

Summary
Immunoglobulin A (IgA) maintains commensal communities in the intestine while preventing dysbiosis. IgA generated against intestinal microbes assures the simultaneous binding to multiple, diverse commensal-derived antigens. However, the exact mechanisms by which B cells mount broadly reactive IgA to the gut microbiome remains elusive. Here we have shown that IgA B cell receptor (BCR) is required for B cell fitness during the germinal center reaction in Peyer’s patches and for generation of gut-homing plasma cells. We demonstrate that IgA BCR drove heightened intracellular signaling in mouse and human B cells and as a consequence, IgA+ B cells received stronger positive selection cues. Mechanistically, IgA BCR signaling offset Fas-mediated death, possibly rescuing low affinity B cells to promote a broad humoral response to commensals. Our findings reveal an additional mechanism linking BCR signaling, B cell fate, and antibody production location which have implications for how intestinal antigen recognition shapes humoral immunity.
Graphical Abstract
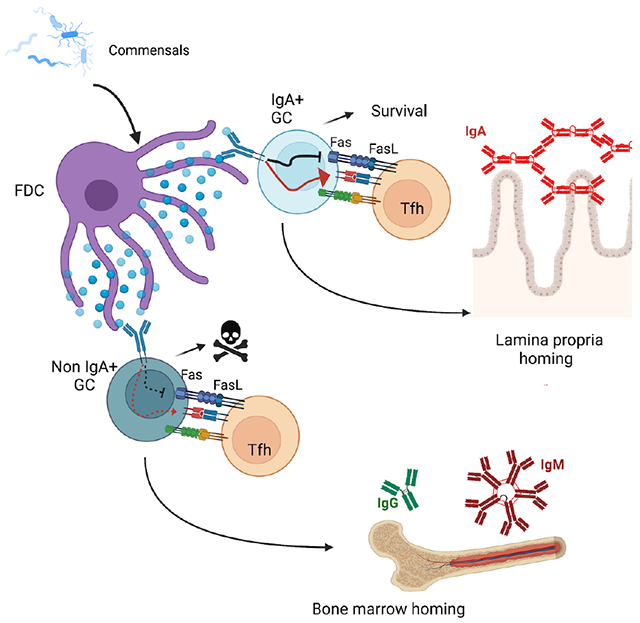
In brief
IgA response is critical for intestinal immunity, however the role of IgA during germinal center (GC) is unknown. Here, Raso et al. reveal that the IgA B cell receptor (BCR) is required for Peyer’s patch (PP) GC B cell survival and selection into memory B cells and gut-homing plasma cells.
INTRODUCTION
Secretion of Immunoglobulin A (IgA) at the intestinal interface is critical to control the ecology of commensal communities: data from both murine and human studies indicate that deficiency in IgA responses alters the composition of the microbiome1–3, leading to increased susceptibility to enteric pathogens and reduced tissue responses to oral vaccines.4,5
In recent years, the specificity of IgA has been the focus of intense investigation: it is accepted that a single IgA can bind multiple unrelated bacterial taxa6–9 a property described as cross-species reactivity10, but species-specific IgA also exist11, and both processes contribute to widen IgA response to commensals. While IgA can coat commensals in the absence of T cells12–14, there is evidence that IgA selection is regulated by T cells.15 Furthermore, IgA-secreting plasma cells in the intestine are highly mutated16–20 and require affinity maturation to properly control commensals.21 Somatic mutations and affinity maturation occur in the germinal center (GC) reaction and depend on T cell help, suggesting that GC may play an important role in establishing a functional intestinal IgA response. Peyer’s patches (PPs)22 are characterized by ongoing GC23, and seminal work has identified them as the main site for the generation of IgA against intestinal antigens.24,25 In PPs, tissue-resident cells produce environmental cues that enforce IgA class switch recombination before B cell entry into the GC26–28, and control differentiation of antigen-specific plasma cells.29 The GC reaction is primarily required for the selection of B cell clones binding antigen with high affinity, that eventually differentiate into antibody secreting plasma cells30–32, a feature which is difficult to reconcile with the known broad reactivity of IgA. However, in contrast to peripheral lymph nodes (LNs) or spleen, GC regulation in PPs has been shown to be less dependent on positive and negative T follicular helper (Tfh) cell signals.33,34 Moreover, recent data have highlighted that the complexity of gut microbiome shapes both affinity and clonality during the GC reaction: under homeostatic conditions, selection of highly dominant B cell clones is rare in PPs, a starkly different situation compared to peripheral LNs upon immunization.30,35,36 Thus, a PP-specific mechanism must exist that modulates the GC reaction and underpins the generation of poly- or cross-species reactive intestinal IgA.
Following acquisition of activation-induced cytidine deaminase (AID)-mediated BCR point mutations, GC B cells test their mutated BCR for binding of antigen displayed by follicular dendritic cells and receive T cell help by cognate Tfh cells. The relative contribution of T cell help and BCR signaling in controlling GC selection has been debated37–41: the current view is that Tfh cells are central in discriminating between B cells carrying BCR with different affinities and they drive differentiation of high affinity B cells into plasma cells.32 Nevertheless, several groups have reported that surface BCR isotypes can intrinsically modulate B cell differentiation, including selection during the GC reaction.42–45
The impact of IgA BCR in the GC reaction of PPs, and its effect on the humoral immune response at the intestinal barrier, remain largely unknown. In the present study, we examined the role of IgA in regulating chronic GC reactions in response to commensal and pathogenic bacteria. We found that IgA facilitated GC B cell fitness and was required for the generation of gut-homing plasma cells. IgA+ B cells were positively selected in the light zone and were resistant to apoptosis. The IgA BCR mediated stronger intracellular signaling and offset FasL-dependent apoptosis. In conclusion, acquisition of an IgA BCR in PPs is not only required for the generation of low inflammatory antibodies that can be translocated into the lumen, but it also lowers the threshold of selection and might contribute to a broad response to commensals.
RESULTS
IgA+ B cells dominate the germinal center reaction and are preferentially selected into the memory compartment.
We initially sought to evaluate whether surface IgA BCR was required for effective participation in the GC reaction. To this end, we used mice with a targeted deletion of the IgA switch region and sequence, which completely abolishes both IgA-expressing B cells and secreted IgA.46 Immunohistochemical analysis of PPs did not reveal altered GC structure in the absence of IgA (Figure 1A), with normal size of the follicle and GC (Figure S1A). We then analyzed PP and mesenteric lymph node (mLN) B cell populations by flow cytometry for the distribution of follicular B cells (B220+ IgD+ GL7−), GC B cells (B220+ IgD- GL7+), memory B cells (MemB, B220+ IgD− GL7− CD38+ CD138−), and plasma cells (PCs, IgD− GL7− CD138+) (Figure 1B, S1B). No difference in B cell subsets was observed in both PPs (Figures 1C–E, S1C) and mLN (Figure S1D) between mice deficient for IgA (Iga−/− and co-housed, littermate controls (Iga+/+), despite an altered distribution of antibody (Ab) isotypes (Figures S1E–J).
Figure 1. IgA+ B cells dominate the germinal center reaction and are preferentially selected into the memory compartment.
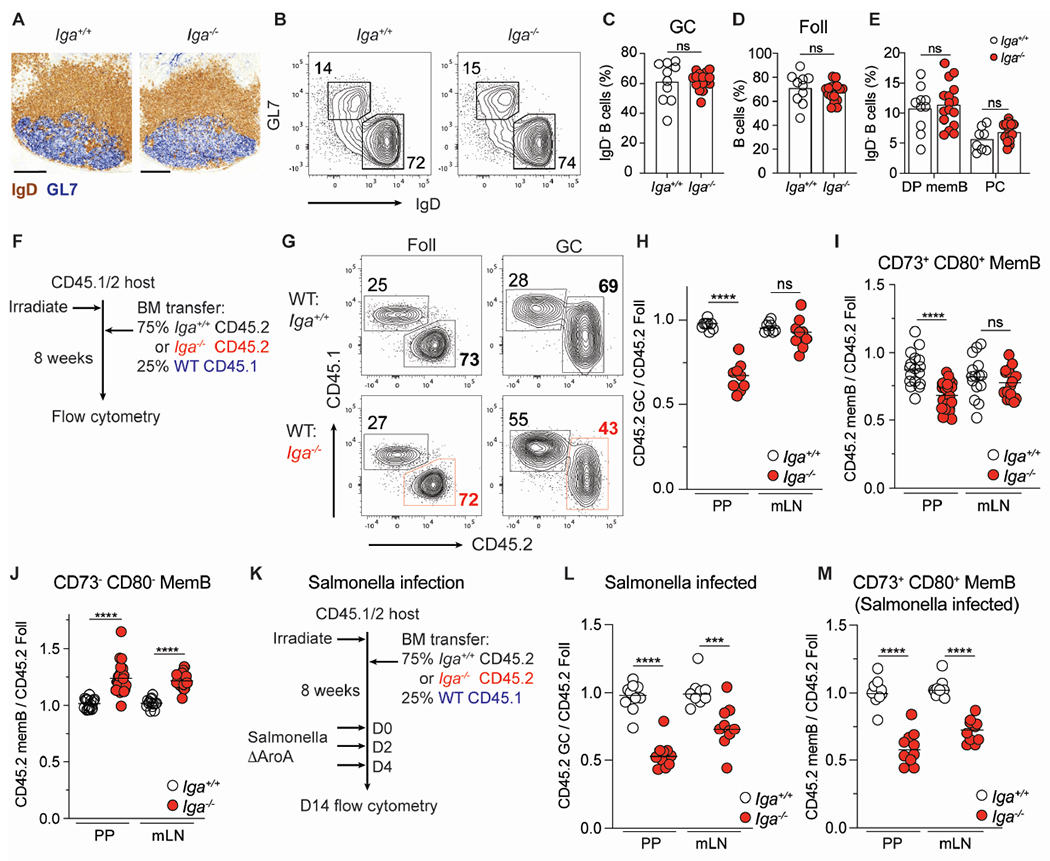
(A-E) Steady state, co-housed, 8–12-week-old Iga+/+ and Iga−/− mice.
(A) Frozen PP sections from Iga+/+ and Iga−/− mice stained with immunohistochemistry antibodies against IgD (brown) and GL7 (blue). Scale bars, 200 μm.
(B) Representative plot of PP GC (GL7+ IgD−) and Foll (GL7− IgD+) B cells in Iga+/+ and Iga−/− mice. Gated on live CD138− B cells.
(C) PP GC B cells as percentage of activated (IgD−) B cells.
(D) PP Foll B cells as percentage of B cells.
(E) CD73+ CD80+ (DP) MemB, PCs as percentage of activated B cells.
(F) Iga−/− mixed BM chimera experimental set up for (G-J).
(G) Representative CD45 staining on Foll and GC B cells in mixed BM chimeras.
(H) Ratio of frequency of CD45.2 GC B cells to CD45.2 Foll B cells in PP and mLN.
(I, J) Ratio of frequency of CD45.2 DP memB cells (I) or CD45.2 CD73− CD80− (DN) MemB cells (J) to CD45.2 Foll B cells in PP and mLN.
(K) Salmonella infection experimental setup in Iga−/− mixed BM chimera for (L-M).
(L) Ratio of frequency of CD45.2 GC B cells to CD45.2 Foll B cells in PP and mLN of Salmonella infected chimeras.
(M) Ratio of frequency of CD45.2 DP MemB cells to CD45.2 Foll B cells in PP and mLN of Salmonella infected chimeras.
Data from at least 3 independent experiments with 2-3 mice per group (A-M). Each symbol represents one mouse. ns=not significant; ***p < 0.0005; ****p < 0.0001; Unpaired two-tailed Student’s t-test.
See also Figure S1.
However, while PP B cells expressing isotypes other than IgA can effectively become GC B cells, suggesting that IgA is not required for GC formation, it does not rule out a role for the IgA BCR during the GC reaction. GC selection is an iterative immune process controlled by competition among B cell clones47, thus, we wanted to test whether IgA was required in a competitive setting. To this end, we generated mixed bone marrow (BM) chimeric mice where most B cells (75%) were from Iga−/− or Iga+/+ CD45.2+ donor and the remaining B cells (25%) were from wild-type (WT) CD45.1+ donor (Figure 1F). Flow cytometry analysis revealed that in PPs, Iga−/− B cells were largely impaired in their ability to participate in the GC reaction compared to WT B cells, while follicular and GC B cells were present at the same ratio in the control mixed BM chimeras (Figures 1G, 1H). No defect was observed in mLN of either mixed BM chimeras, likely due to the low frequency of IgA+ B cells participating in the GC reaction and their peculiar transforming growth factor-β (TGFβ)-independent27, commensal-independent36 nature. The inability of Iga−/− B cells to compete in the GC was also apparent in immunohistochemistry staining of PP frozen sections, with CD45.2 Iga−/− B cells filling the B cell follicle, while inefficiently entering the GC structure, which was instead dominated by CD45.1 WT B cells. This pattern was reversed in control mixed BM, with CD45.2 Iga+/+ B cells being more abundant in both B cell follicle and GC (Figure S1K). GC B cells can give rise to both PCs and MemB cells. MemB cells expressing CD73 and CD80 (double positive, DP) are thought to be directly generated in GCs48. In line with reduced participation in the GC reaction, mixed BM chimeras revealed that Iga−/− B cells also contributed less to GC-derived MemB cells in PPs, but not mLN (Figure 1I). In contrast, IgA deficient B cells were overrepresented in the double-negative (DN, CD73− CD80−) compartment (Figure 1J), suggesting that IgA is required for optimal GC participation and formation of GC-derived MemB cells.
Chronic GC reactions in PPs and mLN are mainly driven by commensals but can also incorporate responses to pathogens22,24. Intestinal inflammation can alter the cytokines milieu and impact GC reaction49,50: to test whether intrinsic IgA-deficiency impacts GC B cell competition upon enteric pathogen encounter, we infected mixed BM chimera with metabolically deficient Salmonella strain (ΔAroA) which is able to induce an IgA response (Figure 1K).49 Similar to homeostatic conditions, Iga−/− B cells were largely outcompeted in the GC of PPs, but enteric infection also preferentially expanded Iga+/+ B cells over Iga−/− B cells in GC reaction in mLN (Figure 1L). In contrast to what was observed during commensal driven responses, DP MemB cells were affected by IgA deficiency in both PPs and mLN upon Salmonella infection (Figure 1M) while DN MemB were largely unchanged (Figure S1L). This discrepancy could be due to enhanced migration of PP-generated DP MemB cells into mLN during Salmonella infection compared to homeostatic situations.
Together our data show that IgA is required for optimal GC persistence and MemB cell formation during both homeostatic responses to commensal bacteria and inflammatory responses driven by pathogenic enteric bacteria.
IgA is required for efficient plasma cell homing to the intestinal lamina propria.
In addition to MemB cells, GC B cells can also differentiate into PCs, and selection during the GC reaction is thought to be critical for the generation of PCs that secrete high affinity Abs32. PP GCs generate most IgA-secreting PCs that egress from lymphatics at the basal side of the PPs using S1PR151 and home to the lamina propria via the integrin alpha 4 beta 7 (α4β7)52,53 and C-C chemokine receptor type 9 (CCR9).54 Administration of the S1PR1 functional antagonist FTY720 traps nascent PCs in the PPs51, therefore allowing full characterization of PCs before they egress and migrate into the lamina propria. Indeed, the vast majority of PCs expressing α4β7 and CCR9 in PPs were IgA+, while PCs that did not express α4β7 and CCR9 (non-gut-homing) were not enriched as IgA expressing cells in PPs (Figure 2A).
Figure 2. IgA is required for efficient plasma cell homing to the intestinal lamina propria.
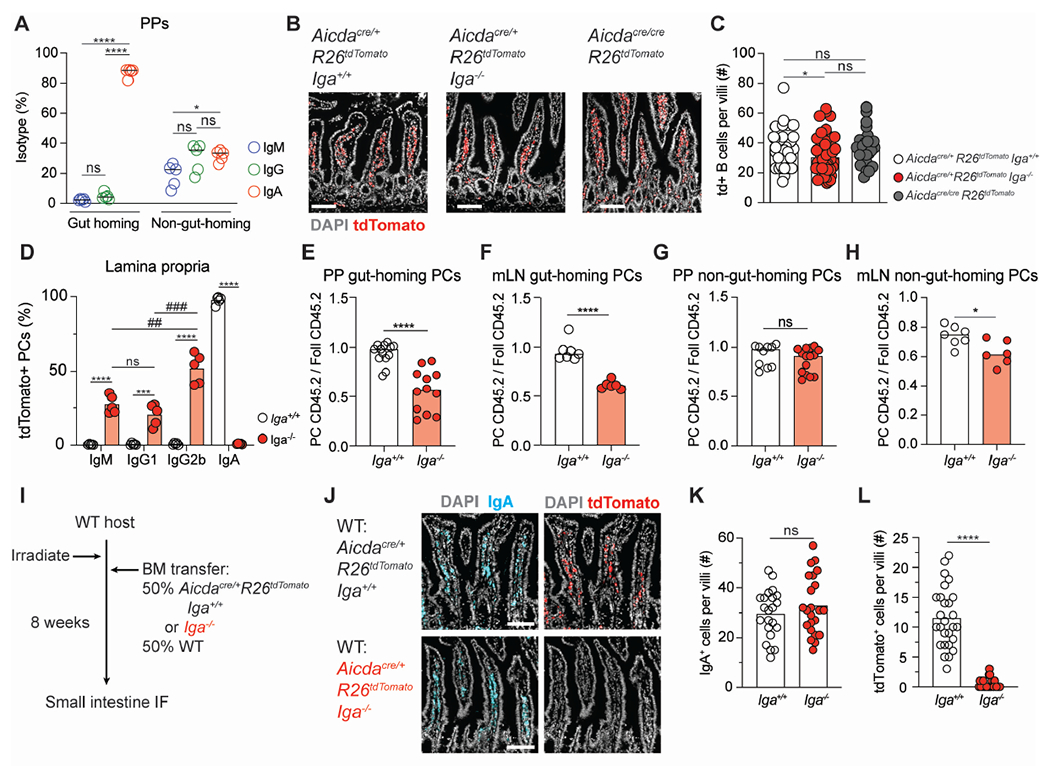
(A) Isotypes of gut-homing (α4β7+ CCR9+) and non-gut homing PCs from PPs of WT mice treated with FTY-720 for seven days. IgG is defined as IgG1+ or IgG2b+.
(B) Representative immunofluorescence of small intestinal villi from Aicdacre/+ Rosa26Stop-tdTomato Iga+/+, Aicdacre/+ Rosa26Stop-tdTomato Iga−/−, Aicdacre/cre Rosa26Stop-tdTomato mice stained with DAPI (gray) and tdTomato (red). Scale bars, 100 μm.
(C) Quantification of multiple images as in (B). Number of tdTom+ cells per villi.
(D) Isotype staining in Aicdacre/+ Rosa26Stop-tdTomato Iga+/+ and Aicdacre/+ Rosa26Stop-tdTomato Iga−/− lamina propria. Gated on CD98+ B220− CD4− PCs.
(E, F) Ratio of frequency of CD45.2 gut-homing PCs to CD45.2 Foll B cells from PPs (E) or mLN (F) of mixed BM chimeras.
(G, H) Ratio of frequency of CD45.2 non-gut-homing PCs to CD45.2 Foll B cells from PPs (G) or mLN (H) of mixed BM chimeras.
(I) Aicdacre/+ Rosa26Stop-tdTomato Iga+/+ mixed BM chimera experimental set up for (J-L).
(J) Representative immunofluorescence of small intestinal villi stained for DAPI (gray), IgA (cyan), and tdTomato (red) in WT: Aicdacre Rosa26Stop-tdTomato Iga+/+ or Iga−/− mixed BM chimeras. Scale bars, 100 μm.
(K, L) Quantification of multiple images as in (J). Number of IgA+ cells (K) or tdTom+ cells (L) per villi.
Data from 2 independent experiments with 2-3 mice per group (A, C, D, K, L). Data from at least 3 independent experiments with 3-5 mice per group (E-H). Each symbol represents one mouse. (C, K, L) Each symbol represents one villi with at least 5 villi quantified per image.
ns=not significant, *p < 0.005; ##p < 0.001; ***, ###p < 0.0005; ****p < 0.0001. Unpaired two-tailed Student’s t-test in E-H, K, L. One way ANOVA was used in A(*), C(*), and D(#) .
See also Figure S2.
PP-generated PCs egress from efferent lymphatics, that connect to the mLN, on their way to reach the lamina propria: similar to PPs, most α4β7 and CCR9 expressing PCs in mLN were IgA+ (Figure S2A). The requirements for IgA BCR in underpinning gut-tropism is not absolute, as analysis of PCs in FTY720-treated Iga−/− mice revealed that IgG1, IgG2b, and IgM B cells were able to upregulate gut-homing receptors (Figure S2B).
In line with this data, absence of IgA does not completely impair gut-homing, as microscopy analysis of fluorescent PCs in the intestinal lamina propria of mice unable to switch to IgA, either because they have selective IgA-deficiency (Aicdacre/+ Rosa26Stop-tdTomato/+ Iga−/−) Or because they lack the molecular machinery required for class switch recombination (Aicdacre/cre Rosa26stop-tdTomato/+) did not show decreased PC compartment compared to IgA-sufficient mice (Aicdacre/+ Rosa26stop-tdTomato/+ Iga+/+) (Figures 2B, 2C, S2C). Analysis of PCs in the lamina propria of Aicdacre/+ Rosa26Stop-tdTomato/+ Iga−/− mice revealed that they are mostly IgG2b, with contributions from IgM and IgG1 as well (Figure 2D).
Nevertheless, fully deficient mice might not sufficiently recapitulate physiological aspects of PC biology, as PC generation from GC B cells is dependent on competition-based selection32. In mixed chimeras while the total Iga−/− PCs were slightly reduced (Figure S2D), a marked reduction was observed in gut-homing Iga−/− PCs in both PPs (Figure 2E) and mLN (Figure 2F) while non-gut-homing PCs were minimally altered (Figure 2G, 2H). To determine whether Iga−/− PCs were nonetheless able to reach the lamina propria, we generated mixed BM chimeras in which half of PCs , either Iga+/+ or Iga−/−, were fluorescently marked (Aicdacre/+ Rosa26stop-tdTomato/+ Iga+/+ or Iga−/−) (Figure 2I). While in the Iga+/+: WT chimera the intestine was repleted with IgA+ PCs, from either WT (non-fluorescent) or Iga+/+ B cells (tdTom+) (Figures 2J, 2K, S2E), Iga−/− PCs (tdTom+) were not observed in the intestine of Iga−/−: WT chimera, suggesting that IgA-deficiency impairs homing to the LP in a competitive setting (Figures 2J, 2L).
Taken together, these data point to a critical role of surface IgA to instruct the generation of gut-homing PCs. It appears that, while non-IgA expressing B cells can differentiate into PCs, they are largely unable to upregulate homing receptors required for efficient migration into the intestinal lamina propria.
Mixed BM chimeras are a robust experimental system to investigate competitive requirements for immunological processes. However, the need for irradiation can introduce alterations due to cell death, especially in the gut55. Therefore, to further expand our findings to a competitive system that does not require irradiation, we took advantage of allelic exclusion, a process by which B cells only transcribe one heavy chain locus during development. As a result of this, IgA heterozygous (Iga+/−) mice normally develop two genetically distinct sets of B cells, one sufficient for IgA and one deficient for IgA, effectively generating a natural WT:Iga−/− chimera (Figure 3A). Since WT and Iga−/− cells carry distinct immunoglobulin heavy chain allotypes, allotypic reagents can be used to distinguish B cells from WT (IgHb) and Iga−/− (IgHa) origin. Due to allelic exclusion, follicular B cells are present at a 50:50 ratio in both WT (IgHa/b) and Iga+/− (IgHa/b) mice: if surface IgA does not preferentially enhance GC fitness, we expect to observe a 50% reduction of total IgA GC B cells in Iga+/− mice compared to WT mice, due to the Iga−/− B cells equal contribution to the GC. However, if surface IgA promotes competitiveness in the GC, we anticipate the total IgA GC B cells to be unaffected in Iga+/− mice. Indeed, we found that totaI IgA GC B cells were indistinguishable between WT and Iga+/− mice, supporting the concept of IgA dominance during GC competition (Figure 3B). In contrast, IgMa B cells (derived from BM that are unable to express IgA) were expanded in activated B cell subsets compared to IgMb WT B cells (Figures 3C, S2F), a finding also observed in mixed BM chimeras (Figure S2G), suggesting that intrinsic IgA deficiency during GC competition allows the persistence of IgM+ GC B cells. Allotypic reagents can be used to track the origin of intestinal Ab by staining intestinal content for Ab-coated intestinal bacteria7,26: Flow cytometry analysis revealed that the mucosal humoral response was dominated by IgA+ cells (Figures 3D, S2H) and unaffected in Iga+/− mice, a finding in accordance with the requirement of IgA for intestinal PC generation (Figures 2I, 2K). Intestinal content can also be stained with serum, to measure the circulating Ab response to gut commensals56. While no changes were observed in commensal-reactive serum IgA between WT and Iga+/− mice (Figure 3E, 3F), Iga+/− PCs showed increased contribution to commensal-reactive IgM in serum (Figures 3G, 3H), suggesting that IgA-deficient B cells were able to respond to commensals and differentiate into PCs, but their response was redirected into systemic circulation. To assess whether a similar mechanism was in place during an antigen-specific response to foreign antigen, we orally immunized Iga+/−, Iga−/−, and WT mice with cholera toxin (CT)57 (Figure 3I). Analysis of Abs from in vitro differentiated MemB cells revealed the anti-CT Ab was dominated by IgA from Iga+/+ B cells (Figure 3J), while Iga−/− B cells increasingly generated anti-CT IgM Abs (Figure 3K) compared to Iga+/+ B cells (Figure 3L). Together our data support a model where surface IgA BCR allows for efficient participation in the PP GC and PC generation, underpinning efficient humoral immune responses at the intestinal barrier.
Figure 3. IgA is dominant in activated B cells of intrinsic chimera mice.
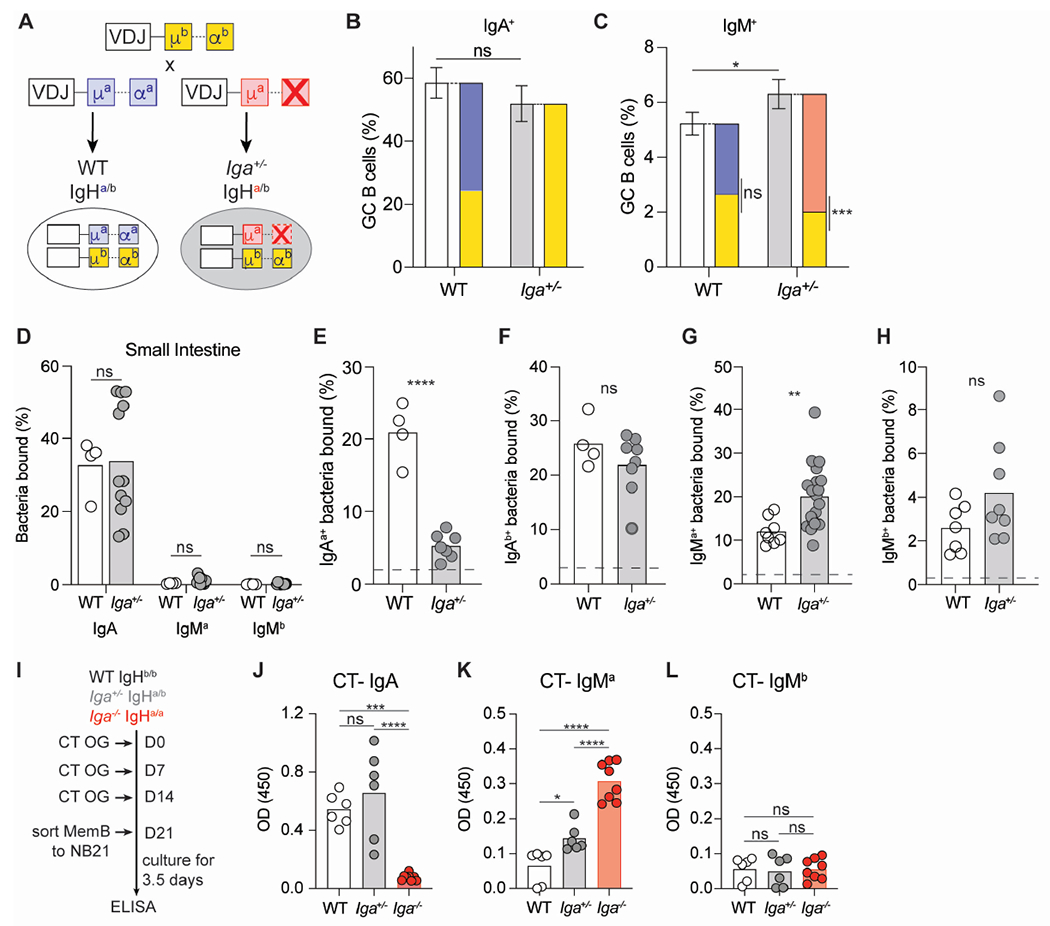
(A) Breeding setup for WT IgHa/b and Iga+/− IgHa/b mice.
(B, C) Percentage of IgA+ (B) and IgM+ (C) GC B cells in PPs. Solid bars represent indicated total isotype in GC; stacked bars represent allotypic isotype in GC of WT and Iga+/− mice.
(D) Percentage of endogenously coated small intestinal bacteria with indicated antibodies.
(E, F) Percentage of intestinal commensal from μMT mice stained by IgAa (E) or IgAb (F) antibodies from Iga+/+ or Iga+/− serum.
(G, H) Percentage of intestinal commensal bacteria from μMT mice stained by IgMa (G) or IgMb (H) antibodies from Iga+/+ or Iga+/− serum. Dashed line represents unstained μMT bacteria in E-H.
(I) Experimental setup for cholera toxin(CT) oral immunization and subsequent MemB cell in vitro culture with NB21 feeder cells.
(J, K, L) ELISA OD for CT specific IgA (J), IgMa (K), and IgMb (L) antibodies detected in MemB supernatant 3.5 days after in vitro culture.
Data from at least 3 independent experiments with 2-3 mice per group (B-H). Data from 4 independent experiments with 1-3 mice per group (J-L). ns=not significant, *p < 0.005; **p < 0.001; ***p < 0.0005; ****p < 0.0001; Unpaired two-tailed Student’s t-test in B-H. One way ANOVA in J-L.
See also Figure S2.
Resistance to cell death and T cell help enhance IgA+ GC B cell persistence.
The GC is divided into anatomically restricted dark zone (DZ) and light zone (LZ) compartments. While flow cytometry analysis of mixed BM chimeras did not show a preferential reduction of Iga−/− B cells in one GC compartment (Figures 4A, 4B, S3A), in situ immunofluorescent microscopy revealed that Iga−/− B cells were predominantly clustered in the LZ (Figures 4C, 4D, 4E). The GC reaction couples massive B cell proliferation with carefully controlled cell death to achieve clonal selection. We therefore initially tested the ability of Iga−/− B cells to undergo proliferation by measuring BrdU incorporation upon short-term labelling. Iga−/− and WT B cells proliferated at similar rates in both GC compartments and in the follicle (Figures 4F, S3B–E). In the DZ, GC B cells undergo cell death in response to the AID-dependent generation of a non-functional BCR.58,59 To test whether Iga−/− B cells were lost at this negative selection stage, we retrovirally expressed Bcl2, an anti-apoptotic gene that drives increased B cell survival (Figure S3F) and can rescue defects in DZ GC survival59 in mixed BM chimeras. Ectopic BCL-2 was unable to rescue Iga−/− B cells in both GC and MemB compartments (Figures 4G, S3G), suggesting IgA BCR does not confer competitive advantage during DZ selection.
Figure 4. IgA+ B cells exhibit higher cell death resistance and experience stronger T cell-dependent help in GC LZ.
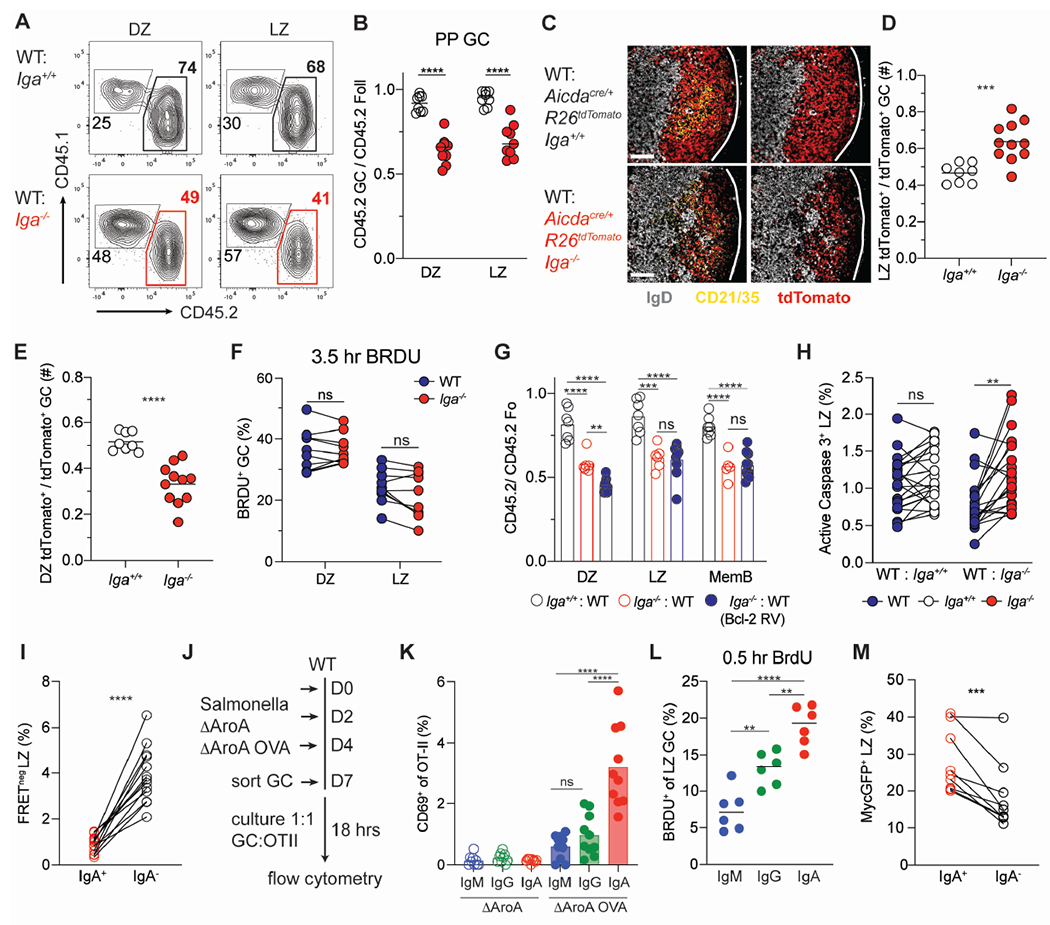
(A) Representative CD45 staining on PP GC LZ (CXCR4lo CD86hi) and DZ (CXCR4hi CD86hi) B cells in mixed BM chimeras.
(B) Ratio of frequency of CD45.2 DZ or LZ B cells to CD45.2 Foll B cells in PP of mixed BM chimeras.
(C) Representative PP immunofluorescence stained for DAPI (gray), tdTomato (red) and CD21/35 (cyan), in WT: Aicdacre/+ Rosa26Stop-tdTomato Iga+/+ or Iga−/− mixed BM chimeras. Scale bars, 100 μm. Lines indicate GC edges with LZ oriented on left, DZ right.
(D, E) Guantification of multiple images as in (C). Ratio of frequency of tdTom+ cells in LZ (D) or DZ (E) to total TdTom+ GC B cells.
(F) Percentage of BrdU+ DZ or LZ GC B cells in PPs of Iga−/− :WT mixed BM chimeras after 3.5 hour BrdU pulse.
(G) Ratio of frequency of indicated CD45.2 B cells to CD45.2 Foll B cells in PP of WT Bcl2: Iga−/− Bcl2 retroviral mixed BM chimeras.
(H) Percentage of active caspase3+ LZ GC B cells in mixed BM chimeras.
(I) Percentage of FRET negative LZ IgA+ and IgA− cells from Rosa26INDIA PPs.
(J) Experimental set up for Salmonella ΔAroA infection and in vitro GC antigen presentation in (K).
(K) Percentage of CD69+ CD4+ OT-II T cells co-cultured with indicated GC B cell isotype. PP GC subsets were sorted according to isotype expression (IgG = IgG1+ or IgG2b+). Mice were infected with Salmonella ΔAroA expressing or not expressing OVA (ΔAroA or ΔAroAOVA). Each symbol is the average of technical replicates from one mouse.
(L) Percentage of BrdU+ LZ GC B cells in PPs for indicated isotypes after 30-minute BrdU pulse. GC subsets were gated according to isotype expression (IgG = IgM− IgA−).
(M) Percentage of cMycGFP+ LZ IgA+ and IgA− B cells from cMyc-GFP PPs
Data are from at least 3 independent experiments with at least 3 mice per group (B, F, H, I, M, K). Data are from 2 experiments with 2-3 mice per group (C-E, G, L). ns=not significant, **p < 0.001; ***p < 0.0005; ****p < 0.0001. Unpaired two-tailed Student’s t-test in B, D, E, F, H, I, M. One-way ANOVA in G, K, L.
See also Figure S3.
In the LZ, GC B cells are deleted due to the absence of positive selection, which integrates both BCR signaling and T cell help. Cell death analysis in mixed BM chimera using an antibody against active caspase 3 that identifies apoptotic GC B cells, revealed a higher frequency of apoptosis among LZ, but not DZ, Iga−/− B cells (Figures 4H, S3H–I), a phenotype restricted to PPs (Figure S3J). Analysis of Rosa26INDIA apoptosis indicator mice which report active caspase 3 via FRET loss58 also revealed that IgA BCR confers protection from cell death in the LZ during the GC reaction in PPs (Figure 4I). To test whether increased apoptosis resistance in LZ IgA+ B cells was associated with higher degree of T cell help, we orally immunized mice with an attenuated Salmonella strain encoding OVA. The same number of GC B cells were sorted according to surface isotypes and co-cultured with naive OVA-specific TCR transgenic CD4+ T cells (Figure 4J). In CD4+ T cells, CD69 is an early activation marker that is rapidly upregulated upon TCR stimulation by cognate peptide-MHCII complex: IgA+ GC B cells induced higher CD69 expression in CD4+ T cells compared to IgM+ or IgG+ GC B cells (Figure 4K). Since no difference was observed in MHCII expression (Figure S3K), our results suggest that IgA expressing GC B cells are either equipped with a more efficient antigen processing and presentation machinery or are better in extracting antigen from follicular dendritic cells. Recently, it has been shown that LZ T cells metabolically refuel LZ GC B cells without controlling their re-entry into S phase60, a process that can be measured by a short pulse of BrdU. We observed that in the PP LZ, IgA+ GC B cells outcompeted other GC isotypes for T cell–derived refueling cues as shown by a higher proportion of IgA+ GC B cells had incorporated BrdU (Figure 4L), with a similar, albeit more subtle, trend also present in mLN (Figure S3L). Correspondingly, using a reporter mouse for c-Myc that can identify GC B cells that received T cell activation signals61,62, we found that IgA+ LZ B cells showed increased c-Myc expression, suggesting an integration of stronger positive selection cues (Figure 4M). Together our results support the concept that the IgA BCR protects GC B cells from cell death in the LZ via enhanced T cell-derived positive interaction that permits preferential selection during GC response.
IgA mediates stronger BCR signaling.
In the GC LZ, B cell clones that bind antigen with higher affinity are preferentially selected for survival58, therefore, we hypothesized that the IgA BCR would mediate stronger BCR signaling. Ex-vivo stimulation of Ca2+ dye-loaded GC B cells with pan anti-BCR revealed that, in contrast to IgM+ or IgG2b+ GC B cells which had undetectable Ca2+ responses, as previously reported37,63, IgA+ GC B cells spiked calcium readily and consistently (Figures 5A and 5D), a feature also observed using isotype-specific Ab stimulation (Figures 5B and 5E), despite no difference in surface expression of the BCR complex (Figure S4A). To increase sensitivity in BCR-dependent Ca2+ release analysis, we also generated mice that express both the red fluorescent protein Tdtomato and the ultrasensitive calcium indicator GCamp6 in GC and post-GC B cells (Aicdacre/+ Rosa26Stop-tdTomato/gCaMP6) and observed a similar IgA-dependent Ca2+ release (Figures 5C, 5F, S4B). In contrast, BCR-dependent Ca2+ release in MemB cells, albeit faster in IgA+ cells, was also observed in MemB cells of other isotypes (Figures S4C–S4G), highlighting the difference between GC and MemB cell BCR signaling. The IgA-dependent Ca2+ spike was BCR dependent as incubation with Ibrutinib, a BTK inhibitor, reduced the calcium flux observed in both IgA+ GC and MemB cells (Figures 5F, S4G- S4K) (22).
Figure 5. Enhanced BCR signaling in murine and human IgA+ GC B cells.
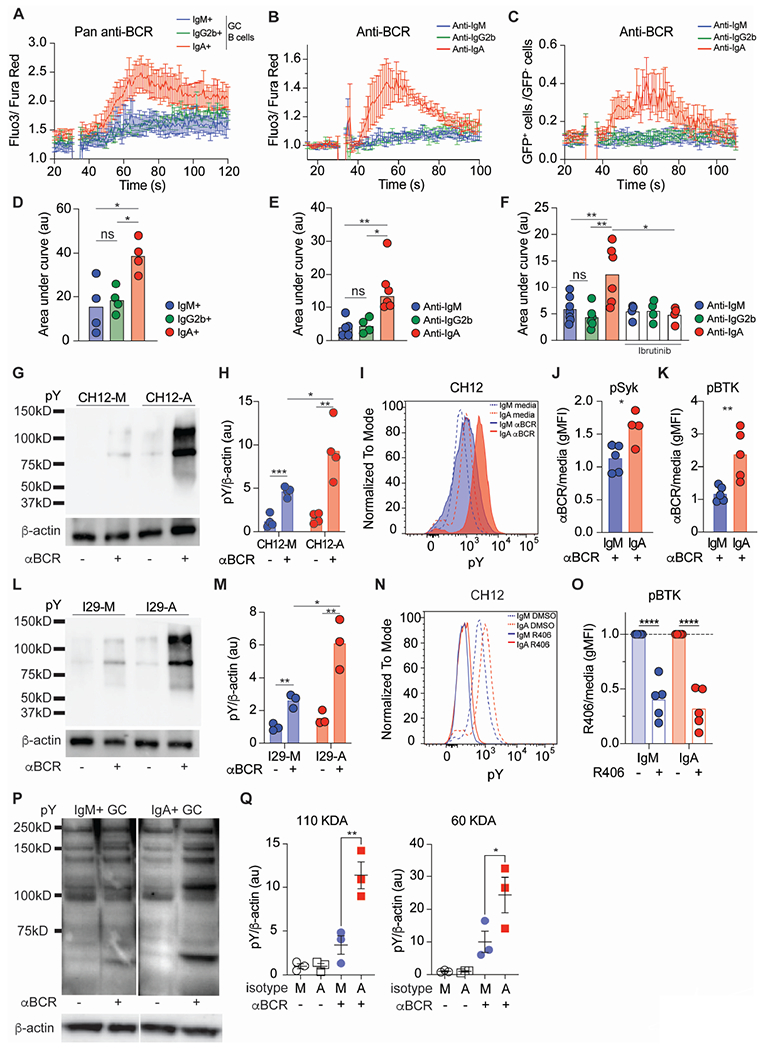
(A) Compiled calcium traces for IgM, IgG2b, and IgA PP GC B cells (defined as isotype negative populations) stimulated with pan anti-BCR.
(B) Compiled calcium traces for Aicdacre/+ Rosa26Stop-tdTomato/+ PP GC B cells stimulated with indicated anti-BCR isotypes.
(C) Compiled calcium traces from Aicdacre/+ Rosa26Stop-tdTomato/gCAMP PP GC B cells stimulated with indicated anti-BCR isotypes. Shown as ratio of frequency of gCAMP-GFP+ to gCAMP-GFP− cells per second.
(D) Area under the curve(AUC) of individual calcium traces calculated between 50 and 100 seconds for (A).
(E) AUC of individual calcium traces calculated between 40 and 100 seconds for (B).
(F) AUC of individual calcium traces calculated between 40 and 100 seconds for (C) Indicated samples were pre-treated with ibrutinib prior to anti-BCR stimulation.
(G) Representative pY and β-actin western blots for CH12 cells stimulated with anti-BCR.
(H) Quantitative analysis from (G) of pY normalized to β-actin.
(I) Representative histogram of pY in CH12 cells after BCR stimulation.
(J, K) pSyk (J) or pBTK (K) gMFI normalized to untreated(media) in CH12 cells after anti-BCR.
(L) Representative pY and β-actin western blot for I29 cells stimulated with anti-BCR.
(M) Quantitative analysis from (L) of pY normalized to β-actin.
(N) Representative pY histogram in CH12 cells after R406 incubation.
(O) pBTK gMFI normalized to untreated(media) after R406 incubation in CH12 cells.
(P) Representative pY and β-actin western blots from GC human tonsils of indicated isotypes after anti-BCR stimulation. Sorted as isotype negative populations.
(Q) Quantitative analysis from (P) of pY normalized to β-actin.
A, D, H are compiled data from 4 independent experiments. B, C, E, F, J, K, O, Q are compiled data from at least 3 independent experiments. ns=not significant, *p < 0.005; **p < 0.001; ***p < 0.0005; ****p < 0.0001. Unpaired two-tailed Student’s t-test in J, K, O. One-way ANOVA in D, E, F, H, M, Q.
BCR signaling via different isotypes has been shown to mediate various biological outcomes42,44,45, but little is known about the impact of the IgA BCR on intracellular signaling. Thus, to investigate IgA BCR signaling with increased granularity, we took advantage of CH12 and I29, two unrelated murine B cell lines with the ability to class switch to IgA64,65, to generate CH12 and I29 as IgM (CH12-M; I29-M) or IgA (CH12-A; I29-A) expressing cell lines (Figures S5A and S5B). Anti-isotype stimulation of CH12 revealed a more profound phosphorylation pattern in CH12-A compared to CH12-M by both western blot and phosphoflow (Figures 5G, 5H, 5I, S5C), including phosphorylation of SYK and BTK (Figures 5J, 5K, S5D). Similar findings were observed in I29 cells (Figures 5L, 5M, S5E).
Both IgA+ B cell lines were characterized by higher basal phosphorylated protein amounts (Figures 5I, S5F, S5G) which was dependent on the BCR signaling, as treatment with the Syk inhibitor, R406, reduced phosphorylated proteins including pBTK while leaving pSYK unchanged (Figures 5N, 5O, S5H–J). To determine whether IgA BCR mediates stronger BCR signaling in human B cells as well, we stimulated sorted GC from tonsils and observed that IgA+ GC B cells had increased total phosphorylated proteins compared to IgM+ GC cells (Figures 5P, 5Q). Our findings highlight that IgA BCR mediates stronger antigen receptor signaling in both basal and activated states, a property also conserved in human.
IgA resistance to FasL-dependent cell death increase GC competitiveness.
In the LZ compartment, in addition to providing help and positively selecting B cells, T cells can eliminate B cells via activation of the FasL-Fas-dependent cell death pathway.34 Previous reports suggest that different B cell isotypes might be differently sensitive to Fas-mediated killing66,67, but the role of IgA BCR in this process has not been investigated. Therefore, we sought to test whether IgA BCR confers resistance to Fas-mediated death using CH12 cells, which express similar surface Fas regardless of isotype (Figure S6A), and FasL expressing vesicles, which express the physiological FasL trimer and can induce apoptosis in Fas-expressing cells68,69. IgA+ CH12 were more protected from Fas dependent death compared to IgM+ CH12, suggesting an IgA intrinsic basal protection, likely driven by tonic, antigen-independent BCR signaling (Figures 6A, S5C).
Figure 6. IgA+ GC B cells are more resistant to Fas-dependent counter selection.
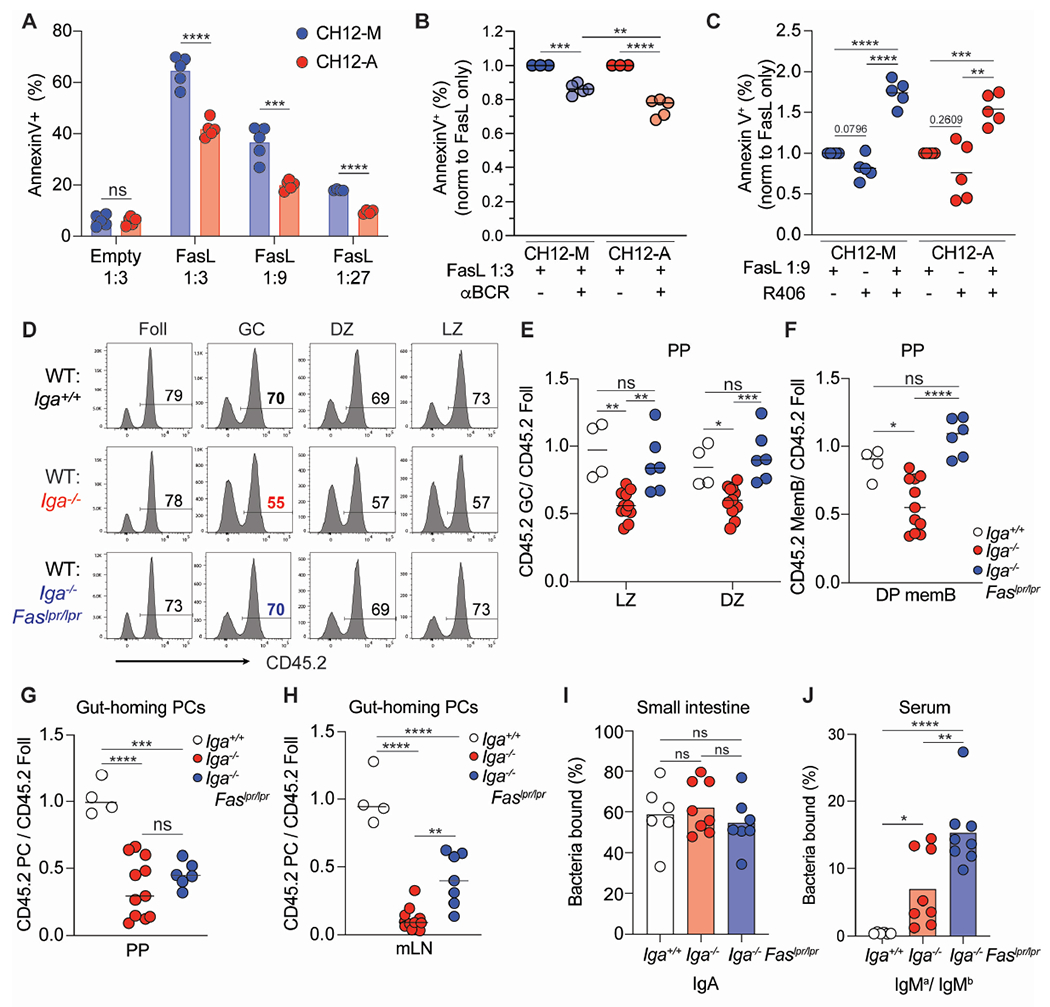
(A) Percentage of dead (defined as annexinV+) CH12 cells 5 hours after incubation with FasL vesicles at indicated dilutions.
(B) Percentage of dead CH12 cells after BCR stimulation and incubation with FasL vesicles. Data is normalized to FasL vesicles alone.
(C) Percentage of dead CH12 cells after incubation with R409 and FasL vesicles. Data is normalized to FasL vesicles alone.
(D) Representative histograms of CD45 staining on mixed BM chimeras for indicated B cell populations.
(E) Ratio of frequency of CD45.2 LZ or DZ B cells to CD45.2 Foll B cells in PPs of mixed BM chimeras.
(F) Ratio of frequency of CD45.2 DP memory B cells to CD45.2 Foll B cells from PPs of mixed BM chimeras.
(G, H) Ratio of frequency of CD45.2 gut-homing PCs to CD45.2 Foll B cells from PPs(G) and mLN(H) of mixed BM chimeras.
(I, J) Mixed BM chimeras using μMT hosts to track antibodies using allotypic isotypes. (I) Percentage of endogenously coated small intestinal bacteria with IgA. (J) Percentage of intestinal commensal from μMT mice stained by IgMa or IgMb antibodies from mixed BM serum. Percentage shown as IgMa/IgMb ratio.
A-C are compiled data from at least 3 independent experiments. E-J are compiled data from 3 independent experiments with 1-3 mice per group (E-H) or 2-3 mice per group (I,J). ns=not significant, *p < 0.005; **p < 0.001; ***p < 0.0005; ****p < 0.0001. Unpaired two-tailed Student’s t-test in A. One-way ANOVA in B, C, E-J.
See also Figure S6.
To test how BCR signaling may offset Fas signaling, CH12 B cells were stimulated immediately prior to incubation with FasL vesicles: while BCR stimulation rescued both CH12-M and CH12-A from Fas-mediated apoptosis, a higher degree of protection was achieved in IgA+ CH12 (Figure 6B). Conversely, inhibition of BCR signaling with the Syk inhibitor R406 enhanced CH12-M and CH12-A sensitivity to FasL vesicles, with CH12-A showing increased propensity to undergo cell death (Figure 6C). Together this data suggests that IgA BCR signaling provides protection against Fas mediated death initiated by membrane FasL, suggesting an intrinsic mechanism that might underpin IgA GC B cell survival in vivo.
Cell-intrinsic suppression of GC B cell survival via FasL-Fas has been shown to be important in preventing outgrowth of GC; inability to undergo FasL-mediated cell death predisposes the host to autoimmunity and B cell lymphoma.34,70–72 Such constraints on GC B cell accumulation in vivo is tissue specific: while Fas-dependent deletion is a dominant mechanism to maintain GC in peripheral lymph node and mLN, in PPs, GC B cells resistance to Fas killing did not confer a competitive advantage.34 In PPs, most GC B cells carry an IgA BCR, thus IgA+ GC B cells might be equipped with molecular machinery that permits a higher threshold for Fas-dependent cell death. Since GC B cells express similar surface Fas independently from the isotype in PPs, (Figure S6B), we speculated that Iga−/− B cells may be preferentially killed via Fas and therefore could be rescued by inactivation of the Fas-induced cell death pathway. To test this hypothesis, we generated mixed BM chimeras with BM from WT and Iga−/−, or Iga resistant to Fas-mediated cell death (Iga−/− FaslPr/lpr). In PPs, we observed that, in contrast to Iga−/− B cells, Iga−/− Faslpr/lpr B cells were able to compete with WT B cells during GC reaction in both LZ and DZ (Figures 6D, 6E), and their restored GC fitness led to physiologic MemB cell compartment formation (Figure 6F). In mLN, where a very small number of GC B cells express IgA BCR, resistance to Fas-mediated cell death allowed Iga−/− Faslpr/lpr B cells outgrowth as GC B cells and MemB cells, supporting previous findings34 (Figures S6C, S6D). No preferential isotype expansion was observed in Iga−/− Faslpr/lpr B cell subsets (Figures S6E, S6F), suggesting that the GC competitive ability was not driven by B cells carrying a specific isotype, but was instead the result of global rescue of GC B cells from Fas-mediated cell death. Finally, although Iga−/− Faslpr/lpr GC B cells could generate PCs at a rate similar to WT B cells (Figure S6G) they were nevertheless unable to differentiate into gut-homing PCs (Figures 6G, 6H) highlighting a previously undescribed role for the IgA BCR in mucosal imprinting that is uncoupled from GC selection. In line with this finding, mixed BM chimeras generated in μMT hosts using allotypic donors (IgHa vs IgHb) to track Ab origins revealed that endogenous intestinal bacteria were coated with IgA coming from WT B cells (Figure 6I), with negligible contribution of IgM from Iga−/− Faslpr/lpr (Figure S6H). In contrast, Iga−− Faslpr/lpr serum gave rise to more IgM specific to commensals (Figure 6J), likely due to B cell outgrowth in mLN (Figures S6C, S6F). These results indicate that Fas counterselection is critical for IgA+ GC B cells’ enhanced competition and point to an IgA-intrinsic resistance to Fas-dependent cell death in vivo. However, rescue of non-IgA GC B cells by Fas deletion does not increase the generation of gut-homing PCs , suggesting that additional mechanisms in IgA GC B cells imprint the ability to direct PCs to the intestinal mucosa.
Discussion
Acquisition of surface IgA upon IgA class switch recombination is routinely described as a default B cell differentiation outcome driven by the abundant active TGFβ available in PP sub-epithelial dome to recently activated B cells.26,27 This process imprints the B cell response to intestinal antigen for the generation of mostly IgA-secreting PCs. The incorporation of J-chain for rapid retrotranslocation via pIgR73,74, the dimeric form for increased avidity75, and the somatic mutation loads16–20 render IgA the most efficient mucosal antibody isotype. Thus, induction of the IgA response was mainly viewed simply as a process geared toward the generation of essential antibodies for intestinal secretion.
Here we described a role of IgA during the GC B cell reaction, providing an additional perspective on the evolutionary requirements of IgA class switch in B cells. IgA is critical both to promote fitness during the GC reaction and to imprint gut-homing properties on nascent PCs. The conflicting demands of broad IgA reactivity and affinity-based GC selection has been partially reconciled by recent works describing tunable affinity maturation in Peyer’s patches35,36, but the mechanism underpinning the GC reaction to intestinal antigens have not been elucidated. Lower microbiome complexity that restores affinity maturation efficiency and leads to enhanced clonal selection in PP GC is associated with reduced IgA+ GC B cells36, suggesting IgA BCR tunes GC selection toward a more permissive reactivity. How, then, are IgA+ B cells selected during the GC reaction to generate a humoral intestinal response that can simultaneously coat several classes of commensals? We propose a model in which the IgA BCR is inherently required for the generation of the broad mucosal antibody repertoire. IgA+ GC B cells display intrinsic protection from apoptosis that is centered around an enhanced BCR intracellular signaling and increased T cell-derived positive cues during selection of antigen-specific clones. Mechanistically, IgA BCR allows GC B cells to offset the Fas-FasL cell death signaling, therefore allowing low affinity B cells to persist and contribute to the PC compartment. While our paper was in revision, a manuscript was published detailing how BCR signaling in non-intestinal GC contributes to the selection of high-affinity clones into the PC pool76. These findings dovetail well with our model, with higher IgA BCR signaling preferentially selecting IgA+ B cell clones for participation to the intestinal humoral response.
The structural basis for the superior positive selection of IgA GC B cells remains unclear. While reports exist that the intracellular tail of IgG1 and IgE do not contribute to their intrinsic effect on GC positive selection, the IgA intracellular tail is distinct from all the other isotypes both in terms of length and sequence and could underpin the specific effect on IgA+ GC B cells during responses in PPs. Additionally, it also conceivable that Ig isotype-specific membrane and constant regions could play a role in IgA expressing cells, by either facilitating BCR clustering and thus lowering the threshold of B cell selection in the GC, or by altering BCR stiffness and thus rendering IgA+ GC B cells superior in extracting antigen displayed on Follicular dendritic cells.77,78
While in our model strong IgA BCR signaling promotes GC competition, IgE BCR mediates autonomous, sustained calcium signaling which constrains GC formation and promotes PC differentiation.42,45 Fas deletion increases IgA-deficient cells competitiveness in PP GC reaction, while Fas-deficient B cells generate a high number of IgE+ PC in periphery.72 One possible explanation for these apparently contradicting results is that BCR signaling is graded in GC B cells carrying different isotype: IgE strongest signaling promotes PC formation in line with a strength of signaling model31,79, while IgA BCR signaling, while dominant over the other BCR isotypes present in PP GC, allows for both GC competition and PC differentiation. IgE B cells do not efficiently participate in the GC reaction and it is unlikely that they are sensitive to FasL-induced death at that stage. Thus, the high IgE titer observed in the Fas or FasL deficient mice might be due to expansion of IgG1+ GC B cells that sequentially switch to IgE and then contribute to the humoral immunity. The stricter FasL-mediated counterselection of low affinity non-IgA GC B cells is an appropriate strategy to avoid the generation of rogue, autoreactive B cell clones34,72
Our results indicate a preferential effect of IgA BCR in PP GC competition compared to mLN and data exist supporting different requirements for IgA GC B cells at these two sites, including dependency on TGFβ signaling for IgA CSR27, on Gα13-coupled receptors for GC formation80,81, on Fas for GC counterselection34, and on commensals for GC response77. Two non-mutually exclusive hypotheses could explain the observed difference between IgA GC in PP and mLN. mLN IgA+ GC B cells could be responding against self-antigen or retroelements and have such low initial affinity that IgA-mediated enhanced BCR signaling only allows for their survival, not for the acquisition of competitive advantage. Additionally, mLN could contain specific environmental factor(s) that modulate GC selection in mLN in a distinct fashion compared to PPs, therefore muting IgA-dependent effect in competition. Additional investigations are required into the nature and origin of IgA+ GC in mLN to mechanistically tease apart their distinct regulation compared to IgA+ GC in PPs.
We also discovered that the IgA BCR was required in a competitive setting for the efficient upregulation of migratory receptors CCR9 and α4β7 and homing of intestinal PCs. This process is independent of Fas-FasL, as restoring competitiveness of non-IgA B cells during GC reaction had no effect on their ability to upregulate CCR9 and α4β7. Thus, additional step(s) that control functional generation of intestinal humoral immunity must take place in PPs during the response to intestinal antigen. While the homing ability could be imprinted during LZ selection in the GC, it is not possible to rule out that BCR signaling could shape PC homing outside of the GC reaction.82,83 IgA PCs isolated from lungs and mediastinal lymph nodes of mice infected with influenza also show marked upregulation of mucosal homing receptors84, suggesting a direct link between IgA BCR and PC migratory capabilities that is independent of the niche that generates PCs. Future investigations will be required to elucidate the cellular and environmental components that which signals, and at which step of PC differentiation, contribute to acquisition of gut tropism in IgA+ B cells. Moreover, whether this process remains in place or is altered during enteric infection or dysbiosis, states that can lead to non-IgA PC homing to the lamina propria,85–87 will require additional studies.
Given the dynamic impact of IgA on gut autoinflammation and enteric infection, understanding how IgA B cells are selected in the GC and give rise to IgA intestinal responses could be exploited therapeutically to develop strategies that restore commensal homeostasis or enhance mucosal vaccine responses.
Limitations of the study
Our data indicate that IgA BCR endows GC B cells with enhanced competitive capacity by offsetting FasL counterselection. In spleen, FasL counterselection removes low affinity GC B cell clones: while we speculated that IgA BCR allows commensal-reactive low affinity clones to persist and to participate in the intestinal humoral response, we have not formally proven it. A high resolution, more comprehensive chart of GC B cell antigen-receptors and the corresponding commensal antigens coupled with direct biophysical affinity measurements will be needed to evaluate the impact of IgA BCR on commensal-reactive GC reaction. Moreover, our ex-vivo and in vitro data suggest that IgA BCR mediates stronger BCR signaling compared to other isotypes in the PP GC. However, in vitro stimulation might not recapitulate the antigen nature, stiffness, and mode of presentation during GC reaction in vivo. In vivo stimulation with physiologic commensal-derived antigens will be needed to carefully evaluate isotype-specific BCR signaling in GC B cell during intestinal humoral response.
STAR METHODS
RESOURCE AVAILABILIY
Lead contact
Additional information and requests for resources and reagents should be directed to and fulfilled by the lead contact, Andrea Reboldi (andrea.reboldi@umassmed.edu).
Materials Availability
Reagents, plasmids, and mouse lines reported in this study are available upon signing a Materials Transfer Agreement.
Data and code availability
No original code has been reported in this paper.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Mice
All mice at UMass Chan Medical School were group housed (up to 5 mice of the same sex) and maintained under standard 12:12 hours light/dark conditions and housed in specific pathogen-free (SPF) conditions. Female and male mice were analyzed at 8-12 weeks of age and littermates of the same sex were cohoused and randomly assigned to groups. CD45.2, CD45.1, R26tdTomato, Aicdacre, Faslpr, gCAMPgfp, OT II, and μMT were purchased from the Jackson Laboratory. Gregory Barton provided Iga−/− mice, IgMa mice were from an internal colony. C.T.M. provided Rosa26INDIA mice.58 O.B. provided cMYC-GFP mice. All procedures were conformed to ethical principles and guidelines approved by the UMass Chan Medical School Institutional Animal Care and Use Committee.
METHOD DETAILS
Bone marrow chimeras, retroviral transduction
CD45.1/2, WT, or μMT mice were lethally irradiated twice with 550 rads gamma-irradiation, 3 hours apart, then intravenously injected with 1-3 x 106 bone marrow cells. Bone marrow was harvested by flushing both tibia and femurs of donor mice, counted, and mixed at ratios indicated in text. Mice were analyzed 8-12 weeks after cell transfer.
For retroviral transduction, PlatE cells were transfected with murine stem cell virus retroviral constructs encoding full length Bcl2 with Lipofectamine 2000 following manufacturer’s protocol. For transduction of BM-derived cells, BM was harvested 4 days after 5-flurouracil injection and cultured in the presence of recombinant IL-3 (20 ug/ml), IL-6 (50ug/ml), and mouse stem cell factor (100 ng/ml). BM cells were spin-infected twice with a retroviral construct expressing Bcl2 with GFP as a reporter. One day after the last spin infection, the cells were injected into lethally irradiated CD45.1/2 recipients. Mice were analyzed 8-10 weeks later.
In vivo treatment, immunization, and infection models
To prevent lymphocyte egress from Peyer’s patches (PPs), the S1PR1 agonist FTY-720 was dissolved in saline solution and administered to mice daily i.p. at 1 mg/kg for 7 days.
For cholera toxin responses, mice were immunized with 10 ug of cholera toxin in PBS by oral gavage. Animals received cholera toxin every 7 days for three consecutive weeks. Memory B cells were sorted and analyzed 7 days after last immunization.
The SL1344 metabolically defective (ΔAroA) S. typhimurium strains were provided by Milena Bogunovic (UMass Chan) and grown at 37°C in Luria broth supplemented with appropriate antibiotics to preserve mutation and plasmid. To induce Ova expression, IPTG was added at an OD600 of 0.4 and allowed to grow for another 2 hours. For the Salmonella infection, wildtype mice were orally gavage three times, on alternate days with 109 CFUs of ΔAroA Salmonella in 200 uL 5% sodium bicarbonate.
For antigen presentation experiments, 3 days after the last dose, follicular and germinal center B cells were sorted from PPs. For mixed BM chimera infections, 8 weeks after BM reconstitution, mice were infected as described above. Seven days after the third dose of Salmonella, mice were scarified, and B cell populations were assessed by flow cytometry.
Sample Preparation and Flow Cytometry
PPs and mesenteric lymph nodes (mLN) were collected into complete media (RPMI, 5% heat inactivated FCS, 10 mM HEPES, 1% Pen/Strep). The tissue was then smashed through a 70 micron cell strainer. Isolated cells were then washed with FACS buffer (DPBS, 2% heat inactivated FCS, 2mM EDTA). Cell suspensions were stained with LIVE/DEAD Fixable Aqua Dead Cell Stain or Fixable Viability Dye eFluor780 in FACS buffer, and Fc receptors blocked with anti-mouse CD16/32. BD Cytofix/Cytoperm kit was used for fixation and intracellular staining. Cells were incubated for 20 min on ice with antibodies described in Key Resource Table. Data were collected on a BD SORP or LSR II and analyzed in FlowJo v10 software.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Purified anti-mouse CD16/32, Clone 93 | Biolegend | CAT# 101330; RRID:AB_2561482 |
| BV711 anti-mouse B220, Clone RA3-6B2 | Biolegend | CAT# 103255; RRID:AB_2563491 |
| AF700 anti-mouse CD19, Clone 6D5 | Biolegend | CAT# 115528; RRID:AB_493735 |
| AF700 anti-mouse CD45.1, Clone A20 | Biolegend | CAT# 110724; RRID:AB_493733 |
| Pacific Blue anti-mouse CD45.1, Clone A20 | Biolegend | CAT# 110722; RRID:AB_492866 |
| BV650 anti-mouse CD45.1, Clone A20 | Biolegend | CAT# 110736; RRID:AB_2562564 |
| AF700 anti-mouse CD45.2, Clone 104 | Biolegend | CAT# 109822; RRID:AB_493731 |
| Pacific Blue anti-mouse CD45.2, Clone 104 | Biolegend | CAT# 109819; RRID:AB_492873 |
| BV650 anti-mouse CD45.2, Clone 104 | Biolegend | CAT# 109836; RRID:AB_2563065 |
| BV510 anti-mouse IgD, Clone 11-26c.2a | Biolegend | CAT# 405723; RRID:AB_2562742 |
| PerCP/Cy5.5 anti-mouse IgD, Clone 11-26c.2a | Biolegend | CAT# 405710; RRID:AB_1575113 |
| Pacific Blue anti-mouse IgD, Clone 11-26c.2a | Biolegend | CAT# 405712; RRID:AB_1937244 |
| Alexa Fluor 647 anti-mouse IgD, Clone 11-26c.2a | Biolegend | CAT# 405708; RRID:AB_893528 |
| BV421 anti-mouse CD138, Clone 281-2 | Biolegend | CAT#142508;RRID:AB_11203544 |
| AF647 anti-mouse GL7, Clone GL7 | Biolegend | CAT# 144606; RRID:AB_2562185 |
| PerCP/Cy5.5 anti-mouse GL7, Clone GL7 | Biolegend | CAT# 144610; RRID:AB_2562979 |
| PE/Cy7anti-mouse CD38, Clone 90 | Biolegend | CAT# 102718; RRID:AB_2275531 |
| PerCP/Cy5.5 anti-mouse CD73, Clone TY/11.8 | Biolegend | CAT#127214;RRID:AB_11219403 |
| BV605 anti-mouse CD80, Clone 16-10A1 | Biolegend | CAT#104729;RRID:AB_11126141 |
| BV605 anti-mouse CD86, Clone GL-1 | Biolegend | CAT#105037;RRID:AB_11204429 |
| FITC anti-mouse CD86, Clone GL-1 | Biolegend | CAT# 105005; RRID:AB_313148 |
| Biotin anti-mouse CXCR4, Clone 2B11/CXCR4 | BD Biosciences | CAT# 551968; RRID:AB_394307 |
| PE anti-mouse CXCR4, Clone 2B11/CXCR4 | BD Biosciences | CAT# 551966; RRID:AB_394305 |
| PE anti-mouse CCR9, Clone 9B1 | Biolegend | CAT# 129708; RRID:AB_2073249 |
| Biotin anti-mouse LPAM-1, Clone DATK32 | Biolegend | CAT#120612;RRID:AB_11203892 |
| APC anti-mouse IgM, Clone II/41 | BD Biosciences | CAT# 550676; RRID:AB_398464 |
| FITC anti-mouse IgM, Clone II/41 | BD Biosciences | CAT# 553437; RRID:AB_394857 |
| Biotin anti-mouse IgM, Clone II/41 | BD Biosciences | CAT# 553436;RRID:AB_394856 |
| FITC anti-mouse IgMa, Clone MA-69 | Biolegend | CAT# 408606; RRID:AB_940541 |
| PE anti-mouse IgMa, Clone MA-69 | Biolegend | CAT# 408608; RRID: AB_940545 |
| FITC anti-mouse IgMb, Clone AF6-78 | Biolegend | CAT# 406206; RRID: AB_315039 |
| PE anti-mouse IgMb, Clone AF6-78 | Biolegend | CAT# 406208; RRID: AB_315041 |
| FITC anti-mouse IgG1, Clone RMG1-1 | Biolegend | CAT# 406605; RRID: AB_493292 |
| PE ant-mouse IgG1, Clone RMG1-1 | Biolegend | CAT# 406607; RRID:AB_10551439 |
| FITC anti-mouse IgG2b, Clone RMG2b-1 | Biolegend | CAT# 406705; RRID: AB_493296 |
| PE anti-mouse IgG2b, Clone RMG2b-1 | Biolegend | CAT# 406708; RRID:AB_2563381 |
| Biotin anti-mouse IgG3, Clone RMG3-1 | Biolegend | CAT# 406803; RRID: AB_315070 |
| FITC anti-mouse IgA | Southern Biotech | CAT# 1040-02; RRID: AB_2794370 |
| PE anti-mouse IgA | Southern Biotech | CAT# 1040-09; RRID: AB_2794375 |
| Biotin anti-mouse IgA | Southern Biotech | CAT# 1040-08; RRID: AB_2794374 |
| Biotin anti-active Caspase 3 | BD Biosciences | CAT# 550557; RRID: AB_393750 |
| AF700 anti-mouse Kappa | Biolegend | CAT# 409508; RRID:AB_2563583 |
| APC anti-mouse CD22, Clone OX-97 | Biolegend | CAT# 126110; RRID:AB_2561630 |
| PerCP/Cy5.5 anti-mouse I-A/I-E, Clone M5/114.15.2 | Biolegend | CAT# 107626; RRID:AB_2191071 |
| PerCP/CY5.5 anti-mouse CD79b, Clone HM79-12 | Biolegend | CAT# 132810; RRID:AB_2632918 |
| PE/Cy7 anti-mouse CD69, Clone H1.2F3 | Biolegend | CAT# 104512; RRID: AB_493564 |
| AF700 anti-mouse CD4, Clone RM4-4 | Biolegend | CAT# 116022; RRID:AB_2715958 |
| PE anti-mouse Valpha2 TCR, Clone B20.1 | BD Biosciences | CAT# 553289; RRID: AB_394760 |
| Pacific Blue anti-mouse TCRbeta, Clone H57-597 | Biolegend | CAT# 109226; RRID:AB_1027649 |
| PE/Cy7anti-mouse Fas, Clone Jo2 | BD Biosciences | CAT# 557653; RRID: AB_396768 |
| FITC Annexin V | BD Biosciences | CAT# 556419; RRID: AB_2665412 |
| PE anti-BTK (pY223), Clone N35-86 | BD Biosciences | CAT# 562753; RRID:AB_2737769 |
| AF647 anti-Syk (pY352), Clone 17A/P-ZAP70 | BD Biosciences | CAT# 557817; RRID: AB_396884 |
| PE anti-phosphotyrosine, Clone PY20 | Biolegend | CAT# 309310; RRID:AB_2572196 |
| AF647 anti-mouse CD98, Clone RL388 | Biolegend | CAT#128210; RRID:AB_2254922 |
| Biotin anti-mouse IgD , Clone 11-26c.2a | Biolegend | CAT# 405734; RRID: AB_2563344 |
| Biotin anti-mouse CD138, Clone 281-2 | Biolegend | CAT# 142512; RRID: AB_2561981 |
| Biotin anti-mouse IgG2b, Clone RMG2b-1 | Biolegend | CAT# 406704; RRID: AB_315067 |
| Biotin anti-mouse CD8a, Clone 53-6.7 | Biolegend | CAT# 100704; RRID: AB_312743 |
| Biotin anti-mouse CD4, Clone GK1.5 | Biolegend | CAT# 100404; RRID: AB_312689 |
| Biotin anti-mouse IgG1, Clone RMG1-1 | Biolegend | CAT# 406604; RRID: AB_315063 |
| Biotin anti-mouse IgMa, Clone MA-69 | Biolegend | CAT# 408603; RRID: AB_940535 |
| Purified anti-mouse IgMa, Clone MA-69 | Biolegend | CAT# 408602; RRID: AB_940549 |
| Biotin anti-mouse IgMb, Clone AF6-78 | Biolegend | CAT# 406204; RRID: AB_315037 |
| Purified anti-mouse IgMb, Clone AF6-78 | Biolegend | CAT# 406202; RRID: AB_315035 |
| Unlabeled Goat anti-mouse IgA | Southern Biotech | CAT# 1040-01; RRID: AB_2314669 |
| Unlabeled Goat anti-mouse IgG2b | Southern Biotech | CAT# 1090-01; RRID: AB_2794517 |
| Unlabeled Goat anti-mouse IgM | Southern Biotech | CAT# 1020-01; RRID: AB_2794197 |
| Biotin anti-mouse GL7, Clone GL7 | Biolegend | CAT# 144616; RRID: AB_2721505 |
| Rabbit Anti-RFP | Rockland | CAT# 600401379; RRID: AB_11182807 |
| AF647 anti-mouse CD21/35, Clone 7E9 | Biolegend | CAT#123424; RRID: AB_2629577 |
| Mouse Anti-Phosphotyrosine, Clone 4g10 | Millipore Sigma | CAT# 05-321; RRID: AB_309678 |
| Mouse Anti beta-Actin, Clone AC-74 | Millipore Sigma | CAT# A5316; RRID: AB_476743 |
| HRP linked anti-mouse IgG | Cell Signaling | CAT# 7076; RRID: AB_330924 |
| HRP conjugated anti-mouse Phospho-Tyrosine (p-Tyr-100) | Cell Signaling | CAT# 5465; RRID: AB_10694719 |
| Purified mouse anti-actin, Clone C4/actin | BD Biosciences | CAT# 612657; RRID: AB_399901 |
| Goat polyclonal anti-mouse IgD | Nordic Immunology | CAT# GAM/IgD(Fc)/7S |
| Rabbit polyclonal anti-RFP | Rockland | CAT# 601-401-379 |
| Donkey anti-rabbit Cy3 | Jackson Immunoresearch | CAT# 711-165-152; RRID: AB 2307443 |
| Donkey anti-goat AF488 | Jackson Immunoresearch | CAT# 705-545-003; RRID: AB_2340428 |
| Human TruStain FcX | Biolegend | CAT# 422302; RRID:AB_2818986 |
| PE/Cy7 anti-human CD19, Clone HIB19 | Biolegend | CAT# 560728; RRID:AB_1727438 |
| BV605 anti-human CD27, Clone O323 | Biolegend | CAT# 302830; RRID:AB_2561450 |
| APC anti-human CD10, Clone HI10a | Biolegend | CAT# 312210; RRID:AB_314921 |
| APC/Cy7 anti-human CD10, Clone HI10a | Biolegend | CAT# 312212; RRID:AB_2146550 |
| PerCP/Cy5.5 anti-human CD138, Clone MI15 | Biolegend | CAT#356509; RRID:AB_2561898 |
| PE Goat F(ab’)2 Anti-human IgM | Southern Biotech | CAT# 2022-09; RRID: AB_2795614 |
| PE Goat F(ab’)2 Anti-human IgA | Southern Biotech | CAT# 2052-09; RRID: AB_2687523 |
| Unlabeled Goat F(ab’)2 Anti-human IgM | Southern Biotech | CAT# 2022-01; RRID: AB_2795610 |
| Unlabeled Goat F(ab’)2 Anti-human IgA | Southern Biotech | CAT# 2052-01; RRID: AB_2795709 |
| FITC Goat F(ab’)2 Anti-human IgG | Thermo Fisher | CAT#AHI1308;RRID:AB_1500723 |
| Bacterial strains | ||
| S. enterica strain SL1344 ΔaroA OVA | Gift from M. Bogunovic | N/A |
| Biological samples | ||
| Frozen human tonsils | Gift from E. Clark | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| AF488 Streptavidin | Biolegend | CAT# 405235 |
| PerCP/Cy5.5 Streptavidin | Biolegend | CAT# 405214 |
| BV605 Streptavidin | Biolegend | CAT# 405229 |
| BV650 Streptavidin | Biolegend | CAT# 405231 |
| PE/Cy7Streptavidin | eBioscience | CAT# 25-4317-82 |
| eFluor780 Live/Dead | BD Biosciences | CAT# 4302687 |
| EF450 Live/Dead | eBioscience | CAT# 65-0863-18 |
| DAPI | Thermo Fisher | CAT# 1306 |
| Cholera Toxin | Sigma | CAT# C8052-2MG |
| 16% PFA | Electrom microscopy sciences | CAT# 15710 |
| DNase I | Sigma | CAT# DN25 |
| Collagenase IV | Worthington | CAT# LS004189 |
| Percoll | GE Healthcare | CAT# 17089101 |
| FTY-720 | Selleck Chemicals | CAT# S5002 |
| Isotonic saline | Ricca Chemical | CAT# 7210-16 |
| Ibrutinib | Selleckchem | CAT# S2680 |
| R406 | Invivogen | CAT# inhr406 |
| Fluo 3 AM | Invitrogen | CAT# F23915 |
| Fura Red | Invitrogen | CAT# F3021 |
| 5-Fluorouracil | Sigma | CAT# F6627 |
| Recombinant Murine IL-3 | Peprotech | CAT# 213-13 |
| Recombinant Murine IL-6 | Peprotech | CAT# 216-16 |
| Recombinant Murine stem cell factor | Peprotech | CAT# 250-03 |
| Lipofectamine 2000 | Invitrogen | CAT# 11668-019 |
| RIPA buffer | Thermo Fisher | CAT# 89900 |
| 4x Laemmli sample buffer | Bio-Rad | CAT# 1610747 |
| Phosphatase inhibitor | Thermo Fisher | CAT# 1861281 |
| BSA | Sigma | CAT# A2153 |
| 10% Mini-PROTEAN® TGX™ Precast Protein Gels | Biorad | CAT# 4561033 |
| Protein Standard | BioRad | CAT# 161-0374 |
| Trans-Blot Turbo Mini 0.2 μm PVDF Transfer Packs | BioRad | CAT# 1704156 |
| ECL prime western blotting detection | GE Healthcare | CAT# RPN2232 |
| SuperSignal West Pico Plus substrate | Thermo Fisher | CAT# 34577 |
| 4 to 12% NuPAGE Bis-Tris gels | Invitrogen | CAT# NP0322BOX |
| Restore PLUS Western blot stripping buffer | Thermo Fisher | CAT# 46430 |
| Napthol AS-MX Phosphate | Sigma | CAT# N4875 |
| N-N-Dimethylformamide | Fisher | CAT# D119-500 |
| Levamisole | Sigma | CAT# L9756 |
| Fast Blue B salt | Sigma | CAT# D9805 |
| SIGMAFAST 3,3’-Diaminobenzidine tablets | Sigma | CAT# D4293 |
| Critical commercial assays | ||
| BRDU FITC kit | BD Biosciences | CAT# 559619 |
| BD Cytofix/Cytoperm | BD Biosciences | CAT# 554714 |
| EasySep Streptavidin RapidSpheres Kit | Stemcell Technologies | CAT# 19860A |
| SYBR green master mix | Biorad | CAT# 1725270 |
| Fluoromount-G | Southern Biotech | CAT# 0100-01 |
| Elisa/Elispot diluent solution | Invitrogen | CAT# 004202 |
| Stop Solution for TMB Substrate | Biolegend | CAT# 423001 |
| Deposited data | ||
| N/A | ||
| Experimental models: Cell lines | ||
| NB-21 | Gift from G. Kelsoe | Kuraoka et al., 2016 |
| I29μ | Gift from C. Schrader | N/A |
| CH12 | Gift from J. Muppidi | N/A |
| Platinum-E (Plat-E) retroviral packaging cell line | Gift from S. Schwab | N/A |
| Experimental models: Organisms/strains | ||
| C57BL/6J (CD45.2) | The Jackson Laboratory | Strain# 000664 ; RRID: IMSR_JAX:000664 |
| Ly5.2 (CD45.1) congenic B6.SJL-Ptprca Pepcb/BoyJ | The Jackson Laboratory | Strain# 002014; RRID: IMSR_JAX:002014 |
| Rosa26-flox-stop-flox-tdTomato | The Jackson Laboratory | Strain# 007914; RRID: IMSR_JAX:007914 |
| Aicda-cre | The Jackson Laboratory | Strain# 007770; RRID: IMSR_JAX:007770 |
| B6.MRL-Faslpr | The Jackson Laboratory | Strain# 000482; RRID: IMSR_JAX:000482 |
| B6J.Cg-Gt(ROSA)26Sortm95.1(CAG-GCaMPgf)Hze/MwarJ (gCAMP) | The Jackson Laboratory | Strain# 028865; RRID: IMSR_JAX:028865 |
| B6.Cg-Tg(TcraTcrb)425Cbn/J (OTII) | The Jackson Laboratory | Strain# 004194; RRID: IMSR_JAX:004194 |
| B6.Cg-Rag2tm1.1Cgn/J | The Jackson Laboratory | Strain# 008449; RRID: IMSR_JAX:008449 |
| B6.129SC-Ighmtm1Cgn/J | The Jackson Laboratory | Strain# 002288; RRID: IMSR_JAX:002288 |
| Iga −/− | Barton Lab | N/A |
| IgMa | Reboldi Lab | N/A |
| Rosa26-INDIA | Mayer Lab | N/A |
| cMYC-GFP | Bannard Lab | N/A |
| Oligonucleotides | ||
| N/A | ||
| Recombinant DNA | ||
| MSCV-IRES-blc2 | Gift from J. Muppidi | |
| Software and algorithms | ||
| FlowJo | Tree Star | https://www.flowjo.com |
| FACSDiva | BD | https://www.bdbiosciences.com/en-us/products/software/instrument-software/bd-facsdiva-software |
| Prism | GraphPad Software | https://www.graphpad.com |
| ZEN 3.1 | Carl Zeiss Microscopy | https://www.zeiss.com/microscopy/en/products/software/zeiss-zen.html |
| ImageJ | NIH | https://imagej.net/ij/index.html |
| Adobe Illustrator | Adobe Systems | https://www.adobe.com |
| Biorender | Biorender | https://www.biorender.com |
| Other | ||
Lamina propria (LP) lymphocytes were isolated as previously described.29,88 Briefly, the small intestine was dissected and flushed with cold PBS and PPs removed. The small intestine was divided into 2 equal parts, opened longitudinally and vortexed in a 50 ml conical tube containing HBSS supplemented with 5% heat-inactivated FBS and 10mM HEPES. Epithelial cells were removed by rotating the tissue in RPMI medium supplemented with 5% heat-inactivated FCS, 10mM HEPES, 10mM EDTA for 30 minutes at 37°C. The intestinal pieces were then washed with complete media (RPMI supplemented with 10% heat-inactivated FCS, 10 mM HEPES, 1% P/S), chopped with scissors, and digested at 37°C for 30 minutes in digestion media (complete media supplemented with 0.5 mg/ml Collagenase type IV and 50 ug/ml DNase I). Digested tissue was passed through 70 micron cell strainer and isolated cells were resuspended in 40% Percoll-RPMI and layered with 80% Percoll-RPMI and subsequently centrifuged for 20 min at 2200 RPM with the break off. The isolated LP cells were then stained for flow cytometry analysis as described above.
For anti–active caspase-3 staining, cells were maintained on ice during harvesting and processing. Cells were stained for with biotin–conjugated anti–active caspase-3 following fixation and permeabilization according to the manufacturer’s instructions and subsequently stained with FITC or AF647 streptavidin.
Bacterial flow cytometry was performed as previously described.56 Briefly, bacteria were washed in sterile-filtered DPBS with 1% BSA and resuspended at approximately 5 x 106 bacteria/mL. Mouse serum was diluted 1:25 in PBS/BSA buffer and 25 uL of this solution was mixed with 25 uL diluted microbes in u-bottom plate. Staining was performed with purified or fluorochrome conjugated antibodies. Cells were washed and resuspended in 1% BSA in DPBS with SYBR Green and analyzed by flow cytometry.
Cell sorting
For sorting, cells were stained as described above and sorted on a BD FACSAria II with an 85 micron nozzle. Cells were maintained at 4°C until sorting. Sorted PP Follicular and germinal center B cells were used for antigen presentation. PP memory B cells were used for NB21 co-culture. Germinal center B cells were sorted as live IgD− GL7+ CD38− CD138− isotype+, Follicular as live IgD+ GL7− CD138−, and memory B cells as live IgD− GL7+ CD38+ CD138− CD73+ CD80+ respectively into complete media for further culture.
BRDU
For BrdU incorporation experiments, animals were given 2.5 mg of BrdU in a single i.p. injection and sacrificed 30 minutes or 3.5 hours later. Staining was performed using the FITC BrdU Flow Kit according to the manufacturer’s instructions. To stain for intracellular antigens, cells were first stained for surface markers and then fixed and permeabilized using the BD Cytofix/Cytoperm Kit per the manufacturer’s instructions.
Cell line culture conditions
Both CH12 and I29 were cultured in B cell media (10% FCS, 100 units/mL penicillin, 100 units/mL streptomycin, MEM nonessential amino acids, 10mM HEPES, 1mM sodium pyruvate, 55mM 2-Mercaptoethanol). To induce class switch to IgA, CH12 and I29 cells were cultured for 48 hours with 2ng/ml TGFβ and 50 ug/ml LPS. The IgA+ or IgM+ population was sorted with a BD FACS Aria II and maintained as CH12 expressing IgM or IgA cell lines and I29 expressing IgM or IgA cell lines.
memB NB21 co-culture
Freshly isolated memory B cells from PPs were FACS sorted as described above and cocultured with NB21 feeder cells as previously described with adjustments.59,89 Briefly 15,000 NB21.2D9 cells/well were seeded into 96-well u bottom plates in 100 uL of B cell media and grown at 37°C and 5% CO2 the morning of B cell sorting and coculturing (Day 0). 2000 memory B cells in 100 uL B cell media were added to each well. Culture plates were centrifuged at day 3.5 and the supernatant collected for antibody detection.
FasL vesicle preparation and killing assay
FasL vesicles were prepared as previously described.90 Briefly, FasL vesicles were collected from N2-mFasL cells that overexpress mouse FasL and empty vesicles from Neo-N2 cells that did not contain FasL used as control. When cells reached ~70% confluence, fresh culture medium without G418 replaced the G418-containing medium. 48 hours later, the supernatants were collected and centrifuged at 4°C, 13,000 rpm in a Sorvall Superspeed centrifuge for 30 min to remove cell debris. Next, the cell-free supernatants were centrifuged for 3 hours at 4°C at 25,000 rpm in a Beckman ultracentrifuge with a SW25 rotor. Following ultracentrifugation, the murine FasL vesicle pellet was suspended with culture medium to 7% of the original volume and passed through a 0.45 micron sterile filter and used for cytotoxicity assays.
For the in vitro FasL killing, 200,000 CH12 cells were plated in 50 uL of warmed calcium media in u-bottom 96 well plates. FasL or empty vesicles were diluted in warm calcium media (DMEM with 2.5% FCS and 10mM HEPES) at the dilutions indicated in the figures and 50 uL were added to each well. Cells were incubated at 37°C with 5% CO2 for 5 hours. Cell death was assessed with annexin V and DAPI. For annexin V staining, cells were stained according to the manufacturer’s instructions. DAPI was added after annexin V staining right before cells were collected by flow cytometry. In R406 and FasL killing assays, CH12 were treated with for 18 hours (overnight) with 1.85uM R406 or DMSO. The next morning, cells were washed and resuspended in new calcium media containing 1.85uM R406 or DMSO and FasL vesicles.
Calcium flux assay
For calcium stimulation, PPs were harvested into warm calcium media (DMEM with 2.5% FCS and 10mM HEPES) and processed as described above. Germinal center and memory B cells were enriched for using EasySept negative B cell enrichment with modifications. Briefly, PPs were incubated with biotinylated antibodies against IgD, CD3, CD4, CD8, and CD138. Following negative enrichment, experiments using calcium dyes were incubated with surface antibodies GL7 and CD38 and 6uM FuraRed and 3uM Fluo-3 AM at 37°C for 20 minutes. Cells were washed and resuspended in warmed calcium media and rested 15 minutes at 37°C prior to stimulation. For gCAMP experiments, after negative B cell enrichment, cells were stained with GL7 and CD38 at room temperature for 10 minutes, washed and rested at 37°C for 15 minutes before stimulation. In some experiments cells were incubated with 100 nM Ibrutinib for 10 minutes at room temperature after surface staining. For all calcium experiments, the first 30 seconds were collected as baseline before cells were stimulated with either individual anti-BCR at 10 ug/mL or pan anti-BCR (10 ug/ml each of goat anti-mouse IgM, IgA, and IgG2b mixed). After 2-3 minutes 1ug/mL Ionomycin was added to measure maximum calcium flux.
Cell line BCR stimulation
For CH12 and I29 BCR stimulation, 200,000 B cells were resuspended in 400 ul of warm calcium media and rested for 30 minutes prior to stimulation. Cells were stimulated with 10ug/mL anti-BCR in warm calcium media for 1 minute then fixed for western blot or phospho-flow. For western blots, cells were spun at max speed room temperature for 10 seconds, the supernatant was removed, and the cell pellet was lysed in RIPA buffer with phosphatase inhibitor. Lysed cells were left on ice for 30 minutes then spun down at max speed for 10 minutes. The supernatant was used as whole cell lysates for western blots. Whole cell lysates were boiled for 5 minutes in Laemmli sample buffer and 2-ME then run on a 10% tris-glycine gel. Proteins were transferred to polyvinylidene fluoride membrane and blocked with 2% BSA in TBS-Tween for one hour at room temperature then incubated with anti-mouse phosphotyrosine-HRP antibody overnight at 4°C. Blots were washed with TBST followed by development with SuperSignal West Pico PLUS chemiluminescent Substate. Actin was detected with mouse anti-actin for 1.5 hours at room temperature followed by anti-mouse HRP for 1.5 hours and detected with ECL Prime Western blotting detection reagent. ImageJ software was used for band densitometry.
For phosphoflow, after anti-BCR stimulation, cells were fixed with 16% PFA at a final concentration of 1% PFA for 10 minutes room temperature. Cells were pelleted and resuspended in 1 mL of ice-cold methanol and left overnight at −20°C. The following day, cells were washed with 3 ml of FACS buffer and then 4 ml of FACS buffer. Cells were blocked in a u-bottom 96 well plate for 20 minutes with Fc block at room temperature, then stained with phosphor-tyrosine or phosphoBTK antibodies for 45 minutes at room temperature and then analyzed by BD SORP or LSR II.
For Syk inhibition, 200,000 I29 or CH12 cells were incubated at 37°C in calcium media containing 2uM R406 Syk inhibitor for 45 minutes. Cells were washed with warm calcium media and pelleted. Cells were resuspended in warm calcium media and immediately fixed for phosphoflow as described above.
In vitro antigen presentation
For antigen presentation assays, follicular, IgM+, IgG+ and IgA+ germinal center B cells were sorted from PPs as described above. OT-II T cells were isolated from spleens of OT-II Rag KO mice. B cells and T cells were co-cultured 1:1 in 200 uL of B cell media in 96 well u-bottom tissue culture plate and incubated at 37°C for 18 hours. CD69 expression on OT-II T cells was assessed by flow cytometry.
Immunohistochemistry and Immunofluorescence
For immunohistochemistry, acetone fixed cryosections were stained goat purified anti-mouse IgD and biotin anti mouse GL7. Then sections were stained with the secondary antibodies anti-goat HRP and streptavidin AP. HRP and AP were developed using DAB reagents and Fastblue (2mg/ml).
For immunofluorescence, tissues were fixed in 4% paraformaldehyde in phosphate buffer overnight at 4°C, washed three times for 30 min in PB, then moved to 30% sucrose in PB overnight. Tissues were flash frozen in cryomolds and the next day, 7 micron sections were cut and then dried for at least 1 hour before staining. Sections were rehydrated in PBS with 1% BSA for 10 min and then stained overnight at 4°C and stained for subsequent steps at room temperature for two hours, all in PBS with 1% BSA, 2% mouse serum, 2% rat serum, and 2% donkey serum. PP sections were stained with primary antibodies: Goat anti-mouse IgD, rabbit anti-mouse RFP, and Alexa647-conjugated anti-CD35/21. Sections were then stained with the secondary antibodies: Cy3 anti-rabbit, AF488 anti-goat, and DAPI. tdTomato cells were counted in the LZ (CD35+) and DZ (CD35−) regions using ImageJ.
Small intestinal villi were first stained with directly conjugated goat anti-mouse IgA FITC and rabbit anti-mouse RFP and subsequently stained with Cy3 anti-rabbit. tdTom+ cells were quantified in both duodenum and ileum villi.
Human tonsils preparation and stimulation
Human tonsil cells were thawed in 37°C IMDM media with 10% Fetal Bovine Serum (FBS), 1% Glutamax, 1% HEPES, 1% Penicillin/Streptomycin and 0.1% 2-Mercaptoethanol. Single cell suspensions were blocked with Fc Block at 4°C for 10min and subsequently, stained with antibodies to identify Germinal Center B cell and Follicular B cells. For sorting of IgA+ GC B cells without stimulating the desired population, cells were incubated IgG FITC and IgM PE, and the double negative population was sorted with FACS Aria. For sorting of IgM+ GC B cells without stimulating the desired population, cells were labeled with additional IgG FITC and IgA PE and sorted as above.
For the western blot signaling assay, sorted cells were rested and stimulated with 10ug/ml anti-IgA or anti-IgM for 5 min. Cells were lysed for 30 min on ice in RIPA buffer containing 0.5% dithiothreitol (DTT) and protease inhibitor cocktail. Lysates were centrifuged for 10 min at 4°C at 14,000g and supernatant was collected. Proteins were separated by electrophoresis using NuPage-Bis-Tris gels and blotted onto polyvinylidene fluoride membranes. Non-specific binding was blocked with 5% BSA in 0.1% TBS-Tween followed by incubation with anti-phosphotyrosine overnight at 4 °C and secondary antibody HRP conjugated antibodies for 1 hour at room temperature. Membranes were washed thoroughly with 0.1% TBS-Tween after antibody incubations and developed using ECL reagents. For re-probing, blots were stripped for 10 min at room temperature with Restore PLUS stripping buffer. Blots were then incubated with β-actin primary antibody and secondary antibody HRP-conjugated antibody for 1 hr at room temperature, washed with TBS-Tween, and developed using ECL reagents. ImageJ software was used for band densitometry.
Statistical analysis and reproducibility
Statistical analyses were performed using GraphPad Prism v10.0 using two-way ANOVA with Bonferroni’s multiple comparisons test or unpaired two tailed Student’s t test. Data are presented as means ± SEM except Figure 3B and 3C are SD. Differences between group means were considered significant at indicated p value. The significance values are stated in all graphs and the number of biological replicates (n) is stated in the figure legends.
Supplementary Material
Highlights.
IgA BCR underpins PP GC competitiveness, memory B cell and plasma cell generation
IgA BCR drives stronger intracellular signaling in GC B cells in vitro and ex vivo
IgA+ GC B cells receive increased T cell help and resist FasL-induced cell death
IgA BCR, not Fas-FasL axis, controls differentiation into gut-homing plasma cells
ACKNOWLEDGMENTS
We thank Carol Schrader for the I29μ cell line, Stephanie Moses for FasL vesicle preparation, Garnett Kelsoe for generously allowing use of NB21 cells, and Kelsey Howley and Carly Burke for mouse genotyping and colony maintenance. We thank E. V. Dang for critical reading of the manuscript.
This work was supported by National Institutes of Health (NIH) grant AI155727 and AI173903 (to A.R.), AI151167 (to M.A.) and NIH training grants AI132152 and AI007349 (to F.R.). J.R.M. is supported by the Intramural Research Program of the National Institutes of Health, Center for Cancer Research, National Cancer Institute.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATIONS OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Catanzaro JR, Strauss JD, Bielecka A, Porto AF, Lobo FM, Urban A, Schofield WB, and Palm NW (2019). IgA-deficient humans exhibit gut microbiota dysbiosis despite secretion of compensatory IgM. Scientific Reports 9, 1–10. 10.1038/s41598-019-49923-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fadlallah J, El-Kafsi H, Sterlin D, Juste C, Parizot C, Dorgham K, Autaa G, Gouas D, Almeida M, Lepage P, et al. (2018). Microbial ecology perturbation in human IgA deficiency. Science Translational Medicine 10, eaan1217. 10.1126/scitranslmed.aan1217. [DOI] [PubMed] [Google Scholar]
- 3.Suzuki K, Meek B, Doi Y, Muramatsu M, Chiba T, Honjo T, and Fagarasan S (2004). Aberrant expansion of segmented filamentous bacteria in IgA-deficient gut. P Natl Acad Sci Usa 101, 1981–1986. 10.1073/pnas.0307317101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hand TW, and Reboldi A (2021). Production and Function of Immunoglobulin A. Annu. Rev. Immunol 39, 695–718. 10.1146/annurev-immunol-102119-074236. [DOI] [PubMed] [Google Scholar]
- 5.Savilahti E, Klemola T, Carlsson B, Mellander L, Stenvik M, and Hovi T (1988). Inadequacy of musocal IgM antibodies in selective IgA deficiency: Excretion of attenuated polio viruses is prolonged. J Clin Immunol 8, 89–94. 10.1007/bf00917895. [DOI] [PubMed] [Google Scholar]
- 6.Bunker JJ, Erickson SA, Flynn TM, Henry C, Koval JC, Meisel M, Jabri B, Antonopoulos DA, Wilson PC, and Bendelac A (2017). Natural polyreactive IgA antibodies coat the intestinal microbiota. Science 358, eaan6619–20. 10.1126/science.aan6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palm NW, Zoete M.R. de, Cullen TW, Barry NA, Stefanowski J, Hao L, Degnan PH, Hu J, Peter I, Zhang W, et al. (2014). Immunoglobulin A Coating Identifies Colitogenic Bacteria in Inflammatory Bowel Disease. Cell 158, 1000–1010. 10.1016/j.cell.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rollenske T, Szijarto V, Lukasiewicz J, Guachalla LM, Stojkovic K, Hartl K, Stulik L, Kocher S, Lasitschka F, Al-Saeedi M, et al. (2018). Cross-specificity of protective human antibodies against Klebsiella pneumoniae LPS O-antigen. Nat Immunol 19, 1–15. 10.1038/s41590-018-0106-2. [DOI] [PubMed] [Google Scholar]
- 9.Sterlin D, Fadlallah J, Adams O, Fieschi C, Parizot C, Dorgham K, Rajkumar A, Autaa G, El-Kafsi H, Charuel J-L, et al. (2019). Human IgA binds a diverse array of commensal bacteria. J Exp Med 217, e20181635. 10.1084/jem.20181635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pabst O, and Slack E (2019). IgA and the intestinal microbiota: the importance of being specific. Mucosal Immunol 13, 1–10. 10.1038/s41385-019-0227-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang C, Chen-Liaw A, Spindler MP, Tortorella D, Moran TM, Cerutti A, and Faith JJ (2022). Immunoglobulin A antibody composition is sculpted to bind the self gut microbiome. Sci Immunol 7, eabg3208. 10.1126/sciimmunol.abg3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tezuka H, Abe Y, Asano J, Sato T, Liu J, Iwata M, and Ohteki T (2011). Prominent Role for Plasmacytoid Dendritic Cells in Mucosal T Cell-Independent IgA Induction. Immunity 34, 247–257. 10.1016/j.immuni.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 13.Bunker JJ, Flynn TM, Koval JC, Shaw DG, Meisel M, McDonald BD, Ishizuka IE, Dent AL, Wilson PC, Jabri B, et al. (2015). Innate and Adaptive Humoral Responses Coat Distinct Commensal Bacteria with Immunoglobulin A. Immunity 43, 1–14. 10.1016/j.immuni.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grasset EK, Chomy A, Casas-Recasens S, Gutzeit C, Bongers G, Thomsen I, Chen L, He Z, Matthews DB, Oropallo MA, et al. (2020). Gut T cell–independent IgA responses to commensal bacteria require engagement of the TACI receptor on B cells. Sci Immunol 5, eaat7117. 10.1126/sciimmunol.aat7117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawamoto S, Maruya M, Kato LM, Suda W, Atarashi K, Doi Y, Tsutsui Y, Qin H, Honda K, Okada T, et al. (2014). Foxp3+ T Cells Regulate Immunoglobulin A Selection and Facilitate Diversification of Bacterial Species Responsible for Immune Homeostasis. Immunity 41, 152–165. 10.1016/j.immuni.2014.05.016. [DOI] [PubMed] [Google Scholar]
- 16.Dunn-Walters DK, Boursier L, and Spencer J (1997). Hypermutation, diversity and dissemination of human intestinal lamina propria plasma cells. Eur. J. Immunol 27, 2959–2964. 10.1002/eji.1830271131. [DOI] [PubMed] [Google Scholar]
- 17.Fischer M, and Küppers R (1998). Human IgA- and IgM-secreting intestinal plasma cells carry heavily mutated VH region genes. Eur. J. Immunol 28, 2971–2977. . [DOI] [PubMed] [Google Scholar]
- 18.Barone F, Vossenkamper A, Boursier L, Su W, Watson A, John S, Walters DKD, Fields P, Wijetilleka S, Edgeworth JD, et al. (2011). IgA-Producing Plasma Cells Originate From Germinal Centers That Are Induced by B-Cell Receptor Engagement in Humans. YGAST 140, 947–956. 10.1053/j.gastro.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kabbert J, Benckert J, Rollenske T, Hitch TCA, Clavel T, Cerovic V, Wardemann H, and Pabst O (2020). High microbiota reactivity of adult human intestinal IgA requires somatic mutations. Journal of Experimental Medicine 217, 612–613. 10.1084/jem.20200275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benckert J, Schmolka N, Kreschel C, Zoller MJ, Sturm A, Wiedenmann B, and Wardemann H (2011). The majority of intestinal IgA+ and IgG+ plasmablasts in the human gut are antigen-specific. J. Clin. Invest 121, 1946–1955. 10.1172/jci44447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wei M, Shinkura R, Doi Y, Maruya M, Fagarasan S, and Honjo T (2011). Mice carrying a knock-in mutation of Aicda resulting in a defect in somatic hypermutation have impaired gut homeostasis and compromised mucosal defense. Nat Immunol 12, 264–270. 10.1038/ni.1991. [DOI] [PubMed] [Google Scholar]
- 22.Reboldi A, and Cyster JG (2016). Peyer’s patches: organizing B-cell responses at the intestinal frontier. Immunological Reviews 271, 230–245. 10.111l/imr.12400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gibbons DL, and Spencer J (2011). Mouse and human intestinal immunity: same ballpark, different players; different rules, same score. Mucosal Immunol 4, 148–157. 10.1038/mi.2010.85. [DOI] [PubMed] [Google Scholar]
- 24.Weinstein PD, and Cebra JJ (1991). The preference for switching to IgA expression by Peyer’s patch germinal center B cells is likely due to the intrinsic influence of their microenvironment. J Immunol 147, 4126–4135. [PubMed] [Google Scholar]
- 25.Craig SW, and Cebra JJ (1971). PEYER’S PATCHES: AN ENRICHED SOURCE OF PRECURSORS FOR IGA-PRODUCING IMMUNOCYTES IN THE RABBIT. Journal of Experimental Medicine 134, 188–200. 10.1084/jem.134.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reboldi A, Arnon TI, Rodda LB, Atakilit A, Sheppard D, and Cyster JG (2016). IgA production requires B cell interaction with subepithelial dendritic cells in Peyers patches. Science 352, aaf4822–aaf4822. 10.1126/science.aaf4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Albright AR, Kabat J, Li M, Raso F, Reboldi A, and Muppidi JR (2019). TGFβ signaling in germinal center B cells promotes the transition from light zone to dark zone. J Exp Med 216, 2531–2545. 10.1084/jem.20181868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roco JA, Mesin L, Binder SC, Nefzger C, Gonzalez-Figueroa P, Canete PF, Ellyard J, Shen Q, Robert PA, Cappello J, et al. (2019). Class-Switch Recombination Occurs Infrequently in Germinal Centers. Immunity 51, 1–37. 10.1016/j.immuni.2019.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trindade BC, Ceglia S, Berthelette A, Raso F, Howley K, Muppidi JR, and Reboldi A (2021). The cholesterol metabolite 25-hydroxycholesterol restrains the transcriptional regulator SREBP2 and limits intestinal IgA plasma cell differentiation. Immunity 54, 2273–2287.e6. 10.1016/j.immuni.2021.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tas JMJ, Mesin L, Pasqual G, Targ S, Jacobsen JT, Mano YM, Chen CS, Weill JC, Reynaud CA, Browne EP, et al. (2016). Visualizing antibody affinity maturation in germinal centers. Science 351, 1048–1054. 10.1126/science.aad3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Phan TG, Paus D, Chan TD, Turner ML, Nutt SL, Basten A, and Brink R (2006). High affinity germinal center B cells are actively selected into the plasma cell compartment. Journal of Experimental Medicine 203, 2419–2424. 10.1084/jem.20061254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Victora GD, and Nussenzweig MC (2022). Germinal Centers. Annu Rev Immunol 40, 1–30. 10.1146/annurev-immunol-120419-022408. [DOI] [PubMed] [Google Scholar]
- 33.Biram A, Winter E, Denton AE, Zaretsky I, Dassa B, Bemark M, Linterman MA, Yaari G, and Shulman Z (2020). B Cell Diversification Is Uncoupled from SAP-Mediated Selection Forces in Chronic Germinal Centers within Peyer’s Patches. Cell Reports 30, 1910–1922.e5. 10.1016/j.celrep.2020.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Razzaghi R, Agarwal S, Kotlov N, Plotnikova O, Nomie K, Huang DW, Wright GW, Smith GA, Li M, Takata K, et al. (2021). Compromised counterselection by FAS creates an aggressive subtype of germinal center lymphoma. J Exp Med 218, 248. 10.1084/jem.20201173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen H, Zhang Y, Ye AY, Du Z, Xu M, Lee C-S, Hwang JK, Kyritsis N, Ba Z, Neuberg D, et al. (2020). BCR selection and affinity maturation in Peyer’s patch germinal centres. Nature 582, 1–23. 10.1038/s41586-020-2262-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nowosad CR, Mesin L, Castro TBR, Wichmann C, Donaldson GP, Araki T, Schiepers A, Lockhart AAK, Bilate AM, Mucida D, et al. (2020). Tunable dynamics of B cell selection in gut germinal centres. Nature 6, 122–126. 10.1038/s41586-020-2865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo W, Weisel F, and Shlomchik MJ (2018). B Cell Receptor and CD40 Signaling Are Rewired for Synergistic Induction of the c-Myc Transcription Factor in Germinal Center B Cells. Immunity 48, 313–326.e5. 10.1016/j.immuni.2018.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davidzohn N, Biram A, Stoler-Barak L, Grenov A, Dassa B, and Shulman Z (2019). Syk degradation restrains plasma cell formation and promotes zonal transitions in germinal centers. Journal of Experimental Medicine 217, 26648–26. 10.1084/jem.20191043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, and Nussenzweig MC (2010). Germinal Center Dynamics Revealed by Multiphoton Microscopy with a Photoactivatable Fluorescent Reporter. Cell 143, 592–605. 10.1016/j.cell.2010.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ersching J, Efeyan A, Mesin L, Jacobsen JT, Pasqual G, Grabiner BC, Dominguez-Sola D, Sabatini DM, and Victora GD (2017). Germinal Center Selection and Affinity Maturation Require Dynamic Regulation of mTORC1 Kinase. Immunity 46, 1045–1058.e6. 10.1016/j.immuni.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gitlin AD, Shulman Z, and Nussenzweig MC (2014). Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature 509, 1–9. 10.1038/nature13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang Z, Robinson MJ, Chen X, Smith GA, Taunton J, Liu W, and Allen CDC (2016). Regulation of B cell fate by chronic activity of the IgE B cell receptor. eLife 5, 409. 10.7554/elife.21238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sundling C, Lau AWY, Bourne K, Young C, Laurianto C, Hermes JR, Menzies RJ, Butt D, Kräutler NJ, Zahra D, et al. (2021). Positive selection of IgG+ over IgM+ B cells in the germinal center reaction. Immunity 54, 988–1001.e5. 10.1016/j.immuni.2021.03.013. [DOI] [PubMed] [Google Scholar]
- 44.Newman R, and Tolar P (2021). Chronic calcium signaling in IgE+ B cells limits plasma cell differentiation and survival. Immunity. 10.1016/j.immuni.2021.11.006. [DOI] [PubMed] [Google Scholar]
- 45.Haniuda K, Fukao S, Kodama T, Hasegawa H, and Kitamura D (2016). Autonomous membrane IgE signaling prevents IgE-memory formation. Nat Immunol 17, 1109–1117. 10.1038/ni.3508. [DOI] [PubMed] [Google Scholar]
- 46.Harriman GR, Bradley A, Das S, Rogers-Fani P, and Davis AC (1996). IgA class switch in I alpha exon-deficient mice. Role of germline transcription in class switch recombination. J Clin Invest 97, 477–485. 10.1172/jci118438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bannard O, and Cyster JG (2017). ScienceDirect Germinal centers: programmed for affinity maturation and antibody diversification. Current Opinion in Immunology 45, 21–30. 10.1016/j.coi.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 48.Taylor JJ, Pape KA, and Jenkins MK (2012). A germinal center–independent pathway generates unswitched memory B cells early in the primary response. J Exp Medicine 209, 597–606. 10.1084/jem.20111696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martinoli C, Chiavelli A, and Rescigno M (2007). Entry Route of Salmonella typhimurium Directs the Type of Induced Immune Response. Immunity 27, 975–984. 10.1016/j.immuni.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 50.Biram A, Liu J, Hezroni EL, Davidzohn N, Schmiedel D, Khatib-Massalha E, Haddad M, Grenov A, Lebon S, Salame TM, et al. (2022). Bacterial infection disrupts established germinal center reactions through monocyte recruitment and impaired metabolic adaptation. Immunity. 10.1016/j.immuni.2022.01.013. [DOI] [PubMed] [Google Scholar]
- 51.Gohda M, Kunisawa J, Miura F, Kagiyama Y, Kurashima Y, Higuchi M, Ishikawa I, Ogahara I, and Kiyono H (2008). Sphingosine 1-phosphate regulates the egress of IgA plasmablasts from Peyer’s patches for intestinal IgA responses. J Immunol 180, 5335–5343. 10.4049/jimmunol.180.8.5335. [DOI] [PubMed] [Google Scholar]
- 52.Wagner N, Löhler J, Kunkel EJ, Ley K, Leung E, Krissansen G, Rajewsky K, and Müller W (1996). Critical role for beta7 integrins in formation of the gut-associated lymphoid tissue. Nature 382, 366–370. 10.1038/382366a0. [DOI] [PubMed] [Google Scholar]
- 53.Farstad IN, Halstensen TS, Lazarovits AI, Norstein J, Fausa O, and Brandtzaeg P (1995). Human intestinal B-cell blasts and plasma cells express the mucosal homing receptor integrin alpha 4 beta 7. Scand J Immunol 42, 662–672. 10.1111/j.1365-3083.1995.tb03709.x. [DOI] [PubMed] [Google Scholar]
- 54.Pabst O, Ohl L, Wendland M, Wurbel M-A, Kremmer E, Malissen B, and Förster R (2004). Chemokine receptor CCR9 contributes to the localization of plasma cells to the small intestine. Journal of Experimental Medicine 199, 411–416. 10.1084/jem.20030996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Crawford PA, and Gordon JI (2005). Microbial regulation of intestinal radiosensitivity. Proc National Acad Sci 102, 13254–13259. 10.1073/pnas.0504830102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koch MA, Reiner GL, Lugo KA, Kreuk LSM, Stanbery AG, Ansaldo E, Seher TD, Ludington WB, and Barton GM (2016). Maternal IgG and IgA Antibodies Dampen Mucosal T Helper Cell Responses in Early Life. Cell 165, 827–841. 10.1016/j.cell.2016.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lycke N, and Bemark M (2010). Mucosal adjuvants and long-term memory development with special focus on CTA1-DD and other ADP-ribosylating toxins. Mucosal Immunol 3, 1–11. 10.1038/mi.2010.54. [DOI] [PubMed] [Google Scholar]
- 58.Mayer CT, Gazumyan A, Kara EE, Gitlin AD, Golijanin J, Viant C, Pai J, Oliveira TY, Wang Q, Escolano A, et al. (2017). The microanatomic segregation of selection by apoptosis in the germinal center. Science 358, eaao2602–14. 10.1126/science.aao2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stewart I, Radtke D, Phillips B, McGowan SJ, and Bannard O (2018). Germinal Center B Cells Replace Their Antigen Receptors in Dark Zones and Fail Light Zone Entry when Immunoglobulin Gene Mutations are Damaging. Immunity 49, 477–489.e7. 10.1016/j.immuni.2018.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Long Z, Phillips B, Radtke D, Meyer-Hermann M, and Bannard O (2022). Competition for refueling rather than cyclic reentry initiation evident in germinal centers. Sci Immunol 7, eabm0775. 10.1126/sciimmunol.abm0775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dominguez-Sola D, Victora GD, Ying CY, Phan RT, Saito M, Nussenzweig MC, and Dalla-Favera R (2012). The proto-oncogene MYC is required for selection in the germinal center and cyclic reentry. Nat Immunol 13, 1083–1091. 10.1038/ni.2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Calado DP, Sasaki Y, Godinho SA, Pellerin A, Köchert K, Sleckman BP, Alborán I.M. de, Janz M, Rodig S, and Rajewsky K (2012). MYC is essential for the formation and maintenance of germinal centers. Nat Immunol 13, 1092–1100. 10.1038/ni.2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Khalil AM, Cambier JC, and Shlomchik MJ (2012). B Cell Receptor Signal Transduction in the GC Is Short-Circuited by High Phosphatase Activity. Science 336, 1178–1181. 10.1126/science.1213368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stavnezer J, Sirlin S, and Abbott J (1985). Induction of immunoglobulin isotype switching in cultured I.29 B lymphoma cells. Characterization of the accompanying rearrangements of heavy chain genes. Journal of Experimental Medicine 161, 577–601. 10.1084/jem.l61.3.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nakamura M, Kondo S, Sugai M, Nazarea M, Imamura S, and Honjo T (1996). High frequency class switching of an lgM+ B lymphoma clone CH12F3 to lgA+ cells. Int Immunol 8, 193–201. 10.1093/intimm/8.2.193. [DOI] [PubMed] [Google Scholar]
- 66.Catlett IM, and Bishop GA (1999). Cutting edge: a novel mechanism for rescue of B cells from CD95/Fas-mediated apoptosis. J Immunol 163, 2378–2381. [PubMed] [Google Scholar]
- 67.Rothstein TL, Wang JKM, Panka DJ, Foote LC, Wang Z, Stanger B, Cui H, Ju S-T, and Marshak-Rothstein A (1995). Protection against Fas-dependent Thl-mediated apoptosis by antigen receptor engagement in B cells. Nature 374, 163–165. 10.1038/374163a0. [DOI] [PubMed] [Google Scholar]
- 68.Xiao S, Jodo S, Sung SJ, Marshak-Rothstein A, and Ju S-T (2002). A Novel Signaling Mechanism for Soluble CD95 Ligand SYNERGY WITH ANTI-CD95 MONOCLONAL ANTIBODIES FOR APOPTOSIS AND NF-κB NUCLEAR TRANSLOCATION*. J Biol Chem 277, 50907–50913. 10.1074/jbc.m206093200. [DOI] [PubMed] [Google Scholar]
- 69.Hohlbaum AM, Gregory MS, Ju ST, and Marshak-Rothstein A (2001). Fas ligand engagement of resident peritoneal macrophages in vivo induces apoptosis and the production of neutrophil chemotactic factors. J Immunol 167, 6217–6224. 10.4049/jimmunol.167.11.6217. [DOI] [PubMed] [Google Scholar]
- 70.Takahashi Y, Ohta H, and Takemori T (2001). Fas Is Required for Clonal Selection in Germinal Centers and the Subsequent Establishment of the Memory B Cell Repertoire. Immunity 14, 181–192. 10.1016/s1074-7613(01)00100-5. [DOI] [PubMed] [Google Scholar]
- 71.Smith KG, Nossal GJ, and Tarlinton DM (1995). FAS is highly expressed in the germinal center but is not required for regulation of the B-cell response to antigen. Proc National Acad Sci 92, 11628–11632. 10.1073/pnas.92.25.11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Butt D, Chan TD, Bourne K, Hermes JR, Nguyen A, Statham A, O’Reilly LA, Strasser A, Price S, Schofield P, et al. (2015). FAS Inactivation Releases Unconventional Germinal Center B Cells that Escape Antigen Control and Drive IgE and Autoantibody Production. Immunity 42, 890–902. 10.1016/j.immuni.2015.04.010. [DOI] [PubMed] [Google Scholar]
- 73.Johansen F-E, Braathen R, and Brandtzaeg P (2000). Role of J chain in secretory immunoglobulin formation. Scandinavian Journal of Immunology 52, 240–248. 10.1046/j.1365-3083.2000.00790.x. [DOI] [PubMed] [Google Scholar]
- 74.Lycke N, Erlandsson L, Ekman L, Schön K, and Leanderson T (1999). Lack of J chain inhibits the transport of gut IgA and abrogates the development of intestinal antitoxic protection. J Immunol Baltim Md 1950 163, 913–919. [PubMed] [Google Scholar]
- 75.Woof JM, and Russell MW (2019). Structure and function relationships in IgA. Mucosal Immunol 4, 1–8. 10.1038/mi.2011.39. [DOI] [PubMed] [Google Scholar]
- 76.Chen ST, Oliveira TY, Gazumyan A, Cipolla M, and Nussenzweig MC (2023). B cell receptor signaling in germinal centers prolongs survival and primes B cells for selection. Immunity 56, 547–561.e7. 10.1016/j.immuni.2023.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nowosad CR, Spillane KM, and Tolar P (2016). Germinal center B cells recognize antigen through a specialized immune synapse architecture. Nat Immunol 17, 870–877. 10.1038/ni.3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wan Z, Zhang S, Fan Y, Liu K, Du F, Davey AM, Zhang H, Han W, Xiong C, and Liu W (2013). B Cell Activation Is Regulated by the Stiffness Properties of the Substrate Presenting the Antigens. J Immunol 190, 4661–4675. 10.4049/jimmunol.1202976. [DOI] [PubMed] [Google Scholar]
- 79.Paus D, Phan TG, Chan TD, Gardam S, Basten A, and Brink R (2006). Antigen recognition strength regulates the choice between extrafollicular plasma cell and germinal center B cell differentiation. Journal of Experimental Medicine 203, 1081–1091. 10.1084/jem.20060087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Muppidi JR, Schmitz R, Green JA, Xiao W, Larsen AB, Braun SE, An J, Xu Y, Rosenwald A, Ott G, et al. (2014). Loss of signalling via Gα13 in germinal centre B-cell-derived lymphoma. Nature 516, 254–258. 10.1038/nature13765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Green JA, Suzuki K, Cho B, Willison LD, Palmer D, Allen CDC, Schmidt TH, Xu Y, Proia RL, Coughlin SR, et al. (2011). The sphingosine 1-phosphate receptor S1P2 maintains the homeostasis of germinal center B cells and promotes niche confinement. Nat Immunol 12, 672–680. 10.1038/ni.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wittner J, Schulz SR, Steinmetz TD, Berges J, Hauke M, Channell WM, Cunningham AF, Hauser AE, Hutloff A, Mielenz D, et al. (2022). Krüppel-like factor 2 controls IgA plasma cell compartmentalization and IgA responses. Mucosal Immunol, 1–15. 10.1038/s41385-022-00503-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pinto D, Montani E, Bolli M, Garavaglia G, Sallusto F, Lanzavecchia A, and Jarrossay D (2013). A functional BCR in human IgA and IgM plasma cells. Blood 121, 4110–4114. 10.1182/blood-2012-09-459289. [DOI] [PubMed] [Google Scholar]
- 84.Price MJ, Hicks SL, Bradley JE, Randall TD, Boss JM, and Scharer CD (2019). IgM, IgG, and IgA Influenza-Specific Plasma Cells Express Divergent Transcriptomes. J Immunol 203, 2121–2129. 10.4049/jimmunol.1900285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Martin JC, Chang C, Boschetti G, Ungaro R, Giri M, Grout JA, Gettler K, Chuang L, Nayar S, Greenstein AJ, et al. (2019). Single-Cell Analysis of Crohn’s Disease Lesions Identifies a Pathogenic Cellular Module Associated with Resistance to Anti-TNF Therapy. Cell 178, 1493–1508.e20. 10.1016/j.cell.2019.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zeng MY, Cisalpino D, Varadarajan S, Hellman J, Warren HS, Cascalho M, Inohara N, and Núñez G (2016). Gut Microbiota-Induced Immunoglobulin G Controls Systemic Infection by Symbiotic Bacteria and Pathogens. Immunity 44, 1–23. 10.1016/j.immuni.2016.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Scheid JF, Eraslan B, Hudak A, Brown EM, Sergio D, Delorey TM, Phillips D, Lefkovith A, Jess AT, Duck LW, et al. (2023). Remodeling of colon plasma cell repertoire within ulcerative colitis patients. J Exp Med 220, e20220538. 10.1084/jem.20220538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ceglia S, Berthelette A, Howley K, Li Y, Mortzfeld B, Bhattarai SK, Yiew NKH, Xu Y, Brink R, Cyster JG, et al. (2023). An epithelial cell-derived metabolite tunes immunoglobulin A secretion by gut-resident plasma cells. Nat Immunol, 1–14. 10.1038/s41590-022-01413-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kuraoka M, Schmidt AG, Nojima T, Feng F, Watanabe A, Kitamura D, Harrison SC, Kepler TB, and Kelsoe G (2016). Complex Antigens Drive Permissive Clonal Selection in Germinal Centers. Immunity, 1–30. 10.1016/j.immuni.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jodo S, Xiao S, Hohlbaum A, Strehlow D, Marshak-Rothstein A, and Ju S-T (2001). Apoptosis-inducing Membrane Vesicles A NOVEL AGENT WITH UNIQUE PROPERTIES*. J Biol Chem 276, 39938–39944. 10.1074/jbc.m107005200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No original code has been reported in this paper.