

Abstract
The immune system plays a crucial role in the regulation of metastasis. Tumor cells systemically change immune functions to facilitate metastatic progression. Through this study, we deciphered how tumoral Galectin-1 (Gal1) expression shapes the systemic immune environment to promote metastasis in head and neck cancer (HNC). In multiple preclinical models of HNC and lung cancer in immunogenic mice, Gal1 fostered the establishment of a pre-metastatic niche through polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs), which altered the local microenvironment to support metastatic spread. RNA sequencing of MDSCs from pre-metastatic lungs in these models demonstrated the role of PMN-MDSCs in collagen and extracellular matrix remodeling in the pre-metastatic compartment. Gal1 promoted MDSC accumulation in the pre-metastatic niche through the NF-κB signaling axis, triggering enhanced CXCL2-mediated MDSC migration. Mechanistically, Gal1 sustained NF-κB activation in tumor cells by enhancing STING protein stability, leading to prolonged inflammation-driven MDSC expansion. These findings suggest an unexpected pro-tumoral role of STING activation in metastatic progression and establish Gal1 as an endogenous positive regulator of STING in advanced-stage cancers.
Introduction
Metastasis is the leading cause of cancer-related morbidity and mortality [1]. Although the cancer survival rate has significantly improved, limited progress has been made in treating metastatic disease. Identifying tumor microenvironmental contribution to specific steps in the metastatic cascade is fundamental to designing effective therapeutics to prevent, inhibit and treat metastases [2]. Malignant cells secrete multiple soluble factors as well as extracellular vesicles to support their growth not just at the primary tumor but also at distant sites. This pre-conditioned secondary tissue, known as pre-metastatic niches, is vital for facilitating homing and subsequent growth of circulating tumor cells [3]. Immune cells are strong partakers in shaping the pre-metastatic niche. Metastasis is known to be a highly inefficient process, largely due to the tumoricidal immune response mediated by cytotoxic NK and CD8+ T cells [4]. Nevertheless, metastatic cancers develop strategies to overcome these inhibitory mechanisms partly through the recruitment of immunosuppressive myeloid cell types, not only at the primary tumor sites but also systemically by altered hematopoiesis [5]. These tumor-driven systemic processes prepare distant sites to become favorable to metastatic seeding and colonization, thereby enhancing metastatic efficiency. Myeloid cells are chief effectors of the pre-metastatic niche function [6]. Myeloid-derived suppressor cells (MDSCs) are known to be the prominent myeloid cell type that causes immunosuppression while also supporting colonization, survival, and outgrowth of disseminated cells at secondary sites [7, 8]. However, tumoral factors influencing their recruitment and mechanistic insights into the process need further investigation. A better understanding of the mechanisms underlying their recruitment and immune-suppressive/ pro-metastatic functions in the metastatic sites preceding metastatic seeding is crucial for guiding more rational drug design to prevent or target metastases. We have previously shown that tumor-secreted Galectin-1 (Gal1) is a strong effector of metastasis in both head and neck and lung cancers [9, 10]. Galectin-1 has emerged as one of the major regulatory pathways, used by the immune system to maintain homeostasis and prevent autoimmune responses. Cancer cells upregulate Gal1 expression to evade anti-tumor immune response either through its intra- or extra-cellular mechanisms, thus nurturing local and systemic immunosuppression. Most studies have highlighted the role of Gal1 in primary tumors site and its regulation of T-cell responses [11–13]. However, the contribution of Gal1 in modulating systemic immunity, in the context of metastasis is poorly understood.
In this study, we investigate how Gal1-induced perturbation of systemic immunity is crucial for its role in metastatic progression. Using multi-color flow cytometry and RNA sequencing (RNA-seq) analyses of tumor/ lung infiltrated MDSCs, we establish how Gal1, through its effects on MDSCs, reshapes the lung microenvironment to support metastatic progression. Unexpectedly, we find that Gal1 enhances the stability of the Stimulator of interferon gene (STING) protein, thereby stimulating the downstream signaling pathway to activate NF-κB-dependent cytokine release that is crucial for MDSC expansion and migration. The cGAS-STING pathway has paradoxical roles depending on the kinetics and strength of its induction. While chronic STING activation has been shown to promote tumor growth and immune suppression in advanced-stage breast cancers [14], its acute activation has been linked to anti-cancer immune responses [15, 16]. However, little is known about the factors that promote chronic STING activation and immune suppression. Here, we show that Galectin-1 is an endogenous modulator of the pro-tumorigenic STING function in cancer cells which can lead to immune suppression and distant metastasis. This has significant clinical implications because our data suggest that tumors with high Gal-1 level show chronic STING activation and thus would not benefit from the use of STING agonists, which are beings tested in the clinic as anti-cancer therapy.
Materials & Methods
Cell lines and culture conditions
HPV- murine oral cancer cells (MOC1 and MOC2) were obtained from Dr. Uppaluri’s lab and maintained in culture in IMDM/F12 (2:1) with 5% FCS, 1% penicillin/streptomycin, amphotericin, 5 ng/mL EGF (Millipore), 40 ng/mL hydrocortisone, and 5 μg/mL insulin. The HPV+ murine oral cancer cells (mEERL) were obtained from Dr. William Spanos (Sanford Research, CA). The cells were grown in IMDM/ Ham’s F-12 (2:1), containing 10% FBS, 25 μg/mL hydrocortisone, 5 μg/mL transferrin, 5 μg/mL insulin, 1.36 ng/mL tri-iodo-thyronine, and 5 ng/mL EGF. LLC and FaDU cells were obtained from ATCC and maintained in DMEM + 10% FCS-containing media. Cells were routinely tested for mycoplasma contamination using the Lonza Mycoplasma Detection Kit. For conditioned media collection experiments, 1% serum-containing media was used. Stable Gal1 knockout cells were generated using CRISPR/ Cas9 targeting as described in Nambiar et al, 2019 [9]. Mouse LLC-Scr and LLC-sh-Gal1 cells were generated as described in Banh et al. [10]. For galectin-1 knockdown in human cells, we used the sh-RNA consist of pools of three target-specific 19-25 nucleotide sequences in length (Cat: SC-35441-SH; Santa Cruz Biotechnology; sequence provided in Supplementary Table 1). For the STING- knockdown in mouse cells, we used a the sh-RNA consist of pools of three target-specific 19-25 nucleotide sequences in length (Cat: SC-15441-SH; Santa Cruz Biotechnology) and scramble control cells were generated using the control plasmid (Cat: SC-108080, Santa Cruz Biotechnology). All experiments were carried out with cells
Tumor models
Seven to nine-week-old male C57BL/6 mice (Strain:000664; RRID: IMSR_ JAX: 000664) or NRG mice (NOD-Rag1null IL2rgnull, NOD rag gamma, Strain 007799; RRID: IMSR_JAX:007799) were purchased from the Jackson Laboratory. For MOC2 tumor experiments, mice were injected either subcutaneously (SC) in the flank region (2.5x105) or orthotopically (4X104) in the buccal cavity of male C57/BL6 mice. For mEERL tumors- 1x106 cells and for LLC tumors, 0.5x 106 cells were injected SC into the flank region of the mice. For any treatment conditions, the mice were randomized into different groups at ~100mm3. Primary tumors were measured two-three times a week and mice were monitored for tumor growth & survival over the period of time. For evaluating myeloid infiltration in the lung, the mice were sacrificed at different time points post-injection of the tumor cells. Metastatic seeding at early intervals was quantified by flow cytometry of the lung by gating for CD45−CD31−EpCam+CK14+ cells in the lung. At late stages of tumor growth, metastasis was quantified by H&E staining of lung and lymph nodes and the number of tumor foci was counted per lung or per node. All mouse tumor studies were repeated at least twice with similar results.
Tissue processing
Tumors were digested using the mouse tumor dissociation kit (Cat: 130-096-730, Miltenyi) per manufacturers guidelines. The lungs were perfused and inflated with PBS and, following which single-cell suspensions were prepared by finely mincing tissues with a scalpel and incubating the tissue on a shaker at 37°C for 20 min (lung) and 40 min (tumor) in 2 mL of digestion media. Tumor tissues was passed through a 100 μm mesh strainer and washed with RPMI media twice. Spleen, lymph nodes, and other tissues were mashed through a 70 μm cell strainer and washed with RPMI media. For blood, tumor, spleen, liver, RBC lysis was performed using ACK lysis buffer for 5 minutes, following which cell suspension was washed with RPMI media twice before proceeding with the staining.
Flow cytometry
Cells were washed with PBS and stained with Live/Dead Zombie Aqua or Live/Dead Zombie NIR™ (ZNIR) Fixable Viability Kit (1:500; Cat: 423105, Biolegend) for 10 minutes at 4°C. Cells were washed in FACS buffer (PBS supplemented with 2% FCS following which Fc receptors were blocked using TrueStain FcX™ [anti-mouse CD16/32, 1:100; Cat: 101319, Biolegend]). The cells were then stained with a panel of antibodies diluted in FACS buffer for 30 minutes at 4°C in the dark. Cell surface staining was performed using fluorophore-conjugated anti-mouse CD45.2 (30-F11, Cat: 103147), TCR-beta (H57-597), CD4 (GK1.5, Cat: 100411), CD8 (53-6.7, Cat: 100712), NK1.1 (PK136), CD11b (M1/70, Cat: 101206), CD11c (N418, Cat: 117333), MHCII (M5/114.15.2), Ly6G (1A8, Cat: 127607), Ly6C (HK1.4, Cat: 128041), F4/80 (BM8, Cat: 123131), CXCR2 (SA044G4, Cat: 149308) from Biolegend. For intracellular staining, cells were fixed using the Fixation-Permeabilization kit protocol (Cat: 88-8824-00, eBioscience) following which intracellular staining was performed for CD206 (MMR, Cat: 141720, Biolegend), Arginase-1 (Cat: 17-3697-80; Invitrogen) and IL-10 (Cat: 505028, Biolegend). Following staining, cells were washed in FACS buffer and analyzed by flow cytometry. Compensations were attained using Anti-Rat and anti-hamster compensation beads (BD Biosciences) and single stained cells. For fixable live/dead staining, compensation was performed using ArC amine reactive compensation beads (Cat: A10346; BD Biosciences). Gating schemes of dissociated tissues was as follows: Epithelial cells (ZNIR−CD45−), Immune cells (ZNIR−CD45+), CD8+ T-cells (ZNIR−CD45+ TCRb+ CD8+), CD4+ T-cells (ZNIR−CD45+TCRb+CD4+), macrophages (ZNIR−CD45+CD11b+F4/80+), PMN-MDSCs (ZNIR−CD45+CD11b+Ly6Clo/intLy6G+), M-MDSC (ZNIR−CD45+CD11b+ Ly6G−Ly6Chi). For positive staining determination FMO controls were used. Flow cytometry was performed on LSR Fortessa (BD Biosciences) in the Dept. of Radiation Oncology FACS facility and analyzed with Cytobank software (Beckman Coulter) and FlowJo (RRID: SCR_008520, Tree Star Inc). tSNE-CUDA/tSNE analyses was carried out using Cytobank Software.
Mouse treatments
For cell depletion studies, mice were randomized at 50mm3 tumor volume and injected with 100 μL antibodies through the intraperitoneal route. An initial depletion with 400 μg of anti-CD8a antibody (Cat: BP0061, clone 2.43, BioXcell) or rat IgG2b isotype control (Cat: BE0090, BioXcell) was administered on days 7, 11, and 15 post-tumor inoculation. Antibody depletion treatment was continued with the administration of 400 μg of antibody every 3-5 days for the duration of the experiment. For depletion of MDSCs, ~1w post tumor inoculation, mice with palpable tumors were injected intraperitoneally (IP) with anti-GR1 antibody 200 μg/dose (Cat: BE0075, clone RB6-8C5, BioXcell) or isotype IgG2b (Cat: BE0090, BioXcell) every 48 hours. On Day 18 after the first anti-GR1 antibody administration, the lungs were harvested to evaluate for metastasis. Tumors & other organs were also harvested to confirm the depletion of MDSC. Depletion efficiency was checked using FACS analysis of blood obtained by retro-orbital bleeding. For conditioned media treatment, concentrated CM from WT-Gal1 or KO-Gal1 cells or similarly concentrated control media was used. When tumors reached ~100mm3 in size, 100μl of CM/control media was injected IP every other day for ~3 weeks until termination.
T-cell suppression assay
Spleens from C57/BL6 mice were employed to isolate CD8+ T cells using the EasySep Mouse T cell isolation kit (Cat: 19853; STEMCELL Technologies). Isolated CD8+ T cells were labeled with 5 μM CSFE and incubated with CD3/28 coated plate for 5 days with or without PMN-MDSCs (sorted from either the tumor or the lung tissues) with MDSC: T cell ratio of 1: 5 in RPMI + 10% FBS containing 50 μM beta-mercapto-ethanol. Flow cytometry was used to measure the loss of CFSE signal in live proliferating CD8+ T-cells. The data were analyzed using ModFit LT software.
MDSC isolation from lungs and tumor
Single-cell suspensions of digested tumor and lung tissues were run on a 40%/80% Percoll gradient. The leukocyte band was harvested and Ly6G+ PMN-MDSCs were sorted using the Anti-Ly6G Microbead Kit (Cat: 130-120-337, Miltenyi Biotec) as per manufacturer recommendations. Purity was determined by running on flow cytometry. For T cell suppression assays, MDSCs were sorted using the Aria FACS sorter at the Stanford Shared FACS Facility (SSFF).
Trichrome staining
Trichrome staining was done at the Histopathology Core at Stanford Medical center, with a trichrome stain kit (American MasterTech). After deparaffinization and rehydration, tissue was incubated in Bouin’s fluid overnight, followed by modified Mayer’s hematoxylin for 7 min and One Step Trichrome Stain for 5 min. Slides were dehydrated with ethanol and xylene and covered with mountant. Images were obtained a Leica microscope, using 40x/0.95 NA and 63x/1.4 NA Plan objectives, following which trichrome (blue) staining was quantified using ImageJ software.
Immunofluorescence
Lungs were harvested and fixed in 10% neutral buffered formalin for 24 hours. Fixed lungs were embedded in paraffin and 5-uM thick sections were prepared for staining using Stanford Pathology core. Briefly, slides were deparaffinized and rehydrated in a series of xylene and ethanol gradients, followed by antigen retrieval using sodium citrate buffer. Slides were washed with water and then TBST (TBS + 0.05% Tween-20) followed by blocking with Blocking Solution/animal serum (Vector Laboratories). Staining with primary and secondary antibodies was performed per the manufacturer’s instructions. The following antibodies were used: Ly6G (Cat: 127601, clone 1A8; Biolegend), CD11b (Cat: ab133357, Abcam). All images were collected on a Leica microscope, using 40x/0.95 NA and 63x/1.4 NA plan objectives.
Immunohistochemistry: Tissue microarray (TMA) staining
Formalin-fixed and paraffin-embedded (FFPE) TMAs were created as previously described [17]. 4 μm-thick sections were cut from the TMAs. For Human MDSC staining, LOX1 (Cat: ab126538, Abcam) and CD15 (Cat: 555400, BD biosciences) were used. For Galectin-1 (Cat: PB9240-1, Boster Bio) and STING (Cat: 13647, Cell Signaling Technologies) were used. LOX1 (OLR1) was scored based on cytoplasmic expression in immune cells infiltrating the tumor and adjacent stroma. Tumor cells and epithelial cells were not included in the scoring. Cases with no or rare LOX1-expressing immune cells were scored as 0, and those with singly scattered LOX1-expressing immune cells were scored as 1. Cases with scores 0 and 1 were combined into the MDSClo group. Cases with frequent LOX1-positive immune cells, either dispersed singly or forming confluent aggregates, were scored as 2 (MDSChi). Tumoral Gal1 and STING were both scored in viable tumor cells based on the intensity of staining. Parakeratosis and immune cells were not included in the scoring. Gal1 was scored by the intensity of cytoplasmic staining, and STING was scored by the intensity of cytoplasmic and membranous staining. Cases with no expression were scored as 0; those with weak or blush staining were scored as 1, and cases with strong staining were scored as 2. Since there were very few cases with a score of 2, the STING staining was grouped as negative (score 0) and positive (score 1-2) for analysis. Similarly, for Gal1 staining, patients were grouped as negative (score 0) and positive (score 1-2) for analysis.
Real-time PCR
RNA was isolated using Trizol (Invitrogen) and treated with DNase I (Cat: EN0525, ThermoFisher Scientific). Quantitative real-time PCR was carried out using Power SYBR Green Master Mix (Cat: 4367659, ThermoFisher Scientific) as described in [9]. For IFNα (Cat: 4331182), IFNβ (Cat: 4331182), and CXCL10 (Cat: 4331182) Taqman probes were used from Applied Biosystems. Reactions were run and analyzed using ABI-7900HT Fast Real-Time PCR System (Applied Biosystems), with normalization to mouse β-actin. The primer sequences for individual genes used for the analyses are listed in Supplementary Table 2.
Immunoblotting
For protein analysis, cells were lysed in RIPA lysis buffer and processed for protein loading as previously published[9]. The following primary antibodies were used to detect specific proteins: Gal1 (Cat: ab138513, Abcam), phospho-STING (Cat: 72971), phospho-TBK1 (ser172, Cat: 5483), STING (Cat:13647), TBK1 (Cat: 3504), IRF-3 (Cat: 4302, RRID: AB_1904036), phospho-IRF-3 (Cat: 29047, RRID: AB_2773013), RelA/p65 (NF-κB (Cat: 8242), phospho-NF-κB (RelA/p65, Cat:3033), NF-κB1 p105/p50 (Cat: 3035), NF-κB2 p100/p52 (Cat: 4882) from Cell Signaling Technologies, and β-Actin (1:5000; Cat: sc-47778 HRP, Santa Cruz Biotechnologies). Secondary antibodies used in this study were HRP-conjugated Donkey anti-Goat IgG (H+L) (1:5000-1:10,000; Cat: A15999 Invitrogen), HRP-conjugated goat anti-rabbit (1:5000; Cat: 7074 Cell Signaling Technologies). Immunoblots were developed with Pierce West Pico (Cat: 35060; Thermo Fisher Scientific) and visualized with ChemiDoc XRS imaging system equipped with Image Lab Software (RRID: SCR_008426; Bio-Rad Laboratories). In each case, experiments were carried out in triplicate and a representative blot is shown unless otherwise stated. Blot densitometry quantification was done using ImageJ (RRID: SCR_003070)
Immunoprecipitation assay
Protein lysates were prepared as described in the immunoblotting section. 500ug protein lysate was pre-incubated with Protein A/G Dynabeads from Thermo Fisher Scientific, following which 2ug of Gal1 antibody (Cat: ab138513, Abcam) was added, and the lysate was incubated with rotation overnight at 4°C. 5% from each immunoprecipitation was set aside for input control, and normal rabbit IgG (Cat: 211-032-17; RRID: AB_2339149, Jackson ImmunoResearch Labs) was utilized as a non-specific IgG control. Lysates were treated with Protein A/G Dynabeads for 3 hours at 4°C. Immunoprecipitated proteins were then collected using a magnetic stand; beads were washed 3-4 times with ice-cold wash buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.05% NP-40) following incubation. Elution of proteins was carried out with IgG elution buffer (Thermo Fisher Scientific) at room temperature for 10 minutes on gentle vortex, boiled in sample loading buffer for 10 minutes, and then analyzed by western blotting for STING and Gal1. To study carbohydrate dependence of Gal1/STING interaction, MOC2 cells were treated with either PBS or lactose (10 mM) for 24 h and used for lysate preparation.
Calculation of MDSC enrichment score
We created an MDSC signature gene set based on marker genes presented in three independent datasets[18–20] as well as genes from our own experimental data. We calculated an enrichment score for each individual patient sample included in TCGA head and neck squamous cell carcinoma (HNSCC) dataset using Gene Set Variation Analysis (GSVA)[21]. In GSVA, for each patient sample, a kernel estimation of the cumulative density function of all the gene expressions was calculated to produce gene expression level statistics and then the gene expression statistic was ranked. For the genes in the MDSC signature gene set, the Kolmogorov-Smirnov-like rank statistic was calculated. The enrichment score for each patient sample was the difference in the statistic between genes inside and outside of the MDSC signature gene set. The higher the score, the patient’s gene expression is more enriched for MDSC gene signatures.
Survival analysis
Survival analysis was performed using the R package “survival”. For the MDSC enrichment score survival analysis, risk groups were defined by using the MDSC enrichment score of 0.1. Patients with MDSC enrichment score > 0.1 were grouped into MDSC high group, otherwise, they were grouped into MDSC low group. Log-rank p-value was used to assess the statistical significance of the survival difference between the two risk groups. Cox proportional hazard model was used to obtain the hazard ratio (HR) and confidence interval (CI). For survival analysis on both MDSC enrichment scores, we used a COX proportional hazard regression model of the MDSC enrichment score. Survival data status was based on the TCGA HNSCC dataset.
Statistical Analysis on MDSC enrichment scores
ANOVA test was used to assess the significance of difference among MDSC enrichment scores for each T stage group (T1, T2, T3, and T4). Student’s t-test was used to assess the significance of the difference between MDSC enrichment scores between tumor stages T1-2 and T3-4. Pearson’s correlation was used to assess the correlation between LGALS1 gene expression and MDSC enrichment scores. Stage information was based on the TCGA HNSCC dataset. Differentially expressed gene (DEG) analysis using the TCGA HNSCC dataset between MDSC high enrichment and low enrichment groups was performed on each gene expression and the p-values were corrected using a false discovery rate. DEGs were defined by a false discovery rate smaller than 0.05 and fold change higher than 1.5.
RNA sequencing (RNA-seq) analysis
RNA was isolated from a minimum of 2x106 sorted PMN-MDSCs from the primary tumors or the lungs using an RNeasy micro Kit (Qiagen). Expression levels of RNA sequencing data were quantified using a quasi-mapping two-phase inference algorithm implemented in Salmon (version 0.9.1) [22]. Transcript sequences for mouse M24 from Gencode (https://www.gencodegenes.org) were used for reference transcriptome indexing. Transcript-per-million (TPM) value was used as a normalized expression level unit. Quantified gene expressions were then log-transformed for downstream analysis. Differentially expressed gene (DEG) analysis between the MDSC tumor group and MDSC lung group was performed on each gene expression and the p-values were corrected using a false discovery rate. DEGs were defined by a false discovery rate smaller than 0.05 and fold change higher than 1.5. To compare the MDSC RNA-level expression with neutrophil expression, publicly available mouse neutrophil RNA-seq data was used (GSE60336). Our MDSC RNA-seq and the public mouse neutrophil RNA-seq data were merged using COMBAT [23]. DEGs for three groups (MDSC lung, MDSC tumor, and neutrophil) were calculated using SAM [24] multiple class comparison with a false discovery rate smaller than 0.05.
Conditioned media (CM) collection
MOC2 WT and MOC2 KO cells were plated at equal numbers 5 x10^6 in a T-225 flask. When the cells were at 70% confluence, the media was changed to 1% serum-containing media and incubated for 24 h. Following the incubation, cells from each of the sets were counted for normalization. The collected media was spun down to remove any dead cells/ floating debris at 2000 rpm for 5 min, filtered with 0.45um filter, and then concentrated to 10x by Amicon Ultra cell centrifugal filters with 3 kDa cutoff (EMD Millipore). The samples were stored at −80°C in aliquots until use. Similarly, concentrated 1% serum-containing media was used as a control.
Luminex Assay
For the Luminex assay, CM collected from the cells in three different batches was used for analyses. This assay was performed by the Human Immune Monitoring Center at Stanford University. Mouse 48 plex Procarta kits (EPX480-20834-901) were purchased from Thermo-Fisher/Life Technologies and used according to the manufacturer’s recommendations with modifications as described. Briefly: Beads were added to a 96-well plate and washed in a BioTek ELx405 washer. Samples were added to the plate containing the mixed antibody-linked beads and incubated overnight at 4°C with shaking. Cold (4°C) and room temperature (RT) incubation steps were performed on an orbital shaker at 500-600 rpm. Following the overnight incubation, plates were washed in a BioTek ELx405 washer and a biotinylated detection antibody was added for 60 minutes at RT with shaking. The plate was washed as described and streptavidin-PE was added for 30 minutes at RT. The plate was washed as above and a reading buffer was added to the wells. Each sample was measured in duplicate. Plates were read using a Luminex 200 or a FM3D FlexMap instrument with a lower bound of 50 beads per sample per cytokine. Custom Assay Chex control beads were purchased from Radix BioSolutions, Georgetown, Texas; and added to all wells.
Isolation of T cells from the spleen
Spleens from tumor-bearing mice were harvested under sterile conditions. Using mechanical disruption, the spleen (in PBS+2% FCS) was dissociated into a cell suspension, which was then filtered through a 70μm filter to obtain a single-cell suspension [9]. Following RBC lysis with ACK lysis buffer, T cells were isolated using Mouse Pan T cell isolation kit from Stem Cell Technologies.
Study Approval
All the animal studies described in the study were conducted as per the APLAC guidelines approved by the Institutional Animal Care and Usage Committee of Stanford University under APLAC protocol number-15106 (Research Compliance Office, Palo Alto, CA 94306). Patient data collection was approved by the Stanford Institutional Review Board (IRB-40425, Palo Alto, CA 94306). Written informed consent was received from participants prior to inclusion in our study.
Statistical analysis
All statistical analysis was performed in Prism version 7.03 or greater (GraphPad Software, RRID:SCR_002798). Power analyses based on preliminary data showed that five mice/ group will provide >93% power to detect a difference of 5 in the mean MDSC level in the lung assuming a standard deviation of 2.0 in a two-sided test with an alpha level of 0.05. Graphs represent mean values ± standard deviation. P values were calculated for bar graphs using unpaired two-tailed Student’s t-test, one-way ANOVA, or Kruskal-Wallis test, and log-rank statistics for survival analyses, as indicated in the figure legends. Comparison of multiple sets of data was achieved with analysis of variance (ANOVA) with Tukey’s multiple comparisons. Survival analysis was determined by Log-Rank (Mantel-Cox) analysis. In boxplots, the center line represents the median, the box limits denote the 25th to the 75th percentile and the whiskers represent the minimum and maximum values. Error bars indicate standard deviation. Statistical significance was set to p<0.05. Analysis was performed using GraphPad Prism v9. All mouse experiments were repeated at least twice with similar results. All in vitro experiments were performed at least two times with similar trends.
Data Availability Statement
The raw RNA-seq data were deposited in the GEO database with accession number GSE234636. All other raw data are available upon request from the corresponding author.
Results
Galectin-1 (Gal1) promotes tumor-induced-MDSC expansion in both local and systemic compartments
We previously showed that Gal1 deletion (KO) or knocking down (KD) in head and neck squamous cell carcinoma (HNSCC) and lung cancer cells significantly reduced metastatic burden [9, 10]. In order to understand the role of the immune system in controlling Gal1-mediated metastasis, we utilized the MOC2 syngeneic HNSCC model, which expresses high levels of Gal1 and undergoes spontaneous metastatic progression as reported previously [9]. We injected MOC2 WT- or KO-Gal1 cells in either immune-competent C57/BL6 mice or immune-deficient Rag2−/− IL2rg−/− mice (T, B, and NK cell deficient) with the same genetic background. As expected, we saw a significant decrease in lung metastases in KO-Gal1 tumor-bearing mice compared to mice with WT-Gal1 tumors (86% reduction in KO-Gal1) (Fig. 1A). In the immune-deficient mice, even though the difference in the primary tumor growth was abrogated (Supplementary Fig. 1A), there continued to be a significant difference in the metastatic burden between the WT- and KO-Gal1 tumors (63% reduction in KO-Gal1). This suggested that the difference in Galectin-1 mediated metastatic burden between the two models to a large extent was independent of T, NK, and B cells. Furthermore, flow cytometric analysis of the lungs revealed a significant reduction in CD11b+ Ly6Clo/int Ly6G+ cells (referred to as MDSCs going forward) in the KO-Gal1 tumor-bearing mice compared to mice with WT-Gal1 tumors in the Rag2−/− IL2rg−/− background (Fig. 1B). Considering the important role that MDSCs play in metastasis, we explored whether Gal1-mediated metastasis was regulated through MDSC recruitment and expansion. Since immune-deficient mice differ significantly in immune response mounted against the primary tumor, we analyzed the difference in myeloid cell infiltration (CD11b+) in WT- and KO-Gal1 primary tumors in immune-competent C57/BL6 mice and found significantly lower myeloid cell levels in the KO- compared to the WT-Gal1 tumors (Fig. 1C, left panel). Since the majority of the myeloid cells in this model are Gr1+MDSCs, we decided to check for the distribution of monocytic (M-MDSC: CD11b+Ly6G−Ly6Chi) and granulocytic MDSCs (PMN-MDSC: CD11b+Ly6Clo/int Ly6G+ cells) (gating-Supplementary Fig. 1B & C). We found a reduced level of both PMN- and M-MDSCs in the KO-Gal1 tumors; however, the largest difference between the MOC2 WT- and KO-Gal1 tumors was seen in the M-MDSC fraction (Fig. 1C, right panel). Interestingly, WT-Gal1 tumor-bearing mice had significant splenomegaly compared to KO-Gal1 counterparts, suggestive of systemic changes in the immune system (Supplementary Fig. 1D). To determine whether Gal1 enhanced MDSC numbers systemically, we analyzed mouse blood samples for MDSC level at Day 14 when the tumors were at similar sizes. We found that the circulating PMN- and M-MDSC levels were both significantly lower in mice bearing KO-Gal1 tumors compared to WT-Gal1 tumors (Fig. 1D). To test whether there is a significant correlation between Gal1 and MDSC levels in patients, we utilized the head and neck squamous cell carcinoma (HNSCC) cohort of the Cancer Genome Atlas (TCGA, N=530). Since there are very few established MDSC gene expression signatures, we curated the gene signature from three different datasets to generate an MDSC enrichment score. Using this enrichment score, we calculated the correlation between LGALS1 mRNA levels and MDSC enrichment for each patient and found it to be significant (R2 = 0.5; p< 2e-16) (Fig. 1E). In addition, MDSC enrichment using our gene signature was related to tumor stage in the TCGA-HNSCC cohort with T3-4 tumors showing increased MDSC enrichment compared to T1-2 tumors (Supplementary Fig. 1E). We then classified each patient to either MDSChigh or MDSClow group based on this enrichment score (Supplementary Fig. 1F). Survival analysis revealed that the MDSChigh group had significantly lower survival compared to the MDSClow group (Log-rank p-value: 0.009) (Fig. 1F). The reduced survival of these patients is in line with studies in other cancers in which MDSC enrichment is associated with poor prognosis [25]. To further assess the relationship between tumor Gal1 and MDSC levels in human HNSCC, we stained a tissue microarray (TMA) of oral cavity cancer (N=81) for Gal1 and PMN-MDSC (CD15+LOX1+) (Fig. 1G, left) and M-MDSC (CD14+HLA-DRneg). We found that 74.5% of the Gal1 high tumors showed high infiltration of PMN-MDSCs compared to 56.6% of Gal1 low tumors; however, the difference did not achieve statistical significance (Fig. 1G; Supplementary Fig. 1G–H). The staining of M-MDSC was very weak in these samples and could not be analyzed. Overall, analysis of both mice and human HNSCC showed an association between Gal1 and tumor MDSC levels, which is linked to poorer survival in HNSCC patients.
Fig. 1: Galectin-1 (Gal1) promotes tumor induced-MDSC both in local and systemic compartments.
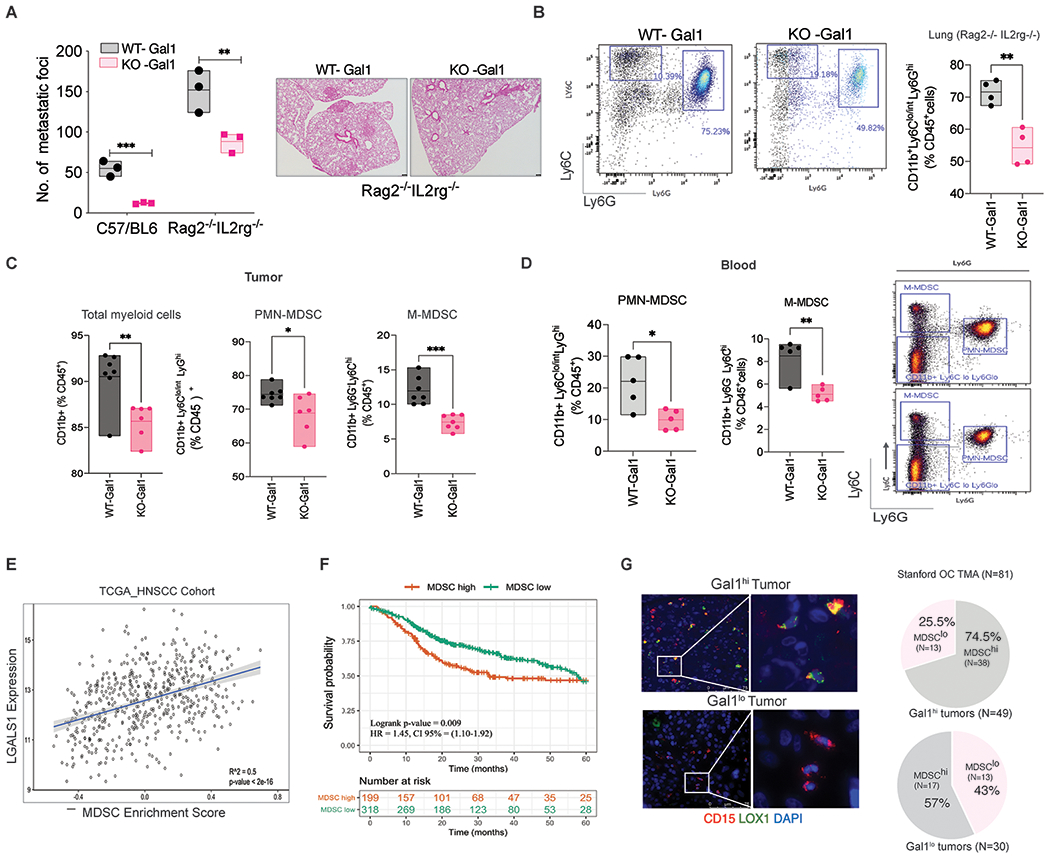
A. Quantification of lung metastases foci after subcutaneous. implantation of MOC2 WT-Gal1 or KO-Gal1 cell lines in immune-competent C57/BL6 or immune deficient (Rag2−/− IL2rg−/−) mice. The number of nodules per lung area was quantified by H&E staining (Right). B. Quantification of CD11b+Ly6Clo/int Ly6G+ cells in lungs of mice implanted with MOC2 WT-Gal1 and KO-Gal1 tumors in Rag2−/− IL2rg−/−, after enzymatic dissociation and flow cytometry analyses. C. Quantification of total myeloid cells (CD11b+), PMN-MDSC (CD11b+Ly6Clo/intLy6G+) and M-MDSC (CD11b+Ly6G− Ly6Chi) cells in MOC2 WT-Gal1 and KO-Gal1 tumors after enzymatic dissociation and flow cytometry analyses. D. Quantification of PMN-MDSC (CD11b+Ly6Clo/intLy6G+) and M-MDSC (CD11b+Ly6G−Ly6Chi) cells in the blood at Day 14 in mouse bearing subcutaneous MOC2 WT/ KO-Gal1 tumors. E. Pearson’s correlation was used to assess the correlation between LGALS1 gene expression and MDSC enrichment scores in the TCGA HNSCC cohort. F. Kaplan Meir survival analyses for the TCGA HNSCC cohort using Gene Set variation analyses depict survival differences between patients with MDSChi and MDSClo (based on the enrichment score). The enrichment score for each patient sample was the difference in the statistic between genes inside and outside of the MDSC signature gene set. For the MDSC enrichment score survival analysis, risk groups were defined by using the MDSC enrichment score of 0.1. G. Immunofluorescence staining of PMN-MDSC in human oral cavity cancer TMA (N=81) using CD15 (red), LOX1 (green), and DAPI (Blue) staining. The white insets represent zoomed area (right image). The Pie chart (right panel) represents the distribution of patients with high or low MDSC burden in the cohort of patients with high Gal1 (N=51) and low Gal1 (N=30) expressing tumors.
Galectin-1 is important for MDSC infiltration into pre-metastatic niches
Previous studies suggest that myeloid cells are the most frequent immune cells in the pre-metastatic niche [26]. To understand if Gal1-induced metastasis was influenced by the enrichment of myeloid cells, specifically MDSCs in pre-metastatic sites, we evaluated the immune changes in the lungs of HNC tumor-bearing mice before overt metastasis was detected. Using tSNE (t-Distributed Stochastic Neighbor Embedding) analysis, we found that both MOC2 (HPV-negative) and mEERL (HPV-positive) models showed significant myeloid infiltration (CD11b+) (70-90% of CD45+ cells) with very few T cells in the lung (Fig. 2A–C). Further analyses of the CD11b+ population in these two models revealed that the majority of the myeloid cells were PMN-MDSCs (~40-65%), M-MDSC (~10-15%), and Ly6C+MO-MØ (monocytes/monocyte-derived macrophages) (~8-12%) (Fig. 2D–E, Supplementary Fig. 2A). The other observed myeloid cells were alveolar (AMs) and interstitial macrophages (IMs) and myeloid dendritic cells. To verify whether tumor Gal1 deletion would reduce PMN-MDSC in the lungs, we analyzed changes in their lung levels following the establishment of subcutaneous tumors. We found that PMN-MDSC level was significantly lower in the lungs of KO-Gal1 tumor-bearing mice compared to the WT-Gal1 counterparts (~46% reduction) (Fig. 2F). To ensure that the reduction in pulmonary PMN-MDSC was not due to the smaller primary tumor size observed for MOC2 KO-Gal1 tumors, we performed immune analyses on the lungs when the KO- and WT-Gal1 tumors were at comparable volumes. We continued to observe significantly lower pulmonary PMN-MDSC levels for KO-Gal1 tumor-bearing mice (Supplementary Fig. 2B); however, there was no difference in the number of CD4+ and CD8+ T cells in the lungs of these mice (Supplementary Fig. 2C). The effect of Gal1 on MDSC number was independent of CD8+ T cells since depleting them did not affect the level of pulmonary MDSCs between the two conditions (Supplementary Fig. 2D–E). We also investigated the Lewis Lung Cancer (LLC) model to see if this effect was generalizable to other cancer types. We saw a significant difference in both PMN-MDSCs (54% reduction) and M-MDSCs (20% reduction) in the lungs of mice bearing knockdown (KD)-Gal1 tumors compared to those with WT-Gal1 tumors before metastatic seeding was observed (Fig. 2G). This data suggest that tumor Gal1 modulates MDSC levels in the lungs and this effect is independent of its role in modulating T cell number.
Fig. 2: Galectin-1 deletion reduces MDSC levels in the lungs in HNC and lung cancer models.
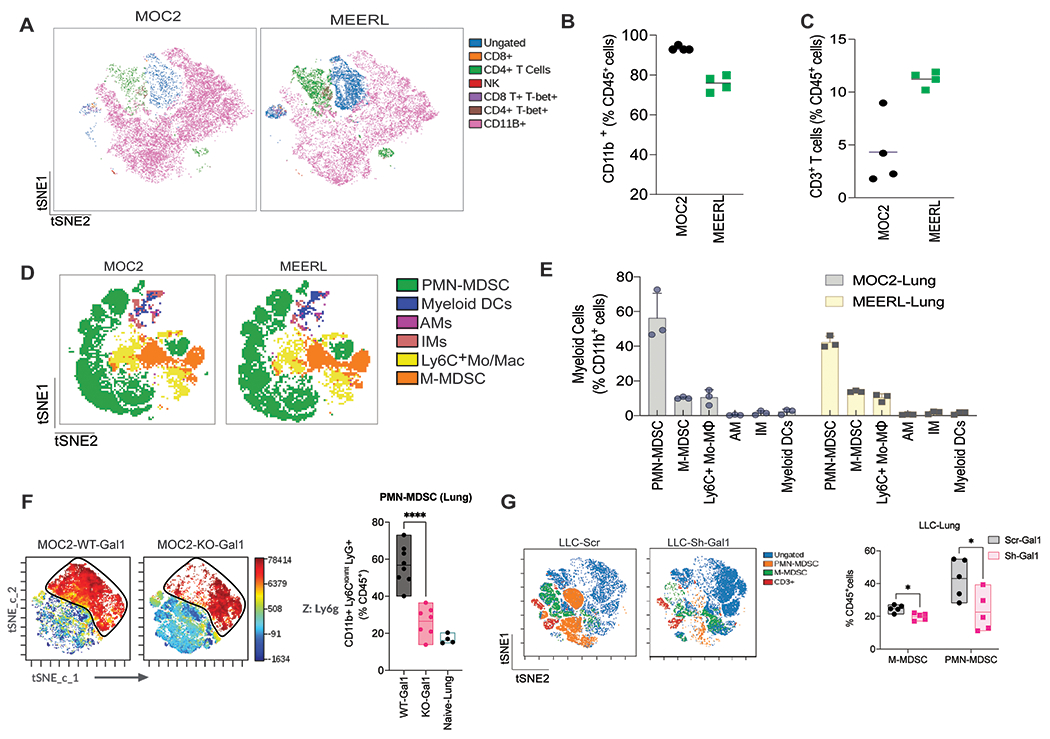
A. tSNE analysis of murine immune cells in lungs of mice bearing 300mm3 subcutaneous MOC2 and mEERL tumors, run on 5000 live CD45+ single cells from each sample. B. Relative proportions of CD11b+ cells in the lungs of tumor-bearing mice. C. Relative proportions of CD3+ T cells in the lungs of tumor-bearing mice. D. tSNE analysis of myeloid cells in lungs of mice bearing 300mm3 subcutaneous MOC2 and mEERL tumors, run on 4000 live CD11b+ cells. E. Quantitation of M-MDSCs, PMN-MDSCs, interstitial macrophages (M), alveolar macrophages (AM), and myeloid DCs as a percentage of CD11b+ cells in the lungs of tumor-bearing mice at Day 14 - a time point before visible metastases are observed. F. tSNE analyses showing the distribution of immune cells in lungs in mice bearing MOC2 WT-Gal1 and KO-Gal1 tumor run on 5000 live CD45+ single cells from each sample (Left), and quantitation of CD11b+Ly6Clo/int Ly6G+ PMN-MDSCs in the lungs (Right) (n = 7-8 mice). G. tSNE analyses of immune cells in lungs (Left) and quantitation of CD11b+Ly6Clo/int Ly6G+ PMN-MDSCs and CD11b+Ly6G−Ly6Chi M-MDSC (Right) in LLC-Scramble and LLC-Sh-Gal1 tumor-bearing mice. Student’s t-test (two-tailed) was used for comparing the two groups. Each dot represents one mouse. Data are shown as mean+/−SD. Experiments have been repeated a minimum of two times with similar results.
Galectin-1-mediated PMN-MDSC expansion promotes lung remodeling and metastasis
Growing experimental evidence indicates that MDSCs are key determinants of pre-metastatic niche formation [26],[27]. MDSCs are known to contribute to diverse features of a pre-metastatic site including induction of vascular leakage, extracellular matrix (ECM) remodeling, and systemic immunosuppression that facilitates metastatic outgrowth[7]. Chemokines, growth factors, and extracellular vesicles released from MDSCs participate in multiple stages of pre-metastatic niche formation and evolution [28, 29]. To better understand the recruitment of MDSCs to the lung, we performed a time course experiment to assess the change in myeloid cell number in the lungs after tumor cell implantation subcutaneously (SC) over a 3-week period. We found that PMN-MDSC levels dramatically increased in the lungs of mice bearing WT-Gal1 tumors between Day 7 and Day 21. In contrast, the pulmonary PMN-MDSC level in mice bearing KO-Gal1 tumors remained largely stable with a small increase around Day 21 (Fig. 3A– 3B). The changes in the lungs paralleled the changes in the blood over time (Fig. 3C, left panel). To ensure that MDSC accumulation in the lungs was not due to the systemic increase in circulating MDSCs, we evaluated the level of MDSCs in the liver and kidneys of these mice where there was no metastasis. We found that only the lungs reflected the difference in the circulating MDSC levels between the two groups (Fig 3C, right panel, and Supplementary Fig. 3A). These data suggest that MDSC accumulation is associated with organotrophic metastasis.
Fig. 3: Galectin-1 mediated PMN-MDSC enrichment in the lung promotes lung remodeling and pre-metastatic niche formation.
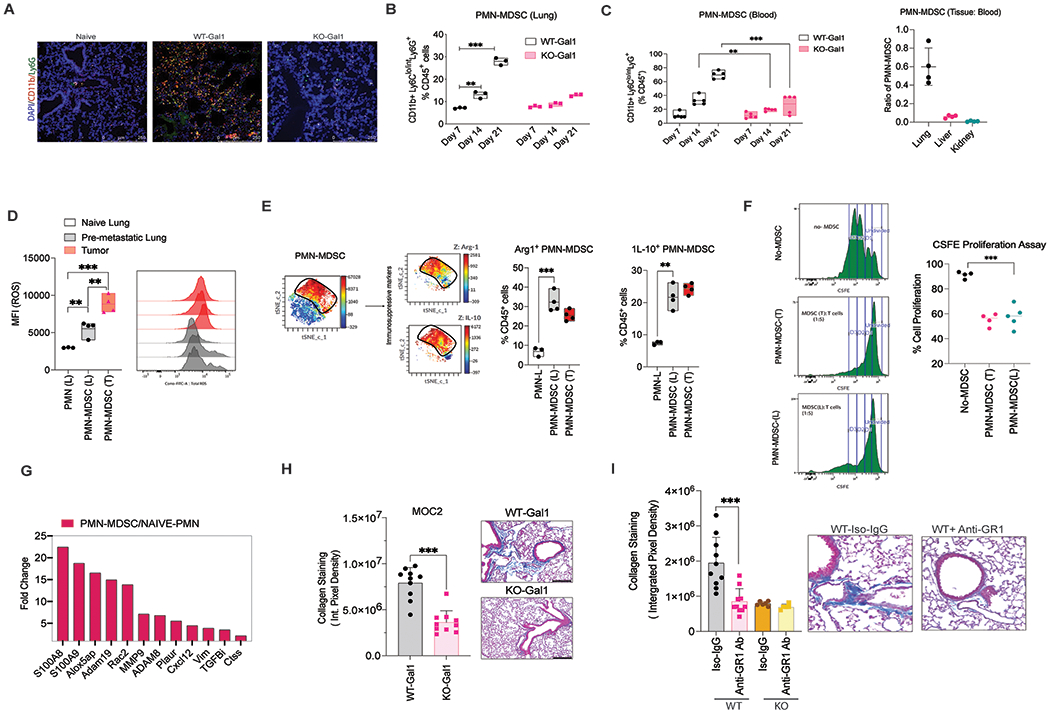
A. Representative images of the lung from Naïve mice or mice bearing either WT-Gal1 or KO-Gal1 tumor showing PMN-MDSC (CD11b+- Green and Ly6G+- red) distribution at Day 14-time point. B. Time-series analyses of PMN-MDSC levels in lungs of mice bearing MOC2 WT-Gal1 and KO-Gal1 tumors at Day 7, 14, and 21 (before metastatic seeding), n=3 (repeated in two independent experiments). C. (Left) Time-series analyses of PMN-MDSC levels in the blood of mice bearing MOC2 WT-Gal1 and KO-Gal1 tumors at Day 7, 14, and 21(n=5 / group); (Right) Ratio of PMN-MDSCs in different tissues (lung, liver, kidney) to the PMN-MDSC levels in blood. D. Graph and histogram showing ROS levels (MFI-DCFDA) in PMNs from Naïve mice or PMN-MDSCs isolated from tumors or corresponding lung tissue (n=4). E. tSNE analysis of immune cells in lungs in mice bearing 300mm3 subcutaneous MOC2 and, run on 5000 live CD45+ single cells from each sample (Left). Quantitation shows a high percentage of Arginase-1+ (Arg-1+) and Interleukin-10+ (IL-10+) PMN-MDSCs in the lungs and the tumor (Right). F. Data showing suppression of T cell proliferation by PMN-MDSCs isolated from the tumor and lung of MOC-2 tumor-bearing mice (n= 4). G. Fold change in gene expression of established pre-metastatic niche components between Naïve PMNs and PMN-MDSCs isolated from tumor-bearing mice using RNA-Seq analyses. H. Representative images and quantification of Trichrome staining intensity in the lungs of MOC2 WT/KO-Gal1 tumor-bearing mice (n=5/group). I. Representative images and quantification of Trichrome staining intensity in lungs of MOC2 WT/KO-Gal1 tumor-bearing mice treated with either Anti-GR1 antibody or the isotype control IgG, n=5/group. Two-tailed unpaired t-test was performed for the comparisons between the two groups. Two-way ANOVA and Tukey’s multiple comparisons test were performed when more than two groups were compared. Each dot represents one mouse, and the bar denotes the mean. Data are shown as mean+/−SD.
To check if pulmonary PMN-MDSCs are immune-suppressive, we first evaluated the expression of reactive oxygen species (ROS), arginase (Arg-1), and interleukin-10 (IL-10) in these cells (Fig. 3D–E). We found that the pulmonary MDSCs from tumor-bearing mice had slightly lower ROS levels compared to tumor MDSCs, however, they had a high level of arginase and IL-10 expression (Fig. 3E; Supplementary Figure 3B) when compared to PMNs from the lungs of naïve mice, indicating a phenotypic difference from naïve neutrophils. However, we did not see a significant difference in the suppressive ability of the lung PMN-MDSCs isolated from either MOC2WT/KO tumor-bearing mice. To validate their immune suppressive function, we performed T cell suppression assays using PMN-MDSCs sorted from the tumor and the lungs of the tumor-bearing mice. We found that these PMN-MDSCs suppressed T cell proliferation at similar levels (Fig. 3F), suggesting that they are indeed suppressive in nature. To tease out potential differences between tumor-resident and pulmonary PMN-MDSCs in tumor-bearing mice, we performed RNA sequencing analyses of these two cell populations isolated on Day 14 (before metastatic seeding in the lung). We also compared the expression patterns in PMN-MDSCs isolated from the tumor (PMN-MDSC-T) and lungs (PMN-MDSC-L) with established naïve neutrophil signatures. We found that the PMN-MDSC-L were significantly enriched in genes associated with migratory chemokines such as S100A8, S100A9, and CXCL2 compared to PMN-MDSC-T and naïve neutrophils (Supplementary Fig. 3C). When we compared the top 100 differentially expressed genes in PMN-MDSC-L compared to naïve neutrophils using STRING cluster analysis, we saw a significant difference in the gene clusters related to collagen remodeling and IL-17 signaling (Supplementary Fig. 3D & 3E). In addition, we also found a significant upregulation of multiple pre-metastatic niche-forming components in PMN-MDSC-L compared to naïve neutrophil RNA expression (Fig. 3G). The Reactome pathway enrichment analysis showed changes in collagen assembly and biosynthesis, ECM degradation in the PMN-MDSC-L compared to naïve neutrophils (Supplementary Figure 3E). These data suggest that PMN-MDSC-L may play a direct role in collagen and ECM remodeling which is important for the homing of disseminated tumor cells, thereby promoting metastasis.
To relate tissue changes with gene expression analyses, we performed Trichrome staining of lungs from WT- and KO-Gal1 tumor-bearing mice on Day 20 (prior to overt metastases). Pulmonary fibrosis is characterized by abnormal alveolar structure, myofibroblast accumulation, and collagen deposition. We found a significant increase in peri-bronchial and peri-vascular fibrosis and increased collagen deposition in WT-Gal1 tumor-bearing mice compared to KO-Gal1 counterparts in both MOC2 (Fig. 3H) and mEERL (Supplementary Fig. 3F) models. To determine that these ECM changes were indeed due to PMN-MDSCs present in the lung, we used an anti-GR1 antibody to deplete these cells (Supplementary Fig. 3G). Depletion of MDSCs led to a decrease in pulmonary collagen staining in MOC2 WT-Gal1 tumor-bearing mice but not in the KO-Gal1 counterpart (Fig. 3I), suggesting that PMN-MDSC in the lungs directly contribute to lung remodeling and ECM changes associated with metastasis.
Galectin-1 promotes MDSC migration through the CXCL2-CXCR2 axis.
Since previous studies have shown that secreted Gal1 from HNSCC mediates immunosuppression through T cells, we sought to determine if the effects on MDSC were dependent on secreted Gal1 and its related secretome using tumor-conditioned media (CM). We injected concentrated CM from either WT- or KO-Gal1 MOC2 cells into the naïve mice by intraperitoneal (IP) injection every 3 days for 3 weeks, after which we harvested the spleen and lungs to evaluate the changes in immune cell populations. As previously reported, we saw a decrease in CD8+ T cells in the spleen of mice injected with WT-Gal1 CM compared to KO-Gal1 CM (Fig. 4A; Supplementary Fig. 4A). However, we did not observe a difference in the number of CD8+ T cells in the lungs of mice receiving either WT- or KO-Gal1 CM. The effect on splenic CD8+ T cells could be due to increased local concentration of secreted Gal1 (via IP injection), which causes T cell apoptosis in the WT-CM injected group. In contrast, the level of CD11b+LyClo/intLy6G+ cells (PMN-MDSCs) was significantly lower in both the lungs and the spleen of KO-Gal1 CM recipients compared to those receiving WT-Gal1 CM (Fig. 4B). Of note, we saw a higher level of both CD8+ T cells and PMD-MDSCs in mice injected with concentrated media alone, likely because the untreated media contained more nutrients and growth factors than tumor-cell CM that have been depleted of these factors during culture. We next performed a similar experiment wherein we injected either WT-Gal1 CM or KO-Gal1 CM in mice bearing KO-Gal1 tumors to determine whether WT-Gal1 CM alone, without tumor cells, could increase PMN-MDSC levels in mice bearing KO-Gal1 tumor-bearing mice. Injection of the WT-Gal1 CM led to a significant increase in lung and spleen PMN-MDSCs, but not T cells, compared to injection of KO-Gal1 CM even though the tumor cells did not express Gal1 (Fig. 4C, Supplementary Fig. 4B).
Fig. 4: Galectin-1 enhances MDSC recruitment to pre-metastatic site by increasing myeloid chemokine production.
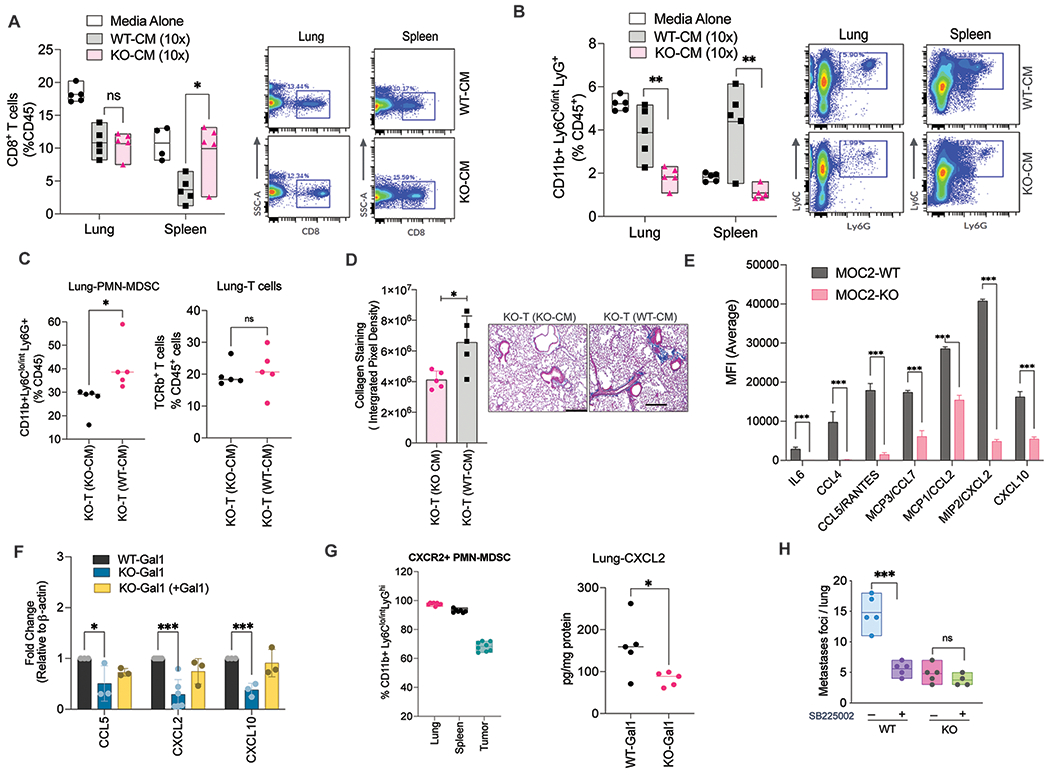
A. Quantification and flow plots showing the percentage of CD8+T cells in the lungs and the spleen of tumor naïve mice after ~3 weeks of treatment with either WT- or KO-Gal1 conditioned media (CM) or control media. B. Quantification and flow plots showing the percentage of PMN-MDSCs in the lung or spleen of tumor naïve mice after ~3 weeks of treatment with either WT- or KO-Gal1 CM or control media. C. Quantitation of PMN-MDSCs and T cells in the lungs of mice bearing KO-Gal1 tumors injected with either WT- or KO-Gal1 CM for 3 weeks. D. Quantitation and representative images showing the amount of collagen staining (Trichrome staining) in the lungs of mice bearing KO-Gal1 tumors injected with either WT- or KO-Gal1 CM for 3 weeks. E. Luminex data showing differentially secreted cytokines between the WT/KO-Gal1 conditioned media from MOC2 cells collected after 24h of culture followed by 10x concentration. F. Relative mRNA expression of CCL5, CXCL2 and CXCL10 in MOC2 WT-Gal1 cells, KO-Gal1, or KO-Gal1 with exogenously reconstituted Gal1. G. (Right panel) Quantification of CXCR2 expression on the PMN-MDSCs isolated from the lung, spleen, and the tumor of MOC2 WT-Gal1 tumor-bearing mice; (Left panel) Quantification of CXCL2 protein levels in the lung lysate from mice bearing MOC2 WT- or KO-Gal1 tumors. H. Quantification of metastatic foci per lung from MOC2 WT/KO-Gal1 tumors treated with vehicle or SB225002 (CXCR2 inhibitor). Two-tailed unpaired t-test was performed for the comparisons between the two groups. Two-way ANOVA and Tukey’s multiple comparisons test were performed when more than two groups were compared. Each dot represents one mouse. Data are shown as mean+/−SD. All experiments were repeated 2 times with similar results.
Further, we observed enhanced lung collagen deposition in the lungs of WT-Gal1 CM injected mice compared to KO-Gal1 CM counterparts (Fig. 4D). These results indicated that there is a significant difference in the secretome between the WT- and KO-Gal1 cells, resulting in the differential response to PMN-MDSC accumulation and lung remodeling.
We next performed the Luminex assay of the two CMs and identified several cytokines that were significantly lower in the KO-Gal1 CM compared to the WT-Gal1 CM (Fig. 4E; Supplementary Fig. 4C). We tested those that have been implicated in MDSC recruitment or function and showed the largest differences between the two CMs (CCL5, CXCL2, CXCL10) to see if the reductions were at the transcriptional level using qPCR. We saw a transcriptional reduction of these three genes in KO-Gal1 cells, which was reversed when these cells were reconstituted with exogenous expression of Gal1 (Fig. 4F).
Previous studies have shown that PMN-MDSCs express CXCR2 [30] [31] and that the CXCL2-CXCR2 axis is important for PMN-MDSC recruitment to pre-metastatic niches in colorectal cancer [28]. Similarly, we found that 92%, 96%, and 68% of the PMN-MDSCs in the spleen, lung, and tumor expressed high cell-surface levels of CXCR2, respectively (Fig. 4G; left panel; Supplementary Fig. 4D). Since CXCL2, a prominent ligand for CXCR2, was one of the most differentially expressed chemokines between WT- and KO-Gal1 cells (Fig. 4E–F), and its level in the lung tissues was higher in mice bearing WT-Gal1 tumor compared to the KO-Gal1 counterparts (Fig. 4G, right panel), we hypothesized that this axis may mediate PMN-MDSC migration to lung to assist metastatic progression. To test this hypothesis, we treated mice bearing either WT- or KO-Gal1 tumors with a selective CXCR2 inhibitor, SB225002. As anticipated, we found that CXCR2 inhibition, compared to vehicle, significantly reduced lung metastases in WT-Gal1 tumor-bearing mice but minimally in KO-Gal1 tumor-bearing mice (Fig. 4H). These data strongly suggest that Gal1-mediated enhanced expression of myeloid-regulating chemokines, especially CXCL2, is driving MDSC migration and promoting metastasis in this model.
Galectin-1 maintains chronic NF-κB activation by preventing auto-phago-lysosomal degradation of STING
Because the noted cytokine changes were at the level of gene expression, we evaluated the activation status of NF-κB, which is a well-known major transcription factor controlling the expression of these chemokines [32]. The canonical wing of the NF-κB pathway was more activated in WT-Gal1 compared to KO-Gal1 or KD-Gal1 cells (Fig. 5A, left). This was also noted in vivo as reflected by enhanced phosphorylation of p65 in the primary WT- compared to KO-Gal1 MOC2 and mEERL tumors (Supplementary Fig. 5A). In contrast, there was no significant difference in the non-canonical NF-κB signaling (Fig. 5A, right).
Fig. 5: Galectin-1 mediates chronic NF-kB activation by preventing autophagolysosomal degradation of STING.
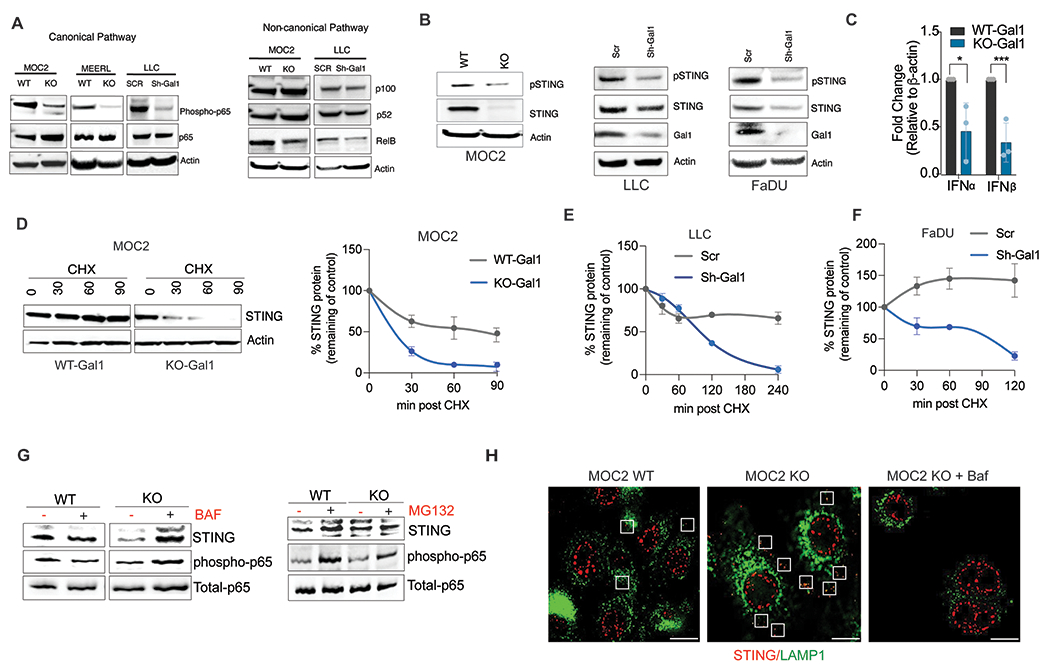
A. Representative immunoblot showing expression of characteristic proteins in the canonical and non-canonical NF-kB signaling pathway in WT- or KO-Gal1 MOC2 or mEERL cells and in WT or Sh-Gal1 LLC cells. B. Representative Immunoblots showing total and phosphorylated (activated) STING protein levels in WT or KO-Gal1 MOC2 cells, WT or Sh-Gal1 LLC cells, and in WT or Sh-Gal1 FaDU cells (human HNSCC). C. q-PCR analysis of IFN-α and IFN-β gene expression levels in MOC2 WT/KO-Gal1 cells. D. Representative Immunoblot of STING in MOC2 WT/KO-Gal1 cells treated with 10 μg/ml translation inhibitor cycloheximide (CHX) for 0 to 90 minutes with β-actin as a loading control (left panel); Quantification of STING level in MOC2 WT/KO-Gal1 cells (right panel, n=3). E. Quantitation of STING level in LLC-Scr and Sh-Gal1 cells treated with 10 μg/ml cycloheximide (CHX) for 0 to 240 minutes. F. Quantitation of STING level in FaDU-Scr and Sh-Gal1 cells treated with 10 μg/ml cycloheximide (CHX) for 0 to 120 minutes. G. Immunoassay of STING in MOC2 WT/KO-Gal1 cells treated with lysosomal fusion inhibitor, 250 nM Bafilomycin (left) or proteasome inhibitor, 5μM MG132 (right) for 6 h. H. Immunofluorescence imaging of STING (red) and LAMP1 (green) in MOC2/KO-Gal1 cells with or without Bafilomycin (Baf) treatment; Boxes indicate points of colocalization between STING and LAMP1. Student’s t-test (two-tailed) was used for comparing a single treatment to the control.
Previous work has shown that the cGAS-STING pathway leads to NF-κB activation in cancer cells [33, 34]. Interestingly, the STING total protein and phosphorylation levels were strongly reduced when Gal1 was deleted in MOC2 cells or downregulated in LLC and human HNSCC FaDu cells (Fig. 5B). The noted change in STING protein level led to a reduction in the mRNA levels of interferon-alpha (IFNα) and interferon-beta (IFNβ) in MOC2 cells (Fig. 5C). However, the mRNA levels of TMEM173 (STING) did not change with Gal1 deletion, suggesting that the difference in STING protein levels is at the post-transcriptional level (Supplementary Fig. 5B). To determine whether the STING protein stability was affected by Gal1 expression, we blocked translation using cycloheximide and followed STING degradation. We found that there was a marked decrease in the half-life of STING when Gal1 is deleted or down-regulated in mouse and human cell lines (Fig. 5D–F; Supplementary Fig. 5C). Acute inhibition of lysosomal degradation using Bafilomycin was sufficient to increase the level of STING in KO-Gal1 cells, whereas proteasomal inhibition with MG132 had a little effect on STING protein levels (Fig. 5G). This result was further supported by immunofluorescent staining showing significant co-localization of STING and LAMP1 (a lysosomal protein) in KO-Gal1 cells but not in WT-Gal1 cells (Fig. 5H; Supplementary Fig. 5D). These results indicate that Gal1 prevents lysosomal degradation of STING, thereby prolonging STING activation. Although it was reported that STING expression is usually suppressed or lost in the majority of cancers, analysis of our HNC TMA revealed that almost 65% of the analyzed HNC samples show medium to high expression of STING in cancer cells (Supplementary Fig. 5E). This is in line with a previous study which reported a significant increase of STING expression in cancer cells in comparison with normal tissue in HNC patients [35]. Even though we did not find a correlation between Gal1 and STING levels in the tested HNSCC cohort, patients with positive Gal1 and STING expression had the wort progression-free survival (PFS) of the entire group (Supplementary Fig. 5F).
Endogenous Galectin-1 interacts with STING and maintains chronic STING activation, enhances MDSC expansion, lung remodeling and metastatic progression.
To formally test if Gal1 affects STING signaling, we treated WT- and KO-Gal1 cells with the STING agonist DMXAA, which robustly induced IFNα and IFNβ in WT, but not in Gal1-KO cells (Fig. 6A). Similarly, treatment with the STING antagonist, H151, suppressed the phosphorylation of IRF3 in WT-Gal1 cells but not in the KO-Gal1 cells, which had an overall reduced basal IRF3 phosphorylation (Fig. 6B). Genetic down-regulation of STING by shRNA also resulted in reduced transcription of IFNα and IFNβ in WT-Gal1 but not in KO-Gal1 cells (Fig. 6C; Supplementary Fig. 6). These data indicate that Gal1-mediated stabilization of STING is important for STING signaling in WT-Gal1 conditions. Furthermore, we show that Gal1 and STING interact with each other. Endogenous STING was co-immunoprecipitated with Gal1 in both MOC2 and FaDu cells when Gal1 was pulled down using an anti-Gal1 antibody (Fig. 6D). In order to further understand if the interaction was dependent on its Gal1’s carbohydrate-binding domain (CBD), we used lactose, which binds to the CBD with very high affinity. We show that the addition of lactose partially inhibits this interaction, suggesting the glycan binding activity is at least in part needed for the interaction (Fig. 6E).
Fig. 6: Endogenous Galectin-1 interacts with STING, maintains chronic activation, leading to MDSC expansion, lung remodeling and metastatic progression.
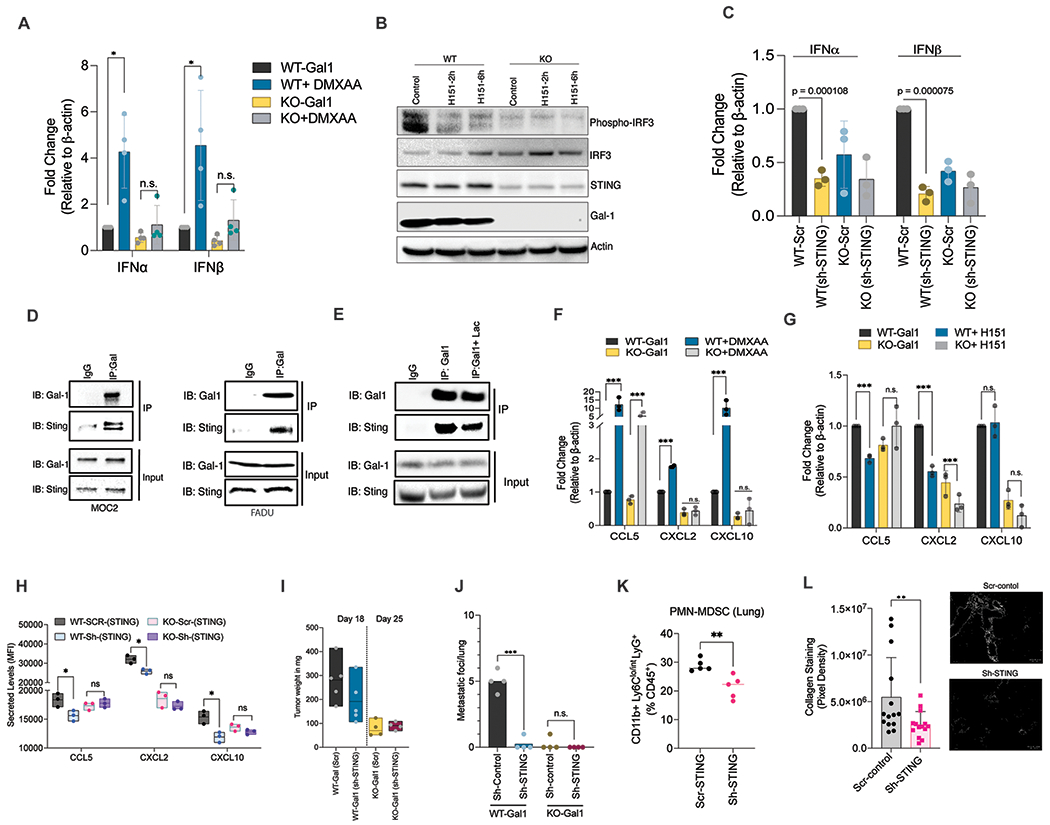
A. q-PCR quantification of IFNα and IFNβ gene expression levels in MOC2 WT/KO-Gal1 cells were treated with 300 μM DMXAA for 16h. B. Immunoblot analyses showing phospho-IRF3 and STING levels in MOC2 WT/KO-Gal1 cells treated with either DMSO or H151 (STING inhibitor) for 2h and 6h. C. q-PCR quantification (left panel) of IFNα and IFNβ gene expression with STING knockdown in MOC2 WT/KO-Gal1 cells using lentiviral delivered shRNAs. D. Extracts of MOC2 and FaDU cells were subjected to immunoprecipitation with an anti-Gal1 antibody and immunoblotted with anti-STING and anti-Gal1 antibodies. E. Protein lysate from either PBS or 10mM lactose (24 h) treated MOC2 cells were subjected to immunoprecipitation with an anti-Gal1 antibody and immunoblotted with anti-STING and anti-Gal1 antibodies. F. q-PCR showing gene expression of CCL5, CXCL2, and CXCL10 following treatment with DMXAA for 24 h in MOC2 WT/KO-Gal1 cells. G. q-PCR showing gene expression of CCL5, CXCL2, and CXCL10 following treatment with H151 for 24 h in MOC2 WT/KO-Gal1 cells. H. Quantification of secreted levels of CCL5, CXCL2, and CXCL10 when STING is knocked down in WT/KO-Gal1 cells using Luminex assay. I. Weights of vector control (Scr) and STING knockdown (shSTING) MOC2 WT/KO-Gal1 tumors following orthotopic buccal injections at Day 20. J. Quantification of metastatic burden in the lungs of mice bearing orthotopic buccal tumor of vector control (Scr) and STING knockdown (shSTING) MOC2 WT/KO-Gal1 tumor cells (n=5/group). K. Quantification of PMN-MDSCs in the lungs after orthotopic buccal implantation of vector control (Scr) or STING knockdown (sh-STING) MOC2 WT/KO-Gal1 tumor cells. L. Quantification of collagen staining in the lungs of mice bearing vector control (scr) or STING knockdown MOC2 WT-Gal1 tumor. Student’s t-test (two-tailed) was used for comparing a single treatment to the control. One-way ANOVA with Tukey’s adjustment was used to compare multiple treatments. Data are shown as mean+/−SD. All experiments were repeated 2 times with similar results.
Treatment with the STING agonist, DMXAA, potently increased the mRNA levels of MDSC-promoting cytokines CCL5, CXCL2 and CXCL10 in MOC2 WT-Gal1 cells. In contrast, in Gal1-KO cells, there was no change in the mRNA levels of CXCL2 and CXCL10 with some induction of CCL5, albeit at a lower level than that seen in WT-Gal1 cells (Fig. 6F). Similarly, treatment with a STING antagonist (H151) suppressed CCL5 expression in WT-Gal1 cells but not in KO-Gal1 cells. In the case of CXCL2, we observed a stronger decrease in WT-Gal1 compared to KO-Gal1 cells. CXCL10 however showed no change with H151 (Fig. 6G). We performed Luminex analyses to check for the change in secreted levels of these cytokines with STING down-regulation in WT/KO cells. We show that STING knockdown significantly reduces the levels of all three cytokines in WT-Gal1 but not in KO-Gal1 cells (Fig. 6H). Together, our results suggest that Gal1-mediated stabilization of STING is important for STING signaling and raised the possibility that Gal1’s effect on MDSC-migratory chemokines depends on STING signaling.
To address this hypothesis, we tested whether STING signaling is required for MDSC expansion in the lung and pulmonary metastasis in WT-Gal1 tumor-bearing mice. While downregulation of STING did not significantly affect the primary tumor size in either WT-or KO-Gal1 tumors (Fig. 6I), it strongly inhibited lung metastases in WT-Gal1 tumor-bearing mice, down to the level of mice-bearing KO-Gal1 tumors (Fig. 6J) with reduced MDSC level in the lungs (Fig. 6K). Trichrome staining of the lungs showed reduced perivascular/ bronchial collagen deposition and lung remodeling in STING knockdown tumor-bearing mice compared to scramble control tumors (Fig. 6L). Thus, chronic STING activation in HNC cells is important for Gal1-mediated MDSC accumulation in the lungs and metastasis progression.
Discussion
Despite multimodality curative treatment, ~30%-40% of HNSCC patients with locoregionally advanced disease develop locoregional recurrences and 20%-30% ultimately progress to metastatic disease [36]. A better understanding of the metastatic process is crucial for the identification of more effective anti-cancer therapies. In this study, we show that Gal1-mediated MDSC recruitment through the CXCL2-CXCR2 axis is crucial for metastatic progression. We also identify the Gal1-STING-NFκB signaling axis in tumor cells as a primary driver of MDSC recruitment to the lungs, leading to metastasis in HNSCC and lung cancer models. Primary tumors have a significant influence on the early steps of the metastatic cascade at secondary sites. It has long been speculated that the metastatic immune microenvironment is an important determinant in the establishment of metastases [37, 38]. However, the exact mechanisms involved in the reprogramming of the immune cell landscape at metastatic sites are poorly defined. Although studies in T and B-cell-deficient mice have provided significant information on myeloid cell function in the pre-metastatic site [39, 40], these studies cannot recapitulate metastatic progression in patients with a functional immune system. Similarly, the use of experimental metastasis models (i.e., cardiac or tail vein tumor cell injection) devoid of the primary tumor pre-conditioning of the immune response at secondary sites does not mimic metastatic spread in patients. Using spontaneous metastasis models in immunocompetent mice, we show that tumor-signaling mechanisms play a direct role in shaping the immune microenvironment at secondary sites. We unravel a novel function of Galectin-1 in establishing lung pre-metastatic niches for cancer cells by recruiting PMN-MDSCs, which in turn support ECM remodeling, maintain immunosuppression, and enhance cancer cell survival in a new microenvironment.
Recruitment of immunosuppressive cells such as MDSCs is one of the classic mechanisms for tumor dissemination and homing to new niches [27]. Chemokines play an important role in MDSC attraction, in which the binding of chemokines to their cell surface receptors triggers the MDSC migration [26, 28]. CXCR2 is one of the dominant CXC chemokine receptors on myeloid cells, and chemokines that signal via this receptor have been shown to play a major role in PMN-MDSC recruitment in other model systems [30]. Elevated levels of systemic and pulmonary CXCL1/2 are associated with increased metastases [41]. Using both a specific CXCR2 inhibitor as well as an anti-GR1 antibody, we show that MDSC depletion is sufficient to reduce Gal-mediated metastasis in mice. Our data support the role of Galectin-1 in modulating chemokines leading to pulmonary MDSC accumulation. However, the reason for specificity in terms of PMN-MDSC enrichment is not fully understood. We believe that this could partially be due to the tissue-specific regulation of neutrophil recruitment which has been well documented. Studies have shown that non-inflamed lungs contain a pool of PMNs that is ~ 2–3 times the number of PMNs in the free circulation [42, 43]. In animal models, it has also been shown that the CXCR2 receptor on neutrophils is one of the most important chemokine receptors involved in neutrophil recruitment into the lung; however, it is not as important for recruitment to the liver or kidney [44]. This may be one reason for the pronounced effect of CXCL2 chemokine blockade in the lung under the Gal1 KO conditions.
Prior studies have also indicated that Galectin-1 can induce epithelial-mesenchymal transition (EMT) in cancer cells that might aid metastasis [45],[46]. However, the results of our experiments using CM isolated from either WT-Gal1 or KO-Gal1 administered in a KO-Gal1 tumor background suggest that Galectin-1-mediated metastasis is at least partially dependent on MDSC-mediated lung remodeling, independent of Gal1’s effects on EMT in tumor cells.
Tumor-induced tissue remodeling involves changes in ECM protein content, posttranslational modification, proteolytic degradation, and reorganization of fibers. Recently, CCR2+ monocytes recruited to the lung were shown to possess pro-fibrotic functions [47]. Moreover, the transfer of PMN-MDSCs induced strong upregulation of mRNA expression of fibrotic markers, fibrosis-inducing agents, and inflammatory cytokines [48]. Our study is therefore in accord with some of these studies showing that MDSCs not only facilitate immunosuppression but also contribute to extensive ECM and vascular remodeling, which aids in the metastatic process.
Most importantly, we establish a link between Galectin-1 and STING that leads to NF–κB activation, resulting in chemokine secretion and recruitment of myeloid cells. Although studies have shown that IFN-independent activities of STING play physiologically important roles in autophagy, and cell proliferation [49, 50], to our knowledge, this study is the first that connects Gal1 to STING and its downstream activity. Recently, it was shown that cells displaying a high level of chromosomal instability exhibited increased levels and perinuclear localization of STING consistent with pathway activation [14],[51]. Chromosomally unstable tumors co-opt the cancer cell-intrinsic cGAS-STING signaling, which then triggers non-canonical NF-κB activation, leading to sustained inflammation, EMT, and metastasis [14]. Interestingly, our studies showed that STING signaling was chronically activated in HNC cells (evident by the presence of phosphorylated STING), leading to sustained canonical activation of NF-κB, resulting in the inflammatory response and enhanced production of myeloid recruiting chemokines. We found that Galectin-1 was required to sustain elevated STING levels in head and neck cancer and lung cancer cells. These results are in line with a recently published study, which showed that the radiation-induced STING pathway in cancer cells could lead to an immunosuppressive effect through the recruitment of myeloid cells, in part via the CCR2 pathway [52]. This is in contrast to the scenario in normal cells, wherein STING-mediated IFN signaling is transient [15] and turned on and off quickly. This avoids the sustained production of STING-dependent proinflammatory genes that may lead to inflammation and autoimmune disorders [53]. Recent studies show that this is possible because STING is targeted to late-stage endo-lysosomes for degradation as soon as it translocates from the endoplasmic reticulum to the vesicles [54]. Overall, these studies suggest that sustained intra-tumoral STING activation could decrease anti-tumor immune response through increased immune-suppressive cytokine/chemokine production in the tumor microenvironment, resulting in an influx of immunosuppressive cells. Here, we identified Gal1 as a critical factor that interacts with STING at the protein level and prevents its lysosomal degradation, thereby increasing the availability of STING to chronically activate NF-κB signaling, resulting in unresolved inflammation. Interestingly Galectin-9, another member of the Galectin family was recently shown to enhance STING degradation through TRIM29-mediated K48-linked ubiquitination of the STING [55]. Although these results are distinct from our findings, they emphasize the role of different galectin family members in regulating STING protein levels. These studies also suggest an intricate connection between two known pathways regulating the innate and adaptive immune response to cancer. In summary, we identify Gal1 as a novel positive regulator of STING in cancer cells, leading to its chronic activation, which in turn promotes metastasis by enhancing inflammation-driven MDSC expansion.
Supplementary Material
Significance.
Galectin-1 increases STING stability in cancer cells that activates NF-κB signaling and CXCL2 expression to promote MDSC trafficking, which stimulates the generation of a pre-metastatic niche and facilitates metastatic progression.
Acknowledgments
We would like to acknowledge the contribution of Pauline Chu & the Stanford Human Histology Core for pathological services. We also thank the Shared Stanford FACS Facility for its services and support. We thank Dr. William Spanos for the mEERL HPV+ cell line and Dr. Ravindra Uppaluri for the HPV− MOC1, and MOC2 cells. We acknowledge the NIH Grant- From the NCI: P01CA257907 (to Q.-T. Le, D. Nambiar, L. Guan, H. Cao, and V. Viswanathan); 2U10CA180868-06 and P30CA124435 (to Q.-T. Le); from the NIDCR: R01DE029672 (to Q.T. Le, D. Nambiar, L. Guan, and H. Cao); R01DE030894 and R01DE031724 (to Q. Le); R01DE029672 and P01CA257907 for the funding support to carry out the project. We thank the Shared Stanford FACS Facility (SSFF) for help with flow cytometry and sorting experiments.
Footnotes
Declaration of Interest: The authors have declared that no conflict of interest exists.
References
- 1.Siegel RL, et al. , Cancer Statistics, 2021. CA Cancer J Clin, 2021. 71(1): p. 7–33. [DOI] [PubMed] [Google Scholar]
- 2.Lambert AW, Pattabiraman DR, and Weinberg RA, Emerging Biological Principles of Metastasis. Cell, 2017. 168(4): p. 670–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peinado H, et al. , Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer, 2017. 17(5): p. 302–317. [DOI] [PubMed] [Google Scholar]
- 4.El-Kenawi A, Hanggi K, and Ruffell B, The Immune Microenvironment and Cancer Metastasis. Cold Spring Harb Perspect Med, 2020. 10(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hiam-Galvez KJ, Allen BM, and Spitzer MH, Systemic immunity in cancer. Nature Reviews Cancer, 2021. 21(6): p. 345–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cole K, Pravoverov K, and Talmadge JE, Role of myeloid-derived suppressor cells in metastasis. Cancer Metastasis Rev, 2021. 40(2): p. 391–411. [DOI] [PubMed] [Google Scholar]
- 7.Marvel D and Gabrilovich DI, Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest, 2015. 125(9): p. 3356–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang F, et al. , Targeting Myeloid-Derived Suppressor Cells for Premetastatic Niche Disruption After Tumor Resection. Ann Surg Oncol, 2021. 28(7): p. 4030–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nambiar DK, et al. , Galectin-1-driven T cell exclusion in the tumor endothelium promotes immunotherapy resistance. Journal of Clinical Investigation, 2019. 129(12): p. 5553–5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banh A, et al. , Tumor galectin-1 mediates tumor growth and metastasis through regulation of T-cell apoptosis. Cancer Res, 2011. 71(13): p. 4423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rubinstein N, et al. , Targeted inhibition of galectin-1 gene expression in tumor cells results in heightened T cell-mediated rejection; A potential mechanism of tumor-immune privilege. Cancer Cell, 2004. 5(3): p. 241–51. [DOI] [PubMed] [Google Scholar]
- 12.Cedeno-Laurent F and Dimitroff CJ, Galectin-1 research in T cell immunity: past, present and future. Clin Immunol, 2012. 142(2): p. 107–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cagnoni AJ, et al. , Galectin-1 fosters an immunosuppressive microenvironment in colorectal cancer by reprogramming CD8(+) regulatory T cells. Proc Natl Acad Sci U S A, 2021. 118(21). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bakhoum SF, et al. , Chromosomal instability drives metastasis through a cytosolic DNA response. Nature, 2018. 553(7689): p. 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corrales L, et al. , Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep, 2015. 11(7): p. 1018–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohkuri T, et al. , STING contributes to antiglioma immunity via triggering type I IFN signals in the tumor microenvironment. Cancer Immunol Res, 2014. 2(12): p. 1199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y, et al. , Identification of hypoxia-regulated proteins in head and neck cancer by proteomic and tissue array profiling. Cancer Res, 2004. 64(20): p. 7302–10. [DOI] [PubMed] [Google Scholar]
- 18.Angelova M, et al. , Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol, 2015. 16: p. 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakamura K, et al. , Dysregulated IL-18 Is a Key Driver of Immunosuppression and a Possible Therapeutic Target in the Multiple Myeloma Microenvironment. Cancer Cell, 2018. 33(4): p. 634–648 e5. [DOI] [PubMed] [Google Scholar]
- 20.Wang G, et al. , Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov, 2016. 6(1): p. 80–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanzelmann S, Castelo R, and Guinney J, GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics, 2013. 14: p. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patro R, et al. , Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods, 2017. 14(4): p. 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson WE, Li C, and Rabinovic A, Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics, 2007. 8(1): p. 118–27. [DOI] [PubMed] [Google Scholar]
- 24.Tusher VG, Tibshirani R, and Chu G, Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A, 2001. 98(9): p. 5116–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hao Z, et al. , Landscape of Myeloid-derived Suppressor Cell in Tumor Immunotherapy. Biomark Res, 2021. 9(1): p. 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keskinov AA and Shurin MR, Myeloid regulatory cells in tumor spreading and metastasis. Immunobiology, 2015. 220(2): p. 236–42. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y, et al. , MDSCs: Key Criminals of Tumor Pre-metastatic Niche Formation. Front Immunol, 2019. 10: p. 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang D, et al. , CXCL1 Is Critical for Premetastatic Niche Formation and Metastasis in Colorectal Cancer. Cancer Res, 2017. 77(13): p. 3655–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arkhypov I, et al. , Myeloid Cell Modulation by Tumor-Derived Extracellular Vesicles. Int J Mol Sci, 2020. 21(17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Highfill SL, et al. , Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med, 2014. 6(237): p. 237ra67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li YM, et al. , Receptor-Interacting Protein Kinase 3 Deficiency Recruits Myeloid-Derived Suppressor Cells to Hepatocellular Carcinoma Through the Chemokine (C-X-C Motif) Ligand 1-Chemokine (C-X-C Motif) Receptor 2 Axis. Hepatology, 2019. 70(5): p. 1564–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richmond A, Nf-kappa B, chemokine gene transcription and tumour growth. Nat Rev Immunol, 2002. 2(9): p. 664–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yum S, et al. , TBK1 recruitment to STING activates both IRF3 and NF-kappaB that mediate immune defense against tumors and viral infections. Proc Natl Acad Sci U S A, 2021. 118(14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abe T and Barber GN, Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-kappaB activation through TBK1. J Virol, 2014. 88(10): p. 5328–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang D, et al. , Activated STING enhances Tregs infiltration in the HPV-related carcinogenesis of tongue squamous cells via the c-jun/CCL22 signal. Biochim Biophys Acta, 2015. 1852(11): p. 2494–503. [DOI] [PubMed] [Google Scholar]
- 36.Specenier P and Vermorken JB, Optimizing treatments for recurrent or metastatic head and neck squamous cell carcinoma. Expert Rev Anticancer Ther, 2018. 18(9): p. 901–915. [DOI] [PubMed] [Google Scholar]
- 37.Massague J and Obenauf AC, Metastatic colonization by circulating tumour cells. Nature, 2016. 529(7586): p. 298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kitamura T, Qian BZ, and Pollard JW, Immune cell promotion of metastasis. Nat Rev Immunol, 2015. 15(2): p. 73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dawson MR, et al. , VEGFR1 activity modulates myeloid cell infiltration in growing lung metastases but is not required for spontaneous metastasis formation. PLoS One, 2009. 4(9): p. e6525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Y and Cao XT, Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell, 2016. 30(5): p. 668–681. [DOI] [PubMed] [Google Scholar]
- 41.Acharyya S, et al. , A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell, 2012. 150(1): p. 165–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hogg JC and Doerschuk CM, Leukocyte traffic in the lung. Annu Rev Physiol, 1995. 57: p. 97–114. [DOI] [PubMed] [Google Scholar]
- 43.Doerschuk CM, et al. , Marginated pool of neutrophils in rabbit lungs. J Appl Physiol (1985), 1987. 63(5): p. 1806–15. [DOI] [PubMed] [Google Scholar]
- 44.Rossaint J and Zarbock A, Tissue-specific neutrophil recruitment into the lung, liver, and kidney. J Innate Immun, 2013. 5(4): p. 348–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bacigalupo ML, et al. , Galectin-1 triggers epithelial-mesenchymal transition in human hepatocellular carcinoma cells. J Cell Physiol, 2015. 230(6): p. 1298–309. [DOI] [PubMed] [Google Scholar]
- 46.Chong Y, et al. , Galectin-1 induces invasion and the epithelial-mesenchymal transition in human gastric cancer cells via non-canonical activation of the hedgehog signaling pathway. Oncotarget, 2016. 7(50): p. 83611–83626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lebrun A, et al. , CCR2(+) monocytic myeloid-derived suppressor cells (M-MDSCs) inhibit collagen degradation and promote lung fibrosis by producing transforming growth factor-beta1. J Pathol, 2017. 243(3): p. 320–330. [DOI] [PubMed] [Google Scholar]
- 48.Sun SN, et al. , G-MDSCs promote aging-related cardiac fibrosis by activating myofibroblasts and preventing senescence. Cell Death Dis, 2021. 12(6): p. 594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Banerjee D, et al. , A non-canonical, interferon-independent signaling activity of cGAMP triggers DNA damage response signaling. Nat Commun, 2021. 12(1): p. 6207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bohnert V, Ritchie C, and Li L, IFN-Independent STING Signaling: Friend or Foe? Immunity, 2020. 53(1): p. 8–10. [DOI] [PubMed] [Google Scholar]
- 51.Bakhoum SF and Cantley LC, The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell, 2018. 174(6): p. 1347–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liang H, et al. , Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat Commun, 2017. 8(1): p. 1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ahn J, et al. , STING manifests self DNA-dependent inflammatory disease. Proc Natl Acad Sci U S A, 2012. 109(47): p. 19386–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gonugunta VK, et al. , Trafficking-Mediated STING Degradation Requires Sorting to Acidified Endolysosomes and Can Be Targeted to Enhance Anti-tumor Response. Cell Rep, 2017. 21(11): p. 3234–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang CX, et al. , Galectin-9 promotes a suppressive microenvironment in human cancer by enhancing STING degradation. Oncogenesis, 2020. 9(7): p. 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw RNA-seq data were deposited in the GEO database with accession number GSE234636. All other raw data are available upon request from the corresponding author.