

Abstract
Fibrinogen is an extraordinary molecule by any estimation. It is large, structurally intricate, and circulates at high concentrations. Its biological end product, insoluble fibrin(ogen) or fibrin, can assume a diverse array of conformations with the ability to interact with numerous plasma proteins and cells and withstand biochemical and biomechanical disruption to facilitate wound healing. Quantitative and qualitative defects in fibrinogen or fibrin are associated with bleeding, thrombosis, inflammation, and diseases affected by these processes. Numerous studies investigating mechanisms by which fibrin(ogen) and fibrin contribute to health and disease have been published in the last 20 years. This review for the 20th anniversary series in the Journal of Thrombosis and Haemostasis summarizes interesting aspects of fibrin(ogen) biology, biochemistry, biophysics, and physiology and highlights exciting findings published in the past 2 decades.
Keywords: Fibrinogen, fibrin, thrombosis, bleeding, inflammation
Introduction.
Fibrinogen and its insoluble products fibrin(ogen) and fibrin are extraordinary molecules. Through their numerous biochemical and biophysical properties, these molecules interact with and activate cells, mediate inflammatory responses, provide structural integrity to blood clots during wound healing, and perform a host of other functions that are central to physiology and instrumental in pathophysiologic mechanisms. The purpose of this review is to summarize important aspects of fibrin(ogen) biology, biochemistry, biophysics, and (patho)physiology and highlight exciting studies published in the past two decades. Additional excellent reviews that have taken deeper dives into specific aspects of fibrinogen synthesis and fibrin formation, structure, and stability, and the contributions of these functions to hemostasis, thrombosis, and disease are also cited within the text.
Fibrinogen synthesis.
Fibrinogen is encoded by 3 genes (FGA, FGB, and FGG) clustered within a 65-kilobase region on human chromosome 4 (4q23–q32) (Fig 1A). CCCTC-binding factor interaction sites flanking the fibrinogen gene cluster direct chromatin looping, enabling coordinate transcription of the fibrinogen genes.1 Over-expression of any one of the fibrinogen genes increases expression of the other two genes. Synthesis occurs primarily in hepatocytes, where the fibrinogen mRNAs are spliced and translated, and the polypeptides are subjected to post-translational modifications. The Aα (67 kDa, 610 residues), Bβ (57 kDa, 461 residues), and γ (47 kDa, 411 residues) polypeptides are then assembled into Bβ-γ and Aα-γ intermediates, then Aα-Bβ-γ half-molecules, and finally into a 340-kDa hexameric glycoprotein containing 2 each of the 3 polypeptide chains (AαBβγ)2 (Fig 1B). Fibrinogen is secreted into the blood where it circulates at high concentrations (2–4 mg/mL, 6–12 μmol/L) with a relatively long half-life (~4 days). Fibrinogen synthesis pathways are reviewed in2,3. Fibrinogen expression increases with age, and is highly responsive to changes in the inflammatory state; circulating fibrinogen is decreased by glucocorticoid administration and increased by interleukin-6-mediated induction of the acute phase response, which can increase circulating fibrinogen ≥2-fold.
Figure 1. Fibrinogen structure.
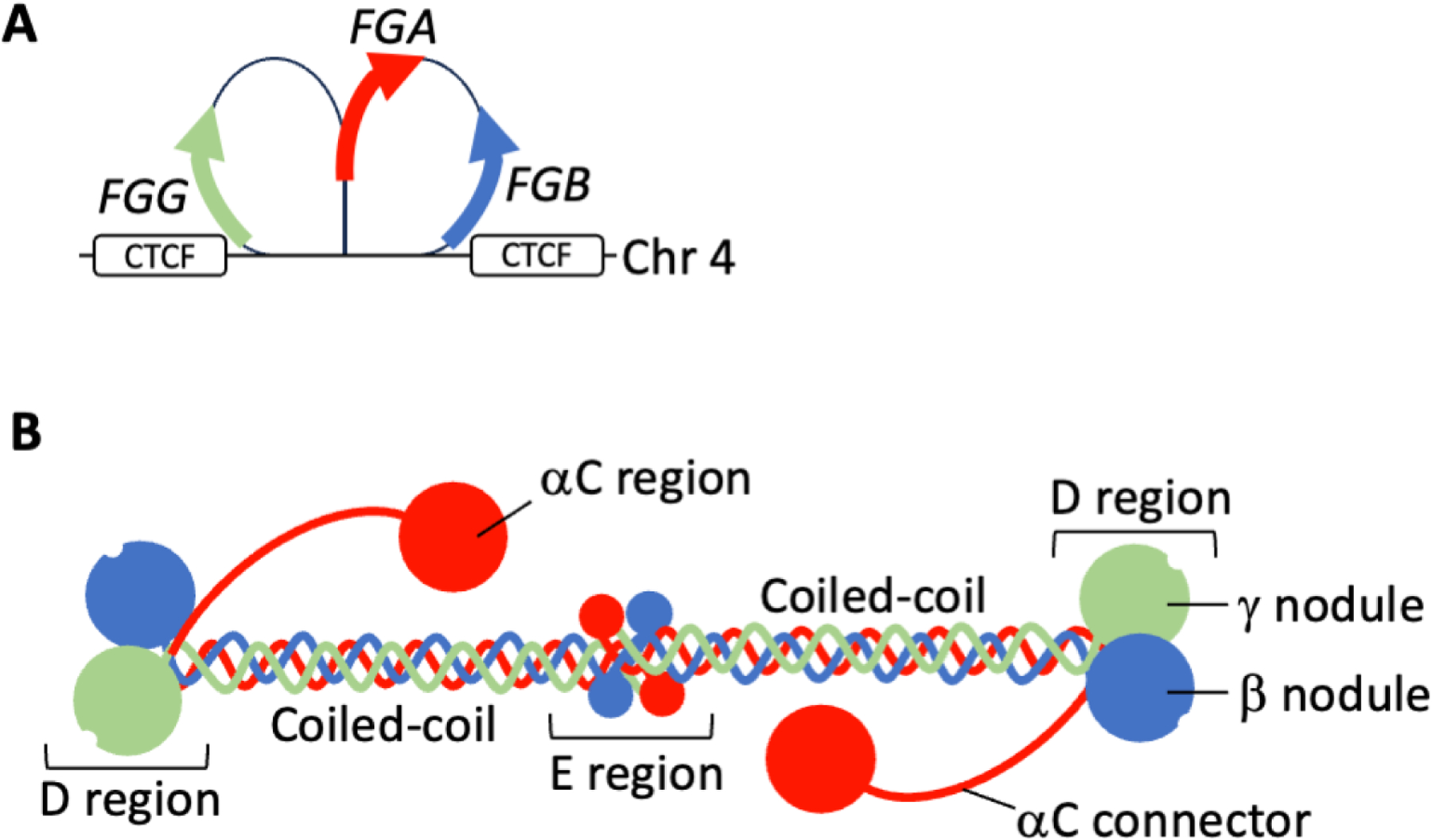
A) Fibrinogen is encoded by 3 genes (FGA, FGB, and FGG) clustered within a 65 kilobase region on human chromosome 4 (4q23-q32). FGA and FGG are in opposite orientation to FGB on the chromosome. CCCTC-binding factor (CTCF) interaction sites flank the fibrinogen gene cluster direct chromatin looping and enable coordinate transcription of the fibrinogen genes. B) Fibrinogen is a 340-kDa hexameric glycoprotein containing 2 each of 3 polypeptide chains: Aα (red), Bβ (blue), and γ (green). The 6 fibrinogen polypeptides are arranged with N-termini in a central E region and C-termini radiating outwards with bilateral symmetry.
Heritability estimates for circulating fibrinogen levels range vary from 25–50%, but only a portion of the genetic regulators have been identified. Variants within the fibrinogen structural genes FGA, FGB, and FGG that are directly associated with fibrinogen disorders have received the most attention.4,5 Briefly, type I deficiencies (congenital hypofibrinogenemia and afibrinogenemia, characterized by the partial or complete loss of fibrinogen, respectively) are associated with mutations within the fibrinogen structural genes (primarily FGA) that eliminate part of the coding sequence, alter transcription, mRNA splicing or stability, and/or protein synthesis, assembly, or secretion. Some patients with FGG mutations have abnormal liver function tests and progressive liver disease due to fibrinogen retention in hepatocytes and formation of hepatocellular cytoplasmic inclusion bodies. Type II mutations cause qualitative changes in fibrinogen (dysfibrinogenemia) and may also present with reduced circulating fibrinogen (hypodysfibrinogenemia).
Notably, however, common polymorphisms within the fibrinogen structural genes explain less than 2% of the variability in plasma fibrinogen concentration. Genomic studies published within the past 20 years have associated circulating fibrinogen levels with loci and molecules outside of the fibrinogen structural genes, revealing intriguing leads for follow-up functional analysis. These include genes involved in inflammatory pathways, as anticipated from biological observations of fibrinogen levels during infection and immune activation, as well as genes with less obvious potential roles. Using a gene silencing-based approach in HepG2 hepatoblastoma cells, Dobson et al functionally tested the contribution of candidate genes within loci associated with circulating fibrinogen levels6 and implicated FGA, IL6R, EEPD1, LEPR, PDIA5, PLEC, and CPS1, in fibrinogen transcriptional regulation and ATXN2L in fibrinogen secretion.7 In an epigenome-wide associate study (EWAS), Hahn et al identified and replicated 83 CpG sites in 61 loci associated with fibrinogen levels.8 This EWAS identified CpG sites near loci previously identified in fibrinogen GWAS that contained IL6R and PLEC, providing strong support that these genes contribute to pathways mediating fibrinogen gene expression. Genes located near the identified CpG sites also include SOCS3 and AIM2, both of which are involved in inflammatory pathways.
By transfecting Huh7 hepatoma cells with a library of human miRNA precursor molecules, Fort et al interrogated post-transcriptional determinants of fibrinogen production and identified 23 miRNAs with down-regulating and 4 with up-regulating effects on fibrinogen synthesis.9 Further studies showed hsa-miR-409–3p overexpression specifically reduced FGB levels. Interestingly, hsa-miR-29 over-expression decreased all 3 fibrinogen mRNAs, consistent with tight, coordinated regulation of fibrinogen expression.
Fibrinogen isoforms in circulation.
Different forms of fibrinogen originating from alternative splicing and/or post-translational modifications can be found in circulation. In humans, fibrinogen containing an alternatively spliced form of the γ chain (γ′) in which the final 4 amino acids (γ408–411, AGDV) encoded by exon 10 of the native γ chain are replaced with 20 amino acids (γʹ408–427, VRPEHPAETEYDSLYPEDDL) encoded by exon 9, has received the most attention (Fig 2A). The biological importance of the fibrinogen γ′ is derived from the substantial negative charge imparted by the alternative sequence of residues including 2 sulfated tyrosine residues. Interestingly, fish and frogs lack the intron necessary for alternative γ chain splicing, and chickens and platypus have the intron but splicing does not generate an extended peptide that could have modified biological functions.10 All mammals appear to have extended γʹ sequences, but the lengths, and therefore the potential unique biochemical functions of these vary.10 In healthy humans, fibrinogen molecules containing the γ′ chain comprise 8–15% of total fibrinogen (~0.3 mg/mL in plasma). However, studies in HepG2 hepatoblastoma cells suggest the inflammatory cytokine interleukin-6 differentially upregulates transcription of γ′ and total fibrinogen 8.3- and 3.6-fold, respectively11, so the relative proportion of γ′-containing molecules may differ in inflammatory settings. A single nucleotide polymorphism in the FGG H2 haplotype within a putative binding site for a polyadenylation factor (10034C>T in the FGG 3ʹ region) has been implicated as a functional variant driving relative γʹ and γA levels. The presence of γ′ residues 414–427 enables a high-affinity binding interaction with thrombin exosite II, which competitively inhibits thrombin-mediated platelet activation, fibrinopeptide B cleavage, and cofactor activation, and sequesters enzymatically active thrombin within the nascent thrombus. The γ′ chain also lacks the platelet integrin αIIbβ3 binding site. These properties of γ′ fibrinogen have been reviewed.12 Early reports suggested the γ′ chain also mediates factor XIII (FXIII) binding to fibrinogen. However, more recent studies showed residues N-terminal to the γ′ chain (γ390–396) comprise the major binding site in both humans and mice.13,14
Figure 2. Fibrinogen FGG and FGA mRNAs undergo alternative splicing.
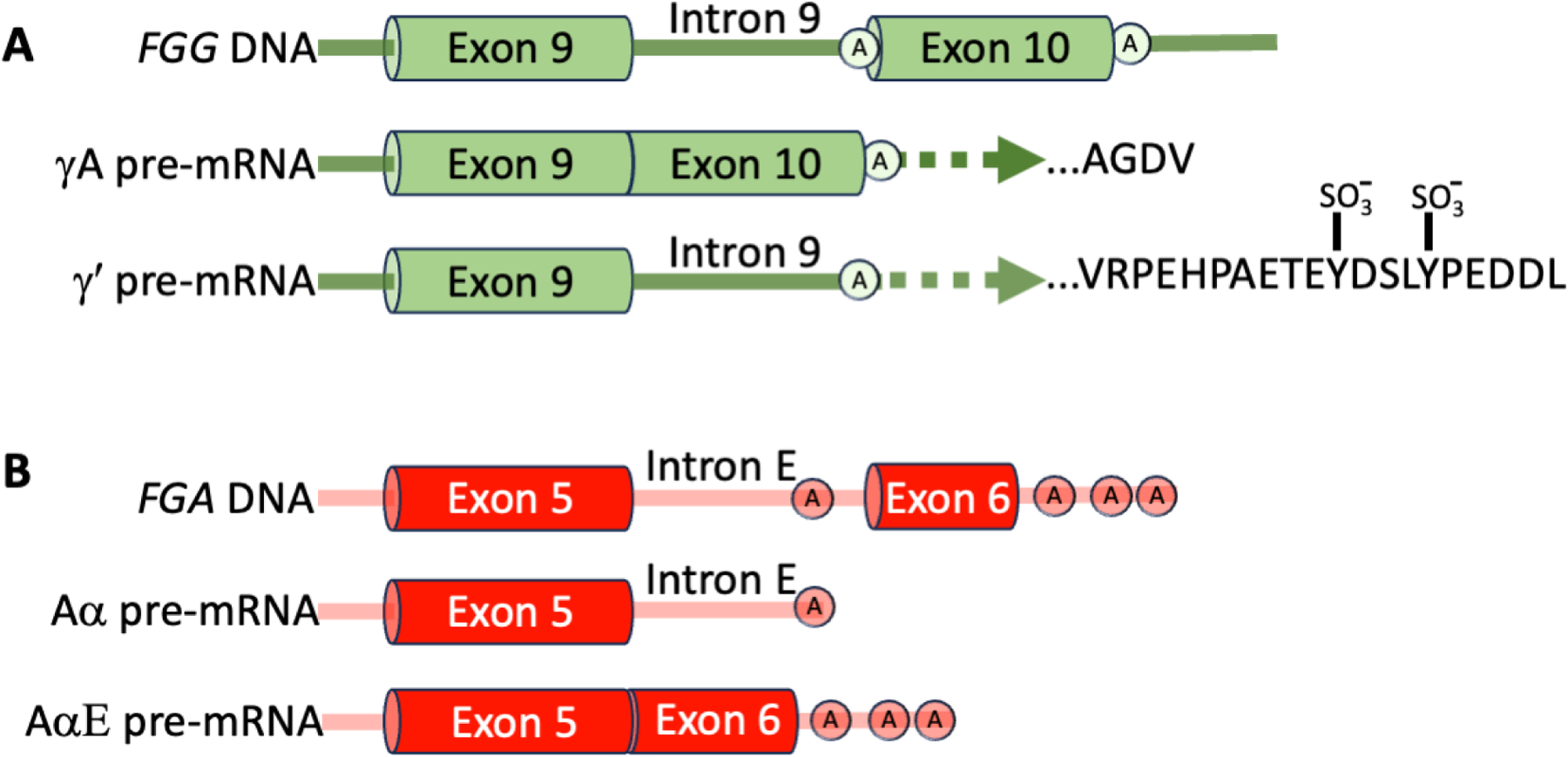
A) Alternatively-spliced form of the γ chain (γ′) in which the final 4 amino acids encoded by exon 10 of the native γ chain (γ408–411) are replaced with 20 amino acids encoded by exon 9 (γʹ408–427). Sulfated tyrosine residues are indicated by SO3−. B) Alternatively-spliced form of the Aα chain that includes an extended αEC C-terminal globular region. Polyadenylation sites are indicated with encircled “A”s.
Alternative splicing also mediates the synthesis of an extended Aα chain containing a C-terminal globular region (αEC) that has homology to the Bβ and γ-chain C-terminal regions (Fig 2B).15 AαE fibrinogen (420 kDa) is only present in ~1% of circulating fibrinogen molecules, but AαE levels are higher in neonates compared to adults. Despite considerable conservation, the function of AαE fibrinogen is unclear but may include enhanced interactions with leukocyte integrins.16 By leveraging zebrafish strains harboring, or not, a single nucleotide deletion in FGA exon 6, Freire et al showed that compared to Aα fibrinogen, expression of the AαE extension shortened the time to vessel occlusion in a laser-induced venous thrombosis model and attributed this to thrombocyte (fish have thrombocytes instead of platelets) adhesion and aggregation.17
Fibrinogen also undergoes post-translational modifications that can occur during synthesis (e.g., disulfide bond formation, glycosylation) and/or after its secretion (e.g., homocysteinylation). Fibrinogen ultrastructure allows for 29 inter- and intra-chain disulfide bonds thought to maintain structural integrity of the central E region N-terminal disulfide knot, the coiled-coil regions extending outward from the E region, and within the Bβ and γ chain globular regions. Using an integrated HPLC and mass spectrometry approach, Butera and Hogg identified 13 disulfide bonds in circulating fibrinogen that are between 10–50% reduced and demonstrated the formation of new disulfide bonds upon fibrin polymerization that are altered by fluid shear forces.18 Thus, even within subspecies of fibrinogen isoforms, variation in the extent of disulfide bonding may tailor fibrinogen structure and function to particular biological and pathobiological settings.
Fibrinogen structure.
The six fibrinogen polypeptides are arranged with N-termini in a central E region and C-termini radiating outwards with bilateral symmetry (Fig 1B).2 The C-terminal globular domains of the Bβ and γ chains (βC and γC nodules, respectively) are present in D regions and the Aα chain extends into an unstructured αC connector and partially structured αC domain that may interact with the E region and/or participate in inter-monomer interactions during fibrin formation. The complexity of this trinodular (D-E-D), ~45 nm hexameric molecule made it a highly sought, but challenging, candidate for structural studies. The earliest X-ray crystallography structure revealed the C-terminal region of the fibrinogen γ chain and fibrinogen fragment D and others soon followed. Collectively, these studies reported considerable structural conservation of fibrinogen regions among humans, cows, chickens, and lamprey. Crystallography has been an experimental workhorse in studies to interrogate natural variants and elucidate structure-function relationships mediating fibrin polymerization. More recent advances in biophysical microscopy techniques uncovered a highly dynamic conformational landscape of fibrin(ogen), including flexible regions that previously escaped resolution in studies that used static methods for structural analysis. Visualization of these mobile regions is particularly important since structural rearrangements facilitate the fibrinogen-to-fibrin transition and expose new epitopes that interact with plasma and cellular proteins. Integrated hydrogen-deuterium exchange mass spectrometry and small-angle X-ray scattering revealed a dynamic model of human fibrinogen in solution and suggested fibrinogen exhibits considerable flexibility.19 The authors detected significant flexions within the coiled-coil connector and within the homodimerization interface in the E-region that may influence fibrinogen macromolecular interactions, as well as its incorporation as fibrin into insoluble networks.19 Visualization of fibrinogen by atomic force microscopy has enabled high-resolution data on the flexible αC connector and globular regions and revealed considerable heterogeneity in length and shape, with implications for their potential role in fibrin formation.20
Fibrin formation, structure, and resistance to lysis.
The thrombin-mediated conversion of fibrinogen to fibrin proceeds via proteolytic removal of fibrinopeptides from the N-termini of the Aα (FpA, 16 amino acids) and Bβ (FpB, 14 amino acids) chains, after which these chains are referred to as α and β (Fig 3). Exposure of the new N-termini permits engagement of these “knobs” into “holes/pockets” in the C-terminal globular domains of the γ and β chains (A and B knobs into a and b pockets, respectively), although interactions between the A knobs and the a pocket and surrounding residues drive the end-to-end extension of monomers into half-staggered (22.5 nm periodicity) protofibrils.21 The A:a interaction strengthens in response to pulling forces up to 40 pN, but decreases above 40 pN.22 This so-called “catch-slip” bond may enhance fibrin polymerization in faster-flowing blood and reduce polymerization in slower-flowing blood. The biological implications of this behavior and whether there is an evolutionary driver of this phenomenon in fibrin is unknown. Subsequent lateral aggregation and branching of nascent fibers produce fibrin polymers in an insoluble fibrin network. Major advances in imaging technologies have enabled elegant visualization of the early stages of fibrin polymerization in the past 20 years. A major “black box” in the fibrinogen structure-function landscape has been the role of the αC regions in fibrin formation and biophysical properties. Due to their inherent flexibility and high susceptibility to cleavage in circulation, challenges in visualizing the αC regions have hindered understanding of their role in fibrin formation and structure. In the absence of functional fibrin knobs, the αC regions cannot independently promote fibrin polymerization. Atomic force microscopy with super high resolution visualized individual αC regions in molecules of fibrinogen and fibrin protofibrils and suggested the αC regions do not substantively alter fibrin structure, but instead enhance fibrin formation kinetics, perhaps by promoting lateral aggregation of protofibrils via a “zipper-like” mechanism.20
Figure 3. Thrombin-mediated conversion of fibrinogen to fibrin.
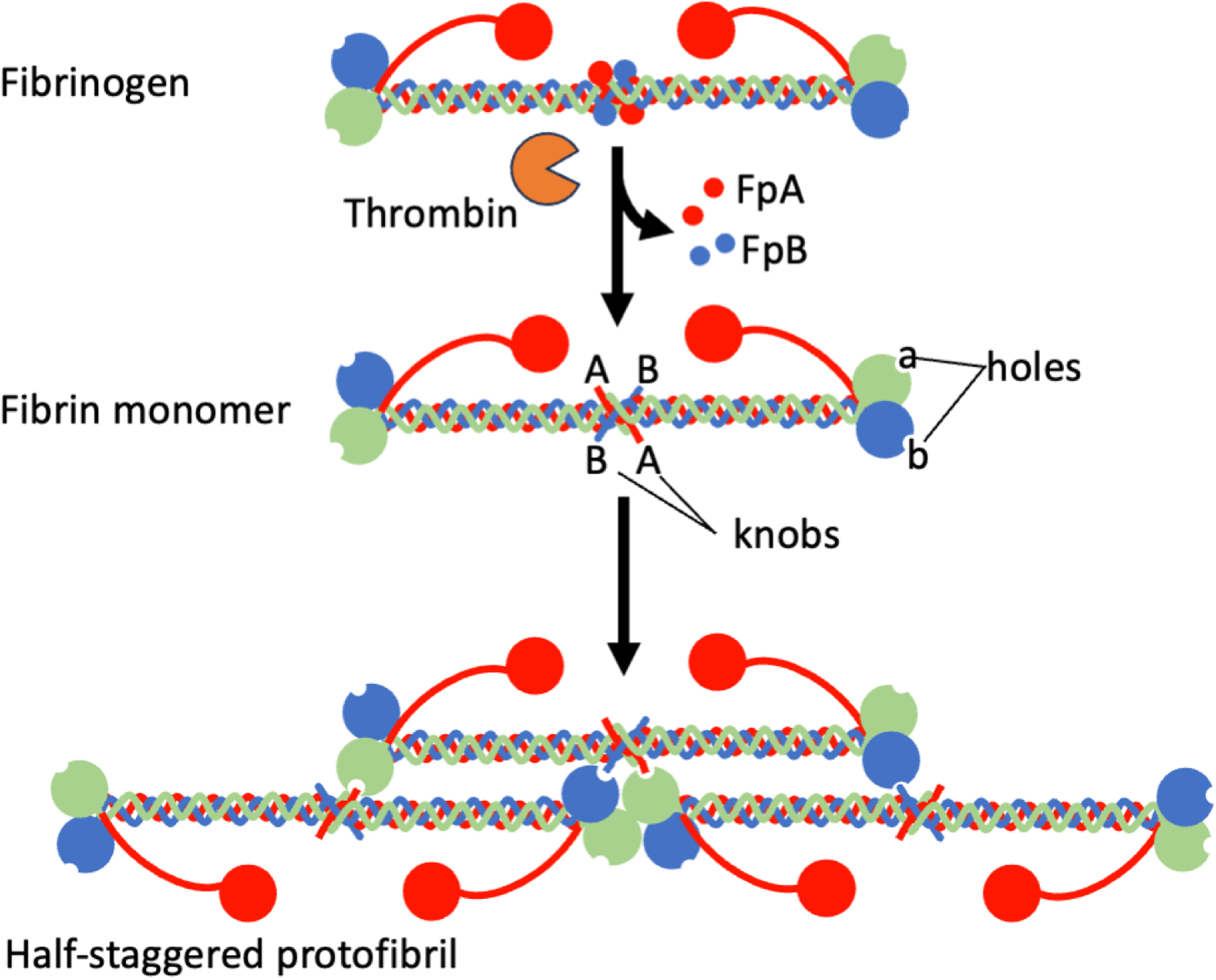
Thrombin-mediated conversation of fibrinogen to fibrin proceeds via proteolytic removal of fibrinopeptides from the N-termini of the Aα (FpA, 16 amino acids) and Bβ (FpB, 14 amino acids) chains. Exposure of the new N-termini on the α and β chains permits engagement of these “knobs” into “holes” (pockets) in the C-terminal globular domains of the γ and β chains, respectively. The αC regions promote the aggregation of half-staggered protofibrils into fibrin fibers integrated into a fibrin network (not shown).
Both genetic and environmental factors can influence the conversion of fibrinogen to fibrin and the resulting structure and function. Modifiers include the presence of alternatively-spliced fibrinogen, post-translational modifications (reviewed in23), and plasma proteins and molecules that interact with fibrinogen and fibrin. The thrombin concentration present during fibrinogen conversion to fibrin is a major determinant of fibrin structure and therefore, its biochemical and biophysical function. Low thrombin concentrations produce thick fibrin fibers arranged in coarse and highly permeable networks that are susceptible to fibrinolysis, whereas higher thrombin concentrations produce thinner fibers in dense networks that are less permeable and more resistant to fibrinolysis (reviewed in24). Counterintuitively, lysis of individual thick fibers is slower than that of thinner fibers, which has been attributed to the strain imposed on fibrin fibers during polymerization; whereas thin fibers are readily lysed by plasmin, thicker fibers are more likely to elongate and lyse more slowly.25 Consequently, changes that alter plasma thrombin generation (e.g., hypercoagulability in hyperprothrombinemia and elevated FVIII or hemostatic insufficiency in factor IX, VIII, or XI deficiency) also alter fibrin network formation, structure, and stability. By controlling the transport rate of zymogens, cofactors, and activated enzymes to and from the injury site, blood flow is a strong determinant of the local thrombin concentration and therefore, fibrin deposition. Blood flow also influences the alignment of fibrin fibers within a clot and the response of the fibrin network to mechanical perturbation, which may protect clots against premature rupture. In both purified systems and in plasma, γA/γ′ fibrinogen interferes with protofibril packing and growth, polymerizes more slowly than γA/γA fibrinogen, and produces clots with altered structure and biophysical properties (reviewed in12), although reported effects differ between studies and may reflect differences in specific experimental conditions, including concentrations of fibrinogen and or thrombin. Importantly, the association of fibrin fiber and network structural properties with clinical pathologies – dense networks of thinner and more compact fibers with thrombotic risk and coarse, porous networks of thicker and less compact fibers with bleeding risk – suggests fibrin structure is both a determinant of hemostasis and thrombosis and a predictive biomarker of coagulation potential (reviewed in26). The potential application of fibrin quality as a biomarker of coagulation dysfunction is underscored by observations of abnormal fibrin structure and function even in patients with bleeding of unknown cause.27
Fibrin biomechanical properties and crosslinking.
Of all fibrin(ogen) features, one of the most intriguing is its essential role as a biophysical entity and determinant of clot mechanical stability. This feature differentiates fibrin from most other clotting factors that participate in coagulation strictly through their biochemical contributions. Combined atomic force-fluorescence microscopy showed individual fibrin fibers exhibit remarkable extensibility and elasticity and can be strained ~330% without rupturing, akin to that seen in spider silk.28 Collectively, these data suggest fibrin monomers within fibers can undergo substantial, reversible structural rearrangements that can accommodate fibrin stretching that may occur in flow and during wound healing. Several regions within fibrinogen and fibrin units in fibers have been proposed to mediate these changes, including the length of tandem repeats in the αC region, and unfolding within the αC region, coiled-coil region, and/or γ chains. The biomechanical properties of fibrin and their physiologic importance are reviewed in29.
Central to fibrin’s biophysical characteristics is its ability to be crosslinked by the transglutaminase factor XIII (FXIII[a]). FXIIIa introduces covalent bonds between γ chains (generating γ-γ dimers), between γ- and α-chains (generating high molecular weight species), and between fibrin chains and other plasma proteins (e.g., α2-antiplasmin) (Fig 4). The nature of intermolecular crosslinks within fibrin monomers has been debated: transverse, between monomers located in adjacent protofibrils, versus longitudinal, between monomers involved in end-to-end interactions between D-regions. Computations from a molecular modeling simulation suggest the distance between crosslinking residues in γ-nodules located in two different strands is not feasible, even if one assumes that the B:b interaction can be reversed in a “pull-out” mechanism, and therefore support the longitudinal model.30 Fibrin crosslinking has major effects on fibrin fiber and network integrity. Crosslinking increases the compaction of fibrin monomers within the fiber31, but has relatively minor effects on fibrin network density. However, crosslinking significantly decreases the extensibility and elasticity of fibrin fibers, and increases fiber stiffness ~2-fold. Crosslinking also stabilizes fibrin branchpoints, protecting these regions from rupture.32 In general, fibrin γ-chain crosslinking modifies clot elasticity by changing protofibril force-extension behavior, and α-chain crosslinking stiffens fibrin fibers, although the distinction between these effects is likely nuanced.33 In vivo, the biophysical properties of fibrin likely reflect contributions of both fibrin fibers and fibrin-binding molecules found in circulation (e.g., von Willebrand factor, neutrophil extracellular traps). Crosslinking of α2-antiplasmin to fibrin enables the retention of α2-antiplasmin in the clot during platelet-mediated clot contraction and is the basis for α2-antiplasmin’s antifibrinolytic activity.34
Figure 4. Fibrin crosslinks introduced by factor XIII.
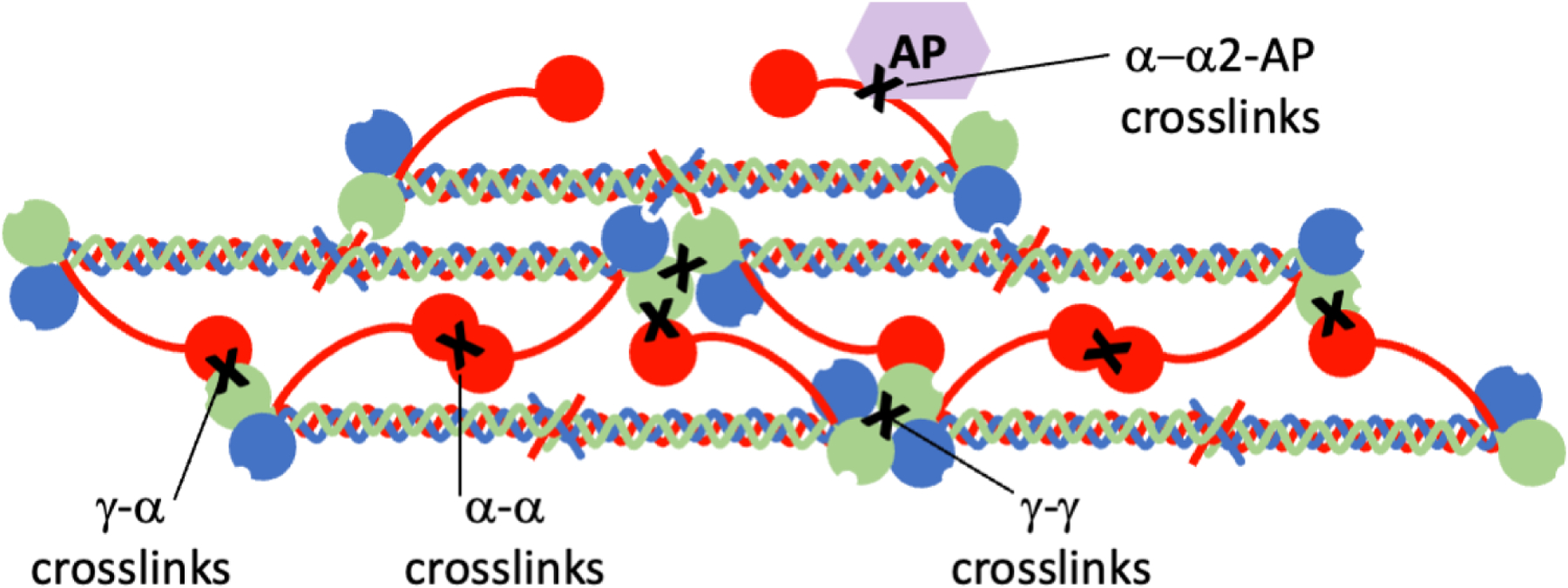
Activated factor XIII (FXIIIa) introduces covalent bonds (“X”) between fibrin γ chains (generating γ-γ dimers), between γ- and α-chains (generating α-chain-rich high molecular weight species), and between fibrin chains and other plasma proteins (e.g., α-chain:α2-antiplasmin [AP]).
Fibrin(ogen) in thrombosis.
Fibrinogen deficiency causes bleeding in humans and animals. However, the contributions of fibrinogen and fibrin to cardiovascular disease and venous thromboembolism in humans have been challenging to elucidate. A brief review of epidemiologic studies on the role of fibrinogen in thrombosis can be found in26. Briefly, elevated total fibrinogen has been generally associated with higher risk of venous thrombosis particularly in the elderly, even after adjusting for potential confounding effects of inflammatory disease. Likewise, plasma fibrinogen levels have also been associated positively with cardiovascular disease risk, including coronary heart disease, stroke, other vascular mortality, and even non-vascular mortality in middle-aged adults. Increased fibrinogen in young adulthood has been associated with the development of subclinical atherosclerosis (coronary artery calcification and carotid intimal-medial thickness) in middle age. Notably, however, findings on the contribution of fibrinogen to thrombotic disease have been discordant, reflecting the complexity of fibrin(ogen) function in complex biological systems. First, fibrinogen levels increase in inflammatory disease, making it difficult to isolate the specific effects of fibrinogen on the disease pathogenesis and outcomes. Second, gene-environment interactions between fibrinogen, FXIII, and other proteins modify the contribution of fibrinogen to thrombosis risk, wherein failure to consider these co-existing modifiers may obscure relevant connections. Third, presence of the alternatively-spliced γ′ chain, which alters risk likely via one or more of its anticoagulant properties, can weaken the prothrombotic effects of increased total fibrinogen. Finally, differences between population ancestries, age, gender, study design (observational, case-control), and outcome (e.g., any ischemic stroke versus cardioembolic stroke) make it difficult to directly compare findings in different reports.
Methods that use genetic variants as inherited, constant instruments can circumvent potential confounding and reverse causation in genetic studies. These methods, which relate genetic polymorphisms, intermediate phenotypes (e.g., fibrinogen level), and clinical disease can be used to demonstrate causation (Fig 5). For example, a case-control study of women aged 18–50 years old carrying the FGA variant leading to the AαThr312Ala polymorphism or the FGB 455G/A variant, correlated with reduced and increased plasma fibrinogen levels, respectively, associated the plasma fibrinogen level with ischemic stroke, but not myocardial infarction.35 The FGG H2 haplotype, which is associated with reduced levels of γ′ but not total fibrinogen and therefore a reduced γʹ-to-total fibrinogen ratio, has been associated with increased risk of VTE and stroke36,37, although this finding has not been unequivocally reproduced. Using Mendelian randomization and genetic instruments for γ′ and total fibrinogen in large-scale GWASs, Maners et al demonstrated a protective effect of increased γ′ fibrinogen and increased total fibrinogen on the risk of VTE and cardioembolic stroke.38 Moreover, whereas increased total fibrinogen increased risk of large artery stroke, γʹ fibrinogen mitigated this effect.38
Figure 5. Principle of Mendelian randomization applied to the fibrinogen FGG H2 haplotype.
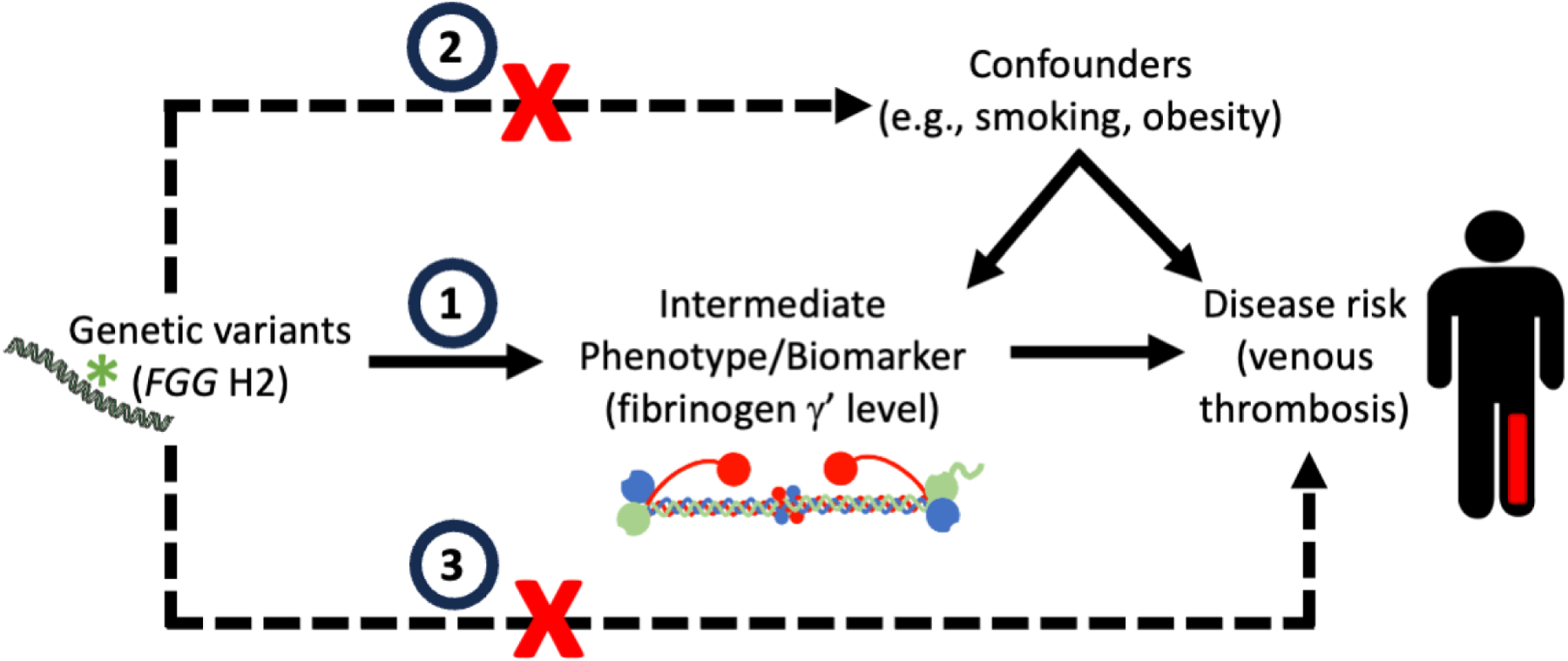
Mendelian randomization relates genetic polymorphisms (e.g., FGG H2 haplotype), intermediate phenotypes (e.g., fibrinogen γʹ level), and clinical disease (e.g., venous thrombosis). Mendelian randomization requires three assumptions: 1) The genetic variant (“instrument”) acts on the intermediate phenotype (“exposure”/biomarker) (e.g., the FGG H2 haplotype determines the fibrinogen γʹ level), (2) The genetic variant does not cause confounding mechanisms (e.g., presence of the FGG H2 haplotype does not influence lifestyle choices that alter disease risk), and 3) the genetic variant mediates its effect solely via the intermediate phenotype and is not independently associated with the disease outcome (e.g., the FGG H2 haplotype does not cause venous thrombosis through a γʹ-independent mechanism). If all of these assumptions are met, genetic variants can be used in well-phenotyped populations with randomized genotypes to demonstrate causal associations between intermediate phenotypes and disease.
Since the fibrinogen genes and protein functions are well conserved between species, animal models have also helped dissect the contributions of total and γʹ fibrinogen to thrombosis risk and determine causation. Mice with genetic hyperfibrinogenemia have increased neointimal hyperplasia in an experimental model of stasis-induced vascular remodeling.39 Mice infused with fibrinogen to 300% of normal levels and subjected to ferric chloride application to the saphenous vein have shortened time to vessel occlusion, increased fibrin deposition within the thrombus, and increased thrombus resistance to thrombolysis.40 Although mice can express an alternatively spliced γ′ chain, the newly introduced residues lack the high-affinity thrombin binding site present in human γ′ fibrinogen. Consequently, murine studies to elucidate the in vivo effects of the γ′ chain have employed creative strategies to introduce the human γ′ sequence into mice. In transgenic mice carrying the prothrombotic factor V Leiden gene mutation, expression of the human γ′ chain reduced thrombus volume following electrolytic injury to the femoral vein.41 In mice infused with human γA/γA or γA/γ′ fibrinogen and then subjected to ferric chloride application to the carotid artery, presence of γA/γ′ led to reduced circulating thrombin-antithrombin complexes, consistent with γ′-mediated thrombin sequestration.42 Collectively, these data identify fibrinogen levels and isoforms as causal determinants of cardiovascular and thrombotic disease, but show that isolated measurements of total or γʹ fibrinogen are unlikely to be useful specific biomarkers for predicting thrombotic risk in humans.
Fibrin structure and crosslinking as a determinant of thrombosis.
Fibrin crosslinking has been implicated as a major determinant of venous thrombus composition and mass. Briefly, following venous thrombus initiation, thrombin converts fibrinogen to fibrin. Fibrin traps resident cells, and activated platelets contract, compressing cells within the thrombus core. The resultant mass, which contains abundant, highly compressed red blood cells (polyhedrocytes) is solid and relatively impermeable to fibrinolytic enzymes. In vitro studies show that increased fibrin network density enhances RBC retention within contracted clots.43 Fibrin crosslinking by FXIII(a) further enhances RBC retention, independent of the fibrin network density.13,43,44 Following ligation of the inferior vena cava, compared to F13a1+/+ mice, F13a1−/− mice produce thrombi with decreased RBC content and consequently, reduced mass. Similarly, contracted clots formed from human or mouse whole blood have reduced RBC retention in the absence of FXIII activity (Fig 6).13 Studies with recombinant fibrinogen variants and with FXIIIa inhibitors associated RBC retention specifically with the formation of α-chain-rich high molecular weight crosslinked species that are produced primarily by plasma, but not platelet-derived, FXIII.43,44 Importantly, the events that mediate this effect occur rapidly after thrombus initiation, when platelet-mediated clot contraction applies force to the fibrin network and its cellular content. Delaying FXIII activation and/or fibrin crosslinking permits clot contraction to occur prior to fibrin stabilization, wherein RBCs are extruded from the clot between extensible fibrin strands.
Figure 6. Fibrin crosslinking by factor XIII promotes retention of red blood cells in contracted clots.
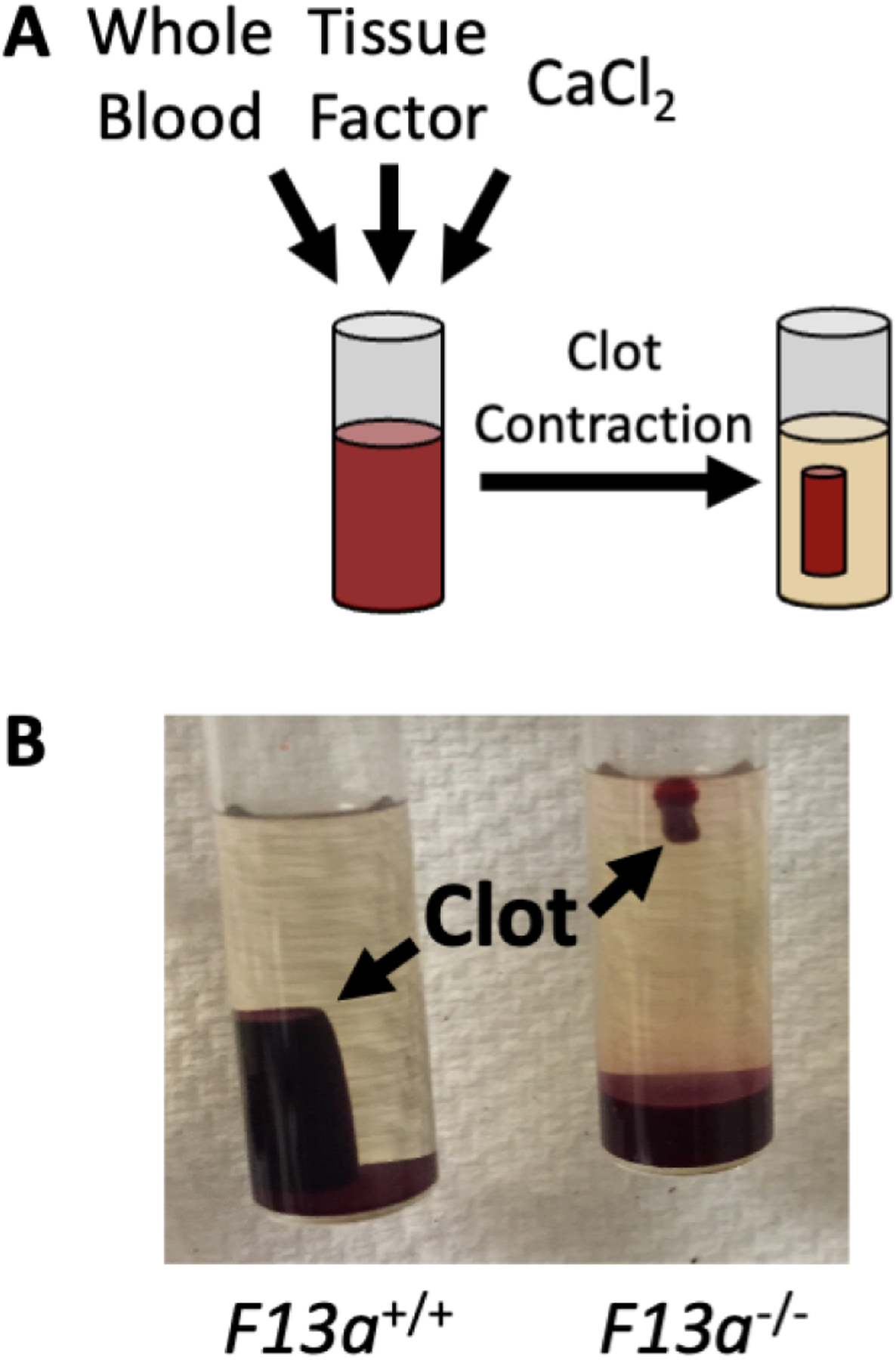
Contracted whole blood clots have reduced red blood cell retention in the absence of factor XIII(a) activity. A) Schematic of a whole blood clot contraction assay. B) Contracted whole blood clots from FXIII-sufficient (F13a1+/+) and -deficient (F13a1−/−) mice.
Interestingly, a common FXIII polymorphism (a valine to leucine substitution at residue 34, Val34Leu) counteracts the effects of high fibrin(ogen) concentrations on RBC retention and clot mass. The FXIII-A Val34Leu polymorphism occurs three amino acids before the thrombin cleavage site and causes ~2.5-fold faster FXIII activation and accelerates fibrin crosslinking.45 In plasmas with low-to-normal fibrinogen levels, the presence of the 34Leu variant produces clots with thin fibrin fibers and low permeability; however, in plasmas with high fibrinogen, this variant produces thicker fibers in clots that have increased permeability and susceptibility to fibrinolysis.45 Accordingly, whereas clots containing a FXIII 34Val allele exhibit an expected fibrinogen concentration-dependent increase in mass, clots with homozygous FXIII 34Leu do not.46 This interesting gene-environment interaction between the FXIII Val34Leu polymorphism (gene) and circulating fibrinogen (environment) is thought to mediate the moderate protective effect of the FXIII 34Leu allele on the risk of venous thrombosis and coronary artery disease (reviewed in47).
Fibrin(ogen) in inflammatory and prothrombotic diseases.
In addition to its role in classical thrombosis, studies in humans and animals have implicated fibrin(ogen) in the pathogenesis of multiple prothrombotic diseases via its contribution to overt thrombosis or as a mediator of tissue injury and inflammation that promote pathogenesis (Fig 7 and reviewed in48). Liver injury is associated with the activation of coagulation and fibrin deposition, which appears to drive liver regeneration and limit fibrosis in part through interactions between the leukocyte αMβ2 integrin and fibrin(ogen).49 Fibrin(ogen) has also been implicated in cancer pathogenesis, where it promotes tumor growth and metastatic potential of circulating tumor cells (reviewed in50). During bacterial infection, fibrin(ogen) has complex and sometimes competing roles that enable microbial function through bacterial-mediated prothrombin activation, provide a physical barrier against bacterial invasion, and enhance the host antimicrobial response. At least part of fibrinogen’s protective effect in mice can be attributed specifically to fibrin formation and the presence of the γ′ isoform.51 Fibrinogen as well as insoluble fibrin(ogen) and fibrin have also been implicated in the pathogenesis of sickle cell disease52 and oral mucosal disease53. Abnormalities in fibrinogen, fibrin, and the fibrin degradation product D-dimer, were most recently implicated in the pathogenesis of the acute respiratory illness coronavirus disease-2019 (COVID-19). COVID-19 patients have significantly elevated fibrinogen, likely secondary to the acute inflammatory response, as well as increased levels of the fibrin breakdown product D-dimer, which correlates with disease severity and mortality risk. Plasma from COVID-19 patients produces abnormally dense clots from increased circulating fibrinogen and/or abnormal post-translational modification (e.g., increased sialic acid54), as well as increased resistance to fibrinolysis.55
Figure 7. Fibrinogen and fibrin are involved in many diseases.
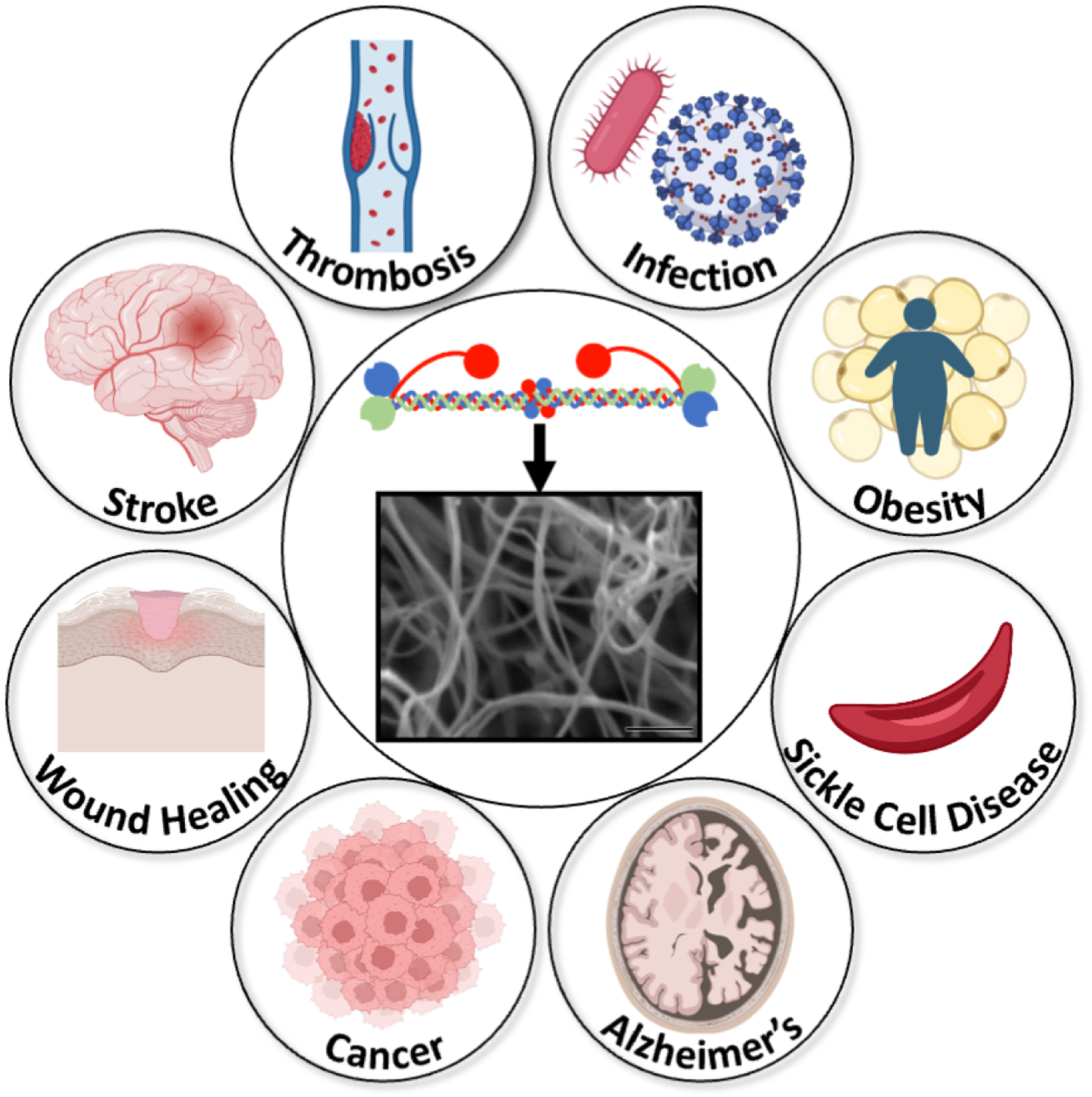
Fibrinogen and/or its insoluble end products fibrin(ogen) or fibrin have been implicated in the pathogenesis of many diseases. Images representing diseases are from Biorender.com.
Fibrin(ogen) in the next 20 years: key outstanding questions.
The studies summarized here provide a high-level view of fibrinogen and fibrin function and highlight a subset of recent, exciting work that has advanced understanding of this remarkable molecule. These discoveries and new technologies will fuel further understanding of fibrinogen and fibrin in the coming decades. Since fibrinogen and fibrin contribute to multiple diseases, algorithms that incorporate genetic and environmental mediators of fibrinogen levels and fibrin clot structure and function may be useful for assessing disease risk, pathogenesis, and outcomes. Since patients with increased thrombotic risk or bleeding of unknown cause exhibit abnormal fibrin quality, assays to quantify fibrin formation, structure, and stability may yield a more “universal metric” for assessing risk than conventional assays. Identifying experimental conditions that maximize the sensitivity of these assays to fibrin(ogen) abnormalities and standardizing these within and between centers is needed to advance the broad application of fibrin assessment tools as clinical diagnostics. There are substantial challenges in imaging and computationally reconstructing large molecules and molecular networks. Obstacles include flexible unstructured regions, numerous potential spatial arrangements of fibrin monomers and protofibrils, and vast computational resources needed to study the dynamic conformation landscape. These challenges have limited structural knowledge of fibrinogen and fibrin. Given the caveats in obtaining images of individual fibrinogen monomers, it is not surprising that visualizing fibrin polymerization has been difficult. Zhmurov et al56 integrated 27 crystal structures to computationally reconstruct an atomic-level model of fibrin oligomers and protofibrils. Their model, which correlates with high-resolution atomic force microscopy images, may enable the in silico interrogation of fibrin’s complex dynamics landscape. Further advances in microscopy and computational simulations, including the use of artificial intelligence and machine learning, will accelerate this line of research. Advancing methods to safely image fibrin directly or via FXIIIa-dependent crosslinking activity in vivo may facilitate the diagnosis of primary and recurrent venous thrombosis and thrombolysis. Therapeutic strategies to modify the circulating fibrinogen level have shown promise in altering the course of hemorrhagic, thrombotic, and inflammatory diseases. Older methods using drugs to decrease the fibrinogen level (e.g., the defibrinogenating agent ancrod or thrombolytics) were associated with bleeding or off-target effects. More recent methods that more precisely modulate fibrinogen level and/or function, including single-stranded antisense oligonucleotides and small interfering RNA (siRNA) to knock down fibrinogen57, or fibrinogen variants that interfere with fibrin polymerization (e.g., fibrinogen that can participate in platelet aggregation but not thrombin-mediated fibrin formation)58 may provide therapeutic benefit with better safety profiles.
ACKNOWLEDGEMENTS
The author thanks Drs. James R. Byrnes and Dre’Von A. Dobson for inspiring several of the figures, Karl C. Desch for a helpful conversation, and Yaqiu Sang and Stéphanie E. Reitsma for reading the manuscript. The author also acknowledges many excellent studies that have contributed substantively to understanding fibrinogen and fibrin that regretfully could not be cited within the imposed reference limits.
FUNDING
This study was supported by funding from the National Institutes of Health, National Heart, Lung, and Blood Institute (R01HL126974 to ASW).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
The author declares no competing financial interests.
REFERENCES
- 1.Espitia Jaimes C, Fish RJ, Neerman-Arbez M. Local chromatin interactions contribute to expression of the fibrinogen gene cluster. J Thromb Haemost. 2018;16(10):2070–2082. [DOI] [PubMed] [Google Scholar]
- 2.Pieters M, Wolberg AS. Fibrinogen and fibrin: An illustrated review. Res Pract Thromb Haemost. 2019;3(2):161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fish RJ, Neerman-Arbez M. Fibrinogen gene regulation. Thromb Haemost. 2012;108(3):419–426. [DOI] [PubMed] [Google Scholar]
- 4.Vu D, Neerman-Arbez M. Molecular mechanisms accounting for fibrinogen deficiency: from large deletions to intracellular retention of misfolded proteins. J Thromb Haemost. 2007;5 Suppl 1:125–131. [DOI] [PubMed] [Google Scholar]
- 5.Casini A, Blondon M, Lebreton A, Koegel J, Tintillier V, de Maistre E, Gautier P, Biron C, Neerman-Arbez M, de Moerloose P. Natural history of patients with congenital dysfibrinogenemia. Blood. 2015;125(3):553–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Vries PS, Chasman DI, Sabater-Lleal M, Chen MH, Huffman JE, Steri M, Tang W, Teumer A, Marioni RE, Grossmann V, Hottenga JJ, Trompet S, Muller-Nurasyid M, Zhao JH, Brody JA, Kleber ME, Guo X, Wang JJ, Auer PL, Attia JR, Yanek LR, Ahluwalia TS, Lahti J, Venturini C, Tanaka T, Bielak LF, Joshi PK, Rocanin-Arjo A, Kolcic I, Navarro P, Rose LM, Oldmeadow C, Riess H, Mazur J, Basu S, Goel A, Yang Q, Ghanbari M, Willemsen G, Rumley A, Fiorillo E, de Craen AJ, Grotevendt A, Scott R, Taylor KD, Delgado GE, Yao J, Kifley A, Kooperberg C, Qayyum R, Lopez LM, Berentzen TL, Raikkonen K, Mangino M, Bandinelli S, Peyser PA, Wild S, Tregouet DA, Wright AF, Marten J, Zemunik T, Morrison AC, Sennblad B, Tofler G, de Maat MP, de Geus EJ, Lowe GD, Zoledziewska M, Sattar N, Binder H, Volker U, Waldenberger M, Khaw KT, McKnight B, Huang J, Jenny NS, Holliday EG, Qi L, McEvoy MG, Becker DM, Starr JM, Sarin AP, Hysi PG, Hernandez DG, Jhun MA, Campbell H, Hamsten A, Rivadeneira F, McArdle WL, Slagboom PE, Zeller T, Koenig W, Psaty BM, Haritunians T, Liu J, Palotie A, Uitterlinden AG, Stott DJ, Hofman A, Franco OH, Polasek O, Rudan I, Morange PE, Wilson JF, Kardia SL, Ferrucci L, Spector TD, Eriksson JG, Hansen T, Deary IJ, Becker LC, Scott RJ, Mitchell P, Marz W, Wareham NJ, Peters A, Greinacher A, Wild PS, Jukema JW, Boomsma DI, Hayward C, Cucca F, Tracy R, Watkins H, Reiner AP, Folsom AR, Ridker PM, O’Donnell CJ, Smith NL, Strachan DP, Dehghan A. A meta-analysis of 120 246 individuals identifies 18 new loci for fibrinogen concentration. Hum Mol Genet. 2016;25(2):358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dobson DA, Holle LA, Lin FC, Huffman JE, Luyendyk JP, Flick MJ, Smith NL, de Vries PS, Morrison AC, Wolberg AS. Novel genetic regulators of fibrinogen synthesis identified by an in vitro experimental platform. J Thromb Haemost. 2023;21(3):522–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hahn J, Bressler J, Domingo-Relloso A, Chen MH, McCartney DL, Teumer A, van Dongen J, Kleber ME, Aissi D, Swenson BR, Yao J, Zhao W, Huang J, Xia Y, Brown MR, Costeira R, de Geus EJC, Delgado GE, Dobson DA, Elliott P, Grabe HJ, Guo X, Harris SE, Huffman JE, Kardia SLR, Liu Y, Lorkowski S, Marioni RE, Nauck M, Ratliff SM, Sabater-Lleal M, Spector TD, Suchon P, Taylor KD, Thibord F, Tregouet DA, Wiggins KL, Willemsen G, Bell JT, Boomsma DI, Cole SA, Cox SR, Dehghan A, Greinacher A, Haack K, Marz W, Morange PE, Rotter JI, Sotoodehnia N, Tellez-Plaza M, Navas-Acien A, Smith JA, Johnson AD, Fornage M, Smith NL, Wolberg AS, Morrison AC, de Vries PS. DNA methylation analysis is used to identify novel genetic loci associated with circulating fibrinogen levels in blood. J Thromb Haemost. 2023;21(5):1135–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fort A, Borel C, Migliavacca E, Antonarakis SE, Fish RJ, Neerman-Arbez M. Regulation of fibrinogen production by microRNAs. Blood. 2010;116(14):2608–2615. [DOI] [PubMed] [Google Scholar]
- 10.Doolittle RF, Hong S, Wilcox D. Evolution of the fibrinogen gamma’ chain: implications for the binding of factor XIII, thrombin and platelets. J Thromb Haemost. 2009;7(8):1431–1433. [DOI] [PubMed] [Google Scholar]
- 11.Rein-Smith CM, Anderson NW, Farrell DH. Differential regulation of fibrinogen gamma chain splice isoforms by interleukin-6. Thromb Res. 2013;131(1):89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uitte de Willige S, Standeven KF, Philippou H, Ariëns RA. The pleiotropic role of the fibrinogen gamma’ chain in hemostasis. Blood. 2009;114(19):3994–4001. [DOI] [PubMed] [Google Scholar]
- 13.Aleman MM, Byrnes JR, Wang J-G, Tran R, Lam WA, Di Paola J, Mackman N, Degen JL, Flick MJ, Wolberg AS. Factor XIII activity mediates red blood cell retention in venous thrombi. J Clin Invest. 2014;124(8):3590–3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Byrnes JR, Wilson C, Boutelle AM, Brandner CB, Flick MJ, Philippou H, Wolberg AS. The interaction between fibrinogen and zymogen FXIII-A2B2 is mediated by fibrinogen residues 390–396 and the FXIII-B subunits. Blood. 2016;128(15):1969–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grieninger G Contribution of the alpha EC domain to the structure and function of fibrinogen-420. Ann N Y Acad Sci. 2001;936:44–64. [DOI] [PubMed] [Google Scholar]
- 16.Lishko VK, Yakubenko VP, Hertzberg KM, Grieninger G, Ugarova TP. The alternatively spliced alpha(E)C domain of human fibrinogen-420 is a novel ligand for leukocyte integrins alpha(M)beta(2) and alpha(X)beta(2). Blood. 2001;98(8):2448–2455. [DOI] [PubMed] [Google Scholar]
- 17.Freire C, Fish RJ, Vilar R, Di Sanza C, Grzegorski SJ, Richter CE, Shavit JA, Neerman-Arbez M. A genetic modifier of venous thrombosis in zebrafish reveals a functional role for fibrinogen AalphaE in early hemostasis. Blood Adv. 2020;4(21):5480–5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Butera D, Hogg PJ. Fibrinogen function achieved through multiple covalent states. Nat Commun. 2020;11(1):5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pinelo JEE, Manandhar P, Popovic G, Ray K, Tasdelen MF, Nguyen Q, Iavarone AT, Offenbacher AR, Hudson NE, Sen M. Systematic mapping of the conformational landscape and dynamism of soluble fibrinogen. J Thromb Haemost. 2023;21(6):1529–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Protopopova AD, Litvinov RI, Galanakis DK, Nagaswami C, Barinov NA, Mukhitov AR, Klinov DV, Weisel JW. Morphometric characterization of fibrinogen’s alphaC regions and their role in fibrin self-assembly and molecular organization. Nanoscale. 2017;9(36):13707–13716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asquith NL, Duval C, Zhmurov A, Baker SR, McPherson HR, Domingues MM, Connell SDA, Barsegov V, Ariens RAS. Fibrin protofibril packing and clot stability are enhanced by extended knob-hole interactions and catch-slip bonds. Blood Adv. 2022;6(13):4015–4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Litvinov RI, Weisel JW. Shear strengthens fibrin: the knob-hole interactions display ‘catch-slip’ kinetics. J Thromb Haemost. 2013;11(10):1933–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Vries JJ, Snoek CJM, Rijken DC, de Maat MPM. Effects of post-translational modifications of fibrinogen on clot formation, clot structure, and fibrinolysis: a systematic review. Arterioscler Thromb Vasc Biol. 2020;40(3):554–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolberg AS. Thrombin generation and fibrin clot structure. Blood Rev. 2007;21(3):131–142. [DOI] [PubMed] [Google Scholar]
- 25.Bucay I, O’Brien ET 3rd, Wulfe SD, Superfine R, Wolberg AS, Falvo MR, Hudson NE. Physical determinants of fibrinolysis in single fibrin fibers. PLoS One. 2015;10(2):e0116350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolberg AS, Sang Y. Fibrinogen and factor XIII in venous thrombosis and thrombus stability. Arterioscler Thromb Vasc Biol. 2022;42(8):931–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hofer S, Ay C, Rejto J, Wolberg AS, Haslacher H, Koder S, Pabinger I, Gebhart J. Thrombin-generating potential, plasma clot formation, and clot lysis are impaired in patients with bleeding of unknown cause. J Thromb Haemost. 2019;17(9):1478–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu W, Jawerth LM, Sparks EA, Falvo MR, Hantgan RR, Superfine R, Lord ST, Guthold M. Fibrin fibers have extraordinary extensibillity and elasticity. Science. 2006;313(5787):634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feller T, Connell SDA, Ariens RAS. Why fibrin biomechanical properties matter for hemostasis and thrombosis. J Thromb Haemost. 2022;20(1):6–16. [DOI] [PubMed] [Google Scholar]
- 30.Zhmurov A, Protopopova AD, Litvinov RI, Zhukov P, Mukhitov AR, Weisel JW, Barsegov V. Structural basis of interfacial flexibility in fibrin oligomers. Structure. 2016;24(11):1907–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurniawan NA, Grimbergen J, Koopman J, Koenderink GH. Factor XIII stiffens fibrin clots by causing fiber compaction. J Thromb Haemost. 2014;12(10):1687–1696. [DOI] [PubMed] [Google Scholar]
- 32.Carlisle CR, Sparks EA, Der Loughian C, Guthold M. Strength and failure of fibrin fiber branchpoints. J Thromb Haemost. 2010;8(5):1135–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Helms CC, Ariëns RA, Uitte de Willige S, Standeven KF, Guthold M. alpha-alpha cross-links increase fibrin fiber elasticity and stiffness. Biophys J. 2012;102(1):168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rijken DC, Abdul S, Malfliet JJ, Leebeek FW, Uitte de Willige S. Compaction of fibrin clots reveals the antifibrinolytic effect of factor XIII. J Thromb Haemost. 2016;14(7):1453–1461. [DOI] [PubMed] [Google Scholar]
- 35.Siegerink B, Rosendaal FR, Algra A. Genetic variation in fibrinogen; its relationship to fibrinogen levels and the risk of myocardial infarction and ischemic stroke. J Thromb Haemost. 2009;7(3):385–390. [DOI] [PubMed] [Google Scholar]
- 36.Uitte de Willige S, de Visser MC, Houwing-Duistermaat JJ, Rosendaal FR, Vos HL, Bertina RM. Genetic variation in the fibrinogen gamma gene increases the risk for deep venous thrombosis by reducing plasma fibrinogen gamma’ levels. Blood. 2005;106(13):4176–4183. [DOI] [PubMed] [Google Scholar]
- 37.Cheung EY, Uitte de Willige S, Vos HL, Leebeek FW, Dippel DW, Bertina RM, de Maat MP. Fibrinogen gamma’ in ischemic stroke: a case-control study. Stroke. 2008;39(3):1033–1035. [DOI] [PubMed] [Google Scholar]
- 38.Maners J, Gill D, Pankratz N, Laffan MA, Wolberg AS, de Maat MPM, Ligthart S, Tang W, Ward-Caviness CK, Fornage M, Debette S, Dichgans M, McKnight B, Boerwinkle E, Group CIW, Consortium I, Consortium McotISG, Smith NL, Morrison AC, Dehghan A, de Vries PS. A Mendelian randomization of gamma’ and total fibrinogen levels in relation to venous thromboembolism and ischemic stroke. Blood. 2020;136(26):3062–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kerlin B, Cooley BC, Isermann BH, Hernandez I, Sood R, Zogg M, Hendrickson SB, Mosesson MW, Lord S, Weiler H. Cause-effect relation between hyperfibrinogenemia and vascular disease. Blood. 2004;103(5):1728–1734. [DOI] [PubMed] [Google Scholar]
- 40.Machlus KR, Cardenas JC, Church FC, Wolberg AS. Causal relationship between hyperfibrinogenemia, thrombosis, and resistance to thrombolysis in mice. Blood. 2011;117(18):4953–4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mosesson MW, Cooley BC, Hernandez I, Diorio JP, Weiler H. Thrombosis risk modification in transgenic mice containing the human fibrinogen thrombin-binding gamma’ chain sequence. J Thromb Haemost. 2009;7(1):102–110. [DOI] [PubMed] [Google Scholar]
- 42.Walton BL, Getz TM, Bergmeier W, Lin F-C, Uitte de Willige S, Wolberg AS. The fibrinogen γA/γ’ isoform does not promote acute arterial thrombosis in mice. J Thromb Haemost. 2014;12(5):680–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Byrnes JR, Duval C, Wang Y, Hansen CE, Ahn B, Mooberry MJ, Clark MA, Johnsen JM, Lord ST, Lam WA, Meijers JC, Ni H, Ariens RA, Wolberg AS. Factor XIIIa-dependent retention of red blood cells in clots is mediated by fibrin alpha-chain crosslinking. Blood. 2015;126(16):1940–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kattula S, Byrnes JR, Martin SM, Cooley BC, Flick MJ, Wolberg AS. Factor XIII in plasma, but not in platelets, mediates red blood cell retention in clots and venous thrombus size in mice. Blood Adv. 2018;2(1):25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ariëns RA, Philippou H, Nagaswami C, Weisel JW, Lane DA, Grant PJ. The factor XIII V34L polymorphism accelerates thrombin activation of factor XIII and affects cross-linked fibrin structure. Blood. 2000;96(3):988–995. [PubMed] [Google Scholar]
- 46.Kattula S, Bagoly Z, Toth NK, Muszbek L, Wolberg AS. The factor XIII-A Val34Leu polymorphism decreases whole blood clot mass at high fibrinogen concentrations. J Thromb Haemost. 2020;18(4):885–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Byrnes JR, Wolberg AS. Newly-recognized roles of factor XIII in thrombosis. Semin Thromb Hemost. 2016;42(4):445–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luyendyk JP, Schoenecker JG, Flick MJ. The multifaceted role of fibrinogen in tissue injury and inflammation. Blood. 2019;133(6):511–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Joshi N, Kopec AK, Ray JL, Cline-Fedewa H, Nawabi A, Schmitt T, Nault R, Zacharewski TR, Rockwell CE, Flick MJ, Luyendyk JP. Fibrin deposition following bile duct injury limits fibrosis through an alphaMbeta2-dependent mechanism. Blood. 2016;127(22):2751–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kwaan HC, Lindholm PF. Fibrin and fibrinolysis in cancer. Semin Thromb Hemost. 2019;45(4):413–422. [DOI] [PubMed] [Google Scholar]
- 51.Negron O, Weggeman M, Grimbergen J, Clark EG, Abrahams S, Hur WS, Koopman J, Flick MJ. Fibrinogen gamma’ promotes host survival during Staphylococcus aureus septicemia in mice. J Thromb Haemost. 2023;21(8):2277–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nasimuzzaman M, Arumugam PI, Mullins ES, James JM, VandenHeuvel K, Narciso MG, Shaw MA, McGraw S, Aronow BJ, Malik P. Elimination of the fibrinogen integrin alpha(M)beta(2)-binding motif improves renal pathology in mice with sickle cell anemia. Blood Adv. 2019;3(9):1519–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Silva LM, Doyle AD, Greenwell-Wild T, Dutzan N, Tran CL, Abusleme L, Juang LJ, Leung J, Chun EM, Lum AG, Agler CS, Zuazo CE, Sibree M, Jani P, Kram V, Martin D, Moss K, Lionakis MS, Castellino FJ, Kastrup CJ, Flick MJ, Divaris K, Bugge TH, Moutsopoulos NM. Fibrin is a critical regulator of neutrophil effector function at the oral mucosal barrier. Science. 2021;374(6575):eabl5450. [DOI] [PubMed] [Google Scholar]
- 54.Moiseiwitsch N, Zwennes N, Szlam F, Sniecinski R, Brown A. COVID-19 patient fibrinogen produces dense clots with altered polymerization kinetics, partially explained by increased sialic acid. J Thromb Haemost. 2022;20(12):2909–2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bouck EG, Denorme F, Holle LA, Middelton EA, Blair AM, de Laat B, Schiffman JD, Yost CC, Rondina MT, Wolberg AS, Campbell RA. COVID-19 and sepsis are associated with different abnormalities in plasma procoagulant and fibrinolytic activity. Arterioscler Thromb Vasc Biol. 2021;41(1):401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhmurov A, Protopopova AD, Litvinov RI, Zhukov P, Weisel JW, Barsegov V. Atomic structural models of fibrin oligomers. Structure. 2018;26(6):857–868 e854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Juang LJ, Hur WS, Silva LM, Strilchuk AW, Francisco B, Leung J, Robertson MK, Groeneveld DJ, La Prairie B, Chun EM, Cap AP, Luyendyk JP, Palumbo JS, Cullis PR, Bugge TH, Flick MJ, Kastrup CJ. Suppression of fibrin(ogen)-driven pathologies in disease models through controlled knockdown by lipid nanoparticle delivery of siRNA. Blood. 2022;139(9):1302–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hur WS, King KC, Patel YN, Nguyen YV, Wei Z, Yang Y, Juang LJ, Leung J, Kastrup CJ, Wolberg AS, Luyendyk JP, Flick MJ. Elimination of fibrin polymer formation or crosslinking, but not fibrinogen deficiency, is protective against diet-induced obesity and associated pathologies. J Thromb Haemost. 2022;20(12):2873–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]