

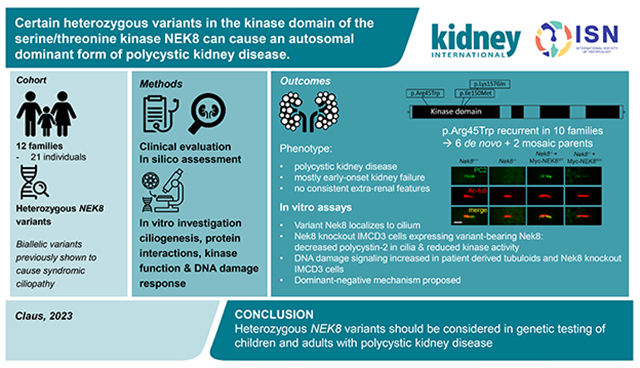
Abstract
Autosomal dominant polycystic kidney disease (ADPKD) resulting from pathogenic variants in PKD1 and PKD2 is the most common form of PKD, but other genetic causes tied to primary cilia function have been identified. Biallelic pathogenic variants in the serine/threonine kinase NEK8 cause a syndromic ciliopathy with extra-kidney manifestations. Here we identify NEK8 as a disease gene for ADPKD in 12 families. Clinical evaluation was combined with functional studies using fibroblasts and tubuloids from affected individuals. Nek8 knockout mouse kidney epithelial (IMCD3) cells transfected with wild type and variant NEK8 were further used to study ciliogenesis, ciliary trafficking, kinase function, and DNA damage responses. Twenty-one affected monoallelic individuals uniformly exhibited cystic kidney disease (mostly neonatal) without consistent extra-kidney manifestations. Recurrent de novo mutations of the NEK8 missense variant p.Arg45Trp, including mosaicism, were seen in ten families. Missense variants elsewhere within the kinase domain (p.Ile150Met and p.Lys157Gln) were also identified. Functional studies demonstrated normal localization of the NEK8 protein to the proximal cilium and no consistent cilia formation defects in patient-derived cells. NEK8-wild type protein or all variant forms of the protein expressed in Nek8 knockout IMCD3 cells were localized to cilia and supported ciliogenesis. However, Nek8 knockout IMCD3 cells expressing NEK8-p.Arg45Trp and NEK8-p.Lys157Gln showed significantly decreased polycystin-2 but normal protein ANKS6 localization in cilia. Moreover, p.Arg45Trp NEK8 exhibited reduced kinase activity in vitro. In patient derived tubuloids and IMCD3 cells expressing NEK8-p.Arg45Trp, DNA damage signaling was increased compared to healthy passage-matched controls. Thus, we propose a dominant-negative effect for specific heterozygous missense variants in the NEK8 kinase domain as a new cause of PKD.
Keywords: NEK8, polycystic kidney disease, kinase, ciliopathy
Graphical Abstract
Lay Summary:
Autosomal dominant polycystic kidney disease (ADPKD) is the most common form of polycystic kidney disease, but other genetic causes tied to primary cilia function have been identified. Heterozygous pathogenic variants in NEK8 were identified in this study as the cause of an autosomal dominant form of PKD in 12 families. Clinical evaluation of patients with variants found within the NEK8 kinase domain was combined with functional studies using fibroblasts and tubuloids from affected individuals, and mouse renal epithelial cells. The study proposes that these heterozygous missense variants act in a dominant-negative fashion to cause kidney disease. The findings further suggest that, NEK8 should be included in gene panel testing for children and adults with PKD, and a causal relationship considered when heterozygous variants in the NEK8 kinase domain are present.
Introduction:
Polycystic kidney diseases (PKD) are the most prevalent inherited kidney diseases.1 Autosomal dominant polycystic kidney disease (ADPKD), resulting from pathogenic variants in PKD1 and PKD2, is the most common form of PKD. This disease usually presents in adulthood and often leads to kidney failure, with kidney failure with replacement therapy (KFRT) typically needed in the 6th decade of life.2 ADPKD-like disease can also be caused by single rare variants in genes such as GANAB, DNAJB11, ALG5, ALG8, ALG9 and IFT140.3-8. The disease is often more attenuated in these patients, with KFRT rare. Autosomal recessive polycystic kidney disease (ARPKD) is mostly caused by biallelic variants in PKHD1. The majority of patients present in utero or in the neonatal period and often the disease progresses to KFRT during childhood.9
PKD is mostly caused by pathogenic variants in genes encoding proteins with roles in the functioning of primary cilia and therefore classified as a ciliopathy. Renal ciliopathies can either present with an isolated kidney phenotype, or can have extensive extra-renal characteristics, since primary cilia are present on nearly all cell types.9,10 Autosomal recessive inheritance is seen in many primary ciliopathies including nephronophthisis, and Bardet-Biedl, Joubert, and Meckel-Gruber syndromes, with multiple, often overlapping, monogenic causes. Autosomal dominant inheritance is rare for ciliopathies, other than ADPKD(-like). Some syndromic ciliopathy genes have both an autosomal recessive and autosomal dominant presentations, include ZNF423.11 For IFT140 and DNAJB11, an autosomal recessive syndromic ciliopathy is seen in addition to the ADPKD(-like) phenotype.5,12,13 Similarly for ALG8 and ALG9, autosomal recessive inheritance results in congenital disorders of glycosylation (CDG), compared to the mild ADPKD(-like) phenotype. Defective maturation and trafficking of the large PKD1 protein, polycystin-1 (PC1), may be a common feature of many of the ADPKD-like diseases.3 4
Biallelic pathogenic variants in NEK8 cause a syndromic ciliopathy with multiorgan developmental defects; referred to as renal-hepatic-pancreatic dysplasia (OMIM entry: 615415)14 The NEK8 protein is one of eleven ‘NEKs’, NimA (Never in mitosis A)-related kinases, with roles in cell cycle control, DNA damage response and other cell functions. In the kidney, NEK8 is required for tubular integrity, thought to regulate the renal tubule epithelial cell cytoskeletal structure, and is involved in ciliary biogenesis/function. It forms a complex with ANKS6, INVS, and NPHP3. ANKS6 likely coordinates the assembly of this complex, linking NPHP3 and INVS to NEK8, and targets the complex to the proximal ciliary axoneme.15,16 NEK8 has been suggested to interact with the polycystin signal transduction pathway, including regulating ciliary protein targeting.17 Functional studies have shown that biallelic NEK8 variants affect ciliogenesis, the DNA damage response, and epithelial morphogenesis.14
In this study we identified NEK8 as a monoallelic disease gene in a large patient cohort that included both recurrent de novo, mosaic, and inherited heterozygous NEK8 variants. We propose a dominant negative-effect for specific heterozygous NEK8 missense variants in the kinase domain.
Materials and Methods:
Patient enrollment and genomic analyses
Informed written consent was obtained, and is on file, from all patients or parents of the patients included in this study, and we adhered to all relevant ethical regulations at the respective institutions. Collaboration between centers was established through GeneMatcher.18 Patients from the Genomics England 100,000 Genomes Project were enrolled as previously described (REC reference 14/EE/1112).19 The procedures for sequencing, including coverage of PKD1, and variant filtering are described in the Supplementary Methods. The following transcripts were used in this study: NM_178170.2 (NEK8), NM_000297.4 (PKD2), NM_025114.4 (CEP290), NM_001009944.2 (PKD1), NM_024876.4 (COQ8B), and NM_000322.5 (PRPH2).
In silico predictions and structural modelling of NEK8 variants
Alphafold sequence AF-Q86SG6-F1 was imported to PyMOL and labelled according to available UniProtKB data due to lack of crystal structure. PyMOL Mutation Wizard was used to model the most likely structural impact of the variants for which AlphaFold produced a per-residue model confidence (pLDDT) >50. pLDDT is a measure for local accuracy of the model and is derived through the local Distance Difference Test (IDDT-Cα).20-22 CADD scores were used for in silico predictions of the identified variants.23 Furthermore, MetaDome’s protein tolerance landscape was used to study genetic tolerance per amino acid position of the current and previous identified variants in the NEK8 kinase domain.24
Analyses of the function of NEK8 in IMCD3 cells
Antibodies and reagents:
Rabbit polyclonal antibodies: Arl13b (17711-1-AP, Proteintech); GPR161 (13398-1-AP, Proteintech); ANKS6 (HPA008355, Sigma); NEK8 antibody is a kind gift from Dr. David Beier25; polycystin-2 antibody is provided by the Baltimore PKD Research and Clinical Core Center. Mouse monoclonal antibodies: acetylated α-tubulin (T7451, Sigma), Myc-4A6 (05724, Sigma); α-Tubulin (T6199, Sigma). Alexa Fluor-conjugated secondary antibodies are from Invitrogen. Primary antibodies were 1:1000 diluted for Western blot, and 1:500 diluted for immunofluorescence except for acetylated α-tubulin.
DNA constructs and CRISPR editing:
Full-length NEK8 was amplified by PCR using the published NEK8 plasmid as a template (pDONR223-NEK8 was a gift from William Hahn & David Root (Addgene plasmid #23418; http://n2t.net/addgene:23418; RRID:Addgene_23418)26 and subcloned into the pcDNA3-myc vector. All NEK8 variants were generated using a QuikChange site-directed mutagenesis kit (Agilent Technologies). The Nek8 guide RNA (gRNA) was designed using an online tool (https://zlab.bio/guide-design-resources, TGTGCACCTGTGCCTGCGAA) and subcloned into pSpCas9(BB)-2A-GFP (px458). All constructs were verified by DNA sequencing. The px458-Nek8 gRNA construct was transfected into IMCD3 cells as described.27 Western blot, immunostaining and kinase assay details, as well as information about statistical analyses, are found in the Supplementary Methods.
Human kidney tubuloid culture, cilia staining and assessment of DNA damage
Details on assessment of DNA damage and cilia staining in tubuloid cultures are described in the Supplementary Methods. 26,27
Assessment of DNA damage
Fibroblast cells (2.5x104) were plated in 6-well plates containing 22x22 coverslips. After 48 h, cells were pulsed with 10 uM EdU for 1 h, rinsed with 1X PBS, and treated with 0.5 mM hydroxyurea (MP Biomedical) for 6 h before fixation. Untreated controls were fixed immediately following the one-hour pulse with EdU. IMCD3 cells (4x104) were plated in a 12-well plate containing circular coverslips 24 h prior to fixation. Further detail on immunostaining and quantification is found in the Supplementary Methods.
Results:
Clinical details
A total of 21 individuals in twelve families with heterozygous NEK8 variants and cystic kidney disease were identified through genetic testing, GeneMatcher18, or in one case in the published literature (Figure 1 and Supplementary Data).28 None of the families were known to be consanguineous, and screening did not reveal a (likely) pathogenic variant in any other PKD related gene. Clinical and genetic information is summarized in Table 1. The age of onset of kidney cysts was variable, with multiple cases diagnosed prenatally and the oldest individual diagnosed in the eighth decade. Initial clinical diagnoses included ARPKD, ADPKD, nephronophthisis, and focal segmental glomerulosclerosis (FSGS: based on biopsy in two patients). None of the patients showed the multiorgan involvement associated with NEK8 biallelic disease. Ten families (14 individuals) had a recurrent variant in NEK8 (c.133C>T, p.Arg45Trp) and these cases, except Family 10, had a consistent phenotype of enlarged kidneys, arterial hypertension, KFRT (before adulthood in non-mosaic cases), and absence of hepatic cysts. The proband from Family 10 had a milder phenotype with slightly enlarged kidneys and at 16 years normal kidney function. Short stature was noted in two of fourteen individuals (Family 7-III-3, Family 7-III-4) with the recurrent p.Arg45Trp variant. The proband with p.Lys157Gln in Family 12 had a comparable phenotype with prenatal onset of enlarged cystic kidneys, severe feeding difficulties, and KFRT at 4 years. Family 11 with p.Ile150Met had a later-onset presentation with only the oldest individual requiring dialysis in her 70s. Two of six individuals (III-1; IV-1) in this family had hypertension and liver cysts and one had borderline enlarged kidneys. No malignancies have been reported in the cohort of 21 cases.
Figure 1. Pedigrees and polycystic kidney images ∣.

a. pedigrees of twelve families with heterozygous NEK8 variants. Solid symbols indicate affected individual with kidney disease. Arrow indicates proband. +/− indicates presence of heterozygous NEK8 variant. −/− indicate biallelic wildtype NEK8. Roman numerals indicate generation. MZ = monozygotic; d. death. b. MRI images of proband of Family 3 at age 16: enlarged kidney with multiple cysts, total kidney volume 1300 ml. c-d. Ultrasound of proband of Family 8: enlarged kidneys with extensive bilateral kidney cysts. At age 2 right kidney measured 13.6 cm and left kidney measured 13.3 cm (c), at age 7 right kidney: 17.5 cm and left kidney: 17.7 cm (d). e-g. CT analysis of Family 9: bilaterally enlarged kidneys with cysts. e. CT at 48 years of II-2, who is mosaic for the NEK8 variant, f-g. CT of proband IV-1 at 23 months (f) and at 5 years old (g). h. Pathology images of right kidney of proband of Family 4 at age 5 (20 x 11 x 10 cm, 1488 grams). i. Pathology images of right kidney of proband of Family 8 at 7 years old. j. Pathology images of left kidney of proband of Family 12 at age 4 (18 x 8 x 9.5 cm). k. Hematoxylin and eosin stain of proband of Family 8 showed multiple larger tubular cysts, with glomerular cysts also noted. l. Hematoxylin and eosin stain of proband of Family 12 shows multiple cysts both glomerular and tubular. Below the capsule cysts are smaller and they are larger at the corticomedullary junction. Some fibrosis is seen towards the medulla.
Table 1 –
Clinical findings
| Fam. - ind. |
Inheritance | NEK8 Variant |
Allelic state |
Gender | Year of birth |
Age PKD diagnosis (years) |
Suspected clinical diagnosis |
Age at KFRT (years) |
Age at KTx (years) |
Enlarged kidneys |
Liver cysts (age in years) |
Hyper -tension |
Other medical diagnoses |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | De novo | Arg45Trp | Het | Male | 2007 | 11 | NPH vs.ADPKD | 11 | 12 | + | − | + | − |
| 2 | De novo | Arg45Trp | Het | Female | 1986 | fetal | ARPKD | 4 | 5 | + | − | + | Alopecia |
| 3 | De novo | Arg45Trp | Het | Male | 2000 | 16 | ADPKD/ARPKD | 19 | 19 | + | − | + | − |
| 4 | De novo | Arg45Trp | Het | Male | 2011 | fetal | ADPKD | 5 | 5 | + | − | + | − |
| 5 | De novo | Arg45Trp | Het | Male | 2000 | neonatal | ADPKD-VEO | 12 | 13 | + | − | + | Left ventricular hypertrophy |
| 6 | Unknown* | Arg45Trp | Het | Female | 2002 | 9 | ARPKD | 11 | 12 | − | − | + | unknown |
| 7 II-8 | Mosaic | Arg45Trp | Mosaic (3%) | Male | ~1975 | unknown | ADPKD | 28 | 32 | unknown | unknown | unknown | unknown |
| 1 III-3 | Familial | Arg45Trp | Het | Male | 2006 | 1.5 | ADPKD | 14 | 14 | + | − | + | Kidney calcifications; short stature |
| 7 III-4 | Familial | Arg45Trp | Het | Male | 2006 | 1.5 | ADPKD | 15 | 15 | + | − | + | Kidney calcifications; short stature |
| 8 | De novo | Arg45Trp | Het | Male | N/A | 2 | ARPKD | 7 | 7 | + | − | + | Left ventricular hypertrophy and dilation; pericardial effusion; microhematuria |
| 9 II-2 | Mosaic | Arg45Trp | Mosaic (6%) | Female | N/A | 3 | ADPKD | 48 | 48 | + | + (48) | − | Melanoma, basal cell carcinoma; migraines; aneurysm; liver hemangioma; diverticulitis |
| 9 III-1 | Familial | Arg45Trp | Het | Female | N/A | 0 | ADPKD | 1 | 3 | + | − | + | Pre-eclampsia; B-cell m-PTLD; leukopenia; migraines; tertiary hyperparathyroidism |
| 9 IV-1 | Familial | Arg45Trp | Het | Female | N/A | fetal | ADPKD | 6 | 6 | + | − | + | Gross hematuria; anemia; small for gestational age |
| 10 | Unknown* | Arg45Trp | Het | Female | 2004 | 3 | NPHP (MSK) | N/A | N/A | +/− | − | in the past | − |
| 11 II-1 | Familial | Ile150Met | Het | Female | 1924 | >70 | ADPKD | 70s | N/A | − | − | + | Retinal dystrophy with PRPH2variant |
| 11 III-1 | Familial | Ile150Met | Het | Female | 1954 | 43 | ADPKD | N/A | N/A | − | + (52) | − | Retinal dystrophy with PRPH2 variant |
| 11 III-2 | Familial | Ile150Met | Het | Male | 1956 | 40 | ADPKD | N/A | N/A | +/− | − | + | Retinal dystrophy with PRPH2 variant |
| 11 III-3 | Familial | Ile150Met | Het | Female | 1961 | 40 | ADPKD | N/A | N/A | − | − | − | − |
| 11 IV-3 | Familial | Ile150Met | Het | Male | 1994 | 14 | FSGS vs. ADTKD | N/A | N/A | − | − | − | Asthma |
| 11 IV-4 | Familial | Ile150Met | Het | Female | 1995 | 18 | FSGS | N/A | N/A | − | + (18) | − | Asthma |
| 12 | De novo | Lys157Gln | Het | Male | 2017 | Fetal | ARPKD | 4 | 4 | + | − | + | Severe feeding difficulties (PEG-J) |
ADPKD = autosomal dominant polycystic kidney disease; ARPKD = autosomal recessive polycystic kidney disease; ADTKD = autosomal dominant tubulointerstitial kidney disease; FSGS = focal segmental glomerulosclerosis; MSK = medullary sponge kidney; KFRT = kidney failure with replacement therapy; KTx = kidney transplantation; NPH = nephronophthisis; m-PTLD = monomorphic post-transplant lymphoproliferative disorder; VEO = very early onset ∣ Roman numerals indicate generation ∣ * DNA from father not available for segregation; mother, and in family 6 also sibling, negative for NEK8 variant ∣ in family 11 three family members had retinal dystrophy which segregated with a likely pathogenic variant in PRPH2, individual III-3 tested negative for the PRPH2 variant and had a normal retinal exam.
In all affected individuals no second NEK8 variant was detected after thorough testing. Three multiplex families illustrated affected parent-to-child transmission (Figure 1a), consistent with autosomal dominant inheritance, while in seven families the variant was de novo; in the remaining cases with a negative family history both parents were not available for screening. Interestingly, two families showed early onset chronic kidney disease in offspring heterozygous for the recurrent p.Arg45Trp variant, which was inherited from a mosaic parent who had later-onset chronic kidney disease (Supplementary Figure S1). Supplementary Table S1 gives an overview of the genetic testing, including CNV and PKD1 analysis, VUS interpretation, and ethnicity and consanguinity for all families.
Imaging and pathology
Radiology findings from Families 3, 8 and 9 are shown in Figure 1b-g. Patients were found to have enlarged kidneys with numerous cysts. In Family 9 CT analysis of the mosaic grandmother at age 48 years old (Figure 1e) and the proband at age 23 months and 5 years old (Figure 1f, 1g) showed enlarged cystic kidneys. Gross and histological analysis of kidneys after bilateral nephrectomy in patients from Families 4, 8, and 12 showed multiple larger tubular and glomerular cysts (Figure 1i-l).
Summary of variants
All three monoallelic NEK8 variants in this study were missense alterations in the kinase domain. In contrast, variants associated with biallelic disease are found in all parts of the gene/protein, including the kinase domain (Figure 2a). None of the variants are present in the gnomAD database that contains more than 140,000 unrelated individuals without known severe pediatric disease.29 The substitutions affect evolutionary conserved residues, and prediction tools predict a damaging effect (Figure 2b-d). The Protein Tolerance Landscape of NEK8 shows that most missense variants lie in a relatively intolerant region of the enzyme, with the notable exception of the p.Thr87Ala alteration previously identified in the biallelic cases (Figure 2b). CADD scores for the three variants studied here predict a significant functional defect and all had a frequency of 0 in gnomAD (Figure 2c). The amino acid positions of these variants are highly conserved across species (Figure 2d).
Figure 2. Position of variants and domain organization of the NEK8 protein.

a. In red novel heterozygous variants described in this paper. In black variants previously published in biallelic cases. b. Protein Tolerance Landscape with identified variants and previously reported missense variants in kinase domain highlighted. Color coding indicates predicted impact of variant on protein function with blue =most tolerant, yellow= neutral, red=most detrimental. c. In silico predictions of the novel heterozygous NEK8 variants. d. Multiple sequence alignment of the human NEK8 full protein zoomed in on well conserved kinase domain. The amino acid positions of the heterozygous NEK8 variants are marked by the red arrows. e. Structural modeling of the NEK8 variants. Inset a. The predicted 3-dimentional structural model of human Serine/threonine-protein kinase NEK8 generated using AlphaFold Protein Structure Database (https://alphafold.ebi.ac.uk) and UniProtKB (https://www.uniprot.org/uniprot/) with associated codes: AF-Q86SG6-F1 and Q86SG6 respectively. The protein contains a protein kinase domain (red) and five ‘Regulator of Chromosome Condensation 1’ (RCC1) repeat domains. Inset b. Missense SNV c.133C>T p.Arg45Trp modelled with the most probable (26.5% likelihood) rotamer demonstrated. Inset c. Missense SNV c.450C>G p.Ile150Met modelled with the most probable (28.5% likelihood) rotamer demonstrated. Inset d. Positions of both variants in b. and c. highlighted in yellow (and shown as 1. and 2., respectively) to demonstrate their juxtaposition in adjacent alpha-helices. The third variant c.469A>G p.Lys157Gln could not be predicted due to a low confidence score, but appears to be in close proximity to the other variants as highlighted by the arrow (shown as 3.). A known active site (4., proton acceptor) and autophosphorylation residue (shown as 5.) are also in close proximity to the variants.
Structural protein modeling
NEK8 contains an N-terminal kinase domain and C-terminal ‘Regulator of Chromosome Condensation 1’ (RCC1) domains (Figure 2a, e inset a.). The RCC1 domains are guanine nucleotide exchange factor (GEF) for the GTP-binding protein Ran and feature a 7-bladed propellor of beta-strands. NEK8 also contains a coiled-coil domain within the protein’s C-terminus thought to be involved in protein-protein interactions. NEK8 is proposed to be auto-phosphorylated at position Thr-210 on the protein kinase domain, followed by phosphorylation occurring at other sites in the molecule. The protein kinase domain also features ATP binding sites and a proton acceptor site.30 The NEK8 variants with high quality structural data (pLDDT > 50) were p.Arg45Trp and p.Ile150Met (Figure 2e, inset b. and c., respectively). The dashed black lines demonstrate likely loss of the canonical geometry between 2 atoms in this loop with overlap of each (numerically labelled in angstroms). The red discs represent significant pairwise overlap of atomic van der Waals radii causing a likely structural ‘clash’. The amino acid volume change in the missense variant p.Arg45Trp (inset b.) is large (190.3 to 226.4 angstroms) with a very small volume change of 163.0 to 165.8 angstroms in missense variant p.Ile150Met (inset c.).31 The two variants are positioned in adjacent alpha-helices (inset d.). Although the crystal structure of the third variant p.Lys157Gln could not be predicted due to a low confidence score, the variant appears to be in close proximity to the others as highlighted by the arrow (inset d.,3).
Analyses of the NEK8 monoallelic variants
To understand the functionality of NEK8 in renal epithelial cells, we inactivated Nek8 in IMCD3 cells using CRISPR/Cas9 (Figure 3a), and then re-expressed the Myc-tagged wild-type (WT) or one of the monoallelic variants of human NEK8 in these Nek8−/− cells. Two other NEK8 mutations in the kinase domain were also included for comparison. One is the approved kinase-dead (KD) mutation p.Lys33Met that was designed based on structural analysis.32 The other is p.Thr87Ala that was found in compound heterozygosity with p.Arg602Trp in a patient with the typical recessive phenotype.14 Immunoblotting showed that the overall expression and stability of these NEK8 mutants were comparable to their wild-type counterpart (Figure 3b), and all mutated NEK8 proteins exhibited a similar incorporation rate and localization patten in cilia as NEK8-WT (Figure 3c). Compared to parental cells, IMCD3 cells with or without expression of endogenous Nek8 or exogenous NEK8 variants displayed a similar percentage of ciliated cells, which are represented by staining with the ciliary membrane marker ARL13B (Figure 3d, upper panel). However, primary cilia in Nek8−/− cells were significantly truncated, which was rescued equally well by NEK8-WT or NEK8-KD (p.Lys33Met) (Figure 3d, lower panel), indicating that the kinase activity of NEK8 is not required for the assembly of primary cilia. Re-expression of p.Arg45Trp, p.Thr87Ala, or p.Ile150Met also significantly rescued the cilia length, although for p.Thr87Ala and p.Ile150Met the length was less than for the WT rescue, whereas NEK8-K157Q failed to correct the cilia length defect. These data are consistent with our findings in patients’ fibroblasts which expressed a monoallelic variant and the wildtype allele, that the overall structure of cilia is not affected by p.Arg45Trp and p.Ile150Met (Supplementary Figure S2a-c), but cilia are shortened by p.Lys157Gln (Supplementary Figure S2d-e). Consistent with the reported role of NEK8 in recruiting ANKS6 to the inversin compartment,16 ANKS6 was undetectable in cilia in Nek8−/− cells (Figure 3e). The recruitment of ANKS6 to cilia was fully recovered by expressing either NEK8-WT or any of the disease variants, but was only rescued to ~30% by p.Lys33Met, the KD mutant (Figure 3e).
Figure 3. Analyses of the function of the NEK8 variants.

(a) Sanger sequencing results of cDNA PCR products showed a nucleotide insertion in Nek8 in the IMCD3 cell line, which causes a frameshift with early termination (45aa). (b-h) Nek8 knockout cells (−/−) were transfected with the empty vector (Mock) or one of the Myc-tagged NEK8 constructs, including the wild type (WT) and indicated single-point mutations. Forty-eight-hr post transfection, cells were treated with G418 (1 μg/mL) for one week. Cells survived the G418 selection were expanded, serum-starved for 24 h, and then subjected to the following analyses. Parental IMCD3 cells (+/+) were used as a positive control for endogenous Nek8, cells were harvested for immunoblotting (IB) with indicated antibodies, and α-tubulin was used as a loading control (b, h). (b) IB with a Myc or NEK8 antibody showed that all NEK8 proteins were expressed. (c-g) Cells were subjected to immunofluorescence (IF) microscopy using the indicated antibodies to visualize Myc-tagged NEK8 variants (c), ARL13B (d), ANKS6 (e), GPR161 (f), or PC2 (g) (scale bar, 2 μm). Primary cilia were labeled with an acetylated α-tubulin (Ac-tub) antibody. Data were collected from >100 cilia in each experimental group. Results from three independent experiments were statistically analyzed and plotted as mean ± SD. P values labeled in black were obtained by comparing to the group of Nek8−/−, mock transfected cells (−/−, Mock). P values labelled in red were obtained by comparing to the Nek8−/− cells stably expressing the wild type NEK8 (−/−, WT). (c) the NEK8 proteins with the various variants exhibited similar localization in cilia. (d, upper panel) Ciliogenesis was quantified by counting the percentage of ciliated (ARL13B and Ac-tub double-positive) cells and all cells were ciliated at a similar level; (d, lower panel) Loss of endogenous Nek8 led to truncated cilia but all the NEK8 variants rescued the ciliary length defect except p.Lys157Gln (K157Q). The cilium length was measured by the length of ARL13B signal in cilia. (e) Loss of Nek8 disrupted the recruitment of ANKS6 into cilia, which was fully rescued by all NEK8 variants, except p.Lys33Met (K33M) that only showed a partial rescue. (f) The percentage of GPR161-positive cilia were not affected by the expression status of NEK8. (g) The percentage of PC2-positive cilia and the fluorescence intensity of PC2 in cilia were both diminished by Nek8 deficiency, although (h) IB showed that PC2 was present at comparable protein levels. (g) Different NEK8 variants exhibited different ability to rescue the ciliary trafficking of PC2. (i) Primary skin fibroblasts were isolated from healthy (WT) and p.Arg45Trp (Fam 1 and Fam 7, III-3) male donors. These cells were analysed by IF to visualize PC2 in cilia, with lower mean intensity seen for the two mutant cells. The percentage of PC2-positive cilia and the mean intensity of ciliary PC2 were quantified in >100 cilia from each group. Results from three independent experiments were statistically analysed and plotted as mean ± SD. (j) HEK293T cells transfected with Myc-tagged wild type NEK8 (WT) or the p.Arg45Trp variant (R45W) were subjected to immunoprecipitation (IP) with anti-Myc antibody or normal mouse IgG (mIgG). The precipitates were then subjected to Phos-tag SDS-PAGE to separate the phosphorylated NEK8 (arrow) from the non-phosphorylated form and analysed by IB. The relative density of phosphorylated NEK8 was quantified from four independent experiments, statistically analysed, and plotted as mean ± SD. (c-g, i, j) ***, P < 0.001; **, P < 0.01; *, P < 0.05; n.s., no statistically significant differences.
When we examined GPR161, a cilium-residing G protein-coupled receptor, we found that its ciliary localization was similar with or without NEK8 and was not affected by the expression of any tested NEK8 mutants (Figure 3f). However, knockout of Nek8 strongly suppressed the ciliary level of polycystin-2 (PC2), the product of PKD2 (Figure 3g). The ciliary targeting of PC2 in Nek8-null cells was fully recovered by re-expressing NEK8-WT, as well as p.Thr87Ala and p.Ile150Met (Figure 3g), while p.Lys33Met and p.Lys157Gln only partially rescued PC2 in cilia. Interestingly, NEK8-p.Arg45Trp completely failed to recover the ciliary level of PC2 in Nek8−/− cells (Figure 3g). Yet, the total protein level of PC2 appeared comparable in IMCD3 cells expressing endogenous Nek8 or exogenous NEK8 variants (Figure 3h). These results suggest that in addition to supporting the assembly of cilia, NEK8 plays a unique role in maintaining the ciliary homeostasis of PC2, and the p.Arg45Trp variant specifically loses this capability. Consistently, fibroblast cells from two patients carrying p.Arg45Trp also exhibited less PC2 in cilia compared to normal fibroblasts (Figure 3i). The PC2 trafficking phenotype in the patient fibroblasts was less severe, however, compared to the IMCD3 cells expressing the variant Nek8, likely because the fibroblasts still have one wild type allele, whereas the IMCD3 cells are Nek8-null and fully expressing NEK8-p.Arg45Trp.
Since p.Arg45Trp is in the kinase domain, we also determined the autophosphorylation of NEK8. Myc-tagged NEK8-WT and NEK8-p.Arg45Trp were overexpressed in 293T cells and immunoprecipitated (IP) by an anti-Myc antibody and Protein-G Sepharose beads. The phosphorylated and non-phosphorylated NEK8 proteins were then separated by Phos-tag SDS-PAGE33 and detected by immunoblotting. As shown in Figure 3j, the autophosphorylation level of NEK8-p.Arg45Trp was significantly lower than NEK8-WT, suggesting that p.Arg45Trp partially loses kinase activity.
Assessment of DNA damage response
NEK8 is a regulator of DNA damage response and NEK8-deficient cells exhibit increased DNA damage14. We examined the DNA damage response in IMCD3 cells by staining cells with histone H2AX phosphorylated at Serine 319, γH2AX, a marker of DNA damage that occurs at double strand breaks and stalled replication forks.34-36 Compared to the parental IMCD3 cells, NEK8 KO cells had a significant increase in γH2AX (Figure 4a). Expression of NEK8-WT, but not NEK8-p.Arg45Trp in the Nek8−/− cells rescued γH2AX accumulation in IMCD3 cells, indicating that partial disruption of the kinase activity results in DNA damage response activation. Next, we examined γH2AX accumulation in cell lines derived from patients. In patient derived tubuloids from Family 12, heterozygous for p.Lys157Gln, we found an increase in gH2AX compared to three healthy passage-matched controls (Figure 4b).
Figure 4. Elevated DNA damage signaling in renal cell lines expressing NEK8 variants.

a. Representative images of γH2AX signaling in IMCD3 cells with the indicated genotypes. Scale bar is 50 μm. Graph represents the mean of three independent experiments. Error bars represent sd. At least 150 nuclei per experiment were analyzed for >10 γH2AX foci, **P <0.01, *P <0.05. b. Representative images of γH2AX signaling in patient derived tubuloids with the indicated genotypes. Scale bar is 50 γm c. The percentage of γH2AX positive cells from three independent experiments was quantified. Line represents the mean. Error bars are sd. **P <0.01, *P <0.05.
In patient-derived fibroblasts, we pulsed cells with EdU to identify S phase cells and γH2AX in EdU-positive cells. In NEK8-deficient cells, γH2AX accumulation occurs primarily in S phase.35 However, we did not detect a significant difference in γH2AX between control, p.Arg45Trp or p.Ile150Met fibroblasts (Supplementary Figure S3). Treatment of NEK8-deficient cells with replication stress inducing drugs leads to a further increase in γH2AX.37 We determined if treating patient fibroblasts with the replication inhibiting drug, hydroxyurea, would lead to an increase in γH2AX activation. In control fibroblasts, hydroxyurea resulted in a significant increase in γH2AX staining intensity indicating activation of the DNA damage response, but this response was similar in p.Arg45Trp or p.Ile150Met fibroblasts (Supplementary Figure S3).
Discussion:
Here we present evidence of monoallelic disease in twelve families associated with three distinct heterozygous NEK8 variants within the kinase domain. Biallelic NEK8 variants cause severe multiorgan developmental defects including renal cystic dysplasia, with primary cilia formation, DNA damage responses, and epithelial morphogenesis implicated.14 Biallelic variants are loss-of-function (LOF) or missense changes that do not have a known phenotypic effect in heterozygous carriers.14 In our families monoallelic inheritance is supported by the demonstrated de novo variants and autosomal dominant inheritance in multiplex families. Assessment of the Protein Tolerance Landscape shows that all three missense variants in this study exist in intolerant regions of the protein (Figure 2b). We believe these variants have a dominant-negative effect, explaining the severe phenotype associated with a single variant. Another distinction of the monoallelic disease is that all the affected individuals primarily have renal phenotypes.
None of the monoallelic variants are present in gnomAD, which contrasts with the previously reported recessive variants for which most can be found as heterozygotes in this database.29 While the genetic tests that were performed differed between the families, whole exome sequencing was completed in at least one individual for every distinct variant (Supplementary Table S1). PKD1 analysis, complicated by the PKD1 pseudogenes38, also differed between families, but long-range PCR and Sanger analysis was performed in at least six patients. Variants of unknown significance in other genes identified in some individuals were interpreted as likely non-causal (Supplementary Table S1). All encountered monoallelic variants were located in the kinase domain, while biallelic, disease-causing variants can be present throughout the gene. Presently, there are not consistent genotype-phenotype correlations for biallelic NEK8 variants.14,15,37-41 In contrast, the kidney phenotype associated with NEK8-p.Arg45Trp and p.Lys157Gln was largely consistent, with enlarged kidneys in infants and limited extra-renal disease, while the p.Ile150Met phenotype was considerably milder.
The monoallelic NEK8 phenotype can be clinically mistaken for either ARPKD or ADPKD. The early onset PKD seen with the p.Arg45Trp and p.Lys157Gln variants, and the childhood kidney failure and requirement for bilateral nephrectomies (5 out of 21 in our cohort), overlaps with ARPKD, although very early onset has been reported in ~1% of ADPKD cases, but often because of biallelic disease.42 The inheritance pattern and localization of cysts in all parts of the nephron (shown in two patients) is more consistent with ADPKD.42 An unusual aspect is the high level of de novo mutations of p.Arg45Trp, similar to whole gene deletions of HNF1B, and recurrent mosaic presentation associated with milder disease. A recent case report also described a de novo p.Arg45Trp variant in a 2-year old patient with infantile nephronophthisis which was characterized by multicystic kidney dysplasia and progressive renal failure.43 There is possible CpG methylation at this site; methylated CpG dinucleotides are more likely to deaminate than other cytosines, making them mutational hot spots.44 45. It will be interesting to see the diagnostic yield of monoallelic NEK8 using genotype first approaches in infantile PKD cohorts.
Functional studies using Nek8 knockout IMCD3 cells identified shorter cilia and defects in the localization of ANKS6 and PC2 to cilia, as previously reported.17 When these cells were transfected with NEK8-WT or the monoallelic NEK8 variants, both NEK8 and ANKS6 localized to the proximal cilium and the cilia length defect was wholly or partially corrected, except for shorter cilia in p.Lys157Gln. However, PC2 localization to the primary cilium was only rescued by re-expressing NEK8-WT, not NEK8-p.Arg45Trp or p.Lys157Gln. The effect of PC2 localization but not some other ciliary components may provide a clue to the cystic kidney specific phenotype we see in the monoallelic cases. Changes at certain positions within the kinase domain with unique functional consequences appear to be crucial. The kinase dead variant, p.Lys33Met had defects in both ANKS6 and PC2 localization. However, a described recessive kinase variant. P.Thr87Ala did not have localization defects for ANKS6 or PC2. This may be because it is a hypomorphic allele (it lies in a highly tolerant region as shown by structural protein modeling) and any defect may be too subtle for our functional assay to detect.
We believe a dominant-negative effect is the most likely disease mechanism. We propose that this dominant-negative effect includes binding of monoallelic mutant NEK8 to wildtype NEK8, and likely other members of the ANKS6 complex, resulting in a complex that is nonfunctional for PC2 trafficking. This will result in a greater than the 50% loss of PC2 in cilia associated with a LOF variant. Our experimental re-expression system is not well suited to analyze interactions between alleles, but we note in cells from two patients heterozygous for p.Arg45Trp a significantly lower level of PC2 in cilia than in normal cells, possibly showing an inactivating interaction with the normal allele.
The location of the variants in the NEK8 kinase domain indicates a mechanism of action related to phosphorylation; NEK8 is known to have autophosphorylation activity.30 With our kinase assay in Nek8 knockout cells transfected with NEK8-p.Arg45Trp we showed a decrease of autophosphorylated NEK8, indicating that kinase function is impacted by this variant. In the juvenile cystic kidney (jck) mouse model, due to a homozygous Nek8 missense variant, abnormal phosphorylation of PC2 was detected which was associated with longer cilia and ciliary accumulation of PC1 and PC2.17 This contrasts to our findings with p.Arg45Trp of normal length but defective ciliary trafficking of PC2, indicating a more specific kinase defect.
We acknowledge several limitations within the present study. We have developed an exogenous expression system for NEK8 variants in a null Nek8 background that has illustrated differences in cilia protein localizations between p.Arg45Trp and p.Lys157Gln and the null cells and a kinase dead mutation. So far because of available constructs/antibodies our analysis has been limited to PC2, but including PC1 and the ARPKD protein, fibrocystin, would also be interesting. In our exogenous expression assay, weaker monoallelic and biallelic variants were not fully informative, likely reflecting its sensitivity and that it is only an approximation of the in vivo situation. A cilia length defect was also detected for p.Lys157Gln but not the other monoallelic variants, suggesting a possible different mode of action that we do not yet fully understand. Patient cells and tubuloid systems were helpful but were not always consistent in their readout, probably because the effects are likely dependent on both cell type and expression level. For example, NEK-pArg45Trp failed to restore DNA damage responses in the IMCD3 cells but no increase in γH2AX staining was observed in the patient fibroblasts. One possibility is the difference in proliferation rates between the two cell lines can be leading to a difference in the replication stress response. A complete understanding of how each missense variant alters kinase function and/or substrate recognition will require longer-term investigation, including the generation of mammalian animal models, phosphoproteomic studies, and the comparative analysis of these variants with the other missense variants identified in the biallelic patients.
To conclude, here we present evidence for a pathogenic effect of specific heterozygous NEK8 variants, provide a new mode of inheritance for NEK8 variants, and expand the phenotype associated with variants in this gene. Heterozygous NEK8 variants within the kinase domain should be considered in patients with infantile PKD. For children and adults with PKD, we suggest NEK8 should be included in gene panel testing and heterozygous variants in the kinase domain evaluated for causality.
Supplementary Material
Supplementary Data: clinical details families
Supplementary Figure S1: mosaicism in parent of later-onset case
Supplementary Table S1: genetic testing overview + details on ethnicity and consanguinity
Supplementary Figure S2: NEK8 localization and ciliation in cells from affected individuals
Supplementary Figure S3: DNA damage response in patient-derived fibroblasts
Acknowledgments
We thank the study participants and their families for their contribution. The full list of funding information and acknowledgements is found in the Supplementary file.
The Genomics England Research Consortium:
John C. Ambrose1; Prabhu Arumugam1; Roel Bevers1; Marta Bleda1; Freya Boardman-Pretty1,2; Christopher R. Boustred1; Helen Brittain1; Mark J. Caulfield1,2; Georgia C. Chan1; Greg Elgar1,2; Tom Fowler1; Adam Giess1; Angela Hamblin1; Shirley Henderson1,2; Tim J. P. Hubbard1; Rob Jackson1; Louise J. Jones1,2; Dalia Kasperaviciute1,2; Melis Kayikci1; Athanasios Kousathanas1; Lea Lahnstein1; Sarah E. A. Leigh1; Ivonne U. S. Leong1; Javier F. Lopez1; Fiona Maleady-Crowe1; Meriel McEntagart1; Federico Minneci1; Loukas Moutsianas1,2; Michael Mueller1,2; Nirupa Murugaesu1; Anna C. Need1,2; Peter O’Donovan1; Chris A. Odhams1; Christine Patch1,2; Mariana Buongermino Pereira1; Daniel Perez-Gil1; John Pullinger1; Tahrima Rahim1; Augusto Rendon1; Tim Rogers1; Kevin Savage1; Kushmita Sawant1; Richard H. Scott1; Afshan Siddiq1; Alexander Sieghart1; Samuel C. Smith1; Alona Sosinsky1,2; Alexander Stuckey1; Melanie Tanguy1; Ana Lisa Taylor Tavares1; Ellen R. A. Thomas1,2; Simon R. Thompson1; Arianna Tucci1,2; Matthew J. Welland1; Eleanor Williams1; Katarzyna Witkowska1,2; Suzanne M. Wood1,2.
1. Genomics England, London, UK
2. William Harvey Research Institute, Queen Mary University of London, London, EC1M 6BQ, UK.
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests. Jennifer Stallworth completed this work as an employee at GGC, and is now a Sanofi employee.
References
- 1.Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. The Lancet. 2007;369(9569):1287–1301. doi: 10.1016/S0140-6736(07)60601-1 [DOI] [PubMed] [Google Scholar]
- 2.Shaw C, Simms RJ, Pitcher D, Sandford R. Epidemiology of patients in England and Wales with autosomal dominant polycystic kidney disease and end-stage renal failure. Nephrol Dial Transplant. 2014;29(10):1910–1918. doi: 10.1093/ndt/gfu087 [DOI] [PubMed] [Google Scholar]
- 3.Besse W, Dong K, Choi J, et al. Isolated polycystic liver disease genes define effectors of polycystin-1 function. J Clin Invest. 2017;127(5):1772–1785. doi: 10.1172/JCI90129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Besse W, Chang AR, Luo JZ, et al. ALG9 Mutation Carriers Develop Kidney and Liver Cysts. J Am Soc Nephrol. 2019;30(11):2091–2102. doi: 10.1681/ASN.2019030298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Senum SR, Li YSM, Benson KA, et al. Monoallelic IFT140 pathogenic variants are an important cause of the autosomal dominant polycystic kidney-spectrum phenotype. Am J Hum Genet. 2022;109(1):136–156. doi: 10.1016/j.ajhg.2021.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cornec-Le Gall E, Olson RJ, Besse W, et al. Monoallelic Mutations to DNAJB11 Cause Atypical Autosomal-Dominant Polycystic Kidney Disease. Am J Hum Genet. 2018;102(5):832–844. doi: 10.1016/j.ajhg.2018.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Porath B, Gainullin VG, Cornec-Le Gall E, et al. Mutations in GANAB, Encoding the Glucosidase IIα Subunit, Cause Autosomal-Dominant Polycystic Kidney and Liver Disease. Am J Hum Genet. 2016;98(6):1193–1207. doi: 10.1016/j.ajhg.2016.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lemoine H, Raud L, Foulquier F, et al. Monoallelic pathogenic ALG5 variants cause atypical polycystic kidney disease and interstitial fibrosis. Am J Hum Genet. 2022;109(8):1484–1499. doi: 10.1016/J.AJHG.2022.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waters AM, Beales PL. Ciliopathies: An expanding disease spectrum. Pediatric Nephrology. 2011;26(7):1039–1056. doi: 10.1007/s00467-010-1731-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. New England Journal of Medicine. 2011;364(16):1533–1543. doi: 10.1056/nejmra1010172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chaki M, Airik R, Ghosh AK, et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell. 2012;150(3):533–548. doi: 10.1016/j.cell.2012.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ateş EA, Turkyilmaz A, Delil K, et al. Biallelic Mutations in DNAJB11 Are Associated with Prenatal Polycystic Kidney Disease in a Turkish Family. Vol 12. Mol Syndromol; 2021:179–185. doi: 10.1159/000513611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jordan P, Arrondel C, Bessières B, et al. Bi-allelic pathogenic variations in DNAJB11 cause Ivemark II syndrome, a renal-hepatic-pancreatic dysplasia. Kidney Int. 2021;99(2):405–409. doi: 10.1016/J.KINT.2020.09.029 [DOI] [PubMed] [Google Scholar]
- 14.Grampa V, Delous M, Zaidan M, et al. Novel NEK8 Mutations Cause Severe Syndromic Renal Cystic Dysplasia through YAP Dysregulation. PLoS Genet. 2016;12(3):e1005894. doi: 10.1371/journal.pgen.1005894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frank V, Habbig S, Bartram MP, et al. Mutations in NEK8 link multiple organ dysplasia with altered Hippo signalling and increased c-MYC expression. Hum Mol Genet. 2013;22(11):2177–2185. doi: 10.1093/hmg/ddt070 [DOI] [PubMed] [Google Scholar]
- 16.Hoff S, Halbritter J, Epting D, et al. ANKS6 is a central component of a nephronophthisis module linking NEK8 to INVS and NPHP3. Nat Genet. 2013;45(8):951–956. doi: 10.1038/ng.2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sohara E, Luo Y, Zhang J, Manning DK, Beier DR, Zhou J. Nek8 regulates the expression and localization of polycystin-1 and polycystin-2. Journal of the American Society of Nephrology. 2008;19(3):469–476. doi: 10.1681/ASN.2006090985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: A Matching Tool for Connecting Investigators with an Interest in the Same Gene. Hum Mutat. 2015;36(10):928–930. doi: 10.1002/humu.22844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smedley D, Smith KR, Martin A, et al. 100,000 Genomes Pilot on Rare-Disease Diagnosis in Health Care - Preliminary Report. N Engl J Med. 2021;385(20):1868–1880. doi: 10.1056/NEJMOA2035790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Varadi M, Anyango S, Deshpande M, et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022;50(D1):D439–D444. doi: 10.1093/NAR/GKAB1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021 596:7873. 2021;596(7873):583–589. doi: 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mariani V, Biasini M, Barbato A, Schwede T. lDDT: a local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics. 2013;29(21):2722. doi: 10.1093/BIOINFORMATICS/BTT473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rentzsch P, Schubach M, Shendure J, Kircher M. CADD-Splice—improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 2021;13(1). doi: 10.1186/s13073-021-00835-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wiel L, Baakman C, Gilissen D, Veltman JA, Vriend G, Gilissen C. MetaDome: Pathogenicity analysis of genetic variants through aggregation of homologous human protein domains. Hum Mutat. 2019;40(8):1030–1038. doi: 10.1002/humu.23798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manning DK, Sergeev M, van Heesbeen RG, et al. Loss of the ciliary kinase Nek8 causes left-right asymmetry defects. J Am Soc Nephrol. 2013;24(1):100–112. doi: 10.1681/ASN.2012050490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johannessen CM, Boehm JS, Kim SY, et al. COT/MAP3K8 drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468(7326):968. doi: 10.1038/NATURE09627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen C, Xu Q, Zhang Y, et al. Ciliopathy protein HYLS1 coordinates the biogenesis and signaling of primary cilia by activating the ciliary lipid kinase PIPKIγ. Sci Adv. 2021;7(26). doi: 10.1126/SCIADV.ABE3401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Obeidova L, Seeman T, Fencl F, et al. Results of targeted next-generation sequencing in children with cystic kidney diseases often change the clinical diagnosis. PLoS One. 2020;15(6):e0235071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–443. doi: 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holland PM, Milne A, Garka K, et al. Purification, cloning, and characterization of Nek8, a novel nima-related kinase, and its candidate substrate Bicd2. Journal of Biological Chemistry. 2002;277(18):16229–16240. doi: 10.1074/jbc.M108662200 [DOI] [PubMed] [Google Scholar]
- 31.Tsai J, Gerstein M. Calculations of protein volumes: Sensitivity analysis and parameters database. Bioinformatics. 2002;18(7):985–995. doi: 10.1093/bioinformatics/18.7.985 [DOI] [PubMed] [Google Scholar]
- 32.Czarnecki PG, Gabriel GC, Manning DK, et al. ANKS6 is the critical activator of NEK8 kinase in embryonic situs determination and organ patterning. Nat Commun. 2015;6. doi: 10.1038/ncomms7023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kinoshita E, Kinoshita-Kikuta E, Koike T. Separation and detection of large phosphoproteins using Phos-tag SDS-PAGE. Nat Protoc. 2009;4(10):1513–1521. doi: 10.1038/NPROT.2009.154 [DOI] [PubMed] [Google Scholar]
- 34.Helt CE, Cliby WA, Keng PC, Bambara RA, O’Reilly MA. Ataxia telangiectasia mutated (ATM) and ATM and Rad3-related protein exhibit selective target specificities in response to different forms of DNA damage. J Biol Chem. 2005;280(2):1186–1192. doi: 10.1074/JBC.M410873200 [DOI] [PubMed] [Google Scholar]
- 35.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273(10):5858–5868. doi: 10.1074/JBC.273.10.5858 [DOI] [PubMed] [Google Scholar]
- 36.Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. 2001;276(51):47759–47762. doi: 10.1074/JBC.C100569200 [DOI] [PubMed] [Google Scholar]
- 37.Choi HJC, Lin JR, Vannier JB, et al. NEK8 Links the ATR-Regulated Replication Stress Response and S Phase CDK Activity to Renal Ciliopathies. Mol Cell. 2013;51(4):423–439. doi: 10.1016/j.molcel.2013.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ali H, Al-Mulla F, Hussain N, et al. PKD1 Duplicated regions limit clinical Utility of Whole Exome Sequencing for Genetic Diagnosis of Autosomal Dominant Polycystic Kidney Disease. Sci Rep. 2019;9(1):4141. doi: 10.1038/s41598-019-40761-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rajagopalan R, Grochowski CM, Gilbert MA, et al. Compound heterozygous mutations in NEK8 in siblings with end-stage renal disease with hepatic and cardiac anomalies. Am J Med Genet A. 2016;170(3):750–753. doi: 10.1002/AJMG.A.37512 [DOI] [PubMed] [Google Scholar]
- 40.Otto EA, Trapp ML, Schultheiss UT, Helou J, Quarmby LM, Hildebrandt F. NEK8 mutations affect ciliary and centrosomal localization and may cause nephronophthisis. J Am Soc Nephrol. 2008;19(3):587–592. doi: 10.1681/ASN.2007040490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Domingo-Gallego A, Pybus M, Bullich G, et al. Clinical utility of genetic testing in early-onset kidney disease: seven genes are the main players. Nephrology, dialysis, transplantation. 2022;37(4):687–696. doi: 10.1093/NDT/GFAB019 [DOI] [PubMed] [Google Scholar]
- 42.Bergmann C. ARPKD and early manifestations of ADPKD: the original polycystic kidney disease and phenocopies. Pediatr Nephrol. 2015;30(1):15–30. doi: 10.1007/s00467-013-2706-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mehawej C, Chouery E, Ghabril R, Tokajian S, Megarbane A. NEK8-Associated Nephropathies: Do Autosomal Dominant Forms Exist? Nephron. Published online 2022. doi: 10.1159/000526841 [DOI] [PubMed] [Google Scholar]
- 44.Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Res. 2002;12(6):996–1006. doi: 10.1101/GR.229102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cooper DN, Mort M, Stenson PD, Ball EV., Chuzhanova NA. Methylation-mediated deamination of 5-methylcytosine appears to give rise to mutations causing human inherited disease in CpNpG trinucleotides, as well as in CpG dinucleotides. Hum Genomics. 2010;4(6):406–410. doi: 10.1186/1479-7364-4-6-406 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data: clinical details families
Supplementary Figure S1: mosaicism in parent of later-onset case
Supplementary Table S1: genetic testing overview + details on ethnicity and consanguinity
Supplementary Figure S2: NEK8 localization and ciliation in cells from affected individuals
Supplementary Figure S3: DNA damage response in patient-derived fibroblasts