

Abstract
The tumor microenvironment possesses mechanisms that suppress anti-tumor immunity. Itaconate is a metabolite produced from the Krebs cycle intermediate cis-aconitate by the activity of immune-responsive gene 1 (IRG1). While it is known to be immune modulatory, the role of itaconate in anti-tumor immunity is unclear. Here, we demonstrate that myeloid-derived suppressor cells (MDSCs) secreted itaconate that can be taken up by CD8+ T cells and suppress their proliferation, cytokine production, and cytolytic activity. Metabolite profiling, stable-isotope tracing, and metabolite supplementation studies indicated that itaconate suppressed biosynthesis of aspartate and serine/glycine in CD8+ T cells to attenuate their proliferation and function. Host deletion of Irg1 in female mice bearing allografted tumors resulted in decreased tumor growth, inhibited the immune suppressive activities of MDSCs, promoted anti-tumor immunity of CD8+ T cells, and enhanced the anti-tumor activity of anti-PD-1 antibody treatment. Furthermore, we found a significant negative correlation between IRG1 expression and response to PD-1 immune checkpoint blockade in melanoma patients. Our findings not only reveal a previously unknown role of itaconate as an immune checkpoint metabolite secreted from MDSCs to suppress CD8+ T cells, but also establish IRG1 as a myeloid-selective target in immunometabolism whose inhibition promotes anti-tumor immunity and enhance the efficacy of immune checkpoint protein blockade.
The ability to suppress anti-tumor immunity within the tumor microenvironment (TME) is critical for tumor development. Globally, this depends on disparate mechanisms that ensure anti-tumor cytolytic cells either fail to recognize or react to tumor cells. These mechanisms take the form of cell-cell contact, i.e., through with immune checkpoint receptors, the production of inhibitory cytokines, and altered extracellular metabolic conditions, all of which act to reduce effector functions in cytotoxic CD8+ T cells and NK cells. Since the activation and effector functions of lymphocytes are closely linked to both their metabolic state and the activities of specific metabolic pathways, this represents a potential point of physiological manipulation for controlling their activity that tumors exploit to suppress their anti-tumor activities1–3. For example, tumor cells secrete immunosuppressive metabolites such as lactate4–7 and 2-hydroxyglutarate (2HG)4,5 that act directly on T cell populations to control their function. While the full scope and contribution of the metabolic crosstalk among cells in the TME is only beginning to be appreciated, it’s becoming clearer that it plays a critical role in tumor development and progression.
Myeloid-derived suppressor cells (MDSCs) constitute a major immunosuppressive population in many tumors required to suppress effector functions in tumor-reactive cytotoxic lymphocytes6. Importantly, MDSCs have also been suggested to contribute to resistance to various cancer therapies, including to anti-CTLA-4 and anti-PD-1/L1 blockade7–9. Thus, targeting their function represents an attractive approach to modulate tumor immunity and improve current cancer immunotherapies. MDSCs exert potent immunosuppressive activities towards T cells and NK cells through a variety of mechanisms, including altering the metabolic makeup of the tumor microenvironment6. However, whether MDSCs secrete specific metabolic products to suppress tumor infiltrating lymphocytes is largely unknown.
Here we report that MDSCs secrete high levels of the TCA cycle derived metabolite, itaconate, and demonstrate that it can be taken up by CD8+ T cells. The production of Itaconate is catalyzed by Immune-Responsive Gene 1 (IRG1; also known as aconitate decarboxylase1, ACOD1; or cis-Aconitate decarboxylase, CAD) from cis-aconitate through a decarboxylation reaction10,11. While itaconate in macrophages has been previously shown to exhibit anti-inflammatory properties10,11, here we show that it suppresses immune effector functions in activated CD8+ T cells. Exposure to itaconate resulted in significant reductions in proliferation and expression of the key cytotoxic effectors granzyme and perforin, and attenuated metabolic flux through aspartate, serine, and glycine synthesis pathways in CD8+ T cells. Importantly, tumors implanted into syngeneic Irg1-deficient mice exhibited marked reduction in growth and improved response to anti-PD-1 therapy. These data reveal a previously unknown metabolic communication mechanism whereby itaconate produced by tumor associated MDSCs suppresses cytotoxic lymphocyte function and provides rationale for targeting IRG1 to enhance the efficacy of immune checkpoint blockade in cancer.
Results
Secretion of Itaconate from MDSCs
To gain better understanding of the metabolites secreted from MDSCs with potential to suppress anti-tumor immunity, we interrogated the metabolite secretome of mouse bone marrow-derived MDSCs (BM-MDSCs) by mass spectrometry. BM-MDSCs were produced by co-culturing bone marrow cells from naïve C57BL/6 mice with D4M3A melanoma cell -conditioned media as we previously described12. After 3 days, CD11b+Ly6G+ polymorphonuclear MDSCs (PMN-MDSCs, also known as granulocytic MDSCs, G-MDSCs) were purified, cultured in fresh medium for 24 hours, and polar metabolites in the supernatants were analyzed by targeted LC-MS-based metabolomics. Supernatants from undifferentiated bone marrow cell culture were collected and analyzed in a similar fashion and used as a control. By comparing the levels of metabolites in the supernatants of MDSC media collected before and after 24 h of culturing, we identified itaconate as a prominent MDSC-secreted metabolite (Figure 1A). Further quantitative analyses demonstrated that the secretion rate of itaconate by MDSCs is approximately 280-fold higher than that of undifferentiation BM cells (Figure 1B, Extended Data Figures 1A and 1B). Several TCA cycle related metabolites (such as malate, succinate, and α-ketoglutarate) were also found to be secreted from MDSCs, but the ratios of the secretion rates from MDSCs versus naïve BM for these metabolites were less significant compared to that of itaconate (Figures 1A, 1B, Extended Data Figures 1A and 1B).
Figure 1. Secretion of itaconate from MDSCs.
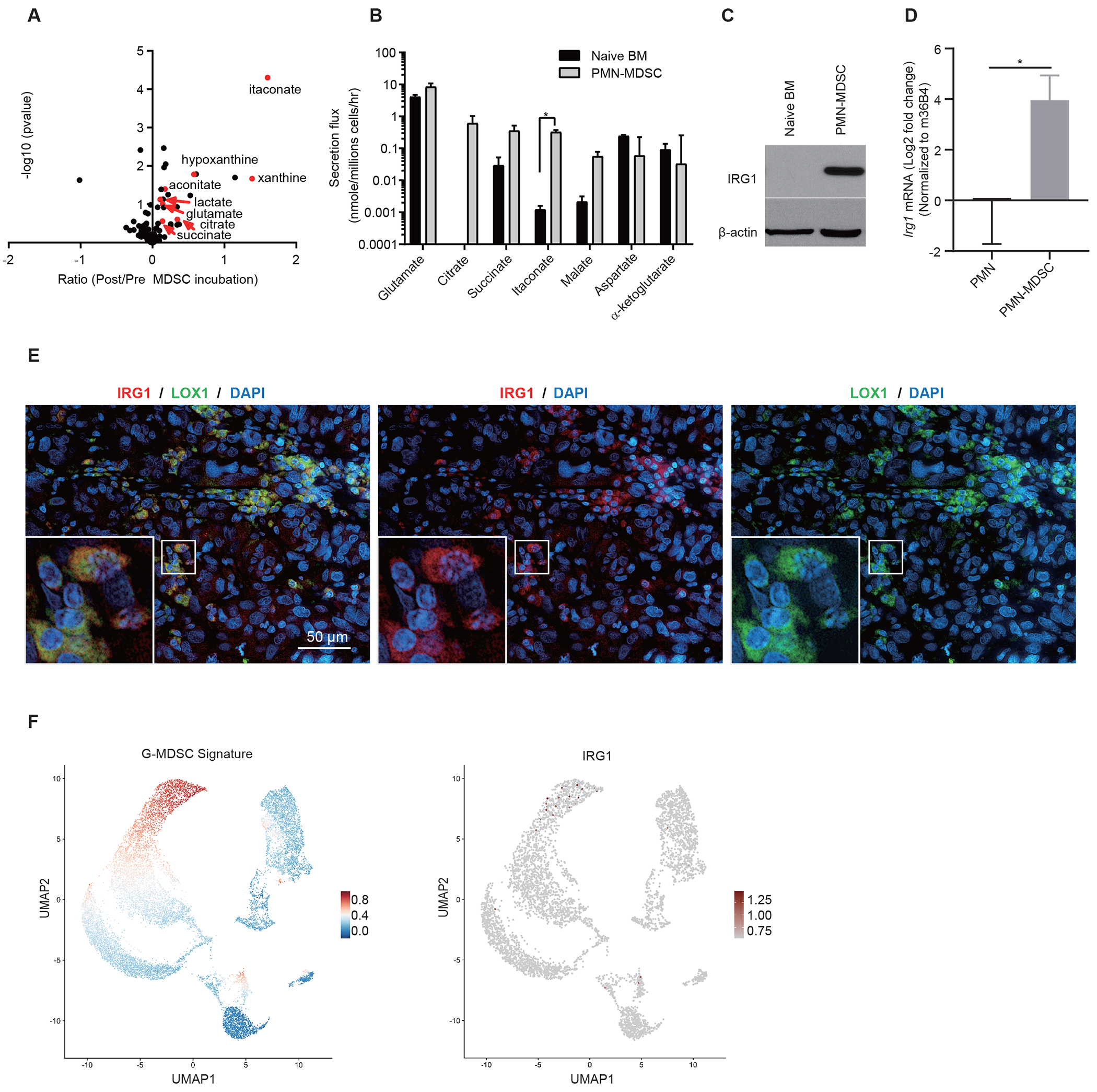
A Mass Spectrometry-based profiling of metabolites secreted by ex vivo differentiated MDSCs revealed increase in itaconate secretion (n=3 biologically independent samples). B Secretion rates of itaconate and various TCA cycle related metabolites from BM-MDSCs versus naïve BM cells (n=3 biologically independent samples). MDSCs or BM cells were reseeded and cultured in fresh medium for 24 hours and polar metabolites in the supernatants were analyzed by targeted LC-MS-based metabolomics. C-D IRG1 protein (C) and mRNA (D) levels were increased upon MDSC differentiation (n=4 biologically independent experiments). E Immunofluorescence analysis of human melanoma tumors revealed IRG1 co-staining with the MDSC marker LOX1. Data shown are representatives of 12 biologically independent human participants. F UMAP showing the gene signature score computed from 390 genes enriched to G-MDSC populations in mouse breast cancer tumors. A sample consist of 14,630 cells was down sampled to include 5,000 cells for better visualization in UMAP, colored by expression of IRG1. P values were calculated by paired (A) or unpaired (B, D) two-tailed Student’s t-tests. Data are represented as mean ± SEM,
Because IRG1 activity is the only known source of itaconate13, we analyzed its expression upon BM-MDSC induction. Western blot analysis revealed markedly more IRG1 in CD11b+Ly6G+ MDSCs, compared to undifferentiated BM cells (Figure 1C). Similarly, qRT-PCR analysis indicated that the mRNA level of Irg1 is ~ 15-fold higher in CD11b+Ly6G+ MDSCs isolated from mouse melanoma tumors, as compared to CD11b+Ly6G+ polymorphonuclear leukocytes isolated from spleens of tumor-free mice (Figure 1D). We next carried out immunofluorescence analysis of IRG1 on human melanoma tumor specimens and found that essentially all IRG1 positive cells were co-stained with Lectin-type oxidized LDL receptor-1 (LOX1), a marker of human PMN-MDSCs14 (Figure 1E and Extended Data Figure 1E), supporting that IRG1 is expressed in polymorphonuclear MDSCs (PMN-MDSCs) in human melanomas. Previously, RAW 264.7 macrophage cells were found to secrete itaconate upon LPS stimulation15. Indeed, we also detected basal level of itaconate secretion in RAW264.7 cells, and treatment of tumor-conditioned media significantly increased Irg1 expression and itaconate secretion in these cells (Extended Data Figures 1C and 1D). However, we did not detect colocalization of IRG1 and CD163, a monocyte/macrophage marker, or with CD3 and B220, markers for T and B cells, respectively, by immunofluorescence analysis of human melanoma tumor specimens (Extended Data Figure 1F), suggesting that itaconate production is limited to PMN-MDSCs within melanoma tumors. We also compared the expression pattern of Irg1 with the PMN-MDSC signature characterized from a recent single-cell transcriptomics study of mouse breast cancer–associated MDSCs16, and found that Irg1 is selectively expressed in a subset of PMN-MDSCs that also expressed Arg2 and Cxcr2 (Figures 1F and Extended Data Figure 2A). Taken together, these results support that Irg1 is selectively expressed in MDSCs and its expression is induced by tumors.
Uptake of Itaconate by CD8+ T cells
We next investigated the biological consequences of itaconate secretion from MDSCs. Since it is well established that MDSCs suppress CD8+ T cell proliferation and cytotoxic effector function, we assessed whether itaconate can be taken up by CD8+ T cells. We activated murine splenic CD8+ T cells via CD3/CD28 cross-linking and incubated them with increasing concentrations of exogenous itaconate for 6 h. Intracellular concentrations of itaconate within the T cells increased in a dose-dependent manner, demonstrating uptake by activated CD8+ cells (Figure 2A). Endogenous itaconate was undetectable in activated CD8+ T cells in the absence of media exposure to itaconate, demonstrating that these cells did not produce itaconate, consistent with their lack of Irg1 expression. We also performed similar experiments in naïve mouse CD8+ cells without activation by CD3/CD28 cross-linking and A549 lung cancer cells, which are known to take up itaconate17. We observed that while all three cell types took up exogenous itaconate with uptake somewhat faster in the CD3/CD28-activated CD8+ T cells (Figure 2B). We also cultured CD3/CD28-activated mouse CD8+ cells with 5 mM [U-13C5]itaconate for 6 h, and found that essentially all itaconate detected in the cellular extracts was labelled as the M+5 species (Figure 2C). Together, these findings show that activated CD8+ T cells, while not able to synthesize itaconate, can take it up upon its secretion by other cells.
Figure 2. Itaconate suppresses CD8+ T cell proliferation and activation.
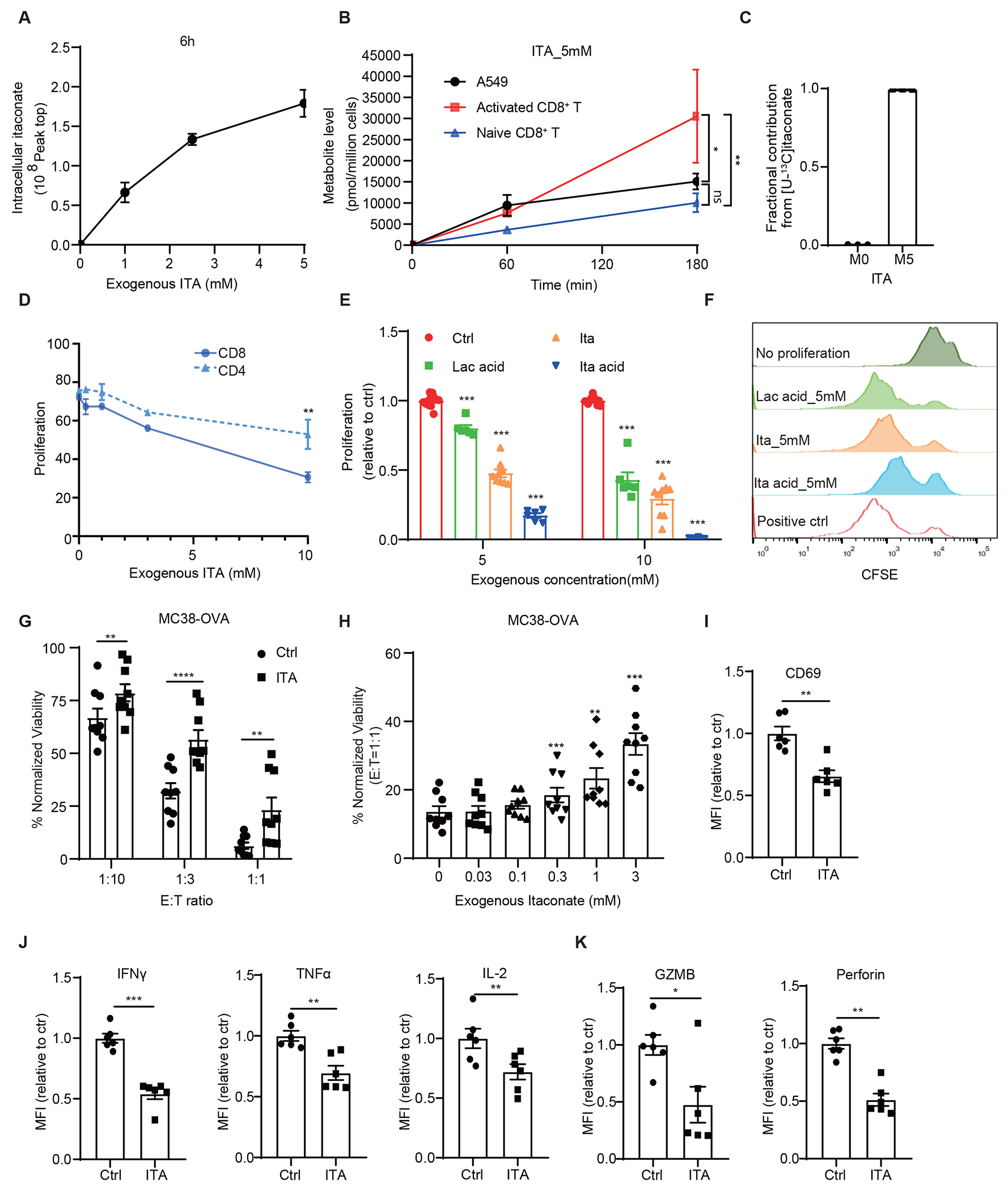
A CD3/CD28 activated CD8+ T cells took up itaconate (ITA) from media in a dose dependent manner measured at 6 h of incubation (n=3 biologically independent samples). B Activated CD8+ T cells took up itaconate at a higher rate than inactivated CD8+ and A549 cancer cell line (n=3 biologically independent samples). C Mass spectrometry analysis of activated CD8+ T cells incubated with [U-13C5]-itaconate for 6 hours revealed that the isotopically labeled species constituted nearly all the detectable itaconate in the lysates (n=3 biologically independent samples). D Effects of itaconate on the proliferation of mouse CD8+ and CD4+ T cells as measured by CFSE dilution and normalized with NaCl control (n=3 biologically independent experiments). E Effects of itaconate, itaconic acid and lactic acid on the proliferation of mouse CD8+ T cells as measured by CFSE dilution (n=12 for NaCl, n=6 for ITA acid & Lac acid, n=9 for ITA, biologically independent experiments). F Representative CFSE intensity histograms from flow cytometry analysis for (E). G Effects of itaconate on CD8+ T to killing of MC38-OVA cells. OT-1 mouse splenic CD8+ T cell were treated with 5 mM NaCl (Ctrl) or Itaconate for 5 days in the presence of OVA peptide and co-cultured with MC38-OVA cells at indicated effector cells : target cells (E:T) ratios for 24 hours, followed by measurement of cell viability of MC38-OVA cells. Data were normalized with no T cell co-culture in each group.(n=3 biologically independent experiments). H MC38-OVA cells co-cultured with itaconate-pretreated CD8+ T cell showed increased viability in an itaconate dose dependent manner. (n=3 biologically independent experiments) I-K Relative median fluorescence intensity (MFI) expression of CD69 (I), IFNγ, TNFα, IL-2, (J), Granzyme B and Perforin (K) in mouse CD8+ T cells treated with 5 mM NaCl (Ctrl) or Itaconate for 3 days in the presence of anti-CD3/CD28, as analyzed by flow cytometry (n = 6 biologically independent experiments). For (B) panels, adjusted p value was calculated using two-sided Tukey’s multiple comparisons test. For other panels, p values were calculated by paired two- tailed t-tests. Data are represented as mean ± SEM.
Itaconate suppresses CD8+ T cell proliferation and activation
We next examined the effects of exogenous itaconate on T cell proliferation. Mouse splenic CD8+ T cells that were activated by CD3/CD28 cross-linking in the presence of increasing concentrations of itaconate exhibited decreased proliferation over a 3-day period in a concentration dependent manner (Figure 2D). In all experiments, equal-molar NaCl was used as a control. In contrast, proliferation of similarly stimulated CD4+ T cells was significantly less sensitive to this effect of itaconate (Figure 2D), despite uptake rates of itaconate by CD4+ and CD8+ T cells appear to be similar (Extended Data Figure 3A). In addition, itaconate treatment reduced the viability of CD8+ T cells (Extended Data Figure 3B) but exhibited no detectable effects on A375 and BP01 melanoma cell (Extended Data Figures 3C and 3D). Notably, the inhibitory effects of itaconate (pH adjusted to 7.4) on CD8+ T cell proliferation was stronger than that of lactic acid, although weaker than itaconic acid (without pH adjustment) (Figures 2E and 2F), suggesting acidity may have an additional effect on inhibiting T cell function. In addition to asking whether itaconate affected the proliferation of CD8+ T cells during CD3/CD28-mediated activation, we also examined whether itaconate affected proliferation in previously activated splenic CD8+ T cells and found that indeed itaconate also attenuated the proliferation of these cells (Extended Data Figure 3E). Similarly, itaconate also inhibited the proliferation of CD3/CD28-activated human CD8+ T cells isolated from PBMCs(Extended Data Figures 3F and 3G).
We next investigated the effects of itaconate on the antigen-directed killing activity of CD8+ T cells against tumor cells. We found that itaconate treatment inhibited the ability of OT-1 transgenic CD8+ T of killing MC38 mouse colon cancer cell expressing ovalbumin (OVA) in a concentration dependent manner (Figures 2G and 2H). In addition, we monitored the expression of various functional markers by flow cytometry to examine the effects of itaconate on activation and function of mouse CD8+ T cells. We found that CD8+ T cells activated with CD3/CD28 antibodies in the presence of itaconate exhibited lower surface expression of the activation marker CD69 than those incubated with control NaCl (Figure 2I, Extended Data Figures 4A and 4D). Intracellular staining and flow cytometry analyses showed that itaconate also significantly reduced the production of cytokines including IL2, TNFα, and IFNγ in CD3/CD28-activated CD8+ T cells (Figure 2J, Extended Data Figures 4B, 4G, 4H and 4I). In addition, itaconate reduced granzyme B and perforin levels (Figure 2K, Extended Data Figures 4C, 4E and 4F).
We next examined the effects of itaconate on JAK-STAT signaling, a key pathway for T cell proliferation and activation, and found itaconate decreased the levels of phospho-STAT3, but not phospho-STAT1 or phospho-JAK1/2, in CD8+ T cells (Extended Data Figures 3H and 3I). Gene set enrichment analysis (GESA) of RNA-seq data obtained from splenic CD8+ T cells also revealed that IFNγ and IFNα response pathways were among the most negatively enriched pathways following itaconate exposure (Extended Data Figures 3J and 3K). In addition, several metabolic pathways were among the top pathways positively enriched for in CD8+ T cell treated with itaconate, including those related to reactive oxygen species pathway, fatty acid metabolism and oxidative phosphorylation (Extended Data Figure 3K). Since itaconate was recently shown to inhibit TET2 DNA dioxygenases leading to suppression of inflammatory gene expression programs in macrophages, we therefore examined the effects of itaconate on 5-hydroxymethylcytosine (5hmC) in CD8+ T cells, which is produced by TET enzyme mediated oxidation of 5-methylcytosine. Indeed, we found that treatment of CD8+ T cells with itaconate significantly reduced the levels of 5hmC, as measured by flow cytometry (Extended Data Figures 3M and 3N). Whether TET2 inhibition by itaconate alters gene expression in T cells and contributes to its effects on proliferation and effector function requires further investigation. Together, these data show that itaconate can exert suppressive effects on the cytotoxic activity and effector functions of CD8+ T cells.
Itaconate impedes aspartate and serine/glycine biosynthesis in CD8+ T cells
Next, we directly examined the effects of exogenous itaconate on CD8+ T cell metabolism by LC/MS-based metabolite profiling analysis. Steady state profiling studies revealed that itaconate treatment led to decreased fumarate and increased succinate levels in T cells (Figure 3A). This is consistent with the inhibition of complex II succinate dehydrogenase by itaconate, as previously reported24,25. Itaconate also caused significant reductions in the levels of several other TCA intermediates and derivatives, including malate, citrate/isocitrate, aconitic acid, asparagine, and aspartate, as well as several glycolytic intermediates (Figure 3A, Extended Data Figure 5A). Since aspartate is a critical amino acid precursor for nucleotide synthesis and a limiting nutrient for cell proliferation18, we further examined the effects of itaconate on the flux from glucose and glutamine to aspartate using stable isotope tracing analyses. As shown in Figure 3B and Extended Data Figure 5C, itaconate treatment of activated CD8+ T cells resulted in a significant decrease in incorporation of carbon from [U-13C]glutamine, but not from [U-13C]glucose, to aspartate.
Figure 3. Itaconate impedes aspartate and serine/glycine biosynthesis in CD8+ T cells.
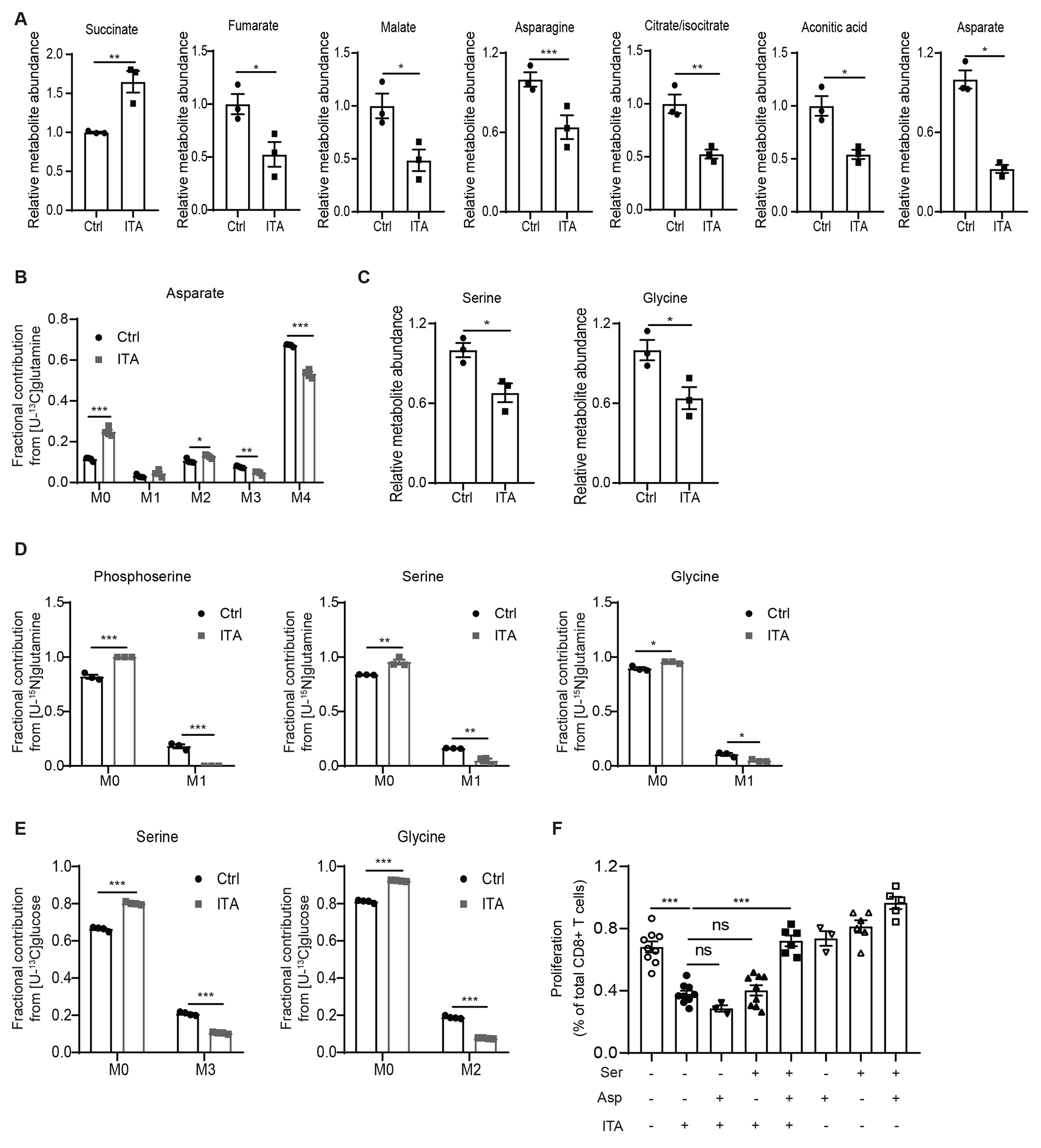
A Levels of various TCA related metabolites in CD8+ T cells exposed to exogenous itaconate (ITA, 5 mM) or NaCl (Ctrl, 5 mM), as revealed by metabolite profiling analyses (n=3 biologically independent samples). B Incorporation of 13C to Aspartate from [U-13C]-glutamine in CD8+ T cells exposed to exogenous itaconate or NaCl (Ctrl) (n=4 biologically independent samples). C Levels of serine and glycine in CD8+ T cells exposed to exogenous itaconate (ITA) or NaCl (Ctrl), as revealed by metabolite profiling analyses (n=3 biologically independent samples). D Incorporation of 15N into phosphoserine, serine, and glycine from [U-15N]-glutamine in CD8+ T cells exposed to exogenous itaconate or NaCl (Ctrl) (n=3 biologically independent samples). E Incorporation of 13C into serine and glycine from [U-13C]-glucose in CD8+ T cells exposed to exogenous itaconate or NaCl (Ctrl) (n=4 biologically independent samples). F Effects of aspartate (Asp) and serine (Ser) supplementation on the proliferation of CD8+ T cells treated with exogenous itaconate (3 mM), as measured by CFSE dilution (at least three independent experiments for each group). For all panels, p values were calculated by paired two- tailed t-tests. Data are represented as mean ± SEM.
Moreover, steady-state metabolite profiling experiments also revealed itaconate-induced decreases in the levels of serine and glycine in CD8+ T cells (Figure 3C), which have been demonstrated to be critical for T cell proliferation and activation, probably through nucleotide synthesis19,20. [U-15N]glutamine tracing analysis revealed that 15N-labeling of phosphoserine, serine and glycine were markedly reduced in CD8+ T cells upon itaconate treatment (Figure 3D). [U-13C]glucose tracing experiments also revealed a similar reduction in 13C labelled serine and glycine (Figure 3E). These findings support that itaconate inhibits serine/glycine biosynthesis flux.
Aspartate, serine, and glycine are important precursors for de novo nucleotide synthesis and hence cell proliferation21. Consistent with this notion, we found that itaconate also caused significant reductions in the levels of various nucleotides (Extended Data Figure 5B). We further investigated whether aspartate and/or serine/glycine metabolism are critical for the inhibitory effects of itaconate on CD8+ T cell proliferation by metabolite supplementation. As shown in Figure 3F, itaconate reduced the proliferation of CD8+ T cells in serine- and glycine- free medium, as measured in CFSE-based proliferation assays. Interestingly, supplementation of aspartate alone or serine/glycine in the medium was not sufficient to reverse the inhibitory effects of itaconate. However, supplementation of aspartate, serine, and glycine in combination restored CD8+ T cells proliferation (Figure 3F). Furthermore, nucleosides supplementation also supported CD8+ T cells proliferation (Extended Data Figures 5D and 5E), consistent with itaconate working on CD8+ T cells in part by depleting the nucleotide precursors serine, glycine, and aspartate.
Loss of Irg1 enhances anti-tumor immunity of CD8+ T cells
To investigate the effects of itaconate on anti-tumor immunity, we examined whether loss of Irg1 in the host impacted tumor growth in mice. Subcutaneous D4M3A syngeneic melanoma tumors were grown in either C57BL/6NJ wildtype control or Irg1 (a.k.a. Acod1)-deficient mice22. We observed that tumor growth rates were lower in Irg1-deficient mice relative to those in the control mice (Figure 4A, Extended Data Figures 6A, 6B and 6C). LC-MS analyses confirmed that the levels of itaconate, but not of lactate, were greatly reduced in mouse tumor tissues and interstitial fluid from Irg1-deficient mice relative to those grown in the WT mice demonstrating that its source was likely host cells within the tumor microenvironment (Extended Data Figures 6D and 6E). Similar effects of host Irg1 deletion on tumor growth was observed in mice bearing MC38 tumors (Extended Data Figure 6I). We also carried out flow cytometry to characterize the effects of Irg1 knockout on immune populations within the tumor microenvironment. This revealed a reduction in the levels of both PMN-MDSCs (CD11b+Ly6G+Ly6Cint) and M-MDSCs (CD11b+Ly6GloLy6Chi) in the D4M3A tumors grown in Irg1 knockout mice (Figure 4B and Extended Data Figure 6F). In addition, D4M3A tumors from Irg1 knockout mice also possessed more CD8+, but not CD4+, T cells when compared to tumors from WT mice, and also exhibited higher proportions of CD8+ T cells expressing IFNγ or granzyme B (Figures 4C, 4D, and Extended Data Figure 6F). Importantly, we found that depletion of CD8+ cells restored D4M3A tumor growth in Irg1 KO mice to a level similar to the WT mice (Extended Data Figure 7A). These observations are consistent with our in vitro observations of itaconate’s effects on CD8+ T cell proliferation and activation (Figure 2).
Figure 4. Loss of Irg1 enhances anti-tumor immunity of CD8+ T cells and the efficacy anti-PD-1 immune checkpoint blockade.
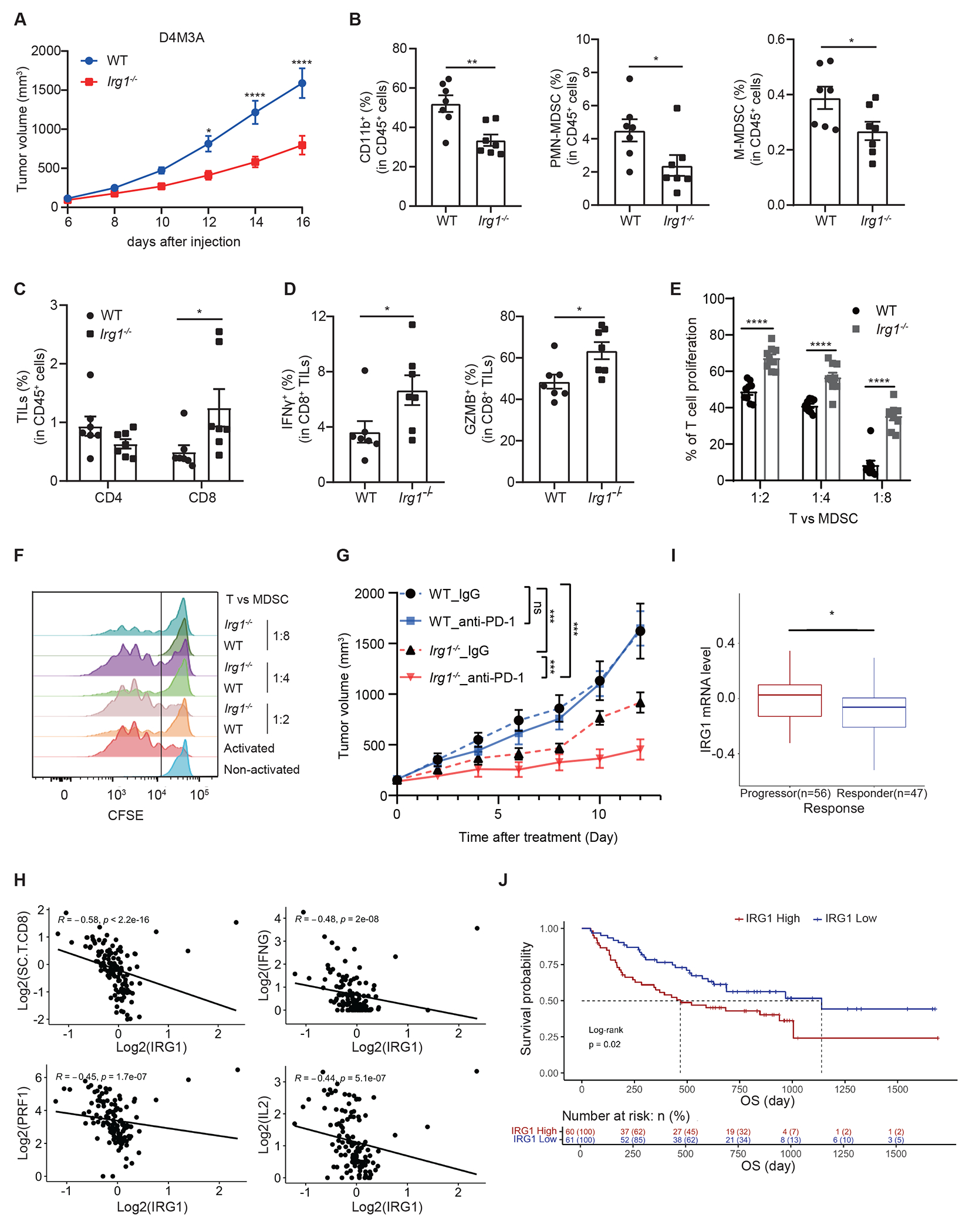
A Syngeneic D4M3A tumors grown in Irg1−/− mice exhibited significantly slower grow compared to those in WT mice (n=7 mice for each group). B FACS analysis revealed that tumors from Irg1−/− mice possessed lower percentages of MDSCs and macrophages (n=7 mice). C Analysis of tumor infiltrating T cells revealed similar representation of CD4+ cells but a significant increase in CD8+ cells in tumors from Irg1−/− mice (n=7 mice). D Tumor infiltrating CD8+ T cells from Irg1−/− mice showed increases in both IFNγ and Granzyme B expression (n=7 mice). E, CFSE-labeled CD8+ T cells were co-cultured at different ratios with splenic MDSCs sorted from WT or Irg1−/− D4M3A tumor-bearing mice. T cell proliferation was monitored 72 hours after co-culture by flow cytometry (n=3 biologically independent experiments). F Representative flow histograms are shown from experiments in (E). G Effects of anti-PD-1 and IgG control treatments on the growth of D4M3A tumors in WT or Irg1−/− mice (n=7 for Irg1−/− IgG group, n=6 mice for the other three groups). H An adjusted IRG1 expression score negatively correlated with Perforin (PRF1)(Spearman, R = −0.45, P = 1.7e-07), IFNG(R= −0.48, P = 2e-08), IL2(R = −0.44, P = 5.1e-07) and CD8+ T cells immune scores(R = −0.58, P < 2.2e-16). Spearman correlation was performed in all cases above. I Adjusted IRG1 expression level in progressors (n = 56 patients) is higher than in responders (n = 47 patients) (two-sided Mann-Whitney test, P=0.023). Data are presented with median values shown in boxplots, where the lower and upper hinges correspond to 25th and 75th percentiles of the data. The lower and upper whisker range from the first quartile – 1.5*IQR to the third quartile + 1.5*IQR. For visualization purposes, outliers (defined as smaller than first quartiles - 1.5*IQR or larger than third quartile + 1.5*IQR) were not shown in the plot, but all data were included in statistical testing. Mann-Whitney test was performed to compare IRG1 expression level between progressors and responders, without multiple comparisons adjustments. The 95% confidence interval is [0.01, 0.14], effect size is 0.224, and p value is 0.0233. J Kaplan Myer analysis of metastatic melanoma patients treated with anti-PD-1 therapy stratified by low vs high expression score of IRG1 revealed significant differences in survival with median OS of 1139 and 468 days, respectively. Overall survival (OS) outcomes stratified by high versus low IRG1 mRNA expression score (split by the median) in a cohort of metastatic melanoma patients treated with anti-PD-1 therapy (n= 121). Tumors with high IRG1 score associated with worse OS (two-sided Kaplan Meier log-rank test, P = 0.02). Adjusted p values were calculated using two-sided Sidak’s multiple comparisons test (A, G), or unpaired two-tailed student’s test (B-E). *p<0.05.. Data are represented as mean ± SEM (A, B-E, G).
To further determine whether IRG1 contributes to the suppressive activity of MDSCs toward CD8+ T cells, we conducted T cell suppression assays using splenic MDSCs isolated from D4M3A-tumor bearing Irg1-deficient or wildtype mice. The capacities of the isolated MDSCs to suppress proliferation of CD3/CD28-activated CD8+ T cells was measured by CFSE dilution-based assays. At T cell/MDSC ratios of 1:2, 1:4 and 1:8, MDSCs from Irg1-deficient mice did not secrete measurable amount of itaconate and exhibited significantly reduced suppressive activity towards T cell proliferation, compared to those from WT animals (Figures 4E, 4F, Extended Data Figures 6G and 6H). Together, these results suggest that inactivation of host Irg1 results in loss of immune suppressive activities of MDSCs toward CD8+ T cells, and thus promotes anti-tumor immunity of CD8+ T cells and impairs syngeneic tumor growth in mice.
Loss of Irg1 enhances anti-PD-1 immune checkpoint blockade
Given the apparent effects of Irg1 loss on anti-tumor immunity of CD8+ T cells, we next examined whether inactivation of Irg1 would influence the sensitivity of tumors to anti-PD-1 immune checkpoint blockade. D4M3A tumor bearing Irg1-deficient, or wildtype control C57BL/6NJ mice were treated with either anti-PD-1 IgG or isotype control IgG. Similar to previously reported data23, anti-PD-1 treatment did not significantly reduced D4M3A tumor growth in WT animals (Figure 4G). However, treatment with anti-PD-1 decreased tumor growth in the Irg1-deficient mice (Figure 4G, Extended Data Figures 7B–F). Therefore, host deletion of Irg1 enhanced the sensitivity of D4M3A tumors to anti-PD-1, strongly supporting a role for Irg1 and itaconate in suppressing anti-tumor immunity.
To explore the clinical relevance of our findings, we examined the potential association of an excess IRG1 expression score (Methods) with CD8+ T cell related gene expression signatures in a large cohort of metastatic melanoma patients that were treated with PD-1 immune checkpoint blockade24. Consistent with the inhibitory effects of itaconate on murine CD8+ T cells ex vivo (Figure 2J and 2K), we observed a significant negative correlation between the IRG1 expression score and IFNγ, IL2, and PRF1 (Perforin) expression in these patient tumors (Figure 4H). More importantly, we found that a lower IRG1 mRNA expression score (median split) was associated with increased response to anti-PD-1 therapy (Wilcox test, p = 0.023) (Figure 4I) and longer overall survival (OS) (median survival days of 1139 vs. 468 days in the IRG1- low group and high group, respectively; two-sided KM log-rank test, p = 0.02) (Figure 4J). This is consistent with the hypothesis that tumors with lower IRG1 levels may be more sensitive to blockade of the PD-L1/PD-1 immune checkpoint pathway.
Discussion
Our findings reveal the potent immunosuppressive properties of MDSC-secreted itaconate and present evidence that it acts as an important immune checkpoint metabolite by mediating intra-tumoral immune suppression. MDSCs use different modalities to suppress anti-tumor immunity including production of ROS, NOS, and adenosine production6. They also promote immune evasion through the manipulation of the metabolic environment within the tumor through nutrient depletion and the removal of certain amino acids necessary for T cell effector function such as arginine via ARG1 expression25 and tryptophan via IDO expression26,27. Considering the highly interconnected nature of metabolic network, it is conceivable that IRG1 could be functionally linked to these other metabolic pathways in MDSCs. However, unlike these other metabolic regulators, IRG1 was selectively expressed in MDSCs and other myeloid cells within the TME and has not been detected in T or NK lymphocytes or cancer cells. IRG1 was previously reported to be expressed in peritoneal residential macrophages from mice harboring ID8 ovarian carcinoma model and in CD14+ monocytes from ascites of human ovarian carcinoma patients28. Similarly, we found that IRG1 is highly expressed in LOX1 positive PMN-MDSCs in human melanoma tumors (Figure 1E). Analysis of publicly available scRNAseq datasets from mouse breast cancer (Figure 1F), melanoma, glioma and small cell lung cancer (Extended Data Figures 2B–2G) revealed a restriction of detectable IRG1 expression to the myeloid compartment, with highest levels in granulocytic lineages, consistent with PMN-MDSCs. It should be noted that the number of Irg1 expressing cells could be under-estimated in scRNAseq analyses due to low sequencing depth and likely drop out and due to the sensitivity of PMN-MDSCs to degradation during sample collection. Irg1 was originally cloned based on its upregulation in response to LPS treatment in in RAW264.7 cells. We found that Irg1 expression levels in MDSCs and RAW264.7 cells can be strongly increased by tumor-conditioned media, suggesting induction by secreted factors from cancer cells. The soluble factors and underlying molecular mechanism by which tumor cells might induce IRG1 in MDSCs require further investigation, which represents a limitation of our current study.
It has been well established that itaconate exerts anti-inflammatory effects in macrophages and plays critical roles in innate immune responses29,10,30. In this study, we revealed a previously unknown role for itaconate in suppressing the proliferation, activation and cytotoxic effector function of CD8+ T cells. However, it remains unclear how itaconate enters these cells, which is a limitation of our study. Previously, itaconate was shown to be taken up by A549 lung cancer cells17 and adipocytes31. The apparently faster rate of accumulation in activated vs. naive CD8+ cells that we observed (Figure 2B) raises the possibility of the upregulation of a specific transporter upon TCR stimulation in CD8+ T cells. The anti-proliferative effects were significant in CD8+ T cells, with a smaller effect on CD4+ T cells and no measured effect on cancer cells. The rates of itaconate uptake by CD8+ and CD4+ T cells appear to be similar (Extended Data Figure 3A), but the gene expression pathways that were impacted by itaconate in CD8+ and CD4+ T cells are quite different (Extended Data Figures 3K and 3L), suggesting that these differential effects of itaconate could be due to downstream intracellular effectors of itaconate action among these different types of cells, which need to be elucidated in the future. The concentrations of itaconate (3-5 mM) we used in our in vitro experiments to examine its effects on CD8+ T cells are comparable to the intracellular concentration of itaconate previously reported in RAW264.7 cells (up to 8 mM)13,15 and are in line with what has been used in the literature for macrophages29–33. Once inside of the cell, itaconate has been shown to inhibit the mitochondrial complex II succinate dehydrogenase in vitro and in macrophages29,31. In addition, itaconate was shown to activate the transcription factors NRF232 and ATF333, and inhibit the activation of inflammasome34,35 and JAK136 in macrophages. In this study, we found that itaconate suppresses STAT3 activation in CD8+ T cells (Extended Data Figures 3H and 3I). However, its connection to itaconate-induced changes in amino acid metabolism remains unclear. Our metabolite supplementation experiments suggest that both serine/glycine and aspartate metabolism pathways are important for the effects of itaconate on the proliferation of CD8+ T cells (Figure 3F), while only aspartate metabolism, but not serine/glycine metabolism, is critical for the effector function of CD8+ T cells in killing tumor cells (Extended Data Figure 5F). Future efforts will be devoted to further characterizing the molecular and metabolic mechanisms underlying the suppressive activities of itaconate on CD8+ T cells.
Our current study establishes IRG1 as a promising immunometabolism target of immune modulatory drugs in MDSCs. Treatment with Irg1 shRNA was previously found to reduce tumor burden in the peritoneum of ID8 and B16 tumor-bearing mice28. Findings from our IRG1 systemic knockout mice confirmed the anti-tumor effects of Irg1 inactivation and further demonstrated that host deletion of Irg1 increased the sensitive of mouse tumors to treatment of anti-PD-1 immune checkpoint blockade. Regarding the identity of the specific cell type(s) of origin of itaconate within the tumors, while the use of a systematic knockout of Irg1 is a weakness in our study, this is ameliorated by the selective expression pattern of Irg1 in MDSCs and potentially other myeloid cells in the tumor microenvironment, but not in T or NK lymphocytes. On the other hand, from a translational perspective the use of systemic knockout of Irg1 may possess advantages over a cell-type specific knockout (i.e., a myeloid specific knockout), since the systemic knockout more closely mimics the action of small molecule inhibitors of IRG1. The unique selective expression pattern of IRG1 would potentially provide a good therapeutic window with low adverse effects for drugs targeting IRG1. Moreover, our findings suggest that the expression level of IRG1 may potentially serve as either a prognostic and/or therapeutic biomarker, which would facilitate future development of IRG1 inhibitors.
Methods
Mouse models
All animal experiments were conducted in accordance with the Institutional Animal Care and Use Committee guidelines at the Massachusetts General Hospital. For allograft models, 8-week-old female C57BL/6NJ-Acod1em1(IMPC)J/J mice (stock no. 029340) (also referred as Irg1−/−) and wild-type control C57BL/6NJ (stock no. 005304) were purchased from Jackson Laboratory (Bar Harbor, Maine). Wild-type C57BL/6 (Female, 6~8 weeks old) were purchased from Taconic Biosciences, Inc (Germantown, NY). All mice were bred and housed under a specific-pathogen-free conditions with 12-h dark/light cycle under 21±1°C temperature and 55–60% humidity and provided ad libitum access to water and food. Animals were injected subcutaneously in the right lateral flank with either D4M3A or MC38 mouse cells (0.5 million cells per mouse). For CD8 T+ cell depletion experiments, 750 μg IgG2b Isotype control (LTF-2) (BioXcell, Cat. #BE0090) or anti-CD8a antibodies (YTS169.4) (BioXCell, Cat. #BE0117) was injected intraperitoneally 3 days prior to tumor cell injection. After that, mice continued to receive 250 μg IgG2b Isotype or anti-CD8a antibodies treatment once weekly. For PD-1 treatment experiment, once tumor sizes reached 120–180 mm3, animals were randomly assigned to the two groups that were administered 100μg of anti-PD-1 or IgG isotype control via intraperitoneal injection every other day for 10 days. Tumor dimensions were calculated from caliper measurements by using the following formula: (D × d2)/2, where D represents the large diameter and d the small diameter of the tumor. Mouse was euthanized when tumor volume reached > 2cm3.
Cell lines and culture conditions
A375, BP0138, D4M3A39, MC38, MC38-OVA and RAW264.7 were cultured in RPMI 1640 (Gibco, Cat. # 21870092) supplemented with 10% FBS, 1% penicillin/streptomycin (Gibco, Cat. # 15140122), 1% glutamine (Life Technologies, Cat. # 25030081). All cells were tested for mycoplasma contamination routinely. Cells were maintained in a humidified, 5% CO2 atmosphere at 37 °C.
Human patient samples
Patients with metastatic melanoma at MGH provided written informed consent for the collection of tissue and blood samples for research and genomic profiling (DF/HCC IRB approved protocol 11-181). Tissue samples were formalin fixed (10% formalin for 48 hours) and embedded in paraffin prior to immunofluorescence. Whole blood was collected in BD Vacutainer CPT tubes (BD362753). Plasma was isolated per manufacturer’s SOP and stored at −80 ºC. Human plasma samples from healthy donors were obtained from Innovative Research.
Bone marrow differentiation into MDSC
Bone marrow (BM) cells were obtained from the femurs and tibias of naive C57/BL6 mice. Two million BM cells per well were cultured in RPMI 1640 medium supplemented with 10% FBS, 5 mM glutamine, 25 mM HEPES, 10 ng/ml GM-CSF, 10 ng/ml IL-6, 10 ng/ml G-CSF and 50 μM 2-mercaptoethanol, with D4M3A melanoma tumor cell conditioned medium (TCCM) in 6 well plates. The cultures were maintained at 37°C in 5% CO2-humidified atmosphere for 72 hours. For TCCM preparation, 800k of D4M3A cells were seeded in 10-cm dish with RPMI 1640 medium containing 3% FBS. After 48 h of culture, the supernatant was collected and centrifuged (500 g, 10 min) to remove cells debris. The supernatant was then filtered through 0.22 μm filter to obtain TCCM.
PMN-MDSC purification
PMN-MDSCs were purified from Bone marrow differentiated MDSC as described in the manual of Myeloid-Derived Suppressor Cell Isolation Kit from Miltenyi (Cat #. 130-094-538). In brief, MDSC suspension was first incubated with an Anti-Ly-6G-Biotin antibody and Anti-Biotin MicroBeads, after Fc block was applied. The cells are subsequently applied to a LS Column (Miltenyi, Cat #. 130-042-401), which retained the magnetically labeled Gr-1highLy-6G+ cells while the unlabeled cells flow through. The magnetically labeled Gr-1highLy-6G+ cells were eluted as the positively selected cell fraction and further purified by applying them to a second LS Column. The population obtained after two positive selection steps was considered as PMN-MDSC.
Measurements of itaconate secretion
Purified PMN-MDSCs were cultured in MDSC medium for 24 h. Supernatants were collected for measuring extracellular levels of various metabolites, including itaconate, by LC-MS. To measure the level of itaconate secretion by MDSCs, WT and Irg1−/− mouse bone marrow cells were differentiated into MDSCs by TCCM for 3 days, replated in RPMI medium for 3 days, following by collection of supernatant for LC-MS. Cell debris was removed from the supernatants by centrifugation of 500 g, 10 min, followed by filtering the supernatants through 0.45 μm filter. Blank medium was saved prior to the initiation of culture as a control. RAW264.7 macrophages were cultured in RPMI:TCCM (7:3) for 48 h, and supernatants were collected for LC-MS analysis in a similar fashion. The secretion rate of metabolite M was calculated as follows: Secretion rate was calculated as (Level of metabolite in cultured medium - Level of metabolite in blank medium)/culture time length/total cells number in end sample.
Western blot analysis
Western blotting was performed as previously described (Zheng et al., 2009) Briefly, cell lysates were prepared using lysis buffer containing HEPES (pH 7.0), 150 mM NaCl, 1% NP-40, 1 mM EDTA, 50 mM NaF, 10 mM β-glycerophosphate, 10 nM calyculin A, 1 mM Na3VO4, and protease inhibitors and normalized by protein concentrations using the Bradford method (Bio-Rad). Protein samples were separated on 8%–12% SDS-PAGE and transferred to PVDF membrane (Millipore). Membranes were blocked in TBST containing 5% nonfat milk, incubated with primary antibodies according to the antibody manufacturer’s instructions, followed by incubation with horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse IgG (Chemicon) and enhanced chemiluminescence detection (Perkin Elmer). The antibodies are listed in Supplementary Table 1.
Real-time PCR
PMN-MDSC cells were prepared from D4M3A tumors. Tumors were shredded into small pieces and incubated for 45 minutes at 37°C in collagenase-containing buffer containing 100 U/ml of collagenase type IV (Worthington, Lakewood, NJ), 50 μg/ml of DNase I (Worthington), and 10% fetal bovine serum in RPMI-1640 medium (Thermo Fisher Scientific, Cambridge, MA). After incubation, cells were treated with ACK lysing buffer and passed through a 70-μm cell strainer to remove debris. Single-cell suspensions PMN cells were prepared from spleens of tumor-free mice by mechanical dissociation, followed by removal of erythrocytes with ACK lysing buffer (Lonza, Walkersville, MD). Cells were labeled with CD11b and Ly6G antibodies and then purified by SH800 cell sorter (Sony).
1x106 flow cytometry-sorted CD11b+ Ly6G+ cells were lysed in 350μl RLT lysis buffer (Qiagen). Total RNA was extracted using the RNeasy Mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol, and cDNAs were prepared using the RevertAid Reverse Transcription Kit (Thermo Fisher Scientific, Cambridge, MA). Quantitative PCR was performed using the SYBR Green I Master (Roche, Basel, Switzerland) reaction mix on a Light Cycler 480 (Roche). Each sample was tested in triplicates, and results were normalized to the expression of m36B4 as an internal control (Forward primer: TCACTGTGCCAGCTCAGAAC; Reverse primer: AATTTCAATGGTGCCTCTGG. Relative gene expression was calculated by the 2ΔΔCt method. Mouse IRG1 Forward primer: AGTTTTCTGGCCTCGACCTG;Reverse primer: AGAGGGAGGGTGGAATCTCT.
Immunofluorescence analysis
For co-immunofluorescent staining, deparaffinized and rehydrated slides from 12 human melanoma tumor specimens were subjected to antigen retrieval via autoclaving in a 10 mM citric acid buffer (pH 6.0). Alexa Fluor 488 Tyramide SuperBoost Kit (Invitrogen) was used for LOX1, CD163, CD3 or B220 staining according to manufacturer’s protocols, then Alexa Fluor® Plus 594 (Invitrogen) secondary antibodies were used for IRG1. Cell nuclei were counterstained with ProLong™ Diamond Antifade Mountant with DAPI (Invitrogen). Images were acquired using Olympus BX53F microscope, Olympus DP80 digital camera, and CellSens Standard software. The positive rates of LOX1+IRG1− and LOX1+IRG1+ samples were calculated. The antibodies are listed in Supplementary Table 1.
T cells isolation
Mouse CD8+T cells were isolated from spleen of naïve C57/BL6 mice with CD8a (Ly-2) MicroBeads, mouse (Miltenyi Biotec, Cat #. 130-117-044). In brief, a single cell suspension was generated by forcing spleens through 70μm filters, followed with incubation with ACK RBC lysis buffer and washing with PBS to remove red blood cells. Then cell suspensions were incubated with anti-CD8a microbeads and sorted using MS columns (Miltenyi Biotec, Cat #. 130-042-201). The magnetically labeled CD8+ cells were eluted as the positively selected cell fraction. Mouse CD4+T cells were isolated in the same fashion using the CD4 (L3T4) MicroBeads, mouse (Miltenyi Biotec, Cat #. 130-117-043).
Human CD8+ T cells were isolated from human PBMCs (Zen-Bio, Inc, Cat # SER-PBMC-P-F) with Naive CD8+ T Cell Isolation Kit, human (Miltenyi Biotec, Cat # 130-093-244). The isolation was performed in a two-step procedure. First, naive T cells were enriched by depletion of non-naive T cells and NK cells, which were indirectly magnetically labeled with a cocktail of biotin-conjugated antibodies and Anti-Biotin MicroBeads. In the second step, the enriched naive T cells were labeled with anti-CD8 MicroBeads for subsequent positive selection of the CD8+ naive T cells.
Itaconate uptake and tracing studies in T cells
To measure the levels of itaconate uptake by activated CD8+ and CD4+ T cells, splenic naive CD8+ or CD4+ T cells from C57/BL6 mice were cultured in RPMI medium with 20 ng/ml IL-2, 10 μg/ml anti-CD3ε, 0.5 μg/ml anti-CD28 for three days. Cells were then reseeded in RPMI medium containing different concentrations of itaconate as indicated (Alfa Aesar, Cat #. AAA1556630) for 6 hours. To measure the rates of itaconate uptake by naïve CD8+ T cells, activated CD8+ T cells, activated CD4+ T cells and A549 cells, cells were cultured in RPMI medium containing 5 mM itaconate for indicated times. Cells were collected, washed with cold PBS twice, and resuspended in the extraction buffer (methanol: acetonitrile: H2O=40:40:20) for LC-MS measurements. Itaconate solution was prepared by dissolving the itaconic acid powder into RPMI medium, followed by pH adjustment to 7.4 with NaOH. All solutions were filtered through 0.22μm filters. To measure the level of itaconate uptake by activated CD8+ T cells from MDSCs cultured medium, activated CD8+ T cells were co-cultured with BM-MDSCs supernatant for indicated time. Cells were collected, washed with cold PBS twice, and resuspended in the metabolite extraction buffer for LC-MS measurements.
For [U-13C]-itaconate tracing, CD8+ T cells activated by anti-CD3e/anti-CD28 were reseeded in RPMI medium containing 2.5 mM of [U-13C]-itaconate (Santa Cruz Biotechnology, Inc, Cat #. SC-495554) and cultured for 6 h before subjected for metabolite extraction of cell pellets. For glucose and glutamine tracing, CD8+ T cells were reseeded in RPMI medium containing isotype labeled metabolites (11 mM of [U13C]-glucose, 2 mM of [U13C]-glutamine, or 2 mM of [U15N]-glutamine) for an additional 2 h of culture before collected for LC-MS analysis.
T cell CFSE Proliferation and viability assays
CD8+ T cells were isolated from spleen of naïve C57/BL6 mice and incubated with 5 μmol CFSE dye (Invitrogen, Cat #. C34554) in pre-warmed 37°C PBS for 15min, protected from light. Five times of the staining volume of RPMI medium was added for 5 minutes incubation at 37°C to stop the staining process. Cells were centrifuged (300 g, 5 min) and resuspended in pre-warmed RPMI-1640 medium containing 20 ng/ml IL-2, 50 μM 2-mercaptoethanol and 0.5 μg/ml anti-CD28, with indicated concentrations of NaCl, itaconate itacononic acid, lactic acid (Sigma, cat no. L6402-1G) added prior to the initiation of culture. Cell density was adjusted to 0.5 million/ml for culturing in 96-well U-bottom plate coated with anti-CD3e for up to 72 hours before flow analysis.
For the CFSE assays to test the effects of serine and aspartate supplementation on T cell proliferation, CD8+ T cells were cultured in serine/glycine-free RPMI medium, supplemented with or without serine (0.3 mM) serine and/or aspartate (7.5 mM) as indicated, and treated with 3 mM itaconate or NaCl dissolved in serine/glycine depleted RPMI medium. Dialyzed FBS (Gibco, Cat #. 26400044) was used to replace regular FBS in this experiment.
For nucleosides supplementation, CD8+ T cells were cultured in RPMI medium with dialyzed FBS, supplemented with or without various nucleosides (Adenosine, Cytidine, Uridine, Guanosine, Thymidine and Inosine, 100μM for each) as indicated and treated with 3 mM itaconate or NaCl.
For cell viability assays, anti-CD28/anti-CD3 activated T cells were reseeded in a density of 0.5 million/ml in the presence of 5 mM itaconate or NaCl for indicated time length. At the end of each timepoint, cells were stained with trypan blue and live cells were counted or stained with Propidium Iodide (PI) and dead cells were measured by Flow Cytometry.
T cell suppression assay
CD8+ T cells were isolated from spleen of naïve C57/BL6 mice and incubated with 5 μmol CFSE dye in pre-warmed 37°C PBS for 15 minutes Cells were plated in triplicates into U-bottom 96-well plates (2x105/well) coated with 10 μg/ml anti-CD3 and 0.5 μg/ml anti-CD28 antibody. Gr-1+ MDSCs were purified using Myeloid-Derived Suppressor Cell Isolation Kit (Miltenyi Biotec, Cat # 130-094-538) from spleens of D4M3A tumor-bearing C57BL/6NJ or Irg1−/− mice. CD8+ T cells were incubated with Gr-1+ cells at 37°C for 72h. On day 3, cells were stained with anti-CD8 antibody and analyzed for CFSE dilution by flow cytometry to measure proliferation of CD8+ T cells.
In Vitro cytotoxicity assay
OT-1 CD8+ T cells were activated by the OVA peptide (1 ng/ml) and treated with NaCl or various concentrations of itaconate for 5 days. MC38-OVA-zsGreen cells (2 x 104) were plated into 24-well plates, cultured overnight, and then co-cultured with activated OT-1 CD8a T cells at the indicated Effector cells: Target cells (E:T) ratios. After 24 hours, nonadherent tumor cells and T cells were washed away by PBS. Tumor cell viability was assessed using flow cytometry. For the effects of serine and aspartate supplementation on T cell cytotoxicity, OT-1 CD8+ cells were cultured in serine/glycine-free RPMI medium, supplemented with or without serine/glycine (0.3 mM) or aspartate (7.5 mM) as indicated, and treated with 3 mM itaconate or NaCl for 5 days in present of OVA peptide. Dialyzed FBS (Gibco, Cat #. 26400044) was used to replace regular FBS in this experiment.
Flow cytometry analysis
All samples were washed and resuspended in FACS buffer (phosphate buffered saline with 2% fetal bovine serum). Single-cell suspensions were incubated with anti-CD16/CD32 mAb (eBioscience, San Diego, CA) to block FcγRII/III receptors for 10 minutes on ice and stained for 30 minutes with various fluorochrome-conjugated monoclonal antibodies. Cells were then washed and resuspended in 7-AAD viability staining solution (Biolegend). Dead cells and doublets were excluded by forward and side scatter. For analysis of intracellular cytokines, cells were treated with brefeldin A solution (Biolegend, Cat #. 420601) for 4 hours prior to sample collection. For staining of intracellular antigens, cells were permeabilized using the Foxp3 Fixation and Permeabilization Kit (eBioscience) before staining. Events were acquired on a Canto Flow Cytometer (Becton Dickinson, Franklin Lakes, NJ). Data were collected for 105–106 cells and analyzed with FlowJo software. The gating strategy is shown in Supplementary Figure 1. The antibodies are listed in Supplementary Table 1.
For 5hmC staining, spleen purified CD8+ T cell was pre-activated by anti-CD3/CD28 for 2 h, washed by PBS twice and cultured with NaCl or Itaconate (3 mM) medium for 24 h. All samples were washed and resuspended in FACS buffer. Single-cell suspensions were incubated with anti-CD16/CD32 mAb for 10 minutes on ice. After washing with FACS buffer, cells were fixed and permeabilized. DNA was denatured by adding 2N HCl for 20 minutes and neutralized with 10 mM Tris-HCl (pH 8.0) for 20 minutes Anti-5hmC (Active Motif, Cat. 39069) and Alexa Fluor 647 Donkey anti-Rabbit IgG (H+L) secondary antibody (Thermo Fisher Scientific) were used for 5-hmC staining. Rabbit IgG was used as an isotype control.
Cancer cell proliferation assay
A375 and BP01 melanoma cells were seeded in 96-well plate (2000 cells per well), and culture media were replaced the next day with fresh medium containing different concentrations of NaCl or itaconate. Cells were cultured for additional 72 hours before subjected to MTS assay. 20 μl of the MTS stock solution was added into each well of the 96 well plate, containing 100 μl of cells in culture medium. The plate was incubated for 2 hours at 37°C in a humidified, 5% CO2 atmosphere. At the end of incubation, the absorbance was recorded at 490 nm using a PerkinElmer plate reader.
LC-MS analysis
For medium analysis, 5 μl medium were mixed with 5 μl of internal standard, and 90 μl of 100% methanol, vortexed for 1 minute and then centrifuged at 16,000g for 30 min at 4 °C. 20 μl of supernatant were diluted with 80 μl of 80% methanol into vials for LC-MS analysis. All cell samples prepared in extraction buffer were centrifuged at 16,000g, for 30 min at 4°C, and supernatants were transferred to vials for LC-MS analysis. Frozen tumor tissue samples were pulverized via cyromill (Retsch). 10 mg of tumor tissue powder were mixed with 10 μl internal standards and 400 μl extraction buffer. Samples were vortexed for 1 min, kept in ice for 10 min and centrifuged at 16,000 g for 30 min at 4 °C. Supernatants were collected for LC-MS analysis. Serum and tumor interstitial fluid samples were prepared as the following. 3 ul of serum/fluid were mixed with 120 ul of 100% ice-cold methanol and internal standards were spiked into the mixture. Samples were vortexed for 10 seconds, kept on ice for 10 min, and centrifuged at 16,000x g for 30 min at 4 °C, and supernatants were subjected for LC-MS analysis.
Water soluble metabolite measurements were obtained by running samples on the Q Exactive PLUS hybrid quadrupole-orbitrap mass spectrometer (Thermo Scientific) coupled with hydrophilic interaction chromatography (HILIC). An XBridge BEH Amide column (150 mm × 2.1mm, 2.5 μM particle size, Waters, Milford, MA) was used. The gradient was solvent A (95%:5% H2O:acetonitrile with 20 mM ammonium acetate, 20 mM ammonium hydroxide, pH 9.4) and solvent B (100% acetonitrile) 0 min, 90% B; 2 min, 90% B; 3 min, 75%; 7 min, 75% B; 8 min, 70%, 9 min, 70% B; 10 min, 50% B; 12 min, 50% B; 13 min, 25% B; 14 min, 25% B; 16 min, 0%B, 20.5 min, 0% B; 21 min, 90% B; 25 min, 90% B. The flow rate was 150 μl/minute with an injection volume of 5 μl and a column temperature of 25°C. The MS scans were in both negative and positive ion mode with a resolution of 140,000 (negative mode) or 70,000 (positive mode) at m/z 200. The automatic gain control (AGC) target was 1 ×106 and the scan range was m/z 75–1000 (negative mode), or 75–1000 (positive mode). All data from isotope labeling experiments were analyzed by El-MAVEN with natural abundance correction.
RNA-seq
Mouse splenic CD8+ or CD4+ T cells were cultured in RPMI medium (in the presence of IL-2, anti-CD28/anti-CD3) with NaCl (5mM) or itaconate (5mM) for 48 hours. Cells pellets were collected for isolation of RNA with RNeasy mini kit from Qiagen (cat# 74104). RNA quality (RIN > 7) was confirmed using the TapeStation 4200 system (Agilent Technologies). Preparation of RNA library and transcriptome sequencing was conducted by Novogene Co., LTD (Sacramento, CA). 250~300 bp insert cDNA library was constructed according to the manufacturer’s instructions ((Illumina Platform, PE150 Q30≥80%)). Pair-end sequencing with reads of 150 bp reads was performed on an Illumina HiSeq 2000. Sequencing reads were mapped to the UCSC mm10 reference genome using STAR version 2.7.0f, and the differential expression analysis was performed with RSEM version 1.3.0. Normalized RNA-seq data were analyzed with GSEA v4.0.3. Enrichment analyses were performed using 1,000 gene set permutations, a weighted enrichment statistic, the Signal2Noise ranking metric, and gene set minimum and maximum sizes of 15 and 500, respectively.
PD-1 Immune Checkpoint Blockade treated melanoma cohort analysis
Patients in Schadendorf cohort had advanced melanoma and had received PD-1 immune checkpoint blockade (ICB) 24. 121 out of 144 patients had RNA-seq data available, and all of these samples were included in survival analysis. Correlations of mRNA expression levels between immune related genes (IFNG, IL2, PRF1, etc.) and adjusted IRG1/ACOD1 score were computed with nonparametric Spearman’s rank correlation coefficient after adjustment for immune infiltrates based on the predicted effects of immune infiltrates on IRG1 in a univariate linear regression. In a naïve analysis, raw IRG1 expression was overall positively associated with total immune cell infiltrate and immune related genes expression levels. We reasoned that clinical outcomes may be associated with excess IRG1 expression relative to the total immune infiltrate, and we calculated an excess IRG1 expression score for each sample adjusting for overall immune infiltrate level for subsequent analyses. Overall immune infiltrate levels and CD8 T cell signatures scores were inferred using scRNA-seq signature scoring as previously described. (Liu et al., 2019). The expression values of immune genes (TPM) were used in the correlative analysis.
Among the 121 patients with bulk RNA-seq data available from Schadendorf cohort, 56 patients with progressed disease (PD) as best response to anti-PD-1 ICB were identified as progressors, 47 patients with complete response (CR) or partial response (PR) were identified as responders. Remaining patients with mixed response (MR) or stable disease (SD) were excluded from downstream analysis. The adjusted mRNA expression levels of IRG1 were compared between progressors and responders, and a Wilcoxon rank sum test was used to evaluate the significance of the difference. Outliers were included in statistical test but were not shown in plot for visualization purposes. Survival analysis was performed utilizing the R packages survminer and survival. For Kaplan–Meier curve survival analysis, a two-sided log-rank test was used to compare overall survival (OS) curves. For Cox proportional hazards regression analysis, a log-rank test was used to evaluate the significance of the model.
Single-Cell RNA-seq Data Processing
scRNA-seq data of MDSC-containing splenic myeloid cells from breast tumor-bearing mice and wildtype controls (GSM4131336_Wild_Type_Expression_Matrix.txt.gz, GSM4131337_PYMT_Expression_Matrix.txt.gz)16 and scRNA-seq data from murine melanoma40, glioma41, and small cell lung cancer42 tumors (GSM4030648_ 15885X1.matrix.mtx.gz, merged GSM4610558_A1_matrix.mtx.gz and GSM4610558 _A1_matrix.mtx.gz, and GSE149179_RPM2_matrix.mtx.gz, respectively) were downloaded from Gene Expression Omnibus (GEO) website. QC filtering removed cells with low gene detection (< 500 genes) and high mitochondrial gene percentage (> 8%). Expression data were then normalized, scaled, and processed under principal components analysis using R package Seurat. Uniform Manifold Approximation and Projection (UMAP) was performed for representation of the data. The data were down sampled to include 5,000 cells for better visualization in UMAP. A G-MDSC (PMN-MDSC) gene signature of 390 genes were curated from supplementary tables in Alshetaiwi et al., 2020. Granulocytic clusters (Csf3r, S100a8/9), monocytic and macrophage clusters (Csf1r, Cd68, Mrc1, Arg1, and Ms4a7), dendritic cells (Itgae, Cx3cr1, and Etv3), monocytes (Ly6c2, Ccr2), microglia (Cx3cr1, C1qa, and Ube216), T cell (Cd3e, Cd4, and Cd8), B cell (Cd19 and MS4a1), NK cell (Gzma, Nkg7, Klr4a), endothelial cell (Mcam, Pecam1), Epithelium (Krt8, Sox2) and tumors (B16F10 (Mc1r, S100a1), Glioma (Sparc, Lgals1, Hmga1), and SCLC (Ncam1 and Insm1)) were identified by the expression of the indicated genes.
Quantification and Statistical Analysis
GraphPad Prism V9 was used for the analysis of the data and graphic representations. Comparisons between two groups were performed with two-tailed unpaired Student’s t test. Comparison among more than two groups were performed with ANOVA analysis followed by Tukey’s test. P values less than 0.05 were considered significant. No statistical methods were used to pre-determine sample sizes but our sample sizes are similar to those reported in previous publications12,38.
Extended Data
Extended Data Figure 1. MDSCs secrete itaconate.
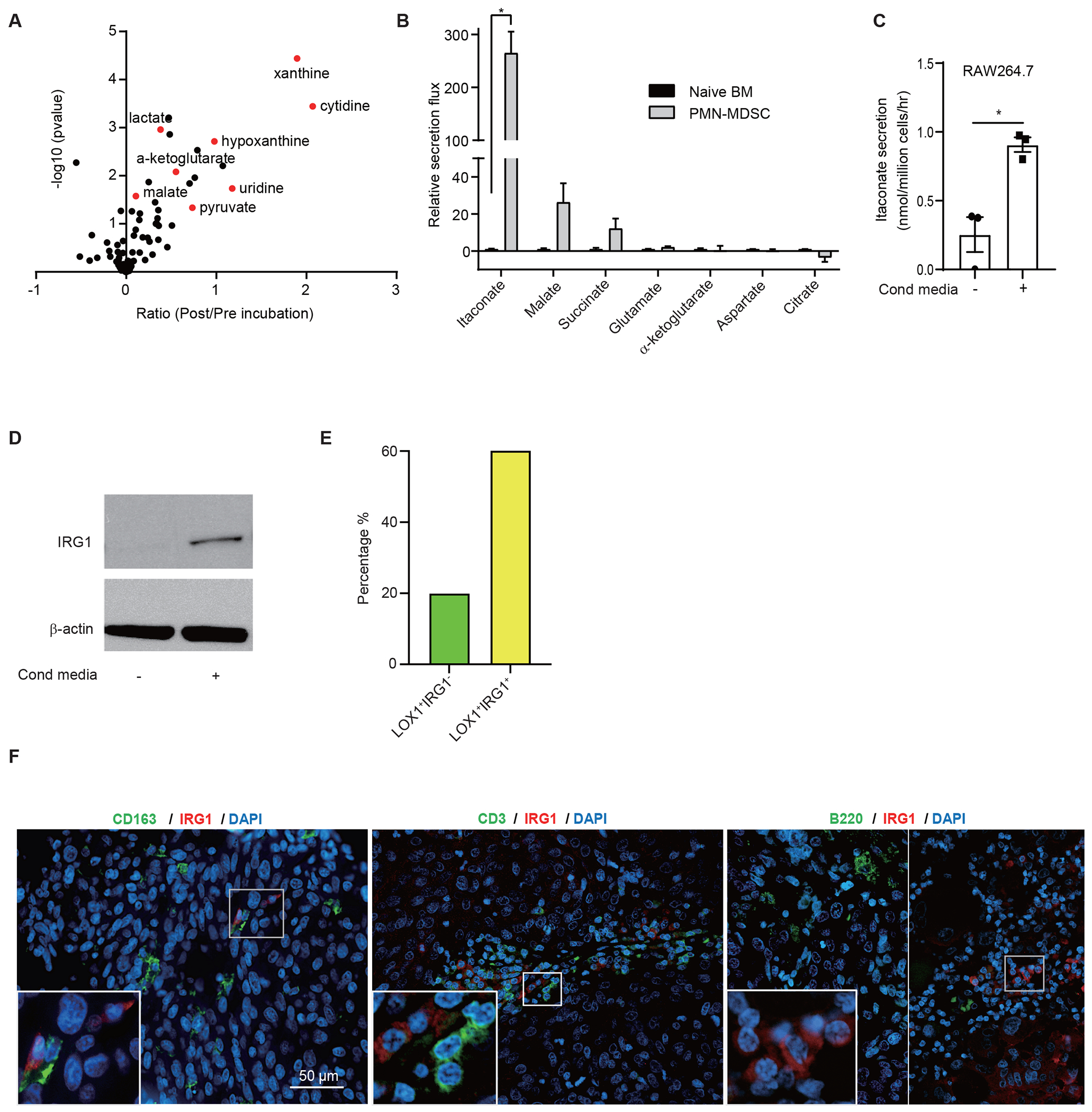
(A) Secretion of various metabolites from naïve BM cells as detected by targeted LC-MS/MS analysis (n=3 biologically independent samples).
(B) Ratios of secretion rates of itaconate and various TCA cycle related metabolites from BM-MDSCs versus naïve BM cells (n=3 biologically independent samples). MDSCs or BM cells were reseeded and cultured in fresh medium for 24 hours and polar metabolites in the supernatants were analyzed by targeted LC-MS-based metabolomics.
(C) Secretion rates of itaconate by RAW264.7 cultured with or without cancer conditioned medium (n=3 biologically independent samples).
(D) Expression of IRG1 protein in RAW264.7 cultured with or without cancer conditioned medium. Blots are representative of 3 independent experiments.
(E) Quantification of the percentage of LOX+IRG1− and LOX1+IRG1+ MDSCs in human melanoma tumor specimens for Fig 1E.
(F) Immunofluorescence analysis of IRG1 and macrophage marker CD163, T cell marker CD3 and B cell marker B220 in human melanoma tumor specimens. Data shown are representatives of 12 biologically independent human participants.
P values were calculated by paired two- tailed t-tests. Data are represented as mean ± SEM.
Extended Data Figure 2. Expression of Irg1 is largely confined to granulocytic myeloid populations within the tumors.
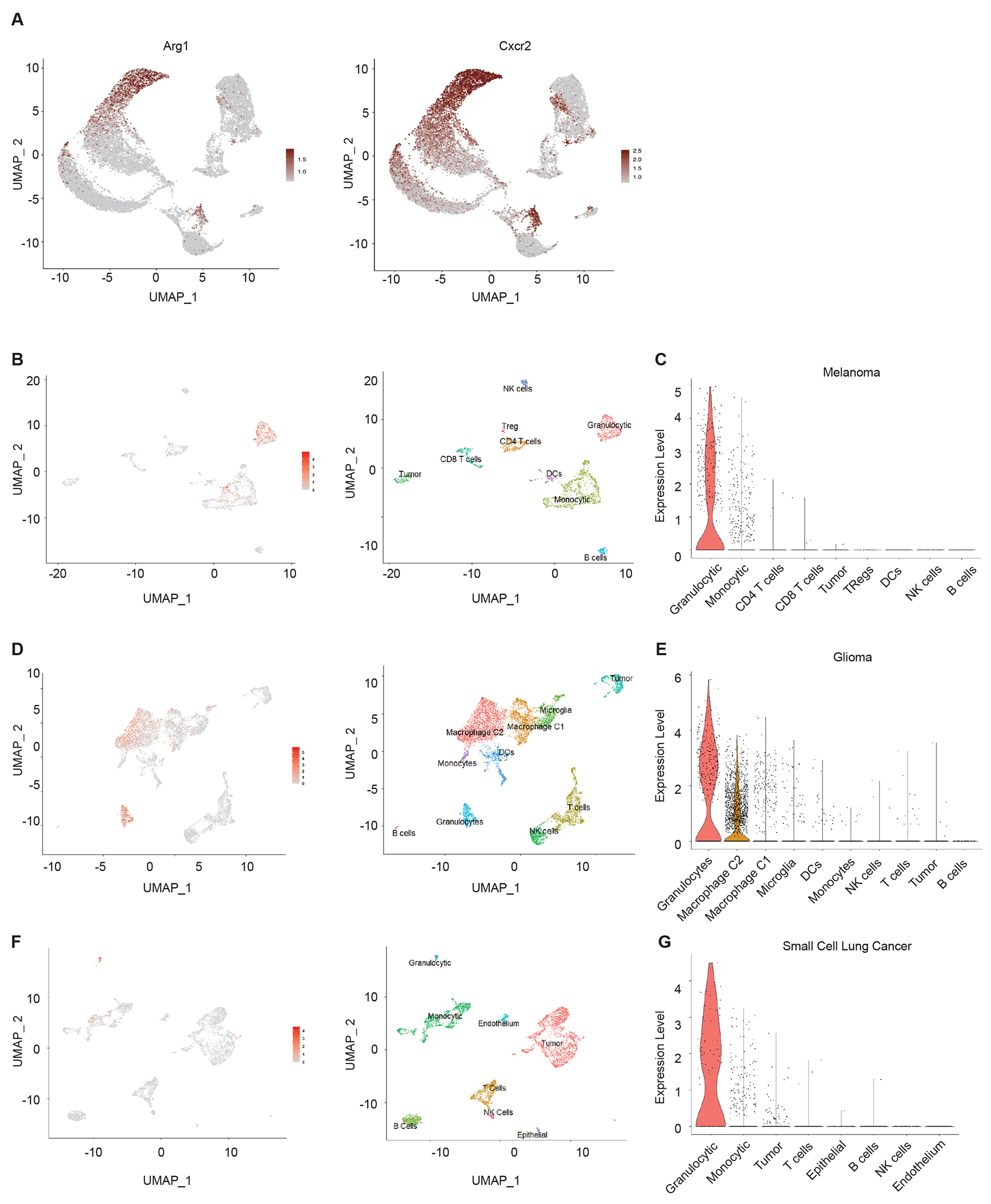
Interrogation of publicly available single cell RNA-Seq data analysis to determine cell specific expression of Irg1 within the indicated experimental murine tumors.
(A) UMAP showing cells expressing Arg1 and Cxcr2 in mouse breast cancer tumors shown in Figure 1F.
(B) UMAP showing cells expressing Irg1 in mouse B16F10 subcutaneous injected tumors.
(C) Violin plots showing levels of Irg1 expression in each cluster from mouse B16F10 subcutaneous injected tumors.
(D) UMAP showing cells expressing Irg1 in an oncogenic transgenic glioma mouse model.
(E) Violin plots showing levels of Irg1 expression in each cluster from an oncogenic transgenic glioma mouse model.
(F) UMAP showing cells expressing Irg1 in a mouse small cell lung cancer model.
(G) Violin plots showing levels of Irg1 expression in each cluster from a mouse small cell lung cancer model.
Extended Data Figure 3. Itaconate suppresses CD8+ T cell proliferation.
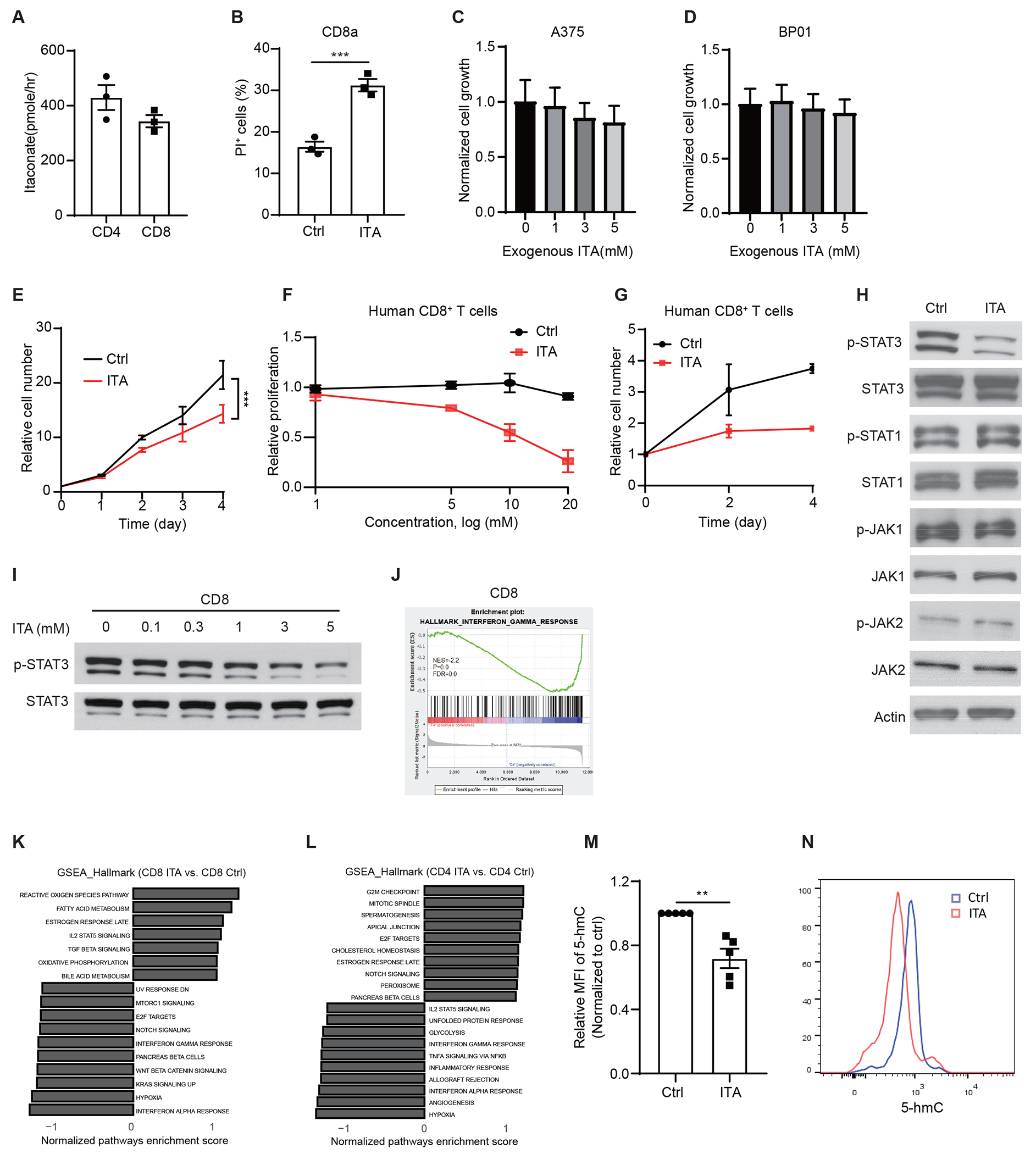
(A) Uptake of Itaconate from media (5 mM) by CD3/CD28 activated CD4+ T or CD8+ T cells at 6 hours of incubation (n=3 biologically independent samples).
(B) Effects of itaconate on the cell viability of CD3/CD28 activated CD8+ T cell death (n=3 biologically independent experiments).
(C) Effects of itaconate on the proliferation of A375 melanoma cells (n=6 biologically independent samples).
(D) Effects of itaconate on the proliferation of BP01 melanoma cells (n=6 biologically independent samples).
(E) Itaconate (5 mM) inhibited the proliferation of mouse CD8+ T cells previously activated by CD3/CD28 as measured by cell number counting (n=3 biologically independent experiments).
(F) Effects of various concentration of itaconate (5 mM) on the proliferation of CD3/CD28-activated human CD8+ T cells as measured by CFSE dilution (n = 5 biologically independent experiments).
(G) Effects of itaconate (5 mM) on the proliferation of CD3/CD28-activated human CD8+ T cells as measured by cell number counting (n = 3 biologically independent experiments).
(H) Effects of itaconate on JAK-STAT signaling in CD3/CD28 activated CD8+ T cells. Cells were treated with NaCl or itaconate (5 mM) for 48 hours before lysates were collected for Western blotting analyses with indicated antibodies. Blots are representative of 3 independent experiments.
(I) Effects of itaconate of various concentrations on the levels p-STAT3 and total STAT3 proteins in CD3/CD28 activated CD8+ T cells. Cells were treated with NaCl or itaconate for 48 hours, before lysates were collected for Western blotting analyses with indicated antibodies. Blots are representative of 3 independent experiments.
(J) GSEA enrichment plots depicting the enrichment of IFNγ-response gene sets in mouse CD8+ T cells treated with itaconate.
(K-L) GSEA of RNA-seq data reveals enriched pathways in mouse CD8+ T cells (K) and CD4+ T cells (L) treated with 5 mM itaconate (ITA) versus NaCl (Ctrl) (n=3 biologically independent samples).
(M) Relative MFI expression of 5-hmC in mouse CD3/CD28-activated CD8+ T cells treated with 3 mM NaCl (Ctrl) or itaconate for 24 hours (n=5 biologically independent experiments).
(N) Representative flow cytometry plots of 5-hmC expression for (M).
For all panels, p values were calculated by paired two- tailed t-tests. Data are mean ± SEM.
Extended Data Figure 4. Itaconate suppresses CD8+ T cell activation.
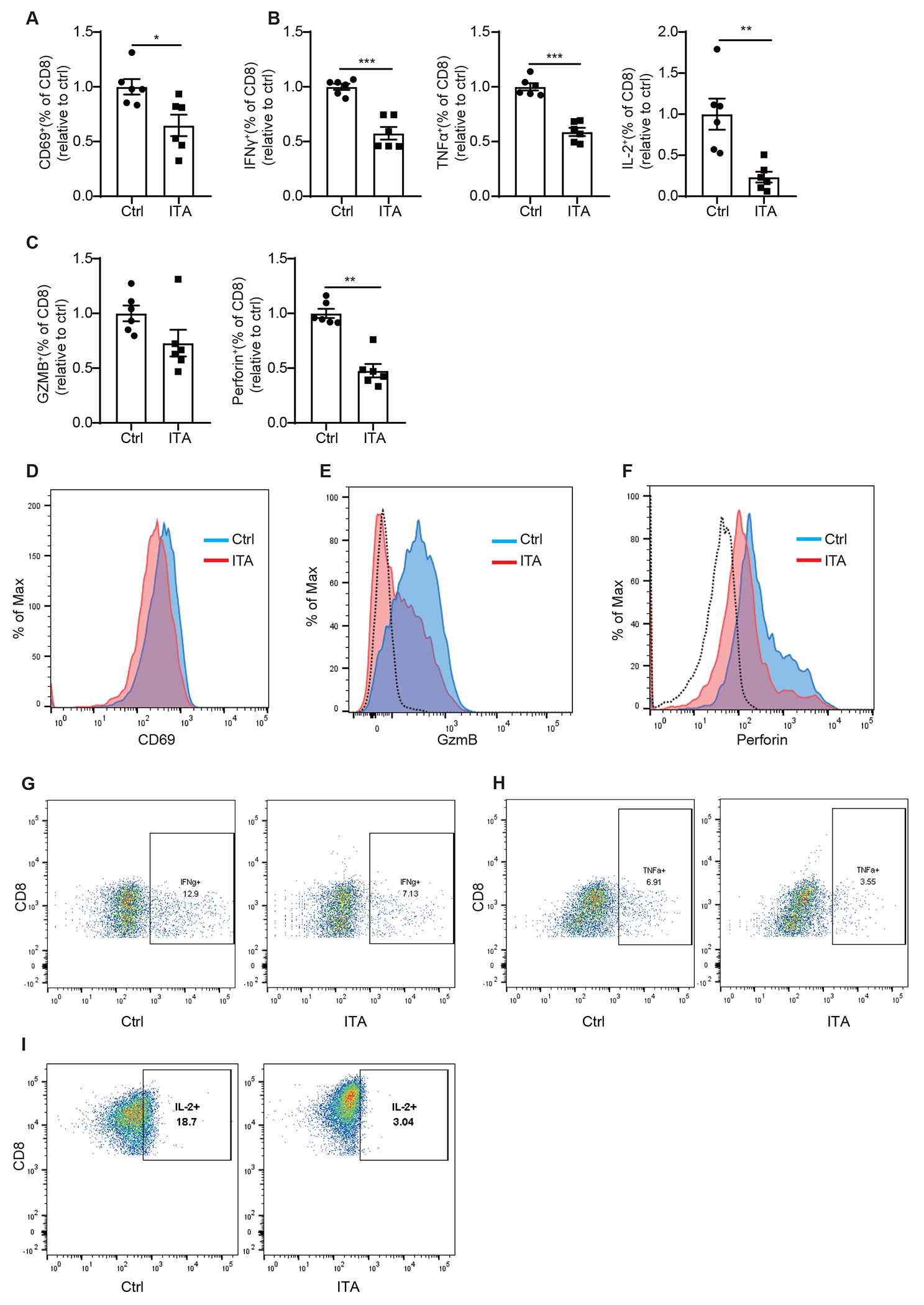
(A-C) Relative percentage of CD69 (A), IFNγ, TNFα, IL-2, (B), Granzyme B and Perforin (C) expressing cells in mouse CD8+ T cells treated with 5 mM NaCl (Ctrl) or itaconate for 3 days in the presence of anti-CD3/CD28, as analyzed by flow cytometry (n= 6 biologically independent experiments).
(D-H) Representative flow cytometry plots for expression of CD69 (D), Granzyme B (E), Perforin (F), IFNγ (G), TNFα(H) and IL-2 (I) in mouse CD8+ T cells treated with 5 mM NaCl (Ctrl) or itaconate for 3 days in the presence of anti-CD3/CD28.
For all panels, p values were calculated by paired two- tailed t-tests. Data are mean ± SEM.
Extended Data Figure 5. Itaconate impedes aspartate and serine/glycine biosynthesis in CD8+ T cells.
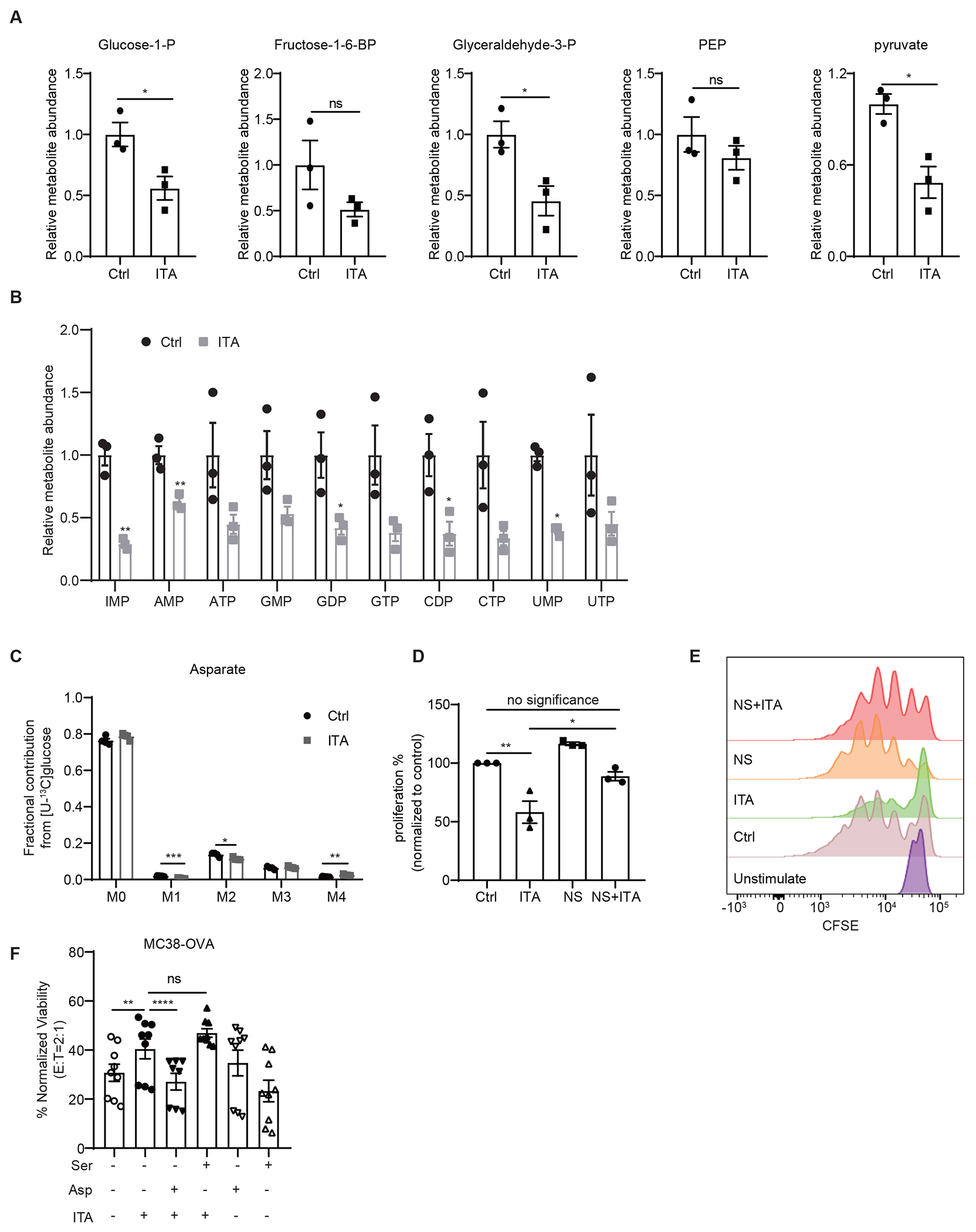
(A) Levels of various glycolytic intermediates in CD8+ T cells exposed to exogenous itaconate (ITA) or NaCl (Ctrl), as revealed by metabolite profiling analyses (n=3 biologically independent samples).
(B) Levels of various nucleotides in CD8+ T cells exposed to exogenous itaconate or NaCl (Ctrl), as revealed by metabolite profiling analyses (n=3 biologically independent samples).
(C) Incorporation of 13C to Aspartate from [U-13C]-glucose in CD8+ T cells exposed to exogenous itaconate or NaCl (Ctrl) (n=4 biologically independent samples).
(D) Effects of nucleosides supplementation on the proliferation of mouse CD8+ T cells treated with 3 mM exogenous itaconate, as measured by CFSE dilution (n=3 biologically independent experiments).
(E) Representative CFSE intensity histograms from flow cytometry analysis for (D).
(F) Effects of aspartate (Asp) and serine (Ser) supplementation on the ability of itaconate (3 mM) treated mouse CD8+ T cells on killing MC38-OVA cells (n=5 biologically independent experiments).
For all panels, p values were calculated by paired two- tailed t-tests. Data are mean ± SEM.
Extended Data Figure 6. Loss of Irg1 enhances anti-tumor immunity of CD8+ T cells.
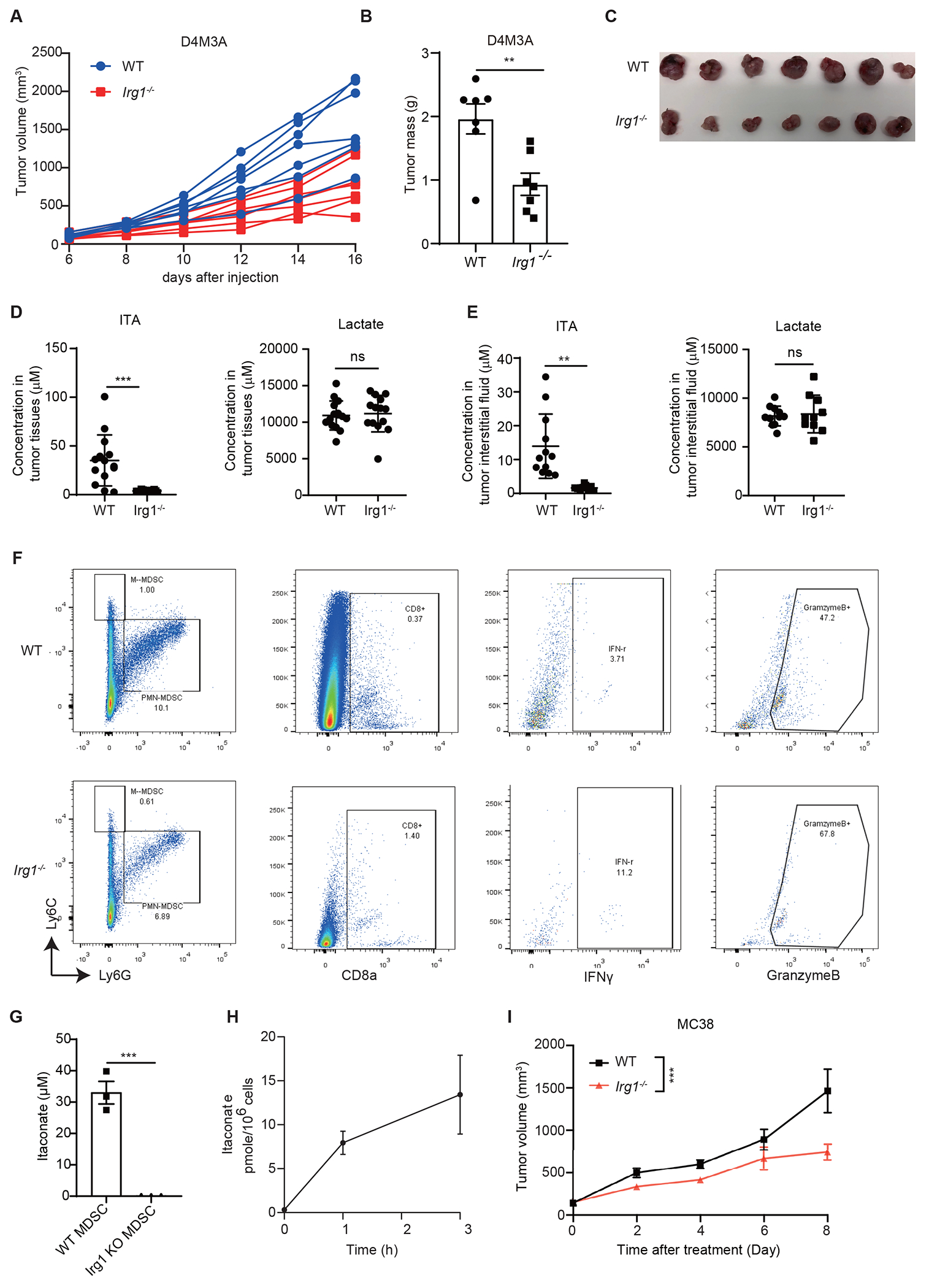
(A) Spider plot of D4M3A syngeneic tumors grown in WT and Irg1−/− mice (n=7 mice).
(B) Tumor mass of D4M3A syngeneic tumors grown in WT and Irg1−/− mice (n=7 mice).
(C) Pictures of D4M3A syngeneic tumors grown in WT and Irg1−/− mice.
(D-E) Measurement of itaconate and lactate in tumor tissues (D) and interstitial fluids (E) showed measurable amounts of itaconate only in tumors grown in WT animals (n=14 for tumor tissues from both groups, n=12 for interstitial fluid samples from WT mice, n=9 interstitial fluid samples from for Irg1−/− mice).
(F) Representative flow plots of MDSCs and CD8+ T cells from D4M3A tumors grown in WT and Irg1−/− mice.
(G) Secretion of itaconate by WT BM-MDSCs versus Irg1−/− BM-MDSCs (n=3 biologically independent samples).
(H) Uptake of itaconate from the culture media of WT BM-MDSCs by CD3/CD28 activated CD8+ T cells (n=3 biologically independent samples).
(I) MC38 syngeneic tumors grown in Irg1−/− mice exhibited significantly slower grow compared to those in WT mice (n=8 mice).
p values were calculated by unpaired two-tailed t-test (B,D,E,G), two-way ANOVA (I). Data are represented as mean ± SEM.
Extended Data Figure 7. Loss of Irg1 enhances the efficacy of anti-PD-1 immune checkpoint blockade.
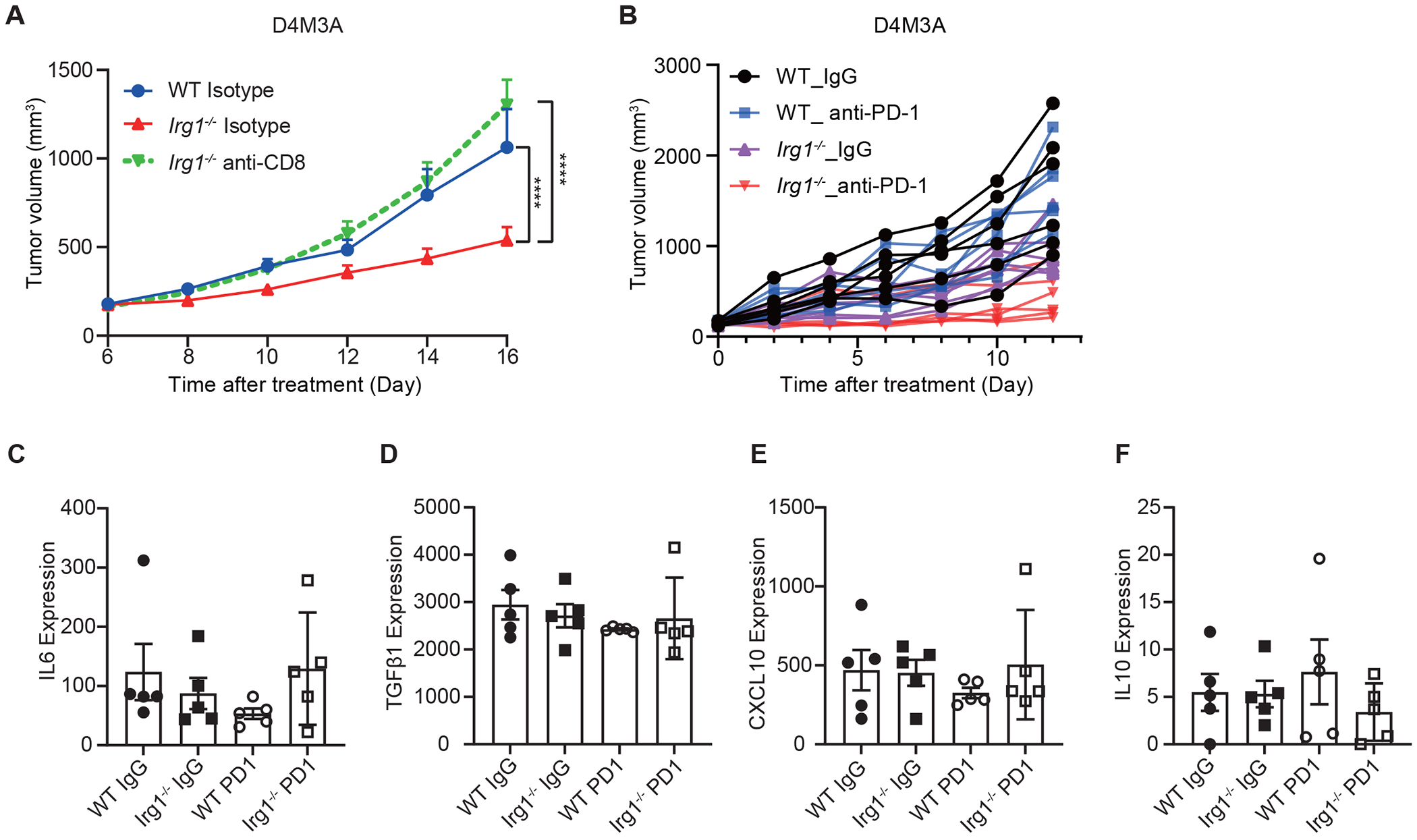
(A) Depletion of CD8+ T cells restored D4M3A tumor growth in Irg1−/− mice (n=7 mice). P value were calculated by two-sided Tukey’s multiple comparisons test.
(B) Spider plot of D4M3A syngeneic tumors grown in WT and Irg1−/− mice and treated with anti-PD-1 or isotype control IgG (n=6 mice).
(C-F) Expression of IL6 (C), TGFβ1(D), CXCL10 (E) and IL10 (F) in D4M3A tumor tissues from WT and Irg1−/− mice that were treated with anti-PD-1 or isotype control IgG (n=5 biologically independent samples).
For all panels, data are represented as mean ± SEM.
Supplementary Material
Acknowledgements
We thank Emily Robitschek and members of the Zheng Lab for helpful discussion on the manuscript. This work is supported by US National Institutes of Health (NIH) R21CA227588, R01CA219814, the Melanoma Research Alliance, the Elsa U. Pardee Foundation and funds from MGH (to B.Z.); and NIH R01CA163591, DP1DK113643 and Stand Up to Cancer (SU2CAACR-DT-20-16) (to J.D.R.). Y.Z. is funded by NCI-R50CA232985. M.P.M is supported by NIH/NCI grants R01CA151588, R01CA198074, U01CA-224145 and the University of Michigan Cancer Center Support Grant (NCI P30CA046592).
Competing Interests
K.T.F. serves on the Board of Directors of Clovis Oncology, Strata Oncology, Kinnate, Checkmate Pharmaceuticals, and Scorpion Therapeutics; Scientific Advisory Boards of PIC Therapeutics, Apricity, Tvardi, ALX Oncology, xCures, Monopteros, Vibliome, and Soley Therapeutics; consultant to Takeda and Transcode Therapeutics. G.M.B has sponsored research agreements with Olink Proteomics, Palleon Therapeutics, InterVenn Biosciences, and Takeda Oncology. She was on scientific advisory boards for Novartis, Merck, Nektar Therapeutics, Iovance, and Ankyra Therapeutics. She consults for InterVenn Biosciences, Ankyra Therapeutics, and Merck. J.D.R. is a paid adviser and/or stockholder in Colorado Research Partners, L.E.A.F. Pharmaceuticals, Rafael Pharmaceuticals and its subsidiaries, Empress, and Agios Pharmaceuticals; a paid consultant of Pfizer; a founder, director, and stockholder of Farber Partners and Serien Therapeutics. The remaining authors declare no competing interests.
Data availability
There are no restrictions as to the availability of materials reported in the manuscript. The data that support the findings of this study are available from the corresponding author upon reasonable request. The RNA-seq data generated in this study have been deposited in the NCBI Gene Expression Omnibus and are accessible through accession number GSE190028 and GSE211255. Source data are provided with this paper.
References
- 1.Chapman NM, Boothby MR & Chi H Metabolic coordination of T cell quiescence and activation. Nature Reviews Immunology 20, 55–70 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Leone RD & Powell JD Metabolism of immune cells in cancer. Nat Rev Cancer 20, 516–531 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaymak I, Williams KS, Cantor JR & Jones RG Immunometabolic Interplay in the Tumor Microenvironment. Cancer Cell 39, 28–37 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tyrakis PA et al. S-2-hydroxyglutarate regulates CD8+ T-lymphocyte fate. Nature 540, 236–41 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bunse L et al. Suppression of antitumor T cell immunity by the oncometabolite ( R )-2-hydroxyglutarate. Nat Med 24, 1192–1203 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Veglia F, Sanseviero E & Gabrilovich DI Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nature Reviews Immunology 67, 1–14 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tarhini AA et al. Immune monitoring of the circulation and the tumor microenvironment in patients with regionally advanced melanoma receiving neoadjuvant ipilimumab. PLoS ONE 9, e87705 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sade-Feldman M et al. Clinical Significance of Circulating CD33+CD11b+HLA-DR- Myeloid Cells in Patients with Stage IV Melanoma Treated with Ipilimumab. Clin. Cancer Res. 22, 5661–5672 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Hou A, Hou K, Huang Q, Lei Y & Chen W Targeting Myeloid-Derived Suppressor Cell, a Promising Strategy to Overcome Resistance to Immune Checkpoint Inhibitors. Front. Immunol. 11, 783 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Neill LAJ & Artyomov MN Itaconate: the poster child of metabolic reprogramming in macrophage function. Nature Reviews Immunology 19, 273–281 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Hooftman A & O’Neill LAJ The Immunomodulatory Potential of the Metabolite Itaconate. Trends in Immunology 40, 687–698 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Kim SH et al. Phenformin Inhibits Myeloid-Derived Suppressor Cells and Enhances the Anti-Tumor Activity of PD-1 Blockade in Melanoma. Journal of Investigative Dermatology 137, 1740–1748 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Michelucci A et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci USA 110, 7820–7825 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Condamine T et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Science Immunology 1, aaf8943–aaf8943 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strelko CL et al. Itaconic acid is a mammalian metabolite induced during macrophage activation. - PubMed - NCBI. J. Am. Chem. Soc. 133, 16386–16389 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alshetaiwi H et al. Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Science Immunology (2020). doi: 10.1126/sciimmunol.aay6017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cordes T et al. Immunoresponsive Gene 1 and Itaconate Inhibit Succinate Dehydrogenase to Modulate Intracellular Succinate Levels. J Biol Chem 291, 14274–14284 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sullivan LB, Gui DY & Vander Heiden MG Altered metabolite levels in cancer: implications for tumour biology and cancer therapy. Nat Rev Cancer 16, 680–693 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Ma EH et al. Serine Is an Essential Metabolite for Effector T Cell Expansion. Cell Metab 25, 345–357 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Ron-Harel N et al. Mitochondrial Biogenesis and Proteome Remodeling Promote One-Carbon Metabolism for T Cell Activation. Cell Metab 24, 104–117 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kelly B & Pearce EL Amino Assets: How Amino Acids Support Immunity. Cell Metab 32, 154–175 (2020). [DOI] [PubMed] [Google Scholar]
- 22.BP D et al. The Nucleotide Sensor ZBP1 and Kinase RIPK3 Induce the Enzyme IRG1 to Promote an Antiviral Metabolic State in Neurons. Immunity 50, 64–76.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lo JA et al. Epitope spreading toward wild-type melanocyte-lineage antigens rescues suboptimal immune checkpoint blockade responses. Science Translational Medicine 13, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu D et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat Med 25, 1916–1927 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodriguez PC et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res 64, 5839–5849 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Smith C et al. IDO is a nodal pathogenic driver of lung cancer and metastasis development. Cancer Discovery 2, 722–735 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu J et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J. Immunol. 190, 3783–3797 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Weiss JM et al. Itaconic acid mediates crosstalk between macrophage metabolism and peritoneal tumors. J Clin Invest 128, 3794–3805 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luan HH & Medzhitov R Food Fight: Role of Itaconate and Other Metabolites in Antimicrobial Defense. Cell Metab 24, 379–387 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zasłona Z & O’Neill LAJ Cytokine-like Roles for Metabolites in Immunity. Mol. Cell 78, 814–823 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Shen H et al. The Human Knockout Gene CLYBL Connects Itaconate to Vitamin B12. Cell 171, 771–782 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mills EL et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 556, 113–117 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bambouskova M et al. Electrophilic properties of itaconate and derivatives regulate the IκBζ–ATF3 inflammatory axis. Nature 42, 1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bambouskova M et al. Itaconate confers tolerance to late NLRP3 inflammasome activation. CellReports 34, 108756 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hooftman A et al. The Immunomodulatory Metabolite Itaconate Modifies NLRP3 and Inhibits Inflammasome Activation. Cell Metab 32, 468–478.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Runtsch MC et al. Itaconate and itaconate derivatives target JAK1 to suppress alternative activation of macrophages. Cell Metab 34, 487–501.e8 (2022). [DOI] [PubMed] [Google Scholar]
- 37.DeNicola GM et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet 47, 1475–1481 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yuan P et al. Phenformin enhances the therapeutic benefit of BRAF(V600E) inhibition in melanoma. Proceedings of the National Academy of Sciences 110, 18226–18231 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jenkins MH et al. Multiple murine BRaf V600Emelanoma cell lines with sensitivity to PLX4032. Pigment Cell & Melanoma Research 27, 495–501 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huffaker TB et al. A Stat1 bound enhancer promotes Nampt expression and function within tumor associated macrophages. Nature Communications 12, 2620 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alghamri MS et al. G-CSF secreted by mutant IDH1 glioma stem cells abolishes myeloid cell immunosuppression and enhances the efficacy of immunotherapy. Science Advances 7, eabh3243 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olsen RR et al. ASCL1 represses a SOX9+neural crest stem-like state in small cell lung cancer. Genes Dev 35, 847–869 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
There are no restrictions as to the availability of materials reported in the manuscript. The data that support the findings of this study are available from the corresponding author upon reasonable request. The RNA-seq data generated in this study have been deposited in the NCBI Gene Expression Omnibus and are accessible through accession number GSE190028 and GSE211255. Source data are provided with this paper.