMSO or 1,4-dioxane as solvents (0.2 M).
First, applying the previously published standard conditions17 with iodo-lenalidomide 2 as starting material led to no reaction after 24 h (Scheme 3). Thus, we decided to start our optimization process by examining various parameters. Increasing the temperature from 80 °C to 100 °C gave a major byproduct. Careful analysis of this byproduct suggests that a Suzuki cross-coupling was taking place under these conditions and a dimerization product 3 was formed as indicated by LC-MS (Scheme 3).
The observation of dimerization product 3 suggests that Suzuki cross-coupling reaction between an aryl iodide and an aryl boronic ester can occur under the above condition with a mild base. Previously, we had to use a strong base such as potassium carbonate and aqueous solution to promote the cross-coupling reaction and significant amount of hydrolysis byproduct was observed (Scheme 4A),14 which has also been reported by others.18 We had to treat the resulting mixture with CDI to re-cyclize the hydrolysis product to imide.14 Not surprisingly, a low yield (~20%) was often obtained for the final product. To test the applicability of the new Suzuki coupling condition with a mild base, we synthesized a model CRBN E3 ligase ligand 4 based on the phenyl linker (Scheme 4B). Since in our previous conditions we had set the temperature to 90 °C, we decided to test our new Suzuki cross-coupling conditions using the same temperature for comparison. Excitingly, our results showed that the new Suzuki coupling conditions improved the yield of product 4 from 20% to 58%. Compared to our previously used Suzuki coupling conditions (Scheme 4A), the new condition with a mild base (Scheme 4B) did not lead to any hydrolysis product.
Scheme 4.
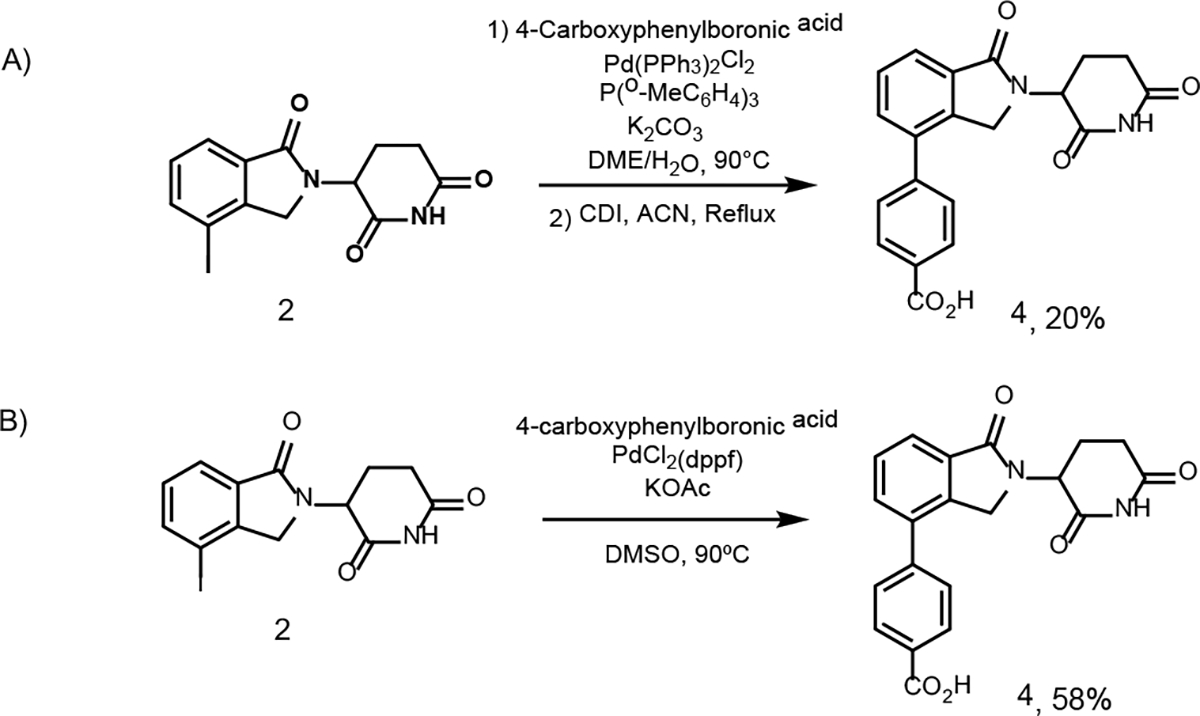
Suzuki cross-coupling model reaction.
Next, we synthesized a small library of twelve lenalidomide derived-compounds with linkers on the ortho-, meta-, or para- positions to the terminal phenyl ring (Scheme 5). Most strategies for PROTAC linker synthesis rely on amines, amides, ethers, or C-C bonds.19 Our strategy would allow the introduction of various linkers to the ortho-, meta-, and para- position of the phenyl ring by incorporating a Boc-amine or tert-butyl ester, phenol, or aldehyde to the phenyl boronic ester or boronic acid motif, which can then be further elaborated to PROTAC linkers. Notably, since most of the twelve compounds were relatively polar, we changed the solvent from DMSO to DMF to avoid any extraction workup procedure to remove the solvent after completing the reaction. Moreover, setting the temperature to 100°C allowed for reaction completion after 12 h.
Scheme 5.
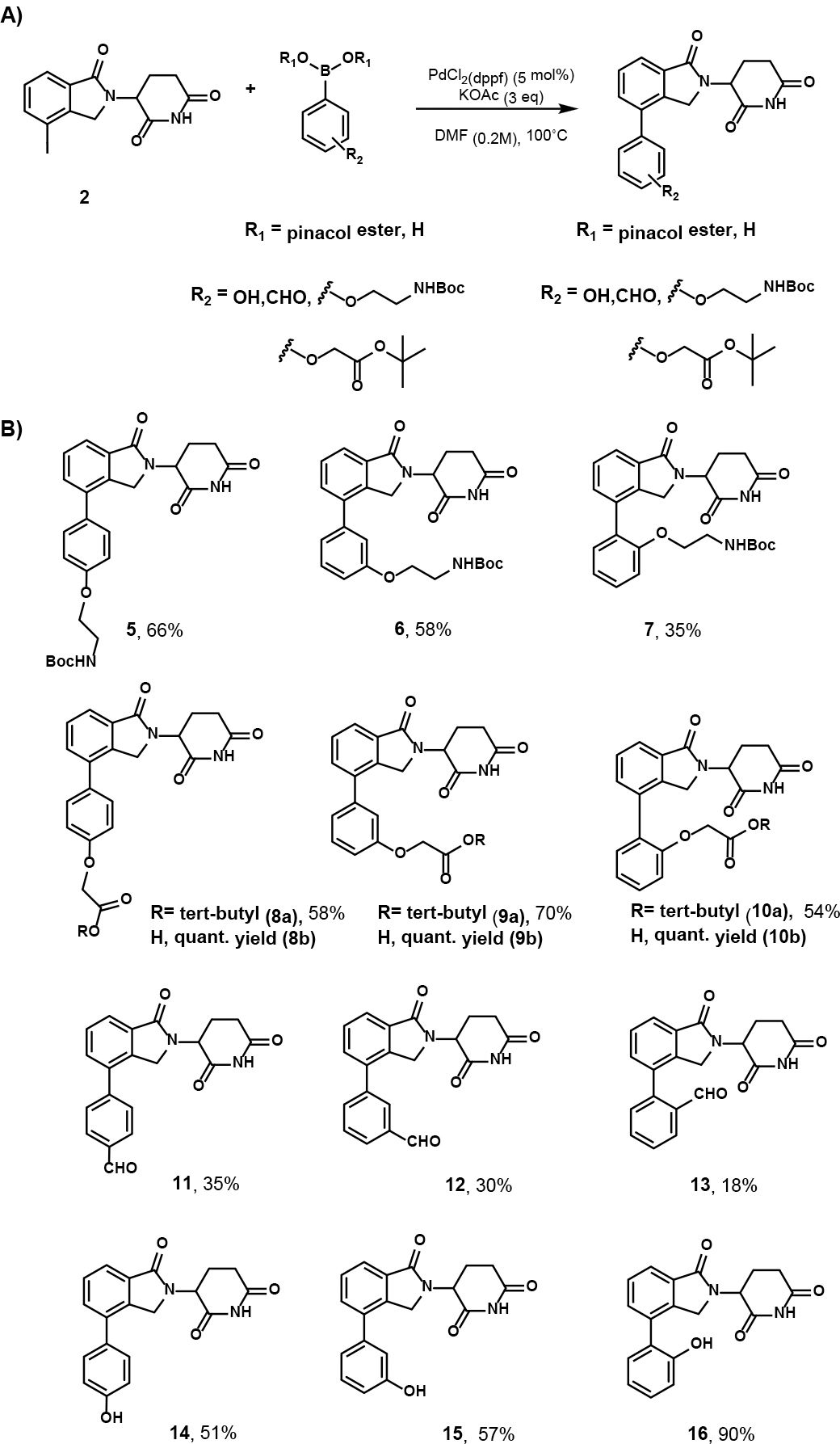
Suzuki cross-coupling conditions and substrate scope of lenalidomide-derived CRBN E3 ligase ligands. [a] Isolated by flash column chromatography.
Finally, we selected three compounds (5, 6, and 7) from the small library to create a partial PROTAC library. Specifically, we designed our partial PROTAC library based on the two-stage strategy recently reported by our group.20 Although many PROTACs have been successfully developed for a wide range of protein targets, the synthesis of these types of bifunctional molecules can be very time-consuming. The two-stage approach can significantly speed up the synthesis process and facilitate the rapid screening of E3 ligase ligands with a variety of different linkers.20 The first stage involves the reaction of a pre-assembled library of E3 ligase ligands with linkers containing a terminal aldehyde in a 1:1 ratio with a POI ligand containing a hydrazide moiety to form PROTACs with an acylhydrazone linkage.20 Importantly, the resulting products in DMSO solution can be used for screening without any further manipulation including purification step since water is the only byproduct.20 The acylhydrazone bond can then be changed to an amide bond for higher stability and more drug-like properties in the second stage.20
Before initiating the synthesis, we confirmed the binding affinity of compounds 5, 6, and 7 to CRBN using a well-established fluorescent polarization assay.21 These three compounds showed Kds of 231 nM for 5, 218 nM for 6, and 100 nM for 7, respectively, while the the thalidomide-based probe has a Kd of 122 nM (Figure 3). It is consistent with our previous results from the cell-based assay.14
Figure 3.
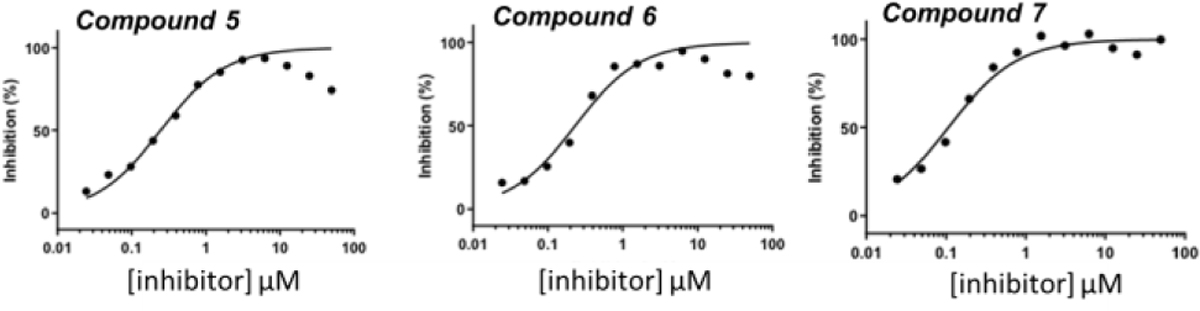
Kds of compounds 5, 6, and 7.
It is well known that the linker plays a significant role in the ternary complex formation and activity of PROTACs.20,22 We sought to choose compounds 5, 6, and 7 for the synthesis of a pre-assembled CRBN E3 ligase partial PROTAC library by systematically examining the length and orientation of the linker. Compounds 5, 6 and 7 contained a terminal Boc-protected amine, which could be easily deprotect and used to attach an alkyl linker containing a terminal aldehyde via an amide coupling reaction. Particularly, we considered that the relatively short partial linker length of these compounds provides the opportunity for systematic examining the substitution pattern on phenyl groups at both ends of the alkyl group. We then removed the Boc group to form a primary amine followed by an amide coupling with a ortho-, meta-, or para- formylbenzoic acid (Scheme 6). We then prepared a small CRBN partial PROTAC library with a relatively short alkyl linker containing a meta or para terminal benzaldehyde that can be further used to react with a hydrazide group in any protein of interest ligand.
Scheme 6.
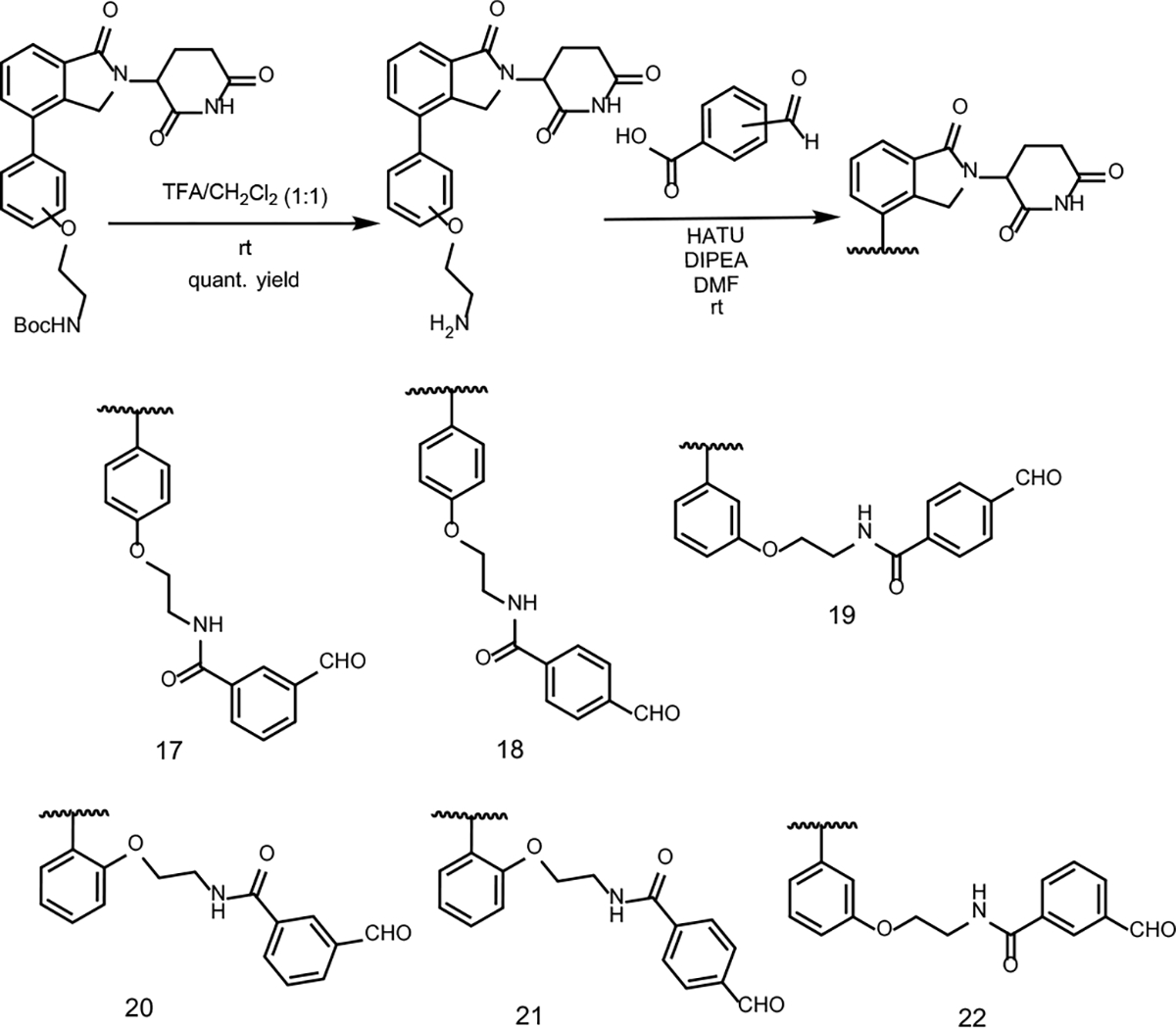
Partial PROTAC library synthesis and substrate scope starting from compounds 5, 6, or 7 obtained from the Suzuki cross-coupling reactions.
Conclusion
In summary, we described a modular chemistry platform for the introduction of substituted phenyls to the C4-position of the lenalidomide-derived analogs under an improved Suzuki cross-coupling condition. We demonstrated that the formation of Suzuki-cross coupling product without hydrolysis of the imide was possible with a mild base. This provides the access to various phenyl substituted lenalidomide analogues. The partial PROTAC library can be used for the synthesis of a full PROTAC library for any protein target and allow us to systematically examine the effect of length and orientation of the linkers with a relatively short distance.
Experimental Section
Example synthesis of 4-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)benzaldehyde
To a solution of 2 (100 mg, 0.270 mmol) in DMF (1.3 mL, 0.2 M) was added 4-formylphenyl boronic acid (80 mg, 0540 mmol), PdCl2(dppf) (10 mg, 5 mol%) and KOAc (80 mg, 0.81 mmol) at room temperature. Then, the reaction mixture was stirred for 12 h at 100 oC. Afterwards, the solvent was concentrated and the sample was diluted with DCM. The crude product was purified by flash column chromatography using an elution gradient of 0–10% MeOH in DCM as mobile phase and a 12 g silica column as stationery phase. Finally, the desired fractions were combined and concentrated to afford the desired compound (33 mg, 35% yield).
1H NMR (400 MHz, DMSO-d6) δ 10.98 (s, 1H), 10.10 (s, 1H), 8.07 – 8.01 (m, 2H), 7.89 – 7.78 (m, 4H), 7.70 (t, J = 7.6 Hz, 1H), 5.16 (dd, J = 13.3, 5.1 Hz, 1H), 2.99 – 2.85 (m, 1H), 2.64 – 2.54 (m, 1H), 2.51 – 2.37 (m, 1H), 2.00 (ddd, J = 10.6, 4.8, 2.6 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 193.32, 173.34, 171.37, 168.17, 144.31, 140.14, 136.06, 135.94, 133.10, 132.33, 130.48, 129.58, 129.41, 123.59, 52.10, 47.68, 31.63, 22.85.
Supplementary Material
Acknowledgements
W.T. thanks the financial support from the University of Wisconsin– Madison Office of the Vice Chancellor for Research and Graduate Education with funding from the Wisconsin Alumni Research Foundation (WARF) through a UW2020 award and National Institutes of Health under the award number R21AI158210. C.M.A.R. was supported in part by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number T32GM008505 (Chemistry–Biology Interface Training Program). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This study made use of the Medicinal Chemistry Center at UW-Madison instrumentation funded by the Lachman Institute for Pharmaceutical Development and UW Carbone Cancer Center (NIH P30 CA014520).
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supporting Information
The authors have cited additional references within the Supporting Information. [23–26]
Data Availability Statement
The data that supports the findings of this study are available from the corresponding author upon reasonable request.
References
- [1].Qiu X, Sun N, Kong Y, Li Y, Yang X, Jiang B, Org. Lett. 2019, 21 (10), 3838–3841. [DOI] [PubMed] [Google Scholar]
- [2].Sun X, Gao H, Yang Y, He M, Wu Y, Song Y, Tong Y, Rao Y, Signal Transduct. Target. Ther. 2019, 4 (1), 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Sosič I, Bricelj A, Steinebach C, Chem. Soc. Rev. 2022, 51 (9), 3487–3534. [DOI] [PubMed] [Google Scholar]
- [4].Yao T, Xiao H, Wang H, Xu Int X. J. Mol. Sci. 2022, 23 (18), 10328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bricelj A, Steinebach C, Kuchta R, Gütschow M, Sosič I, Front. Chem. 2021, 9, 707317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kazantsev A, Krasavin M, Expert Opin. Ther. Pat. 2022, 32 (2), 171–190. [DOI] [PubMed] [Google Scholar]
- [7].Kong NR, Liu H, Che J, Jones LH, ACS Med. Chem. Lett. 2021, 12 (11), 1861–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Min J, Mayasundari A, Keramatnia F, Jonchere B, Yang SW, Jarusiewicz J, Actis M, Das S, Young B, Slavish J, Yang L, Li Y, Fu X, Garrett SH, Yun M, Li Z, Nithianantham S, Chai S, Chen T, Shelat A, Lee RE, Nishiguchi G, White SW, Roussel MF, Potts PR, Fischer M, Rankovic Z, Angew. Chem. Int. Ed. 2021, 60 (51), 26663–26670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Xie H, Li C, Tang H, Tandon I, Liao J, Roberts BL, Zhao Y, Tang W, J. Med. Chem. 2023, 66 (4), 2904–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hayhow TG, Borrows REA, Diène CR, Fairley G, Fallan C, Fillery SM, Scott JS, Watson DW, Chem. Eur. J. 2020, 26 (70), 16818–16823. [DOI] [PubMed] [Google Scholar]
- [11].Liu H, Sun R, Ren C, Qiu X, Yang X, Jiang B, Org. Biomol. Chem. 2021, 19 (1), 166–170. [DOI] [PubMed] [Google Scholar]
- [12].Bricelj A, Dora Ng YL, Ferber D, Kuchta R, Müller S, Monschke M, Wagner KG, Krönke J, Sosič I, Gütschow M, Steinebach C, ACS Med. Chem. Lett. 2021, 12 (11), 1733–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hoffmann M, Kasserra C, Reyes J, Schafer P, Kosek J, Capone L, Parton A, Kim-Kang H, Surapaneni S, Kumar G, Cancer Chemother. Pharmacol. 2013, 71 (2), 489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yang K, Zhao Y, Nie X, Wu H, Wang B, Almodovar-Rivera CM, Xie H, Tang W, Cell Chem. Biol. 2020, S2451945620301458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Xiao D, Wang Y, Wang H, Zhou Y, Li J, Lu W, Jin J, Arch. Pharm. (Weinheim, Ger.) 2020, 353 (7), 1900376. [DOI] [PubMed] [Google Scholar]
- [16].Li Y, Yang J, Aguilar A, McEachern D, Przybranowski S, Liu L, Yang C-Y, Wang M, Han X, Wang S, J. Med. Chem. 2019, 62 (2), 448–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ishiyama T, Murata M, Miyaura N, J. Org. Chem. 1995, 60 (23), 7508–7510. [Google Scholar]
- [18].Brown J, Su SCK, Shafer JA, J. Am. Chem. Soc. 1966, 88 (19), 4468–4474. [DOI] [PubMed] [Google Scholar]
- [19].Zagidullin A, Milyukov V, Rizvanov A, Bulatov E, Explor. Targeted Anti-Tumor Ther. 2020, 1 (5), 381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Roberts BL, Ma Z-X, Gao A, Leisten ED, Yin D, Xu W, Tang W, ACS Chem. Biol. 2020, 15 (6), 1487–1496. [DOI] [PubMed] [Google Scholar]
- [21].Du Y, Protein-Protein Interactions, Springer, New York, 2015, p. 529–544. [Google Scholar]
- [22].Troup RI, Fallan C, Baud MGJ, Explor. Targeted Anti-Tumor Ther. 2020, 1 (5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Li Y, J. Yang, A. Aguilar, D. McEachern, S. Przybranowski, L. Liu, C.-Y. Yang, M. Wang, X. Han, S. Wang, J. Med. Chem 2019, 62, 2, 448–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Vazquez-Rodriguez S, Wright M, Rogers CM, Cribbs AP, Velupillai S, Philpott M, Lee H, Dunford JE, Huber KVM, Robers MB, Vasta JD, Thezenas M-L, Bonham S, Kessler B, Bennett J, Fedorov O, Raynaud F, Donovan A, Blagg J, Bavetsias V, Oppermann U, Bountra C, Kawamura A, Brennan PE, Angew. Chem. Int. Ed. 2019, 58, 515–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Newton P, Harrison P, Clulow S, J. Biomol. Screen. 2008, 13, 674–682. [DOI] [PubMed] [Google Scholar]
- [26].Fischer ES, Böhm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, Nagel J, Serluca F, Acker V, Lingaraju GM, Tichkule RB, Schebesta M, Forrester WC, Schirle M, Hassiepen U, Ottl J, Hild M, Beckwith REJ, Harper JW, Jenkins JL, Thomä NH, Nature 2014, 512, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that supports the findings of this study are available from the corresponding author upon reasonable request.