

Abstract
The extreme polymorphisms of human leukocyte antigen (HLA) class I proteins result in structural variations in their peptide binding sites to achieve diversity in antigen presentation. External factors could independently constrict or alter HLA class I peptide repertoires. Such effects of the assembly factor tapasin were assessed for HLA-B*44:05 (Y116) and a close variant, HLA-B*44:02 (D116), which have low and high tapasin dependence, respectively, for their cell surface expression. Analyses of the HLA-B*44:05 peptidomes in the presence and absence of tapasin reveal that peptides with C-terminal tryptophans and higher predicted affinities are preferentially selected by tapasin, coincident with reduced frequencies of peptides with other C-terminal amino acids, including leucine. Comparisons of the HLA-B*44:05 and HLA-B*44:02 peptidomes indicate the expected structure-based alterations near the peptide C-termini, but also C-terminal amino acid frequency and predicted affinity changes among the unique and shared peptide groups for B*44:02 and B*44:05. Overall, these findings indicate that the presence of tapasin and the tapasin-dependence of assembly alter HLA class I peptide binding preferences at the peptide C-terminus. The particular C-terminal amino acid preferences that are altered by tapasin are expected to be determined by the intrinsic peptide binding specificities of HLA class I allotypes. Additionally, the findings suggest that tapasin deficiency and reduced tapasin dependence expand the permissive affinities of HLA class I bound peptides, consistent with prior findings that HLA class I allotypes with low tapasin dependence have increased breadth of CD8+ T cell epitope presentation and are more protective in HIV infections.
Keywords: HLA class I, MHC class I, HLA-B*44:02, HLA-B*44:05, Immunopeptidome, Tapasin, Peptide loading complex, Tryptophan, Half-life, peptide binding affinity
Introduction
Human Leukocyte Antigen (HLA) class I molecules present peptide antigens to CD8+ T cells (1) to trigger cytokine production and cytotoxicity. Thousands of HLA class I polymorphic variants exist in the human population (2), with each variant containing a unique peptide binding site that determines its peptide repertoire specificity (3). Most humans express 6 different HLA class I molecules on the surface of nucleated cells. The peptide binding specificities of individual HLA class I variants determine their abilities to induce CD8+ T cell responses and contribute to adaptive immunity against viruses and cancers.
Following the processing of cytosolic proteins via the proteasome, the assembly of major histocompatibility complex (MHC) class I molecules with peptides occurs in the endoplasmic reticulum (ER) lumen with the aid of the peptide-loading complex (PLC) (4, 5). Tapasin, a key component of the PLC, has multiple roles in MHC class I assembly. Together with the chaperone calreticulin and the oxidoreductase ERp57, tapasin interacts with peptide-deficient versions of MHC class I molecules in the ER lumen and stabilizes this conformation (5–11). In doing so, the PLC increases peptide binding to MHC class I molecules. Tapasin also optimizes the MHC class I peptide repertoire towards the binding of higher-affinity peptides and higher affinity peptides more readily dissociate tapasin-MHC class I complexes (6–9).
HLA class I allotypes are known to vary in their requirements for tapasin, exhibiting a wide spectrum of tapasin-dependencies (12–15). Noteworthy for such differences are two closely related HLA-B allotypes- HLA-B*44:05 and HLA-B*44:02, which differ by a single amino acid at position 116 within their heavy chain sequences. B*44:02 and B*44:05 are at the extreme ends of the tapasin-dependency spectrum (6, 13–15). These two allotypes allow for an understanding of precisely how tapasin influences HLA class I peptidome compositions, which thus far remains uncharacterized on a global scale. Recent advances in mass spectrometry of HLA class I peptidomes from monoallelic cell lines have allowed for the generation of peptidome datasets for a large number of individual HLA class I allotypes (16, 17). We applied a similar approach to examine the key differences between HLA-B*44:05 peptidomes isolated from B*44:05 monoallelic cells expressing or lacking tapasin. The marked reduction in HLA-B*44:02 expression in cells lacking tapasin precluded an analysis of the B*44:02 peptidome from cells lacking tapasin. However, the peptidomes of HLA-B*44:05 and B*44:02 from tapasin-sufficient cells were compared, revealing key insights into the effects of tapasin dependence of an allotype on peptide selection.
Materials and methods
Materials
PureProteome Protein A magnetic beads were obtained from EMD Millipore (LSKMAGA10). The RPMI-1640 media with L-glutamine, 1X PBS, L-glutamine, fetal bovine serum (FBS) and Antibiotic-Antimycotic (100X) were procured from Gibco (ThermoFisher Scientific). Several lab chemicals including brefeldin A, polybrene, phenylmethylsulfonyl fluoride (PMSF), sodium deoxycholate, octyl-beta-D glucopyranoside, iodoacetamide, triton X-100, triethanolamine (TEA), tris-base, glycine, protease inhibitor cocktail (catalog # P8340) and sodium azide were obtained from Sigma Aldrich. Dimethyl pimelimidate (DMP, catalog # 21667), FITC, acetonitrile, blasticidin S-HCl and trifluoroacetic acid were purchased from ThermoFisher Scientific. Bio-Rad 4–20% precast polyacrylamide gels (catalog # 4561096) were used for SDS-PAGE wherever manual gels were not used. Accugene 0.5 M EDTA solution was obtained from Lonza Bioscience.
Cell lines and vector constructs
The 721.221 cell line (obtained from R. DeMars) lacks endogenous HLA-A, B, and C expression (18). The 721.221 cell lines expressing HLA-B*44:02 or HLA-B*44:05 (gifted by Dr. Wilfredo F. Garcia-Beltran, Ragon Institute) were grown in R10 media (RPMI 1640 (L-Glutamine containing media) supplemented with 10 % (v/v) FBS, 2 mM L-glutamine, and 1X antibiotic-antimycotic). The plasmids plentiCRISPRv2-BLAST, psPAX2 and pMD2.G were procured from Addgene.
Crosslinking of Protein A magnetic beads to W6/32 antibody
The W6/32 antibody (19) was purified from mouse ascites and crosslinked to PureProteome Protein A magnetic beads as previously described (20). Briefly, following equilibration with 200 mM TEA pH 8.3, Protein A magnetic beads were incubated with the W6/32 antibody (2 μg antibody/μL of beads slurry) in 1X PBS for 1h at 4 °C. The unbound antibody was removed by washing the beads thrice with 200 mM TEA pH 8.3. Beads were crosslinked to the bound antibody using freshly prepared 5 mM DMP in 200 mM TEA pH 8.3 for 1h at ambient temperature. The crosslinking reaction was quenched using 5% (v/v) 1 M Tris-HCl pH 8.0 for 30 min at ambient temperature. Non-crosslinked antibody was further removed by incubating the beads with 0.1 M glycine-HCl pH 2.7 for 1 min. The beads were washed thrice with ice-cold PBS and stored at 4 °C in 1X PBS with 0.01% sodium azide.
CRISPR/Cas9-based gene editing, single-cell cloning, and immunoblotting
To create 721.221-B*44:05 cells with tapasin knockout, the single guide RNA (sgRNA) targeting a sequence 5’-AAGCGGCTCATCTCGCAGTG-3’ within exon 3 of the tapasin gene was used. The oligos for sgRNA synthesis (ordered from IDT) were cloned into the pLentiCRISPRv2-BLAST (pLB) vector. HEK293T cells were transfected with pLB-tapasin sgRNA vector together with the packaging plasmid psPAX2 and VSV-G envelope expressing plasmid pMD2.G. The virus particles were harvested, filtered, and used for infection of 721.221-B*44:05 cells by spinoculation at 2500 rpm for 2h at ambient temperature in the presence of 8 μg/mL polybrene. Following the selection of transduced cells with 5 μg/mL blasticidin, single-cell cloning was performed by plating the cells at a density of 5 cells/mL in 96 well plates.
For detection of tapasin knockout, single-cell clones were harvested and lysed in 1X tris buffered Saline (TBS) with 1% triton X-100. Tapasin was detected in the lysates following 12% SDS-PAGE and western transfer using anti-tapasin antibody (MABF249 from Sigma-Aldrich).
HLA-B heavy chain and β2-microglobulin (β2m) were probed using HC10 (mouse hybridoma procured from University of Michigan Hybridoma Core) and purified anti-β2m (Biolegend; catalog # 316302) antibodies.
Flow cytometry staining and antibody binding capacity determination
Before each immunoaffinity purification experiment discussed in the following sections, the cells were harvested and stained for surface HLA class I using W6/32-FITC antibody (FITC conjugation was performed in the lab following the manufacturer’s protocol). The staining was done in 1X PBS containing 2% FBS (staining buffer) for 30 min at 4 °C. The cells were washed twice with the staining buffer and stained with 7-AAD viability dye. Unfixed cells were analyzed by flow cytometry using BD LSRFortessa™ cell analyzer.
For quantitative flow cytometry experiments Quantum™ simply cellular anti-mouse IgG beads (Bangs Laboratory, Inc.) were stained concurrently with the cells using the same antibody dilution and analyzed at the same time as the cells.
The analysis of flow cytometry data was performed using FlowJo™. Live cells were gated based on negative 7-AAD staining and W6/32 geometric mean fluorescence intensity (gMFI) values were determined. Standard curves were plotted using the geometric mean fluorescence intensity (gMFI) values for various bead populations vs. the known number of Fc receptors on the respective beads as given by the manufacturer. The antibody binding capacity (ABC) of cells was determined by interpolation from standard curves. The W6/32-ABC values of cells represent the approximate number of HLA class I molecules expressed on individual cells.
Stability and half-life calculations of cell surface peptide- HLA-B complexes
Cell surface stability of peptide-HLA-B complexes was measured as described previously (21, 22). Briefly, the 721.221-B*44:05 cells (wild-type or tapasin-KO) and 721.221-B*44:02 cells were counted and added to a 96-well plate in triplicates for each time point in RPMI+10% FBS media. Brefeldin A was added at 0.5 μg/mL concentration and the cells were harvested after incubation in brefeldin A for indicated time periods. The cells were washed with 1X PBS and incubated with W6/32-FITC antibody for 30 minutes at 4 °C in the staining buffer mentioned above followed by two washes in the staining buffer. Cells were stained with viability dye 7-AAD and analyzed by flow cytometry using BD LSRFortessa™ cell analyzer.
The geometric MFI values for cells at various time points were analyzed in GraphPad Prism using one phase decay model constraining the plateau to a constant value of zero to determine the half-life values. Average half-lives from multiple experiments were analyzed by two-tailed unpaired t-tests to determine significance values.
Immunoaffinity (IA) purification of peptide-HLA class I complexes
For immunoaffinity purification, as published previously (23), 721.221-B*44:02 and 721.221-B*44:05 (wild type and tapasin-KO cells) were grown to ~2 to 4 × 108 cell density. The cells were lysed in 10 mL of lysis buffer (1 mM EDTA, 1 mM PMSF, 1% octyl-beta-D glucopyranoside and 0.25 % sodium deoxycholate in 1X PBS supplemented with protease inhibitor cocktail (1:200) and freshly prepared 0.2 mM iodoacetamide). The lysis was performed at 4 °C for 1 h on rotation. The lysates were cleared by centrifugation at 14000 rpm for 30 min at 4 °C. The cleared lysates were incubated with 2 mL blank protein A beads for pre-clearing at 4 °C for 30 min on rotation. The pre-cleared lysates were incubated with 2 mL of protein A beads crosslinked to W6/32 antibody (W6/32 beads) on rotation for 3h at 4 °C. The beads were harvested and washed with 8 column volumes of 1X PBS followed by 1 column volume each of 0.1X PBS and water. HLA class I complexes were eluted in six serial fractions with 0.2 mL of freshly prepared 0.2% (v/v) trifluoracetic acid. For reuse, the beads were regenerated with 1 column volume of water and 3 column volumes of 1X PBS before storing the beads in 1X PBS with 0.01% sodium azide. All the wash buffers as well as the elution buffer were kept ice-cold.
Isolation of peptides
The peptides were further isolated from the immunoprecipitated peptide-HLA class I complexes using either Sep-Pak C18 cartridges (Waters Corp, Catalog # WAT020515) or stage tips packed manually using C18 extraction disks (20 stacked disks, 3M™ Empore™). The stage tips or the cartridges were activated with 80% acetonitrile in 0.1% TFA followed by a wash with 0.1% TFA. The eluates were loaded on the stage tips or Sep-Pak cartridges followed by washing with 0.1% TFA. Peptides were eluted using 30% acetonitrile in 0.1% TFA and dried using vacuum centrifugation.
Mass spectrometry
The peptides were resuspended in 2% ACN, 0.1% formic acid for mass spectrometry. The mass spectrometry was performed in Proteomics Resource Facility in the department of pathology at the University of Michigan on Thermo Q Exactive HF. For MS/MS, 2 μL of peptide solution was resolved on a nano-capillary reverse phase column (Acclaim PepMap C18, 2 microns, 50 cm, ThermoScientific) using 0.1% formic acid/acetonitrile gradient at 300 nL/min (2–25% acetonitrile for 105 min; 25–40% acetonitrile for 20 min followed by a 90% acetonitrile wash for 10 min and a further 30 min re-equilibration with 2% acetonitrile) and directly introduced into Q Exactive HF mass spectrometer (Thermo Scientific, San Jose CA). MS1 scans were acquired at 60K resolution (AGC target=3e6, max injection time=50ms). Data-dependent high-energy C-trap dissociation MS/MS spectra were acquired for the 20 most abundant ions (Top20) following each MS1 scan (15K resolution; AGC target=1e5; relative collision energy ~28%).
Mass spectrometry data analysis
All MS raw files (.raw files) were analyzed using PEAKS studio software. MS/MS spectra were searched against a sequence database created from the human sequences of UniprotKB/Swiss-Prot (Download date: 2015-06-19) appended with reversed protein sequences as decoys and common contaminants. The mass tolerance was set at 10 ppm for precursor ions and 0.02 Da for the fragment ions. Isotopic error correction and common variable modifications of methionine oxidation, N-terminal acetylation, and cysteine carbamidomethylation were enabled. The enzyme specificity was set as ‘none’. The false discovery rate (FDR) of 1% was used filtration of peptide-spectra matches (PSMs). MS/MS data from six runs of peptides eluted each from the B*44:02, B*44:05 -WT, and B*44:05-tapasin-KO cells were used for analyses. The peptides of lengths 8–14 were included in further analysis and the selected peptides were further filtered to remove 1) common contaminant peptides, 2) peptides derived from bovine proteins, and 3) peptides present in the MS/MS run of HLA class I null 721.221 cells.
Analyses of peptidome datasets
The mass spectrometry outputs provided by the PEAKS studio software were analyzed using an R language-based script that converted individual cells with numerical outputs into “TRUE” and “FALSE” labels, such that any peptide that was detected in a mass spectrometry run appeared as “TRUE”, whereas undetected peptides appeared as “FALSE.”
The peptides were then grouped based on the number of runs across which each peptide was detected. For comparisons described in Figures 2 and 3 and Supplemental Figure 1, total peptides detected in ≥2 independent MS/MS runs of a specific condition are used. For comparisons described in Figure 4 and Supplemental Figures 3 and 4, peptides were divided into shared and unique groups. Peptides unique to a specific allotype or condition were those detected in ≥2 independent MS/MS runs of only that allotype or condition (not detected in any run of the other allotype/condition). Peptides shared between any two conditions were those detected in ≥2 independent MS/MS runs of each of the two conditions under consideration.
Shannon entropy plots, Seq2Logo plots, and prediction of binding affinities of peptides
Shannon Entropy (SE) was used as a diversity index to determine the amino acid variabilities at specific positions within a peptide sequence for each allele or condition (21, 24). The peptide sequences from each dataset were assessed using equation “ here E(i) is the SE for position ‘i’ within the peptide sequence and qi is the probability of finding an amino acid at position ‘i’. The maximum SE = 4.3 indicates maximum flexibility when all the 20 amino acids are found at a position with equal probability. The smaller the value of SE for a position, the higher the restrictiveness of that position. Individual SE values were calculated for each position within a subset of shared or unique peptides grouped by length.
The consensus Seq2Logo motifs (25) for individual peptide subsets, for example, shared and unique peptides for each allele or condition were plotted using a web-based sequence logo generation tool (https://services.healthtech.dtu.dk/service.php?Seq2Logo-2.0).
The binding affinities of peptides within the shared and unique subsets were predicted using the NetMHCpan-4.1 (26) server (https://services.healthtech.dtu.dk/service.php?NetMHCpan-4.1).
Amino Acid Distribution Calculations
The fractional distribution of amino acids at the PC and PC-2 positions for the 9-mer, 10-mer, and 11-mer peptides was calculated from the fraction of total peptides in a subset with a particular amino acid at a specific position. These fractional distributions were plotted using Prism GraphPad Prism 9.
Statistical analysis
Statistical analyses were performed in GraphPad Prism version 9.4.1 or using R studio via the specific tests/methods mentioned in the respective figure legends.
Results
Influences of tapasin deficiency and tapasin dependence upon HLA-B44 expression and stability
The contribution of tapasin to shaping the HLA-B*44 peptidomes was determined by using CRISPR/Cas9-based gene editing to knockdown the expression of tapasin in HLA class I-deficient 721.221 cells that had been transduced to generate monoallelic 721.221-HLA-B*44:05 cells. Single-cell cloning using the limiting dilution method resulted in the generation of 721.221-B*44:05 clones with a complete tapasin knockout (B*44:05-tapasin-KO) (Figure 1A). Quantitative flow cytometry assays following surface staining by the peptide-HLA class I complex-specific W6/32 antibody (19, 27) showed a significant reduction in the number of HLA-B*44:05 molecules on the surface of 721.221-B*44:05-tapasin-KO (B*44:05-TPN-KO) cells when compared to wild type 721.221-B*44:05 (B*44:05-WT) cells, approximating the levels observed in 721.221-B*44:02 (B*44:02-WT) cells, which were also generated (Figure 1B). There was a strong decrease in surface HLA class I expression in 721.221-B*44:02-tapasin-KO cells compared to that in 721.221-B*44:02-WT cells, consistent with prior findings (Figure 1C) (6, 13, 14). The extremely low surface HLA class I expression prevented the use of 721.221-B*44:02-tapasin-KO cells for mass spectrometry-based peptidome analysis. By measuring the rate of loss of cell surface HLA class I expression in the presence of brefeldin A, which blocks protein secretory trafficking, we observed a small non-significant decrease in the cell surface stabilities of HLA-B*44:05 in the tapasin knockout cells relative to the corresponding tapasin-expressing cells (Figure 1D). Despite higher surface expression, B*44:05 also exhibited a small non-significant decrease in cell surface stability compared with B*44:02 in the parental 721.221 cells expressing tapasin (Figure 1E).
Figure 1: Influences of tapasin deficiency and tapasin dependence upon HLA-B44 expression and stability:
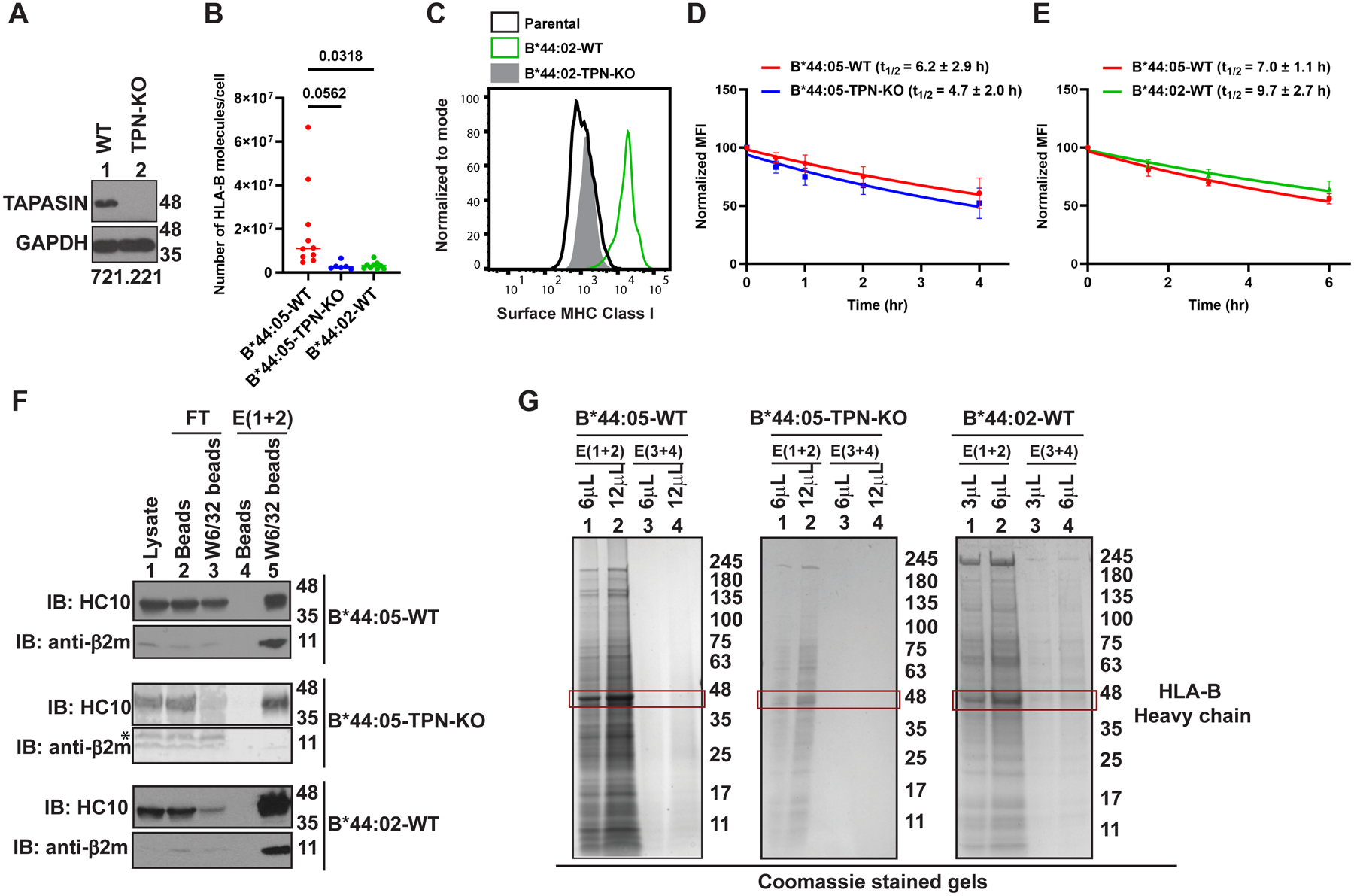
A) Representative blots showing tapasin (TPN) expression in the lysates of 721.221-B*44:05 WT (lane 1) and tapasin knockout (lane 2) cells. The sgRNA sequence is given in the materials and methods. B) The graph shows number of HLA-B molecules expressed on the surface of WT (n=10), TPN-KO (n=6) 721.221-B*44:05 cells and 721.221-B*44:02 cells (n=9) measured by quantitative flow cytometry following staining with W6/32-FITC antibody. Statistical significance was calculated using an ordinary one-way ANOVA with Tukey’s correction for multiple comparisons. C) Representative histograms show HLA-B*44:02 expression on WT or TPN-KO cells measured by flow cytometry following staining with W6/32-FITC antibody. A representative histogram for surface MHC Class I levels on 721.221-parental cells is shown as a control. D and E) Plots show averaged changes in the expression (normalized relative to the 0 h condition) of HLA class I on the surface of B*44:05-WT vs B*44:05-TPN-KO 721.221cells (C, n=4), and B*44:05-WT vs B*44:02-WT cells (D, n=5) assessed using the W6/32 antibody, at different time points following brefeldin A treatment. For each condition, calculated half-lives are indicated as t1/2. F) Representative blots of HLA-B*44 purifications from B*44:05-WT, B*44:05-TPN-KO and B*44:02-WT 721.221 cells. “Beads” indicates protein A beads used for pre-clearing of lysates and “W6/32 beads” indicates protein A beads crosslinked to W6/32 antibody. HLA-B heavy chain and β2m are probed using HC10 and anti-β2m antibodies, respectively. The low signal for β2m in the B*44:05-TPN-KO eluates from W6/32 beads is likely due to the short exposure of the representative blot. Follow up experiments verified that heavy chain: β2m ratios are similar in the eluates from B*44:05-WT and B*44:05-TPN-KO cells. G) Representative Coomassie brilliant blue stained gels of HLA-B*44 purifications from B*44:05-WT, B*44:05-TPN-KO and B*44:02-WT 721.221 cells. The red outlined box marks the HLA-B heavy chain bands in the eluates from W6/32 beads. E, Elution; FT, Flow through; IB, Immunoblot.
For the immunopeptidome analyses, we adopted an immunoaffinity (IA)-based approach for the purification of peptide-HLA-B complexes followed by LC-MS/MS-based identification of HLA-B binding peptides (16, 17, 23). The W6/32 antibody was covalently crosslinked to Protein A beads and used for the purification of peptide-HLA class I complexes. The presence of heavy chain and light chain (β2-microglobulin; β2m) signals in the elutions from W6/32 beads (Figure 1F; lanes 5) in contrast to the elutions from blank beads (Figure 1F; lanes 4) confirmed the successful purification of HLA class I. In the Coomassie blue stained SDS-PAGE gels, we observed a band at a size consistent with that of HLA class I heavy chains (~48 kDa) in the elutions from W6/32 beads (Figure 1G). The overall amount of HLA class I heavy chain recovered was higher in the elution fractions from B*44:05-WT cells compared to B*44:05-tapasin-KO cells and also compared to that from 721.221-B*44:02 cells (Figure 1G, lanes 1 and 2). The HLA-B*44 bound peptides from the three cell lines were further fractionated from the purified proteins using C18 reverse-phase chromatography and used for mass spectrometric analyses. For further analyses, six sets of MS/MS data corresponding to HLA-B purifications undertaken around the same timeframe were included for the B*44:05-tapasin-KO vs B*44:05-WT comparisons (Figure 2). Similarly, six sets of MS/MS data from separate but overlapping HLA-B purifications were used for the B*44:05-WT vs B*44:02-WT comparisons (Figure 3).
Figure 2: Characteristics of HLA-B*44:05 peptides purified from wild type or tapasin-KO cells:
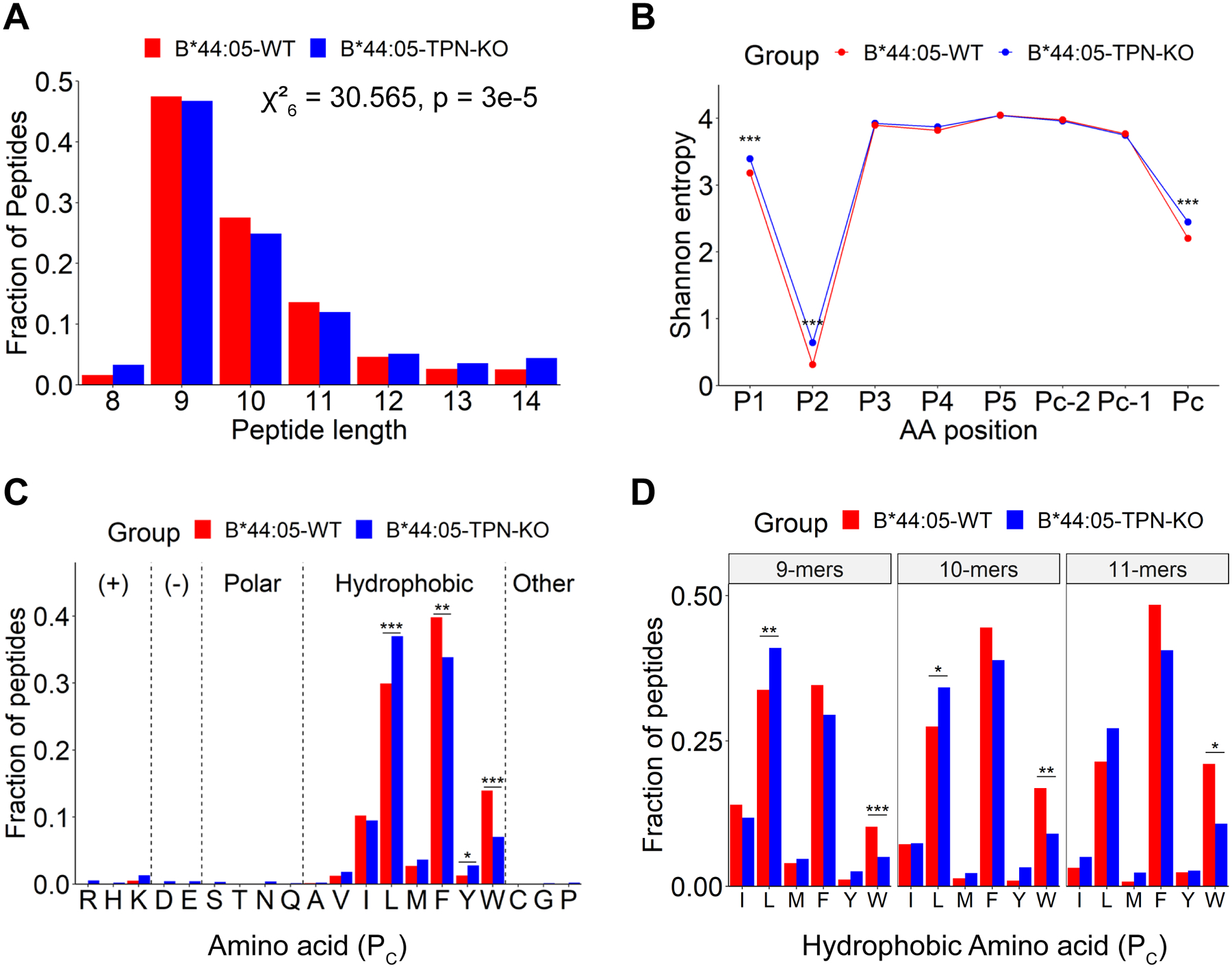
A) Length distributions of peptides identified for the B*44:05-WT and B*44:05-TPN-KO conditions, indicated as a fraction of total number of peptides. Significant differences in distribution were determined using the Chi-squared test of independence. B) Shannon Entropy (SE) plots for indicated positions within the 9-mer, 10-mer, and 11-mer peptides from (A). The PC, PC-1 and PC-2 represent the C-terminal, and the −1 and −2 positions relative to the C-terminus, respectively. Significant differences in SE at each position were determined using a bootstrap hypothesis test with Bonferroni corrected p-values. C and D) The fractional distribution of indicated amino acids at the PC position of combined 9–11-mer peptides (C) or 9-mer, 10-mer and 11-mer peptides as indicated (D), grouped according to the side chain properties of individual amino acids (C) or length (D). Significant differences in the amino acid distributions were determined using the Chi-squared test of independence, with standardized residual post-hoc tests using Benjamini-Hochberg corrections. *: p < 0.05; **: p < .01; ***: p < .001. Peptides that were overlapping across two or more runs for each condition (WT or TPN-KO) were included in the analyses.
Figure 3: Characteristics of peptides purified from B*44:02 compared with those from B*44:05 and the molecular basis for the specificity differences:
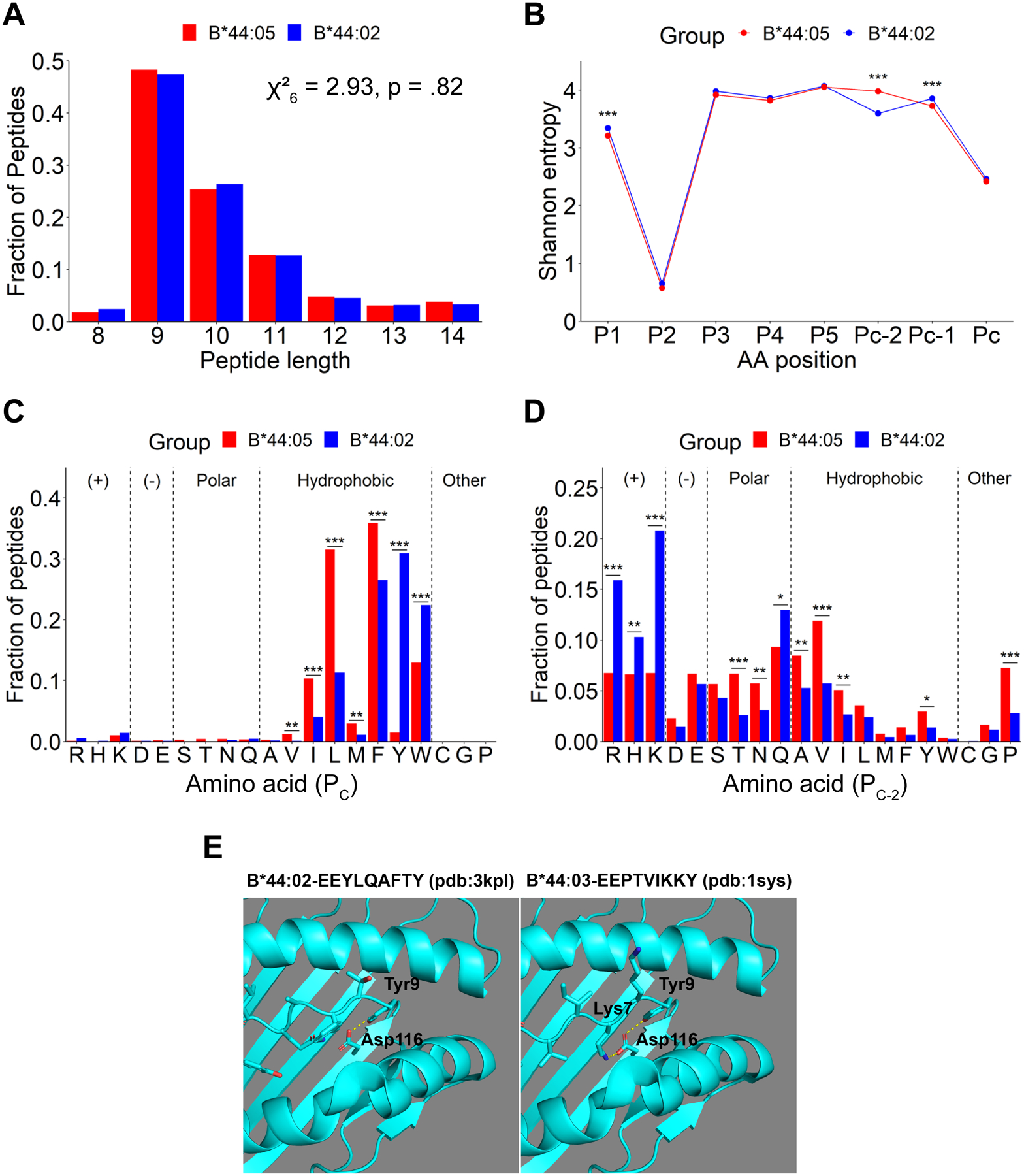
A) Length distributions of identified peptides, indicated as a fraction of total number of peptides. Non-significant differences in distributions were determined using the Chi-squared test of independence. B) SE plots for indicated positions within 9-mer, 10-mer, and 11-mer peptides from (A). The PC, PC-1 and PC-2 represent the C-terminal, and the −1 and −2 positions relative to the C-terminus, respectively. Significant differences in SE at each position were determined using a bootstrap hypothesis test with Bonferroni corrected p-values. C and D) The fractional distribution of all amino acids at the PC (C) and PC-2 (D) positions of 9-mer, 10-mer, and 11-mer peptides. Peptides are grouped by side chain properties. Significant differences in distributions were determined using the Chi-squared test of independence, with standardized residual post-hoc tests using Benjamini-Hochberg corrections. *: p < 0.05; **: p < .01; ***: p < .001. Peptides that were overlapping across two or more runs for each condition (B*44:05 and B*44:02) were included in the analyses. E) Structure of B*44:02 and B*44:03 with indicated peptides to depict the molecular basis for the tyrosine at PC and basic residue at PC-2 specificities. The Figure was prepared using the PyMOL Molecular Graphics System (version 1.8.4.2) Schrödinger, LLC.
Greater non-canonical characteristics of B*44:05 peptides from tapasin-KO cells
Peptides identified in the six independent LC-MS/MS runs of B*44:05 eluates from WT and tapasin-KO cells were first compared. The peptides that were overlapping across ≥ 2 runs were further analyzed (Figure 2), comprising a total of 1852 and 2492 peptides for the B*44:05-WT and B*44:05-tapasin-KO conditions, respectively. The most frequent peptide lengths observed are 9–11-mers for both B*44:05-WT and B*44:05-tapasin-KO cells, as expected for HLA class I-binding peptides (Figure 2A). However, 9–11-mers constitute a smaller fraction of B*44:05 peptides under the tapasin-KO condition when compared to the wild type condition (Figure 2A). Non-canonical lengths are more prevalent in the tapasin-KO condition and the overall length distribution differences are statistically significant (Figure 2A).
Peptide diversity analysis using Shannon entropy (SE) calculations (21, 24) indicates the lowest entropy and strongest restriction at the P2 position for both the B*44:05-WT and B*44:05-tapasin-KO peptide groups (Figure 2B), with a very strong enrichment of glutamic acid (Supplemental Figures 1A and 1B). The PC position is the second most restrictive position across different peptide lengths for both the groups, followed by the P1 position (Figure 2B and Supplemental Figures 1A and 1B). The strong restrictions observed at the P2 and PC positions are consistent with the previous reports that have described P2 and PC amino acids as anchor residues for binding of peptide ligands to the members of the HLA-B44 supertype (28). Comparative analyses of SE plots of 9–11-mers suggest that the peptides identified under B*44:05-tapasin-KO condition are significantly less restrictive at the P1, P2 and PC positions compared to the peptides identified under B*44:05-WT condition (Figure 2B). Overall, these data suggest that tapasin deficiency reduces the restrictiveness of peptide length and sequence for binding to B*44:05. However, the possibility that a higher non-specific peptide content contributes to the increased non-canonical characteristics of peptides identified in the tapasin-KO condition cannot be ruled out (Figures 2A and 2B).
Reduced tryptophan prevalence in the PC positions of B*44:05 peptides purified from tapasin-KO cells
Seq2Logo plots (25) illustrate the very strong preference for glutamic acid (E) at the P2 position among all the B*44 peptide groups (Supplemental Figure 1). Comparative analysis of the amino acid compositions at the PC positions of 9–11-mer peptides in the wild type and tapasin-KO groups reveals a significant increase in the fraction of peptides with leucine at the PC position in the B*44:05-tapasin-KO group when compared to the B*44:05-WT group (Figure 2C). On the other hand, there is a significant decrease in the representation of peptides with C- terminal tryptophan and phenylalanine in the tapasin-KO condition compared to those in the B*44:05-WT group (Figure 2C). There is also a significant increase in the tyrosine content in the tapasin-KO condition, although peptides with C-terminal tyrosines represent a minor fraction of peptides in both groups (Figure 2C). Deconvolution of the total peptide sets into individual lengths of 9–11-mers reveals a significant reduction in C-terminal tryptophan frequency across all three lengths in the tapasin-KO condition, an increase in leucine frequency that is significant among 9-mer and 10-mer peptides, but no additional C-terminal residue preference differences that are significant (Figure 2D). A small increase in tryptophan preference in the B*44:05-WT condition relative to the tapasin-KO condition is apparent across each length in the Seq2Logo plots (Supplemental Figures 1A compared to 1B).
Distinct restrictiveness and C-terminal residue preferences among peptides bound to B*44:02 and B*44:05
Peptides identified in six-independent LC-MS/MS runs of eluates from B*44:05 and B*44:02 cells were compared next. Peptides overlapping across ≥ 2 runs were further analyzed (Figure 3), comprising a total of 1915 and 1775 peptides for the B*44:05 and B*44:02 conditions, respectively. The most common peptide lengths observed among the peptides identified for both B*44:05 and B*44:02 conditions are 9–11-mers. There are no significant differences in the length distributions (Figure 3A).
Peptidome diversity analysis using SE calculations indicate the lowest entropy and strongest restriction for glutamic acid at the P2 position for both B*44:05 and B*44:02 peptides (Figure 3B and Supplemental Figures 1C and 1D). The PC position is the second most restricted position across the two peptide groups. Comparative analyses of the SE plots of 9–11-mers among B*44:02 and B*44:05 peptides show no significant differences in the restrictiveness of peptide binding to B*44:02 versus B*44:05 at either P2 or PC positions. Interestingly, a significantly higher restriction is seen at the PC-2 position for B*44:02 peptides (Figure 3B) which is not observed for B*44:05 peptides. On the other hand, compared to B*44:02 peptides, a small but significant decrease in SE value for P1 and PC-1 positions is also observed among B*44:05 peptides (Figure 3B).
Analysis of the fractional distribution of all twenty amino acids reveals a strong preference for hydrophobic amino acids at PC position among peptides identified for both B*44:02 and B*44:05 allotypes. There is a significantly higher prevalence of tryptophan and tyrosine at the PC position among B*44:02 peptides when compared to B*44:05 peptides (Figure 3C). The high C-terminal tryptophan prevalence among B*44:02 peptides is consistent with the B*44:02 data from a recently published large dataset (17) (Table I). Compared to B*44:02 peptides, B*44:05 peptides display a significantly higher preference for the less bulky hydrophobic amino acids, phenylalanine, leucine, isoleucine, methionine, and valine at PC position (Figure 3C). The Seq2Logo plots also illustrate the increased B*44:02 preference for C-terminal tyrosine and tryptophan across all lengths, compared to the generally greater dominance of C-terminal leucine or phenylalanine among B*44:05 peptides (Supplemental Figures 1C and 1D).
Table I: Analysis of C-terminal tryptophan preferences among common HLA class I allotypes:
The table shows 90 HLA class I allotypes for which C-terminal tryptophan prevalence among 9–11-mer peptides was analyzed using previously published peptidome datasets (1,7). Tapasin dependencies derived from a previous study (15) are shown.
| HLA Class I allotype | Tapasin dependence (15) | PC tryptophan content (in percentage) (17) | HLA Class I allotype | Tapasin dependence (15) | PC tryptophan content (in percentage) (17) |
|---|---|---|---|---|---|
| A*01:01 | 32.11 | 0.26 | B*35:03 | 2.10 | 0.06 |
| A*02:01 | 2.02 | 0.00 | B*35:07 | ND | 0.86 |
| A*02:02 | 1.45 | 0.03 | B*37:01 | 3.13 | 0.00 |
| A*02:03 | ND | 0.00 | B*38:01 | 6.39 | 2.01 |
| A*02:04 | ND | 0.00 | B*38:02 | ND | 3.25 |
| A*02:05 | 1.49 | 0.03 | B*40:01 | 1.90 | 0.00 |
| A*02:06 | ND | 0.00 | B*40:02 | 1.71 | 0.08 |
| A*02:07 | ND | 0.03 | B*40:06 | ND | 0.00 |
| A*02:11 | ND | 0.09 | B*42:01 | 1.72 | 0.31 |
| A*03:01 | 3.14 | 0.06 | B*44:02 | 190.40 | 36.24 |
| A*11:01 | 4 | 0.02 | B*44:03 | 274.16 | 29.93 |
| A*11:02 | 3.99 | 0.00 | B*45:01 | 16.31 | 0.13 |
| A*23:01 | 2.36 | 4.40 | B*46:01 | 60.66 | 0.00 |
| A*24:02 | 3.61 | 6.69 | B*49:01 | 82.40 | 0.05 |
| A*24:07 | ND | 6.82 | B*50:01 | 286.17 | 0.45 |
| A*25:01 | 3.91 | 38.58 | B*51:01 | 4.92 | 0.18 |
| A*26:01 | 8.33 | 0.27 | B*52:01 | 17.51 | 0.17 |
| A*29:02 | 4.38 | 0.00 | B*53:01 | 1.96 | 34.05 |
| A*30:01 | 6.52 | 0.00 | B*54:01 | ND | 0.09 |
| A*30:02 | 4.54 | 0.00 | B*55:01 | 6.46 | 0.07 |
| A*31:01 | 3.67 | 0.00 | B*55:02 | ND | 0.07 |
| A*32:01 | 2.92 | 37.73 | B*56:01 | 4.60 | 0.00 |
| A*33:01 | 5.34 | 0.00 | B*57:01 | 28.43 | 59.30 |
| A*33:03 | 4.20 | 0.00 | B*57:03 | 2.46 | 23.69 |
| A*34:01 | ND | 0.04 | B*58:01 | 4.30 | 64.52 |
| A*34:02 | 4.66 | 0.06 | B*58:02 | 73.28 | 5.08 |
| A*36:01 | 16.33 | 0.11 | C*01:02 | 3.67 | 0.00 |
| A*66:01 | 3.77 | 0.00 | C*02:02 | 4.17 | 0.00 |
| A*68:01 | 1.17 | 0.17 | C*03:02 | 3.19 | 0.20 |
| A*68:02 | 1.19 | 0.00 | C*03:03 | 3.01 | 0.57 |
| A*74:01 | 2.31 | 0.04 | C*03:04 | 2.32 | 0.74 |
| B*07:02 | 2.37 | 0.31 | C*04:01 | 13.59 | 0.96 |
| B*07:04 | ND | 0.14 | C*04:03 | ND | 0.81 |
| B*08:01 | 23.78 | 0.17 | C*05:01 | 1.68 | 0.52 |
| B*13:01 | ND | 12.62 | C*06:02 | 7.43 | 0.06 |
| B*13:02 | 12.35 | 0.04 | C*07:01 | 1.38 | 0.48 |
| B*14:02 | 7.68 | 0.09 | C*07:02 | 1.63 | 0.19 |
| B*15:01 | 6.10 | 0.14 | C*07:04 | 1.69 | 0.00 |
| B*15:02 | ND | 0.42 | C*08:02 | 1.97 | 0.18 |
| B*15:03 | 7.52 | 0.11 | C*12:02 | 2.83 | 0.00 |
| B*15:10 | 3.72 | 0.13 | C*12:03 | 3.76 | 2.74 |
| B*15:17 | ND | 3.20 | C*14:02 | 3.19 | 0.53 |
| B*18:01 | 2.40 | 0.92 | C*15:02 | 3.31 | 0.12 |
| B*27:05 | 3.38 | 0.12 | C*16:01 | 5.98 | 1.72 |
| B*35:01 | 1.26 | 1.79 | C*17:01 | 2.92 | 0.12 |
ND, not determined
The fractional amino acid distributions at the PC-2 position also vary considerably between B*44:02 and B*44:05 peptides (Figure 3D). B*44:02 peptides exhibit a significantly higher fraction of positively charged amino acids; histidine, arginine and lysine at the PC-2 position compared to B*44:05 peptides (Figure 3D). In contrast to B*44:02 peptides, B*44:05 peptides display a more significant prevalence of valine, alanine, threonine, proline, asparagine, and isoleucine at PC-2 position (Figure 3D).
Thus, HLA-B*44:02 and HLA-B*44:05 allotypes display distinct preferences for amino acids at PC and PC-2 positions of bound peptides, consistent with structural differences in the respective peptide-binding sites (D116 and Y116, respectively) (13). D116 can form hydrogen bonds with tyrosine at the PC position and salt bridges with positively charged residues at the PC-2 position, accounting for the observed peptide specificity differences (Figure 3E; similar to B*44:02, D116 is also present in B*44:03). On the other hand, the F pocket in B*44:05 is hydrophobic (pdb:1SYV) (13), and steric hinderance posed by the bulky aromatic ring of Y116 and side chains of nearby heavy chain residues 114, 156 and 97 allow accommodation of peptides with less bulky hydrophobic residues, valine, isoleucine, leucine and methionine at the PC position (Figure 3C and 3D). Besides, Y116 forms hydrogen bonds with aspartate 114 and arginine 97 residues in the B*44:05 heavy chain and an additional water-mediated hydrogen bond with the PC-2 residue of bound peptide (13). This might explain the significantly higher prevalence of hydrogen bonding amino acids such as threonine and asparagine at the PC-2 positions of B*44:05 peptides (Figure 3D).
Variable C-terminal tryptophan prevalence and affinities among different B*44:05 peptide groups
The identified peptides were further categorized into unique and shared groups for B*44:05 vs. B*44:05-tapasin-KO and for B*44:05 vs. B*44:02 comparisons (Supplemental Figure 2A and 2B, respectively). Among these groups, 272 peptides were unique to the B*44:05-WT, 779 peptides were unique to the B*44:05-tapasin-KO condition, while 1287 peptides were shared between the two conditions (Figure 4A and Supplemental Figure 3). The tapasin-KO group has a larger pool of unique peptides, suggesting that this condition is tolerant of more diverse sequences. Additionally, 1049 peptides were unique to B*44:05, 853 peptides were unique to the B*44:02, while 673 peptides were shared between the two conditions (Figure 4B and Supplemental Figure 4). In this comparison, the B*44:05 group has a slightly larger pool of unique peptides.
Figure 4. Tapasin and tapasin-dependent assembly facilitate the acquisition of B*44:05 peptides with enhanced C-terminal tryptophan content and higher affinities.
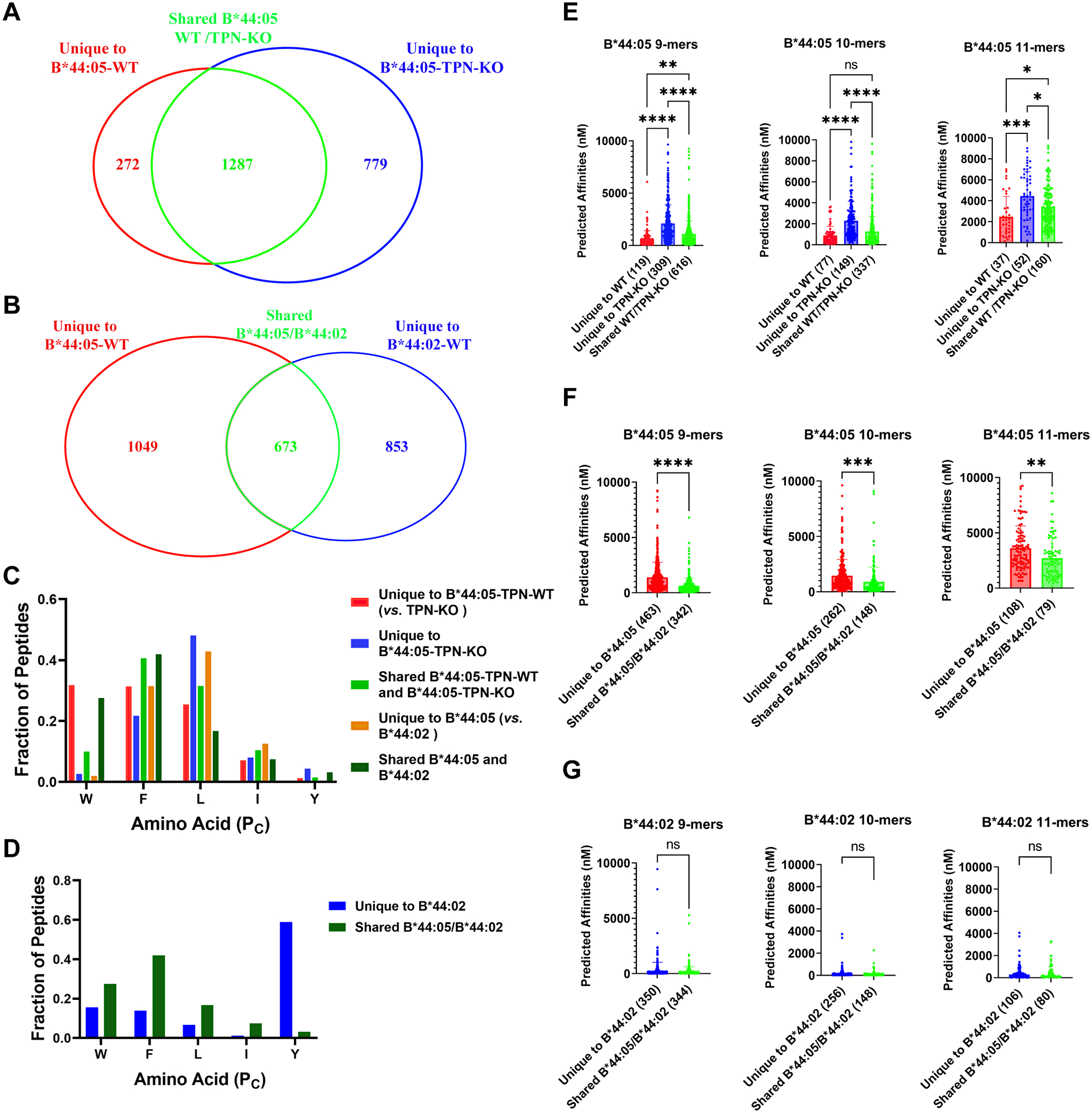
A) The Venn diagram shows the total number of peptides grouped into the unique to B*44:05-TPN-KO, unique to B*44:05-WT, or shared categories, as described in Supplemental Figure 2A. B) The Venn diagram shows the total number of peptides grouped into those unique to B*44:02-WT, unique to B*44:05-WT, or shared between the two categories, as described in Supplemental Figure 2B. C and D) Fraction of 9–11-mer peptides with indicated C-terminal amino acids for the different groups of B*44:05 peptides (C) and B*44:02 peptides (D). E-G) The predicted binding affinities (nM) of B*44:05 (E and F) or B*44:02 (G) peptides were calculated for the indicated peptide groups and plotted separately for the 9-mer, 10-mer, and 11-mer peptides. Peptides with predicted affinities >10,000 nM are excluded in all cases. Statistical analyses are based on one-way Anova analyses (E) or unpaired t-tests (F and G). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. ns, non significant.
Relative to the peptides unique to the B*44:05-tapasin-KO condition or the shared peptides, peptides unique to the B*44:05-WT condition display a higher tryptophan prevalence at the PC position (Figure 4C and Supplemental Figure 3). Tryptophan prevalence is also reduced at the PC position among peptides unique to the B*44:05-tapasin-KO condition compared to the shared peptides (Figure 4C and Supplemental Figure 3). In contrast, tryptophan is largely excluded among the peptides unique to B*44:05 in the B*44:02/B*44:05 comparisons (Figure 4C and Supplemental Figure 4A). Conversely, tryptophan is enriched among shared B*44:02/B*44:05 peptides (Figure 4C and Supplemental Figure 4C). These findings indicate enrichment of tryptophan among B*44:05 peptides unique to cells expressing tapasin, and those shared with an allotype that can only assemble in the presence of tapasin (B*44:02). We suggest that B*44:05 assembles with peptides via both tapasin-dependent and tapasin-independent routes. Among all possible B*44:05 peptides, tapasin-dependent assembly and assembly in the presence of tapasin promote the selection of peptides with C-terminal tryptophans.
C-terminal tyrosine is dominant among peptides unique to B*44:02 and absent or under-represented among peptides unique to B*44:05 or the shared peptides (Figures 4C, 4D and Supplemental Figure 4). Additionally, peptides unique to B*44:02 show an enrichment of the positively charged amino acids at their PC-2 positions (Supplemental Figure 4B), as discussed previously (Figures 3D and 3E).
Next, we predicted the binding affinities of peptides using NETMHCpan 4.1 (26) for 9-mer, 10-mer, and 11-mer peptides observed in the different unique and shared groups. Interestingly, 11-mer peptides, across all the peptide groups, have the lowest predicted binding affinities (Figures 4E–G, a higher number on Y-axis corresponds to a lower predicted binding affinity). Across all three peptide lengths, the peptides unique to the B*44:05-tapasin-KO condition yield the lower predicted binding affinities compared to peptides unique to the B*44:05-WT condition, and the shared peptides have intermediate predicted affinities relative to the two unique groups (Figure 4E). Lower predicted binding affinities of peptides unique to the B*44:05-tapasin-KO condition further substantiate the reduced restrictiveness of peptide binding to B*44:05 in the tapasin-KO cells. Importantly and notably, the predicted binding affinities of B*44:05 peptides shared with B*44:02 are significantly higher than those of the peptides unique to B*44:05 (Figure 4F). On the other hand, despite the altered specificities of the peptides unique to B*44:02 compared to those shared between B*44:02 and B*44:05, no significant differences were observed in the predicted binding affinities (Figure 4G). These findings suggest that the peptidomes of allotypes such as HLA-B*44:05 with low tapasin dependence may include both optimized and suboptimal peptides, whereas the peptides bound to a strictly tapasin-dependent allotype such as HLA-B*44:02 are more significantly optimized.
DISCUSSION
In this study, we compared the peptidomes of two members of the B44 supertype, HLA-B*44:02 and HLA-B*44:05, that differ significantly in their tapasin dependencies, ER retention and thermostabilities (6, 13–15, 29, 30), despite just a single amino acid difference within their heavy chain sequences. Lower surface levels of B*44:05 detected under tapasin-deficient conditions compared with the wild type conditions (Figure 1B) could result from increased intracellular accumulation of sub-optimally assembled peptide-HLA-B*44:05 complexes and the presence of epitope-free unstable B*44:05 heavy chains at the cell surface not detected by W6/32 antibody. Since tapasin-dependent peptides represent a small fraction of the overall B*44:05 peptidome (Figure 4A, unique to B*44:05-WT), the absence of such peptides may not per se explain the low cell surface HLA class I expression in the tapasin-KO condition. On the other hand, the lower predicted B*44:05 binding affinities for peptides unique to the tapasin-KO condition (Figure 4E) support the model of suboptimal assembly of B*44:05 with lower affinity peptides in the absence of tapasin. Our results, supported by earlier findings, clearly demonstrate that despite the high “tapasin independence” of HLA-B*44:05, the presence of tapasin facilitates high expression of HLA-B*44:05 complexes at the cell surface and induces an altered peptidome. The findings indicate that even the highly “tapasin-independent” allotypes are not truly “tapasin-independent”, but rather “facultative” and have lower tapasin dependence. The presence of tapasin promotes binding of high-affinity peptides and high cell surface expression.
Structural differences between HLA class I allotypes are clearly mediators of their peptide binding specificity differences (for example, Figure 3E), as is well known. Additionally, we suggest that tapasin’s influences can determine the prevalence of some C-terminal residues, such as tryptophan among B*44:05 peptides. This is indicated by the loss of the C-terminal tryptophan preference among B*44:05 peptides under tapasin-deficient conditions (Figures 2C and 2D). Furthermore, peptides unique to the tapasin-KO condition are strongly depleted for C-terminal tryptophan (Figure 4C and Supplemental Figure 3). On the other hand, there is increased prevalence of C-terminal tryptophans among B*44:05 peptides shared with B*44:02, compared to those unique to B*44:05, in the B*44:05 vs B*44:02 comparison group (Figure 4C and Supplemental Figure 4). Among HLA-B44 allotypes, the peptide PC tyrosine and PC-2 basic residue specificities are imposed by structural constraints, whereas the PC tryptophan specificity is at least partly determined by the additional presence of tapasin and by the tapasin-dependence of an allotype. Thus, in addition to its well-characterized function in optimizing the peptide repertoire, our study demonstrates that tapasin and tapasin-dependent assembly alter the C-terminal amino acid preferences among peptides bound to HLA-B44 allotypes.
Recent structural studies indicate the presence of a relatively open conformation of the MHC class I peptide binding groove in the tapasin-MHC class I complexes compared to the peptide-filled MHC class I complexes (10, 11). The editing loop at the N-terminus of tapasin sits on top of the F pocket, while a β hairpin element of tapasin contacts the base of the F pocket, resulting in a “clamp” like binding of the tapasin to the α2–1 domain of the MHC class I peptide binding groove (10, 11). The editing loop of tapasin has been suggested to stabilize the widening of the F pocket creating a peptide-receptive conformation (11). The increased detection of B*44:05 peptides with C-terminal tryptophans in the presence of tapasin suggests that tapasin-mediated widening of the F pocket is important to facilitate the binding of bulky amino acids such as tryptophan. Whether this is generally true for other HLA class I proteins that have high tryptophan content (Table I) at the peptide-C-termini remains to be studied. The effects of tapasin on the peptidome composition and peptide-binding specificities of individual HLA class I proteins are undoubtedly linked both to the intrinsic specificity of peptide-binding to the particular HLA class I protein, as well as to the intrinsic flexibility and “openness” of the peptide-binding site in the peptide-free conformation. Tryptophan incorporation might only be facilitated by tapasin for those HLA class I allotypes that are both sterically permissive for peptide C-terminal tryptophan and that have binding grooves that lack an intrinsic openness sufficient for tryptophan accommodation. Approximately 21% and 10% of allotypes that were included in a recent large dataset of HLA class I peptidomes (17) bind peptides with C-terminal tryptophans at frequencies greater than 1% and 10% respectively (Table I, 3rd column). These include allotypes that rank high and low on the tapasin dependence spectrum (15) (Table I, 2nd column). Both proteasomes and immunoproteasomes contain chymotryptic activity (31–33), which cleaves C-terminal to tryptophan and other hydrophobic amino acid residues (https://enzyme.expasy.org/EC/3.4.21.1). The relatively low C-terminal tryptophan frequency within HLA class I peptidomes (Table I) is likely dominantly imposed by steric constraints of their peptide binding sites, and also by the relative low frequency of occurrence of tryptophan residues within protein structures (https://www.nimbios.org/~gross/bioed/webmodules/aminoacid.htm). By promoting binding or inhibiting dissociation, tapasin could promote the capture of peptides with rare C-terminal amino acids. Overall, based on the findings of this study, we predict that tapasin will generally influence the C-terminal amino acid compositions of HLA class I-bound peptides, although the specific influences will be HLA class I allotype-dependent.
The predicted binding affinities of B*44:02 peptides are similar among the unique and shared (with B*44:05) groups, indicating that the two groups are similarly optimized (Figure 4G). In contrast, the predicted affinities of B*44:05 peptides are significantly higher in the shared (with B*44:02) group compared to peptides unique to B*44:05 (Figure 4F). These findings suggest that B*44:05, in the presence of tapasin, acquires peptides via both the tapasin-dependent and tapasin-independent mechanisms. Peptides shared with B*44:02 have higher predicted affinities, consistent with a tapasin-dependent mode of loading. The presence of tapasin thus partially optimizes peptide binding to HLA class I allotypes with low tapasin dependence, whereas optimization is applied more stringently to highly tapasin-dependent allotypes. We used NETMHCpan4.1 (26) binding affinity predictions that are trained on both the binding affinity (BA) datasets from the immune epitope database (IEDB) and eluted ligands (EL) datasets from various mass spectrometry studies. While integration of BA and EL datasets makes NETMHC pan4.1 more robust than earlier prediction servers, there may be differences between predicted and actual affinities for some peptides in the analyses shown in Figure 4. The large number of peptides that are used for the predictions is expected to average errors across the different conditions being compared. Additionally, there may be variations in the number of peptides used in training datasets for each allotype, which could affect the relative prediction accuracies. To mitigate the effects of such variations, all comparisons shown in Figure 4 are performed for peptides identified in association with just one of the B44 allotypes.
Molecular dynamics simulations indicate that the peptide-binding grooves of allotypes with low tapasin dependence are in a partially closed conformation even in their peptide-free states, stabilized by the intrinsic structural characteristics of these proteins (34, 35). In contrast, the peptide-binding grooves of highly tapasin-dependent allotypes exist in more unstable and open conformations in the absence of a peptide ligand (34, 35). Tapasin-binding near the F-pocket of the peptide binding grooves stabilizes the peptide-receptive open conformations (11). A partially open conformation for allotypes with low tapasin dependence could facilitate the intrinsic binding of some peptides, but exclude the binding of other peptides, particularly those with bulky hydrophobic residues such as tryptophans, which require tapasin’s assistance for loading.
Overall, our study provides important insights into how tapasin shapes the peptidomes of HLA class I allotypes with high or low tapasin dependence, altering both the affinities and specificities of peptide binding. It is likely that additional low-abundance and low-affinity peptides are present under the tapasin-deficient conditions, which are undetectable using the current approaches. These studies also explain why patients expressing HLA class I allotypes with low tapasin dependence are more protected in the context of infections (15). The ability of allotypes with low tapasin dependence to load peptides under suboptimal assembly conditions and to expand the repertoire to include lower affinity peptides could confer an important means of maintaining CD8+ T cell immunity in disease. While a high affinity peptide repertoire is considered a key outcome of MHC class I assembly, these findings illustrate the intrinsic resilience of the antigen presentation system related to their ligand affinity distributions that is also encoded within the allelic variants.
Supplementary Material
Key Points:
Tapasin alters HLA class I peptide binding preferences at peptide C-termini.
Tapasin-independent assembly expands affinity range of peptide-HLA class I binding.
Acknowledgements
We thank Elizabeth Smith from the University of Michigan Hybridoma Core for antibody production. We thank Corey Powell from CSCAR (Consulting for Statistics, Computing and Analytics Research) at University of Michigan for help with the statistical analysis. We thank Drs. Laura Santambrogio, Leonard Foster and Queenie Chan for many helpful discussions and Dr. Jie Geng for assistance with Fig. 3E.
This work was supported by the National Institutes of Health Grants to MR (R01AI044115 and R21AI164025), AJZ (T32AI007528 and T32AI007413) and AIN (R01GM94231), and National Cancer Institute grant to AIN (U24CA271037).
Footnotes
Conflicts of Interest
All the authors have no conflicts of interest to declare.
Disclaimer: This project has been funded in whole or in part with federal funds from the Frederick National Laboratory for Cancer Research, under Contract No.75N91019D00024. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This Research was supported in part by the Intramural Research Program of the NIH, Frederick National Lab, Center for Cancer Research.
REFERENCES
- 1.Jiang J, Natarajan K, and Margulies DH. 2019. MHC Molecules, T cell Receptors, Natural Killer Cell Receptors, and Viral Immunoevasins-Key Elements of Adaptive and Innate Immunity. Adv Exp Med Biol 1172: 21–62. [DOI] [PubMed] [Google Scholar]
- 2.Robinson J, Barker DJ, Georgiou X, Cooper MA, Flicek P, and Marsh SGE. 2020. IPD-IMGT/HLA Database. Nucleic Acids Res 48: D948–D955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zaitoua AJ, Kaur A, and Raghavan M. 2020. Variations in MHC class I antigen presentation and immunopeptidome selection pathways. F1000Res 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blum JS, Wearsch PA, and Cresswell P. 2013. Pathways of antigen processing. Annu Rev Immunol 31: 443–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blees A, Januliene D, Hofmann T, Koller N, Schmidt C, Trowitzsch S, Moeller A, and Tampe R. 2017. Structure of the human MHC-I peptide-loading complex. Nature 551: 525–528. [DOI] [PubMed] [Google Scholar]
- 6.Williams AP, Peh CA, Purcell AW, McCluskey J, and Elliott T. 2002. Optimization of the MHC class I peptide cargo is dependent on tapasin. Immunity 16: 509–520. [DOI] [PubMed] [Google Scholar]
- 7.Rizvi SM, and Raghavan M. 2006. Direct peptide regulatable interactions between tapasin and MHC class I molecules. Proc. Natl. Acad. Sci 103: 18220–18225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen M, and Bouvier M. 2007. Analysis of interactions in a tapasin/class I complex provides a mechanism for peptide selection. Embo J 26: 1681–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wearsch PA, and Cresswell P. 2007. Selective loading of high-affinity peptides onto major histocompatibility complex class I molecules by the tapasin-ERp57 heterodimer. Nat Immunol 8: 873–881. [DOI] [PubMed] [Google Scholar]
- 10.Jiang J, Taylor DK, Kim EJ, Boyd LF, Ahmad J, Mage MG, Truong HV, Woodward CH, Sgourakis NG, Cresswell P, Margulies DH, and Natarajan K. 2022. Structural mechanism of tapasin-mediated MHC-I peptide loading in antigen presentation. Nat Commun 13: 5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muller IK, Winter C, Thomas C, Spaapen RM, Trowitzsch S, and Tampe R. 2022. Structure of an MHC I-tapasin-ERp57 editing complex defines chaperone promiscuity. Nat Commun 13: 5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peh CA, Burrows SR, Barnden M, Khanna R, Cresswell P, Moss DJ, and McCluskey J. 1998. HLA-B27-restricted antigen presentation in the absence of tapasin reveals polymorphism in mechanisms of HLA class I peptide loading. Immunity 8: 531–542. [DOI] [PubMed] [Google Scholar]
- 13.Zernich D, Purcell AW, Macdonald WA, Kjer-Nielsen L, Ely LK, Laham N, Crockford T, Mifsud NA, Bharadwaj M, Chang L, Tait BD, Holdsworth R, Brooks AG, Bottomley SP, Beddoe T, Peh CA, Rossjohn J, and McCluskey J. 2004. Natural HLA class I polymorphism controls the pathway of antigen presentation and susceptibility to viral evasion. J Exp Med 200: 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rizvi SM, Salam N, Geng J, Qi Y, Bream JH, Duggal P, Hussain SK, Martinson J, Wolinsky SM, Carrington M, and Raghavan M. 2014. Distinct Assembly Profiles of HLA-B Molecules. J Immunol 192: 4967–4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bashirova AA, Viard M, Naranbhai V, Grifoni A, Garcia-Beltran W, Akdag M, Yuki Y, Gao X, O’HUigin C, Raghavan M, Wolinsky S, Bream JH, Duggal P, Martinson J, Michael NL, Kirk GD, Buchbinder SP, Haas D, Goedert JJ, Deeks SG, Fellay J, Walker B, Goulder P, Cresswell P, Elliott T, Sette A, Carlson J, and Carrington M. 2020. HLA tapasin independence: broader peptide repertoire and HIV control. Proc Natl Acad Sci U S A 117: 28232–28238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abelin JG, Keskin DB, Sarkizova S, Hartigan CR, Zhang W, Sidney J, Stevens J, Lane W, Zhang GL, Eisenhaure TM, Clauser KR, Hacohen N, Rooney MS, Carr SA, and Wu CJ. 2017. Mass Spectrometry Profiling of HLA-Associated Peptidomes in Mono-allelic Cells Enables More Accurate Epitope Prediction. Immunity 46: 315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sarkizova S, Klaeger S, Le PM, Li LW, Oliveira G, Keshishian H, Hartigan CR, Zhang W, Braun DA, Ligon KL, Bachireddy P, Zervantonakis IK, Rosenbluth JM, Ouspenskaia T, Law T, Justesen S, Stevens J, Lane WJ, Eisenhaure T, Lan Zhang G, Clauser KR, Hacohen N, Carr SA, Wu CJ, and Keskin DB. 2020. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat Biotechnol 38: 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shimizu Y, and DeMars R. 1989. Production of human cells expressing individual transferred HLA-A,-B,-C genes using an HLA-A,-B,-C null human cell line. J Immunol 142: 3320–3328. [PubMed] [Google Scholar]
- 19.Barnstable CJ, Bodmer WF, Brown G, Galfre G, Milstein C, Williams AF, and Ziegler A. 1978. Production of monoclonal antibodies to group A erythrocytes, HLA and other human cell surface antigens-new tools for genetic analysis. Cell 14: 9–20. [DOI] [PubMed] [Google Scholar]
- 20.Lamoliatte F, McManus FP, Maarifi G, Chelbi-Alix MK, and Thibault P. 2017. Uncovering the SUMOylation and ubiquitylation crosstalk in human cells using sequential peptide immunopurification. Nat Commun 8: 14109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yarzabek B, Zaitouna AJ, Olson E, Silva GN, Geng J, Geretz A, Thomas R, Krishnakumar S, Ramon DS, and Raghavan M. 2018. Variations in HLA-B cell surface expression, half-life and extracellular antigen receptivity. Elife 7: e34961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zarling AL, Luckey CJ, Marto JA, White FM, Brame CJ, Evans AM, Lehner PJ, Cresswell P, Shabanowitz J, Hunt DF, and Engelhard VH. 2003. Tapasin is a facilitator, not an editor, of class I MHC peptide binding. J Immunol 171: 5287–5295. [DOI] [PubMed] [Google Scholar]
- 23.Bassani-Sternberg M, Chong C, Guillaume P, Solleder M, Pak H, Gannon PO, Kandalaft LE, Coukos G, and Gfeller D. 2017. Deciphering HLA-I motifs across HLA peptidomes improves neo-antigen predictions and identifies allostery regulating HLA specificity. PLoS Comput Biol 13: e1005725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rao X, Costa AI, van Baarle D, and Kesmir C. 2009. A comparative study of HLA binding affinity and ligand diversity: implications for generating immunodominant CD8+ T cell responses. J Immunol 182: 1526–1532. [DOI] [PubMed] [Google Scholar]
- 25.Thomsen MC, and Nielsen M. 2012. Seq2Logo: a method for construction and visualization of amino acid binding motifs and sequence profiles including sequence weighting, pseudo counts and two-sided representation of amino acid enrichment and depletion. Nucleic Acids Res 40: W281–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reynisson B, Alvarez B, Paul S, Peters B, and Nielsen M. 2020. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res 48: W449–W454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parham P, Barnstable CJ, and Bodmer WF. 1979. Use of a monoclonal antibody (W6/32) in structural studies of HLA-A,B,C, antigens. J Immunol 123: 342–349. [PubMed] [Google Scholar]
- 28.DiBrino M, Parker KC, Margulies DH, Shiloach J, Turner RV, Biddison WE, and Coligan JE. 1995. Identification of the peptide binding motif for HLA-B44, one of the most common HLA-B alleles in the Caucasian population. Biochemistry 34: 10130–10138. [DOI] [PubMed] [Google Scholar]
- 29.Thammavongsa V, Raghuraman G, Filzen TM, Collins KL, and Raghavan M. 2006. HLA-B44 polymorphisms at position 116 of the heavy chain influence TAP complex binding via an effect on peptide occupancy. J Immunol 177: 3150–3161. [DOI] [PubMed] [Google Scholar]
- 30.Thammavongsa V, Schaefer M, Filzen T, Collins KL, Carrington M, Bangia N, and Raghavan M. 2009. Assembly and intracellular trafficking of HLA-B*3501 and HLA-B*3503. Immunogenetics 61: 703–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, and Huber R. 1997. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 386: 463–471. [DOI] [PubMed] [Google Scholar]
- 32.Harshbarger W, Miller C, Diedrich C, and Sacchettini J. 2015. Crystal structure of the human 20S proteasome in complex with carfilzomib. Structure 23: 418–424. [DOI] [PubMed] [Google Scholar]
- 33.Unno M, Mizushima T, Morimoto Y, Tomisugi Y, Tanaka K, Yasuoka N, and Tsukihara T. 2002. The structure of the mammalian 20S proteasome at 2.75 A resolution. Structure 10: 609–618. [DOI] [PubMed] [Google Scholar]
- 34.Sieker F, Springer S, and Zacharias M. 2007. Comparative molecular dynamics analysis of tapasin-dependent and -independent MHC class I alleles. Protein Sci 16: 299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sieker F, Straatsma TP, Springer S, and Zacharias M. 2008. Differential tapasin dependence of MHC class I molecules correlates with conformational changes upon peptide dissociation: a molecular dynamics simulation study. Mol Immunol 45: 3714–3722. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.