

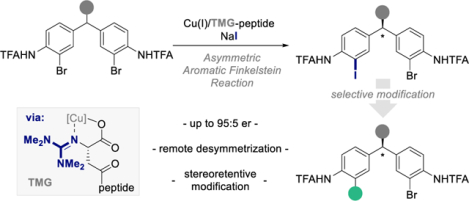
Abstract
A first-of-its-kind enantioselective aromatic Finkelstein reaction is disclosed for the remote desymmetrization of diarylmethanes. The reaction operates through a copper-catalyzed C‒I bond forming event and high levels of enantioselectivity are achieved through the deployment of a tailored guanidinylated peptide ligand. Strategic use of transition-metal mediated reactions enables the chemoselective modification of the aryl iodide products, thus, the synthesis of a diverse set of otherwise difficult-to-access diarylmethanes in excellent levels of selectivity is realized from a common intermediate. A mixed experimental/computational analysis of steric parameters and substrate conformations identifies the importance of remote conformational effects as a key to achieving high enantioselectivity in this desymmetrization reaction.
Graphical Abstract
Since its discovery in 1910, the Finkelstein reaction has been synonymous with halide exchange for the preparation of primary alkyl iodides.1–3 The substitution of alkyl bromides and chlorides under a well-defined SN2 regime4–5 and the elegant exploitation of Le Chatelier’s principle to drive the reaction by precipitation of NaBr or NaCl have rendered the Finkelstein reaction a classic in introductory organic chemistry textbooks and a reliable tool for organic synthesis (Figure 1A).6–8 While halide exchange in aryl halides is also precedented,9–10 it took nearly a century following Finkelstein’s discovery, until Buchwald’s report on the copper-catalyzed aromatic Finkelstein reaction appeared, realizing a synthetically useful protocol for C‒Br to C‒I exchange at Csp2 centers (Figure 1B).11 Despite a renewed interest in the development of milder methods,12–16 Buchwald’s method remains state-of-the-art. Indeed, the traditional copper mediated approach that operates via an efficient oxidative addition / halide exchange / reductive elimination sequence has shown utility in various synthetic campaigns.17–19
Figure 1.
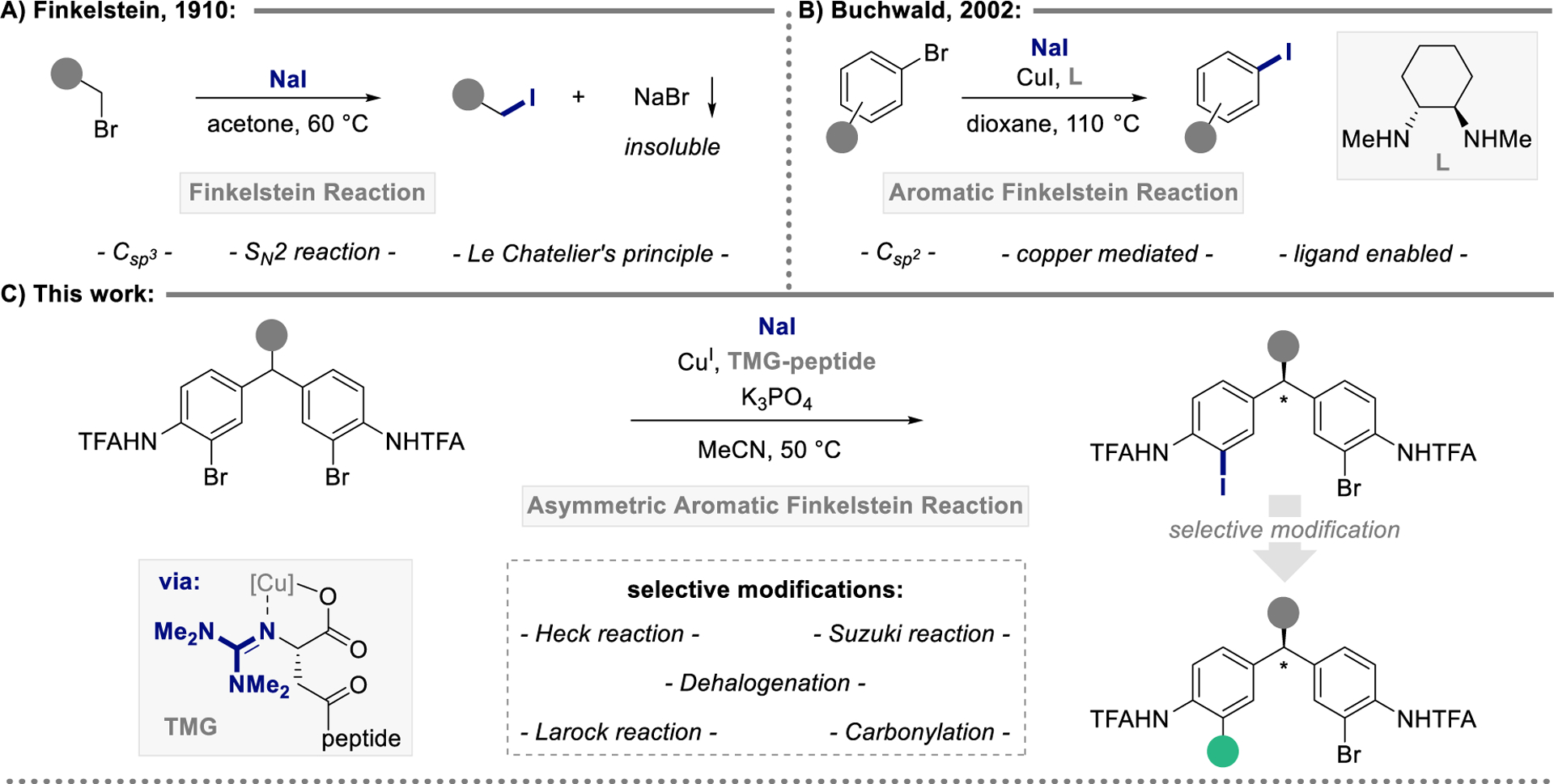
Development of an asymmetric aromatic Finkelstein reaction.
Notably, implementation of the enantioselective aromatic Finkelstein reaction has not yet been reported in the literature.
Motivated by recent observations applying guanidinylated peptide-based ligands in asymmetric copper-based cross-couplings, we sought to establish a synthetic platform that would allow for the development of an enantioselective aromatic Finkelstein reaction for remote desymmetrization of diarylmethanes.20–24 The generation of stereocenters removed from the center of reaction remains a major challenge in contemporary asymmetric catalysis.25–27 In this field, peptide-based catalysts have found particular utility, stimulating the present study of their capacity to mediate remote aryl bromide to aryl iodide substitution. We further hypothesized that leveraging the enhanced reactivity of the C‒I bond towards venerable cross-coupling reactions would allow for translation of the installed stereoinformation into chemoselective transition metal-catalyzed transformations (Figure 1C).28 Consequently, the asymmetric aromatic Finkelstein reaction would, facilitate the streamlined synthesis of a structurally diverse library of enantio-enriched diarylmethanes, negating the need to identify chiral catalysts and ligands for each individual cross-coupling reaction.
The selection of 1 as a model substrate was motivated by the privileged role of diarylmethanes in drug discovery.29–35 We began by subjecting 1 to a CuI/TMG-Asp-d-Pro-OLi catalyst system using sodium iodide as iodide source and we were gratified to observe the formation of 2 in 49% NMR-yield with 90:10 er, alongside achiral bis-substituted product 3 (10% yield) (see Supporting Information). Encouraged by this result, ligand optimization was initiated (Table 1). While utilization of monomeric, dimeric, and trimeric tetramethylguanidine N-capped peptides furnished 2 in promising levels of enantioselectivity (up to 91:9 er, L3-L7), tetrameric β-turn peptides with a Li-Di+1-Li+3 sequence proved superior, generating 2 in good yields (up to 64%) and excellent selectivities (up to 96:4 er) (L8-L16). In general, the nature of the i+3 position had only a minor influence on selectivity (L9-L12), yet the presence of a C-terminal carboxylate proved essential (L8).
Table 1.
Ligand optimization.
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| entry | ligand | i | i+1 | i+2 | i+3 | 1 [%] | 2 [%] | 3 [%] | er (2) |
| 1 | L1 | TMG-Asp | d-Pro | Aib-OLi | 15 | 62 | 20 | 6 : 94 | |
| 2 | L2 | TMG-Gly-OLi | 14 | 37 | 29 | 50 : 50 | |||
| 3 | L3 | TMG-1NaI-OH | 13 | 38 | 30 | 58 : 42 | |||
| 4 | L4 | TMG-Neo-OH | 25 | 44 | 18 | 66 : 34 | |||
| 5 | L5 | TMG-d-Asp | Pro-OLi | 23 | 47 | 7 | 90 : 10 | ||
| 6 | L6 | TMG-d-Asp | αMe-Pro-OLi | 24 | 52 | 10 | 91 : 9 | ||
| 7 | L7 | TMG-Asp | d-Pro | Acpc-OLi | 51 | 40 | 4 | 10 : 90 | |
| 8 | L8 | TMG-Asp | d-Pro | Acpc | 2NaI-NHMe | 42 | 48 | 9 | 19 : 81 |
| 9 | L9 | TMG-Asp | d-Pro | Acpc | Leu-OLi | 13 | 62 | 19 | 5 : 95 |
| 10 | L10 | TMG-Asp | d-Pro | Acpc | tLeu-OLi | 8 | 62 | 22 | 4 : 96 |
| 11 | L11 | TMG-Asp | d-Pro | Acpc | Chg-OLi | 14 | 64 | 14 | 4 : 96 |
| 12 | L12 | TMG-Asp | d-Pro | Acpc | Phe-OLi | 8 | 55 | 16 | 4 : 96 |
| 13 | L13 | TMG-Asp | d-Pro | Aic | Phe-OLi | 8 | 54 | 17 | 5 : 95 |
| 14 | L14 | TMG-Asp | d-Pro | Aib | Val-OLi | 6 | 56 | 20 | 4 : 96 |
| 15 | L15 | TMG-Asp | d-Pro | Aib | d-Leu-OLi | 31 | 52 | 8 | 8 : 92 |
| 16 | L16 | TMG-d-Asp | d-Pro | Acpc | Phe-OLi | 45 | 30 | 4 | 72 : 28 |
Reaction conditions: 1 (0.10 mmol), Cu(MeCN)4BF4 (10 mol%), ligand (15 mol%), K3PO4 (0.40 equiv.), NaI (0.12 mmol), MeCN (0.2 mL), 50 °C, 16 h. Yield determined by 1H NMR using dibromomethane as internal standard. Enantiomeric ratio (er) determined by chiral HPLC. Abbreviations: TMG, tetramethylguanidine; 1NaI, 1-naphtylalanine; Neo, neopentylglycine; αMe-Pro, α-methyl proline; Aib, 2-aminoisobutyric acid; Acpc, 1-Aminocyclopropane-1-carboxylic acid; Aic, 2-Aminoindane-2-carboxylic acid; Chg, cyclohexylglycine.
Similarly, alteration of the catalyst τ-angle through incorporation of disubstituted amino acids (Acpc, Aic, Aib) at the i+2 position did not impact the reaction outcome and 2 was isolated in good yield and excellent er (L12-L14).36 Finally, we investigated the effect of stereochemical alteration of the amino acid sequence. Employing L15, which contains an optically inverted i+3 position had a negligible effect on the reaction outcome. In contrast, substitution of Asp for d-Asp (i-position) resulted in an inversion of the sense of enantio-induction with 2 obtained in 72:28 er. While L9-L13 furnished the product in similar yield and selectivity, L10 performed better on a larger scale and was therefore used going forward.
While separation of 2 from remaining starting material 1 and side-product 3 was not possible, a crystal structure of 2 could be obtained, allowing for assignment of the absolute configuration as (S) (Figure 2B, see Supporting Information). Our overall strategic vision included the use of the aromatic Finkelstein reaction to set up subsequent reactions based on the canonical selective transformation of the C‒I over the C‒Br bond, rendering purification of 2 obsolete. We therefore turned our attention to product derivatization.
Figure 2.
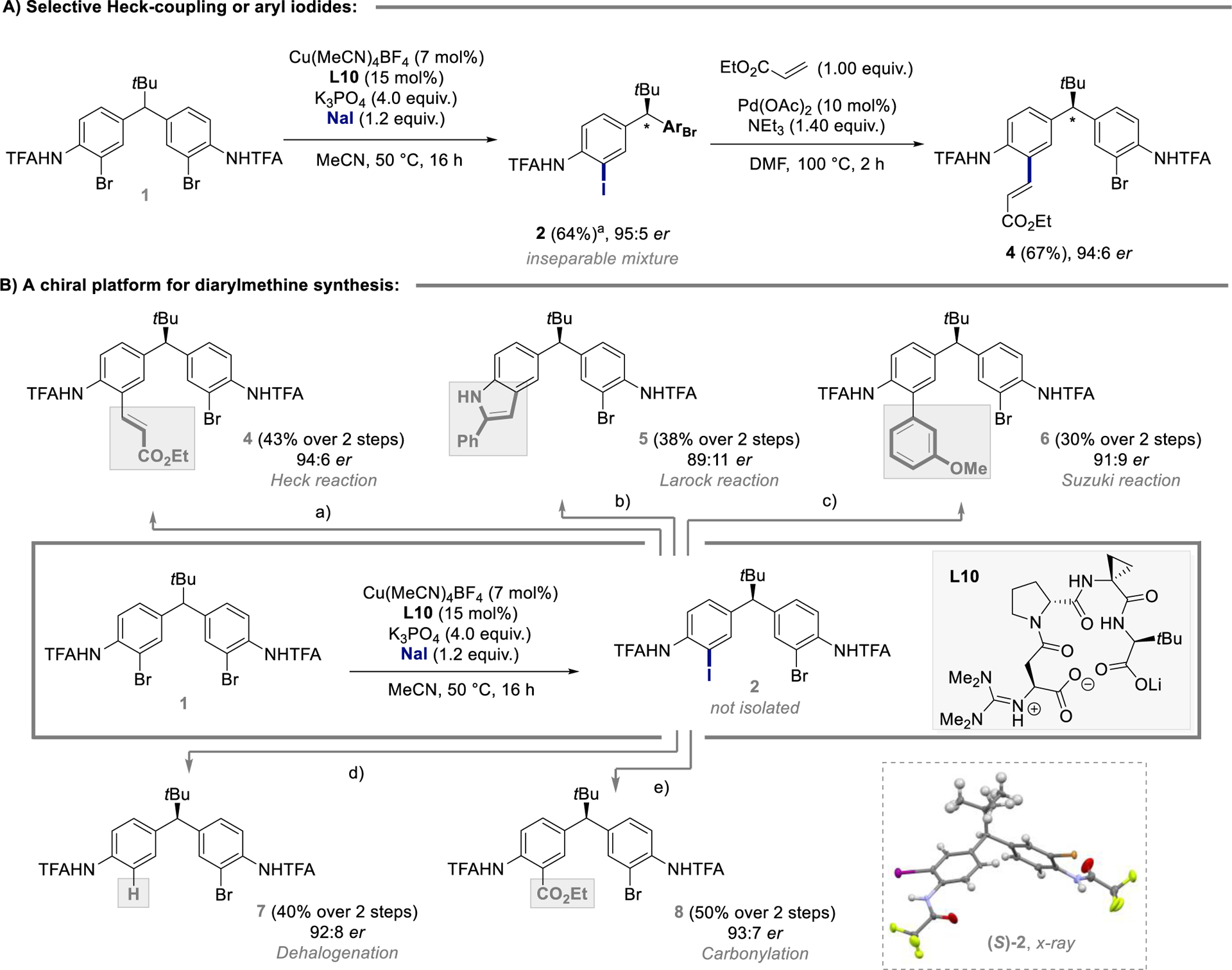
A) Identification of an asymmetric aromatic Finkelstein / Heck reaction sequence. aYield determined by 1H NMR using dibromomethane as internal standard. B) Asymmetric aromatic Finkelstein reaction as platform for the synthesis of enantioenriched diarylmethanes. Reaction conditions (0.2 mmol scale): a) Pd(OAc)2, NEt3, ethyl acrylate, DMF; b) CuI, PPh3, K3PO4, phenylacetylene, 1,4-dioxane; c) Pd(OAc)2, NEt3, 3-methoxyphenylboronic acid, toluene; d) Pd(OAc)2, NaBH4, TMEDA, DMF; e) Pd(OAc)2, NEt3, EtOH/DMF (1:4), CO. X-ray structure of 2 is shown with atomic thermal parameters calculated at 50% probability levels.
Initial attempts targeted chemoselective Heck-reactions. While the use of phosphine ligands resulted in complex product mixtures, a Pd(OAc)2/NEt3-catalyst system enabled the desired conversion of 2 (purified mixture containing 1 and 3) to 4 in 67% yield with retention of enantioselectivity (Figure 2A). Stimulated by this result, we aimed to advance the asymmetric aromatic Finkelstein / cross-coupling strategy to a more general derivatization platform, enabling the synthesis of diverse diarylmethanes over two steps with a single chromatographic purification.
Pleasingly, the same strategy was compatible with various transition-metal catalyzed transformations (Figure 2B).
Heck-product 4 could be isolated in 43% over 2 steps with excellent enantioselectivity (94:6 er). Our strategy was furthermore compatible with a Cu(I)-catalyzed Larock-type indole formation, enabling the synthesis of 5 in 38% over 2 steps, despite an observable drop in selectivity (89:11 er), which in this case, can be attributed to background reaction of the remaining starting material. Implementation of a Suzuki-reaction using 3-methoxyphenyl-boronic acid proved fruitful with 6 being isolated in 30% over 2 steps and 91:9 er. Enantioenriched mono-brominated diarylmethane 7 could be synthesized via a palladium-catalyzed dehalogenation using NaBH4 as reductant (40% over 2 steps, 92:8 er). Finally, insertion of carbon monoxide into the newly installed C‒I bond in the presence of a palladium catalyst resulted in the formation of 8 in 50% over 2 steps with 93:7 er. It is notable, from a strategic standpoint, that each of these transformations is enabled by desymmetrization of a simple, and common starting material, circumventing individual campaigns for new chiral ligands and catalysts.
Having established the synthetic potential of the asymmetric aromatic Finkelstein reaction as a platform for chiral diarylmethane synthesis, we explored mechanistic and structural requirements to achieve high selectivity. Diarylmethanes have been successfully utilized in several methodology and drug discovery campaigns.26–35 In particular, our lab has established a high degree of compatibility with small-peptide catalysis over a broad range of mechanistically distinct transformations.20–24, 36–37 A series of experiments was therefore undertaken to identify parameters that govern the privileged role of diarylmethanes in peptide catalysis.
The involvement of a secondary kinetic resolution in the enhancement of selectivity is often characterized by an increase of enantioselectivity over time. We thus monitored the reaction progress and indeed observed a continuous increase in er from 88:12 to 95:5 (Figure 3A). Furthermore, the reaction features an induction period of around 6 h, which results from the required deprotonation of the - NHTFA moiety to reveal the active substrate for catalysis (see Supporting Information).
Figure 3.
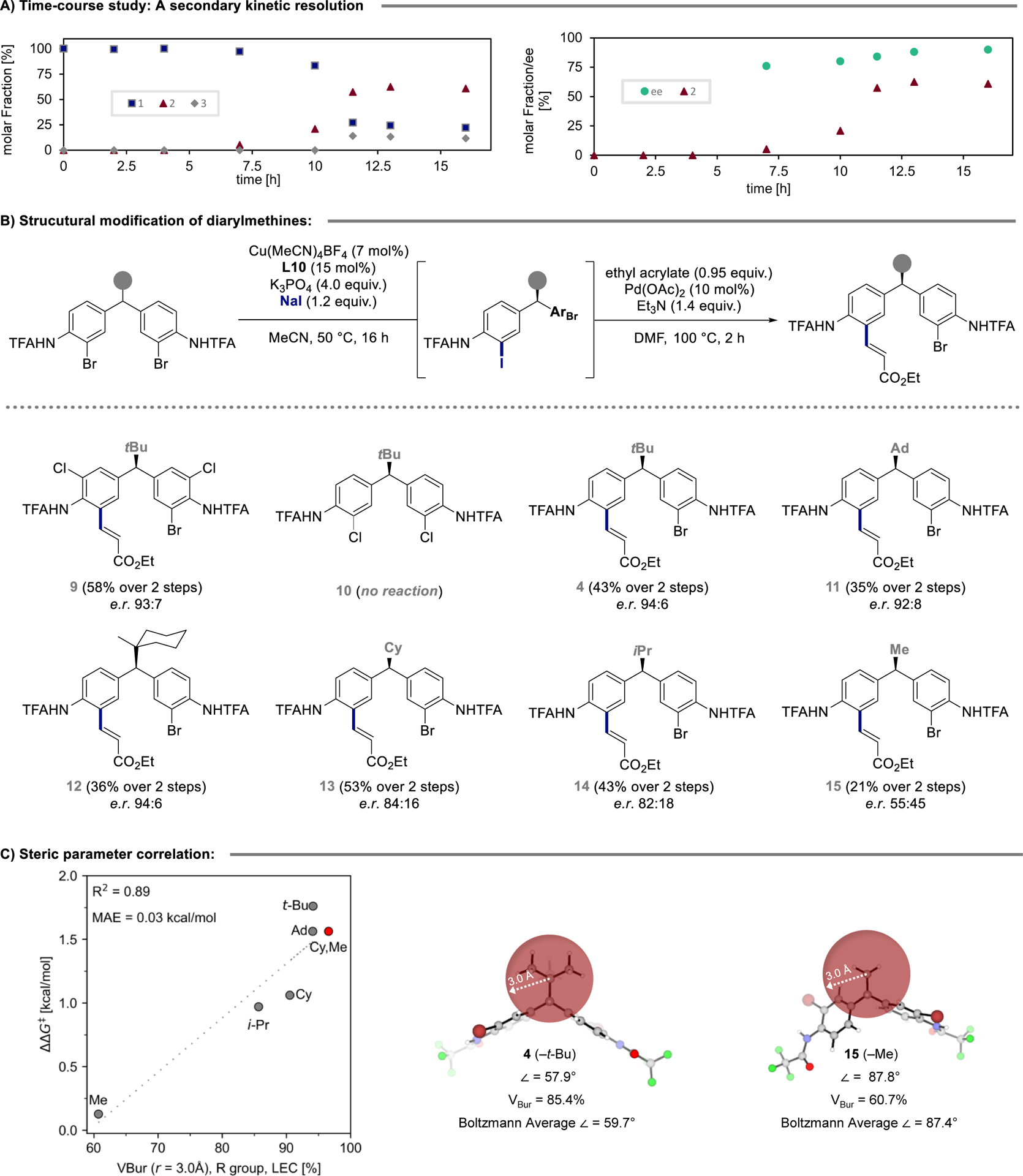
Diarylmethane scope and correlation of steric demand with enantioselectivity.
Next, we investigated the influence of diarylmethane structure on reaction outcome (Figure 3B). Evaluation of the impact of structural modification was possible by isolation of the corresponding Heck-reaction products. Initially, the role and nature of the halide substituent was investigated. Addition of chloro-substituents to the aryl-core was well-tolerated furnishing diarylmethane 9 in 58% yield over two steps in 94:6 er. In contrast, no conversion to product was observed upon subjecting 10 to the reaction conditions, highlighting the relatively poor reactivity of aryl chlorides compared to aryl bromides.
Perhaps most interesting in the context of remote substituents is the bridge between the two aromatic rings of the substrate. Considering the excellent enantioselectivity obtained for tBu-substituted substrate 4 (94:6 er), the very good levels of selectivity upon installation of an adamantyl (11, 92:8 er) and a methylcyclohexyl group (12, 94:6 er) are unsurprising. The presence of a tertiary carbon-center in the α-position to the stereogenic center, however, resulted in a notable drop in selectivity (cyclohexyl, 13, 84:16 er; isopropoyl, 14, 82:18 er). Further decrease of the steric profile upon installation of a methyl group (15), yielded a near racemic product (55:45 er).
In previous studies, we noted linear free energy relationships between empirical steric parameters and the observed enantioselectivity in the catalytic desymmetrization reaction of diarylmethane-bis(phenol) substrates.37–38 Here we show that a computed steric descriptor, buried volume (VBur) computed at 3.0 Å sphere centered at the substituent group carbon, correlates well with the measured enantioselectivities (R2 = 0.89, Figure 3C).39–40 Computed steric descriptors provide advantages over empirically derived parameters, particularly because they can be readily computed for uncommon substituents (e.g., methylcyclohexyl group in 12). To showcase this advantage, we used 12 as a test case to evaluate the accuracy of the correlation and found that it accurately predicted the enantioselectivity within 0.03 kcal/mol of the measured value. Given the nature of this remote functionalization, we sought to understand how the change in a distal group (as measured by VBur) induces conformational changes that imbue high enantioselectivity. Inspection of the conformational ensemble of each diarylmethane revealed that the identity of the substituent group influences the adopted conformation of the substrate; specifically, the Boltzmann averaged plane angle (∠) between the two aryl groups varies ca. 30° depending on the size of the R group (59.7° for 4 and 87.4° for 15, Figure 3C).41 The conformational change manifests itself in the 13C NMR shift of the central methine carbon signal, which correlates well with ∠ and er (see Supporting Information). This suggests that the structural organization of the diarylmethine is determining enantioselectivity and sheds light on the mechanistic effect of a distal substituent.
In summary, we disclose the first report of a highly enantioselective, copper-catalyzed aromatic Finkelstein reaction. Guanidinylated peptide ligands serve as enabling tool for the desymmetrization of diarylmethanes via stereoselective bromide to iodide substitution at Csp2-centers. Subsequent stereoretentive transition metal-catalyzed transformations enable a platform for the generation of chiral diarylmethane libraries. To elucidate the privileged nature of diarylmethanes in desymmetrization reactions, this study identified key parameters that govern selectivity, establishing underlying principles for future studies. A secondary kinetic resolution was identified as a crucial contributor to the excellent levels of enantioselectivity and a computed steric parameter led to insight into the preorganization required for selective catalysis. The features of the diarylmethane scaf-fold remain of great interest in not only asymmetric catalysis, but also in the study of ligand receptor interactions in medicinal chemistry. It seems plausible that the determinants of selectivity in one field may be related to selectivity in the other, which may justify further exploration of this analogy.
Supplementary Material
ACKNOWLEDGMENT
We are grateful to Juan Serviano for help with collecting IR spectroscopic data. This work is supported by the National Institute of General Medical Sciences of the United States National Institutes of Health (R35 GM 132092 to S.J.M. and R35 GM 136271 to M.S.S.). T.M. is grateful for a Walter Benjamin Fellowship (DFG, MO 4495/1-1). A National Science Foundation Graduate Research Fellowship (DGE-2139841) for T.E.M, and a Roaring Fork Valley Research Circle Postdoctoral Fellowship for M.A.H. (American Cancer Society, PF-21-089-01-ET) are kindly acknowledged. L.J.K, M.A.H, and M.S.S are thankful to the Center for High Performance Computing (CHPC) at the University of Utah for the support and resources provided.
Footnotes
The Supporting Information is available free of charge via the Internet at http://pubs.acs.org.”
Detailed experimental procedures and analytical data; X-ray crystallographic data for 2 (CCDC 2286887); computational data.
The authors declare no competing financial interest.
REFERENCES
- (1).Finkelstein H Darstellung organischer Jodide aus den entsprechenden Bromiden und Chloriden. Ber. Dtsch. Chem. Ges 1910, 43, 1528–1532. [Google Scholar]
- (2).Sharts CM; Sheppard WA Modern methods to prepare monofluoroaliphatic compounds. Org. React 1974, 21, 125–406. [Google Scholar]
- (3).Miller JA; Nunn MJ Synthesis of alkyl iodides. J. Chem. Soc., Perkin Trans 1 1976, 416–420. [Google Scholar]
- (4).Hayami J; Hihara N; Kaji A SN2 reactions in dipolar aprotic solvents. IX. An estimation of nucleophilicities and nucleofugicities of anionic nucleophiles studied in the reversible Finkelstein reactions of benzyl derivatives in acetonitrile - dissociative character of the reaction as studied by the nucleofugicity approach. Chem. Lett 1979, 413–414. [Google Scholar]
- (5).Chalk CD; McKenna J; Williams IH NPE effects in bimolecular nucleophilic substitution. J. Am. Chem. Soc 1981, 103, 272–281. [Google Scholar]
- (6).Kürti L; Czakó B Strategic Applications of Named Reactions in Organic Synthesis. Elsevier, Amsterdam: 2005. [Google Scholar]
- (7).Smith MB; March J Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (6th Ed.), Wiley-Interscience, New York: 2007. [Google Scholar]
- (8).Olah GA; Narang SC; Field LD Synthetic methods and reactions. 103. Preparation of alkyl iodides from alkyl fluorides and chlorides with iodotrimethylsilane or its in situ analogs. J. Org. Chem 1981, 46, 3727–3728. [Google Scholar]
- (9).Takagi K; Hayama N; Okamoto T Synthesis of aryl iodides from aryl halides and potassium iodide by means of Nickel catalyst. Chem. Lett 1978, 7, 191–192. [Google Scholar]
- (10).Lindley J Copper assisted nucleophilic substitution of aryl halogen. Tetrahedron 1984, 40, 1433–1456. [Google Scholar]
- (11).Klapars A; Buchwald SL Copper-Catalyzed Halogen Exchange in Aryl Halides: An Aromatic Finkelstein Reaction. J. Am. Chem. Soc 2002, 124, 14844–14845. [DOI] [PubMed] [Google Scholar]
- (12).Li L; Liu W; Zeng H; Mu X; Cosa G; Mi Z; Li C-J Photo-induced Metal-Catalyst-Free Aromatic Finkelstein Reaction. J. Am. Chem. Soc 2015, 137, 8328–8331. [DOI] [PubMed] [Google Scholar]
- (13).Evano G; Nitelet A; Thilmany P; Dewez DF Metal-Mediated Halogen Exchange in Aryl and Vinyl Halides: A Review. Front. Chem 2018, 6, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Meyer-Eppler G; Küchler L; Tenten C; Benkhäuser C; Brück S; Lützen A Cheap and Easy Synthesis of Highly Functionalized (Het)aryl Iodides via the Aromatic Finkelstein Reaction. Synthesis 2014, 46, 1085–1090. [Google Scholar]
- (15).Jin X; Davies RP Copper-catalysed aromatic-Finkelstein reactions with amine-based ligand systems. Cat. Sci. Technol 2017, 7, 2110–2117. [Google Scholar]
- (16).Andrews MJ; Carpentier A; Slawin AMZ; Cordes DB; Macgregor SA; Watson AJB Mechanism of Cu-Catalyzed Iododeboronation: A Description of Ligand-Enabled Transmetalation, Disproportionation, and Turnover in Cu-Mediated Oxidative Coupling Reactions ACS Catal. 2023, 13, 11117–11126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Fürstner A; Kennedy JW Total syntheses of the tylophora alkaloids cryptopleurine, (–)-antofine, (–)-tylophorine, and (–)-ficuseptine, C. Chem. Eur. J 2006, 12, 7398–7410. [DOI] [PubMed] [Google Scholar]
- (18).Kim CH; An HJ; Shin WK; Yu W; Woo SK; Jung SK; Lee E Total Synthesis of (–)-Amphidinolide E. Angew. Chem. Int. Ed 2006, 45, 8019–8021. [DOI] [PubMed] [Google Scholar]
- (19).Jones CP; Anderson KW; Buchwald SL Sequential Cu-catalyzed amidation-base-mediated camps cyclization: a two-step synthesis of 2-aryl-4-quinolones fromo-halophenones. J. Org. Chem 2007, 72, 7968–7973. [DOI] [PubMed] [Google Scholar]
- (20).Kim B; Chinn AJ; Fandrick DR; Senanayake CH; Singer RA; Miller SJ Distal Stereocontrol Using Guanidinylated Peptides as Multifunctional Ligands: Desymmetrization of Diarylmethanes via Ullman Cross-Coupling. J. Am. Chem. Soc 2016, 138, 7939–7945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Chinn AJ; Kim B; Kwon Y; Miller SJ Enantioselective Intermolecular C–O Bond Formation in the Desymmetrization of Diarylmethines Employing a Guanidinylated Peptide-Based Catalyst. J. Am. Chem. Soc 2017, 139, 18107–18114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kwon Y; Chinn A; Kim B; Miller SJ Divergent Control of Point and Axial Stereogenicity: Catalytic Enantioselective C−N Bond-Forming Cross-Coupling and Catalyst-Controlled Atroposelective Cyclodehydration. Angew. Chem. Int. Ed 2018, 57, 6251–6255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Yoon H; Galls A; Rozema SD; Miller SJ Atroposelective Desymmetrization of Resorcinol-Bearing Quinazolines via Cu-Catalyzed C–O Bond Formation. Org. Lett 2022, 24, 762–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Li B; Chao Z; Li C; Gu Z Cu-Catalyzed Enantioselective Ring Opening of Cyclic Diarylaiodoniums toward the Synthesis of Chiral Diarylmethanes. J. Am. Chem. Soc 2018, 140, 9400–9403. [DOI] [PubMed] [Google Scholar]
- (25).Chu L; Wang X-C; Moore CE; Rheingold AL; Yu J-Q Pd-Catalyzed Enantioselective C–H Iodination: Asymmetric Synthesis of Chiral Diarylmethylamines. J. Am. Chem. Soc 2013, 135, 16344–16347. [DOI] [PubMed] [Google Scholar]
- (26).Schmidt F; Stemmler RT; Rudolph J; Bolm C Catalytic asymmetric approaches towards enantiomerically enriched diarylmethanols and diarylmethylamines. Chem. Soc. Rev 2006, 35, 454–470. [DOI] [PubMed] [Google Scholar]
- (27).Wei J; Gandon V; Zhu Y Amino Acid-Derived Ionic Chiral Catalysts Enable Desymmetrizing Cross-Coupling to Remote Acyclic Quaternary Stereocenters. J. Am. Chem. Soc 2023, 145, 16796–16811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Toto P; Gesquière J-C; Cousaert N; Deprez B; Willand N UFU (“Ullmann–Finkelstein–Ullmann”): a new multicomponent reaction. Tetrahedron Lett. 2006, 47, 4973–4978. [Google Scholar]
- (29).Chu W-D; Zhang L-F; Bao X; Zhao X-H; Zeng C; Du J-Y; Zhang G-B; Wang F-X; Ma X-Y; Fan C-A Asymmetric Catalytic 1,6-Conjugate Addition/Aromatization of para-Quinone Methides: Enantioselective Introduction of Functionalized Diarylmethine Stereogenic Centers. Angew. Chem. Int. Ed 2013, 52, 9229–9233. [DOI] [PubMed] [Google Scholar]
- (30).Cheng X-F; Li Y; Su Y-M; Yin F; Wang J-Y; Sheng J; Vora HU; Wang X-S; Yu J-Q Pd(II)-Catalyzed Enantioselective C–H Activation/C–O Bond Formation: Synthesis of Chiral Benzofuranones. J. Am. Chem. Soc 2013, 135, 1236–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lee T; Wilson TW; Berg R; Ryberg P; Hartwig JF Rhodium-Catalyzed Enantioselective Silylation of Arene C–H Bonds: Desymmetrization of Diarylmethanols. J. Am. Chem. Soc 2015, 137, 6742–6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Guduguntla S; Hornillos V; Tessier R; Fananas-Mastral M; Feringa BL Chiral Diarylmethanes via Copper-Catalyzed Asymmetric Allylic Arylation with Organ-olithium Compounds. Org. Lett 2016, 18, 252–255. [DOI] [PubMed] [Google Scholar]
- (33).Lu S; Song X; Poh SB; Yang H; Wong MW; Zhao Y Access to Enantiopure Triarylmethanes and 1,1-Diarylalkanes by NHC-Catalyzed Acylative Desymmetrization. Chem. Eur. J 2017, 23, 2275–2281. [DOI] [PubMed] [Google Scholar]
- (34).Bishop MJ; McNutt RW An efficient synthesis of the benzhydrylpiperazine delta opioid agonist (+)-BW373U86. Bioorg. Med. Chem. Lett 1995, 5, 1311–1314. [Google Scholar]
- (35).Wada S; Hitomi T; Tokuda H; Tanaka R Anti-Tumor-Initiating Effects of Spiro-biflavonoids from Abies sachalinensis. Chem. Biodivers 2010, 7, 2303–2308. [DOI] [PubMed] [Google Scholar]
- (36).Hurtley AE; Stone EA; Metrano AJ; Miller SJ Desymmetrization of Diarylmethylamido Bis(phenols) through Peptide-Catalyzed Bromination: Enantiodivergence as a Consequence of a 2 amu Alteration at an Achiral Residue within the Catalyst. J. Org. Chem 2017, 82, 11326–11336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Gustafson J; Sigman MS; Miller SJ Linear Free Energy Relationship Analysis of a Catalytic Desymmetrization Reaction of a Diarylmethane-Bis(phenol). Org. Lett 2010, 12, 2794–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Sigman MS; Harper KC; Bess EN; Milo A The Development of Multidimensional Analysis Tools for Asymmetric Catalysis and Beyond. Acc. Chem. Res 2016, 49, 1292–1301. [DOI] [PubMed] [Google Scholar]
- (39).Poater A; Cosenza B; Correa A; Giudice S; Ragone F; Scarano V; Cavallo L SambVca: A web application for the calculation of the buried volume of N-heterocyclic carbene ligands. Eur. J. Inorg. Chem 2009, 13, 1759–1766. [Google Scholar]
- (40).Clavier H; Nolan SP Percent buried volume for phosphine and N-heterocyclic carbeneligands: steric properties in organometallic chemistry. Chem. Commun 2010, 46, 841–861. [DOI] [PubMed] [Google Scholar]
- (41).The plane angle range within each conformational ensemble is less than 3.4° for all compounds, see full data in the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.