

Abstract
Bioinks for 3D bioprinting of tumor models should not only meet printability requirements but also accurately maintain and support phenotypes of tumor surrounding cells to recapitulate key tumor hallmarks. Collagen is a major extracellular matrix protein for solid tumors, but low viscosity of collagen solution has made 3D bioprinted cancer models challenging. This work produces embedded, bioprinted breast cancer cells and tumor organoid models using low‐concentration collagen I based bioinks. The biocompatible and physically crosslinked silk fibroin hydrogel is used to generate the support bath for the embedded 3D printing. The composition of the collagen I based bioink is optimized with a thermoresponsive hyaluronic acid‐based polymer to maintain the phenotypes of both the noninvasive epithelial and invasive breast cancer cells, as well as cancer‐associated fibroblasts. Mouse breast tumor organoids are bioprinted using optimized collagen bioink to mimic in vivo tumor morphology. A vascularized tumor model is also created using a similar strategy, with significantly enhanced vasculature formation under hypoxia. This study shows the great potential of embedded bioprinted breast tumor models utilizing a low‐concentration collagen‐based bioink for advancing the understanding of tumor cell biology and facilitating drug discovery research.
Keywords: 3D cancer models, collagen, extracellular matrix, freeform reversible embedding of suspended hydrogels, hyaluronic acid, vascularization
The physically crosslinked silk hydrogel is used as a support bath for bioprinting breast tumor cells and organoid‐laden tumor microenvironment models. The optimized low‐concentration collagen I based bioink maintains the phenotypes of breast tumor cell and organoids and enables the generation of organotypic and vascularized breast tumor models.
1. Introduction
Breast cancer is the leading cause of cancer‐related mortality in women.[ 1 ] The traditional 2D drug screening models have failed in developing an effective therapy against cancers, including breast cancers, due to the inherent limitations of 2D models, particularly the lack of 3D extracellular matrix (ECM) and cell–cell interaction resulting in altered tumor cell growth and drug response.[ 2 ] 3D tumor models that can replicate the cell–ECM and cell–cell interactions have shown to better represent the tumor microenvironment (TME) and have become powerful tools to study tumor biology and therapy.[ 2 ] The TME is comprised of cancer cells and stromal cells, such as fibroblast, endothelial, and immune cells, and their residing ECM.[ 3 ] The interaction among these components shape the TME and thus contribute to cancer progression, drug resistance and metastasis.[ 4 ] 3D bioprinting, which allows layer‐by‐layer deposition of multiple cells and biomatrix in the predefined architecture, can produce complex, heterogeneous and physiologically relevant TME models that better predict the in vivo drug response.[ 5 ] Thus, 3D bioprinted tumor models have become a powerful and reproducible platform to study tumor growth, angiogenesis, and metastasis and enable high‐throughput along with more accurate patient specific drug screening.[ 5 , 6 ] Many bioprinted solid tumor models were initially focused on characterizing the behavior and drug response of only the cancer cells in a 3D ECM.[ 7 ] However, more recent bioprinted TME models include other key players such as the stromal cells in TME together with the cancer cells, which demonstrated that the stromal cells appreciably affected the therapeutic efficacy.[ 8 , 9 ] Cancer associated fibroblasts (CAFs) and endothelial cells are the two most abundant cells in the tumor stroma.[ 3 ] The stromal cells arrange around the cancerous lesion and remarkably influence the cancer development.[ 10 ] The CAFs can remodel the ECM structure and component, and thus promote tumor migration.[ 11 ] The tumor recruited endothelial cells form the vasculature system provide nutrients and oxygen to assist cancer growth and spreading to distant sites.[ 12 ] CAF can also drive tumor angiogenesis by activating endothelial cells through its secretome.[ 13 ] In terms of 3D bioprinting, the selection of the bioink, which constitutes a biomaterial matrix containing a heterogeneous population of cells, is one of the most crucial prerequisites to reconstitute the TME.[ 14 ] The bioink for each tumor model should be carefully optimized as it provides the ECM components influencing the cell adhesion, migration, proliferation, differentiation, and vascularization.[ 15 ]
Matrigel is the basement‐membrane matrix extracted from the Engelbreth–Holm–Swarm mouse sarcoma and is frequently used in tumor cell culture and considered as the gold standard material for culturing tumor organoids.[ 16 ] However, the composition of Matrigel is not well‐defined, as it consists of a mixture of various laminins, collagen type IV, and entactin. This lack of standardization results in batch‐to‐batch variability, leading to uncertainties in cell studies and a lack of reproducibility.[ 17 ] Moreover, the mouse sarcoma‐based ECM may not provide the breast tissue specific microenvironment for breast cancer. Consequently, it becomes challenging to fine‐tune the matrix to precisely accommodate the desired cell behavior.[ 18 ]
Various component defined natural polymers with tunable properties such as gelatin, alginate, and hyaluronic acid (HA) have been successfully used to bioprint tumor models with each polymer having distinct strengths and limitations for 3D bioprinting application.[ 8 , 19 ] Another important natural polymer, collagen type I, is the most abundant structural component of the tumor ECM.[ 5 ] Collagen I plays an essential role in breast cancer development through cell–matrix motif interaction.[ 20 ] It can promote tumor metastasis through the epithelial to mesenchymal (EMT) transition.[ 21 ] Aligned collagen fibrils also provides a niche to facilitate the migration of breast cancer cells.[ 22 ] While several tumor models have been fabricated with collagen I,[ 23 ] 3D bioprinting is minimally used in developing collagen‐based tumor models. This is because the extrusion‐based 3D bioprinting, the most popular printing method due to its convenience and cost‐effectiveness, requires a viscoelastic and shear‐thinning property of the bioink.[ 24 ] The viscosity of collagen solution, especially at low concentration, is typically too low, thus 3D bioprinting of collagen‐based bioink is challenging. Many extrusion‐based 3D printing studies have used a 3% collagen concentration as the ink[ 25 , 26 , 27 ] and it has been demonstrated that a minimum concentration of 17.5 mg mL−1 is required to achieve optimal printability.[ 28 ] Previous works on the low‐concentration collagen bioinks always required additives such as Pluronic F‐127, tannic acid, or nanocellulose to increase the printability of collagen, but the additives may potentially affect the tumor cell viability and phenotype.[ 23 ] Another recent approach comes from a technique called freeform reversible embedding of suspended hydrogels (FRESH) or embedded printing.[ 29 ] The suspension bath used in FRESH can support the printing of very low viscosity bioinks inside the bath and prevent their spreading to maintain the scaffold morphology and allow bioinks crosslinking in situ to ensure good cell viability. For example, human heart components have been successfully bioprinted with collagen I bioink alone using FRESH.[ 30 ] A single report in the literature used FRESH in cancer models to build a high‐throughput drug screening platform but the study lacks information regarding the development of organotypic cancer models.[ 31 ]
In the present study (Figure 1 ), we developed an optimized formulation of low‐concentration collagen I based bioink that was compatible with both breast tumor cells and CAFs. We confirmed good printability of the bioink in a silk fibroin (SF) hydrogel‐based support bath and then evaluated how matrix components affect breast tumor cells and CAF phenotype and oncogenic traits. Further, we cultured organoids from transgenic mice tumors and then determined if the bioprinted tumor organoids can preserve a morphology, similar to primary breast tumor. Finally, we bioprinted organotypic breast tumor models that reconstructed several features of the TME, including angiogenesis, EMT, and invasion. Taken together, we demonstrate the embedded bioprinting strategy with an optimized, component defined collagen I based bioink can be applied to fabricate biomimetic tumor models. These models represent technical advances for future drug discovery research.
Figure 1.
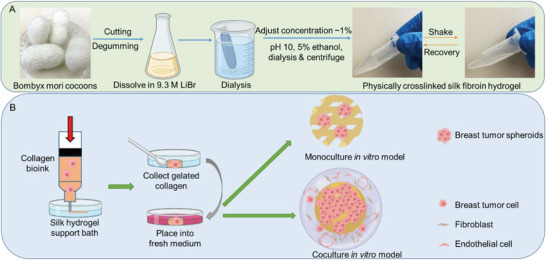
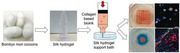
Schematic illustration of SF hydrogel based supporting bath for 3D bioprinting of organotypic breast tumor models. A) Preparation of physically crosslinked silk fibroin hydrogel. B) Development of embedded bioprinting strategy of breast tumor models using collagen‐based bioink.
2. Results
2.1. SF Hydrogel Provided a Good Support Bath for Low‐Concentration Collagen Bioprinting
The SF molecules self‐assembled and easily formed physically crosslinked hydrogel due to conformation transition of SF induced by ethanol (Figure S1A, Supporting Information). Time sweeps for SF hydrogel showed a time‐independent behavior of solutions under shearing at 22 °C, indicating its stability within the time frame of analysis (Figure 2A). Moreover, after disruption by an external large strain stimulus (500%), the SF hydrogel was able to rapidly recover from a flow state to an elastic state when the strain was reduced to 10%, demonstrating the self‐healing ability (Figure 2B). A good suspension material should also possess the shear thinning property and a certain yield stress to meet the requirements for embedded bioprinting.[ 32 ] The physically crosslinked SF hydrogel was found to be a good support bath. The shear‐thinning property of SF hydrogel was first validated. When the shear rate was increased from 1 to 5 s−1, the SF hydrogel viscosity dropped markedly from 8.5 to 2 Pa s (Figure 2C). The yield stress was determined by applying a range of small shear rates from 1 to 5 s−1 and interpolating the shear stress value at zero shear rate based on a linear fitting.[ 33 ] The obtained yield stress value of 7.5 Pa was close to some reported support baths, such as Carbopol and agarose,[ 32 , 33 ] implying its fitness for embedded printing.
Figure 2.
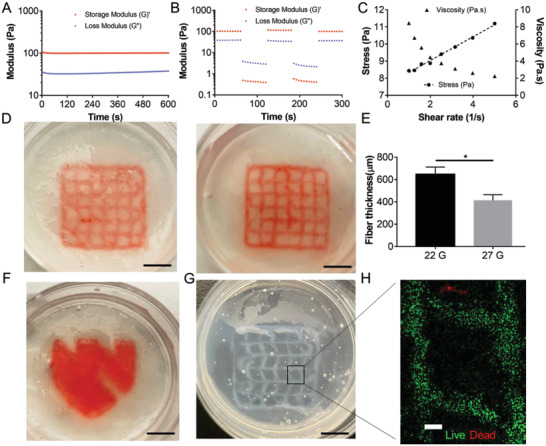
Evaluation of rheological properties and embedded printing performance in the SF hydrogel support bath. A) Time sweep of modulus of SF hydrogel. B) Recovery of modulus under alternating strain (10% and 500%) of SF hydrogel. C) Shear rate dependent change of viscosity and stress of SF hydrogel. D&E) Embedded printing of collagen I precursor (3 mg mL−1 with a red‐colored dye) into a grid structure in the SF hydrogel support bath using 22G and 25G needles (n = 7, *p < 0.05). F) Embedded printing of collagen I precursor (3 mg mL−1 with dye) into a solid structure (UNMC logo) in the SF hydrogel support bath using 22G needle. G) Polymerized collagen with the lattice structure printed in SF hydrogel support bath. H) Live and dead staining of 21PT cells in the bioprinted scaffold after the collagen was polymerized. Scale bar: black, 5 mm; white, 500 µm.
Due to the low viscosity of the 3 mg mL−1 collagen I solution, traditional extrusion printing is rather challenging. However, the problem was solved in the presence of the SF support bath (Figure S1B, Supporting Information). Constructs with a grid structure (15 × 15 × 1.5 mm, distance between adjacent strands ≈2.5 mm) were first printed using the 3 mg mL−1 collagen solution to assess the influence of nozzle size on the printing fidelity and strand diameter. No major visible distortion was seen on the collagen filaments printed in the SF support bath (Figure 2D), especially when using the small size nozzle. The small size nozzle (25G) also resulted in a significantly smaller strand diameter (Figure 2E). A solid and more complex structure (UNMC logo) was later successfully printed in the SF support bath (Figure 2F). The polymerized collagen hydrogel was observable inside the translucent SF support bath (Figure 2G). Live and dead staining of 21PT breast cancer cells in the collagen bioink showed the strand outline of the printed grid and revealed over 90% viability immediately after embedded printing (Figure 1H), indicating minimal damage to the cells from the embedded printing process.
2.2. Collagen–HA–pNIPAM (CH) Bioink Maintained Noninvasive Epithelial Breast Cancer Cell Phenotype
Both collagen I and HA are important ECM components of breast cancer TME. Previous studies in the literature have demonstrated that HA interacts with breast cancer cells via CD44 receptors[ 34 ] and the addition of HA can better mimic the biophysical and biochemical microenvironments in breast cancer ECM.[ 35 , 36 ] Therefore, we attempted to prepare a bioink composing both HA and collagen. However, physically mixing HA with collagen precursor could not retain HA in the collagen network long enough. Chemically crosslinking HA with collagen is not straightforward and may raise toxicity issues. Inspired by the thermoresponsive property of the poly(N‐isopropylacrylamide) (pNIPAM) polymer, whose hydrodynamic size is substantially increased due to the enhanced inter‐molecular interaction when the temperature (T) is above the lower critical solution temperature (LCST),[ 37 ] we presumed that a thermosensitive HA molecule should retain in the collagen network longer than native HA at T > LCST. We have successfully conjugated the amine terminated pNIPAM to the HA backbone using DMTMM as the crosslinking agent (Figure 3A). Proton nuclear magnetic resonance (1H NMR) analysis indicated a 5% grafting ratio (Figure S2, Supporting Information). The resulting HA–pNIPAM was cyto‐compatible and exhibited a thermoresponsive property; it became cloudy when the solution temperature was above 34 °C. The HA–pNIPAM solution could form a thermoresponsive hydrogel when the polymer concentration was at least 8% (w/v) (Figure 3B). The novel bioink was prepared by mixing the collagen I (3 mg mL−1) precursor solution with the HA–pNIPAM (16 mg mL−1) solution before its polymerization at 37 °C. The collagen I precursor was a clear, transparent, and colorless solution when it was incubated at 37 °C but the collagen I with HA–pNIPAM (CH) quickly (<15 s) showed a white and cloudy appearance at 37 °C. The relative stiffness or storage modulus of the polymerized CH hydrogel was close to that of the native collagen (C) hydrogel (Figure S3, Supporting Information). The two collagen‐based hydrogels presented similar morphologies under Scanning electron microscope (SEM) imaging (Figure 3D). However, it was found that the HA–pNIPAM incorporation resulted in a significant reduction in the fiber thickness but did not significantly change the pore cross section size (Figure 3E). We also prepared a rhodamine labeled HA and conjugated it with pNIPAM following the same procedure to monitor the HA release profile from the collagen hydrogel at 37 °C. At all tested time points, the HA–pNIPAM showed at least 2.5–3‐fold higher amount remained in the collagen hydrogel (Figure S4, Supporting Information), confirming our hypothesis.
Figure 3.
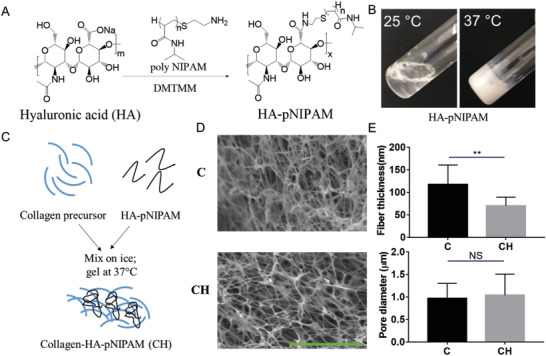
Preparation and characterization of an engineered collagen‐based hydrogel containing HA‐poly(N‐isopropylacrylamide) (HA–pNIPAM). A) Synthesis of HA–pNIPAM conjugate using the crosslinking agent DMTMM. B) Thermosensitive HA–pNIPAM solution (8% w/v) switched from a solution state at 25 °C into a hydrogel form at 37 °C. C) Schematic illustration of the preparation of collagen I hydrogel containing HA–pNIPAM (CH). D) SEM images of collagen I hydrogel (C) and CH hydrogel. E) Comparison of fiber diameter and cross‐section pore sizes between the collagen and CH (n = 10, **p < 0.01; NS, not significant). Scale bar: 5 µm.
After establishing the bioink formulation and the bioprinting strategy, the cancer cell behavior and phenotype were successively determined in the bioprinted models. 21PT cells, a noninvasive human breast cancer cell line derived from primary breast cancer, was used as the model cells.[ 38 ] Three bioprinted 21PT models with different materials (i.e., Matrigel, collagen and collagen–HA–pNIPAM; denoted as M, C, and CH, respectively) were prepared. Significantly, the tumor cells displayed different morphologies and phenotypes after 7 days of culture (Figure 4A). In both Matrigel and CH models, most 21PT cells (>85%) formed acinar colonies with the median spheroid size of 60 µm (Figure 4C,D). No significant differences in the acinar colony ratio or the spheroid size were found between Matrigel and the CH. The immunofluorescent (IF) staining also revealed a noninvasive epithelial phenotype in both models (Figure 4B). However, in the bioprinted collagen model, less than 25% of the cells formed acinar colony and the spheroid size tended to be smaller with a median size of 25 µm. The majority of 21PT cells in the bioprinted collagen model were either isolated or loosely clumped together (Figure 4A). Many of these cells also showed a tail structure, suggesting an invasive mesenchymal‐like phenotype (Figure 4B). We compared the cell proliferation in three models and found that 21PT cells proliferated faster in Matrigel and CH models than in the collagen model (Figure 4E). Altogether, our results are consistent with the reported data that collagen I alone could induce the EMT transition of epithelial breast cancer cells.[ 21 ] On the contrary, the CH hydrogel with the addition of HA–pNIPAM in collagen I was able to maintain the noninvasive epithelial phenotype of 21PT cells without affecting the stiffness of the collagen hydrogel itself. Further, we analyzed the influence of HA–pNIPAM concentration in the hybrid collagen hydrogel on the cell behavior. For these experiments, we prepared bioprinted CH models at lower concentration of HA–pNIPAM (Figure S6A, Supporting Information) and found that the decreased amount of HA–pNIPAM correlated with the reduced acinar colony ratio (Figure S6B,C, Supporting Information), underlying the critical role of HA–pNIPAM as the bioink component in regulating the 21PT cell behavior and phenotype.
Figure 4.
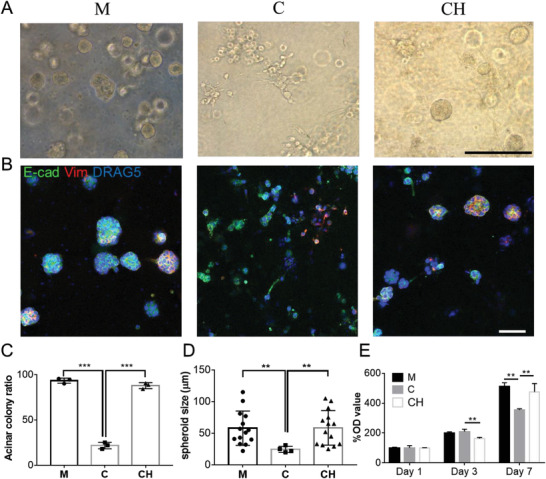
Behavior and morphology of 21PT human breast cancer cells in three models with different bioinks: Matrigel (M), collagen (C), and CH at day 7. A) 21PT cells formed mostly noninvasive acinar colonies in Matrigel and CH models but mostly nonacinar colonies in the collagen model. B) IF staining of E‐cadherin (E‐cad, green) and vimentin (Vim, red) proteins on 21PT cells in three bioprinted models. C) Quantification 21PT cells forming the acinar colony in three bioprinted models (n = 3, ***p < 0.001). D) Comparison of spheroid size formed in three bioinks (n = 14, **p < 0.01). E) Proliferation of 21PT cells in three bioprinted models determined by MTT study (n = 4, **p < 0.01; NS, not significant). Scale bar: black, 250 µm; white, 100 µm.
To validate different morphology of 21PT cells in three conditions, we performed the RNA‐seq analysis of 21PT cells from three models and compared them with the 2D cultured counterparts (Figure S7, Supporting Information). We first focused on the EMT relevant gene expression. Many typical EMT promoting genes, such as CDH2, TWIST1, EPCAM and VIM etc. were upregulated in the collagen and 2D culture conditions (Figure 5A) compared to those in the Matrigel and CH models. In contrast, many EMT suppressive genes such as CD82, CSTA, KRT17, etc. were downregulated in the collagen and 2D culture groups. We confirmed a downregulation of CDH1 and an upregulation of Vim in the collagen group (Figure S8, Supporting Information), which aligns well with previous IF staining results. We also determined the top‐ranking cancer relevant canonical pathways in these three conditions of culture using IPA analysis. Significantly, we found cells in Matrigel and CH models shared many similar pathways including HIF1α, tumor microenvironment, GP6 signaling, breast cancer regulation by Stathmin1, ephrin receptor signaling, and protein kinase A signaling (Figure 5B). However, cells in the collagen model displayed many other pathways such as basal cell carcinoma signaling, calcium signaling, mitochondrial dysfunction, and estrogen receptor signaling. These results further validated that the 21PT cells in the CH model had similar behavior to those in Matrigel model and maintained their noninvasive phenotype.
Figure 5.
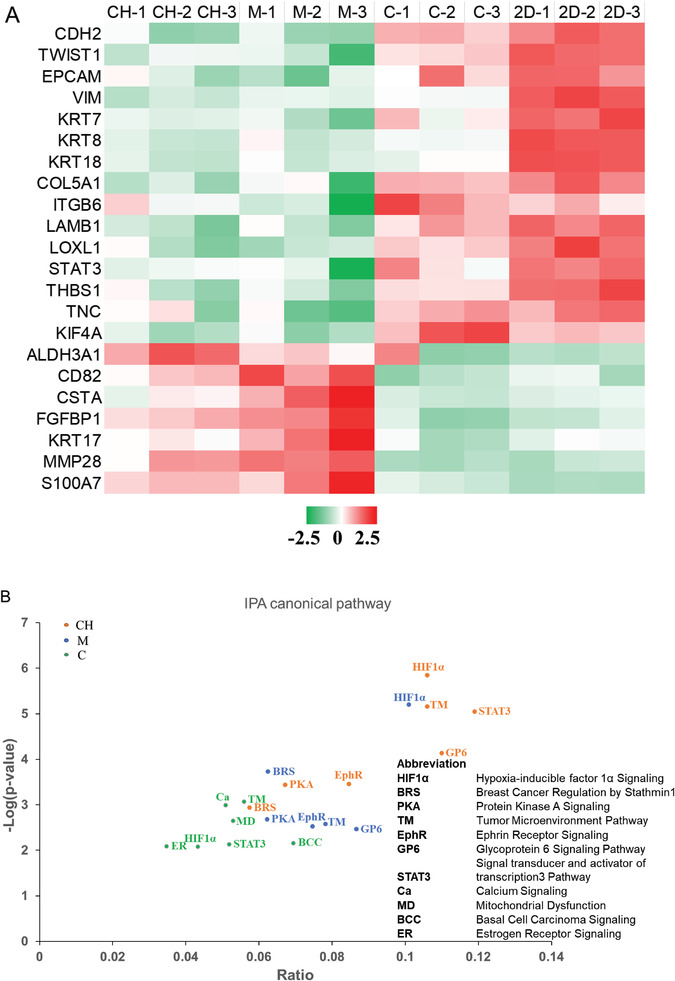
Transcriptomic analysis of gene expression changes of 21PT cells cultured in three models as compared to 2D culture. A) Heatmap of representative EMT‐related genes in three models and 2D plate. The color code represents the row z‐score, where a red color indicates higher expression of a gene, while a green color indicates lower expression of a gene. B) Canonical pathway identified by IPA showed similar enrichment between 21PT cells cultured in Matrigel and CH models but different from cells cultured in the collagen model.
2.3. Bioprinted Constructs Modeled Distinct Tumor Cell Subtype and CAF Phenotype
To further evaluate our model using triple negative breast cancer subtype, we used MDA‐MB‐231 cell line, which is widely used and exhibits invasive and metastatic properties in in vitro 2D and 3D cultures, as well as in in vivo setting. In all three bioprinted models using different bioinks, i.e., Matrigel, collagen, and CH, the cells showed a mixture of epithelial like and mesenchymal like phenotypes after a 7 day culture (Figure 6A). The cells with the mesenchymal phenotype formed a satellite structure in the bioprinted Matrigel model while in the bioprinted collagen and CH models the cells with the mesenchymal phenotype were single isolated cells with an elongated tail structure. The mesenchymal phenotype ratio was comparable in all three models (Figure S9, Supporting Information). In contrast, the cells with the noninvasive epithelial phenotype presented spheroid structure in the Matrigel model and isolated round structure in the collagen and CH models. After staining the cells with E‐cadherin and vimentin antibodies, as expected MDA‐MB‐231 cells showed no staining with E‐cadherin in all three models. The satellite structure part from the Matrigel model and the elongated tail structure part from the collagen and CH models were positive with vimentin staining, consistent with their mesenchymal phenotype (Figure 6B). The tail structure of these cells from the collagen and CH models was also evident from the live/dead staining. We speculated that by measuring the ratio of these mesenchymal cells (determined from the ratio of cells with the tail structure), we could evaluate the EMT process of the cells. For this purpose, we bioprinted two modified MDA‐MB‐231 cell lines using the CH bioink. One cell line was transfected with the EHD2 short hairpin RNA (shRNA) to knockdown EHD2 and the other cell line was transfected with a scrambled shRNA. We have previously reported EHD2 influenced the metastasis potential of breast cancer cells.[ 39 ] By staining the bioprinted cells using live/dead staining method, we simultaneously determined the viability of the cells and the mesenchymal cell ratio (Figure 6C). Both types of cells showed high viability (>95%) in the bioprinted CH model at day 7. Meanwhile, the mesenchymal cell ratio was significantly reduced for the EHD2 shRNA transfected cells (Figure 6D), implying reduced metastasis potential which is consistent with our previous finding.[ 39 ] We also evaluated the phenotype of noncancerous breast epithelial cells (76NTERT) in three bioprinted models, but they all presented a spheroid structure, suggesting the role of an oncogene in directing the phenotype difference in each model (Figure S10, Supporting Information).
Figure 6.
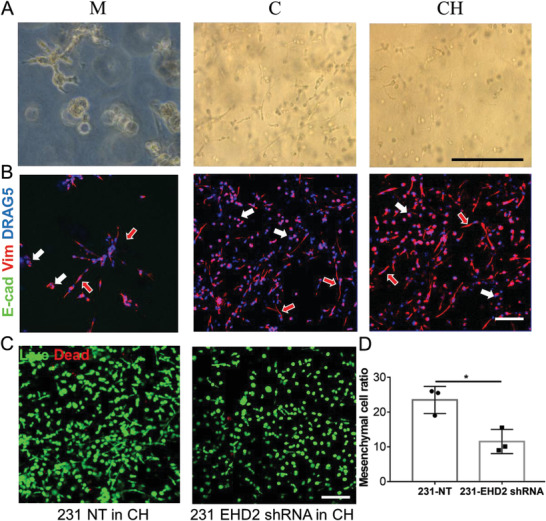
Evaluation of MDA‐MB‐231 human breast cancer cells behavior and phenotype in embedded bioprinted models of three different bioinks. A) MDA‐MB‐231 showed a mixture of noninvasive epithelial and mesenchymal phenotypes in all three bioinks at day 7. B) IF staining of E‐cadherin (not detected) and vimentin proteins on MDA‐MB‐231 cells in three bioinks. The cells or cell clusters with epithelial phenotypes were indicated by the all‐white arrow, while the red‐in‐white arrow represented the mesenchymal phenotypes. C) Live and dead staining of MDA‐MB‐231 cells transfected with NT and EHD2 shRNA in CH model. D) Quantification of mesenchymal phenotype ratio of two modified MDA‐MB‐231 cells in the bioprinted CH model (n = 3, *p < 0.05). Scale bar: black, 250 µm; white, 100 µm.
The CAFs, a major player in the TME process,[ 40 ] are known to have a crosstalk with tumor cells to influence the tumor progression and drug resistance.[ 11 ] Next, we investigated the phenotypes of a CAF (39VTF, previously derived in our laboratory) in three models. CAFs in the Matrigel model presented a spheroid structure with little α‐smooth muscle actin (α‐SMA) expression, distinguished from the elongated structure in the collagen and CH model with strong α‐SMA expression (Figure S11A,B, Supporting Information). Most importantly, in the CH model coculturing 21PT and CAFs, both types of cells were able to preserve their own phenotype (Figure S11C, Supporting Information).
2.4. Bioprinted Tumor Organoids Recapitulated In Vivo Tumor Morphology
Given that breast cancer cells of different subtypes as well as CAFs grew and maintained their phenotypes in the bioprinted CH model, we next determined the possibility of bioprinting primary tumor tissues. Patient‐derived organoids (PDO) are considered important tools for precision medicine due to their ability to preserve characteristics of the original tumor.[ 41 ] In this study, we used mouse tumor tissue derived organoids from the C3(1)‐tag transgenic mice.
C3(1)‐tag (FVB/N strain), is a genetically engineered murine model in which the expression of SV40 T‐antigen in mammary epithelial cells is driven by rat prostatic steroid binding protein C3(1) promoter. 90% of C3(1)‐tag mammary tumors represent human basal‐like triple negative breast cancer (lack progesterone, estrogen, and human epidermal growth factor receptor 2 receptors) with 5%–10% claudin‐low features.[ 42 , 43 ] The C(3)1‐tag animals spontaneously develop mammary epithelial atypia at week 8 of age and progress into mammary intraepithelial neoplasia (close to human ductal carcinoma in situ) at week 12 of age and finally become invasive after week of 16–20 of age, which correlates well with the progression of human breast cancer.[ 44 ] The mouse model has been used in many studies to test novel therapeutics toward breast cancer and has also been applied in tissue engineering models to reproduce tumor behavior in vivo.[ 45 , 46 ] The organoids derived from C3(1)‐tag mice (12 w) were encapsulated within bioinks and bioprinted into the support bath (Figure 7A). The embedded bioprinting process allowed high‐throughput generation of tumor organoid models in the ECM bioink in a reproducible manner (Figure 7B). Three hydrogel materials (Matrigel, collagen and CH) were used as the bioink matrix for fabricating the bioprinted tumor organoid models. Bioprinted tumor organoids in the Matrigel and CH models remained in the spheroid structure from day 1 to day 7, however, in the collagen, cells became more invasive and quickly spread to the surrounding area on day 1, leading to the loss of the original spheroid structure (Figure 7C). Bioprinted tumor organoids were then harvested after 7 days of culture. The hematoxylin and eosin (H&E) staining of the organoid samples showed consistent morphologies with those found under microscope in each model (Figure 7D). IF staining with E‐cadherin and the stromal cell marker, α‐SMA, demonstrated a similar morphology to the bioprinted tumor organoids in the CH model when compared to primary tumor tissue based on the cell phenotype and ratio (Figure 7E,F). However, the morphologies were altered in both Matrigel and collagen models. No obvious staining of α‐SMA was detected in the Matrigel model; in comparison, an increased level of α‐SMA expression was found in the collagen model (Figure 7E,G). These results suggested the bioprinted tumor organoids using the CH bioink enabled a better spatial organization and phenotype maintenance of both the cancer cells and the stromal cells from the organoids, resembling the in vivo tumor tissue.
Figure 7.
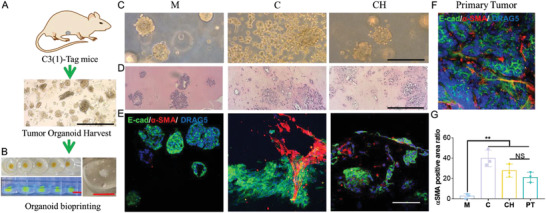
The bioprinted tumor organoids in CH bioink recapitulated in vivo tumor morphology. A) Breast tumor organoids were harvested from mice tumors. B) Left: Top view of fluorescein isothiocyanate (FITC) labeled bioink printed in the SF hydrogel support bath and side view under UV light. Right: zoomed view of organoid loaded CH bioink printed in the SF hydrogel support bath. C) Morphology of breast tumor organoids in three bioinks at day 1. D) H&E staining of breast tumor organoids at day 7 in three bioinks. E&F) IF staining of E‐cadherin and α‐SMA proteins on breast tumor organoids and mice tumor sample. G) Quantification of α‐SMA positive area ratio (area of α‐SMA positive region divided by total area of α‐SMA and E‐cadherin positive region) from organoid samples in three bioinks and the primary tumor (n = 3, *p < 0.05; NS, not significant). Scale bar: red, 5 mm; black, 250 µm; white, 100 µm.
2.5. Bioprinted Organotypic Tumor Models Emulated Key TME Characteristics
Hypoxia is known to play a vital role in cancer angiogenesis and progression.[ 47 ] Hypoxia condition (5% O2) was implemented to avoid further generating complex hypoxia mimicking structure.[ 48 ] We first confirmed the hypoxia responsiveness of the human breast cancer cells in the bioprinted CH model. Many characteristic hypoxia‐related genes were shown to have increased regulation when the bioprinted 21PT cells in CH model were cultured under a hypoxia environment (Figure 8A). Specifically, many of those upregulated genes such as VEGFA, MMP1, MMP7, MMP9, and LOXL2 could contribute to both the EMT and tumor angiogenesis process. This prompted us to develop a bioprinted vascularized breast tumor model. The organotypic model was comprised of two regions, the outside surrounding region was bioprinted with the human umbilical vein endothelial cells (HUVECs) and normal fibroblast cells (stroma region) and the inner core region was bioprinted with the 21PT cells only (tumor region, Figure 8B). To enhance the endothelial cell differentiation and vessel formation, fibroblast cells were included and the bioink for the stromal cells consisted of 3 mg mL−1 collagen I and 3 mg mL−1 of fibrin. Fibrin was included in our study based on the preliminary results that revealed unsatisfactory small vessel formation in the CH bioink alone, but significant improvement was observed after the addition of fibrin. Subsequently, we discovered that vessel formation was comparable between the CH bioink plus fibrin group and the collagen plus fibrin group. To simplify the experimental design, we opted to utilize collagen plus fibrin bioink for the fabrication of the angiogenesis region. The bioink for the tumor region only consisted of the CH hydrogel as we have evaluated. Due to the printability of low‐concentration collagen solution in the SF hydrogel support bath, the two‐region construct was successfully printed with the stroma region tightly connected with the tumor region (Figure 8C). IF staining of the neighboring region showed E‐cadherin positive cancer cells, and the vimentin positive stromal cells were adjacent with each other (Figure 8D). The bioprinted organotypic models were then cultured in normoxia and hypoxia conditions for up to 7 days. Afterward, the samples were fixed and stained with the vasculature marker CD31 and the cancer cell marker E‐cadherin; the stroma region next to the tumor region was imaged. Endothelial cells in the bioprinted model formed more capillary‐like networks under hypoxia compared to normoxia. Bioprinted organotypic model in hypoxia showed a nearly two times (p < 0.001) total vessel tube length as compared to the bioprinted model in normoxia (Figure 8F). In addition, the hypoxia condition promoted EMT, with more E‐cadherin positive cancer cells found in the stroma region under hypoxia compared to normoxia (Figure 8E). The amount of migrated tumor cells in hypoxia were about 1.5 times (p < 0.01) higher than that in normoxia (Figure 8G). Thus, many TME features, such as tumor stroma, tumor vasculature, EMT and invasion were emulated in the embedded bioprinted organotypic models using the low‐concentration collagen I based bioink.
Figure 8.
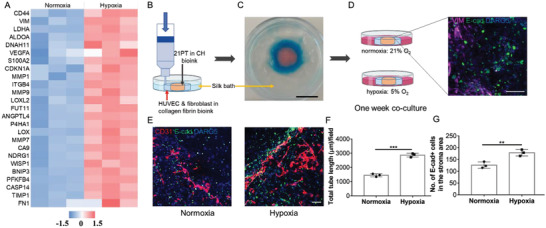
Evaluation of hypoxia influence on cancer associated angiogenesis in a vascularized breast tumor model. A) Transcriptomic analysis of 21PT cells cultured in CH bioink under normoxia and hypoxia condition. The color code represents the row z‐score, where a red color indicates higher expression of a gene, while a blue color indicates lower expression of a gene. B) Embedded bioprinting of a vascularized breast tumor model in the silk bath. C) Illustration of two distinguished stroma and tumor areas in the bioprinted vascularized breast tumor model after printing. D) After the collagen polymerization, bioprinted models were transferred to normoxia and hypoxia culture conditions. The inserted image showed the border region between fibroblast cells (vimentin positive) and 21PT cells (E‐cadherin positive) at day 1. E) IF staining of CD31 and E‐cadherin in the stroma area under normoxia and hypoxia condition at day 7. F) Quantification of total capillary tube length per field after 7 day normoxia and hypoxia culture (n = 3, ***p < 0.001). G) Quantification of migrated 21PT cells after 7 day normoxia and hypoxia culture (n = 3, **p < 0.01). Scale bar: black, 1 cm; white, 100 µm.
3. Discussion
In this study, we present evidence that the biocompatible and physically crosslinked silk fibroin hydrogel can be used for embedded 3D printing. Using collagen I based bioink, we present evidence that the phenotypes of both the noninvasive epithelial and invasive breast cancer cells were maintained. Furthermore, a vascularized tumor model under hypoxia condition, mimicked tumor angiogenesis process.
Our bioprinted breast tumor models used collagen, the most abundant structural protein in the breast cancer ECM. Collagen I has a key role in breast tumor development and metastasis.[ 20 ] Increased collagen I deposition and linearization are correlated with the breast cancer subtype and invasion.[ 49 ] The crosslinking of the collagen fibrils, mediated by the lysyl oxidase (LOX) enzyme, results in the ECM stiffening, which further drives many cellular pathways relevant to tumor proliferation and aggressiveness.[ 50 , 51 ] In recognition of the role of collagen in breast cancer development, collagen hydrogels are considered the naturally born and versatile platforms for creating breast tumor models. In addition, collagen I can support the growth and/or differentiation of many other types of cells, such as fibroblasts, endothelial cells, and mesenchymal stem cells (MSC), which makes it an ideal matrix to accommodate the heterogenous population of cells in TME.[ 52 , 53 ] Recently, 3D bioprinted tumor models have become more popular as they present more closer in vivo microarchitecture, defined organization of different cells in the spatial dimension.[ 19 ] Another advantage of 3D bioprinting is that it also allows easy manipulation of the cells and biomaterials (bioink) with unlimited geometry design at high resolution.[ 54 ] Although previous studies could develop two‐compartment scaffolds with different collagen concentration or components, their methods always relied on sequential polymerization of two collagen gels, which was time consuming and not flexible.[ 55 , 56 ] In addition, molding was required to accommodate the shape and dimension of the individual collagen compartment. The intrinsic feature of 3D bioprinting overcomes these limitations through rapid prototyping and printing. It enables fast and flexible design and fabrication of complex structures such as three or more compartmental scaffolds that replicate the complexity in the tumor microenvironment. However, 3D bioprinted collagen‐based tumor models are less frequently achieved, which is mainly attributed to the low viscosity of the collagen solution. The development of FRESH and embedded printing technology provides a strategy without any viscosity additives.[ 57 ] However, we found that the gelatin bath, which is first used in FRESH, is not suitable for low‐concentration collagen bioink as it is dissolved quickly, in less than 15 min at 37 °C, but the low‐concentration collagen would require several hours for complete polymerization. Pluronic F‐127 and Carbopol bath could absorb water easily, thereby damaging the structure of collagen hydrogel. Agarose gel bath has been reported with significant distortion on the print bed path.[ 58 ] Herein, we provide evidence that the physically crosslinked SF hydrogel can be applied as a good support bath for collagen bioprinting. It had a relatively small yield stress and good self‐healing ability. It allowed the printing of low‐concentration collagen I with good fidelity and further complete polymerization of the collagen bioink in the support bath. The only disadvantage is the laborious process to prepare SF hydrogel bath from the cocoons, which could be possibly solved by purchasing the commercially available SF solution.
HA is another important component of the breast tumor ECM as it regulates cancer stem cell niche and its drug resistance.[ 59 ] We incorporated an HA molecule with 290 kDa size in our model, as several studies have demonstrated a significantly higher concentration of low molecular weight (<500 kDa) and oligo‐HA (<10 kDa) in the tumor microenvironment. Conversely, in nonpathological settings, newly synthesized HA exhibits relatively high molecular weight (>1000 kDa).[ 60 , 61 , 62 ] Our bioink was designed with the purpose of retaining HA in the collagen network longer without chemical crosslinking. We utilized the thermoresponsive property of the pNIPAM modified HA polymer. The cyto‐compatible HA–pNIPAM based hydrogels have been used for drug delivery and stem cell culture.[ 63 , 64 ] The HA–pNIPAM incorporation resulted in an almost 3 times higher increase in the amount of HA component retaining in the collagen compared to the native HA itself. Although it was not comparable to the covalently linked HA, our method brought in much more convenience, consistency, and minimal toxicity concern. It will be interesting to investigate the influence of HA molecular weight on the modified HA–pNIPAM retention in future studies.
The CH bioink was successfully embedded bioprinted along with the collagen bioink. The CH bioink led not only to the higher amount of HA in the collagen (Figure S4, Supporting Information), but also to the phenotype maintenance of various breast cancer cell lines. To better replicate breast cancer heterogeneity, we evaluated three breast cell lines: nontumorigenic 76NTERT cells, a noninvasive primary patient‐tumor 21PT cells, and the invasive and mesenchymal MDA‐MB‐231 cells. While 76NTERT cells showed the noninvasive epithelial phenotype in all three models, 21PT cell line formed spheroids with similar sizes in the CH bioink and the Matrigel bioink, keeping a major noninvasive phenotype, but displayed an invasive phenotype in the collagen model. Lastly, MDA‐MB‐231 cells showed a mixture of invasive mesenchymal phenotype and round epithelial phenotype in all three bioinks. RNA‐seq data showed EMT gene expression and Ingenuity Pathway Analysis (IPA) further confirmed the similarity between 21PT cultured in Matrigel model and CH model (Figure 5). Although it seems both Matrigel and CH bioinks could maintain the phenotype of all three cells, the batch‐to‐batch variability and undefined composition associated with Matrigel can affect the models’ reproducibility.[ 65 ] Another problem regarding Matrigel is the altered phenotype of embedded CAFs (Figure S11, Supporting Information). The CH bioink has defined components and maintained the phenotypes for both breast tumor cells and CAF. It was not quite clear why CH bioink had such ability, especially for the noninvasive epithelial breast cancer cells. It is well known that the microarchitecture and mechanical property of collagen I hydrogel in terms of the fiber thickness, pore sizes and stiffness could elicit different cellular responses of the encapsulated cells.[ 66 ] For example, thicker collagen fiber increased the invasiveness of MDA‐MB‐231 cells with mesenchymal migratory phenotype and the MCF‐7 cells with ameboid migratory phenotype.[ 67 ] Small pores inside the collagen could impede the MMP independent cell migration.[ 68 ] In our case, after HA–pNIPAM incorporation, the pore size was not changed significantly but the fiber thickness was decreased. Interestingly, previous studies revealed that collagen fibers in physical condition, such as the basement membrane of epithelial tissue showed fiber diameters of 20–40 nm compared to 30–100 nm in stroma region of carcinomas.[ 69 , 70 ] We postulate that the reduction in fiber thickness can be attributed to the collapse of the thermoresponsive HA–pNIPAM at elevated temperatures, which consequently leads to the shrinkage of the associated collagen fibril precursor. Those changes resulted in the spheroid formation for the noninvasive 21PT cells but did not change the migratory behavior of MDA‐MB‐231 cells. More detailed mechanisms should be explored in future work. The stiffness of our CH hydrogel is lower than that of breast tissues/tumors, which has been reported to vary based on factors such as measurement methods, species, location, and disease status.[ 71 , 72 , 73 ] It should be noted that, upon cell encapsulation, the stiffness of the constructs tends to undergo dynamic increases over time due to the formation of tissue‐like structures and compaction of the hydrogel.[ 74 , 75 , 76 ] In our current study, the primary focus does not revolve around the examination of the effects of modulus or stiffness on cell behaviors. Therefore, specific experiments or an in‐depth discussion on this particular issue were not designed or provided. The importance of investigating the influence of modulus/stiffness on cell behaviors is acknowledged and will be explored in future studies.
PDO are generated from tumor biopsies or surgical procedures and can serve as models to study cancer and facilitate personalized therapy.[ 77 ] Most organoid models are established in Matrigel, which does not well support the phenotypes of stromal cells and thus fails to replicate the native TME. Encouraged by the successful coculture of 21PT cells and cancer associated fibroblasts, we continued to develop the tumor organoid models in the CH bioink. We used mouse breast tumor organoids generated from transgenic mice tumors which contained stromal cells. We bioprinted the mouse tumor organoids with three bioinks and compared the organoid morphology and cell phenotype in the three models. The stroma cell growth/differentiation was disfavored in the Matrigel model but overwhelmed in the collagen model. The tumor cells lost the acini‐like structure in the collagen model but kept the acinar morphology in the Matrigel model. Only the bioprinted CH model reconstituted the in vivo tumor morphology and the phenotypes of both epithelial tumor cells and CAF. Although embedded printing of tumor organoids has been studied before in a gelatin bath for drug screening purposes, the stromal component was still lacking.[ 31 ] Our results are consistent with a recent study using a polyethylene glycolhydrogel based pancreatic tumor model for coculturing PDO and stromal cells.[ 78 ] Thus, our 3D bioprinted CH model serves as a useful model for development of a platform for high‐throughput drug screening using PDO. For our future studies, we plan to coculture PDOs with stromal cells/CAF using the 3D printed format. The optimization process will involve mixing PDOs and CAFs at various ratios to evaluate the impact on spheroid growth and CAF behavior. Furthermore, we intend to perform a transcriptome analysis to compare the gene expression profiles of patient‐derived cancer tumor cells and the bioprinted models. Finally, we bioprinted a more complex two‐zone organotypic breast tumor model comprising breast tumor cells in the core and endothelial cells and fibroblast cells in the surrounding zone, further validating the potential of embedded printing in SF bath using the low‐concentration collagen‐based bioink. For this purpose, instead of using CH bioink, we applied a 3 mg mL−1 collagen and 3 mg mL−1 fibrin as the matrix to encapsulate endothelial cells and fibroblasts. The addition of fibroblasts and fibrin was critical for the formation of 3D vascularized tumor models[ 79 ] and our results also showed the modified 3D printed bioink supported endothelial network formation even in normoxia condition. As the development of breast tumor is closely linked to hypoxia, we cultured the organotypic model in a hypoxia condition and found that both the breast tumor extravasation and vessel formation were significantly enhanced, recapitulating the in vivo response of the breast tumors.[ 80 ] One possible reason is the increased secretion of pro‐angiogenic factors from the tumor cells under hypoxia. These factors are known to permeate the hydrogel network and the transcriptome study of the 21PT cells revealed many factors such as VEGFA, MMP and LOXL2 were upregulated under hypoxia.
Our strategy should also facilitate the development of a bioprinted cancer‐on‐a‐chip model.[ 81 ] Many other cells, such as the immune cells and adipocytes, MSC cells can be further included in the collagen‐based bioinks as collagen I has already been widely used for the 3D culture of macrophages, T cells, adipocytes and MSC cells.[ 53 , 82 , 83 , 84 ] For example, a bioprinted breast cancer and CAR T‐cell model can be used for high‐throughput evaluation of cell therapy response in the solid tumor.[ 85 ] One feature of the cancer‐on‐a‐chip system is the perfusable vasculature, which can be potentially achieved by embedded printing.[ 58 , 86 ] We conducted a preliminary study by first printing the 15% Pluronic F127 as a sacrificial ink in the SF bath and then infusing the channel with a dye solution (Figure S12, Supporting Information). The versatility of this platform should have huge potential in 3D tumor modeling.
4. Conclusion
In summary, we developed a SF hydrogel support bath that allowed embedded bioprinting of low viscosity collagen‐based bioinks at good shape fidelity. The addition of HA–pNIPAM within collagen I bioink maintained the original phenotypes of breast cancers of different subtypes and CAF. This biofabrication technique further provides significant advantage of high‐throughput manufacturing of tumor organoid models that replicated the in vivo tumor morphology and cell phenotype. Additionally, organotypic models with complex structures and at least three types of cells can be fabricated by simply adjusting the collagen‐based bioink components and printing patterns. The embedded bioprinted tumor models with the collagen‐based bioink should be a promising platform for studying tumor microenvironment and patient specific drug screening.
5. Experimental Section
Preparation of SF Hydrogel and Rheological Characterization
Following the previous method, The Bombyx mori cocoons (from Mulberry Farms) were boiled in the 0.02 m Na2CO3 aqueous solution (Fisher) for 30 min at 100 °C.[ 87 ] The boiled cocoons were rinsed with deionized water three times to remove the sericin from the surface of silk fiber. The rinsed cocoons were then dried at room temperature overnight. To prepare the purified aqueous SF solution, the degummed and dried silk fibers were dissolved in 9.3 m lithium bromide aqueous solution at 90 °C for 3 h. Then, the dissolved SF solution with an approximate 20% concentration (w/v) was dialyzed with deionized water at 4 °C for 3 days in a 6–8 kDa molecular weight cut‐off (MWCO) dialysis bag. The purified SF solution was centrifuged at 4500 rounds per min (rpm) for 10 min at 4 °C to remove insoluble aggregates. The final SF concentration was determined by weighing the mass before and after a complete drying process. To prepare the physically crosslinked silk hydrogel, the SF solution was first diluted to 1% concentration (w/v) using deionized water and the solution pH was adjusted to 9.5–10 using 1N NaOH aqueous solution. The solution was then mixed with pure ethanol to reach a final concentration of ethanol at 5% (v/v).[ 88 ] The final solution was kept in a capped tube and incubated for at least 24 h at 37 °C. After gelation, the residual ethanol was removed by dialysis against water for one day at 4 °C and the hydrogel was collected and stored at 4 °C.
Synthesis of HA‐Poly(N‐isopropylacrylamide) (pNIPAM) Conjugate
The HA–pNIPAM polymer conjugate was prepared by conjugating the amine terminated pNIPAM (Sigma, 5.5 kDa) to the HA backbone (290 kDa, Bloomage Biotech) using coupling agent 4‐(4,6‐dimethoxy‐1,3,5‐triazin‐2‐yl)−4‐methyl‐morpholinium chloride (DMTMM, TCI America) (Figure 2A ). Typically, 100 mg of HA (0.25 mmol) was dissolved in 20 mL de‐ionized (DI) water. Next, 100 mg of pNIPAM (0.02 mmol) and 140 mg DMTMM (0.5 mmol) were added to the HA solution, respectively. After all the agents were dissolved, the pH of the solution was adjusted to 6.5 using 1N HCl (Fisher Chemical) solution. The reaction mixture was stirred at room temperature for 72 h. After that, the mixture was transferred to a 50 kDa MWCO dialysis bag (Spectrum) and dialyzed against DI water for 5 days at room temperature, with the water changed twice every day. The dialyzed solution was freeze‐dried in a benchtop lyophilizer (FreeZone from Labconco) to obtain the HA–pNIPAM polymer conjugate. The conjugate was stored at −20 °C before use.
1H NMR Analysis of HA–pNIPAM
1H NMR analysis was conducted using a 500 MHz Bruker NMR system and then analyzed with the Topspin 4.0 software. The polymer conjugate was dissolved in D2O (Acros Organics) at 5 mg mL−1 for NMR acquisition with the chemical shifts referred to the solvent peak of D2O at 4.78 ppm at 25 °C. Annotation and assignment of the chemical shifts as well as the grafting ratio calculations were performed following the reference method.[ 89 ]
Preparation and Characterization of Collagen and Collagen–HA–pNIPAM Bioinks
Bovine collagen type I (10 mg mL−1, Advanced Biomatrix) was first neutralized with 1N sodium hydroxide solution and then mixed with one ninth of its total volume of 10X phosphate buffered saline (PBS) to prepare a collagen precursor solution (9 mg mL−1). It was then diluted with another two times volume of PBS to prepare a collagen precursor solution at 3 mg mL−1. Following that, HA–pNIPAM stock solution was prepared by dissolving the conjugate in PBS at 80 mg mL−1 with solution pH adjusted to 7, using 1N sodium hydroxide buffer. Different amounts of HA–pNIPAM stock solution and PBS were then added to the 9 mg mL−1 collagen precursor solution to achieve the collagen–HA–pNIPAM (CH) precursor solution with a final collagen concentration at 3 mg mL−1 with varying concentration of HA–pNIPAM (at 16, 8, and 4 mg mL−1, respectively). All the above steps were conducted on ice to prevent the early gelation of collagen precursor. The precursor solution was polymerized in a 37 °C environment. The relative stiffness or storage modulus of the collagen hydrogels were measured using the Discovery HR‐2 rheometer (TA Instruments) at 37 °C following the previous method.[ 90 ] In each test, 200 µL of precursor solution was placed between the 20 mm parallel plates with the geometry gap set at 500 µm and the strain set at a constant 10%.
SEM Imaging
The polymerized hydrogel was fixed with 2% paraformaldehyde at room temperature for 15 min, washed with water, and then lyophilized. After mounting the lyophilized sample on a stub, surfaces of the cross‐section of each hydrogel were coated with a thin layer of gold in the sputter coater. The structure and pore sizes of each sample were studied by the SEM (FEI Quanta 200).
Cell Culture
Breast cancer cell line, 21PT was cultured in normal 2D culture conditions, as previously described.[ 91 , 92 ] The MDA‐MB‐231 breast cancer cell line was cultured in Roswell Park Memorial Institute (RPMI) 1640 (Invitrogen) medium with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin–streptomycin (Gibco). The HUVECs were cultured in the endothelial basal medium including all the necessary supplements provided by the supplier (Lonza). For normoxia culture condition, cells were maintained at 37 °C with 5% CO2 and 21% O2. For hypoxia culture condition, cells were maintained at 37 °C with 5% CO2 and 5% O2 in trigas incubator (Thermo Fisher Scientific). The medium was changed every two days.
Isolation of Organoids from Primary Murine Tumor C3(1)‐Tag
Mouse tumor organoids were derived from tumors from 12 weeks C3(1)‐tag mice (originally obtained from the Jackson Laboratory). All animal procedures conducted have been approved by the IACUC at UNMC (approval# 18‐103‐08 FC). The isolation and process methods were described previously.[ 93 ] Briefly, tumors were physically minced and enzymatically digested using a digestion medium containing 2 mg mL−1 collagenase (Sigma), 2 mg mL−1 trypsin (Life Technologies), 5% v/v fetal bovine serum (Gibco), 5 µg mL−1 gentamycin (Life Technologies), and 5 µg mL−1 insulin (Sigma) in 20 mL of Dulbecco's Modified Eagle Medium/Nutrient Mixture F‐12 (DMEM/F12, Life Technologies) media for 1 h at 180 rpm in 37 °C shaker. The digested tissues were palleted by spinning down at 400g for 10 min. The palleted organoids were transferred to bovine albumin (BSA) coated (2.5% BSA solution) 15 mL falcon tube using BSA coated pipette to maximize organoid yield. Spinning down at 400g for 10 min. was repeated. The pallet was resuspended with 10 mL DMEM/F12 media and 80 µL of DNase (Sigma) to get rid of extracellular DNA released from stromal cells and necrotic tissues. A series of fast and short differential centrifugation steps (1700 rpm for 15 s for three times) were followed to deplete single cancer cell or stromal cell and enrich for epithelial organoids. The number of tumor organoids were counted and embedded in hydrogels/bioinks in presence of 2.5 × 10−9 m fibroblast growth factor 2 (Peprotech) growth factor and 1% v/v ITS (insulin, transferrin, and selenite) (Bio‐Techne Corporation).
Embedded Bioprinting Process
To illustrate the feasibility of embedded 3D bioprinting using the low‐concentration collagen I solution, 21PT cells were suspended in the collagen precursor solution (3 mg mL−1) at 1 × 105 mL−1 on ice. The bioink was then transferred into the deposition syringe (Nordson EFD) and loaded into the Bioplotter 3D (EnvisionTEC) extrusion printer. The syringes were capped with printing nozzles with either 22 gauge (G) or 27 G size metal tips. The printing parameters and patterns were adjusted by VisualMachines software (EnvisionTEC). Printing pressure for the bioinks was 0.4 ± 0.1 bar. Typical printing speed was 4 mm s−1 using the 22 G nozzle and 7 mm using the 27 G nozzle. The bioink was printed into a 5 mm thick SF support bath at room temperature in a 5 cm cell culture dish. The printed collagen together with the SF bath was incubated at 37 °C for at least 2 h for complete gelation.
For the evaluation of cell behavior and phenotype, 21PT and MDA‐MB‐231 cell lines and cancer associated fibroblasts (39VTF) were suspended in three types of hydrogel precursor solutions at 1 × 105 mL−1 on ice. Following the same printing parameters as described above, a solid circle shape scaffold (8 mm diameter and ≈1.5 mm thickness) was printed in the SF support bath with each bioink to generate different bioprinted models. After the hydrogel was fully polymerized, each scaffold was mildly taken out from the bath using a spatula and placed in one well of the 6‐well plate with 4 mL of medium for a long‐term culture. In terms of the coculture model, 21PT and 39VTF cells at 1 × 105 mL−1 were mixed in the CH bioink on ice and then bioprinted.
For mouse tumor organoids bioprinting, about 400 tumor organoids were mixed in each 0.5 mL of three types of hydrogel precursor solutions and the bioink was printed following the same method described above. To test the high‐throughput printing feasibility, an FITC dye at 1 mg mL−1 was added into the bioink and the bioink was printed into the SF bath in each well of a 48‐well plate. The bioprinted organoids were cultured in the same medium used for 21PT cells. In order to evaluate how different matrices affect cancer cell behavior and phenotype, the above‐mentioned cells/organoids with the same density were encapsulated within Matrigel (denoted as M, obtained from Corning) and collagen I (denoted as C) for comparison with CH bioink.
For the fabrication of an organotypic tumor model, 21PT cells at 4 × 106 mL−1 were suspended in the CH bioink and printed into the SF bath to prepare the core region (8 mm diameter and ≈1.5 mm thickness) while the endothelial cells HUVEC at 2×106 mL−1 and immortal human fibroblasts H16NF[ 94 ] at 5 × 105 mL−1 were suspended in the bioink consisting of 3 mg mL−1 collagen I and 3 mg mL−1 human fibrin (3 mg mL−1 human fibrinogen plus 0.5 U mL−1 thrombin, EMD Millipore) and printed as the surrounding region (0.8 and 1.2 mm as the inner and outside diameter, ≈1.5 mm thickness). The bioprinted organotypic tumor model was cultured in a medium containing 50% 21PT culture medium and 50% endothelial cell culture medium.
Live/Dead Staining and Cell Proliferation Study
A live/dead assay was used to evaluate the cell viability inside the bioink after bioprinting following the published protocol.[ 92 ] The cancer cell proliferation in different bioinks were tested at three different time points (days 1, 3, and 7) by the MTT ([3‐(4,5‐dimethylthiazol‐2‐yl)−2,5‐diphenyltetrazolium bromide, Sigma) assay.[ 8 ]
IF and Histological Staining
3D cultured cancer cells with or without stromal cells in different matrix were washed with PBS and then fixed in 4% PFA at 4 °C for 3 h, respectively. The samples were then washed twice in PBS and permeabilized with 0.2% Triton X‐100 (Sigma) for 10 min at room temperature (RT), followed by blocking with 1% BSA solution overnight at 4 °C. Subsequently, the 21PT or MDA‐MB‐231 samples were incubated overnight at 4 °C with primary antibodies anti‐E‐cadherin (1:100 dilutions in blocking buffer, Cell Signaling) or antivimentin (1:100, Sigma). The cancer associated fibroblast cells were incubated with antialpha smooth muscle actin (α‐SMA, 1:100, Sigma) primary antibody. The tumor organoid samples were labeled with the anti‐E‐cadherin (1:100) and anti‐α‐SMA (1:100) antibodies and the vascularized tumor model samples were labeled with the anti‐E‐cadherin (1:100) and anti‐CD31 (1:100, Cell Signaling) or antivimentin (1:100) antibodies. On the next day, after rinsing three times with PBS, samples were incubated with secondary fluorescent antibodies at room temperature for 2 h and further incubated with DRAQ5 PBS buffer (1:1000, Thermo Scientific) for 30 min before confocal imaging. All IF staining images in the study were acquired using a Zeiss 880 confocal microscope with layer‐by‐layer scanning, capturing at least ten layers of images. The acquired images were then reconstructed by stacking all the scanned images together using Zeiss Zen blue software. ImageJ software was used to analyze the IF images to determine the ratio between the epithelial and mesenchymal phenotypes. Cell counts were manually adjusted based on the sizes and shapes of the clusters when cell clusters were observed.
Each sample of tumor organoids in the bioprinted model of respective bioink after 7 day culture was used for histological evaluation. The sample was washed with PBS and fixed in 4% paraformaldehyde solution at 4 °C for 3 h and then paraffin‐embedded and sectioned using a microtome. Each 10 µm thick section was deparaffinized in xylene and stepwise rehydrated in baths of reduced concentrations of ethanol prior to staining.[ 95 ] H&E staining was carried out following the standard protocol in UNMC tissue core facility.
Transcriptomic Analysis
21PT cells cultured for 7 days in different 3D matrices, followed by washing with PBS, and then homogenized by a bead mill homogenizer (Fisher Scientific) in the cell lysis buffer (Qiagen). 2D cultured 21PT cells on day 7 were directly lysed with the lysis buffer in the plate. The total RNA was isolated from the cell lysate using QIA‐Shredder and RNeasy mini‐kits (Qiagen) following the manufacturer's protocols. RNA‐seq was performed at the Genomic Service Core of UNMC. The quality of the extracted RNA was evaluated using an Agilent fragment analyzer. RNA with an RNA integrity score over 8.0 was used for RNA‐seq library preparation with the sample preparation kit TruSeq RAN Sample Preparation v2 and sequenced on an Illumina NovaSeq 6000 instrument.
IPA Canonical Pathway Analysis
The differentially expressed genes from RNAseq analysis were further analyzed using Ingenuity Pathway Analysis (IPA; QIAGEN Inc.).
Statistical Analysis
All experiments were repeated at least thrice. All quantitative data are expressed as the mean ± the standard deviation (SD). Statistical analysis was performed using an unpaired Student's t‐test for comparisons between two groups, while one way ANOVA with a Neuman‐Keuls post‐hoc analysis was employed for comparisons involving three or more groups. The analysis was carried out using GraphPad Prism 7 software. The significance was represented as follows: *p < 0.05; **p < 0.01; ***p < 0.001.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
This work was supported by Mary and Dick Holland Regenerative Medicine Program start‐up grant, Nebraska Research Initiative Funding, NIH (R01 AR073225) (B.D.); Mary and Dick Holland Regenerative Medicine Program pilot grant (B.D. and V.B.); Cancer and Smoking Disease Research Funds Grant from The Nebraska Department of Health and Human Services (B.D., H.B., and V.B.), grants from DOD (W81XWH‐17‐1‐0616 and W81XWH‐20‐1‐0058 to H.B. and W81XWH‐20‐1‐0546 to V.B.). The authors thank the UNMC tissue facility for conducting the section and H&E staining of mouse tumor samples.
Shi W., Mirza S., Kuss M., Liu B., Hartin A., Wan S., Kong Y., Mohapatra B., Krishnan M., Band H., Band V., Duan B., Embedded Bioprinting of Breast Tumor Cells and Organoids Using Low‐Concentration Collagen‐Based Bioinks. Adv. Healthcare Mater. 2023, 12, 2300905. 10.1002/adhm.202300905
Contributor Information
Vimla Band, Email: vband@unmc.edu.
Bin Duan, Email: bin.duan@unmc.edu.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Arnold M., Morgan E., Rumgay H., Mafra A., Singh D., Laversanne M., Vignat J., Gralow J. R., Cardoso F., Siesling S., Soerjomataram I., Breast 2022, 66, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bahcecioglu G., Basara G., Ellis B. W., Ren X., Zorlutuna P., Acta Biomater. 2020, 106, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rodrigues J., Heinrich M. A., Teixeira L. M., Prakash J., Trends Cancer 2021, 7, 249. [DOI] [PubMed] [Google Scholar]
- 4. Ramamonjisoa N., Ackerstaff E., Front. Oncol. 2017, 7, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kang Y., Datta P., Shanmughapriya S., Ozbolat I. T., ACS Appl. Bio. Mater. 2020, 3, 5552. [DOI] [PubMed] [Google Scholar]
- 6. Germain N., Dhayer M., Dekiouk S., Marchetti P., Int. J. Mol. Sci. 2022, 23, 3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sánchez‐Salazar M. G., Álvarez M. M., Trujillo‐de Santiago G., Int. J. Bioprint. 2021, 21, e00120. [Google Scholar]
- 8. Wang Y., Shi W., Kuss M., Mirza S., Qi D., Krasnoslobodtsev A., Zeng J., Band H., Band V., Duan B., ACS Biomater. Sci. Eng. 2018, 4, 4401. [DOI] [PubMed] [Google Scholar]
- 9. Chen H., Cheng Y., Wang X., Wang J., Shi X., Li X., Tan W., Tan Z., Theranostics 2020, 10, 12127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bremnes R. M., Dønnem T., Al‐Saad S., Al‐Shibli K., Andersen S., Sirera R., Camps C., Marinez I., Busund L. T., J. Thorac. Oncol. 2011, 6, 209. [DOI] [PubMed] [Google Scholar]
- 11. Asif P. J., Longobardi C., Hahne M., Medema J. P., Cancers 2021, 13, 4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Forster J. C., Harriss‐Phillips W. M., Douglass M. J., Bezak E., Hypoxia 2017, 5, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Joshi R. S., Kanugula S. S., Sudhir S., Pereira M. P., Jain S., Aghi M. K., Cancers 2021, 13, 1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Datta P., Dey M., Ataie Z., Unutmaz D., Ozbolat I. T., NPJ Precis. Oncol. 2020, 4, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Henke E., Nandigama R., Ergün S., Front. Mol. Biosci. 2020, 6, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Habanjar O., Diab‐Assaf M., Caldefie‐Chezet F., Delort L., Int. J. Mol. Sci. 2021, 22, 12200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aisenbrey E. A., Murphy W. L., Nat. Rev. Mater. 2020, 5, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim S., Min S., Choi Y. S., Jo S.‐H., Jung J. H., Han K., Kim J., An S., Ji Y. W., Kim Y.‐G., Cho S.‐W., Nat. Commun. 2022, 13, 1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shukla P., Yeleswarapu S., Heinrich M. A., Prakash J., Pati F., Biofabrication 2022, 14, 032002. [DOI] [PubMed] [Google Scholar]
- 20. Papanicolaou M., Parker A. L., Yam M., Filipe E. C., Wu S. Z., Chitty J. L., Wyllie K., Tran E., Mok E., Nadalini A., Skhinas J. N., Lucas M. C., Herrmann D., Nobis M., Pereira B. A., Law A. M. K., Castillo L., Murphy K. J., Zaratzian A., Hastings J. F., Croucher D. R., Lim E., Oliver B. G., Mora F. V., Parker B. L., Gallego‐Ortega D., Swarbrick A., O'Toole S., Timpson P., Cox T. R., Nat. Commun. 2022, 13, 4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Carey S. P., Martin K. E., Reinhart‐King C. A., Sci. Rep. 2017, 7, 42088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Conklin M. W., Eickhoff J. C., Riching K. M., Pehlke C. A., Eliceiri K. W., Provenzano P. P., Friedl A., Keely P. J., Am. J. Pathol. 2011, 178, 1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Redmond J., McCarthy H., Buchanan P., Levingstone T. J., Dunne N. J., Mater. Sci. Eng., C 2021, 122, 111944. [DOI] [PubMed] [Google Scholar]
- 24. Askari M., Afzali Naniz M., Kouhi M., Saberi A., Zolfagharian A., Bodaghi M., Biomater. Sci. 2021, 9, 535. [DOI] [PubMed] [Google Scholar]
- 25. Osidak E. O., Kozhukhov V. I., Osidak M. S., Domogatsky S. P., Int. J. Bioprint. 2020, 6, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shim J.‐H., Jang K.‐M., Hahn S. K., Park J. Y., Jung H., Oh K., Park K. M., Yeom J., Park S. H., Kim S. W., Wang J. H., Kim K., Cho D.‐W., Biofabrication 2016, 8, 014102. [DOI] [PubMed] [Google Scholar]
- 27. Shim J.‐H., Kim J. Y., Park M., Park J., Cho D.‐W., Biofabrication 2011, 3, 034102. [DOI] [PubMed] [Google Scholar]
- 28. Rhee S., Puetzer J. L., Mason B. N., Reinhart‐King C. A., Bonassar L. J., ACS Biomater. Sci. Eng. 2016, 2, 1800. [DOI] [PubMed] [Google Scholar]
- 29. Hinton T. J., Jallerat Q., Palchesko R. N., Park J. H., Grodzicki M. S., Shue H.‐J., Ramadan M. H., Hudson A. R., Feinberg A. W., Sci. Adv. 2015, 1, e1500758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee A., Hudson A. R., Shiwarski D. J., Tashman J. W., Hinton T. J., Yerneni S., Bliley J. M., Campbell P. G., Feinberg A. W., Science 2019, 365, 482. [DOI] [PubMed] [Google Scholar]
- 31. Maloney E., Clark C., Sivakumar H., Yoo K., Aleman J., Rajan S. A. P., Forsythe S., Mazzocchi A., Laxton A. W., Tatter S. B., Strowd R. E., Votanopoulos K. I., Skardal A., Micromachines 2020, 11, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang B., Liu T., Gao G., Zhang X., Wu B., Micromachines 2022, 13, 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ning L., Mehta R., Cao C., Theus A., Tomov M., Zhu N., Weeks E. R., Bauser‐Heaton H., Serpooshan V., ACS Appl. Mater. Interfaces 2020, 12, 44563. [DOI] [PubMed] [Google Scholar]
- 34. Carvalho A. M., Soares da Costa D., Reis R. L., Pashkuleva I., Adv. Healthcare Mater. 2022, 11, 2101309. [DOI] [PubMed] [Google Scholar]
- 35. Unnikandam Veettil S. R., Hwang D., Correia J., Bartlett M. D., Schneider I. C., Acta Biomater. 2021, 130, 183. [DOI] [PubMed] [Google Scholar]
- 36. Brigham M. D., Bick A., Lo E., Bendali A., Burdick J. A., Khademhosseini A., Tissue Eng., Part A 2009, 15, 1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hou L., Wu P., Soft Matter 2015, 11, 2771. [DOI] [PubMed] [Google Scholar]
- 38. Band V., Zajchowski D., Swisshelm K., Trask D., Kulesa V., Cohen C., Connolly J., Sager R., Cancer Res. 1990, 50, 7351. [PubMed] [Google Scholar]
- 39. Luan H., Bielecki T., Mohapatra B., Islam N., Mushtaq I., Bhat A., Mirza S., Chakraborty S., Raza M., Storck M., Toss M., Meza J., Thoreson W., Coulter D., Rakha E., Band V., Band H., eLife 2023, 12, e81288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ping Q., Yan R., Cheng X., Wang W., Zhong Y., Hou Z., Shi Y., Wang C., Li R., Cancer Gene Ther. 2021, 28, 984. [DOI] [PubMed] [Google Scholar]
- 41. Veninga V., Voest E. E., Cancer Cell 2021, 39, 1190. [DOI] [PubMed] [Google Scholar]
- 42. Gupta V., Carey J. L., Kawakubo H., Muzikansky A., Green J. E., Donahoe P. K., MacLaughlin D. T., Maheswaran S., Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Maroulakou I. G., Anver M., Garrett L., Green J. E., Aagaard P., Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 11236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Green J. E., Shibata M.‐A., Yoshidome K., Liu M.‐L., Jorcyk C., Anver M. R., Wigginton J., Wiltrout R., Shibata E., Kaczmarczyk S., Wang W., Liu Z.‐Y., Calvo A., Couldrey C., Oncogene 2000, 19, 1020. [DOI] [PubMed] [Google Scholar]
- 45. Wigginton J. M., Park J.‐W., Gruys M. E., Young H. A., Jorcyk C. L., Back T. C., Brunda M. J., Strieter R. M., Ward J., Green J. E., Wiltrout R. H., J. Immunol. 2001, 166, 1156. [DOI] [PubMed] [Google Scholar]
- 46. Su C. Y., Burchett A., Dunworth M., Choi J. S., Ewald A. J., Ahn E. H., Kim D. H., Biomaterials 2021, 275, 120922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liao D., Johnson R. S., Cancer Metastasis Rev. 2007, 26, 281. [DOI] [PubMed] [Google Scholar]
- 48. Yi H. G., Jeong Y. H., Kim Y., Choi Y. J., Moon H. E., Park S. H., Kang K. S., Bae M., Jang J., Youn H., Paek S. H., Cho D. W., Nat. Biomed. Eng. 2019, 3, 509. [DOI] [PubMed] [Google Scholar]
- 49. Acerbi I., Cassereau L., Dean I., Shi Q., Au A., Park C., Chen Y. Y., Liphardt J., Hwang E. S., Weaver V. M., Integr. Biol. 2015, 7, 1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liverani C., Mercatali L., Cristofolini L., Giordano E., Minardi S., Porta G. D., De Vita A., Miserocchi G., Spadazzi C., Tasciotti E., Amadori D., Ibrahim T., Cell Mol. Bioeng. 2017, 10, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xiao Q., Ge G., Cancer Microenviron. 2012, 5, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lee J. W., Choi Y. J., Yong W. J., Pati F., Shim J. H., Kang K. S., Kang I. H., Park J., Cho D. W., Biofabrication 2016, 8, 015007. [DOI] [PubMed] [Google Scholar]
- 53. Skardal A., Mack D., Kapetanovic E., Atala A., Jackson J. D., Yoo J., Soker S., Stem Cells Transl. Med. 2012, 1, 792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gu B. K., Choi D. J., Park S. J., Kim Y. J., Kim C. H., Adv. Exp. Med. Biol. 2018, 1078, 15. [DOI] [PubMed] [Google Scholar]
- 55. Bordeleau F., Tang L. N., Reinhart‐King C. A., Phys. Biol. 2013, 10, 065004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sapudom J., Rubner S., Martin S., Pompe T., Adv. Healthcare Mater. 2016, 5, 1861. [DOI] [PubMed] [Google Scholar]
- 57. Shiwarski D. J., Hudson A. R., Tashman J. W., Feinberg A. W., APL Bioeng. 2021, 5, 010904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bakht S. M., Gomez‐Florit M., Lamers T., Reis R. L., Domingues R. M. A., Gomes M. E., Adv. Funct. Mater. 2021, 31, 2104245. [Google Scholar]
- 59. Saha T., Lukong K. E., Front. Oncol. 2022, 12, 856974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kim P. K., Halbrook C. J., Kerk S. A., Radyk M., Wisner S., Kremer D. M., Sajjakulnukit P., Andren A., Hou S. W., Trivedi A., Thurston G., Anand A., Yan L., Salamanca‐Cardona L., Welling S. D., Zhang L., Pratt M. R., Keshari K. R., Ying H., Lyssiotis C. A., eLife 2021, 10, e62645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schmaus A., Klusmeier S., Rothley M., Dimmler A., Sipos B., Faller G., Thiele W., Allgayer H., Hohenberger P., Post S., Sleeman J. P., Br. J. Cancer 2014, 111, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Monslow J., Govindaraju P., Puré E., Front. Immunol. 2015, 6, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Luckanagul J. A., Ratnatilaka Na Bhuket P., Muangnoi C., Rojsitthisak P., Wang Q., Rojsitthisak P., Polymers 2021, 13, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ekerdt B. L., Fuentes C. M., Lei Y., Adil M. M., Ramasubramanian A., Segalman R. A., Schaffer D. V., Adv. Healthcare Mater. 2018, 7, 1800225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Blanco‐Fernandez B., Rey‐Vinolas S., Bağcı G., Rubi‐Sans G., Otero J., Navajas D., Perez‐Amodio S., Engel E., ACS Appl. Mater. Interfaces 2022, 14, 29467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Xie J., Bao M., Bruekers S. M. C., Huck W. T. S., ACS Appl. Mater. Interfaces 2017, 9, 19630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sapudom J., Rubner S., Martin S., Kurth T., Riedel S., Mierke C. T., Pompe T., Biomaterials 2015, 52, 367. [DOI] [PubMed] [Google Scholar]
- 68. Wolf K., te Lindert M., Krause M., Alexander S., te Riet J., Willis A. L., Hoffman R. M., Figdor C. G., Weiss S. J., Friedl P., J. Cell Biol. 2013, 201, 1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Prince E., Kumacheva E., Nat. Rev. Mater. 2019, 4, 99. [Google Scholar]
- 70. Prince E., Cruickshank J., Ba‐Alawi W., Hodgson K., Haight J., Tobin C., Wakeman A., Avoulov A., Topolskaia V., Elliott M. J., McGuigan A. P., Berman H. K., Haibe‐Kains B., Cescon D. W., Kumacheva E., Nat. Commun. 2022, 13, 1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Berg W. A., Mendelson E. B., Cosgrove D. O., Doré C. J., Gay J., Henry J. P., Cohen‐Bacrie C., Eagles D. A., Am. J. Roentgenol. 2015, 205, 448. [DOI] [PubMed] [Google Scholar]
- 72. Yoo J., Seo B. K., Park E. K., Kwon M., Jeong H., Cho K. R., Woo O. H., Song S. E., Cha J., Cancer Imaging 2020, 20, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Plodinec M., Loparic M., Monnier C. A., Obermann E. C., Zanetti‐Dallenbach R., Oertle P., Hyotyla J. T., Aebi U., Bentires‐Alj M., Lim R. Y. H., Schoenenberger C. A., Nat. Nanotechnol. 2012, 7, 757. [DOI] [PubMed] [Google Scholar]
- 74. Kim J. E., Reynolds D. S., Zaman M. H., Mak M., Cabane E., Integr. Biol. 2018, 10, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Duan B., Yin Z., Kang L. H., Magin R. L., Butcher J. T., Acta Biomater. 2016, 36, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Koorman T., Jansen K. A., Khalil A., Haughton P. D., Visser D., Rätze M. A. K., Haakma W. E., Sakalauskaitè G., van Diest P. J., de Rooij J., Derksen P. W. B., Oncogene 2022, 41, 2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Liu L., Yu L., Li Z., Li W., Huang W., J. Transl. Med. 2021, 19, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Below C. R., Kelly J., Brown A., Humphries J. D., Hutton C., Xu J., Lee B. Y., Cintas C., Zhang X., Hernandez‐Gordillo V., Stockdale L., Goldsworthy M. A., Geraghty J., Foster L., O'Reilly D. A., Schedding B., Askari J., Burns J., Hodson N., Smith D. L., Lally C., Ashton G., Knight D., Mironov A., Banyard A., Eble J. A., Morton J. P., Humphries M. J., Griffith L. G., Jørgensen C., Nat. Mater. 2022, 21, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Meng F., Meyer C. M., Joung D., Vallera D. A., McAlpine M. C., Panoskaltsis‐Mortari A., Adv. Mater. 2019, 31, 1806899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Song J., Miermont A., Lim C. T., Kamm R. D., Sci. Rep. 2018, 8, 17949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Monteiro M. V., Zhang Y. S., Gaspar V. M., Mano J. F., Trends Biotechnol. 2022, 40, 432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sridharan R., Ryan E. J., Kearney C. J., Kelly D. J., O'Brien F. J., ACS Biomater. Sci. Eng. 2019, 5, 544. [DOI] [PubMed] [Google Scholar]
- 83. Sadjadi Z., Zhao R., Hoth M., Qu B., Rieger H., Biophys. J. 2020, 119, 2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Louis F., Piantino M., Liu H., Kang D.‐H., Sowa Y., Kitano S., Matsusaki M., Cyborg Bionic. Syst. 2021, 2021, 1412542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Dey M., Kim M. H., Dogan M., Nagamine M., Kozhaya L., Celik N., Unutmaz D., Ozbolat I. T., Adv. Funct. Mater. 2022, 32, 2203966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Neufeld L., Yeini E., Reisman N., Shtilerman Y., Ben‐Shushan D., Pozzi S., Madi A., Tiram G., Eldar‐Boock A., Ferber S., Grossman R., Ram Z., Satchi‐Fainaro R., Sci. Adv. 2021, 7, eabi9119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wei L., Wu S., Kuss M., Jiang X., Sun R., Reid P., Qin X., Duan B., Bioact. Mater. 2019, 4, 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Liu Y., Ling S., Wang S., Chen X., Shao Z., Biomater. Sci. 2014, 2, 1338. [DOI] [PubMed] [Google Scholar]
- 89. D'Este M., Eglin D., Alini M., Carbohydr. Polym. 2014, 108, 239. [DOI] [PubMed] [Google Scholar]
- 90. Shi W., Fang F., Kong Y., Greer S. E., Kuss M., Liu B., Xue W., Jiang X., Lovell P., Mohs A. M., Dudley A. T., Li T., Duan B., Biofabrication 2022, 14, 014107. [DOI] [PubMed] [Google Scholar]
- 91. Souter L. H., Andrews J. D., Zhang G., Cook A. C., Postenka C. O., Al‐Katib W., Leong H. S., Rodenhiser D. I., Chambers A. F., Tuck A. B., Lab. Invest. 2010, 90, 1247. [DOI] [PubMed] [Google Scholar]
- 92. Wang Y., Mirza S., Wu S., Zeng J., Shi W., Band H., Band V., Duan B., OncoTargets Ther. 2018, 9, 32191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Padmanaban V., Grasset E. M., Neumann N. M., Fraser A. K., Henriet E., Matsui W., Tran P. T., Cheung K. J., Georgess D., Ewald A. J., Nat. Protoc. 2020, 15, 2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Shi W., Hass B., Kuss M. A., Zhang H., Ryu S., Zhang D., Li T., Li Y.‐L., Duan B., Carbohydr. Polym. 2020, 233, 115803. [DOI] [PubMed] [Google Scholar]
- 95. Shi W., Fang F., Kong Y., Greer S. E., Kuss M., Liu B., Xue W., Jiang X., Lovell P., Mohs A. M., Dudley A. T., Li T., Duan B., Biofabrication 2021, 14, 014107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.