

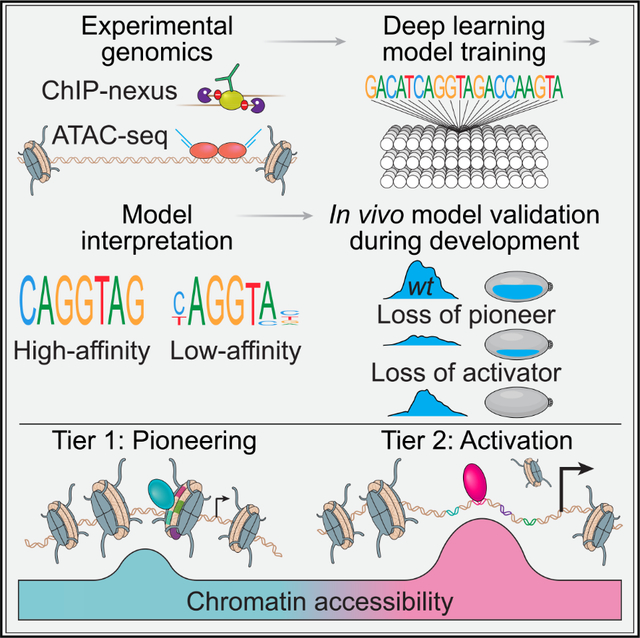
SUMMARY
Chromatin accessibility is integral to the process by which transcription factors (TFs) read out cis-regulatory DNA sequences, but it is difficult to differentiate between TFs that drive accessibility and those that do not. Deep learning models that learn complex sequence rules provide an unprecedented opportunity to dissect this problem. Using zygotic genome activation in Drosophila as a model, we analyzed high-resolution TF binding and chromatin accessibility data with interpretable deep learning and performed genetic validation experiments. We identify a hierarchical relationship between the pioneer TF Zelda and the TFs involved in axis patterning. Zelda consistently pioneers chromatin accessibility proportional to motif affinity, whereas patterning TFs augment chromatin accessibility in sequence contexts where they mediate enhancer activation. We conclude that chromatin accessibility occurs in two tiers: one through pioneering, which makes enhancers accessible but not necessarily active, and the second when the correct combination of TFs leads to enhancer activation.
In brief
Brennan and Weilert et al. combine experimental and computational genomics, deep learning models, and developmental genetics to investigate how transcription factors open chromatin during embryogenesis. They identify which DNA sequences predict chromatin accessibility in Drosophila and reveal that pioneers and activators modulate accessibility according to distinct mechanisms during enhancer activation.
Graphical abstract
INTRODUCTION
Cellular transitions during development are driven by enhancers, cis-regulatory DNA sequences that instruct genes to become expressed at the right time and place. Each enhancer contains a distinct combination and arrangement of sequence recognition motifs for transcription factors (TFs), such that only a specific combination of TFs, present at the right time and place in development, can stimulate activation.1,2 How exactly combinations of TFs read out the cis-regulatory code to mediate enhancer activation is a fundamental question in biology.
An important layer of the cis-regulatory code is chromatin accessibility.3 Chromatin accessibility both informs and is impacted by the binding of TFs and is thus an integral part of the process by which enhancers become activated. Before activation, developmental enhancers are maintained in a state of intrinsically high nucleosome occupancy, such that they are inaccessible to most TFs.4–8 In the first step toward activation, so-called “pioneer” TFs make enhancers accessible. Pioneer TFs are typically expressed early during cellular transitions and can bind their motifs within nucleosomal DNA.9–11 Once accessible, additional TFs may bind to and activate enhancers, leading to the expression of target genes. However, TFs frequently cooperate in modulating accessibility,12–16 making it hard to differentiate between pioneer TFs and non-pioneer TFs and raising the possibility that any TF may function as a pioneer TF.17–19
Distinguishing between motifs of TFs that actively drive chromatin accessibility and those that follow it more passively is computationally challenging. A motif may be statistically over-represented in accessible regions, but whether it facilitates accessibility or contributes to enhancer activation once the region is accessible is not clear. Identifying pioneer TFs experimentally is also challenging. In in vitro experiments, pioneer TFs have an affinity for nucleosomes and tend to be structurally capable of binding their motif on nucleosomal DNA,20–24 but the general rules by which pioneers may read out nucleosomal DNA sequences are unknown.
To distinguish pioneer TFs from non-pioneer TFs, one possibility is to model chromatin accessibility data in a high-resolution and quantitative fashion while taking motif combinations and arrangements into account.19 This approach is very powerful when combined with interpretable convolutional neural networks (CNNs), which can learn complex DNA sequence rules embedded in the cis-regulatory code de novo.25 In this learning paradigm, the CNN learns to predict the experimental data directly from genomic sequences and thus learns motifs in their combinatorial contexts. The rules are general since the performance is evaluated based on a withheld subset of the data that the model does not train on. Once the model accurately predicts these test data, the learned sequence rules are extracted from the model using interpretation tools.26
This approach has been successfully used to predict assay for transposase-accessible chromatin with sequencing (ATAC-seq) data,27–31 revealing TF motifs predicted to contribute to chromatin accessibility in different experimental systems. However, since not all TFs and their binding motifs are known under these conditions, it is difficult to evaluate whether the discovered motifs belong to known TFs with characterized properties.32 Likewise, the models can predict synergistic effects between TF motifs,29,30 but the exact rules and the underlying mechanisms are not known. This makes it very challenging to connect the rules extracted from deep learning models with known biology.
To better leverage this approach, we decided to learn both TF binding data and chromatin accessibility data in the early Drosophila embryo, a well-studied model system with a wide range of data from genetics, biochemistry, and imaging experiments. Studying early embryogenesis has the advantage that chromatin accessibility is established de novo as the zygotic genome is activated, and the first gene expression programs are established along the anteroposterior and dorsoventral axes.33–35 Although the emerging heterogeneity of the cells could make it more challenging for the CNN to learn the sequence rules from bulk data, it is easy to test and validate the learned rules because of the available mutants and detailed knowledge of TFs and enhancers.36
The major driver of Drosophila zygotic genome activation is the maternally provided zinc-finger TF Zelda.37 Within one hour into development, or by the embryo’s eighth nuclear cycle, Zelda binds the majority of its motifs genome-wide, which are highly enriched among developmental enhancers.38–40 At these regions, Zelda binding is required for nucleosome depletion and chromatin accessibility6,41,42 and facilitates the binding of patterning TFs, including the binding of the dorsoventral patterning TFs Dorsal43,44 and Twist,45 and the anteroposterior patterning TFs Bicoid46–48 and Caudal.5 Furthermore, in vitro experiments suggest that Zelda can bind to nucleosomes.20,49 Taken together, Zelda has the characteristics of a pioneer TF.
Although Zelda is well studied, whether it cooperates with other early-acting TFs in the embryo to induce chromatin accessibility is not known. GAGA factor (GAF) and CLAMP are additional pioneers important for zygotic genome activation, but whether they synergize with Zelda is not clear. They regulate largely distinct sets of regions from Zelda and tend to be more promoter-specific.50–54 Patterning TFs, on the other hand, strongly overlap in binding with Zelda, but it is unknown whether they cooperate with Zelda and can function as pioneer TFs.38–40,55,56 Bicoid has been reported to play a pioneering role in a subset of regions,57 but the underlying sequence rules have not been characterized. Likewise, whether other patterning TFs can increase accessibility is unknown.
To learn DNA sequence rules at the highest possible resolution in the early Drosophila embryo, we used ChIP-nexus, a chromatin immunoprecipitation technique that maps genome-wide TF binding footprints at base-resolution by virtue of a strand-specific exonuclease,58 and employed a CNN called BPNet, which directly predicts these data at base-resolution, allowing it to learn precise rules of TF cooperativity in vivo.59 We then generated time course chromatin accessibility measurements and applied a modified BPNet model, ChromBPNet,29 to predict ATAC-seq data bias-free at base-resolution. This allowed us to leverage the same CNN approach for both data types in a system where we could validate the learned rules experimentally. We identified a clear directional relationship in binding between Zelda and the patterning TFs and found that Zelda and the patterning TFs both increase accessibility but through distinct modes. Although Zelda acts as a bona fide pioneer TF, even at low-affinity motifs, the patterning TFs increase accessibility through transactivation. These results show that chromatin accessibility during zygotic genome activation follows complex sequence rules and is driven both by pioneers and transcriptional activators in distinct steps.
RESULTS
Neural networks predict Zelda’s role in helping other TFs bind
To determine the binding and cooperativity of TFs in the early embryo, we performed high-quality ChIP-nexus experiments on staged embryos (Figure 1A). We chose the two best-known pioneers, Zelda and GAF, the main dorsoventral patterning TFs Dorsal and Twist, and the main anteroposterior patterning TFs Bicoid and Caudal. We then trained a BPNet model to predict ChIP-nexus data from DNA sequence and interpreted the sequence rules as previously described.59 This approach models cis-regulatory sequences in their native genomic contexts and learns TF binding in an inherently combinatorial motif space. Motifs are learned de novo, and the genomic instances to which they match are defined not only by a sequence match but also by a contribution score toward the binding predictions. To maximize the accuracy of the model’s learned sequence rules, we optimized the model to achieve high prediction accuracy and confirmed the results through cross-validation (Figures S2A–S2C).
Figure 1. BPNet predicts a hierarchical relationship between Zelda and patterning TFs in the early Drosophila embryo.
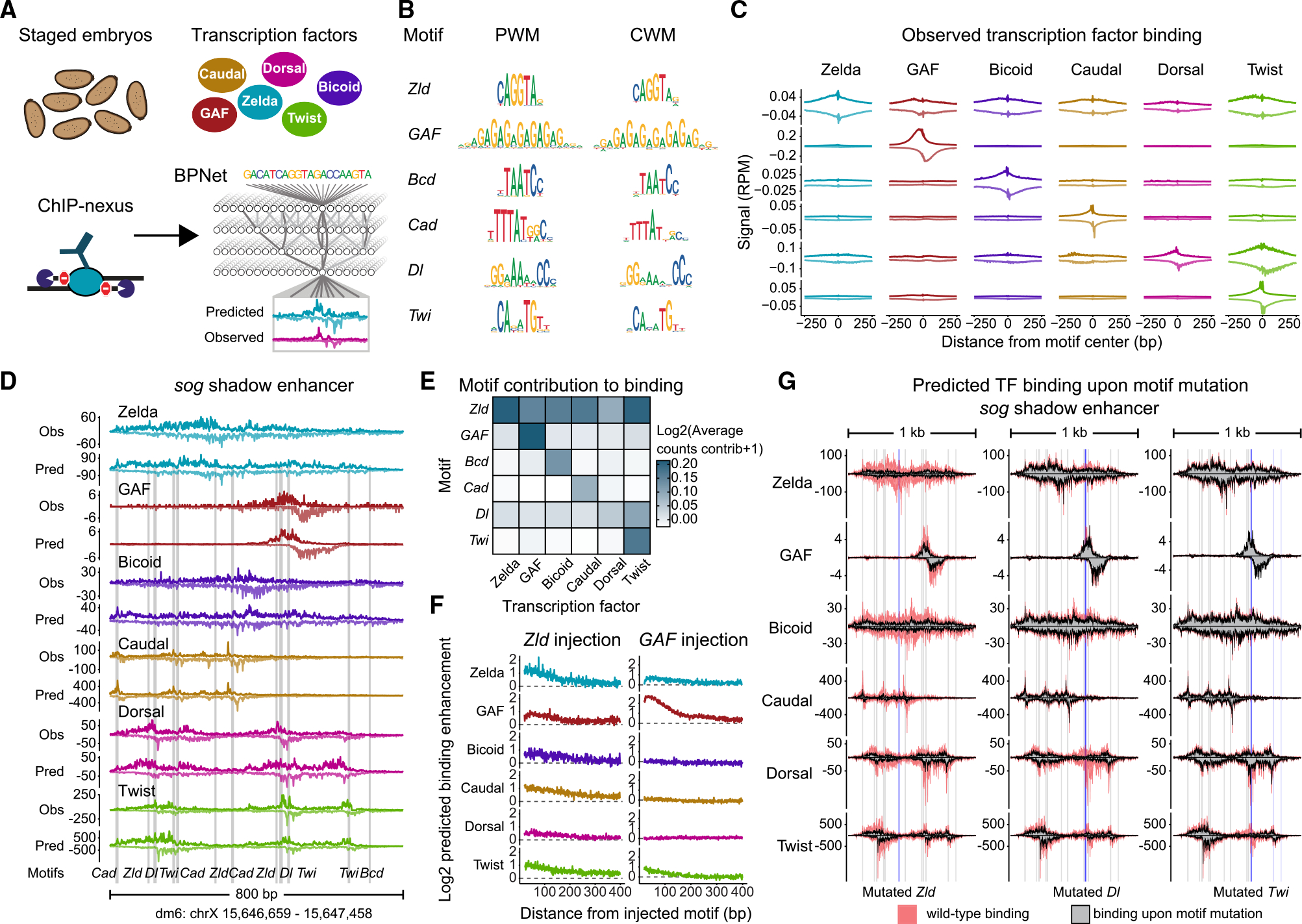
(A) ChIP-nexus produced high-resolution, strand-specific binding of Zelda (Zld), GAGA factor (GAF), Bicoid (Bcd), Caudal (Cad), Dorsal (Dl), and Twist (Twi) in stage 5 embryos. A multi-task BPNet model was trained to predict TF binding from DNA sequence. See also Figures S1A and S2A–S2B.
(B) Identified motifs are shown as a frequency-based position weight matrix (PWM) and as a contribution weight matrix (CWM), which are highly similar for all TFs. See also Figure S2C.
(C) Average ChIP-nexus TF binding footprints show that motifs directly bound by a TF have sharp footprints. Strand-specific data (+ strand on top; − strand at bottom) in reads per million (RPM) were averaged centered on each motif.
(D) BPNet’s predictive accuracy illustrated at the sog shadow enhancer, which was withheld during training. Observed (Obs) ChIP-nexus data are shown above the BPNet-predicted (Pred) data. Motifs contributing to the predictions are found below. Additional enhancers are provided in Figure S3.
(E) The average counts contribution score for all mapped motifs toward the binding of each TF reveals that the Zelda motif contributes to the binding of all TFs, but not vice versa, indicating a hierarchical relationship. Darker colors indicate that a motif (y axis) has a higher contribution score (shown on log scale) to the binding of a TF (x axis).
(F) In silico injections of motifs into randomized sequences confirm that the Zelda motif is predicted to boost the binding of all TFs, while the GAF motif boosts only GAF’s binding. TF binding was predicted by BPNet when each motif was alone and when a Zelda motif (left), or a GAF motif (right), was injected at a given distance, up to 400 bp away (x axis). The average fold-change binding enhancement in the presence of Zelda/GAF is shown on the y axis.
(G) When mutating a Zelda motif in the sog shadow enhancer, BPNet predicts reduced binding of all TFs, while mutating a Dorsal motif has a smaller but notable effect. Predicted binding at the wild-type sequence (red) is overlaid with the predicted binding when individual motifs are computationally mutated (gray). Blue bars highlight the mutated motifs; gray bars are all other mapped motifs. See also Figure S3.
The discovered motifs included all known motifs for the BPNet-modeled TFs (Figure 1B), represented either as a frequency-based position weight matrix (PWM) or as the novel contribution weight matrix (CWM), which is the model’s extracted contribution of each base for TF binding (motif instances provided in Data S1). As expected, these motifs showed sharp ChIP-nexus binding footprints by the corresponding TFs, indicating direct TF-DNA interactions (Figure 1C). We manually inspected well-studied enhancers to ensure that the ChIP-nexus predictions matched the experimental data and that experimentally validated motifs were mapped accurately (Figures 1D and S3). For example, the neuroectodermal sog shadow enhancer had the expected motifs for Zelda, Dorsal, Twist, and Bicoid.43,44,60–65 This enhancer is part of a withheld region set that was never seen by the model during training, highlighting how the model correctly predicts TF binding from DNA sequence alone (Figure 1D).
We then extracted the rules of TF cooperativity from the model. We first measured the average contribution of each motif toward the binding of each TF (Figure 1E). As expected, all motifs strongly contributed toward their own TF’s binding, but some motifs also contributed to the binding of other TFs, suggesting cooperativity between TFs. Most prominently, the Zelda motif is predicted to be important for the binding of all other TFs (Figure 1E), including Bicoid, Caudal, Dorsal, and Twist, which have been shown in previous genetic experiments to depend on Zelda.5,6,41,43,45,46 Additionally, BPNet predicts that Twist binding depends on the Dorsal motif. Dorsal and Twist have previously been reported to cooperate,61,66–69 but our result suggests that this cooperativity is directional, i.e., the Dorsal motif is more important for Twist binding than the Twist motif is important for Dorsal binding. This is also reflected in the experimental ChIP-nexus data, which show Twist accumulation over the Dorsal motif but not vice versa (Figure 1C). Interestingly, the motif for GAF did not strongly contribute to the binding of TFs other than GAF itself, although GAF is known to promote chromatin accessibility.50,53,54,70,71
To internally validate that BPNet learned different rules of cooperativity for Zelda and GAF, we used the trained model to predict TF binding when motif pairs are injected into randomized sequences (Figure 1F). For each TF, we measured the in silico binding enhancement when the motif is flanked by a Zelda or GAF motif at a given distance (up to 400 bp). Consistent with our initial results, injecting a Zelda motif generally boosted the binding of all TFs, whereas the GAF motif only had a strong boosting effect on another GAF motif (Figure 1F). Notably, all observed cooperativity occurred when the motifs were spaced within nucleosome-range distances, consistent with an effect on nucleosomes.
To test whether these rules also apply to the enhancers critical for embryonic patterning, we computationally mutated the sequence of TF motifs at the well-known enhancers and predicted the effects on TF binding using BPNet (Figures 1G and S3). As expected, mutating Zelda motifs consistently had a strong effect on the binding of other TFs, in agreement with experimental evidence.5,6,38,43,45,46 In contrast, the effects of mutating patterning TF motifs were more enhancer-specific. At the dpp enhancer, mutating Dorsal motifs affected Dorsal and Twist binding, as expected (Figure S3A, right). However, at the sog shadow enhancer, mutating a Dorsal motif also had an effect on the binding of other TFs (Figure 1G). These results suggest more complex rules at some enhancers and raise the question of whether chromatin accessibility plays a role in the observed cooperativity.
The sequence rules for chromatin accessibility reveal motif-driven pioneer TFs
To understand the relationship between TF binding and chromatin accessibility, we performed ATAC-seq experiments72,73 in a developmental time course of four 30-min intervals during the maternal-to-zygotic transition. This allowed us to measure how enhancers transition from a naturally closed state within a homogeneous cell population in the embryo to a more accessible, primed, or active state during pattern formation.51,74–78 The first embryo collection (1–1.5 h after egg laying [AEL]) covers the time when Zelda binds throughout the genome in the eighth nuclear cycle.39 In later stages, zygotic transcription begins, and the patterning TFs become active.35,79,80 In agreement with previous studies, we find that genome-wide chromatin accessibility increases over the four time points51 (Figure 2A).
Figure 2. ChromBPNet reveals distinct contributions from pioneers and patterning TFs in early Drosophila embryos.
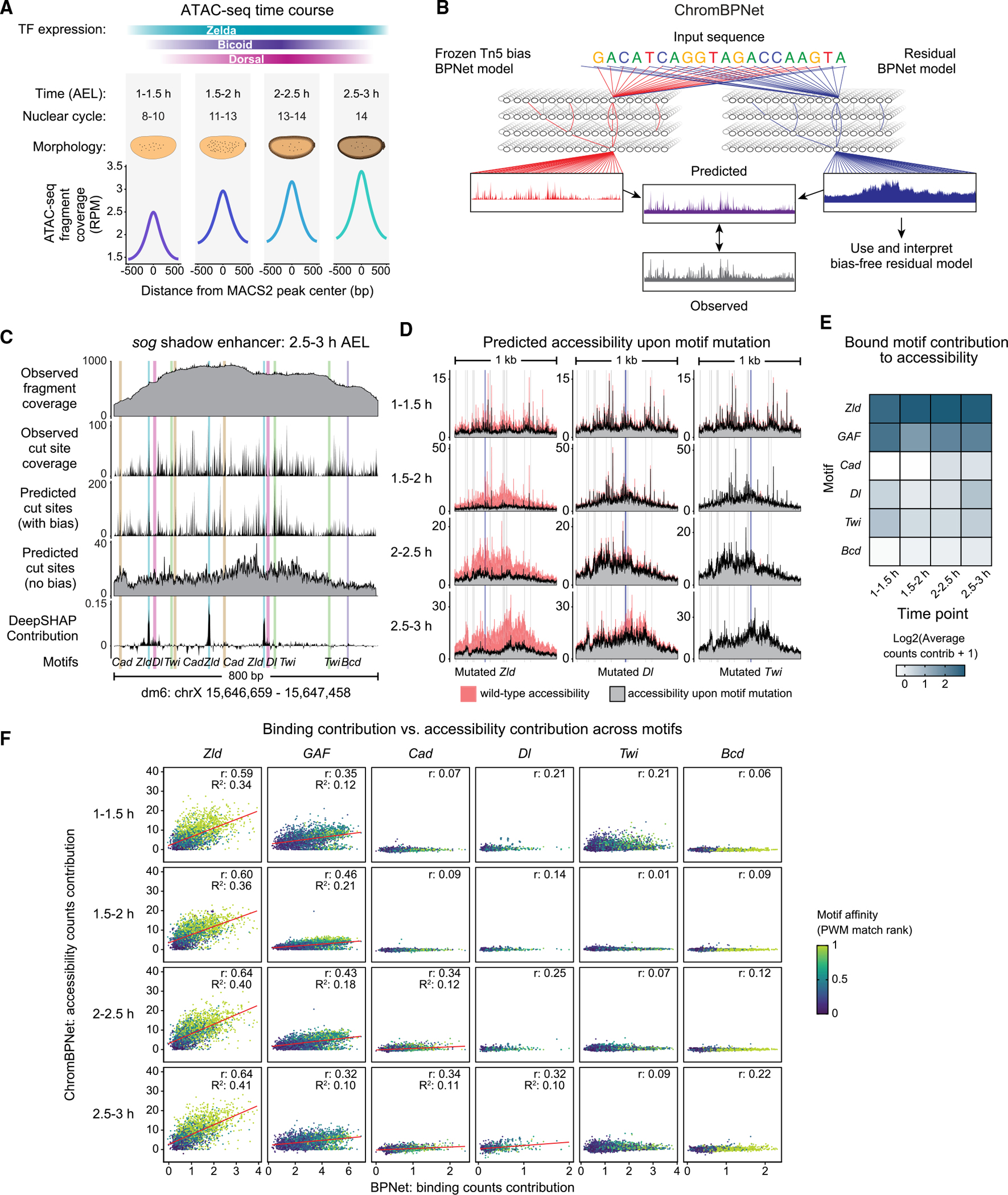
(A) ATAC-seq experiments were performed in four 30-min windows on hand-sorted embryos. See also Figure S1B.
(B) ChromBPNet predicts bias-free chromatin accessibility at base-resolution. A bias model is first trained on ATAC-seq data at closed genomic regions to learn baseline Tn5 sequence bias, then frozen and used for training alongside a second, residual BPNet model on open ATAC-seq regions. When the bias model is removed, the residual model predicts the bias-removed ATAC-seq data. See also Figures S2D–S2H.
(C) ChromBPNet accurately predicts accessibility at the sog shadow enhancer (2.5–3 h data). Experimentally generated ATAC-seq data are shown as conventional fragment coverage (first track) and Tn5 cut site coverage (second track), which closely mirrors ChromBPNet’s prediction from the combined model (third track). After removing the bias model, ChromBPNet’s predicted profile is more evenly distributed (fourth track). The counts contribution scores for each base across the enhancer (fifth track) shows spikes at BPNet-mapped motifs. Additional enhancers provided in Figures S4A–S4D.
(D) ChromBPNet predicts the effect of mutating a Zelda (left), Dorsal (middle), and Twist (right) motif at the sog shadow enhancer for each time point (same motifs as in Figure 1G). Mutating the Zelda motif had the largest effect on chromatin accessibility, while the Dorsal motif mutation lowered accessibility to a lesser extent and only at later time points. See also Figures S4E–S4H.
(E) Average counts contribution scores for each BPNet-mapped motif (y axis) for all time points (x axis) show that pioneering motifs contribute to chromatin accessibility at all time points, whereas patterning TF motifs have a lesser contribution that is limited to later time points. See also Figure S2I–S2K.
(F) Pioneer TF motifs show a three-way correlation between binding contribution, accessibility contribution, and motif strength. Patterning TFs show much weaker, time point-specific relationships, suggesting context-dependent behavior. For each bound and accessible motif for all TFs, the binding counts contribution scores (x axis) and accessibility counts contribution scores (y axis) are plotted. The motif strength (color scale) represents the rank percentile of the PWM match scores. Pearson correlation values (r) and coefficient of determination R2 values were calculated. Red lines are shown for plots with an r > 0.3.
Chromatin accessibility is generated by multiple TFs and could differ in different parts of the embryo. To precisely learn the cis-regulatory sequence rules underlying these complex data, we adapted ChromBPNet, a variation of BPNet that predicts ATAC-seq data at the highest resolution.29 Rather than training on whole fragment coverage, the model predicts the cut sites made by the Tn5 transposase, which more accurately represent accessibility measured by ATAC-seq (Figure 2B). Since Tn5 transposase possesses a strong sequence preference in its cut sites,81,82 ChromBPNet first explicitly learns the Tn5 bias rules by training on closed genomic regions (i.e., with low counts and non-peak ATAC-seq signals) (Figure 2B). In a second training step alongside the now-frozen bias model, an additional BPNet model learns the residual sequence rules of the ATAC-seq accessible regions beyond the Tn5 bias (Figures S2D and S2E). After the second training step, the bias model is removed, and the residual model is interpreted to extract the biologically relevant sequence rules that predict chromatin accessibility.
We trained separate ChromBPNet models for each of the ATAC-seq time points, omitting regions with annotated promoters to ensure that the sequence rules learned were specific for enhancers and not strongly driven by core promoter motifs. As with BPNet, we computed performance metrics, conducted hyperparameter tuning, and trained cross-validation models to ensure that model training was successful (Figures S2F–S2H).
To visually inspect ChromBPNet’s predictions, we used the sog shadow enhancer as an example (Figure 2C; additional enhancers in Figure S4). The observed cut site coverage from the original ATAC-seq data closely matched the combined model’s prediction (Figure 2C), consistent with the high-performance metrics (Figures S2F–S2H). When using only the residual model, the predicted chromatin accessibility was more evenly distributed over the entire enhancer, suggesting that the Tn5 cut site bias was successfully removed (Figure 2C).
As with BPNet, we extracted base-resolution contribution scores for all sequences and summarized the de novo-learned motifs. The motifs for Zelda and GAF were robustly rediscovered at all time points, consistent with them being pioneer TFs that open chromatin (Figure S2I). The motifs for the patterning TFs were, however, not as clear-cut. We discovered Caudal-like, Dorsal-like, and Twist-like motifs, which deviated from those learned by the TF binding model but nevertheless showed the expected ChIP-nexus binding footprints, confirming their identity (Figure S2I). It did not return the Bicoid motif despite previous evidence that Bicoid plays a role in chromatin accessibility.57 This points to limitations either in the sequence rules learned by the model or in our ability to extract the rules. For example, multiple TFs often compete for binding to similar motifs, including Bicoid,83,84 which could make it difficult to correctly discover and aggregate motifs for individual TFs.
To evaluate how well the sequence rules were learned, we first inspected the contribution scores at known enhancers. Although Zelda motifs consistently stood out with high scores, the motifs for the patterning TFs showed a much smaller contribution and only in some instances (Figures 2C and S4, top). This nevertheless confirmed that the motifs were learned and suggested that the Bicoid motif may also weakly contribute to chromatin accessibility in context-specific instances (Figures S2K and S4). In silico mutagenesis confirmed these results (Figures 2D and S4, bottom). Mutating a Zelda motif in the sog shadow enhancer strongly reduced the predicted accessibility for all time points, but mutating a Dorsal motif also weakly reduced the predicted accessibility (Figure 2D), especially at the later time points when patterning TFs bind most strongly.5,79 Likewise, mutating the Bicoid motif weakly decreased chromatin accessibility at the Kr enhancer (Figure S4H). Taken together, the interpretations suggest that patterning TFs contribute to accessibility in a manner consistent with the TF binding model and previous knowledge.
We next systematically compared the rules of binding with those of accessibility. We selected regions that are accessible and contain TF motifs mapped by the binding model, which ensures that the motifs are of high quality and unambiguously mapped to the TF through a direct sequence-to-binding relationship. We confirmed that the Zelda and GAF motif instances had a high contribution to accessibility at all time points, whereas those of the patterning TFs had a much smaller contribution (Figure 2E). Similar effect sizes were predicted when each TF motif was injected into randomized sequences (Figure S2J). Using these mapped motif instances, we then plotted the predicted contribution to accessibility as a function of the predicted binding contribution (Figure 2F).
Strikingly, we observed a strong correlation for both Zelda and GAF motifs between accessibility and binding contributions (Pearson correlations for Zelda 0.59–0.64), despite being learned by different models on different types of data (Figure 2F). When we derive a simple score for motif strength (rank percentile of the PWM match scores), we see that with increasing motif strength, binding and accessibility contributions also increase. This three-way association suggests that the accessibility generated by Zelda and GAF is motif-driven and not strongly reliant on the surrounding enhancer context, which agrees with the conventional model that pioneer TFs come first and mediate the initial step in enhancer activation.
In contrast, the same plots for the patterning TFs show weaker correlations between TF binding and chromatin accessibility (Figure 2F). Here, stronger measures of motif strength are associated with stronger binding contributions but not accessibility contributions. One exception is Dorsal, where the binding and accessibility contributions correlate more highly at the last time point (with a Pearson correlation value of 0.32) and show an association with motif strength. Taken together, our binding and accessibility models suggest an operational definition of pioneer TFs in which pioneer TFs open chromatin in a motif-driven fashion, whereas other TFs may also play a role in increasing chromatin accessibility but do so in a weaker and more context-dependent manner.
Zelda’s effect on opening chromatin depends on motif affinity
The correlation between motif strength, TF binding, and chromatin accessibility suggests that pioneer TFs read out motif affinities. This is surprising since the thermodynamic differences between high and low-affinity sequences are very small at approximately −3 kcal/mol,85 and pioneering is expected to occur through TF binding on nucleosomes, where sequence recognition is structurally more constrained than on naked DNA.10,20,21,23,24,86–88 Furthermore, this suggests that ChromBPNet learned relative motif affinities quite accurately despite being trained on data with complex sequence rules. This would be consistent with previous studies showing that relative motif affinities can be extracted from CNN models.89–91
To validate that our models learned motif affinities for Zelda, we first took all bound Zelda motifs mapped by BPNet and plotted their sequences ordered by contribution to Zelda binding (Figure 3A). The motif that contributed most to binding was the canonical CAGGTAG motif, whereas low-affinity binding motifs included motifs where the last base was not a G (CAGGTAH) or the first base was a T (TAGGTAG). These results are consistent with the Zelda motif affinities determined previously by gel shift studies and mutant data37–39,92,93 and correlate with the observed chromatin accessibility across these motifs (Figure S5A).
Figure 3. The pioneer TF Zelda reads out motif affinity to drive chromatin accessibility.
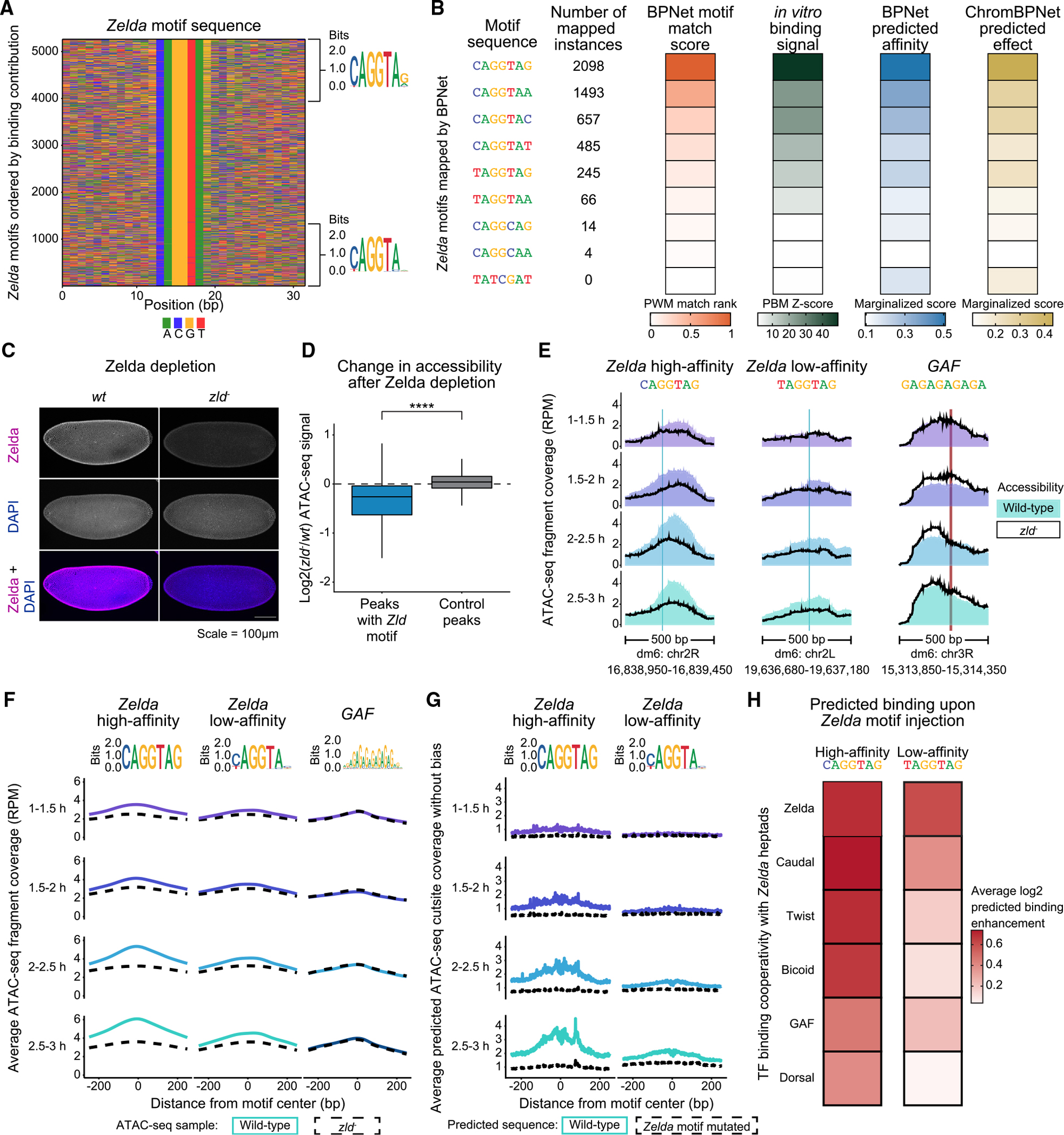
(A) The Zelda-binding contributions from the BPNet model reflect the known Zelda motif affinities. Zelda motif sequences, ordered by their counts contribution scores to Zelda binding, are shown from high (top) to low (bottom). Motif logos for the highest and lowest quartiles mainly differ in the first and last base of the 7-mer sequence. See also Figure S5A.
(B) The model-derived motif strengths strongly correlate with experimentally measured Zelda motif affinities. Shown for all mapped Zelda motif 7-mer sequences and a negative control (TATCGAT) are: the rank percentile of their PWM match scores (orange), the median Z scores from Zelda protein-binding microarray (PBM) experiments (green), and the marginalized effects predicted by the trained BPNet (blue) and ChromBPNet (gold). See also Figure S5B.
(C) Confocal images of stage 5 embryos show strong Zelda protein depletion in zld− versus wt embryos.
(D) Chromatin accessibility is significantly reduced at ATAC-seq peaks containing mapped Zelda motifs. Using DESeq2, the log2-fold changes between wt and zld− embryos were calculated for each peak region over time, and the median values among the four time points were plotted. Peaks containing Zelda motifs are significantly different from control peaks without Zelda motifs (Wilcoxon rank-sum test, p < 2e−16). See also Figures S1C and S5C.
(E) Zelda motif strength determines the reduction in chromatin accessibility in zld− embryos. Individual examples of normalized accessibility in wt (shaded profile) and zld− (black line) embryos are shown at a high-affinity Zelda motif (CAGGTAG, left) and a low-affinity Zelda motif (TAGGTAG, middle), with the GAF motif (right) as a control. No other BPNet-mapped motifs are found within these regions.
(F) Average chromatin accessibility profiles for wt and zld− embryos show that high- and low-affinity motifs both facilitate Zelda’s pioneering, but low-affinity motifs do so to a lesser extent. Among regions that only contain a single Zelda motif, those with the 250 highest- and 250 lowest-affinity motifs were selected (summarized as motif logos). GAF motifs were used as control. Anchored on these Zelda motifs, the average profiles of normalized ATAC-seq data are shown for wt (colored lines) and zld− embryos (dotted black lines). Motifs mapping to promoters were excluded, as in ChromBPNet training. See also Figures S5D–S5E.
(G) Average ChromBPNet-predicted chromatin accessibility (bias-corrected cut site coverage) at the same high- and low-affinity Zelda motif regions for the wt sequences and after computationally mutating the Zelda motifs. The results confirm that ChromBPNet has learned the effects of Zelda motif affinity.
(H) BPNet has also learned that low-affinity Zelda motifs boost TF binding less than high-affinity motifs. TF motifs were injected into randomized sequences with either a high-affinity Zelda motif (CAGGTAG) or a low-affinity Zelda motif (TAGGTAG) at a given distance away for up to 200 bp, and the average TF binding enhancement over no added Zelda was predicted (y axis). See also Figures S5G–S5H.
To more comprehensively test how well relative Zelda motif affinities were learned, we performed in vitro protein-binding microarray (PBM) experiments94,95 for Zelda (Figure 3B). PBM-extracted affinities have been shown to correlate with Kd affinity measurements.91,96–98 We calculated the median Z score of the binding signal and its corresponding median E score for all relevant Zelda motif 7-mers, as well as a negative control sequence (TATCGAT) used previously in gel shift experiments.38 Strikingly, the simple BPNet-derived motif strength scores used earlier closely matched the PBM data (Figures 3B and S5B). For example, both the PBM and BPNet-derived motif strength scores show on average a 3-fold difference in affinity between the CAGGTAG and TAGGTAG sequences.
Relative motif affinities for genomic motif instances can also be extracted from models without deriving their motif representations first. This is done by predicting TF binding on individual motif instances stripped from the surrounding genomic context.89–91 To test this approach, we “marginalized” each Zelda motif by injecting it into randomized sequences and measuring the effects on binding and chromatin accessibility. The log-transformed measurements were very similar to our previous BPNet-derived motif strength scores and closely matched with the PBM-binding Z scores (Figure 3B). These results collectively confirm that the BPNet and ChromBPNet models have accurately learned relative Zelda-binding affinities.
We next performed experiments on Zelda-depleted embryos6 to test whether the pioneering effect of Zelda depends on motif affinity. We confirmed that the zld− embryos had no detectable Zelda by immunostaining (Figure 3C) and performed ATAC-seq time course experiments. Consistent with previous observations,6,41,57 Zelda-bound regions showed a global decrease in accessibility in zld− embryos compared with wild type, whereas regions without a Zelda motif remained unchanged (Figures 3D and S5C).
We then asked whether individual low-affinity Zelda motifs by themselves influence chromatin accessibility. We selected regions with either a single high-affinity (CAGGTAG) or a single low-affinity (TAGGTAG) Zelda motif and no other BPNet-mapped motif nearby. At regions with the high-affinity Zelda motif, a clear reduction in chromatin accessibility was observed in zld− embryos (Figure 3E, left). At regions with the low-affinity TAGGTAG motifs, we observed the same effect but weaker (Figure 3E, middle). To quantify this difference, we selected the genomic regions with the 250 highest- and lowest-affinity Zelda motifs. To minimize confounding effects, these regions had no other mapped motifs nearby and did not overlap promoters. As expected, the regions with the high-affinity Zelda motifs had more Zelda binding in the ChIP-nexus data than those with the low-affinity motifs (Figure S5D). Using these regions, we found that the low-affinity Zelda motifs had on average a 5-fold weaker effect on chromatin accessibility than the high-affinity Zld motifs, although control regions with a single GAF motif were unchanged (Figures 3F and S5E). These differences were strikingly similar to those predicted by ChromBPNet upon mutating the Zelda motifs (Figure 3G). These results demonstrate that low-affinity Zelda motifs can promote accessibility, but to a lesser extent than high-affinity CAGGTAG motifs.
Since low-affinity Zelda motifs have a smaller effect on chromatin accessibility, we expected them to also have a weaker effect on promoting the binding of patterning TFs. To test this, we performed in silico motif injections and measured the average predicted binding of each TF with and without the presence of different Zelda motif variants. For all TFs, the resulting fold-change binding enhancement was indeed higher for the high-affinity CAGGTAG motif than for the low-affinity TAGGTAG motif, but the latter still had a measurable effect (Figure 3H). Likewise, ChromBPNet predicted that both high- and low-affinity Zelda motifs boosted the effect of patterning TF motifs on chromatin accessibility, but to a different extent (Figures S5G and S5H). These effects corroborate the role of Zelda’s motif affinity in opening chromatin and helping patterning TFs bind.
Patterning TFs contribute to chromatin accessibility
Thus far, the results suggest that patterning TFs do not have the same pioneering capabilities as Zelda but could increase chromatin accessibility in some contexts, perhaps depending on which other motifs are present within that region. To systematically investigate motif combinations, we used a “motif island” approach in which genomic regions are grouped according to their motif combinations (Figure 4A). An island is initially defined as a 200-bp region centered on a TF-bound motif, but if there is an overlap with other islands, the islands get merged and classified by their motif combinations (islands provided in Data S2). Most of these multi-motif islands are between 200 and 300 bp wide and thus are the size of typical enhancers99 (Table S1).
Figure 4. Patterning TFs increase chromatin accessibility in a context-dependent manner.
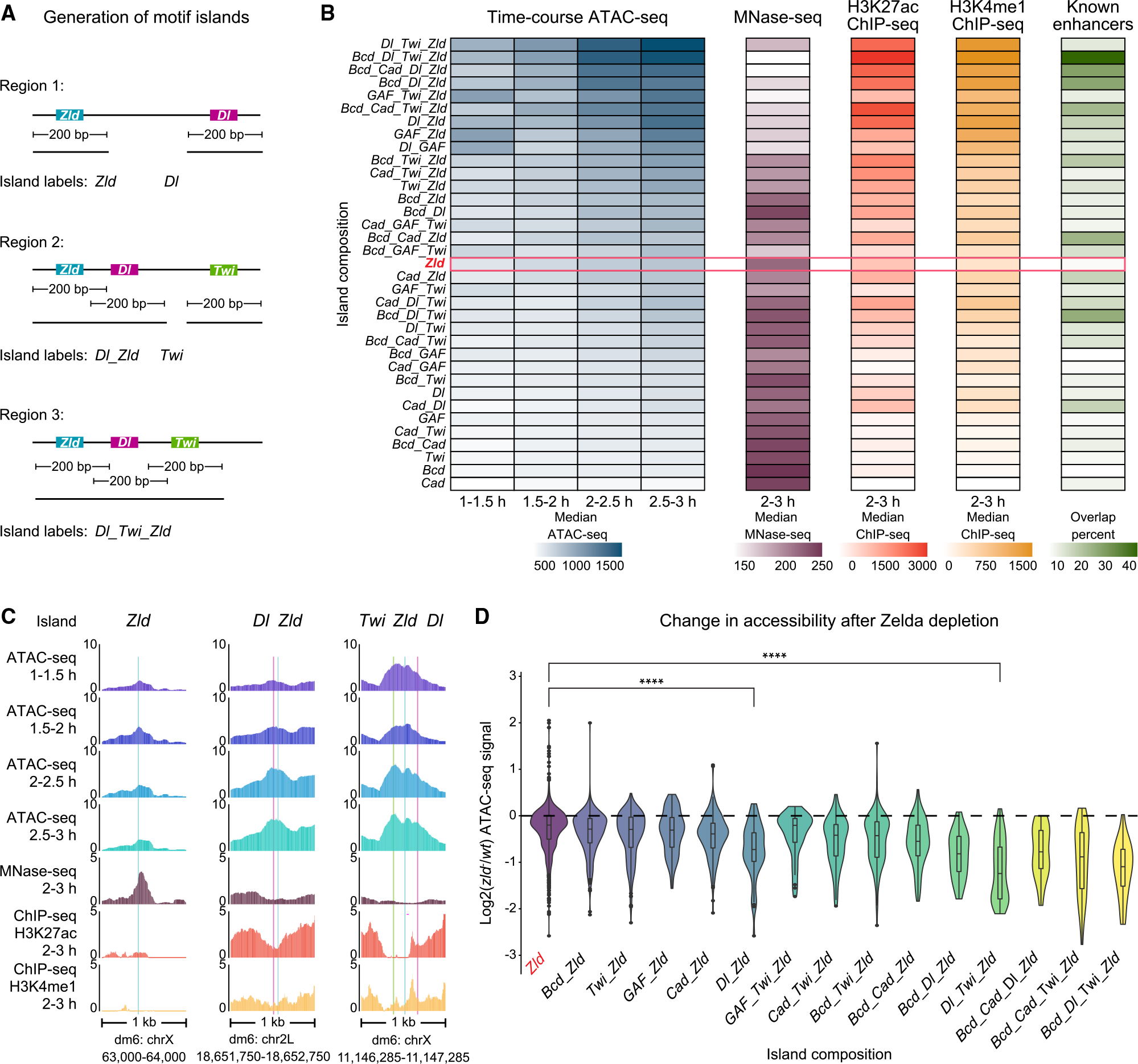
(A) Schematic summary of motif islands. Motif islands are generated by first resizing all BPNet-mapped and bound motifs to 200 bp wide. Next, overlapping regions are merged and classified based on the motifs that compose them. See also Table S1.
(B) Islands with combinations of Zelda and patterning TF motifs contain the highest chromatin accessibility, nucleosome depletion, active enhancer histone modifications, and known enhancer overlap. For each motif island type with a specific motif composition (y axis), the median normalized ATAC-seq fragment coverage, MNase-seq signal, H3K27ac ChIP-seq signal, H3K4me1 ChIP-seq signal and the overlap with enhancers active in 2–4 h AEL74 embryos are shown via the color scale. The red bar highlights islands that contain only Zelda motifs, and islands are ordered by total ATAC-seq signal. See also Figures S1D–S1E and S5F.
(C) Individual island examples, where colored bars indicate BPNet-mapped motifs (blue = Zld, magenta = Dl, green = Twi).
(D) Chromatin accessibility is most strongly reduced in zld− embryos at islands containing Zelda and patterning TF motifs. Using DESeq2, log2-fold changes in ATAC-seq signal between wt and zld− embryos were calculated for each island, and their median changes across the time points are shown. Islands that contain patterning TF motifs in addition to Zelda motifs show significantly more changes than those with Zelda motifs only, e.g., the difference between Zld and Dl_Zld islands (p = 8.3e−11, Wilcoxon rank-sum test) and Zld and Dl_Twi_Zld islands (p < 2.22e−16, Wilcoxon rank-sum test).
To better characterize the enhancer states for different motif combinations, we used staged embryos and performed micrococcal nuclease digestion with sequencing (MNase-seq) and ChIP-seq experiments for the histone modifications H3K27ac and H3K4me1. We then analyzed the properties of each island combination and calculated their overlap with a curated list of enhancers that have been identified as being active in the early embryo74 (Figure 4B; individual examples in Figure 4C).
The results are not only consistent with Zelda’s role in pioneering but also reveal the role of patterning TFs. Islands without a Zelda motif typically have very low accessibility and histone modifications, coupled with higher nucleosome occupancy. Islands that only have Zelda motifs and no other motif (Figure 4B, red box) show an increase in chromatin accessibility over time, with an effect proportional to the number of Zelda motifs (Figure S5F). Nevertheless, these regions overall show a modest effect on chromatin accessibility, have low levels of histone modifications, and are not enriched for active enhancers.74 By contrast, the highest levels of enhancer accessibility and activity are found at islands that also have motifs for patterning TFs. Islands containing motifs for both Zelda and patterning TFs show much higher levels of accessibility, nucleosome depletion, and histone modifications than Zelda-only islands. Taken together, these results suggest that it is the combination of Zelda motifs and patterning TF motifs that generates the highest levels of accessibility, which would explain why it has been challenging to causally link individual TFs such as Bicoid to increased levels of chromatin accessibility beyond those generated by pioneer TFs.57
To detect the effect of patterning TFs on chromatin accessibility experimentally, we used our zld− ATAC-seq data. Upon Zelda depletion, the patterning TFs are still expressed38,43,46 but show strongly reduced binding to the genome.6,46 If the patterning TFs contribute to chromatin accessibility, then their effect should also be lost in zld− embryos, in addition to the loss of accessibility mediated by Zelda. Indeed, we found that depleting Zelda had a stronger effect on regions with motifs for both Zelda and patterning TFs compared with those with only Zelda motifs (Figure 4D). For example, islands with Zelda, Dorsal, and Twist motifs had a much more pronounced fold-change loss in accessibility than Zelda-only islands. These experimental results support the model in which high levels of chromatin accessibility are established in a hierarchical manner by a combination of motifs for the pioneer Zelda and downstream patterning TFs.
Patterning TFs contribute to accessibility when mediating activation
Our results suggest that patterning TFs increase chromatin accessibility when their motifs are present in specific combinations that include Zelda motifs. Enhancers with such motif combinations also tend to be active enhancers, raising the question of whether enhancer activity and accessibility are directly functionally coupled. This would be consistent with previous observations that the highest levels of accessibility and TF binding are often found at active enhancers.74–76,78,100,101 Alternatively, it is possible that the binding of patterning TFs also consistently contributes to the accessibility, but that their dependence on Zelda motifs for binding creates the requirement for motif combinations. To distinguish between these possibilities, we focused on Dorsal since this allowed us to leverage available mutants and extensive existing knowledge on bona fide Dorsal target enhancers that mediate transcriptional activation.
Dorsal is present in the early embryo as a ventral-to-dorsal nuclear concentration gradient. At high levels of nuclear Dorsal, the nuclei acquire mesodermal identity; at low levels of Dorsal, they acquire neuroectodermal identity; and in the absence of Dorsal, they acquire dorsal ectodermal identity61 (Figure 5A). The key to Dorsal’s ability to specify three tissue types is its ability to function as a dual TF that can activate mesoderm and neuroectoderm genes and repress dorsal ectoderm genes. This switch in function is possible because the repressed enhancers have Dorsal motifs that are flanked by low-affinity motifs for the repressor Capicua (Cic).102–105
Figure 5. Patterning transcription factors increase chromatin accessibility through transcriptional activation.
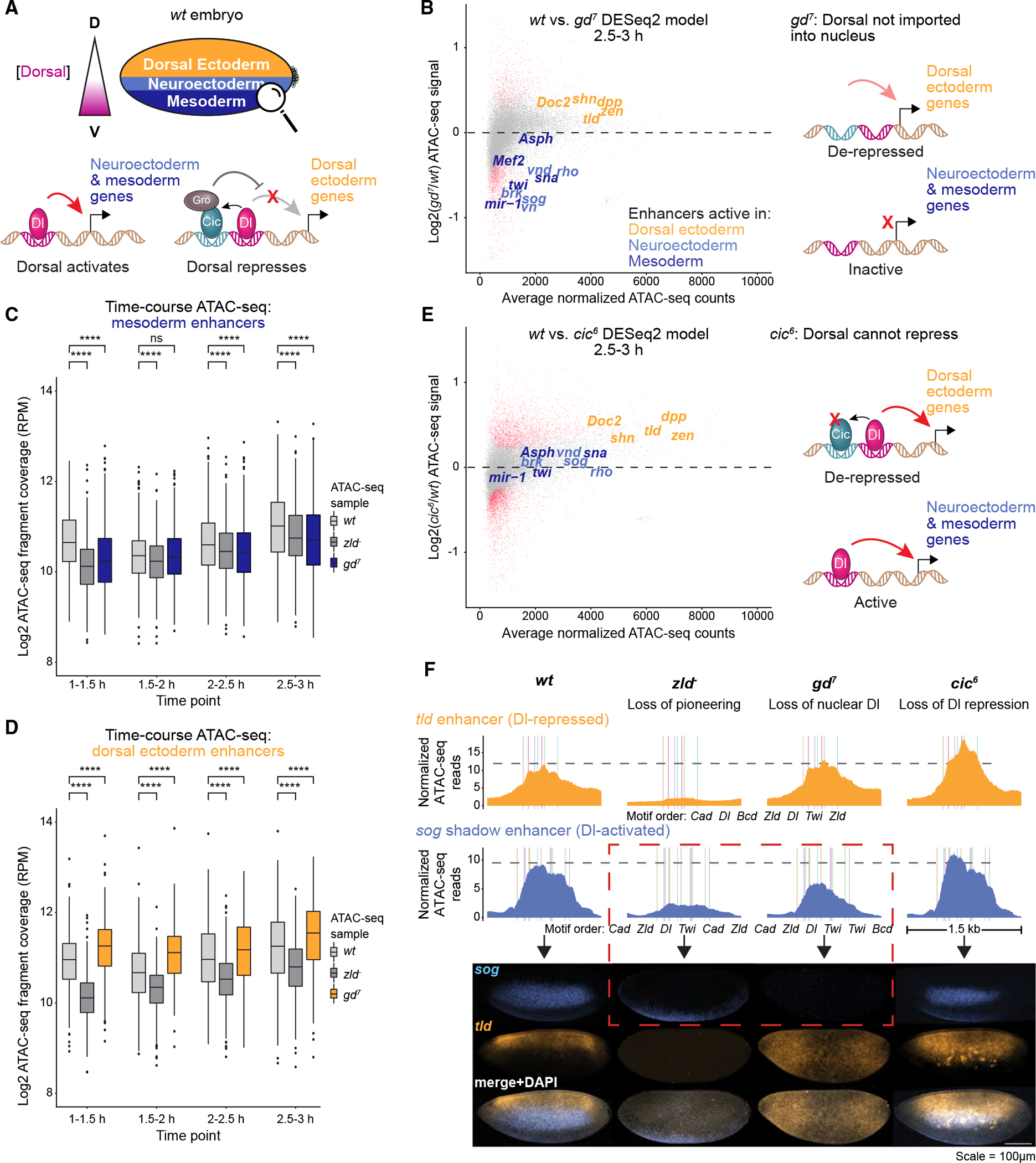
(A) Dorsoventral patterning in the early Drosophila embryo occurs through a nuclear concentration gradient of the Dorsal TF, which activates mesodermal and neuroectodermal target genes but represses dorsal ectodermal genes. Dorsal repression occurs through Capicua, whose binding at these regions depends on Dorsal and which recruits the co-repressor Groucho.
(B) In embryos lacking nuclear Dorsal (gd7), chromatin accessibility is specifically reduced at Dorsal-activated enhancers but not at Dorsal-repressed enhancers. Differential accessibility was calculated between wt and gd7 embryos for all time points and the MA plot for the 2.5–3 h AEL time point is shown. Red dots represent statistically significant differences (false discovery rate [FDR] = 0.05). Known dorsoventral enhancers are colored by the tissue type in which they are active. See also Figures S1F and S6A–S6B.
(C) Mesoderm enhancers, as characterized previously108 (n = 416), have significantly reduced chromatin accessibility in gd7 embryos when they are inactive (Wilcoxon rank-sum tests, four asterisks: p < 0.0001). Normalized ATAC-seq fragment coverage was calculated across 1 kb centered on each enhancer. See also Figure S6C.
(D) Dorsal ectoderm enhancers108 (n = 380) gain chromatin accessibility in gd7 embryos where they are not repressed by Dorsal.
(E) In cic6 embryos, where Capicua’s interaction with Groucho is abrogated and Dorsal can no longer repress, chromatin accessibility is increased at Dorsal-repressed enhancers. Differential accessibility analysis between wt and cic6 embryos was performed as in (B). See also Figures S1G and S6D–S6E.
(F) Chromatin accessibility and target gene activation do not always correlate (dashed red box). ATAC-seq data at a Dorsal-repressed enhancer (tld) and Dorsal-activated enhancer (sog shadow) upon loss of Zelda (zld−), nuclear Dorsal (gd7), and Dorsal-mediated repression (cic6) are shown on top as normalized ATAC-seq fragment coverage from the 2.5–3 h AEL time point across 1.5 kb windows: dm6 coordinates chr3R:24,748,748–24,750,248 (tld) and chrX:15,646,300–15,647,800 (sog shadow). The wt ATAC-seq maximum value is marked as a dotted gray line. Colored bars are BPNet-mapped motifs listed below. Multiplexed hybridization chain reaction experiments show sog and tld expression in stage 5 wt, zld−, gd7, and cic6 mutant embryos (scale is 100 um). Note that sog expression is partially reduced upon loss of Zelda’s pioneering, but completely gone upon loss of Dorsal. Meanwhile, tld expression is ablated in the absence of Zelda but expands upon loss of Dorsal or Dorsal-mediated repression. See also Figures S6F–S6G.
If Dorsal consistently contributes to chromatin accessibility by binding to target enhancers, we would expect that the loss of Dorsal would lead to decreased accessibility at all its target genes. To test this, we performed ATAC-seq time course experiments on gastrulation defective (gd7) mutant embryos, where Dorsal remains cytoplasmic and inactive in the entire embryo (Figure S6A), resulting in dorsal ectoderm fate throughout the embryo.78,106–108 We then used DESeq2109 to analyze the differential accessibility upon loss of Dorsal (gd7) compared with wild type (Figures 5B and S6B).
When we examined well-characterized Dorsal target enhancers, we noticed a striking difference in accessibility between enhancers that are activated by Dorsal and those that are repressed. Mesoderm enhancers (e.g., twi and sna) and neuroectoderm enhancers (e.g., sog and brk), which are activated by Dorsal, show significantly decreased accessibility upon loss of Dorsal (purples in Figure 5B). Conversely, the Dorsal-repressed enhancers do not show decreased accessibility and even show a slight increase, despite losing Dorsal binding (orange in Figure 5B). These results suggest that Dorsal’s ability to increase chromatin accessibility is tied to its role as a transcriptional activator.
To confirm this effect more broadly, we used a validated set of dorsoventral enhancers.108 We plotted the ATAC-seq signal for each time point and found that the mesoderm enhancers showed decreased chromatin accessibility in both zld− and gd7 embryos (Figure 5C), as did neuroectodermal enhancers (Figure S6C). Dorsal ectoderm enhancers also lose accessibility in zld− embryos but gain accessibility in gd7 embryos from the earliest time points on, suggesting that this is mediated by the loss of Dorsal repression (Figure 5D). This further corroborates that the loss of Dorsal binding does not always lead to the loss of accessibility but rather depends on the Dorsal’s ability to activate these enhancers.
To test this hypothesis more directly, we specifically manipulated the ability of Dorsal to repress without affecting its ability to activate. In cic6 mutant embryos, Cic has a small deletion in its interaction domain (N2) with the co-repressor Groucho and no longer functions as a repressor102 (Figure 5E). As a result, Dorsal can still activate mesoderm and neuroectoderm enhancers, but it can no longer repress dorsal ectodermal enhancers, where it is now expected to function as a weak activator.102 Thus, in cic6 embryos, the Dorsal-activated enhancers should be unchanged compared with wild type, whereas enhancers normally repressed by Dorsal (e.g., tld, zen, and dpp102,104,105) should have higher accessibility. Indeed, when we performed ATAC-seq experiments in cic6 mutant embryos (Figure S6D), we found that dorsal ectoderm enhancers showed statistically significant increased accessibility (Figure 5E, orange), whereas mesoderm and neuroectoderm enhancers not regulated by Cic generally remained unchanged (Figure 5E, purples). These results demonstrate that the chromatin accessibility at Dorsal target enhancers depends on the activation state induced by Dorsal rather than the binding of Dorsal.
The finding that Dorsal’s effect on chromatin accessibility is dependent on the sequence context and coupled to its transactivation effect suggests that Bicoid might operate in the same way. In support of this, we found the regions where Bicoid is required for accessibility57 to be strongly bound by Bicoid, to have a higher predicted contribution to accessibility in the ChromBPNet model, and to have the histone marks of active enhancers (Figures S2L–S2N). In addition, we found that Bicoid-regulated enhancers that are repressed by high-affinity Cic motifs (e.g., hkb, tll, and hb)102,110–112 also increased in accessibility in cic6 mutant embryos (Figures S6E and S6F), further supporting the model that the accessibility of Bicoid target enhancers depends on their activation state.
In summary, our results suggest that chromatin accessibility levels depend on both the consistent pioneering effect of Zelda and the combinatorial effect that patterning TFs have on enhancer activation. Since the patterning TFs depend on Zelda for binding, this could mean either that Zelda’s effect is much stronger than that of patterning TFs, perhaps due to its high concentration, or that the patterning TFs mainly function at a step downstream of pioneering. In support of the latter model, patterning TFs such as Dorsal and Bicoid do not have a weak effect but play a critical role in the activation of their target genes in a manner that is different from Zelda.38,43,46,60,113,114 To illustrate this difference, we directly compared the accessibility of the known tld and sog shadow enhancers with their target gene expression across various mutants (Figure 5F). The target gene expression was visualized by multiplexed in situ hybridization experiments using hybridization chain reaction.115
These results confirmed that chromatin accessibility and target gene expression do not always correlate (Figure 5F). In zld− embryos, accessibility is dramatically reduced at both the tld and sog enhancers due to the loss of pioneering. Compared with this strong and consistent effect upon the loss of Zelda, the loss of Dorsal (gd7) led to a modest decrease in accessibility, only at the Dorsal-activated enhancer sog and not at the Dorsal-repressed enhancer tld or in cic6 mutants where Dorsal can still be activated (Figures 5F and S6G for more enhancers). Thus, Dorsal’s effect on accessibility is weaker than that of Zelda and depends on its transactivation effect. The reverse is true for gene activation: Dorsal’s effect on gene expression is stronger than that of Zelda (red box, Figure 5F). In zld− embryos, sog is still expressed after some delay in cells with high concentrations of Dorsal (red box, Figure 5F). This effect is specific to the examined sog enhancer since the same expression pattern is obtained when the enhancer is part of a reporter.43,60 In contrast, sog expression is completely abolished in the absence of Dorsal, consistent with previous results.113 Thus, Zelda has a stronger effect on accessibility, whereas Dorsal has a stronger effect on activation, arguing that they involve functionally separable processes that both have effects on chromatin accessibility.
DISCUSSION
Here, by combining TF binding and chromatin accessibility data with deep learning models and using Drosophila genetics as a validation tool, we asked how TFs mediate chromatin accessibility in the Drosophila embryo. We investigated whether the role of opening chromatin is restricted to TFs axiomatically classified as pioneers or if TFs more generally contribute to chromatin accessibility. We find that there is a clear hierarchical relationship between the pioneer Zelda and the patterning TF and that both contribute to accessibility through distinct cis-regulatory sequence rules.
We therefore propose a model in which chromatin accessibility is governed by two distinct processes: pioneering and activation (Figure 6). Pioneers like Zelda consistently bestow basal accessibility by reading out motif affinity, whereas patterning TFs require an already accessible state for their binding and increase chromatin accessibility in a context-dependent manner. For example, when Dorsal motifs are flanked by motifs for the repressor Cic, no increase in accessibility is observed. This demonstrates that the increase in accessibility is not dependent on Dorsal binding per se but on the total effect that the TFs have on the activation of the enhancer and thus is governed by the cis-regulatory rules of activation. In contrast, Zelda consistently increases chromatin accessibility even in the absence of enhancer activation.
Figure 6. Pioneering and enhancer activation increase chromatin accessibility.
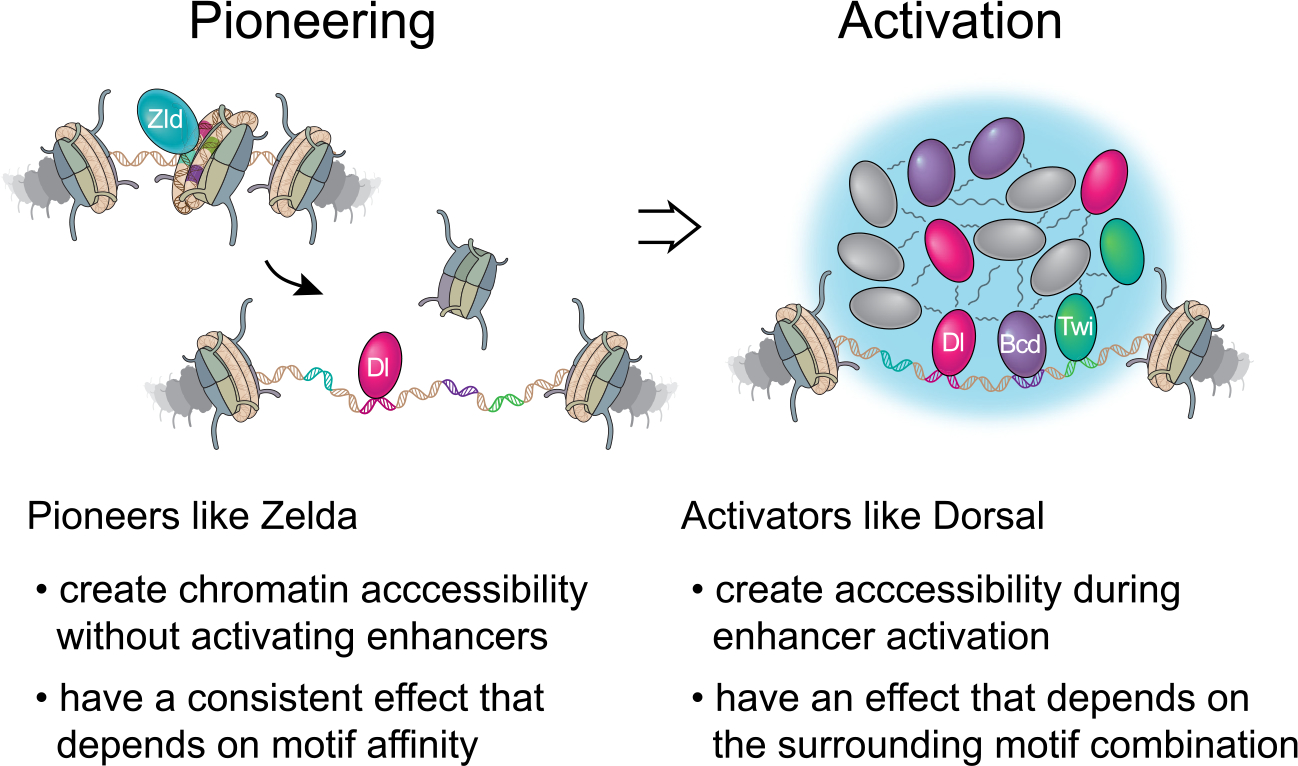
Chromatin accessibility at enhancers is established in a two-tier process that involves pioneering and activation. The pioneer Zelda bestows basal chromatin accessibility at enhancers without necessarily activating them. It does so by reading out its motif affinity on nucleosomal DNA and producing a consistent effect that is not dependent on the surrounding motif combination. The accessible DNA then allows the binding of patterning TFs such as Dorsal. Activation occurs when patterning TFs bind at high concentrations and enable the formation of hubs through multivalent weak interactions with each other and cofactors such as histone acetyltransferases. Whether or not Zelda is present in these hubs is unclear. Since enhancer activation through hubs is DNA-templated, it is inherently dependent on the motif combination within the enhancer. How enhancer activation increases chromatin accessibility further is not clear, possibly due to histone acetylation and the highly dynamic nature of hubs.
The functional separation between pioneering and activation is consistent with previous observations in the early Drosophila embryo. Zelda unambiguously generates chromatin accessibility very early on but is insufficient for the activation of most developmental enhancers and functions together with patterning TFs during zygotic genome activation.41,77,114,116,117 Although Zelda is essential for a small subset of genes that are expressed early and have Zelda motifs at their promoter,38,80,93 many patterning genes such as sog do not require Zelda for activation and eventually become expressed in zld− embryos.38 Zelda is, however, a strong potentiator of transcription.5,43,45,46,60 This suggests that Zelda’s effect on chromatin accessibility is not required for activation but boosts the effect of activators. A similar potentiating effect of Zelda has been observed at the level of transcriptional bursting. Dorsal mainly affects burst frequency, whereas Zelda has additional effects on burst size.114
These functional differences are consistent with pioneering and activation being physically separate processes. Zelda binds its motifs in the presence of nucleosomes,20,49 whereas Dorsal, Twist, Caudal, and Bicoid require accessible DNA for binding.5,6,43,45,46,48,60 Although Zelda could also bind to accessible regions, this may not occur to a large extent since Zelda binds to chromatin transiently on the order of seconds47 and does not co-localize with RNA Pol II or at the sites of active transcription.47,116 Thus, pioneering appears to be the process associated with nucleosome removal, whereas enhancer activation occurs on accessible DNA.
Remarkably, both processes appear to read out cis-regulatory information very precisely. Our deep learning models and experimental validations revealed that pioneering by Zelda depends on the motif’s affinity. However, how Zelda rapidly recognizes its motifs on nucleosomal DNA and opens chromatin is not clear. Zelda’s DNA binding domain is insufficient for pioneering in vivo,118 and although in vivo studies point to a constant involvement of ATP-dependent chromatin remodeling,119,120 how Zelda interacts with chromatin remodelers is not known. Furthermore, in vitro studies suggest that TF binding to nucleosomes is structurally constrained and may be preferred at certain positions on the nucleosome,20,21,86,87 which is at odds with Zelda’s ability to consistently read out motif affinity. We therefore speculate that pioneer TFs recognize their motifs in vivo more efficiently than in vitro, perhaps aided by chromatin remodelers.
Another interesting observation was that the pioneer GAF was not predicted to play the same role as Zelda. Although the GAF motif was correctly identified to play a strong role in chromatin accessibility and boost GAF binding to another GAF motif nearby, it was not predicted to strongly promote the binding of the patterning TFs. An explanation for the difference may be that GAF multimerizes on DNA and remains on chromatin on the order of minutes.121–124 Such stable binding makes sense in the light of GAF’s role in genome structure124–128 and transcriptional memory.122,129,130 GAF could generate accessible chromatin, but by binding to the newly opened DNA itself, it could partially occlude the binding of additional TFs.
How chromatin accessibility increases further when an activating combination of patterning TFs bind is also not clear. An attractive model is that the right cis-regulatory motif combination on accessible DNA seeds the formation of hubs.131–134 This would explain why this part of the cis-regulatory code is inherently context-specific and dependent on the balance between activators and repressors, their concentrations, and the motif affinities (Figure 6). Supporting this model, hubs have been observed via imaging studies for multiple TFs in the early Drosophila embryo, including Zelda, Dorsal, and Bicoid.47,48,60,116 Hubs containing either Dorsal or Bicoid were dependent on Zelda,48,60 which is consistent with DNA accessibility being a requisite for hub formation. Moreover, if hubs regulate transcriptional bursting, this could explain why Dorsal and Zelda have different effects.114 Dorsal may determine the burst frequency by regulating the speed of hub formation on already accessible DNA, whereas Zelda also facilitates chromatin accessibility and thus may affect the burst size by providing more time and space for hub formation.
Taken together, our results suggest that TFs read out cis-regulatory sequences during two processes, pioneering and activation, and those follow distinct sequence rules. We likely discovered this in the early Drosophila embryo because the TFs there have distinct roles in each process. The pioneer Zelda creates basal chromatin accessibility throughout the embryo, which then allows the patterning TFs to activate genes in specific parts of the embryo. However, having two interdependent regulatory processes that both read out motif affinities could be a general principle of the cis-regulatory code, even if the same TFs mediate both pioneering and activation. From a theoretical perspective, having a process with an energy-expending step such as ATP-dependent chromatin remodeling and having TFs read out the same motifs twice represents an appealing explanation for the dynamic nature and high specificity of transcriptional regulation.135,136
Limitations of the study
Since we only examined one developmental context, it remains to be shown how the cis-regulatory sequence rules change when the TFs are present at different concentrations or with different TFs. The ability of Zelda and other Drosophila and mammalian TFs is concentration-dependent,17,18,118 and Zelda may not function as a strong pioneer in other developmental contexts.137 Furthermore, the distinction between pioneering and activation may not always be clear-cut, even in our system. At high concentrations, Dorsal could also function as a pioneer since the Dorsal motif contributed more consistently to chromatin accessibility at our last time point (Figure 2F). Indeed, it has been shown in mammals that a TF can function as both pioneer and activator with different concentration threshold requirements.138 Nevertheless, separate contributions of pioneering and enhancer activation toward chromatin accessibility are likely a general feature of the cis-regulatory code. In both Drosophila and mammals, the highest accessibility is typically found at active enhancers75,76,139–142; however, chromatin accessibility is often only a mediocre predictor for enhancer activity.143–147
Another limitation of the study is that the deep learning models are only as good as we can accurately train them and extract the learned sequence rules. Although such models are ideally suited for discovering cis-regulatory sequence rules de novo without prior biological assumptions, we may miss the learned features of the model. For example, it is unclear whether the models learned subtle sequence rules that contribute to nucleosome occupancy or positioning. Future studies will have to more specifically address additional layers of the cis-regulatory code.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Julia Zeitlinger (jbz@stowers.org).
Materials availability
Antibodies generated in this study are available upon request.
Data and code availability
The raw and processed data for ChIP-nexus, ChIP-seq, ATAC-seq, MNase-seq, and protein binding microarray experiments have been deposited at GEO under series accession number GSE218852 (GEO: GSE218852) and are publicly available as of the date of publication. Original data, including microscopy images, can be accessed from the Stowers Original Data Repository (ODR: http://www.stowers.org/research/publications/libpb-2357). Trained BPNet and ChromBPNet models are available at Zenodo (Zenodo: https://zenodo.org/record/8075860). All original code has been deposited at GitHub (GitHub: https://github.com/zeitlingerlab/Brennan_Zelda_2023) and is publicly available as of the date of publication. The DOI is listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit polyclonal anti-Zelda | Koenecke et al.108 | 366735–1 |
| Rabbit polyclonal anti-Bicoid | This paper | U9982EL040–1 |
| Rabbit polyclonal anti-Caudal | This paper | U4197EL190–1 |
| Rabbit polyclonal anti-Dorsal | He et al.58 | 126740–44 |
| Rabbit polyclonal anti-Twist | He et al.58 | 131424–2 |
| Rabbit polyclonal anti-GAF | This paper | 163185–42 |
| Rabbit polyclonal anti-H3K27ac | Active Motif | 39133; RRID: AB_2561016 |
| Mouse monoclonal anti-H3K4me1 | Active Motif | 39635; RRID: AB_2793284 |
| Anti-rabbit IgG Alexa Fluor 568 secondary antibody | ThermoFisher | A10042; RRID: AB_2534017 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| 37% formaldehyde solution | VWR | Cat# 50–00-0 |
| Dynabeads Protein A | ThermoFisher | Cat# 10008D |
| phi29 DNA polymerase | New England Biolabs | Cat# M0269S |
| Lambda exonuclease | New England Biolabs | Cat# M0262S |
| Q5 High-Fidelity 2x Master Mix | New England Biolabs | Cat# M0492S |
| dNTP solution mix | New England Biolabs | Cat# N0447S |
| MNase | New England Biolabs | Cat# M0247S |
| RNase A | ThermoFisher | Cat# EN0531 |
| Phenol:chloroform:isoamyl alcohol (25:24:1) (v/v/v) | VWR | Cat# 136112–00-0 |
| Proteinase K | ThermoFisher | Cat# 25530049 |
| Western Blocking Reagent | Millipore Sigma | Cat# 11921681001 |
| ProLong Gold Antifade Mountant with DAPI | ThermoFisher | Cat# P36931 |
| OptiPrep Density Gradient Medium | Millipore Sigma | Cat# D1556 |
| ProLong Glass Antifade Mountant | ThermoFisher | Cat# P36980 |
|
| ||
| Critical commercial assays | ||
|
| ||
| End Repair Module | New England Biolabs | Cat# E6050S |
| dA-Tailing Module | New England Biolabs | Cat# E6053S |
| Quick Ligation Kit | New England Biolabs | Cat# M2200S |
| High Throughput Library Prep Kit | KAPA Biosystems | Cat# KK8234 |
| Monarch DNA Gel Extraction Kit | New England Biolabs | Cat# T1020 |
| Monarch PCR & DNA Cleanup Kit | New England Biolabs | Cat# T1030 |
| PURExpress In Vitro Protein Synthesis Kit | New England Biolabs | Cat# E6800 |
| Hybridization Chain Reaction (HCR) v3.0 | Molecular Instruments | N/A |
|
| ||
| Deposited data | ||
|
| ||
| Raw and analyzed NGS and PBM data | This paper | GEO: GSE218852 |
| Trained deep learning models (Zenodo) | This paper | Zenodo: https://zenodo.org/recore/8075860 https://doi.org/10.5281/zenodo.8118135 |
| Raw images | This paper | ODR: http://www.stowers.org/research/publications/libpb-2357 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Drosophila melanogaster: Oregon-R | Koenecke et al.108 | FlyBase: FBsn0000276 |
| Drosophila melanogaster: UAS-shRNA-zld: P{UAS-zld.shRNA} | Sun et al.6 | FlyBase: FBtp0147479 |
| Drosophila melanogaster: Maternal Triple Driver (MTD)-Gal4: P{COG-GAL4:VP16}; P{Gal4-nos.NGT}40; P{nos-Gal4-VP16} | Bloomington Stock Center | BSC: 31777 |
| Drosophila melanogaster: gd7: gd7/winscy, P{hs-hid}5 | Koenecke et al.108 | N/A |
| Drosophila melanogaster: cic6: cic6/TM3, Sb1 | Papagianni et al.101 | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Oligonucleotides for ChIP-nexus, see Table S2 | IDT | https://research.stowers.org/zeitlingerlab/protocols.html |
| Illumina Index primer 1: 5’-CAAGCAGAAGAC GGCATACGAGAT[i7]GTCTCGTGGGCTCGG-3’ | IDT | https://support-docs.illumina.com/SHARE/AdapterSeq/1000000002694_17_illumina_adapter_sequences.pdf |
| Illumina Index primer 2: 5’-AATGATACGGCGACC ACCGAGATCTACAC[i5]TCGTCGGCAGCGTC-3’ | IDT | https://support-docs.illumina.com/SHARE/AdapterSeq/1000000002694_17_illumina_adapter_sequences.pdf |
| Illumina Transposase adapter read 1 (Nextera A): 5’- TCGTCGGCAGCGTCAGATGTGTATAA GAGACAG-3’ | IDT | https://support-docs.illumina.com/SHARE/AdapterSeq/1000000002694_17_illumina_adapter_sequences.pdf |
| Illumina Transposase adapter read 2 (Nextera B): 5’- GTCTCGTGGGCTCGGAGATGTGTATA AGAGACAG-3’ | IDT | https://support-docs.illumina.com/SHARE/AdapterSeq/1000000002694_17_illumina_adapter_sequences.pdf |
| Mosaic end primer: /5Phos/CTGTCTCTTATAC A/3ddC/ | IDT | Tn5mC1.1-A1block |
| gd7 heat shock forward primer: 5’-GGAGCGAC AATTCAATTCAAACAAGC-3’ | IDT | N/A |
| gd7 heat shock reverse primer: 5’-GTAGCTGTG GCTGCAGTGCATCG-3’ | IDT | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pETM11-Sumo3-Tn5 plasmid | Hennig et al.148 | E54K,L372P |
| His6-tagged SenP2 protease plasmid | Hennig et al.148 | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| FIJI | Schindelin et al.149 | https://fiji.sc/ |
| Cutadapt v.2.5 | Martin154 | https://cutadapt.readthedocs.io/en/v2.5/ |
| Bowtie2 v.2.3.5.1 | Langmead and Salzberg155 | https://bowtie-bio.sourceforge.net/bowtie2/manual.shtml |
| MACS2 v.2.2.7.1 | Zhang et al.156 | https://github.com/macs3-project/MACS |
| Irreproducible Discovery Rate framework v.2.0.3 | Li et al.157 | https://github.com/nboley/idr |
| Picard v.2.23.8 | Broad Institute of MIT and Harvard158 | http://broadinstitute.github.io/picard |
| deepTools2 v.3.5.1 | Ramírez et al.159 | https://deeptools.readthedocs.io/en/latest/ |
| BPNet software | Avsec et al.59 | https://github.com/kundajelab/bpnet/ |
| Keras v.2.2.4 & v.2.5.0 | Chollet et al.160 | https://pypi.org/project/keras/ |
| TensorFlow1 backend v.1.7 & v.2.5.1 | Abadi et al.161 | https://www.tensorflow.org/install/pip |
| Adam optimizer | Kingma and Ba162 | N/A |
| DeepLIFT v.0.6.9.0 | Shrikumar et al.163 | https://github.com/kundajelab/DeepExplain |
| TF-MoDISco v.0.5.3.0 & v.0.5.16.0 | Shrikumar et al.164 | https://github.com/kundajelab/tfmodisco |
| ChromBPNet software | Anshul Kundaje’s lab, Stanford University | https://github.com/kundajelab/chrombpnet |
| DeepLIFT v.0.6.13.0 | Shrikumar et al.163 | https://github.com/kundajelab/shap |
| DESeq2 v.1.36.0 | Love et al.109 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| R v.4.2.0 | R core team | https://www.r-project.org/ |
| Rstudio | RStudio | https://rstudio.com |
| ggplot2 v.3.3.6 | Wickham168 | https://ggplot2.tidyverse.org/ |
|
| ||
| Other | ||
|
| ||
| All code and analyses that contributed to this work | This paper | https://github.com/zeitlingerlab/Brennan_Zelda_2023 https://doi.org/10.5281/zenodo.8118135 |
| Bioruptor Pico sonication device | Diagenode | https://www.diagenode.com/en/p/bioruptor-pico-sonication-device |
| Point scanning confocal microscope | Zeiss | 780 |
| Spinning disk microscope | Nikon | Eclipse Ti2 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Drosophila strains
Oregon-R flies were used as the wild type (wt) strain in all experiments. Embryos depleted for maternal Zelda (zld−) were generated by crossing UAS-shRNA-zld females to MTD-Gal4 males as previously described6 and tested for embryonic lethality37 and Zelda depletion using immunostaining (Figure 3). Embryos lacking nuclear Dorsal were laid by gd7/gd7 mothers generated from a gd7/winscy, P{hs-hid}5 stock that was heat-shocked at the larval stage at 37°C for 1 h on two consecutive days to eliminate heterozygous mothers.6 Loss of the hs-hid sequence was confirmed using PCR with gd7 heat shock primers on genomic DNA extracted from heat-shock survivors. The cic6/TM3, Sb1 stock was generated using CRISPR/Cas9 as previously described.102 Cic6 embryos were collected from cic6/cic6 mothers identified by wt bristles and were confirmed to be embryonic lethal.
Drosophila embryo collections, fixation, and sorting
All embryos were collected from population cages using apple juice plates with yeast paste, following two pre-clearings as previously described.58,80 For ChIP-nexus, ChIP-seq, and MNase-seq experiments, embryos were collected for 1 h and aged for 2 h at 25°C, yielding collections of 2–3 h after egg laying (AEL). For ATAC-seq, embryos were collected in 30-min windows and aged accordingly to generate the 1–1.5, 1.5–2, 2–2.5, and 2.5–3 h AEL time points. All embryos were dechorionated using 50% bleach for 2 min and sufficiently rinsed with water afterwards. For ATAC-seq, embryos were hand-sorted based on morphology in ice-cold PBT immediately following dechorionation using an inverted contrasting microscope (Leica DMIL) as described.80 For ChIP-nexus, ChIP-seq, and MNase-seq, embryos were first fixed with 1.8% formaldehyde (final concentration in water phase) in heptane and embryo fix buffer (50 mM HEPES, 1 mM EDTA, 0.5 mM EGTA, 100 mM NaCl) while vortexing for 15 min. For ChIP-nexus and ChIP-seq, the vitelline membrane was removed using methanol/heptane and embryos were stored in methanol at −20°C until use. For these experiments, embryos were rehydrated using PBT and sorted to remove out-of-stage embryos using either hand-sorting or cytometry (Copas Plus, macroparticle sorter, Union Biometrica). For MNase-seq, embryos were spun down at 500 × g, 4°C, for 1 min, and fixation was quenched by adding 10 mL PBT-glycine (125 mM glycine in PBT) and vortexing for 2 min. Embryos were hand-sorted based on morphology in ice-cold PBT and then used in MNase-seq experiments.
METHOD DETAILS
ChIP-nexus and ChIP-seq experiments
For each ChIP, 10 μg of antibody was coupled to 50 μL of Protein A Dynabeads (ThermoFisher, 10008D) and incubated overnight at 4°C prior to ChIP. All ChIP-nexus experiments were performed using antibodies custom generated by Genscript: Zelda (aa 1117–1327), Dorsal (aa 39–346), Twist (C-terminus), Bicoid (C-terminus), Caudal (aa 1–214), GAF (aa 1–382). ChIP-seq experiments were performed with the following commercially available antibodies: H3K27ac (Active motif, 39133) and H3K4me1 (Active motif, 39635). For all TFs, at least three biological replicates were performed using embryos from different collections. For ChIP-seq, at least two biological replicates were performed in the same way. Approximately 0.2–0.4 grams of fixed 2–3 h AEL embryos were used for all ChIP experiments. Chromatin extracts were prepared by douncing embryos in Lysis Buffer A1 (15 mM HEPES pH 7.5, 15 mM NaCl, 60 mM KCl, 4 mM MgCl2, 0.5% Triton X-100, 0.5 mM DTT (add fresh)), washing nuclei with ChIP Buffer A2 (15 mM HEPES pH 7.5, 140 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100, 0.5% N-lauroylsarcosine, 0.1% sodium deoxycholate, and 0.1% SDS), and sonicating with a Bioruptor Pico (Diagenode) for six cycles of 30 s on and 30 s off. ChIP-nexus library preparation steps include end repair, dA-tailing, adapter ligation, barcode extension, and lambda exonuclease digestion and was performed as previously described,58 except that the ChIP-nexus adapter mix contained four fixed barcodes and PCR library amplification was performed directly after circularization of the purified DNA fragments (without addition of the oligo and BamHI digestion). ChIP-seq was performed as previously described and included a whole cell extract (WCE).69,78 Single-end sequencing was performed on an Illumina NextSeq 500 instrument (75 or 150 cycles). Replicates for each TF and histone modification were generated and showed high concordance (Figures S1A and S1E). The full ChIP-nexus protocol can be found on the Zeitlinger lab website at https://research.stowers.org/zeitlingerlab/protocols.html.
ATAC-seq experiments
For ATAC-seq time course experiments, the following amounts of hand-sorted embryos were used: 400 embryos (1–1.5 h AEL); 100 embryos (1.5–2 h AEL); 40 embryos (2–2.5 h AEL, 2.5–3 h AEL). Following sorting, embryos were immediately dounced in ATAC Resuspension Buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2) with 0.1% IGEPAL CA-630 and nuclei were harvested by centrifugation. Tn5 transposition was performed as previously described.72,73 Briefly, the nuclear pellet was incubated for 3 min on ice in ATAC resuspension buffer supplemented with 0.1% IGEPAL CA-630, 0.1% Tween-20, and 0.01% Digitonin (Promega, G9441). The reaction was stopped by adding ATAC Resuspension Buffer with 0.1% Tween-20 followed by centrifugation. Tagmentation took place at 37°C for 30 min at 1000 rpm in a 50 μL reaction volume containing 10 μL of 5x Tagment DNA Buffer (50 mM Tris-HCl pH 7.4, 25 mM MgCl2, 50% DMF) 16.5 μL 1x PBS, 0.5 μL 10% Tween-20, 0.5 μL 1% Digitonin,1–2 μM of Tn5 transposase loaded with oligonucleotides, and water. Tn5 transposase was purified in-house using pETM11-Sumo3-Tn5 and His6-tagged SenP2 protease plasmids as previously described.148 The resulting fragments were purified using the Monarch PCR & DNA Cleanup Kit (NEB). Libraries were constructed using Illumina Nextera Dual Indexing, and qPCR was used to prevent over-amplification as described.73 All ATAC-seq experiments were performed in triplicate, with highly correlated replicates (Figures S1B, S1C, S1F, and S1G), and paired-end sequencing was performed on an Illumina NextSeq 500 instrument (2× 75 bp cycles).
MNase-seq experiments
For each MNase digestion, 100 hand-sorted 2–3 h AEL Drosophila embryos were used. Nuclei were extracted by douncing in PBS with 0.1% IGEPAL CA-630. The nuclei were harvested by centrifugation and resuspended gently in MNase Digestion Buffer (PBS with 0.1% Triton X-100 and 1 mM CaCl2). MNase digestion was performed with 100 U MNase (NEB, M0247S) for 30 min at 37°C. The reaction was stopped with 20 mM EGTA. The nuclei were treated with 50 μg/ml RNase A (ThermoFisher, EN0531) for 1 h at 37 °C and 1000 rpm, and subsequently incubated overnight at 65 °C and 1000 rpm with 200 μg/ml Proteinase K (ThermoFisher, 25530049) and 0.5% SDS for reverse cross-linking. DNA was extracted using phenol-chloroform (VWR, K169). Libraries were constructed from 10 ng purified DNA using the High Throughput Library Prep Kit from KAPA Biosystems (KK8234) according to the manufacturer’s instructions. Three experimental replicates were performed, and replicates were highly correlated (Figure S1D). Paired-end sequencing was performed on an Illumina NextSeq 500 instrument (2× 75 bp cycles).
Antibody staining and microscopy experiments
Embryos were collected and aged to be 2–3 h old, fixed with 1.8% formaldehyde, and stored in 100% methanol at −20°C prior to immunostaining. Embryo aliquots were rehydrated in an ethanol:PBT gradient and blocked for 30 min using the Roche Western blocking reagent (Millipore Sigma, 11921681001) and PBT. Primary antibody incubation occurred at 4°C overnight with a 1:200 antibody dilution in PBT/blocking reagent with the same Zelda, Dorsal, and Twist antibodies used for ChIP-nexus experiments. Embryos were then washed six times with PBT, blocked again, and incubated with a donkey anti-rabbit IgG Alexa Fluor 568 secondary antibody (ThermoFisher, A10042), 1:500, at 4°C overnight. After eight washes with PBT, embryos were mounted with ProLong Gold Antifade Mountant with DAPI (Invitrogen, P36931). Images were acquired on a Zeiss LSM-780 point scanning confocal microscope with a 32 channel GaAsP detector and a plan-apochromat 10x objective lens, N.A. 0.45, using the ZEN Black 2.3 SP1 software by Zeiss. The Alexa Fluor 568 track used a DPSS 561 nm laser excitation at 6.5%, and the DAPI track used a Diode 405 nm laser excitation at 6.0%. Images were collected using a frame size of 1024 × 1024, a zoom of 1.5, and a pixel dwell time of 3.15 μs. Confocal z-stacks were maximum intensity projected and all image processing steps were performed using FIJI.149 All microscopy and processing settings were kept the same when comparing wt to zld− or gd7 embryos.
Protein binding microarray experiments
For all PBM experiments, the C-terminal region of Zelda, which includes the four zinc fingers (#3–6) that are known to bind CAGGTAG motifs, were used.37,38 These zinc fingers were cloned into a T7-driven GST expression vector, pTH6838. The TF sample was expressed by using a PURExpress In Vitro Protein Synthesis Kit (New England BioLabs) and analyzed in duplicate on two different PBM arrays (HK and ME) with differing probe sequences. The ME array was designed by Julian Mintseris and Mike Eisen,150 and the HK array by Hilal Kazan, following methodology described by Philippakis et al.151 Each array consists of ~41,000 60-base probe sequences (each containing 35 unique bases); the two array types have completely different probe sequences. Each PBM is designed using de Bruijn sequences, such that all possible 10-mers, and 32 copies of every non-palindromic 8-mer are contained on each array, offering an unbiased survey of TF binding preferences. PBM laboratory methods including data analysis followed the procedures previously described.152,153 PBM data were generated with motifs derived using Top10AlignZ.95 Z-scores and E-scores were calculated for each 8-mer as previously described.94,95 Octamers were grouped together based on their 7-mer sequences while also considering reverse complements, and the median E-score and Z-score was calculated for each 7-mer. The 7-mer sequences matching BPNet-mapped Zelda motifs were then extracted and the two PBM replicates were averaged for each Zelda motif.
In situ hybridization by hybridization chain reaction (HCR) experiments
Embryos were collected and developed to a final age of 2–3 h AEL. Embryos were fixed in 4.5% formaldehyde fixation solution for 25 min, devitellinized, and stored in 100% methanol at −20°C. HCR probes were designed against entire transcripts by Molecular Instruments to detect NM_001272649.2 (sog) and NM_079763.4 (tld).115 Embryos were rehydrated and HCR was performed according to Molecular Instruments’ HCR RNA-FISH protocol for whole-mount fruit fly embryos with the following exceptions. Embryos were not treated with xylene and proteinase K. Samples were rocked gently during all steps of the detection and amplification stages. During the detection stage, probe input was increased to 3 μL of1 μM stock and probe hybridization volume increased to 500 μL per sample. During the amplification stage, hairpin input was increased to 10 μL of3 μM stock and hairpin solution volume increased to 500 μL per sample. Embryos were allowed to incubate with hairpin solution containing 0.4 μg/mL DAPI for 44 h. Following HCR, embryos were cleared in OptiPrep (Millipore Sigma, D1556) and mounted in ProLong Glass Antifade Mountant (ThermoFisher, P36980). Images were acquired with an Orca Flash 4.0 sCMOS on a confocal Nikon Eclipse Ti2 microscope equipped with a Yokagawa CSU W1 Spinning Disk. Samples were illuminated with 405 nm, 561 nm, and 640 nm lasers to image DAPI, AlexaFluor546 and AlexaFluor647 respectively. A Nikon Plan-Apo 20x objective, N.A. 0.75, was used to acquire the images along with appropriate emission filters. Maximum intensity Z projections and adjustments to the brightness and contrast were performed in ImageJ/FIJI.149
ChIP-nexus data processing
ChIP-nexus single-end sequencing reads were pre-processed by trimming off fixed and random barcodes and reassigning them to FASTQ read names. ChIP-nexus adapter fragments were trimmed from the 3’ end of the fragments using cutadapt (v.2.5154). ChIP-nexus reads were aligned using bowtie2 (v.2.3.5.1155) to the Drosophila melanogaster genome assembly dm6. Aligned ChIP-nexus BAM files were deduplicated based on unique fragment coordinates and barcode assignments. Normalized ChIP-nexus coverage was acquired through reads-per-million (RPM) normalization, where the ChIP-nexus sample coverage was scaled by the total number of reads divided by 106. ChIP-nexus peaks were mapped using MACS2 (v.2.2.7.1156) with parameters designed to resimulate the full fragment length coverage rather than the single stop base coverage (–keep-dup=all -f=BAM –shift=−75 –extsize=150). ChIP-nexus peaks were filtered for pairwise reproducibility using the Irreproducible Discovery Rate framework (IDR) (v.2.0.3157). Peaks used for downstream analysis were selected from the largest pairwise comparison using the IDR framework.
ATAC-seq data processing
ATAC-seq paired-end sequencing reads were aligned using bowtie2 (v.2.3.5.1155) to the Drosophila melanogaster genome assembly dm6. Aligned ATAC-seq BAM files were marked for duplicates using Picard (v.2.23.8158) based on unique fragment coordinates, deduplicated, reoriented according to a Tn5 enzymatic cut correction of −4/+4 on fragment ends, filtered to contain fragment lengths no greater than 600 bp, and corrected for dovetailed reads. Normalized ATAC-seq coverage was acquired through reads-per-million (RPM) normalization, where the ATAC-seq sample coverage was scaled by the total number of reads divided by 106, as performed previously.51,77 Cut site ATAC-seq coverage was acquired by treating each of the fragment ends as a “cut event” and generating coverage based on only these “cut events”. ATAC-seq peaks were mapped using MACS2 (v.2.2.7.1156) with default paired-end parameters using ATAC-seq fragment coverage. ATAC-seq peaks were filtered for pairwise reproducibility using the Irreproducible Discovery Rate framework (IDR) (v.2.0.3157). Peaks used for downstream analysis were selected from the largest pairwise comparison using the IDR framework.
ChIP-seq data processing
ChIP-seq single-end sequencing reads were aligned using bowtie2 (v.2.3.5.1155) to the Drosophila melanogaster genome assembly dm6. Aligned ChIP-seq BAM files were deduplicated based on unique fragment coordinates and fragments extended based on the average experiment fragment length as determined with an Agilent 2100 Bioanalyzer. Normalized ChIP-seq coverage was acquired using the deepTools subfeature bamCompare (v.3.5.1159) using parameters to generate log2 fold-change scaling (–scaleFactorsMethod=readCount –operation=log2 –binSize=1). ChIP-seq peaks were mapped using MACS2 (v.2.2.7.1156) with default parameters and an applied background coverage using the associated WCE ChIP-seq control experiment. ChIP-seq peaks were filtered for pairwise reproducibility using the Irreproducible Discovery Rate framework (IDR) (v.2.0.3157).
MNase-seq data processing
MNase-seq paired-end sequencing reads were aligned using bowtie2 (v.2.3.5.1155) to the Drosophila melanogaster genome assembly dm6. Aligned MNase-seq BAM files were deduplicated based on unique fragment coordinates and filtered to contain fragment lengths no greater than 600 bp. Normalized MNase-seq coverage was acquired through reads-per-million (RPM) normalization, where the MNase-seq sample coverage was scaled by the total number of reads divided by 106.
BPNet model training and optimization
BPNet architecture and software was applied as previously described.59 Model inputs were 1000 bp genomic sequences centered on the ChIP-nexus peaks of TFs of interest. Model outputs were the predicted counts (total reads across each region) and predicted profile (coverage signal across each region) for Zelda, Dorsal, Twist, Caudal, Bicoid, and GAF ChIP-nexus experiments. 95,282 IDR-reproducible peaks from Zelda, Dorsal, Twist, Caudal, Bicoid, and GAF ChIP-nexus experiments were pooled and used as model inputs. Validation datasets were peaks located across chr2L (~18% of peaks), test datasets were peaks located across chrX (~19% of peaks), and peaks located across chrY and nonstandard chromosome contigs were excluded from analysis. The remaining regions were used for model training. Hyper-parameters were optimized by selected testing of parameter values deviating from the default BPNet architecture (number of dilational convolutional layers, number of filters in each convolutional layer, filter length of the first convolutional layer, filter length of the deconvolutional layer, learning rate, and counts-to-profile loss balancing). Model optimality was assessed based on counts and profile performance of each task, with a focused emphasis on the Zelda task performance, as this was our key TF of interest. After optimization, the final BPNet model architecture contained 9 dilated convolutional layers, 256 filters in each convolutional layer, a filter length of 7bp for both the input convolutional layer and output deconvolutional layer, a learning rate of 0.004, and a counts-to-profile weighting value (lambda) of 100. Final optimized model performance was assessed through comparing (1) area under the Precision-Recall Curves (auPRC) for profiles over different bins of resolution between observed ChIP-nexus profiles and predicted BPNet profiles (Figure S2A) and (2) counts correlations of observed ChIP-nexus signals to predicted BPNet signals for each TF (Figure S2B) as previously described.59 The auPRC values were benchmarked alongside replicate-replicate, observed-random, and observed-average observed profile comparisons to establish an in-context understanding of predicted profile accuracy. In order to test the stability of this optimized model architecture (fold 1), we trained two additional models with shuffled training, validation, and test sets (three-fold validation). The stability of the performance metrics as well as the stability of the returned downstream motif grammar was compared to the original optimized model training event (Figure S2C). All BPNet models were implemented and trained using Keras (v2.2.4160), TensorFlow1 backend (v.1.7161), the Adam optimizer.162 Training was performed using a NVIDIA® TITAN RTX GPU with CUDA v9.0 and cuDNN v7.0.5 drivers.
Motif extraction, motif curation, and motif island generation
DeepLIFT (v0.6.9.0, derived from the Kundaje Lab fork of DeepExplain (https://github.com/kundajelab/DeepExplain)163 was applied to the trained BPNet model to generate the contribution of each base across a given input sequence to the predicted output counts and profile signals. Contribution scores for counts and profile outputs were generated for all 6 TF tasks. TF-MoDISco (v.0.5.3.0164) was then applied across each TF separately. For each TF, regions of high counts contribution were identified, clustered based on within-group contribution and sequence similarity, and consolidated into motifs. The Zelda, Dorsal, Twist, Caudal, Bicoid, and GAF motifs were manually identified based on similarity to previous literature and validation of ChIP-nexus binding from the pertinent TF. Once motifs were characterized and confirmed, they were remapped back to their TF-specific peaks based on both Jaccardian similarity to the TF-MoDISco contribution weight matrix (CWM) and sufficient total absolute contribution across the mapped motif. This mapping approach is previously described.59 However, as we were interested in lower affinity motif representations than were previously identified by BPNet, mapping thresholds were lowered to mapping the motif if the CWM Jaccard similarity percentile was equal to or greater than 10% and if the total absolute contribution percentile was equal to or greater than 0.5%. After mapping, motifs were filtered for redundant assignment of palindromic sequences and overlapping peaks. Mapped and bound motifs were next clustered into ‘motif islands’ based on their proximity. Each island initially starts as a 200 bp region centered on the motif and gets clustered and merged with another nearby motif island if they overlap. In this manner, islands get extended as long as there is a motif within less than 200 bp. In the end, the vast majority of islands are still between 200–400 bp in width, while single-motif islands are 200 bp wide (Table S1). Island types with fewer than 30 genomic instances were filtered out. ATAC-seq and MNase-seq coverage was calculated across 250 bp windows centered on the island, while the H3K27ac and H3K4me1 signals were calculated across 1.5 kb windows centered on the island since these marks are typically on the enhancer flanks.
ChromBPNet model training and optimization
ChromBPNet is a modification of BPNet, designed to explain the relationship between genomic sequence and base-resolution ATAC-seq cut site coverage.29 ChromBPNet possesses similar model architecture to BPNet, but the training process contains extra steps to accommodate for the Tn5 sequence bias that influences the positions of the ATAC-seq cut sites. If the Tn5 sequence is not accounted for, the positional information of the cut sites cannot be reliably interpreted. The details of ChromBPNet’s bias correction will be published in a separate manuscript as part of ENCODE. Briefly, ChromBPNet corrects the bias during the training step by simultaneously passing sequence information through (1) a frozen, pre-trained model that has already learned Tn5 sequence bias and (2) an unfrozen, randomly-initialized residual model that will learn the unbiased sequence rules associated with ATAC-seq cut site coverage. During training, the sequence information will pass through both of these models and their respective outputs will be added together to represent training loss. By adding the two model outputs, ChromBPNet is evaluating both Tn5 sequence bias and sequence rules of accessibility, which can be compared to the actual ATAC-seq cut site coverage (which also possesses both of these features). After the training step has been completed, we remove the frozen Tn5 bias model and apply downstream interpretations only to the second model which contains the unbiased sequence rules that explain accessibility coverage of ATAC-seq cut sites.
To train the highest-quality set of models in the Drosophila genome, we trained a custom Tn5 bias model to represent the Tn5 sequence bias in our data. The Tn5 bias model architecture followed ChromBPNet defaults. The Tn5 bias model output was the pooled coverage of the 2.5–3 h ATAC-seq experiments. This time point was chosen for the bias model because it was the most likely time in which this model could have learned underlying sequence grammar of interest and therefore the most optimal to validate against. The Tn5 bias model inputs were genomic regions that met the following criteria: (1) closed (non-peak ATAC-seq regions across all time points), (2) unbound (non-peak ChIP-nexus regions across all TFs described above), (3) low-coverage regions (containing less than five times the cut sites as the lowest coverage 2.5–3 h ATAC-seq IDR-reproducible peak region), (4) 2114 bp in width, and (5) at least 750bp away from an annotated fly TSS. These criteria were applied in order to ensure that Tn5 sequence bias was only learned at regions that were closed, inactive, and representative of noise-based cut site coverage. After application of these criteria, the Tn5 bias model was trained on 2,326 training regions and 883 validation regions. Training, validation, and test regions were determined based on the chromosomes reported above for BPNet. In order to validate that the Tn5 bias model learned only Tn5 sequence bias and no other grammar rules, particularly motif-driven rules, we collected Tn5 counts and Tn5 profile contribution scores using the DeepSHAP implementation of DeepLIFT (https://github.com/kundajelab/shap163) and ran TF-MoDISco (v.0.5.16.0164). For profile contribution, the Tn5 sequence bias was returned (Figure S2E), but no motif consensus logos were returned. For counts contribution, neither Tn5 nor motif consensus logos were returned. This confirmed that our Tn5 bias model was only learning positional Tn5 sequence bias information. In order to follow-up this validation, we injected the sequences of likely canonical motifs into 256 genomic sequences from the test chromosome (chrX) and averaged the effects to observe that the Tn5 bias model did not predict an increase in coverage magnitude (Figure S2D).
After Tn5 bias model training, ChromBPNet architecture and software was applied (https://github.com/kundajelab/chrombpnet). Model inputs were 2114 bp genomic sequences centered on IDR-reproducible ATAC-seq peaks. To fairly compare the results between four ChromBPNet models for each developmental time point measured using ATAC-seq (1–1.5 h, 1.5–2 h, 2–2.5 h, 2.5–3 h), we sought to train each of the models with the pooled IDR-reproducible ATAC-seq peaks from every time point measured. Additionally, because we wished to characterize enhancer accessibility rules, we removed peaks that were within 750 bp of an annotated TSS, as we know that accessibility at promoters can be dictated by different sequence rules than at enhancers. After the time points were pooled and promoter-proximal peaks removed, 41,497 ATAC-seq peaks were included. In order to train more robust models, we also included curated non-peak regions (described above) sampled to 10% of the ATAC-seq peaks for training (4,150 non-peak regions). The inclusion of both peak and non-peak ATAC-seq regions allows the model to better differentiate between accessible and inaccessible sequences. In total, 45,647 regions were used as ChromBPNet model inputs. Validation datasets were peaks located across chr2L (~16% of peaks), test datasets were peaks located across chrX (~19% of peaks), and peaks located across chrY and nonstandard chromosome contigs were excluded from analysis. The remaining regions were used for model training. In addition to shared peaks across different ChromBPNet models to maintain inter-model stability, we also sought to train each of the models with the same ChromBPNet architecture. For this, an optimization search was required, and we again decided to optimize on the pooled coverage of the 2.5–3 h ATAC-seq experiments through selected testing of parameter values deviating from the default ChromBPNet architecture (number of filters in each convolutional layer, filter length of the first convolutional layer, and filter length of the deconvolutional layer). Model optimality was assessed based on the counts and profile performance of the bias-removed predictions, as well as prioritizing model depth to avoid over-distribution of motif grammar within sequence representations. After optimization, the final ChromBPNet model architecture contained 128 filters in each convolutional layer and a filter length of 7 bp for both the input convolutional layer and 75 bp for the output deconvolutional layer. We then trained ChromBPNet models on the pooled cut site coverage of the four developmental time point ATAC-seq experiments (1–1.5 h, 1.5–2 h, 2–2.5 h, 2.5–3 h). Final optimized model performance was assessed through comparing (1) the ability of the model to differentiate peak and non-peak regions using area under the receiver operating characteristic curve (ROC AUC) (Figure S2F), (2) counts correlations of observed ATAC-seq cut sites to ChromBPNet predictions (Figure S2G), and (3) profile prediction accuracy of observed ATAC-seq cut sites to ChromBPNet predictions using Jensen-Shannon distances benchmarked by randomly shuffled region profiles (Figure S2H). In order to test the stability of these different ChromBPNet models, we trained two additional models across each ATAC-seq time point with shuffled training, validation, and test sets (three-fold validation). The stability of the performance metrics as well as the stability of the returned downstream motif grammar was compared to the original optimized model training event (fold 1). All ChromBPNet models were implemented and trained using Keras (v2.5.0160), TensorFlow2 backend (v.2.5.1161), and the Adam optimizer.162 Training was performed using a NVIDIA® TITAN RTX GPU with CUDA v11.0 and cuDNN v8.3.0 drivers.
ChromBPNet contribution score generation and validation
DeepLIFT (v0.6.13.0, derived from the Kundaje Lab fork of DeepSHAP (https://github.com/AvantiShri/shap)163) was applied to the trained ChromBPNet model to generate the contribution of each base across a given input sequence to the predicted output counts and profile signals. Contribution scores for counts and profile outputs were generated for each trained ChromBPNet model across all time points (1–1.5 h, 1.5–2 h, 2–2.5 h, 2.5–3 h). TF-MoDISco (v.0.5.16.0164) was then applied for each trained ChromBPNet model in order to identify regions of high counts contribution, cluster based on within-group contribution and sequence similarity, and consolidate these clusters into motifs. Pertinent motifs (Zelda, GAF, Caudal, Twist-like, and Dorsal-like) were manually identified based on similarity to previous literature and ChIP-nexus binding was measured across these accessibility-identified motifs to validate that they were indeed relevant binding sites that also contribute towards explaining the ChromBPNet models across the designated time points (Figure S2I).
Using binding and accessibility models to examine motif effects in silico
In order to internally measure the “marginalized” effects of motifs without the surrounding genomic context, we adopted an in silico approach by which we injected motifs into many seed-controlled randomized sequences and generated BPNet and ChromBPNet predictions of these sequences with and without the motifs. We used 64 randomized sequences for BPNet predictions and 512 for ChromBPNet predictions (accessibility predictions contain greater sequence complexity and therefore required more trials to establish stable predictions across randomly generated sequences), averaging predictions across each of these randomized sequence sets. After performing in silico injections of a single motif, we visualized the output profiles generated from randomized sequence alone or motif-injected sequences for the Tn5 bias model, ChromBPNet models, and BPNet across all TF motifs.
It has been previously described that accurate predictions of relative motif affinities can be extracted from a BPNet model trained on ChIP-nexus data.89–91 We then summarized the “marginalized” effects of motifs above to compare how motif affinity changes Zelda’s influence at the level of both binding and accessibility. After performing in silico injections of a single motif described above, we summed the values of the output profiles generated from randomized sequence alone or motif-injected sequences for both ChromBPNet and BPNet. These sums were then subtracted in log-space and referred to as “marginalized” scores, characterized as:
where is the predicted sum of the counts when a motif is injected into the random sequence and is the predicted sum of the counts of the averaged random sequences without injections. These “marginalized” scores were computed for each Zelda motif variant for all ChromBPNet models and BPNet.
In order to test the effects of motif pairs on cooperativity for binding and accessibility without surrounding genomic context, in silico motif interaction analysis was performed to measure “binding enhancement” as described previously.59 In brief, this involved injecting two motif sequences (motif A and motif B) across motif pair distances ranging up to 400 bp into random sequences. Binding predictions and accessibility predictions were measured in these different simulation scenarios from BPNet (where represents the sum of the counts predicted across a 200 bp window, centered on motif A) and ChromBPNet (where represents the sum of the counts predicted across the entire 1000 bp window), respectively. We measured four different cases: (1) neither motif A nor motif B were injected into the sequence , (2) motif A only was injected into the sequence , (3) motif B only was injected into the sequence , and (4) motif A and motif B were both injected into the sequence at a designated distance . These cases were measured and averaged across 64 trials for BPNet predictions and 512 trials for ChromBPNet predictions (accessibility predictions contain greater sequence complexity and therefore required more trials to establish stable predictions across randomly generated sequences). After all measurements were collected across all motif combinations and distances, then averaged across trials, the in silico motif pair cooperativity for each was calculated using the following equation:
where is the predicted pseudocounts represented by the 20th percentile quantile cutoff value for both binding and accessibility predictions across each window when motif A and motif B are present and when only motif A is present (case 4 and 2, respectively, described above). The motif pairs considered were combinations of the highest affinity representations of Zelda (CAGGTAG), Dorsal (GGGAAAACCC), Twist (AACACATGTT), Caudal (TTTTATGGCC), Bicoid (TTAATCC), and GAF (GAGAGAGAGAGAGAGAG). For both BPNet and all ChromBPNet models, these high-affinity motifs were also tested alongside an additional lower affinity representation of Zelda (TAGGTAG) in a pairwise fashion with all other motifs to investigate Zelda’s changing influence on other TFs based on motif affinity.
Using binding and accessibility models to examine motif effects in genomic sequences
In order to measure the in-context effects of a motif within its surrounding genomic sequence, we computationally generated genomic sequences with this motif’s sequence mutated by randomly shuffling the bases that belong to this motif. We generated 16 randomized mutation sequences per motif instance to establish mutation stability, averaging predictions across each of these randomized mutation sets. We performed this genomic perturbation for all mapped TF motifs across our curated set of genomic enhancers (described above) and visualized the output profiles generated for both BPNet and all ChromBPNet models.
In order to summarize the accessibility effects of mutating high- and low-affinity Zelda motifs, the 250 highest- and lowest-affinity Zelda motif-containing-only islands were identified. Using the procedure described above for all Zelda motifs in these genomic islands, accessibility profiles from unmodified island sequences and Zelda-mutated island sequences were predicted using the ChromBPNet models. After generating the profiles for each island, we summed the profiles into a single scalar value for WT sequences and Zelda-mutated sequences . Relative accessibility effects of high- and low-affinity Zelda motifs were characterized by the log2 fold-change measured effect, represented as .
Differential chromatin accessibility analysis
To determine the differential chromatin accessibility between wt embryos with mutant zld−, gd7, and cic6 embryos, we used DESeq2 with default parameters and FDR = 0.05.109 Briefly, for each comparison between wt and mutant ATAC-seq data sets, we calculated ATAC-seq cut site coverage at the same pooled IDR-reproducible ATAC-seq peaks from all time points that were used for ChromBPNet prior to promoter removal (see “ChromBPNet model training and optimization”). For all time points we used three replicates and built one DESeq model encompassing ATAC-seq counts from all time points. To compute the differential chromatin accessibility, we then used each DESeq2 model to conduct pairwise comparisons between between wt and mutant conditions within each time point and computed the log2(mutant/wt) values. In this way, log2(mutant/wt) < 0 represent a loss in chromatin accessibility in the mutant, while log2(mutant/wt) > 0 represent a gain in chromatin accessibility in the mutant, while p-adjusted < 0.05 loci are highlighted. We performed this differential chromatin accessibility approach for all wt-to-mutant comparisons.
Enhancer collection
The bulk set of mesodermal and dorsal ectodermal enhancers used in this study were previously defined based on differential histone acetylation and have been validated three-fold using 1) Vienna tiles,165 2) TF binding and motif enrichment analysis, and 3) differential RNA-seq expression of nearby target genes across the dorsoventral axis.108 More limited sets of validated neuroectodermal enhancers were collected from previous work.78,166 All anterior-posterior patterning enhancers were collected from earlier studies.75,76 We additionally used a bulk, highly curated set of enhancers that were previously characterized as active in blastoderm embryos based on 1) in situ hybridization images, 2) transgenic reporters, 3) Vienna tiles, and 4) the REDfly167 database when calculating motif island overlaps with active enhancers.74
QUANTIFICATION AND STATISTICAL ANALYSIS
All computational and statistical analyses performed, software used, and data processing steps are described in their respective methods sections. No further statistical analyses were conducted. Figure legends describe the details of the data plotted, including what statistical tests were performed, significance, and sample size. All code used to analyze and plot the data has been deposited at https://github.com/zeitlingerlab/Brennan_Zelda_2023 and software information is presented in the key resources table.
Supplementary Material
Highlights.
Deep learning identifies DNA sequence rules of TFs in the early Drosophila embryo
Zelda consistently pioneers chromatin accessibility proportional to motif affinity
Activators depend on Zelda and augment accessibility when mediating activation
Chromatin accessibility comes from pioneering and sequence context-dependent activation
ACKNOWLEDGMENTS
We thank Žiga Avsec, Robb Krumlauf, Kausik Si, Vikki Weake, and Zeitlinger lab members for helpful comments and suggestions. We thank Martha Weilert and Mark Miller for help with illustrations; and Beth Canfield for help with Drosophila husbandry. We also thank the Stowers Technology Centers for support: Sequencing and Discovery Genomics (Anoja Perera, Michael Peterson, and Amanda Lawlor), Lab Services (Stacey Walker), Cytometry (Jeff Haug and KyeongMin Bae), and Computational Biology (Hua Li, Madelaine Gogol, and Jonathon Russell). This research was funded by the Stowers Institute for Medical Research, the Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health under the F31 award number F31HD108901 to K.J.B, the Stanford BioX Fellowship to A.P., the NIH grants RO1GM63024 and R35GM148241 to C.A.R., and the Canadian Institutes of Health Research grant FDN-148403 to T.R.H. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
J.Z. owns a patent on ChIP-nexus (no. 10287628). A.K. is on the scientific advisory board of PatchBio, SerImmune, AINovo, TensorBio and OpenTargets, was a consultant with Illumina, and owns shares in Illumina, Deep Genomics, Immunai, and Freenome Inc. All other authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.devcel.2023.07.007.
REFERENCES
- 1.Spitz F, and Furlong EEM (2012). Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet. 13, 613–626. 10.1038/nrg3207. [DOI] [PubMed] [Google Scholar]
- 2.Levine M, and Davidson EH (2005). Gene regulatory networks for development. Proc. Natl. Acad. Sci. USA 102, 4936–4942. 10.1073/pnas.0408031102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeitlinger J (2020). Seven myths of how transcription factors read the cis-regulatory code. Curr. Opin. Syst. Biol. 23, 22–31. 10.1016/j.coisb.2020.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barozzi I, Simonatto M, Bonifacio S, Yang L, Rohs R, Ghisletti S, and Natoli G (2014). Coregulation of transcription factor binding and nucleosome occupancy through DNA features of mammalian enhancers. Mol. Cell 54, 844–857. 10.1016/j.molcel.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li X-Y, and Eisen MB (2018). Zelda potentiates transcription factor binding to zygotic enhancers by increasing local chromatin accessibility during early Drosophila melanogaster embryogenesis. 10.1101/380857. [DOI] [Google Scholar]
- 6.Sun Y, Nien CY, Chen K, Liu HY, Johnston J, Zeitlinger J, and Rushlow C (2015). Zelda overcomes the high intrinsic nucleosome barrier at enhancers during Drosophila zygotic genome activation. Genome Res. 25, 1703–1714. 10.1101/gr.192542.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tillo D, Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Field Y, Lieb JD, Widom J, Segal E, and Hughes TR (2010). High nucleosome occupancy is encoded at human regulatory sequences. PLoS One 5, e9129. 10.1371/journal.pone.0009129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Veil M, Yampolsky LY, Grüning B, and Onichtchouk D (2019). Pou5f3, SoxB1, and Nanog remodel chromatin on high nucleosome affinity regions at zygotic genome activation. Genome Res. 29, 383–395. 10.1101/gr.240572.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iwafuchi-Doi M, and Zaret KS (2014). Pioneer transcription factors in cell reprogramming. Genes Dev. 28, 2679–2692. 10.1101/gad.253443.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zaret KS (2020). Pioneer transcription factors initiating gene network changes. Annu. Rev. Genet. 54, 367–385. 10.1146/annurev-genet-030220-015007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larson ED, Marsh AJ, and Harrison MM (2021). Pioneering the developmental frontier. Mol. Cell 81, 1640–1650. 10.1016/j.molcel.2021.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Swinstead EE, Paakinaho V, Presman DM, and Hager GL (2016). Pioneer factors and ATP-dependent chromatin remodeling factors interact dynamically: A new perspective: multiple transcription factors can effect chromatin pioneer functions through dynamic interactions with ATP-dependent chromatin remodeling factors. BioEssays 38, 1150–1157. 10.1002/bies.201600137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao Y, Vartak SV, Conte A, Wang X, Garcia DA, Stevens E, Kyoung Jung S, Kieffer-Kwon K-R, Vian L, Stodola T, et al. (2022). “Stripe” transcription factors provide accessibility to co-binding partners in mammalian genomes. Mol. Cell 82, 3398–3411.e11. 10.1016/j.molcel.2022.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mirny LA (2010). Nucleosome-mediated cooperativity between transcription factors. Proc. Natl. Acad. Sci. USA 107, 22534–22539. 10.1073/pnas.0913805107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eck E, Liu J, Kazemzadeh-Atoufi M, Ghoreishi S, Blythe SA, and Garcia HG (2020). Quantitative dissection of transcription in development yields evidence for transcription-factor-driven chromatin accessibility. eLife 9, e56429. 10.7554/eLife.56429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adams CC, and Workman JL (1995). Binding of disparate transcriptional activators to nucleosomal DNA is inherently cooperative. Mol. Cell. Biol. 15, 1405–1421. 10.1128/MCB.15.3.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hansen JL, Loell KJ, and Cohen BA (2022). A test of the pioneer factor hypothesis using ectopic liver gene activation. eLife 11, e73358. 10.7554/eLife.73358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hansen JL, and Cohen BA (2022). A quantitative metric of pioneer activity reveals that HNF4A has stronger in vivo pioneer activity than FOXA1. Genome Biol. 23, 221. 10.1186/s13059-022-02792-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sherwood RI, Hashimoto T, O’Donnell CW, Lewis S, Barkal AA, van Hoff JP, Karun V, Jaakkola T, and Gifford DK (2014). Discovery of directional and nondirectional pioneer transcription factors by modeling DNase profile magnitude and shape. Nat. Biotechnol. 32, 171–178. 10.1038/nbt.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fernandez Garcia M, Moore CD, Schulz KN, Alberto O, Donague G, Harrison MM, Zhu H, and Zaret KS (2019). Structural features of transcription factors associating with nucleosome binding. Mol. Cell 75, 921–932.e6. 10.1016/j.molcel.2019.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu F, Farnung L, Kaasinen E, Sahu B, Yin Y, Wei B, Dodonova SO, Nitta KR, Morgunova E, Taipale M, et al. (2018). The interaction landscape between transcription factors and the nucleosome. Nature 562, 76–81. 10.1038/s41586-018-0549-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sekiya T, Muthurajan UM, Luger K, Tulin AV, and Zaret KS (2009). Nucleosome-binding affinity as a primary determinant of the nuclear mobility of the pioneer transcription factor FoxA. Genes Dev. 23, 804–809. 10.1101/gad.1775509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meers MP, Janssens DH, and Henikoff S (2019). Pioneer factor-nucleosome binding events during differentiation are motif encoded. Mol. Cell 75, 562–575.e5. 10.1016/j.molcel.2019.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soufi A, Garcia MF, Jaroszewicz A, Osman N, Pellegrini M, and Zaret KS (2015). Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 161, 555–568. 10.1016/j.cell.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eraslan G, Avsec Ž, Gagneur J, and Theis FJ (2019). Deep learning: new computational modelling techniques for genomics. Nat. Rev. Genet. 20, 389–403. 10.1038/s41576-019-0122-6. [DOI] [PubMed] [Google Scholar]
- 26.Novakovsky G, Dexter N, Libbrecht MW, Wasserman WW, and Mostafavi S (2023). Obtaining genetics insights from deep learning via explainable artificial intelligence. Nat. Rev. Genet. 24, 125–137. 10.1038/s41576-022-00532-2. [DOI] [PubMed] [Google Scholar]
- 27.Maslova A, Ramirez RN, Ma K, Schmutz H, Wang C, Fox C, Ng B, Benoist C, and Mostafavi S; Immunological; Genome Project (2020). Deep learning of immune cell differentiation. Proc. Natl. Acad. Sci. USA 117, 25655–25666. 10.1073/pnas.2011795117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Atak ZK, Taskiran II, Demeulemeester J, Flerin C, Mauduit D, Minnoye L, Hulselmans G, Christiaens V, Ghanem G-E, Wouters J, et al. (2021). Interpretation of allele-specific chromatin accessibility using cell state-aware deep learning. Genome Res. 31, 1082–1096. 10.1101/gr.260851.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trevino AE, Müller F, Andersen J, Sundaram L, Kathiria A, Shcherbina A, Farh K, Chang HY, Pașca AM, Kundaje A, et al. (2021). Chromatin and gene-regulatory dynamics of the developing human cerebral cortex at single-cell resolution. Cell 184, 5053–5069.e23. 10.1016/j.cell.2021.07.039. [DOI] [PubMed] [Google Scholar]
- 30.Kim DS, Risca VI, Reynolds DL, Chappell J, Rubin AJ, Jung N, Donohue LKH, Lopez-Pajares V, Kathiria A, Shi M, et al. (2021). The dynamic, combinatorial cis-regulatory lexicon of epidermal differentiation. Nat. Genet. 53, 1564–1576. 10.1038/s41588-021-00947-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Minnoye L, Taskiran II, Mauduit D, Fazio M, Van Aerschot L, Hulselmans G, Christiaens V, Makhzami S, Seltenhammer M, Karras P, et al. (2020). Cross-species analysis of enhancer logic using deep learning. Genome Res. 30, 1815–1834. 10.1101/gr.260844.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Minnoye L, Marinov GK, Krausgruber T, Pan L, Marand AP, Secchia S, Greenleaf WJ, Furlong EEM, Zhao K, Schmitz RJ, et al. (2021). Chromatin accessibility profiling methods. Nat. Rev. Methods Primers 1, 10. 10.1038/s43586-020-00008-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schulz KN, and Harrison MM (2019). Mechanisms regulating zygotic genome activation. Nat. Rev. Genet. 20, 221–234. 10.1038/s41576-018-0087-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vastenhouw NL, Cao WX, and Lipshitz HD (2019). The maternal-to-zygotic transition revisited. Development 146, dev161471. 10.1242/dev.161471. [DOI] [PubMed] [Google Scholar]
- 35.Kwasnieski JC, Orr-Weaver TL, and Bartel DP (2019). Early genome activation in Drosophila is extensive with an initial tendency for aborted transcripts and retained introns. Genome Res. 29, 1188–1197. 10.1101/gr.242164.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Small S, and Arnosti DN (2020). Transcriptional enhancers in drosophila. Genetics 216, 1–26. 10.1534/genetics.120.301370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liang H-L, Nien C-Y, Liu H-Y, Metzstein MM, Kirov N, and Rushlow C (2008). The zinc-finger protein Zelda is a key activator of the early zygotic genome in Drosophila. Nature 456, 400–403. 10.1038/nature07388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nien C-Y, Liang H-L, Butcher S, Sun Y, Fu S, Gocha T, Kirov N, Manak JR, and Rushlow C (2011). Temporal coordination of gene networks by Zelda in the early Drosophila embryo. PLoS Genet. 7, e1002339. 10.1371/journal.pgen.1002339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harrison MM, Li XY, Kaplan T, Botchan MR, and Eisen MB (2011). Zelda binding in the early Drosophila melanogaster embryo marks regions subsequently activated at the maternal-to-zygotic transition. PLoS Genet. 7, e1002266. 10.1371/journal.pgen.1002266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Satija R, and Bradley RK (2012). The TAGteam motif facilitates binding of 21 sequence-specific transcription factors in the Drosophila embryo. Genome Res. 22, 656–665. 10.1101/gr.130682.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schulz KN, Bondra ER, Moshe A, Villalta JE, Lieb JD, Kaplan T, McKay DJ, and Harrison MM (2015). Zelda is differentially required for chromatin accessibility, transcription factor binding, and gene expression in the early Drosophila embryo. Genome Res. 25, 1715–1726. 10.1101/gr.192682.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li X-Y, Harrison MM, Villalta JE, Kaplan T, and Eisen MB (2014). Establishment of regions of genomic activity during the Drosophila maternal to zygotic transition. eLife 3, e03737. 10.7554/eLife.03737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Foo SM, Sun Y, Lim B, Ziukaite R, O’Brien K, Nien C-Y, Kirov N, Shvartsman SY, and Rushlow CA (2014). Zelda potentiates morphogen activity by increasing chromatin accessibility. Curr. Biol. 24, 1341–1346. 10.1016/j.cub.2014.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kanodia JS, Liang H-L, Kim Y, Lim B, Zhan M, Lu H, Rushlow CA, and Shvartsman SY (2012). Pattern formation by graded and uniform signals in the early Drosophila embryo. Biophys. J. 102, 427–433. 10.1016/j.bpj.2011.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yáñez-Cuna JO, Dinh HQ, Kvon EZ, Shlyueva D, and Stark A (2012). Uncovering cis-regulatory sequence requirements for context-specific transcription factor binding. Genome Res. 22, 2018–2030. 10.1101/gr.132811.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu Z, Chen H, Ling J, Yu D, Struffi P, and Small S (2014). Impacts of the ubiquitous factor Zelda on bicoid-dependent DNA binding and transcription in Drosophila. Genes Dev. 28, 608–621. 10.1101/gad.234534.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mir M, Stadler MR, Ortiz SA, Hannon CE, Harrison MM, Darzacq X, and Eisen MB (2018). Dynamic multifactor hubs interact transiently with sites of active transcription in Drosophila embryos. eLife 7, e40497. 10.7554/eLife.40497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mir M, Reimer A, Haines JE, Li X-Y, Stadler M, Garcia H, Eisen MB, and Darzacq X (2017). Dense bicoid hubs accentuate binding along the morphogen gradient. Genes Dev. 31, 1784–1794. 10.1101/gad.305078.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McDaniel SL, Gibson TJ, Schulz KN, Fernandez Garcia M, Nevil M, Jain SU, Lewis PW, Zaret KS, and Harrison MM (2019). Continued activity of the pioneer factor Zelda is required to drive zygotic genome activation. Mol. Cell 74, 185–195.e4. 10.1016/j.molcel.2019.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gaskill MM, Gibson TJ, Larson ED, and Harrison MM (2021). GAF is essential for zygotic genome activation and chromatin accessibility in the early Drosophila embryo. eLife 10, e66668. 10.7554/eLife.66668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blythe SA, and Wieschaus EF (2016). Establishment and maintenance of heritable chromatin structure during early Drosophila embryogenesis. eLife 5, e20148. 10.7554/eLife.20148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Duan J, Rieder L, Colonnetta MM, Huang A, Mckenney M, Watters S, Deshpande G, Jordan W, Fawzi N, and Larschan E (2021). CLAMP and Zelda function together to promote Drosophila zygotic genome activation. eLife 10, e69937. 10.7554/eLife.69937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fuda NJ, Guertin MJ, Sharma S, Danko CG, Martins AL, Siepel A, and Lis JT (2015). GAGA factor maintains nucleosome-free regions and has a role in RNA polymerase II recruitment to promoters. PLoS Genet. 11, e1005108. 10.1371/journal.pgen.1005108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moshe A, and Kaplan T (2017). Genome-wide search for Zelda-like chromatin signatures identifies GAF as a pioneer factor in early fly development. Epigenetics Chromatin 10, 33. 10.1186/s13072-017-0141-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.MacArthur S, Li X-Y, Li J, Brown JB, Chu HC, Zeng L, Grondona BP, Hechmer A, Simirenko L, Keränen SVE, et al. (2009). Developmental roles of 21 Drosophila transcription factors are determined by quantitative differences in binding to an overlapping set of thousands of genomic regions. Genome Biol. 10, R80. 10.1186/gb-2009-10-7-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Combs PA, and Eisen MB (2017). Genome-wide measurement of spatial expression in patterning mutants of Drosophila melanogaster. [version 1; peer review: 2 approved]. F1000Res 6, 41. 10.12688/f1000research.9720.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hannon CE, Blythe SA, and Wieschaus EF (2017). Concentration dependent chromatin states induced by the bicoid morphogen gradient. eLife 6, e28275. 10.7554/eLife.28275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.He Q, Johnston J, and Zeitlinger J (2015). ChIP-nexus enables improved detection of in vivo transcription factor binding footprints. Nat. Biotechnol. 33, 395–401. 10.1038/nbt.3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Avsec Ž, Weilert M, Shrikumar A, Krueger S, Alexandari A, Dalal K, Fropf R, McAnany C, Gagneur J, Kundaje A, et al. (2021). Base-resolution models of transcription-factor binding reveal soft motif syntax. Nat. Genet. 53, 354–366. 10.1038/s41588-021-00782-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yamada S, Whitney PH, Huang S-K, Eck EC, Garcia HG, and Rushlow CA (2019). The drosophila pioneer factor Zelda modulates the nuclear microenvironment of a dorsal target enhancer to potentiate transcriptional output. Curr. Biol. 29, 1387–1393.e5. 10.1016/j.cub.2019.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hong J-W, Hendrix DA, Papatsenko D, and Levine MS (2008). How the Dorsal gradient works: insights from postgenome technologies. Proc. Natl. Acad. Sci. USA 105, 20072–20076. 10.1073/pnas.0806476105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dunipace L, Ákos Z, and Stathopoulos A (2019). Coacting enhancers can have complementary functions within gene regulatory networks and promote canalization. PLOS Genet. 15, e1008525. 10.1371/journal.pgen.1008525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shin D-H, and Hong J-W (2017). The short gastrulation shadow enhancer employs dual modes of transcriptional synergy. Int. J. Dev. Biol. 61, 73–80. 10.1387/ijdb.160165jh. [DOI] [PubMed] [Google Scholar]
- 64.Crocker J, Tamori Y, and Erives A (2008). Evolution acts on enhancer organization to fine-tune gradient threshold readouts. PLoS Biol. 6, e263. 10.1371/journal.pbio.0060263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hong J-W, Hendrix DA, and Levine MS (2008). Shadow enhancers as a source of evolutionary novelty. Science 321, 1314. 10.1126/science.1160631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shirokawa JM, and Courey AJ (1997). A direct contact between the dorsal rel homology domain and Twist may mediate transcriptional synergy. Mol. Cell. Biol. 17, 3345–3355. 10.1128/MCB.17.6.3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bhaskar V, and Courey AJ (2002). The MADF-BESS domain factor Dip3 potentiates synergistic activation by Dorsal and Twist. Gene 299, 173–184. 10.1016/s0378-1119(02)01058-2. [DOI] [PubMed] [Google Scholar]
- 68.Jiang J, and Levine M (1993). Binding affinities and cooperative interactions with bHLH activators delimit threshold responses to the dorsal gradient morphogen. Cell 72, 741–752. 10.1016/0092-8674(93)90402-c. [DOI] [PubMed] [Google Scholar]
- 69.He Q, Bardet AF, Patton B, Purvis J, Johnston J, Paulson A, Gogol M, Stark A, and Zeitlinger J (2011). High conservation of transcription factor binding and evidence for combinatorial regulation across six Drosophila species. Nat. Genet. 43, 414–420. 10.1038/ng.808. [DOI] [PubMed] [Google Scholar]
- 70.Judd J, Duarte FM, and Lis JT (2021). Pioneer-like factor GAF cooperates with PBAP (SWI/SNF) and NURF (ISWI) to regulate transcription. Genes Dev. 35, 147–156. 10.1101/gad.341768.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tsukiyama T, Becker PB, and Wu C (1994). ATP-dependent nucleosome disruption at a heat-shock promoter mediated by binding of GAGA transcription factor. Nature 367, 525–532. 10.1038/367525a0. [DOI] [PubMed] [Google Scholar]
- 72.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cusanovich DA, Reddington JP, Garfield DA, Daza RM, Aghamirzaie D, Marco-Ferreres R, Pliner HA, Christiansen L, Qiu X, Steemers FJ, et al. (2018). The cis-regulatory dynamics of embryonic development at single-cell resolution. Nature 555, 538–542. 10.1038/nature25981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Haines JE, and Eisen MB (2018). Patterns of chromatin accessibility along the anterior-posterior axis in the early Drosophila embryo. PLoS Genet. 14, e1007367. 10.1371/journal.pgen.1007367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bozek M, Cortini R, Storti AE, Unnerstall U, Gaul U, and Gompel N (2019). ATAC-seq reveals regional differences in enhancer accessibility during the establishment of spatial coordinates in the Drosophila blastoderm. Genome Res. 29, 771–783. 10.1101/gr.242362.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Calderon D, Blecher-Gonen R, Huang X, Secchia S, Kentro J, Daza RM, Martin B, Dulja A, Schaub C, Trapnell C, et al. (2022). The continuum of Drosophila embryonic development at single-cell resolution. Science 377, eabn5800. 10.1126/science.abn5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Koenecke N, Johnston J, He Q, Meier S, and Zeitlinger J (2017). Drosophila poised enhancers are generated during tissue patterning with the help of repression. Genome Res. 27, 64–74. 10.1101/gr.209486.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Irizarry J, and Stathopoulos A (2021). Dynamic patterning by morphogens illuminated by cis-regulatory studies. Development 148, dev196113. 10.1242/dev.196113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen K, Johnston J, Shao W, Meier S, Staber C, and Zeitlinger J (2013). A global change in RNA polymerase II pausing during the Drosophila midblastula transition. eLife 2, e00861. 10.7554/eLife.00861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bentsen M, Goymann P, Schultheis H, Klee K, Petrova A, Wiegandt R, Fust A, Preussner J, Kuenne C, Braun T, et al. (2020). ATAC-seq footprinting unravels kinetics of transcription factor binding during zygotic genome activation. Nat. Commun. 11, 4267. 10.1038/s41467-020-18035-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li Z, Schulz MH, Look T, Begemann M, Zenke M, and Costa IG (2019). Identification of transcription factor binding sites using ATAC-seq. Genome Biol. 20, 45. 10.1186/s13059-019-1642-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jung C, Bandilla P, von Reutern M, Schnepf M, Rieder S, Unnerstall U, and Gaul U (2018). True equilibrium measurement of transcription factor-DNA binding affinities using automated polarization microscopy. Nat. Commun. 9, 1605. 10.1038/s41467-018-03977-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Datta RR, Ling J, Kurland J, Ren X, Xu Z, Yucel G, Moore J, Shokri L, Baker I, Bishop T, et al. (2018). A feed-forward relay integrates the regulatory activities of bicoid and Orthodenticle via sequential binding to suboptimal sites. Genes Dev. 32, 723–736. 10.1101/gad.311985.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Maerkl SJ, and Quake SR (2007). A systems approach to measuring the binding energy landscapes of transcription factors. Science 315, 233–237. 10.1126/science.1131007. [DOI] [PubMed] [Google Scholar]
- 86.Dodonova SO, Zhu F, Dienemann C, Taipale J, and Cramer P (2020). Nucleosome-bound SOX2 and SOX11 structures elucidate pioneer factor function. Nature 580, 669–672. 10.1038/s41586-020-2195-y. [DOI] [PubMed] [Google Scholar]
- 87.Michael AK, Grand RS, Isbel L, Cavadini S, Kozicka Z, Kempf G, Bunker RD, Schenk AD, Graff-Meyer A, Pathare GR, et al. (2020). Mechanisms of OCT4-SOX2 motif readout on nucleosomes. Science 368, 1460–1465. 10.1126/science.abb0074. [DOI] [PubMed] [Google Scholar]
- 88.Michael AK, and Thomä NH (2021). Reading the chromatinized genome. Cell 184, 3599–3611. 10.1016/j.cell.2021.05.029. [DOI] [PubMed] [Google Scholar]
- 89.Horton CA, Alexandari AM, Hayes MGB, Marklund E, Schaepe JM, Aditham AK, Shah N, Shrikumar A, Afek A, Greenleaf WJ, et al. (2022). Short tandem repeats bind transcription factors to tune eukaryotic gene expression. 10.1101/2022.05.24.493321. [DOI] [PubMed] [Google Scholar]
- 90.Koo PK, Majdandzic A, Ploenzke M, Anand P, and Paul SB (2021). Global importance analysis: an interpretability method to quantify importance of genomic features in deep neural networks. PLoS Comput. Biol. 17, e1008925. 10.1371/journal.pcbi.1008925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Alexandari AM, Horton CA, Shrikumar A, Shah N, Li E, Weilert M, Pufall MA, Zeitlinger J, Fordyce PM, and Kundaje A (2023). De novo distillation of thermodynamic affinity from deep learning regulatory sequence models of in vivo protein-DNA binding. 10.1101/2023.05.11.540401. [DOI] [Google Scholar]
- 92.Harrison MM, Botchan MR, and Cline TW (2010). Grainyhead and Zelda compete for binding to the promoters of the earliest-expressed Drosophila genes. Dev. Biol. 345, 248–255. 10.1016/j.yd-bio.2010.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.ten Bosch JR, Benavides JA, and Cline TW (2006). The TAGteam DNA motif controls the timing of Drosophila pre-blastoderm transcription. Development 133, 1967–1977. 10.1242/dev.02373. [DOI] [PubMed] [Google Scholar]
- 94.Berger MF, Philippakis AA, Qureshi AM, He FS, Estep PW, and Bulyk ML (2006). Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat. Biotechnol. 24, 1429–1435. 10.1038/nbt1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, et al. (2014). Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443. 10.1016/j.cell.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bulyk ML, Huang X, Choo Y, and Church GM (2001). Exploring the DNA-binding specificities of zinc fingers with DNA microarrays. Proc. Natl. Acad. Sci. USA 98, 7158–7163. 10.1073/pnas.111163698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Siggers T, Duyzend MH, Reddy J, Khan S, and Bulyk ML (2011). Non-DNA-binding cofactors enhance DNA-binding specificity of a transcriptional regulatory complex. Mol. Syst. Biol. 7, 555. 10.1038/msb.2011.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Badis G, Berger MF, Philippakis AA, Talukder S, Gehrke AR, Jaeger SA, Chan ET, Metzler G, Vedenko A, Chen X, et al. (2009). Diversity and complexity in DNA recognition by transcription factors. Science 324, 1720–1723. 10.1126/science.1162327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li L, and Wunderlich Z (2017). An enhancer’s length and composition are shaped by its regulatory task. Front. Genet. 8, 63. 10.3389/fgene.2017.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shlyueva D, Stelzer C, Gerlach D, Yáñez-Cuna JO, Rath M, Boryń ŁM, Arnold CD, and Stark A (2014). Hormone-responsive enhancer-activity maps reveal predictive motifs, indirect repression, and targeting of closed chromatin. Mol. Cell 54, 180–192. 10.1016/j.molcel.2014.02.026. [DOI] [PubMed] [Google Scholar]
- 101.McKay DJ, and Lieb JD (2013). A common set of DNA regulatory elements shapes Drosophila appendages. Dev. Cell 27, 306–318. 10.1016/j.devcel.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Papagianni A, Forés M, Shao W, He S, Koenecke N, Andreu MJ, Samper N, Paroush Z, González-Crespo S, Zeitlinger J, et al. (2018). Capicua controls Toll/IL-1 signaling targets independently of RTK regulation. Proc. Natl. Acad. Sci. USA 115, 1807–1812. 10.1073/pnas.1713930115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Reeves GT, and Stathopoulos A (2009). Graded dorsal and differential gene regulation in the Drosophila embryo. Cold Spring Harb. Perspect. Biol. 1, a000836. 10.1101/cshperspect.a000836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jiménez G, Guichet A, Ephrussi A, and Casanova J (2000). Relief of gene repression by Torso RTK signaling: role of capicua in Drosophila terminal and dorsoventral patterning. Genes Dev. 14, 224–231. 10.1101/gad.14.2.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kirov N, Zhelnin L, Shah J, and Rushlow C (1993). Conversion of a silencer into an enhancer: evidence for a co-repressor in dorsal-mediated repression in Drosophila. EMBO J. 12, 3193–3199. 10.1002/j.1460-2075.1993.tb05988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ing-Simmons E, Vaid R, Bing XY, Levine M, Mannervik M, and Vaquerizas JM (2021). Independence of chromatin conformation and gene regulation during Drosophila dorsoventral patterning. Nat. Genet. 53, 487–499. 10.1038/s41588-021-00799-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stathopoulos A, Van Drenth M, Erives A, Markstein M, and Levine M (2002). Whole-genome analysis of dorsal-ventral patterning in the Drosophila embryo. Cell 111, 687–701. 10.1016/s0092-8674(02)01087-5. [DOI] [PubMed] [Google Scholar]
- 108.Koenecke N, Johnston J, Gaertner B, Natarajan M, and Zeitlinger J (2016). Genome-wide identification of Drosophila dorso-ventral enhancers by differential histone acetylation analysis. Genome Biol. 17, 196. 10.1186/s13059-016-1057-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Löhr U, Chung H-R, Beller M, and Jäckle H (2009). Antagonistic action of bicoid and the repressor Capicua determines the spatial limits of Drosophila head gene expression domains. Proc. Natl. Acad. Sci. USA 106, 21695–21700. 10.1073/pnas.0910225106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jiménez G, Shvartsman SY, and Paroush Z (2012). The Capicua repressor–a general sensor of RTK signaling in development and disease. J. Cell Sci. 125, 1383–1391. 10.1242/jcs.092965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ajuria L, Nieva C, Winkler C, Kuo D, Samper N, Andreu MJ, Helman A, González-Crespo S, Paroush Z, Courey AJ, et al. (2011). Capicua DNA-binding sites are general response elements for RTK signaling in Drosophila. Development 138, 915–924. 10.1242/dev.057729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Boija A, and Mannervik M (2016). Initiation of diverse epigenetic states during nuclear programming of the Drosophila body plan. Proc. Natl. Acad. Sci. USA 113, 8735–8740. 10.1073/pnas.1516450113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Keller SH, Jena SG, Yamazaki Y, and Lim B (2020). Regulation of spatiotemporal limits of developmental gene expression via enhancer grammar. Proc. Natl. Acad. Sci. USA 117, 15096–15103. 10.1073/pnas.1917040117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Choi HMT, Schwarzkopf M, Fornace ME, Acharya A, Artavanis G, Stegmaier J, Cunha A, and Pierce NA (2018). Third-generation in situ hybridization chain reaction: multiplexed, quantitative, sensitive, versatile, robust. Development 145, dev165753. 10.1242/dev.165753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dufourt J, Trullo A, Hunter J, Fernandez C, Lazaro J, Dejean M, Morales L, Nait-Amer S, Schulz KN, Harrison MM, et al. (2018). Temporal control of gene expression by the pioneer factor Zelda through transient interactions in hubs. Nat. Commun. 9, 5194. 10.1038/s41467-018-07613-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Larson ED, Komori H, Fitzpatrick ZA, Krabbenhoft SD, Lee CY, and Harrison M (2022). Premature translation of the Drosophila zygotic genome activator Zelda is not sufficient to precociously activate gene expression. G3 (Bethesda) 12, jkac159. 10.1093/g3journal/jkac159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gibson TJ, and Harrison MM (2023). Protein-intrinsic properties and context-dependent effects regulate pioneer-factor binding and function. 10.1101/2023.03.18.533281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Markert J, and Luger K (2021). Nucleosomes meet their remodeler match. Trends Biochem. Sci. 46, 41–50. 10.1016/j.tibs.2020.08.010. [DOI] [PubMed] [Google Scholar]
- 120.Iurlaro M, Stadler MB, Masoni F, Jagani Z, Galli GG, and Schübeler D (2021). Mammalian SWI/SNF continuously restores local accessibility to chromatin. Nat. Genet. 53, 279–287. 10.1038/s41588-020-00768-w. [DOI] [PubMed] [Google Scholar]
- 121.Tang X, Li T, Liu S, Wisniewski J, Zheng Q, Rong Y, Lavis LD, and Wu C (2022). Kinetic principles underlying pioneer function of GAGA transcription factor in live cells. Nat. Struct. Mol. Biol. 29, 665–676. 10.1038/s41594-022-00800-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bellec M, Dufourt J, Hunt G, Lenden-Hasse H, Trullo A, Zine El Aabidine A, Lamarque M, Gaskill MM, Faure-Gautron H, Mannervik M, et al. (2022). The control of transcriptional memory by stable mitotic bookmarking. Nat. Commun. 13, 1176. 10.1038/s41467-022-28855-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Espinás ML, Jiménez-García E, Vaquero A, Canudas S, Bernués J, and Azorín F (1999). The N-terminal POZ domain of GAGA mediates the formation of oligomers that bind DNA with high affinity and specificity. J. Biol. Chem. 274, 16461–16469. 10.1074/jbc.274.23.16461. [DOI] [PubMed] [Google Scholar]
- 124.Katsani KR, Hajibagheri MA, and Verrijzer CP (1999). Co-operative DNA binding by GAGA transcription factor requires the conserved BTB/POZ domain and reorganizes promoter topology. EMBO J. 18, 698–708. 10.1093/emboj/18.3.698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Batut PJ, Bing XY, Sisco Z, Raimundo J, Levo M, and Levine MS (2022). Genome organization controls transcriptional dynamics during development. Science 375, 566–570. 10.1126/science.abi7178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mahmoudi T, Katsani KR, and Verrijzer CP (2002). GAGA can mediate enhancer function in trans by linking two separate DNA molecules. EMBO J. 21, 1775–1781. 10.1093/emboj/21.7.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ogiyama Y, Schuettengruber B, Papadopoulos GL, Chang JM, and Cavalli G (2018). Polycomb-dependent chromatin looping contributes to gene silencing during drosophila development. Mol. Cell 71, 73–88.e5. 10.1016/j.molcel.2018.05.032. [DOI] [PubMed] [Google Scholar]
- 128.Hug CB, Grimaldi AG, Kruse K, and Vaquerizas JM (2017). Chromatin architecture emerges during zygotic genome activation independent of transcription. Cell 169, 216–228.e19. 10.1016/j.cell.2017.03.024. [DOI] [PubMed] [Google Scholar]
- 129.Raff JW, Kellum R, and Alberts B (1994). The Drosophila GAGA transcription factor is associated with specific regions of heterochromatin throughout the cell cycle. EMBO J. 13, 5977–5983. 10.1002/j.1460-2075.1994.tb06943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Matharu NK, Yadav S, Kumar M, and Mishra RK (2021). Role of vertebrate GAGA associated factor (vGAF) in early development of zebrafish. Cells Dev. 166, 203682. 10.1016/j.cdev.2021.203682. [DOI] [PubMed] [Google Scholar]
- 131.Shrinivas K, Sabari BR, Coffey EL, Klein IA, Boija A, Zamudio AV, Schuijers J, Hannett NM, Sharp PA, Young RA, et al. (2019). Enhancer features that drive formation of transcriptional condensates. Mol. Cell 75, 549–561.e7. 10.1016/j.molcel.2019.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sharma R, Choi K-J, Quan MD, Sharma S, Sankaran B, Park H, LaGrone A, Kim JJ, MacKenzie KR, Ferreon ACM, et al. (2021). Liquid condensation of reprogramming factor KLF4 with DNA provides a mechanism for chromatin organization. Nat. Commun. 12, 5579. 10.1038/s41467-021-25761-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Morin JA, Wittmann S, Choubey S, Klosin A, Golfier S, Hyman AA, Julicher F, and Grill SW (2020). Surface condensation of a pioneer transcription factor on DNA. 10.1101/2020.09.24.311712. [DOI] [Google Scholar]
- 134.Treen N, Shimobayashi SF, Eeftens J, Brangwynne CP, and Levine M (2021). Properties of repression condensates in living Ciona embryos. Nat. Commun. 12, 1561. 10.1038/s41467-021-21606-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Shelansky R, and Boeger H (2020). Nucleosomal proofreading of activator-promoter interactions. Proc. Natl. Acad. Sci. USA 117, 2456–2461. 10.1073/pnas.1911188117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Estrada J, Wong F, DePace A, and Gunawardena J (2016). Information integration and energy expenditure in gene regulation. Cell 166, 234–244. 10.1016/j.cell.2016.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Larson ED, Komori H, Gibson TJ, Ostgaard CM, Hamm DC, Schnell JM, Lee C-Y, and Harrison MM (2021). Cell-type-specific chromatin occupancy by the pioneer factor Zelda drives key developmental transitions in Drosophila. Nat. Commun. 12, 7153. 10.1038/s41467-021-27506-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Xiong L, Tolen EA, Choi J, Velychko S, Caizzi L, Velychko T, Adachi K, MacCarthy CM, Lidschreiber M, Cramer P, et al. (2022). Oct4 differentially regulates chromatin opening and enhancer transcription in pluripotent stem cells. eLife 11, e71533. 10.7554/eLife.71533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Russ BE, Olshansky M, Li J, Nguyen MLT, Gearing LJ, Nguyen THO, Olson MR, McQuilton HA, Nüssing S, Khoury G, et al. (2017). Regulation of H3K4me3 at transcriptional enhancers characterizes acquisition of virus-specific CD8+ T cell-lineage-specific function. Cell Rep. 21, 3624–3636. 10.1016/j.celrep.2017.11.097. [DOI] [PubMed] [Google Scholar]
- 140.Vierbuchen T, Ling E, Cowley CJ, Couch CH, Wang X, Harmin DA, Roberts CWM, and Greenberg ME (2017). AP-1 transcription factors and the BAF complex mediate signal-dependent enhancer selection. Mol. Cell 68, 1067–1082.e12. 10.1016/j.molcel.2017.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Stavreva DA, Coulon A, Baek S, Sung MH, John S, Stixova L, Tesikova M, Hakim O, Miranda T, Hawkins M, et al. (2015). Dynamics of chromatin accessibility and long-range interactions in response to glucocorticoid pulsing. Genome Res. 25, 845–857. 10.1101/gr.184168.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hoffman JA, Trotter KW, Ward JM, and Archer TK (2018). BRG1 governs glucocorticoid receptor interactions with chromatin and pioneer factors across the genome. eLife 7, e35073. 10.7554/eLife.35073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Shashikant T, Khor JM, and Ettensohn CA (2018). Global analysis of primary mesenchyme cell cis-regulatory modules by chromatin accessibility profiling. BMC Genomics 19, 206. 10.1186/s12864-018-4542-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bozek M, and Gompel N (2020). Developmental transcriptional enhancers: A subtle interplay between accessibility and activity: considering quantitative accessibility changes between different regulatory states of an enhancer deconvolutes the complex relationship between accessibility and activity. BioEssays 42, e1900188. 10.1002/bies.201900188. [DOI] [PubMed] [Google Scholar]
- 145.Bravo González-Blas C, Quan X-J, Duran-Romañ a R, Taskiran II, Koldere D, Davie K, Christiaens V, Makhzami S, Hulselmans G, de Waegeneer M, et al. (2020). Identification of genomic enhancers through spatial integration of single-cell transcriptomics and epigenomics. Mol. Syst. Biol. 16, e9438. 10.15252/msb.20209438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Reddington JP, Garfield DA, Sigalova OM, Karabacak Calviello A, Marco-Ferreres R, Girardot C, Viales RR, Degner JF, Ohler U, and Furlong EEM (2020). Lineage-resolved enhancer and promoter usage during a time course of embryogenesis. Dev. Cell 55, 648–664.e9. 10.1016/j.devcel.2020.10.009. [DOI] [PubMed] [Google Scholar]
- 147.Jacobs J, Atkins M, Davie K, Imrichova H, Romanelli L, Christiaens V, Hulselmans G, Potier D, Wouters J, Taskiran II, et al. (2018). The transcription factor Grainy head primes epithelial enhancers for spatiotemporal activation by displacing nucleosomes. Nat. Genet. 50, 1011–1020. 10.1038/s41588-018-0140-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hennig BP, Velten L, Racke I, Tu CS, Thoms M, Rybin V, Besir H, Remans K, and Steinmetz LM (2018). Large-scale low-cost NGS library preparation using a robust Tn5 purification and tagmentation protocol. G3 (Bethesda) 8, 79–89. 10.1534/g3.117.300257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mintseris J, and Eisen MB (2006). Design of a combinatorial DNA microarray for protein-DNA interaction studies. BMC Bioinformatics 7, 429. 10.1186/1471-2105-7-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Philippakis AA, Qureshi AM, Berger MF, and Bulyk ML (2008). Design of compact, universal DNA microarrays for protein binding microarray experiments. J. Comput. Biol. 15, 655–665. 10.1089/cmb.2007.0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lam KN, van Bakel H, Cote AG, van der Ven A, and Hughes TR (2011). Sequence specificity is obtained from the majority of modular C2H2 zinc-finger arrays. Nucleic Acids Res. 39, 4680–4690. 10.1093/nar/gkq1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Weirauch MT, Cote A, Norel R, Annala M, Zhao Y, Riley TR, Saez-Rodriguez J, Cokelaer T, Vedenko A, Talukder S, et al. (2013). Evaluation of methods for modeling transcription factor sequence specificity. Nat. Biotechnol. 31, 126–134. 10.1038/nbt.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10. 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 155.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Li Q, Brown JB, Huang H, and Bickel PJ (2011). Measuring reproducibility of high-throughput experiments. Ann. Appl. Stat. 5, 1752–1779. 10.1214/11-AOAS466. [DOI] [Google Scholar]
- 158.Institute Broad. Picard tools. http://broadinstitute.github.io/picard/faq.html. [Google Scholar]
- 159.Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165. 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Chollet F (2015). Keras. https://keras.io. [Google Scholar]
- 161.Abadi M, Agarwal A, Barham P, Brevdo E, Chen Z, Citro C, Corrado GS, Davis A, Dean J, Devin M, et al. (2015). TensorFlow: large-scale machine learning on heterogeneous systems. 10.48550/arXiv.1603.04467. [DOI] [Google Scholar]
- 162.Kingma DP, and Ba J (2014). Adam: a method for stochastic optimization. 10.48550/arXiv.1412.6980. [DOI] [Google Scholar]
- 163.Shrikumar A, Greenside P, and Kundaje A (2017). Learning important features through propagating activation differences. 10.48550/arXiv.1704.02685. [DOI] [Google Scholar]
- 164.Shrikumar A, Tian K, Avsec Z, Shcherbina A, Banerjee A, Sharmin M, Nair S, and Kundaje A (2020). Technical note on transcription factor motif discovery from importance scores (TF-MoDISco), version 0.5.6.5. 10.48550/arXiv.1811.00416. [DOI] [Google Scholar]
- 165.Kvon EZ, Kazmar T, Stampfel G, Yáñez-Cuna JO, Pagani M, Schernhuber K, Dickson BJ, and Stark A (2014). Genome-scale functional characterization of Drosophila developmental enhancers in vivo. Nature 512, 91–95. 10.1038/nature13395. [DOI] [PubMed] [Google Scholar]
- 166.Zeitlinger J, Zinzen RP, Stark A, Kellis M, Zhang H, Young RA, and Levine M (2007). Whole-genome ChIP-chip analysis of Dorsal, Twist, and Snail suggests integration of diverse patterning processes in the Drosophila embryo. Genes Dev. 21, 385–390. 10.1101/gad.1509607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gallo SM, Gerrard DT, Miner D, Simich M, Des Soye B, Bergman CM, and Halfon MS (2011). REDfly v3.0: toward a comprehensive database of transcriptional regulatory elements in Drosophila. Nucleic Acids Res. 39, D118–D123. 10.1093/nar/gkq999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wickham H (2016). ggplot2: elegant graphics for data analysis (Springer). 10.1007/978-3-319-24277-4?trk=public_post_comment-text. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw and processed data for ChIP-nexus, ChIP-seq, ATAC-seq, MNase-seq, and protein binding microarray experiments have been deposited at GEO under series accession number GSE218852 (GEO: GSE218852) and are publicly available as of the date of publication. Original data, including microscopy images, can be accessed from the Stowers Original Data Repository (ODR: http://www.stowers.org/research/publications/libpb-2357). Trained BPNet and ChromBPNet models are available at Zenodo (Zenodo: https://zenodo.org/record/8075860). All original code has been deposited at GitHub (GitHub: https://github.com/zeitlingerlab/Brennan_Zelda_2023) and is publicly available as of the date of publication. The DOI is listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit polyclonal anti-Zelda | Koenecke et al.108 | 366735–1 |
| Rabbit polyclonal anti-Bicoid | This paper | U9982EL040–1 |
| Rabbit polyclonal anti-Caudal | This paper | U4197EL190–1 |
| Rabbit polyclonal anti-Dorsal | He et al.58 | 126740–44 |
| Rabbit polyclonal anti-Twist | He et al.58 | 131424–2 |
| Rabbit polyclonal anti-GAF | This paper | 163185–42 |
| Rabbit polyclonal anti-H3K27ac | Active Motif | 39133; RRID: AB_2561016 |
| Mouse monoclonal anti-H3K4me1 | Active Motif | 39635; RRID: AB_2793284 |
| Anti-rabbit IgG Alexa Fluor 568 secondary antibody | ThermoFisher | A10042; RRID: AB_2534017 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| 37% formaldehyde solution | VWR | Cat# 50–00-0 |
| Dynabeads Protein A | ThermoFisher | Cat# 10008D |
| phi29 DNA polymerase | New England Biolabs | Cat# M0269S |
| Lambda exonuclease | New England Biolabs | Cat# M0262S |
| Q5 High-Fidelity 2x Master Mix | New England Biolabs | Cat# M0492S |
| dNTP solution mix | New England Biolabs | Cat# N0447S |
| MNase | New England Biolabs | Cat# M0247S |
| RNase A | ThermoFisher | Cat# EN0531 |
| Phenol:chloroform:isoamyl alcohol (25:24:1) (v/v/v) | VWR | Cat# 136112–00-0 |
| Proteinase K | ThermoFisher | Cat# 25530049 |
| Western Blocking Reagent | Millipore Sigma | Cat# 11921681001 |
| ProLong Gold Antifade Mountant with DAPI | ThermoFisher | Cat# P36931 |
| OptiPrep Density Gradient Medium | Millipore Sigma | Cat# D1556 |
| ProLong Glass Antifade Mountant | ThermoFisher | Cat# P36980 |
|
| ||
| Critical commercial assays | ||
|
| ||
| End Repair Module | New England Biolabs | Cat# E6050S |
| dA-Tailing Module | New England Biolabs | Cat# E6053S |
| Quick Ligation Kit | New England Biolabs | Cat# M2200S |
| High Throughput Library Prep Kit | KAPA Biosystems | Cat# KK8234 |
| Monarch DNA Gel Extraction Kit | New England Biolabs | Cat# T1020 |
| Monarch PCR & DNA Cleanup Kit | New England Biolabs | Cat# T1030 |
| PURExpress In Vitro Protein Synthesis Kit | New England Biolabs | Cat# E6800 |
| Hybridization Chain Reaction (HCR) v3.0 | Molecular Instruments | N/A |
|
| ||
| Deposited data | ||
|
| ||
| Raw and analyzed NGS and PBM data | This paper | GEO: GSE218852 |
| Trained deep learning models (Zenodo) | This paper | Zenodo: https://zenodo.org/recore/8075860 https://doi.org/10.5281/zenodo.8118135 |
| Raw images | This paper | ODR: http://www.stowers.org/research/publications/libpb-2357 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Drosophila melanogaster: Oregon-R | Koenecke et al.108 | FlyBase: FBsn0000276 |
| Drosophila melanogaster: UAS-shRNA-zld: P{UAS-zld.shRNA} | Sun et al.6 | FlyBase: FBtp0147479 |
| Drosophila melanogaster: Maternal Triple Driver (MTD)-Gal4: P{COG-GAL4:VP16}; P{Gal4-nos.NGT}40; P{nos-Gal4-VP16} | Bloomington Stock Center | BSC: 31777 |
| Drosophila melanogaster: gd7: gd7/winscy, P{hs-hid}5 | Koenecke et al.108 | N/A |
| Drosophila melanogaster: cic6: cic6/TM3, Sb1 | Papagianni et al.101 | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Oligonucleotides for ChIP-nexus, see Table S2 | IDT | https://research.stowers.org/zeitlingerlab/protocols.html |
| Illumina Index primer 1: 5’-CAAGCAGAAGAC GGCATACGAGAT[i7]GTCTCGTGGGCTCGG-3’ | IDT | https://support-docs.illumina.com/SHARE/AdapterSeq/1000000002694_17_illumina_adapter_sequences.pdf |
| Illumina Index primer 2: 5’-AATGATACGGCGACC ACCGAGATCTACAC[i5]TCGTCGGCAGCGTC-3’ | IDT | https://support-docs.illumina.com/SHARE/AdapterSeq/1000000002694_17_illumina_adapter_sequences.pdf |
| Illumina Transposase adapter read 1 (Nextera A): 5’- TCGTCGGCAGCGTCAGATGTGTATAA GAGACAG-3’ | IDT | https://support-docs.illumina.com/SHARE/AdapterSeq/1000000002694_17_illumina_adapter_sequences.pdf |
| Illumina Transposase adapter read 2 (Nextera B): 5’- GTCTCGTGGGCTCGGAGATGTGTATA AGAGACAG-3’ | IDT | https://support-docs.illumina.com/SHARE/AdapterSeq/1000000002694_17_illumina_adapter_sequences.pdf |
| Mosaic end primer: /5Phos/CTGTCTCTTATAC A/3ddC/ | IDT | Tn5mC1.1-A1block |
| gd7 heat shock forward primer: 5’-GGAGCGAC AATTCAATTCAAACAAGC-3’ | IDT | N/A |
| gd7 heat shock reverse primer: 5’-GTAGCTGTG GCTGCAGTGCATCG-3’ | IDT | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pETM11-Sumo3-Tn5 plasmid | Hennig et al.148 | E54K,L372P |
| His6-tagged SenP2 protease plasmid | Hennig et al.148 | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| FIJI | Schindelin et al.149 | https://fiji.sc/ |
| Cutadapt v.2.5 | Martin154 | https://cutadapt.readthedocs.io/en/v2.5/ |
| Bowtie2 v.2.3.5.1 | Langmead and Salzberg155 | https://bowtie-bio.sourceforge.net/bowtie2/manual.shtml |
| MACS2 v.2.2.7.1 | Zhang et al.156 | https://github.com/macs3-project/MACS |
| Irreproducible Discovery Rate framework v.2.0.3 | Li et al.157 | https://github.com/nboley/idr |
| Picard v.2.23.8 | Broad Institute of MIT and Harvard158 | http://broadinstitute.github.io/picard |
| deepTools2 v.3.5.1 | Ramírez et al.159 | https://deeptools.readthedocs.io/en/latest/ |
| BPNet software | Avsec et al.59 | https://github.com/kundajelab/bpnet/ |
| Keras v.2.2.4 & v.2.5.0 | Chollet et al.160 | https://pypi.org/project/keras/ |
| TensorFlow1 backend v.1.7 & v.2.5.1 | Abadi et al.161 | https://www.tensorflow.org/install/pip |
| Adam optimizer | Kingma and Ba162 | N/A |
| DeepLIFT v.0.6.9.0 | Shrikumar et al.163 | https://github.com/kundajelab/DeepExplain |
| TF-MoDISco v.0.5.3.0 & v.0.5.16.0 | Shrikumar et al.164 | https://github.com/kundajelab/tfmodisco |
| ChromBPNet software | Anshul Kundaje’s lab, Stanford University | https://github.com/kundajelab/chrombpnet |
| DeepLIFT v.0.6.13.0 | Shrikumar et al.163 | https://github.com/kundajelab/shap |
| DESeq2 v.1.36.0 | Love et al.109 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| R v.4.2.0 | R core team | https://www.r-project.org/ |
| Rstudio | RStudio | https://rstudio.com |
| ggplot2 v.3.3.6 | Wickham168 | https://ggplot2.tidyverse.org/ |
|
| ||
| Other | ||
|
| ||
| All code and analyses that contributed to this work | This paper | https://github.com/zeitlingerlab/Brennan_Zelda_2023 https://doi.org/10.5281/zenodo.8118135 |
| Bioruptor Pico sonication device | Diagenode | https://www.diagenode.com/en/p/bioruptor-pico-sonication-device |
| Point scanning confocal microscope | Zeiss | 780 |
| Spinning disk microscope | Nikon | Eclipse Ti2 |