

Abstract
FGFR3 and PIK3CA are among the most frequently mutated genes in bladder tumors. We hypothesized that recurrent mutations in these genes might be caused by common carcinogenic exposures such as smoking and other factors. We analyzed 2,816 bladder tumors with available data on FGFR3 and/or PIK3CA mutations, focusing on the most recurrent mutations detected in ≥10% of tumors. Compared to tumors with other FGFR3/PIK3CA mutations, FGFR3-Y375C was more common in tumors from smokers than never-smokers (p=0.009), while several APOBEC-type driver mutations were enriched in never-smokers: FGFR3-S249C (p=0.013) and PIK3CA-E542K/PIK3CA-E545K (p=0.009). To explore possible causes of these APOBEC-type mutations, we analyzed RNA-seq data from 798 bladder tumors and detected several viruses, with BK polyomavirus (BKPyV) being the most common. We then performed immunohistochemical (IHC) staining for polyomavirus (PyV) Large T-antigen (LTAg) in an independent set of 211 bladder tumors. Overall, by RNA-seq or IHC-LTAg, we detected PyV in 26 out of 1,010 bladder tumors with significantly higher detection (p=4.4 × 10−5), 25/554 (4.5%) in non-muscle-invasive bladder cancers (NMIBC) versus 1/456 (0.2%) of muscle-invasive bladder cancers (MIBC). In the NMIBC subset, the FGFR3/PIK3CA APOBEC-type driver mutations were detected in 94.7% (18/19) of PyV-positive vs. 68.3% (259/379) of PyV-negative tumors (p=0.011). BKPyV tumor positivity in the NMIBC subset with FGFR3- or PIK3CA-mutated tumors was also associated with a higher risk of progression to MIBC (p=0.019). In conclusion, our results support smoking and BKPyV infection as risk factors contributing to bladder tumorigenesis in the general patient population through distinct molecular mechanisms.
Keywords: Bladder cancer, smoking, polyomavirus, FGFR3 mutations, PIK3CA mutations, APOBEC
INTRODUCTION
Urinary bladder cancer (BC) is the tenth most common malignancy worldwide (1). The majority of patients present with non-muscle-invasive bladder cancer (NMIBC, 75%), which is highly recurrent (~65%) (2) but has a high 5-year survival rate of 70–96% (3). In contrast, the less common but more advanced form, muscle-invasive bladder cancer (MIBC, 25%), has a high metastatic potential with a 5-year survival rate of <50% (3,4). Some NMIBC (10–25%) progress to MIBC (4).
Risk factors for BC include environmental exposures such as cigarette smoke, which explains over 50% of BC risk (5), and various occupational chemicals (6). Additionally, BC risk is significantly increased in carriers of certain germline genetic variants (7). Carcinogenic exposures may generate recurrent somatic mutations that can be studied individually or as mutational signatures (8). Thus, mutational analysis can help establish the causality of various risk factors. The mutation spectrum in bladder tumors differs significantly between NMIBC and MIBC. Specifically, the oncogenes FGFR3 and PIK3CA are the most frequently mutated genes in NMIBC, with mutations occurring in 65% and 25% of tumors, respectively (4). In contrast, tumor suppressor genes are most commonly mutated in MIBC, with only ~15% of MIBC carrying an FGFR3 or PIK3CA mutation (4).
We hypothesized that understanding the etiologies of common driver mutations can pinpoint causal risk factors for BC. In turn, these findings can help elucidate oncogenic mechanisms and inform prevention, outcomes and treatment options for BC. Here, we explored the association between BC risk factors and recurrent FGFR3 and PIK3CA mutations in tumors.
MATERIALS & METHODS
Bladder cancer samples
Mutation data for a total of 2,816 BC patients (1,573 NMIBC and 1,243 MIBC) with available smoking history and tumor stage were pooled from 7 independent studies: Spanish Bladder Cancer Study (SBCS) (9,10), UROMOL (11), New England Bladder Cancer Study (NEBCS) (12), Dana-Farber (13), MSK-IMPACT (14), TCGA-BLCA (15), and Solit (16). The per-cohort summary of the data is presented in Table S1. Several studies used targeted sequencing panels: NEBCS (44 genes), Dana-Farber (237 genes), MSK-IMPACT (341- and 410-gene panels), and Solit (~240 genes). SBCS profiled FGFR3 mutations with a SNAPshot assay or targeted sequencing (9,17). Mutations in UROMOL were analyzed based on RNA-seq and in TCGA-BLCA based on whole-exome sequencing data. Data for MSK-IMPACT, TCGA-BLCA and Solit were retrieved from cBioPortal (18) in 2019; Dana-Farber data was downloaded from published supplementary materials (13), and data for SBCS, UROMOL (European Genome-Phenome Archive (EGAD00001002717)), and NEBCS were provided by the co-authors of the paper.
Data on lifetime occupational history was available only for NEBCS (19). Somatic mutation data for FGFR3 and PIK3CA genes was available in all studies except for SBCS, for which PIK3CA mutation data was unavailable. All somatic mutations, including synonymous, non-synonymous and indels, were used in the analyses.
Analysis of viral transcriptome in RNA-seq of bladder tumors
The unaligned RNA-seq reads for the UROMOL and TCGA-BLCA tumors were de novo assembled using MegaHit (version 1.2.9, RRID:SCR_018551) (20). The non-human contigs were annotated using BLAST search (blastn, RRID:SCR_001598) against the total NCBI nucleotide (nt) database as of November 2019. RNA-seq reads were also aligned against a fusion reference genome that included human genome (hg38) and representative GenBank sequences of 1,126 human viruses (Table S2) using STAR version 2.5.3ab (RRID:SCR_004463) (21). The total numbers of viral reads were counted from the STAR alignments and by aligning the raw reads back to the contigs using bowtie2 (RRID:SCR_016368) and picard BamIndexStats (http://broadinstitute.github.io/picard, v2.20.8). Coverage plots normalized against all human reads were generated from these alignments using bedtools genomecov (v2.29.0) and the R (v3.6.2) package ggplot2. All genomic analyses were done using the NIH Biowulf high-performance computational cluster (http://hpc.nih.gov). Samples were called positive for a specific virus if they contained assembled viral contigs greater than 300 bp with at least 8 reads mapping to this virus (Table S3).
BKPyV infection in HBLAK cells
The spontaneously immortalized bladder cell line HBLAK was purchased in 2018 from CELLnTEC and cultured in CnT-Prime Epithelial Culture Medium (CELLnTEC), according to the manufacturer’s protocol. Cells were authenticated by microsatellite fingerprinting (AmpFLSTR Identifiler PCR Amplification Kit, Thermo Fisher) by the Cancer Genomics Research Laboratory/NCI, and regularly tested for mycoplasma contamination (MycoAlert Mycoplasma Detection Kit, Lonza). HBLAK cells were seeded at 100,000 cells per well in 12-well plates and infected in triplicates with rearranged BKPyV (strain Gardner, NCBI accession: LC029411.1) at 0.5 multiplicity of infection (MOI) and mock-infected, as previously described (22). Cells were harvested 1-, 3-, 5-, and 7-days post-infection (dpi). For BKPyV immunofluorescent staining with pAb416 (EMD Millipore), HBLAK cells were seeded onto glass slides and stained as previously described (23). Low-molecular-weight DNA was isolated using the Hirt protocol (24). DNA copies of the BKPyV genome were quantified using qPCR and normalized against human mitochondrial (MT) DNA using previously described primers and methods (23). RNA was extracted with TRI reagent (Invitrogen) and assessed for quality using spectrophotometry and the TapeStation (Agilent). RNA-seq libraries for all infected samples and 5 dpi mock-infected samples were prepared using the Takara Pico Input Mammalian kit and sequenced on the Illumina NextSeq 550. The generated RNA-seq reads were aligned against a fusion reference genome using the same pipelines as for tumor RNA-seq samples. BKPyV mapped reads were counted using picard BamIndexStats and normalized versus the length of the BKPyV genome (~5.1 kb) and the total number of reads per sample to calculate RPKM. Gene counts were generated with STAR-count and analyzed using the DESeq2 package in the R statistical environment (25). Count data were normalized using blind variance stabilizing transformation. Normalized count data and qPCR data were plotted using GraphPad Prism 8.
LTAg-IHC staining in bladder tumor microarrays
The previously described NEBCS bladder tumor microarray (TMA) (26) was serially sectioned at 5 μm thickness on Sakura Autosection microtome and mounted on charged slides. The first slide of each block was stained with H&E (hematoxylin and eosin), and two consecutive slides were stained with antibody against Large T Antigen (LTAg), clone PAb416 (#DP02, EMD Millipore), which detects LTAg from multiple polyomaviruses. Slides were baked at 60°C for one hour before immunostaining on Ventana Discovery Ultra automated stainer with the following conditions: antigen retrieval for 64 min at 96°C and pH 9; incubation with the antibody at 0.5 μg/ml concentration in antibody diluent (#S3022, Agilent) for 32 min at 36°C; incubation with anti-mouse HQ-AntiHQ HRP detection system for 12 min with DAB chromogen and Hematoxylin II counterstain. Negative and positive controls were represented by FFPE blocks of HeLa cells mock-transfected or transfected with a construct for truncated TAg of BKPyV (27). The cells were transfected with trunc-TAg expression construct using Lipofectamine 2000, pelleted, mounted in an FFPE block and processed alongside TMA blocks. Slides were scanned for digital image analysis on AT2 slide scanner (Leica Biosystems) at a 40x magnification. De-arraying of TMA cores and marker analysis was done using HALO software (Indica Labs, Albuquerque, NM). A random forest classifier was utilized to separate exogenous debris from tissue content, followed by training the Immune Cell module v1.3 to detect rare events of true positive LTAg staining. Staining was considered positive if detected on both replica slides, with at least 2 dots on each slide, without a significant technical background. All positive slides were independently reviewed by three pathologists, including one not associated with this study.
Statistical analyses
The relationship between mutation status (FGFR3 or PIK3CA) and smoking was evaluated using multivariate polytomous (multinomial) logistic regression models to calculate odds ratios (ORs) and 95% confidence intervals (CIs). To account for mutational processes affecting multiple genes, we compared tumors with specific FGFR3 and PIK3CA mutations to other mutations in these genes and tumors designated as wild-type (without any FGFR3 and PIK3CA mutations). Thus, logistic regression analysis was performed in tumors divided into the following groups: i) positive for the mutation of interest, ii) negative for the mutation of interest but positive for another mutation in the gene, and iii) tumors without any mutations in the gene (wild-type, WT). Smoking status was coded as a categorical variable either in five levels (never, former, current, occasional, and unknown) or three levels (never, ever, unknown). Ever-smokers included former, current and occasional smokers. Patients with unknown smoking status made up <4% of the total dataset, <5% of all NMIBC and <3% of all MIBC (Table S1). The exclusion of samples with missing values for smoking status did not affect the association results and conclusions of the paper. Occupational exposure information available in NEBCS was classified into two categorical levels (high or low risk) based on lifetime occupational history (19). Other variables such as sex, race, age, smoking status, and tumor stage were treated as categorical variables, as described in Table S1. Final multivariable regression models included age, sex, smoking status, and tumor stage; the variables were included in the model when parameter estimates changed by more than 10% compared to unadjusted and forward stepwise models. Model estimates were calculated with R v3.5.1, package nnet. Validation analysis was conducted in R by 5,000 permutations of the main independent variable (cigarette smoking status). Random sample distributions were generated from the data using the R functions sample() and replicate(), without sample replacement. Fisher’s exact tests for 2×2 contigency tables were used to compare the counts of APOBEC-type mutations per each virus category and in BKPyV-positive vs BKPyV-negative tumors, as well as to determine if the risk factors, smoking status and PyV positivity were independent. Pearson Chi-square tests were used to compare PyV-positivity rates between NMIBC and MIBC sets. Progression-free survival (PFS, months) was evaluated in the UROMOL study as progression to MIBC, using Kaplan-Meier analysis (R packages survminer and survival).
Data availability
RNA-seq data for HBLAK cells were deposited in the NCBI Sequence Read Archive (SRA) under project PRJNA643143.
RESULTS
We analyzed data for 2,816 BC patients from seven studies (9–16) with available smoking history, tumor stage, and mutation status for FGFR3 and PIK3CA (Table S1). We chose these genes because they are frequently mutated in BC and thus included in all sequencing panels (whole-exome and targeted) used in these studies. Of the 2,816 tumors analyzed, 1,310 (47%) had at least one FGFR3 or PIK3CA mutation. To ensure sufficient statistical power, we focused on mutations in these genes detected in ≥10% of tumors: FGFR3-S249C (detected in 57% of tumors), FGFR3-Y375C (20%), PIK3CA-E545K (37%), and PIK3CA-E542K (19%) (Table S4). Our conclusions on the significance of these associations between smoking status and specific mutations were further supported by permutation resampling analysis (with 5,000 permutations of smoking status). We found FGFR3-Y375C (TAT>TGT) to be more common in tumors from ever-smokers than never-smokers, both when compared to tumors with other FGFR3 mutations (OR=1.84 (1.17–2.91), p=0.009) and FGFR3-WT tumors (OR=1.86 (1.19–2.92), p=0.007, Table 1).
Table 1.
Analysis of associations between smoking status and FGFR3-Y375C, FGFR3-S249C, and PIK3CA- E542K/E545K mutations
| FGFR3 Mutation | Smoking Status* | Total | FGFR3-Y375C, N=217† | FGFR3 other mutations, N=832† | FGFR3-WT, N=1758† | FGFR3-Y375C | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compared to other FGFR3 mutations | Compared to FGFR3-WT | |||||||||||
| % | % | % | P | P | ||||||||
| Y375C | Never | 647 | 4.2 | 26.4 | 69.1 | |||||||
| Ever | 2056 | 8.8 | 29.7 | 61.3 | 0.009 | 0.007 | ||||||
| Never | 647 | 4.2 | 4.2 | 26.4 | ||||||||
| Former | 973 | 9.2 | 9.2 | 32.0 | 0.014 | 0.016 | ||||||
| Current | 665 | 11.1 | 11.1 | 34.4 | 0.008 | 0.008 | ||||||
| FGFR3 Mutation | Smoking Status* | Total | FGFR3-S249C, N=622† | FGFR3 other mutations, N=427† | FGFR3-WT, N=1758† | FGFR3-S249C | ||||||
| Compared to other FGFR3 mutations | Compared to FGFR3-WT | |||||||||||
| % | % | % | P | P | ||||||||
| S249C | Never | 647 | 20.4 | 10.2 | 69.1 | 0.013 | 0.685 | |||||
| Ever | 2056 | 22.1 | 16.3 | 61.3 | ||||||||
| Never | 647 | 20.4 | 20.4 | 10.2 | 0.020 | 0.755 | ||||||
| Former | 973 | 23.9 | 23.9 | 17.3 | 0.581 | 0.985 | ||||||
| Current | 665 | 25.7 | 25.7 | 19.8 | ||||||||
| PIK3CA Mutation | Smoking Status* | Total | PIK3CA- E542K/E545K, N=280† | PIK3CA other mutations, N=193† | PIK3CA-WT, N=1499† | PIK3CA-E542K/E545K | ||||||
| Compared to other PIK3CA mutations | Compared to PIK3CA-WT | |||||||||||
| % | % | % | P | P | ||||||||
| E542K or E545K | Never | 499 | 17.6 | 8.8 | 73.3 | 0.009 | 0.004 | |||||
| Ever | 1369 | 12.8 | 10.4 | 76.6 | ||||||||
| Never | 499 | 17.6 | 17.6 | 8.8 | 0.010 | 0.044 | ||||||
| Former | 619 | 13.9 | 13.9 | 10.0 | 0.298 | 0.624 | ||||||
| Current | 332 | 13.6 | 13.6 | 10.8 | ||||||||
WT, wild-type;
Ever-smokers include former, current and occasional smokers;
See Table S1 for detailed information;
Odds ratios (ORs) and 95% confidence intervals (CIs) are calculated using polytomous logistic regression models, adjusting for study, tumor stage (Ta, T1, T2+), and sex.
In contrast, the three other common mutations—FGFR3-S249C (TCC>TGC), PIK3CA-E545K (TGA>TAA), and PIK3CA-E542K (TGA>TAA)—were enriched in never-smokers: FGFR3-S249C (OR=1.54 (1.10–2.18), p=0.013) and PIK3CA-E545K or PIK3CA-E542K (OR=1.81 (1.17–2.82), p=0.009, Table 1), compared to tumors with other FGFR3 or PIK3CA mutations. These three mutations have been previously attributed to the activities of APOBEC3A/B enzymes, which catalyze cytosine deamination (28,29). Similar trends with smoking status were observed in stratified analyses across all seven datasets (Table S5). The associations were stronger in studies with a higher proportion of NMIBC cases, likely attributable to increased statistical power due to higher mutation frequencies in NMIBC compared to MIBC. Other known BC risk factors, such as male sex, age, and employment in a high-risk occupation, were not associated with the four common mutations analyzed (Table S5).
Because APOBEC3A/B are interferon-stimulated genes (30), we hypothesized that viral infections might contribute to the observed enrichment of APOBEC-type driver mutations in tumors of never-smokers. We thus conducted an RNA-seq analysis of 798 bladder tumors (390 from the UROMOL study and 408 from TCGA-BLCA). By querying the tumor RNA-seq data for the presence of viral sequences, we detected several viruses (Figure 1A, Table S3, Table S6), with BK polyomavirus (BKPyV) being the most common. In NMIBC, BKPyV was detected in 5.3% (20/376) of all tumors and 5.0% (14/279) of tumors with FGFR3 or PIK3CA mutations (Figure 1A, Table S6). In contrast, only one of 423 MIBC (0.2%) was BKPyV-positive; this tumor had a previously annotated BKPyV integration (31) and no FGFR3 or PIK3CA mutations. Among NMIBC tumors with FGFR3 or PIK3CA mutations, BKPyV detection was associated with a higher risk of progression to MIBC (p=0.019, Figure 1B).
Figure 1. BKPyV infection as a possible cause of APOBEC-mediated mutagenesis in NMIBC.
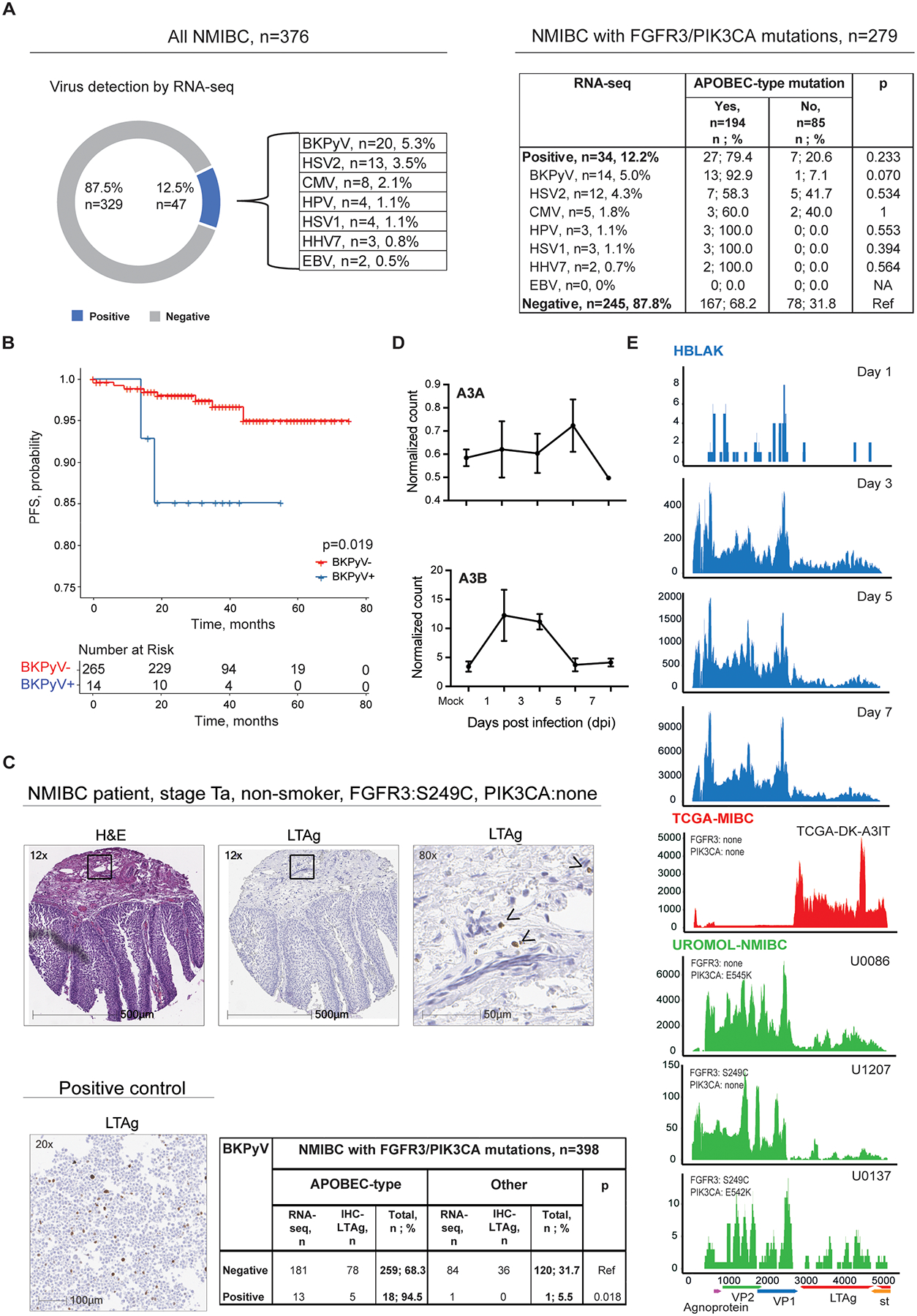
A) Several viruses were detected by RNA-seq analysis of 376 UROMOL-NMIBC tumors: BK Polyomavirus (BKPyV), Herpes Simplex Virus 2 (HSV2), Cytomegalovirus (CMV), Human papillomavirus (HPV), Herpes Simplex Virus 1 (HSV1), Human Betaherpesvirus 7 (HHV7), and Epstein-Barr Virus (EBV). Within the subset of 279 tumors with FGFR3/PIK3CA mutations, APOBEC-type driver mutations in these two genes were most common in the BKPyV-positive tumors. P-values are for Fisher’s exact test. B) Time to progression to MIBC in NMIBC patients with FGFR3 or PIK3CA mutations from the UROMOL study. Progression-free survival (PFS) was evaluated by Kaplan-Meier analysis in 279 patients with FGFR3 or PIK3CA mutations according to BKPyV status (yes/no) by RNA-seq. C) Representative IHC images demonstrating H&E and Large T antigen (LTAg) staining in one of the 5 positive NMIBC tumors; images from additional tumors are shown in Figure S1A. Marked areas are shown at a higher magnification. Arrows point to cells positive for LTAg staining (brown dots). NMIBC patient 1 is a 71-yr old never-smoking male with stage Ta, grade 1 bladder tumor with the APOBEC-type FGFR3-S249C mutation. Positive control for LTAg antibody: HeLa cells transfected with an expression construct for truncated T antigen. The table shows results for NMIBC tumors with FGFR3/PIK3CA mutations based on BKPyV status, as determined by RNA-seq or IHC-LTAg. Tumors positive for BKPyV were more likely to harbor an APOBEC-type FGFR3/PIK3CA mutation than BKPyV-negative tumors. P-values are for Fisher’s exact test. D) Variance stabilized read counts for APOBEC3A (A3A) and APOBEC3B (A3B) RNA expression corresponding to mock-infection and 1, 3, 5, and 7 days post-BKPyV infection. E) Representative coverage plots for BKPyV RNA-seq on different days post-infection in HBLAK cells (n=3) (blue), NMIBC tumors from UROMOL (green) and the only BKPyV-positive MIBC tumor (TCGA-BLCA), in which virus is integrated (red). The y-axis shows the read depth per million human reads. Positions and open reading frames within the 5-Kb BKPyV genome are shown under the X-axis. Agnoprotein – regulates viral proliferation; VP1 and VP2 - encode structural proteins for virion capsids during viral proliferation; LTAg and st - encode large and small tumor (T) antigens involved in the initiation of viral replication that causes an oncogenic transformation of the host genome.
We also conducted immunohistochemical (IHC) staining in 211 bladder tumors from an independent study (NEBCS) using an antibody for SV40 Large T Antigen (LTAg), which detects several polyomaviruses (PyV), including BKPyV. LTAg was detected in five of 178 NMIBC tumors but none of 33 MIBC tumors. All five LTAg-positive tumors harbored an APOBEC-type mutation in FGFR3 or PIK3CA (Figure 1C, Figure S1A, Table S7).
Overall, by RNA-seq or IHC-LTAg analyses, we detected 26 PyV-positive tumors: 4.5% (25/554) of NMIBC vs. 0.2% (1/456) of MIBC (p=4.4 × 10−5, Table 2). Among NMIBC tumors with FGFR3 or PIK3CA mutations, 94.7% (18/19) of PyV-positive tumors carried at least one APOBEC-type driver mutation vs. 68.3% (259/379) of PyV-negative tumors (p=0.011, Figure 1C, Table 2). As expected for independent factors, tumor PyV positivity was uniformly distributed across smoking groups. In the total dataset (n=944), tumor PyV positivity was 1.5% in never-smokers and 2.4% in ever-smokers (p=0.470) (Table S7).
Table 2.
Summary of polyomavirus positivity and APOBEC-type driver mutation positivity among BC samples
| Subset | Total | Samples positive for PyV | ||
|---|---|---|---|---|
| n | n | % | p-val | |
| Total BC | 1010 | 26 | 2.6 | na |
| NMIBC | 554 | 25 | 4.5 | 4.4E-05 |
| NMIBC with FGFR3 or PIK3CA mutation | 398 | 19 | 4.8 | na |
| MIBC | 456 | 1 | 0.2 | Ref |
| Subset: NMIBC with FGFR3 or PIK3CA mutation | Total | Samples positive for APOBEC-type driver mutation (FGFR3-S249C, PIK3CA-E545K, or PIK3CA-E542K) |
||
| n | n | % | p-val | |
| BKPyV+ | 19 | 18 | 94.7 | 0.011 |
| BKPyV− | 379 | 259 | 68.3 | Ref |
BC, bladder cancer; BKPyV, BK polyomavirus; NMIBC, non-muscle invasive bladder cancer; PyV, polyomavirus.
To explore why these APOBEC-type driver mutations were enriched in BKPyV-positive NMIBC tumors, we performed total RNA-seq in BKPyV-infected immortalized human uroepithelial cells (HBLAK). We observed increased APOBEC3B (but not APOBEC3A) mRNA expression early post-infection without changes in cell viability (Figure 1D, Figure S1B, Figure S1C. Both BKPyV-infected HBLAK cells and BKPyV-positive NMIBC exhibited similar viral expression patterns: strong expression of the viral capsid genes VP1 and VP2 (Figure 1E), which encode structural proteins expressed late in the viral cycle, indicating productive viral replication. In contrast, in the MIBC tumor from TCGA-BLCA with previously annotated BKPyV integration (31), we observed nearly undetectable VP1 and VP2 but abundant LTAg expression (Figure 1E).
DISCUSSION
Understanding the molecular causes of mutations detected in tumors can help refine known cancer risk factors and identify new ones. Here, we showed that different risk factors were associated with some of the most recurrent mutations in bladder tumors.
We demonstrated that one of the most common mutations (FGFR3-Y375C) detected in bladder tumors is likely caused by tobacco smoking. These results are consistent with the previously reported link between TAT>TGT mutations and defective activity of the nucleotide excision repair (NER) pathway in smokers (12,32). Tobacco carcinogens induce DNA damage, such as bulky lesions, which are excised and may be erroneously repaired through compensatory low-fidelity pathways.
In contrast, never-smokers were more likely to harbor three other common mutations— all APOBEC-type: FGFR3-S249C, PIK3CA-E542K, and PIK3CA-E545K. This finding agrees with a report that in vitro exposure to smoking-related chemicals does not induce APOBEC-type mutations (33). PyV infection instead may contribute to this enrichment of APOBEC-type mutations. Additionally, in the FGFR3/PIK3CA-mutated set, NMIBC that were PyV-positive were more likely to progress to MIBC than PyV-negative NMIBC. Our RNA-seq results also revealed that BKPyV-positive NMIBC tumors have distinct viral expression profiles compared to the only MIBC tumor (TCGA-BLCA) with documented BKPyV integration. IHC-LTAg also demonstrated a distinct staining pattern in BKPyV-positive NMIBC, characterized by isolated positive cells instead of the diffuse, clonal staining commonly seen in BKPyV-positive MIBC with viral integration (34).
By adulthood, >80% of individuals are seropositive for BKPyV, which subclinically persists in the urinary tract (35,36). While low levels of intermittent BKPyV shedding can be detected in the urine (7%) or stool (18%) of immunocompetent individuals (37,38), the reactivation of BKPyV is naturally controlled by the immune response. In immunosuppressed individuals (e.g., organ transplant recipients (OTRs), and kidney transplant recipients, specifically), BKPyV reactivation is common and manifests as abundant viruria, viremia, strong IHC-LTAg tissue staining, and increased risk of BC (39). Specifically, BKPyV viruria and PyV-associated nephropathy can develop in as high as 51–64% and 4–10% of kidney transplant recipients, respectively (40). However, the consequences of BKPyV reactivation on BC risk in individuals without apparent immunodeficiency (non-OTRs) have not been well-studied due to technical challenges of detection and the transient nature of reactivation that might also be triggered by various environmental exposures.
In NMIBC from the general population, we found limited numbers of IHC-LTAg-positive tumor cells in a pattern distinct from the strong clonal staining observed in MIBC tumors of OTRs, which often present with BKPyV integration in the host genome (34,41). The sparse staining pattern in BKPyV-positive NMIBC may be harder to detect and considerably depend on sample quality. However, our detection methods (RNA-seq and IHC-LTAg) likely improved sensitivity and specificity compared to standard DNA PCR-based assays. RNA-seq might detect additional and more variable viral sequences missed by sequence-specific qPCR assays. Moreover, the detection of transcribed PyV sequences indicates active protein-producing infection, in contrast with qPCR detection in DNA, which does not discriminate between active and latent infections. Another study examined PyV positivity in non-OTR BC patients using DNA PCR-based assays and found that PyV positivity rates were similar to our findings (42). One key difference is that we additionally found that BKPyV positivity is enriched in NMIBC compared to MIBC. Our more sensitive detection approaches (RNA-seq and LTAg-IHC) may have helped us capture this association. As LTAg expression varies throughout the viral lifecycle, we suggest RNA-seq is more sensitive than IHC-LTAg for BKPyV detection.
The BKPyV expression patterns from both non-OTR NMIBC and infected HBLAK cells in vitro are consistent with that of episomal (non-integrated) virus. In contrast, integrated BKPyV is common in MIBC tumors of OTRs (41) and was observed only in one non-OTR tumor (MIBC from TCGA-BLCA). Thus, episomal and integrated PyV forms might be characteristic of NMIBC vs. MIBC, respectively. Episomal BKPyV suggests productive infection, while integration into the host genome is not part of the normal viral life cycle and irreversibly disrupts virus replication (34,43). The integration of BKPyV can promote the expression of LTAg, a viral oncogene implicated in tumorigenesis through cellular transformation partly by inhibiting tumor suppressor proteins (44,45).
BKPyV-positive MIBC is more common in OTRs (21% of post-transplant BC patients) (41) compared to non-OTRs, with only one BKPyV-positive MIBC in the whole TCGA-BLCA dataset (0.2%). This could depend on the impaired ability to eliminate viruses in immunocompromised OTR patients. Genomically integrated BKPyV may also be more easily targeted by immune surveillance than episomal forms, potentially from the increased expression of the novel or foreign antigens. The predominance of BKPyV integration in the MIBCs of OTRs (41) compared to non-OTRs could indicate that integrated viral forms are more readily eliminated in immunocompetent environments. The effects of non-integrated, productive BKPyV infections in urothelial oncogenesis are less understood.
BKPyV reactivation that might occur even in transiently immunosuppressed conditions may upregulate APOBEC enzymes and subsequently generate APOBEC-type driver mutations, thus initiating or promoting BC. This is consistent with the hit-and-run hypothesis (46), which proposes that transient oncogenic virus infections can provide a survival benefit in premalignant cells or early-stage tumors (i.e., NMIBC) before being cleared by host responses. Supporting this paradigm, an in vitro study (47) showed that BKPyV infection of urothelial cells stimulates APOBEC3B-mediated accumulation of abasic sites, which can be erroneously repaired, leading to mutations. As immune surveillance can eradicate virus-infected cells, BKPyV infection confers disadvantages to tumors. However, as tumors advance to highly genomically unstable states like MIBC, they may be able to thrive and progress independently of virus-mediated support. Thus, the BKPyV positivity in tumors of non-OTRs could reflect their reliance on viral factors: high dependency in NMIBC (BKPyV is detectable) and no or low dependency in MIBC (BKPyV is no longer needed and thus eliminated).
We also found an increased risk of progressing to MIBC in BKPyV-positive NMIBC tumors, even though BKPyV might be immunologically cleared by the time of progression. The risk of progression with BKPyV may be due to increased expression of APOBEC3B and APOBEC-mediated mutagenesis, which has been previously shown to increase genomic instability and shorten the progression-free survival of NMIBC patients (11,48), or through other virus-mediated mechanisms.
Genotoxic injury caused by PyV infections and cigarette smoking may trigger distinct repair pathways and types of base substitutions (32,47). The enrichment of APOBEC activity in never-smokers in our and previous studies (12,49) supports this idea. APOBEC mutagenesis is a major mutational process in BC, which could be induced by multiple factors such as genotoxic drug exposures, inflammation, and intrinsic genomic stress that activate the expression of APOBEC enzymes (30,50). The proportion of APOBEC-type FGFR3 and PIK3CA mutations caused entirely by hit-and-run PyV infection is difficult to estimate. While further research is needed, our study supports the idea that BKPyV infection contributes to the emergence of the highly recurrent APOBEC-type FGFR3-S249C, PIK3CA-E545K, and PIK3CA-E542K mutations in BC.
The strength of our study is the combination of tumor and epidemiological data, allowing us to identify potential links between environmental factors and common mutations. However, the tumors in our study were predominately analyzed by targeted sequencing panels, which precluded genome-wide mutational signature analysis. Despite this limitation, using RNA-seq and IHC-LTAg staining for BC patients from the general population (non-OTRs), we detected PyV in 2.6% of all BC tested, including 4.5% of NMIBC (25/554) and 0.2% of MIBC (1/456). PyV positivity in non-OTRs could be underestimated for several reasons, such as spatially dispersed expression patterns in tissue samples and viral clearance by the host response.
In conclusion, we report that two common exposures—tobacco smoke and BKPyV infection—contribute to the etiology of several common FGFR3 and PIK3CA mutations through distinct molecular mechanisms. Our results also highlight potential differences in PyV-related oncogenic effects in NMIBC and MIBC, warranting further investigation into the mechanistic and translational relevance of these findings. Specifically, the role of transient BKPyV infection in non-OTR BC needs to be further explored using tumor screening with high-sensitivity detection methods. As many studies support the link between BKPyV and renourinary cancers, the potential of vaccination against BKPyV to protect from BC is also worth exploring.
Supplementary Material
Prevention Relevance Statement:
Tobacco smoking likely causes one of the most common mutations in bladder tumors (FGFR3-Y375C), while viral infections might contribute to three others (FGFR3-S249C, PIK3CA-E542K, and PIK3CA-E545K). Understanding the causes of these mutations may lead to new prevention and treatment strategies, such as viral screening and vaccination.
ACKNOWLEDGEMENTS
We thank all the patients and investigators from the contributing studies – UROMOL (Europe), NEBCS (USA), and SBCS/EPICURO Study Investigators (Spain). We thank Dr. Petra Lentz (Molecular Digital Pathology Laboratory/DCEG/NCI) for providing an independent evaluation of IHC-LTAg staining of bladder tumors, Dr. Oscar Florez-Vargas (Laboratory of Translational Genomics Branch, DCEG/NCI) for advising on statistical analyses and Dr. Eric Engels (Infections and Immunoepidemiology Branch, DCEG/NCI) for critical comments and suggestions. This study was supported by the Intramural Research Program of the National Cancer Institute, with M. Garcia-Closas, D. Baris, A. Johnson, M. Schwenn, N. Rothman, D.T. Silverman, and S. Koutros additionally supported by funding for NEBCS (ZIA CP010125-24). The results are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Conflict of interest:
CBB is an inventor on patents issued to NCI and receives licensing royalties for BKPyV vaccine technologies. LD has sponsored research agreements with C2i Genomics, Photocure, Natera, AstraZeneca and Ferring, has advisory/consulting roles at Ferring, UroGEN, and has received speaker/video honoraria from Roche, Pfizer and Astellas. Other authors have no conflicts of interest to report in relation to this work.
REFERENCES
- 1.IARC. [cited 2021 Mar 20]. Available from: <https://gco.iarc.fr/today/online-analysis-table>.
- 2.Babjuk M, Burger M, Capoun O, Cohen D, Comperat EM, Dominguez Escrig JL, et al. European Association of Urology Guidelines on Non-muscle-invasive Bladder Cancer (Ta, T1, and Carcinoma in Situ). Eur Urol 2022;81(1):75–94 doi 10.1016/j.eururo.2021.08.010. [DOI] [PubMed] [Google Scholar]
- 3.Cancer Statistics Center. [cited 2023 Mar 11]. Available from:. <https://cancerstatisticscenter.cancer.org/>.
- 4.Knowles MA, Hurst CD. Molecular biology of bladder cancer: new insights into pathogenesis and clinical diversity. Nat Rev Cancer 2015;15(1):25–41 doi 10.1038/nrc3817. [DOI] [PubMed] [Google Scholar]
- 5.Freedman ND, Silverman DT, Hollenbeck AR, Schatzkin A, Abnet CC. Association between smoking and risk of bladder cancer among men and women. JAMA 2011;306(7):737–45 doi 10.1001/jama.2011.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Silverman D, Koutros S, Figueroa J, Prokunina-Olsson L, Rothman N. Bladder Cancer. Schottenfeld and Fraumeni Cancer Epidemiology and Prevention. Fourth ed. New York, NY: Oxford University Press; 2017. p 977–96. [Google Scholar]
- 7.Koutros S, Kiemeney LA, Pal Choudhury P, Milne RL, Lopez de Maturana E, Ye Y, et al. Genome-wide Association Study of Bladder Cancer Reveals New Biological and Translational Insights. Eur Urol 2023:127–37 doi 10.1016/j.eururo.2023.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013;500(7463):415–21 doi 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amaral AF, Mendez-Pertuz M, Munoz A, Silverman DT, Allory Y, Kogevinas M, et al. Plasma 25-hydroxyvitamin D(3) and bladder cancer risk according to tumor stage and FGFR3 status: a mechanism-based epidemiological study. J Natl Cancer Inst 2012;104(24):1897–904 doi 10.1093/jnci/djs444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia-Closas M, Malats N, Silverman D, Dosemeci M, Kogevinas M, Hein DW, et al. NAT2 slow acetylation, GSTM1 null genotype, and risk of bladder cancer: results from the Spanish Bladder Cancer Study and meta-analyses. Lancet 2005;366(9486):649–59 doi 10.1016/S0140-6736(05)67137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hedegaard J, Lamy P, Nordentoft I, Algaba F, Hoyer S, Ulhoi BP, et al. Comprehensive Transcriptional Analysis of Early-Stage Urothelial Carcinoma. Cancer Cell 2016;30(1):27–42 doi 10.1016/j.ccell.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 12.Koutros S, Rao N, Moore LE, Nickerson ML, Lee D, Zhu B, et al. Targeted Deep Sequencing of Bladder Tumors Reveals Novel Associations between Cancer Gene Mutations and Mutational Signatures with Major Risk Factors. Clin Cancer Res 2021;27(13):3725–33 doi 10.1158/1078-0432.CCR-20-4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nassar AH, Umeton R, Kim J, Lundgren K, Harshman L, Van Allen EM, et al. Mutational Analysis of 472 Urothelial Carcinoma Across Grades and Anatomic Sites. Clin Cancer Res 2019;25(8):2458–70 doi 10.1158/1078-0432.CCR-18-3147. [DOI] [PubMed] [Google Scholar]
- 14.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 2017;23(6):703–13 doi 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robertson AG, Kim J, Al-Ahmadie H, Bellmunt J, Guo G, Cherniack AD, et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017;171(3):540–56 e25 doi 10.1016/j.cell.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim PH, Cha EK, Sfakianos JP, Iyer G, Zabor EC, Scott SN, et al. Genomic predictors of survival in patients with high-grade urothelial carcinoma of the bladder. Eur Urol 2015;67(2):198–201 doi 10.1016/j.eururo.2014.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balbas-Martinez C, Sagrera A, Carrillo-de-Santa-Pau E, Earl J, Marquez M, Vazquez M, et al. Recurrent inactivation of STAG2 in bladder cancer is not associated with aneuploidy. Nat Genet 2013;45(12):1464–9 doi 10.1038/ng.2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6(269):pl1 doi 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Colt JS, Karagas MR, Schwenn M, Baris D, Johnson A, Stewart P, et al. Occupation and bladder cancer in a population-based case-control study in Northern New England. Occup Environ Med 2011;68(4):239–49 doi 10.1136/oem.2009.052571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015;31(10):1674–6 doi 10.1093/bioinformatics/btv033. [DOI] [PubMed] [Google Scholar]
- 21.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29(1):15–21 doi 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verhalen B, Justice JL, Imperiale MJ, Jiang M. Viral DNA replication-dependent DNA damage response activation during BK polyomavirus infection. J Virol 2015;89(9):5032–9 doi 10.1128/JVI.03650-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang M, Entezami P, Gamez M, Stamminger T, Imperiale MJ. Functional reorganization of promyelocytic leukemia nuclear bodies during BK virus infection. mBio 2011;2(1):e00281–10 doi 10.1128/mBio.00281-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirt B Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol 1967;26(2):365–9 doi 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- 25.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lenz P, Pfeiffer R, Baris D, Schned AR, Takikita M, Poscablo MC, et al. Cell-cycle control in urothelial carcinoma: large-scale tissue array analysis of tumor tissue from Maine and Vermont. Cancer Epidemiol Biomarkers Prev 2012;21(9):1555–64 doi 10.1158/1055-9965.EPI-12-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Starrett GJ, Serebrenik AA, Roelofs PA, McCann JL, Verhalen B, Jarvis MC, et al. Polyomavirus T Antigen Induces APOBEC3B Expression Using an LXCXE-Dependent and TP53-Independent Mechanism. mBio 2019;10(1) doi 10.1128/mBio.02690-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi MJ, Meng XY, Lamy P, Banday AR, Yang J, Moreno-Vega A, et al. APOBEC-mediated Mutagenesis as a Likely Cause of FGFR3 S249C Mutation Over-representation in Bladder Cancer. Eur Urol 2019;76(1):9–13 doi 10.1016/j.eururo.2019.03.032. [DOI] [PubMed] [Google Scholar]
- 29.Rheinbay E, Nielsen MM, Abascal F, Wala JA, Shapira O, Tiao G, et al. Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature 2020;578(7793):102–11 doi 10.1038/s41586-020-1965-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Middlebrooks CD, Banday AR, Matsuda K, Udquim KI, Onabajo OO, Paquin A, et al. Association of germline variants in the APOBEC3 region with cancer risk and enrichment with APOBEC-signature mutations in tumors. Nat Genet 2016;48(11):1330–8 doi 10.1038/ng.3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cancer Genome Atlas Research N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014;507(7492):315–22 doi 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim J, Mouw KW, Polak P, Braunstein LZ, Kamburov A, Kwiatkowski DJ, et al. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat Genet 2016;48(6):600–6 doi 10.1038/ng.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baker SC, Mason AS, Southgate J. Procarcinogen Activation and Mutational Signatures Model the Initiation of Carcinogenesis in Human Urothelial Tissues In Vitro. Eur Urol 2020;78(2):143–7 doi 10.1016/j.eururo.2020.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Papadimitriou JC, Randhawa P, Rinaldo CH, Drachenberg CB, Alexiev B, Hirsch HH. BK Polyomavirus Infection and Renourinary Tumorigenesis. Am J Transplant 2016;16(2):398–406 doi 10.1111/ajt.13550. [DOI] [PubMed] [Google Scholar]
- 35.Doerries K Human polyomavirus JC and BK persistent infection. Adv Exp Med Biol 2006;577:102–16 doi 10.1007/0-387-32957-9_8. [DOI] [PubMed] [Google Scholar]
- 36.Gossai A, Waterboer T, Nelson HH, Michel A, Willhauck-Fleckenstein M, Farzan SF, et al. Seroepidemiology of Human Polyomaviruses in a US Population. Am J Epidemiol 2016;183(1):61–9 doi 10.1093/aje/kwv155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vanchiere JA, Abudayyeh S, Copeland CM, Lu LB, Graham DY, Butel JS. Polyomavirus shedding in the stool of healthy adults. J Clin Microbiol 2009;47(8):2388–91 doi 10.1128/JCM.02472-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Husseiny MI, Anastasi B, Singer J, Lacey SF. A comparative study of Merkel cell, BK and JC polyomavirus infections in renal transplant recipients and healthy subjects. J Clin Virol 2010;49(2):137–40 doi 10.1016/j.jcv.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gupta G, Kuppachi S, Kalil RS, Buck CB, Lynch CF, Engels EA. Treatment for presumed BK polyomavirus nephropathy and risk of urinary tract cancers among kidney transplant recipients in the United States. Am J Transplant 2018;18(1):245–52 doi 10.1111/ajt.14530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nickeleit V, Singh HK. Polyomaviruses and disease: is there more to know than viremia and viruria? Curr Opin Organ Transplant 2015;20(3):348–58 doi 10.1097/MOT.0000000000000192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Starrett GJ, Yu K, Golubeva Y, Lenz P, Piaskowski ML, Petersen D, et al. Evidence for virus-mediated oncogenesis in bladder cancers arising in solid organ transplant recipients. Elife 2023;12 doi 10.7554/eLife.82690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Llewellyn MA, Gordon NS, Abbotts B, James ND, Zeegers MP, Cheng KK, et al. Defining the frequency of human papillomavirus and polyomavirus infection in urothelial bladder tumours. Sci Rep 2018;8(1):11290 doi 10.1038/s41598-018-29438-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kenan DJ, Mieczkowski PA, Latulippe E, Cote I, Singh HK, Nickeleit V. BK Polyomavirus Genomic Integration and Large T Antigen Expression: Evolving Paradigms in Human Oncogenesis. Am J Transplant 2017;17(6):1674–80 doi 10.1111/ajt.14191. [DOI] [PubMed] [Google Scholar]
- 44.Wendzicki JA, Moore PS, Chang Y. Large T and small T antigens of Merkel cell polyomavirus. Curr Opin Virol 2015;11:38–43 doi 10.1016/j.coviro.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gjoerup O, Chang Y. Update on human polyomaviruses and cancer. Adv Cancer Res 2010;106:1–51 doi 10.1016/S0065-230X(10)06001-X. [DOI] [PubMed] [Google Scholar]
- 46.Starrett GJ, Buck CB. The case for BK polyomavirus as a cause of bladder cancer. Curr Opin Virol 2019;39:8–15 doi 10.1016/j.coviro.2019.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baker SC, Mason AS, Slip RG, Skinner KT, Macdonald A, Masood O, et al. Induction of APOBEC3-mediated genomic damage in urothelium implicates BK polyomavirus (BKPyV) as a hit-and-run driver for bladder cancer. Oncogene 2022;41(15):2139–51 doi 10.1038/s41388-022-02235-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rouf Banday A, Onabajo OO, Lin SH, Obajemu A, Vargas JM, Delviks-Frankenberry KA, et al. Targeting natural splicing plasticity of APOBEC3B restricts its expression and mutagenic activity. Commun Biol 2021;4(1):386 doi 10.1038/s42003-021-01844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fantini D, Seiler R, Meeks JJ. Molecular footprints of muscle-invasive bladder cancer in smoking and nonsmoking patients. Urol Oncol 2019;37(11):818–25 doi 10.1016/j.urolonc.2018.09.017. [DOI] [PubMed] [Google Scholar]
- 50.Kanu N, Cerone MA, Goh G, Zalmas LP, Bartkova J, Dietzen M, et al. DNA replication stress mediates APOBEC3 family mutagenesis in breast cancer. Genome Biol 2016;17(1):185 doi 10.1186/s13059-016-1042-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data for HBLAK cells were deposited in the NCBI Sequence Read Archive (SRA) under project PRJNA643143.