

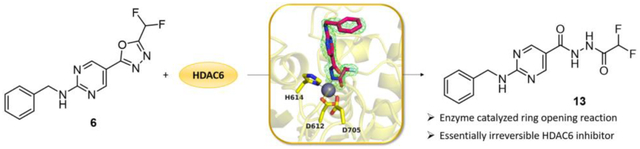
Abstract
Histone deacetylase 6 (HDAC6) is an important drug target in oncology and non-oncological diseases. Most available HDAC6 inhibitors (HDAC6i) utilize a hydroxamic acid as zinc-binding group which limits the therapeutic opportunities due its genotoxic potential. Recently, difluoromethyl-1,3,4-oxadiazoles (DFMOs) were reported as potent and selective HDAC6i, but their mode of inhibition remained enigmatic. Herein, we report that DFMOs act as mechanism-based and essentially irreversible HDAC6i. Biochemical data confirm that DFMO 6 is a tight-binding HDAC6i capable of inhibiting HDAC6 via a two-step slow-binding mechanism. Crystallographic and mechanistic experiments suggest that the attack of 6 by the zinc-bound water at the sp2 carbon closest to the difluoromethyl moiety followed by a subsequent ring opening of the oxadiazole yields the deprotonated difluoroacetylhydrazide 13 as active species. The strong anionic zinc coordination of 13 and the binding of the difluoromethyl moiety in the P571 pocket finally results in an essentially irreversible inhibition of HDAC6.
Keywords: Histone deacetylase, epigenetics, cancer, enzyme kinetics, HDAC6
Graphical Abstract
INTRODUCTION
Histone deacetylases (HDACs) are epigenetic drug targets that have originally been assumed to modify histone modifications by removing acetyl groups from lysine residues. Meanwhile, however, it has turned out that the substrate spectrum of the enzyme family is more complex.1 In agreement with the usual division into four classes, it is now clear that only class I HDACs (HDACs 1, 2, 3, and 8) actually regulate histones.1 Class III HDACs differ from the other zinc-dependent isoforms by the fact that they are NAD+-dependent, whereas class IV consists of no more than one isoform, HDAC11, whose biological role is yet unclear.2,3 A more versatile class of HDACs is class II with class IIa enzymes (HDACs 4, 5, 7, 9) playing a crucial role in gene expression, despite their poor deacetylase qualities.1,4–6 Class IIb, on the other hand, includes HDAC6 and the polyamine deacetylase HDAC10 which are both mainly located in the cytosol.1,7,8 Tailored to fit the highly-conserved active sites of the different isoforms, HDAC inhibitors (HDACi) typically consist of a zinc-binding group (ZBG), a variably sized cap group and a suitable linker connecting the two units.9 In contrast to unselective or class I-specific HDAC inhibition by HDACi such as vorinostat, belinostat, panobinostat, and romidepsin, which have been introduced as FDA-approved anticancer drugs in the past two decades, HDAC6 inhibition has no effect on histones and is thus presumed to cause less severe adverse effects.10,11 Originally considered to be a tubulin deacetylase, HDAC6 has since been found to regulate a range of other proteins as well; most notably cortactin, the Alzheimer-related tau, and the chaperone Hsp90.7,12–15 Serving this particular range of substrates, HDAC6 regulation has been investigated as a promising treatment option for non-oncological conditions, for example neurodegenerative diseases13,14,16,17, several rare disorders, like Rett syndrome and Charcot-Marie-Tooth disease18,19, autoimmune diseases, and other chronic conditions including idiopathic pulmonary fibrosis and inflammasome-mediated disorders.20–22 Through enabling aggresome formation, HDAC6 is further involved in cellular protein degradation which makes it a prominent target for synergistic drug combination approaches with proteasome inhibitors.23–25 On the clinical level, this synergism is already being addressed by the combination of bortezomib, dexamethasone, and the pan-HDACi panobinostat for the treatment of multiple myeloma while further combination studies using the HDAC6-preferential inhibitor ricolinostat are ongoing.26 Other promising targets for synergistic activities with HDAC6i that are currently being investigated include BET-proteins27,28, topoisomerases29, the lysine-specific demethylase 1 (LSD1)30–33, and Hsp9034–38. In consequence, and despite having limited clinical anticancer potential on its own, HDAC6 has turned out as a prominent drug target for combination therapies but only few of the many selective HDAC6i presented so far have yet entered clinical trials.39–42 One major limitation in this regard seems to be the fact that most HDAC6i incorporate hydroxamate ZBGs which affect the drug’s tolerability and overall performance by promoting off-target interactions and the appearance of toxic metabolites. In fact, hydroxamate groups have long been suspected of releasing hydroxylamine or undergoing the Lossen rearrangement yielding isocyanates under physiological conditions.11,43 Given that both species are highly mutagenic and thus unsuitable for long-term therapy, there is an urgent need for alternative ZBGs but even after several years of intensive research, there are only few candidates with pleasing chelating properties and low toxicity levels.44 Beside ethyl hydrazides45 and several non-hydroxamate compounds of yet undisclosed structures that are currently in phase II trials, the most promising HDAC6-selective binding motif seems to be the difluoromethyl-1,3,4-oxadiazole (DFMO) group that has been discovered by Kim et al.46 According to their study, the DFMO group exhibited excellent HDAC6 inhibition in the low nanomolar concentration range with a high selectivity over HDAC1.46 Despite frequently appearing in patents46–50, this ZBG is relatively underrepresented in research manuscripts. However, in 2022, the DFMO derivative SE-7552 was used as a selective HDAC6 inhibitor to overcome leptin resistance in obesity, marking its first appearance in scientific literature.51 In the same year, we successfully incorporated the DFMO warhead into proteolysis-targeting chimeras (PROTACs) to selectively degrade HDAC6.52 However, the mechanism by which DFMOs inhibit or degrade HDAC6 remained enigmatic. In a conference abstract published in 2022, we disclosed that a DFMO derivative underwent an enzyme-catalyzed ring opening reaction, resulting in an acylhydrazide that was co-crystallized in an extended conformation within the active site of HDAC6.53 More recently, Barinka and co-workers54 conducted a comparative assessment of a hydroxamate-based HDAC6 inhibitor and its corresponding DFMO analog. Biochemical and cell-based assays unequivocally demonstrated the superior potency and selectivity of the DFMO ZBG.54 Similarly, the high HDAC6 selectivity of DFMOs compared to related hydroxamic acid-derived HDAC6i was confirmed in a recent patent.55
In 2023, Steinkühler and co-workers56 reported the structure of the HDAC6 complex with a hydrazide inhibitor resulting from a double hydrolysis of a related oxadiazole inhibitor. The authors speculated that the crystallized hydrazide may not be solely responsible for the remarkable HDAC6 inhibition observed. Instead, they proposed the existence of a high-affinity intermediate that forms a tight and long-lived enzyme-inhibitor complex. This intermediate may take the form of a closed hydrated intermediate or a protonated acylhydrazide, both of which were proposed as possible active species. However, the nature of the active species could not be conclusively confirmed.56
Herein, we report the full experimental details of our 2022 conference abstract53 demonstrating that DFMOs act as selective, mechanism-based, and essentially irreversible inactivators capable of inhibiting HDAC6 via a two-step slow-binding mechanism. Our findings reveal that that the zinc-bound water attacks the sp2 carbon nearest to the difluoromethyl moiety of the DFMO group followed by a subsequent ring opening of the oxadiazole, thereby yielding a deprotonated difluoroacetylhydrazide as active species.
RESULTS AND DISCUSSION
Design, synthesis, and HDAC inhibition of difluoromethyloxadiazole-based HDAC6 inhibitors.
To identify key structural requirements for selective HDAC6 inhibition by DFMO-derived inhibitors, we decided to pursue a fragment-based approach. In the first step, to investigate the influence of the (hetero)aromatic linker, HDAC6i fragments containing phenyl (1), pyridinyl (2), and pyrimidinyl (3) linkers attached to the DFMO ZBG were included in the design and synthesis of initial prototypic compounds. For the synthesis of the fragments 1, 2, and 3 the respective carbonitriles were transformed into the corresponding tetrazoles by the treatment with sodium azide, followed by the reaction with difluoroacetic anhydride (DFAA) to generate the DFMO group via a Huisgen 1,3,4-oxadiazole synthesis (see Scheme S1, Supporting Information).57 The three synthesized fragments were screened for their inhibition of HDAC6 and HDAC1–4 using biochemical HDAC inhibition assays. The pyrimidinyl derivative 3 displayed the highest inhibitory potency against HDAC6, while all fragments were inactive against the control isoforms HDAC1–4 (Table 1).
Table 1.
Inhibitory activities of the synthesized difluoromethyl-1,3,4-oxadiazoles against HDAC6 and the control isoforms HDAC1–4; IC50 values [μM, mean ± SD] or percent inhibition at 10 μM; n. e.: no effect = < 15% inhibition at 10 μM; n. d.: not determined.a
| |||||
|---|---|---|---|---|---|
| Cpd | HDAC6 | HDAC1 | HDAC2 | HDAC3 | HDAC4 |
| 1 | n. e. | n. e. | n. e. | n. e. | n. e. |
| 2 | 39% | n. e. | n. e. | n. e. | n. e. |
| 3 | 56% | n. e. | n. e. | n. e. | n. e. |
| 6 | 0.193 ± 0.006 μM | n. e. | n. e. | n. e. | n. e. |
| 7 | 0.337 ± 0.026 μM | n. e. | n. e. | n. e. | n. e. |
| 9 | 76% | n. e. | n. e. | n. e. | n. e. |
| 10 | 75% | n. e. | n. e. | n. e. | n. e. |
| 12 | 27% | n. e. | n. e. | n. e. | n. e. |
| Vorinostat | 0.039 ± 0.005 μM | 0.128 ± 0.009 μM | 0.158 ± 0.033 μM | 0.079 ± 0.016 μM | n. d. |
| TMP-269 | n. d. | n. d. | n. d. | n. d. | 0.753 ± 0.010 μM |
preincubation of HDAC1–4 or 6 and inhibitor: 1 h at 25 °C.
Due to the initial activity of the pyrimidinyl fragment 3, we designed full sized HDACi including a benzyl as well as a para-methoxy benzyl cap group, an aminopyrimidinyl linker, and the DFMO ZBG. To obtain 6 and 7 the respective benzylamines were subjected to a nucleophilic aromatic substitution reaction with 2-chloropyrimidine-5-carbonitrile. The resulting carbonitrile intermediates were converted into the corresponding DFMO derivatives as described above (Scheme 1A). In subsequent HDAC inhibition assays we observed submicromolar inhibitory activities against HDAC6 for both full-sized HDAC6i (6 and 7), with IC50 values of 0.193 μM and 0.337 μM, respectively, and no activity against HDAC1–4 (Table 1).
Scheme 1.
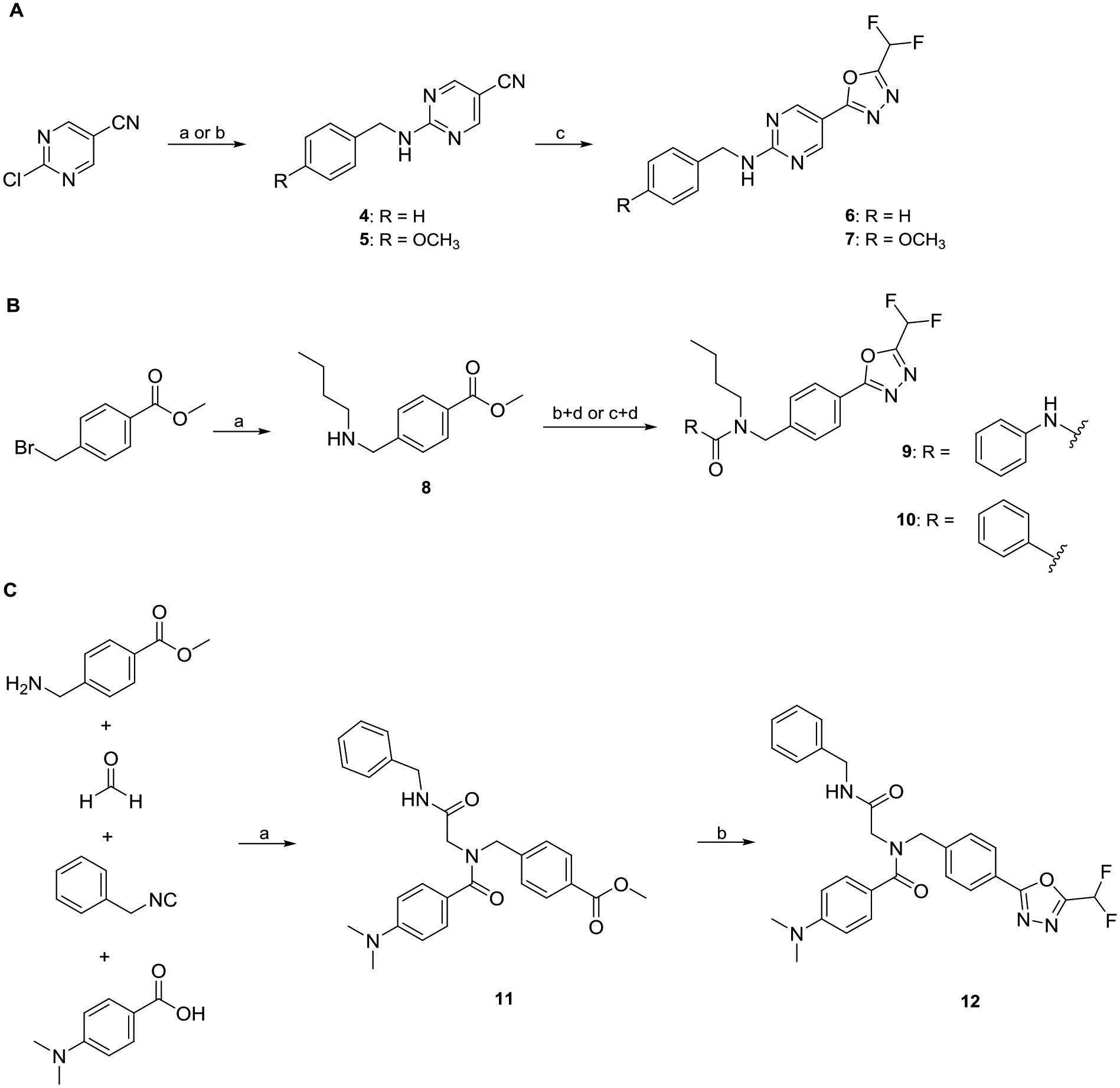
A: Synthesis of full sized HDAC6i 6 and 7. a) Benzylamine, DIPEA, EtOH, 90 °C, 18 h (4); b) 4-methoxybenzylamine, DIPEA, EtOH, 90 °C, 18 h (5); c) i: NaN3, NH4Cl, LiCl·H2O, DMF, 100 °C, 18 h; ii: DFAA, 70 °C, 18 h. B: Synthesis of nexturastat analogs 9 and 10. a) n-Butylamine, THF, rt., 3 h; b) phenyl phenylcarbamate, TEA, THF, 66 °C, 2 h (9); c) benzoyl chloride, CH2Cl2, rt., 2 h (10); d) i: hydrazine monohydrate, MeOH, 70 °C, 3 h; ii: DFAA, DMF, 70 °C, 1 h; iii: Burgess reagent, THF, 60 °C, 18 h. C: Synthesis of the peptoid-based HDACi 12. a) TEA, MeOH, rt., 72 h; b) i: hydrazine monohydrate, MeOH, 70 °C, 3 h; ii: DFAA, TEA, DMF, 70 °C, 1 h; iii: Burgess reagent, TEA, THF, 60 °C, 18 h.
Additionally, the DFMO ZBG was introduced in potent well-established HDAC6i such as nexturastat A and our previously published peptoid-based HDAC6i.58,59 For the synthesis of the nexturastat derivatives (9, 10), n-butylamine was alkylated with methyl 4-(bromomethyl)benzoate. Next, the resulting intermediate 8 was treated with phenyl phenylcarbamate or benzoyl chloride to provide the corresponding urea and carboxamide derivatives. The respective products were subjected to a hydrazinolysis followed by a difluoroacetylation reaction with DFAA. The resulting acylhydrazides were converted into the desired DFMO ZBG via a dehydrative cyclization reaction using Burgess reagent (Scheme 1B). The peptoid derivative (12) was synthesized starting from an Ugi four-component reaction.59,60 The formation of the DFMO moiety was accomplished in three steps from methyl ester intermediate 11 via the hydrazinolysis, difluoroacetylation, and dehydrative cyclization sequence described above (Scheme 1C). Interestingly, the nexturastat A analogs 9 and 10 and the peptoid-based HDACi 12 displayed only moderate inhibitory activity against HDAC6 and were inactive against HDAC1–4. The typical structural features of selective hydroxamate-based HDAC6i include a benzyl linker in combination with a bulky or branched cap group.61 Our results for the DFMO derivatives 9, 10, and 12 indicate that this HDAC6 pharmacophore cannot be directly translated to DFMO-based HDAC6i. A possible explanation for this phenomenon could be a different binding mode in the active site of HDAC6. Due to the highest HDAC6 inhibitory activity in this set of compounds, we selected 6 for elucidating its binding mode in the second catalytic domain 2 (CD2) of Danio rerio (zebrafish) HDAC6.
Compound 6 is a substrate analog of HDAC6 that undergoes an enzyme-catalyzed ring opening reaction.
Oxadiazole 6 was cocrystallized with HDAC6, and crystals diffracted X-rays to 2.00 Å resolution. The initial electron density map of the enzyme-inhibitor complex was phased by molecular replacement using the structure of the unliganded enzyme (PDB 5EEM)62 as a search probe for rotation and translation function calculations. After a molecular replacement solution was achieved and initial rounds of crystallographic structure refinement were completed, we attempted to fit oxadiazole 6 into strong |Fo|–|Fc| difference electron density in the active site (Figure 1A, B). Surprisingly, the intact oxadiazole would not fit satisfactorily in this electron density map (Figure 1C). After studying the electron density map and considering the possible reactivity of the oxadiazole moiety, we concluded that the oxadiazole ring had undergone nucleophilic attack by the zinc-bound water to yield a ring-opened form – acylhydrazide 13 – which fits the initial, unbiased electron density map perfectly (Figure 1D). The structure of the HDAC6–13 complex was refined to convergence with R/Rfree = 0.185/0.223.
Figure 1.
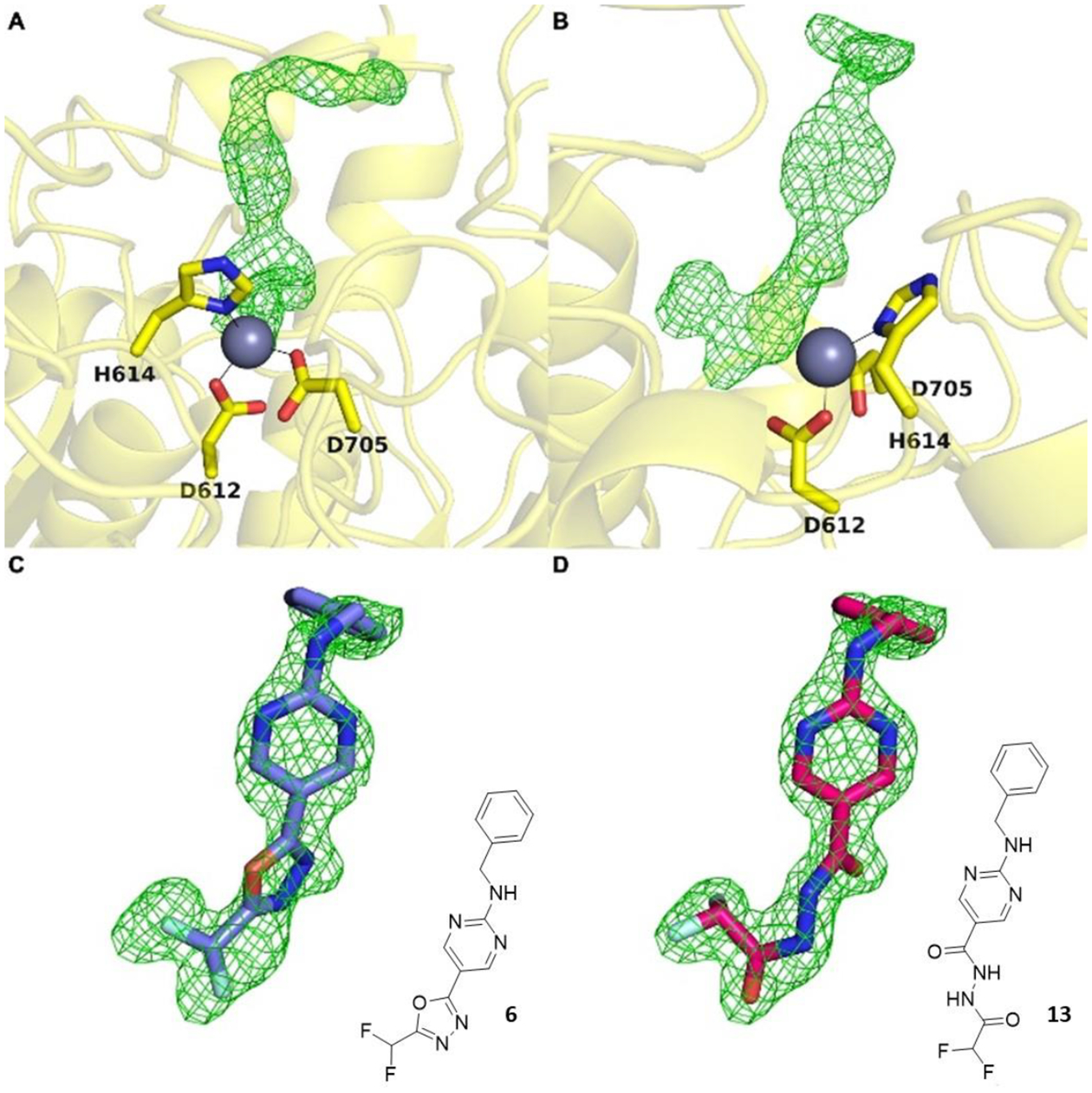
The initial |Fo|–|Fc| map (two orientations (A) and (B)) calculated from X-ray diffraction data collected from crystals of HDAC6 cocrystallized with inhibitor 6 reveals strong, unbiased electron density for the bound inhibitor in the active site. Surprisingly, this difference density could not be fit satisfactorily with intact oxadiazole 6 (C); instead, it could be fit perfectly with acylhydrazide 13 resulting from hydrolysis and ring-opening of the oxadiazole (D).
A Polder omit map of the final enzyme-inhibitor complex is shown in Figure 2A. Inhibitor binding does not trigger any major structural changes in the protein, and the root-mean-square deviation is 0.177 Å for 315 Cα atoms between the inhibitor-bound and unliganded enzyme (PDB 5EEM). Interestingly, the structure reveals an extensive array of intermolecular interactions that stabilize the bound inhibitor (Figure 2B). Key among these interactions is coordination of an acylhydrazide nitrogen to the catalytic zinc ion (N•••Zn2+ distance = 2.0 Å). This interaction requires deprotonation of the acylhydrazide NH group closest to the difluoromethyl group to form a nitrogen anion – this could result directly from the mechanism of ring-opening, or it could result from deprotonation of the neutral acylhydrazide (as discussed later).
Figure 2.
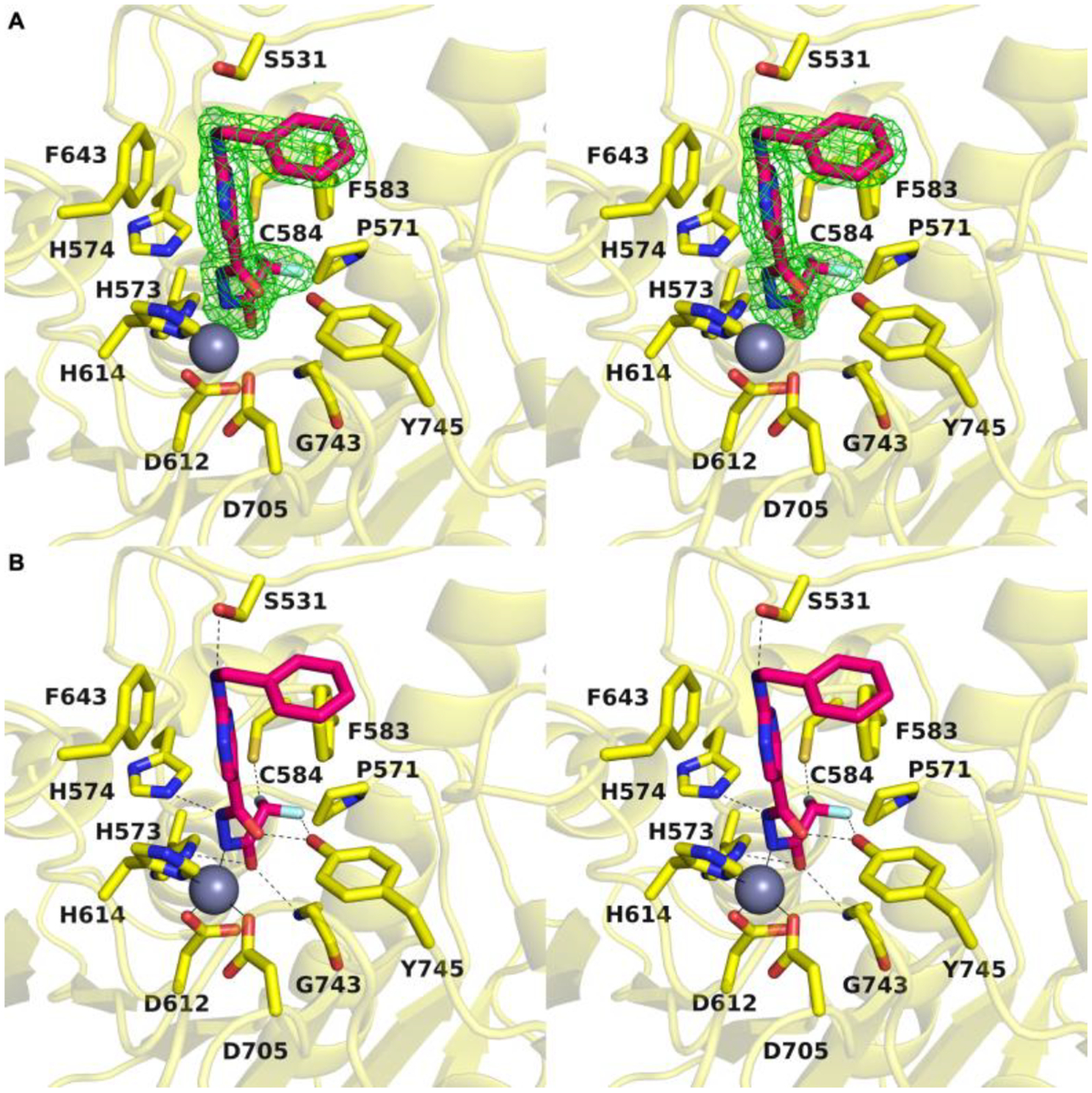
A: Stereoview of the Polder omit map of 13 contoured at 3σ (PDB 8GD4). B: Stereoview highlighting intermolecular interactions in the enzyme-inhibitor complex in the active site of HDAC6. The catalytic zinc ion is shown as a gray sphere; metal coordination and hydrogen bond interactions are shown as solid and dashed black lines, respectively (PDB 8GD4).
The bound inhibitor makes numerous hydrogen bond interactions with active site residues. One carbonyl group of the acylhydrazide forms hydrogen bonds with H574 and the backbone NH group of G743, and the other carbonyl group forms a hydrogen bond with Y745. Interestingly, both C–F groups engage in hydrogen bond interactions: one C–F group forms a hydrogen bond with C584 and the other forms a hydrogen bond with Y745. Finally, the benzylamino NH group forms a hydrogen bond with S531, and both aromatic rings of the inhibitor engage in offset stacked and edge-to-face aromatic interactions with F583 and F643.
Overlay of the structure of the HDAC6 complex with acylhydrazide 13 and the recently-reported structure of the HDAC6 complex with the hydrazide Cmpd3 resulting from hydrolysis of another oxadiazole inhibitor (ITF5924, PDB 8A8Z)56 reveals slight shifts of 0.9 Å in the orientation of the zinc-binding group and the aromatic linker region; unlike the situation for zinc coordination by an amide NH group, the primary amino group of the hydrazide does not have to be deprotonated to coordinate to zinc (Figure 3). Other differences between the binding of acylhydrazide and hydrazide inhibitors include the hydrogen bond with catalytic tyrosine Y745, which at 2.1 Å is 0.4 Å shorter in the complex with the hydrazide (making this a very short hydrogen bond), and the binding of a water molecule in the P571 pocket.
Figure 3.
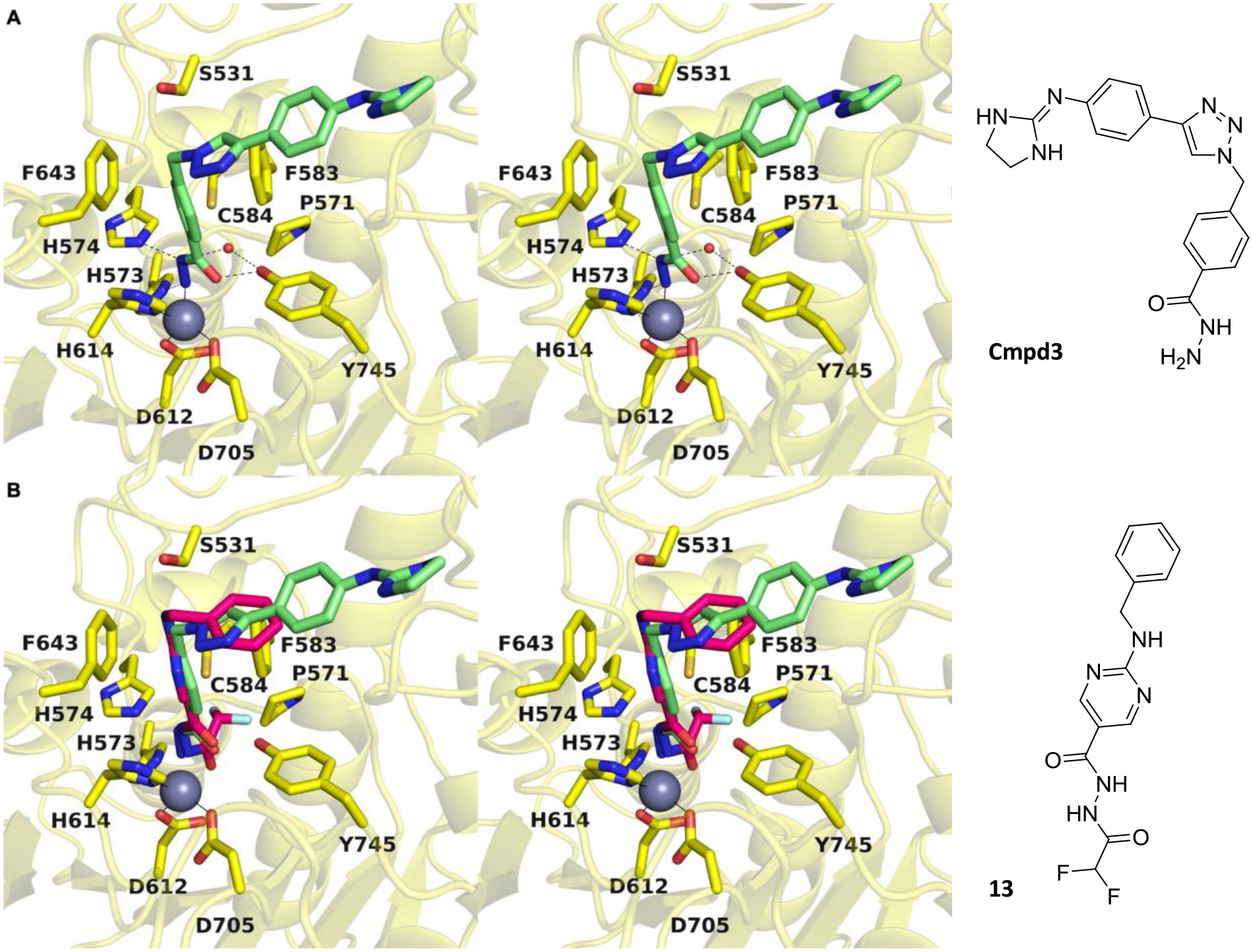
A: Stereoview of the oxadiazole-derived hydrazide inhibitor Cmpd3 bound in the active site of HDAC6 (PDB 8A8Z). B: Overlay of the oxadiazole-derived acylhydrazide 13 and hydrazide inhibitor Cmpd3 bound in the active site of HDAC6.
Difluoromethyl-1,3,4-oxadiazoles are mechanism-based and essentially irreversible HDAC6 inhibitors.
The enzyme-catalyzed ring opening reaction observed for 6 in the presence of HDAC6 prompted us to investigate the structural requirements for this unique mode of action in detail. To this end, we synthesized the cocrystallized acylhydrazide 13 as a reference compound as well as the corresponding methyl-1,3,4-oxadiazole (15), monofluoromethyl-1,3,4-oxadiazole (16) and trifluoromethyl-1,3,4-oxadiazole (17) analogs of 6 (Figure 4; see Scheme S3 and S4 for synthetic details, Supporting Information). Furthermore, due to the crystal structure reported by Steinkühler et al.56 we synthesized the hydrazide 14 (Figure 4, see Scheme S4 for synthetic details, Supporting Information). The subsequent HDAC6 inhibition assays revealed that the protonated acylhydrazide 13 and the methyl-1,3,4-oxadiazole derivative 15 displayed only very weak inhibitory properties with less than 15% inhibition at the highest concentration tested (10 μM). Furthermore, the hydrazide 14 and the monofluoromethyl-1,3,4-oxadiazole 16 showed only weak HDAC6 inhibitory activity with 30% and 54% inhibition at 10 μM. These results suggest that the oxadiazole C=N bond is insufficiently activated for nucleophilic attack in methyl- and monofluoromethyl-oxadiazoles 15 and 16 to yield a zinc-bound nitranion comparable to deprotonated 13. The low inhibitory potency of 13 itself suggests that there is a higher energetic barrier for deprotonation to yield a zinc-bound nitranion compared with the hydrolysis of oxadiazole 6 which would directly yield a zinc-bound nitranion. The nitranion makes a strong charge-charge interaction with zinc, whereas the amino group of 14 would make a weaker dipole-charge interaction once deprotonated. This may account for the weaker inhibitory potency observed for 14.
Figure 4.
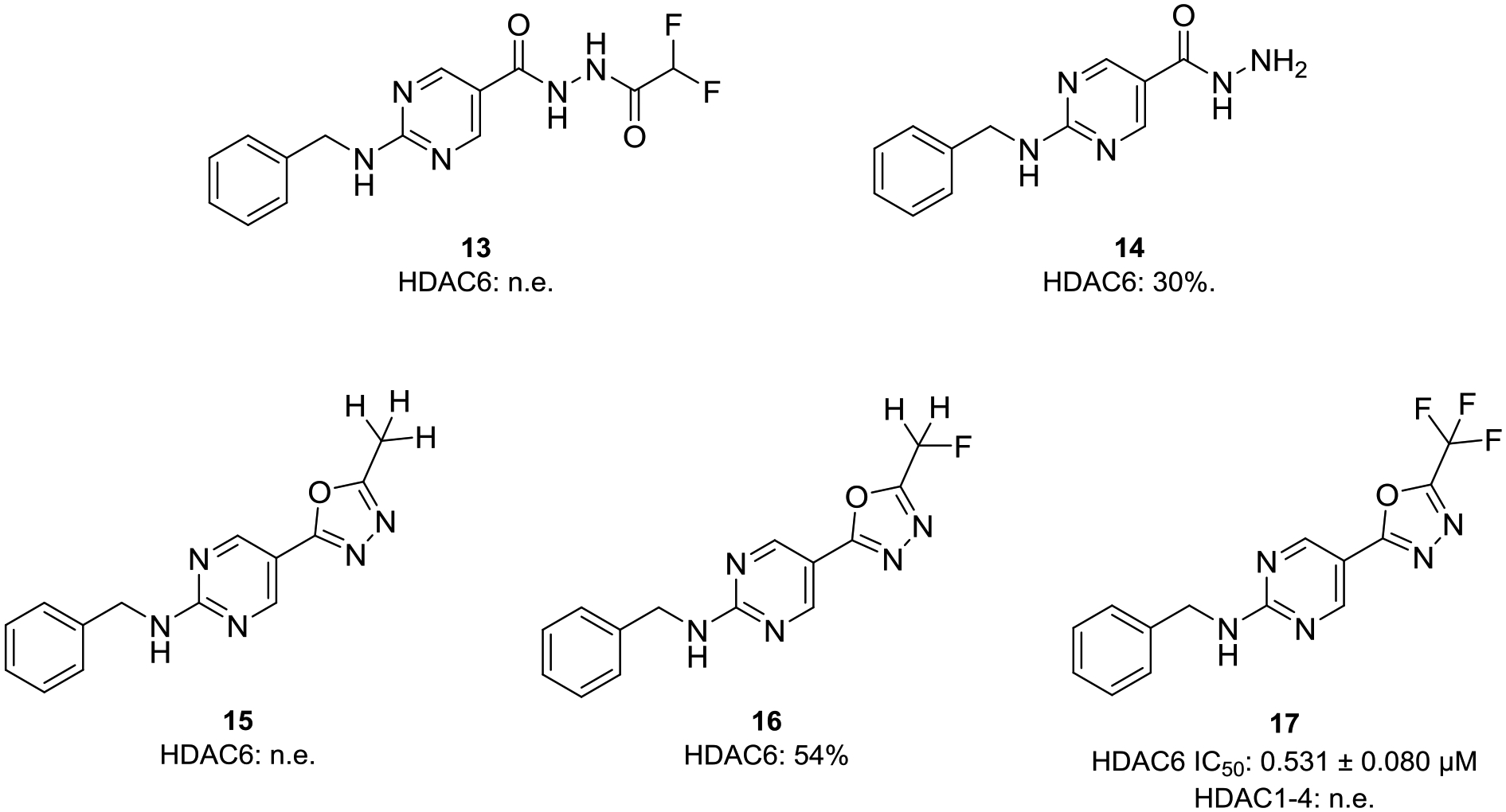
Structures of the acylhydrazide (13), hydrazide (14), methyl-1,3,4-oxadiazole (15), monofluoromethyl-1,3,4-oxadiazole (16) and trifluoromethyl-1,3,4-oxadiazole (17) analogs. Inhibitory activities of prepared compounds against HDAC1–4 and HDAC6; IC50 [μM, mean ± SD] or percent inhibition at 10 μM; n.e.: no effect = < 15% inhibition at 10 μM.
In contrast, the trifluoromethyl-1,3,4-oxadiazole (TFMO) 17 displayed submicromolar HDAC6 inhibitory activity against HDAC6 (IC50: 0.531 μM) and no inhibition of the four control isoforms HDAC1–4. Consequently, we focused on the difluoromethyl-1,3,4-oxadiazole 6 and the trifluoromethyl-1,3,4-oxadiazole 17 in our in-depth evaluation of the binding kinetics. Most hydroxamates are HDACi with fast-on and fast-off binding kinetics, while HDACi with alternative ZBGs such as aminoanilides and alkyl hydrazides are often slow- and tight-binding inhibitors.63–65 To investigate whether 6 and 17 display slow-on binding properties we performed HDAC6 inhibition assays with different preincubation times using vorinostat as control; the results are summarized in Figure 5A. As expected, the HDAC6 inhibition by vorinostat did not depend on the preincubation time. In contrast, the observed concentration-effect curves of 6 and thus the IC50 values were highly dependent on the preincubation time (5 min IC50: 0.347 μM; 1 h IC50: 0.193 μM; 2 h IC50: 0.129 μM), thereby indicating a slow-binding profile. Similarly, 17 also showed a substantial decrease in the HDAC6 IC50 values upon preincubation (5 min IC50: 0.840 μM; 1 h IC50: 0.531 μM; 2 h IC50: 0.601 μM).
Figure 5.
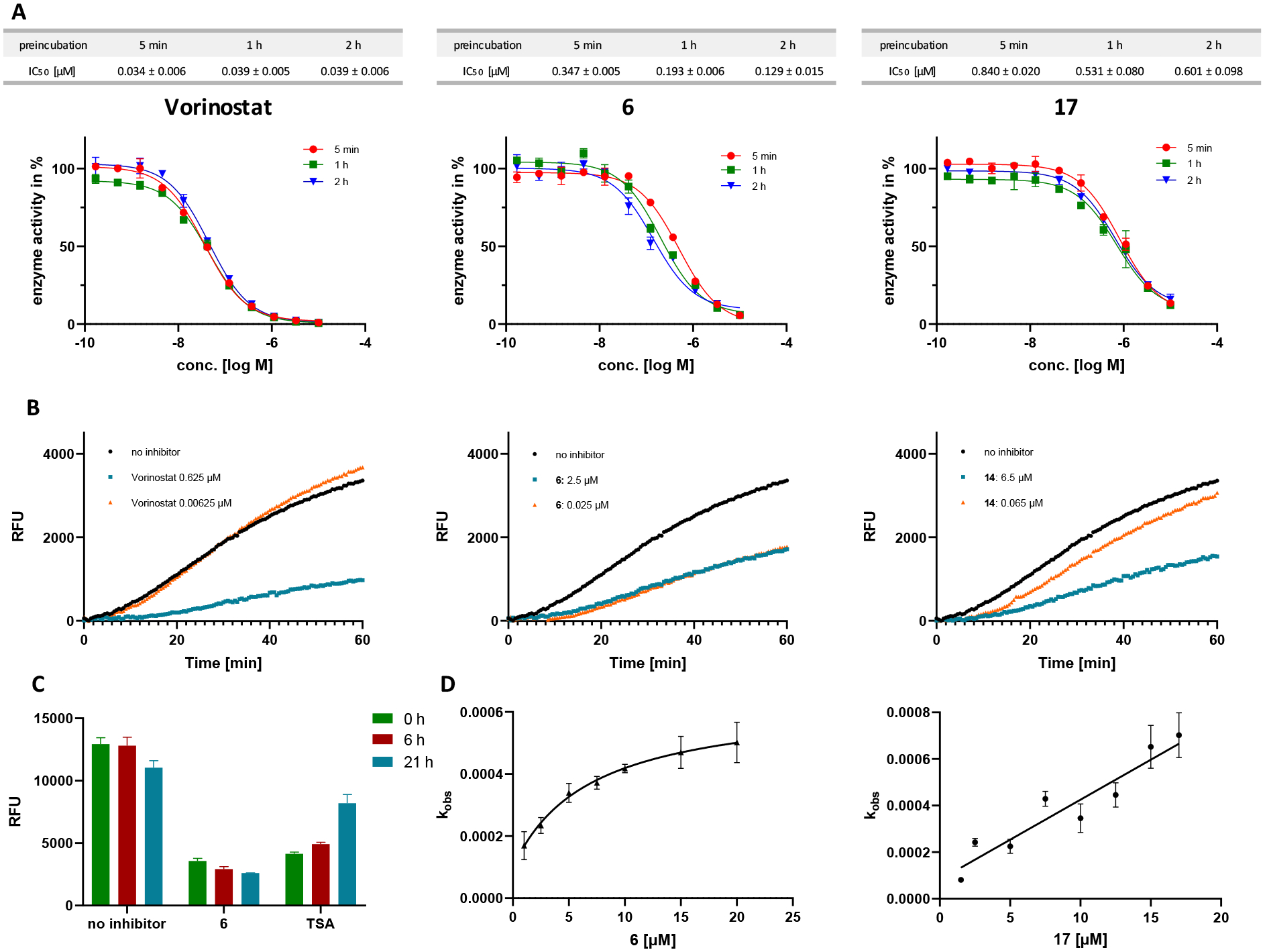
Analysis of the association and dissociation behavior of 6 and 17 at HDAC6. A: Representative dose-response curves and IC50 values of vorinostat (left, control), 6 (middle), and 17 (right) after preincubation with HDAC6 for 5, 60, and 120 min. B: Progression curves of 100-fold jump dilution experiments with vorinostat (left, control), 6 (middle), and 17 (right) at HDAC6. Inhibitor concentrations are indicated on the left. Fluorescence of cleaved AMC is measured in relative fluorescence units (RFU). C: Recovered HDAC6 activity from samples incubated with DMSO, 6, and trichostatin A (TSA, control) after dialysis against 10.000-fold excess fresh buffer. D: The apparent first-order rate constant kobs (mean ± SD) was plotted against the corresponding inhibitor concentrations [I]. The resulting curves were fitted into Equation 2 or 3 (see Supporting Information). (left) 6: the hyperbolic relationship between kobs and [I] indicates slow-binding, “induced-fit” mechanism II; (right) 17: the linear relationship between kobs and [I] indicates slow-binding mechanism I.63
To determine whether 6 and 17 are tight-binding inhibitors of HDAC6, we analyzed the dissociation behavior of both compounds by 100-fold jump dilution experiments using vorinostat as control. Briefly, HDAC6 in assay buffer was incubated with an excess of the respective inhibitor (at least 10-fold IC50) or with blank (DMSO 1%) for 1 hour at room temperature. Subsequently, this mixture was diluted 100-fold either with the respective inhibitor at the original concentration or assay buffer. The substrate (Z-Lys(Ac)-AMC (ZMAL)) and trypsin were added to all samples and the time-dependent in situ AMC release was monitored continuously following our previously reported protocol.63 In the case of the 100-fold jump dilution of vorinostat (Figure 5B, left), HDAC6 regained full deacetylase activity compared to blank (DMSO 1%) which is in excellent agreement with the fast-on/fast-off binding behavior of vorinostat. Conversely, the HDAC6 activity could not be restored after 100-fold dilution of 6 (Figure 5B, middle), hence indicating that 6 or the ring-opened deprotonated acylhydrazide 13 disengages very slowly from HDAC6. Dialysis experiments with 10,000-fold excess of buffer over 21 hours confirmed the tight-binding properties of 6 (Figure 5C), suggesting that the unique binding mode of 6 leads to essentially irreversible inhibition of HDAC6. In contrast, the deacetylase activity of HDAC6 was nearly completely restored after the 100-fold jump dilution of 17 (Figure 5B, right). These results indicate that the closely related analogs 6 (tight-binding, essentially irreversible inhibitor) and 17 (fast-off binding properties) differ in their dissociation behavior and might therefore act via different modes of action.
The trifluoromethyl analogue 17 similarly undergoes HDAC6-catalyzed ring opening based on LC-MS analysis of the product mixture resulting from incubation with HDAC6 (Figure S3 and S4, Supporting Information). However, in contrast with DFMO analogue 6, jump dilution experiments with 17 revealed that it binds to HDAC6 reversibly. The crystal structure of the HDAC6–13 complex shows that the difluoromethyl moiety binds in a small pocket defined in part by P571. We hypothesize that the trifluoromethyl group of 17 is sufficiently larger than the difluoromethyl group of 13 so as to destabilize binding of the trifluoromethyl group in the P571 pocket, which results in reversible rather than irreversible inhibition. Substitution of the CHF2 group of 13 with a CF3 group to generate a model of 17 bound in the HDAC6 active site suggests a steric clash as well as an unfavorable electrostatic interaction between the C–F group and the backbone carbonyl of G582 (Figure 6).
Figure 6.
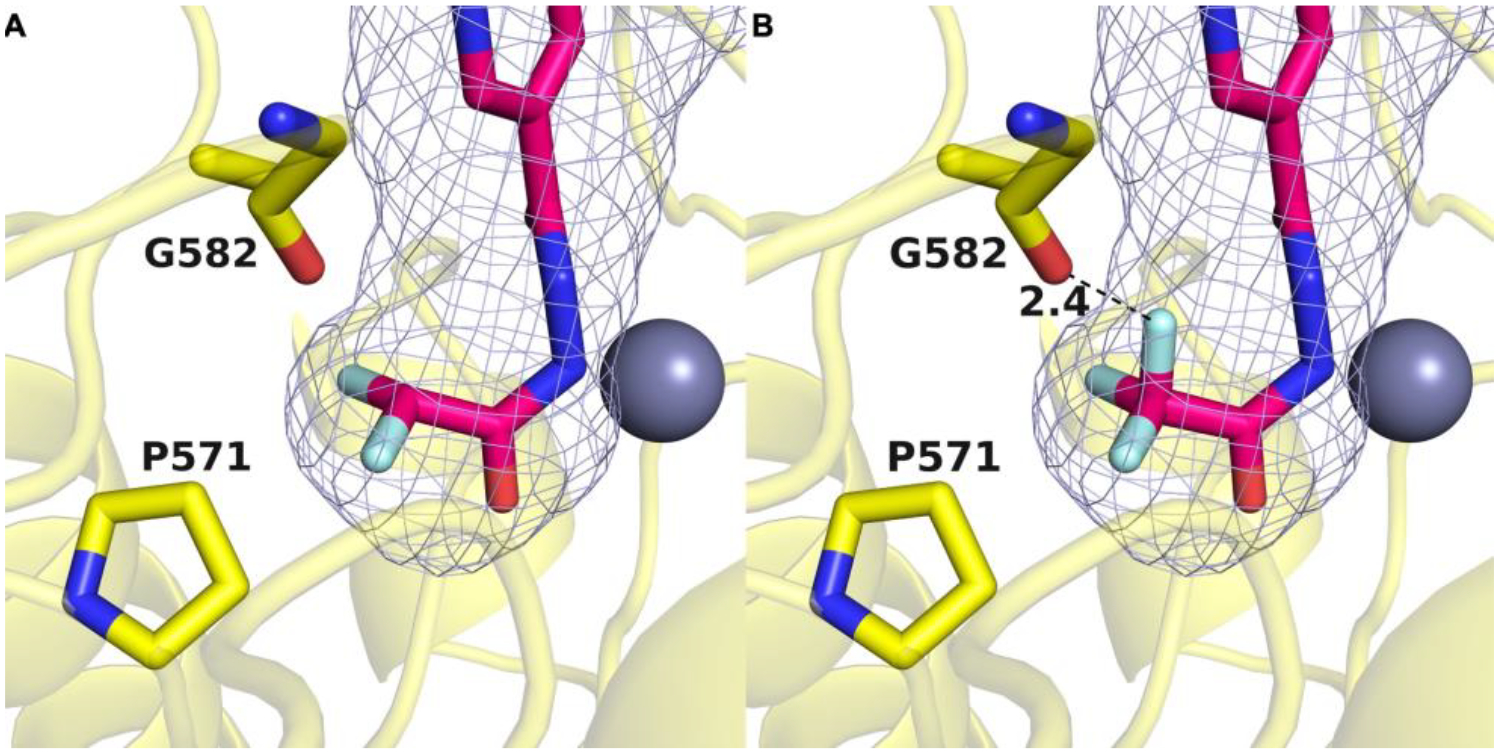
A: Binding pocket of HDAC6 in purple mesh generated by GetCleft showing the orientation of the difluoromethyl group determined in the crystal structure of the complex with 13 (PDB 8GD4). B: Substitution of the CHF2 group with a CF3 group yields a model of the complex with hydrolyzed 17 in the ring-opened form. The additional fluorine atom results in a clash with the backbone carbonyl of G582.
Quantitative LC-MS experiments were conducted with compounds 6 and 17 (Figure S3, Supporting Information) revealing that both compounds can undergo a second hydrolysis reaction, thereby resulting in the formation of the hydrazide 14. Notably, the concentration of the generated hydrazide differs by a factor of two. When HDAC6 was incubated with 100 μM of 6, 9 μM of 14 was produced, while 17 yielded 18 μM of 14. The second hydrolysis reaction from the respective acylhydrazide to hydrazide 14 requires the restoration of the Zn2+ coordination sphere by the entry of a second water molecule. The fast-off binding kinetics of 17 may facilitate the entry of a second water molecule, potentially explaining the increased concentration of 14 when HDAC6 is incubated with 17.
To confirm the hypothesis of different binding mechanisms for the difluoromethyl and the trifluoromethyl analogs, we performed HDAC6 kinetic studies. Our preincubation experiments demonstrated that 6 and 17 are slow-binding inhibitors of HDAC6. The most common types of slow-binding mechanisms are “simple slow-binding” (mechanism I) and “induced-fit” (mechanism II) (see Figure S1, Supporting information).63, 66 While mechanism I represents a single-step slow-binding mode of inhibition, mechanism II is characterized as a two-step slow-binding inhibition mode.63, 66 The slow-binding mechanisms I and II can be distinguished by their respective relationships between the rate constant for the onset of inhibition (kobs) and the inhibitor concentration.63, 66 To determine the binding mechanism of 6 and 17 at HDAC6, we utilized the Progression-Method63, 66 and measured a series of progression curves using fixed concentrations of enzyme, substrate, and different inhibitor concentrations. Subsequently, the generated data were fitted into the Equation 1 (see Supporting Information) to calculate the kobs values for the different inhibitor concentrations. The resulting kobs vs inhibitor concentration plots are depicted in Figure 5D. In the case of 6 we observed a hyperbolic relationship between kobs and the inhibitor concentration (Figure 5D, left), suggesting that 6 inhibits HDAC6 via the slow-binding “induced-fit” mechanism II. For 17 the relationship between inhibitor concentration and kobs was linear (Figure 5D, right), indicating binding via the single-step slow-binding mechanism I. As discussed above, the DFMO 6 is hydrolyzed by HDAC6 and afterwards essentially trapped in the active site in a small pocket defined in part by P571, while the trifluoromethyl analog 17 is hydrolyzed and dissociates fast out of the enzyme. These differences might explain the different slow-binding mechanisms observed for 6 and 17.
Proposed reaction mechanism.
DFMO 6 is an essentially irreversible inhibitor of HDAC6, even though deprotonated 13 does not form a covalent bond with any residues in the enzyme active site. We have determined that the generation of deprotonated acylhydrazide 13 requires the enzyme, since 6 does not undergo hydrolysis in the absence of enzyme to yield 13. This implicates the reactive zinc-bound water molecule as the nucleophile for oxadiazole hydrolysis. Moreover, a mass shift of 2 is observed by mass spectrometry for acylhydrazide 13 when HDAC6 is incubated with 6 in H218O instead of H216O. Subsequent hydrolysis of the 18O-labeled acylhydrazide yields hydrazide 14 without the 18O label as determined by mass spectrometry (see Figure S5, Supporting Information). These results indicate that the 18O label was contained in the second hydrolysis product, difluoroacetate, which further implies that the initial nucleophilic attack of zinc-bound water at oxadiazole 6 occurs exclusively at the C=N bond closest to the difluoromethyl moiety. This result supports our proposed reaction mechanism depicted in Figure 7, in which the described C=N bond undergoes nucleophilic attack by the zinc-bound water molecule to form a tetrahedral intermediate. Electron rearrangement results in the ring opening of the oxadiazole, directly forming the deprotonated acylhydrazide. The negatively charged nitrogen can strongly coordinate to the zinc ion, supporting the assumption for essential irreversible inhibition. Due to the crucial strong anionic zinc coordination, the proposed mechanism is in agreement with our results, that the synthetic acylhydrazide 13, bearing a protonated nitrogen, does not show any inhibitory activity. In other words, it is easier to generate the nitrogen anion of 13 through oxadiazole hydrolysis, captured and stabilized by Zn2+ coordination during the ring opening mechanism, rather than by direct deprotonation of synthetic 13. While this manuscript was under review, a similar mechanism for the generation of zinc-bound 13 was proposed on the basis of computational chemistry calculations.67
Figure 7.
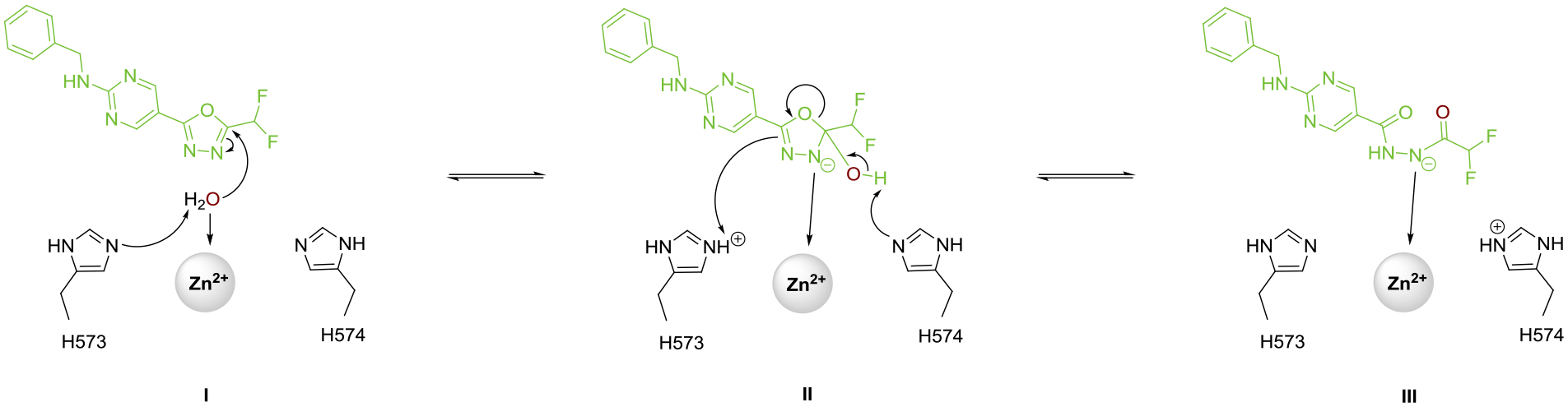
Proposed reaction mechanism of Zn2+ catalyzed ring opening reaction of DFMO compound 6.
The histidine dyad H573/H574 plays a crucial role as proton shuttle in the deacetylation of native acetyllysine peptide substrates of HDAC6 and, presumably, in the enzyme-catalyzed ring opening reaction of DFMOs/TFMOs.62 Furthermore, recent reports by Steinkühler and co-workers56 as well as Barinka and co-workers54 highlighted the importance of Y745 for the kinetics of the second hydrolysis reaction. Both groups observed a significant increased conversion rate into the respective hydrazide derivative when Y745 was mutated into phenylalanine. To gain further insights into the reaction mechanism, we prepared H573A, H574A, and Y745F mutants of HDAC6 and conducted quantitative LC-MS analysis after incubation of wild-type HDAC6 and the respective mutants with DFMO 6 and TFMO 17 (see Figure S6, Supporting Information). For the histidine mutants H573A and H574A, we observed a notable reduction in the formation of the acylhydrazides 13 and 20. In addition, both mutants were unable to hydrolyze 6 and 17 to the hydrazide 14. These results clearly confirm the critical role of the histidine dyad H573/H574 in the ring opening reaction of DFMOs and TFMOs as well as the second hydrolysis reaction to the corresponding hydrazide. When incubating the Y745F mutant of HDAC6 with 6 and 17, we observed an increased formation of the hydrazide 14 compared to wild-type. In fact, in the case of incubation with the TFMO 17, the hydrazide was detected as the main reaction product. Our crystal structure of the HDAC6–13 complex revealed that one carbonyl group and one C-F group of deprotonated 13 form hydrogen bonds with Y745 (Figure 2). Due to the absence of two important hydrogen bonds with Y745, deprotonated 13 may disengage from the catalytic center of the Y745F mutant, thereby facilitating a second hydrolysis reaction after the restoration of the Zn2+ coordination sphere by the entry of a second water molecule. Based on our crystallographic data (Figure 2) and LC-MS experiments with wild-type and Y745F HDAC6 (Figure S6, Supporting Information), we conclude that Y745 is critically important for the binding mode of deprotonated 13 and significantly contributes to the essentially irreversible binding of this active species.
CONCLUSION
In summary, we have characterized the DFMO derivative 6 as potent and selective HDAC6 inhibitor. In IC50 shift experiments with various preincubation times compound 6 showed slow-on binding properties with decreasing IC50 values. By analysis of the slow-binding characteristics we found that 6 inhibits HDAC6 via a two-step slow binding mechanism. To investigate the dissociation characteristics, we performed jump dilution experiments that revealed an essentially irreversible binding mode of DFMO 6 to its target. Additional dialysis experiments further confirmed the tight-binding properties of 6. The trifluoromethyl analog 17, on the other hand, acts as a slow-binding inhibitor following a single-step slow binding mechanism. In contrast to 6, compound 17 was observed to disengage from the enzyme with fast-off binding properties in jump dilution assays, thus confirming that the two derivatives act via different modes of inhibition.
Steinkühler et al. recently reported the structure of the HDAC6 complex with a hydrazide inhibitor resulting from hydrolysis of a related oxadiazole inhibitor.56 The authors postulated that the remarkable HDAC6 inhibition is not solely attributed to the crystallized hydrazide, but rather to a high-affinity intermediate that forms a stable and long-lasting enzyme-inhibitor complex.56 They suggested a closed hydrated intermediate and a protonated acylhydrazide as potential active species.56 Using our crystallographic and mechanistic data, we are now able to confirm that DFMOs serve as substrate analogs and therefore as mechanism-based inhibitors undergoing an HDAC6-catalyzed ring-opening reaction which is initiated by the attack of the zinc-bound water at the sp2 carbon closest to the difluoromethyl moiety. Ultimately, this leads to the formation of the deprotonated acylhydrazide 13 as the active species. The analysis of the resulting HDAC6-13 complex reveals an extensive array of intermolecular interactions that stabilize the bound inhibitor, particularly the strong anionic zinc coordination of 13 in combination with the binding of the difluoromethyl moiety in the P571 pocket. These structural features contribute to an exceptionally tight enzyme−inhibitor complex, thereby leading to an essentially irreversible inhibition of HDAC6.
EXPERIMENTAL SECTION
Chemistry.
Chemicals were obtained from abcr GmbH, Acros Organics, BLDpharm, Carbolution Chemicals, Carl Roth, Fluorochem, Sigma-Aldrich, TCI Chemicals or VWR and used without further purification. Technical grade solvents were distilled prior to use. For all HPLC purposes, acetonitrile in HPLC-grade quality (HiPerSolv CHROMANORM, VWR) was used. Water was purified with a PURELAB flex® (ELGA VEOLIA). Air-sensitive reactions were carried out under argon atmosphere utilizing standard Schlenk techniques. Thin-layer chromatography (TLC) was carried out on prefabricated plates (silica gel 60, F 254, Merck). Components were visualized either by irradiation with ultraviolet light (254 nm or 366 nm) or by staining appropriately. Column chromatography was carried out on silica gel (NORMASIL 60®, 40–63 μm, VWR or MACHEREY-NAGEL silica gel 60 ®, 40–63 μm). Mixtures of two or more solvents are specified as (“solvent A”/”solvent B”, 67/33, v/v) meaning that 100 mL of the respective mixture consists of 67 mL of “solvent A” and 33 mL of “solvent B”. Preparative silica gel flash column chromatography was performed on an Interchim puriFlash XS520Plus with diode-array detection (DAD) from 200 to 400 nm. The uncorrected melting points were determined using a Büchi Melting Point M-565 apparatus or Barnstead Electrothermal 9100 apparatus. Nuclear Magnetic Resonance Spectroscopy (NMR): Proton (1H), carbon (13C) and fluorine (19F) NMR spectra were recorded either on Bruker Avance DRX 500 (500 MHz 1H NMR, 126 MHz 13C NMR), a Bruker Avance III 600 (600 MHz 1H NMR, 151 MHz 13C NMR), a Bruker Avance III HD 400 (400 MHz 1H NMR, 101 MHz 13C NMR, 377 MHz 19F NMR), a Varian/Agilent Mecury-plus-400 (400 MHz 1H NMR, 101 MHz 13C NMR, 376 MHz 19F NMR) or a Varian/Agilent Mecury-plus-300 (300 MHz 1H, 75 MHz 13C NMR, 282 MHz NMR 19F NMR). The chemical shifts are given in parts per million (ppm). Deuterated chloroform (CDCl3) and deuterated dimethyl sulfoxide (DMSO-d6) were used as solvents. The residual solvent signal (CDCl3: 1H NMR: 7.26 ppm, 13C NMR: 77.1 ppm; DMSO-d6: 1H NMR: 2.50 ppm, 13C NMR: 39.52 ppm) was used for calibration. The multiplicity of each signal is reported as singlet (s), doublet (d), triplet (t), quartet (q), pentet (p), multiplet (m) or combinations thereof. Multiplicities and coupling constants are reported as measured and might disagree with the expected values. 19F NMR spectra were recorded proton-decoupled if not stated otherwise. Mass Spectrometry: High resolution electrospray ionisation mass spectra (HRMS-ESI) were acquired either with a Bruker Daltonik GmbH micrOTOF coupled to a LC Packings Ultimate HPLC system and controlled by micrOTOFControl3.4 and HyStar 3.2-LC/MS, a Bruker Daltonik GmbH ESI-qTOF Impact II coupled to a Dionex UltiMateTM 3000 UHPLC system and controlled by micrOTOFControl 4.0 und HyStar 3.2-LC/MS, a Bruker micrOTOF-Q mass spectrometer coupled with a HPLC Dionex UltiMate 3000 or a LTQ Orbitrap XL. Low resolution electrospray ionisation mass spectra (LRMS ESI) were acquired either with an Advion expression® compact mass spectrometer (CMS) coupled with an automated TLC plate reader Plate Express® (Advion), an API 2000 mass spectrometer coupled with an Agilent HPLC HP 1100 using an EC50/2 Nucleodur C18 Gravity 3 μm column or on an Agilent Infinity Lab LC/MSD-system coupled with an Agilent HPLC 1260 Infinity II using an EC50/2 Nucleodur C18 Gravity 3 μm column. High Performance Liquid Chromatography (HPLC): For analytical purposes HPLC measurements were performed on a Thermo Fisher Scientific UltiMateTM 3000 UHPLC system with a Nucleodur 100–5 C18 (250 × 4.6 mm, Macherey Nagel), using a flow rate of 1 mL/min and a temperature of 25 °C with an appropriate gradient. Detection was implemented by UV absorption measurement at a wavelength of λ = 220 nm and λ = 250 nm. Bidest. H2O (A) and MeCN (B) were used as eluents with an addition of 0.1% TFA for eluent A. For preparative purposes a AZURA Prep. 500/1000 gradient system with a Nucleodur 110–5 C18 HTec (150 × 32 mm, Macherey Nagel) column was used with a flow rate of 20 mL/min and an appropriate gradient. Detection was implemented by UV absorption measurement at a wavelength of λ = 220 nm and λ = 250 nm. Bidest. H2O (A) and MeCN (B) were used as eluents with an addition of 0.1% TFA for eluent A. Purity: The purity of all final compounds was 95% or higher. Purity was determined via HPLC at 250 nm using the protocols described above, if not stated otherwise.
General procedure A.
The respective carbonitrile (1.0 eq), NaN3 (2.0 eq), NH4Cl (2.0 eq), and LiCl (0.8 eq) were suspended in DMF (2 mL, 1 M) and the resulting mixture was subjected to microwave irradiation at 150 W and 100 °C under vigorous stirring for 24 h. The reaction mixture was then filtered over a 5 cm layer of silica (CH2Cl2/MeOH, 9/1, v/v). Upon removal of the solvent under reduced pressure, the crude tetrazole intermediate was dissolved in CH2Cl2 (30 mL) and difluoroacetic anhydride (DFAA, 6.1 eq) was added dropwise at 0 °C. The resulting solution was allowed to warm to rt and stirred for 24 h. After completion of the reaction, the mixture was diluted with CH2Cl2 (30 mL) and washed with 1 M NaOH (2 × 10 mL), water (1 × 10 mL), and brine (1 × 10 mL). Drying of the organic layer over Na2SO4 and subsequent evaporation of the solvent afforded the desired product.
General procedure B.
The respective methyl ester (1.0 eq) was dissolved in MeOH (0.2 M). Hydrazine monohydrate (10.0 eq.) was added and the reaction mixture was stirred at 70 °C for 3 h. After completion of the reaction, the solvent was removed under reduced pressure. The crude product was used directly in the next step. The resulting hydrazide (1.0 eq) was dissolved in DMF (0.1 M). DFAA (1.3 eq) and Et3N (2.0 eq) were added and the reaction mixture was stirred at 70 °C for 1 h. The mixture was concentrated under reduced pressure and used without further purification in the next reaction. The resulting difluoromethylacylhydrazide (1.0 eq) was taken up in dry THF (0.1 M). Et3N (3.0 eq) and Burgess reagent (3.0 eq) were added, and the reaction mixture was stirred at 60 °C for 18 h. The solvent was removed under reduced pressure and the crude was purified by preparative HPLC.
General procedure C.
The respective carbonitrile (1.0 eq) was dissolved in DMF (0.25 M). NaN3 (2.0 eq), NH4Cl (1.3 eq), and LiCl (0.5 eq) were added and the reaction mixture was stirred at 100 °C for 18 h. After completion of the reaction, the mixture was quenched with ice water (8 mL) and acidified with 1 M HCl to pH = 2. The precipitated solid was filtered and washed with cold water. The tetrazole product was used without further purification in the next step. The tetrazole derivative (1.0 eq) was dissolved in toluene (0.05 M). DFAA (3.0 eq) was added and the solution was stirred at 70 °C for 18 h. The reaction mixture was concentrated under reduced pressure and purified by preparative HPLC.
2‐(Difluoromethyl)‐5‐phenyl‐1,3,4‐oxadiazole (1).
Benzonitrile (206 mg, 2.00 mmol, 1.0 eq), NaN3 (260 mg, 4.00 mmol, 2.0 eq), NH4Cl (213 mg, 4.00 mmol, 2.0 eq), and LiCl (66 mg, 1.56 mmol, 0.8 eq) were suspended in DMF (2 mL) and the resulting mixture was subjected to microwave irradiation at 150 W and 100 °C under vigorous stirring for 24 h. After evaporation of the solvent, the mixture was dissolved in 1 M NaOH (20 mL) and washed with EtOAc (2 × 10 mL) to remove excess benzonitrile. The aqueous layer was acidified using 10% HCl (pH 4) and extracted with CH2Cl2 (3 × 50 mL). The combined organic layers were dried over MgSO4 and concentrated under reduced pressure. The intermediate 5-phenyl-1H-tetrazole was obtained as a white solid (216 mg, 1.47 mmol) and used without analytical characterization for the next reaction step. A solution of 5-phenyl-1H-tetrazole (180 mg, 1.22 mmol, 1.0 eq) in CH2Cl2 (18 mL) was cooled to 0 °C and DFAA (0.75 mL, 6.10 mmol, 5.0 eq) was added dropwise. The resulting solution was allowed to warm to rt and stirred for 24 h before water (20 mL) was added. The mixture was extracted with CH2Cl2 (3 × 30 mL) and the combined organic layers were washed with brine (10 mL) and dried over MgSO4. Removal of the solvent under reduced pressure and recrystallisation from EtOAc (2 mL) and petrol ether (20 mL) afforded the desired product 1 as a white solid (193 mg, 0.98 mmol). Yield: 81%; mp. 101–105 °C; 1H NMR (300 MHz, CDCl3): δ 8.19–8.07 (m, 2H), 7.67–7.46 (m, 3H), 6.92 (t, J (H, F) = 51.7 Hz, 1H); 13C NMR (101 MHz, CDCl3): δ 166.3, 158.3 (t, 2J (C, F) = 28.8 Hz), 132.9, 129.4, 127.6, 122.8, 106.0 (t, 1J (C, F) = 240.9 Hz; 19F NMR (377 MHz, CDCl3) δ −127.0, −127.1; HRMS (ESI) m/z [M+H]+ calcd for C9H7F2N2O+ 197.0521, found 197.0516; HPLC (95% H2O 1 min, then to 95% MeCN in 7 min, then 100% MeCN to 17 min, 254 nm), tR = 8.86 min, 95% purity.
2-(Difluoromethyl)-5-(6-methylpyridin-3-yl)-1,3,4-oxadiazole (2).
The product was synthesized according to the general procedure B, using methyl 6-methylnicotinate (453 mg, 3.00 mmol, 1.0 eq) as starting material. The reaction mixture was purified by silica gel column chromatography (DCM/MeOH, 97/3, v/v) to afford 2 as yellow oil (146 mg, 0.70 mmol). Yield: 23%; Rf = 0.58 (DCM/MeOH, 95/5, v/v) ; 1H NMR (600 MHz, DMSO-d6) δ 9.08 (d, J = 2.3 Hz, 1H), 8.31 (dd, J = 8.1, 2.3 Hz, 1H), 7.56 (t, J (H, F) = 51.4 Hz, 1H), 7.53 (d, J = 8.2 Hz, 1H), 2.59 (s, 3H); 13C NMR (151 MHz, DMSO-d6) δ 164.0, 162.8, 158.6 (t, 2J (C, F) = 29.2 Hz), 147.3, 135.0, 123.9, 116.6, 106.7 (t, 1J (C, F) = 238.8 Hz), 24.4; 19F NMR (565 MHz, DMSO-d6) δ −121.5, −121.6; HRMS (ESI) m/z [M+H]+ calcd for C9H8F2N3O+ 212.0630, found 212.0620; HPLC (95% H2O 5 min, then to 95% MeCN in 5 min, then 100% MeCN to 20 min, 254 nm), tR = 10.98 min, 95% purity.
2-(Difluoromethyl)-5-(pyrimidin-5-yl)-1,3,4-oxadiazole (3).
Synthesis according to the general procedure A starting from 5-cyanopyrimidine (208 mg, 2.00 mmol, 1.0 eq) afforded 3 as a yellow solid (80 mg, 0.41 mmol). Yield: 20%; mp. 104–107 °C; 1H NMR (400 MHz, CDCl3): δ 9.49–9.40 (m, 3H), 6.97 (t, J (H, F) = 51.6 Hz, 1H); 13C NMR (75 MHz, CDCl3): δ 162.1, 161.5, 159.2 (t, 2J (C, F) = 29.3 Hz), 155.4, 118.4, 105.7 (t, 1J (C, F) = 241.9 Hz); 19F NMR (377 MHz, CDCl3) δ −119.2, −119.3. HRMS (ESI) m/z [M+H]+ calcd for C7H5F2N4O+ 199.0426, found 199.0425; HPLC (95% H2O 1 min, then to 95% MeCN in 7 min, then 100% MeCN to 17 min, 254 nm), tR = 6.59 min, 97% purity.
2-(Benzylamino)pyrimidine-5-carbonitrile (4).
Benzylamine (0.328 mL, 3.00 mmol, 1.0 eq) and 2-chlorpyrimidine-5-carbonitrile (837 mg, 6.00 mmol, 2.0 eq) were dissolved in EtOH (4 mL, 0.3 M). DIPEA (1.56 mL, 6.00 mmol, 3.0 eq) was added and the reaction mixture was stirred at 90 °C for 18 h. After completion of the reaction, the solvent was removed under reduced pressure. EtOAc was added and the organic phase was washed with brine, dried over Na2SO4, filtered and evaporated. The crude was purified by silica gel column using cyclohexane/EtOAc gradient (16% to 25% EtOAc) to obtain the desired product as a yellow solid (598 mg, 2.85 mmol). Yield: 95%; mp. 170–172 °C; Rf = 0.5 (cyclohexane/EtOAc, 3/1, v/v); 1H NMR (600 MHz, DMSO-d6) δ 8.80 (t, J = 6.4 Hz, 1H), 8.71 – 8.65 (m, 2H), 7.33 – 7.29 (m, 1H), 7.31 – 7.25 (m, 3H), 7.25 – 7.19 (m, 1H), 4.55 (d, J = 6.4 Hz, 2H); 13C NMR (151 MHz, DMSO-d6) δ 161.9, 161.6, 139.1, 128.5, 127.2, 127.0, 117.2, 95.6, 44.1; LRMS (ESI) m/z [M+H]+ calcd for C12H11N4+ 211.1, found 210.9.
2-[(4-Methoxybenzyl)amino]pyrimidine-5-carbonitrile (5).
4-Methoxybenzylamine (0.235 mL, 1.80 mmol, 1.0 eq) and 2-chlorpyrimidine-5-carbonitrile (502 mg, 3.60 mmol, 2.0 eq) were dissolved in EtOH (6 mL, 0.3 M). DIPEA (0.943 mL, 5.40 mmol, 3.0 eq) was added and the reaction mixture was stirred at 90 °C for 18 h. After completion of the reaction, the solvent was removed under reduced pressure. EtOAc was added and the organic phase was washed with brine, dried over Na2SO4, filtered and evaporated. The crude was purified by silica gel column using cyclohexane/EtOAc gradient (16% to 25% EtOAc) to obtain the desired product as a yellow solid (455 mg, 1.70 mmol). Yield: 94%; mp. 177–178 °C; Rf= 0.18 (cyclohexane/EtOAc, 3/1, v/v); 1H NMR (600 MHz, CDCl3) δ 8.53 (d, J = 2.9 Hz, 1H), 8.18 – 8.14 (m, 1H), 7.26 – 7.21 (m, 2H), 6.90 – 6.85 (m, 2H), 6.42 (s, 1H), 4.56 (d, J = 5.6 Hz, 2H), 3.79 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 161.6, 161.4, 160.6, 159.4, 129.2, 129.2, 116.3, 114.2, 97.0, 55.3, 45.2; LRMS (ESI) m/z [M+H]+ calcd for C13H13N4O+ 241.1, found 240.9.
N-Benzyl-5-[5-(difluoromethyl)-1,3,4-oxadiazol-2-yl]pyrimidin-2-amine (6).
The product was synthesized according to general procedure C, using 4 (55 mg, 0.26 mmol, 1.0 eq) as starting material. The crude was purified by silica gel column chromatography (cyclohexane/ EtOAc, 4/1, v/v) to afford 6 as a white solid (31 mg, 0.10 mmol). Yield: 38%; mp. 174–176 °C; Rf = 0.28 (cyclohexane/ EtOAc, 3/1, v/v); 1H NMR (600 MHz, DMSO-d6) δ 8.87 (s, 2H), 8.71 (t, J = 6.4 Hz, 1H), 7.51 (t, J (H, F) = 51.4 Hz, 1H), 7.34 – 7.28 (m, 4H), 7.26 – 7.19 (m, 1H), 4.60 (d, J = 6.4 Hz, 2H); 13C NMR (151 MHz, DMSO-d6) δ 163.3, 163.1, 157.7 (t, 2J (C, F) = 29.2 Hz), 157.3, 157.2, 139.3, 128.5, 127.2, 127.0, 106.7, 106.5 (t, 1J (C, F) = 237.4 Hz), 44.2; 19F NMR (565 MHz, DMSO-d6) δ −121.1, −121.2; HRMS (ESI) m/z [M+H]+ calcd for C14H12F2N5O+ 304.1004, found 304.0995; HPLC (95% H2O 5 min, then to 95% MeCN in 5 min, then 100% MeCN to 20 min, 254 nm), tR = 12.71 min, 99% purity.
5-[5-(Difluoromethyl)-1,3,4-oxadiazol-2-yl]-N-(4-methoxybenzyl)pyrimidin-2-amine (7).
The product was synthesized according to general procedure C, using 5 (336 mg, 1.40 mmol, 1.0 eq) as starting material. Purification by RP flash column chromatography using a water/ACN gradient (5% to 95% ACN) yielding 7 as a white solid (225 mg, 0.70 mmol). Yield: 50%; mp. 165–166 °C; 1H NMR (600 MHz, DMSO-d6) δ 8.89 – 8.83 (m, 2H), 8.65 (t, J = 6.4 Hz, 1H), 7.51 (t, J (H, F) = 51.4 Hz, 1H), 7.27 – 7.22 (m, 2H), 6.89 – 6.84 (m, 2H), 4.52 (d, J = 6.3 Hz, 2H), 3.71 (s, 3H); 13C NMR (151 MHz, DMSO-d6) δ 163.3, 163.0, 157.7 (t, 2J (C, F) = 29.2 Hz), 157.3, 157.2, 131.2, 128.7, 113.9, 107.5 (t, 1J (C, F) = 238.4 Hz), 106.4, 55.2, 43.7; 19F NMR (565 MHz, DMSO-d6) δ −121.1, −121.2; HRMS (ESI) m/z [M+H]+ calcd for C15H14F2N5O2+ 334.1110, found 334.1113; HPLC (95% H2O 5 min, then to 95% MeCN in 5 min, then 100% MeCN to 20 min, 254 nm), tR = 12.66 min, 98% purity.
Methyl 4-[(butylamino)methyl]benzoate (8).
Methyl 4-(bromomethyl)benzoate (263 mg, 1.15 mmol, 1.0 eq) and butylamine (0.568 mL, 5.75 mmol, 5.0 eq) were dissolved in THF (3 mL, 0.4 M) and the reaction was stirred at room temperature for 1 h. After completion of the reaction, the mixture was diluted with NaHCO3 (30 mL) and extracted with DCM (3 × 30 mL), dried over Na2SO4, filtered and the solvent was removed under reduced pressure. The crude product was purified by silica gel column chromatography (DCM/MeOH, 95/5, v/v) to afford 8 as a colourless oil (221 mg, 1.00 mmol). Yield: 87%; Rf = 0.28 (DCM/MeOH, 95/5, v/v); 1H NMR (600 MHz, DMSO-d6) δ 7.92 – 7.87 (m, 2H), 7.48 – 7.44 (m, 2H), 3.83 (s, 3H), 3.75 (s, 2H), 2.47 (t, J = 7.3 Hz, 2H), 1.40 (dq, J = 8.6, 7.0 Hz, 2H), 1.34 – 1.24 (m, 2H), 0.84 (t, J = 7.3 Hz, 3H); NH-proton was not detectable; 13C NMR (151 MHz, DMSO-d6) δ 166.4, 146.9, 129.2, 128.2, 128.1, 52.7, 52.1, 48.5, 31.7, 20.1, 14.0; LRMS (ESI) m/z [M+H]+ calcd for C13H20NO2+ 222.1, found 222.1.
1-Butyl-1-{4-[5-(difluoromethyl)-1,3,4-oxadiazol-2-yl]benzyl}−3-phenylurea (9).
The product was synthesized according to general procedure B, using 19 (340 mg, 1.00 mmol, 1.0 eq) as starting material, affording 9 as a white lyophilized solid (29 mg, 0.071 mmol). Yield: 9%; 1H NMR (600 MHz, DMSO-d6) δ 8.38 (s, 1H), 8.06 – 8.02 (m, 2H), 7.53 (t, J (H, F) = 51.4 Hz, 1H), 7.51 (d, J = 8.1 Hz, 2H), 7.45 (dd, J = 8.2, 6.9 Hz, 2H), 7.25 – 7.19 (m, 2H), 6.96 – 6.90 (m, 1H), 4.68 (s, 2H), 3.36 – 3.32 (m, 2H), 1.49 (p, J = 7.5 Hz, 2H), 1.26 (h, J = 7.4 Hz, 2H), 0.86 (t, J = 7.4 Hz, 3H); 13C NMR (151 MHz, DMSO-d6) δ 165.4, 158.4 (t, 2J (C, F) = 29.5 Hz), 155.4, 144.7, 140.6, 128.4, 128.3, 127.4, 122.0, 121.1, 120.3, 106.8 (t, 1J (C, F) = 238.3 Hz), 49.4, 46.5, 30.2, 19.6, 13.9; 19F NMR (565 MHz, DMSO-d6) δ −121.4, −121.5; HRMS (ESI) m/z [M+H]+ calcd for C21H23F2N4O2+ 401.1784, found 401.1791; HPLC (95% H2O 5 min, then to 95% MeCN in 5 min, then 100% MeCN to 20 min, 254 nm), tR = 17.05 min, 96% purity.
N-Butyl-N-{4-[5-(difluoromethyl)-1,3,4-oxadiazol-2-yl]benzyl}benzamide (10).
8 (211 mg, 1.00 mmol, 1.0 eq) was dissolved in DCM (2.5 mL, final concentration 0.2 M). Benzoyl chloride (280 mg, 2 mmol, 2 eq) was separately dissolved in DCM (2.5 mL) and added dropwise over 10 mins to the reaction. The mixture was stirred at room temperature for 2 h. After completion, the reaction was quenched by the addition of water (10 mL) and extracted with DCM (3 × 10 mL), dried over Na2SO4, filtered and the solvent was removed under reduced pressure. The crude intermediate methyl 4-((N-butylbenzamido)methyl)benzoate was purified by silica gel column chromatography (cyclohexane/EtOAc, 3/1, v/v) to afford a yellow oil (187 mg, 0.57 mmol). This intermediate was used without analytical characterization for the next reaction step. 10 was synthesized in the next steps according to the general procedure B using methyl 4-((N-butylbenzamido)methyl)benzoate (187 mg, 0.57 mmol, 1.0 eq) as starting material, affording 10 as a white lyophilized solid (32 mg, 0.084 mmol). Yield: 15%; 1H NMR (600 MHz, CDCl3, mixture of 2 rotamers in 0.6:0.4 ratio) δ 8.11 (d, J = 7.8 Hz, 2H), 7.54 (d, J = 7.8 Hz, 1H), 7.42 (d, J = 19.2 Hz, 6H), 6.92 (t, J (H, F) = 51.7 Hz, 1H), 4.93 – 4.79 (m, 1.2H), 4.68 – 4.54 (m, 0.8H), 3.55 – 3.43 (m, 0.8H), 3.28 – 3.13 (m, 1.2H), 1.75 – 1.58 (m, 0.8H), 1.55 – 1.44 (m, 1.2H), 1.42 – 1.31 (m, 0.8H), 1.16 – 1.06 (m, 1.2H), 1.01 – 0.90 (m, 1.2H), 0.79 – 0.67 (m, 1.8H); 13C NMR (151 MHz, CDCl3, mixture of 2 rotamers) δ 172.5, 166.0, 158.3, 158.2 (t, 2J (C, F) = 29.1 Hz), 142.9, 142.6, 136.0, 129.7, 128.7, 128.6, 127.9, 127.6, 126.5, 121.7, 115.7, 113.8, 105.8 (t, 1J (C, F) = 240.9 Hz), 52.4, 48.7, 47.6, 45.0, 30.4, 29.1, 20.2, 19.6, 13.8, 13.5; 19F NMR (565 MHz, CDCl3) δ −120.0, −120.1; HRMS (ESI) m/z [M+H]+ calcd for C21H22F2N3O2+ 386.1675, found 386.1674; HPLC (95% H2O 5 min, then to 95% MeCN in 5 min, then 100% MeCN to 20 min, 254 nm), tR = 13.40 min, 98% purity.
Methyl 4-({N-[2-(benzylamino)-2-oxoethyl]-4-(dimethylamino)benzamido}methyl)benzoate (11).
Compound 11 was synthesized as described.59
N-[2-(Benzylamino)-2-oxoethyl]-N-{4-[5-(difluoromethyl)-1,3,4-oxadiazol-2-yl]benzyl}−4-(dimethylamino)benzamide (12).
The product was synthesized according to general procedure B, using 11 (135 mg, 0.30 mmol, 1.0 eq) as starting material, affording 12 as a white lyophilized solid (13.7 mg, 0.026 mmol). Yield: 9%; 1H NMR (600 MHz, DMSO-d6) δ 8.44 (t, J = 5.9 Hz, 1H), 8.04 (d, J = 8.0 Hz, 2H), 7.59 (t, J (H, F) = 51.7 Hz, 1H), 7.55 (d, J = 7.8 Hz, 2H), 7.35 – 7.29 (m, 4H), 7.24 (t, J = 7.3 Hz, 3H), 6.65 (d, J = 8.5 Hz, 2H), 4.73 (s, 2H), 4.30 (d, J = 5.9 Hz, 2H), 3.94 (s, 2H), 2.93 (s, 6H); 13C NMR (151 MHz, DMSO-d6) δ 172.0, 168.3, 165.4, 158.4 (t, 2J (C, F) = 29.5 Hz), 151.4, 143.1, 139.4, 128.8, 128.4, 127.4, 127.0, 121.3, 111.2, 106.8 (t, 1J (C, F) = 238.3 Hz), 42.3; 19F NMR (565 MHz, DMSO-d6) δ −121.4, −121.5; HRMS (ESI) m/z [M+H]+ calcd for C28H28F2N5O3+ 520.2155, found 520.2075; HPLC (95% H2O 5 min, then to 95% MeCN in 5 min, then 100% MeCN to 20 min, 254 nm), tR = 15.29 min, 99% purity.
2-(Benzylamino)-N’-(2,2-difluoroacetyl)pyrimidine-5-carbohydrazide (13).
18 (371 mg, 1.50 mmol, 1.0 eq) was dissolved in MeOH (10 mL, 0.2 M). Hydrazine monohydrate (0.75 mL, 10.00 mmol, 10.0 eq) was added and the reaction mixture was stirred at 70 °C for 3 h. After completion of the reaction, the solvent was removed under reduced pressure. The crude was used directly in the next step. The resulting hydrazide (1.0 eq) was dissolved in DMF (15 mL, 0.1 M). DFAA (0.205 mL, 1.65 mmol, 1.1 eq) was added and it was stirred at 70 °C for 1 h. The solvent was removed under reduced pressure and the crude was purified by preparative HPLC to afford 13 as a white lyophilized solid (82 mg, 0.26 mmol). Yield: 17%; 1H NMR (600 MHz, DMSO-d6) δ 10.90 (s, 1H), 10.45 (s, 1H), 8.73 (s, 2H), 8.45 (t, J = 6.4 Hz, 1H), 7.31 (d, J = 4.8 Hz, 4H), 7.26 – 7.20 (m, 1H), 6.42 (t, J (H, F) = 53.0 Hz, 1H), 4.58 (d, J = 6.4 Hz, 2H); 13C NMR (151 MHz, DMSO-d6) δ 163.3, 163.1, 161.7 (t, 2J (C, F) = 25.8 Hz), 158.4, 158.0, 139.7, 128.4, 127.2, 126.9, 114.6, 108.3 (t, 1J (C, F) = 246.6 Hz), 44.2; 19F NMR (565 MHz, DMSO-d6) δ −127.1, −127.2; HRMS (ESI) m/z [M+H]+ calcd for C14H14F2N5O2+ 322.1110, found 322.1110; HPLC (95% H2O 5 min, then to 95% MeCN in 5 min, then 100% MeCN to 20 min, 254 nm), tR = 11.28 min, 98% purity.
2-(Benzylamino)pyrimidine-5-carbohydrazide (14).
18 (243 mg, 1.00 mmol, 1.0 eq) was dissolved in MeOH (5 mL, 0.2 M). Hydrazine monohydrate (1.00 mL, 10.00 mmol, 10.0 eq) was added and the reaction mixture was stirred at 70 °C for 3 h. After completion of the reaction, the solvent was removed under reduced pressure. The solvent was removed under reduced pressure and the crude was purified by preparative HPLC to afford 14 as a yellow lyophilized solid (203 mg, 0.83 mmol). Yield: 83%; 1H NMR (600 MHz, DMSO-d6) δ 9.53 (s, 1H), 8.65 (s, 2H), 8.24 (t, J = 6.4 Hz, 1H), 7.31 – 7.27 (m, 4H), 7.24 – 7.18 (m, 1H), 4.54 (d, J = 6.4 Hz, 2H), 4.38 (s, 2H).; 13C NMR (151 MHz, DMSO-d6) δ 163.9, 163.0, 157.8, 157.3, 139.9, 128.4, 127.2, 126.8, 115.8, 44.1; HRMS (ESI) m/z [M+H]+ calcd for C12H14N5O+ 244.1193, found 244.1186; HPLC (95% H2O 5 min, then to 95% MeCN in 5 min, then 100% MeCN to 20 min, 254 nm), tR = 10.56 min, 96% purity.
N-Benzyl-5-(5-methyl-1,3,4-oxadiazol-2-yl)pyrimidin-2-amine (15).
4 (105 mg, 0.50 mmol, 1.0 eq) was dissolved in DMF (0.25 M). NaN3 (65 mg, 1.00 mmol, 2.0 eq), NH4Cl (35 mg, 0.65 mmol, 1.3 eq) and LiCl (11 mg, 0.25 mmol, 0.5 eq) were added and the reaction mixture was stirred at 100 °C for 18 h. After completion of the reaction, the mixture was quenched with ice water (4 mL) and acidified with 1 M HCl. The precipitated solid was filtered and washed with cold water. The tetrazole product was used without further purification in the next step. The tetrazole derivative (126 mg, 0.50 mmol, 1.0 eq) was dissolved in toluene (0.05 M). acetic anhydride (6 mL, 63 mmol, 126.0 eq) was added and it was stirred at 70 °C for 18 h. The reaction mixture was concentrated under reduced pressure and conducted without purification in the next reaction step. The mixture was dissolved (MeOH/H2O, 0.15 M, 1/1, v/v), K2CO3 (207 mg, 1.50 mmol, 3.0 eq) was added and the reaction mixture was stirred at room temperature for 12 h. The crude product was concentrated under reduced pressure and purified by RP flash column chromatography using a water/ACN gradient (5% to 95% ACN) yielding 15 as a white solid (72 mg, 0.27 mmol). Yield: 54%; mp. 189–192 °C; 1H NMR (500 MHz, DMSO-d6) δ 8.78 (s, 2H), 8.53 (t, J = 6.3 Hz, 1H), 7.34 – 7.26 (m, 4H), 7.26 – 7.18 (m, 1H), 4.58 (d, J = 6.3 Hz, 2H), 2.53 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ 163.1, 162.9, 161.5, 156.6, 156.2, 139.5, 128.4, 127.2, 126.9, 107.6, 44.2, 10.6; HRMS (ESI) m/z [M+H]+ calcd for C14H14N5O+ 268.1193, found 268.1189; HPLC (95% H2O 5 min, then to 95% MeCN in 5 min, then 100% MeCN to 20 min, 254 nm), tR = 12.29 min, 96% purity.
N-Benzyl-5-(5-(fluoromethyl)-1,3,4-oxadiazol-2-yl)pyrimidin-2-amine (16).
Synthesis according to the general procedure A starting from 14 (100 mg, 0.41 mmol, 1.0 eq.), using monofluoroacetic acid (0.028 mL, 0.49 mmol, 1.2 eq.) to afford 16 as a white lyophilized solid (4.6 mg, 0.016 mmol). Yield: 4%; 1H NMR (500 MHz, DMSO-d6): δ 8.85 (s, 2H), 8.64 (t, J = 6.4 Hz, 1H), 7.34 – 7.27 (m, 4H), 7.27 – 7.18 (m, 1H), 5.71 (d, J = 46.7 Hz, 2H), 4.60 (d, J = 6.2 Hz, 2H); 13C NMR (126 MHz, DMSO-d6): δ 163.0, 162.9, 160.9 (d, 2J (C, F) = 19.5 Hz), 157.1, 156.8, 139.4, 128.4, 127.2, 127.0, 107.0, 73.6 (d, 1J (C, F) = 165.3 Hz), 44.2; 19F NMR (471 MHz, DMSO-d6) δ −74.2. HRMS (ESI) m/z [M+H]+ calcd for C14H13FN5O+ 286.1099, found 286.1101; HPLC (95% H2O 1 min, then to 95% MeCN in 7 min, then 100% MeCN to 17 min, 254 nm), tR = 12.44 min, 96% purity.
N-Benzyl-5-[5-(trifluoromethyl)-1,3,4-oxadiazol-2-yl]pyrimidin-2-amine (17).
The product was synthesized according to general procedure C, using 4 (105 mg, 0.50 mmol, 1.0 eq) and trifluoracetic anhydride (TFAA, 0.139 mL, 1.00 mmol, 2.0 eq) as starting material. The crude was purified by silica gel column chromatography (cyclohexane/EtOAc, 4/1, v/v) to afford 17 as a white solid (88 mg, 0.27 mmol). Yield: 54%; mp. 185–187 °C; Rf = 0.4 (cyclohexane/EtOAc, 4/1, v/v); 1H NMR (600 MHz, DMSO-d6) δ 8.89 (d, J = 1.5 Hz, 2H), 8.78 (t, J = 6.4 Hz, 1H), 7.34 – 7.28 (m, 4H), 7.23 (ddd, J = 8.6, 5.3, 3.3 Hz, 1H), 4.61 (d, J = 6.2 Hz, 2H); 13C NMR (151 MHz, DMSO-d6) δ 164.2, 163.2, 157.6, 157.5, 153.4 (q, 2J (C, F) = 43.8 Hz), 139.3, 128.5, 127.3, 127.0, 117.4 (q, 1J (C, F) = 270.3 Hz), 106.1, 44.2; 19F NMR (565 MHz, DMSO-d6) δ −65.0; HRMS (ESI) m/z [M+H]+ calcd for C14H11F3N5O+ 322.0910, found 322.0481; HPLC (95% H2O 5 min, then to 95% MeCN in 5 min, then 100% MeCN to 20 min, 254 nm), tR = 13.21 min, 97% purity.
Methyl 2-(benzylamino)pyrimidine-5-carboxylate (18).
Benzylamine (0.220 mL, 2.00 mmol, 1.0 eq) and methyl 2-chlorpyrimidin-5-carboxylate (345 mg, 2.00 mmol, 1.0 eq) were dissolved in EtOH (10 mL, 0.2 M). DIPEA (0.524 mL, 3.00 mmol, 1.5 eq) was added and the reaction mixture was stirred at 80 °C for 18 h. After completion of the reaction, the solvent was removed under reduced pressure. EtOAc was added and the organic phase was washed with brine, dried over Na2SO4, filtered and evaporated. The product was obtained as a yellow solid (371 mg, 1.50 mmol) and used without further purification. Yield: 76%; mp. 151–153 °C; 1H NMR (600 MHz, DMSO-d6) δ 8.74 (d, J = 3.1 Hz, 2H), 8.61 (t, J = 6.4 Hz, 1H), 7.32 – 7.29 (m, 4H), 7.25 – 7.21 (m, 1H), 4.58 (d, J = 6.4 Hz, 2H), 3.78 (s, 3H); 13C NMR (151 MHz, DMSO-d6) δ 164.7, 163.5, 159.8, 159.8, 139.4, 128.4, 127.2, 126.9, 112.6, 51.7, 44.2; LRMS (ESI) m/z [M+H]+ calcd for C13H14N3O2+ 244.1, found 243.9.
Methyl 4-[(1-butyl-3-phenylureido)methyl]benzoate (19).
Phenyl phenylcarbamate (149 mg, 0.70 mmol, 1.0 eq) and 8 (197 mg, 0.84 mmol, 1.2 eq) were dissolved in THF (7 mL, 0.1 M). Et3N (0.195 mL, 1.40 mmol, 2.0 eq) was added and the reaction mixture was stirred at 60 °C for 2 h. The reaction was allowed to cool down to room temperature and quenched by the addition of water (10 mL). The mixture was extracted wit EtOAc (3 × 5 mL). The combined organic layers were washed with brine, dried over Na2SO4, filtered and the solvent was removed under reduced pressure. The crude product was purified by silica gel column chromatography (cyclohexane/EtOAc, 3/1, v/v) to afford 19 as colourless oil (207 mg, 0.60 mmol). Yield: 87%; Rf = 0.36 (cyclohexane/EtOAc, 3/1, v/v); 1H NMR (600 MHz, CDCl3) δ 8.05 – 7.98 (m, 2H), 7.36 (d, J = 8.2 Hz, 2H), 7.31 – 7.27 (m, 2H), 7.25 – 7.22 (m, 2H), 7.00 (tt, J = 7.1, 1.3 Hz, 1H), 6.29 (s, 1H), 4.61 (s, 2H), 3.90 (s, 3H), 3.34 – 3.30 (m, 2H), 1.65 – 1.56 (m, 2H), 1.35 (h, J = 7.4 Hz, 2H), 0.92 (t, J = 7.3 Hz, 3H); 13C NMR (151 MHz, CDCl3) δ 166.8, 155.3, 143.1, 138.8, 130.2, 129.5, 129.5, 128.9, 127.1, 123.2, 119.9, 52.1, 50.5, 47.8, 30.5, 20.2, 13.8; LRMS (ESI) m/z [M+H]+ calcd for C20H25N2O3+ 341.2, found 341.2.
Biological experiments
HDAC Inhibition Assays
Preincubation assay for HDAC1–4 and HDAC6.
In vitro inhibitory activity assays against HDAC1–3 and HDAC6 were performed using a modified protocol based on our previously published assays.63 In vitro inhibitory activities against HDAC4 were measured using a previously published protocol with slight modifications.68 For compounds and controls, 3fold serial dilutions of the respective DMSO-stock solution in assay buffer (50 mM Tris−HCl, pH 8.0, 137 mM NaCl, 2.7 mM KCl, 1.0 mM MgCl2•6 H2O, 5 mg/mL BSA), were prepared and 5.0 μL of this serial dilutions were transferred into OptiPlate-96 black micro-plates (PerkinElmer). Then, 25 μL assay buffer and 10 μL enzyme solution (human recombinant HDAC1 (BPS Bioscience, Catalog# 50051); HDAC2 (BPS Bioscience, Catalog# 50052); HDAC3/NcoR2 (BPS Bioscience, Catalog# 50003); HDAC4 (BPS Bioscience, Catalog# 50004); HDAC6 (BPS Bioscience, Catalog# 50006) were added. Enzyme and inhibitor were preincubated at 25 °C for 60 minutes. Afterwards, the fluorogenic substrate ZMAL (Z-Lys(Ac)-AMC69; 10 μL; 75 μM in assay buffer) was added. In the case of HDAC4 the fluorogenic substrate Boc-Lys(Tfa)-AMC (Bachem, Catalog# 4060676, 10 μL; 42.86 μM in assay buffer) was added. The total assay volume (50 μL, max. 1% DMSO) was incubated at 37 °C for 90 min. Subsequently, 50 μL trypsin solution (0.4 mg/mL trypsin in buffer: 50 mM Tris−HCl, pH 8.0, 100 mM NaCl) was added, followed by additional 30 minutes of incubation at 37 °C. Fluorescence (excitation: 355 nm, emission: 460 nm) was measured using an FLUOstar OPTIMA microplate reader (BMG LABTECH). Compounds were tested at least twice in duplicates; the 50% inhibitory concentration (IC50) was determined by plotting normalized dose response curves using nonlinear regression (Prism 8).
IC50-shift experiments at HDAC6.
For IC50-shift experiments we used the preincubation assay for HDAC6 as stated above and varied the preincubation period as follows: enzyme and the respective inhibitor dilutions were preincubated at 25°C for 5 to 120 minutes. Afterwards, the assay protocol was continued as stated above. Compounds were tested at least twice in duplicates.
Determination of binding kinetics via the Progression Method.63, 66
HDAC6 deacetylase activity was evaluated at varying inhibitor concentrations. Appropriate inhibitor concentrations were chosen based on the previously determined IC50 values. To ensure substrate excess during the experiment, the substrate concentration was set to five times KM. KM was determined using a series of substrate concentrations (see Figure S2). The respective steady-state velocities were plotted against the corresponding substrate concentrations [S] and fitted to the Michaelis-Menten equation (KM HDAC6 = 19.27 μM). For the progression curves, the enzyme was incubated with the fluorogenic substrate and inhibitor in assay buffer (50 μL; 50 mM Tris−HCl, pH 8.0, 137 mM NaCl, 2.7 mM KCl, 1.0 mM MgCl2•6 H2O, 5 mg/mL BSA) and 50 μL trypsin solution (40 ng/μL; buffer: 50 mM Tris−HCl, pH 8.0, 100 mM NaCl). The total assay volume (100 μL) contained the following final concentrations: HDAC6 (Lot #: 220419-GC; 220 pg/μL), ZMAL (93.0 μM) and trypsin (20 ng/μL). In situ AMC release was monitored continuously by fluorescence readings (excitation: 360 nm, emission: 460 nm; TECAN Spark® multimode microplate reader) recorded every 0.5 min for 45 minutes at 37 °C. The relationship between AMC concentration and relative fluorescence units (RFU) was determined and the measured RFU were transformed into the respective AMC concentration in μM. The data of each progression curve were fitted to obtain the apparent first-order rate constant kobs (Eq. 1). The apparent first-order rate constants kobs were replotted against the corresponding inhibitor concentrations [I] and the curves were either fitted into Eq. 2 or Eq. 3. Compounds were tested in triplicates. Data were fitted to the relevant equations using Prism 8.
100-fold Jump-Dilution experiments.
HDAC6 (22 ng/μL) in assay buffer (50 mM Tris−HCl, pH 8.0, 137 mM NaCl, 2.7 mM KCl, 1.0 mM MgCl2•6 H2O, 5 mg/mL BSA) was incubated with an inhibitor-concentrate (at least 10fold IC50) or blank (DMSO 1%) for 1 hour at room temperature. Afterwards, this “incubation-mix” was diluted 100fold either in the presence of the respective inhibitors at their original concentrations or solely with assay buffer. The substrate ZMAL (25 μL; 372 μM in assay buffer) and trypsin (50 μL; 40 ng/μL; buffer: 50 mM Tris−HCl, pH 8.0, 100 mM NaCl) were added to all samples. The total assay volume (100 μL) contained the following final concentrations: HDAC6 (Lot #: 220419-GC; 220 pg/μL), ZMAL (93 μM) and trypsin (20 ng/μL). The time-dependent in situ AMC release was monitored continuously by fluorescence readings (excitation: 360 nm, emission: 460 nm; TECAN Spark® multimode microplate reader) recorded every 0.5 min for 60 minutes at 37°C. Compounds were tested in triplicates.
LCMS Experiments
General information.
Danio rerio HDAC6 catalytic domain 2 (zCD2) was expressed and purified as previously described, with the modification of 0 mM imidazole in Buffer A.70
zCD2 Mutagenesis.
Two mutants were generated of zCD2, Y745F and H573A were prepared using the Q5 Site-Directed Mutagenesis Kit (New England Biolabs) with the addition of 5% DMSO. PCR primer sequences and annealing temperatures were generated using NEBaseChanger and listed in Table 2. Following sequence confirmation, the plasmids were transformed into BL21(DE3) cells expressed and purified by the same method as wild-type zCD2. Mutant H574A was generated previously, and expressed then purified by the same method as wild-type zCD2.62
Table 2.
Primers for zCD2 Mutagenesis
| Primer: | Sequence: | Annealing Temperature: |
|---|---|---|
| Y745F Fwd | 5’-GGAAGGCGGTTTTAACCTGACCA-3’ | 63 °C |
| Y745F Rev | 5’-AGGATAATCAGCACACGAC-3’ | |
| H573A Fwd | 5’-TCCGCCGGGCGCGCACGCAGAAAAAGATACCGC-3’ | 72 °C |
| H574A Rev | 5’-CGCACGATGGCCACCGCA-3’ |
LC-MS to study oxadiazole hydrolysis.
To study the zHDAC6 CD2-catalyzed ring-opening reaction of 6 to yield 13, 50 μM enzyme in size exclusion buffer (50 mM HEPES (pH 7.5), 100 mM KCl, 1 mM TCEP, 5% glycerol (v/v)) was incubated with 100 μM inhibitor overnight, total volume 100 μL. Control experiments were also performed with 100 μM inhibitor in size exclusion buffer only, to prove that the ring-opening reaction only occurs in the presence of enzyme. Following overnight incubation, protein was precipitated using 100 μL of methanol followed by filtering through a 22-μm GV DURAPORE filter. A 2 μL aliquot was injected over a C18 reverse phase column on a Waters Acquity UPLC-MS using a 2-min gradient of 95:5 H2O:MeCN to 5:95 H2O:MeCN. Mass spectra were analyzed using Mestrenova.
LC-MS to determine H218O incorporation.
To ascertain the utilization of H218O in the zHDAC6 CD2-catalyzed hydrolysis of 6 to yield 13, 100-μL samples of 50-μM enzyme in size exclusion buffer (50 mM HEPES (pH 7.5), 100 mM KCl, 1 mM TCEP, 5% glycerol (v/v)) were dialyzed against size exclusion buffer lacking glycerol overnight (50 mM HEPES (pH 7.5), 100 mM KCl, 1 mM TCEP). Samples were then lyophilized for 2 h before being resolubilized in 100 μL buffer including 100 μM inhibitor in H216O and H218O. Samples were then incubated overnight and subject to LC-MS analysis as described above.
Irreversibility of inhibition.
To determine the irreversibility of binding, 300-μL samples of 1 μM of zCD2 were incubated with 100-μM inhibitor for 1 h before being subject to dialysis. Initial measurements of inhibition were made through the standard discontinuous assay using the commercially available HDAC substrate RHKK(Ac)-AMC. A 25 μL sample was taken from dialysis and incubated with 25 μL of 250 μM substrate for 30 minutes. Following this 50 μL of developer solution was added consisting of 1 μM trypsin to cleave the AMC group, and 10 μM TSA to stop the reaction. Fluorescence of the AMC group was measured using an Infinite M1000Pro plate reader at excitation 360nM and emission 460 nM.
Samples were dialyzed against 3 L of size exclusion buffer [50 mM HEPES (pH 7.5), 100 mM KCl, 5% glycerol (v/v), and 1 mM TCEP] supplemented with 0.25% DMSO (v/v) at 4° C. Samples were taken at 6 hours post incubation and subjected to the standard discontinuous assay as above. The remaining 200 μL was subjected to dialysis overnight in 2 L of size exclusion buffer supplemented with 0.25% DMSO (v/v) and a sample taken at 21 hours before being measured as described above. Experiment was performed in triplicates.
X-Ray Crystallography
Crystal structure determination.
The zHDAC6 CD2–6 complex was crystallized by the sitting-drop vapor diffusion method. A 100-nL drop of protein solution [10 mg/mL HDAC6 CD2, 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 7.5), 100 mM KCl, 5% glycerol (v/v), 1 mM tris(2-carboxyethyl)phosphine (TCEP), and 2 mM 6] was combined with 100 nL of precipitant solution [0.04 M citric acid, 0.06 M bis-Tris propane (pH 5.0), 16% (w/v) polyethylene glycol 3,350] and equilibrated against 80 μL of precipitant solution in the well reservoir at 4° C. Plate-like crystals formed within 24 hours and were harvested after 48 hours. Crystals were flash-cooled in mother liquor supplemented with 20% ethylene glycol prior to data collection.
X-ray diffraction data were collected on the NSLS-II AMX beamline at Brookhaven National Laboratory.71 Data were indexed and scaled on the AMX automated fast-dp pipeline. The electron density map was phased from these data by molecular replacement using the program Phaser and the structure of unliganded HDAC6 CD2 less water molecules (PDB 5EEM).62 Atomic coordinates were built and manipulated in Coot72 and refined using Phenix.73
The Patterson map calculated for these data revealed a substantial peak (49.4% of the origin peak height) indicating severe translational non-crystallographic symmetry. Therefore, we indexed and scaled the data in space group P1 using iMOSFLM and Aimless to validate the space group.74, 75 This data were phased with Phaser76 and the program Zanuda77 was used to validate the space group (P212121) and crystallographic origin with 2 molecules in the asymmetric unit.
Upon discovering that 6 had undergone hydrolysis to yield 13 in the enzyme active site, the atomic coordinates of 13 were built into the electron density map during the later stages of refinement. All data collection and refinement statistics are recorded in Table S1.
PAINS analysis
We filtered all compounds for pan-assay interference compounds (PAINS) using the online filter http://zinc15.docking.org/patterns/home/.78
Supplementary Material
ACKNOWLEDGMENTS
B.K. acknowledges the Bonn International Graduate School of Drug Sciences (BIGS DrugS) for financial support of a research stay at the D.W.C. lab. We are grateful to Dr. Charles W. Ross III and Prof. Monica McCallum for their assistance with mass spectrometry experiments at the University of Pennsylvania. This research utilized the AMX beamline of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. The Center for BioMolecular Structure (CBMS) is primarily supported by the National Institutes of Health, National Institute of General Medical Sciences (NIGMS) through a Center Core P30 Grant (P30GM133893), and by the DOE Office of Biological and Environmental Research (KP1607011).
Funding
We thank the US National Institutes of Health for grant GM49758 in support of this research.
ABBREVIATIONS
- CDCl3
chloroform-d
- DFMO
difluoromethyl-1,3,4-oxadiazole
- DMSO
dimethylsulfoxide
- DCM
dichloromethane
- EtOAc
ethyl acetate
- HDAC
histone deacetylase
- HDACi
histone deacetylase inhibitor
- MeOH
methanol
- min
minutes
- rt
room temperature
- TFA
trifluoroacetic acid
- TFMO
trifluoromethyl-1,3,4-oxadiazole.
Footnotes
Supplementary Information
Electronic Supplementary Information (ESI) available: Supplementary Figures, Schemes, Equations and Tables, 1H NMR, 13C NMR, 19F NMR and HPLC data. (PDF)
Molecular strings formula. (CSV)
Accession Code
The atomic coordinates and crystallographic structure factors of the HDAC6-13 complex have been deposited in the Protein Data Bank (www.rcsb.org) with accession code 8GD4. Authors will release the atomic coordinates upon article publication.
The authors declare no competing financial interest.
REFERENCES
- (1).Ho TCS; Chan AHY; Ganesan A Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem 2020, 63, 12460–12484. [DOI] [PubMed] [Google Scholar]
- (2).Gregoretti IV; Lee YM; Goodson HV Molecular Evolution of the Histone Deacetylase Family: Functional Implications of Phylogenetic Analysis. J. Mol. Biol 2004, 338, 17–31. [DOI] [PubMed] [Google Scholar]
- (3).Roche J; Bertrand P Inside HDACs with More Selective HDAC Inhibitors. Eur. J. Med. Chem 2016, 121, 451–483. [DOI] [PubMed] [Google Scholar]
- (4).Wang P; Wang Z; Liu J Role of HDACs in Normal and Malignant Hematopoiesis. Mol. Cancer 2020, 19, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Mielcarek M; Zielonka D; Carnemolla A; Marcinkowski JT; Guidez F HDAC4 as a Potential Therapeutic Target in Neurodegenerative Diseases: A Summary of Recent Achievements. Front. Cell. Neurosci 2015, 9, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Parra M; Verdin E Regulatory Signal Transduction Pathways for Class IIa Histone Deacetylases. Curr. Opin. Pharmacol 2010, 10, 454–460. [DOI] [PubMed] [Google Scholar]
- (7).Hubbert C; Guardiola A; Shao R; Kawaguchi Y; Ito A; Nixon A; Yoshida M; Wang XF; Yao TP HDAC6 Is a Microtubule-Associated Deacetylase. Nature 2002, 417, 455–458. [DOI] [PubMed] [Google Scholar]
- (8).Hai Y; Shinsky SA; Porter NJ; Christianson DW Histone Deacetylase 10 Structure and Molecular Function as a Polyamine Deacetylase. Nat. Commun 2017, 8, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Bertrand P Inside HDAC with HDAC Inhibitors. Eur. J. Med. Chem 2010, 45, 2095–2116. [DOI] [PubMed] [Google Scholar]
- (10).Kalin JH; Bergman JA Development and Therapeutic Implications of Selective Histone Deacetylase 6 Inhibitors. J. Med. Chem 2013, 56, 6297–6313. [DOI] [PubMed] [Google Scholar]
- (11).Shen S; Kozikowski AP Why Hydroxamates May Not Be the Best Histone Deacetylase Inhibitors - What Some May Have Forgotten or Would Rather Forget? ChemMedChem 2016, 11, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zhang X; Yuan Z; Zhang Y; Yong S; Salas-burgos A; Koomen J; Olashaw N; Parsons JT; Yang X; Dent SR; Yao T; Lane WS; Seto E HDAC6 Modulates Cell Motility by Altering the Acetylation Level of Cortactin. Mol. Cell. Biol 2007, 27, 197–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Cohen TJ; Guo JL; Hurtado DE; Kwong LK; Mills IP; Trojanowski JQ; Lee VMY The Acetylation of Tau Inhibits Its Function and Promotes Pathological Tau Aggregation. Nat. Commun 2011, 2, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ding H; Dolan PJ; Johnson GVW Histone Deacetylase 6 Interacts with the Microtubule-Associated Protein Tau. J. Neurochem 2008, 106, 2119–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Karagöz GE; Rüdiger SGD Hsp90 Interaction with Clients. Trends Biochem. Sci 2015, 40, 117–125. [DOI] [PubMed] [Google Scholar]
- (16).Simões-Pires C; Zwick V; Nurisso A; Schenker E; Carrupt PA; Cuendet M HDAC6 as a Target for Neurodegenerative Diseases: What Makes It Different from the Other HDACs? Mol. Neurodegener 2013, 8, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Dompierre JP; Godin JD; Charrin BC; Cordelières FP; King SJ; Humbert S; Saudou F Histone Deacetylase 6 Inhibition Compensates for the Transport Deficit in Huntington’s Disease by Increasing Tubulin Acetylation. J. Neurosci 2007, 27, 3571–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Brindisi M; Saraswati AP; Brogi S; Gemma S; Butini S; Campiani G Old but Gold: Tracking the New Guise of Histone Deacetylase 6 (HDAC6) Enzyme as a Biomarker and Therapeutic Target in Rare Diseases. J. Med. Chem 2020, 63, 23–39. [DOI] [PubMed] [Google Scholar]
- (19).Adalbert R; Kaieda A; Antoniou C; Loreto A; Yang X; Gilley J; Hoshino T; Uga K; Makhija MT; Coleman MP Novel HDAC6 Inhibitors Increase Tubulin Acetylation and Rescue Axonal Transport of Mitochondria in a Model of Charcot-Marie-Tooth Type 2F. ACS Chem. Neurosci 2020, 11, 258–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Korfei M; Mahavadi P; Guenther A Targeting Histone Deacetylases in Idiopathic Pulmonary Fibrosis: A Future Therapeutic Option. Cells 2022, 11, 1–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Campiani G; Cavella C; Osko JD; Brindisi M; Relitti N; Brogi S; Saraswati AP; Federico S; Chemi G; Maramai S; Carullo G; Jaeger B; Carleo A; Benedetti R; Sarno F; Lamponi S; Rottoli P; Bargagli E; Bertucci C; Tedesco D; Herp D; Senger J; Ruberti G; Saccoccia F; Saponara S; Gorelli B; Valoti M; Kennedy B; Sundaramurthi H; Butini S; Jung M; Roach KM; Altucci L; Bradding P; Christianson DW; Gemma S; Prasse A Harnessing the Role of HDAC6 in Idiopathic Pulmonary Fibrosis: Design, Synthesis, Structural Analysis, and Biological Evaluation of Potent Inhibitors. J. Med. Chem 2021, 64, 9960–9988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Magupalli VG; Negro R; Tian Y; Hauenstein AV; Di Caprio G ; Skillern W; Deng Q; Orning P; Alam HB; Maliga Z; Sharif H; Hu JJ; Evavold CL; Kagan JC; Schmidt FI; Fitzgerald KA; Kirchhausen T; Li Y; Wu H HDAC6 Mediates an Aggresome-like Mechanism for NLRP3 and Pyrin Inflammasome Activation. Science 2020, 369, eaas8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Hideshima T; Bradner JE; Wong J; Chauhan D; Richardson P; Schreiber SL; Anderson KC Small-Molecule Inhibition of Proteasome and Aggresome Function Induces Synergistic Antitumor Activity in Multiple Myeloma. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 8567–8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Hideshima T; Richardson PG; Anderson KC Mechanism of Action of Proteasome Inhibitors and Deacetylase Inhibitors and the Biological Basis of Synergy in Multiple Myeloma. Mol. Cancer Ther 2011, 10, 2034–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Bhatia S; Krieger V; Groll M; Osko JD; Reßing N; Ahlert H; Borkhardt A; Kurz T; Christianson DW; Hauer J; Hansen FK Discovery of the First-in-Class Dual Histone Deacetylase-Proteasome Inhibitor. J. Med. Chem 2018, 61, 10299–10309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Vogl DT; Raje N; Jagannath S; Richardson P; Hari P; Orlowski R; Supko JG; Tamang D; Yang M; Jones SS; Wheeler C; Markelewicz RJ; Lonial S Ricolinostat, the First Selective Histone Deacetylase 6 Inhibitor, in Combination with Bortezomib and Dexamethasone for Relapsed or Refractory Multiple Myeloma. Clin. Cancer Res 2017, 23, 3307–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Atkinson SJ; Soden PE; Angell DC; Chung C; Giblin KA; Smithers N; Furze RC; Gordon L; Drewes G; Rioja I; Witherington J; Parr NJ; Prinjha RK The Structure Based Design of Dual HDAC / BET Inhibitors as Novel Epigenetic Probes. Med. Chem. Commun 2014, 18, 342–351. [Google Scholar]
- (28).Tang F; Yang Z; Tan Y; Li Y Super-Enhancer Function and Its Application in Cancer Targeted Therapy. npj Precis. Oncol 2020, 4, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Guerrant W; Patil V; Canzoneri JC; Oyelere AK Dual Targeting of Histone Deacetylase and Topoisomerase II with Novel Bifunctional Inhibitors. J. Med. Chem 2012, 55, 1465–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zhijun H; Shusheng W; Han M; Jianping L; Li-sen Q; Dechun L Pre-Clinical Characterization of 4SC-202, a Novel Class I HDAC Inhibitor, against Colorectal Cancer Cells. Tumor Biol 2016, 37, 10257–10267. [DOI] [PubMed] [Google Scholar]
- (31).Kalin JH; Wu M; Gomez AV; Song Y; Das J; Hayward D; Adejola N; Wu M; Panova I; Chung HJ; Kim E; Roberts HJ; Roberts JM; Prusevich P; Jeliazkov JR; Roy Burman SS; Fairall L; Milano C; Eroglu A; Proby CM; Dinkova-Kostova AT; Hancock WW; Gray JJ; Bradner JE; Valente S; Mai A; Anders NM; Rudek MA; Hu Y; Ryu B; Schwabe JWR; Mattevi A; Alani RM; Cole PA Targeting the CoREST Complex with Dual Histone Deacetylase and Demethylase Inhibitors. Nat. Commun 2018, 9, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Morera L; Lübbert M; Jung M Targeting Histone Methyltransferases and Demethylases in Clinical Trials for Cancer Therapy. Clin. Epigenetics 2016, 8, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Wobser M; Weber A; Glunz A; Tauch S; Seitz K; Butelmann T; Hesbacher S; Goebeler M; Bartz R; Kohlhof H; Schrama D; Houben R Elucidating the Mechanism of Action of Domatinostat (4SC-202) in Cutaneous T Cell Lymphoma Cells. J. Hematol. Oncol 2019, 12, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Ojha R; Huang HL; HuangFu WC; Wu YW; Nepali K; Lai MJ; Su CJ; Sung TY; Chen YL; Pan SL; Liou JP 1-Aroylindoline-Hydroxamic Acids as Anticancer Agents, Inhibitors of HSP90 and HDAC. Eur. J. Med. Chem 2018, 150, 667–677. [DOI] [PubMed] [Google Scholar]
- (35).Mehndiratta S; Lin MH; Wu YW; Chen CH; Wu TY; Chuang KH; Chao MW; Chen YY; Pan SL; Chen MC; Liou JP N-Alkyl-Hydroxybenzoyl Anilide Hydroxamates as Dual Inhibitors of HDAC and HSP90, Downregulating IFN-γ Induced PD-L1 Expression. Eur. J. Med. Chem 2020, 185, 111725. [DOI] [PubMed] [Google Scholar]
- (36).Liu T; Wan Y; Xiao Y; Xia C; Duan G Dual-Target Inhibitors Based on HDACs: Novel Antitumor Agents for Cancer Therapy. J. Med. Chem 2020, 63, 8977–9002. [DOI] [PubMed] [Google Scholar]
- (37).de Lera AR; Ganesan A Two-Hit Wonders: The Expanding Universe of Multitargeting Epigenetic Agents. Curr. Opin. Chem. Biol 2020, 57, 135–154. [DOI] [PubMed] [Google Scholar]
- (38).He S; Dong G; Li Y; Wu S; Wang W; Sheng C Potent Dual BET/HDAC Inhibitors for Efficient Treatment of Pancreatic Cancer. Angew. Chem. Int. Ed 2020, 59, 3028–3032. [DOI] [PubMed] [Google Scholar]
- (39).Gaisina IN; Tueckmantel W; Ugolkov A; Shen S; Hoffen J; Dubrovskyi O; Mazar A; Schoon RA; Billadeau D; Kozikowski AP Identification of HDAC6-Selective Inhibitors of Low Cancer Cell Cytotoxicity. ChemMedChem 2016, 11, 81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Depetter Y; Geurs S; De Vreese R; Goethals S; Vandoorn E; Laevens A; Steenbrugge J; Meyer E; de Tullio P; Bracke M; D’hooghe M; De Wever O Selective Pharmacological Inhibitors of HDAC6 Reveal Biochemical Activity but Functional Tolerance in Cancer Models. Int. J. Cancer 2019, 145, 735–747. [DOI] [PubMed] [Google Scholar]
- (41).Zhang X-H; Qin-Ma; Wu H-P; Khamis MY; Li Y-H; Ma L-Y; Liu H-M A Review of Progress in Histone Deacetylase 6 Inhibitors Research: Structural Specificity and Functional Diversity. J. Med. Chem 2021, 64, 1362–1391. [DOI] [PubMed] [Google Scholar]
- (42).Jenke R; Reßing N; Hansen FK; Aigner A; Büch T Anticancer Therapy with HDAC Inhibitors: Mechanism-Based Combination Strategies and Future Perspectives. Cancers 2021, 13, 634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Wang CY; Lee LH Mutagenicity and Antibacterial Activity of Hydroxamic Acids. Antimicrob. Agents Chemother 1977, 11, 753–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Zhang L; Zhang J; Jiang Q; Zhang L; Song W Zinc Binding Groups for Histone Deacetylase Inhibitors. J. Enzyme Inhib. Med. Chem 2018, 33, 714–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Yue K; Sun S; Jia G; Qin M; Hou X; Chou CJ; Huang C; Li X First-in-Class Hydrazide-Based HDAC6 Selective Inhibitor with Potent Oral Anti-Inflammatory Activity by Attenuating NLRP3 Inflammasome Activation. J. Med. Chem 2022, 65, 12140–12162. [DOI] [PubMed] [Google Scholar]
- (46).Kim Y; Lee CS; Oh JT; Song H; Choi J; Lee J Oxadiazole Amine Derivative Compounds as Histone Deacetylase 6 Inhibitor, and the Pharmaceutical Composition Comprising the Same WO 2017/065473 A1, 2017.
- (47).Lee J; Han Y; Kim Y; Min J; Bae M; Kim D; Jin S; Kyung J 1,3,4-Oxadiazole Sulfamide Derivative Compounds as Histone Deacetylase 6 Inhibitor, and the Pharmaceutical Composition Comprising the Same WO 2017/018805 A1, 2017.
- (48).Ito M; Sugiyama H; Kubo O; Kikuchi F; Yasu T; Kakegawa K; Ikeda Z; Miyazaki T; Arikawa Y; Okawa T; Yonemori J; Toita A; Kojima T; Asano Y; Sato A; Maezaki H; Sasaki S; Kokubo H; Homma M; Sasaki M; Imadea Y Heterocyclic Compound WO 2019/027054 A1, 2019.
- (49).Lee CK; Ko MS; Yun SH; Kim HM 1,3,4-Oxadiazole Derivative Compounds as Histone Deacetylase 6 Inhibitor, and the Pharmaceutical Composition Comprising the Same WO 2021/210857 A1, 2021.
- (50).Mandegar MA; Patel S; Ding P; Bhatt U; Holan M; Lee J; Li Y; Medina J; Nerurkar A; Seidl F; Sperandino D; Widjaja T Fluoroalkyl-Oxadiazoles and Uses Thereof WO 2021/127643 A1, 2021.
- (51).Çakır I; Hadley CK; Pan PL; Bagchi RA; Ghamari-Langroudi M; Porter DT; Wang Q; Litt MJ; Jana S; Hagen S; Lee P; White A; Lin JD; McKinsey TA; Cone RD Histone Deacetylase 6 Inhibition Restores Leptin Sensitivity and Reduces Obesity. Nat. Metab 2022, 4, 44–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Keuler T; König B; Bückreiß N; Kraft FB; König P; Schäker-Hübner L; Steinebach C; Bendas G; Gütschow M; Hansen FK Development of the First Non-Hydroxamate Selective HDAC6 Degraders. Chem. Commun 2022, 58, 11087–11090. [DOI] [PubMed] [Google Scholar]
- (53).Cragin A; Watson PR; König B; Hansen FK; Christianson DW A Novel Zinc Binding Group for HDAC6 Inhibition. FASEB J 2022, 36, DOI: 10.1096/fasebj.2022.36.S1.R3604. [DOI] [Google Scholar]
- (54).Ptacek J; Snajdr I; Schimer J; Kutil Z; Mikesova J; Baranova P; Havlinova B; Tueckmantel W; Majer P; Kozikowski A; Barinka C Selectivity of Hydroxamate- and Difluoromethyloxadiazole-Based Inhibitors of Histone Deacetylase 6. In Vitro and in Cells. Int. J. Mol. Sci 2023, 24. 10.3390/ijms24054720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Yates CM Metalloenzyme Inhibitor Compounds US 2018/0256572 A1, 2018.
- (56).Cellupica E; Caprini G; Cordella P; Cukier C; Fossati G; Marchini M; Rocchio I; Sandrone G; Vanoni MA; Vergani B; Źrubek K; Stevenazzi A; Steinkühler C Difluoromethyl-1,3,4-Oxadiazoles Are Slow-Binding Substrate Analog Inhibitors of Histone Deacetylase 6 with Unprecedented Isotype Selectivity. J. Biol. Chem 2023, 299, 102800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Huisgen R; Sauer J; Sturm HJ Acylierung 5-Substituierter Tetrazole zu 1.3.4-Oxdiazolen. Angew. Chemie 1958, 70, 272–273. [Google Scholar]
- (58).Bergman JA; Woan K; Perez-Villarroel P; Villagra A; Sotomayor EM; Kozikowski AP Selective Histone Deacetylase 6 Inhibitors Bearing Substituted Urea Linkers Inhibit Melanoma Cell Growth. J. Med. Chem 2012, 55, 9891–9899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Krieger V; Hamacher A; Cao F; Stenzel K; Gertzen CGW; Schäker-Hübner L; Kurz T; Gohlke H; Dekker FJ; Kassack MU; Hansen FK Synthesis of Peptoid-Based Class I-Selective Histone Deacetylase Inhibitors with Chemosensitizing Properties. J. Med. Chem 2019, 62, 11260–11279. [DOI] [PubMed] [Google Scholar]
- (60).Diedrich D; Hamacher A; Gertzen CGW; Alves Avelar LA; Reiss GJ; Kurz T; Gohlke H; Kassack MU; Hansen FK Rational Design and Diversity-Oriented Synthesis of Peptoid-Based Selective HDAC6 Inhibitors. Chem. Commun 2016, 52, 3219–3222. [DOI] [PubMed] [Google Scholar]
- (61).Sippl W; Melesina J; Simoben CV; Bülbül EF; Praetorius L; Robaa D Strategies to Design Selective Histone Deacetylase Inhibitors. ChemMedChem 2021, 16, 1336–1359. [DOI] [PubMed] [Google Scholar]
- (62).Hai Y; Christianson DW Histone Deacetylase 6 Structure and Molecular Basis of Catalysis and Inhibition. Nat. Chem. Biol 2016, 12, 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Schäker-Hübner L; Haschemi R; Büch T; Kraft FB; Brumme B; Schöler A; Jenke R; Meiler J; Aigner A; Bendas G; Hansen FK Balancing Histone Deacetylase (HDAC) Inhibition and Drug-Likeness: Biological and Physicochemical Evaluation of Class I Selective HDAC Inhibitors. ChemMedChem 2022, 17, e202100755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Jiang Y; Xu J; Yue K; Huang C; Qin M; Chi D; Yu Q; Zhu Y; Hou X; Xu T; Li M; Chou CJ; Li X Potent Hydrazide-Based HDAC Inhibitors with a Superior Pharmacokinetic Profile for Efficient Treatment of Acute Myeloid Leukemia in Vivo. J. Med. Chem 2022, 65, 285–302. [DOI] [PubMed] [Google Scholar]
- (65).Yue K; Sun S; Jia G; Qin M; Hou X; Chou CJ; Huang C; Li X First-in-Class Hydrazide-Based HDAC6 Selective Inhibitor with Potent Oral Anti-Inflammatory Activity by Attenuating NLRP3 Inflammasome Activation. J. Med. Chem 2022, 65, 12140–12162. [DOI] [PubMed] [Google Scholar]
- (66).Copeland RA Evaluation of Enzyme Inhibitors - A Guide for Medical Chemists and Pharmacologists, 2nd ed.; John Wiley and Sons, Inc., Hoboken: New Jersey, 2013. [Google Scholar]
- (67).Motlová L; Snajdr I; Kutil Z; Andris E; Ptacek J; Novotna A; Novakova Z; Havlinova B; Tueckmantel W; Draberova H; Majer P; Schutkowski M; Kozikowski A; Rulisek L; Barinka C Comprehensive Mechanistic View of the Hydrolysis of Oxadiazole- Based Inhibitors by Histone Deacetylase 6 (HDAC6). ACS Chem. Biol 2023, 18, 1594–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Reßing N; Schliehe-Diecks J; Watson PR; Sönnichsen M; Cragin AD; Schöler A; Yang J; Schäker-Hübner L; Borkhardt A; Christianson DW; Bhatia S; Hansen FK Development of Fluorinated Peptoid-Based Histone Deacetylase (HDAC) Inhibitors for Therapy-Resistant Acute Leukemia. J. Med. Chem 2022, 65, 15457–15472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Kraft FB; Hanl M; Feller F; Schäker-Hübner L; Hansen FK Photocaged Histone Deacetylase Inhibitors as Prodrugs in Targeted Cancer Therapy. Pharmaceuticals 2023, 16, 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Osko JD; Christianson DW Methods for the Expression, Purification, and Crystallization of Histone Deacetylase 6–Inhibitor Complexes. Methods Enzymol 2019, 626, 447–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Schneider DK; Soares AS; Lazo EO; Kreitler DF; Qian K; Fuchs MR; Bhogadi DK; Antonelli S; Myers SS; Martins BS; Skinner JM; Aishima J; Bernstein HJ; Langdon T; Lara J; Petkus R; Cowan M; Flaks L; Smith T; Shea-McCarthy G; Idir M; Huang L; Chubar O; Sweet RM; Berman LE; McSweeney S; Jakoncic J AMX - the Highly Automated Macromolecular Crystallography (17-ID-1) Beamline at the NSLS-II. J. Synchrotron Radiat 2022, 29, 1480–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and Development of Coot. Acta Crystallogr. Sect. D 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Adams PD; Afonine PV; Bunkóczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung L-W; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. Sect. D 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Battye TGG; Kontogiannis L; Johnson O; Powell HR; Leslie AGW IMOSFLM: A New Graphical Interface for Diffraction-Image Processing with MOSFLM. Acta Crystallogr. Sect. D 2011, 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Evans PR; Murshudov GN How Good Are My Data and What Is the Resolution? Acta Crystallogr. Sect. D 2013, 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser Crystallographic Software. J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Lebedev AA; Isupov MN Space-Group and Origin Ambiguity in Macromolecular Structures with Pseudo-Symmetry and Its Treatment with the Program Zanuda. Acta Crystallogr. Sect. D 2014, 70, 2430–2443. [DOI] [PubMed] [Google Scholar]
- (78).Baell JB; Holloway GA New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem 2010, 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.