

SUMMARY
Mechanisms underlying distinct specification, commitment, and differentiation phases of cell fate determination remain undefined due to difficulties capturing these processes. Here, we interrogate the activity of ETV2, a transcription factor necessary and sufficient for hematoendothelial differentiation, within isolated fate intermediates. We observe transcriptional upregulation of Etv2 and opening of ETV2-binding sites, indicating new ETV2 binding, in a common cardiac-hematoendothelial progenitor population. Accessible ETV2-binding sites are active at the Etv2 locus but not at other hematoendothelial regulator genes. Hematoendothelial commitment coincides with the activation of a small repertoire of previously accessible ETV2-binding sites at hematoendothelial regulators. Hematoendothelial differentiation accompanies activation of a large repertoire of new ETV2-binding sites and upregulation of hematopoietic and endothelial gene regulatory networks. This work distinguishes specification, commitment, and sublineage differentiation phases of ETV2-dependent transcription and suggests that the shift from ETV2 binding to ETV2-bound enhancer activation, not ETV2 binding to target enhancers, drives hematoendothelial fate commitment.
In brief
Mechanisms driving the specification, commitment, and differentiation phases of fate determination remain elusive. Steimle et al. investigate the activity of the hematoendothelial master transcription factor ETV2 and identify that the shift from ETV2 binding to ETV2-bound enhancer activation, not ETV2 binding to target enhancers, drives hematoendothelial fate commitment.
Graphical Abstract
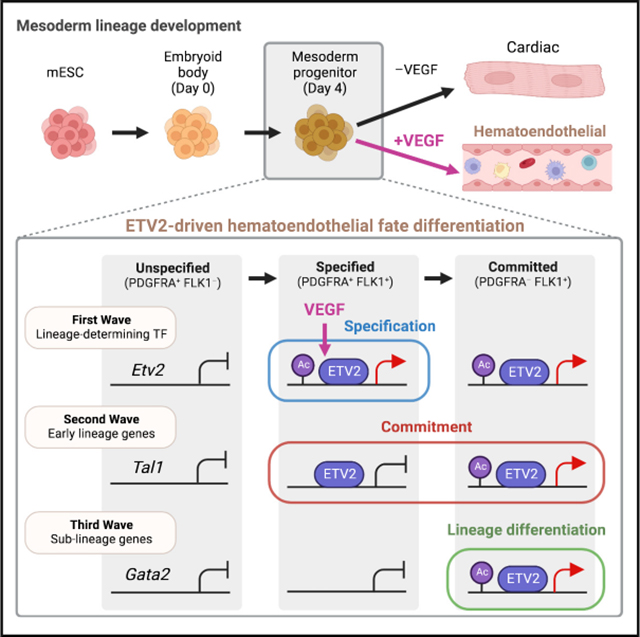
INTRODUCTION
Successive cell fate decisions underpin the development of multicellular organisms.1–3 Transcription factors (TFs) play a central role in fate decisions by binding cis-regulatory elements and directing fate-specific transcriptional and chromatin states.4–6 The transcriptional mechanisms that drive cell fate transitions are an area of active investigation.7–9 Individual cells undergoing fate transition often display a mixture of progenitor and destination transcriptional and chromatin states.7,10–12 One example is a phenomenon described as lineage priming, in which progenitor cells exhibit a subset of transcriptional and chromatin states characteristic of destination cells.13–16 However, the mechanisms that drive transcriptional and chromatin state changes during fate transition processes remain unresolved, in part due to difficulties isolating and interrogating specific intermediates during cell fate decisions.
An early common mesoderm progenitor generates both the cardiac lineage for heart development and the hematoendothelial lineage for hematopoiesis and vasculogenesis.17–28 Vascular endothelial growth factor (VEGF) reception by FLK1 (VEGFR2, Kdr) induces hematoendothelial fate specification by transcriptionally activating Etv2,29–31 an Ets-family, lineage-determining TF for the vertebrate hematoendothelial lineage.29,32–41 Etv2 expression in FLK1+ mesoderm specifies the hematoendothelial lineage at the expense of the cardiac lineage cell autonomously.42,43
In this study, we investigate dynamics of transcription, chromatin accessibility, and enhancer activity in specification, commitment, and differentiation phases of hematoendothelial development by examining fate intermediates originating from single time points of mouse embryonic stem cell (mESC) differentiation. This allows the distinct transcriptional mechanisms distinguishing specification, commitment, and sublineage differentiation phases of hematoendothelial gene expression to be delivered.
RESULTS
Sequential derivation of PDGFRα - and FLK1-expressing mesoderm populations
During vertebrate development, PDGFRα+ FLK1+ (PF) cells are defined as multipotent cardiovascular progenitors with cardiac and hematoendothelial potential.24,25 Previous work delineated the fates derived from PDGFRα+ and/or FLK1+ populations from mESC differentiation using a serum-free medium containing activin A, BMP4, and VEGF (ABV) (Figure 1A).24,25 We observed that 100% of the PDGFRα+ populations were mesoderm, only the PF population generated cardiomyocytes, and only the PDGFRα− FLK1+ (F) population formed blood colonies (Figures 1A–1D and S1A–S1D).24,25 These observations confirmed that the PF population defined a cardiomyogenic progenitor and the F population a hematoendothelial progenitor and that the PDGFRα+ FLK1− (P) population lacked both potentials.24,25,29
Figure 1. Two waves of hematoendothelial gene expression.
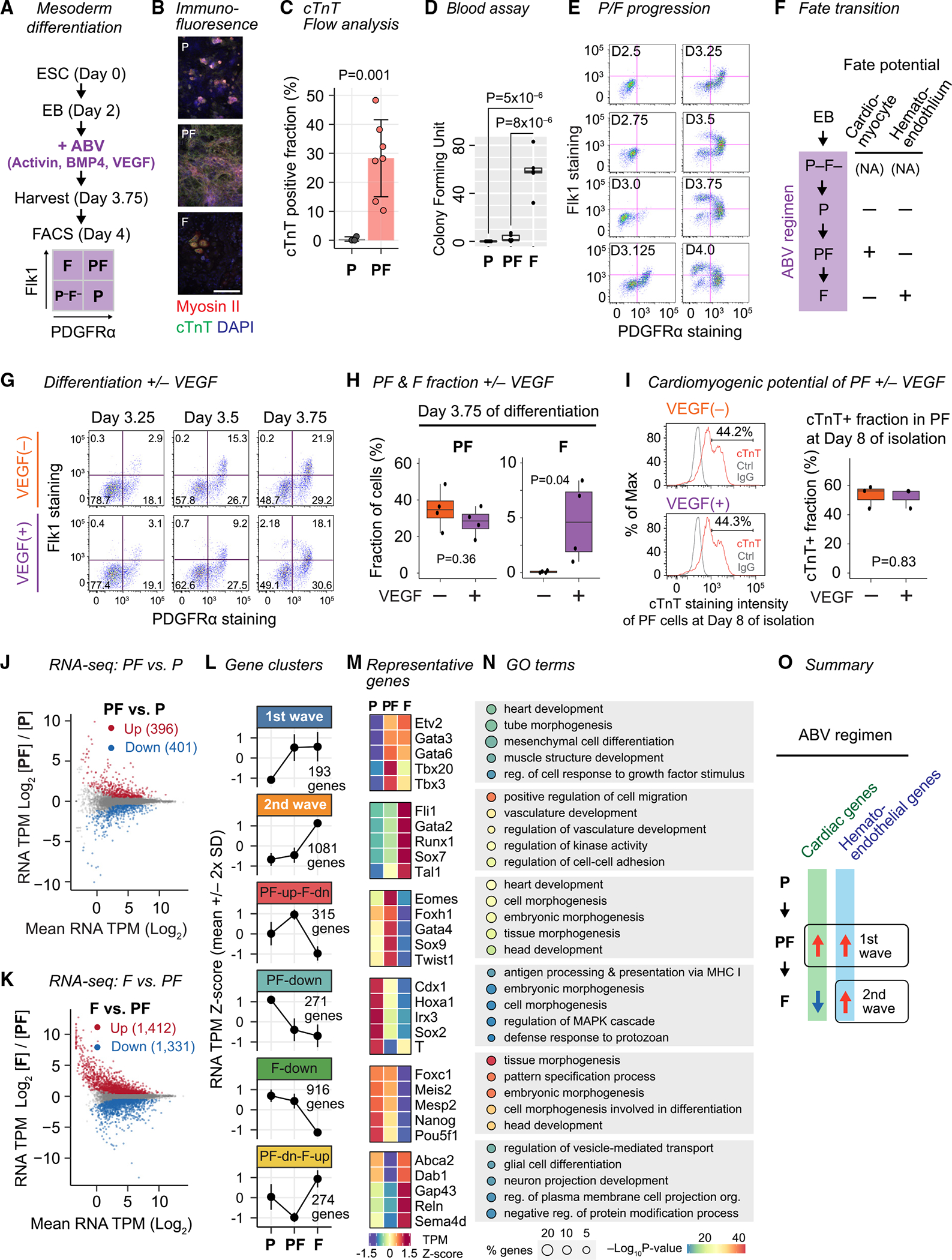
(A) mESC differentiation in the ABV regimen and isolation of PDGFRα (P)/Flk1 (F) populations. See Figure S1 for additional characterization.
(B) Immunofluorescence for cardiac myosin II and cardiac troponin (cTnT) in day 4 P, PF, and F cells. All images same magnification; scale bar: 100 μm.
(C) Fraction of cTnT+ cells in day 8 P (n = 3) or PF (n = 6) cells by flow cytometry. p, generalized linear model (GLM) P value accounting for experiment batches. Data, mean ± SD.
(D) Blood colony-forming units from 12,000 P, PF, or F cells 10 days after isolation. Data are represented by boxplot.
(E) P/F flow cytometry analysis at the indicated time.
(F) Temporal relationship and fate potential of P/F populations at day 4.
(G) P/F flow cytometry at the indicated time with or without VEGF.
(H) Fraction of PF and F populations at day 3.75 with or without VEGF. p, GLM P value accounting for treatment series batches. n = 4. Data are represented by boxplot.
(I) Flow cytometry plot (left) and quantification (right) for cTnT-stained PF cells at day 8. p, GLM P value. n = 3. Data are represented by boxplot.
(J) Transcriptome comparison between P and PF at day 4.
(K) Transcriptome comparison between PF and F at day 4.
(L) The unions of differentially expressed dynamic genes identified in (J) and (K) are clustered into 6 groups.
(M) mRNA levels of representative genes in each group from (L).
(N) Top 5 Gene Ontology (GO) terms by P value overrepresented in each group from (L).
(O) Two waves of gene expression toward hematoendothelial fate commitment in F.
We performed a time course study to define the emergence of PDGFRα/FLK1 subpopulations. Cells were initially PDGFRα− FLK1− (P–F–) at day 2.5. The P population emerged at day 3, followed by PF at day 3.125, and finally F at day 3.5 (Figures 1E and S1E). Differentiation of isolated PDGFRα/FLK1 subpopulations confirmed this order of differentiation (Figure S1F). Thus, the P population was an early multipotent mesodermal progenitor that gave rise to PF, a common cardiomyogenic/hematoendothelial progenitor, which could differentiate into cardiomyocytes upon isolation from ABV or into the hematoendothelial F population if retained in ABV (Figure 1F).
We confirmed previous observations that VEGF signaling is required for hematoendothelial commitment, driving the transition from PF to F (Figures 1G and 1H).21–23,29 However, mESC differentiation with or without VEGF generated the PF population or PF-derived cardiomyocytes with comparable efficiency (Figures 1G–1I). Thus, VEGF was dispensable for generating the common cardiac/hematoendothelial PF population but was required for the hematoendothelial commitment in the PF-to-F transition.
Two distinct waves of hematoendothelial gene activation
The resolution of PDGFRα/FLK1 fate potentials allowed us to investigate mechanisms driving the sequential phases of cardiomyogenic/hematoendothelial fate commitment. We first investigated gene expression differences between the P, PF, and F populations at day 4 of differentiation using RNA sequencing (RNA-seq) (Figures 1J, 1K, S1G, and S1H; Table S1). We identified a union set of 3,050 genes that were differentially expressed either between P and PF, between PF and F, or both and clustered them based on their expression dynamics (Figures S1I and S1J; Table S1). This process identified six distinct patterns of expression and revealed two waves of gene expression affiliated with hematoendothelial development (Figure 1L). The “first-wave” cluster contained genes upregulated during the P-to-PF transition and remained high in F (193 genes) (Figure 1M). This cluster was overrepresented for Gene Ontology (GO) terms related to hematoendothelial and cardiomyogenic fates (Figure 1N; Table S2) and TFs implicated in hematopoietic and cardiac development such as Tbx3, Hand2, and Gata6.44,45 Interestingly, Etv2, the lineage-determining TF for the hematoendothelial fate, was included in this first-wave cluster (Figure 1M), despite the PF population not yet being committed to the hematoendothelial fate.
The “second-wave” cluster contained genes whose expression was low in P and PF, and upregulated during the PF-to-F transition, accompanying hematoendothelial commitment and differentiation (1,081 genes) (Figure 1L). This cluster was overrepresented by hematoendothelial GO terms (Figure 1N) and included regulators of hematoendothelial, hematopoietic, and endothelial differentiation (Figures 1M, S1K, and S1L). Overall, the dynamic changes in transcription were associated with phenotypic cell fates and a pattern of differentiation mirroring development in vivo, lending credence to the sequential emergence of fate intermediates inferred from their temporal emergence in vitro (Figure 1O).
VEGF induces Etv2 in the PF population without large-scale hematoendothelial gene expression
The unexpected observation that Etv2 is upregulated in PF led us to further interrogate Etv2 expression dynamics (Figure 1M). Etv2 was 26-fold upregulated in PF relative to P and then only 2-fold upregulated in F relative to PF (Figure 2A). Etv2 was among the most upregulated TF genes during the P-to-PF transition and among the highest expressed TFs in PF (98th percentile; Figure S2A). Etv2 was the only ETS-family TF upregulated in PF and affiliated with the first-wave gene cluster (Figures 2B and S2B). Using the published single-cell transcriptome for mESCs undergoing spontaneous differentiation,46 we confirmed that Etv2-expressing individual cells increased from 35% among P to 71% among PF and plateaued at 74% among F (Figures 2C and S2C) and that Etv2 mRNA abundance in individual cells increased from P to PF and then plateaued in F (Figure S2D). We further confirmed ETV2 protein reporter expression in 72% of the PF cells, increased from 33% of P and 8% of P–F– cells (Figure 2D). Together, these observations established that Etv2 was a first-wave hematoendothelial gene that was strongly induced in PF and remained highly expressed in F.
Figure 2. VEGF induces Etv2 without inducing second-wave genes in PF.
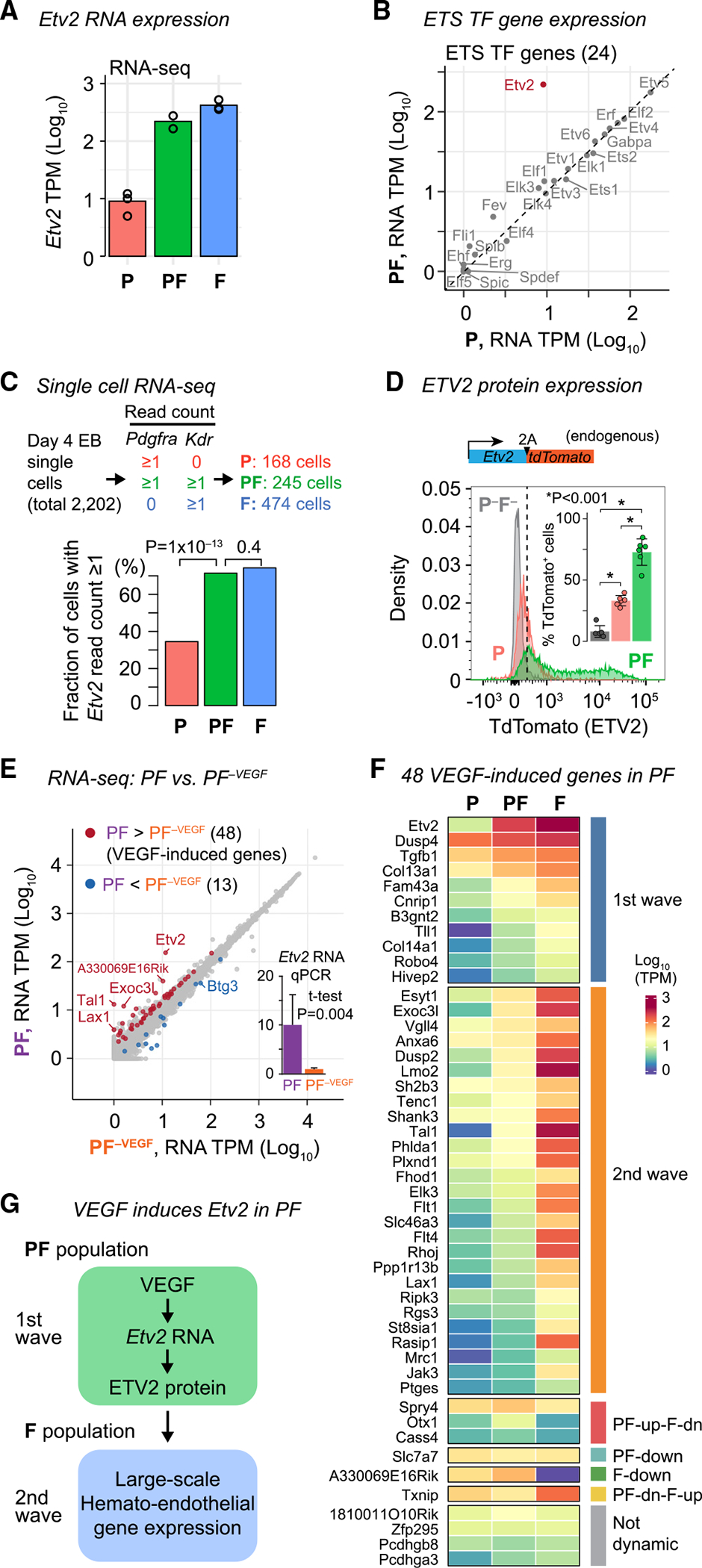
(A) Etv2 mRNA levels in P, PF, and F. See Figure S2 for additional characterization. Data, mean with all replicates shown.
(B) Transcripts per million (TPMs) for all 24 ETS-family genes in P and PF.
(C) (Top) Cells from single-cell RNA-seq data46 are grouped in P, PF, and F by pdgfra and Flk1 counts. (Bottom) Fraction of P, PF, or F single cells expressing Etv2. p, Fisher’s exact test p value.
(D) Histogram of tdTomato expression levels of P–F–, P, and PF cells at day 4 of differentiation of ETV2–2A-tdTomato transgenic mESC. (Inset) Quantification of tdTomato+ cells. p, GLM p value accounting for experiment batches. Data, mean ± SD.
(E) Transcriptome comparison between day 4 PF derived with VEGF (PF) and PF without VEGF (PF−VEGF). Inset, quantitative RT-PCR for Etv2. p, t test p value. Data, mean ± SD.
(F) mRNA levels for the 48 VEGF-induced genes in P, PF, and F with VEGF. Genes grouped by dynamic clusters (Figure 1L).
(G) VEGF induces the first-wave gene Etv2 without causing large-scale second-wave hematoendothelial gene expression in PF; second-wave expression occurs in F.
We tested the hypothesis that VEGF drove Etv2 transcription in PF by RNA-seq of PF derived with or without VEGF. Only 61 genes were differentially expressed between the treatments, including 48 upregulated “VEGF-induced” genes (Figure 2E; Table S1). The strongest VEGF-induced gene was Etv2 (Figure 2E). Only 11 of the 48 VEGF-induced genes were first-wave genes, and Etv2 was the only known early regulator of hematoendothelial specification (Figure 2F). In contrast, 27 of the 48 VEGF-induced genes were second-wave genes and were only weakly induced by VEGF in PF but strongly upregulated in F, including several well-known regulators of hematoendothelial specification (e.g., Tal1, Lmo2, and Flt1) (Figure 2F). This analysis indicated that VEGF strongly upregulated Etv2 without activating other hematoendothelial genes in the PF population (Figure 2G).
Pervasive gains of chromatin accessibility at ETS TF motif sites in PF and F
We hypothesized that Etv2 expression in PF primed the hematoendothelial gene regulatory network in PF for the subsequent activation in F. We sought to distinguish between two models: that the transition from Etv2 expression to ETV2 binding at target genes coincides with the PF-to-F transition or that ETV2 binds and poises at the target genes in PF until the transition to F. To distinguish between these hypotheses, we measured genomewide chromatin accessibility in P, PF, and F by assay for transposase-accessible chromatin (ATAC)-seq (Figures 3A and 3B; Table S3). We identified a union set of 41,383 sites that were differentially accessible either between P and PF, between PF and F, or both. These sites were predominantly distal to gene transcription start sites (96% ≥ 2 kb transcription start site [TSS]; Figure S3A). We clustered them based on their accessibility dynamics into 5 clusters (Figures S3B and S3C; Table S3). The enriched TF motifs within each cluster were consistent with the developmental gene expression patterns of the TFs observed in analogous gene expression clusters (Figures 3C and 3D). Specifically, we identified first- and second-wave alterations in chromatin accessibility that mirrored the sequential waves of hematoendothelial gene expression. First-wave ATAC sites (3,223) opened in the P-to-PF transition and remained open in F, whereas second-wave ATAC sites (19,534) were closed in P and PF and opened in the PF-to-F transition (Figure 3C). Both the first- and second-wave ATAC sites were most strongly overrepresented for the ETS motif (Figure 3D). Overrepresentation of the ETS motif in the first-wave ATAC sites supported the hypothesis that Etv2 expression resulted in first-wave ETV2 binding in PF, despite the absence of large-scale hematoendothelial gene activation. Second-wave ETS motif enrichment suggested that first- and second-wave ETV2-binding events may distinguish different stages of hematoendothelial development.
Figure 3. Accessible ETV2-binding sites emerge in PF and expand in F.
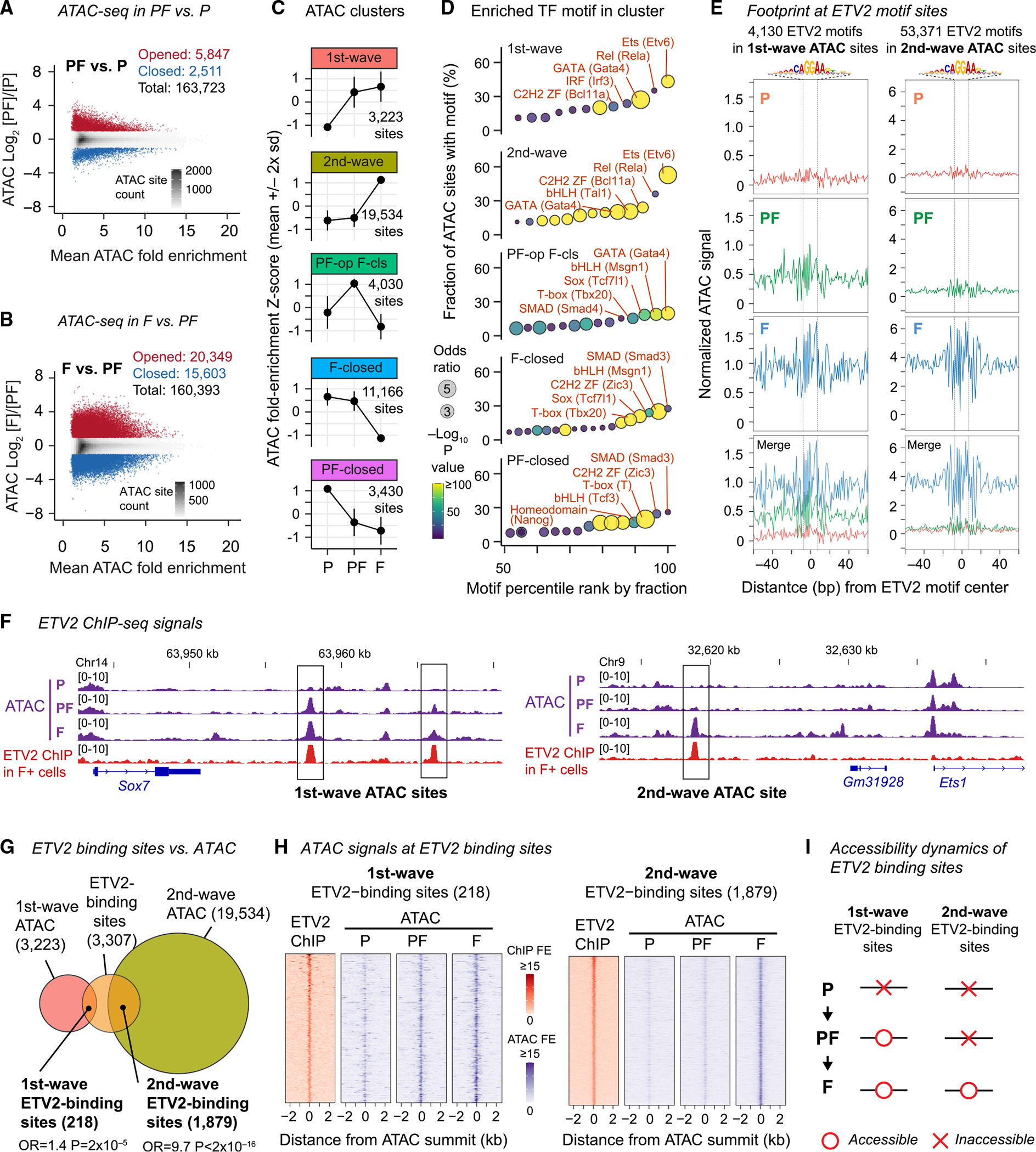
(A) MA plot comparing ATAC-seq signals in PF vs. P. See Figure S3 for additional characterization.
(B) Same as (A) but shown for F vs. PF.
(C) The unions of differentially accessible sites identified in (A) and (B) are clustered into 5 groups. PF-opened (PF-op); F-closed (F-cls) sites.
(D) TF family motifs overrepresented (odds ratio >1) within clusters. Parentheses, most overrepresented TF within family.
(E) Mean base-resolution profile of 120 bp centered around ETV2 motif center in first-wave (left) or second-wave (right) ATAC sites.
(F) ATAC and ETV2 ChIP-seq fold enrichment signal tracks showing first-wave (left) and second-wave (right) ETV2-binding sites. ETV2 ChIP-seq data are fromFLK1+ cells.41
(G) ETV2-binding sites overlapping the first-wave and second-wave ATAC sites. p, Fisher’s exact test p value.
(H) ATAC-seq fold enrichment signals at first-wave (left) and second-wave (right) ETV2-binding sites.
(I) The first-wave and second-wave ETV2-binding sites differ in the timing of accessibility gains at PF or F.
VEGF dependence and TF footprinting support differential ETV2 binding in PF and F
We hypothesized that the observed gains of chromatin accessibility at ETS motif sites in PF indicated ETV2 binding induced by VEGF, as VEGF induced Etv2 expression in PF (Figure 2E). To test this hypothesis, we profiled the chromatin accessibility by ATAC-seq in PF derived with (PF) or without VEGF (PF–VEGF). Only 318 sites showed higher and 388 sites showed lower accessibility in PF relative to PF–VEGF, respectively (Figure S3D; Table S3). The 318 sites with higher accessibility, but not the 388 sites with lower accessibility, were overrepresented for the ETS-family motif (Figures S3E and S3F), supporting the hypothesis that VEGF-driven Etv2 expression resulted in ETV2 binding in PF.
We next investigated whether ETS motif sites in first- and second-wave ATAC sites exhibited TF footprinting.47,48 At the ETV2 motif sites within the first-wave ATAC sites, footprints were undetectable in P but emerged in PF and remained present in F (Figure 3E). Therefore, at the first-wave ATAC sites, an ETS TF likely bound the ETV2 motif in PF and remained bound in F. At the second-wave ATAC sites, footprints were absent in both P and PF, but strongly emerged in F, suggesting de novo TF binding at ETV2 motifs in F. Because ETV2 is the only VEGF-dependent ETS TF that was transcriptionally induced in PF (Figures 2B and 2F), we infer that the footprinting analysis reflects specific ETV2 binding. These observations indicated two waves of ETV2 binding events: the first wave starting in PF, and the second wave starting in F.
We utilized the distinction between first- and second-wave ATAC clusters to refine available ETV2 chromatin immunoprecipitation (ChIP)-seq datasets generated in mESC-derived ETV2-overexpressing FLK1+ cells, which would include both PF and F populations (Table S4).41 Of the 3,307 ETV2-binding sites identified in the FLK1+ population, 218 ETV2-binding sites overlapped the first-wave ATAC sites (“first-wave ETV2-binding sites”), whereas 1,879 ETV2-binding sites overlapped the second-wave ATAC sites (“second-wave ETV2-binding sites”) (Table S3; Figures 3F–3H). ETV2-binding sites were underrepresented in other types of dynamic ATAC sites (Figure S3G). These results suggested that ETV2 binds a select set of first-wave ETV2-binding sites in PF, prior to the hematoendothelial commitment, followed by a large set of second-wave ETV2 binding in F, upon hematoendothelial fate commitment (Figure 3I).
Three patterns of accessibility and activation dynamics at ETV2-binding sites
We hypothesized that ETV2 binding primed first-wave ETV2-binding sites in PF that became active only upon transition to the F population. We performed ChIP-seq for histone H3 lysine 27 acetylation (H3K27ac), a modification associated with active regulatory elements, in PF and PF–VEGF from day 4 of differentiation and in F from day 5 of differentiation (Table S4, Figures S4A and S4B). Only 15 of the 218 first-wave ETV2-binding sites (7%), representing “early-active” first-wave ETV2-binding sites (Figures 4A and 4B), were marked with H3K27ac in PF (Figure 4A). This included one ETV2-binding site located at the Etv2 locus itself (Figure 4C). VEGF promoted H3K27ac of these sites in PF, as all 15 sites showed higher H3K27ac levels in PF compared with PF–VEGF (Figure S4C). H3K27ac-marked first-wave ETV2-binding sites increased to 63% in F (137 sites; Figures 4A–4C). We termed the 123 first-wave ETV2-binding sites that gained H3K27ac in F but not in PF “delayed-active” ETV2-binding sites. These results identified a highly selective set of early-active first-wave ETV2-binding sites that were accessible and marked with H3K27ac in PF and a large number of delayed-active ETV2-binding sites that were active in PF but acquired H3K27ac later in F (Figure 4B). This result supported the hypothesis that most first-wave ETV2-binding sites were poised and accessible in PF but only became active upon the transition to F.
Figure 4. PF-opened ETV2-binding sites become activated in F and accompany transcriptional activation of early hematoendothelial regulators.
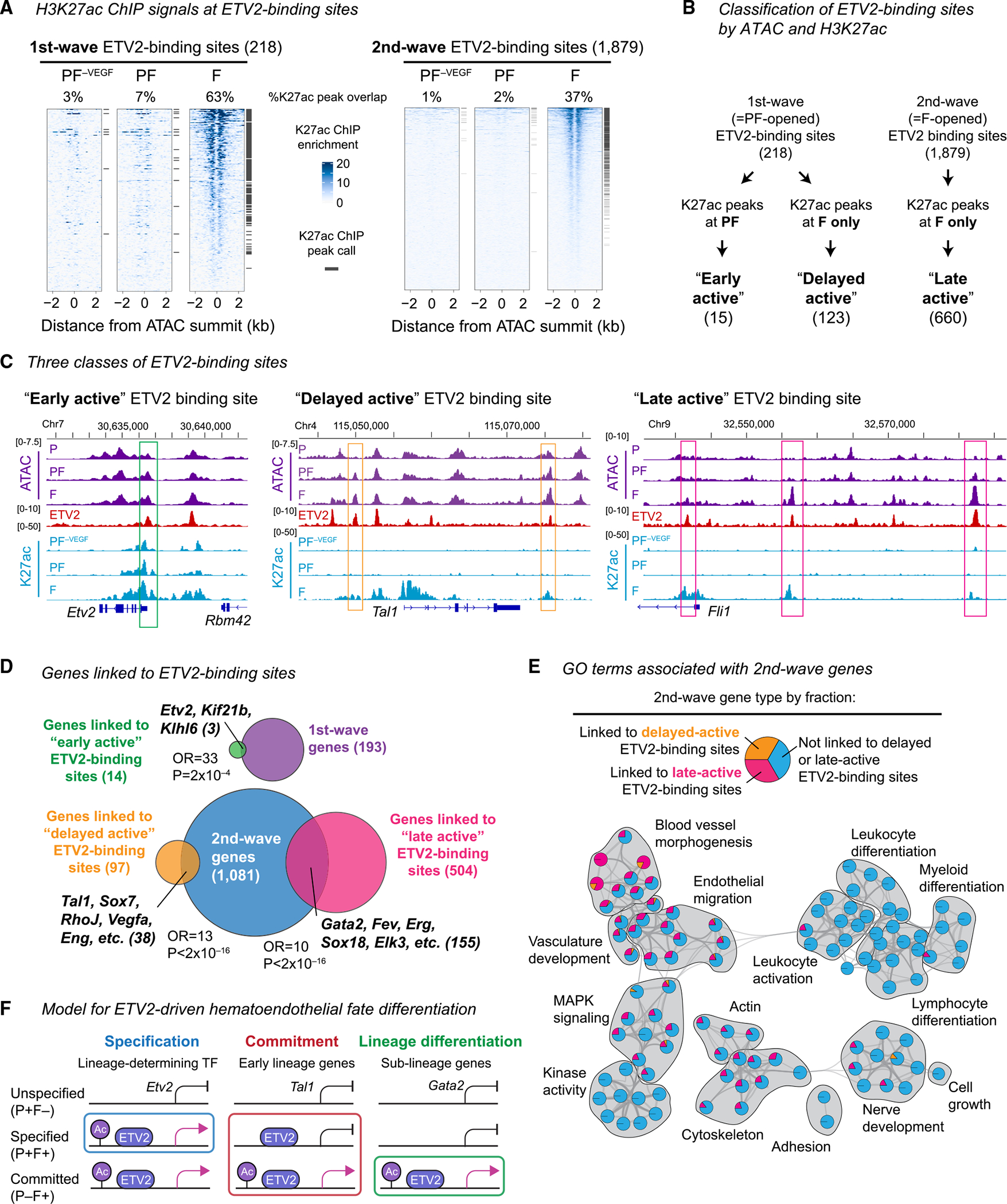
(A) H3K27ac ChIP-seq fold enrichment signals at first-wave (left) and second-wave (right) ETV2-binding sites. H3K27ac ChIP-seq is performed in PF–VEGF and PF isolated at day 4 and F isolated at Day 5. See Figure S4 for additional characterization.
(B) Classification of the first- and second-wave ETV2-binding sites into early-active, delayed-active, and late-active ETV2-binding sites.
(C) H3K27ac ChIP-seq fold enrichment signal tracks, along with ATAC-seq and ETV2 ChIP-seq tracks.
(D) (Top) Relationship between genes linked to early-active ETV2-binding sites and the first-wave genes. (Bottom) Relationship between genes linked to delayed-active or genes linked to late-active ETV2-binding sites and the second-wave genes. Parentheses, number of genes. p, Fisher’s exact test p value.
(E) GO terms overrepresented in the second-wave genes. For each GO term (circle), the fraction of the GO-associated genes linked to delayed-active (yellow) or late-active (magenta) ETV2-binding sites or not linked to delayed or late-active ETV2-binding sites (blue) are shown as a pie chart. Lines link GO terms by similarity.
(F) Timing of ETV2 binding and the timing of ETV2-bound enhancer activation distinguish different phases of hematoendothelial gene expression.
The 1,644 second-wave ETV2-binding sites were almost all devoid of H3K27ac signals in PF (99%) (Figure 4A). However, in F, the H3K27ac-marked fraction increased to 37% (689 sites; Figures 4A–4C). This identified a large subset of second-wave ETV2-binding sites activated selectively in F, in synchrony with accessibility gains. We termed the 660 second-wave ETV2-binding sites that gained H3K27ac only in F “late-active” ETV2-binding sites (Figure 4B). Together, H3K27ac dynamics identified early-active first-wave ETV2-binding sites that became open and active in PF, delayed-active first-wave sites that became open in PF but active in F, and late-active second-wave ETV2-binding sites that became open and active in F (Figure 4B).
Activation dynamics of ETV2-binding sites correlates with the hematoendothelial gene regulatory hierarchy
We hypothesized that the sequential ETV2 binding and activation of ETV2-binding sites might underlie the regulatory hierarchy governing hematoendothelial fate differentiation. To test this hypothesis, we linked the distinct dynamics of accessible ETV2-binding sites and gained H3K27ac with sequential waves of gene expression, specifically linking early-active ETV2-binding sites to first-wave genes and delayed-active and late-active ETV2-binding sites to second-wave genes (Figure S4D). Only 3 first-wave genes, including Etv2, linked to early-active ETV2-binding sites (Figure 4D).
The 38 second-wave genes linked to delayed-active ETV2-binding sites included early upstream regulators of the hematoendothelial gene regulatory network, including Tal1 and Sox7 (Figure 4D).18,33,49,50 The delayed-active ETV2-binding sites at second-wave genes suggested selective priming of important early regulators of hematoendothelial development in PF. Other well-characterized Etv2 targets were absent from this cohort.
The 155 second-wave genes linked to late-active ETV2-binding sites included many TFs for endothelial development such as Fli1, Gata2, and Sox18 (Figure 4D). Furthermore, these genes were enriched for GO terms related to endothelial development, but not hematopoietic development (Figures 4E and S4E), suggesting that late-active ETV2 binding selectively activated the endothelial branch of the hematoendothelial gene regulatory network, as suggested previously.33,51 Delayed-active and late-active ETV2-binding sites were also found at some first-wave genes (Figure S4F) and might contribute to the secondary upregulation or maintenance of first-wave gene expression in F. These data suggested that late-active ETV2-binding sites were associated with the upregulation of genes downstream of commitment but essential for sublineage-specific differentiation within the hematoendothelial lineage. Overall, this analysis identified three different stages of hematoendothelial development promoted by ETV2, specification in PF, commitment during PF-to-F transition, and differentiation in F, distinguished by the sequential regulation of ETV2 binding and ETV2-bound enhancer activation (Figure 4F).
DISCUSSION
ETV2 has been identified as a master regulator TF for the hematoendothelial lineage development.40,52,53 Our observations that Etv2 is strongly expressed and that ETV2-binding sites gain accessibility in the multipotent PF mesoderm progenitor prior to hematoendothelial fate commitment were unexpected. A recent study reported a pioneering activity of ETV2 capable of binding to nucleosomal DNA for chromatin opening.54 We propose that ETV2 opens and primes the ETV2-target hematoendothelial gene enhancers in the multipotent PF progenitor prior to hematoendothelial lineage commitment (Figure 4F).
How ETV2 activates different classes of genes for hematoendothelial development has been an outstanding question. Our data suggest that a combination of the timing of ETV2 binding and the timing of ETV2-bound enhancer activation enables sequential ETV2-dependent gene activation. Etv2 itself becomes activated early upon ETV2 binding. This observation is consistent with the previously described feedforward activation of Etv2,55 through an early-active ETV2-binding site, which may contribute to strong Etv2 expression in the PF population. Subsequent early activation of other essential early activators of hematoendothelial commitment, such as Tal1 and Sox7, requires a transition from bound to bound and activated enhancers. Finally, a late wave of ETV2 binding occurs at several TFs essential for endothelial development, such as Fli1, Gata2, and Sox18. This work provides a mechanistic model for how a single lineage-determining TF can differentially regulate distinct phases of specification, commitment, and sublineage differentiation during cell fate differentiation.
Limitations of the study
By investigating fate intermediates originating from a single time point of mESC differentiation, this study captures molecular dynamics during fate transitions. This approach contrasts with the conventional approach that compares samples from different time points.4,56 A limitation of our approach is that our data represent averages of transcriptional states, chromatin states, or fate potentials of cells within isolated populations. Single-cell approaches can resolve transcriptional and chromatin states of individual cells but can only map these states along inferred developmental time scales and do not allow for direct evaluation of the fate potentials of the cells under investigation.10,57–59 Our approach enables unambiguous determination of temporal relationships between isolated populations and evaluation of their fate potential. Combining fate intermediate isolation with single-cell approaches may address the potential heterogeneity of cells within intermediate states.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Kohta Ikegami (kohta.ikegami@cchmc.org).
Materials availability
Cell lines used in this study will be available upon request with a completed Materials Transfer Agreement.
Data and code availability
RNA-seq and ATAC-seq data are available at GEO with an accession number GSE136692.
This paper does not report original code.
All data reported in this paper will be shared by the lead contact upon request. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Mouse embryonic stem cells
We used the ZX1 mouse embryonic stem cell (ESC) line85 for transcriptome and chromatin analyses. We used the Bry-GFP mESC line86 for characterization of mESC differentiation. These two mESC lines were indistinguishable in their pluripotent states and differentiation potentials. We used the mESC line mESC with a C-terminal TdTomato insertion at the Etv2 locus and a heterozygous GFP knockin at the Brachyury locus for the analysis of ETV2 and BRACHYURY protein expression. mESCs were maintained in a serum-free and feeder-free culture system.25,87–89 For ESC maintenance, Neurobasal medium (ThermoFisher Scientific Cat. 21103049) and DMEM/F12 (ThermoFisher Scientific Cat. 10565018) (1:1) were supplemented with 0.5x N-2 supplement (ThermoFisher Scientific Cat. 17502048), 0.5x B27 supplement (ThermoFisher Scientific Cat. 17504044), 1x penicillin-streptomycin (ThermoFisher Scientific Cat. 15140148), 2mM Glutamine (ThermoFisher Scientific Cat. A2916801), 150μM mono-thioglycerol (MilliporeSigma, Cat. M6145), 0.05% BSA (MilliporeSigma, Cat. A9576), 1000 unit Mouse LIF (MilliporeSigma Cat. ESG1106), 1 μM PD0325901 (Procell Cat. 04–0006-10), 3 μM CHIR99021 (Procell Cat. 04–0004-10). PD0325901 and CHIR99021 were removed 2 days before the initiation of differentiation.
METHOD DETAILS
Mesoderm induction
To initiate differentiation, ESCs were dissociated with TrypLE express (ThermoFisher Scientific) and cultured in a 3:1 mixture of IMDM (ThermoFisher Scientific Cat. 12440053) and Ham’s F12 (ThermoFisher Scientific Cat. 11765054) medium supplemented with 0.5x N-2 supplement, 0.5x B27 supplement, 1x penicillin-streptomycin, 2mM Glutamine, 0.5 mM ascorbic acid (MilliporeSigma Cat. A4544), 450 μM mono-thioglycerol, 0.05% BSA at the density of 0.1 million cells per mL in a 10-cm Petri dish (Becton Dickenson) for inducing embryoid bodies.24,25 After 48 h, the embryoid bodies (EBs) were dissociated with TrypLE express. For mesoderm induction, the dissociated EBs were re-aggregated in the ABV regimen, defined as the StemPro-34 SFM medium (ThermoFisher Scientific Cat. 10639011) supplemented with 2mM Glutamine, 0.5 mM ascorbic acid, 450 μM mono-thioglycerol, 200 μg/mL human transferrin (MilliporeSigma Cat. T8158), 6 ng/mL human bFGF (R&D systems Cat. 233FB), 1 ng/mL human BMP4 (R&D systems Cat. 314BP), 8 ng/mL human Activin A (R&D systems Cat. 338AC), 5 ng/mL mouse VEGF (R&D systems Cat. 494MV). For mesoderm induction in the AB regimen, the culture medium without 5 ng/mL VEGF was used. FLK1 inhibitor, 1.2 μM ZD6474 (SelleckChem Cat. S1046) was treated from day 2 to day 3.75.
Hematopoietic and cardiac lineage induction
For hematopoietic lineage and cardiac lineage induction, mesodermal cells were sorted (see flow cytometry section) and cultured in the StemPro-34 SF medium supplemented with 2 mM Glutamax-I (ThermoFisher Scientific Cat. 35050–061), 1 mM ascorbic acid, and 30 ng/mL bFGF24,25 for indicated duration at the density of 0.2 million cells per ml in the individual wells of a 48-well flat bottom plate (Becton Dickenson) coated with gelatin (MilliporeSigma).
Flow cytometry
For cell sorting based on PDGFRα and FLK1 expression levels, embryoid bodies under mesoderm induction in the ABV or AB regimen were harvested and dissociated at the indicated time and incubated with the PE-conjugated anti-PDGFRα antibody (ThermoFisher Scientific Cat. 12–1401-81) and the PE/Cy7-conjugated anti-FLK1 antibody (Biolegend Cat. 359911) for 1 h on ice. Cells were sorted by the PDGFRα and FLK1 staining levels into the StemPro34 medium using the FACSAria II sorting instrument (BD Bioscience). For cardiac troponin (cTnT) staining, Day 8 cells were harvested and fixed with cytofix/cytoperm fixation/permeabilization solution (BD Bioscience, Cat. 554714) following manufacturer’s instructions. Cells were incubated with BV421-labeled anti-cTnT antibody (BD Bioscience Cat. 565618) for 1.5 h and analyzed by LSRFortessa cell analyzer (BD Bioscience). Flowjo software was used to visualize flow cytometry data.
Immunofluorescence
For immunohistochemistry, the Day 8 cells were fixed with 4% formaldehyde (ThermoFisher Scientific) for 24 h and washed with PBS. Cells were permeabilized in PBS containing 0.25% Triton X-100 for 10 min. The anti-cTnT antibody (Thermo Fisher Scientific, Cat. MS-295) and anti-myosin heavy chain class II (Abcam Cat. ab55152) antibodies were used.
Blood colony-forming assay
Twelve thousand cells were plated at 2 days after cell sorting (Day 6 post differentiation) into a methylcellulose-based medium containing hematopoietic cytokines (MethoCult M3434, StemCell Technologies, Cat. 03434). Hematopoietic colonies were counted by manufacturer’s instructions at 10 days post-plating (Day 16 of differentiation).
Quantitative RT-PCR
Quantitative real-time RT-PCR was performed using an ABI Prism 7500 Fast SDS (Applied Biosystems). Total RNA was extracted using NucleoSpin RNAII kit (Takara, Cat#740955.50). qRT-PCR was performed on 96-well optical reaction plates with one-step SYBR Green PCR master mix (Bio-Rad Laboratories, Cat#1725150).
RNA-seq
Library preparation was performed by the University of Chicago Genomics Facility using the Illumina TruSeq RNA Sample prep kit v2 (Part #RS-122–2001). Library fragments were approximately 275bps in length and were quantitated using the Agilent Bio-analyzer 2100 and pooled in equimolar amounts. Single-ended, 51bp sequencing was performed on the Illumina HiSeq2500 in Rapid Run Mode by the University of Chicago Facility.
RNA-seq data preprocessing
Transcripts were aligned to the indexed reference for mm10 with default settings using TopHat2 v2.1.1.60,61 Reads were filtered using bamtools using the following settings: -isDuplicate false -mapQuality “>10”.90 Transcript reads (TPMs) were counted post-alignment using StringTie.62,91 TPMs are listed in Table S1. Raw and processed RNA-seq data are available at GEO with an accession number GSE136692.
Differentially expressed genes
Differential expression testing was performed using edgeR v3.16.563–67 and limma v3.30.1368 packages in R v3.3.2. Low level genes were removed within each condition using median log2-transformed counts per gene per million mapped reads (cpm) of 1 and a union generated from those lists. Differential expression testing was performed using a general linear model (GLM) framework. For comparing PF and PF–VEGF, a covariate for replicate was included to correct for batch effect. Genes with absolute log2 fold change greater than 0.5 and false discovery rate (FDR) smaller than 5% were defined as differentially expressed genes. The human TF annotation92 was downloaded from http://humantfs.ccbr.utoronto.ca/download.php and incorporated into the mouse gene list based on the gene symbols. Differentially expressed genes are listed in Table S1.
Clustering of genes using RNA-seq data
First, the union of differentially expressed genes in the P-vs-PF and PF-vs-F comparisons was selected. Second, for each of the union differentially expressed genes, mean TPMs (mTPM) across replicates were computed. Third, for each gene, the Z score of Log10(1+mTPM) was computed across conditions (P, PF, and F). Forth, the Z score matrix was processed using the kmeans function in R with k = 6 and set.seed(109). Finally, the Z score matrix was processed in the fviz_nbclust function in the factoextra package in R to compute the total within sum of square for k = 1 to k10, which supported that the choice of k = 6 was reasonable based on the total within-sum of square approaching the minimum.
ATAC-seq
ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing) was performed as previously described.93 Libraries were amplified and normalized with the Illumina Nextera DNA Library prep kit (FC-121–1031) according to the manufacturer’s protocols. Libraries were quantitated using the Agilent Bioanalyzer, pooled in equimolar amounts, and sequenced with 50-bp single-end reads on the Illumina HiSeq following the manufacturer’s protocols through the Genomics Core Facility at the University of Chicago.
ATAC-seq data preprocessing
Sequencing reads were aligned to the mm10 genome using Bowtie2 v2.3.069 and SAMtools v0.1.19.70,71 Peak calling was performed using MACS2 callpeak72,73 using the settings –nomodel –shift 100 –extsize 200 -q 0.1 after pooling biological replicates. A fold-enrichment track was generated using MACS2 using the bdgcmp function (-m FE) for visualization on the genome browser. Following removal of ENCODE blacklist sites,94,95 a union set of sites was generated by identifying summits that overlapped within 200bp and arbitrarily selecting the summit of highest positional value. Summits were then extended by 200bp in both directions to create the set of union sites. The union set of sites utilizes biological groups not explicitly described in this manuscript, but are relevant and publicly accessible through GEO. Fold-enrichment scores were assigned to each site using the multiBigwigSummary function from deepTools.74
Differentially accessible sites
For differentially accessible site analysis, we first extracted ATAC sites that were identified in either of the two datasets under comparison or both. The fold-enrichment scores for this set of ATAC sites were processed using a general linear model in edgeR v3.16.563–67 and limma v3.30.1368 packages in R v3.3.2 to identify differentially accessible sites. Differentially accessible sites with the p-value less than 10−3 and absolute log2 fold change greater than 1 were selected for downstream analyses. The list of differentially accessible sites are listed in Table S3.
Clustering of accessible sites using ATAC-seq data
First, the union of differentially accessible sites in the P-vs-PF and PF-vs-F comparisons was selected. Second, for each of the union differentially accessible sites, the Z score of ATAC fold-enrichment score across conditions (P, PF, and F) was computed. Third, the Z score matrix was processed using the kmeans function in R with k = 5 and set.seed(109). Finally, the Z score matrix was processed in the fviz_nbclust function in the factoextra package in R to compute the total within sum of square for k = 1 to k10, which supported that the choice of k = 5 was reasonable based on the total within-sum of square approaching the minimum.
H3K27ac ChIP-seq
Cells sorted at Day 4 or Day 5 of differentiation and frozen with liquid nitrogen were crosslinked in 0.5% formaldehyde for 5 min at room temperature, and then the reaction was quenched with 125 mM glycine. Cross-linked cells were rinsed with PBST (PBS with 0.1% Tween 20) and then resuspended in LB3-Triton (1 mM EDTA, 0.5 mM EGTA, 10 mM Tris-HCl pH 8, 100 mM NaCl, 0.1% Na-Deoxycholate, 0.5% N-lauroyl sarcosine, 1% Triton) supplemented with a protease inhibitor cocktail (Calbiochem 539131). Chromatin was extracted by sonication using Bioruptor (Diagenode). Cell extract was cleared by centrifugation and an aliquot was saved for input DNA sequencing. Cell extract from 0.5 million cells was incubated with mouse monoclonal anti-H3K27ac antibody (Wako MABI0309, Lot 14007; 2 μL per IP) in A 200 μL reaction for 12 h or longer at 4°C. Immunocomplex was captured by Protein G-conjugated magnetic dynabeads (ThermoFisher) and washed. Immunoprecipitated DNA was reverse-crosslinked and used to construct high-throughput sequencing libraries using NEBNext Ultra II DNA Library Prep Kit (New England Biolabs). DNA libraries were processed on an Illumina HiSeq machine for paired-end sequencing.
H3K27ac ChIP-seq data processing and analysis
Paired-end ChIP-seq reads were aligned to the mouse reference genome mm10 using Bowtie2 with the “–no-mixed –no-discordant -X 1000” parameter set,69 and then aligned reads with MAPQ >20 were retained. H3K27ac-enriched peaks were identified using MACS72 with two biological replicates of ChIP and one biological replicate of the input data, with the default narrow-peak option. An ATAC site was considered overlapping with a H3K27ac region, if the ATAC site (400 bp in size) overlaps the H3K27ac peak region by at least 1 bp, as determined by the intersect function of BedTools.75 For data visualization, input-normalized per-base fold-enrichment scores were computed using MACS2 with two biological replicates of ChIP and one biological replicate of the input data. We then computed the sum of the signal coverage of the fold-enrichment data within 50-bp windows across the genome, and the 50-bp window data were quantile-normalized across all experimental conditions (Day4 PF–VEGF, Day4 PF, and Day 5 F) using normalize.-quantiles function in the preprocessCore package (v1.36.0) in software R.76 The list of H3K27ac enriched regions are listed in Table S4.
ETV2-binding sites
We used the previously published ETV2 ChIP-seq data performed in an FLK-positive cell population derived from mESC differentiation,41 available at GEO under the accession ID GSE59402. This dataset used a mESC cell line that carries a V5-epitope tagged ETV2 transgene under the doxycycline-inducible promoter.41 In the experiment, the V5-epitope tagged ETV2 was expressed from Day 2 to Day 3.5 of differentiation by doxycycline, and ETV-associated chromatin was immunoprecipitated using an anti-ETV2 antibody or an anti-V5 tag antibody.41 We aligned sequencing reads to the mm10 mouse reference genome using Bowtie269 with the default “–sensitive” parameter. Reads with MAPQ scores greater than 20 were used in downstream analyses. Reads from biological replicates of ChIP and the corresponding input were processed by MACS2 (v2.1.0).72,73 Aligned reads from three ChIP-seq replicates (one replicate from anti-ETV2 ChIP and two replicates from anti-V5 ChIP; Short Read Archive IDs SRR1514692, SRR1514695, SRR1514696) and aligned reads from two control ChIP experiments (one replicate from non-ETV2 induced cells and one replicate from IgG control; Short Read Archive IDs SRR1514691 and SRR1514694) were used in MACS2 program72,73 to identify statistically overrepresented peak regions and peak summits and to produce fold-enrichment scores. The parameters used in MACS2 were [call-peak -g hg –nomodel –extsize 200 –call-summits]. Identified ETV2-enriched regions with the p value <10−10 and the fold enrichment score greater than 4 that did not overlap ENCODE mm10 blacklisted regions (https://www.encodeproject.org/annotations/ENCSR636HFF/) or the mitochondrial genome were selected. This yielded 3,868 ETV2-binding sites (Table S4). An ATAC site was considered possessing ETV2-binding sites when the 400-bp region centered around the ATAC site summit overlapped the summit of at least one ETV2-binding site.
TF footprint analysis
We used the TOBIAS package for the TF footprint analysis using ATAC-seq data.77 First, replicate-combined ATAC-seq read data were processed by TOBIAS ATACorrect function to generate signal tracks corrected for the estimated Tn5 transposase sequence bias. Second, the bias-corrected data were processed by TOBIAS ScoreBigwig function to calculate footprint scores across the union of ATAC sites identified in P, PF, and F. Third, the footprint scores were processed by TOBIAS BINDetect function to detect footprints at ETV2 motifs within the first-wave ATAC sites or the second-wave ATAC sites with the “–motif-pvalue 0.01 –bound-pvalue 0.01” parameter set. The ETV2 motif sequence (M09067_2.00_Etv2) was acquired from the CisBP database (Database build Version 2.00) at http://cisbp.ccbr.utoronto.ca/.96 Finally, the output files of the BINDetect function were processed in TOBIAS PlotAggregate function to generate the aggregate signal plots.
Gene ontology analysis
Gene ontology (GO) analyses were performed using Metascape.78,79 Gene Symbols of the genes of interest were used to examine overrepresentation of Biological Process and Molecular Function GO terms with default parameters (minimum gene count 3, p < 0.01, enrichment over background >1.5). p-values were derived from cumulative hypergeometric statistical tests and computed in Metascape.79 Reported GO terms in the figures are the “Summary” GO terms of all associated GO terms, and the number of genes represent the union of genes affiliated with the associated GO terms. In the network of GO terms, which was generated as a part of the Metascape analysis, all reported GO terms are shown.
DNA motif analysis
We used FIMO in the MEME suite (v5.0.5)80 to scan for the presence of TF motifs in each ATAC site (+/−100 bp from ATAC summit). We used the Mus musculus CIS-BP TF motif database (v2.0.0),96 which contains Position Weight Matrix and TF family annotation for each TF. For each ATAC site and for each TF, we determined whether at least one motif is present or not. We then performed, for each TF motif, Fisher’s exact test with a contingency table with counts for ATAC sites with motif presence or not and differentially accessible or not. This resulted in, for each motif for each ATAC group, a p-value, odds ratio, and motif-containing fraction of ATAC sites. To find overrepresented motifs, we selected motifs with odds ratio greater than 1 and the 95% confidence interval of the odds ratio not overlapping 1. We then selected a TF motif with the highest score defined by log2(odds ratio) × fraction for each motif family. The TF name, TF family name, fraction, and the p-value are plotted in figures.
Association between ATAC sites and genes
One ATAC site was linked to one gene when the ATAC site summit resided in the gene body or 100 kb upstream of the transcription start site (TSS) of that gene. ATAC sites that did not fulfill this condition were not linked to genes. We used the following algorithm to select a single gene when multiple genes could be linked: a) ATAC sites located in the gene body of one gene and within 100 kb upstream of another gene were assigned to the gene whose gene body contains the ATAC sites; b) ATAC sites located in the gene body of two different genes were assigned to the gene whose TSS was closer to the ATAC sites; and c) ATAC sites located within 100 kb upstream of two different genes were assigned to the gene whose TSS was closer to the ATAC sites.
Single-cell RNA-seq analysis
Single-cell RNA-seq data was downloaded from GEO (GSE130146)46 and imported into R using Seurat package version 4.0.0.81–84 In preprocessing, genes expressed in at least three cells were kept, and cells with less than 5% mitochondria read and greater than 2000 unique genes were kept. This preprocessing resulted in 16,249 unique genes and 2,202 cells. Genes with read counts greater than 0 were considered expressed in a cell, and genes with read counts equal to 0 were considered not expressed in a cell. To obtain expression levels, read counts were first normalized within each cell by dividing by the total counts of each cell, then multiplied by 10000, and then natural-log transformed, following the “LogNormalization” method in the Seurat package. The single-cell P population was defined as pdgfra -expressed, Kdr-not-expressed cells; the single-cell PF population was defined as pdgfra -expressed, Kdr-expressed cells; and the single-cell F population was defined as Kdr-expressed, pdgfra-not-expressed cells. To compute statistical significance of Etv2-expressed cell count differences between P and PF, Fisher’s exact test examined the association between Kdr-expressed cells and Etv2-expressed cells within pdgfra-expressed cells. For the comparison between PF and F, Fisher’s exact test examined the association between pdgfra-expressed cells and Etv2-expressed cells within Kdr-expressed cells. Wilcox rank-sum test examined the statistical significance of Etv2 expression level differences between P and PF and between PF and F, using normalized read counts.
Reference genome
We used mouse reference genome mm10 for all data analyses.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| PE-conjugated anti-PDGFRα antibody | ThermoFisher Scientific | Cat# 12–1401-81; RRID:AB_657615 |
| PE/Cy7-conjugated anti-Flk1 antibody | Biolegend | Cat# 359911; RRID:AB_2563551 |
| BV421-labeled anti-cTnT antibody | BD Bioscience | Cat# 565618; AB_2739306 |
| anti-cTnT antibody | Thermo Fisher Scientific | Cat#MS-295; RRID:AB_61810 |
| anti-myosin heavy chain class II | Abcam | Cat#ab55152; RRID:AB_944199 |
| anti-H3K27ac antibody | Wako | Cat#MABI0309; RRID:AB_11126964 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Penicillin-streptomycin | ThermoFisher Scientific | Cat#15140148 |
| Mono-thioglycerol | ThermoFisher Scientific | Cat#M6145 |
| N-2 supplement | ThermoFisher Scientific | Cat#17502048 |
| B27 supplement | ThermoFisher Scientific | Cat# 17504044 |
| L-Glutamine | ThermoFisher Scientific | Cat# A2916801 |
| Glutamax-I | ThermoFisher Scientific | Cat# 35050–061 |
| Paraformaldehyde Solution | ThermoFisher Scientific | Cat# J19943.K2 |
| Triton X-100 | ThermoFisher Scientific | Cat#A16046-AE |
| Neurobasal medium | ThermoFisher Scientific | Cat# 21103049 |
| DMEM/F12 medium | ThermoFisher Scientific | Cat# 10565018 |
| Iscove’s modified Dubelcco’s medium (IMDM) | ThermoFisher Scientific | Cat# 31980030 |
| Ham’s F12 medium | ThermoFisher Scientific | Cat# 31765035 |
| Stempro-34 SF medium | ThermoFisher Scientific | Cat# 10639011 |
| Propidium Iodide | ThermoFisher Scientific | Cat# P3566 |
| TrypLE Express | ThermoFisher Scientific | Cat# 12605010 |
| Fetal bovine serum, heat inactivated | ThermoFisher Scientific | Cat# 10438026 |
| Bovine serum albumin | MilliporeSigma | Cat# A9576 |
| Mouse Leukemia Inhibitory Factor | MilliporeSigma | Cat# ESG1106 |
| Human transferrin | MilliporeSigma | Cat#T8158 |
| L-Ascorbic acid | MilliporeSigma | Cat#S4544 |
| EmbryoMax 0.1% Gelatin Solution | MilliporeSigma | Cat#ES-006-B |
| CHIR99021 | Reprocell | Cat# 04–0004-10 |
| PD0325901 | Reprocell | Cat# 04–0006-10 |
| Human basic FGF | R&D systems | Cat#233FB |
| Human BMP4 | R&D systems | Cat#314BP |
| Human Activin A | R&D systems | Cat#338AC |
| Mouse VEGF | R&D systems | Cat#484MV |
| ZD6474 | SelleckChem | Cat#S1046 |
| Normocin | InvivoGen | Cat# ant-nr-1 |
|
| ||
| Critical commercial assays | ||
|
| ||
| MethoCult M3434 | StemCell Technologies | Cat#03434 |
| NucleoSpin RNAII kit | Takara | Cat#740955.50 |
| one-step SYBR Green PCR master mix | Bio-Rad Laboratories | Cat#1725150 |
| Illumina TruSeq RNA Sample prep kit v2 | Illumina | Cat #RS-122–2001 |
| Fixation/permeabilization kit | BD Biosciences | Cat# 554714 |
|
| ||
| Deposited data | ||
|
| ||
| RNA- and ATAC-seq data | This paper | GSE136692 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Mouse: ZX1 embryonic stem cell | Laboratory of Michael Kyba | N/A |
| Mouse: Bry-GFP embryonic stem cell | Laboratory of Gordon Keller | N/A |
| Mouse: Etv2-tdTomato embryonic stem cell | Laboratory of Kyunghee Choi | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| qPCR primer: Mus musculus Nkx2–5 F: 5’- ACATTTTACCCGGGAGCCTA-3’ |
This paper | N/A |
| qPCR primer: Mus musculus Nkx2–5 R: 5’- GGCTTTGTCCAGCTCCACT-3’ |
This paper | N/A |
| qPCR primer: Mus musculus Bra (T) F: 5’- CCGGTGCTGAAGGTAAATGT-3’ |
This paper | N/A |
| qPCR primer: Mus musculus Bra (T) R: 5’- CCCCGTTCACATATTTCCAG-3’ |
This paper | N/A |
| qPCR primer: Mus musculus Isl1 F: 5’- TCATCCGAGTGTGGTTTCAA-3’ |
This paper | N/A |
| qPCR primer: Mus musculus Isl1 R: 5’- TTCCTGTCATCCCCTGGATA-3’ |
This paper | N/A |
| qPCR primer: Mus musculus Etv2 F: 5’- GCCGGGAATGAATTATGAGA-3’ |
This paper | N/A |
| qPCR primer: Mus musculus Etv2 R: 5’- CCCGAAGCGGTATGTGTACT-3’ |
This paper | N/A |
| qPCR primer: Mus musculus Scl/Tal1 F: 5’- gaggtcctaaccagccgagt-3’ |
This paper | N/A |
| qPCR primer: Mus musculus Scl/Tal1 R: 5’- cgtcctgtccctctagttgc-3’ |
This paper | N/A |
| qPCR primer: Mus musculus Runx1 F: 5’- TTTTCGAAAGGAAACGATGG-3’ |
This paper | N/A |
| qPCR primer: Mus musculus Runx1 R: 5’- TGGCATCTCTCATGAAGCAC-3’ |
This paper | N/A |
| qPCR primer: Mus musculus Gapdh F: 5’- TGTGTCCGTCGTGGATCTGA-3’ |
This paper | N/A |
| qPCR primer: Mus musculus Gapdh R: 5’- GATGCCTGCTTCACCACCTT-3’ |
This paper | N/A |
| qPCR primer: Mus musculus Mesp1 F: 5’- GCTTCACACCTAGGGCTCAG-3’ |
This paper | N/A |
| qPCR primer: Mus musculus Mesp1 R: 5’- GACTCAGGATCCAGGACTCG-3’ |
This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Flowjo | Flowjo, LLC | N/A |
| TopHat2 v2.1.1 | Kim et al., 2013; Trapnell et al., 200960, 61 |
https://ccb.jhu.edu/software/tophat/index.shtml |
| StringTie | Perteaetal., 2015, 201662 | https://ccb.jhu.edu/software/stringtie/ |
| edgeR V3.16.5 | McCarthy et al., 2012; Robinson and Smyth, 2007, 2008; Robinson et al., 2010; Zhou et al., 201463–67 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| limma v3.30.13 | Ritchie etal.,201568 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| R V3.3.2 | R Core Team | https://www.r-project.org/ |
| Bowtie2 v2.3.0 | Langmead and Salzberg, 201269 | https://github.com/BenLangmead/bowtie2 |
| SAMtools v0.1.19 | Li et al., 2009; Li, 201170, 71 | https://github.com/samtools/ |
| MACS2 | Feng et al., 2012; Zhang et al., 200972, 73 |
https://pypi.org/project/MACS2/ |
| deepTools | Ramirez et al., 201674 | deeptools.readthedocs.io |
| BedTools | Quinlan and Hall, 201075 | bedtools.readthedocs.io |
| preprocessCore v1.36.0 | Bolstad et al., 200376 | bioconductor.org/packages/release/bioc/html/preprocessCore.html |
| TOBIAS | Bentsen et al., 202077 | https://github.com/loosolab/TOBIAS |
| Metascape | Tripathi et al., 2015; Zhou etal., 201978, 79 |
metascape.org |
| MEME suite v5.0.5 | Grant et al., 201180 | meme-suite.org |
| Seurat v4.0.0 | Haoetal., 2021; Stuart et al., 2019; Butler et al., 2018; Satijaetal., 201581–84 |
satijalab.org/seurat |
Highlights.
Accessible ETV2-binding sites emerge in multipotent progenitors prior to commitment
Almost no ETV2-binding sites are activated until hematoendothelial fate commitment
Second set of ETV2-binding sites are activated in committed hematoendothelial cells
ETV2 binding to binding-site activation drives hematoendothelial commitment
ACKNOWLEDGMENTS
We are thankful for technical assistance from the University of Chicago Functional Genomics, Cytometry and Antibody Technology, and Light Microscopy Cores and the Cincinnati Children’s Hospital Medical Center DNA Core. The graphical abstract was created with BioRender.com. This work was funded by NIH grants R01HL55337 to K.C.; R01DK119621 and P01HL146366 to B.L.B.; R21/R33AG054770 and R21HG012423 to K.I.; and R01HD111938, R01HL147571, and R01HL163523 to I.P.M. Support was also provided by NIH grants F30HL131298 to R.D.N.; F32HL156465 to J.D.S.; T32GM007183 to J.D.S. and E.H.; T32HL139430 to J.D.S.; T32HL007381 to J.D.S., C.K., R.D.N., and A.D.H.; and T32GM007197 to A.D.H. and American Heart Association 16POST30740016 for T.S.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112665.
REFERENCES
- 1.Ohno S (1978). Major sex-determining genes. Monogr. Endocrinol. 11, 1–140. 10.1007/978-3-642-81261-3. [DOI] [PubMed] [Google Scholar]
- 2.Waddington CH (2014). The Strategy of the Genes (Routledge; ). [Google Scholar]
- 3.Sulston JE, and Horvitz HR (1977). Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev. Biol. 56, 110–156. 10.1016/0012-1606(77)90158-0. [DOI] [PubMed] [Google Scholar]
- 4.Stergachis AB, Neph S, Reynolds A, Humbert R, Miller B, Paige SL, Vernot B, Cheng JB, Thurman RE, Sandstrom R, et al. (2013). Developmental fate and cellular maturity encoded in human regulatory DNA landscapes. Cell 154, 888–903. 10.1016/j.cell.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spitz F, and Furlong EEM (2012). Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet. 13, 613–626. 10.1038/nrg3207. [DOI] [PubMed] [Google Scholar]
- 6.Levine M, and Davidson EH (2005). Gene regulatory networks for development. Proc. Natl. Acad. Sci. USA 102, 4936–4942. 10.1073/pnas.0408031102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moris N, Pina C, and Arias AM (2016). Transition states and cell fate decisions in epigenetic landscapes. Nat. Rev. Genet. 17, 693–703. 10.1038/nrg.2016.98. [DOI] [PubMed] [Google Scholar]
- 8.Miroshnikova YA, Shahbazi MN, Negrete J, Chalut KJ, and Smith A (2023). Cell state transitions: catch them if you can. Development 150, dev201139. 10.1242/dev.201139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wagner DE, and Klein AM (2020). Lineage tracing meets single-cell omics: opportunities and challenges. Nat. Rev. Genet. 21, 410–427. 10.1038/s41576-020-0223-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buenrostro JD, Corces MR, Lareau CA, Wu B, Schep AN, Aryee MJ, Majeti R, Chang HY, and Greenleaf WJ (2018). Integrated single-cell analysis maps the continuous regulatory Landscape of human hematopoietic differentiation. Cell 173, 1535–1548.e16. 10.1016/j.cell.2018.03.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olsson A, Venkatasubramanian M, Chaudhri VK, Aronow BJ, Salomonis N, Singh H, and Grimes HL (2016). Single-cell analysis of mixed-lineage states leading to a binary cell fate choice. Nature 537, 698–702. 10.1038/nature19348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Velten L, Haas SF, Raffel S, Blaszkiewicz S, Islam S, Hennig BP,Hirche C, Lutz C, Buss EC, Nowak D, et al. (2017). Human haematopoietic stem cell lineage commitment is a continuous process. Nat. Cell Biol. 19, 271–281. 10.1038/ncb3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu M, Krause D, Greaves M, Sharkis S, Dexter M, Heyworth C, and Enver T (1997). Multilineage gene expression precedes commitment in the hemopoietic system. Genes Dev. 11, 774–785. 10.1101/gad.11.6.774. [DOI] [PubMed] [Google Scholar]
- 14.Brunskill EW, Park J-S, Chung E, Chen F, Magella B, and Potter SS (2014). Single cell dissection of early kidney development: multiline-age priming. Development 141, 3093–3101. 10.1242/dev.110601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyamoto T, Iwasaki H, Reizis B, Ye M, GraF T, Weissman IL, and Akashi K (2002). Myeloid or lymphoid promiscuity as a critical step in hematopoietic lineage commitment. Dev. Cell 3, 137–147. 10.1016/s1534-5807(02)00201-0. [DOI] [PubMed] [Google Scholar]
- 16.Kontaraki J, Chen HH, Riggs A, and Bonifer C (2000). Chromatin fine structure profiles for a developmentally regulated gene: reorganization of the lysozyme locus before trans-activator binding and gene expression. Genes Dev. 14, 2106–2122. 10.1101/gad.14.16.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi K, Kennedy M, Kazarov A, Papadimitriou JC, and Keller G(1998). A common precursor for hematopoietic and endothelial cells. Development 125, 725–732. 10.1242/dev.125.4.725. [DOI] [PubMed] [Google Scholar]
- 18.Ema M, Faloon P, Zhang WJ, Hirashima M, Reid T, Stanford WL, Orkin S, Choi K, and Rossant J (2003). Combinatorial effects of Flk1 and Tal1 on vascular and hematopoietic development in the mouse. Genes Dev. 17, 380–393. 10.1101/gad.1049803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kataoka H, Takakura N, Nishikawa S, Tsuchida K, Kodama H, Kunisada T, Risau W, Kita T, and Nishikawa S-I (1997). Expressions of PDGF receptor alpha, c-Kit and Flk1 genes clustering in mouse chromosome 5 define distinct subsets of nascent mesodermal cells. Dev. Growth Differ. 39, 729–740. 10.1046/j.1440-169x.1997.t01-5-00009.x. [DOI] [PubMed] [Google Scholar]
- 20.Park C, Afrikanova I, Chung YS, Zhang WJ, Arentson E, Fong Gh G.h., Rosendahl A, and Choi K (2004). A hierarchical order of factors in the generation of FLK1- and SCL-expressing hematopoietic and endothelial progenitors from embryonic stem cells. Development 131, 2749–2762. 10.1242/dev.01130. [DOI] [PubMed] [Google Scholar]
- 21.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, and Schuh AC (1995). Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 376, 62–66. 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 22.Shalaby F, Ho J, Stanford WL, Fischer KD, Schuh AC, Schwartz L, Bernstein A, and Rossant J (1997). A requirement for Flk1 in primitive and definitive hematopoiesis and vasculogenesis. Cell 89, 981–990. 10.1016/s0092-8674(00)80283-4. [DOI] [PubMed] [Google Scholar]
- 23.Yamashita J, Itoh H, Hirashima M, Ogawa M, Nishikawa S, Yurugi T, Naito M, Nakao K, and Nishikawa S (2000). Flk1-positive cells derived from embryonic stem cells serve as vascular progenitors. Nature 408, 92–96. 10.1038/35040568. [DOI] [PubMed] [Google Scholar]
- 24.Kattman SJ, Huber TL, and Keller GM (2006). Multipotent flk-1+ cardiovascular progenitor cells give rise to the cardiomyocyte, endothelial, and vascular smooth muscle lineages. Dev. Cell 11, 723–732. 10.1016/j.devcel.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 25.Kattman SJ, Witty AD, Gagliardi M, Dubois NC, Niapour M, Hotta A, Ellis J, and Keller G (2011). Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell 8, 228–240. 10.1016/j.stem.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 26.Moretti A, Caron L, Nakano A, Lam JT, Bernshausen A, Chen Y, Qyang Y, Bu L, Sasaki M, Martin-Puig S, et al. (2006). Multipotent embryonic isl1+ progenitor cells lead to cardiac, smooth muscle, and endothelial cell diversification. Cell 127, 1151–1165. 10.1016/j.cell.2006.10.029. [DOI] [PubMed] [Google Scholar]
- 27.Motoike T, Markham DW, Rossant J, and Sato TN (2003). Evidence for novel fate of Flk1+ progenitor: contribution to muscle lineage. Genesis 35, 153–159. 10.1002/gene.10175. [DOI] [PubMed] [Google Scholar]
- 28.Yang L, Soonpaa MH, Adler ED, Roepke TK, Kattman SJ, Kennedy M, Henckaerts E, Bonham K, Abbott GW, Linden RM, et al. (2008). Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature 453, 524–528. 10.1038/nature06894. [DOI] [PubMed] [Google Scholar]
- 29.Kataoka H, Hayashi M, Nakagawa R, Tanaka Y, Izumi N, Nishikawa S, Jakt ML, Tarui H, and Nishikawa S-I (2011). Etv2/ER71 induces vascular mesoderm from Flk1+PDGFRα+ primitive mesoderm. Blood 118, 6975–6986. 10.1182/blood-2011-05-352658. [DOI] [PubMed] [Google Scholar]
- 30.Rasmussen TL, Shi X, Wallis A, Kweon J, Zirbes KM, Koyano-Nakagawa N, and Garry DJ (2012). VEGF/Flk1 signaling cascade transactivates Etv2 gene expression. PLoS One 7, e50103. 10.1371/journal.pone.0050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao H, and Choi K (2017). A CRISPR screen identifies genes controlling Etv2 threshold expression in murine hemangiogenic fate commitment. Nat. Commun. 8, 541. 10.1038/s41467-017-00667-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee D, Park C, Lee H, Lugus JJ, Kim SH, Arentson E, Chung YS, Gomez G, Kyba M, Lin S, et al. (2008). ER71 acts downstream of BMP, Notch, and Wnt signaling in blood and vessel progenitor specification. Cell Stem Cell 2, 497–507. 10.1016/j.stem.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wareing S, Mazan A, Pearson S, Göttgens B, Lacaud G, and Kouskoff V (2012). The Flk1-Cre-mediated deletion of ETV2 defines its narrow temporal requirement during embryonic hematopoietic development. Stem Cell. 30, 1521–1531. 10.1002/stem.1115. [DOI] [PubMed] [Google Scholar]
- 34.Elcheva I, Brok-Volchanskaya V, Kumar A, Liu P, Lee J-H, Tong L, Vodyanik M, Swanson S, Stewart R, Kyba M, et al. (2014). Direct induction of haematoendothelial programs in human pluripotent stem cells by transcriptional regulators. Nat. Commun. 5, 4372. 10.1038/ncomms5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Val S, Chi NC, Meadows SM, Minovitsky S, Anderson JP, Harris IS, Ehlers ML, Agarwal P, Visel A, Xu S-M, et al. (2008). Combinatorial regulation of endothelial gene expression by ets and forkhead transcription factors. Cell 135, 1053–1064. 10.1016/j.cell.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferdous A, Caprioli A, Iacovino M, Martin CM, Morris J, Richardson JA, Latif S, Hammer RE, Harvey RP, Olson EN, et al. (2009). Nkx2–5 transactivates the Ets-related protein 71 gene and specifies an endothelial/endocardial fate in the developing embryo. Proc. Natl. Acad. Sci. USA 106, 814–819. 10.1073/pnas.0807583106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morita R, Suzuki M, Kasahara H, Shimizu N, Shichita T, Sekiya T, Kimura A, Sasaki K-I, Yasukawa H, and Yoshimura A (2015). ETS transcription factor ETV2 directly converts human fibroblasts into functional endothelial cells. Proc. Natl. Acad. Sci. USA 112, 160–165. 10.1073/pnas.1413234112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Veldman MB, Zhao C, Gomez GA, Lindgren AG, Huang H, Yang H, Yao S, Martin BL, Kimelman D, and Lin S (2013). Transdifferentiation of fast skeletal muscle into functional endothelium in vivo by transcription factor Etv2. PLoS Biol. 11, e1001590. 10.1371/journal.pbio.1001590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Val S, and Black BL (2009). Transcriptional control of endothelial cell development. Dev. Cell 16, 180–195. 10.1016/j.devcel.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koyano-Nakagawa N, and Garry DJ (2017). Etv2 as an essential regulator of mesodermal lineage development. Cardiovasc. Res. 113, 1294–1306. 10.1093/cvr/cvx133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu F, Li D, Yu YYL, Kang I, Cha M-J, Kim JY, Park C, Watson DK, Wang T, and Choi K (2015). Induction of hematopoietic and endothelial cell program orchestrated by ETS transcription factor ER71/ETV2. EMBO Rep. 16, 654–669. 10.15252/embr.201439939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Palencia-Desai S, Kohli V, Kang J, Chi NC, Black BL, and Sumanas S (2011). Vascular endothelial and endocardial progenitors differentiate as cardiomyocytes in the absence of Etsrp/Etv2 function. Development 138, 4721–4732. 10.1242/dev.064998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rasmussen TL, Kweon J, Diekmann MA, Belema-Bedada F, Song Q, Bowlin K, Shi X, Ferdous A, Li T, Kyba M, et al. (2011). ER71 directs mesodermal fate decisions during embryogenesis. Development 138, 4801–4812. 10.1242/dev.070912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stennard FA, and Harvey RP (2005). T-box transcription factors and their roles in regulatory hierarchies in the developing heart. Development 132, 4897–4910. 10.1242/dev.02099. [DOI] [PubMed] [Google Scholar]
- 45.Tremblay M, Sanchez-Ferras O, and Bouchard M (2018). GATA transcription factors in development and disease. Development 145, dev164384. 10.1242/dev.164384. [DOI] [PubMed] [Google Scholar]
- 46.Zhao H, and Choi K (2019). Single cell transcriptome dynamics from pluripotency to FLK1 mesoderm. Development 146, dev182097. 10.1242/dev.182097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zaret K (1997). Identifying specific protein-DNA interactions within living cells, or in “in vivo footprinting. Methods 11, 149–150. 10.1006/meth.1996.0400. [DOI] [PubMed] [Google Scholar]
- 48.Neph S, Vierstra J, Stergachis AB, Reynolds AP, Haugen E, Vernot B, Thurman RE, John S, Sandstrom R, Johnson AK, et al. (2012). An expansive human regulatory lexicon encoded in transcription factor footprints. Nature 489, 83–90. 10.1038/nature11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lilly AJ, Mazan A, Scott DA, Lacaud G, and Kouskoff V (2017). SOX7 expression is critically required in FLK1-expressing cells for vasculogenesis and angiogenesis during mouse embryonic development. Mech. Dev. 146, 31–41. 10.1016/j.mod.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Behrens AN, Zierold C, Shi X, Ren Y, Koyano-Nakagawa N, Garry DJ, and Martin CM (2014). Sox7 is regulated by ETV2 during cardiovascular development. Stem Cells Dev. 23, 2004–2013. 10.1089/scd.2013.0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sinha T, Lammerts van Bueren K, Dickel DE, Zlatanova I, Thomas R, Lizama CO, Xu S-M, Zovein AC, Ikegami K, Moskowitz IP, et al. (2022). Differential Etv2 threshold requirement for endothelial and erythropoietic development. Cell Rep. 39, 110881. 10.1016/j.celrep.2022.110881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lammerts van Bueren K, and Black BL (2012). Regulation of endothelial and hematopoietic development by the ETS transcription factor Etv2. Curr. Opin. Hematol 19, 199–205. 10.1097/MOH.0b013e3283523e07. [DOI] [PubMed] [Google Scholar]
- 53.Zhao H, Xu C, Lee T-J, Liu F, and Choi K (2017). ETS transcription factor ETV2/ER71/Etsrp in hematopoietic and vascular development, injury, and regeneration. Dev. Dyn. 246, 318–327. 10.1002/dvdy.24483. [DOI] [PubMed] [Google Scholar]
- 54.Gong W, Das S, Sierra-Pagan JE, Skie E, Dsouza N, Larson TA, Garry MG, Luzete-Monteiro E, Zaret KS, and Garry DJ (2022). ETV2 functions as a pioneer factor to regulate and reprogram the endothelial lineage. Nat. Cell Biol. 24, 672–684. 10.1038/s41556-022-00901-3. [DOI] [PubMed] [Google Scholar]
- 55.Koyano-Nakagawa N, Shi X, Rasmussen TL, Das S, Walter CA, and Garry DJ (2015). Feedback mechanisms regulate ets variant 2 (Etv2) gene expression and hematoendothelial lineages. J. Biol. Chem. 290, 28107–28119. 10.1074/jbc.M115.662197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paige SL, Thomas S, Stoick-Cooper CL, Wang H, Maves L, Sandstrom R, Pabon L, Reinecke H, Pratt G, Keller G, et al. (2012). A temporal chromatin signature in human embryonic stem cells identifies regulators of cardiac development. Cell 151, 221–232. 10.1016/j.cell.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bendall SC, Davis KL, Amir E-AD, Tadmor MD, Simonds EF, Chen TJ, Shenfeld DK, Nolan GP, and Pe’er D (2014). Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell 157, 714–725. 10.1016/j.cell.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386. 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ma S, Zhang B, LaFave LM, Earl AS, Chiang Z, Hu Y, Ding J, Brack A, Kartha VK, Tay T, et al. (2020). Chromatin potential identified by shared single-cell profiling of RNA and chromatin. Cell 183, 1103–1116.e20. 10.1016/j.cell.2020.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pertea M, Kim D, Pertea GM, Leek JT, and Salzberg SL (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667. 10.1038/nprot.2016.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McCarthy DJ, Chen Y, and Smyth GK (2012). Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297. 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Robinson MD, and Smyth GK (2007). Moderated statistical tests for assessing differences in tag abundance. Bioinformatics 23, 2881–2887. 10.1093/bioinformatics/btm453. [DOI] [PubMed] [Google Scholar]
- 65.Robinson MD, and Smyth GK (2008). Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics 9, 321–332. 10.1093/biostatistics/kxm030. [DOI] [PubMed] [Google Scholar]
- 66.Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhou X, Lindsay H, and Robinson MD (2014). Robustly detecting differential expression in RNA sequencing data using observation weights. Nucleic Acids Res. 42, e91. 10.1093/nar/gku310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li H (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Feng J, Liu T, Qin B, Zhang Y, and Liu XS (2012). Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 7, 1728–1740. 10.1038/nprot.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-seq (MACS). Genome Biol. 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165. 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bolstad BM, Irizarry RA, Astrand M, and Speed TP (2003). A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19, 185–193. 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 77.Bentsen M, Goymann P, Schultheis H, Klee K, Petrova A, Wiegandt R, Fust A, Preussner J, Kuenne C, Braun T, et al. (2020). ATAC-seq footprinting unravels kinetics of transcription factor binding during zygotic genome activation. Nat. Commun. 11, 4267. 10.1038/s41467-020-18035-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tripathi S, Pohl MO, Zhou Y, Rodriguez-Frandsen A, Wang G, Stein DA, Moulton HM, DeJesus P, Che J, Mulder LCF, et al. (2015). Meta- and orthogonal integration of influenza “OMICs” data defines a role for UBR4 in virus budding. Cell Host Microbe 18, 723–735. 10.1016/j.chom.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, and Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523. 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Grant CE, Bailey TL, and Noble WS (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495–502. 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420. 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Iacovino M, Bosnakovski D, Fey H, Rux D, Bajwa G, Mahen E, Mitanoska A, Xu Z, and Kyba M (2011). Inducible cassette exchange: a rapid and efficient system enabling conditional gene expression in embryonic stem and primary cells. Stem Cell. 29, 1580–1588. 10.1002/stem.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fehling HJ, Lacaud G, Kubo A, Kennedy M, Robertson S, Keller G, and Kouskoff V (2003). Tracking mesoderm induction and its specification to the hemangioblast during embryonic stem cell differentiation. Development 130, 4217–4227. 10.1242/dev.00589. [DOI] [PubMed] [Google Scholar]
- 87.Ying Q-L, Nichols J, Chambers I, and Smith A (2003). BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell 115, 281–292. 10.1016/s0092-8674(03)00847-x. [DOI] [PubMed] [Google Scholar]
- 88.Ying Q-L, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, Cohen P, and Smith A (2008). The ground state of embryonic stem cell self-renewal. Nature 453, 519–523. 10.1038/nature06968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gadue P, Huber TL, Paddison PJ, and Keller GM (2006). Wnt and TGF-β signaling are required for the induction of an in vitro model of primitive streak formation using embryonic stem cells. Proc. Natl. Acad. Sci. USA 103, 16806–16811. 10.1073/pnas.0603916103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Barnett DW, Garrison EK, Quinlan AR, Strömberg MP, and Marth GT (2011). BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 27, 1691–1692. 10.1093/bioinformatics/btr174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, and Salzberg SL (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, Chen X, Taipale J, Hughes TR, and Weirauch MT (2018). The human transcription factors. Cell 175, 598–599. 10.1016/j.cell.2018.09.045. [DOI] [PubMed] [Google Scholar]
- 93.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Amemiya HM, Kundaje A, and Boyle AP (2019). The ENCODE blacklist: identification of problematic regions of the genome. Sci. Rep. 9, 9354. 10.1038/s41598-019-45839-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, et al. (2014). Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443. 10.1016/j.cell.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq and ATAC-seq data are available at GEO with an accession number GSE136692.
This paper does not report original code.
All data reported in this paper will be shared by the lead contact upon request. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.