

Abstract
The Plasmodium proteasome is a promising antimalarial drug target due to its essential role in all parasite lifecycle stages. Furthermore, proteasome inhibitors have synergistic effects when combined with current first-line artemisinin and related analogues. Linear peptides that covalently inhibit the proteasome are effective at killing parasites and have a low propensity for inducing resistance. However, these scaffolds generally suffer from poor pharmacokinetics and bioavailability. Here we describe the development of covalent, irreversible, macrocyclic inhibitors of the Plasmodium falciparum proteasome. We identified compounds with excellent potency and low cytotoxicity; however, the first generation suffered from poor microsomal stability. Further optimization of an existing macrocyclic scaffold resulted in an irreversible covalent inhibitor carrying a vinyl sulfone electrophile that retained high potency and low cytotoxicity and had acceptable metabolic stability. Importantly, unlike the parent reversible inhibitor that selected for multiple mutations in the proteasome, with one resulting in a 5,000-fold loss of potency, the irreversible analogue only showed a 5-fold loss in potency for any single point mutation. Furthermore, an epoxyketone analogue of the same scaffold retained potency against a panel of known proteasome mutants. These results confirm that macrocycles are optimal scaffolds to target the malarial proteasome and that the use of a covalent electrophile can greatly reduce the ability of the parasite to generate drug resistance mutations.
Keywords: Plasmodium falciparum, cyclic peptides, antimicrobial resistance
Graphical Abstract
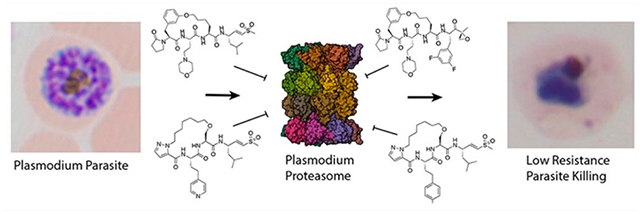
Plasmodium falciparum causes the most severe and lethal cases of malaria in humans. The disease remains a threat to 40% of the world’s population and causes over 600,000 deaths per year, mainly among children under the age of five.1 The emergence of both artemisinin (ART) partial resistance and of resistance to partner drugs used in ART-based combination therapies (ACTs) is particularly concerning, as these drugs are widely used in endemic populations.2–6 There is an urgent need to develop drugs with novel mechanisms of action and broad therapeutic potential to improve interventions and overcome multidrug resistance.
An attractive target for new drugs is the Plasmodium proteasome, which is essential throughout the parasite’s life cycle. Inhibitors of the proteasome synergize with ART to enhance killing of the parasite in vivo.7,8 A significant challenge for the development of Plasmodium proteasome inhibitors is optimizing selectivity over the human enzyme. We have previously used substrate screening and cryo-electron microscopy to evaluate differences in ligand binding preferences between the human and parasite proteasomes.9 This has enabled the discovery of potent, irreversible covalent inhibitors that are selective for the Plasmodium proteasome (Figure 1). These selective inhibitors enabled validation of the proteasome as a viable antimalarial target using mouse models of infection.7 Building on this foundation, we and others developed various peptidic scaffolds and incorporated non-natural amino acids to increase overall selectivity and potency for the malaria proteasome.10–12 These parasite-selective compounds have enabled more detailed studies of the parasite’s potential to generate resistance mechanisms against proteasome inhibitors.13 Though promising, linear peptide inhibitors suffer from poor bioavailability and stability in vivo.11
Figure 1.
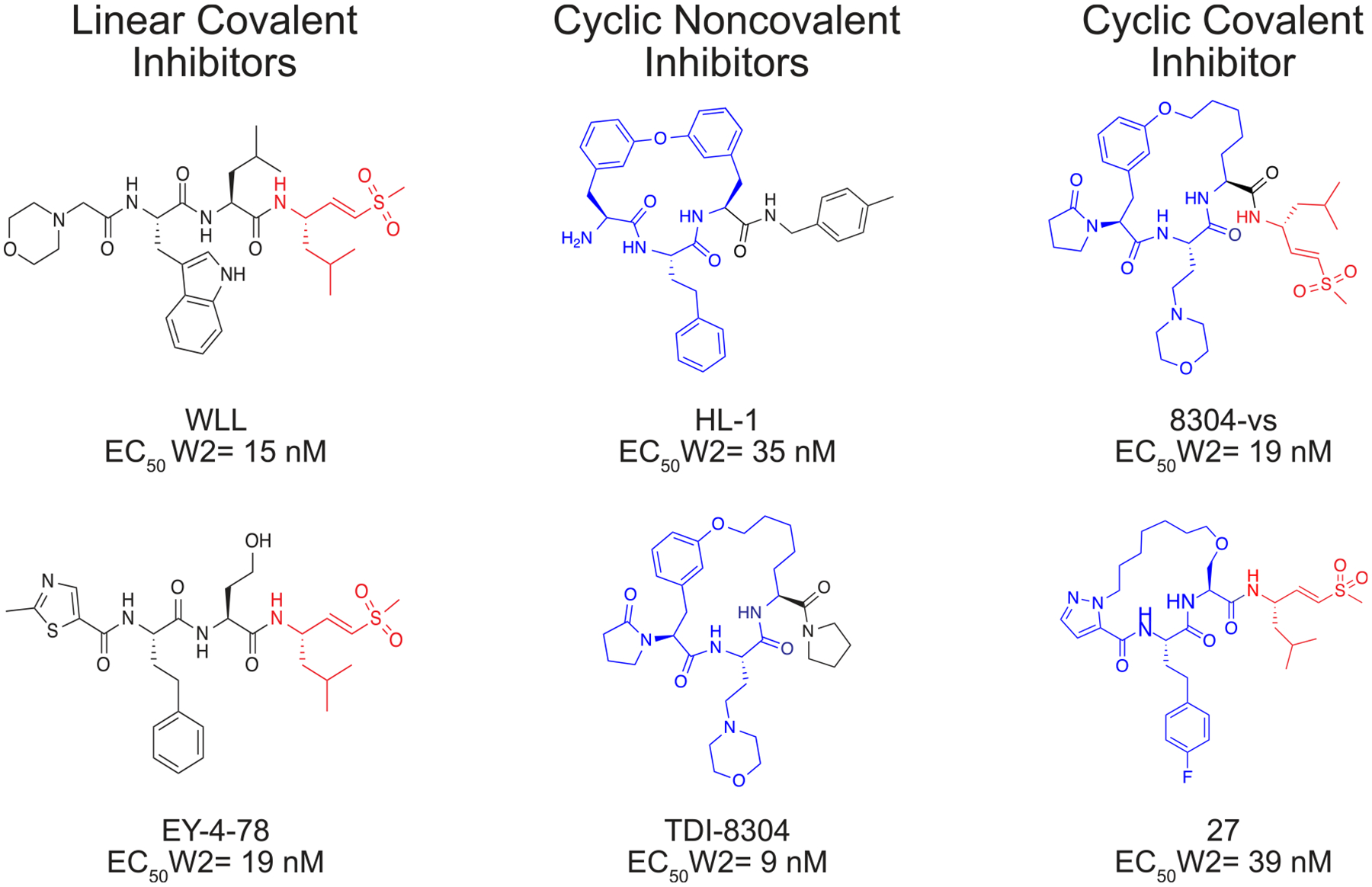
Chemical structures of representative macrocyclic covalent proteasome inhibitors are shown on the right. Previously reported linear covalent or noncovalent macrocyclic inhibitors are shown on the left as a reference. The covalent electrophilic warhead leucine vinyl sulfone is colored red on each structure and the macrocycles are colored blue. EC50 values represent the mean concentration required to inhibit P. falciparum asexual blood stage parasite growth by 50%.9,11,16,17
Peptide macrocycles have the potential to overcome many of the limitations of linear peptides as therapeutic agents.14 The former are generally more stable than their linear counterparts because cyclization drastically reduces the number of rotatable bonds, can mask hydrogen bond donors through internal hydrogen bonding, and can reduce proteolytic metabolism.15 Our prior screen of a small library of reversibly binding proteasome inhibitors identified a macrocyclic peptide containing a biphenyl ether with low nanomolar potency against intraerythrocytic parasites and low toxicity in human cells.16 However, this molecule has poor solubility and rapid clearance. This scaffold was further developed to increase its potency and to optimize its pharmacokinetic properties, resulting in the promising lead molecule TDI-8304.17,18 However, we recently observed that this class of noncovalent cyclic peptide inhibitors can induce relatively high levels of resistance compared to similar classes of linear covalent irreversible proteasome inhibitors.19
We set out to assess the potential of various macrocyclic scaffolds as potent, selective, and metabolically stable proteasome inhibitors. We also sought to evaluate how switching from a reversible inhibitor to a covalent inhibitor impacts resistance generation in the parasite. We describe herein the synthesis and characterization of a series of covalent macrocyclic inhibitors that explore the effects of ring size, capping elements, use of non-natural amino acids, and overall hydrophobicity on potency, selectivity, and metabolic stability. While many of the compounds are potent and selective inhibitors of Plasmodium growth, some of the most potent molecules suffered from instability in both mouse and human microsomes. Therefore, we synthesized a covalent irreversible analogue of the optimized macrocyclic scaffold TDI-8304 by adding a vinyl sulfone electrophile (8304-vs). Both TDI-8304 and 8304-vs showed selective and potent inhibition of the parasite and favorable metabolic stability with exceptionally low toxicity to host cells. Resistance studies demonstrated that both compounds induced a similar point mutation in the proteasome that altered the inhibitory potency of the molecules. However, the covalent binding version of the compound encountered only mild resistance with a 5-fold drop in potency, whereas the reversible binding molecule had a 5,000 drop in potency for this same proteasome mutant. We further assessed the importance of covalency by synthesizing an epoxyketone analogue of TDI-8304 (8304-epoxy) and demonstrated that both the vinyl sulfone and the epoxyketone derivatives showed low cross resistance with known proteasome mutations. These results suggest that cyclic peptides are potentially optimal scaffolds for proteasome inhibitors and that the use of covalent binding functional groups can help to reduce resistance liabilities.
RESULTS
Structure–Activity Relationship.
Previously we used substrate profiling methods to define the specificity of the parasite proteasome relative to the human counterpart.11 We determined that a bulky aromatic group at the P3 position could produce a high level of selectivity and potency for the P. falciparum enzyme over the human enzyme. We also found that the adjacent P2 position could present various natural and unnatural amino acids without compromising potency. Therefore, we theorized that cyclic peptides could be synthesized via a terminal alkene on the N-terminal capping group linked through Grubbs-mediated cross metathesis to various P2 amino acids. Our first set of compounds was based on our most potent and selective linear peptide EY-4–7811 and contained a homophenylalanine at the P3 position with either a 2-Aminohex-5-enoic acid or an allyl protected serine at P2 to cyclize the peptides (Figure 2A). The linear versions of the compounds (1, 2) showed equal potency against the parasite (EC50 < 3 nM) as compared with previously explored linear proteasome inhibitors (WLL EC50 = 15 nM; Figure 2B). The corresponding cyclized versions (3, 4) showed similar potency to the parent molecules, but the cyclization dramatically increased toxicity toward human foreskin fibroblasts (HFFs) (Figure S1). We altered the size of the macrocycle in the third pair of peptides by increasing the length of the N-terminal alkene and found that changing the length of the capping group had no impact on the potency of 3 compared to linear peptides 1 and 2. However, the 15-member macrocycle of peptide 6 had a 10-fold decrease in potency for the parasite and a 4-fold reduction in selectivity when compared with peptide 3. This finding suggested that differences in the linear and cyclized peptides may become greater as the ring size increases (Figure 2C).
Figure 2.
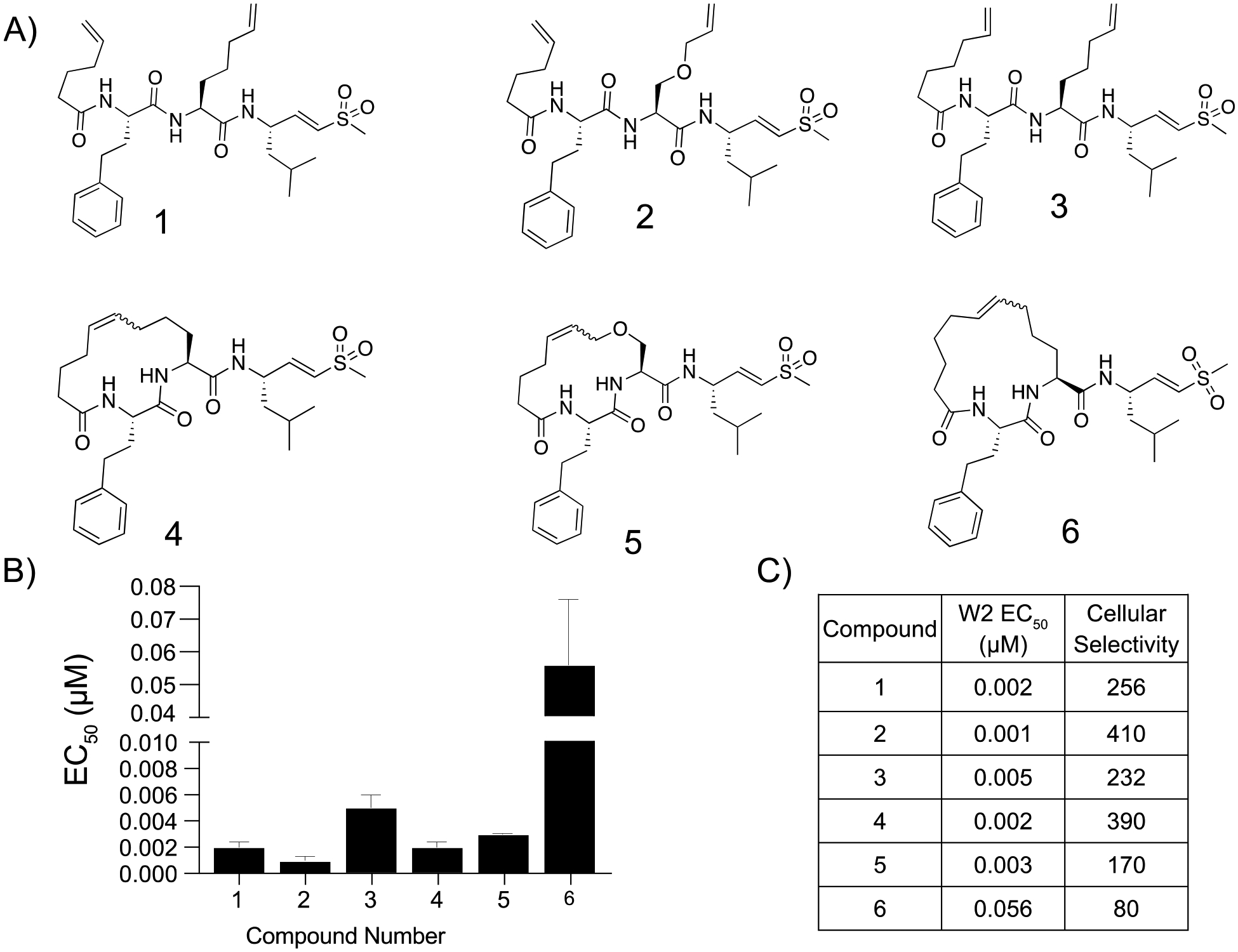
Potency and selectivity of macrocyclic peptide inhibitors containing an alkyl backbone and their linear counterparts. (A) Structures of the macrocyclic peptide inhibitors and their linear counterparts. (B) Compound mean ± SEM EC50 values against P. falciparum asexual blood stage W2 parasites of each inhibitor. Data were generated using 72 h dose–response assays (N, n = 2,2). (C) EC50 values against W2 parasites and cellular selectivity indexes for P. falciparum parasites compared to primary mammalian HFF cells (listed as the ratio of HFF EC50 to parasite EC50 values).
We hypothesized that because parasitized red blood cells tend to be more permeant to peptide-based inhibitors compared to other cell types, the increased host cell toxicity in the HFFs may be due to increased lipophilicity and thus greater permeability of the cyclic peptides into human cells.20,21 We evaluated this hypothesis by comparing the biochemical selectivity of our compounds to purified Plasmodium and human proteasomes. To test the importance of lipophilicity in potency and cytotoxicity, we synthesized a second set of cyclic peptides using a diether linkage to increase the hydrophilicity of the backbone (Figure 3A; Scheme S1). Interestingly, linear peptide 7 showed micromolar inhibition of parasite growth (EC50 = 2.7 μM) and very low HFF toxicity (EC50 ≥ 50 μM), while the extension of the capping group by one methylene group in peptide 8 led to both substantially more potent antiparasitic activity (EC50 = 0.016 μM), but also higher cytotoxicity (EC50 = 1.1 μM) (Figure 3B,C; Figure S2). Peptide 8 showed modest inhibition of the P. falciparum proteasome β2 subunit, while peptide 7 had no inhibitory effects, and neither peptide had any inhibitory activity against the human β2 subunit (Figure 3D). Peptide 7 showed comparable inhibition of the β5 subunits of each proteasome, and peptide 8 was 6-fold more selective for the Plasmodium β5 subunit over the human counterpart. Macrocycle 9 was 19-fold more potent than its linear counterpart 8 and maintained a low cytotoxicity. The cyclization of 8 led to a significant decrease in cytotoxicity for macrocycle 10 while maintaining similar parasite growth inhibition, potentially due to its 3-fold decrease in potency against the purified human β5 subunit. Nevertheless, it was difficult to achieve high degrees of selectivity for purified proteasomes, and it is likely that the combination of cell permeability and biochemical selectivity between the two proteasomes led to differences in antiparasitic effects and cytotoxicity. We also reduced the double bond within the macrocycles to test the importance of unsaturation within the backbone, resulting in a 2-fold decrease in potency in the saturated macrocycle 11 compared to parent compound 9 and a 15-fold reduction in potency in compound 12 compared to the parent 10. Both saturated macrocycles had negligible changes in cytotoxicity, suggesting that an unsaturated double bond increases the potency but not the cytotoxicity for the ether linked macrocycles.
Figure 3.
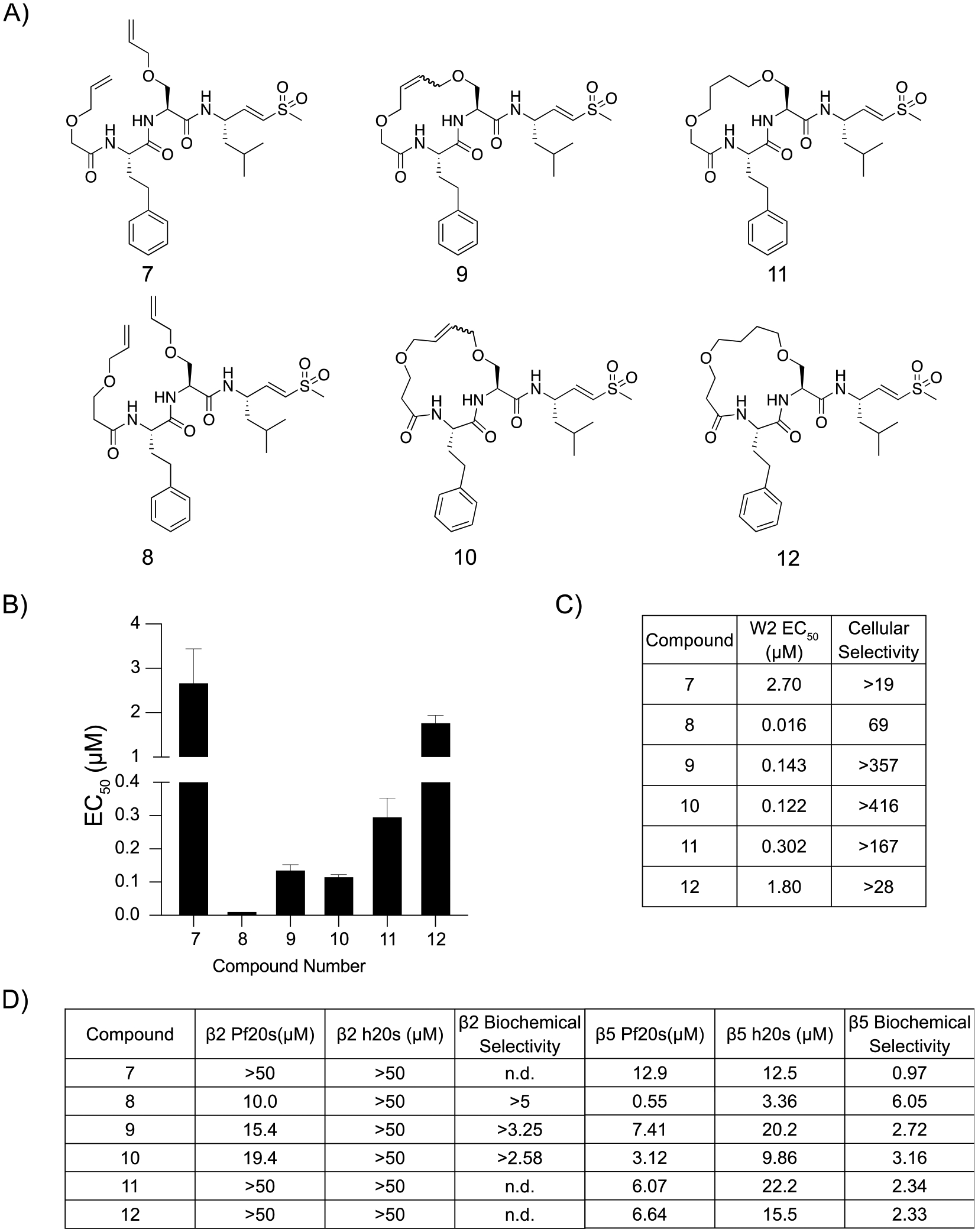
Potency and selectivity of ether linked macrocyclic peptide inhibitor and their linear counterparts. (A) Structures of ether linked cyclic peptides and corresponding linear counterparts. (B) Compound mean ± SEM EC50 values against P. falciparum asexual blood stage W2 parasites for each inhibitor. Data were generated using 72 h dose–response assays (N, n = 2,2). (C) EC50 values against W2 parasites and cellular selectivity indexes for P. falciparum parasites compared to primary mammalian HFF cells (listed as the ratio of HFF EC50 to parasite EC50 values). (D) Potencies (IC50) of compounds assayed against either the β2 or the β5 subunits of the purified Plasmodium or human 20s proteasomes in 1 h inhibition assays. Biochemical selectivity was calculated as the ratio of potency for the two proteasomes (h20s/Pf20s).
Although our previous set of compounds showed promise, the acyl capping group could be metabolized, similar to N-acyl amino acids.22 Therefore, we synthesized a third set of compounds with either imidazole or pyrazole capping groups that have previously been successfully used for orally bioavailable macrocyclic proteasome inhibitors (Figure 4A; Scheme S2).23 Our pyrazole capped linear peptides 13 and 14 both showed greater potency and reduced off-target cytotoxicity when compared to their imidazole-containing complements (Figure 4B,C; Figure S3). Both the saturated and unsaturated macrocycles followed the same trend as the linear peptides, as the pyrazole capped inhibitors had greater potency when compared to the same molecules capped with an imidazole. In each case, the imidazole capped macrocycles showed a more potent inhibition of the β2 subunit of the P. falciparum proteasome than the corresponding pyrazole inhibitors. No compound in this set had any inhibitory effect against the β2 subunit of the human proteasome at concentrations of up to 50 μM (Figure 4D). Notably, compounds with the larger 17-member macrocycles were the most potent inhibitors of Plasmodium growth (18, EC50 = 19 nM and 22, EC50 = 5 nM) and were at least 200-fold more potent for the parasite than the HFFs in 72 h dose–response assays (Figure 4B).
Figure 4.
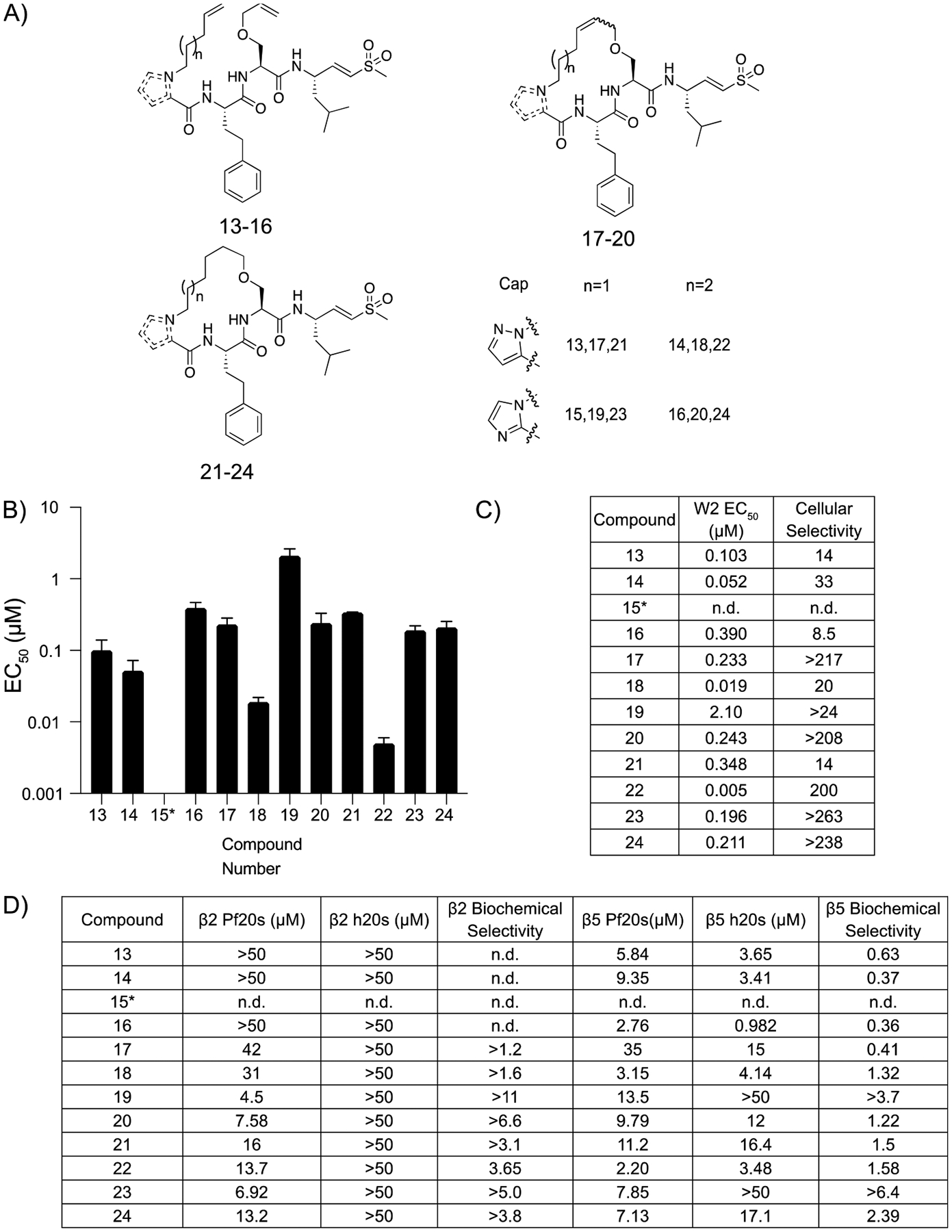
Potency and selectivity of imidazole and pyrazole capped macrocyclic peptides and their linear counterparts. (A) Structures of imidazole and pyrazole cyclic peptides and corresponding linear counterparts. The inset shows the capping group and length of alkyl chain for each compound. (B) Compound mean ± SEM EC50 values for each inhibitor assayed against P. falciparum asexual blood stage W2 parasites. Data were generated using 72 h dose–response assays (N, n = 2,2). (C) EC50 values against W2 parasites and cellular selectivity indexes comparing P. falciparum parasites to mammalian HFF cells (listed as the ratio of HFF EC50 to parasite EC50 values). (D) Potencies (IC50) of compounds assayed against either the β2 or the β5 subunits of the purified Plasmodium or human 20s proteasomes in 1 h inhibition assays. Biochemical selectivity was calculated by taking a ratio of potency for the two proteasomes (h20s/Pf20s).
Although 22 was slightly more cytotoxic than 18, it was chosen to continue our structure–activity relationship study as its synthesis does not result in isomers. Previous work had shown that the P3 homophenylalanine (hfe) is oxidized to the corresponding phenol and that the resulting phenolic metabolite has greatly reduced antiparasitic activity.11 To overcome this liability, we synthesized electron-deficient aromatic ring analogues of 22 to decrease the likelihood of oxidation (Figure 5A; Scheme S3).24 Compound 26 replaced the hfe with a 4-pyridine, which resulted in a slight decrease in antiparasitic activity (EC50 = 75 nM), and a large improvement in cytotoxicity (EC50 = 20 μM) (Figure 5B,C; Figure S4). Similarly, the introduction of a 4-fluorophenyl (27) resulted in a slight reduction in potency (EC50 = 39 nM) but a greater than 400-fold increase in selectivity over HFFs.
Figure 5.
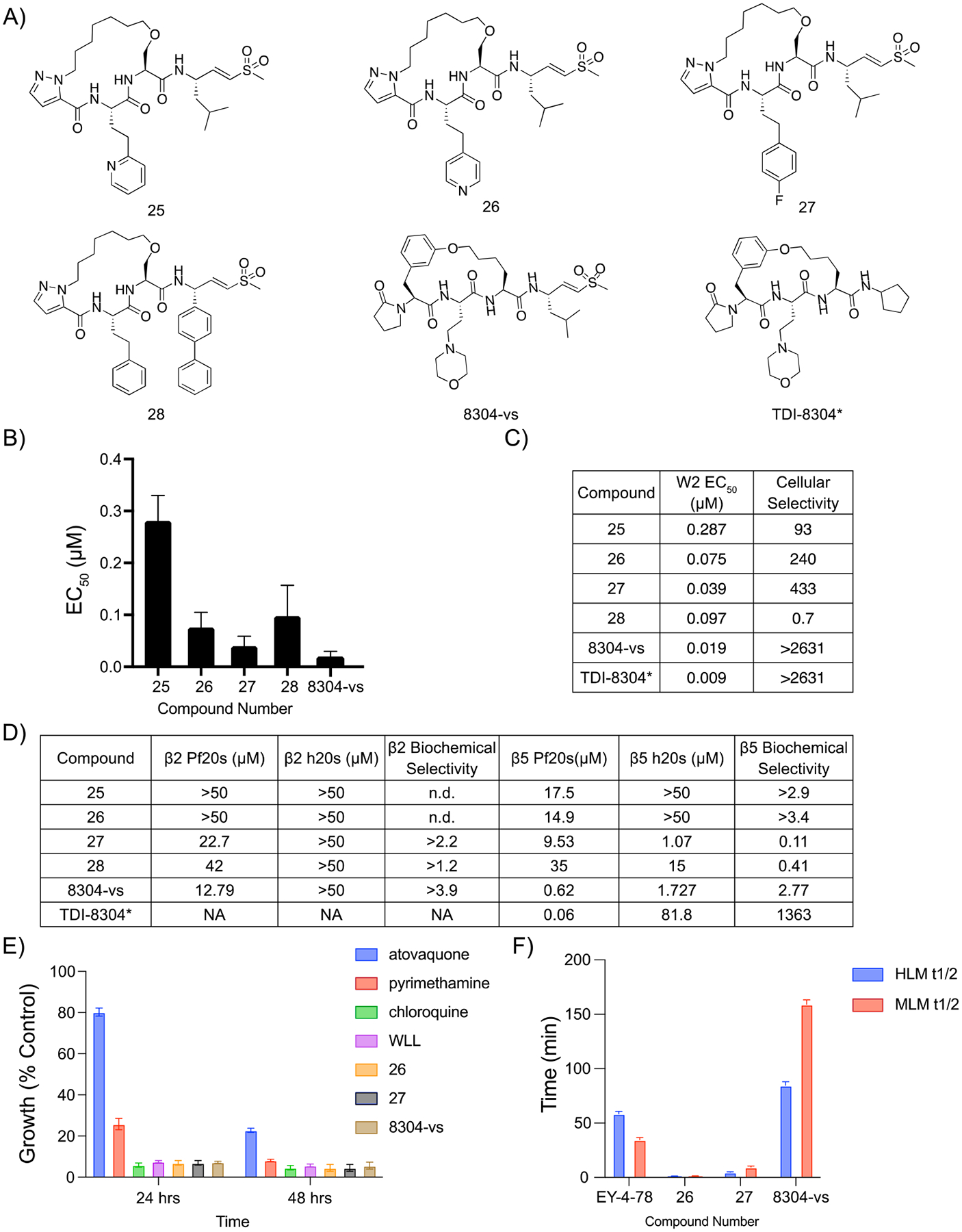
Covalent irreversible proteasome inhibitor 8304-vs is a potent, selective, and stable inhibitor of the P. falciparum proteasome. (A) Structures of pyrazole capped inhibitors with varying P3 or P1 groups and 8304-vs, a covalent version of a previously described noncovalent macrocyclic inhibitor of the proteasome. (B) Compound mean ± SEM EC50 values for each inhibitor assayed against P. falciparum asexual blood stage W2 parasites. Data were generated using 72 h dose–response assays (N, n = 2,2). (C) EC50 values against W2 parasites and cellular indexes comparing P. falciparum parasites to mammalian HFF cells (listed as the ratio of HFF EC50 to parasite EC50 values). (D) Potencies (IC50) of compounds assayed either the β2 or the β5 subunits of the purified Plasmodium or human 20s proteasomes in 1 h inhibition assays. Biochemical selectivity was calculated as the ratio of potency for the two proteasomes (h20s/Pf20s). (E) Plot of rate of kill assay, in which compounds were dosed at their EC50 and parasite viability was monitored over time and compared against known fast (chloroquine), medium (pyrimethamine), and slow (atovaquone) acting inhibitors of parasite growth ± SEM. N,n = 6,2. (F) Bar graphs depicting the half-life of compounds in either human (blue) or mouse (red) microsomes. Compound metabolism was measured by LC/MS/MS. *TDI-8304 data were previously reported.17
We then tested the importance of the P1 leucine moiety in potency and selectivity. Based on recent reports of potent orally available peptide boronates containing bulky P1 residues, we replaced the P1 leucine of 22 with a biphenyl alanine (28) (Figure 5A; Scheme S4).12 Interestingly, this modification substantially increased the overall toxicity of the compounds toward the HFFs (Figure S5). This is likely due to the ability of the biphenyl analogue to inhibit both the human β2 and β5 subunits (Figure 5D). We therefore did not advance this compound for further study.
To further explore the antiplasmodial effects and stability of our two most potent and selective covalent macrocyclic inhibitors (26 and 27) we tested them in a parasite rate of kill assay.25 Both inhibitors showed kill rates similar to those of the fast-acting antimalarial drug chloroquine, which suggests that our inhibitors could rapidly clear infections (Figure 5E). Parasites were treated with inhibitor for 1 h, washed to remove compound, and cultured a further 72 h prior to quantifying parasite growth. Both 26 and 27 showed parasite killing at concentrations over 1 μM (Figure S5). To assess metabolic stability, compounds were incubated in both human and mouse microsomes and metabolite formation was monitored over time using LC/MS. Surprisingly, 27 was metabolized quickly, with a half-life of only 1.2 min in both sets of microsomes. Peptide 26 was modestly better with a 4.7 min half-life in human microsomes and 9.4 min in mouse microsomes (Figure 5F).
Given the overall poor metabolic stability of our initial lead molecules, we turned our attention to an established cyclic peptide scaffold that was highly effective at inhibiting the Plasmodium proteasome. This scaffold is based on a screening hit that we identified from a library of reversible inhibitors of the proteasome.16 The core macrocyclic scaffold was further optimized to yield a reversible inhibitor TDI-8304.17 We theorized that we could convert TDI-8304 into a covalent inhibitor by replacing the C-terminal cyclopentyl functional group with our optimal P1 leucine vinyl sulfone electrophilic warhead, which has already been shown to covalently react with the active site tyrosine in both the human and malarial proteasome (Figure 1; Figure 5A; Scheme S5). Our rationale was to generate an irreversible inhibitor with increased stability compared to our original linear peptide vinyl sulfone. Furthermore, this compound allowed us to directly compare the impact of switching to a covalent inhibition mechanism on acquisition of resistance. Importantly, the resulting compound (8304-vs) had only a 2-fold reduction in potency compared to the parent (EC50 = 19 nM compared to 9 nM in TDI-8304; Figure 1; Figure 5B). In addition, the irreversible 8304-vs showed no measurable cell toxicity within the solubility limits of the compound, suggesting that it has a >5,000-fold selectivity for parasites over the human HFFs (Figure 5C). The vinyl sulfone compound also performed as well as compounds 26 and 27 in the rate of kill assays. However, 8304-vs did not show killing at 100× EC50 in a 1 h pulse assay (Figure S5). This suggests that 8304-vs may have reduced cellular uptake compared to those of 26 and 27, which both have lower potency against the purified proteasome. The 8304-vs inhibitor is also stable for almost 1.5 h in human microsomes and over 2.5 h in mouse microsomes, suggesting that it has favorable pharmacological properties and was therefore worthy of further development (Figure 5F).
Resistance Selections.
We profiled both 27 and 8304-vs for their propensity to select for parasite resistance, using a minimum inoculum of resistance (MIR) platform.26 P. falciparum Dd2-B2 parasites were exposed to 3× EC50 concentrations of either compound at various starting inocula (four wells of 2.5 × 106 plus three wells of 3 × 107 parasites), and cultures were monitored for recrudescence for 60 days. No recrudescence was observed for either compound. In a repeat experiment, each compound was tested against triplicate wells of 3.3 × 107 parasites each. One of three wells became parasite-positive for compound 27 (MIR of 1 × 108 in this experiment), whereas no recrudescence was observed for 8304-vs. We then performed an additional selection experiment with 8304-vs, using two flasks of 1 × 109 parasites each, which yielded one positive flask (MIR: 2 × 109). An additional selection was performed with one flask of 1 × 109 parasites from the hypermutable P. falciparum line, Dd2-Polδ, which also yielded recrudescent parasites. This line harbors two mutations introduced into its DNA polymerase δ and is ~5 to 8-fold more mutable than Dd2-B2.27 Recrudescent parasites from selections with 27 and 8304-vs were cloned by limiting dilution, and whole-genome sequencing of resistant clones revealed mutations in the chymotrypsin like β5 subunit and in the β6 proteasome subunit. Resistance selections of 8304-vs yielded clones with the β6 subunit mutation S157L, which was previously selected by TDI-8304 pressure (Table 1).19 Selected clones were phenotyped and showed EC50 increases of 4- to 6-fold (Figure S6). Compound 27 was selected for the β5 subunit mutation M45I, which has previously been selected for by MPI-12.12 Selected clones had a 14.1- to 22.3-fold increase in their EC50 to 27, compared with the parental line (Figure S7).
Table 1.
Plasmodium falciparum Asexual Blood Stage Minimal Inoculum for Resistance (MIR) Selection Summary
| name | chemical class | starting selective pressure | resistant parasites in 4 wells of 2.5 × 106 | resistant parasites in 3 wells of 3 × 107 | resistant parasites in 2 flasks of 1 × 109 | MIR | selected proteasome mutation | EC50 fold shift against selection compoundc |
|---|---|---|---|---|---|---|---|---|
| WLL | vinyl sulfone | 3 × EC50 (50 nM) | 0/4 wells | none | NDa | >1 × 109 | N/A | N/A |
| 27 | vinyl sulfone | 3 × EC50 (23 nM) | 0/4 wells | 1/3 wells | ND | 1 × 108 | β5 M45I | 14.1–22.3× |
| 8304- vs | vinyl sulfone | 3 × EC50 (72 nM) | 0/4 wells | 0/3 wells | 1/2 flasksb | 2 × 109 | β6 S157L | 4.0–6.0× |
| TDI-8304 | macrocyclic peptide | 3 × EC50 (33 nM) | 0/4 wells | 3/3 wells | ND | 3×107 | β6 S157L | 2621× |
| β6 N151Y | 1.7× |
Prior studies with WLL selections obtained 0/3 recrudescent flasks inoculated with 3.3×108 each, yielding a predicted MIR >1 × 109.
One flask of the hypermutable Dd2-B2 Poldline inoculated at 1 × 109 parasites and pressured with 8304-vs also yielded positive parasites, as reported herein.
EC50 fold shift calculated as a ratio between the EC50 of the selected mutant over the EC50 of the Dd2-B2 parental line. N,n = 4–5,2. ND, not done. N/A, not applicable. MIRs are calculated as the total number of parasites inoculated divided by the total number of positive cultures (when obtained). The formula extends up to the largest inoculum at which parasites were obtained (or tested in the case of negative cultures) and includes lower inocula (see Methods).
Mutant Panel.
Because the addition of the vinyl sulfone to TDI-8304 greatly improved its resistance profile, we synthesized a 2,4-difluorophenylalanine epoxyketone to assess the importance of specific covalent warheads for overcoming resistance. We profiled 8304-vs, 8304-epoxy, and 27 against a panel of proteasome mutants selected for in previous studies,19 comprising several β5 and β6 mutations on a Dd2-B2 background. Compound 27 had a significant increase in EC50 against all proteasome mutant lines assessed, with the exception of β5 A50V and β6 N151Y (the latter of which saw a slight but significant decrease in EC50 (Figure S8). Overall, 8304-vs showed a modest increase in EC50 in parasites with the β5 M45I and M45V mutations (1.8-fold and 1.5-fold, respectively) and a slightly higher 8.8-fold increase in EC50 in parasites with the β6 S157L mutation (Figure 6). Interestingly, this mutation was also present in previous selections with TDI-8304, to which it mediated a more than 2,900-fold increase in EC50.19 None of the mutants profiled had significantly increased EC50 values to 8304-epoxy, and a slight decrease in the EC50 value for this compound was observed with β5 M45 V mutant parasites.
Figure 6.
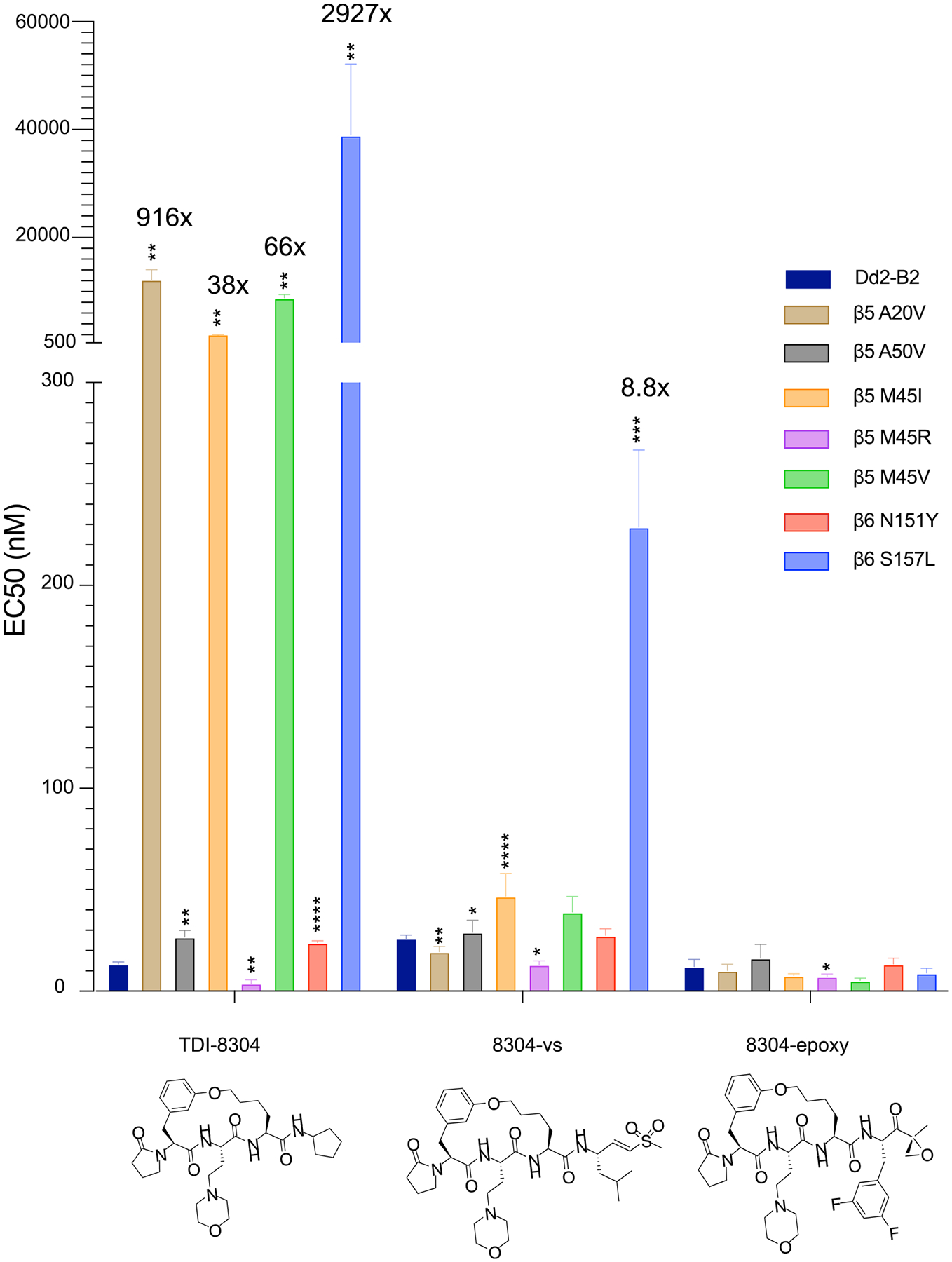
Profiling of potency of macrocyclic reversible and irreversible proteasome inhibitor based on the TDI-8304 scaffold against P. falciparum 20S β5 and β6 subunit mutants. Dd2-B2 parasites containing previously selected proteasome mutations were assayed against TDI-830419 and the irreversible analogues 8304-vs and 8304-Epoxy. Statistical significance, compared to Dd2-B2, was calculated using unpaired t tests with Welch’s correction. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. N, n = 3–5,2. TDI-8304 data were previously reported.19
DISCUSSION
Resistance to ART and ACT treatments is a major challenge to the control and mitigation of malaria. The Plasmodium proteasome has emerged as a high-value drug target due to its necessity for parasite development and lifecycle progression and for the essential role it plays in reducing ART-induced proteotoxic stress. Proteasome inhibitors also have the very desirable feature of synergizing with ART derivatives, including against ART-resistant parasites.9,13 Combination therapies utilizing proteasome inhibitors in tandem with ART are therefore a promising strategy to both treat malaria and combat the dissemination of ART resistance. Nevertheless, the challenge remains to design Plasmodium proteasome-specific inhibitors that do not cross react with human proteasomes. Moreover, although our results with covalent inhibitors of the malaria proteasome are promising, there are challenges in developing covalent drugs due to their inherent reactivity and possible toxicity caused by the formation of adducts with off-target proteins. Regardless, we remain optimistic that optimization of the selectivity of the inhibitor and tuning the reactivity of the electrophile can sufficiently mitigate off-target toxicity, as has been demonstrated for several clinically approved drugs such as Ibrutinib.28
Previous work in our lab leveraged the use of natural and unnatural amino acids to design selective irreversible covalent P. falciparum proteasome inhibitors.9,11 Having previously defined the P1 and P3 peptide positions as the determinants of selectivity, we describe here the synthesis and characterization of irreversible covalent macrocycles linked via the P2 side chain and the N-terminal capping group. We were able to determine the impact of various capping groups and several types of cycle linkages on the antiparasitic potency and off-target mammalian cytotoxicity. Notably, decreasing the lipophilicity of our cycles resulted in reduced cytotoxicity. We also found that substituting the capping group of our cycles with a pyrazole greatly increased the potency toward the parasite, but it also increased cytotoxicity. Interestingly, exchange of our P1 leucine with a biphenyl alanine resulted in a complete inversion of selectivity, both phenotypically and biochemically, which underscores the importance of the P1 leucine for Plasmodium selectivity. This observation suggests that moieties similar to the bulky biphenyl alanine vinyl sulfone at the P1 position could be used to develop better inhibitors of the human proteasome. We sought to optimize our inhibitors for metabolic stability by varying electron-deficient aromatic side chains on the P3. While these analogues proved to be potent inhibitors of parasite growth with low cytotoxicity, they ultimately showed poor microsomal stability, suggesting that cyclization alone cannot be used to increase metabolic stability.
We subsequently focused our attention on developing an irreversible covalent inhibitor, based on a structure previously explored for noncovalent inhibition of the Plasmodium proteasome.17 This strategy has proven to be powerful in developing covalent kinase inhibitors by appending an electrophilic reactive group to noncovalent compounds.29 We posited that adding a vinyl sulfone onto the P1 position of the cycle could allow it to react with the active site threonine and convert the reversible inhibitor TDI-8304 into the covalent irreversible inhibitor 8304-vs. While we did not directly demonstrate a covalent inhibition mechanism for 8304-vs, its high potency and low induction of resistance combined with the propensity of the leu-vs electrophile to form covalent bonds with the active site threonine of both human and malaria proteasomes suggested that this compound is indeed a covalent inhibitor. The resulting compound showed potency comparable to that of TDI-8304, with no cytotoxicity to HFFs, a fast rate of kill, and substantially more stability than previous linear peptide inhibitors and the cyclic peptides tested herein.
Converting the previously tested noncovalent proteasome inhibitor TDI-8304 into the irreversible covalent inhibitor 8304-vs, via the addition of the vinyl sulfone reactive group, enabled us to compare their resistance liabilities as a function of the inhibition mechanism. The irreversible 8304-vs showed an MIR of 2 × 109, compared to our earlier value of 3 × 107 obtained with TDI-8304.19 This new vinyl sulfone therefore yielded a very similar resistance profile to WLL-vs, for which resistance was only obtained with 2 × 109 parasites.13,19 These data provide evidence for a significantly improved resistance liability associated with covalent binding. For the reversible inhibitor TDI-8304, mutations in the proteasome likely increase the off rate of the compound, leading to reduced active site occupancy and lower overall inhibition as the molecule is cleared. While the off rate for the irreversibly binder 8304-vs can also be increased by mutations within the proteasome, covalent inhibitors work via a two-step inhibitory reaction—a reversible association followed by an irreversible covalent bond formation.30 The covalent 8304-vs may therefore experience off rates similar to its noncovalent counterpoint, but over time, it will irreversibly inactivate the proteasome. This is clearly demonstrated with the S157L mutation, which was selected for by both TDI-8304 and 8304-vs, yet showed a 2,900-fold increase in EC50 for the former, contrasting with a 4- to 6-fold shift for the latter. This mutation likely leads to the loss of a hydrogen bond donor and acceptor from the serine hydroxyl group required for TDI-8304 binding to the proteasome active site, and 8304-vs is presumably less impacted by this mutation because of its electrophilic warhead. 8304-vs also compared favorably to TDI-8304 against a panel of parasite lines with mutations in the P. falciparum proteasome subunits β5 and β6, showing increases in EC50 that were orders of magnitude lower than the fold shifts seen against TDI-8304.
To further demonstrate the importance of covalency to overcome resistance, we synthesized an epoxyketone derivative, 8304-epoxy. This compound performed extremely well against the panel of proteasome mutant lines, with no significant increase in EC50 against any of the lines tested. However, it was much less stable than 8304-vs in both human and mouse microsomes (Figure S9). The instability of 8304-epoxy is likely due to the susceptibility of the epoxy ring opening as well as the epoxyketone being a substrate for other enzymes such as serine hydrolases, while the vinyl sulfone is much more stable due to its lower general reactivity.31,32 These results reinforce our previous work in suggesting that the vinyl sulfone is the optimal electrophilic warhead for the development of an irreversible covalent proteasome drug.
CONCLUSIONS
Novel treatments and strategies are needed to combat the spread of artemisinin partial resistance. The P. falciparum proteasome, an enzyme essential in all parts of the parasite’s life cycle, continues to be a promising target for such therapies. Our data provide compelling evidence that optimized properties for both covalent and noncovalent macrocyclic inhibitors of the Plasmodium proteasome can be combined to create more potent and selective inhibitors of parasite growth with better stability. Moreover, our efforts reinforce the importance of covalency in overcoming parasite resistance and specifically the difficulty in developing resistance against covalent inhibitors of the proteasome. While 8304-vs stands out as a lead compound, further testing is needed to determine bioavailability and in vivo efficacy. This underscores the importance of further synthetic and medicinal chemistry efforts to advance proteasome inhibitors as promising new drugs to partner with ART derivatives and leverage their synergy against ART-resistant parasites.
METHODS
Mammalian Cytotoxicity Assays.
Human foreskin fibroblasts (HFFs) were kept in DMEM supplemented with 5% FBS. HFFs were seeded at 2000 cells per well for 4 h, and then compound was added for 72 h. Cell viability was measured using the CellTiter-Blue Assay (Promega) as per the manufacturer’s instructions.
Biochemical Inhibition of the Proteasome.
Plasmodium falciparum proteasomes were purified as previously described.7 Purified Pf20S or h20S proteasomes (R&D Systems) were tested for subunit specific inhibition in 20 mM HEPES and 0.5 mM EDTA pH 7.4. For β2 inhibition, purified proteasome (1 nM final concentration) was activated with PA28 (12 nM final concentration; E380 Boston Biochem) followed by addition of Boc-LRR-AMC (50 μM final concentration) and inhibitor simultaneously. Cleavage was measured using fluorescence (EX 380 nm/EM 460 nm) with Cytation 3 imaging reader (BioTek, Winooski, VT, USA) for 60 min, and slope was used to calculate the inhibition curve. For β5 inhibition, the Suc-LLVY-AMC (10 μM final concentration) was used as the substrate.
In Vitro Drug Susceptibility Assays.
In the Bogyo lab, P. falciparum W2 parasites, obtained from the Malaria Research and Reference Reagent Resource Center, were cultured in human erythrocytes purchased from the Stanford Blood Center (No blood type discrimination) and maintained as previously described.7 Ring stage P. falciparum was inoculated at 1% parasitemia and 0.5% hematocrit in a 96-well plate spotted with compounds. The cultures were incubated in a 10-point 2-fold serial dilution dose–response for 72 h and then fixed with a final concentration of 1% paraformaldehyde (in PBS) for 30 min at room temperature. The nuclei were stained with YOYO-1 at a final concentration of 50 nM and incubated at room temperature overnight. The percentage of YOYO-1 positive parasitized erythrocytes was quantified per well by a BD Accuri C6 automated flow cytometer. For in vitro rate of kill assays, 3D7 ring-stage parasites at 0.5% parasitemia and 2% hematocrit were exposed to each inhibitor at concentrations corresponding to 10 × EC50. Inhibitors were washed out at 24 or 48 h, and drug-free parasites were then cultured in fresh erythrocytes and culture media until readout was performed at 72 h post-treatment. In pulse assays, plates were incubated with compound for 1 h. Parasites were then washed twice with media before fresh media was added, and growth continued for 71 h prior to quantifying parasitemia as above. Fosmidomycin was used as a positive control. In the Fidock lab, the sensitivity of Dd2-B2 Pf20S WT and mutant lines to our compounds was determined by exposing asexual blood stage parasites to a 10-point 2-fold serial dilution of drug in a dose–response assay. Parasites were seeded at 0.2% parasitemia and 1% hematocrit and plated in 96-well plates with a final volume of 200 μL. Plates were incubated at 37 °C for 72 h under normal culturing conditions. Final parasitemia was determined through flow cytometry on an iQue Plus flow cytometer following staining with 1× SYBR Green and 100 nM MitoTracker Deep Red (ThermoFisher) for 30 min at 37 °C. Statistical significance was calculated using unpaired t-tests with Welch’s correction.
Parasite Culture for Resistance.
P. falciparum asexual blood stage parasites were cultured at 3% hematocrit in human O+ RBCs in RPMI-1640 media, supplemented with 25 mM HEPES, 50 mg/l-hypoxanthine, 2 mM l-glutamine, 0.21% sodium bicarbonate, 0.5% (wt/vol) AlbuMAXII (Invitrogen), and 10 μg/mL gentamycin, in modular incubator chambers (Billups- Rothenberg) at 5% O2, 5% CO2, and 90% N2 at 37°C. Dd2 parasites were obtained from T. Wellems (NIAID, NIH). Dd2-B2 is a genetically homogeneous line that was cloned from Dd2 by limiting dilution in the Fidock lab.
Minimum Inoculum of Resistance Studies.
Resistance selections were performed by culturing Dd2-B2 or Dd2 Pol-δ parasites at 3 × EC50, as previously described.19 Media was changed daily for the first 6 days of selections, and cultures were monitored by Giemsa staining and microscopy until parasites were cleared. Thereafter, media was replenished every 2 days, and cultures were monitored twice weekly for recrudescence by Giemsa staining and microscopy. Cultures were maintained under drug pressure for 60 days or until recrudescent parasites were observed. Bulk resistant lines were cloned by limited dilution cloning. The MIR value is defined as the minimum number of parasites used to obtain resistance and calculated as follows: total number of parasites inoculated ÷ total number of positive cultures.
Whole-Genome Sequencing.
P. falciparum parasites were lysed in 0.05% saponin and washed with 1× PBS, and genomic DNA (gDNA) was purified using the QIAamp DNA Blood Midi Kit (Qiagen). gDNA concentrations were quantified by Qubit using the dsDNA HS Assay (Invitrogen). 200 ng of gDNA was used to prepare sequencing libraries using the Illumina DNA Prep kit with Nextera DNA CD Indexes (Illumina). Samples were multiplexed and sequenced on an Illumina MiSeq using the MiSeq Reagent Kit V3 600 (Illumina) to obtain 300 base pair paired-end reads at an average of 30× depth of coverage. Sequence reads were aligned to the P. falciparum 3D7 reference genome (PlasmoDB ver. 48) using Burrow-Wheeler Alignment. PCR duplicates and unmapped reads were filtered out using Samtools and Picard. Reads were realigned around indels using GATK RealignerTargetCreator, and base quality scores were recalibrated using GATK BaseRecalibrator. GATK HaplotypeCaller (version 4.2.2) was used to identify all single nucleotide polymorphisms (SNPs). SNPs were filtered based on quality scores (variant quality as a function of depth QD > 1.5, mapping quality > 40, min base quality score > 18) and read depth (>5) to obtain high-quality SNPs, which were annotated using snpEFF. Integrated Genome Viewer was used to visually verify the presence of SNPs. BIC-Seq was used to check for copy number variations using the Bayesian statistical model. Copy number variations in highly polymorphic surface antigens and multigene families were removed as these are prone to stochastic changes during in vitro culture.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the NIH (R21/R33 AI125781 to M.B. and D.A.F.; R01 AI109023 to D.A.F.; and R01 AI143714 to G.L.). Additional funding was provided by the Department of Defense (W81XWH2210520 to D.A.F., M.B., G.L., and W.H.G.).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.3c00310.
HFF cell viability for compounds (Figures S1–S4), pulse killing assays (Figure S5), minimum inoculum of resistance selections (Figures S6–S8), microsome stability (Figure S9), compound characterization and compound synthesis (Supplementary Schemes), and mass spectrometry data (PDF)
Mass spectrometry analysis (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acsinfecdis.3c00310
The authors declare no competing financial interest.
Contributor Information
John M. Bennett, Department of Chemistry, Stanford University, Stanford, California 94305, United States
Kurt E. Ward, Department of Microbiology and Immunology, Columbia University Medical Center, New York, New York 10032, United States; Center for Malaria Therapeutics and Antimicrobial Resistance, Columbia University Medical Center, New York, New York 10032, United States;.
Ryan K. Muir, Department of Pathology, Stanford University School of Medicine, Stanford, California 94305, United States.
Stephanie Kabeche, Department of Biochemistry, Stanford University School of Medicine, Stanford, California 94305, United States.
Euna Yoo, Department of Pathology, Stanford University School of Medicine, Stanford, California 94305, United States; Chemical Biology Laboratory, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Frederick, Maryland 21702, United States;.
Tomas Yeo, Department of Microbiology and Immunology, Columbia University Medical Center, New York, New York 10032, United States; Center for Malaria Therapeutics and Antimicrobial Resistance, Columbia University Medical Center, New York, New York 10032, United States.
Grace Lam, Department of Medicine, Division of Gastroenterology and Hepatology, Stanford University School of Medicine, Stanford, California 94304, United States.
Hao Zhang, Department of Microbiology and Immunology, Weill Cornell Medicine, New York, New York 10065, United States;.
Jehad Almaliti, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California San Diego, La Jolla, California 92093, United States.
Gabriel Berger, Department of Pathology, Stanford University School of Medicine, Stanford, California 94305, United States.
Franco F. Faucher, Department of Chemistry, Stanford University, Stanford, California 94305, United States
Gang Lin, Department of Microbiology and Immunology, Weill Cornell Medicine, New York, New York 10065, United States;.
William H. Gerwick, Scripps Institution of Oceanography, University of California San Diego, La Jolla, California 92037, United States; Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California San Diego, La Jolla, California 92093, United States
Ellen Yeh, Department of Pathology, Stanford University School of Medicine, Stanford, California 94305, United States; Department of Microbiology and Immunology, Stanford University School of Medicine, Stanford, California 94304, United States.
David A. Fidock, Department of Microbiology and Immunology, Columbia University Medical Center, New York, New York 10032, United States; Center for Malaria Therapeutics and Antimicrobial Resistance and Division of Infectious Diseases, Columbia University Medical Center, New York, New York 10032, United States
Matthew Bogyo, Department of Pathology, Stanford, University School of Medicine, Stanford, California 94305, United States; Scripps Institution of Oceanography, University of California San Diego, La Jolla, California 92037, United States;.
REFERENCES
- (1).World Malaria Report 2022; World Health Organization: Geneva, 2022. [Google Scholar]
- (2).Noedl H; Socheat D; Satimai W Artemisinin-Resistant Malaria in Asia. N. Engl. J. Med 2009, 361 (5), 540–541. [DOI] [PubMed] [Google Scholar]
- (3).Uwimana A; Legrand E; Stokes BH; Ndikumana J-LM; Warsame M; Umulisa N; Ngamije D; Munyaneza T; Mazarati J-B; Munguti K; Campagne P; Criscuolo A; Ariey F; Murindahabi M; Ringwald P; Fidock DA; Mbituyumuremyi A; Menard D Emergence and Clonal Expansion of in vitro Artemisinin-Resistant Plasmodium falciparum Kelch13 R561H Mutant Parasites in Rwanda. Nat. Med 2020, 26 (10), 1602–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Balikagala B; Fukuda N; Ikeda M; Katuro OT; Tachibana S-I; Yamauchi M; Opio W; Emoto S; Anywar DA; Kimura E; Palacpac NMQ; Odongo-Aginya EI; Ogwang M; Horii T; Mita T Evidence of Artemisinin-Resistant Malaria in Africa. N. Engl. J. Med 2021, 385 (13), 1163–1171. [DOI] [PubMed] [Google Scholar]
- (5).Straimer J; Gnädig NF; Witkowski B; Amaratunga C; Duru V; Ramadani AP; Dacheux M; Khim N; Zhang L; Lam S; Gregory PD; Urnov FD; Mercereau-Puijalon O; Benoit-Vical F; Fairhurst RM; Ménard D; Fidock DA K13-Propeller Mutations Confer Artemisinin Resistance in Plasmodium falciparum Clinical Isolates. Science 2015, 347 (6220), 428–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ashley EA; Dhorda M; Fairhurst RM; Amaratunga C; Lim P; Suon S; Sreng S; Anderson JM; Mao S; Sam B; Sopha C; Chuor CM; Nguon C; Sovannaroth S; Pukrittayakamee S; Jittamala P; Chotivanich K; Chutasmit K; Suchatsoonthorn C; Runcharoen R; Hien TT; Thuy-Nhien NT; Thanh NV; Phu NH; Htut Y; Han K-T; Aye KH; Mokuolu OA; Olaosebikan RR; Folaranmi OO; Mayxay M; Khanthavong M; Hongvanthong B; Newton PN; Onyamboko MA; Fanello CI; Tshefu AK; Mishra N; Valecha N; Phyo AP; Nosten F; Yi P; Tripura R; Borrmann S; Bashraheil M; Peshu J; Faiz MA; Ghose A; Hossain MA; Samad R; Rahman MR; Hasan MM; Islam A; Miotto O; Amato R; MacInnis B; Stalker J; Kwiatkowski DP; Bozdech Z; Jeeyapant A; Cheah PY; Sakulthaew T; Chalk J; Intharabut B; Silamut K; Lee SJ; Vihokhern B; Kunasol C; Imwong M; Tarning J; Taylor WJ; Yeung S; Woodrow CJ; Flegg JA; Das D; Smith J; Venkatesan M; Plowe CV; Stepniewska K; Guerin PJ; Dondorp AM; Day NP; White NJ Spread of Artemisinin Resistance in Plasmodium falciparum Malaria. N. Engl. J. Med 2014, 371 (5), 411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Li H; Ponder EL; Verdoes M; Asbjornsdottir KH; Deu E; Edgington LE; Lee JT; Kirk CJ; Demo SD; Williamson KC; Bogyo M Validation of the Proteasome as a Therapeutic Target in Plasmodium Using an Epoxyketone Inhibitor with Parasite-Specific Toxicity. Chem. Biol 2012, 19 (12), 1535–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bridgford JL; Xie SC; Cobbold SA; Pasaje CFA; Herrmann S; Yang T; Gillett DL; Dick LR; Ralph SA; Dogovski C; Spillman NJ; Tilley L Artemisinin Kills Malaria Parasites by Damaging Proteins and Inhibiting the Proteasome. Nat. Commun 2018, 9 (1), 3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Li H; O’Donoghue AJ; van der Linden WA; Xie SC; Yoo E; Foe IT; Tilley L; Craik CS; da Fonseca PCA; Bogyo M Structure- and Function-Based Design of Plasmodium-Selective Proteasome Inhibitors. Nature 2016, 530 (7589), 233–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).LaMonte GM; Almaliti J; Bibo-Verdugo B; Keller L; Zou BY; Yang J; Antonova-Koch Y; Orjuela-Sanchez P; Boyle CA; Vigil E; Wang L; Goldgof GM; Gerwick L; O’Donoghue AJ; Winzeler EA; Gerwick WH; Ottilie S Development of a Potent Inhibitor of the Plasmodium Proteasome with Reduced Mammalian Toxicity. J. Med. Chem 2017, 60 (15), 6721–6732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Yoo E; Stokes BH; de Jong H; Vanaerschot M; Kumar T; Lawrence N; Njoroge M; Garcia A; Van der Westhuyzen R; Momper JD; Ng CL; Fidock DA; Bogyo M Defining the Determinants of Specificity of Plasmodium Proteasome Inhibitors. J. Am. Chem. Soc 2018, 140 (36), 11424–11437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Xie SC; Metcalfe RD; Mizutani H; Puhalovich T; Hanssen E; Morton CJ; Du Y; Dogovski C; Huang S-C; Ciavarri J; Hales P; Griffin RJ; Cohen LH; Chuang B-C; Wittlin S; Deni I; Yeo T; Ward KE; Barry DC; Liu B; Gillett DL; Crespo-Fernandez BF; Ottilie S; Mittal N; Churchyard A; Ferguson D; Aguiar ACC; Guido RVC; Baum J; Hanson KK; Winzeler EA; Gamo F-J; Fidock DA; Baud D; Parker MW; Brand S; Dick LR; Griffin MDW; Gould AE; Tilley L Design of Proteasome Inhibitors with Oral Efficacy in vivo against Plasmodium falciparum and Selectivity over the Human Proteasome. Proc. Natl. Acad. Sci. U. S. A 2021, 118 (39), No. e2107213118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Stokes BH; Yoo E; Murithi JM; Luth MR; Afanasyev P; Da Fonseca PCA; Winzeler EA; Ng CL; Bogyo M; Fidock DA Covalent Plasmodium falciparum-Selective Proteasome Inhibitors Exhibit a Low Propensity for Generating Resistance in vitro and Synergize with Multiple Antimalarial Agents. PLOS Pathog. 2019, 15 (6), No. e1007722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lau JL; Dunn MK Therapeutic Peptides: Historical Perspectives, Current Development Trends, and Future Directions. Bioorg. Med. Chem 2018, 26 (10), 2700–2707. [DOI] [PubMed] [Google Scholar]
- (15).Nielsen DS; Shepherd NE; Xu W; Lucke AJ; Stoermer MJ; Fairlie DP Orally Absorbed Cyclic Peptides. Chem. Rev 2017, 117 (12), 8094–8128. [DOI] [PubMed] [Google Scholar]
- (16).Li H; Tsu C; Blackburn C; Li G; Hales P; Dick L; Bogyo M Identification of Potent and Selective Non-Covalent Inhibitors of the Plasmodium falciparum Proteasome. J. Am. Chem. Soc 2014, 136 (39), 13562–13565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zhan W; Zhang H; Ginn J; Leung A; Liu YJ; Michino M; Toita A; Okamoto R; Wong T; Imaeda T; Hara R; Yukawa T; Chelebieva S; Tumwebaze PK; Lafuente-Monasterio MJ; Martinez-Martinez MS; Vendome J; Beuming T; Sato K; Aso K; Rosenthal PJ; Cooper RA; Meinke PT; Nathan CF; Kirkman LA; Lin G Development of a Highly Selective Plasmodium falciparum Proteasome Inhibitor with Anti-malaria Activity in Humanized Mice. Angew. Chem., Int. Ed 2021, 60 (17), 9279–9283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zhang H; Ginn J; Zhan W; Liu YJ; Leung A; Toita A; Okamoto R; Wong T-T; Imaeda T; Hara R; Yukawa T; Michino M; Vendome J; Beuming T; Sato K; Aso K; Meinke PT; Nathan CF; Kirkman LA; Lin G Design, Synthesis, and Optimization of Macrocyclic Peptides as Species-Selective Antimalaria Proteasome Inhibitors. J. Med. Chem 2022, 65 (13), 9350–9375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Deni I; Stokes BH; Ward KE; Fairhurst KJ; Pasaje CFA; Yeo T; Akbar S; Park H; Muir R; Bick DS; Zhan W; Zhang H; Liu YJ; Ng CL; Kirkman LA; Almaliti J; Gould AE; Duffey M; O’Donoghue AJ; Uhlemann A-C; Niles JC; Da Fonseca PCA; Gerwick WH; Lin G; Bogyo M; Fidock DA Mitigating the Risk of Antimalarial Resistance via Covalent Dual-Subunit Inhibition of the Plasmodium Proteasome. Cell Chem. Biol 2023, 30, 470–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Desai SA Why Do Malaria Parasites Increase Host Erythrocyte Permeability? Trends Parasitol. 2014, 30 (3), 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Naccache P; Sha’afi RI Patterns of Nonelectrolyte Permeability in Human Red Blood Cell Membrane. J. Gen. Physiol 1973, 62 (6), 714–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Battista N; Bari M; Bisogno T N-Acyl Amino Acids: Metabolism, Molecular Targets, and Role in Biological Processes. Biomolecules 2019, 9 (12), 822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Li D; Zhang X; Ma X; Xu L; Yu J; Gao L; Hu X; Zhang J; Dong X; Li J; Liu T; Zhou Y; Hu Y Development of Macrocyclic Peptides Containing Epoxyketone with Oral Availability as Proteasome Inhibitors. J. Med. Chem 2018, 61 (20), 9177–9204. [DOI] [PubMed] [Google Scholar]
- (24).Lazzara PR; Moore TW Scaffold-Hopping as a Strategy to Address Metabolic Liabilities of Aromatic Compounds. RSC Med. Chem 2020, 11 (1), 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Sanz LM; Crespo B; De-Cózar C; Ding XC; Llergo JL; Burrows JN; García-Bustos JF; Gamo F-J aru Killing Rates Allow to Discriminate between Different Antimalarial Mode-of-Action. PLoS One 2012, 7 (2), No. e30949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Duffey M; Blasco B; Burrows JN; Wells TNC; Fidock DA; Leroy D Assessing Risks of Plasmodium falciparum Resistance to Select Next-Generation Antimalarials. Trends Parasitol. 2021, 37 (8), 709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kümpornsin K; Kochakarn T; Yeo T; Okombo J; Luth MR; Hoshizaki J; Rawat M; Pearson RD; Schindler KA; Mok S; Park H; Uhlemann A-C; Jana GP; Maity BC; Laleu B; Chenu E; Duffy J; Moliner Cubel S; Franco V; Gomez-Lorenzo MG; Gamo FJ; Winzeler EA; Fidock DA; Chookajorn T; Lee MCS Generation of a Mutator Parasite to Drive Resistome Discovery in Plasmodium falciparum. Nat. Commun 2023, 14 (1), 3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Broccoli A; Argnani L; Morigi A; Nanni L; Casadei B; Pellegrini C; Stefoni V; Zinzani PL Long-Term Efficacy and Safety of Ibrutinib in the Treatment of CLL Patients: A Real Life Experience. J. Clin. Med 2021, 10 (24), 5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Abdeldayem A; Raouf YS; Constantinescu SN; Moriggl R; Gunning PT Advances in Covalent Kinase Inhibitors. Chem. Soc. Rev 2020, 49 (9), 2617–2687. [DOI] [PubMed] [Google Scholar]
- (30).Strelow JM A Perspective on the Kinetics of Covalent and Irreversible Inhibition. SLAS Discovery 2017, 22 (1), 3–20. [DOI] [PubMed] [Google Scholar]
- (31).Almaliti J; Alzweiri M; Alhindy M; Al-Helo T; Daoud I; Deknash R; Naman CB; Abu-Irmaileh B; Bustanji Y; Hamad I Discovery of Novel Epoxyketone Peptides as Lipase Inhibitors. Molecules 2022, 27 (7), 2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Joyce JA; Baruch A; Chehade K; Meyer-Morse N; Giraudo E; Tsai F-Y; Greenbaum DC; Hager JH; Bogyo M; Hanahan D Cathepsin Cysteine Proteases Are Effectors of Invasive Growth and Angiogenesis during Multistage Tumorigenesis. Cancer Cell 2004, 5 (5), 443–453. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.