

Abstract
Background & Aims:
Lipid accumulation induced by alcohol consumption is not only an early pathophysiological response but also a prerequisite for the progression of alcohol-associated liver disease (ALD). Alternative splicing regulates gene expression and protein diversity; dysregulation of this process is implicated in human liver diseases. However, how the alternative splicing regulation of lipid metabolism contributes to the pathogenesis of ALD remains undefined.
Approach & Results:
Serine-arginine-rich protein kinase 2 (SRPK2), a key kinase controlling alternative splicing, is activated in hepatocytes in response to alcohol, in mice with chronic-plus-binge alcohol feeding, and in patients with ALD. Such induction activates sterol regulatory element binding protein 1 (SREBP-1) and promotes lipogenesis in ALD. Overexpression of fibroblast growth factor 21 (FGF21) in transgenic mice abolishes alcohol-mediated induction of SRPK2 and its associated steatosis, lipotoxicity, and inflammation; these alcohol-induced pathologies are exacerbated in FGF21 knockout mice. Mechanistically, SRPK2 is required for alcohol-mediated impairment of serine-arginine splicing factor 10 (SRSF10), which generates exon 7 inclusion in lipin 1, and triggers concurrent induction of lipogenic regulators—lipin 1β and SREBP-1. FGF21 suppresses alcohol-induced SRPK2 accumulation through mTORC1 inhibition-dependent degradation of SRPK2. Silencing SRPK2 rescues alcohol-induced splicing dysregulation and liver injury in FGF21 knockout mice.
Conclusions:
These studies reveal that 1) The regulation of alternative splicing by SRPK2 is implicated in lipogenesis in humans with ALD; 2) FGF21 is a key hepatokine that ameliorates ALD pathologies largely by inhibiting SRPK2; and 3) Targeting SRPK2 signaling by FGF21 may offer potential therapeutic approaches to combat ALD.
Keywords: alcoholic liver injury, alternative splicing, fibroblast growth factor 21, serine-arginine rich splicing factor 10, sterol regulatory-element binding protein-1, Lipin 1, lipogenesis
Graphical Abstract
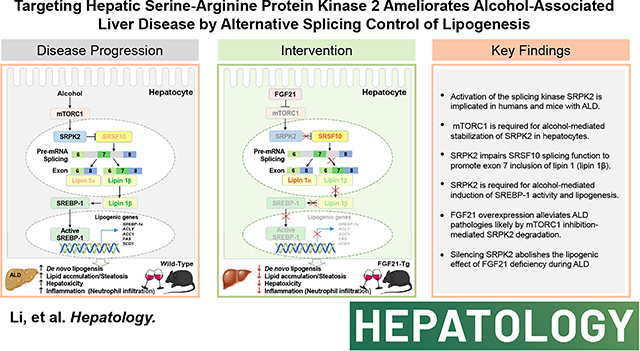
Introduction
Alcohol-associated liver disease (ALD) encompasses a spectrum of liver abnormalities that range from hepatic steatosis (lipid accumulation) to steatohepatitis (hepatocyte death and inflammation), fibrosis, cirrhosis, and hepatocellular carcinoma(1, 2). Among patients with ALD, 90% are found to have hepatic steatosis, and 35% are found to have alcoholic hepatitis. To date, there are no US Food and Drug Administration (FDA)-approved drugs for ALD(1, 2). A better understanding of the pathogenic mechanism of ALD is urgently needed to develop new therapies(1, 2). De novo lipogenesis, which provides newly synthesized fatty acids in hepatocytes, is a critical metabolic feature in fatty liver disease (3–5). De novo lipogenesis is controlled by several core lipogenic enzymes, including ATP citrate lyase (ACLY), acetyl-CoA carboxylase (ACC), and fatty acid synthase (FAS). Inhibition of fatty acid synthesis has been postulated to offer a therapeutic opportunity for fatty liver disease since ACLY, ACC1, and FAS are all induced in humans with this disease(3, 5). However, despite decades of pharmaceutical efforts to design orally bioavailable small molecule inhibitors for lipogenic enzymes, such as ACC and FAS inhibitors, these targets remain intractable due to the lack of potent and specific compounds with desirable drug-like properties(5). A proposed alternative approach is to suppress the transcription regulators, such as sterol regulatory element-binding protein 1 (SREBP-1), which controls its target genes governing the lipogenic process(4, 6). Our studies demonstrate that the mammalian target of rapamycin complex 1 (mTORC1) promotes SREBP-1-dependent lipogenesis in mice and humans with ALD(4). However, whether alcohol-induced lipogenesis is regulated by alternative splicing is poorly understood.
Post-transcriptional mechanisms that fine-tune mRNAs and generate proteomic diversity include alternative pre-mRNA splicing, capping, polyadenylation, methylation, nuclear export, and mRNA stability(7–9). Alternative splicing is regulated by serine-arginine rich protein kinases (SRPKs), RNA-binding proteins including serine-arginine rich (SR) proteins, and heterogeneous nuclear ribonucleoproteins (hnRNPs). The serine-arginine rich splicing factors (SRSFs) contain RNA recognition motifs and arginine-serine repeat domains, which regulate mRNA processing by binding to exons in pre-mRNAs and recruiting other SR proteins(7). Importantly, SRPK2 plays an important role in regulating alternative splicing by phosphorylating SRSFs and modulating their splicing activity, which leads to their conformation changes, subcellular localization, and interactions with other regulators(7–9). While SRPK2 has been implicated in cancer cell processes (8), the effect of SRPK2 on alternative splicing events associated with ALD remains unknown.
Fibroblast growth factor 21 (FGF21) is a fasting-induced hormone that is secreted predominantly by hepatocytes (10, 11). Unlike the classical members of the FGF family, the hepatokine FGF21 lacks heparin-binding properties and can be released into circulation(10). We have demonstrated that FGF21 exerts effects on fatty acid oxidation in fasted mice and systemic energy expenditure in mice(11). In Phase 2 clinical trials, FGF21 administration improves metabolic status and liver pathologies in patients with diabetes and NASH (12, 13). Given its anti-diabetic effects (11, 13), we sought to investigate the role of FGF21 in the pathogenesis of ALD. Whether alternative splicing mechanisms are involved in the metabolic action of FGF21 remains undefined.
Here we characterize activation of SRPK2 as a novel lipogenic regulator in hepatocytes and as a pathologic feature in patients with alcoholic steatohepatitis. Our findings demonstrate that: 1) SRPK2 triggers alcohol-induced pre-mRNA splicing of lipin 1 by binding to the RNA-binding protein SRSF10 and promoting the inclusion of exon 7 in lipin 1β, leading to lipin 1β production, SREBP-1 activation, and triglyceride overproduction; 2) FGF21 blocks alcohol-induced activation of mTORC1 and promotes the degradation of SRPK2; 3) Overexpression of FGF21 in transgenic mice ameliorates alcohol-induced steatosis, lipotoxicity, and inflammation, whereas deletion of FGF21 in mice aggravates the severity of ALD; and 4) Silencing SRPK2 rescues persistent pathological phenotypes in alcohol-fed FGF21 knockout mice. Therefore, FGF21 serves as a hepatokine that delays and alleviates ALD by inhibiting SRPK2.
Results
Hepatic SRPK2 induction is linked to SREBP-1-driven lipogenesis in a pre-clinical mouse model and in humans with ALD—
SRPK2 phosphorylates and regulates activity of SR proteins, which are essential components of alternative splicing (9). As shown in Fig. 1A–C and Fig. S1A–B, chronic-binge alcohol feeding caused an approximately 3-fold increase in SRPK2 abundance over the basal level of pair-fed mice, without affecting its mRNA levels. Similarly, phosphorylation of SR proteins, such as pSRp55 and pSRp20, was increased by ~2-fold in alcohol-fed mice. As shown in Fig. 1D–K and Fig. S1C–D, chronic-binge alcohol feeding led to excessive fat accumulation within hepatocellular vacuoles, as assessed by hematoxylin and eosin (H&E) staining. Additionally, hepatic triglyceride levels were elevated by ~6-fold, and this increase was positively correlated with induction of SRPK2. Furthermore, plasma levels of triglyceride and cholesterol were significantly increased by ~2-fold and 1.5-fold, respectively. Therefore, a functional link between uncontrolled activation of SRPK2 and lipid accumulation may be a key determinant of early-stage ALD. Our studies have demonstrated that activation of sterol regulatory element-binding protein-1 (SREBP-1), a master lipogenic transcription factor, plays a critical role in promoting steatosis in both mice and humans with ALD (4). To decipher the potential mechanism mediating the role of SRPK2 in alcoholic steatosis, we initially found that proteolytic cleavage of SREBP-1, as assessed by the active, nuclear form of SREBP-1, was increased ~2-fold in alcohol-fed mice. Immunohistochemical staining showed elevated immunoreactivity for rate-limiting enzymes involved in fatty acid synthesis, such as ACLY, FAS, and SCD1, in lipid-laden hepatocytes of perivascular areas in alcohol-fed mice. Likewise, immunoblots also showed protein abundance of FAS and SCD1 was elevated by nearly 2- and 4-fold, respectively. The elevation of nuclear SREBP-1, FAS, and SCD1 was positively correlated with SRPK2 activation. In support of these findings, mRNA levels of SREBP-1 target lipogenic genes (4), including SREBP-1c, ACLY, ACC1, FAS, elongation of long-chain fatty acids family member 6 (ELOVL6), SCD1, and acyl-CoA: diacylglycerol acyltransferase 2 (DGAT2), were elevated by more than 2-fold in alcohol-fed mice. Taken together, alcohol consumption induces SRPK2 activity and stimulates SREBP-1-dependent lipogenesis, contributing to the accumulation of newly synthesized lipids in hepatocytes.
Fig. 1. Chronic alcohol consumption increases hepatic SRPK2 levels and activity, stimulates SREBP-1 cleavage, and upregulates transcription of SREBP-1-dependent lipogenic genes in a pre-clinical mouse model and in human liver samples.
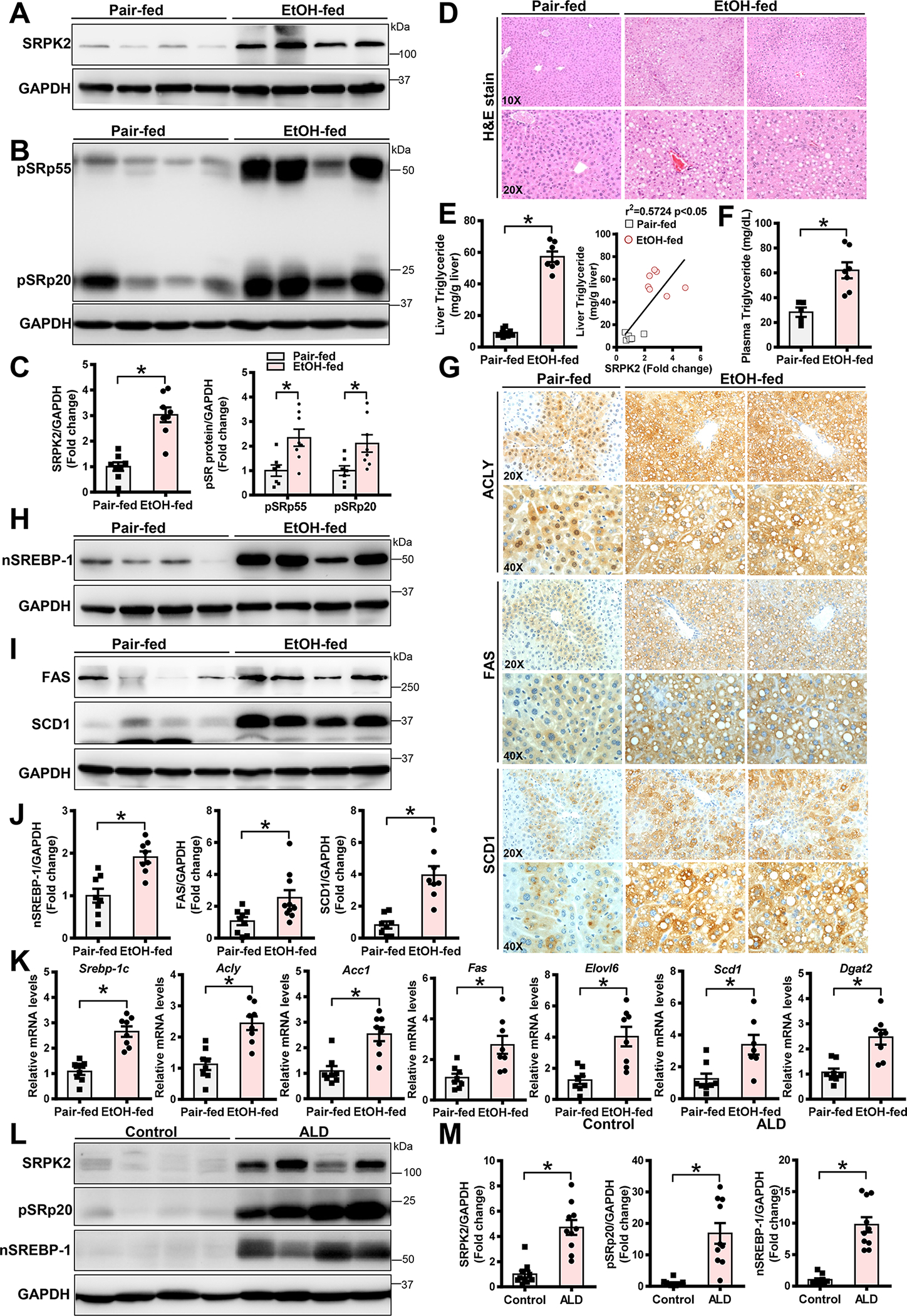
A-C. Representative immunoblots for SRPK2 levels (A) and kinase activity (B), as assessed by phosphorylation of its downstream targets, including SR proteins (pSRp55) and (pSRp20), in livers from pair-fed mice and chronic-binge alcohol-fed mice. Levels of SRPK2 and SR protein phosphorylation (C) were quantified by densitometry, normalized to those of GAPDH, and presented as the fold change relative to the control.
D. Representative Hematoxylin & Eosin (H&E) staining of liver sections in mice.
E-F. Measurements of liver and plasma triglyceride concentrations. Liver lipids were extracted, and triglyceride concentrations were measured and expressed as mg of lipid/g of liver tissue. Hepatic triglyceride levels were positively correlated with SRPK2 levels in pair-fed and chronic-binge alcohol-fed mice.
G. Representative immunohistochemistry staining of liver sections for lipogenic enzymes including ACLY, FAS, and SCD1. Notably, positive staining for ACLY was much higher in the cytoplasm and nucleus of lipid-rich hepatocytes of chronic-binge alcohol-fed mice. Positive staining for FAS and SCD1 was primarily located in the cytoplasm of lipid-rich hepatocytes in chronic-binge alcohol-fed mice.
H-J. Representative immunoblots for nuclear SREBP-1 (nSREBP-1), FAS, and SCD1. The active, nuclear form of SREBP-1 and expression of its target genes, FAS and SCD1, were increased in chronic-binge alcohol-fed mice.
K. Real-time qRT-PCR analysis of mRNA levels of lipogenic genes, including SREBP-1c, ACLY, ACC1, FAS, ELOVL6, SCD1, and DGAT2.
The data are presented as the mean ± S.E.M., n=6–8 per group. *P<0.05, vs pair-fed mice.
All images were acquired using 10X, 20X, or 40X objectives.
L-M. Representative immunoblots for SRPK2, pSRp20, and nuclear SREBP-1 in livers of healthy human controls and in patients with ALD.
The data are presented as the mean ± S.E.M., n=10 per group. *P<0.05, vs. healthy human controls.
To investigate the clinical relevance of SRPK2 to the pathology of human ALD, we found that SRPK2 protein levels and pSRp20 were upregulated by over 5-fold in patients with ALD when compared with normal liver controls (Fig. 1L–M). Nuclear SREBP-1 was increased by ~10-fold, which was positively correlated with hyperactivation of SRPK2 in patients with ALD (Fig. 1M and Fig. S1E), consistent with mice fed a chronic-binge alcohol diet. Together, these results highlight the pathological impact of SRPK2 on human ALD.
Ethanol exposure induces SRPK2 activity and promotes lipid accumulation in cultured hepatocytes—
As shown in Fig. 2A and 2F, protein levels of SRPK2 were elevated 1.5-fold by ethanol exposure at 50 mM and up to ~2-fold at a higher concentration (100 mM) in primary mouse hepatocytes. Ethanol exposure dose-dependently increased pSRp20 in hepatocytes or AML12 cells, consistent with SRPK2 activation in mice and humans with ALD (Fig. 1). However, alcohol did not significantly alter mRNA levels of SRPK2 in primary mouse hepatocytes and AML 12 cells compared with controls (Fig. S2A–B). Since SRPK2 has been shown to translocate to the nucleus and phosphorylate its downstream effectors (SR proteins) (8), we determined whether ethanol exposure might regulate the subcellular distribution of SRPK2. Immunofluorescence showed ethanol exposure enhanced SRPK2 staining intensity and led to its translocation from the cytoplasm into the nucleus of hepatocytes, particularly in nuclear speckles (Fig. 2B). Consequently, exposing hepatocytes to ethanol increased expression of SREBP-1c, FAS, and SCD1 and elevated intracellular triglyceride levels in a dose-dependent manner (Fig. 2C–G). Taken together, ethanol not only increases SRPK2 abundance but also triggers its nuclear translocation and stimulates SR protein phosphorylation, thereby promoting newly synthesized triglyceride production in hepatocytes.
Fig. 2. Ethanol-induced lipogenesis and triglyceride overproduction are mimicked by SRPK2 overexpression and diminished by SRPK2 knockdown in cultured hepatocytes.
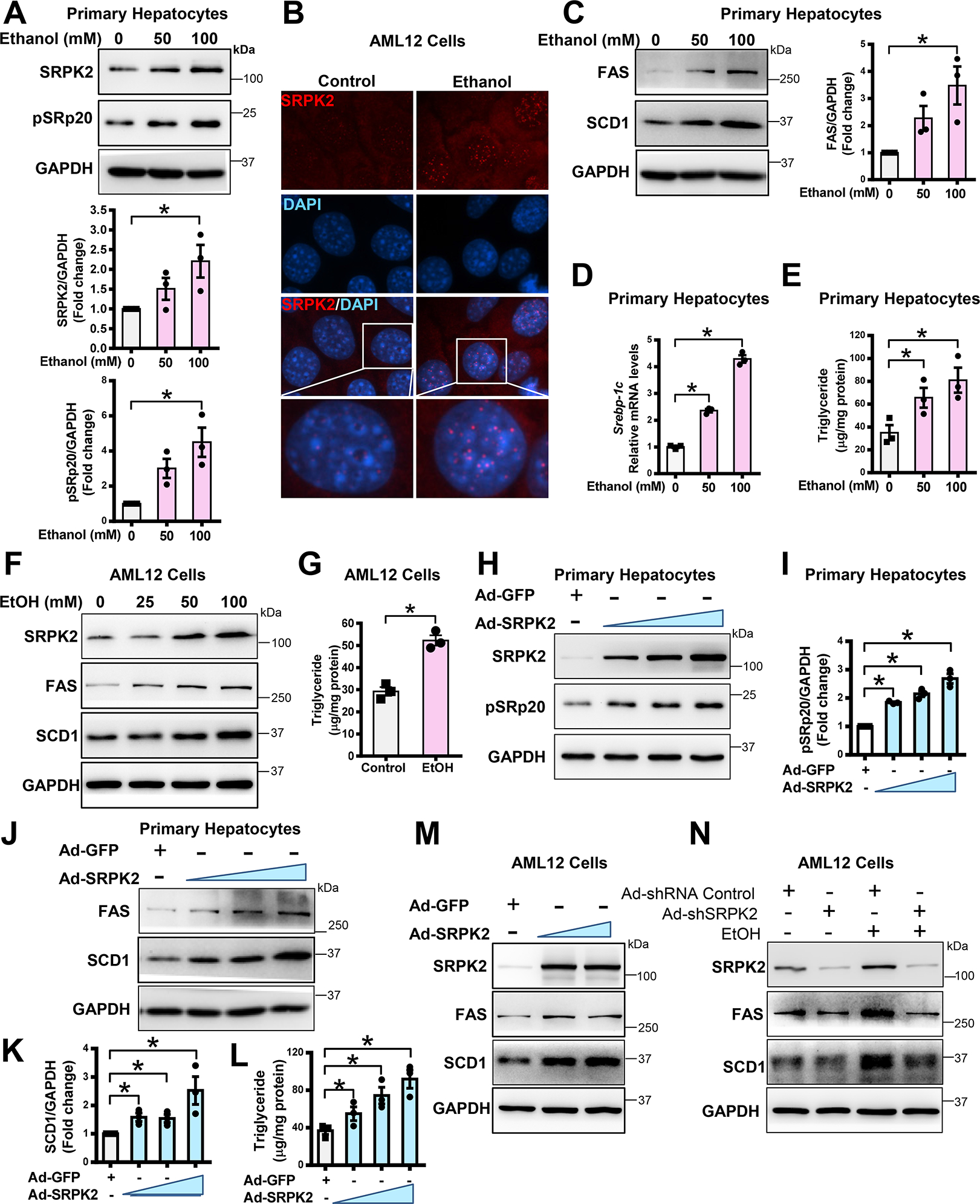
A. Immunoblotting analysis and densitometric quantification of SRPK2 and pSRp20 in primary mouse hepatocytes exposed to ethanol. Primary mouse hepatocytes were incubated with increasing doses of ethanol (50 – 100 mM) for 24 h.
B. Immunofluorescent staining of SRPK2 (red) and nuclear staining with DAPI (blue) in AML12 mouse hepatocytes exposed to ethanol (100 mM) for 24 h.
C. Immunoblotting analysis and densitometric quantification of lipogenic enzymes including FAS and SCD1 in primary mouse hepatocytes exposed to various doses of ethanol for 24 h.
D-E. mRNA levels of SREBP-1c and intracellular triglyceride levels were dose-dependently increased by ethanol exposure in primary mouse hepatocytes.
F. Immunoblotting analysis of SRPK2, FAS, and SCD1 in AML12 hepatocytes exposed to increasing doses of ethanol (25 – 100 mM) for 24 h.
G. Triglyceride concentrations were increased by exposure of AML12 hepatocytes to ethanol (100 mM) for 24 h.
H-L. Overexpression of SRPK2 is sufficient to increase expression of lipogenic enzymes and elevate lipid content in primary mouse hepatocytes. Primary mouse hepatocytes were transduced with an adenovirus expressing either GFP (Ad-GFP) at a multiplicity of infection (MOI) of 5 or an adenovirus encoding SRPK2 (Ad-SRPK2) at different MOIs (1–5). Immunoblotting analysis and densitometric quantification of SRPK2, pSRp20, FAS, or SCD1 were performed.
M. Overexpression of SRPK2 is sufficient to increase expression of lipogenic enzymes in AML12 hepatocytes. AML12 cells were transduced with Ad-GFP at an MOI of 5 or Ad-SRPK2 at different MOIs of 1–5.
N. Adenovirus-mediated knockdown of SRPK2 abolishes the ability of ethanol to promote lipogenic enzyme expression in AML12 cells. AML12 cells were transduced for 24 h with adenoviruses encoding either short hairpin RNA targeting the SRPK2 gene (Ad-shSRPK2) at an MOI of 10 or shRNA control (Ad-shRNA Control) at an MOI of 10, followed by incubation with ethanol (100 mM) for an additional 24 h.
The data are presented as the mean ± S.E.M., n=3, *P<0.05 between two groups.
All images were acquired using 40X objectives.
SRPK2 is essential for ethanol-induced expression of lipogenic enzymes and lipid accumulation in cultured hepatocytes—
As shown in Fig. 2H–M, adenoviral overexpression of SRPK2 was confirmed by increases in SRPK2 and pSRp20 levels. Ectopic expression of SRPK2 was sufficient to stimulate key lipogenic enzyme expression and elevate lipid content in cultured hepatocytes, mimicking the lipogenic effect of alcohol. To further illustrate the role of SRPK2 in lipogenesis, we observed that ethanol-induced increase in FAS and SCD1 expression was abolished by SRPK2 knockdown (Fig. 2N), suggesting that alcohol-induced lipogenesis depends on SRPK2 in hepatocytes.
Knockdown of SRPK2 ameliorates chronic-binge alcohol-induced hepatic fatty acid synthesis and steatosis in mice—
Given that SRPK2 activation acts as a determinant of lipid accumulation in humans and mice with ALD (Fig. 1), we assessed the relative contribution of endogenous SRPK2 to alcoholic fatty liver in vivo. Protein levels of SRPK2 and phosphorylation of SR protein were reduced by ~80% in mice that received adenoviral gene transfer of Ad-shSRPK2 (Fig. 3A). After chronic-binge alcohol feeding, mice with SRPK2 knockdown (SRPK2 KD) exhibited reduced liver injury and lower levels of hepatic and plasma triglycerides and cholesterol (Fig. 3B–D and Fig. S3A–B). Therefore, inhibition of SRPK2 can ameliorate steatosis and hyperlipidemia caused by alcohol feeding. Next, we sought to delineate the mechanism underlying the protective effect of SRPK2 inhibition on alcoholic steatosis. SREBP-1c has been indicated to be transcriptionally upregulated by nuclear SREBP-1 via a feed-forward mechanism, as nuclear SREBP-1 directly binds to the sterol regulatory element motif present on the promoters of the SREBP-1c gene (6). As illustrated in Fig. 3E–H and Fig. S3C, the accumulation of nuclear SREBP-1 caused by alcohol was nearly abolished in SRPK2 KD mice, and mRNA levels of SREBP-1c were suppressed by ~50%, suggesting that the autoregulation of SREBP-1c transcription is blocked. Notably, the transcription of SREBP-1c target lipogenic genes, including ACLY, ACC1, FAS, ELOVL6, SCD1, and DGAT2, was significantly downregulated in SRPK2 KD livers. Accordingly, protein levels of lipogenic enzymes, FAS and SCD1, were reduced by over 60% upon SRPK2 knockdown. Positively stained hepatocytes for ACLY, FAS, and SCD1 were predominantly present in lipid droplet-rich hepatocytes of alcohol-fed mice, but this positive staining was eliminated in SRPK2 KD livers. Our data indicate that SRPK2 inhibition suppresses SREBP-1-driven lipogenesis and ameliorates alcoholic steatosis.
Fig. 3. Adenovirus-mediated knockdown of hepatic SRPK2 ameliorates chronic-binge alcohol feeding-induced fatty acid synthesis, steatosis, and liver injury in mice.
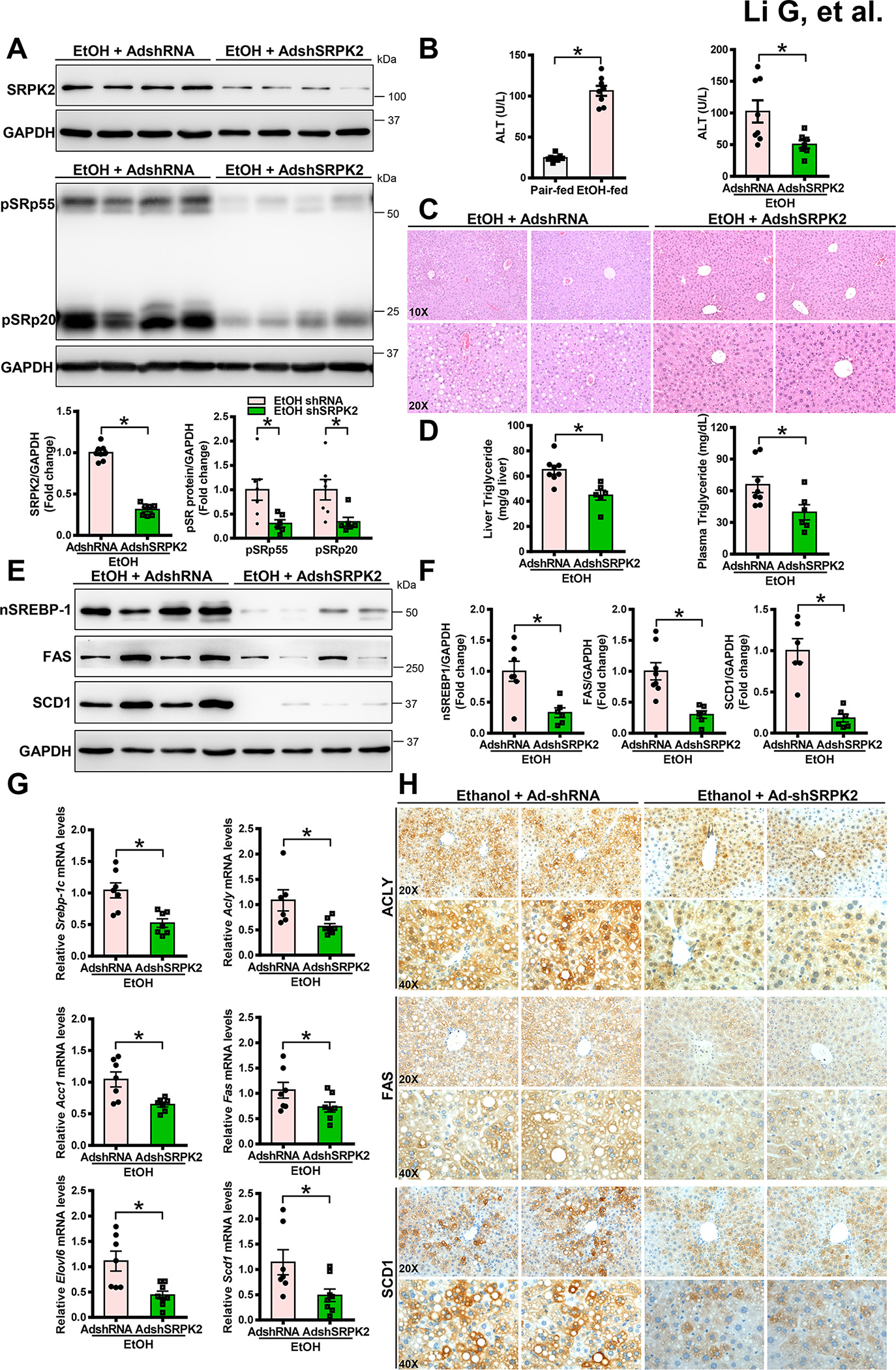
Mice were administered via tail vein injections of adenoviral vectors encoding shRNA targeting the SRPK2 gene (AdshSRPK2) or shRNA control (AdshRNA). After injection, the mice were fed a Lieber-DeCarli alcohol liquid diet for 10 days, plus one binge of alcohol at the end of experiments. All mice were subsequently sacrificed 9 hours post-binge.
A. Immunoblots and densitometric quantification for the abundance of SRPK2 and phosphorylation of SR proteins. Adenovirus-mediated knockdown of hepatic SRPK2 in mice was confirmed by a dramatic decrease in SRPK2 levels and SR protein phosphorylation.
B. Alcohol-induced elevation of liver injury, as assessed by plasma ALT levels, was reduced in mice upon SRPK2 knockdown.
C. H&E staining for hepatic steatosis in mice.
D. Chronic-binge alcohol feeding-induced elevation of hepatic and plasma triglyceride levels in mice were lowered by silencing SRPK2.
E-F. Immunoblots (E) and densitometric quantification (F) for nSREBP-1, FAS, and SCD1.
G. Real-time qRT-PCR analysis of hepatic lipogenic gene expression. Ad-shSRPK2-injected mice exhibited downregulation of SREBP-1c and its target genes under conditions of chronic-binge alcohol feeding.
H. Immunohistochemistry staining of liver sections using antibodies against ACLY, FAS, or SCD1. Upon chronic-binge alcohol feeding, strong positive staining for ACLY, FAS, and SCD1 was visualized mainly in hepatocytes of Ad-shRNA control-injected mice; however, the number and intensity of ACLY+, FAS+, and SCD1+ hepatocytes were reduced by silencing hepatic SRPK2.
The data are presented as the mean ± S.E.M., n=6–8 per group. *P<0.05 between two groups.
Images were acquired using 20X and 40X objectives.
Overexpression of SRPK2 mimics the effect of alcohol on lipotoxicity and the production of hepatocyte-derived inflammatory mediators in vitro—
As shown in Fig. S2C–H, ethanol exposure dose-dependently increased cleaved caspase-3, a hallmark of apoptosis, in primary mouse hepatocytes likely owing to triglyceride overproduction. Interestingly, cleaved caspase-3 was also elevated by overexpressing SRPK2, which coincided with hepatocellular triglyceride elevation (Fig. 2L). It has been indicated that hepatocytes produce chemokines that attract innate immune cells, e.g., macrophages and neutrophils, in response to liver injury(14). C-X-C motif ligand 1 (CXCL1) is a key inflammatory chemokine that is predominantly derived from hepatocytes and stimulates neutrophil recruitment(15). Interestingly, mRNA levels of CXCL1 were increased in ethanol-treated hepatocytes, suggesting that the alcoholic inflammatory response is potentially mediated by hepatocyte-derived CXCL1 in an autocrine/paracrine manner. Overexpression of SRPK2 was sufficient to upregulate expression of hepatocyte derived CXCL1, recapitulating ethanol’s action in hepatocytes.
Knockdown of SRPK2 attenuates chronic-binge alcohol feeding-induced liver injury and inflammation in mice—
Hepatocyte apoptosis is a major characteristic of liver injury (2, 4). As shown in Fig. S3D–G, cleaved caspase-3 was minimally expressed in normal mice but was increased by ~4-fold in alcohol-fed mice, which may explain the alcohol-induced hepatocyte death. Conversely, the protective effect of SRPK2 inhibition on lipotoxicity was associated with cleaved caspase-3 reduction in SRPK2 KD livers. In patients with alcoholic hepatitis, liver inflammation is characterized by increased pro-inflammatory chemokines, such as CXCL1, and neutrophil infiltration (16). As shown in Fig. S3H–I, expression of pro-inflammatory cytokines, such as interleukin 1β (IL-1β) and tumor necrosis factor-alpha (TNFα), was ~3- to 5-fold higher in alcohol-fed mice, but this elevation was decreased by 50–70% upon SRPK2 knockdown. The mRNA levels of the monocyte/macrophage marker CD11b were increased by alcohol feeding, which was diminished in SRPK2 KD livers. Interestingly, alcohol-induced elevation of CXCL1 and neutrophil markers, such as myeloperoxidase (MPO) and (Ly6G), and the number of MPO+ neutrophils were also lowered in SRPK2 KD livers. MPO+ neutrophil infiltration was seen in alcohol-fed mice (Fig. S3J), possibly due to SRPK2-induced CXCL1 elevation in hepatocytes (Fig. S2G–H). Accumulated evidence demonstrates that caspase-1, the key effector protein of the inflammasome NLRP3, is required for IL-1β production(17). To uncover a mechanism underlying the anti-inflammatory role of SRPK2 inhibition, caspase-1 activity was analyzed by assessing cleavage fragments (p10 and p20) of caspase-1 as described previously(17). Interestingly, caspase-1 activity was increased in alcohol-fed mice but was diminished by ~60% upon SRPK2 knockdown (Fig. S3K–L). Thus, SRPK2 inhibition mitigates progression of alcoholic steatosis to steatohepatitis by suppressing hepatocyte damage and neutrophil infiltration.
Overexpression of FGF21 in transgenic mice inhibits SRPK2 activity and protects against ALD pathologies—
We next explore the effect of the hepatocyte-derived factor FGF21 on alcohol-induced SRPK2 and liver pathologies. We used an FGF21 transgenic mouse model (FGF21-Tg), in which human FGF21 was overexpressed predominantly in the liver under the control of the human ApoE promoter (18). As shown in Fig. 4A–C and Fig. S4A–B, FGF21-Tg mice were resistant to chronic-binge alcohol feeding-induced fatty liver and metabolic disorders, as evidenced by reduced hepatic triglyceride overproduction and normalized hyperlipidemia. These results closely resembled the phenotype observed in SRPK2 KD mice (Fig. 3). To dissect the mechanism accounting for the protective phenotype of FGF21-Tg mice, we tested the hypothesis that FGF21 is functionally connected to SRPK2 in the regulation of metabolic homeostasis. As illustrated in Fig. 4D–I and Fig. S4C–E, SRPK2 abundance, pSRp55, and pSRp20 were increased by alcohol administration in WT mice but were reduced by ~50% in FGF21-Tg mice, returning to nearly normal levels. Furthermore, levels of nuclear SREBP-1 as well as expression of SREBP-1c and its target genes were increased ~2-fold in alcohol-fed WT mice but were decreased by approximately 50% in FGF21-Tg mice. Analogous to reduced steatosis severity in SRPK2 KD mice, the accumulation of positively stained hepatocytes for ACLY, FAS, or SCD1 during ALD was markedly lowered in FGF21-Tg mice, suggesting that SRPK2 inhibition may represent a mechanism underlying the anti-lipogenic effect of FGF21. Interestingly, the beneficial effects of FGF21 overexpression on alcoholic liver injury and lipotoxicity were evidenced by reduced plasma ALT levels and disrupted caspase-3-dependent apoptosis (Fig. 4J and Fig. S4F). Likewise, cleaved forms (p20 and p10) of caspase-1, an inflammatory mediating caspase, were diminished by ~60% in FGF21-Tg mice (Fig. 4K). Expression of pro-inflammatory cytokines and mediators, such as IL-1β, TNF-α, and Cd11b, was blunted by ~50%. The neutrophil-mediated inflammatory response, as reflected by induction of CXCL1, MPO, and Ly6g, was effectively normalized in FGF21-Tg mice (Fig. 4L–M).
Fig. 4. Overexpression of FGF21 in transgenic mice suppresses alcohol-mediated induction of SRPK2 and protects against the progression of ALD.
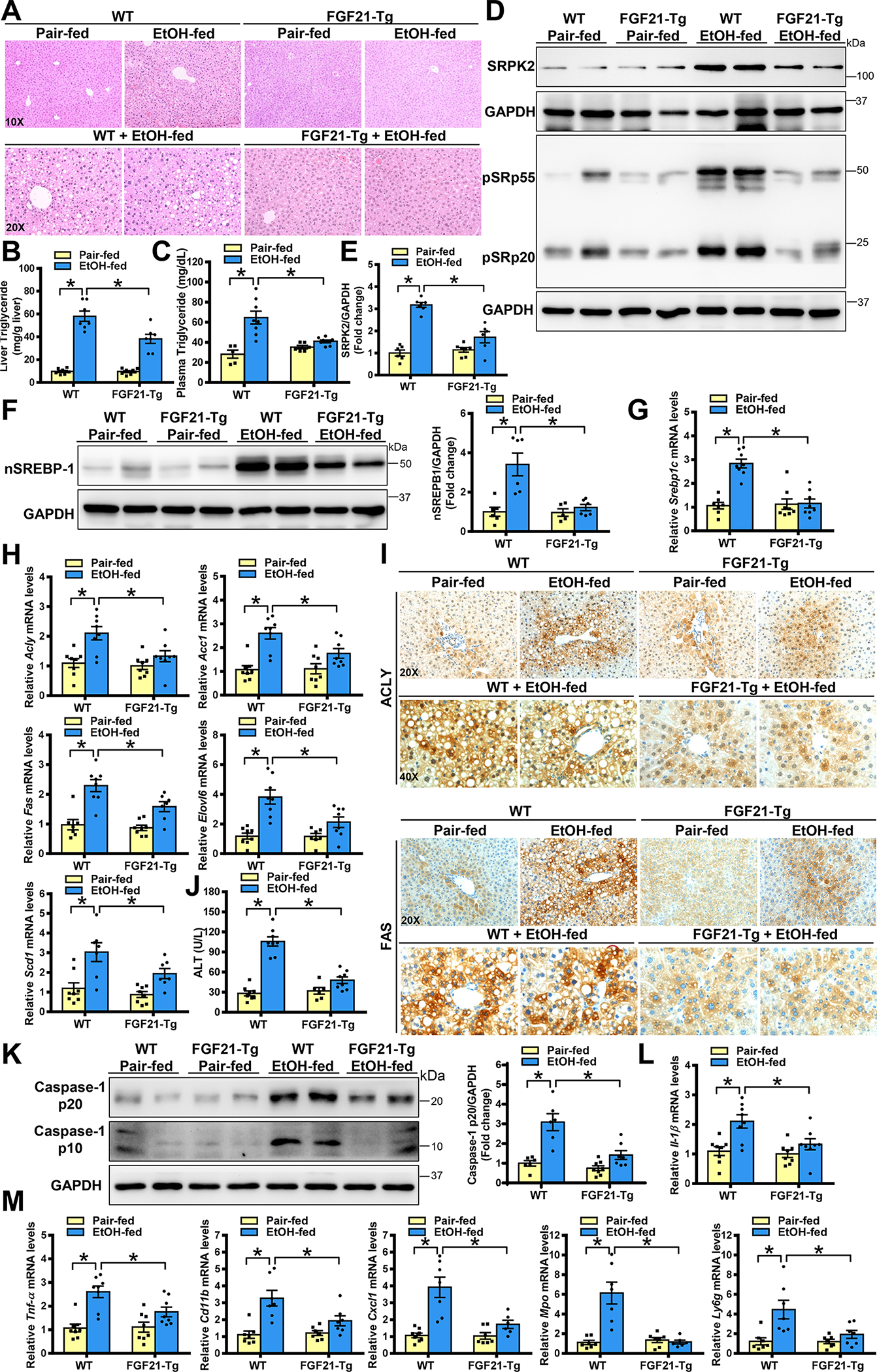
WT mice and FGF21 overexpression in transgenic (FGF21-Tg) mice were subjected to a Lieber-DeCarli alcohol liquid diet for 10 days plus one binge alcohol feeding at the end of experiments, or a pair-fed control diet. All mice were sacrificed 9 hours post-binge.
A. Representative H&E staining of live sections in WT and FGF21-Tg mice.
B-C. Hepatic and plasma triglyceride concentrations were lowered in FGF21-Tg mice after chronic-binge alcohol feeding.
D-F. Immunoblots and densitometric quantification for SRPK2, phosphorylation of SR proteins, and nSREBP-1.
G-H. Real-time qRT-PCR analysis of mRNA levels of SREBP-1 and its target lipogenic genes. FGF21-Tg mice exhibited much lower expression of lipogenic genes than that in WT mice after chronic-binge alcohol feeding.
I. Immunohistochemistry staining for ACLY and FAS. Notably, the number and distribution of ACLY+ and FAS+ hepatocytes were decreased in FGF21-Tg mice following chronic-binge alcohol feeding.
J. ALT assays to measure liver injury.
K. Immunoblots and densitometric quantification for p10 and p20 fragments of caspase-1.
L-M. Real-time qRT-PCR analysis showed that expression of pro-inflammatory regulators and mediators (IL-1β, TNF-α, Cdc11b, CXCL1, MPO, and Ly6g) was much lower in FGF21-Tg mice than in WT mice after chronic-binge alcohol feeding.
The data are presented as the mean ± S.E.M., n=6–8 per group. *P<0.05 between two groups.
Images were acquired using 20X and 40X objectives.
Alcohol-mediated induction of SRPK2 stabilization is mediated by mTORC1 in hepatocytes—
Our recent studies demonstrate that mTORC1 signaling contributes to alcohol-induced lipogenesis. To decipher the potential mechanism responsible for induction of SRPK2 by alcohol, we tested the hypothesis that aberrant activation of SRPK2 by alcohol is linked to mTORC1 signaling. To this end, we first examined the role of the mTORC1 inhibitor, rapamycin, in regulating SRPK2. As illustrated in Fig. 5A–G and Fig. S5A, rapamycin inhibited mTORC1 activity in AML12 hepatocytes under conditions of nutrient-induced mTORC1 activation, as indicated by decreased phosphorylation of mTORC1 downstream effectors, such as S6 and 4E-BP1, and a mobility shift of 4E-BP1. Rapamycin treatment also inhibited mTORC1 activation in response to ethanol. Intriguingly, rapamycin repressed SRPK2 protein levels and FAS induction in cultured hepatocytes exposed to ethanol. However, the decrease in SRPK2 protein levels did not appear to be attributed to the downregulation of SRPK2 mRNA. Next, we tested the possibility that rapamycin reduces SRPK2 protein levels by regulating its degradation. Chase studies using cycloheximide (CHX), a protein synthesis inhibitor, revealed that SRPK2 protein levels dropped more rapidly in the presence of rapamycin treatment than in the absence of rapamycin. Collectively, mTORC1 inhibition promotes SRPK2 degradation in hepatocytes.
Fig. 5. FGF21 overexpression mimics the inhibitive effect of rapamycin on alcohol-induced accumulation of SRPK2 protein in hepatocytes in vitro and in vivo.
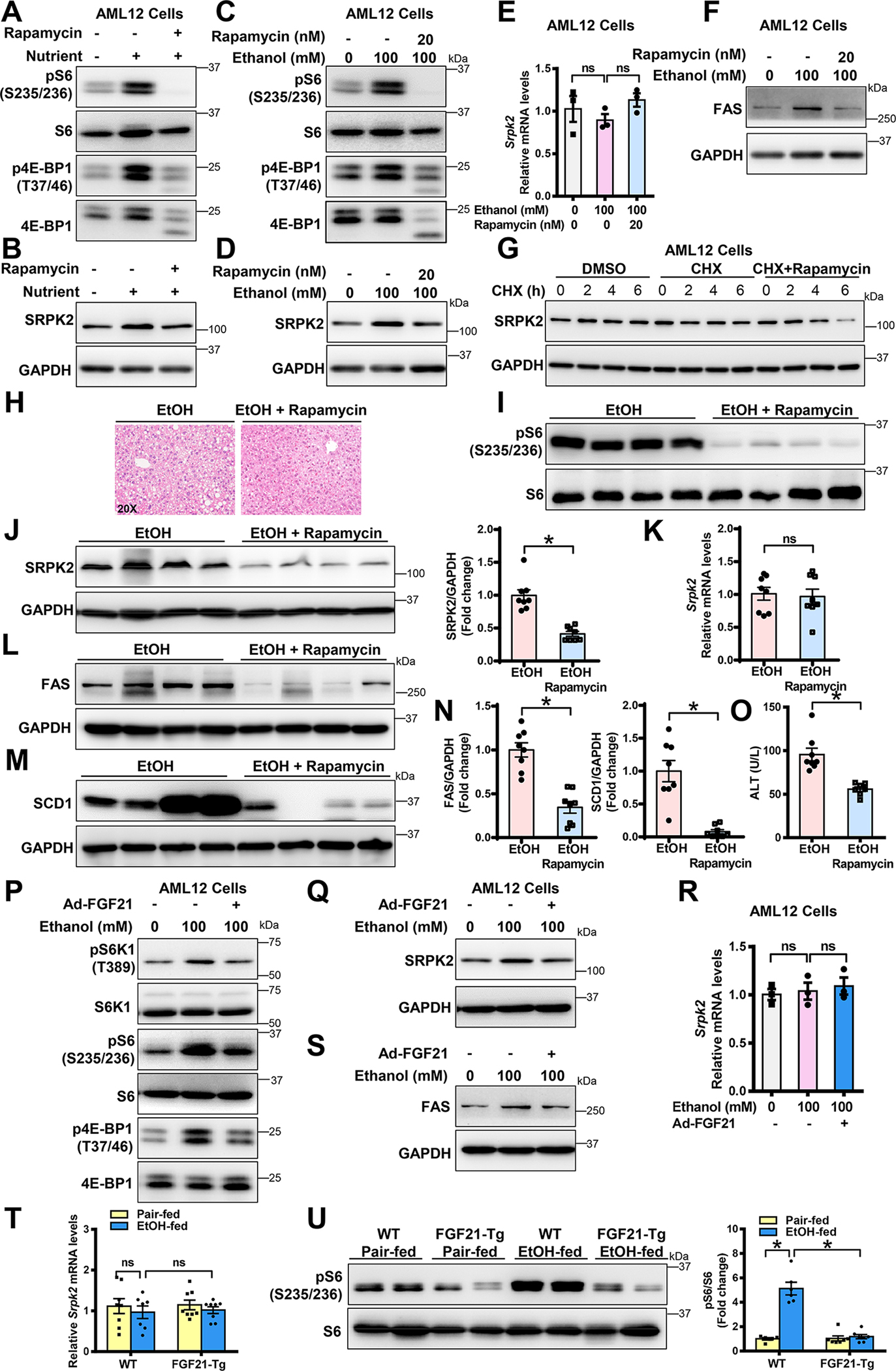
A-B. mTORC1 inhibition by rapamycin treatment decreased SRPK2 abundance in AML12 hepatocytes under conditions of nutrient-stimulated activation of mTORC1. AML12 cells were incubated for 24 h in medium containing 2% fetal bovine serum (FBS) or in medium containing 10% FBS (Nutrient-rich condition) in the absence or presence of rapamycin (20 nM). Cell lysates were immunoblotted with antibodies against SRPK2, as well as the phosphorylated forms of mTORC1 downstream effectors, including S6 at Ser235/236 (pS6) and 4E-BP1 at Thr37/46 (p4E-BP1).
C-D. mTORC1 inhibition by rapamycin protects hepatocytes from ethanol-induced accumulation of SRPK2 protein. AML12 cells were incubated for 24 h with or without ethanol (100 mM) in the absence and presence of rapamycin (20 nM) under cultured conditions in medium containing 2% FBS.
E. Real-time qRT-PCR revealed no significant changes in SRPK2 mRNA when protein levels were decreased in rapamycin-treated AML12 cells. Cells were incubated for 24 h with increasing doses of ethanol (100 mM) in medium containing 2% FBS. The decrease in SRPK2 protein did not appear to be due to a decrease in SRPK2 mRNA levels in rapamycin-treated hepatocytes.
F. mTORC1 inhibition by rapamycin reduced ethanol-induced FAS in AML12 cells.
G. mTORC1 inhibition by rapamycin promotes the degradation of SRPK2 in AML12 hepatocytes. Cells were chased for the indicated durations with the protein synthesis inhibitor cycloheximide (CHX, 50 μg/ml) in the absence or presence of rapamycin (20 nM).
H. Rapamycin treatment ameliorates hepatic steatosis in chronic-binge alcohol-fed mice. Mice fed the alcohol diet were administered daily intraperitoneal injections with either vehicle or rapamycin (1mg/kg/day) for 10 days. The mice were gavaged with a single dose of ethanol (5 g/kg) and then sacrificed 9 hours post-gavage. H&E staining for hepatic steatosis was shown.
I. Representative immunoblots of phosphorylation of S6 (pS6) in rapamycin-treated mice.
J-K. Immunoblots and densitometric quantification (J) as well as real-time qRT-PCR analysis (K) for SRPK2. Rapamycin treatment decreased SRPK2 induction via chronic-binge alcohol feeding but it did not affect its mRNA expression.
L-N. Immunoblots (L-M) and densitometric quantification (N) for FAS and SCD1.
O. Rapamycin treatment reduced alcohol-induced liver injury in mice, as assessed by plasma ALT levels.
P. FGF21 inhibits alcohol-induced mTORC1 signaling in hepatocytes. AML-12 cells were transduced with or without an adenoviral vector expressing FGF21 at an MOI of 5 and were subsequently incubated for 24 h with ethanol in medium containing 2% FBS. Cell lysates were immunoblotted with antibodies against SRPK2, as well as the phosphorylated forms of mTORC1 downstream effectors, including S6K1 at Thr389, S6 at Ser235/236, and 4E-BP1 at Thr37/46.
Q-R. Inhibition of mTORC1 by FGF21 attenuates alcohol-mediated induction of SRPK2 and FAS in AML-12 cells, without affecting SRPK2 mRNA levels.
T. Extent of SRPK2 mRNA levels was comparable in the livers of alcohol-fed WT and FGF21-Tg mice.
U. FGF21 overexpression in transgenic mice inhibits alcohol-induced activation of mTORC1, as indicated by reduced phosphorylation of S6 at Ser235/236.
The data are presented as the mean ± S.E.M., n=6–8 per group. *P<0.05 between two groups.
Images were acquired using 20X objectives.
mTORC1 inhibition by rapamycin suppresses SRPK2 accumulation and ameliorates ALD pathologies in mice—
As shown in Fig. 5H–O and Fig. S5B, rapamycin repressed hepatic mTORC1 activity, as evidenced by decreased phosphorylation of S6, in alcohol-fed mice. Rapamycin-treated mice exhibited slightly smaller lipid droplets in hepatocytes, compared to untreated mice following alcohol feeding. Moreover, rapamycin treatment resulted in a ~50% reduction in SRPK2 abundance, without affecting its mRNA levels. This reduction was accompanied by a 60% - 80% decrease in expression of FAS and SCD1, suggesting that mTORC1 inhibition renders hepatocytes insensitive to alcohol–mediated SRPK2 accumulation and lipogenesis. Furthermore, plasma ALT levels and hepatic cleavage of caspase-3 were significantly reduced in rapamycin-treated mice, indicative of attenuated hepatocyte death.
FGF21 inhibits accumulation of SRPK2 protein caused by alcohol possibly by repressing mTORC1 signaling in vitro and in vivo—
SRPK2 induction by alcohol was reduced in FGF21-Tg mouse livers (Fig. 4D). To elucidate the mechanisms by which FGF21 attenuates alcohol-induced SRPK2, we found that mRNA levels of SRPK2 were comparable between the livers of WT and FGF21-Tg mice (Fig. 5T), suggesting that FGF21 may regulate SRPK2 via a posttranslational mechanism. Given that mTORC1 is required for alcohol-mediated stability of SRPK2 in hepatocytes, it raised the possibility that FGF21 abolishes the ability of alcohol to stimulate mTORC1 signaling and SRPK2 accumulation. In support of this, adenoviral overexpression of FGF21 suppressed mTORC1 activity, lowered SRPK2 abundance, and blocked FAS induction in AML-12 cells exposed to ethanol (Fig. 5P–R and Fig. S5C). Consistently, FGF21-Tg mice showed lower basal levels of mTORC1 activity and reduced mTORC1 signaling caused by alcohol (Fig. 5U), analogous to the downregulation of SRPK2 in rapamycin-treated mice (Fig. 5I). These observations suggest that like rapamycin, FGF21 represses alcohol-mediated accumulation of SRPK2 through mTORC1 inhibition-mediated degradation of SRPK2.
Genetic deficiency of FGF21 in mice promotes SRPK2 induction and exacerbates alcohol-associated hepatic steatosis, lipotoxicity, and inflammation—
To further establish a role of FGF21 loss in regulating SRPK2 signaling, a genetically engineered FGF21 knockout (FGF21 KO) mouse model(20) was used. As shown in Fig. 6A–C and Fig. S6A–B, WT mice had moderate alcoholic steatosis, whereas FGF21 KO mice developed more severe steatosis and dyslipidemia, as evident by an ~ 2-fold elevation in hepatic triglyceride levels and an ~30% increase in plasma triglyceride and cholesterol concentrations. Moreover, the abundance of SRPK2 and phosphorylation of SR proteins were markedly increased in WT mice after alcohol administration. Notably, SRPK2 protein levels were increased by ~2-fold in FGF21 KO mice, without affecting SRPK2 mRNA expression (Fig. 6D–F). Furthermore, severe steatosis and triglyceride overproduction were correlated with pronounced accumulation of SRPK2 in FGF21 KO mice. Nuclear SREBP-1 and transcriptional activity of SREBP-1c target genes were at their highest levels in alcohol-fed FGF21 KO mice. Parallel with their steatosis severity, the number of ACLY+, FAS+, and SCD1+ hepatocytes were much higher in FGF21 KO livers than in WT livers (Fig. 6G–J and Fig. S6C–E), suggesting that the susceptibility of mice lacking FGF21 to developing ALD is likely attributed to SRPK2-mediated lipogenesis. Consequently, FGF21 deficiency enhanced alcohol-induced caspase-3 cleavage and liver injury (Fig. 6K and Fig. S6F). As shown in Fig. 6L–N, cleaved forms of caspase-1 and mRNA levels of prominent pro-inflammatory regulators were increased in WT livers; the induction was markedly potentiated by FGF21 deficiency, demonstrating that FGF21 loss amplifies lipotoxicity and inflammatory liver injury. Moreover, the alcohol-induced neutrophil inflammatory response was further enhanced in FGF21 KO mice.
Fig. 6. FGF21 knockout mice exhibit enhanced SRPK2 activity and develop more severe ALD pathologies.
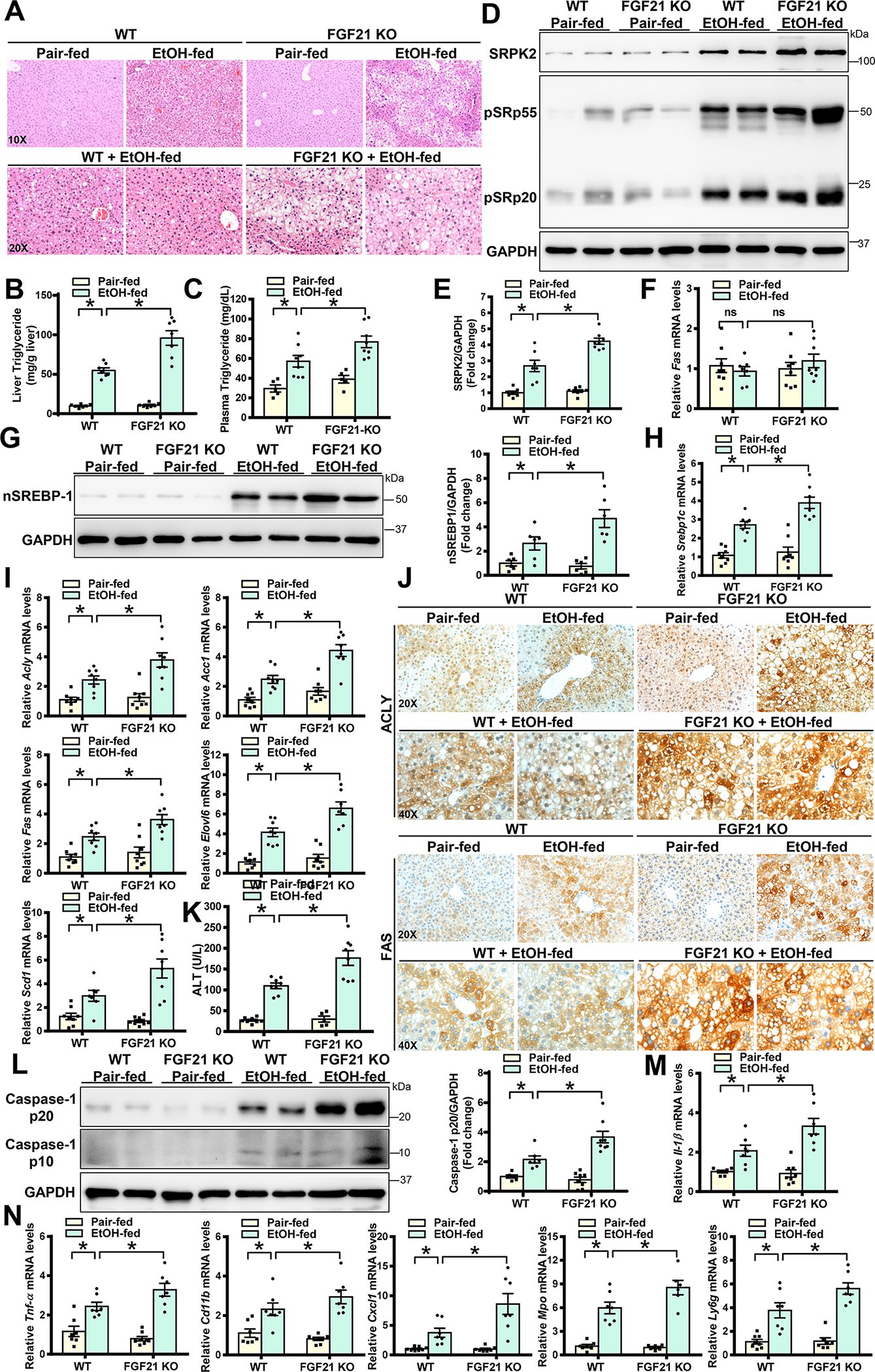
WT and FGF21 knockout (FGF21 KO) mice were subjected to a Lieber-DeCarli alcohol liquid diet for 10 days plus one binge alcohol feeding at the end of experiments, or a pair-fed control liquid diet. All mice were sacrificed 9 hours post-binge.
A. H&E staining of liver sections in both WT and FGF21 KO mice.
B-C. Liver and plasma triglyceride levels were higher in FGF21 KO mice than those in WT mice after chronic-binge alcohol feeding.
D-E. Immunoblots and densitometric quantification for SRPK2, phosphorylation of SR proteins, and nSREBP-1.
F. Real-time qRT-PCR analysis of mRNA levels of SRPK2.
G. Immunoblots and densitometric quantification for nSREBP-1.
H-I. Real-time qRT-PCR analysis showed that mRNA levels of SREBP-1 and its target genes were increased in FGF21 KO mice after chronic-binge alcohol feeding.
J. Immunohistochemistry for ACLY and FAS. Upon chronic-binge alcohol feeding, positive staining for ACLY and FAS was visualized mainly in hepatocytes of WT mice; the distribution and intensity of positively stained hepatocytes were higher in FGF21 KO mice than in WT mice.
K. ALT assays to measure liver injury.
L. Immunoblots and densitometric quantification for p10 and p20 fragments of caspase-1.
M-N. Real-time qRT-PCR analysis of the expression of pro-inflammatory cytokines (IL-1β and TNF-α), as well as neutrophil chemokines (CXCL1) and neutrophil markers (MPO and Ly6g). An inflammatory response was increased in WT mice following chronic-binge alcohol feeding; this induction was further enhanced in FGF21 KO mice.
The data are presented as the mean ± S.E.M., n=6–8 per group. *P<0.05 between two groups.
Images were acquired using 20X and 40X objectives.
Knockdown of SRPK2 rescues metabolic dysfunction and ameliorates ALD pathologies in FGF21 KO mice—
We tested the hypothesis that the ability of FGF21 ablation to aggravate ALD is mediated by SRPK2. As shown in Fig. 7A–E and Fig. S7A, hepatic knockdown of SRPK2 in FGF21 KO mice was verified by a marked reduction of SRPK2 protein levels and SR protein phosphorylation, reaching levels seen in WT controls. Excessive hepatic lipid deposition, triglyceride overproduction, and elevated plasma lipid levels seen in AdshRNA control-injected FGF21 KO mice were largely reversed by silencing SRPK2. As shown in Fig. 7F–H and Fig. S7B–C, alcohol-induced nuclear accumulation of SREBP-1 as well as protein and mRNA levels of its target genes were further increased up to ~4-fold in FGF21 KO mice that received AdshRNA control, but decreased ~50% upon SRPK2 knockdown, returning to levels of WT mice. Meanwhile, positively stained hepatocytes for ACLY, FAS, and SCD1 and excessive lipid deposits observed in FGF21 KO mice were remarkably lowered by SRPK2 knockdown. As shown in Fig. 7I–K and Fig. S7D–E, the protective effect of SRPK2 knockdown against liver injury was likely attributed to the reduction of caspase-3 signaling. Moreover, the anti-inflammatory effects of SRPK2 inhibition in FGF21 KO mice were evident by a reduction of cleaved caspase-1, IL-1β, and neutrophil mediators. To our knowledge, this is the first evidence that FGF21 deficiency promotes the progression of ALD in an SRPK2-dependent manner.
Fig. 7. Silencing hepatic SRPK2 rescues aberrant regulation of lipid metabolism and alleviates ALD pathologies in FGF21 KO mice.
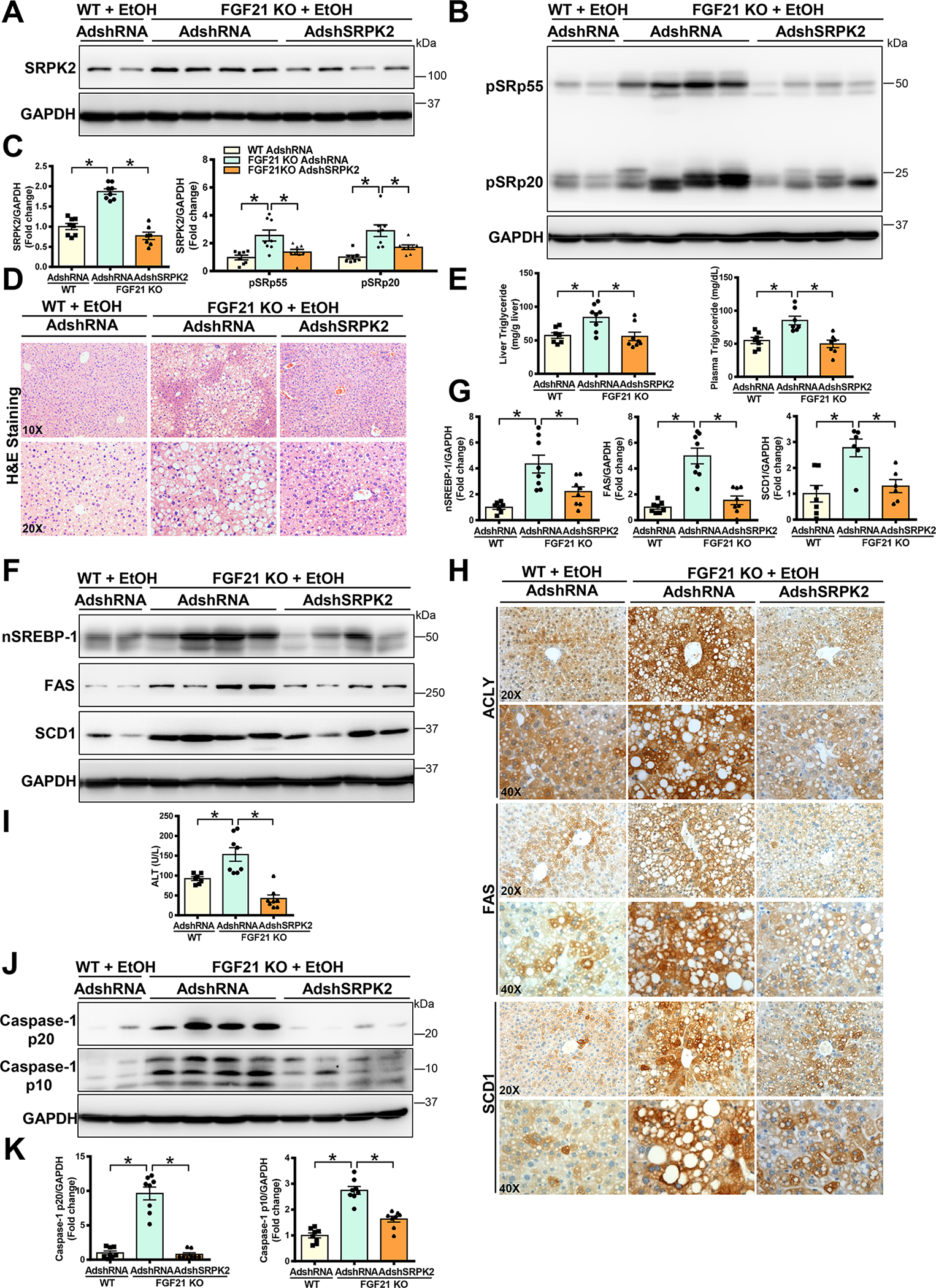
WT or FGF21 KO mice were injected with either Ad-shSRPK2 or Ad-shRNA control via tail vein injection. Subsequently, adenovirus-injected mice were subjected to a Lieber-DeCarli alcohol liquid diet for 10 days plus one time of alcohol binge feeding at the end of experiments. The mice were sacrificed 9 hours post-binge.
A-C. Immunoblots (A-B) and densitometric quantification (C) for SRPK2 and phosphorylation of SR proteins. Adenovirus-mediated hepatic knockdown of SRPK2 in FGF21 KO mice was verified by dramatically decreased SRPK2 and phosphorylation of SR proteins.
D. H&E staining showed that alcohol-induced hepatic steatosis was dramatically increased in FGF21 KO mice; However, severe steatosis in FGF21 KO mice was attenuated by silencing SRPK2.
E. Hepatic and plasma triglyceride levels in mice.
F-G. Immunoblots (F) and densitometric quantification (G) for nSREBP-1, FAS, and SCD1. Upon chronic-binge alcohol feeding, hepatic levels of nSREBP-1 and its target lipogenic enzymes were increased in FGF21 KO mice but these effects were diminished by silencing SRPK2.
H. Immunohistochemistry reveals that upon chronic-binge alcohol feeding, the number and distribution of ACLY+ and FAS+ hepatocytes were higher in FGF21 KO mice than in WT mice. However, the number and intensity of ACLY+ and FAS+ hepatocytes in FGF21 KO mice were lowered by silencing SRPK2.
I. Liver injury was measured by ALT assays. Alcohol-induced liver injury in FGF21 KO mice given an injection of AdshRNA control was restored to normal levels upon SRPK2 knockdown.
J-K. Immunoblots (J) and densitometric quantification (K) for cleaved caspase-1.
The data are presented as the mean ± S.E.M., n=6–8 per group. *P<0.05 between two groups.
Images were acquired using 20X and 40X objectives.
Knockdown of SRPK2 reverses abnormal pre-mRNA splicing involved in lipid metabolism during ALD—
We sought to determine whether the splicing kinase SRPK2 is functionally linked to dysregulated pre-mRNA splicing in ALD. Several spliced pre-mRNAs are involved in lipid metabolism, such as the lipogenic regulator lipin 1. Lipin 1 is implicated in humans with fatty liver and obesity (21). The lipin 1 gene generates two major functional mRNA transcripts through alternative splicing of exon 7(22, 23). One transcript (lipin 1α) skips exon 7 to encode lipin 1α protein, and another transcript (lipin 1β) includes exon 7 to encode lipin 1β protein (22). Unlike lipin 1α, lipin 1β upregulates the lipogenic transcriptional factor SREBP-1 in cultured cells(21). We investigated whether lipin 1 splicing may be involved in alcohol-induced lipid biosynthesis. Alcohol feeding caused a ~3-fold increase in lipin 1β and no significant changes in lipin 1α. Consequently, expression of total lipin 1 and the ratio of lipin 1β/α were increased by ~3-fold, suggesting that the lipin 1β transcript constitutes an inducible isoform in response to ethanol challenge. The induction of lipin 1β was positively correlated with SRPK2 activity and hepatic triglyceride levels (Fig. 8A–C and Fig. S8A). While lipin 1 has been indicated as a known splicing target of serine-arginine rich splicing factor 10 (SRSF10) (23), the effect of SRPK2 on SRSF10 splicing functions has not been investigated. As shown in Fig. 8D–I and Fig. S8B, expression of SRSF10 was decreased by nearly 50% in alcohol-fed mice but was restored by SRPK2 knockdown. Likewise, the number of SRSF10+ hepatocytes increased in SRPK2 KD mice. Consequently, mRNA levels of lipin 1β and the ratio of lipin 1β/α were downregulated by approximately 50% in SRPK2 KD mice, with minimal changes in lipin 1α. RT-PCR analysis also revealed a decrease in lipin 1β spliced isoform in SRPK2 KD mice (Fig. S8C). Mechanistically, co-immunoprecipitation showed that ethanol exposure increased the interaction between SRPK2 and SRSF10 in AML12 hepatocytes (Fig. 8J), suggesting the critical role of SRSF10 in the regulation of lipin 1 splicing by SRPK2. Moreover, the splicing function of SRSF10, as assessed by RT-PCR analysis of splicing of other target pre-mRNAs of SRSF10, such as pyruvate kinase isoforms (PKM), ATPase phospholipid transporting 8A1 (Atp8a1), and inner membrane mitochondrial protein (Immt1)(23), appeared to be restored upon SRPK2 knockdown (Fig. S8C). Taken together, SRPK2 acts as a key component of the lipogenic splicing machinery that disrupts SRSF10 splicing function and favors lipin 1β isoform production in ALD.
Fig. 8. Manipulation of SRPK2 and FGF21 modulates dysregulation of SRSF10-mediated lipogenic pre-mRNA splicing associated with ALD.
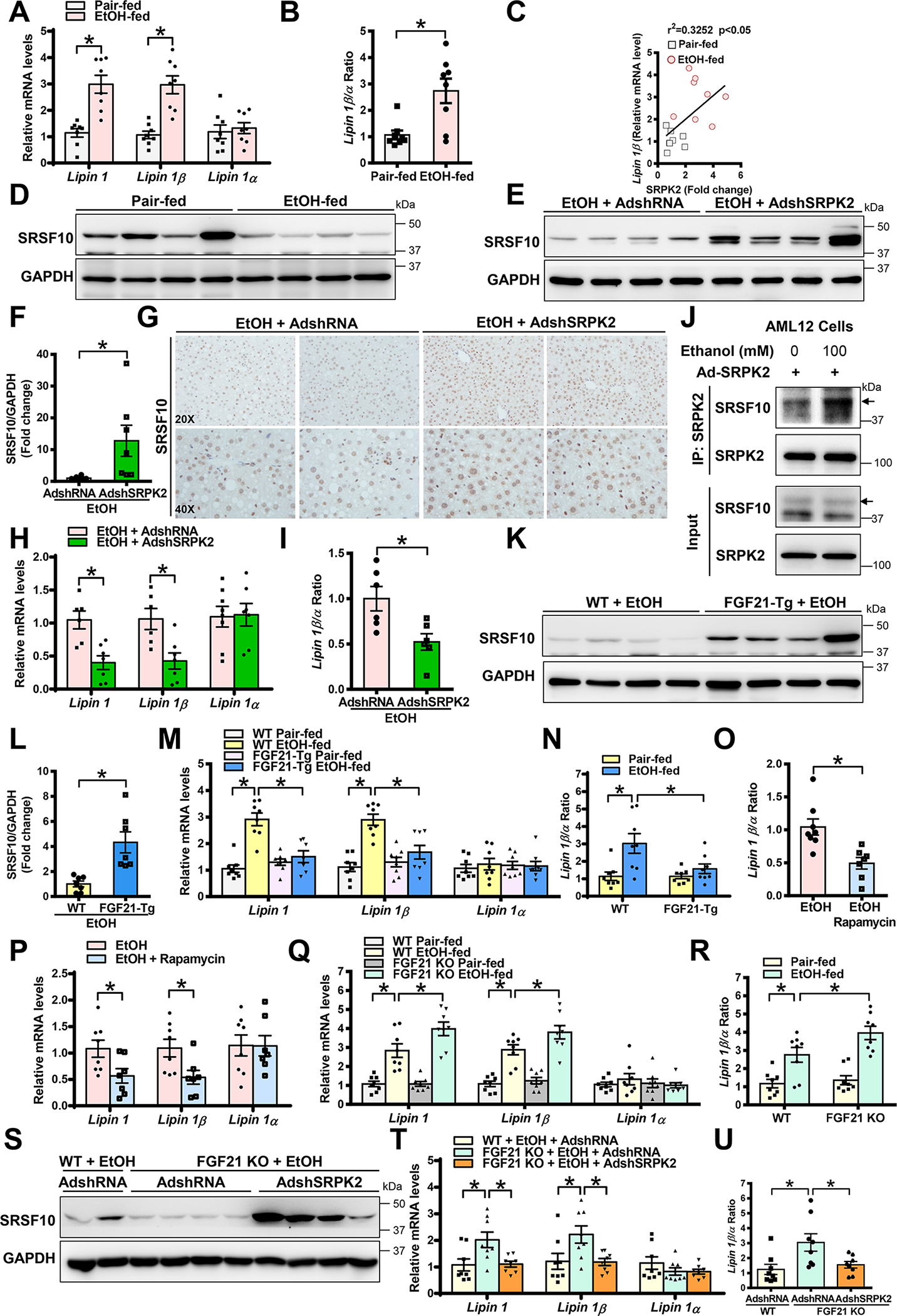
A-B. Real-time qRT-PCR analysis of mRNA levels of total lipin 1 and different lipin 1 splicing isoforms in pair-fed and alcohol-fed mice. Lipin 1β/α ratio was calculated using their relative mRNA levels.
C. Positive correlation between hepatic SRPK2 and lipin 1β levels in pair-fed and alcohol-fed mice.
D. Immunoblots for SRSF10 in pair-fed and alcohol-fed mice.
E-F. Immunoblots (E) and densitometric quantification (F) for SRSF10 in alcohol-fed mice that were injected with either AdshRNA control or AdshSRPK2.
G. Immunohistochemistry for SRSF10. Notably, upon chronic-binge alcohol feeding, positive staining for SRSF10 was visualized mainly in the nucleus of hepatocytes in Ad-shRNA control-injected control mice, but the intensity and distribution of SRSF10+ hepatocytes were increased in SRPK2-knockdown mice.
H-I. Real-time qRT-PCR analysis of lipin 1, lipin 1β, lipin 1α, as well as lipin 1 β/α ratio in control and SRPK2-knockdown mice.
J. Ethanol exposure enhanced the interaction between SRPK2 and SRSF10 in AML-12 hepatocytes. AML12 cells were transduced with Ad-SRPK2 at an MOI of 5 and subsequently incubated for 24 h with or without ethanol (100 mM) in medium containing 2% FBS. Cell lysates were subjected to immunoprecipitation with an anti-SRPK2 antibody; immunoprecipitated proteins and cell lysates were immunoblotted with indicated antibodies.
K-L. Immunoblots (K) and densitometric quantification (L) for SRSF10 in WT and FGF21-Tg mice.
M-N. Real-time qRT-PCR analysis showed that mRNA levels of lipin 1 and lipin 1β, as well as the lipin 1β/α ratio, were increased by alcohol administration in WT mice, and this induction was attenuated in FGF21-Tg mice.
O-P. Real-time qRT-PCR analysis for skipping of exon 7 (lipin 1α) and inclusion of exon 7 (lipin 1β) revealed that rapamycin treatment decreased lipin 1β induction caused by chronic binge alcohol feeding but did not affect mRNA expression levels of lipin 1α.
Q-R. Real-time qRT-PCR analysis of mRNA levels of lipin 1 and lipin 1 spliced isoforms as well as the lipin 1β/α ratio in WT and FGF21 KO mice.
S. Immunoblots for SRSF10 showed that upon alcohol administration, hepatic SRSF10 was downregulated in AdshRNA-injected FGF21 KO mice and this impairment was rescued by silencing SRPK2.
T-U. Real-time qRT-PCR analysis of mRNA levels of lipin 1, lipin 1β, and lipin 1α, as well as the ratio of lipin 1β/lipin 1α.
The data are presented as the mean ± S.E.M., n=6–8 per group. *P<0.05 between two groups.
Images were acquired using 20X and 40X objectives.
Manipulating FGF21 modulates SRPK2-controlled alternative splicing events associated with ALD—
Since the above data indicate that silencing SRPK2 restores SRSF10-dependent splicing of lipin 1, we asked whether manipulating FGF21 might affect pre-mRNA splicing events controlled by SRPK2. As shown in Fig. 8K–N and Fig. S8C, impairment of SRSF10 and its target gene splicing patterns were restored in the livers of FGF21-Tg mice, an effect that was also seen in SRPK2 KD livers. Remarkably, the exon 7 inclusion in lipin 1 and lipin1β/α ratio were decreased by ~60% in FGF21-Tg mice. Similar results were obtained in rapamycin-treated mice (Fig. 8O–P), suggesting that FGF21 or mTORC1 inhibition restores SRSF10-mediated splicing of lipin 1 by repressing SRPK2. As shown in Fig. 8Q–U and Fig. S8D–E, alcohol-mediated downregulation of SRSF10 and induction of lipin 1β in WT livers were further potentiated in FGF21 KO livers. Intriguingly, in FGF21 KO mice given Ad-shRNA control injection, FGF21 deficiency-mediated splicing dysregulation was counteracted by SRPK2 knockdown. Our studies uncover an alternative splicing mechanism by which FGF21 protects against lipogenesis and ALD, at least in part, by inhibiting SRPK2.
Discussion
The present study illustrates the alternative splicing mechanism by which SRPK2 is necessary for alcohol-induced hepatocellular fatty acid synthesis and lipid accumulation in a preclinical mouse model and in ALD patients. Our proposed model depicts that alcohol triggers SRPK2 protein stability in hepatocytes in a mTORC1-dependent manner. Such induction diminishes SRSF10 splicing function, induces exon 7 inclusion in lipin 1, and generates the lipin 1β spliced isoform. This splicing process promotes SREBP-1-dependent lipogenesis and contributes to the progression of ALD. We have discovered a post-transcriptional mechanism for the protective effect of FGF21 against ALD. Specifically, FGF21 suppresses alcohol-induced accumulation of SRPK2, possibly by disrupting mTORC1 signaling and promoting SRPK2 degradation. This consequently enhances the splicing function of SRSF10 on lipin 1 and blocks SREBP-1-driven lipogenesis. Thus, targeting SRPK2 signaling with FGF21 intervention may constitute a potentially effective approach for managing ALD.
Hyperactivation of SRPK2 represents an alternative splicing mechanism responsible for alcohol-induced lipogenesis—
Since lipogenesis is a hallmark of fatty liver disease(5), understanding the signaling pathways controlling lipogenesis is critical for developing a new therapeutic approach to combat fatty liver. One of our most important findings is that alcohol-induced lipogenesis is linked to hyperactivation of SRPK2 in mice, closely resembling the observations in patients with ALD. Alcohol-induced lipogenesis is dependent on SRPK2 in hepatocytes and in vivo; inhibition of SRPK2 ameliorates alcoholic steatosis and liver injury. Alcohol consumption triggers de novo lipogenesis in the liver(4), in which carbon precursors of acetyl-CoA are converted into fatty acids(24). However, how alcohol intake generates hepatic acetyl-CoA to promote lipid production remains largely unknown. ATP citrate lyase (ACLY), the enzyme that cleaves cytosolic citrate to generate acetyl-CoA and fatty acids, is upregulated by fructose consumption (24). Clinical trials are currently pursuing the inhibition of ACLY as a potential treatment for metabolic disease(5). Consistent with these findings, the present study reveals increased numbers of ACLY-positive hepatocytes, as well as activation of SREBP-1 and its target lipogenic genes, including ACLY, ACC1, FAS, and SCD1, in alcohol-fed mice. Strikingly, silencing SRPK2 diminishes alcohol-mediated induction of lipogenesis. Our studies have uncovered the previously unrecognized mechanism by which alcohol-activated SRPK2 is hepatocyte-specific; it contributes to the progression of ALD by upregulating rate-limiting enzymes involved in acetyl-CoA generation and lipogenesis.
The hepatokine FGF21 protects against ALD pathologies, at least in part, by inhibiting SRPK2—
One of our most interesting findings is that mice lacking FGF21 develop more severe steatosis, lipotoxicity, and liver inflammation upon chronic-binge alcohol feeding. Strikingly, worsened pathological changes associated with FGF21 deficiency are diminished upon SRPK2 knockdown, providing a functional link between FGF21 and its downstream effector SRPK2. Conversely, overexpression of FGF21 represses induction of SRPK2 and expression of lipogenic genes caused by alcohol in hepatocytes. Overexpression of FGF21 in mice protects hepatocytes from alcohol-mediated accumulation of SRPK2 and attenuates ALD pathologies in mice. FGF21 possesses anti-inflammatory effects by inhibiting macrophage infiltration in the vascular wall (25). In line with these reports, FGF21 improves alcoholic inflammation, such as neutrophil infiltration, via a mechanism which inhibits hepatocyte-derived neutrophil chemokines such as CXCL1. We have demonstrated that DEPTOR suppresses alcohol-induced activation of mTORC1 and ameliorates alcoholic steatohepatitis (4). To elucidate the mechanism by which FGF21 regulates SRPK2, the present study shows that mTORC1 inhibition by rapamycin promotes SRPK2 degradation in hepatocytes and suppresses both SRPK2 accumulation and lipogenesis in alcohol-fed mice. These findings are also supported by a recent study showing that mTORC1 activates S6K1 to phosphorylate SRPK2 at Ser494, which in turn promotes cancer cell growth (8). Likewise, overexpression of FGF21 represses alcohol-induced mTORC1 signaling in mice. FGF21 also blunts alcohol-mediated induction of both mTORC1 and SRPK2 in a hepatocyte-autonomous manner. While other mechanisms are involved, we suspect that FGF21 promotes SRPK2 degradation, at least in part, by inhibiting mTORC1. FGF21-mediated inhibition of SRPK2 functions as a checkpoint for controlling lipogenesis and alcoholic liver injury. As such, FGF21 may provide a new therapeutic avenue for halting and reversing ALD. Since SRPK2 induction occurs in later-stage ALD in humans, it would be of interest to further define the possible role of the FGF21-SRPK2 axis in the progression of liver fibrosis associated with late-stage ALD.
The hepatokine FGF21 downregulates hepatic lipogenesis through an SRPK2-mediated alternative splicing mechanism—
Accumulating evidence indicates that SRSF10 modulates alternative splicing of its target transcripts, such as lipin 1, the key regulator of lipogenesis (22, 26). Lipin 1β knockdown abolishes the lipogenic effects of SRSF10 knockdown in hepatocytes(21). Consistent with these findings, the present study shows that hepatic lipin 1β, but not lipin 1α, is an inducible isoform in alcohol-fed mice likely due to defective SRSF10. This induction promotes SREBP-1-driven lipogenesis. At the mechanistic levels, alcohol promotes the interaction between SRPK2 and SRSF10. Furthermore, the splicing function of SRSF10 on lipin 1 pre-mRNA is restored in FGF21-Tg mice. Alcohol-mediated impairment of SRSF10 is persistently disrupted in FGF21 KO mice but rescued by silencing SRPK2. Consequently, restoration of SRSF10 decreases lipid 1 exon 7 inclusion and lipin 1β. The splicing of exon 7 in lipin 1 is controlled by SRSF10-regulated cis-elements located in the constitutive exon 8(23). It is conceivable that FGF21 antagonizes the ability of SRPK2 to promote exon 7 inclusion by modulating the SRPK2-SRSF10 interaction and the binding of SRSF10 to lipin 1 exon 8. These studies demonstrate that in the control of lipid homeostasis, FGF21 functions as a ‘molecular switch’ by turning off SRPK2 and facilitating SRSF10 function, which maintains relatively low levels of lipin 1β and SREBP-1.
In conclusion,
SRPK2 acts as a critical splicing kinase that contributes to aberrant alternative splicing events in hepatocytes exposed to alcohol and in humans with ALD. Studies with gain- and loss-of-function strategies illuminate the therapeutic mechanism by which FGF21, as the key hepatokine, alleviates ALD pathologies by targeting SRPK2.
Materials and Methods
Animal Model for Chronic-Plus-Binge Alcohol Feeding—
The Bin-binge mouse model of ALD was developed as described previously (4, 27). This pre-clinical mouse model of chronic-plus-binge alcohol feeding was used. This mouse model recapitulates drinking patterns and key pathological changes seen in humans with ALD, such as steatosis and liver inflammation(27). All mice were housed in a temperature-controlled environment with a 12-h light/dark cycle. All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Texas Health San Antonio (UTHSA).
Animal Models with FGF21 Gain-of-function and Loss-of-function Strategies—
We used an FGF21 transgenic mouse model (FGF21-Tg), in which human FGF21 was predominantly overexpressed in the liver under the control of the human ApoE promoter including its hepatic control region(18). We also used a genetically engineered FGF21 knockout (FGF21 KO) mouse model, as previously described (20). These mice were provided by Eli Lilly and Company, Indianapolis, Indiana. After 5 days of acclimation on a control liquid diet, these mice were subjected to either a Lieber-DeCaril alcohol liquid diet for 10 days, along with one dose of alcohol binge feeding, or a pair-fed control diet plus one dose of an isocaloric maltose dextrin solution at the end of experiments.
Human Liver Tissue Samples—
Normal human liver samples (n =10) and alcoholic liver disease tissues (n =10) with steatosis, inflammation, and severe fibrosis (F3–4) were obtained from donor livers and recipient’s livers, respectively, during liver transplantations from the Liver Tissue Procurement and Distribution System (LTPDS) at the University of Minnesota as described previously(4). Particularly, normal healthy liver samples were obtained from the part of donor livers that were not used for transplantation. Individuals with both liver cancer and an alcohol-drinking history (2 to 3 drinks/day) were excluded from the study. Prior to this analysis, all normal controls and patient records were anonymized and de-identified.
Supplementary Material
Acknowledgment:
We are grateful to Dr. Ricardo J Samms at Eli Lilly and Company for his invaluable help and scientific input. We also thank the members of the Zang laboratory for their technical assistance and insightful discussion.
Grant support:
This work was supported in part by the National Institutes of Health Grants RO1 DK100603, RO1 DK121527, and R21 AA026922 (to MZ). MZ is also supported in part by the Distinguished Chair Endowment Fund in Research from the Ewing Halsell Foundation at the University of Texas Health San Antonio.
The Abbreviations used are:
- ALD
Alcohol-associated liver disease
- FGF21
fibroblast growth factor 21
- SRPK2
serine-arginine rich protein kinase 2
- SR proteins
serine-arginine rich proteins
- SRSF10
serine-arginine rich splicing factor 10
- mTORC1
mammalian target of rapamycin complex 1
- S6K1
the 70-kDa ribosomal protein S6 kinase 1
- S6
Ribosomal Protein S6
- 4E-BP1
the eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1
- CHX
cycloheximide
- lipin 1
a protein to perform a key reaction in the synthesis of triglycerides and phospholipids
- SREBP-1
sterol regulatory element binding protein 1
- ACLY
ATP citrate lyase
- ACC1
acetyl CoA carboxylase 1
- FAS
fatty acid synthase
- SCD1
stearoyl CoA desaturase1
- ELOVL6
elongation of Long-Chain fatty acid family member 6
- DGAT2
diacylglycerol acyltransferase-2
- ALT
Alanine Aminotransferase
- IL-1β
Interleukin 1β
- TNF-α
Tumor Necrosis Factor α
- CXCL1
chemokine (C-X-C motif) ligand 1
- MPO
myeloperoxidase
- Ly6g
lymphocyte antigen 6 complex locus G6D
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- AML12
hepatocytes, alpha mouse liver 12 hepatocytes.
Footnotes
Declaration of Interests: Andrew C. Adams owns stock in and is employed by Eli Lilly & Company. The remaining authors have nothing to report.
References
- 1.Seitz HK, Bataller R, Cortez-Pinto H, Gao B, Gual A, Lackner C, Mathurin P, et al. Alcoholic liver disease. Nat Rev Dis Primers 2018;4:16. [DOI] [PubMed] [Google Scholar]
- 2.Gao B, Ahmad MF, Nagy LE, Tsukamoto H. Inflammatory pathways in alcoholic steatohepatitis. J Hepatol 2019;70:249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Svensson RU, Parker SJ, Eichner LJ, Kolar MJ, Wallace M, Brun SN, Lombardo PS, et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat Med 2016;22:1108–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen H, Shen F, Sherban A, Nocon A, Li Y, Wang H, Xu MJ, et al. DEP domain-containing mTOR-interacting protein suppresses lipogenesis and ameliorates hepatic steatosis and acute-on-chronic liver injury in alcoholic liver disease. Hepatology 2018;68:496–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Batchuluun B, Pinkosky SL, Steinberg GR. Lipogenesis inhibitors: therapeutic opportunities and challenges. Nat Rev Drug Discov 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab 2011;13:376–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fu XD, Ares M, Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet 2014;15:689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee G, Zheng Y, Cho S, Jang C, England C, Dempsey JM, Yu Y, et al. Post-transcriptional Regulation of De Novo Lipogenesis by mTORC1-S6K1-SRPK2 Signaling. Cell 2017;171:1545–1558 e1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang HY, Lin W, Dyck JA, Yeakley JM, Songyang Z, Cantley LC, Fu XD. SRPK2: a differentially expressed SR protein-specific kinase involved in mediating the interaction and localization of pre-mRNA splicing factors in mammalian cells. J Cell Biol 1998;140:737–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geng L, Lam KSL, Xu A. The therapeutic potential of FGF21 in metabolic diseases: from bench to clinic. Nat Rev Endocrinol 2020;16:654–667. [DOI] [PubMed] [Google Scholar]
- 11.Li Y, Wong K, Giles A, Jiang J, Lee JW, Adams AC, Kharitonenkov A, et al. Hepatic SIRT1 attenuates hepatic steatosis and controls energy balance in mice by inducing fibroblast growth factor 21. Gastroenterology 2014;146:539–549 e537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harrison SA, Ruane PJ, Freilich BL, Neff G, Patil R, Behling CA, Hu C, et al. Efruxifermin in non-alcoholic steatohepatitis: a randomized, double-blind, placebo-controlled, phase 2a trial. Nat Med 2021;27:1262–1271. [DOI] [PubMed] [Google Scholar]
- 13.Gaich G, Chien JY, Fu H, Glass LC, Deeg MA, Holland WL, Kharitonenkov A, et al. The Effects of LY2405319, an FGF21 Analog, in Obese Human Subjects with Type 2 Diabetes. Cell Metab 2013;18:333–340. [DOI] [PubMed] [Google Scholar]
- 14.Zhou Z, Xu M-J, Gao B. Hepatocytes: a key cell type for innate immunity. Cellular & Molecular Immunology 2016;13:301–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang B, Xu MJ, Zhou Z, Cai Y, Li M, Wang W, Feng D, et al. Short- or long-term high-fat diet feeding plus acute ethanol binge synergistically induce acute liver injury in mice: an important role for CXCL1. Hepatology 2015;62:1070–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dominguez M, Miquel R, Colmenero J, Moreno M, García-Pagán JC, Bosch J, Arroyo V, et al. Hepatic expression of CXC chemokines predicts portal hypertension and survival in patients with alcoholic hepatitis. Gastroenterology 2009;136:1639–1650. [DOI] [PubMed] [Google Scholar]
- 17.Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, Barrieau M, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest 2012;122:3476–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ, Sandusky GE, et al. FGF-21 as a novel metabolic regulator. J Clin Invest 2005;115:1627–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen M, Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol 2009;10:741–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fisher FM, Chui PC, Nasser IA, Popov Y, Cunniff JC, Lundasen T, Kharitonenkov A, et al. Fibroblast growth factor 21 limits lipotoxicity by promoting hepatic fatty acid activation in mice on methionine and choline-deficient diets. Gastroenterology 2014;147:1073–1083.e1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pihlajamäki J, Lerin C, Itkonen P, Boes T, Floss T, Schroeder J, Dearie F, et al. Expression of the splicing factor gene SFRS10 is reduced in human obesity and contributes to enhanced lipogenesis. Cell Metab 2011;14:208–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Péterfy M, Phan J, Reue K. Alternatively spliced lipin isoforms exhibit distinct expression pattern, subcellular localization, and role in adipogenesis. J Biol Chem 2005;280:32883–32889. [DOI] [PubMed] [Google Scholar]
- 23.Li H, Cheng Y, Wu W, Liu Y, Wei N, Feng X, Xie Z, et al. SRSF10 regulates alternative splicing and is required for adipocyte differentiation. Mol Cell Biol 2014;34:2198–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao S, Jang C, Liu J, Uehara K, Gilbert M, Izzo L, Zeng X, et al. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature 2020;579:586–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin Z, Pan X, Wu F, Ye D, Zhang Y, Wang Y, Jin L, et al. Fibroblast growth factor 21 prevents atherosclerosis by suppression of hepatic sterol regulatory element-binding protein-2 and induction of adiponectin in mice. Circulation 2015;131:1861–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei N, Cheng Y, Wang Z, Liu Y, Luo C, Liu L, Chen L, et al. SRSF10 Plays a Role in Myoblast Differentiation and Glucose Production via Regulation of Alternative Splicing. Cell Rep 2015;13:1647–1657. [DOI] [PubMed] [Google Scholar]
- 27.Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc 2013;8:627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.