

Abstract
The meninges are densely innervated by nociceptive sensory neurons that mediate pain and headache1,2. Bacterial meningitis causes life-threatening infections of the meninges and central nervous system, affecting more than 2.5 million people a year3–5. How pain and neuroimmune interactions impact meningeal antibacterial host defences are unclear. Here we show that Nav1.8+ nociceptors signal to immune cells in the meninges through the neuropeptide calcitonin gene-related peptide (CGRP) during infection. This neuroimmune axis inhibits host defences and exacerbates bacterial meningitis. Nociceptor neuron ablation reduced meningeal and brain invasion by two bacterial pathogens: Streptococcus pneumoniae and Streptococcus agalactiae. S. pneumoniae activated nociceptors through its pore-forming toxin pneumolysin to release CGRP from nerve terminals. CGRP acted through receptor activity modifying protein 1 (RAMP1) on meningeal macrophages to polarize their transcriptional responses, suppressing macrophage chemokine expression, neutrophil recruitment and dural antimicrobial defences. Macrophage-specific RAMP1 deficiency or pharmacological blockade of RAMP1 enhanced immune responses and bacterial clearance in the meninges and brain. Therefore, bacteria hijack CGRP–RAMP1 signalling in meningeal macrophages to facilitate brain invasion. Targeting this neuroimmune axis in the meninges can enhance host defences and potentially produce treatments for bacterial meningitis.
The meninges consist of three membranes (dura, arachnoid and pia) that protect the central nervous system (CNS) from injury and infection1. Bacterial pathogens can invade the meninges and brain to cause severe pathology. Acute bacterial meningitis has mortality rates of up to 30%, and survivors often show post-infectious neurological sequelae3–5. S. pneumoniae is a leading cause of bacterial meningitis in children, in immunocompromised adults and in the elderly3,5. S. agalactiae (group B Streptococcus) is a leading cause of meningitis in neonates6. There is a need to better understand host responses to these bacterial pathogens and to define factors that protect the CNS against pathogen invasion. Nociceptors are peripheral somatosensory neurons that detect noxious and harmful stimuli, and whose activation leads to pain7. Nociceptors can communicate with the vasculature and immune cells through neuropeptides to induce neurogenic inflammation or immunomodulation8. A repertoire of innate and adaptive immune cells reside within the dural meninges that have roles in tissue repair, antigen sampling and host defence9–13. However, the impact of neuronal signals on these meningeal immune cells in host defence has not been clearly defined.
Nociceptors control bacterial meningitis
Bacterial meningitis is accompanied by severe headache3,14. The meninges are innervated by trigeminal ganglia (TG) nociceptors that drive headache1. Nav1.8 (encoded by SCN10A) is a voltage-gated sodium channel expressed by nociceptors that mediates mechanical, cold and inflammatory pain15. CGRP is stored in dense core vesicles in nociceptor nerve terminals16,17. We performed confocal imaging of mouse meninges and confirmed the occurrence of a dense network of Nav1.8+ and CGRP+ nerves in the dura (Fig. 1a). In humans, the main route of CNS invasion by bacterial pathogens is through haematogenous spread to the brain18. To model this infection, we intravenously injected mice with S. pneumoniae or S. agalactiae. We next isolated meningeal and brain tissue to analyse the time course of bacterial invasion (Fig. 1b–d). During S. pneumoniae infection in mice, bacteria reached the dura mater by 6 h and the inner meningeal layers (pia and arachnoid), choroid plexus (ChP) and the brain by 24 h (Fig. 1c and Extended Data Fig. 1a). During S. agalactiae infection in mice, bacteria invaded the dura mater, pia and arachnoid by 12 h and the brain by 24 h (Fig. 1d).
Fig. 1 |. Nociceptors regulate bacterial CNS invasion by S. pneumoniae and S. agalactiae.
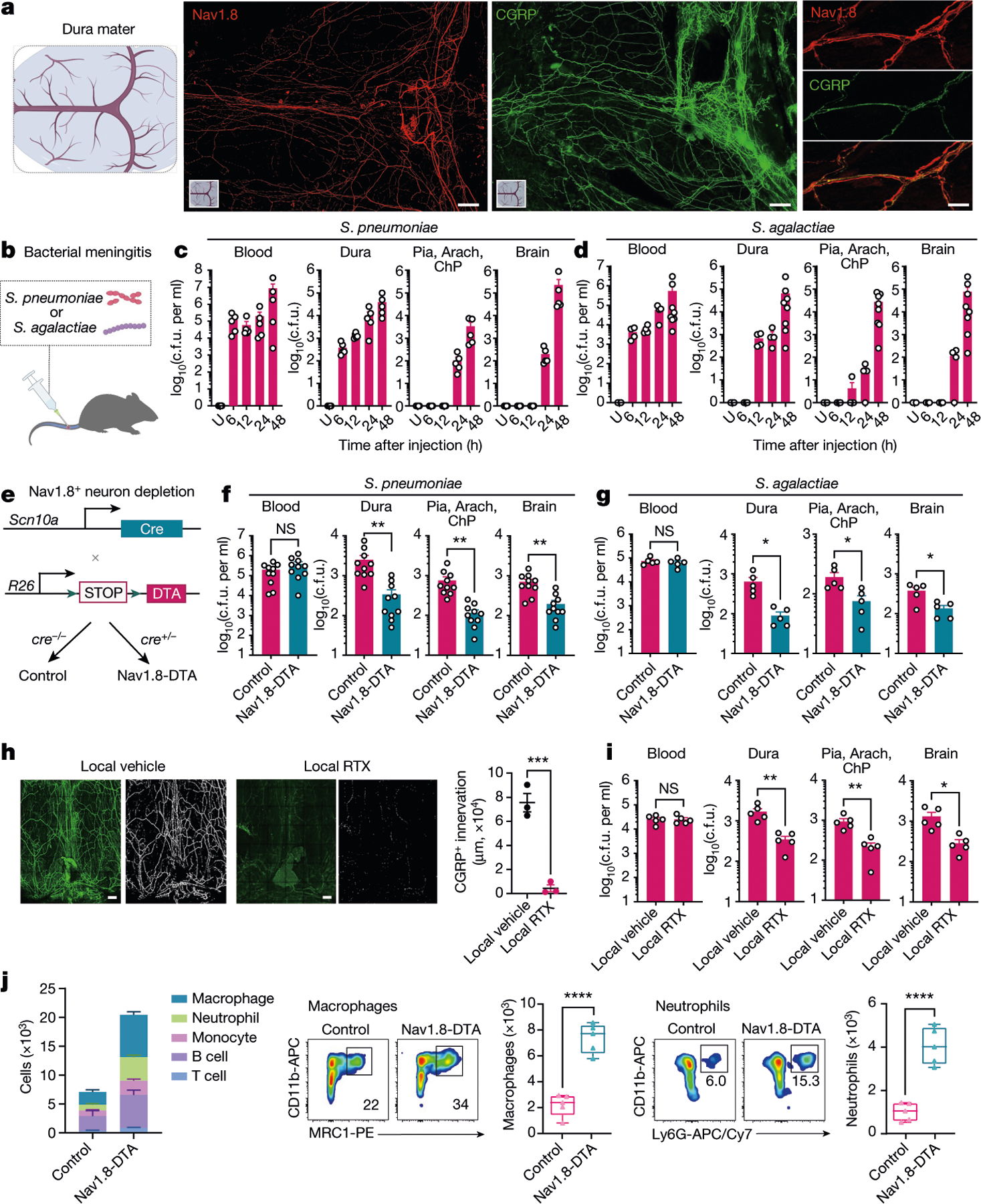
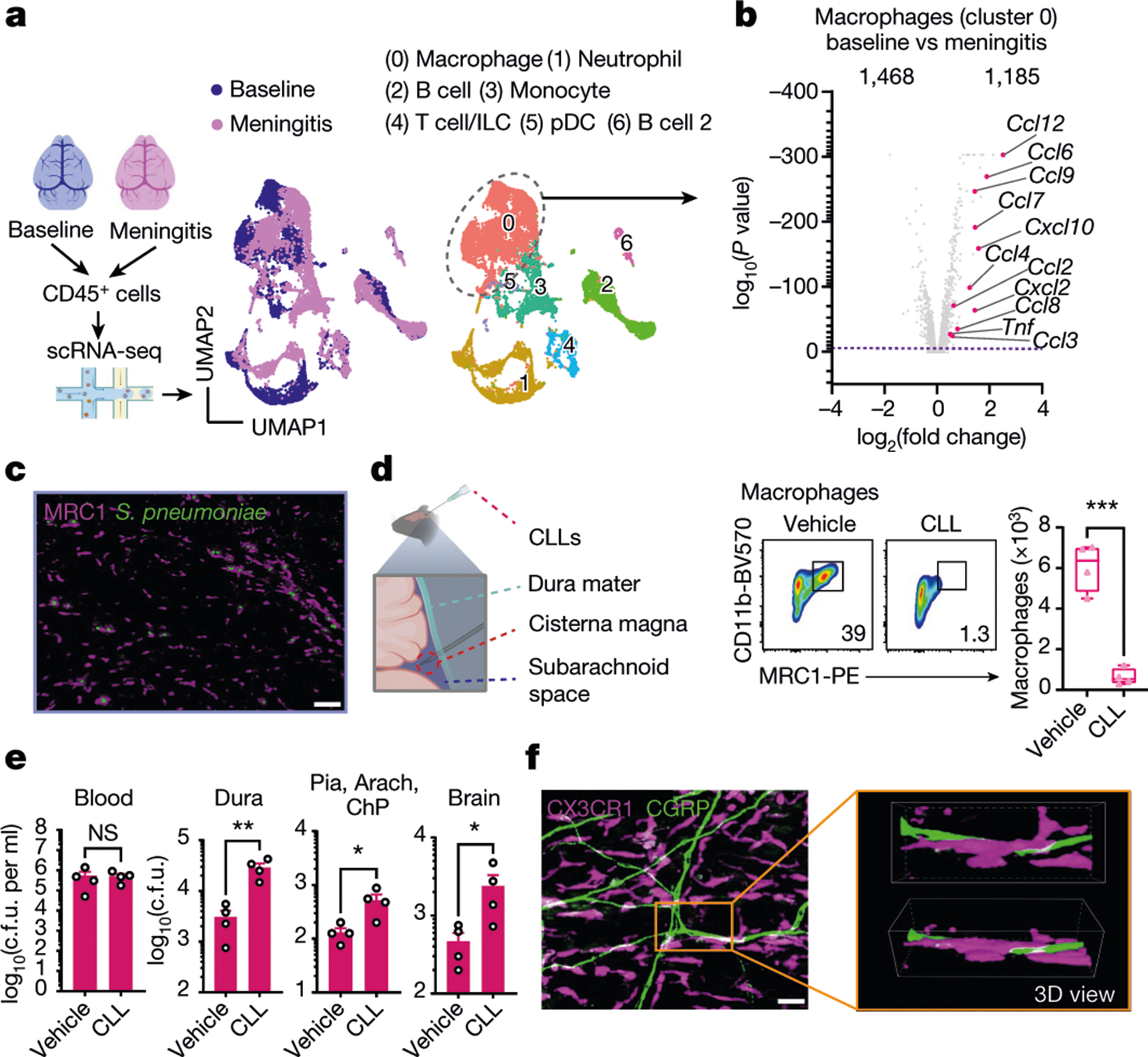
a, Whole-mount confocal images of mouse brain meninges (dura mater) showing tissue innervation by Nav1.8+CGRP+ nociceptors. Scale bars, 300 μm (low magnification) and 15 μm (high magnification). b, Scheme of the haematogenous bacterial meningitis model. Mice received an intravenous injection of the bacterial pathogen S. pneumoniae (3 × 107 colony forming units (c.f.u.)) or S. agalactiae (1 × 108 c.f.u.). c,d, Bacterial load recovery from blood, dura mater (dura), pia mater, arachnoid mater and choroid plexus (Pia, Arach, ChP), and brain samples collected at different time points after infection determined by quantitative culture (log(c.f.u.) plotted). c, After injection with S. pneumoniae. n = 3 (uninfected (U)) or n = 5 (6 h, 12 h, 24 h, 48 h). d, After injection with S. agalactiae. n = 3 (uninfected), n = 4 (6 h, 12 h, 24 h) or n = 8 (48 h). e, Generation of Nav1.8+ nociceptor-deficient (Nav1.8-DTA) and control mice. f,g, Bacterial load in samples collected from Nav1.8-DTA and control mice 24 h after injection with S. pneumoniae (n = 10 per group) (f) or S. agalactiae (n = 5 per group) (g). h, Images and quantification of meningeal innervation by CGRP+ neurons (green) was quantified (white) after local application of RTX or vehicle above skull suture sites (n = 3 per group). Scale bar, 300 μm. i, Bacterial load in samples collected 24 h after injection with S. pneumoniae from mice treated with a localized injection of vehicle or RTX (n = 5 per group). j, Flow cytometry quantification of meningeal immune cells in the dura mater of Nav1.8-DTA and control mice 24 h after S. pneumoniae infection. Left, combined numbers of immune cell subsets analysed. Centre, representative flow cytometry plots and quantification of meningeal macrophages (Cd11b+MRC1+ gates). Right, representative flow cytometry plots and quantification of meningeal neutrophils (Cd11b+Ly6G+ gates). n = 5 per group for all analysis in j. Statistical analysis: unpaired two-sided t-tests (f–j). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, NS, not significant. n = biologically independent samples from mouse tissues. Error bars indicate the mean ± s.e.m. Box plots show the median, interquartile range (IQR), and minimum and maximum values. Each experiment was repeated at least twice, and results presented are representative of 2 or more replicates. Exact P values are provided in Supplementary Table 1. Illustrations in a and b were created with BioRender.com.
To determine the role of nociceptors in host defence, we bred Nav1.8-Cre mice with Cre-dependent diphtheria toxin A (DTA) mice to create Nav1.8-DTA (Nav1.8-Cre+DTA+) nociceptor ablated mice and Cre− control littermates (Fig. 1e). When these mice were infected with S. pneumoniae or S. agalactiae, Nav1.8-DTA mice showed decreased CNS infection. In detail, reduced S. pneumoniae bacterial loads were recovered from meninges and brain at 24 h and 48 h after infection in Nav1.8-DTA mice (Fig. 1f and Extended Data Fig. 1b). By contrast, bacterial loads did not differ in blood, spleen, lung or skin (Fig. 1f and Extended Data Fig. 1b,c). Nav1.8-DTA mice also showed decreased meningeal and brain infection by S. agalactiae compared with control mice (Fig. 1g). Cleaved caspase-3 and histopathological analyses showed that Nav1.8-DTA mice had decreased staining levels and brain damage, respectively, compared with control littermates following S. pneumoniae infection (Extended Data Fig. 1d,e). As a second approach to target nociceptors, mice were treated systemically with resiniferatoxin (RTX), a high-affinity agonist for TRPV1, which can be used for chemical ablation of nociceptors19,20. We confirmed that meningeal nociceptor innervation was ablated in Nav1.8-DTA and RTX-treated mice compared with control mice (Extended Data Fig. 1f,g). RTX-treated mice also showed less S. pneumoniae invasion of the dura, pia, arachnoid, ChP and brain compared with vehicle-treated mice (Extended Data Fig. 1h). Given that the dura was an early site of infection, we proposed that nociceptors regulate invasion before bacterial entry into the brain. When bacteria were intracisternally injected (to bypass the dura mater), Nav1.8-DTA mice and control mice did not show differences in bacterial loads (Extended Data Fig. 1i).
We next asked whether locally targeting meningeal nociceptive nerves affects outcomes of bacterial meningitis. RTX or vehicle was injected above skull suture sites, which are innervated by extracranial branches of meningeal nociceptors2. Localized RTX injection led to loss of CGRP+ nerves in meninges but not CGRP+ nerves in back skin (Fig. 1h and Extended Data Fig. 2a). Following S. pneumoniae intravenous infection, RTX-treated mice showed decreased bacterial invasion of the meninges and brain compared with vehicle-treated mice (treatments given through localized injections above skull sutures) (Fig. 1i). As a second approach for meningeal nerve ablation, Nav1.8-Cre mice were bred with CGRPα–GFP-DTRflox mice21, in which Cre induces diphtheria toxin receptor (DTR) expression under Calca and enables DTX-mediated ablation of Nav1.8+CGRP+ neurons. In these mice, DTX injection locally above skull sutures depleted CGRP+ nerves in meninges but not back skin (Extended Data Fig. 2b,c). After S. pneumoniae infection, mice ablated of Nav1.8+CGRP+ meningeal nerves showed less bacterial invasion of the dura, pia, ChP and brain compared with littermate controls (Extended Data Fig. 2d).
We hypothesized that nociceptors may regulate meningeal immune cell responses. S. pneumoniae infection increased dural immune cell populations over time (Extended Data Fig. 3a). Compared with control littermates, Nav1.8-DTA mice showed increased meningeal leukocyte numbers following S. pneumoniae infection, with greater myeloid (macrophages, neutrophils and monocytes) and lymphocyte (B cells and T cells) recruitment (Fig. 1j and Extended Data Fig. 3b). In the dura, blood and spleen, immune populations did not differ between Nav1.8-DTA and control mice at baseline (Extended Data Fig. 3c,d). Mannose receptor C-type 1 (MRC1) is a marker for meningeal macrophages12, and CD11b+MRC1+ dural macrophages were increased in Nav1.8-DTA mice compared with control mice following S. pneumoniae infection (Fig. 1j). Neutrophils are key responders to infection and are increased in the cerebrospinal fluid of patients with meningitis18. CD11b+Ly6G+ neutrophils were also increased in Nav1.8-DTA mice compared with control littermates following infection (Fig. 1j).
Bacteria induce meningeal CGRP release
Bacterial meningitis is characterized by headache in humans3,14, but pain during bacterial meningitis has not been characterized in animals. Grimace scores measure headache-like pain behaviours in mice22. Following S. pneumoniae and S. agalactiae infection, mice displayed increased grimace scores compared with uninfected mice (Extended Data Fig. 4a). CGRP injection also induced increased grimace scores (Extended Data Fig. 4b). Nociceptor activation causes the release of CGRP from nerve terminals in the dura that can drive headache16. We asked whether bacterial meningitis induces CGRP release. Using meningeal explants, we observed increased levels of soluble CGRP over the course of S. pneumoniae infection (Fig. 2a,b). Nav1.8-DTA mice displayed decreased CGRP release compared with control mice, which shows that nociceptors are a major source of CGRP (Fig. 2c). S. agalactiae infection also induced CGRP release in the meninges (Fig. 2d). When mice were infected with fluorescently labelled bacteria, CGRP+ nerve fibres were found juxtaposed with S. pneumoniae and S. agalactiae in dural meninges (Fig. 2e). The dura is innervated by nociceptors, and the somas of these receptors are in TG. We found that S. pneumoniae accumulated in TG in proximity to CGRP+ cell bodies after infection (Extended Data Fig. 4c).
Fig. 2 |. Bacteria activate nociceptors that release CGRP in meninges.
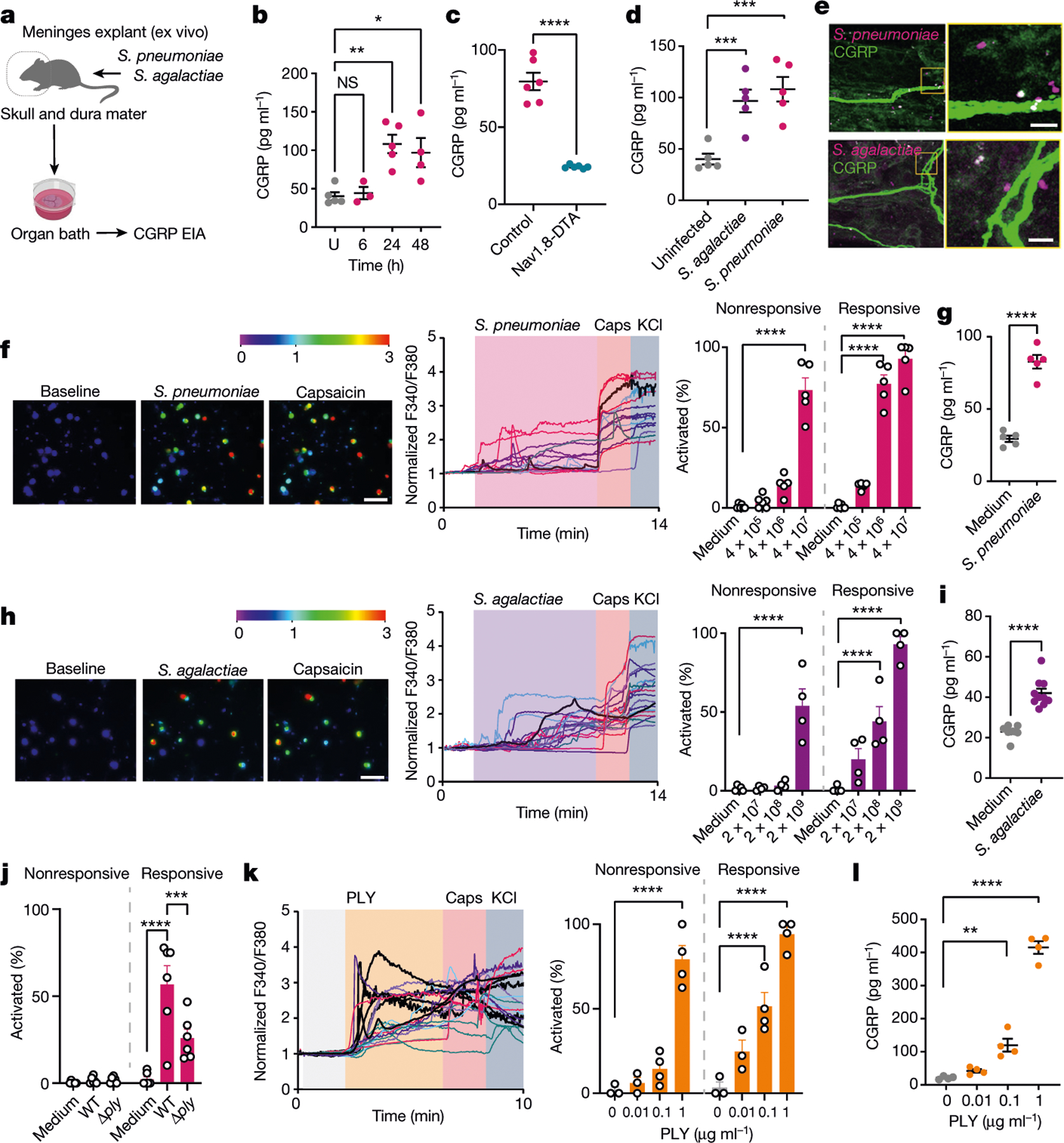
a, Illustration of meninges explant CGRP assays from mice infected with S. pneumoniae (3 × 107 c.f.u.) or S. agalactiae (1 × 108 c.f.u.). EIA, enzyme immunoassay. b, CGRP release induced by S. pneumoniae. n = 5 (uninfected), n = 3 (6 h), n = 5 (24 h), n = 4 (48 h). c, CGRP released from Nav1.8-DTA and control mice 24 h after S. pneumoniae injection (n = 6 per group). d, CGRP released 24 h after S. pneumoniae or S. agalactiae injection (n = 5 per group). e, Dura mater showing bacteria near CGRP+ nociceptors 24 h after S. pneumoniae (3 × 107 c.f.u.) or S. agalactiae (1 × 108 c.f.u.) infection. Scale bar, 5 μm. f, Representative fields (left) and calcium traces (centre) of trigeminal neurons responding to S. pneumoniae (4 × 107 c.f.u.), capsaicin (Caps; 1 μM) and/or KCl (40 mM). Color bar, F340/F380 ratio. Scale bar, 100 μm. Right, proportions of capsaicin-nonresponsive and capsaicin-responsive neurons responding to S. pneumoniae. n = 6 (medium, 4 × 105 c.f.u.) or n = 5 (4 × 106, 4 × 107 c.f.u.). g, CGRP concentrations in trigeminal neuron supernatants incubated with S. pneumoniae (4 × 107 c.f.u.) for 30 min. n = 5 per group. h, Representative fields (left) and calcium traces (centre) of trigeminal neurons responding to S. agalactiae (2 × 109 c.f.u.), capsaicin (1 μm) and/or KCl (40 mM). Color bar, F340/F380 ratio. Scale bar, 100 μm. Right, capsaicin-nonresponsive and capsaicin-responsive neurons responding to S. agalactiae (2 × 107 to 2 × 109 c.f.u.). n = 4 per group. i, CGRP levels from trigeminal neurons incubated for 30 min with S. agalactiae (2 × 109 c.f.u.). n = 7 (medium) or n = 10 (S. agalactiae). j, Capsaicin-nonresponsive and capsaicin-responsive neurons to WT or Δply S. pneumoniae (4 × 106 c.f.u.). n = 6 per group. k, Left, calcium traces of neurons responding to PLY (1 μg ml−1), capsaicin (1 μM) and/or KCl (40 mM). Right, proportions of capsaicin-nonresponsive and capsaicin-responsive neurons to PLY. n = 3 (0–0.01 μg ml−1) or n = 4 (0.1–1 μg ml−1). l, CGRP levels from trigeminal neurons stimulated with PLY (0.01–1 μg ml−1) for 30 min. n = 4 per group. Statistical analysis: one-way ANOVA with Tukey post-tests (b,d,f,h,j–l) or unpaired two-sided t-tests (c,g,i). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. n = biologically independent samples from mouse tissues (b–d) and cells (f–l). Mean ± s.e.m. shown. Exact P values are provided in Supplementary Table 1. Illustration in a was created with BioRender.com.
Bacteria activate trigeminal nociceptors
We next asked whether bacterial pathogens can directly activate nociceptors to release CGRP. Measurement of intracellular calcium levels showed that TG neurons responded to S. pneumoniae, with increasing bacterial concentrations activating greater neuronal proportions (Fig. 2f). Many bacteria-responsive neurons also responded to capsaicin, a ligand for TRPV1 (ref.7). Furthermore, S. pneumoniae application induced CGRP release from TG neurons (Fig. 2g). S. agalactiae also induced TG neuron calcium influx and CGRP release (Fig. 2h,i). We previously found that pore-forming toxins can activate nociceptors to drive pain20,23. S. pneumoniae utilizes pneumolysin (PLY), a cholesterol-dependent cytolysin, for CNS invasion24,25. Wild-type (WT) S. pneumoniae induced more calcium responses compared with isogenic mutant bacteria lacking PLY (Δply) (Fig. 2j). After S. pneumoniae infection, we detected PLY+ bacteria localized close to CGRP+ nerves in dural meninges (Extended Data Fig. 4d). Recombinant PLY induced calcium responses and CGRP release in TG neurons in a dose-dependent manner (Fig. 2k–l). We next cultured TG neurons in microfluidic chips to compartmentally isolate axons from soma and non-neuronal cells to determine whether PLY can act directly on nerves. PLY applied to the axonal chamber induced calcium responses (Extended Data Fig. 4e,f). S. agalactiae produces a β-haemolysin/cytolysin (β-H/C), which mediates its pathogenesis, including development of meningitis26. WT S. agalactiae induced more calcium responses in TG neurons compared with ΔcylE bacteria, which lack β-H/C (Extended Data Fig. 4g). Serum CGRP levels did not differ after S. pneumoniae infection in Nav1.8-DTA mice compared with control mice (Extended Data Fig. 4h), which indicated the potential specificity of this bacteria for the meninges. Taken together, these data show that bacterial pathogens can activate nociceptors and CGRP release in the meninges through secreted toxins.
CGRP suppresses meningeal host defences
We next proposed that CGRP signalling could play a part in antibacterial host defences. RAMP1 forms a receptor complex with its co-receptor calcitonin-receptor-like receptor (CALCRL) to bind CGRP16,17. Ramp1−/− mice showed less bacterial invasion of the meninges and brain following S. pneumoniae infection compared with Ramp1+/+ littermate controls (Fig. 3a). We observed increased meningeal immune cells, including macrophages and neutrophils, in infected Ramp1−/− mice compared with Ramp1+/+ mice (Fig. 3b and Extended Data Fig. 5a). By contrast, systemic CGRP injection into mice potentiated S. pneumoniae CNS invasion and decreased immune cell numbers in the meninges (Fig. 3c,d and Extended Data Fig. 5b). Treatment with CGRP also potentiated S. agalactiae CNS invasion (Extended Data Fig. 5c). CGRP has been shown to induce anti-inflammatory cytokine interleukin-10 (IL-10)27, and IL-10 expression increases during bacterial meningitis28. However, treatment of mice with anti-IL-10 antibody did not affect S. pneumoniae infection caused by CGRP injection (Extended Data Fig. 5d). We next treated mice with BIBN4096S (also known as olcegepant), an antagonist of RAMP1 signalling29. Pre-treatment with BIBN4096 reduced bacterial CNS invasion compared with vehicle treatment (Fig. 3e). We next determined the effect of treatment after infection, whereby BIBN4096 was given at 6 h, when we had observed S. pneumoniae meningeal invasion (Fig. 1c), followed by daily dosing. Treatment after infection delayed the onset of clinical symptoms of S. pneumoniae infection in mice, and reduced weight loss and mortality (Extended Data Fig. 6a). Bacterial load recovery from meninges and brain was also decreased (Extended Data Fig. 6b). Bacterial loads in lung, spleen and skin did not differ after BIBN4096 treatment (Extended Data Fig. 6c).
Fig. 3 |. CGRP and RAMP1 signalling contribute to bacterial meningitis.
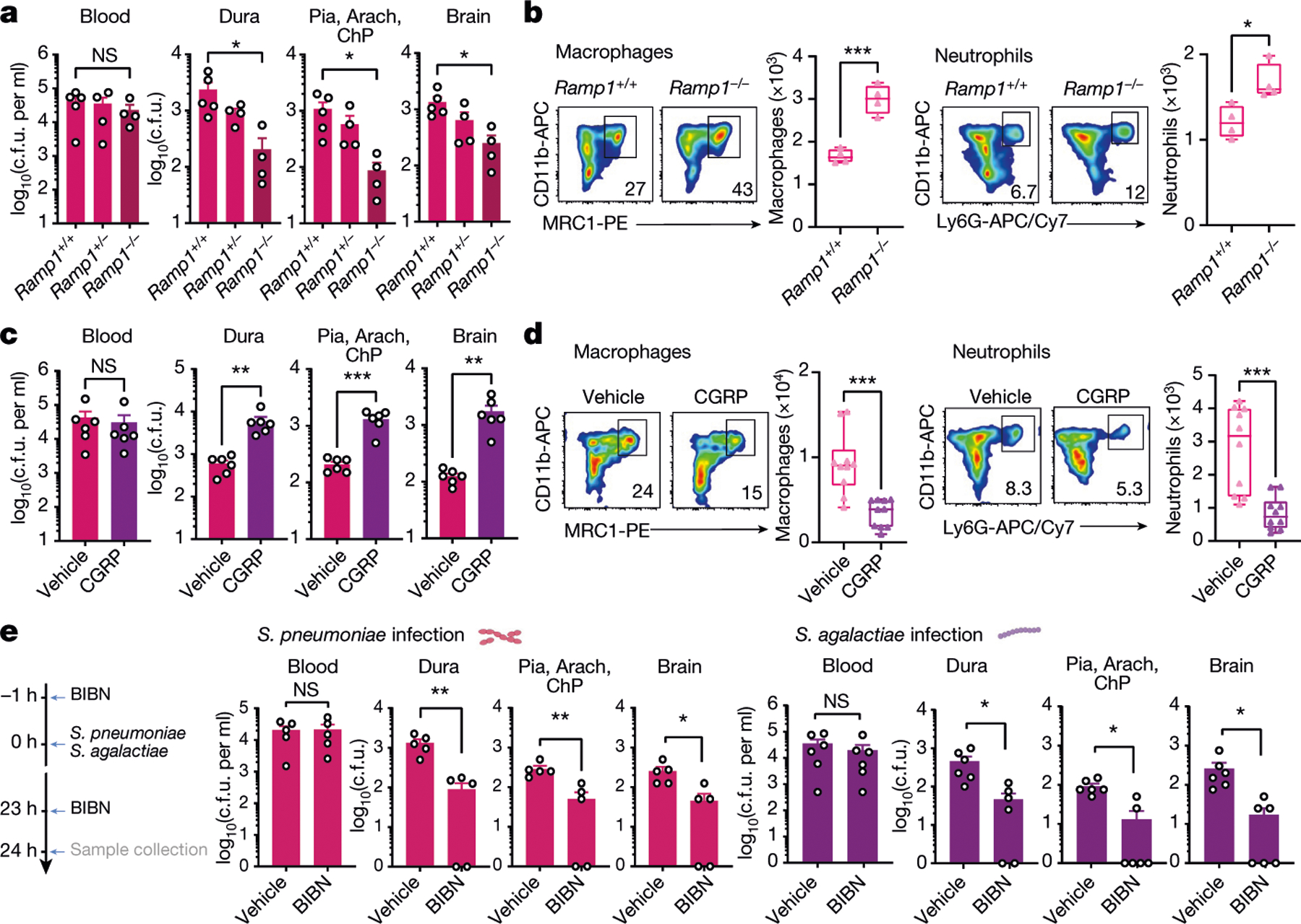
a,b, Role of the CGRP receptor RAMP1 in bacterial meningitis. a, Bacterial load in samples collected from Ramp1 knockout (Ramp1−/−) and control littermate (Ramp1+/−, Ramp1+/+) mice 24 h after injection with S. pneumoniae (3 × 107 c.f.u.). n = 5 (Ramp1+/+) or n = 4 (Ramp1−/−, Ramp1+/−). b, Flow cytometry quantification of meningeal macrophages (Cd11b+MRC1+ gates) and neutrophils (Cd11b+Ly6G+ gates) in Ramp1 knockout (Ramp1−/−) and control (Ramp1+/+) mice 24 h after injection with S. pneumoniae (3 × 107 c.f.u.). n = 4 per group. c,d, Impact of CGRP (2 μg, intraperitoneally (i.p.)) treatment on bacterial meningitis. c, Bacterial load in samples collected 24 h after injection with S. pneumoniae (3 × 107 c.f.u.) in mice treated with CGRP or vehicle. n = 6 per group. d, Flow cytometry quantification of meningeal macrophages (Cd11b+MRC1+ gates) and neutrophils (Cd11b+Ly6G+ gates) 24 h after injection with S. pneumoniae (3 × 107 c.f.u.) in mice treated with CGRP or vehicle. n = 10 per group. e, Left, scheme of the experiment to examine the protective effects of the RAMP1 antagonist BIBN4096 (BIBN) on bacterial meningitis. Right, bacterial load in samples collected 24 h after injection with S. pneumoniae (3 × 107 c.f.u.; n = 5 per group) or S. agalactiae (1 × 108 c.f.u.; n = 6 per group) from mice treated with BIBN4096 (300 μg kg−1, i.p.) or vehicle. Statistical analysis: one-way ANOVA with Tukey post-tests (a) or unpaired two-sided t-tests (b–e). *P < 0.05, **P < 0.01, ***P < 0.001. n = biologically independent samples from mouse tissues. Error bars indicate the mean ± s.e.m. Box plots show the median, IQR, and minimum and maximum values. Each experiment was performed at least twice, and results presented are representative of 2 or more replicates. Exact P values are provided in Supplementary Table 1.
We next performed single-cell RNA-sequencing (scRNA-seq) analysis of CD45+ meningeal cells to determine whether meningeal immune cells can respond to neurons (Fig. 4a). After defining cluster identities by comparing transcriptional markers with published meningeal single-cell datasets10,30, we observed a repertoire of myeloid and lymphoid cells (Fig. 4a and Extended Data Fig. 7a). We next analysed these immune populations for expression of neuropeptide receptors. Ramp1 and Calcrl ranked at the top of the list of neuropeptide receptors expressed in meningeal immune cell clusters (Fig. 4b). Other neuropeptide receptors expressed included Ramp2, Ramp3, Vipr1, Tacr1, Mrgbrb2 and Adcyap1r1, although at much lower levels compared with Ramp1 and Calcrl (Fig. 4b). Ramp1 was highly expressed across myeloid immune cells (neutrophils, monocytes and macrophages) that were also marked by the antimicrobial enzyme lysozyme M (encoded by Lyz2) (Fig. 4c).
Fig. 4 |. Loss of myeloid Ramp1 expression improves the meningeal response against infection.
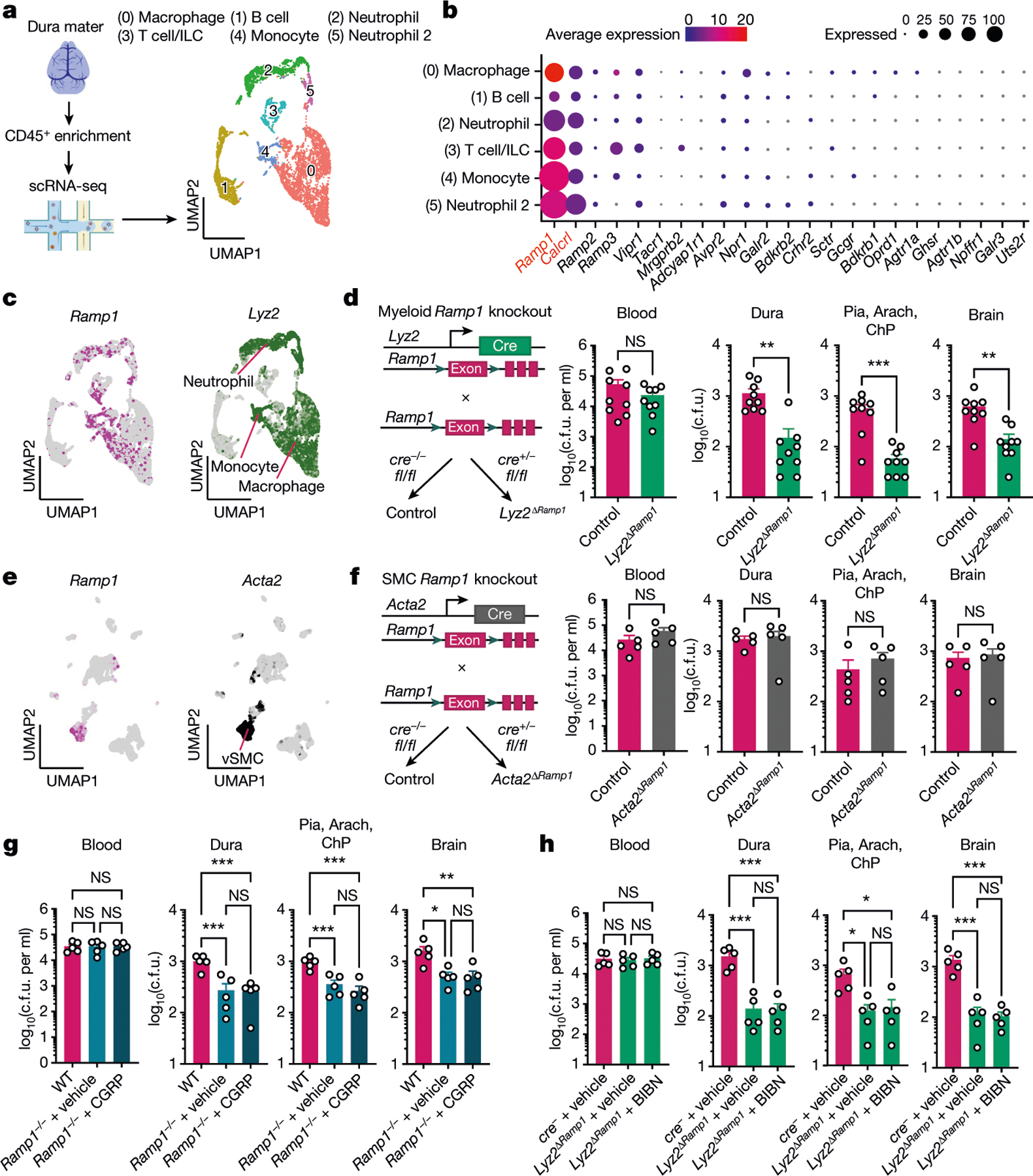
a,b, scRNA-seq analysis of CD45+ meningeal cells. a, Left, illustration of scRNA-seq analysis. Right, uniform manifold approximation and projection (UMAP) visualizations of CD45+ cell types in dural meninges at baseline. ILC, innate lymphoid cells. b, Dot plots showing the average expression levels per cluster and the percentage of cells from each cluster expressing genes for neuropeptide receptors. Ramp1 and Calcrl, which form the CGRP receptor complex, are highlighted in red font. n = 10 pooled meninges. c, Expression of the CGRP receptor Ramp1 and the myeloid marker Lyz2 by meningeal CD45+ cells. d, Left, schematic of the generation of myeloid-specific Ramp1 knockout (Lyz2ΔRamp1) mice. Right, bacterial load in samples collected from Lyz2ΔRamp1 and control mice 24 h after injection with S. pneumoniae (3 × 107 c.f.u.). n = 9 per group. e, UMAP visualization of CD45− meningeal cells expressing Ramp1 and the vascular smooth muscle cell (vSMC) marker Acta2. n = 10 pooled meninges. f, Left, schematic of the generation of SMC-specific Ramp1 knockout (Acta2ΔRamp1) mice. Right, bacterial load in samples collected from Acta2ΔRamp1 and control mice 24 h after injection with S. pneumoniae (3 × 107 c.f.u.). n = 5 per group. g, Bacterial load in samples collected from global Ramp1 knockout (Ramp1−/−) and control (WT) mice 24 h after injection with S. pneumoniae (3 × 107 c.f.u.). Ramp1 knockout mice were treated with a RAMP1 agonist (CGRP, 2 μg, i.p.) or vehicle. n = 5 per group. h, Bacterial load in samples collected from Lyz2ΔRamp1 and control mice 24 h after injection with S. pneumoniae (3 × 107 c.f.u.). Lyz2ΔRamp1 mice were treated with a RAMP1 antagonist (BIBN4096, 300 μg kg−1 i.p.) or vehicle. n = 5 per group. Statistical analysis: unpaired two-sided t-tests (d,f) or one-way ANOVA with Tukey post-tests (g,h). *P < 0.05, **P < 0.01, ***P < 0.001. n = biologically independent samples from mouse tissues. Each experiment was performed at least twice, and results presented are representative of 2 or more replicates. Data shown as the mean ± s.e.m. Exact P values are provided in Supplementary Table 1. Illustration in a was created with BioRender.com.
We next investigated which meningeal cell types mediate RAMP1 signalling to affect bacterial meningitis. To this end, we bred tissue-specific Cre lines with Ramp1fl/fl mice to generate conditional knockout strains. First, we bred Lyz2cre mice with Ramp1fl/fl mice to ablate the receptor in myeloid immune cells (Fig. 4c,d). Loss of Ramp1 in myeloid cells (Lyz2ΔRamp1) led to significantly reduced bacterial loads in the meninges and brain following S. pneumoniae infection compared with control Ramp1fl/fl mice (Fig. 4d). scRNA-seq analysis of CD45− non-immune cells of the meninges showed that Acta2+ vascular smooth muscle cells represented the main non-immune population expressing Ramp1 and Calcrl (Fig 4e and Extended Data Fig. 7b). We bred Acta2cre mice with Ramp1fl/fl mice to determine the role of RAMP1 in vascular smooth muscle cells during S. pneumoniae infection. In contrast to Lyz2cre-mediated Ramp1 ablation, Acta2ΔRamp1 mice did not show differences in bacterial recovery from the meninges or brain compared with control cre− mice (Fig 4f). We next ascertained whether CGRP and BIBN4096 may have effects independent of RAMP1 signalling in our infection models. Ramp1−/− mice showed decreased bacterial loads in the meninges and brain compared with WT controls. By contrast, CGRP did not potentiate S. pneumoniae infection in Ramp1−/− mice (Fig. 4g). BIBN4096S treatment of Lyz2ΔRamp1 mice did not further enhance bacterial clearance in the meninges and brain compared with vehicle-treated Lyz2ΔRamp1 mice. This result indicates that the antagonist probably regulates infection through myeloid immune RAMP1 signalling (Fig. 4h). Therefore, RAMP1 signalling in myeloid immune cells suppress effective host defences during bacterial meningitis.
Macrophages drive antibacterial immunity
Macrophages and other professional phagocytes are important mediators of bacterial ingestion, killing and resolution of infections. To better understand leukocyte responses against bacterial meningitis, we performed scRNA-seq of CD45+ immune cells collected from the dural meninges 24 h after S. pneumoniae infection and of immune cells collected from uninfected animals (Fig. 5a and Extended Data Fig. 8a). Infection induced significant transcriptional changes, with the greatest number of differentially expressed genes in myeloid immune cell clusters composed of macrophages, monocytes and neutrophils (Extended Data Fig. 8b). Pathway enrichment analysis further highlighted the roles of these cells in host responses to infection (Fig. 5b and Extended Data Fig. 8c–e).
Fig. 5 |. Meningeal macrophages are required for host defence against S. pneumoniae infection.
a,b, scRNA-seq analysis of meningeal immune responses to bacterial infection. a, Left, illustration of single-cell analysis of S. pneumoniae infection. Right, UMAP visualizations of CD45+ cell types in the meninges at baseline and 24 h after injection of S. pneumoniae (meningitis, 3 × 107 c.f.u.). pDC, plasmacytoid dendritic cells. b, Volcano plot showing genes that are differentially expressed (1,468 downregulated genes; 1,185 upregulated genes) in the cluster of Mrc1+ macrophages in response to infection (baseline versus meningitis), highlighting the upregulation of chemotaxis-related genes (n = 10 pooled meninges per group). c, Whole-mount confocal images of mouse meninges (dura mater) showing meningeal macrophages (MRC1+ cells) associated with CMTPX-labelled S. pneumoniae 24 h after injection of bacteria. Scale bar, 25 μm. d,e, Depletion of meningeal MRC1+ macrophages by intracisternal injection of CLLs 3 days before infection. d, Left, illustration of experimental scheme. Right, flow cytometry analysis of meningeal macrophages. n = 4 per group. e, Bacterial loads 24 h after injection with S. pneumoniae (3 × 107 c.f.u.) in mice treated with CLLs or vehicle. n = 4 per group. f, Whole-mount confocal images of meninges (dura mater) showing the presence of macrophages (CX3CR1+ cells) near CGRP+ nerve fibres. Scale bar, 10 μm. Statistical analysis: Wilcoxon rank-sum test, dashed line, P = 0.01 (b) or unpaired two-sided t-tests (d,e). *P < 0.05, **P < 0.01, ***P < 0.001. n = biologically independent samples from mouse tissues. Each experiment was performed at least twice, and results presented are representative of 2 or more replicates. Error bars indicate the mean ± s.e.m. Box plots show the median, IQR, and minimum and maximum values. Exact P values provided in Supplementary Table 1. Illustrations in a and d were created with BioRender.com.
Macrophages are the most abundant immune cell type in the meninges and therefore may be the first responders to bacteria. We detected S. pneumoniae associated with meningeal MRC1+ macrophages 24 h after infection (Fig. 5c and Extended Data Fig. 9a). In macrophages, biological processes related to leukocyte recruitment, including increased expression of mediators that promote chemotaxis such as Ccl12, Ccl2, Ccl3 and Tnf, were enriched (Fig. 5b and Extended Data Fig. 7c). We proposed that macrophages may orchestrate meningeal protection through the recruitment of immune cells that exert antimicrobial functions such as neutrophils and monocytes (Extended Data Fig. 8d,e). Intracisternal injection of clodronate-laden liposomes (CLLs) to target CNS-associated phagocytes, including meningeal macrophages, depleted meningeal MRC1+ macrophages but not dendritic cells, monocytes or neutrophils at baseline (Fig. 5d and Extended Data Fig. 9b). Following S. pneumoniae infection, CLL treatment increased bacterial loads in the meninges and brain compared with mice treated with vehicle liposomes (Fig. 5e). Moreover, CLL treatment resulted in an overall reduction in recruited immune cells in the meninges but not in liver tissues (Extended Data Fig. 9c,d). Although we cannot rule out effects of CLLs on other cells, these data suggest a key role for meningeal macrophages in host defence. Dural CX3CR1+ macrophages were also often found associated with CGRP+ nerve fibres (Fig. 5f). We therefore next investigated how CGRP–RAMP1 signalling affects meningeal macrophages during infection.
RAMP1 regulates meningeal macrophages
Because Lyz2 is expressed in all myeloid cells, we sought to identify a marker to target Ramp1 in meningeal macrophages without affecting other phagocytes. A recent study31 found that CNS border-associated macrophages, including meningeal macrophages, express Pf4 (which encodes CXCL4) and can be labelled using Pf4cre mice; Pf4 was also absent in microglia. Our scRNA-seq analysis of meningeal cells confirmed Pf4 expression by CD11b+MRC1+ dural macrophages but not in neutrophils or monocytes (Fig. 6a and Extended Data Fig. 8a). Pf4cre mice were bred with TdTomato reporter mice, which resulted in the labelling of macrophages associated with CGRP+ nerves and CD31+ blood vessels in dural meninges (Fig. 6b). We next bred Pf4cre mice with Ramp1fl/fl mice to generate mice lacking Ramp1 specifically in Pf4+ cells (Pf4ΔRamp1). Similar to results observed in myeloid-specific Ramp1 knockout mice (Lyz2ΔRamp1), Pf4ΔRamp1 mice were less susceptible to meningeal and CNS infections by S. pneumoniae compared with control mice (Fig. 6c). Of note, Pf4 is also expressed by megakaryocytes, although these cells are absent in meningeal populations. Taken together, these data indicate that RAMP1 signalling in macrophages regulates meningeal host defences.
Fig. 6 |. CGRP suppresses meningeal macrophage immunity.
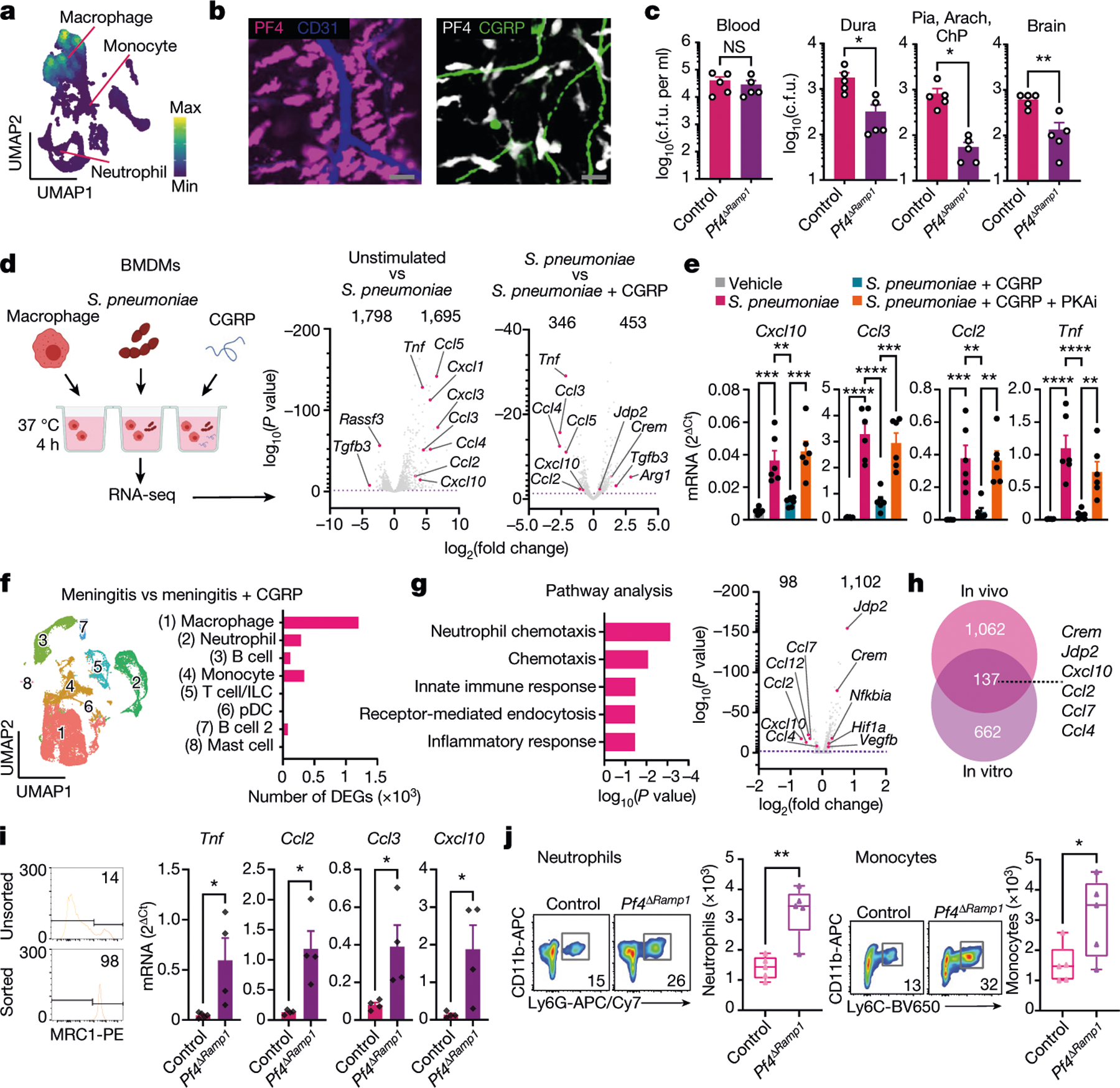
a, UMAP visualization of Pf4 expression in meningeal macrophages. b, Whole-mount meningeal images of PF4+ macrophages, CD31+ blood vessels and CGRP+ nerves. Scale bar, 15 μm. c, Bacterial load from Pf4ΔRamp1 and control mice 24 h after S. pneumoniae injection (3 × 107 c.f.u.). n = 5 per group. d,e, In vitro assay using BMDMs. d, Left, illustration of experimental scheme. Right, volcano plots showing differentially expressed genes (DEGs) in unstimulated macrophages, S. pneumoniae-infected macrophages, and with CGRP treatment (S. pneumoniae versus S. pneumoniae + CGRP), highlighting chemotaxis-related genes. n = 4 per group. e, qPCR analysis of chemotaxis-related genes in macrophages exposed to S. pneumoniae, CGRP and a PKAi. n = 6 per group. f,g, scRNA-seq analysis of meningeal immune responses to infection and CGRP treatment in vivo. f, Left, UMAP visualizations of CD45+ cells in meninges from uninfected mice, infected with S. pneumoniae and/or treated with CGRP. Right, number of differentially expressed genes induced by CGRP treatment after infection in each population. g, Gene ontology (GO) biological processes of macrophages (cluster 0) (left) and volcano plot (right) of macrophage genes affected by CGRP treatment in vivo. h, Venn diagram highlighting the overlap of genes induced by CGRP in vitro and in vivo. i, qPCR of macrophages sorted from meninges of control mice (n = 5 for Tnf; n = 4 for Ccl2, Ccl3, Cxcl10) and Pf4ΔRamp1 mice (n = 4 for Tnf, Ccl2, Ccl3, Cxcl10) 24 h after S. pneumoniae injection. j, Flow cytometry of neutrophils (Cd11b+Ly6G+ gates) and monocytes (Cd11b+Ly6G−Ly6Chi gates) in meninges of Pf4ΔRamp1 and control mice 24 h after S. pneumoniae injection. n = 5 per group. Statistical analysis: Wilcoxon rank-sum test, dashed line, P = 0.01 (d,g), unpaired two-sided t-tests (c,i,j) or one-way ANOVA with Tukey post-tests (e). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. n = biologically independent samples from mouse tissues (c,i,j) and primary cells (d,e). Each experiment was performed at least twice, and results presented are representative of 2 or more replicates. Numbers at the top of the plots in d and g indicate the number of downregulated and upregulated genes. Error bars indicate the mean ± s.e.m. Box plots show the median, IQR, and minimum and maximum values. Exact P values are provided in Supplementary Table 1. Illustration in d was created with BioRender.com.
To gain insight into how CGRP affects macrophage responses to S. pneumoniae, bone-marrow-derived macrophages (BMDMs) were incubated with serum-opsonized S. pneumoniae in the presence of CGRP or vehicle. We did not observe defects in macrophage antimicrobial activity when exposed to CGRP (Extended Data Fig. 10a). We next asked whether CGRP induces transcriptional changes in macrophages in response to bacterial infection. To this end, we performed RNA-seq analysis of cells treated with vehicle, with bacteria alone or with CGRP and bacteria (Fig. 6d). Macrophages exposed to S. pneumoniae had upregulated levels of cytokines (Fig. 6d). By contrast, treatment with CGRP polarized bacteria-stimulated macrophage transcriptional phenotypes to a suppressed expression of cytokines, including TNF, CCL3 and CCL4 (Fig. 6d). We confirmed that protein levels of these cytokines were also reduced by CGRP treatment during S. pneumoniae infection of macrophages (Extended Data Fig. 10b). RAMP1–CALCRL forms a G protein-coupled receptor that signals through Gαs, which leads to increased levels of the second messenger cyclic AMP and activation of the cAMP-dependent protein kinase PKA17. To investigate the role of this pathway, macrophages were treated with the cAMP analogue Rp-8-CPT-cAMP, a PKA inhibitor (PKAi), before application of CGRP and S. pneumoniae. Quantitative PCR with reverse transcription (RT–qPCR) analysis showed that CGRP downregulated the expression of multiple chemotactic mediators (Fig. 6e and Extended Data Fig. 10c). This cytokine suppression activity of CGRP was blocked when cells were pre-treated with the PKAi (Fig. 6e). To determine whether CGRP affects meningeal macrophages in vivo, we performed scRNA-seq analysis of dural CD45+ cells from mice treated with CGRP or vehicle during S. pneumoniae infection (Fig. 6f). CGRP treatment substantially altered the transcriptional profile of meningeal immune cell populations, with the greatest number of differentially expressed genes in Mrc1+Pf4+ macrophages (Fig. 6f). Pathway enrichment showed that CGRP treatment suppressed chemotactic gene expression (Fig. 6g). In line with our hypothesis, we identified multiple chemokines that were downregulated by CGRP in vivo, including Ccl2 and Cxcl10 (Fig. 6g), which overlapped with CGRP-downregulated genes in vitro (Fig. 6h). We also observed upregulation of Crem (which encodes ICER) and Jdp2 (which encodes Jun dimerization protein 2) (Fig. 6d,g,h and Extended Data Fig. 10c), transcription factors previously found to downregulate NF-κB and cytokine expression in macrophages32,33. These data indicate that the CGRP–RAMP1 axis induces a transcriptional programme in macrophages that blunts cytokine expression.
We next determined the role of meningeal macrophage RAMP1 signalling in S. pneumoniae infection using Pf4ΔRamp1 mice. Compared with control mice, macrophages isolated from the meninges of Pf4ΔRamp1 mice showed increased Tnf, Ccl2, Ccl3 and Cxcl10 levels (Fig. 6i). By contrast, expression of Crem, Jdp2 and Ramp1 were downregulated in Pf4ΔRamp1 mice (Extended Data Fig. 10d). At baseline, macrophages isolated from Nav1.8-DTA mice did not show changes in chemokines (Extended Data Fig. 10e). In support of the impact of CGRP and RAMP1 signalling on macrophage-mediated recruitment of immune cells, the meninges of Pf4ΔRamp1 mice exhibited higher numbers of neutrophils and monocytes than control mice (Fig. 6j and Extended Data Fig. 10f). Overall, our data uncover a nociceptive neuroimmune axis in the meninges and reveal how bacterial pathogens exploit this signalling pathway to evade antimicrobial immunity (Extended Data Fig. 10g).
Discussion
Headache is a frequent early symptom of bacterial meningitis3,5,14. However, the mechanisms of pain and the role of neuroimmune signalling in host defence were unknown. Here we showed that S. pneumoniae and S. agalactiae, two bacterial pathogens, activate nociceptors to promote meningeal and brain invasion, thereby linking neurons to the pathogenesis of bacterial meningitis. In this neuroimmune axis, CGRP+ neurons signal to PF4+MRC1+ macrophages through RAMP1 to inhibit antibacterial immune responses and blunt host defences.
Barrier tissues are innervated by nociceptors that mediate pain8. The CNS has its own protective barrier tissue: the meninges. Unlike the CNS parenchyma, the dura mater contains a rich repertoire of immune cells that mediate antimicrobial host defences10,12,13,34. We found that intracranial nociceptors directly sense bacterial pathogens and control the activity of meningeal immune cells and leukocyte trafficking. We showed that both systemic and local depletion of nociceptors protected mice against CNS bacterial invasion. A positive correlation between pain intensity and severity of bacterial meningitis has been described3,5,14. Treatment with triptans, which reduces CGRP release, can protect mice against bacterial meningitis35. Our findings link nociceptor activation to infection pathogenesis due to neuronal suppression of immunity through a meningeal CGRP–RAMP1 axis. Nociceptor ablation resulted in reduced brain invasion and pathology, and was associated with an increased number of meningeal immune cells.
The physiological role of this meningeal neuroimmune axis remains unclear, although it may represent a strategy to limit inflammation and CNS pathology. Nociceptors limit the intensity and the duration of inflammation in the lungs and joints, protecting these tissues from inflammation and loss of function36,37. Nociceptors also support wound healing in the skin38,39. CGRP has been shown to protect the CNS against the deleterious effects of inflammation, blood–brain barrier injury, brain oedema and cognitive decline that occurs from cerebral ischaemic injury40,41. These data indicate that CGRP has a role in promoting CNS protection. By contrast, tumour cells and pathogens can benefit from the immunomodulatory activity of sensory innervation19,20,42,43. Herein, we described two human bacterial pathogens that seem to exploit the immunomodulatory activity of nociceptors in the meninges to evade immunity and promote CNS invasion.
Depending on the inflammatory context, nociceptors positively or negatively regulate leukocyte trafficking in other tissues such as the skin and lungs8,19,20,38,44,45. In our current study, nociceptors only regulated bacterial load in the meninges and brain and not in peripheral tissues after intravenous infection with S. pneumoniae or S. agalactiae. A possible explanation for this effect is the topology of infection, whereby nociceptors innervating epithelial layers of skin and gut are more tuned to external pathogen invasion. By contrast, nociceptors innervating the meninges may better interface with blood-borne pathogens invading the CNS.
We showed that S. pneumoniae and S. agalactiae can directly activate nociceptors and induce CGRP release through PLY and β-H/C, respectively. It remains to be determined how PLY and β-H/C bind to and target nociceptors. PLY is a cholesterol-dependent cytolysin and mediates the pathogenesis of bacterial meningitis24,25. Nociceptors may also be targeted through cholesterol or other membrane lipids. During skin infections, Streptococcus pyogenes activates nociceptors through streptolysin S20, and Staphylococcus aureus through α-haemolysin23, to produce pain. A common feature of all these toxins is their ability to form membrane pores, leading to ion influx. For nociceptors, cation influx induces SNARE-dependent release of neuropeptides, including CGRP. Human patients with bacterial meningitis show increased CGRP levels in the cerebrospinal fluid and blood35,46. Here we confirmed the release of CGRP in mouse models of bacterial meningitis.
We also found that nociceptors signal through CGRP to meningeal macrophages, which regulated bacterial meningitis outcome. The CGRP receptor RAMP1 and its co-receptor CALCRL form a G protein-coupled receptor that signals through adenylyl cyclase-mediated production of cAMP and PKA activation17. In the context of bacterial infection, upregulation of cAMP through virulence factors such as pertussis toxin and cholera toxin suppress leukocyte recruitment47. We showed that CGRP signalling through this cAMP–PKA pathway polarizes macrophage responses to S. pneumoniae, inhibiting the production of chemotactic factors (for example, Ccl2, Ccl3 and Tnf) and upregulating the expression of immunosuppressive transcriptional factors (for example, Crem and Jdp2). Increased cAMP levels also counteract chemokine signalling through Gαi, which results in the inhibition of chemotaxis and contributes to increased pathogenesis during bacterial infections47.
Neuron–macrophage crosstalk has key roles in host defence in other barrier sites. In the gut, sympathetic neurons maintain muscularis macrophages in an anti-inflammatory transcriptional state and a tissue-protective phenotype through the release of noradrenaline and the activation of β2-adrenergic receptors expressed by the macrophages48,49. The β2-adrenergic receptor, like RAMP1, signals through Gαs, and cAMP mediates macrophage polarization towards a tissue-protective phenotype47. In the skin, the neuropeptide TAFA4, produced by GINIP+ sensory neurons, stimulate the production of IL-10 by dermal macrophages to promote tissue healing38. It is possible that CGRP+ meningeal nociceptors signal to RAMP1+ macrophages to promote wound healing and resolution of CNS inflammation. In the context of bacterial meningitis, we found that this neuron–macrophage crosstalk impaired bacterial clearance. Therefore, some pathogens may hijack early activation of this neuroimmune axis to evade immunity.
RAMP1 antagonists and antibodies against CGRP are currently used for the prevention and treatment of migraine17. Our observation that treatment with a RAMP1 antagonist ameliorated bacterial meningitis in mice holds potential for therapeutic translation. Recent studies have identified immune cell niches in skull bone marrow that traffic to meninges through channels30,50. It would be interesting to determine whether nociceptors affect these bone marrow immune populations and their trafficking to the meninges.
The meninges are classically defined as tissues that protect the CNS on the basis of the anatomorphological characteristics of this structure that surrounds the brain and spinal cord. Despite this assumption, our knowledge about how the cellular and molecular components of the meninges interact to mediate tissue protection or host defence is limited. We uncovered a neuroimmune axis in the meninges whereby nociceptors modulate the activity of meningeal immune cells and bacterial pathogens exploit this axis to facilitate CNS invasion. This result highlights the complexities of host–pathogen interactions and neuroimmune crosstalk. In light of recent findings of the role of meningeal immune cells in health and disease9,11–13,30,50, our study suggests that nociceptors could affect the function of the CNS and play parts beyond bacterial meningitis. Future research could lead to the development of treatments for infections and other CNS diseases by targeting the somatosensory nervous system.
Methods
Bacterial strains and culture
All procedures related to pathogenic bacteria were approved by the Committee on Microbiological Safety of Harvard Medical School and conducted under Biosafety Level 2 protocols and guidelines. The S. pneumoniae clinical isolate WU2 (serotype 3), the S. pneumoniae isogenic PLY mutant (ΔPly) strain WU2-PLA51 and S. agalactiae clinical isolate COH1 (serotype III)52 and its isogenic mutant strain lacking β-H/Cin (ΔcylE)53 were used in this study. S. pneumoniae and S. agalactiae were grown in tryptic soy broth (Sigma) and Todd–Hewitt broth (Sigma), respectively, supplemented with 0.5% yeast extract (Sigma). Bacteria were grown at 37 °C with 5% CO2 to mid-log phase, and growth was evaluated by monitoring at an optical density of 600 nm. Frozen stocks of bacteria in 20% glycerol (Sigma) were prepared and kept at −80 °C until use.
Animals
All experiments and procedures using mice were approved by the Institutional Animal Care and Use Committee at Harvard Medical School and were conducted in accordance with National Institutes of Health animal research guidelines. Mice were bred and housed in individually ventilated micro-isolator cages within a full barrier, specific pathogen-free animal facility with temperature (22 ± 2 °C) and humidity (55 ± 5%) control at Harvard Medical School under a 12 h light–dark cycle with ad libitum access to food and water. C57BL/6 mice were purchased from Jackson Laboratories or Charles River. C57BL/6-Tg (Pf4-icre) Q3Rsko/J (Jax 008535), B6.FVB-Tg(Acta2-cre)1Rkl/J (Jax 029925), B6.129P2-Lyz2tm1(cre)Ifo/J (Jax 004781), B6.129P2-Gt(ROSA)26Sortm1(DTA)Lky/J (Jax 009669), B6.129P2(Cg)-Cx3cr1tm1Litt/J (Jax 005582), which harbour a GFP knock-in allele, and B6.129S2(Cg)-Ramp1tm1.1Tsuj/WkinJ mice (Jax 031560) were purchased from Jackson Laboratories. Calca (CGRPα)-GFP-DTRflox reporter mice21 were provided by M. Zylka (UNC Chapel Hill). Nav1.8-Cre knock-in mice15 were provided by J. Wood (University College London). Ramp1fl/fl (C57BL/6N-Ramp1tm1c(EUCOMM)Wtsi/H) mice were purchased from MRC Harwell Institute. B6.129S2(Cg)-Ramp1tm1.1Tsuj/WkinJ heterozygous mice were bred together to produce WT and knockout littermates. Nav1.8-Cre heterozygous mice were bred with B6.129P2-Gt(ROSA)26Sortm1(DTA)Lky/J homozygous mice to generate nociceptor-ablated Nav1.8-DTA (Nav1.8-Cre+/−;DTA+/−) mice and control littermates (Nav1.8-Cre−/−;DTA+/−). Nav1.8-Cre heterozygous mice were bred with Rosa26 CAG-LSL-ReaChR::mCitrine (heterozygous) mice (Jax 026294) to generate Nav1.8–mCitrine reporter mice. Nav1.8-Cre homozygous mice were bred with Calca-GFP-DTRflox heterozygous mice to generate Nav1.8-Calca-DTR (Nav1.8-Cre+/−Calca-DTR+/−) mice or into littermate control mice (Nav1.8-Cre+/−Calca-DTR−/−). For conditional knockout experiments, Pf4ΔRamp1 (C57BL/6-Tg(Pf4-icre)Q3Rsko/J+/−; Ramp1fl/fl), Lyz2ΔRamp1 (B6.129P2-Lyz2tm1(cre)Ifo/J+/−;Ramp1fl/fl) and Acta2ΔRamp1 (B6.FVB-Tg(Acta2-cre)1Rkl/J+/−;Ramp1fl/fl) mice were generated and bred to Ramp1fl/fl mice to generate mice with specific depletion of RAMP1 in meningeal macrophages (Pf4ΔRamp1), myeloid cells (Lyz2ΔRamp1), vascular smooth muscle cells (Acta2ΔRamp1) and control littermates (cre−/−; Ramp1fl/fl). Both male and female age-matched mice from 8 to 14 weeks of age were used for all experiments in this study.
Haematogenous bacterial meningitis
Bacterial strains were grown to mid-log phase as described in the section ‘Bacterial strains and culture’. Bacteria were centrifuged at 5,000g for 15 min, resuspended in saline solution to a final concentration of 3 × 108 colony-forming units (c.f.u.) per ml for S. pneumoniae and 1 × 109 c.f.u. per ml for S. agalactiae. The bacterial suspension was kept on ice until use. A total volume of 100 μl of inoculum containing S. pneumoniae (3 × 107 c.f.u.) or S. agalactiae (1 × 108 c.f.u.) was injected into the tail vein (intravenous injection) of 7–14-week-old mice using a 0.5 cc syringe fitted with a 31-gauge needle (BD Biosciences). In some cases, bacteria were labelled with CellTracker Red CMTPX dye (C34552, Invitrogen) 30 min before injection into mice. An aliquot of the final suspension was used to confirm the concentration of bacteria in the inoculum by plating serial dilutions on blood agar plates.
Bacterial load recovery analysis
Mice were deeply anaesthetized by an intraperitoneal injection of tribromoethanol solution (Avertin, 500 mg kg−1) and transcardially perfused with saline solution. Blood was sampled immediately before perfusion. Other tissue samples (meninges, choroid plexus, brain, lung, spleen and skin) were dissected and weighed. Tissue samples were then transferred into 2 ml Eppendorf tubes containing 5 mm stainless steel beads (Qiagen) and 1 ml of ice-cold sterile saline. Samples were homogenized in TissueLyser II (Qiagen) for 10 min at 30 Hz. To determine bacterial load recovery, serial dilutions were made and plated on TSA plates with 5% sheep blood plates (BD Biosciences). The plates were incubated overnight at 37 °C with 5% CO2 and the number of c.f.u. in each sample determined.
Systemic or local meningeal ablation of nociceptors using RTX or DTX
RTX (Sigma-Aldrich) was used to deplete TRPV1+ nociceptors throughout the body as previously described19,20. Male and female 4-week-old C57BL/6 mice were lightly anaesthetized by inhalation of isoflurane (Patterson Veterinary), 3% in oxygen using a precision vaporizer. Mice received subcutaneous injections of escalating doses of RTX (30 μg kg−1, 70 μg kg−1 and 100 μg kg−1) or vehicle control (PBS with 1.2% DMSO and 0.06% Tween-80) on three consecutive days. Mice were used for experiments 4 weeks after the injections. Local depletion of meningeal nociceptors was performed by injecting 5 μl of RTX (100 ng) or vehicle control subcutaneously above the cranial suture of WT mice. The skin was partially opened on the scalp to reveal the cranial suture, injections were given and the suture closed with wound clips (Autoclip, 7 mm) and tissue adhesive (Vetbond, 3M). Mice were used for experiments 3 weeks after local injection of RTX or vehicle. As a second approach to locally ablate meningeal nociceptors, we injected 5 μl of DTX (10 ng) above the cranial suture as described above in Nav1.8-Cre+/−Calca-DTR+/− mice or littermate control mice (Nav1.8-Cre+/−Calca-DTR−/−). These mice were used for experiments 3 weeks after DTX injection. Dural meninges or back skin was stained for CGRP with rabbit anti-CGRP followed by Dylight 488 anti-rabbit antibody and imaged. Innervation was quantified by confocal microscopy (see ‘Immunostaining and microscopy’ section for more details). Quantification of CGRP+ nerve fibre density was performed using ImageJ software, specifically the ImageJ package tools ‘Skeletonize’ and ‘Analyze Skeleton (2D/3D)’.
Isolation and culture of trigeminal neurons
TG were dissected immediately after euthanasia by CO2 inhalation. Trigeminal cells were enzymatically dissociated in 2 ml of HEPES-buffered saline (Sigma) containing collagenase A (1 mg kg−1, Sigma) and dispase II (2.4 U ml−1, Roche) for 40 min at 37 °C. Cells were centrifuged for 5 min at 300g at 4 °C and resuspended in 800 μl of DMEM/10% FBS containing DNase I (150 U ml−1, Thermo Fisher). Trigeminal cells were dissociated by gently pipetting with decreasing tip diameters to create single-cell suspensions. Cells were resuspended in 2 ml neurobasal medium (Life Technologies), and the cell suspension was added to the top of a 10% BSA gradient in neurobasal medium. Cells were centrifuged at 260g for 10 min, the supernatant containing cell debris was discarded and the resulting pellet was resuspended in neurobasal medium for cell counting and plating. For calcium imaging experiments, 2,000 trigeminal neurons were plated onto a culture dish previously coated with laminin and incubated overnight in neurobasal A medium supplemented with 50 ng ml−1 nerve growth factor (Thermo Fisher). In a separate experiment, 2,000 trigeminal neurons were plated onto one side of a microgroove plate (XC150 XonaChip) and cultured for 6 days in neurobasal A medium (no growth factor), whereas medium supplemented with 50 ng ml−1 nerve growth factor (Thermo Fisher) was applied on the other well to stimulate sprouting of nerve fibres through the microwells. For CGRP release experiments, 5,000 trigeminal neurons were transferred to each well of a flat bottom 96-well plate previously coated with laminin and incubated with neurobasal A medium plus 50 ng ml−1 nerve growth factor (Thermo Fisher) and cytosine arabinoside (10 μM, Sigma) for 6 days.
Intracellular calcium levels and CGRP release in trigeminal neuron cultures
Cultures of mouse trigeminal neurons were prepared as described in ‘Isolation and culture of trigeminal neurons’. For calcium measurements, neurons were loaded with the calcium indicator Fura-2-AM (5 μM, Thermo Fisher) for 30 min at 37 °C, washed twice with Krebs–Ringer solution (Boston BioProducts) and immediately imaged using an Eclipse Ti-S/L100 inverted microscope (Nikon) and Zyla sCMOS camera. Excitation of Fura-2-AM was induced by an ultraviolet light source (Lambda XL lamp, Sutter Instrument) at 340 nm and 380 nm wavelengths. The 340/380 ratiometric images were acquired and analysed using NIS-elements software (Nikon). After recording the baseline calcium levels for 2 min, cells were stimulated with S. pneumoniae (4 × 105 to 4 × 107 c.f.u. per ml), S. agalactiae (2 × 107 to 2 × 109 c.f.u. per ml) or PLY (0.01–1 μg ml−1), followed by capsaicin (1 μM) and KCl (40 mM). An increase of 15% or more from baseline calcium levels was considered a positive response to a ligand. Cells that did not respond to the positive controls (capsaicin and KCl) were excluded from quantification analyses. For CGRP release assays, neurons were incubated with S. pneumoniae (4 × 107 c.f.u. per ml), S. agalactiae (2 × 109 c.f.u. per ml) or PLY (0.01–1 μg ml−1) for 30 min (37 °C, 5% CO2). After incubation, the supernatant was collected and used to quantify the concentration of CGRP using an enzyme linked immunosorbent kit (Cayman Chemical) according to the manufacturer’s instructions.
Isolation and culture of BMDMs
Mouse BMDMs were obtained as previously described54 with minor modifications. Progenitor cells were collected from the bone marrow of femurs and tibia and cultured in DMEM supplemented with 10% FBS, 1% penicillin–streptomycin solution (15140122, Gibco) and macrophage-colony stimulating factor (20 ng ml−1; 15140122, Pepro-Tech) for 8 days (37 °C, 5% CO2). Culture medium was replaced with fresh supplemented DMEM at day 4. Fully differentiated macrophages (>90% of cells CD11b+F4/80+ determined by flow cytometry) were collected at day 8 and immediately used for experiments.
Phagocytic killing assay
Cultures of mouse macrophages (BMDMs) were prepared as described in the section ‘Isolation and culture of BMDMs’. S. pneumoniae (5 × 105 c.f.u. per well) was co-incubated with mouse macrophages (5 × 105 cells per well) in DMEM supplemented with 10% mouse serum for 1–24 h (37 °C, 5% CO2) with gentle shaking (150 r.p.m.). CGRP (100 nM, GenScript) or vehicle (DMEM) was added to the cells immediately before adding the bacteria. The number of bacteria in each well was determined by serial-dilution plating on TSA plates with sheep blood agar (BD Biosciences), and bacterial colonies were counted after overnight incubation at 37 °C in 5% CO2.
ELISA
Mouse macrophages (5 × 105 cells per well) were obtained as described in the section ‘Isolation and culture of BMDMs’ and incubated with S. pneumoniae or vehicle (DMEM + 10% mouse serum) for 24 h (37 °C, 5% CO2). Cells were treated with CGRP (100 nM, GenScript) or vehicle (DMEM) immediately before adding the bacteria. After incubation, supernatants were collected, filtered and stored at −80 °C until ELISA was performed. The concentrations of CXCL10, CCL3, CCL2 and TNF in the supernatants were determined using ELISA kits (R&D Systems) following the manufacturer’s protocols.
Magnetic-activated cell sorting of meningeal cells
Mice were deeply anaesthetized by intraperitoneal injection of tribromoethanol solution (Avertin, 500 mg kg−1) and transcardially perfused with 30 ml saline solution. Samples from the meninges (cortical dura mater) were dissected as previously described11,55,56 and incubated for 30 min at 37 °C in 0.5 ml of DMEM/F12 medium containing Dispase (1 U ml−1; StemCell Technologies) and Liberase TL (0.25 mg ml−1; Sigma). After incubation, cells were centrifuged at 400g for 10 min, resuspended in 1 ml ice-cold cell wash buffer (BioLegend), gently dissociated using a 1 ml pipette and filtered through a 40 μm cell strainer (Flowmi, Scienceware). The resulting cell suspension was then used for magnetic-activated cell sorting (MACS)-based purification using CD45 MicroBeads (Miltenyi Biotec) or F4/80 MicroBeads UltraPure (Miltenyi Biotec) and MS MACS columns (Miltenyi Biotec) following the manufacturer’s directions.
Intracisterna magna injections
Injections into the cisterna magna were used to deliver bacteria into the subarachnoid space and to deplete meningeal macrophages. The procedure was performed as previously described55,56 with small modifications. Mice were first anaesthetized with isoflurane (4%) in an induction chamber and treated with carprofen (20 mg kg−1, subcutaneous) and buprenorphine (0.1 mg kg−1, subcutaneous) immediately before the surgery. Lubrication of corneas was maintained using Puralube, and anaesthesia by isoflurane was maintained through a nose cone during the surgery. Animals were transferred to a stereotaxic frame, and the skin of the head was shaved and aseptically prepared by swabbing betadine followed by ethanol (three times each). The cranium was exposed by making a surgical anterior–posterior incision with a scalpel blade, and the subcutaneous tissue and muscles of the neck were gently separated to access the dura mater of the cisterna magna. S. pneumoniae (103 c.f.u., 5 μl), mannosylated liposomes containing clodronate (m-Clodrosome Encapsula Nano Sciences, 5 μl) or empty mannosylated liposomes (m-Encapsome Encapsula Nano Sciences, 5 μl) were injected into the subarachnoid space using a 30-gauge 0.5-inch needle mounted on a 25- μl Hamilton syringe. After injection, muscles were re-aligned, and the incision was closed using wound clips (Autoclip, 7 mm) and tissue adhesive (Vetbond, 3M). After surgery, animals were placed in a cage containing a warming pad and monitored for 1 h after surgery. Additional doses of carprofen (every 24 h) were administered for 72 h after surgery, and wounds were monitored for adequate healing.
CGRP release assay from meninges explants
Mice were euthanized by CO2 inhalation, and the skullcap containing meninges was dissected and rapidly transferred to 24-well plates containing 1 ml DMEM. The explants were incubated for 30 min at 32 °C with gentle shaking (150 r.p.m.). After incubation, the medium from the organ cultures was collected and used to determine the levels of CGRP using a CGRP EIA kit (Cayman Chemical) according to the manufacturer’s instructions.
In vivo CGRP and BIBN4096 treatment
We evaluated the impact of CGRP signalling on the outcome of bacterial meningitis by treating mice with CGRP injection or with the CGRP antagonist BIBN4096 (Tocris). For these experiments, mice were treated with CGRP (0.1 mg kg−1), BIBN4096 (0.3 mg kg−1) or vehicle by intraperitoneal injection. Treatments were performed 2 h before induction of bacterial meningitis as described in the section ‘Haematogenous bacterial meningitis’ and again 24 h later. Additionally, the role of IL-10 on the effects of CGRP was determined by treating mice with vehicle or CGRP and either isotype control antibody or neutralizing anti-IL-10 antibody (Bio-X-Cell, 200 μg in 100 μl, intraperitoneally, daily). Treatment doses were selected on the basis of previous publications using these compounds22,57.
Meningeal flow cytometry
Mice were deeply anaesthetized with an intraperitoneal injection of tribromoethanol solution (Avertin, 500 mg kg−1) and transcardially perfused with 30 ml saline solution. Tissue samples were dissected, minced and incubated for 30 min at 37 °C in 0.5 ml of DMEM/F12 medium containing Dispase (1 U ml−1; StemCell Technologies) and Liberase TL (0.25 mg ml−1; Sigma). Samples from blood and spleen were processed without incubation in Liberase. After incubation, cells were centrifuged at 400g for 10 min, resuspended in 1 ml ice-cold cell wash buffer (BioLegend), gently dissociated using a 1 ml pipette and filtered through a 40 μm cell strainer (Flowmi, Scienceware). The resulting cell suspension was incubated with mouse FcR blocking reagent (Miltenyi Biotec) for 10 min, and then incubated with the following reagents: DAPI (4′,6-diamidino-2-phenylindole, Dilactate, 3 μM, BioLegend); anti-mouse CD45-PE-Cy7 clone 30-F11 (1:200, BioLegend); anti-mouse CD11b Brilliant Violet 570 or APC clone M1/70 (1:200, BioLegend); anti-mouse Ly-6C Brilliant Violet 650 clone HK1.4 (1:200, BioLegend); anti-mouse CD206 (Mrc1) PE clone C068C2 (1:200, BioLegend); anti-mouse Ly-6G PE-cyanine5 or APC-cyanine7 clone 1A8 (1:200, Thermo Fisher Scientific); anti-mouse CD3 FITC or Alexa Fluor 700 clone 17A2 (1:200, BioLegend); and anti-mouse CD19 PE-Cyaine5.5 clone 1D3 (1:200, Thermo Fisher Scientific). After incubation, cells were centrifuged for 5 min at 300g and resuspended in 500 μl of cell wash buffer, then centrifuged again and resuspended in wash buffer containing 2% paraformaldehyde (PFA). Flow cytometry was performed on a FACSymphony A5 flow cytometer (BD Biosciences). Flow cytometry data were collected and exported using BD FACSDiva software (BD Biosciences) and analysed using FlowJo software (v.10). Representative flow cytometry gating schemes for meningeal immune cells are shown in Supplementary Fig. 1. A small aliquot (20 μl) was used to count the total number of cells in each sample, and the results were used to convert the percentage of immune cell populations into cell numbers.
Blood and spleen flow cytometry
For flow cytometry of blood and splenic cells, cells from blood and spleen were collected and treated with 1× RBC lysis buffer (eBioscience, 00–4333-57) to remove red cells. Immune cells were then stained with combinations of the following reagents: Live/Dead Near-IR (Thermo Fisher, L34975; 1:2,000); anti-mouse CD45 conjugated with Pacific Blue (BioLegend, 103126, clone 30-F11; 1:500); anti-mouse CD19 conjugated with APC (BioLegend, 115511, clone 6D5; 1:300); anti-mouse CD3 conjugated with APC A700 (BioLegend, 100215, clone 17A2; 1:300); anti-mouse CD4 conjugated with BV650 (BioLegend, 100469, clone GK1.5; 1:300); anti-mouse CD8 conjugated with Percp-Cy5.5 (BioLegend, 100733, clone 53–6.7; 1:300); anti-mouse CD44 conjugated with PE-Cy7 (Invitrogen, 25–0441-82, clone IM7; 1:300); anti-mouse CD62L conjugated with BV605 (BioLegend, 104437, clone MEL-14; 1:300); anti-mouse NK1.1 conjugated with PE (BioLegend, 108707, clone PK136; 1:300); anti-mouse CD11B conjugated with BV605 (BioLegend, 101237, clone M1/70; 1:300); anti-mouse CD172a conjugated with FITC (BioLegend, 144005, clone P84; 1:300); anti-mouse Ly6G conjugated with PE-Cy7 (BioLegend, 127617, clone 1A8; 1:300); anti-mouse Siglec F conjugated with PE (BD, 552126, clone E50–2440; 1:300); anti-mouse Ly6C conjugated with Percp-Cy5.5 (BioLegend, 128011 clone HK1.4; 1:300). Samples were incubated on ice for 20 min in flow buffer (2% FBS, 2 mM EDTA in PBS) and then washed twice with flow buffer. For transcription factor staining (mouse anti-FOXP3 Alexa Fluor 488), a transcription factor staining buffer set (eBioscience) was used according to the manufacturer’s instructions. Data were acquired using a Cytoflex S flow cytometer (Beckman Coulter) and analysed using FlowJo software (v.10). Representative flow cytometry gating schemes for blood and spleen immune cells are shown in Supplementary Fig. 2.
RT–qPCR
Mouse macrophages were obtained as described in the section ‘Isolation and culture of BMDMs’. Macrophages (5 × 105 cells per well) were incubated with S. pneumoniae (5 × 105 c.f.u. per well) and CGRP (100 nM) or vehicle (DMEM) in DMEM supplemented with 10% mouse serum for 4 h (37 °C, 5% CO2). In some wells, the cells were pre-treated with the selective PKAi Rp-8-CPT-cAMP (10 μM, Cayman Chemical) 1 h before incubation. After incubation, the supernatant was removed and replaced by 500 μl TRIzol, and plates were frozen at −80 °C until RNA extraction. For total RNA extraction, samples were thawed at room temperature, mixed with 100 μl chloroform, and centrifuged at 12,000g for 15 min at 4 °C. The aqueous phase was transferred to a new tube and combined with equal parts of isopropyl alcohol, mixed well and centrifuged at 12,000g for 10 min at 4 °C. After discarding the supernatant, 500 μl of 75% ethanol was added to the samples, vortexed well and centrifuged at 7,500g for 5 min. Supernatant was then discarded and the pellet containing RNA was resuspended in 50 μl nuclease-free water. mRNA in the samples was reverse transcribed into cDNA using an iScript cDNA Synthesis kit (Bio-Rad). Relative gene expression was determined using gene-specific primers (PrimerBank) and SYBR Green master mix (Life Technologies) on a QuantStudio 5 RT–PCR system (Applied Biosystems). Expression data were collected and exported using the QuantStudio Design & Analysis Software (v.1.5.1, Applied Biosystems). Expression levels were normalized to β-actin expression (Actb) using the 2−ΔCt method. The following primer sequences (5′-to-3′) were used: Actb forward: AGCTGCGTTTTACACCCTTT, Actb reverse AAGCCATGCCAATGTTGTCT; Ramp1 forward: GGATGAGA GTCCCATAGTCAGG, Ramp1 reverse GGGGCTCTGCTTGCCAT; Jdp2 forward: CTCCTCCTGCTATGATGCCT, Jdp2 reverse CTCTTGCC CAGTTTCACCTC; Crem forward: TGGACTGTGGTACGGCCAAT, Crem reverse CAGTTTCATCTCCAGTTACA; Tnf forward: CAGGCGGTGC CTATGTCTC, Tnf reverse CGATCACCCCGAAGTTCAGTAG; Ccl2 forward: TTAAAAACCTGGATCGGAACCAA; Ccl2 reverse GCATTAGCTTCAGATT TACGGGT; Ccl3 forward: TTCTCTGTACCATGACACTCTGC, Ccl3 reverse CGTGGAATCTTCCGGCTGTAG; Cxcl10 forward: CCAAGTGCTGCCGT CATTTTC, Cxcl10 reverse GGCTCGCAGGGATGATTTCAA; and Ccl7 forward: GCTGCTTTCAGCATCCAAGTG, Ccl7 reverse CCAGGGACAC CGACTACTG.
RNA-seq of macrophages
Macrophages were cultured as described in the section ‘Isolation and culture of BMDMs’. Cells (1 × 106 per well) were incubated with 10% mouse serum-opsonized S. pneumoniae (1 × 106 c.f.u.) and CGRP (100 nM) or vehicle (PBS) for 4 h. After incubation, the supernatant was removed and replaced by 500 μl of TRIzol, and plates were frozen at −80 °C until RNA extraction as described in the section ‘RT–qPCR’. RNA-seq library preparation (Kapa mRNA Hyperprep, Roche) and sequencing (NovaSeq SP, 2× 50 bp, Illumina) was conducted by the Bauer Sequencing Core at Harvard University.
scRNA-seq of meninges
Samples containing meningeal immune cells (CD45-enriched) or meningeal non-immune cells (CD45-depleted) were obtained as described in the section ‘Magnetic-activated cell sorting of meningeal cells’ and used for droplet-based scRNA-seq (10x Genomics). Library preparation (Chromium Next GEM Single Cell 3′ Reagent kit v3.1) and sequencing (Illumina NovaSeq 6000 System) were conducted by the Bauer Sequencing Core at Harvard University following the manufacturer’s instructions. The quality of the single-cell suspensions (viability >80%, concentration = 1,000 cells per μl) was confirmed immediately before encapsulation using acridine orange–propidium iodide stain (Logos Biosystems) and a LUNA-FX7 counter (Logos Biosystems). Encapsulation of cells was performed in a Chromium Controller (10x Genomics) targeting 10,000 cells per sample. Following encapsulation and mRNA barcoding, cDNA was synthesized, isolated and amplified (11 cycles) using a Single Cell 3ʹ GEM kit (10x Genomics) and a SPRIselect reagent kit (Beckman Coulter). The quality of the amplified cDNA (concentration, size and purity) was verified using a High Sensitivity D5000 ScreenTape and 4200 TapeStation system (Agilent Technologies). Next, amplified cDNA was used for library construction. cDNA fragmentation, end repair, A-tailing, adaptor ligation and sample index PCR amplification were performed using Chromium Next GEM Single Cell 3ʹ Library Kit v.3.1 reagents (10x Genomics). After library construction, quality control was performed using a High Sensitivity D5000 ScreenTape and 4200 TapeStation system (Agilent Technologies). Quantification was performed using a Kapa qPCR Complete Universal kit (Roche Sequencing Solutions) and a CFX96 RT–PCR detection system (Bio-Rad Laboratories). For sequencing, CD45+ sample libraries were pooled and equally distributed across the two lanes of an Illumina NovaSeq S1 flow cell (Read 1: 28 bp; i7 index: 8 bp; Read 2: 90 bp). The CD45− library was sequenced in a single lane of an Illumina NovaSeq S4 flow cell (Read 1: 28 bp; i7 index: 8 bp; Read 2: 90 bp). After sequencing, the quality control summary report confirmed that each library contained a minimum of 10,000 cells (10,545–12,185) and an average sequencing depth of 50,178 reads per cell. Cell Ranger (10x Genomics) analysis pipelines were used to demultiplex raw sequencing data and to perform alignment, filtering and counting of barcodes and unique molecular identifiers. The reference mouse genome mm10 v.2020-A was used. Count matrices generated by Cell Ranger imported into R (v.4.0.3) were used for further analysis using Seurat package (v.4.1.0)58,59.
Analysis was performed using only high-quality cells that had unique molecular identifier counts between 4,000 and 70,000 and <25% of their genes corresponding to the mitochondrial genome. Samples were normalized and scaled using Seurat’s SCTransform function followed by principal component analysis. Clustering and visualization were performed by running uniform manifold approximation and projection dimensional reduction of 15 principal components for each cluster resolution of 0.3. Cell types were defined by comparing cluster marker genes with previously published scRNA-seq datasets of mouse meninges10,11,12,60. Visualization of genes illustrating expression levels was performed using R/Seurat commands (DoHeatmap, FeaturePlot and DotPlot)58,59 and the R/Nebulosa package59,61.
Differential gene expression analysis
Differential gene expression analysis was performed in sequencing datasets using the R package DESeq2 (bulk RNA-seq of BMDMs) or Seurat (scRNA-seq of meninges)55,56,58,59,62. Genes were considered differentially expressed when the adjusted P value was lower than 0.05. The list of differentially expressed genes was used in pathway enrichment analysis and to create volcano plots and Venn diagrams.
Pathway enrichment analysis
Pathway enrichment analysis of the list of differentially expressed genes was performed using the database for annotation, visualization, and integrated discovery (DAVID) tool (http://david.abcc.ncifcrf.gov). Gene ontology (GO) terms in the Biological Processes category with P < 0.05 were considered significant. Statistically significant, non-redundant GO-enriched terms were plotted.
Mouse grimace scale
The mouse grimace scale was used to quantify spontaneous pain-like behaviours as previously described63–65. Animals were acclimated in clear acrylic chambers (8 × 8 × 8 cm) 1 day before baseline testing. Measurements were taken at day 0 (baseline, before injection) and 1–2 days after injection with S. pneumoniae, S. agalactiae or vehicle. Mice were individually recorded for 10 min with high-definition cameras (GoPro). From these 10-min videos, the first image with a clear view of the animal’s face from every minute of the video was extracted using iMovie (Apple). The selected images were randomized, and blinded scoring was performed by investigators who were unaware of the groups and time points. As described in the original method, for each image, orbital tightening (white arrowhead), nose bulge (black arrowhead), cheek bulge (white arrow) and ear position (black arrow) were scored (0, not present; 1, moderately visible; and 2, severely visible) and the total score for each image was averaged. To investigate the role of pain signalling in bacterial meningitis, mice were treated with long-lasting buprenorphine (1.5 mg kg−1 subcutaneously) or at the same time with S. pneumoniae infection.
Immunostaining and microscopy
Dissection and whole-mount immunofluorescence staining of meninges were performed as previously described with minor modifications10,11,55,66. Mice were euthanized and intracardially perfused with 30 ml PBS followed by 30 ml PBS/4% PFA. The cortical meninges were dissected and post-fixed in PBS/4% PFA solution at 4 °C for 24 h. Before immunostaining, samples were transferred to PBS and incubated for 24 h at 4 °C to remove PFA. Free-floating samples were incubated with blocking solution (PBS with 0.1% Triton X-100 and 5% donkey serum) in 24-well plates for 2 h at room temperature with agitation. Blocking solution was then replaced by staining solution (PBS with 0.1% Triton X-100 and 2% donkey serum) containing the primary antibodies rabbit anti-rat CGRP (1:500; C8198, Sigma) or goat anti-mouse Mrc1 (5 μg ml−1; AF2535, R&D Systems) and incubated for 24 h at 4 °C with agitation. Samples were washed five times with PBS to remove unbound primary antibodies and then incubated (24 h at 4 °C with agitation) with staining solution containing the secondary antibodies donkey anti-rabbit IgG conjugated with DyLight 488 (1:500; ab98488, Abcam) or donkey anti-goat IgG conjugated with Alexa Fluor 488 (1:500; ab150129, Abcam). In some cases, bacteria were labelled with Cell-Tracker Red CMTPX dye (C34552, Invitrogen) 30 min before injection into mice. For the staining of blood vessels, the primary antibody rat anti-mouse CD31 conjugated with Alexa Fluor 647 (5 μg; MEC13.3 Bio-Legend) was injected into mice 5 min before perfusion. Stained samples were washed five times with PBS and mounted in Prolong Gold Antifade reagent (Cell Signaling). For skin immunostaining for CGRP, skin punch biopsies (12 mm) were fixed in 4% PFA solution at 4 °C for 24 h, embedded in OCT (Sakura Finetek, 4583) and cryosectioned (20 μm) onto Superfrost Plus slides (Thermo Fisher). Slides were stained with rabbit ant-rat CGRP (1:500; C8198, Sigma) followed by anti-rabbit IgG conjugated with Alexa Fluor 647 (1:500; Abcam) and Hoechst 33342 DNA staining solution (1 μM; Thermo Scientific).
In a separate set of experiments, Calca-GFP-DTR mice were infected with CMTPX-labelled S. pneumoniae, stained using anti-PLY antibody (1:200), followed by whole-mount imaging. Fluorescence imaging was performed using a Leica Stellaris 8 confocal microscope (Leica) and LAS X software (Leica). System-optimized settings were used to acquire the full thickness of the tissue (z axis) and for tile-stitching (xy axes). Merged maximum projection images were exported.
Brain histopathology
Brain samples were collected from Nav1.8-DTA and control mice 24 h after injection of S. pneumoniae (3 × 107 c.f.u. in 100 μl, intravenously). Mice were deeply anaesthetized with tribromoethanol solution (Avertin, 500 mg kg−1, intravenously) and transcardially perfused with 30 ml saline solution followed by 10 ml of 4% PFA in saline. After dissection, brain samples were maintained in 4% PFA for 3 days at 4 °C. Fixed brains were embedded in paraffin, sectioned (10 μm thick), and half of the slides were stained using haematoxylin and eosin by the Rodent Histopathology Core at Harvard Medical School. Pictures of the brain samples were taken automatically using a Leica DMi8 microscope (sCMOS camera, ×40 NA 0.85 objective) and Thunder software (Leica). A total of 96 pictures (12 pictures × 4 samples × 2 groups) of brain cortex images were collected and randomized for blinded scoring. Each field was assigned a score from 0 to 3 based on neuronal morphology: grade 0 (not altered); grade 1 (no vacuolation with small numbers of pyknotic cells); grade 2 (moderate vacuolation and pyknosis); and grade 3 (extensive vacuolation, pyknosis and tissue loss or liquefactive necrosis)67,68. For the analysis of caspase-3 activity, slides were deparaffinized and rehydrated before immunostaining. Slides were heated at 60 °C for 10 min, washed twice with xylene for 10 min and incubated with a series of graded ethanol solutions (100%, 95%, 70%, 50% and 30% in PBS) for 5 min each. Slides were incubated with blocking solution (PBS with 0.1% Triton X-100 and 5% donkey serum) for 1 h at room temperature. After incubation, blocking solution was removed and slides were incubated with staining solution (PBS with 0.1% Triton X-100 and 2% donkey serum) containing rabbit anti-human cleaved caspase-3 antibody (1:400; 9661, Cell Signaling) for 24 h at 4 °C. Slides were rinsed five times with PBS and incubated for 2 h with staining solution containing donkey anti-rabbit IgG conjugated with Alexa Fluor 647 (1:500; Abcam) and Hoechst 33342 DNA staining solution (1 μM; Thermo Scientific). After incubation, slides were rinsed five times with PBS and coverslip mounted with Prolong Gold Antifade reagent (Cell Signaling). Fluorescence imaging was performed using a Leica Stellaris 8 confocal microscope (Leica) and LAS X software (Leica). System-optimized settings were used to acquire the full thickness of the tissue (z axis) and for tile-stitching (xy axes). Merged maximum projection images were exported. Fluorescence intensity was calculated using Fiji software69 by measuring the integrated density of the of cleaved caspase-3 staining in the brain. Results are expressed as the fold change of control (uninfected) brain.
Clinical score
The progression of bacterial meningitis was determined as previously described with a few adaptations70. Scoring was performed based on the previously described scale of weight loss (0–3), activity (0–4), time to return to upright position (0–4), coat (0–3), posture (0–2), eyes (0–4), breathing (0–4), limb paresis or ataxia (0–4). The pre-symptomatic period was defined as the time from inoculation until clinical score ≤ 4).
General experimental design
All in vivo experiments were performed in both male and female age-matched mice and littermates when possible. Treatment groups of mice were randomized and evenly distributed across both male and female littermates and/or cagemates. Grimace scores and histopathological analysis were performed in a blinded manner. In experiments involving transgenic mice, littermates with different genotypes were housed together for the duration of experiments. Animal numbers were estimated on the basis of pilot studies of S. agalactiae and S. pneumoniae infections in our laboratory and published work51,52,66.
Statistical analysis and reproducibility
Statistical analysis was performed using GraphPad Prism Software. One-way analysis of variance (ANOVA) with appropriate multiple comparisons tests was used to compare three independent groups. Two-group comparisons were made using two-tailed unpaired Student’s t-test. For comparisons of multiple factors, two-way ANOVA with appropriate multiple comparisons tests was used. Microscopy pictures (micrographs) and infection datasets are representative of at least two independent repetitions. Exact P values for specific group comparisons in all the figures are supplied in Supplementary Table 1.
Figure experimental and illustration details
For Fig. 1a, the left micrograph shows whole-mount meninges from Nav1.8–mCitrine reporter mice, with mCitrine fluorescence in red. The central micrograph shows whole-mount meninges from CalcaGFP-DTR mice, with GFP fluorescence in green. The right micrograph shows Nav1.8-Cre–mCitrine reporter mice meninges stained with rabbit anti-CGRP followed by Dylight 488 anti-rabbit antibody. Shown are CGRP immunostaining (green) and Nav1.8-mCitrine (red) and overlayed images (yellow).
For Fig. 1h, dural meninges whole-mount tissues from vehicle or RTX-treated mice were stained with rabbit anti-CGRP followed by Dylight 488 anti-rabbit antibody, and imaged by confocal microscopy as described in the Methods. The left images show CGRP staining in green. For the right images, ImageJ software was used for skeletonization of CGRP+ nerves as described in the Methods. ImageJ quantification of nerve density is plotted on the right graph.
Figure 2a shows an illustration of mouse infection by S. pneumoniae or S. agalactiae, followed by dural meninges dissection and organ explant bath. CGRP levels were subsequently analysed by enzyme immunoassay (EIA).
In Fig. 2e, WT mice were infected with CellTracker Red CMTPX Dye labelled bacteria (S. pneumoniae or S. agalactiae). Whole-mount dural meninges were stained with rabbit anti-CGRP followed by Dylight 488 anti-rabbit antibody. Confocal microscopy images show fluorescent bacteria (red) close by CGRP+ nerve fibres (green).
Figure 4a shows an illustration of the experiment, with dural meningeal enrichment of CD45+ cells by MACS bead sorting followed by 10x Genomics based scRNA-seq analysis.
Figure 5a shows an illustration of the experiment showing dura mater enrichment of CD45+ cells at baseline and 24 h after S. pneumoniae infection followed by 10x Genomics based scRNA-seq analysis.
In Fig. 5c, WT mice were infected with CMTPX-labelled S. pneumoniae. After 24 h of infection, whole-mount dural meninges was dissected and stained for meningeal macrophages (MRC1+ cells) with anti-mouse MRC1 (5 μg ml−1; AF2535 R&D Systems) and donkey anti-goat IgG conjugated with Alexa Fluor 488 (1:500; ab150129, Abcam) as described in the Methods. Confocal microscopy images show meningeal macrophages (MRC1 signal) in magenta and S. pneumoniae (CMTPX signal) in green.
Figure 5d shows an illustration on the left of injection of mice with CLLs through the intracisternal magna route. Also shown are magnified images of the dura mater, cisterna magna and subarachnoid space.
In Fig. 5f, whole-mount dural meninges from CX3CR1–GFP mice were stained with rabbit anti-CGRP followed by Alexa Fluor 647 anti-rabbit antibody. Confocal microscopy images show meningeal macrophages (magenta) close by CGRP+ nerve fibres (green).
For Fig. 6b, left, Pf4cre mice were bred with Rosa26-TdTomato reporter mice (Ai14 line). Rat anti-mouse CD31 conjugated with Alexa Fluor 647 was intravenously injected into these mice 30 min before imaging. Mice were euthanized and whole-mount meninges imaged by confocal microscopy. Images show PF4-Tdtomato+ macrophages (red) and CD31+ blood vessels (blue). On the right, whole-mount meninges from PF4-Cre;TdTomato reporter mice were stained with rabbit anti-CGRP and Dylight 488 anti-rabbit antibody. Confocal microscopy image shows PF4-Tdtomato+ macrophages (white) and CGRP+ nerves (green).
In Fig. 6d, left, schematic of incubation of unstimulated BMDMs with S. pneumoniae alone or with S. pneumoniae and CGRP for 4 h, followed by RNA-seq analysis. Right, volcano plots showing differential gene expression of stimulated versus S. pneumoniae, and S. pneumoniae versus S. pneumoniae + CGRP-treated macrophages.
In Fig. 6h, Venn diagram created in Adobe illustrator showing numbers of genes differentially induced only in vivo by CGRP injection (1,062 genes), only in vitro by CGRP treatment (662 genes) and overlapping among the two conditions (137 genes). In the overlapping subset, six genes are highlighted (Crem, Jdp2, Cxcl10, Ccl2, Ccl7 and Ccl4).
In Extended Data Fig. 1a, mice were injected with CellTracker Red CMTPX Dye labelled S. pneumoniae. At 24 h after injection, rat anti-mouse CD31 conjugated with Alexa Fluor 647 (5 μg) was injected into mice 5 min before perfusion. Whole-mount dural meninges was dissected and imaged by confocal microscopy. In the micrograph, CD31 staining is in white and S. pneumoniae in green.
In Extended Data Fig. 1d, control or Nav1.8-DTA mice were infected with CMTPX-labelled S. pneumoniae. At 24 h after injection, mice were perfused with 4% PFA, brains embedded in paraffin, sectioned and stained as described in the Methods for cleaved caspase-3 and nuclei with Hoechst dye. Confocal microscopy images show cleaved caspase-3 in red, S. pneumoniae in green and Hoechst staining of nuclei in blue. Left, schematic showing brain section.
In Extended Data Fig. 1e, control or Nav1.8-DTA mice were infected with S. pneumoniae. At 24 h after the injection, mice were perfused with 4% PFA, brains embedded in paraffin, sectioned and stained with haematoxylin and eosin. Pictures from the brain samples were taken using a Leica DMi8 microscope and histopathology scores analysed as described in the Methods.
In Extended Data Figs. 1f,g and 2c, dural meninges whole-mount tissues were stained for rabbit anti-CGRP followed by Dylight 488 anti-rabbit antibody and imaged by confocal microscopy as described in the Methods. Left, images show CGRP staining in green. Right, for these images, ImageJ software was used for skeletonization of CGRP+ nerves as described in the Methods. ImageJ quantification of nerve density is plotted and analysed in the graph on the right.
In Extended Data Fig. 2a,b, back skin sections were stained with rabbit anti-CGRP followed by rabbit IgG conjugated with Alexa Fluor 647 (1:500; Abcam) and Hoechst 33342 DNA staining solution (1 μM; Thermo Scientific) and imaged by confocal microscopy as described in the Methods. Left, images show CGRP staining in green. Right, for these images, ImageJ software was used for skeletonization of CGRP+ nerves as described in the Methods. ImageJ quantification of nerve density is plotted and analysed in the graph on the right.
In Extended Data Fig. 4c, CalcaGFP-DTR mice were infected with CMTPX-labelled S. pneumoniae. After 24 h of infection, TG were dissected and imaged by confocal microscopy. Calca-GFP signal is shown in green and CMTPX-labelled S. pneumoniae in red.
In Extended Data Fig. 4d, CalcaGFP-DTR mice were infected with CMTPX-labelled S. pneumoniae. After 24 h of infection, whole-mount dural meninges was dissected and stained for anti-PLY. The CALCA-GFP signal is shown in green, anti-PLY is shown in blue and fluorescent S. pneumoniae in red.
In Extended Data Fig. 4e, the schematic on the left shows the micro-fluidic chambers used to culture TG neurons and axons, which extend through the middle chamber and grow into the right chamber. Cells were loaded with Fura-2 dye for ratiometric imaging. Calcium imaging was performed and PLY added to the right chamber. Analysis shows increased fluorescence in individual axons.
In Extended Data Fig. 7b, the schematic on the left shows the process for scRNA-seq of CD45− non-immune cells from the dura mater. Dura mater was dissected from mice, underwent CD45 MACS bead depletion, and negative cells were then subjected to 10x Genomics based scRNA-seq analysis.
In Extended Data Fig. 9a, WT mice were infected with CMTPX-labelled S. pneumoniae. After 24 h of infection, whole-mount dural meninges was dissected and stained for meningeal macrophages (MRC1+ cells) with anti-mouse MRC1 (5 μg ml−1; AF2535, R&D Systems) and donkey anti-goat IgG conjugated with Alexa Fluor 488 (1:500; ab150129, Abcam) as described in the Methods. Confocal microscopy images show meningeal macrophages (MRC1 signal) in magenta and S. pneumoniae (CMTPX signal) in green.
Extended Data
Extended Data Fig. 1 |. Nociceptors suppress meninges-mediated protection of CNS to infection.
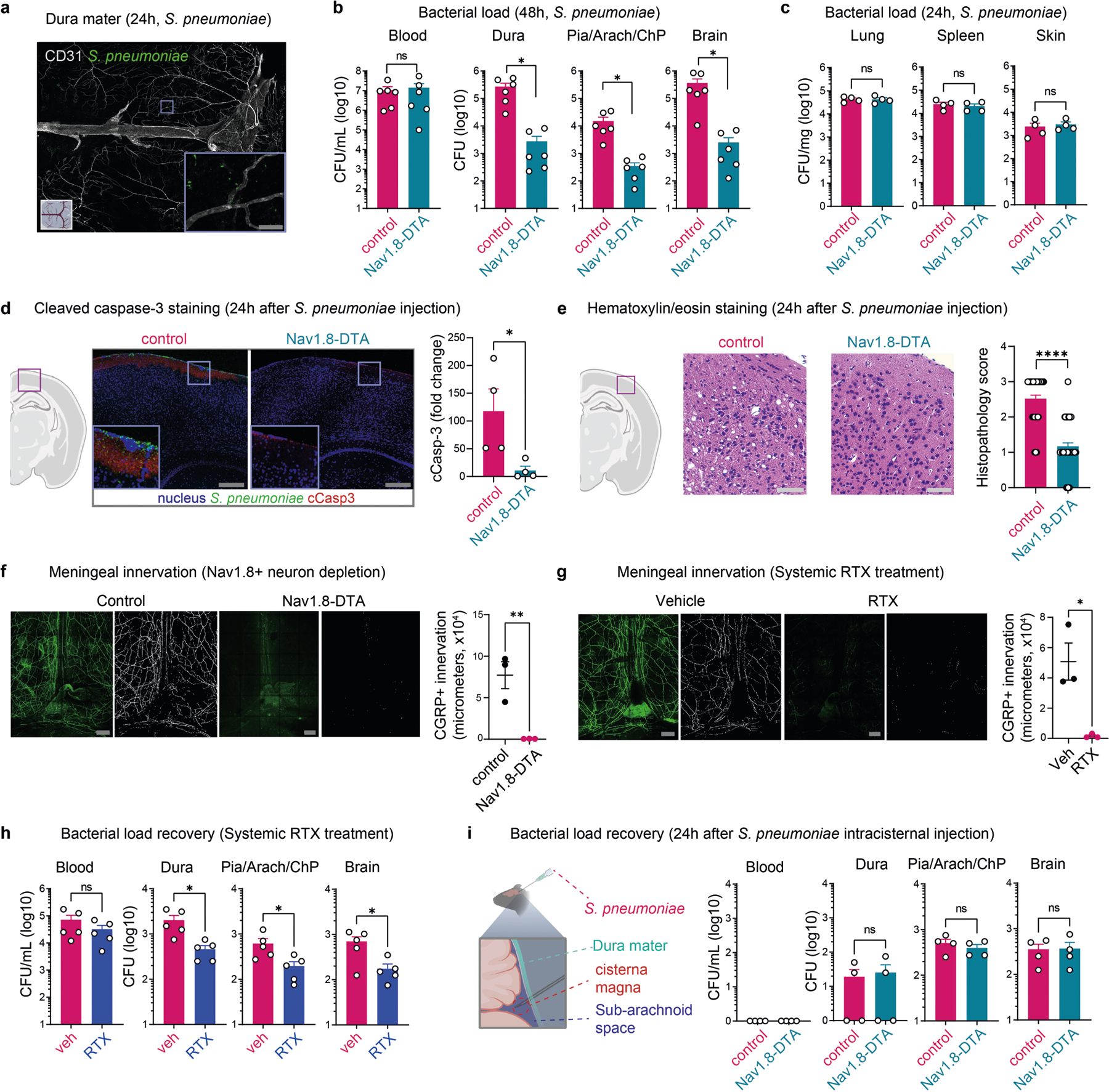
a, Whole-mount confocal images of mouse meninges (dura mater) showing extravascular localization of S. pneumoniae 24 h post-injection of CMTPX-labeled bacteria. Scale bar = 100 μm. b, Bacterial load in samples collected from Nav1.8-DTA and control mice 48h after injection of S. pneumoniae (n = 6/group). c, Bacterial load in samples collected from Nav1.8-DTA and control mice 24 h after injection of S. pneumoniae (n = 4/group). d, Left, Illustration created with BioRender.com (https://biorender.com). Right, Imaging and quantification of cleaved caspase-3 staining in brain samples collected from Nav1.8-DTA and control mice 24 h after injection of CMTPX-labeled S. pneumoniae. Results are presented as fold-change relative to cCasp-3 staining of brain samples from uninfected mice (n = 4/group). Scale bar = 200 μm. e, Left, Illustration created with BioRender.com (https://biorender.com). Hematoxylin and eosin staining of brain sections, and blinded histopathology scores of brain samples from Nav1.8-DTA and control mice 24 h after injection of S. pneumoniae. n = 48/group (12 fields/sample, 4 samples/group). Scale bar = 50 μm. f, g, Meningeal innervation by CGRP+ neurons (green = CGRP staining; white = skeletonization) in (f) Nav1.8-DTA and control mice, and in (g) WT mice treated with systemic injection of resiniferatoxin (RTX) or vehicle (n = 3/group). Scale bar = 300 μm. h, Bacterial load in samples collected 24 h after injection of S. pneumoniae from mice treated with RTX or vehicle (n = 5/group). i, Left, illustration created with BioRender.com (https://biorender.com). Right, Bacterial load in samples collected from Nav1.8-DTA and control mice 24 h after intracisternal injection of S. pneumoniae (n = 4/group). Statistical analysis: (b—i) Unpaired t-tests. *p < 0.05, **p < 0.01, ****p < 0.0001. n = biologically independent samples from mouse tissues. Each experiment was performed at least twice, and results presented are representative of 2 or more replicates. ns = not significant. Mean ±SEM. Exact p-values in Supplementary Table 1.
Extended Data Fig. 2 |. Local depletion of meningeal nociceptors reduces CNS infection by bacteria.
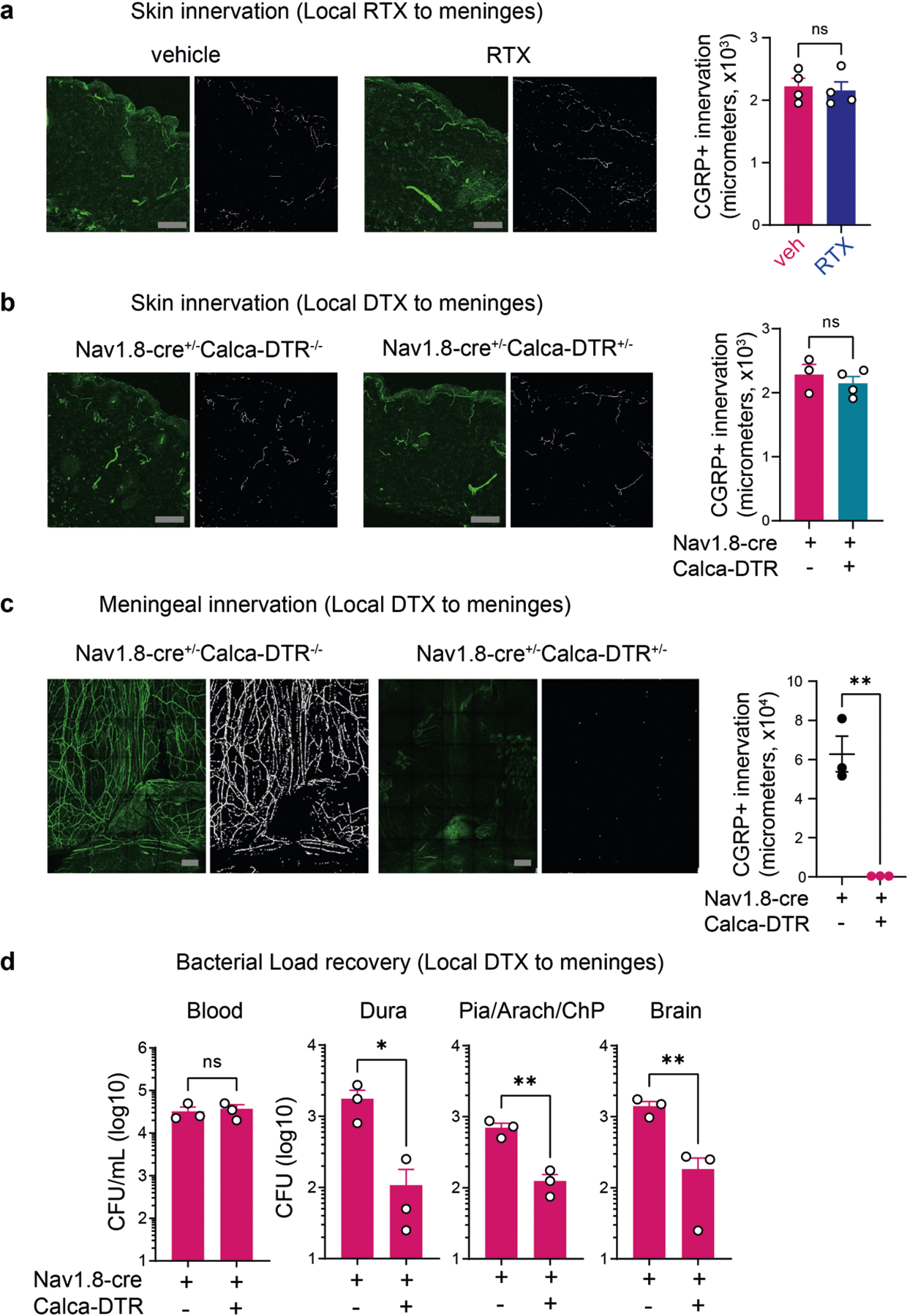
a, b, Back skin innervation by CGRP+ neurons quantified 3 weeks after either local injection of (a) resiniferatoxin (RTX) or vehicle into WT mice (n = 4/group), or (b) diphtheria toxin (DTX) into Nav1.8-Calca-DTR (Nav1.8-cre+/−Calca-DTR+/−) mice (n = 4) or into littermate control (Nav1.8-cre+/−Calca-DTR−/−) mice (n = 3). Scale bar = 100 μm. c, Meningeal innervation by CGRP+ neurons after local application of DTX in Nav1.8-Calca-DTR mice or control littermates (n = 3/group). Scale bar = 300 μm. d, Bacterial load in samples collected 24h after injection of S. pneumoniae from Nav1.8-Calca-DTR mice or control littermates treated locally with DTX (n = 3/group). a, b, c, green = CGRP staining; white = skeletonization. Statistical analysis: (a, b, c, d) Unpaired two-sided t-tests. *p < 0.05, **p < 0.01. n = biologically independent samples from mouse tissues. Each experiment was performed at least twice, and results presented are representative of 2 or more replicates. ns = not significant. Mean ±SEM. Exact p-values in Supplementary Table 1.
Extended Data Fig. 3 |. Nociceptors regulate meningeal immunity against bacterial infection.
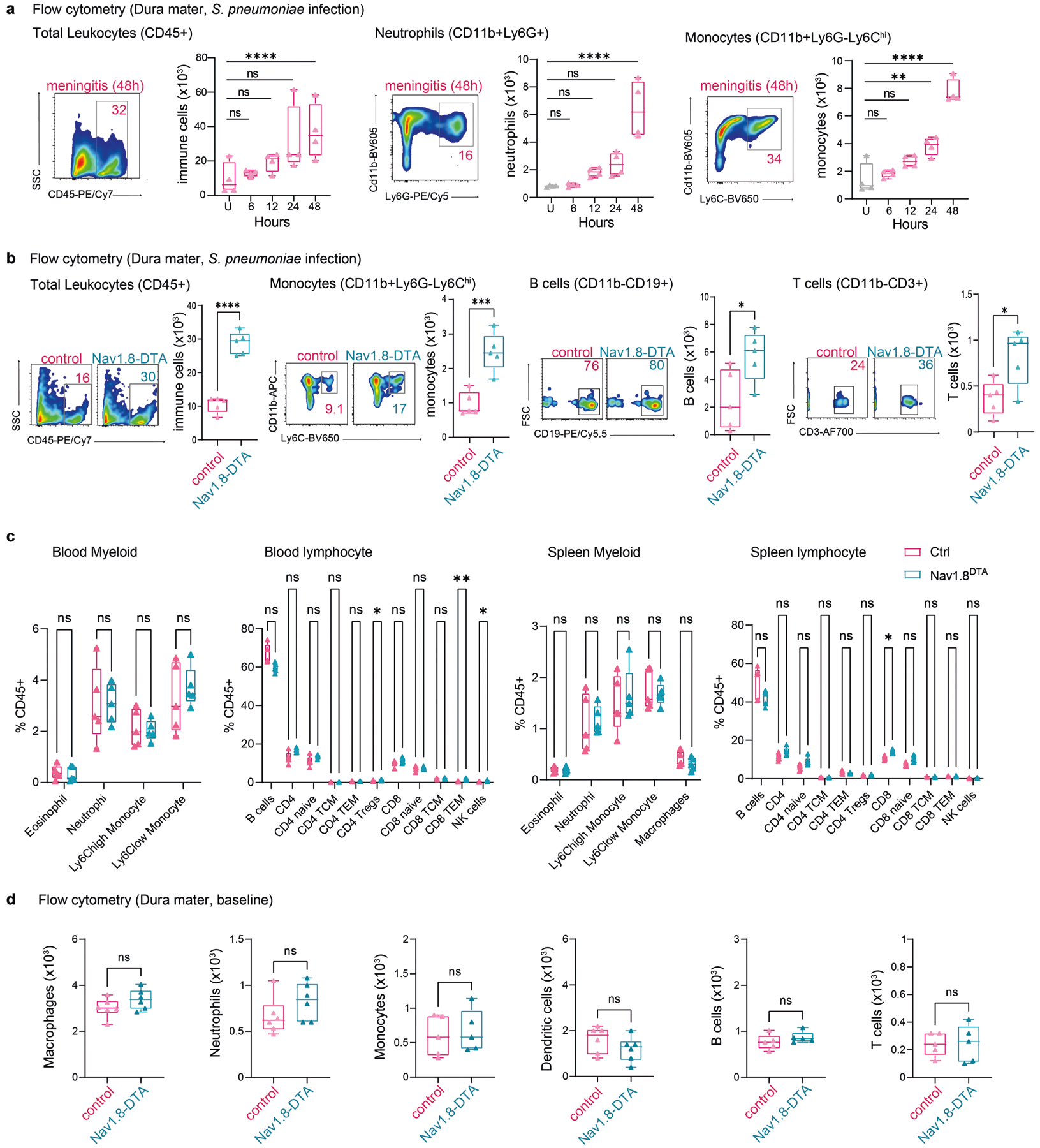
a, Representative flow cytometry plots and quantification of total leukocytes (CD45+ gate), neutrophils (CD11b+Ly6G+ gates), and monocytes (CD11b+Ly6G−Ly6Chi gates) in the meninges at baseline and different time points after injection of S. pneumoniae (n = 4/group). b, Representative flow cytometry plots and quantification of total leukocytes (CD45+ gate), monocytes (CD11b+Ly6G−Ly6Chi gates), B cells (CD11b−CD19+ gates), and T cells (CD11b−CD3+) in the meninges of Nav1.8-DTA mice or control littermates 24 h after S. pneumoniae injection (n = 5/group). c, Flow cytometric quantification of myeloid and lymphoid immune cells in the blood and spleen of Nav1.8-DTA mice and littermate controls (n = 4/group). d, Quantification of immune cells in the meninges from Nav1.8-DTA mice and wt controls by flow cytometry (n = 5/group for monocytes, B cells, and T cells; n = 6/group for macrophages, neutrophils, and DCs). Statistical analysis: (a) One-way ANOVA with Tukey post-tests. (b, c, d) Unpaired two-sided t-tests. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. n = biologically independent samples from mouse tissues. Each experiment was performed at least twice, and results presented are representative of 2 or more replicates. ns = not significant. Box plots = median, IQR, min/max. Exact p-values in Supplementary Table 1.
Extended Data Fig. 4 |. S. pneumoniae and S. agalactiae induce pain and act on trigeminal ganglion neurons.
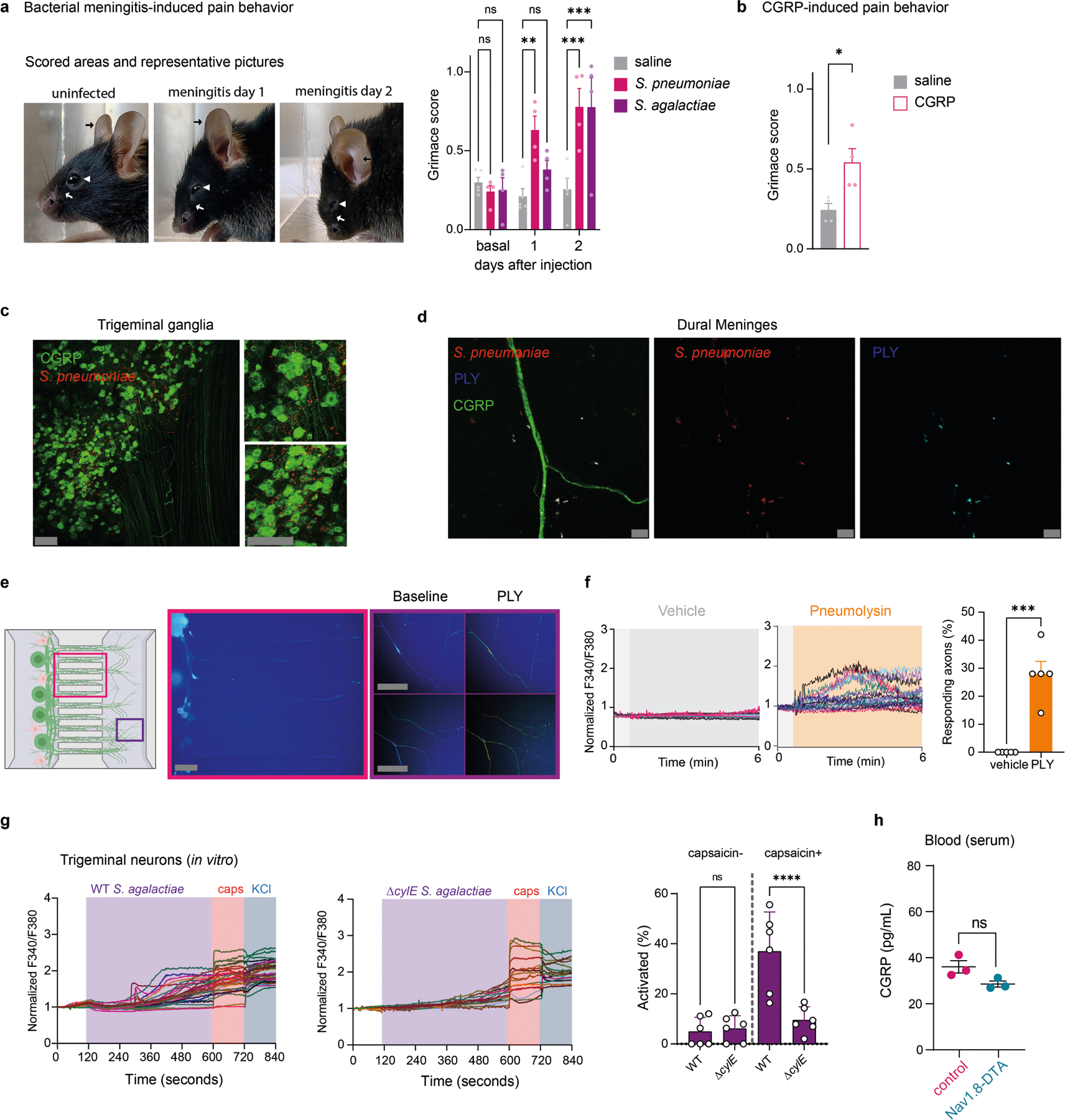
a, Left, Representative pictures of mice at baseline (uninfected), 1 day, or 2 days after S. pneumoniae infection. Right, Grimace scores of mice at baseline (uninfected), 1 day, and 2 days after injection of S. pneumoniae (n = 4), S. agalactiae (n = 4), or saline (n = 5). Orbital tightening (white arrowhead), nose bulge (black arrowhead), cheek bulge (white arrow), and ear position (black arrow) were scored. b, Grimace scores of mice injected with CGRP (2 μg, i.p.) or vehicle (n = 4/group). c, Trigeminal ganglion from Calca-GFP (Green) mice 24 h after injection of dye-labelled S. pneumoniae (red). Scale bar = 100 μm. d, Whole-mount staining of meninges from Calca-GFP mice (CGRP) collected 24 h after injection of dye-labelled S. pneumoniae (red) and co-stained with anti-Pneumolysin antibody (cyan). Scale bar = 10 μm. e, f, Trigeminal ganglion neurons plated in microfluidic chambers with cell bodies on left and axons growingto right chamber. Vehicle or PLY was added to right chamber during Fura-2 calcium imaging. (e) Left, illustration created with BioRender.com (https://biorender.com). Right, representative fields of neurons; (f) Traces and quantification (f) of intracellular calcium levels in individual axons stimulated with vehicle or S. pneumoniae toxin pneumolys in (n = 4/group). Scale bar = 50 μm. g, Left, Representative Fura-2 calcium traces of individual trigeminal ganglion neurons stimulated with wildtype S. agalactiae or the isogenic ΔcylE toxin-deficient mutant bacteria (2×107 c.f.u.), followed by capsaicin (1uM) and KCl (40 mM). Right, proportions of capsaicin non-responsive and capsaicin-responsive neurons that responded to wild-type or ΔcylE S. agalactiae (n = 6/group). h, Blood CGRP levels from Nav1.8-DTA mice and control littermates 24 h after injection of S. pneumoniae (3×107 c.f.u.) (n = 3/group). Statistical analysis: (b, f, g, h) Unpaired two-sided t-tests. (a) One-way ANOVA with Tukey post-tests. *p < 0.05, **p < 0.01, ***p < 0.001. n = individual mice (a, b) and biologically independent samples from mouse primary cells (f-g) and tissues (h). Each experiment was performed at least twice, and results presented are representative of 2 or more replicates. ns = not significant. Mean ±SEM. Exact p-values in Supplementary Table 1.
Extended Data Fig. 5 |. CGRP and RAMP1 signaling impair host response against bacterial meningitis.
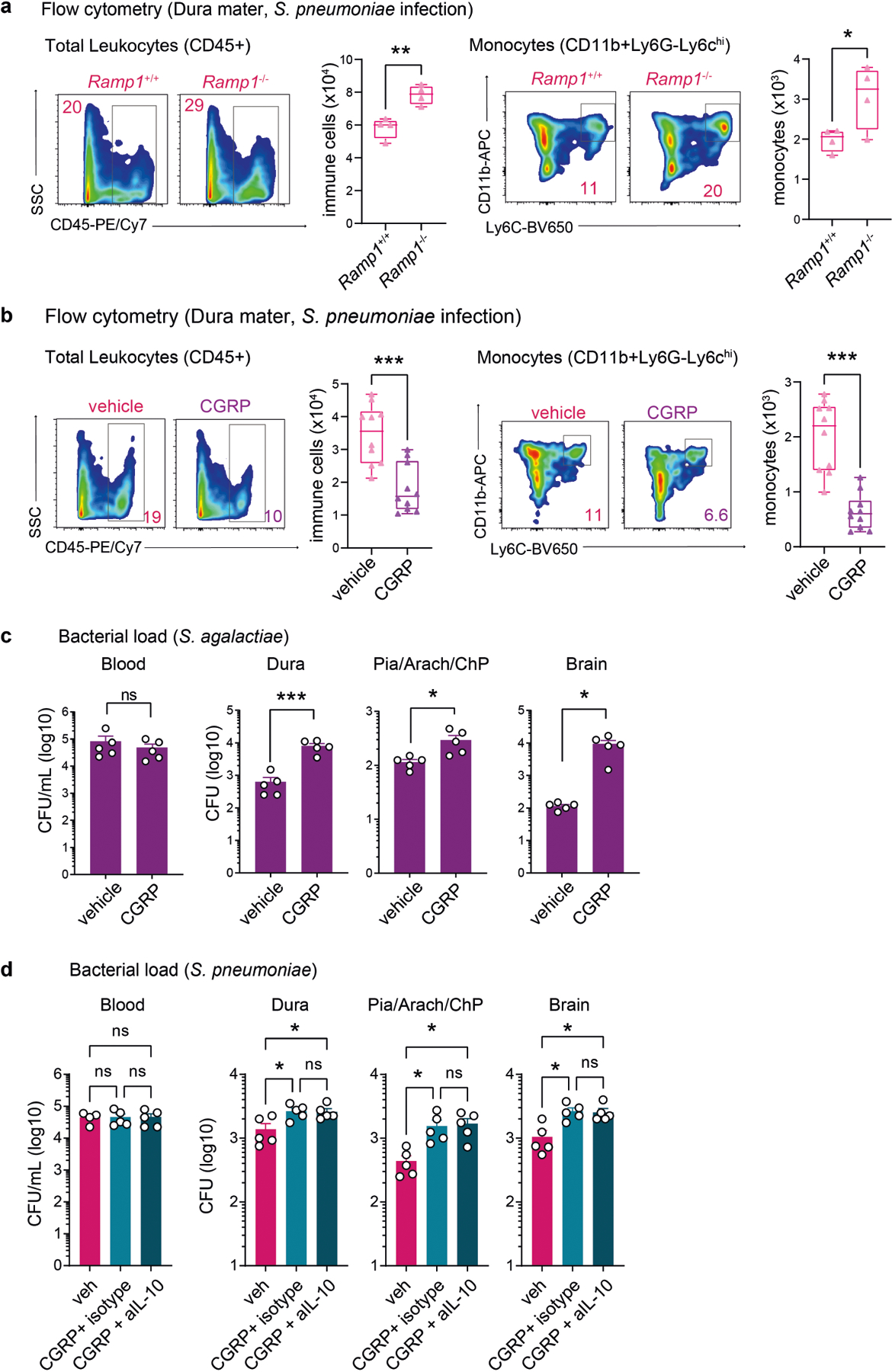
a, Representative flow cytometry plots and quantification of total leukocytes (CD45+ gate) and monocytes (CD11b+ Ly6G−Ly6Chi gates) in the meninges of Ramp1 knockout (Ramp1−/−) and control (Ramp1+/+) mice 24 h after S. pneumoniae injection (n = 4/group). b, Representative flow cytometry plots and quantification of total leukocytes (CD45+ gate) and monocytes (CD11b+Ly6G−Ly6Chi gates) 24h after S. pneumoniae injection in the meninges of mice treated with CGRP or vehicle (n = 10/group). c, Bacterial load 24h after injection of S. agalactiae in samples from mice treated with CGRP or vehicle (n = 5/group). d, Bacterial load in samples collected from mice treated with vehicle (n = 4 for blood; n = 5 for meninges and brain) or CGRP (CGRP 2 μg i.p., daily) and either isotype control (n = 5) or neutralizing anti-IL-10 (200 μg i.p., daily) (n = 5). Statistical analysis: (a, b, c) Unpaired two-sided t-tests. (d) One-way ANOVA with Tukey post-tests. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. ns = not ANOVA with Tukey post-tests. *p < 0.05, **p < 0.01, ***p < 0.001. n = biologically independent samples from mouse tissues. Each experiment was performed at least twice, and results presented are representative of 2 or more replicates. ns = not significant. Error bars = mean ±SEM. Box plots = median, IQR, min/max. Exact p-values in Supplementary Table 1.
Extended Data Fig. 6 |. Blockade of CGRP signaling affects bacterial meningitis.
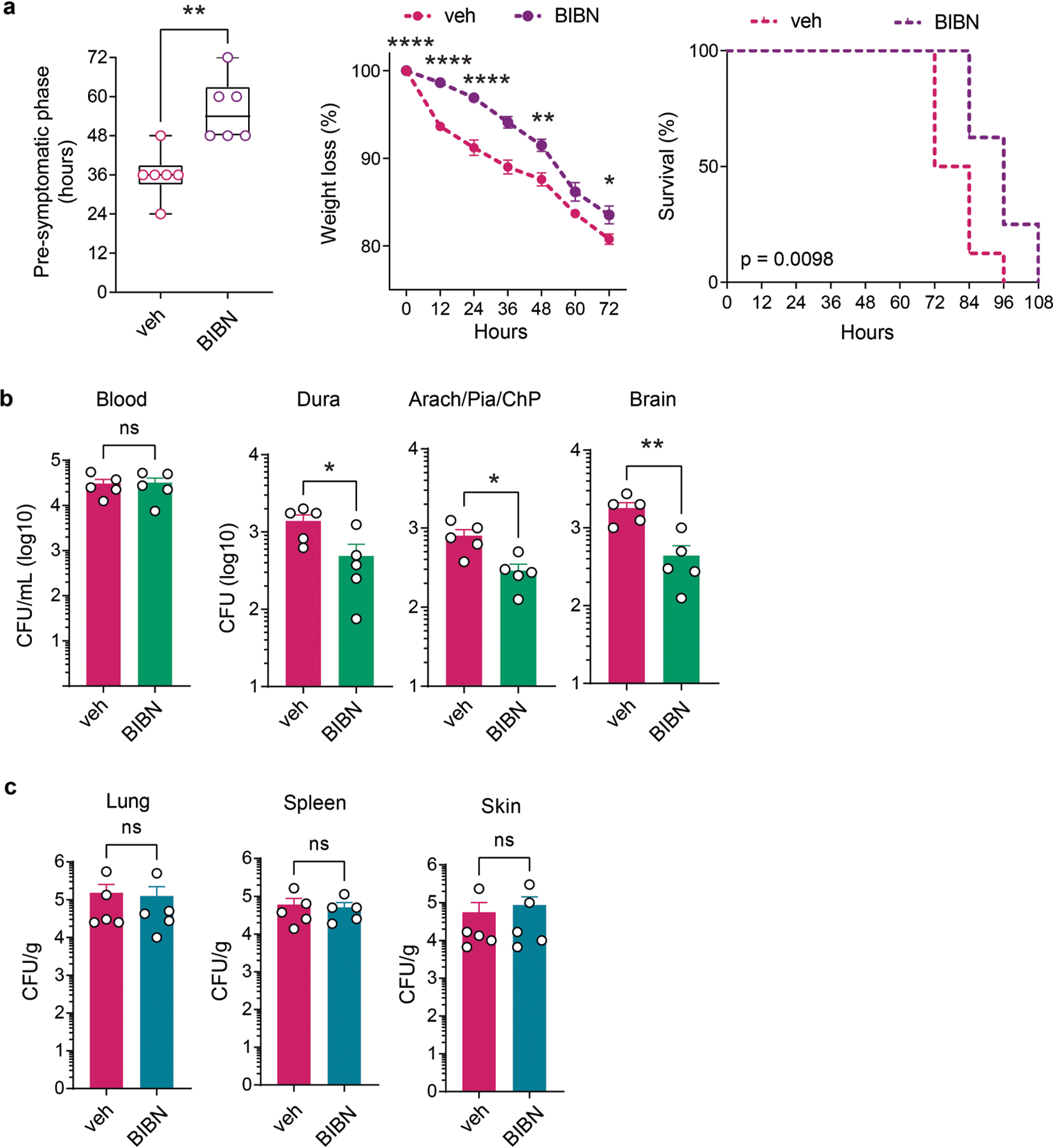
a, Analysis of disease progression in mice treated with BIBN (300 μg/kg, i.p.) or vehicle control, starting at 6 h post injection with S. pneumoniae (n = 6/group). b, Bacterial load recovery from samples at 24 h post infection in mice treated with BIBN (300 μg/kg, i.p.) or vehicle control, starting at 6 h post injection with S. pneumoniae (n = 5/group). c, Bacterial load in samples collected from peripheral tissues 24 h after injection of S. pneumoniae in mice treated with BIBN (300 μg/kg, i.p.) or vehicle (n = 5/group). Statistical analysis: (a, b, c) Unpaired two-sided t-tests. (a, weight loss) One-way ANOVA with Tukey post-tests. (a, survival) Kaplan Meier with Mantel-Cox comparison. *p < 0.05, **p < 0.01, ****p < 0.0001. n = individual mice (a) and biologically independent samples from mouse tissues (b-c). Each experiment was performed at least twice, and results presented are representative of 2 or more replicates. ns = not significant. Error bars = mean±SEM. Box plots = median, IQR, min/max. Exact p-values in Supplementary Table 1.
Extended Data Fig. 7 |. Single-cell RNA-sequencing analysis of meningeal cells.
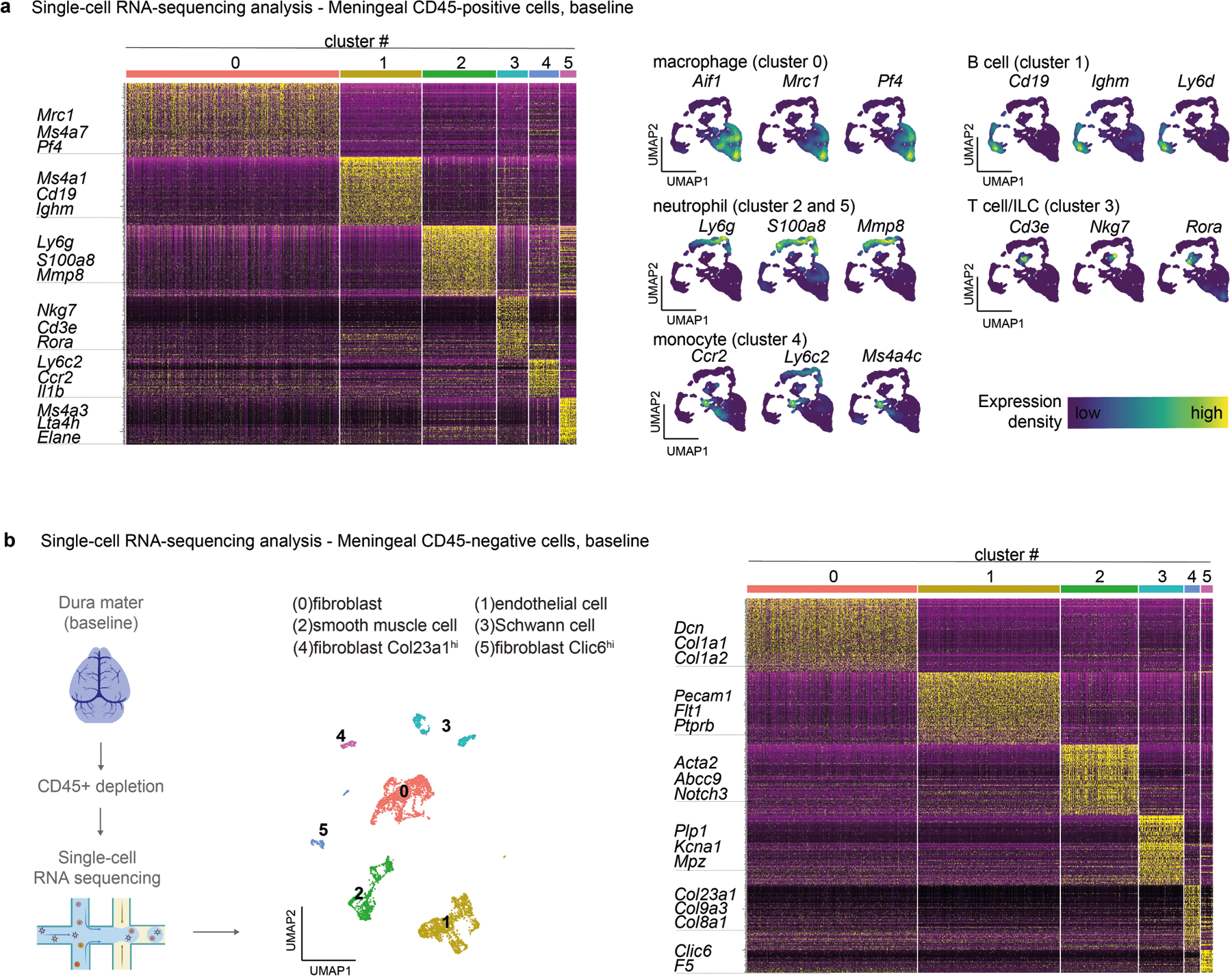
a, Single-cell RNA-sequencing analysis of meningeal immune cells (CD45-positive cells). Left, heatmap showing normalized expression of 100 top cluster marker genes in meningeal immune cells of the meninges, with key marker genes highlighted. Right, UMAP visualization of the expression of key marker genes for each immune cell cluster (n = 10 pooled mouse meninges). b, Left, Illustration created with BioRender.com (https://biorender.com). showing single-cell RNA-sequencing analysis of meningeal nonimmune cells (CD45-negative cells). Center, Uniform Manifold Approximation and Projection (UMAP) visualizations of CD45-negative cell types in the meninges at baseline. Right, heatmap showing normalized expression of 100 top cluster marker genes in nonimmune cells of the meninges, with key marker genes highlighted (n = 10 pooled mouse meninges).
Extended Data Fig. 8 |. Transcriptional responses of meningeal immune cells to bacterial meningitis.
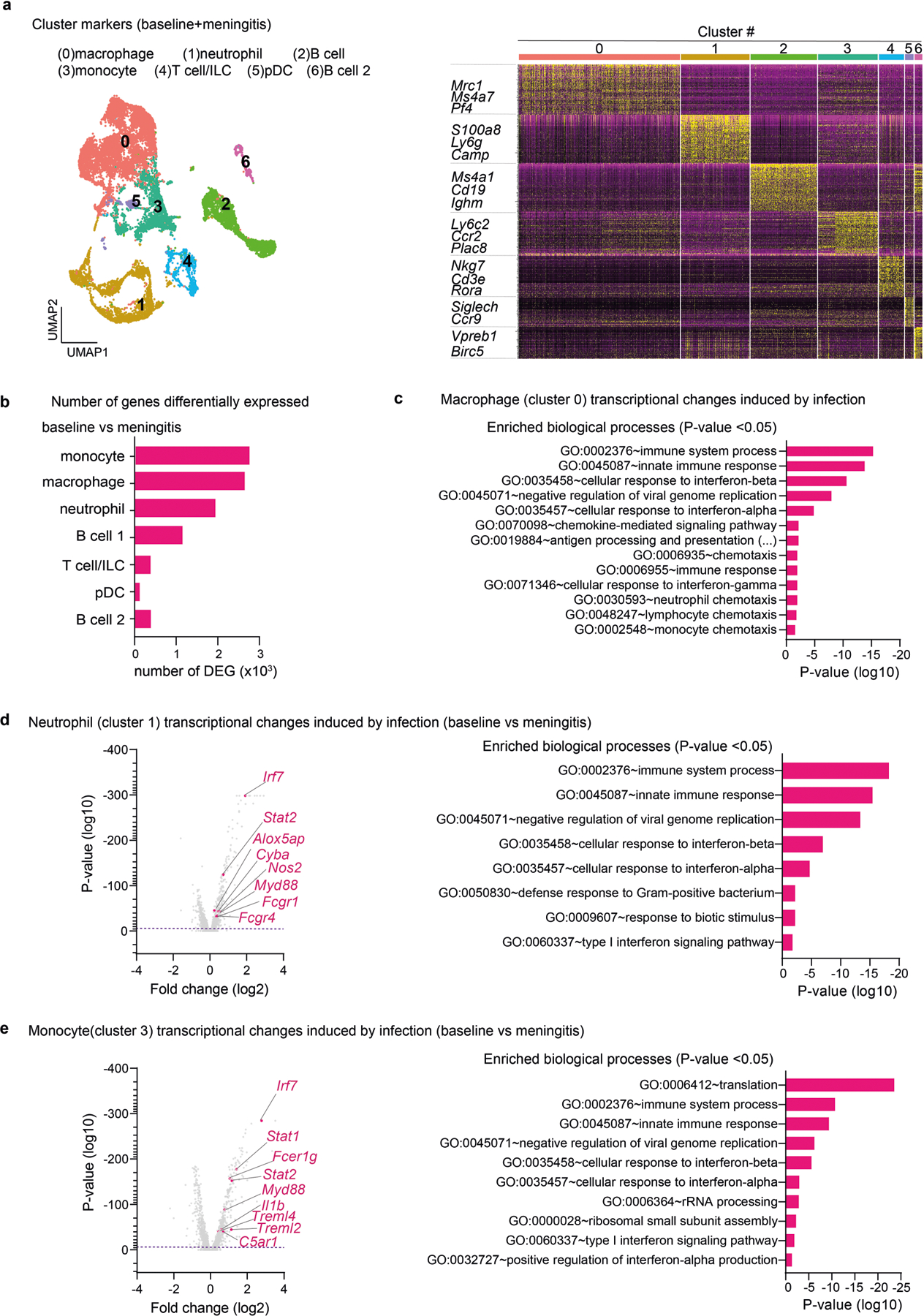
a, Single-cell RNA-sequencing analysis of meningeal immune responses to bacterial infection. Left, Uniform Manifold Approximation and Projection (UMAP) visualizations of CD45-positive cell types in the meninges at baseline and 24 h after injection of S. pneumoniae (meningitis). Right, heatmap showing normalized expression of 100 top cluster marker genes with key immune marker genes highlighted. b, Number of genes that were differentially expressed in each immune cell population during infection (baseline vs meningitis). c, Annotated GO biological processes of genes differentially expressed by the cluster of macrophages in response to infection (baseline vs meningitis), highlighting the enrichment of processes related to chemotaxis. d, Annotated GO biological processes and volcano plot of genes differentially expressed by the cluster of neutrophils in response to infection (baseline vs meningitis), highlighting upregulation of processes and genes related to antimicrobial activity. e, Annotated GO biological processes and volcano plot of genes differentially expressed by the cluster of monocytes in response to infection (baseline vs meningitis). (n = 10 pooled mouse meninges/group). Statistical analysis: (b, c, d, e) Fisher’s Exact score (enriched biological processes) and Wilcoxon rank-sum test (volcano plots of DEG), dashed purple line = p < 0.01.
Extended Data Fig. 9 |. Meningeal macrophages engulf bacteria and regulate immune responses against bacterial invasion.
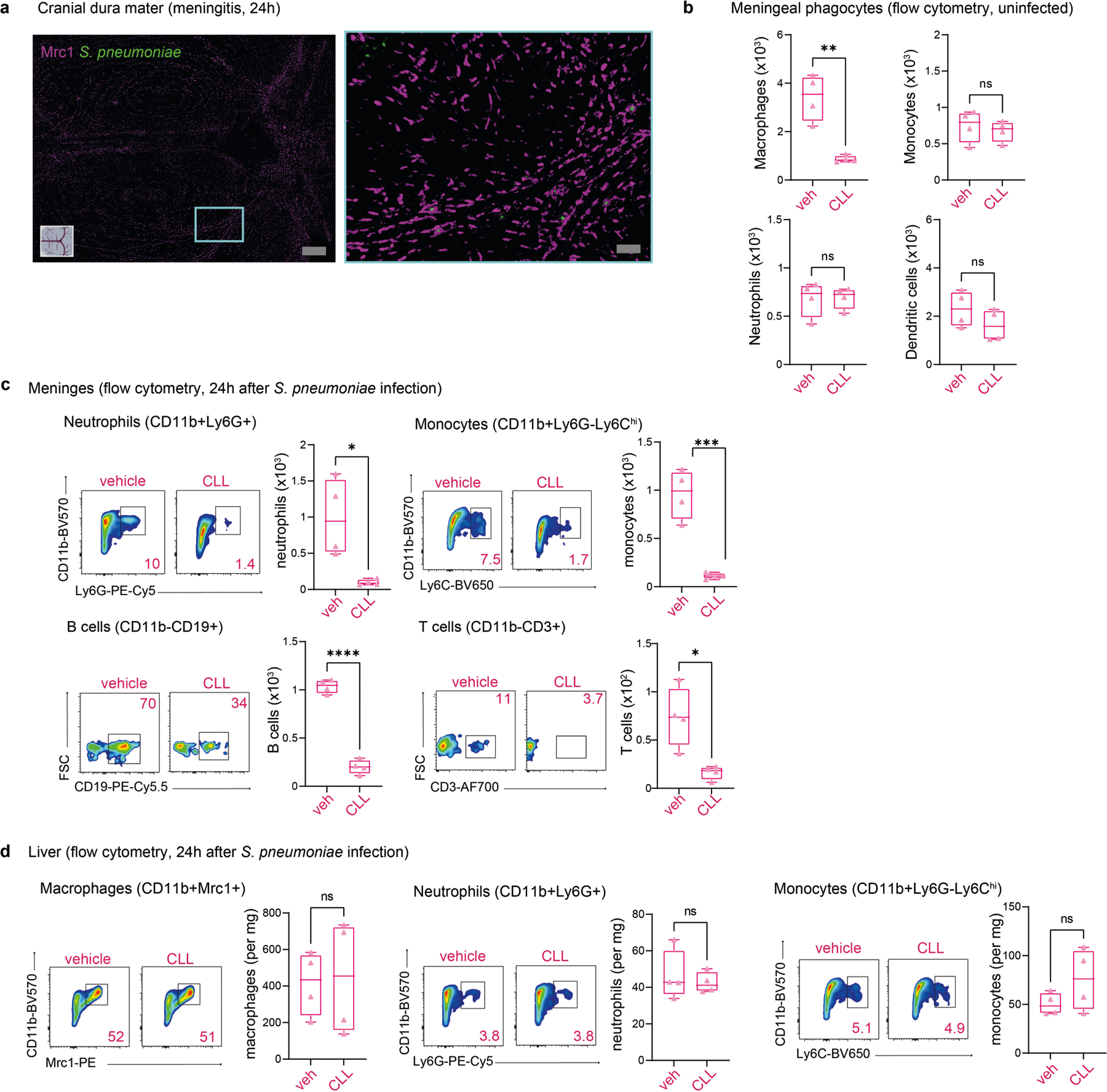
a, Whole-mount confocal images of mouse meninges (dura mater) showing meningeal macrophages (Mrc1+ cells) associated with S. pneumoniae 24 h post-injection of CMTPX-labeled bacteria. Scale bar = 500 μm (left) and 50 μm (right). b, c, d, Tissue-specific impact of depletion of meningeal Mrc1+ macrophages by intracisternal injection of clodronate liposomes (CLL). b, Flow cytometric quantification of meningeal macrophages, monocytes, neutrophils, and dendritic cells 3 days after intracisternal injection with clodronate liposomes (CLL, 5 mL) or vehicle (5 mL) (n = 4/group). c, Representative flow panels and quantification of neutrophils (CD11b+Ly6G+ gates), monocytes (CD11b+Ly6G−Ly6Chi gates), B cells (CD11b–CD19+ gates), and T cells (CD11b–CD3+ gates) 24 h after injection of S. pneumoniae in mice treated with CLL (5 μL) or vehicle. d, Representative flow panels and quantification of macrophages (CD11b+Mrc1+ gates), neutrophils (CD11b+Ly6G+ gates), and monocytes (Cd11b+Ly6G−Ly6Chi gates) in the liver of mice treated with CLL (5 μL) or vehicle. (n = 4/group). Statistical analysis: (b, c, d) Unpaired two-sided t-tests. *p < 0.05, ***p < 0.001, ****p < 0.0001. n = biologically independent samples from mouse tissues. Each experiment was performed at least twice, and results presented are representative of 2 or more replicates. ns = not significant. Box plots = median, IQR, min/max. Exact p-values in Supplementary Table 1.
Extended Data Fig. 10 |. CGRP and RAMP1 polarization of macrophage responses.
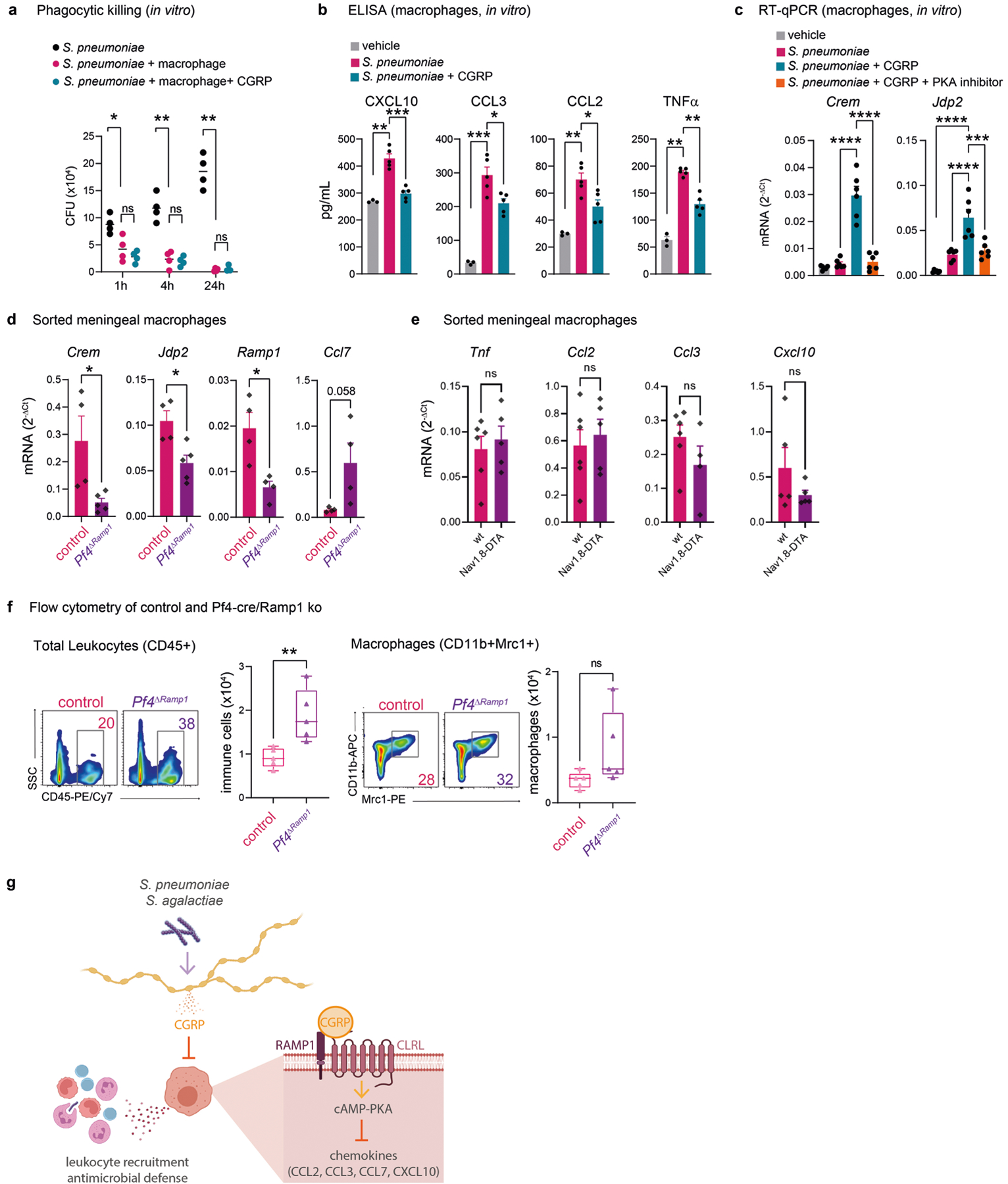
a, Phagocytic killing assay showing amount of S. pneumoniae recovered after incubation with macrophages (BMDM) in presence of CGRP or vehicle (n = 4/group). b, Concentration of chemotaxis-related mediators in macrophage supernatants after 24 h of incubation with vehicle (n = 3), S. pneumoniae (n = 5), or S. pneumoniae+CGRP (n = 5). c, Crem and Jdp2 expression determined by qPCR in macrophages after 4 h incubation with S. pneumoniae together with CGRP, PKA inhibitor (PKAi), or vehicle. Results are normalized to Beta-actin gene expression (n = 6/group). d, Crem, Jdp2, Ramp1, and Ccl7 transcript levels determined by qPCR in FACS purified macrophages from meninges of Pf4ΔRamp1 mice (n = 5 for Crem and Jdp2; n = 4 for Ramp1 and Ccl7) or control mice (n = 4 for Crem, Jdp2, Ramp1, Ccl7) 24 h after injection of S. pneumoniae. Results are normalized to Beta-actin expression. e, Expression of chemotactic mediators determined by qPCR in meningeal macrophages from Nav1.8-DTA (n = 5) and control mice (n = 6). f, Flow cytometric quantification of total leukocytes (CD45+) and macrophages (Cd11b+Mrc1+) in meninges of Pf4ΔRamp1 and control mice 24 h after S. pneumoniae injection (n = 5/group). g, Illustration created with BioRender.com (https://biorender.com). Bacterial pathogens S. pneumoniae and S. agalactiae invade the meninges, activating trigeminal nociceptors to induce release of CGRP. CGRP acts through its receptor RAMP1 on Pf4+Mrc+ meningeal macrophages, downregulating expression of chemokines, suppressing leukocyte recruitment and antimicrobial defenses. Nociceptor ablation or blockade of CGRP signaling enhances host defense against meningitis. Statistical analysis: (a, b, c) One-way ANOVA with Tukey post-tests. (d, e, f) Unpaired two-sided t-tests. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. n = biologically independent samples from mouse tissues (d-f) and cells (a-c). Each experiment was performed at least twice, and results presented are representative of 2 or more replicates. ns = not significant. Error bars = mean ±SEM. Box plots = median, IQR, min/max. Exact p-values in Supplementary Table 1.
Supplementary Material
Acknowledgements
We thank R. Malley for providing the S. pneumoniae strains used in this study; N. C. El-Ali, N. Weeks and staff at the Harvard University Bauer Core Facility for technical support; M. R. Wessels, D. E. Higgins, M. Lehtinin, H. Xu, T. M. Cunha and members of the Chiu Lab for helpful discussions; J. Buenrostro and R. Shrestha for computational advice; S. Lin, N. M. Gilette, K. Lezgiyeva, S. J. Sannajust, E. Erhard, O. Clancy and A. Prystupa for technical help and analysis; A. Frey for manuscript feedback; and J. L. Gibbs for mentorship. This work was supported by National Institutes of Health grants R01AI130019 and R01DK127257 to I.M.C.; Burroughs Wellcome fund, the Kenneth Rainin Foundation, the Food Allergy Science Initiative, Fairbairn Lyme Initiative to I.M.C.; 2R01NS078263 and 5R01NS115972 to D.L.; P50MH112491 to the Conte Center; R01NS116716 to K.S.D; and T32GM007753 to D.V.N. K.H. was supported by the Harvard Medical School Undergraduate Immunology Summer Program.
Competing interests
I.M.C. and F.A.P-R. are named inventors on US patent application 2021/0145937A1, ‘Methods and compositions for treating a microbial infection’, which includes targeting CGRP and its receptors to treat infections. The Chiu Lab receives research support from Abbvie/Allergan and Moderna.
Footnotes
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-023-05753-x.
Additional information Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-023-05753-x.
Data availability
All scRNA-seq datasets and bulk RNA-seq datasets generated and analysed during this study have been deposited into the NCBI Gene Expression Omnibus database under accession number GSE221681. The reference mouse genome mm10 v.2020-A can be accessed under the assembly number GRCm38. Raw imaging or other datasets from this paper will be made available upon request to the corresponding author.
References
- 1.Levy D, Labastida-Ramirez A & MaassenVanDenBrink A Current understanding of meningeal and cerebral vascular function underlying migraine headache. Cephalalgia 39, 1606–1622 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Burstein R, Zhang X, Levy D, Aoki KR & Brin MF Selective inhibition of meningeal nociceptors by botulinum neurotoxin type A: therapeutic implications for migraine and other pains. Cephalalgia 34, 853–869 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van de Beek D et al. Clinical features and prognostic factors in adults with bacterial meningitis. N. Engl. J. Med 351, 1849–1859 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Schiess N, Groce NE & Dua T The impact and burden of neurological sequelae following bacterial meningitis: a narrative review. Microorganisms 9, 900 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ostergaard C, Konradsen HB & Samuelsson S Clinical presentation and prognostic factors of Streptococcus pneumoniae meningitis according to the focus of infection. BMC Infect. Dis 5, 93 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doran KS & Nizet V Molecular pathogenesis of neonatal group B streptococcal infection: no longer in its infancy. Mol. Microbiol 54, 23–31 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Basbaum AI, Bautista DM, Scherrer G & Julius D Cellular and molecular mechanisms of pain. Cell 139, 267–284 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pinho-Ribeiro FA, Verri WA Jr. & Chiu IM Nociceptor sensory neuron-immune interactions in pain and inflammation. Trends Immunol. 38, 5–19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fitzpatrick Z et al. Gut-educated IgA plasma cells defend the meningeal venous sinuses. Nature 587, 472–476 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Hove H et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat. Neurosci 22, 1021–1035 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Rustenhoven J et al. Functional characterization of the dural sinuses as a neuroimmune interface. Cell 184, 1000–1016.e27 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rua R et al. Infection drives meningeal engraftment by inflammatory monocytes that impairs CNS immunity. Nat. Immunol 20, 407–419 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rebejac J et al. Meningeal macrophages protect against viral neuroinfection. Immunity 55, 2103–2117.e10 (2022). [DOI] [PubMed] [Google Scholar]
- 14.Lampl C, Yazdi K, Buzath A & Klingler D Migraine-like headache in bacterial meningitis. Cephalalgia 20, 738–739 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Abrahamsen B et al. The cell and molecular basis of mechanical, cold, and inflammatory pain. Science 321, 702–705 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Strassman AM & Levy D Response properties of dural nociceptors in relation to headache. J. Neurophysiol 95, 1298–1306 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Arkless K, Argunhan F & Brain SD CGRP discovery and timeline. Handb. Exp. Pharmacol 255, 1–12 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Dando SJ et al. Pathogens penetrating the central nervous system: infection pathways and the cellular and molecular mechanisms of invasion. Clin. Microbiol. Rev 27, 691–726 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baral P et al. Nociceptor sensory neurons suppress neutrophil and γδ T cell responses in bacterial lung infections and lethal pneumonia. Nat. Med 24, 417–426 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pinho-Ribeiro FA et al. Blocking neuronal signaling to immune cells treats streptococcal invasive infection. Cell 173, 1083–1097.e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCoy ES et al. Peptidergic CGRPα primary sensory neurons encode heat and itch and tonically suppress sensitivity to cold. Neuron 78, 138–151 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rea BJ et al. Peripherally administered calcitonin gene-related peptide induces spontaneous pain in mice: implications for migraine. Pain 159, 2306–2317 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blake KJ et al. Staphylococcus aureus produces pain through pore-forming toxins and neuronal TRPV1 that is silenced by QX-314. Nat. Commun 9, 37 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Braun JS et al. Pneumococcal pneumolysin and H2O2 mediate brain cell apoptosis during meningitis. J. Clin. Invest 109, 19–27 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wellmer A et al. Decreased virulence of a pneumolysin-deficient strain of Streptococcus pneumoniae in murine meningitis. Infect. Immun 70, 6504–6508 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doran KS, Liu GY & Nizet V Group B streptococcal β-hemolysin/cytolysin activates neutrophil signaling pathways in brain endothelium and contributes to development of meningitis. J. Clin. Invest 112, 736–744 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jusek G, Reim D, Tsujikawa K & Holzmann B Deficiency of the CGRP receptor component RAMP1 attenuates immunosuppression during the early phase of septic peritonitis. Immunobiology 217, 761–767 (2012). [DOI] [PubMed] [Google Scholar]
- 28.van Furth AM et al. High levels of interleukin 10 and tumor necrosis factor α in cerebrospinal fluid during the onset of bacterial meningitis. Clin. Infect. Dis 21, 220–222 (1995). [DOI] [PubMed] [Google Scholar]
- 29.Olesen J et al. Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N. Engl. J. Med 350, 1104–1110 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Brioschi S et al. Heterogeneity of meningeal B cells reveals a lymphopoietic niche at the CNS borders. Science 373, eabf9277 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McKinsey GL et al. A new genetic strategy for targeting microglia in development and disease. eLife 9, e54590 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harzenetter MD et al. Negative regulation of TLR responses by the neuropeptide CGRP is mediated by the transcriptional repressor ICER. J. Immunol 179, 607–615 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Maruyama K et al. Nociceptors boost the resolution of fungal osteoinflammation via the TRP channel–CGRP–Jdp2 axis. Cell Rep. 19, 2730–2742 (2017). [DOI] [PubMed] [Google Scholar]
- 34.De Vlaminck K et al. Differential plasticity and fate of brain-resident and recruited macrophages during the onset and resolution of neuroinflammation. Immunity 55, 2085–2102.e9 (2022). [DOI] [PubMed] [Google Scholar]
- 35.Hoffmann O et al. Triptans reduce the inflammatory response in bacterial meningitis. J. Cereb. Blood Flow Metab 22, 988–996 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Strausbaugh HJ et al. Painful stimulation suppresses joint inflammation by inducing shedding of l-selectin from neutrophils. Nat. Med 5, 1057–1061 (1999). [DOI] [PubMed] [Google Scholar]
- 37.Sterner-Kock A et al. Neonatal capsaicin treatment increases the severity of ozone-induced lung injury. Am. J. Respir. Crit. Care Med 153, 436–443 (1996). [DOI] [PubMed] [Google Scholar]
- 38.Hoeffel G et al. Sensory neuron-derived TAFA4 promotes macrophage tissue repair functions. Nature 594, 94–99 (2021). [DOI] [PubMed] [Google Scholar]
- 39.Smith PG & Liu M Impaired cutaneous wound healing after sensory denervation in developing rats: effects on cell proliferation and apoptosis. Cell Tissue Res. 307, 281–291 (2002). [DOI] [PubMed] [Google Scholar]
- 40.Liu Z et al. Calcitonin gene-related peptide prevents blood–brain barrier injury and brain edema induced by focal cerebral ischemia reperfusion. Regul. Pept 171, 19–25 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Zhai L et al. Endogenous calcitonin gene-related peptide suppresses ischemic brain injuries and progression of cognitive decline. J. Hypertens 36, 876–891 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Saloman JL et al. Ablation of sensory neurons in a genetic model of pancreatic ductal adenocarcinoma slows initiation and progression of cancer. Proc. Natl Acad. Sci. USA 113, 3078–3083 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Balood M et al. Nociceptor neurons affect cancer immunosurveillance. Nature 611, 405–412 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Riol-Blanco L et al. Nociceptive sensory neurons drive interleukin-23-mediated psoriasiform skin inflammation. Nature 510, 157–161 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang S et al. Nonpeptidergic neurons suppress mast cells via glutamate to maintain skin homeostasis. Cell 184, 2151–2166.e16 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berg RM et al. Circulating levels of vasoactive peptides in patients with acute bacterial meningitis. Intensive Care Med. 35, 1604–1608 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Serezani CH, Ballinger MN, Aronoff DM & Peters-Golden M Cyclic AMP: master regulator of innate immune cell function. Am. J. Respir. Cell Mol. Biol 39, 127–132 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gabanyi I et al. Neuro-immune interactions drive tissue programming in intestinal macrophages. Cell 164, 378–391 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matheis F et al. Adrenergic signaling in muscularis macrophages limits infection-induced neuronal loss. Cell 180, 64–78.e16 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cugurra A et al. Skull and vertebral bone marrow are myeloid cell reservoirs for the meninges and CNS parenchyma. Science 373, eabf7844 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Malley R et al. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc. Natl Acad. Sci. USA 100, 1966–1971 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mu R et al. Identification of CiaR regulated genes that promote group B streptococcal virulence and interaction with brain endothelial cells. PLoS ONE 11, e0153891 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Doran KS, Chang JC, Benoit VM, Eckmann L & Nizet V Group B streptococcal β-hemolysin/cytolysin promotes invasion of human lung epithelial cells and the release of interleukin-8. J. Infect. Dis 185, 196–203 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Toda G, Yamauchi T, Kadowaki T & Ueki K Preparation and culture of bone marrow-derived macrophages from mice for functional analysis. STAR Protoc. 2, 100246 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alves de Lima K et al. Meningeal γδ T cells regulate anxiety-like behavior via IL-17a signaling in neurons. Nat. Immunol 21, 1421–1429 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Louveau A et al. Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Argunhan F et al. Calcitonin gene-related peptide protects against cardiovascular dysfunction independently of nitric oxide in vivo. Hypertension 77, 1178–1190 (2021). [DOI] [PubMed] [Google Scholar]
- 58.Hafemeister C & Satija R Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hao Y et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jordao MJC et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 363, eaat7554 (2019). [DOI] [PubMed] [Google Scholar]
- 61.Alquicira-Hernandez J & Powell JE Nebulosa recovers single cell gene expression signals by kernel density estimation. Bioinformatics 10.1093/bioinformatics/btab003 (2021). [DOI] [PubMed] [Google Scholar]
- 62.Yang NJ et al. Anthrax toxins regulate pain signaling and can deliver molecular cargoes into ANTXR2+ DRG sensory neurons. Nat. Neurosci 25, 168–179 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hohlbaum K, Corte GM, Humpenoder M, Merle R & Thone-Reineke C Reliability of the mouse grimace scale in C57BL/6JRj mice. Animals (Basel) 10, 1648 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Langford DJ et al. Coding of facial expressions of pain in the laboratory mouse. Nat. Methods 7, 447–449 (2010). [DOI] [PubMed] [Google Scholar]
- 65.Whittaker AL, Liu Y & Barker TH Methods used and application of the mouse grimace scale in biomedical research 10 years on: a scoping review. Animals (Basel) 11, 673 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Deng L et al. The group B streptococcal surface antigen I/II protein, BspC, interacts with host vimentin to promote adherence to brain endothelium and inflammation during the pathogenesis of meningitis. PLoS Pathog. 15, e1007848 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ekici MA et al. Effect of etanercept and lithium chloride on preventing secondary tissue damage in rats with experimental diffuse severe brain injury. Eur. Rev. Med. Pharmacol. Sci 18, 10–27 (2014). [PubMed] [Google Scholar]
- 68.Zille M et al. Visualizing cell death in experimental focal cerebral ischemia: promises, problems, and perspectives. J. Cereb. Blood Flow Metab 32, 213–231 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mook-Kanamori B, Geldhoff M, Troost D, van der Poll T & van de Beek D Characterization of a pneumococcal meningitis mouse model. BMC Infect. Dis 12, 71 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All scRNA-seq datasets and bulk RNA-seq datasets generated and analysed during this study have been deposited into the NCBI Gene Expression Omnibus database under accession number GSE221681. The reference mouse genome mm10 v.2020-A can be accessed under the assembly number GRCm38. Raw imaging or other datasets from this paper will be made available upon request to the corresponding author.