

Abstract
Pediatric and adult autoimmune encephalitis (AE) are often associated with antibodies to the NR1 subunit of the N-methyl-D-aspartate receptor (NMDAR). Very little is known regarding the cerebrospinal fluid (CSF) humoral immune profile and antibody genetics associated with pediatric αNMDAR-AE. Using a combination of cellular, molecular and immunogenetics tools, we collected CSF from pediatric subjects and generated 1) flow cytometry data to calculate the frequency of B cell subtypes in the CSF of pediatric subjects with αNMDAR-AE and controls, 2) a panel of recombinant human antibodies from a pediatric case of αNMDAR-AE that was refractory to treatment and 3) a detailed analysis of the antibody genes that bound the NR1 subunit of the NMDAR. Antigen-experienced B cells including memory, plasmablast and antibody-secreting cells were expanded in the pediatric αNMDAR-AE cohort, but not in the controls. These antigen-experienced B cells in the CSF of a pediatric case of NMDAR-AE that was refractory to treatment had expanded use of VH2 heavy chain genes with high somatic hypermutation that all bound to the NR1 subunit of the NMDAR. A CDR3 motif was identified in this refractory case that likely drove early-stage activation and expansion of naïve B cells to antibody-secreting cells, facilitating autoimmunity associated with pediatric αNMDAR-AE through the production of antibodies that bind NR1. These features of humoral immune responses in the CSF of pediatric αNMDAR-AE patients may be relevant for clinical diagnosis and treatment.
INTRODUCTION
Encephalitis is a potentially devastating inflammatory disease of the brain caused by infection or autoimmune conditions.(1–4) The most common form of autoimmune encephalitis (AE) in adults and children is associated with antibodies against N-methyl-d-aspartate glutamate receptors (NMDARs), termed αNMDAR-AE (anti-NMDAR-autoimmune encephalitis).(5, 6) The NR1 subunit of the NMDAR (also known as GluN1) is the primary target of antibodies against the NMDAR.(7–9) Anti-NMDAR antibodies can be detected in the serum and CSF, albeit misdiagnosis based in part on overinterpretation of serum positivity has been reported.(10) Prognosis in αNMDAR-AE is dependent on early recognition, prompt reduction of αNMDAR antibodies, and removal of associated tumors (when identified).(11, 12) Even with these treatments, relapses occur in up to 25% of patients.(13, 14) and outcomes range from full/partial recovery to death.(15, 16)
IgG purified from CSF of adult and pediatric αNMDAR-AE patients(17) and monoclonal αNMDAR antibodies expressed by B cells from the cerebrospinal fluid of adult NMDAR-AE patients(18, 19) bind, cross-link, and internalize the NR1 subunit of the NMDAR, leading to altered neuronal synaptic function. In a separate case, a human monoclonal antibody cloned from an 18-year old patient with αNMDAR-AE was shown to rapidly localize to synaptic NMDAR’s of primary rat neurons but did not alter synaptic calcium flux.(20) The mechanism of αNMDAR antibody emergence has recently been clarified with evidence that the NR1 subunit of the NMDAR is present in germinal centers of patients with αNMDAR-AE.(21, 22) In addition, cervical lymph nodes and ovarian teratomas in patients with NMDAR-AE harbor both the NR1 protein and germinal centers where B cells are activated to produce IgA and IgG antibody against the NR1 subunit of the NMDAR.(21) This data substantiates the observation of B cell removal as an effective second-line treatment for pediatric αNMDAR-AE(23) and αNMDAR-AE in general.(24, 25)
There have been no focused studies to understand humoral immune response and associated antibody genetics among pediatric αNMDAR-AE patients. Thus, our lab focused on profiling the humoral immune response to NR1, the obligatory subunit of NMDA receptors in pediatric αNMDAR-AE patients and pediatric OND controls.(26) To do this, we used flow cytometry to determine the frequency of B cell subsets including ASCs in the CSF of pediatric patients diagnosed with αNMDAR-AE and pediatric OND controls. Next, we used electrophysiology to evaluate whether antibody pools from a pediatric patient with severe αNMDAR-AE impacted neuron function. Finally, we isolated ASCs and used single cell PCR technology to determine the frequency of CSF derived B cells expressing antibodies that bind to the NR1 subunit of the NMDA receptor. Profiling the humoral immune response could aid in our understanding of why attacks on the nervous system occur among pediatric patients diagnosed αNMDAR-AE. Study samples came from multiple pediatric patients, including one severe case that resulted in death. This patient’s sample was included to understand the immunologic profile of severe disease. Our hope is that these data will aid clinicians’ ability to prognosticate and treat patients in the future.
METHODS
Subjects
All pediatric subjects and/or their legally authorized study partners signed the written informed consent approved by the Institutional Review Board of the UT Southwestern Medical Center (UTSW), in accordance with the Federal-wide Assurance on file with the Department of Health and Human Services (USA). Samples were collected and processed through the Neuroscience Biorepository at UTSW(27, 28) under authorization by the IRB. This cohort includes 7 pediatric patients diagnosed with autoimmune encephalitis with detectable αNMDAR antibodies in either the cerebrospinal fluid (CSF) or serum and met criteria for αNMDAR-AE.(29) An additional 13 pediatric patients with no detectable anti-NMDAR antibodies were included as an Other Neurological Disease (OND) control population. See Table 1 for a summary of the cohort.
TABLE 1.
Subject information
| NMDAR-AE Subject Code | Age | Sex |
|---|---|---|
| 0972 | 8 | F |
| 1306 | 15 | F |
| 1463 | 2 | F |
| 0978 | 8 | M |
| 1547 | 3 | M |
| 1712 | 13 | F |
| 1783 | 17 | M |
| OND Subject Code1 | ||
| 1282 | 5 | F |
| 1295 | 16 | M |
| 1506 | 2 | M |
| 1515 | 15 | M |
| 1470 | 18 | M |
| 1739 | 14 | M |
| 1792 | 4 | M |
| 1819 | 15 | F |
| 1839 | 13 | F |
| 0800 | 8 | M |
| 1904 | 2 | F |
OND category included subjects diagnosed with demyelinating disease, psychosis, cancer, or physical trauma
Cell-based assay for NMDAR autoreactivity by serum and CSF supernatant
Serum and CSF supernatant were used to determine binding of IgG antibodies to the NR1 subunit of the NMDAR(30, 31) using the commercially available cell based assay (CBA) from EurImmun (https://www.euroimmun.com/products/indications/autoantikrper-diagnostik/neurology/autoimmune-encephalitis/aak-gegen-nmdar.html) and following manufacturer recommendations. Images were acquired using an inverted Zeiss LSM780 microscope.
Electrophysiology
Acute brain slice preparation:
Four to seven days following the injection procedure, mice were anesthetized with the inhalation anesthetic, isoflurane, then were rapidly decapitated and their brains removed into ice-cold dissection artificial cerebral spinal fluid (ACSF). Acute coronal slices 300–350μm thick containing hippocampus were made on a vibrating microtome (Leica Biosystems, Wetzlar, Germany) in ice-cold dissection ACSF. Immediately after slicing, a cut was made between CA3 and CA1 to prevent recurrent excitation, and a second cut was made between the left and right hemispheres. Slices from each hemisphere were held for 30–45 minutes at 35°C in separate recovery chambers, each containing ACSF with 2mM MgCl2 and 1mM CaCl2. Slices were cooled to room temperature (RT, 24°C) and maintained at RT prior to recording.
Extracellular “field” electrophysiology:
All recordings were performed at 32–33°C in Warner Instruments (Hamden, CT) low-profile PH-1 recording chambers continuously perfused with ACSF containing 2.5mM CaCl2 and 1mM MgCl2. 0.1ms current pulses were delivered via A-M Systems (Carlsborg, WA) Model 2100 stimulators through concentric bipolar stimulating electrodes (Model #CBARB75, FHC, Bowdoin, ME). Stimulating and recording electrodes (1–2 MΩ) were placed laterally in the stratum radiatum 400–500μm apart. Postsynaptic responses were measured with A-M Systems Model 1800 dual-channel amplifiers, highpass filtered at 1–5kHz, and digitized at 10kHz with a Molecular Devices (Sunnyvale, CA) Digidata 1440 digitizer. Signals were acquired with Clampex (v 10.7, Molecular Devices). All experiments were preceded by a stable baseline for at least 20-minutes.
Input/Output (I/O) curves:
Stimulus intensity was increased from 0–10mA in 2mA steps at a rate of 0.05Hz. The fEPSP slope and fiber volley amplitude of 5 consecutive traces is averaged at each stimulus intensity per slice.
Analysis and statistics:
fEPSP slope fitting and fiber volley measurements were made using Clampfit (v 10.7, Molecular Devices). Data cleaning and descriptive statistics were determined using custom Python 3.0 scripts, and hypothesis testing and posthoc analysis was performed using Statistica (Dell, Round Rock, TX).
Solutions:
Dissection ACSF contained (in mM):
87 NaCl, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 20 dextrose, 75 sucrose, 7 MgSO4, 0.5 CaCl2 and saturated with 5%CO2/95%O2 at pH 7.3. Recording (standard) ACSF contained (in mM): 126 NaCl, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 10 dextrose, 1 MgCl2, 2.5 CaCl2 and saturated with 5%CO2/95%O2 at pH 7.3. Slice maintenance ACSF contained (in mM): 126 NaCl, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 10 dextrose, 2 MgCl2, 1 CaCl2 and saturated with 5%CO2/95%O2 at pH 7.3.
Flow Cytometry
CSF was centrifuged and cells were stained with a panel of antibodies that identify B cell subsets including total CD19+ B cells, naïve B cells, memory B cells, plasmablasts (PBs) and antibody-secreting cells (ASCs). Individual antigen-experienced CD19+CD27+ B cells (which include memory B cells, plasmablast B cells and antibody-secreting B cells) from the CSF of Subject 1547 were isolated by fluorescence-activated cell-sorting into 96-well plates with 4 μL of 40 mM DTT and 14 U RNasin Ribonuclease Inhibitors in 0.5x PBS. Cells were flash frozen on dry ice after sorting and stored at −80°C.
Single-cell Antibody PCR
Individually sorted antigen experienced CD19+CD27+ B cells from the CSF were lysed, mRNA was reverse-transcribed, and immunoglobulin variable regions were amplified with multiple rounds of PCR as previously described.(32) Sanger sequencing was performed at the UTSWMC (University of Texas Southwestern Medical Center) sequencing core to generate the antibody variable domain reads.
Sequence Analysis
Sequences were analyzed using the VDJServer analysis portal.(33) V, D, J gene calls, FR and CDR regions were annotated using IgBlast v1.14.1(34) with VDJServer IMGT 2019.01.23 germline database. Unproductive antibody rearrangements and truncated sequence reads (did not extend from the beginning of CDR1 to the first two codons of the J gene) were filtered out. CDR3 charge properties and mutational analysis was performed using Alakazam in the Immcantation suite v4.2.0.(35) GraphPad Prism software was used to determine the statistical significance of differences between groups and build graphs for figures. Non-parametric ANOVA was used with a post-hoc analysis to do pairwise comparison of patient groups with the healthy controls using the Dunnett multiple comparison method.(36) Replacement frequency in CDR:FR was analyzed excluding near-zero ratios.
VH2+ Clone Cluster Identification
Of the 31 VH2–70 rearrangements, 28 were identified with a CDR3 motif of VH2–70-N-DH6-N-DH2-N-JH6. Clustering of these rearrangements was defined by the CDR3 amino acid sequence having the same CDR3 length and discordance in 2 or fewer amino acids.
Recombinant human antibody cloning, expression, and isolation
HEK293T cells (ATCC, Manassas, VA) were maintained in HyClone Dulbeccos Modified Eagles Medium (DMEM) (GE Healthcare Life Sciences). All recombinant human antibodies (rhAbs) were transiently transfected into HEK293T cells with the lipid transfection reagent JetPEI (PolyPlus Transfection) as done previously.(37, 38) Supernatants from these cultures were collected on days 3, 5, 7 and 10. The cell pellets were spun down and supernatants were passed through 0.2um filters and subjected to antibody purification on the NGC QUEST 10 system. The concentration of the antibodies were determined by sandwich ELISA as done previously.(37, 38)
Cell-based assay for NMDAR and AQP4 autoreactivity by rhAbs
Autoreactivity for the NMDA receptor (the NR1 subunit)(30, 31) and AQP4 was detected using a commercially-available cell-based assay (Euroimmun, FA 112d-1010–51 and FA 1128-1010-50). Cells were incubated with 20 μg/mL rhAbs in PBS + 0.1% Tween-20 (Sigma-Aldrich) + 1% Goat Serum (Life Technologies) for 30 min in a humidified chamber. After washing with PBS + 0.2% Tween-20, cells were incubated with the provided secondary antibody for 30 min in a humidified chamber. The wash was repeated, then cells were incubated with 1 μg/mL DAPI (Invitrogen) for 5 min. After washing, a coverslip was added to each slide and visualized on a Zeiss LSM780 confocal microscope using a 20x/0.80NA air objective lens. AQP-4 slides were visualized on a Zeiss Axioscope.A1 widefield microscope using a 20x/0.45NA air objective lens. Fluorescence intensity was measured using FIJI (https://fiji.sc/).
Cell-based assay for Hep2 autoreactivity by rhAbs
Hep2 autoreactivity was detected using a commercially-available cell-based assay (Bion Enterprises, ANK-120). Cells were incubated with 20 μg/mL rhAbs in PBS + 0.1% Tween-20 + 1% Goat Serum for 30 min in a humidified chamber. After washing twice with PBS, cells were incubated with the provided secondary antibody for 30 min in a humidified chamber. The washes were repeated, and cells were incubated with 1 μg/mL DAPI in PBS for 5 min. After washing, the slide was coverslipped and visualized on a Zeiss Axioscope.A1 widefield microscope using a 20x/0.45NA air objective lens. Fluorescence intensity was measured through FIJI. Positive staining of a rhAb was defined as fluorescence intensity greater than or equal to the mean fluorescent intensity of the negative control plus 6 standard deviations.
Quantification of autoreactivity by rhAbs
Autoreactivity was quantified using Fiji.(39) Cells were identified by using the Threshold function to detect nuclei and were added to the Region of Interest (ROI) manager. The mean fluorescence intensity (MFI) of positively-stained cells was measured, and the MFI of the background (regions where no cells were detected) was subtracted from this value. The MFI of NR1-expressing cells were normalized to that of control cells that do not express NR1.
RESULTS
We had previously utilized a panel of flow cytometry markers to immune-profile B cell subsets in the CSF of patients with neurodegenerative diseases.(37, 38, 40) Because αNMDAR-AE patients present with high titers of αNMDAR IgG1 antibody in the CSF or blood, we asked whether the B cell subset profile in pediatric αNMDAR-AE patients was different from pediatric OND controls. Indeed, while 43% of B cells in the CSF of pediatric OND controls were antigen-naïve B cells (Figure 1A), 75% of B cells in the CSF of pediatric αNMDAR-AE patients were antigen-experienced B cells including memory B cells, plasmablasts (PBs) and antibody-secreting B cells (Figure 1B). One of the pediatric αNMDAR-AE patients was refractory to treatment and displayed features typical of this diagnosis including serum and CSF binding to NR1 of the NMDAR and reduction of stimulus-evoked NMDAR-mediated synaptic responses in area CA1 of mouse hippocampal slices using extracellular “field” recordings. (Supplemental Figure 1). This treatment refractory pediatric αNMDAR-AE patient case also had a significant expansion of antibody-secreting B cells in the CSF compared to pediatric OND controls (42% vs19%, Figure 1A and 1C) and the pediatric αNMDAR-AE patient cohort (42% vs 15%, Figure 1B and 1C). In addition, the ASC:PB ratio was 14.00 in this treatment refractory pediatric αNMDAR-AE patient case compared to 1.12 in the pediatric OND control cohort and 1.50 in the pediatric αNMDAR-AE patient cohort (Figure 1D).
FIGURE 1. B cell profile in the CSF of pediatric Autoimmune Encephalitis.
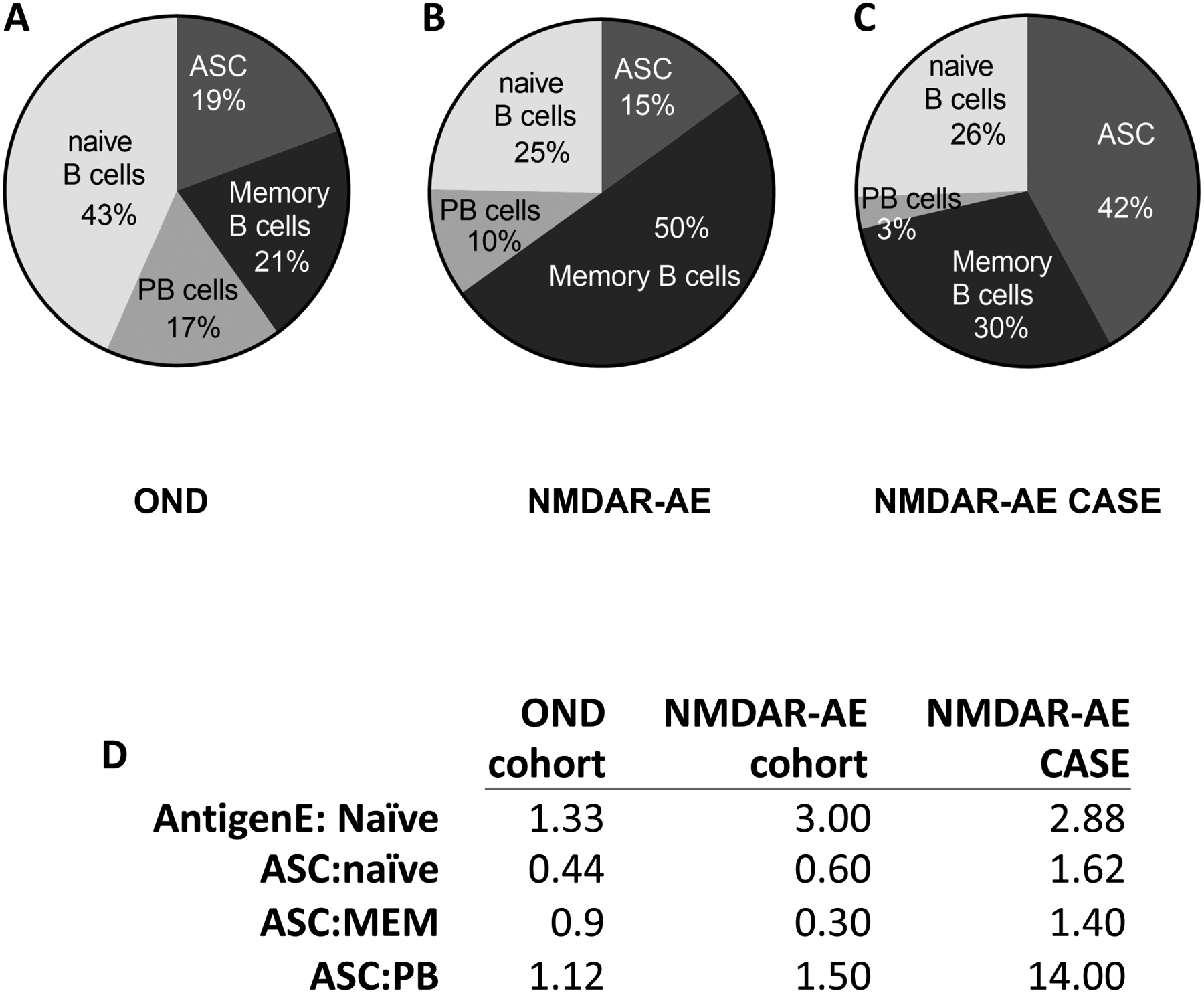
Cerebrospinal fluid (CSF) was collected from pediatric patients with autoimmune encephalomyelitis (AE). Shown are B cell subset frequencies by flow cytometry for (A) pediatric patients who tested negative for antibodies against the NMDAR (OND), (B) pediatric patients who tested positive for antibodies against the NMDA receptor and met diagnostic criteria for NMDAR-AE (NMDAR-AE) and (C) one pediatric NMDAR-AE case refractory to treatment (NMDAR-AE CASE). (D) Ratios of B cell subsets in each group. In D, abbreviations are as follows: AntigenE, Antigen Experienced; ASC, antibody secreting cell; MEM, memory; PB, plasmablast.
To investigate this treatment refractory pediatric αNMDAR-AE patient’s humoral immune response to the NMDAR, we used single-cell PCR to amplify heavy chain genes from antigen-experienced CD19+CD27+ B cells in the CSF. Heavy chain antibody gene usage was analyzed (Figure 2A) and revealed that this subject had an expansion of antibody-experienced B cells in the CSF using variable heavy chain family 2 genes (VH2+) to 39.7% (expected frequency of 9.8%). In fact, 83.3% of VH2+ B cells in the CSF of this patient displayed heavy somatic hypermutation accumulation with an average of 3.6% mutation frequency overall (Figure 2B) and 2.3% replacement frequency overall (Figure 2C) indicating an antigen-driven response. Of note, VH3+ and VH4+ B cells in the CSF of this same patient accumulate codon replacement mutations in the CDRs as a mechanism of increasing antigen-affinity with CDR:FR ratios >2.0 (VH3; 2.6 and VH4; 2.3). VH2+ B cells in the CSF of this patient, however, had a replacement frequency CDR:FR ratio of 0.8 (Figure 2D).
FIGURE 2. Antibody variable heavy chain use dysregulated in pediatric NMDAR-AE.
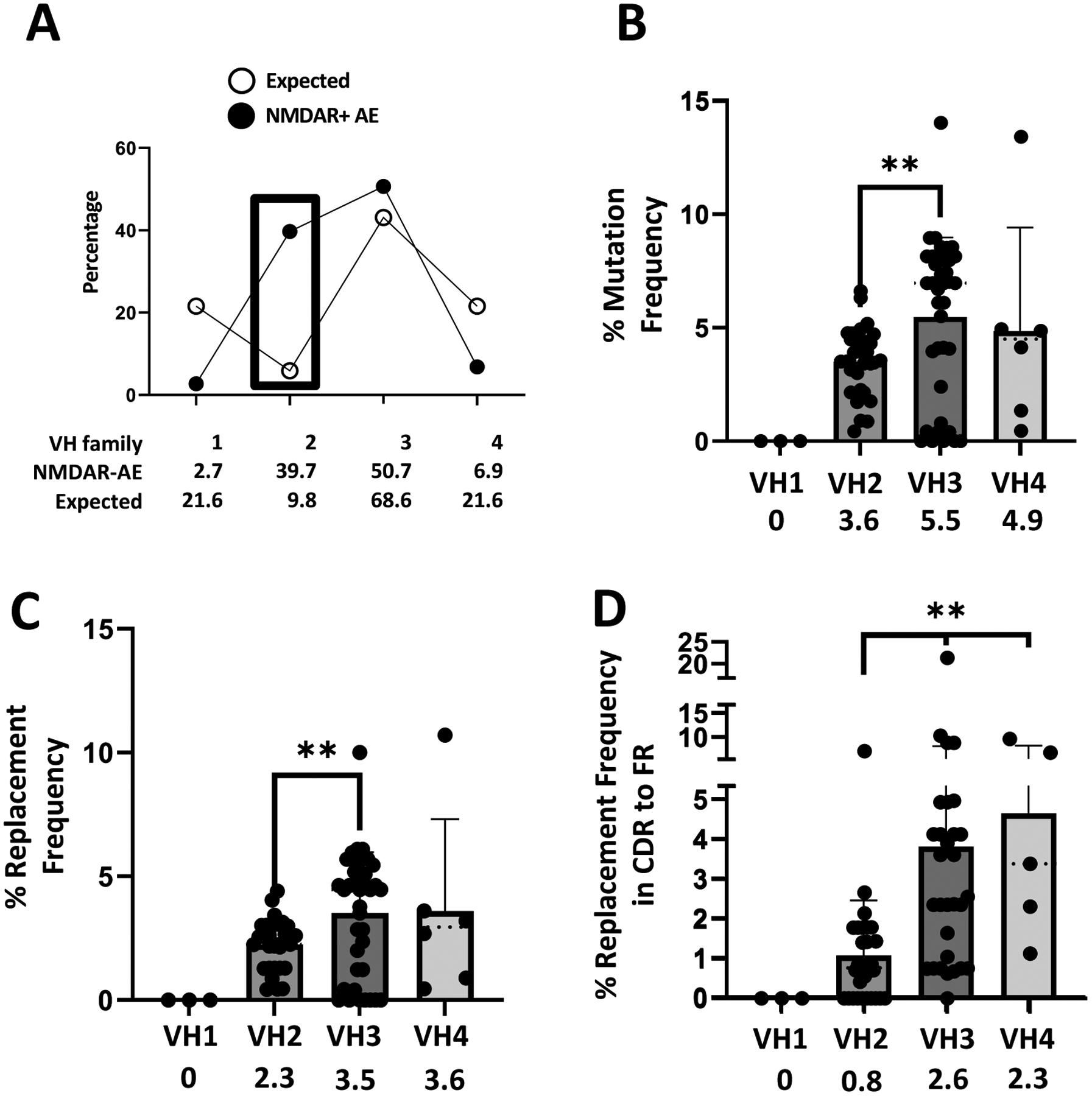
(A) Single B cells from the cerebrospinal fluid of a pediatric NMDAR-AE patient were queried for their antibody variable heavy chain usage and compared to the expected frequency in healthy controls. (B) Mutational frequencies within each variable antibody heavy chain family. (C) Replacement frequencies within each variable antibody heavy chain family. (D) CDR:FR ratio of replacement frequency within each variable antibody heavy chain family.
Further investigation of these thirty-one VH2+ B cells revealed that all 31 used the VH2–70 gene, two DH segments (DH6–13 and DH2–2) and the JH6 gene with a total CDR3 length of sixty-three nucleotides. Clonal clusters were identified for fifteen of these sequences. Six of these clones had three sequences per clone, and four clone clusters had two sequences. We cloned five of the antibodies from CSF-derived B cells of this pediatric αNMDAR-AE patient using the VH2–70/DH6–13/DH2–2/JH6 CDR3 motif. The CDR3 motif of all five VH2+ B cells that we cloned was: AR-X-RG-XQQX-ED-PNXY-XXDV and all five used the VL1–51 light chain. We also cloned five antibodies expressed by non-VH2+ CSF-derived B cells of this same pediatric αNMDAR-AE patient (Table 2).
TABLE 2.
rhAb gene information
| rhAb Name | VH gene | JH gene | CDR3 AA length |
#RM | VL gene | #RM |
|---|---|---|---|---|---|---|
| AG01 | 5–51 | 4 | 30 | 8 | VL2–14 | 8 |
| AG02 | 4–31 | 3 | 54 | 24 | VL2–14 | 8 |
| AG03 | 4–39 | 4 | 57 | 2 | VK2–30 | 1 |
| AG04 | 3–74 | 4 | 42 | 14 | VL1–51 | 2 |
| AG05 | 3–49 | 4 | 30 | 12 | VL2–14 | 8 |
| AG06 | 2–70 | 6 | 63 | 1 | VL1–51 | 2 |
| AG07 | 2–70 | 6 | 63 | 10 | VL1–51 | 3 |
| AG08 | 2–70 | 6 | 63 | 9 | VL1–51 | 1 |
| AG09 | 2–70 | 6 | 63 | 7 | VL1–51 | 5 |
| AG10 | 2–70 | 6 | 63 | 6 | VL1–51 | 1 |
Abbreviations: VH, variable heavy; JH, junction heavy; CDR3, complementarity determining region 3; AA, amino acid; #RM, number of replacement mutations; VL, variable lambda light chain; VK, variable kappa light chain
Binding of these 10 recombinant human antibodies (rhAbs) to the NR1 subunit of the NMDAR is presented in Figure 3 (Panel A, images; Panel B, quantification). Of the 10 rhAbs produced, 5 of them bound to the NR1 subunit of the NMDAR (AG06, AG07, AG08, AG09 and AG10). Of note, all 5 that bound NR1 utilized the VH2–70 heavy chain antibody gene with the AR-X-RG-XQQX-ED-PNXY-XXDV CDR3 motif. None of the 10 rhAbs bound to the control antigen, AQP4 (Supplemental Figure 2) or the control cell line, Hep2 (Supplemental Figure 3). Of note, both the heavy and light chains of the 5 rhAbs that bound NR1 were heavily mutated (Table 2), indicating an antigen-driven response.
FIGURE 3. Binding of recombinant human antibodies to the NR1 subunit of the NMDAR.
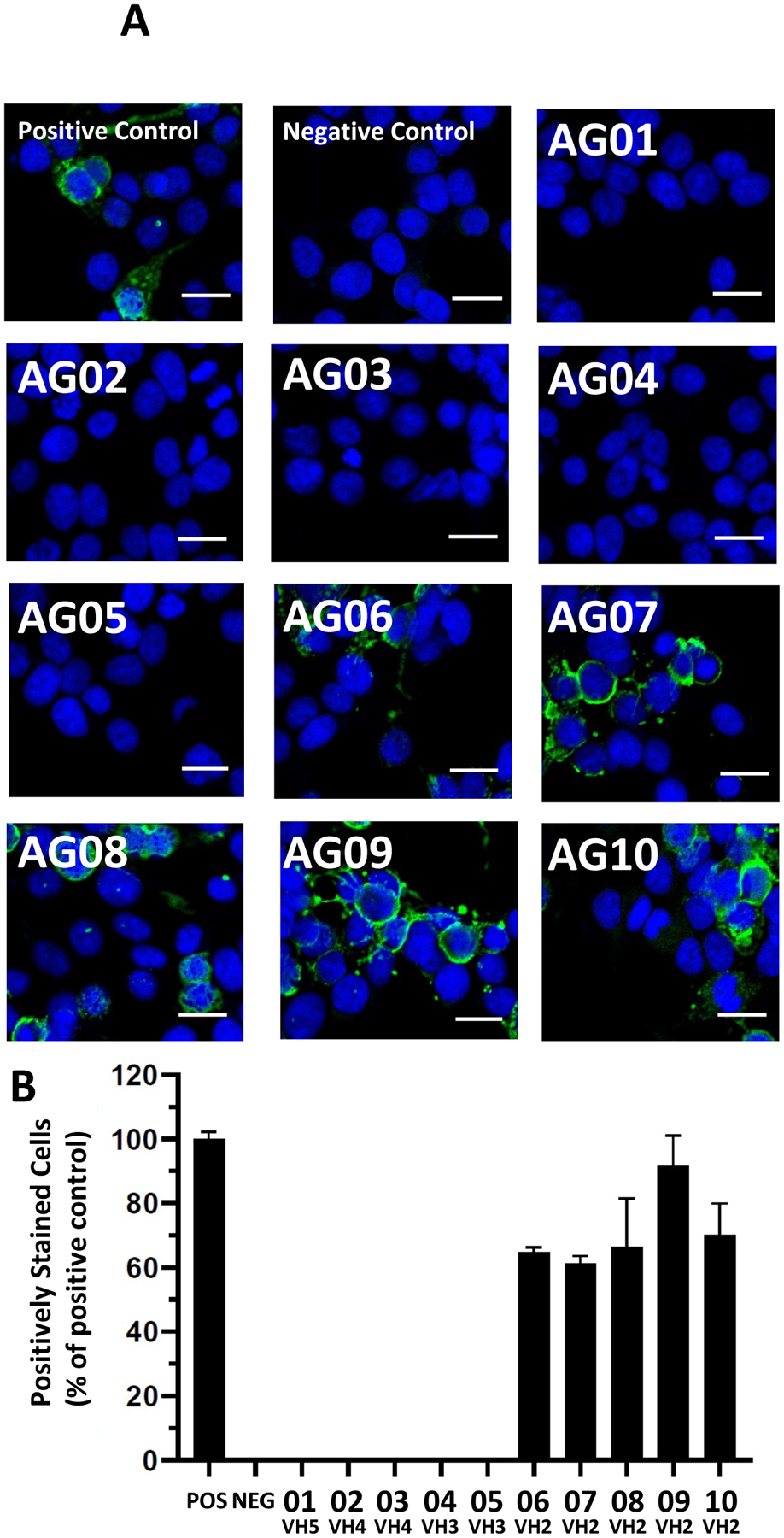
(A) Antibodies expressed by individual B cells from the CSF of a pediatric NMDAR-AE patient refractory to treatment were expressed and tested for binding to the NR1 subunit of the NMDAR using a commercially available kit as described in the methods. Blue: DAPI staining. Green: NMDAR. Scale bar: 20 μm. (B) Frequency of positively-stained cells.
Finally, we sought to determine if the germline configuration of the antibody rearrangement prior to encounter with the driving antigen could also bind the NR1 subunit of the NMDAR. To do this, we identified one clonally related cluster (AG06 and AG09, Figure 4A) and reverted the heavy chain antibody variable genes of these two rhAbs to their germline configuration. AG08 was also reverted to germline configuration because this rearrangement maintained the AR-X-RG-XQQX-ED-PNXY-XXDV CDR3 motif, but did not meet criteria to cluster with any other VH2+ rearrangements due to 4 AA substitutions at the DH2–2 to JH6 junction (Figure 4A). The three reverted rhAbs were designated AG06gL, AG08gL and AG09gL to indicate that the antibody heavy chain no longer contained the somatic hypermutations of their highly mutated counterparts (Figure 4B). These new germline rhAbs (AG06gL, AG08gL and AG09gL) and their highly mutated counterparts (AG06, AG08 and AG09) were tested for binding to a cell line expressing the NR1 subunit of the NMDAR (Figure 5A). The frequency of cells that were positively stained with AG06gL and AG09gL were reduced compared to their highly mutated counterparts, while no cells were positively stained with AG08gL (Figure 5B). The mean fluorescence intensity of AG06gL binding to NR1, however, was similar to AG06, while the mean fluorescence intensity of AG09gL binding to NR1 was 4.4-fold less than AG09 (Figure 5C). AG08gL binding to NR1 was below the detection threshold. Thus, accumulation of somatic hypermutations increases NR1 binding intensity, but NR1 binding by the original antibody rearrangements prior to somatic hypermutation accumulation is frequently detectable. Of note, exact matches of the CDR3 amino acid sequences from these 3 rhAbs are not in the AIRR Data Commons,(41) Immune Epitope Database,(42) or Genbank(43) and CDR3 motif queries are not accommodated on these platforms.
FIGURE 4. Clonal Expansion of VH2+ antigen experienced B cells in the CSF of a pediatric NMDAR-AE patient refractory to treatment.
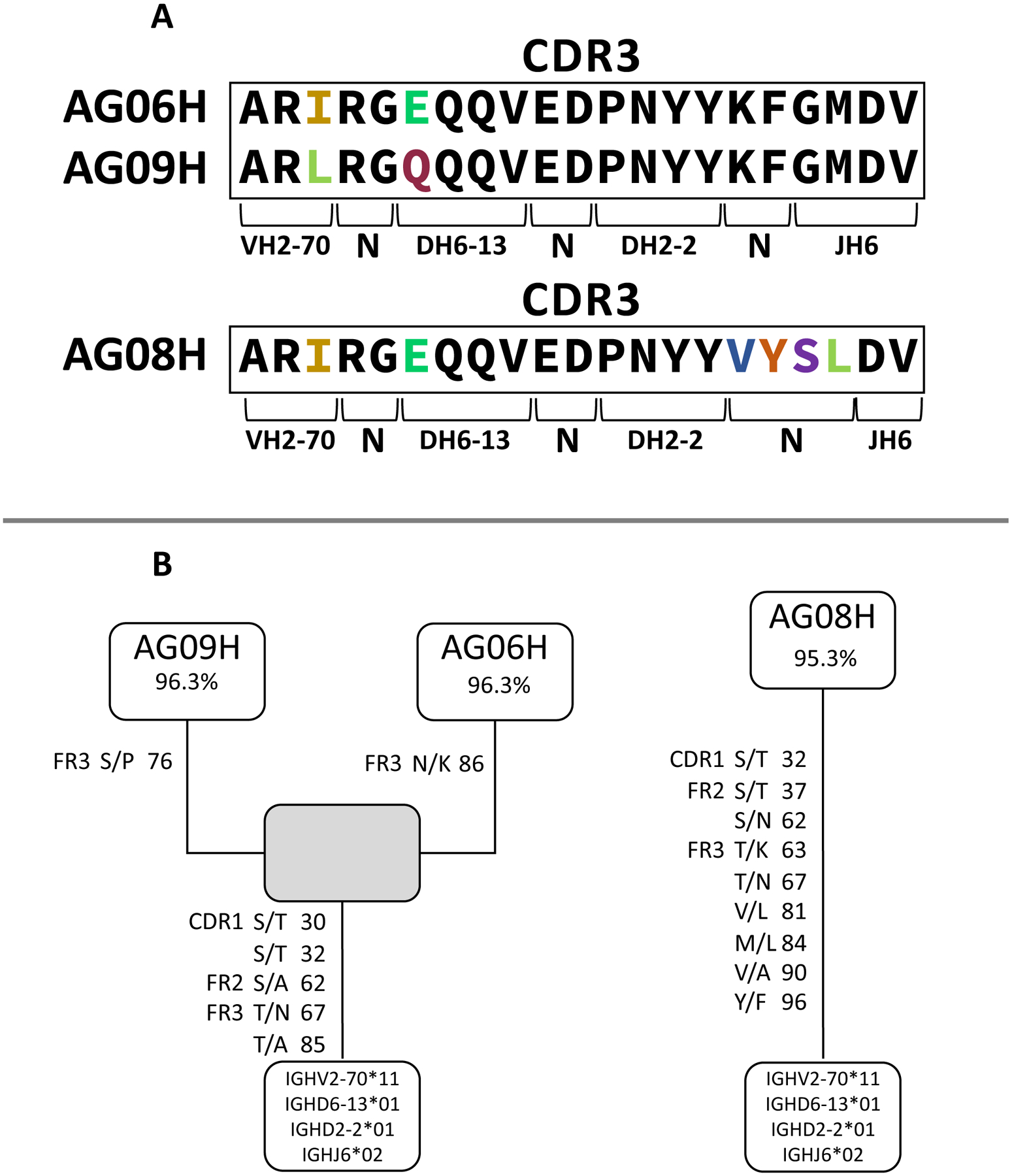
(A) The CDR3 amino acid sequences of AG06, AG08 and AG09 are in the boxes with differences indicated in color. (B) Clonal relationship of AG06, AG08 and AG09 using a Hamming distance of 2. Replacement mutations are indicated by region, codon number and the amino acid replacement. The gray box indicates a common clone member that was not detected in the repertoire. Germline V, D and J genes are indicated at the root of the clone.
FIGURE 5: Comparison of rhAbs to germline configuration.
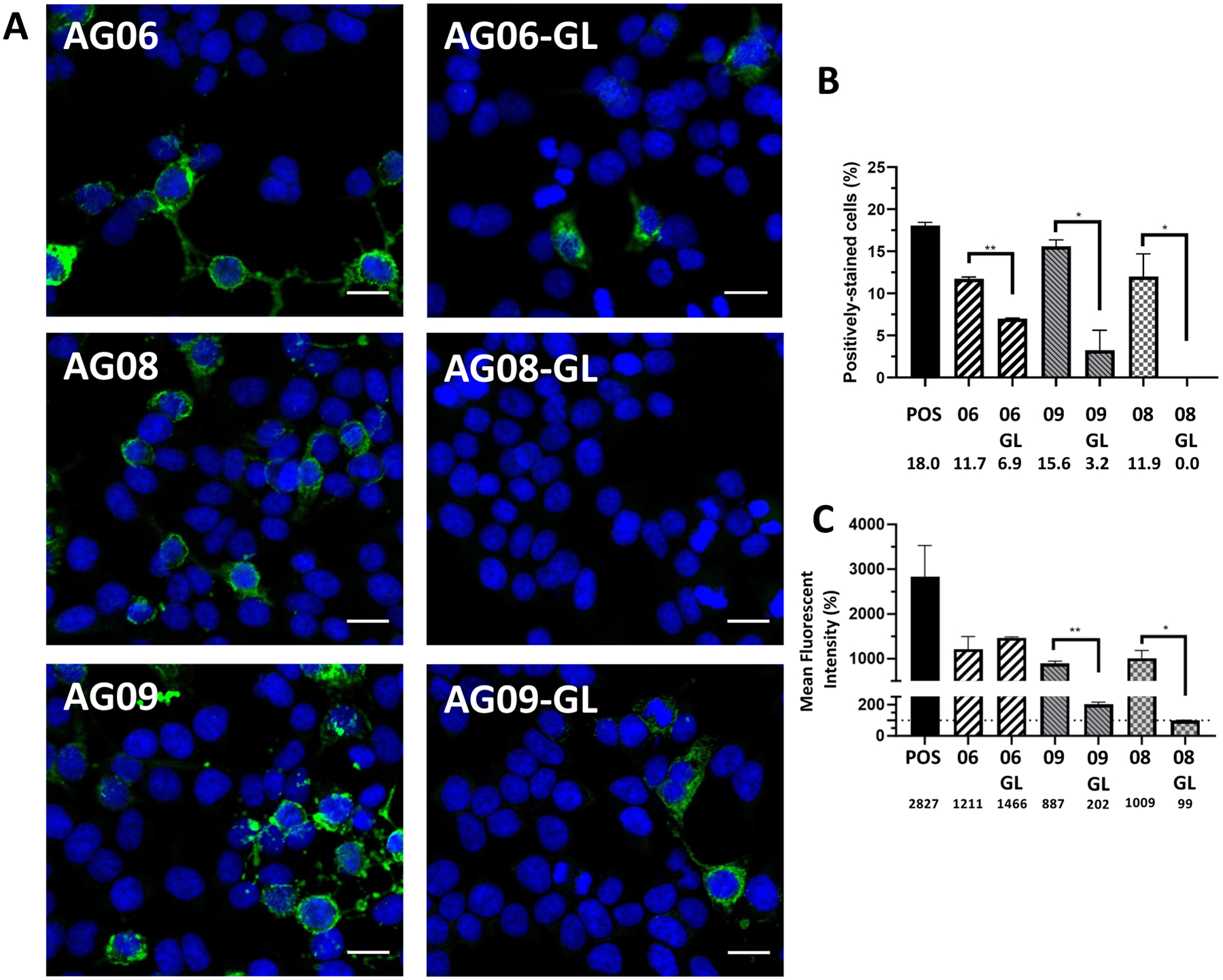
(A) Antibodies expressed by individual B cells from the CSF of a pediatric NMDAR-AE patient refractory to treatment were reverted to their germline configuration and tested for binding to the NR1 subunit of the NMDAR using a commercially available kit as described in the methods. Blue: DAPI staining. Green: NMDAR. Scale bar: 20 μm. (B) Frequency of positively-stained cells. (C) Mean fluorescence intensity of cells. The horizontal dotted line represents the MFI of control cells as the threshold of detection.
DISCUSSION
This study investigates binding to the NR1 subunit of the NMDAR by highly expanded antigen-experienced B cells using the VH2–70 gene isolated from the CSF of a pediatric αNMDAR-AE patient refractory to treatment. Indeed, here we show that only those B cells utilizing the VH2–70 gene bind NR1. This is a novel finding that may be subject-specific but may also be pediatric-specific, as others have not detected any VH2+ antigen-experienced B cells in the CSF of adult αNMDAR-AE cases.(18, 19) In fact, it is evident that certain sub-motifs within the CDR3 gene-sequence of these VH2+ antigen-experienced B cells drive continued NR1 binding in the absence of somatic hypermutation and have not been previously reported in other databases including the current AIRR Data Commons which contains ~1 billion CDR3 AIRR-seq derived sequences.(41)
The first indication of this unexpected finding of VH2+ enrichment was discordance in CSF-derived B cell subtypes between a cohort of pediatric αNMDAR-AE patients and OND controls. Antigen-experienced B cells in the CSF of the pediatric αNMDAR-AE patients were 3-fold greater than what was observed in the pediatric OND controls. In fact, the pediatric OND cohort maintained an expected balance of naïve B cells and antigen-experienced B cells (memory, plasmablasts and antibody-secreting cells). Within the three subtypes of antigen-experienced B cells, we expected an expansion of memory B cells in the pediatric αNMDAR-AE patients since they constitute the B cell pool from which ASCs emerge. Indeed, the memory B cell frequency was increased in the pediatric αNMDAR-AE patients. Expansion of memory B cells, however, did not result in a subsequent increase in the frequency of plasmablasts or ASCs in the pediatric αNMDAR-AE patients, despite sampling in the acute phase of disease in these cases. Of note, others have reported expansion of antibody secreting cells (“plasma cells”) in the CSF of four adult αNMDAR-AE patients compared to 25 adult controls,(19) but this is the first report focused on pediatric αNMDAR-AE patients.
The pediatric αNMDAR-AE case that was refractory to treatment displayed an extensive enrichment of long-lived antibody-secreting cells (ASC’s) in the CSF, such that 42% of all B cells in the CSF were ASCs. This particular B cell subtype (ASC’s) is incredibly difficult to target by anti-CD20 B cell depletion therapy because they do not express CD20.(44, 45) Furthermore, as ASCs are long-lived,(46) they do not require replenishment from the memory B cell pool, and thus depletion of CD20+ memory B cells would not impact ASC frequency. Even steroids would not be a successful therapeutic as ASCs are less focused on proliferation and more focused on antibody production.(46) Antibodies that target CD19,(47, 48) which is expressed at low levels on ASCs,(47, 48) might be successful, if penetrance into CNS could be overcome. Proteasome inhibitors target protein production,(49) and may be useful in the treatment of αNMDAR-AE(50) but one side effect is the development of peripheral neuropathy.(51) Future efforts must be made to identify patients with αNMDAR-AE who demonstrate expansion of ASCs in the CSF and develop therapeutic options targeting this antigen-experienced B cell subtype with careful consideration of current recommendations.(23–25)
Immunogenetic studies revealed an unexpected expansion of VH2+ antigen-experienced B cells in the CSF of the pediatric αNMDAR-AE case that was refractory to treatment. There are only 2 germline genes within the VH2 family(52) and it is seldom used by B cells even at the expected frequency of 9.8%.(53–59) Yet in this treatment-refractory pediatric αNMDAR-AE case, there was a 4-fold increase in VH2 usage over the expected frequency. In the adult αNMDAR-AE studies reported by others,(18, 19) no VH2+ CSF-derived B cells were detected. This raises the possibility that the immunogenetics of pediatric αNMDAR-AE are discordant from adult αNMDAR-AE since essentially the same approaches were used to generate the repertoires. We used somatic hypermutation analyses to investigate whether this expansion of VH2+ B cells was an antigen-driven response.(60, 61) To do this, we calculated the overall mutation-frequency (MF) and replacement-frequency (RF) of the VH2+ antigen-experienced B cells. The data showed that while well above baseline, the MF and RF were significantly reduced compared to VH3+ antigen-experienced B cells in the CSF of this same patient. It should be noted, however, that all of the VH2+ B cells were using the same VH, DH(2) and JH segments; thus, there could be skewing of the MF and RF due to low diversity. Within the VH2+ B cells, there was also a lack of targeting of replacement mutations to the CDRs as evidenced by the low percentage of replacements in CDR versus FR. This is in contrast to VH3+ and VH4+ B cells from this same αNMDAR-AE case, which demonstrated high skewing of replacement mutations towards CDR1 and CDR2 as evidenced by the high percentage of replacements in CDR versus FR. It is thought that this bias towards replacement mutation accumulation in the CDRs generates greater antigen binding diversity and affinity in the CDRs of the antibody while bias against replacement mutation accumulation in the FRs facilitates structural integrity in the FRs of the antibody.(62–65) However, none of these studies featured analysis of human VH2+ antigen-experienced B cells.
Since antigen-experienced B cells from the CSF of the αNMDAR-AE case refractory to treatment were expanded as evaluated by flow cytometry and were highly enriched for VH2 usage with unusual replacement mutation features, we asked whether they bound to the NR1 subunit of the NMDAR. We included 5 VH2+ antigen-experienced B cells, 2 using VH3, 2 using VH4 and 1 using VH5. Others have shown that 5.9% of ASC’s from the CSF of adult αNMDAR-AE patients bind the NR1 subunit of the NMDAR using VH3, VH4 or VH5 antibody genes, but they did not have any candidates to test using VH2.(18, 19) We found that 5 of 5 VH2+ B cells bound the NR1 subunit, but none of those using VH3, VH4 or VH5 genes bound NR1 despite compelling evidence of antigen-driven selection. Since others have not detected VH2+ ASCs in the CSF of adult αNMDAR-AE patients, we suspect that this could be a phenotype of pediatric αNMDAR-AE refractory to treatment. Furthermore, VH2–70+ antigen-experienced B cells in the CSF may be grossly enriched for binding to the NR1 subunit of the NMDAR, which may also explain the near non-existent detection (<1%) of the VH2–70 gene in large healthy human antibody rearrangement datasets.(41, 66, 67) Whether these NR1-binding VH2+ B cells in the CSF matriculated from germinal centers or extrafollicular sites remains unknown, although others have provided evidence that B cell activation in response to NR1 occurs in the cervical lymph nodes.(21) Of note, affinity maturation occurs at both sites.(68) Understanding the role of NR1-binding B cells in refractory cases of pediatric αNMDAR-AE, and the tissue structures from which they matriculate is of critical importance.
We further suspect that this propensity towards NR1-binding by VH2+ antibodies is primarily driven by a CDR3 motif we identified in the NR1-binding VH2+ antibodies. Indeed, it has been well-established that long CDR3 lengths, use of tryptophan and tyrosines, and positively charged residues (arginine, lysine and histidine) can infer autoreactivity.(69–75) The NR1-binding VH2+ antibodies we investigated here all have these features. For example, the CDR3 length of the VH2+ antibodies is 63 nucleotides due to use of the longest J-segment (JH6) and an unusual incorporation of two D-segments in tandem (DH6–13 and DH2–2). All NR1-binding VH2+ antibodies had arginine (R) at positions 2 and 4, and tyrosine (Y) at position 15. AG06, AG09, AG07 and AG08 also had a tyrosine at position 14, and AG10, AG07 and AG08 had a tyrosine at position 17. In addition, the majority of SHM within the NR1-binding VH2+ antibodies were focused in frameworks and by convention do not contribute to antigen binding. Due to these features, we expected that removing the SHM from the NR1-binding VH2+ antibodies would not completely abrogate binding. Indeed, of the 3 NR1-binding VH2+ antibodies we reduced to germline configurations, only AG08gL displayed complete abrogation of positively stained NR1-expressing cells. Of note, AG08 had a considerable deviation from AG06 and AG09 in the DH2–2 to JH6 junction which may have required compensation by SHM to maintain NR1 binding. SHM accumulation in AG06 and AG09 may also support higher affinity binding to NR1 or may be carry over mutations due to high B cell proliferation.(68) It is also possible that NR1 is not the driving antigen as we suggest. For example, others have demonstrated that NR1 antibodies emerge following neurotropic virus infection.(76–79)
We have demonstrated that pediatric αNMDAR-AE is characterized by an expansion of antigen-experienced B cells in the CSF by flow cytometry. Immunogenetics support the notion that VH2 expansion within the antigen-experienced B cell repertoire of the CSF is driven by a gross enrichment of NR1 binding. These VH2+ antibodies also display features of a unique CDR3 motif that may drive initial NR1 reactivity prior to SHM incorporation. These results are limited by the inclusion and sequence data of only one pediatric case of αNMDAR-AE refractory to treatment and examination of antibodies from 5 VH2+ antigen-experienced B cells. Nevertheless, expansion of VH2+ antigen-experienced B cells in the CSF of pediatric αNMDAR-AE may be an indicator of NR1-associated autoimmunity.
Supplementary Material
KEY POINTS.
Antibody Secreting Cell expansion in the CSF of pediatric NMDAR-AE
VH2 overuse in treatment refractory pediatric NMDAR-AE case
Identification of a CDR3 motif of NR1-binding VH2–70+ B cells
ACKNOWLEDGEMENTS
The authors thank the patients and their families who donated samples for our molecular, cellular and immunogenetics studies of pediatric αNMDAR-AE. We thank all the clinical staff at UTSW for their assistance in sample acquisition and transport. The authors also thank the Children’s Medical Research flow core for use of their flow cytometry instrumentation and expertise and the Whole Brain Microscopy Facility for use of their imaging technology.
REDCap was used to archive clinical data associated with samples and was funded by NIH to UTSW (Grant UL1 TR003163). This work was funded by the NIH to NM (National Institute of Neurological Disorders and Stroke award numbers NS098229 and NS102417).
REFERENCES
- 1.Lancaster E 2022. Autoantibody Encephalitis: Presentation, Diagnosis, and Management. J Clin Neurol 18: 373–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nguyen L, and Wang C. 2023. Anti-NMDA Receptor Autoimmune Encephalitis: Diagnosis and Management Strategies. Int J Gen Med 16: 7–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pruss H 2021. Autoantibodies in neurological disease. Nat Rev Immunol 21: 798–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Uy CE, Binks S, and Irani SR. 2021. Autoimmune encephalitis: clinical spectrum and management. Pract Neurol 21: 412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gable MS, Sheriff H, Dalmau J, Tilley DH, and Glaser CA. 2012. The frequency of autoimmune N-methyl-D-aspartate receptor encephalitis surpasses that of individual viral etiologies in young individuals enrolled in the California Encephalitis Project. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 54: 899–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guasp M, and Dalmau J. 2018. Encephalitis associated with antibodies against the NMDA receptor. Med Clin (Barc) 151: 71–79. [DOI] [PubMed] [Google Scholar]
- 7.Moscato EH, Peng X, Jain A, Parsons TD, Dalmau J, and Balice-Gordon RJ. 2014. Acute mechanisms underlying antibody effects in anti-N-methyl-D-aspartate receptor encephalitis. Ann Neurol 76: 108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Makuch M, Wilson R, Al-Diwani A, Varley J, Kienzler AK, Taylor J, Berretta A, Fowler D, Lennox B, Leite MI, Waters P, and Irani SR. 2018. N-methyl-D-aspartate receptor antibody production from germinal center reactions: Therapeutic implications. Ann Neurol 83: 553–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gastaldi M, Waters P, and Vincent A. 2017. Detection of NMDARs Antibodies in Encephalitis. Methods Mol Biol 1677: 117–126. [DOI] [PubMed] [Google Scholar]
- 10.Flanagan EP, Geschwind MD, Lopez-Chiriboga AS, Blackburn KM, Turaga S, Binks S, Zitser J, Gelfand JM, Day GS, Dunham SR, Rodenbeck SJ, Clardy SL, Solomon AJ, Pittock SJ, McKeon A, Dubey D, Zekeridou A, Toledano M, Turner LE, Vernino S, and Irani SR. 2023. Autoimmune Encephalitis Misdiagnosis in Adults. JAMA neurology 80: 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balu R, McCracken L, Lancaster E, Graus F, Dalmau J, and Titulaer MJ. 2019. A score that predicts 1-year functional status in patients with anti-NMDA receptor encephalitis. Neurology 92: e244–e252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nissen MS, Orvik MS, Nilsson AC, Ryding M, Lydolph M, and Blaabjerg M. 2022. NMDA-receptor encephalitis in Denmark from 2009 to 2019: a national cohort study. J Neurol 269: 1618–1630. [DOI] [PubMed] [Google Scholar]
- 13.Zhong R, Chen Q, Zhang X, Zhang H, and Lin W. 2022. Relapses of Anti-NMDAR, Anti-GABABR and Anti-LGI1 Encephalitis: A Retrospective Cohort Study. Frontiers in immunology 13: 918396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zografou C, Vakrakou AG, and Stathopoulos P. 2021. Short- and Long-Lived Autoantibody-Secreting Cells in Autoimmune Neurological Disorders. Frontiers in immunology 12: 686466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Datta AK, Pandit A, Biswas S, Biswas A, Roy BK, and Gangopaddhyay G. 2021. Spectrum of Anti-NMDA Receptor Antibody Encephalitis: Clinical Profile, Management and Outcomes. Ann Indian Acad Neurol 24: 383–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Titulaer MJ, McCracken L, Gabilondo I, Armangue T, Glaser C, Iizuka T, Honig LS, Benseler SM, Kawachi I, Martinez-Hernandez E, Aguilar E, Gresa-Arribas N, Ryan-Florance N, Torrents A, Saiz A, Rosenfeld MR, Balice-Gordon R, Graus F, and Dalmau J. 2013. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 12: 157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hughes EG, Peng X, Gleichman AJ, Lai M, Zhou L, Tsou R, Parsons TD, Lynch DR, Dalmau J, and Balice-Gordon RJ. 2010. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J Neurosci 30: 5866–5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kreye J, Wenke NK, Chayka M, Leubner J, Murugan R, Maier N, Jurek B, Ly LT, Brandl D, Rost BR, Stumpf A, Schulz P, Radbruch H, Hauser AE, Pache F, Meisel A, Harms L, Paul F, Dirnagl U, Garner C, Schmitz D, Wardemann H, and Pruss H. 2016. Human cerebrospinal fluid monoclonal N-methyl-D-aspartate receptor autoantibodies are sufficient for encephalitis pathogenesis. Brain 139: 2641–2652. [DOI] [PubMed] [Google Scholar]
- 19.Malviya M, Barman S, Golombeck KS, Planaguma J, Mannara F, Strutz-Seebohm N, Wrzos C, Demir F, Baksmeier C, Steckel J, Falk KK, Gross CC, Kovac S, Bonte K, Johnen A, Wandinger KP, Martin-Garcia E, Becker AJ, Elger CE, Klocker N, Wiendl H, Meuth SG, Hartung HP, Seebohm G, Leypoldt F, Maldonado R, Stadelmann C, Dalmau J, Melzer N, and Goebels N. 2017. NMDAR encephalitis: passive transfer from man to mouse by a recombinant antibody. Ann Clin Transl Neurol 4: 768–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dean CA, Metzbower SR, Dessain SK, Blanpied TA, and Benavides DR. 2022. Regulation of NMDA Receptor Signaling at Single Synapses by Human Anti-NMDA Receptor Antibodies. Front Mol Neurosci 15: 940005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Al-Diwani A, Theorell J, Damato V, Bull J, McGlashan N, Green E, Kienzler AK, Harrison R, Hassanali T, Campo L, Browne M, Easton A, Soleymani Majd H, Tenaka K, Iorio R, Dale RC, Harrison P, Geddes J, Quested D, Sharp D, Lee ST, Nauen DW, Makuch M, Lennox B, Fowler D, Sheerin F, Waters P, Leite MI, Handel AE, and Irani SR. 2022. Cervical lymph nodes and ovarian teratomas as germinal centres in NMDA receptor-antibody encephalitis. Brain 145: 2742–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang XY, Lei S, Zhang L, Liu X, Lin MT, Blumcke I, Piao YS, Zhou D, and Li JM. 2020. Co-expression of NMDA-receptor subunits NR1, NR2A, and NR2B in dysplastic neurons of teratomas in patients with paraneoplastic NMDA-receptor-encephalitis: a retrospective clinico-pathology study of 159 patients. Acta Neuropathol Commun 8: 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nosadini M, Thomas T, Eyre M, Anlar B, Armangue T, Benseler SM, Cellucci T, Deiva K, Gallentine W, Gombolay G, Gorman MP, Hacohen Y, Jiang Y, Lim BC, Muscal E, Ndondo A, Neuteboom R, Rostasy K, Sakuma H, Sharma S, Tenembaum SN, Van Mater HA, Wells E, Wickstrom R, Yeshokumar AK, Irani SR, Dalmau J, Lim M, and Dale RC. 2021. International Consensus Recommendations for the Treatment of Pediatric NMDAR Antibody Encephalitis. Neurology(R) neuroimmunology & neuroinflammation 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abboud H, Probasco JC, Irani S, Ances B, Benavides DR, Bradshaw M, Christo PP, Dale RC, Fernandez-Fournier M, Flanagan EP, Gadoth A, George P, Grebenciucova E, Jammoul A, Lee ST, Li Y, Matiello M, Morse AM, Rae-Grant A, Rojas G, Rossman I, Schmitt S, Venkatesan A, Vernino S, Pittock SJ, Titulaer MJ, and N. Autoimmune Encephalitis Alliance Clinicians. 2021. Autoimmune encephalitis: proposed best practice recommendations for diagnosis and acute management. J Neurol Neurosurg Psychiatry 92: 757–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abboud H, Probasco J, Irani SR, Ances B, Benavides DR, Bradshaw M, Christo PP, Dale RC, Fernandez-Fournier M, Flanagan EP, Gadoth A, George P, Grebenciucova E, Jammoul A, Lee ST, Li Y, Matiello M, Morse AM, Rae-Grant A, Rojas G, Rossman I, Schmitt S, Venkatesan A, Vernino S, Pittock SJ, Titulaer M, and N. Autoimmune Encephalitis Alliance Clinicians. 2021. Autoimmune encephalitis: proposed recommendations for symptomatic and long-term management. J Neurol Neurosurg Psychiatry 92: 897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wollmuth LP, Chan K, and Groc L. 2021. The diverse and complex modes of action of anti-NMDA receptor autoantibodies. Neuropharmacology 194: 108624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Estrada K, Whelan CW, Zhao F, Bronson P, Handsaker RE, Sun C, Carulli JP, Harris T, Ransohoff RM, McCarroll SA, Day-Williams AG, Greenberg BM, and MacArthur DG. 2018. A whole-genome sequence study identifies genetic risk factors for neuromyelitis optica. Nat Commun 9: 1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J, Bazzi SA, Schmitz F, Tanno H, McDaniel JR, Lee CH, Joshi C, Kim JE, Monson N, Greenberg BM, Hedfalk K, Melamed E, and Ippolito GC. 2021. Molecular Level Characterization of Circulating Aquaporin-4 Antibodies in Neuromyelitis Optica Spectrum Disorder. Neurology(R) neuroimmunology & neuroinflammation 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T, Cortese I, Dale RC, Gelfand JM, Geschwind M, Glaser CA, Honnorat J, Hoftberger R, Iizuka T, Irani SR, Lancaster E, Leypoldt F, Pruss H, Rae-Grant A, Reindl M, Rosenfeld MR, Rostasy K, Saiz A, Venkatesan A, Vincent A, Wandinger KP, Waters P, and Dalmau J. 2016. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol 15: 391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suh-Lailam BB, Haven TR, Copple SS, Knapp D, Jaskowski TD, and Tebo AE. 2013. Anti-NMDA-receptor antibody encephalitis: performance evaluation and laboratory experience with the anti-NMDA-receptor IgG assay. Clin Chim Acta 421: 1–6. [DOI] [PubMed] [Google Scholar]
- 31.Bien CG, Rohleder C, Mueller JK, Bien CI, Koethe D, and Leweke FM. 2021. Neural Autoantibodies in Cerebrospinal Fluid and Serum in Clinical High Risk for Psychosis, First-Episode Psychosis, and Healthy Volunteers. Front Psychiatry 12: 654602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ligocki AJ, Rounds WH, Cameron EM, Harp CT, Frohman EM, Courtney AM, Vernino S, Cowell LG, Greenberg B, and Monson NL. 2013. Expansion of CD27high plasmablasts in transverse myelitis patients that utilize VH4 and JH6 genes and undergo extensive somatic hypermutation. Genes Immun 14: 291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Christley S, Scarborough W, Salinas E, Rounds WH, Toby IT, Fonner JM, Levin MK, Kim M, Mock SA, Jordan C, Ostmeyer J, Buntzman A, Rubelt F, Davila ML, Monson NL, Scheuermann RH, and Cowell LG. 2018. VDJServer: A Cloud-Based Analysis Portal and Data Commons for Immune Repertoire Sequences and Rearrangements. Frontiers in immunology 9: 976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ye J, Ma N, Madden TL, and Ostell JM. 2013. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res 41: W34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gupta NT, Vander Heiden JA, Uduman M, Gadala-Maria D, Yaari G, and Kleinstein SH. 2015. Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics 31: 3356–3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zar J 2010. Biostatistical Analysis.
- 37.AJ L, R. JR, R. WH, AA G, L. M, S. M, L. L, C. D, H. PM, G. D, G. BM, F. EM, W. ES, R. W, M. E, W. CE, S. AM, and M. NL. 2015. A distinct class of antibodies may be an indicator of gray matter autoimmunity in early and established RRMS patients. ASN neuro pii: 1759091415609613. doi: 10.1177/1759091415609613. Print 2015 Sep-Oct. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rivas JR, Ireland SJ, Chkheidze R, Rounds WH, Lim J, Johnson J, Ramirez DM, Ligocki AJ, Chen D, Guzman AA, Woodhall M, Wilson PC, Meffre E, White C 3rd, Greenberg BM, Waters P, Cowell LG, Stowe AM, and Monson NL. 2017. Peripheral VH4+ plasmablasts demonstrate autoreactive B cell expansion toward brain antigens in early multiple sclerosis patients. Acta Neuropathol 133: 43–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, and Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Joshi C, Sivaprakasam K, Christley S, Ireland S, Rivas J, Zhang W, Sader D, Logan R, Lambracht-Washington D, Rosenberg R, Cullum M, Hitt B, Li QZ, Barber R, Greenberg B, Cowell L, Zhang R, Stowe A, Huebinger R, Kelley B, and Monson N. 2022. CSF-Derived CD4(+) T-Cell Diversity Is Reduced in Patients With Alzheimer Clinical Syndrome. Neurology(R) neuroimmunology & neuroinflammation 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Christley S, Aguiar A, Blanck G, Breden F, Bukhari SAC, Busse CE, Jaglale J, Harikrishnan SL, Laserson U, Peters B, Rocha A, Schramm CA, Taylor S, Vander Heiden JA, Zimonja B, Watson CT, Corrie B, and Cowell LG. 2020. The ADC API: A Web API for the Programmatic Query of the AIRR Data Commons. Front Big Data 3: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR, Wheeler DK, Sette A, and Peters B. 2019. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res 47: D339–D343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, and Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sanz I 2014. Rationale for B cell targeting in SLE. Semin Immunopathol 36: 365–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen D, Gallagher S, Monson NL, Herbst R, and Wang Y. 2016. Inebilizumab, a B Cell-Depleting Anti-CD19 Antibody for the Treatment of Autoimmune Neurological Diseases: Insights from Preclinical Studies. J Clin Med 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nutt SL, Hodgkin PD, Tarlinton DM, and Corcoran LM. 2015. The generation of antibody-secreting plasma cells. Nat Rev Immunol 15: 160–171. [DOI] [PubMed] [Google Scholar]
- 47.Bluml S, McKeever K, Ettinger R, Smolen J, and Herbst R. 2013. B-cell targeted therapeutics in clinical development. Arthritis Res Ther 15 Suppl 1: S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mei HE, Schmidt S, and Dorner T. 2012. Rationale of anti-CD19 immunotherapy: an option to target autoreactive plasma cells in autoimmunity. Arthritis Res Ther 14 Suppl 5: S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dinoto A, Cheli M, Bratina A, Sartori A, and Manganotti P. 2021. Bortezomib in anti-N-Methyl-d-Aspartate-Receptor (NMDA-R) encephalitis: A systematic review. J Neuroimmunol 356: 577586. [DOI] [PubMed] [Google Scholar]
- 50.Scheibe F, Pruss H, Mengel AM, Kohler S, Numann A, Kohnlein M, Ruprecht K, Alexander T, Hiepe F, and Meisel A. 2017. Bortezomib for treatment of therapy-refractory anti-NMDA receptor encephalitis. Neurology 88: 366–370. [DOI] [PubMed] [Google Scholar]
- 51.Yamamoto S, and Egashira N. 2021. Pathological Mechanisms of Bortezomib-Induced Peripheral Neuropathy. Int J Mol Sci 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cook GP, Tomlinson IM, Walter G, Riethman H, Carter NP, Buluwela L, Winter G, and Rabbitts TH. 1994. A map of the human immunoglobulin VH locus completed by analysis of the telomeric region of chromosome 14q. Nat Genet 7: 162–168. [DOI] [PubMed] [Google Scholar]
- 53.Bennett JL, Haubold K, Ritchie AM, Edwards SJ, Burgoon M, Shearer AJ, Gilden DH, and Owens GP. 2008. CSF IgG heavy-chain bias in patients at the time of a clinically isolated syndrome. J Neuroimmunol 199: 126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Campbell MJ, Zelenetz AD, Levy S, and Levy R. 1992. Use of family specific leader region primers for PCR amplification of the human heavy chain variable region gene repertoire. Mol Immunol 29: 193–203. [DOI] [PubMed] [Google Scholar]
- 55.Hansen A, Jacobi A, Pruss A, Kaufmann O, Scholze J, Lipsky PE, and Dorner T. 2003. Comparison of immunoglobulin heavy chain rearrangements between peripheral and glandular B cells in a patient with primary Sjogren’s syndrome. Scand J Immunol 57: 470–479. [DOI] [PubMed] [Google Scholar]
- 56.Rouziere AS, Kneitz C, Palanichamy A, Dorner T, and Tony HP. 2005. Regeneration of the immunoglobulin heavy-chain repertoire after transient B-cell depletion with an anti-CD20 antibody. Arthritis Res Ther 7: R714–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Walter MA, Gibson WT, Ebers GC, and Cox DW. 1991. Susceptibility to multiple sclerosis is associated with the proximal immunoglobulin heavy chain variable region. J Clin Invest 87: 1266–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brezinschek HP, Foster SJ, Brezinschek RI, Dorner T, Domiati-Saad R, and Lipsky PE. 1997. Analysis of the human VH gene repertoire. Differential effects of selection and somatic hypermutation on human peripheral CD5(+)/IgM+ and CD5(−)/IgM+ B cells. J Clin Invest 99: 2488–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aguilera I, Melero J, Nunez-Roldan A, and Sanchez B. 2001. Molecular structure of eight human autoreactive monoclonal antibodies. Immunology 102: 273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harp C, Lee J, Lambracht-Washington D, Cameron E, Olsen G, Frohman E, Racke M, and Monson N. 2007. Cerebrospinal fluid B cells from multiple sclerosis patients are subject to normal germinal center selection. J Neuroimmunol 183: 189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dorner T, Foster SJ, Brezinschek HP, and Lipsky PE. 1998. Analysis of the targeting of the hypermutational machinery and the impact of subsequent selection on the distribution of nucleotide changes in human VHDJH rearrangements. Immunol Rev 162: 161–171. [DOI] [PubMed] [Google Scholar]
- 62.Yaari G, Benichou JI, Vander Heiden JA, Kleinstein SH, and Louzoun Y. 2015. The mutation patterns in B-cell immunoglobulin receptors reflect the influence of selection acting at multiple time-scales. Philos Trans R Soc Lond B Biol Sci 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shlomchik MJ, Marshak-Rothstein A, Wolfowicz CB, Rothstein TL, and Weigert MG. 1987. The role of clonal selection and somatic mutation in autoimmunity. Nature 328: 805–811. [DOI] [PubMed] [Google Scholar]
- 64.Schramm CA, and Douek DC. 2018. Beyond Hot Spots: Biases in Antibody Somatic Hypermutation and Implications for Vaccine Design. Frontiers in immunology 9: 1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jolly CJ, Wagner SD, Rada C, Klix N, Milstein C, and Neuberger MS. 1996. The targeting of somatic hypermutation. Semin Immunol 8: 159–168. [DOI] [PubMed] [Google Scholar]
- 66.DeWitt WS, Lindau P, Snyder TM, Sherwood AM, Vignali M, Carlson CS, Greenberg PD, Duerkopp N, Emerson RO, and Robins HS. 2016. A Public Database of Memory and Naive B-Cell Receptor Sequences. PLoS One 11: e0160853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Briney B, Inderbitzin A, Joyce C, and Burton DR. 2019. Commonality despite exceptional diversity in the baseline human antibody repertoire. Nature 566: 393–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Elsner RA, and Shlomchik MJ. 2020. Germinal Center and Extrafollicular B Cell Responses in Vaccination, Immunity, and Autoimmunity. Immunity 53: 1136–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Crouzier R, Martin T, and Pasquali JL. 1995. Heavy chain variable region, light chain variable region, and heavy chain CDR3 influences on the mono- and polyreactivity and on the affinity of human monoclonal rheumatoid factors. J Immunol 154: 4526–4535. [PubMed] [Google Scholar]
- 70.Ichiyoshi Y, and Casali P. 1994. Analysis of the structural correlates for antibody polyreactivity by multiple reassortments of chimeric human immunoglobulin heavy and light chain V segments. J Exp Med 180: 885–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Luning Prak ET, Monestier M, and Eisenberg RA. 2011. B cell receptor editing in tolerance and autoimmunity. Ann N Y Acad Sci 1217: 96–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Martin T, Crouzier R, Weber JC, Kipps TJ, and Pasquali JL. 1994. Structure-function studies on a polyreactive (natural) autoantibody. Polyreactivity is dependent on somatically generated sequences in the third complementarity-determining region of the antibody heavy chain. J Immunol 152: 5988–5996. [PubMed] [Google Scholar]
- 73.Mian IS, Bradwell AR, and Olson AJ. 1991. Structure, function and properties of antibody binding sites. J Mol Biol 217: 133–151. [DOI] [PubMed] [Google Scholar]
- 74.Radic MZ, Mackle J, Erikson J, Mol C, Anderson WF, and Weigert M. 1993. Residues that mediate DNA binding of autoimmune antibodies. J Immunol 150: 4966–4977. [PubMed] [Google Scholar]
- 75.Zhang Z, Burrows PD, and Cooper MD. 2004. The molecular basis and biological significance of VH replacement. Immunol Rev 197: 231–242. [DOI] [PubMed] [Google Scholar]
- 76.Schabitz WR, Rogalewski A, Hagemeister C, and Bien CG. 2014. VZV brainstem encephalitis triggers NMDA receptor immunoreaction. Neurology 83: 2309–2311. [DOI] [PubMed] [Google Scholar]
- 77.Pruss H, Finke C, Holtje M, Hofmann J, Klingbeil C, Probst C, Borowski K, Ahnert-Hilger G, Harms L, Schwab JM, Ploner CJ, Komorowski L, Stoecker W, Dalmau J, and Wandinger KP. 2012. N-methyl-D-aspartate receptor antibodies in herpes simplex encephalitis. Ann Neurol 72: 902–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Armangue T, Leypoldt F, Malaga I, Raspall-Chaure M, Marti I, Nichter C, Pugh J, Vicente-Rasoamalala M, Lafuente-Hidalgo M, Macaya A, Ke M, Titulaer MJ, Hoftberger R, Sheriff H, Glaser C, and Dalmau J. 2014. Herpes simplex virus encephalitis is a trigger of brain autoimmunity. Ann Neurol 75: 317–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Alexopoulos H, and Dalakas MC. 2019. The immunobiology of autoimmune encephalitides. J Autoimmun 104: 102339. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.