

Abstract
Spectrins are cytoskeletal proteins that are expressed ubiquitously in the mammalian nervous system. Pathogenic variants in SPTAN1, SPTBN1, SPTBN2 and SPTBN4, four of the six genes encoding neuronal spectrins, cause neurological disorders. Despite their structural similarity and shared role as molecular organizers at the cell membrane, spectrins vary in expression, subcellular localization and specialization in neurons, and this variation partly underlies non-overlapping disease presentations across spectrinopathies. Here, we summarize recent progress in discerning the local and long-range organization and diverse functions of neuronal spectrins. We provide an overview of functional studies using mouse models, which, together with growing human genetic and clinical data, are helping to illuminate the aetiology of neurological spectrinopathies. These approaches are all critical on the path to plausible therapeutic solutions.
Introduction
Neuronal spectrins coordinate the positioning and stabilization of multifunctional nanodomains and microdomains of ion channels, cell adhesion molecules, membrane transporters and scaffolding proteins1–3. Together with actin, spectrin forms a submembrane cytoskeleton thought to impart mechanical resilience to neuronal processes and mediate signalling events4. Additionally, spectrins promote vesicle and organelle transport2. Given the multifaceted roles of spectrins in neurons, it is not surprising that pathogenic variants in spectrin genes lead to neurodevelopmental disorders. Clinical variants in four (SPTAN1, SPTBN1, SPTBN2 and SPTBN4) of the six spectrin genes expressed in the nervous system have been genetically and functionally linked to neurological disorders whose clinical presentations include intellectual disability (ID), developmental delay (DD), seizures, movement disorders and behavioural abnormalities (Table 1). The advent of accessible whole-exon sequencing for clinical diagnosis has enabled the identification of genes and variants underlying new neurological spectrinopathies and measurably expanded the list of individuals affected by these rare disorders.
Table 1 |.
Disorders associated with spectrin gene variants
| Gene | Protein | Variant type | Inheritance | Common clinical diagnoses | OMIM entry |
|---|---|---|---|---|---|
| SPTAN1 | αII-Spectrin | Missense, nonsense, in-frame insertions, in-frame deletions, duplications, frameshifts | De novo, inherited | Cerebellar ataxia, DD, DEE-5, ID, neuropathy, SP | 182810 |
| SPTBN1 | βII-Spectrin | Missense, nonsense, in-frame deletions, frameshifts, splice site | De novo | ASD, ADHD, DDISBA, dysmorphisms, hypertonia or hypotonia, hearing impairments, ID, S | 182790 |
| SPTBN2 | βIII-Spectrin | Missense, nonsense, in-frame deletion | De novo, inherited | Autosomal dominant spinocerebellar ataxia 5 (SCA5) | 600224 |
| Spinocerebellar ataxia, autosomal recessive 14 (SCAR14) | 615386 | ||||
| Cerebellar ataxia, DD, ID | –a | ||||
| SPTBN4 | βIV-Spectrin | Missense, nonsense, in-frame deletion, frameshifts, canonical splice site | Inherited | Choreoathetosis, DD, ID, NEDHND, nystagmus, S | 617519 |
ADHD, attention deficit hyperactivity disorder; ASD, autism spectrum disorder; DD, developmental delay; DDISBA, developmental delay, impaired speech and behavioural abnormalities; DEE, developmental and epileptic encephalopathy; ID, intellectual disability; NEDHND, neurodevelopmental disorder with congenital hypotonia, neuropathy and deafness; S, infantile seizures and epilepsy; SP, spasticity, paraplegia or quadriplegia; OMIM, Online Mendelian Inheritance in Man (https://www.omim.org).
No OMIM record.
Spectrinopathies of the nervous system diverge in their primary clinical diagnoses but overlap in their syndromic presentations, underscoring both the functional similarities and the distinct cellular and subcellular specializations of spectrins (Fig. 1). Spectrins are broadly expressed in the nervous system and form elongated, rod-like polypeptides directly coupled to the actin cytoskeleton to form remarkably regular networks that line the cell membrane spanning the neuron. These arrays form by assembling a basic motif that comprises α-spectrin and β-spectrin heterodimers, which then form head-to-head tetramers that crosslink F-actin (Fig. 2). This meshwork integrates into the cytosolic side of the plasma membrane by direct association with membrane lipids and ankyrins1,2 (Fig. 2a). These structural hubs bring together other molecular partners, whose specific identities determine local function, and their disruption drives the underlying pathophysiology of the different spectrinopathies.
Fig. 1 |. Cellular localization and organization of neuronal spectrins.
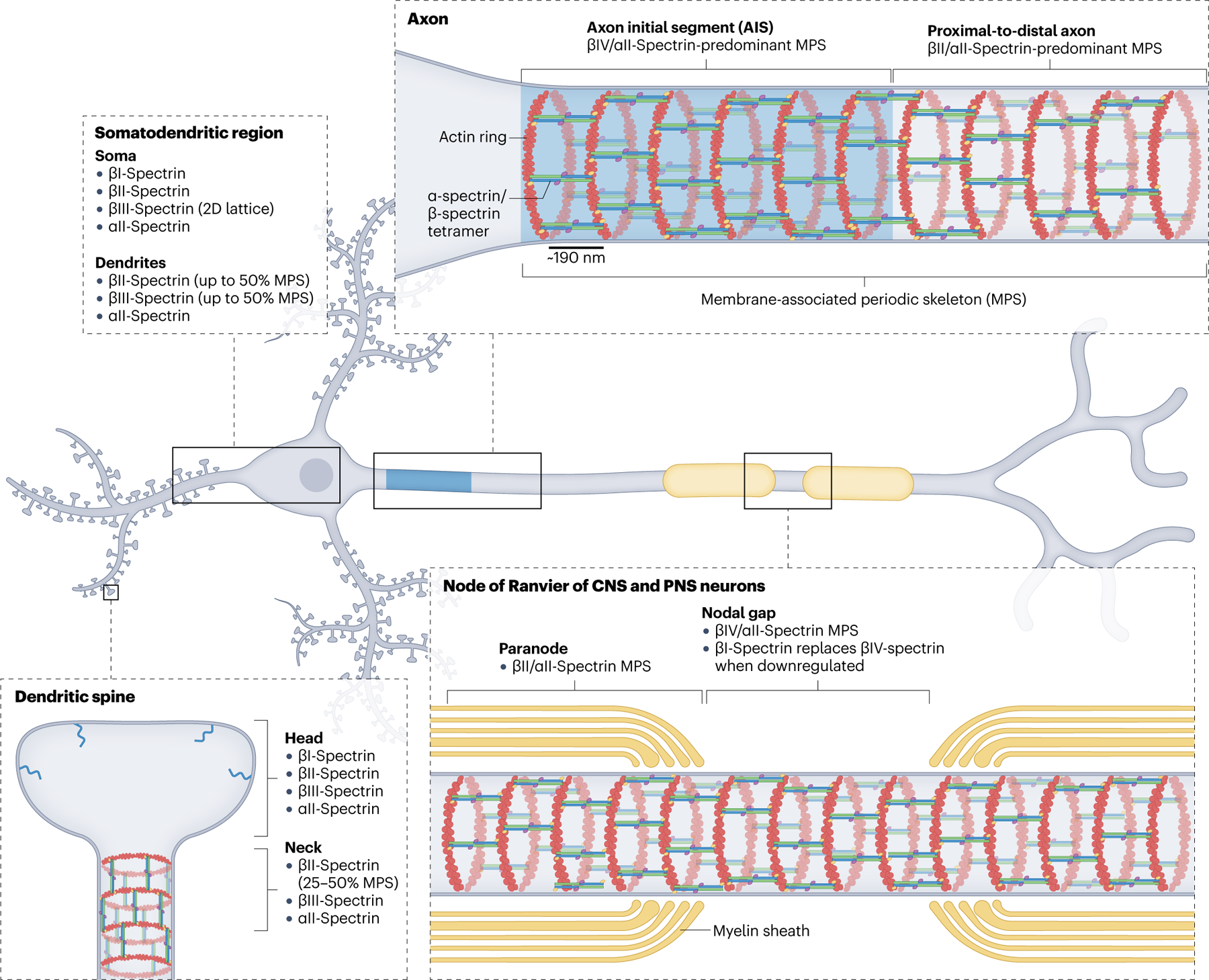
Mammalian neurons express six of the seven spectrins, which follow a general pattern of domain localization and organization across different neuron types. βIV-spectrin is enriched at the axon initial segment (AIS), where, together with αII-spectrin, actin and other key molecules, it forms a membrane-associated periodic skeleton (MPS)34. The MPS is best characterized by actin rings enwrapping the circumference of neuronal processes with a ~190 nm periodicity, which is determined by their cross-linking by spectrin tetramers in their fully elongated conformation. The proximal-to-distal axon, including axonal branches, expresses βII-spectrin and αII-spectrin in high abundance together with relatively less abundant βIII-spectrin, all integrated into the MPS that spans the full axon. In myelinated axons of both the CNS and the PNS, βIV/αII-spectrins are localized and periodically organized in the nodal gap of nodes of Ranvier (NoR), flanked by βII/αII-spectrins in the paranode, also periodically distributed60. Upon loss of βIV-spectrin, βI-spectrin localizes to NoR and rescues βIV-spectrin function. This redundancy is not available at the AIS, probably because βI-spectrin localization depends on its molecular partner ankyrin-R, which is not recruited to the AIS59. Unlike in the axon, the probability of detecting the MPS in dendritic shafts of mature neurons, which includes βII-spectrin and βIII-spectrin, is about 50%33. In addition to the quasi-1D organization of the MPS, βIII-spectrin can form 2D polygonal lattices in the soma and dendritic shaft. In dendritic spines, βII-spectrin and βIII-spectrin adopt MPS periodicity in the neck, but not in the head.
Fig. 2 |. Tetrameric assembly and structural domains of neuronal spectrins.
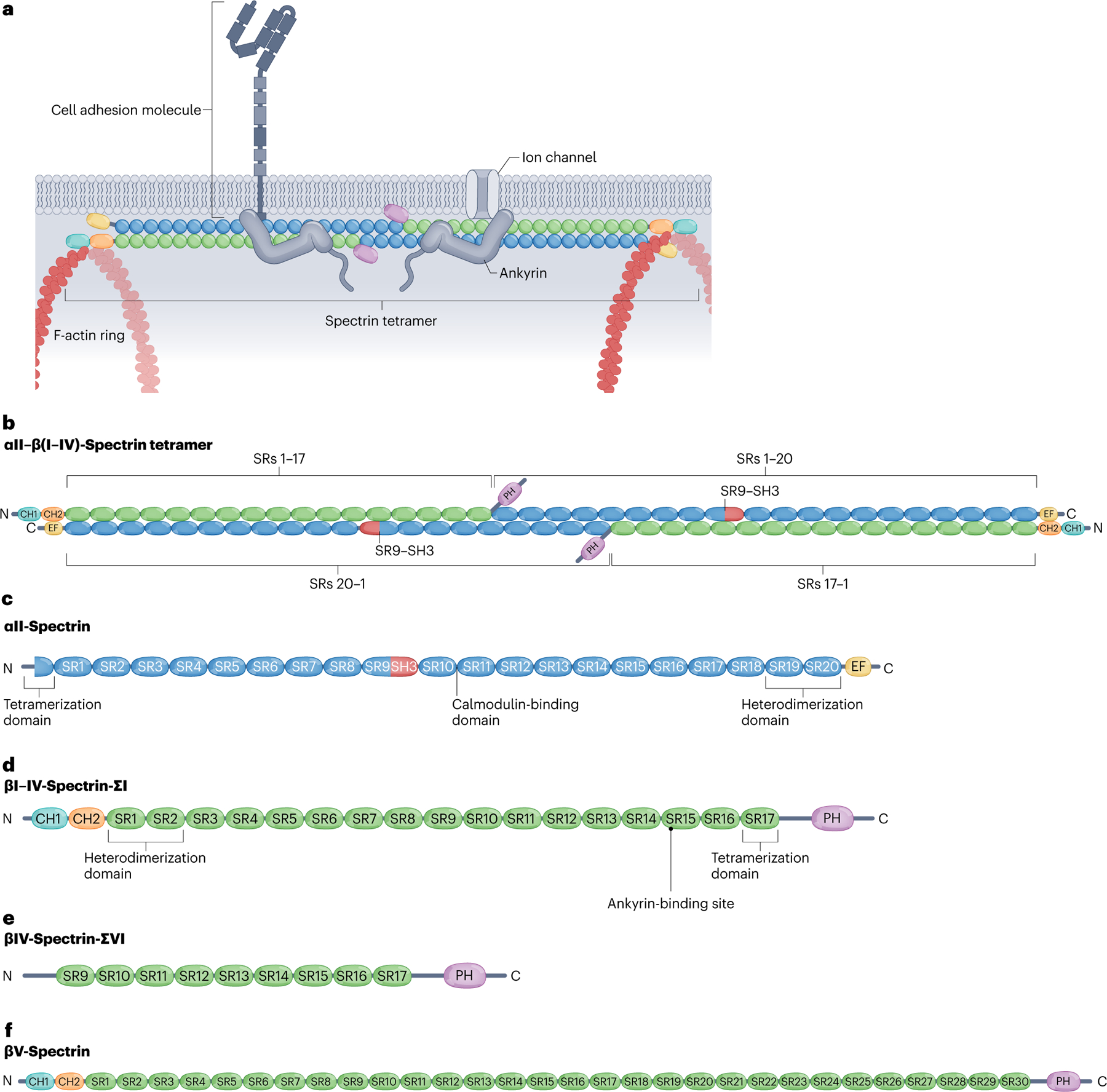
a, Canonical spectrins form heterotetramers of two α-units and two β-units that crosslink F-actin rings along the neuronal membrane. Spectrins bind ankyrins, which in turn stabilize membrane-spanning proteins such as cell adhesion molecules and ion channels. b, Spectrin tetramers assemble by linking heterodimers head-to-head via non-covalent association between the partial spectrin repeats (SRs) in the N terminus adjacent to SR1 in the α-spectrin subunits (blue) and partial SR17 at the N terminus of the β-spectrin subunits (green). Complementary motifs in SR1 and SR2 of βI–IV spectrins and SR19 and SR20 of αII-spectrin bind covalently to enable the antiparallel lateral assembly of α–β-spectrin heterodimers. c, αII-Spectrin spans 20 modular SRs (blue), a calcium-binding EF hand domain (yellow) close to the C terminus, an Src-homology 3 (SH3) domain (red) in SR9 and a calmodulin (CaM)-binding loop in SR10. d, Canonical βI–βIV-spectrins contain 16 full SRs and a partial 17th SR (green), two N-terminal tandem calponin homology (CH) domains (teal and orange), an ankyrin-binding site in SR15 and a C-terminal pleckstrin homology (PH) domain (purple). The CH domains enable binding to actin and the PH domain binds membrane lipids. e, The alternatively spliced βIV-spectrin-ΣVI isoform, which is important for maintenance of the axon initial segment (AIS), lacks the CH domains and the first eight full SRs, but retains ankyrin-binding activity. f, Giant βV-spectrin contains 29 full SRs plus a partial 30th SR. Whether βV-spectrin associates with αII-spectrin is not clear.
In this Review, we describe the function of spectrins in mammalian neurons and summarize recent advances in delineating their cell-type-specific and neuronal-domain-specific localization and functional specialization. We examine how impaired expression and pathogenic variants in spectrin genes lead to altered protein function, the physiological and behavioural consequences of these changes in mouse models, and their relationship to clinical presentations in humans. Lastly, we discuss potential mechanistic overlap across spectrinopathies of the nervous system and future directions that may inform the rational design of therapeutic approaches.
Spectrins in neuron architecture and function
The basic set of a single α-spectrin, one giant β-spectrin and one canonical β-spectrin with ankyrin-binding activity (Fig. 2) already present in bilaterians expanded markedly in vertebrates through whole-genome duplication events3. Non-mammalian vertebrates express four β-spectrin genes. SPTB encodes βI-spectrin, the red blood cell β-spectrin5,6 also found in neurons; SPTBN1 encodes βII-spectrin, first characterized in the brain7,8 and expressed in all tissues; SPTBN4 encodes βIV-spectrin, found in the nervous system, pancreatic islets9 and cardiomyocytes10; and SPTBN5 encodes the giant βV-spectrin, which lacks the ankyrin-binding sequence and is expressed at modest levels in the cerebellum and in auditory hair and photoreceptor cells11,12 (Fig. 2d,f). Mammals also express βIII-spectrin, encoded by SPTBN2, which was first identified in the brain13,14 (Fig. 2d) and detected at high levels in the pancreas, kidney, reproductive tissues and skin. In addition, vertebrates express αII-spectrin, which is encoded by SPTAN1 and associates ubiquitously with β-spectrins15 (Fig. 2c); and αI-spectrin, encoded by SPTA1, which is found exclusively in mammalian erythrocytes16. Alternative splicing expands the spectrin complement17. For example, βIV-spectrin is also expressed as isoforms lacking portions of the N or C termini9,18 (Fig. 2e).
Molecular architecture of spectrins
Spectrins are elongated molecules formed by in-tandem spectrin repeats (SRs), each containing 99–114 residues and extending approximately 100 nm in length. Crystal structures show that SRs adopt a left-handed, anti-parallel, three-helix coiled-coil topology, and are connected by short α-helical linkers19. Canonical βI–βIV-spectrins contain 16 full SRs and a partial 17th SR20, two N-terminal tandem calponin homology (CH) domains, an ankyrin-binding site in SRs 14 and 15, and a C-terminal pleckstrin homology (PH) domain (Fig. 2d). βIV-spectrin has an additional sequence between the final SR and the PH domain9 (Fig. 2d). Giant βV-spectrin contains 29 full SRs plus a partial 30th SR11 (Fig. 2f). By contrast, αII-spectrin, the obligatory partner of neuronal β-spectrins, contains 20 SRs, an Src-homology 3 (SH3) domain in SR9, a calmodulin (CaM)-binding loop in SR10 and calcium-binding EF hand domains near the C terminus (Fig. 2c). Complementary motifs in SRs 1 and 2 of βI–βIV-spectrins and SRs 20 and 21 of αII-spectrin enable the antiparallel lateral assembly of α–β-spectrin heterodimers21 (Fig. 2b). Spectrin dimers assemble into tetramers via head-to-head non-covalent association between partial repeats in each α-spectrin and β-spectrin subunit (Fig. 2b–d). Atomic force microscopy studies show that the tertiary structure of SRs imparts elasticity to the molecules22. This spring-like property facilitates elastic recovery of spectrin molecules when subjected to shear stress during circulation in erythrocytes23 or mechanical tension in axons during growth and fasciculation24,25, and offers a rationale for the formation of long-range ordered spectrin–actin assemblies in neurons26.
Spectrins and the neuronal cytoskeleton
The ability of spectrins and actin to form long-range ordered networks was first observed via electron microscopy in erythrocytes, where they organize as a hexagonal lattice, in which six spectrin tetramers about 60–80 nm in length crosslink short actin protofilaments27 capped by adducin28,29 and tropomodulin30,31. Visualization using 3D-stochastic optical reconstruction microscopy (3D-STORM) confirmed that native erythrocytic spectrin tetramers adopt a relaxed conformation32 and also revealed a similar 2D polygonal spectrin lattice in the somatodendritic compartment of cultured rodent neurons33
(Fig. 1). This 2D assembly progressively develops both in the soma and in dendrites in vitro and depends on actin polymerization and βII-spectrin for its formation33. Images obtained with 3D-STORM suggest that the somatodendritic spectrin lattice contains tetramers of αII/βIII-spectrin in their extended conformation. However, whether βIII-spectrin is an essential component of this structure and the functional role of the 2D lattice are not known. In accordance with spectrin’s canonical role, it is possible that one function is to stabilize protein complexes in those neuronal regions or to modulate endocytosis; this remains to be clarified.
The membrane-associated periodic skeleton (MPS), consisting of actin, spectrins and binding partners, is conserved across organisms and neuron tyes34–37. The MPS is composed of submembrane actin rings periodically spaced at ~190 nm throughout axons and mature dendrites, which corresponds to the extended conformation of spectrin tetramers33–35 (Fig. 1). This remarkable periodicity of the actin lattice is established very early in neuronal development and is likely to be conserved across neuron types and species. In mouse neurons, the MPS is detected in the proximal axon of cultured neurons as early as day in vitro 2 (DIV2) and propagates towards distal axon regions as neurons mature38. However, a recent study suggests that the MPS nucleates from multiple periodic patches along the growing axon that expand and coalesce into a single scaffold39. That the youngest part of the axon with the lowest actin–spectrin periodicity has the greatest axonal diameter implicates the gradual MPS assembly in the progressive constriction of the growing axon39,40. The assembly and integrity of the MPS depend on both actin and spectrin. Perturbation of actin using destabilizing drugs and depletion of βII-spectrin, the most ubiquitous β-spectrin in neurons, by in vitro short hairpin RNA knockdown or in vivo genetic knockout prevents the assembly of the MPS or disrupts its stability. These disruptions correlate with deficits in the structural integrity of the axon initial segment (AIS), the growth of axons in vitro and in vivo, and the formation of long-range axonal tracts in mouse brains33,34,38,41,42.
Early studies in Caenorhabditis elegans showed that worms lacking β-spectrin are more prone to axon breakage upon movement43, which suggests that spectrins and the MPS promote the integrity and mechanical stability of axons under mechanical stress26. This protection could be due, in part, to the unfolding properties and intrinsic flexibility of the SRs22,44, which probably confer tension-buffering properties on the MPS and allow axons to stretch reversibly without compromising their structural or functional properties45. Several studies also support the idea that spectrins and the MPS are critical regulators of axon diameter. Loss of either βII-spectrin or the actin-capping protein α-adducin, a component of the axonal MPS, results in axon enlargement and degeneration in mouse models40,41. The ability of the MPS to regulate axonal radial contractility is facilitated by the actin-binding protein non-muscle myosin II, which associates with periodic F-actin rings via its head domains46,47.
Other functions attributed to the actin and spectrin-based MPS include serving as a signalling platform (discussed below) and acting as a diffusion barrier at the AIS that selectively filters proteins to contribute to neuronal polarity48,49. In addition, direct crosstalk between the MPS and microtubules, in which these two cytoskeletal networks depend on each other for their formation, stability and regulation, has been proposed50.
Subcellular localization and functional specialization
Despite their remarkable structural and functional domain similarities, β-spectrins are differentially expressed across neuronal types and preferentially localized to specific functional compartments (for example, the AIS, nodes of Ranvier (NoR), dendrites and spines, and the postsynapse) (Fig. 1). We next discuss the preferential segregation of neuronal spectrins and what is known about how this spatial distribution is molecularly codified.
Axonal spectrins: axon initial segment.
αII-Spectrin and βIV-spectrin are the most abundant spectrin tetramers at the AIS, where they incorporate into the MPS with a ~190 nm periodicity51–53 (Fig. 1). αII-Spectrin associates with both βIV-spectrin-ΣI and βIV-spectrin-ΣVI isoforms, which depend on ankyrin-G for their recruitment to this axonal domain54,55. Right after axonal specification and before AIS formation, periodic αII/βII-spectrin tetramers can already be detected in the proximal axon38. However, as neurons mature, both the βII-spectrin signal and its periodicity at the AIS diminish, whereas ankyrin-G and βIV-spectrin levels increase to become highly periodic by DIV12 (ref. 38). Early βII-spectrin expression promotes the formation of a periodic βIV-spectrin assembly at the AIS and proper AIS development38. Consistent with these roles, neurons cultured in vitro and mouse brains lacking βII-spectrin exhibit fragmented AIS41,42,48 and expanded localization of βIV-spectrin beyond the normal AIS boundaries48. Similarly, loss of αII-spectrin, which lowers βII-spectrin levels by more than 70%, results in a reduction in the number of AISs in the mouse cortex52. The remaining AISs are fragmented, with lower levels and less periodic distribution of βIV-spectrin52. Impaired AIS formation in either αII-spectrin- or βII-spectrin-deficient neurons occurs despite a significant increase in levels of the βIV-spectrin-ΣVI isoform41,52. These results are surprising given that βIV-spectrin-ΣVI, which lacks actin binding but interacts with ankyrin-G, restores AIS morphology and the clustering of ankyrin-G and other critical AIS components of neurons lacking βIV-spectrin56. Unexpectedly, βIV-spectrin periodicity at the AIS is not affected by chemical perturbation of the submembrane actin and microtubule lattices52. Thus, the organization and function of βIV-spectrin in the AIS do not require the submembrane cytoskeleton but, rather, depend on ankyrin-G binding53,57. Targeting of βIV-spectrin to the AIS can be further modulated by phosphorylation-dependent conformational changes in 480 kDa ankyrin-G, the giant isoform that localizes at the AIS and promotes its development56,57. βI-Spectrin, the major β-spectrin in erythrocytes, is not normally found at the AIS but re-localizes to this domain in parvalbumin-positive interneurons lacking βIV-spectrin58. However, βI-spectrin cannot compensate for loss of AIS βIV-spectrin. Unlike βII-spectrin and βIV-spectrin, βI-spectrin preferentially binds ankyrin-R59, which cannot be recruited to the AIS; therefore, βI-spectrin is unable to stabilize ion channels and other AIS components58. Together, spectrins regulate the morphology, structural integrity and macromolecular composition of the AIS, which is required for efficient action potential initiation and propagation in the nervous system.
Axonal spectrins: nodes of Ranvier.
Spectrins collaborate with ankyrins to position macromolecular complexes that are essential for the ultrastructural organization and function of NoR60. Ankyrin-G and βIV-spectrin are confined to the nodal gap and organized with a ~190 nm periodicity61,62 (Fig. 1). Early in development, βIV-spectrin-ΣI is the predominant isoform at NoR in mouse neurons; however, βIV-spectrin-ΣVI levels increase robustly after birth, quickly exceeding βIV-spectrin-ΣI in abundance63. Loss of ankyrin-G or βIV-spectrin does not disrupt the clustering of voltage-gated sodium channels (NaV) at NoR of sensory neurons owing to a compensatory mechanism whereby ankyrin-R and βI-spectrin concentrate at the nodes to stabilize these channels59. Studies in conditional knockout mouse models that selectively lack either βI-spectrin or βIV-spectrin, or both, only in peripheral sensory neurons (PSNs) demonstrated a hierarchy of nodal spectrins. βIV-Spectrin is the main nodal spectrin; however, βI-spectrin can fully compensate for its loss at NoR64. Although nodal β-spectrins are not required for NaV clustering during development, they are essential for maintaining NaV assemblies at NoR and the structural integrity of sensory axons, and their loss leads to axonal degeneration64. βII-Spectrin localizes to the paranodal region of NoR, where it is periodically organized62,65 (Fig. 1) and required for paranode-dependent clustering of nodal NaV66. Loss of βII-spectrin disrupts the paranode–juxtaparanode membrane barrier and leads to diffusion of voltage-activated KV1.2 potassium channels into paranodes and nodal gaps67. αII-Spectrin also forms a periodic cytoskeleton at nodes and paranodes68 (Fig. 1). Loss of αII-spectrin disrupts the periodicity of βIV-spectrin at the nodal gap and of βII-spectrin at paranodes, impairs NoR assembly and maintenance, disrupts the restricted juxtaparanode localization of KV1.2 and causes axon degeneration68. These studies underscore the critical roles of nodal spectrins in organizing key ion channels and cytoskeletal components at NoR, which are essential for fast action potential propagation in myelinated axons and for maintaining axon integrity.
Somatodendritic spectrins.
As in axons, αII/βII-spectrin tetramers organize the MPS in dendritic shafts and in a subset of spine necks of mature (DIV16-21) cultured mouse neurons, and probably in vivo33,38,41,69,70 (Fig. 1). The formation of the dendritic MPS depends on the expression and local concentration of βII-spectrin33,38. Ankyrin-B, a βII-spectrin binding partner, is a critical regulator of βII-spectrin dendritic levels and dendritic MPS formation38. Loss of ankyrin-B results in a twofold increase in dendritic βII-spectrin in DIV10 neurons without overall changes in its total brain expression or in the organization of the axonal MPS38,71. Although the MPS is very irregular in DIV10 control dendrites, at this timepoint βII-spectrin and α-adducin exhibit a highly periodic distribution in all dendrites of neurons lacking ankyrin-B, quantitatively similar to their axonal periodicity38. The periodic distribution of βII-spectrin at spine necks is less penetrant and detected in 25–50% of spines, depending on culture conditions and super-resolution imaging modalities33,69. By contrast, F-actin rings are periodically organized at the neck of every spine69, suggesting that other β-spectrins may selectively help to assemble the dendritic MPS in a subset of spines.
Tetramers containing βIII-spectrin are likely to contribute to the periodic organization of actin and other MPS components in spines. βIII-Spectrin is highly enriched in dendrites and at the neck of spines, as shown by confocal and platinum replica electron microscopy72,73 (Fig. 1). A periodic βIII-spectrin signal has also been detected by STORM in dendritic shafts of mature neurons33. Consistent with a functional role for βIII-spectrin in spines, its knockdown decreases the density of dendritic spines and alters the formation of the constricted necks of spines in hippocampal neuronal cultures73. Deficits in βIII-spectrin also reduce the number of synapses and impair the localization of critical postsynaptic density (PSD) components, including metabotropic glutamate receptor 1α (mGluR1α) and delta-2 glutamate receptor (GluRδ2) within spines of cerebellar Purkinje neurons72,74,75. These deficits, which are likely to arise from a failure of βIII-spectrin’s essential role in localizing and scaffolding membrane proteins at dendritic domains, alter cerebellar excitability and cause motor impairments in mouse models72,74.
Multiple spectrins localize at the spine head, including βII-spectrin and βIII-spectrin (Fig. 1), whose signals do not appear to co-localize with PSD markers70. This localization pattern contrasts with previous reports that βII-spectrin selectively interacts with NMDA receptor subunits through binding sites distinct from those of members of the PSD95/SAP90 family76. βI-Spectrin is localized throughout dendrites77,78 and enriched at the PSD79–81. Interestingly, the βI-spectrin signal at the PSD does not co-localize with αII-spectrin, which is found mostly at the spine neck81. Whether βI-spectrin functions in its monomeric form in this subsynaptic domain, is integrated into spectrin tetramers or is periodically organized remains to be determined. βI-Spectrin may have both structural and functional roles in dendritic spines through its interaction with F-actin and the small GTPase RAC3 (ref. 82). Expression of the actin-binding domain of βI-spectrin stabilizes actin filaments in dendritic spines by reducing its depolymerization rate82. Functionally, these changes disrupt the dynamic regulation of actin in spines and the morphological and electrophysiological plasticity of spines, as evidenced by increases in the sizes of spine heads, in the trafficking of AMPA receptors into spines and in AMPA receptor-mediated synaptic responses82. The inability of a mutant ankyrin-G that lacks βIV-spectrin binding to rescue deficits in spine development associated with ankyrin-G loss prompted the suggestion that βIV-spectrin may localize to spines and participate in the modulation of spine morphology83. However, the binding site for βIV-spectrin in ankyrin-G is shared with other β-spectrins. Thus, the specific β-spectrin(s) that modulate the roles of ankyrin-G in spines, and whether βIV-spectrin localizes to spines, remain to be determined through super-resolution microscopy, biochemical and functional studies.
Spectrins in intracellular transport.
The capacity of spectrins to act as transport facilitators has critical physiological consequences41,42,84. The implication of spectrin in organelle transport was first suggested by the co-migration of βII-spectrin with axonal organelles in the optic nerve85, and by the formation of complexes between βII-spectrin and kinesin motors KIF3A, KIF5B and KIF1A and the p150Glued dynactin subunit41. Other spectrins also associate with motor proteins or their adaptors. For instance, αII-spectrin interacts and travels with similar velocity to KIF3 in rat optic nerves86. βIII-Spectrin binds the dynactin actin-related protein 1 (Arp1) subunit87. Lastly, βV-spectrin binds KIF3A and myosin VIIa motors and the dynactin subunit dynamitin (p50) in photoreceptor cells12. Spectrins also interact with various organelles12,41,85,88 and probably operate as adaptors or accessory proteins that promote the recruitment of microtubule-based motors to cargoes. For example, βII-spectrin associates with synaptic vesicles and lysosomes in mouse brain lysates, and its loss impairs their bidirectional axonal transport in hippocampal and cortical neurons41,42. Similarly, βV-spectrin forms complexes with opsin-containing vesicles in vivo and promotes their recruitment to myosin VIIa and transport to the outer segments of photoreceptor cells12.
β-Spectrins are recruited to intracellular membranes via coupling of their PH domains to membrane phospholipids. Expression of the K2207Q βII-spectrin PH domain mutant, which is incapable of binding phosphoinositide89, cannot restore axonal transport in neurons lacking βII-spectrin41,90. Similarly, reconstituted motility assays using cytoplasmic material and liposomes from squid axons showed that expression of a dominant-negative construct containing the PH domain of βII-spectrin or βIII-spectrin impaired organelle and liposome retrograde motility88. These studies indicate that the association of β-spectrins with membrane lipids is required for axonal transport driven by dynein or dynactin88. β-Spectrins also bind integral membrane proteins on vesicles, as suggested by the direct interaction of βII-spectrin with synapsin I91. The association of β-spectrins with vesicles could be mediated by ankyrin-B, which binds surface phosphoinositol trisphosphate lipids in organelles and regulates axonal transport71. However, the Y1874A mutant form of βII-spectrin, which does not bind ankyrin-B, rescues synaptic vesicle dynamics in neurons lacking βII-spectrin, which suggests that ankyrin-B and βII-spectrin promote axonal transport through independent pathways41.
How can spectrins simultaneously integrate into the axonal MPS and associate with motors and cargoes? Are the axonal transport deficits in neurons lacking β-spectrins secondary to MPS disruption? It is possible that high F-actin concentrations combined with the low-affinity association between F-actin and the CH domains of β-spectrins partition spectrin molecules between motor protein-bound and MPS-associated pools. Interestingly, βII-spectrin regulates axonal cargo transport from early axonal development in the absence of the MPS, which forms a few days later41. However, the secondary effects of acute MPS disassembly on axonal organelle trafficking warrant further investigation.
Spectrins in signal transduction.
The ability of spectrins to periodically organize functionally related membrane proteins suggests that the MPS may serve as a structural platform that regulates signalling events spatially and temporally. Multiple signalling molecules, including G-protein-coupled receptors (GPCRs), cell adhesion molecules and receptor tyrosine kinases (RTKs), are periodically distributed in axons and co-localize with the MPS near the ankyrin-binding region of the spectrin tetramer4. The incorporation of a subset of signalling molecules into the MPS is potentiated (or only detected) following extracellular stimulation4. Thus, the initial association of these molecules with the MPS probably increases their local concentration, amplifies their recruitment through multivalent interactions with ankyrins and spectrins, and promotes the formation of signalling hubs4. In support of this view, periodically localized cannabinoid type 1 receptor (CB1) and neural cell adhesion molecule 1 increased their association with the MPS upon treatment with a CB1 agonist, which resulted in RTK transactivation and activation of the ERK cascade in neurons4. Interestingly, ERK signalling causes degradation of spectrin and the MPS by calpain, which provides a feedback loop mechanism for neuronal signal attenuation4.
Insights from mouse models
The high evolutionary conservation of spectrin genes across mammals has made knock-in, global and conditional knockout mouse models of spectrins valuable tools for discerning their roles in the nervous system and investigating the pathogenic mechanisms of nervous system spectrinopathies (Fig. 3; see Supplementary Table S1).
Fig. 3 |. Deficiencies in mouse models of neuronal spectrin dysfunction.
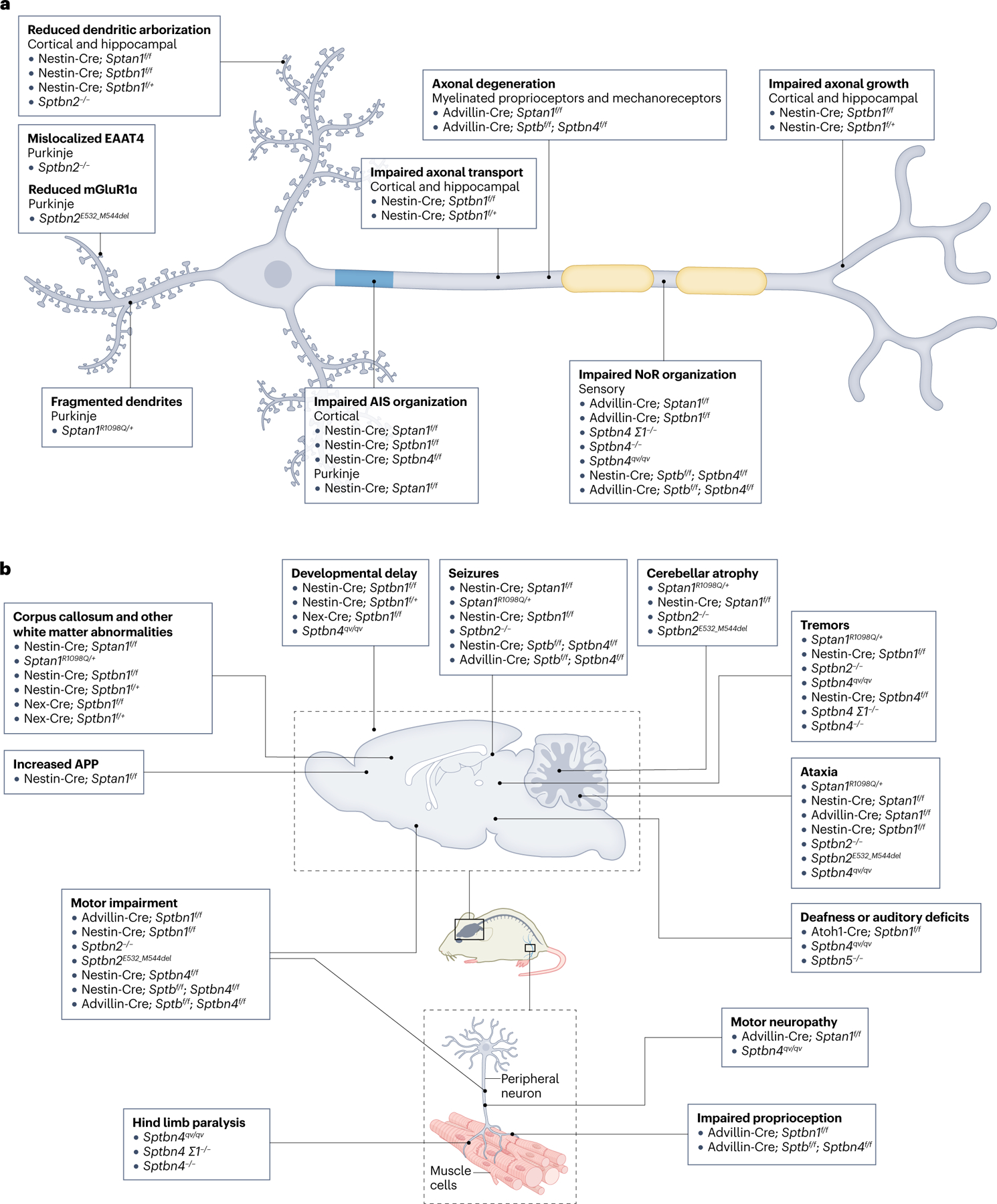
a, Loss, haploinsufficiency and mutations in spectrins in mice induce global, region and functional domain-specific neuronal defects in vivo and in vitro, including reduced dendritic arborization, axonal degeneration, protein mislocalization and reduced axonal transport. Neuron type and mouse model source (see Supplementary Table S1) indicated in parentheses. b, Major anatomical and functional phenotypes observed in mouse models of spectrin deficits in the CNS and PNS. Mouse model source (see Supplementary Table S1) indicated in parentheses. AIS, axon initial segment; APP, amyloid precursor protein; EAAT4, excitatory amino acid transporter 4; mGluR1α, metabotropic glutamate receptor type 1α; NoR, nodes of Ranvier.
Sptan1 mouse models
Although global loss of αII-spectrin causes early lethality around mouse embryonic day 12.5–16.5 and craniofacial, neural tube and cardiac anomalies, haploinsufficiency does not affect the lifespan or lead to obvious abnormalities92. Mice with conditional loss of αII-spectrin in all neural progenitors (Nestin-Cre; Sptan1f/f) survive for the first month of life, providing a powerful tool to define the in vivo roles of αII-spectrin during early CNS development52. Juvenile Nestin-Cre; Sptan1f/f mice exhibit generalized seizures with nearly continuous limb movements and abnormal EEG discharges. These epileptic presentations are in line with cellular and anatomical brain abnormalities that can alter neuronal excitability, including fewer and fragmented AISs in the cerebral cortex, disrupted cortical lamination (probably owing to arrested neuronal migration) and reduced complexity of dendritic arborization in the mouse cortex and in hippocampal neuron cultures52. Loss of αII-spectrin also resulted in fewer Purkinje neurons, which show fragmented and shorter AISs, disrupted formation of Pinceaux terminals from basket cells, thinning of their dendrites and altered organization of axonal projections. Brains from Nestin-Cre; Sptan1f/f mice exhibited increased immunostaining for β-amyloid precursor protein in Purkinje neurons, the thalamus and the cortex, suggesting widespread neuronal degeneration52. CRISPR-mediated deletion of Sptan1 in embryonic rat forebrain via in utero electroporation disrupted neuronal polarity, AIS organization and dendritic development93. Loss of αII-spectrin also impaired callosal axon growth and guidance, and resulted in corpus callosum dysgenesis, defective GABAergic innervation and reduced frequency of miniature inhibitory postsynaptic currents in cortical pyramidal neurons, all consistent with hyperexcitability and epileptic presentations.
αII-Spectrin also promotes the organization of excitable axonal domains in PSNs. Young adult Advillin-Cre; Sptan1f/f mice with selective αII-spectrin loss in PSNs exhibit severe ataxia, motor impairments and reduced nerve conduction velocity associated with preferential degeneration of myelinated proprioceptor and mechanoreceptor axons68. Affected sensory axons show a reduced number of NoR, disrupted paranodal junctions and mislocalized KV1.2 channels, suggesting mechanical weakening of the axons at NoR68. Degeneration of peripheral axons is consistent with motor neuropathy phenotypes reported for some affected individuals carrying disease-linked SPTAN1 variants (described below)94–96.
Models of αII-spectrin deficits in selective neurons have provided insights into its roles in nervous system function. However, the in vivo characterization of morphological and behavioural phenotypes associated with pathogenic SPTAN1 variants predicted to operate via dominant or dominant-negative mechanisms has lagged. The identification of a mouse line carrying the spontaneous p.R1098Q mutation97 allows us to study how heterozygous expression of the p.R1098C or p.R1098S variants (Fig. 4a), in the same residue, causes combined ataxia, spasticity, ID and seizures in humans98. Initial evaluations show that heterozygous p.R1098Q mice develop progressive ataxia with tremors, cerebellar degeneration and extensive neuronal loss in the neocortex and hippocampus97. Substitutions at p.R1098 are predicted to intrinsically enhance proteolysis of αII-spectrin by calpain, which may destabilize the submembrane spectrin cytoskeleton, disrupt axon and dendritic development and integrity, and cause global neurodegeneration97.
Fig. 4 |. Spectrin variants associated with neurological disorders.
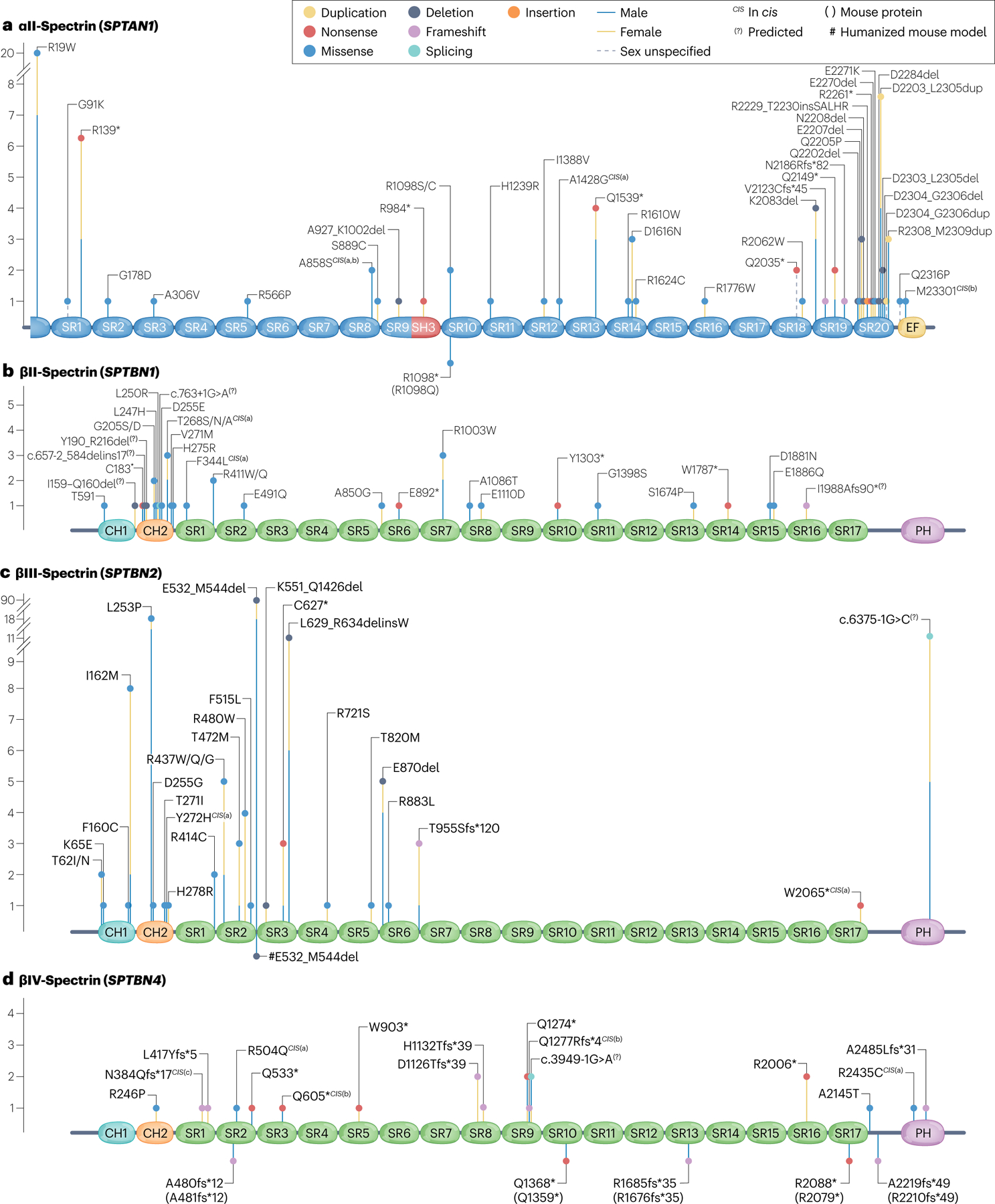
a, Multiple reported αII-spectrin variants have been associated with neurological disorders. Variant types include missense (blue), nonsense (red), duplication (yellow), deletion (dark grey), splicing (teal), insertion (orange) and frameshift (violet). The cis superscript indicates compound heterozygous (in cis), with the letter in parentheses indicating the corresponding variant pair for a single individual. The sex of the reported individual is indicated by the lines below the dots (male, blue line; female, yellow line; unknown, discontinuous line). Number of individuals of each sex for each variant is indicated by the length of the corresponding line below the oval-shaped dot measured relative to the y-axis. Variants are distributed throughout the spectrin repeats (SRs; blue), with a cluster in the heterodimerization region (SRs 19–20). b, βII-Spectrin variants associated with a neurodevelopmental syndrome. These variants emerge largely de novo and are spread throughout the SRs (green), with a strong cluster in the second calponin homology (CH2; orange) domain. c, βIII-Spectrin variants associated with ataxia, developmental delay (DD) and intellectual disability (ID). d, Reported human βIV-spectrin variants associated with disorders of the CNS and PNS. Only homozygous and compound heterozygous carriers manifest clinical presentations. The carrier of the N384Qfs*17CIS(c) variant also bears a maternally inherited deletion with a breakpoint spanning [chr19.g.(?_41,001,394)_ (41,011,375_?)del (GRCh37)], which is predicted to delete exons 6–11 (ref. 160). Knock-in mouse models are indicated in the lower part of the protein schematic, with the corresponding mutated site in the mouse spectrin homologue shown in parentheses. PH, pleckstrin homology; SH, Src-homology. Part b adapted from ref. 42, Springer Nature Limited.
Sptbn mouse models
Conditional loss of βI-spectrin in all neural progenitors (Nestin-Cre; Sptbnf/f) does not lead to any discernible alterations in the structural organization of the AIS of cortical neurons or in motor performance58. Similarly, mice selectively lacking βI-spectrin in PSNs (Advillin-Cre; Sptbnf/f) show normal proprioceptive responses, nerve conduction velocity and nodal NaV channel clustering64. Thus, although βI-spectrin is highly expressed in multiple neuronal types, it does not seem to be essential for central or PSN function, probably owing to compensation by other neuronal β-spectrins.
Sptbn1 mouse models
Whole-body loss of the embryonic liver fodrin (ELF) isoform of βII-spectrin, which lacks the PH domain, results in mid-gestational embryonic lethality with severe growth delay and aberrant development of the heart, gut, liver and brain99. Mice with complete loss of βII-spectrin in all neural progenitors (Nestin-Cre; Sptbn1f/f) exhibit early lethality at around 5 weeks, accompanied by global DD, overt hyperactivity and motor deficits, tremors and seizures41,42. Brain βII-spectrin haploinsufficiency also leads to global DD and social interaction deficits in young adult mice42. DD was also observed upon selective loss of βII-spectrin in cortical projection neurons (Nex-Cre; Sptbn1f/f), suggesting neuronal-autonomous mechanisms42. Selective loss of βII-spectrin in auditory hair cells (Atoh1-Cre; Sptbn1f/f) causes deafness, probably owing to its critical roles in organizing the actin cytoskeleton around the base of hair cell stereocilia rootlets100. Behavioural effects of brain βII-spectrin deficiency in mice are consistent with autism spectrum disorder (ASD), attention deficit and hyperactivity disorder (ADHD), and learning, motor and hearing deficits reported in carriers of SPTBN1 variants (discussed below)42,101.
βII-Spectrin is required for the establishment of long-range cerebellar axons and of axonal tracts connecting the cerebral hemispheres in the mouse brain, and for the proper formation of neocortical layers that give rise to callosal-projecting neurons41,42. That βII-spectrin regulates brain cytoarchitecture is demonstrated by the presence of corpus callosum hypoplasia in humans with SPTBN1 variants and in mice with complete loss or haploinsufficiency of βII-spectrin in the entire brain or only in cortical projection neurons41,42. βII-Spectrin also has critical roles in myelinated PNS axons. At paranodes, βII-spectrin provides a barrier that promotes the polarized assembly of macromolecular complexes67. Mice lacking βII-spectrin in PSNs (Advillin-Cre; Sptbn1f/f) exhibit impairments in motor coordination and proprioceptive responses67.
Sptbn2 mouse models
Studies in whole body βIII-spectrin hypomorph mice have demonstrated its role in stabilizing surface excitatory amino acid transporter 4 (EAAT4) in dendrites of Purkinje neurons102–104. Loss of βIII-spectrin also reduces dendritic levels of the glutamate transporter GLAST and ankyrin-R, and sodium currents in Purkinje neurons103,104. In addition, expression of the p.E532_M544del βIII-spectrin variant associated with spinocerebellar ataxia type 5 (SCA5) impairs localization of mGluR1α in Purkinje neuron dendritic spines and long-term potentiation in knock-in mice74 (Fig. 3). It is likely that combined deficits in cerebellar glutamatergic transmission and in the firing of Purkinje neurons in βIII-spectrin-deficient mice may intrinsically diminish Purkinje neuron output and cause their degeneration. Beyond progressive deficits in motor function, βIII-spectrin-deficient mice exhibit myoclonic seizures, underperform in a subset of cognitive and memory tasks, and show thinning of dendrites of prefrontal cortex neurons72,104,105. These behavioural phenotypes correlate with cognitive deficits of probands homozygous for the recessive human C627* variant, which is predicted to eliminate most βIII-spectrin expression105. Interestingly, neither βIII-spectrin haploinsufficient mice nor individuals hemizygous for the C627* variant show obvious cerebellar or cortical dysfunction72,105,106, which suggests a total loss-of-function mechanism. By contrast, multiple individuals heterozygous for de novo missense SPTBN2 variants develop severe childhood-onset cerebellar ataxia and ID107–113. These findings point to strong gain-of-function or dominant-negative effects, which may not be recapitulated by loss-of-function models and warrants further investigation.
Sptbn4 mouse models
Autosomal recessive variants in Sptbn4 are associated with auditory and motor neuropathies, progressive ataxia with hindlimb paralysis and tremors in six lines of homozygous ‘quivering’ (Sptbn4qv/qv) mice114 (Fig. 3). These Sptbn4 variants result in different truncations of βIV-spectrin and in its reduced expression, which correlates with the severity of the neurological phenotypes. For example, Sptbn4qv3J/qv3J mice, which carry a frameshift mutation at G2210 that is predicted to eliminate the PH domain and add a novel 49 amino acid extension in the C-terminal region, have disrupted NoR in the CNS but mostly normal PNS nodes114–116. By contrast, the nonsense Q1359* mutation in SR10 found in Sptbn4qv4J/qv4J mice114 impairs the localization of nodal, paranodal and juxtaparanodal proteins at NoR in the PNS115,116. The more penetrant effect of the Sptbn4qv4J/qv4J mutation may be explained by the loss of the ankyrin-binding region of βIV-spectrin-ΣI and an extensively truncated βIV-spectrin-ΣVI isoform, combined with their reduced expressions114,116. Global loss of the βIV-spectrin-ΣI isoform recapitulates the quivering mice phenotypes18,51, which underscores its roles in the organization and function of the AIS and NoR in vivo. Interestingly, a gene trap insertion mouse model that lacks full-length βIV-spectrin-ΣVI and βIV-spectrin-ΣI but might express N-terminal truncation fragments of βIV-spectrin-ΣI shows the hallmarks of the quivering mice phenotype, including a more severe progression than the isoform-specific knockout mice54.
Functional evidence from mouse models suggests that pathogenic SPTBN4 variants in the CNS operate principally through destabilization of the AIS followed by progressive neuronal dysfunction and degeneration. Conditional knockout of βIV-spectrin in the brain (Nestin-Cre; Sptbn4f/f) causes tremors and poor motor performance, collateral to reduced expression of ankyrin-G and NaV channels in the AIS of cortical neurons58. By contrast, NaV clustering at CNS NoR is not affected because of compensation by βI-spectrin64. βIV-spectrin is also dispensable for normal clustering of NaV channels at NoR in the PNS (Advillin-Cre; Sptbn4f/f) through the functional compensation of the partners βI-spectrin and ankyrin-R59,64. Interestingly, a nerve biopsy from a proband with compound heterozygous p.R504Q/p.R2435C βIV-spectrin variants showed normal NoR morphology and NaV channel clustering, but weaker KCNQ2 immunoreactivity, an observation replicated in myelinated axons of Sptbn4qv3J/qv3J and ankyrin-G conditional knockout mice115,117,118. It is possible that the redundant ankyrin-R–βI-spectrin complex, which is important for the localization and maintenance of KV3.1b at NoR118, does not rescue KCNQ2. KCNQ2 deficiency may contribute to sensory nerve dysfunction and peripheral neuropathy.
Sptbn5 mouse models
The contribution of βV-spectrin to cochlear outer hair cell (OHC) function and auditory responses was recently investigated using a global knockout mouse model of Sptbn5 (Sptbn5−/−) generated by a targeted exon deletion119. These mice exhibit normal OHC electromechanical activity and auditory thresholds, probably owing to the undetectable expression of Sptbn5 transcripts in OHCs. By contrast, Sptbn5 transcripts are present in spiral ganglion neurons and βV-spectrin loss is associated with decreases in the number of afferent and efferent nerve fibres and in auditory brainstem response wave 1 amplitudes119. These data suggest that βV-spectrin promotes the maintenance and function of nerves in the peripheral auditory system but has no critical role in OHCs.
Spectrinopathies of the nervous system
Although pathogenic variants in spectrins are categorized as rare, their identification in individuals with neurological disorders is rapidly growing. Here we describe the spectrinopathies of the nervous system (Table 1) and summarize current knowledge regarding their clinical presentation (Fig. 5) and pathogenic mechanisms.
Fig. 5 |. Major phenotypes in humans with spectrinopathies of the nervous system.
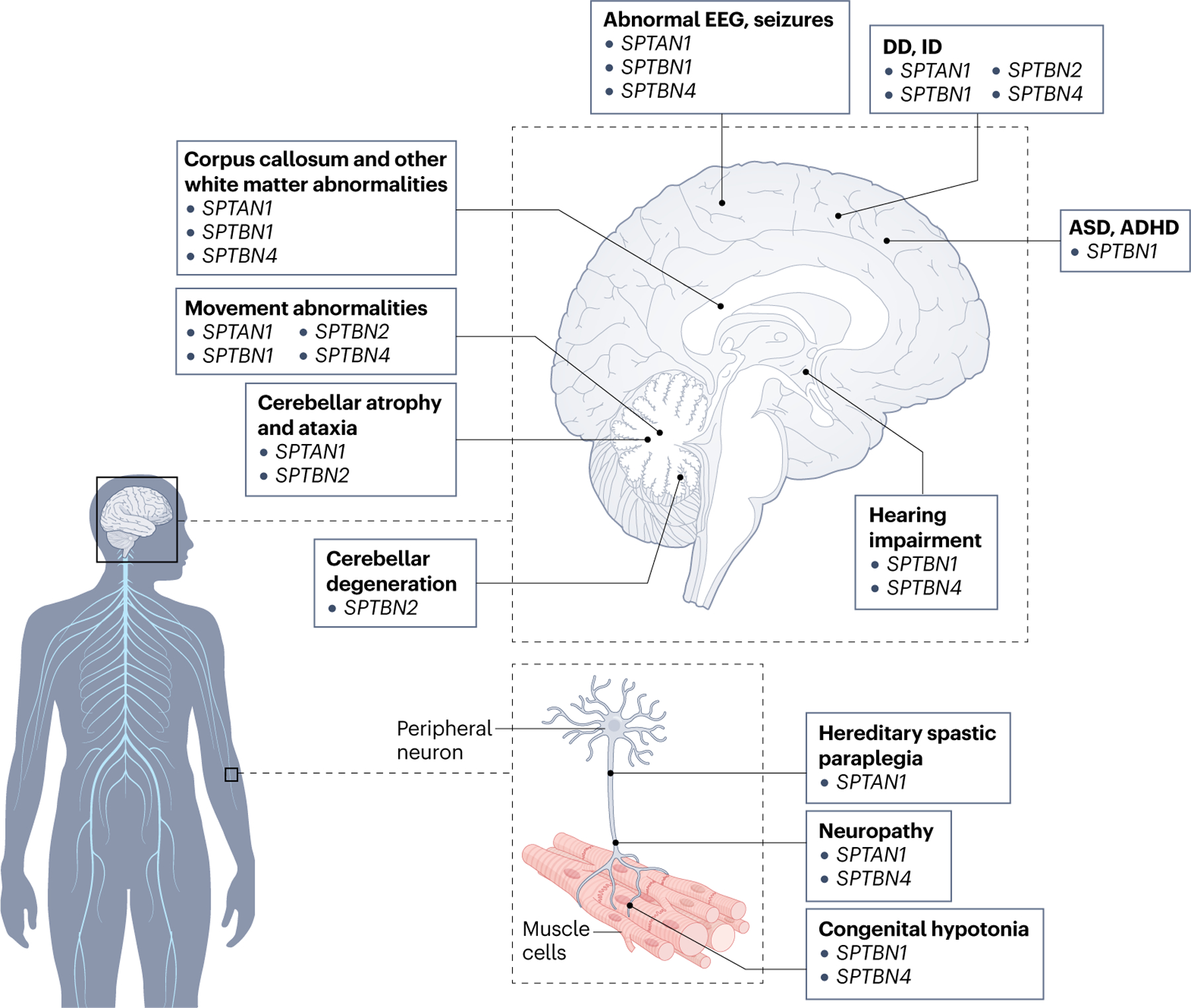
Pathogenic variants in spectrins cause complex neurological syndromes in both the brain and periphery that have overlapping pathologies and clinical presentations across spectrin genes. Affected spectrin genes indicated in parentheses. ADHD, attention deficit hyperactivity disorder; ASD, autism spectrum disorder; DD, developmental delay; ID, intellectual disability.
SPTAN1 spectrinopathy: epilepsy, ID and motor deficits
More than 40 pathogenic de novo and inherited SPTAN1 variants have been linked to a wide range of neurological presentations94–98,120–137 (Table 1 and Figs. 4a and 5; see Supplementary Table S2). The first association of αII-spectrin with human neurological diseases was the identification of de novo heterozygous in-frame deletion (p.E2207del) and in-frame duplication (p.R2308_M2309dup) variants in SR20 in two patients with West syndrome120. In addition to early-infantile epileptic encephalopathy (EIEE) with frequent severe seizures, these individuals presented with abnormal cortical and white matter development, corpus callosum thinning, hypomyelination, cerebellar atrophy, severe DD, ID and spastic quadriplegia120. Other studies have implicated de novo in-frame duplications and deletions in SPTAN1 in EIEE and epilepsy120–123,125,127,130,131,136. More recently, heterozygous nonsense SPTAN1 variants have been linked to hereditary motor neuropathy94,95, a de novo heterozygous nonsense variant to sensorimotor peripheral neuropathy with DD134, homozygous and biallelic missense variants to hereditary spastic paraplegia133,134, and both de novo and dominantly inherited missense variants to cerebellar ataxia with ID, often accompanied by spasticity and seizures98,136.
The corresponding amino acid position affected by pathogenic SPTAN1 variants does not predict the presence or severity of specific neurological presentations. However, the emerging picture points to a segregation of EIEE and West syndrome diagnosis with a cluster of variants in the spectrin dimerization domain in SR20 (Fig. 4a). The variability in variant type, disease onset and phenotypic spectrum suggests that SPTAN1 variants operate through different mechanisms. For instance, aggregates of αII-spectrin have been detected in cortical mouse neurons expressing p.E2207del and p.R2308_M2309dup mutants120, and in patient-derived induced pluripotent stem cell-derived glutamatergic neurons expressing a p.D2303_L2305dup variant93, all of which are EIEE-linked variants, which suggests gain-of-function or dominant-negative effects. By contrast, αII-spectrin haploinsufficiency probably contributes to hereditary motor neuropathy presentations, given that hereditary motor neuropathy-linked nonsense SPTAN1 variants cause degradation of the mutant transcript by nonsense-mediated decay94. SPTAN1 variants associated with cerebellar ataxia and mild ID (p.K2083del), hereditary spastic paraplegia (p.R19W) and combined ataxia, spasticity, ID and seizure phenotypes (p.R1624C, p.R1098C and p.Q2205P) possibly destabilize SR folding through the loss of electrostatic interactions and hydrogen bonds98. Interestingly, the spontaneous p.R1098Q mutation in mouse αII-spectrin leads to progressive ataxia with tremors and seizures in mice97. Substitutions at p.R1098, near the start of SR10, which contains the calpain cleavage and CaM-binding sites, are predicted to alter αII-spectrin’s CaM affinity and sensitivity to calpain proteolysis97. Overall, disease-linked SPTAN1 variants impair the organization and maintenance of the neuronal actin–spectrin cytoskeleton, which has functional consequences in axonal development and connectivity, the formation and function of excitable axonal and synaptic domains, and neuronal excitability.
SPTBN1 spectrinopathy: DD, ID, epilepsy, ADHD and ASD
SPTBN1 variants cause an autosomal-dominant syndrome of early onset characterized by global developmental language and motor delays42,101 (Table 1 and Figs. 4b and 5; see Supplementary Table S2). Affected individuals also co-present with mild to severe ID, seizures, movement abnormalities, hypertonia and hypotonia, and hearing impairments. Behavioural diagnoses include ASD, ADHD, sleep disturbances, anxiety, emotional liability and aggressive or self-injurious behaviours. Notably, ASD and ADHD are concurrent in a subset of probands, which supports the identification of SPTBN1 as a top risk gene among genes with rare truncating variants that co-occur in these disorders138. Thirty-four unique heterozygous variants in SPTBN1 have been identified in 33 affected individuals from 32 families, including 1 pair of siblings and 1 proband with 2 variants in cis42,101 (Fig. 4b). Of those, 24 variants are missense, 4 are nonsense and 3 are canonical splice site variants, with 2 variants predicted to lead to in-frame deletions and 1 to a frameshift that introduces a premature stop codon. Three additional variants are predicted to cause protein frameshifts and loss of function42,101. Parental studies suggest that most individuals in these studies carry de novo variants. However, two maternal half-siblings inherited the p.R1003W variant from their unaffected mother, who shows low levels of mosaicism42. In addition, the carrier of the c.763+1G>A splice variant inherited it from his mother, who exhibits learning disabilities but not ID101.
Approximately half of the variants cluster in the CH domains, which show a higher degree of missense variant constraint in the population than the rest of the protein (ExAC v.10)139, indicating its critical function. CH domain variants show deleterious effects on βII-spectrin’s interactions with F-actin and αII-spectrin, and on cytoskeleton dynamics and neuronal morphology. Interestingly, variants within the proximal region of the second CH domain induce destabilizing structural effects on βII-spectrin that reduce its solubility and cause its aberrant accumulation within cytosolic aggregates together with F-actin and αII-spectrin42. Other variants affect βII-spectrin’s interactions with ankyrins or membrane lipids because they result in truncated polypeptides that lack the ankyrin-binding motif and/or the PH domain.
The pathogenicity of clinically relevant SPTBN1 variants is also supported by structure–function studies in which they failed to rescue deficits in axonal length, axonal organelle transport, AIS organization and dendritic development of cortical neurons lacking mouse βII-spectrin42. These results demonstrate the functional conservation between human and mouse βII-spectrin and highlight their multifaceted roles. Expression and functional studies in mouse neurons lacking βII-spectrin and patient-derived induced pluripotent stem cells suggest that loss-of-function mechanisms contribute to neuronal dysfunction. However, changes in the binding affinity for F-actin, in neuronal morphology and in the formation of cytosolic aggregates, which are not observed upon reduction in βII-spectrin levels, indicate that CH domain variants are likely to contribute to neurological deficits through dominant or dominant-negative effects that affect the submembrane cytoskeleton.
SPTBN2 spectrinopathy: spinocerebellar ataxia with and without ID
Inherited autosomal dominant variants in SPTBN2 cause late-onset SCA5 (refs. 75,140–142) (Table 1 and Figs. 4c and 5; see Supplementary Table S2). In addition, de novo and autosomal recessive SPTBN2 variants have been associated with early childhood ataxia, which often co-segregates with ID and DD104,106,108–113,143–152 (Fig. 4c; see Supplementary Table S2). SCA5, the first brain spectrinopathy reported, was described in three unrelated families in the United States, France and Germany153–155. Symptoms appear anytime between early childhood and the seventh decade of life, but most frequently during adulthood. MRI and autopsy evaluations from these families show cerebellar cortical atrophy without other brain alterations, consistent with a pure form of cerebellar degeneration. Affected members of the American and French families are hemizygous for a 13 amino acid (p.E532_M544del) and a 5 amino acid (L629_R634delinsW) in-frame-insertion deletion in SR3, respectively. The German family carries the p.L253P variant in the second CH domain75. In addition, the SR2 p.T472M variant co-segregates with late-onset pure cerebellar ataxia in members of a family of Norwegian descent140. Interestingly, de novo variants that affect the same residues have been identified in unrelated patients with early childhood ataxia, DD and cognitive deficits, strongly supporting their pathogenic effects107–111,143–145. The growing number of cases also indicates that the association of SPTBN2 variants with ataxia and various neurological presentations is more common than previously appreciated156.
Evaluation of post-mortem samples from patients with SCA5 points to significant degeneration of the cerebellum with Purkinje neuron loss, thinning of the molecular layer and changes in the distribution of the EAAT4 and the glutamate receptor GluRδ2, probably owing to their reduced stability at the surfaces of Purkinje neuron dendrites75. Changes in glutamate signalling secondary to deficits in βIII-spectrin levels and/or function could contribute to Purkinje neuron death and to motor and cognitive deficits. The p.L253P SCA5 variant significantly increases binding affinity for F-actin, suggesting that altered modulation of F-actin dynamics could also contribute to disease pathology157.
SPTBN4 spectrinopathy: congenital hypotonia, neuropathy and deafness, with and without ID
Sixteen SPTBN4 variants have been linked to a neurodevelopmental disorder with congenital hypotonia, neuropathy and deafness (NEDHND)115,158–163 (Table 1 and Figs. 4d and 5; see Supplementary Table S2). These include 11 nonsense, 4 missense and 1 canonical splice site variants as well as 1 multi-exon deletion (Fig. 4d). Most affected individuals carry recessive homozygous SPTBN4 variants; three have compound heterozygous variants. This suggests that variants in both SPTBN4 alleles are necessary to induce pathogenicity. Common presentations among probands include lack of head control and ability to sit, stand and walk, congenital muscular hypotonia and axonal neuropathy, often accompanied by severe DD and ID. More than 50% of probands also experience seizures or have abnormal EEG recordings115,158–163.
The first association of SPTBN4 variants with NEDHND was the identification of a homozygous p.Q533* variant in a 10-year-old boy born to healthy heterozygous carrier parents158. A muscle biopsy revealed an absence of sarcolemma βIV-spectrin and the presence of demyelinating axonal motor neuropathy, probably owing to loss of sodium channels at neuromuscular junction sites. Six additional individuals with homozygous or compound heterozygous SPTBN4 variants were reported to present with NEDHND, central vision impairment and ID115. Functional characterization of this subset of SPTBN4 variants in rat hippocampal neurons indicated changes in the organization of the AIS, which are likely to alter neuronal excitability. In addition, a homozygous canonical splice site variant predicted to generate an in-frame βIV-spectrin polypeptide missing the protein sequence encoded by exon 19 was found in a sibling pair also born to heterozygous carrier parents159. Although these siblings shared severe hypotonia and axonal neuropathy, they did not exhibit ID or deafness, suggesting that the resulting βIV-spectrin, which is predicted to lack 49 amino acids at the beginning of SR7, is partially functional. The clinical spectrum associated with SPTBN4 variants was recently expanded with the identification of five individuals carrying biallelic variants, who in addition to the common neurological presentations also developed horizontal nystagmus, abnormal EEG without seizures and choreoathetosis160. This broad nervous system phenotype underscores the critical functions of βIV-spectrin in the organization and maintenance of key neuronal domains in both the CNS and the PNS. Interestingly, the absence of symptoms in heterozygous carriers of pathogenic SPTBN4 variants is consistent with the apparent lack of neurological phenotypes in mice with partial loss of βIV-spectrin54. It is possible that these partial deficiencies in βIV-spectrin are rescued by βI-spectrin58,64.
Overlapping and diverging pathogenic mechanisms in spectrinopathies
Spectrinopathies of the nervous system are largely syndromic and display a wide range of clinical presentations within and across the affected spectrin genes (Table 1 and Fig. 5; see Supplementary Table S2). Underlying this variety is the intrinsic multifunctionality of spectrins, together with their cell-specific expression in the nervous system and their domain-specific localization in neurons (Fig. 1). Consequently, the penetrance and degree of pathogenicity of a spectrin variant is likely to be determined by the extent to which it affects some, or all, spectrin functions and its resulting effects at the cellular and circuit levels. Within this complexity, certain overlapping clinical features of brain spectrinopathies indicate similarities in the underlying molecular mechanisms and/or converging pathways (Fig. 5). For example, a large group of individuals affected by any of the four nervous system spectrinopathies present with DD, ID, seizures or movement abnormalities.
A convergent pathogenic mechanism may operate through αII-spectrin deficiency, either directly caused by αII-spectrin variants or through effects of β-spectrin variants on αII-spectrin function, or in the stability of the corresponding spectrin tetramers. For example, SPTBN1 variants that cause βII-spectrin aggregation in cortical neurons sequester endogenous αII-spectrin within the aggregates42. Co-aggregation of αII-spectrin and βII-spectrin is also observed in patient-derived glutamatergic neurons expressing clinically relevant SPTAN1 variants93. αII/βII-Spectrin aggregates interfere with normal protein function and may be inherently toxic to neurons. These aggregates also sequester F-actin and further disrupt cytoskeleton organization and dynamics, which underlie observed aberrant neuronal morphologies42.
Several disease-linked βII-spectrin variants cluster in the CH domains and dysregulate binding to F-actin42. Molecular modelling suggests that their range of effects on F-actin binding affinity is probably due to both local and CH domain-wide conformational changes caused by modified intramolecular interactions that affect contacts at the βII-spectrin–F-actin interface. Modelling also predicts structural instability due to substantial charge changes and steric hindrance introduced by amino acid substitution in the CH domain, which probably underlies protein aggregation in cells42. Interestingly, the site of the pathogenic p.L250R βII-spectrin variant is conserved in βIII-spectrin (p.L253), and the p.L253P βIII-spectrin change causes SCA5 (refs. 42,75). The L250/L253 site modulates βII-spectrin/βIII-spectrin affinity for F-actin42,164, suggesting that the deficits in this function as a shared pathogenic mechanism. However, given that very few human variants have been reported in the CH domains of βIV-spectrin and that their effects on F-actin binding are unknown, it is not clear whether disruption of β-spectrin/F-actin is a universal pathogenic mechanism shared by all β-spectrinopathies.
Macro-scale disruption of the spectrin–actin cytoskeleton resulting from spectrin deficits disturbs the organization of functional neuronal macrodomains. Deficits in αII-spectrin, βII-spectrin and βIV-spectrin are associated with AIS loss, fragmentation or reduction in key AIS components such as ion channels42,48,52,93,115 (Fig. 3a). These structural and macromolecular changes alter neuronal polarity, AP initiation and kinetics, and ion channel function, which may contribute to seizures and epileptic phenotypes in multiple patients across these syndromes. Similarly, the roles of αII-spectrin, βII-spectrin and βIV-spectrin in organizing macromolecular complexes at NoR of PSN axons (Fig. 3a) likely underlie hypotonia and hypertonia, and neuropathy presentations associated with their selective disruptions42,94–96,98,120,133,134,158–160.
Although failure to properly traffic or stabilize neuronal membrane proteins appears to be a shared mechanism of spectrinopathies, the individual spectrin partners, cell type expression and neuronal domain localization likely confer specificity (Fig. 1). For example, βIII-spectrin interacts with EAAT4 in the soma and dendrites of Purkinje neurons and with mGluR1α in dendritic spines74,75,102. Loss of βIII-spectrin or SCA5-linked variants cause deficient mGluR1-mediated long-term potentiation, Purkinje cell degeneration and cerebellar dysfunction through combined and cumulative effects of glutamate excitotoxicity and disrupted synaptic function74 (Fig. 3a). Loss of βII-spectrin, or its pathogenic variants, affects axonal organelle transport, which probably contributes to altered protein distribution, diminished axonal growth in vitro and in vivo, and impairments in brain-wide axonal connectivity observed in probands and mouse models41,42 (Fig. 3).
Deficits associated with spectrin dysfunction might also result from alterations in their binding partners, ankyrins, which are regulators of neuronal transport, membrane organization and long-range axonal connectivity1–3,71,165. For example, pathogenic SPTBN4 variants impair βIV-spectrin binding to ankyrin-G and its clustering at the AIS159. Likewise, multiple βII-spectrin variants associated with the SPTBN1 syndrome truncate the polypeptide prior to the ankyrin-binding domain and impair its binding to ankyrin-B42. In addition, SCA5-linked SPTBN2 variants reduce ankyrin-R localization in Purkinje neuron dendrites, which is required for regulating NaV levels and intrinsic excitability103. Dendritic underdevelopment has also been commonly observed across mouse and cellular models of either neuronal spectrin deficiency or SPTAN1, SPTBN1 and SPTBN2 spectrinopathies42,72,74,97,104 (Fig. 3a). These effects suggest an additional pathogenic mechanism that can affect synaptic function; however, the functional roles of spectrin in dendrites and postsynaptic domains have not been extensively studied.
Concluding remarks and future directions
Pathogenic variants in four of the six spectrin genes expressed in the nervous system have been genetically and functionally associated with complex neurological syndromes. Although conditional knockout models of βI-spectrin (SPTB) did not show apparent neuronal or behavioural phenotypes58,64, whether variants in this gene result in neurological deficits remains to be determined. A recent report linked de novo βV-spectrin (SPTBN5) variants to ID, aggressive behaviours and variable presentations including facial dysmorphisms and autistic behaviours in four unrelated individuals166. However, this study only evaluated the functional impact of putative pathogenic variants using in silico prediction tools. Thus, whether these variants are indeed pathogenic and the extent to which they affect βV-spectrin expression, localization or specialized functions in neurons remain to be established. Future studies that focus on discerning the neuronal types and domains in which βV-spectrin is expressed in the nervous systems, its functional roles and the impact of putative disease variants using human samples and mouse models will shed light on the likelihood of pathogenicity of these and other SPTBN5 variants.
Various reports have also linked non-genetic spectrin deficits to neurological dysfunction. For instance, βIV-spectrin autoantibodies have been selectively detected in three unrelated individuals with paraneoplastic neuropathy, suggesting that they can either contribute to the development of neuropathic symptoms or serve as a biomarker167,168. In addition, βIV-spectrin levels are silenced through DNA methylation in the cerebral cortex of patients with Alzheimer disease, suggesting that impaired AIS and/or NoR function contributes to disease pathology169. The importance of maintaining proper βIV-spectrin levels during brain development is further demonstrated by significant associations between DNA methylation at the SPTBN4 locus and severe delays in language and motor skills170. Spectrin levels have also been associated with neurodegeneration. As an example, increased levels of αII-spectrin and βII-spectrin breakdown products, generated by aberrant activation of calpain-dependent proteolysis, correlate with amyloid-β deposits and neurofibrillary tangles in the brains of individuals with Alzheimer disease171,172. αII-Spectrin and βII-spectrin are also enriched in Lewy bodies in the brains of individuals with Parkinson disease173,174. Interestingly, spectrin preferentially binds α-synuclein phosphorylated at Ser129, which promotes aggregation and neurotoxicity. This enhanced interaction might reorganize the actin cytoskeleton and contribute to mitochondrial dysfunction in Parkinson disease175.
Although our understanding of the neuronal roles of spectrins continues to advance, their biology in other cell types in the nervous system is largely understudied, and whether or how deficiency of spectrins in these cells may contribute to spectrinopathies is largely unknown. One study in mice lacking βII-spectrin in myelinating glial cells in both the CNS and the PNS showed that loss of the protein affected the formation and maintenance of NoR, and also altered action potential conduction velocities176. Given the marked importance of glial cells in neurodevelopmental disorders177–179, this gap in knowledge deserves attention. We also lack a comprehensive understanding of the cell types and developmental stages that are vulnerable to disruptions in each spectrin. Therefore, cell-specific, spatiotemporal maps of spectrin expression in the brain and in the PNS in humans and in animal models used to study spectrinopathies will refine our knowledge of disease progression and define potential entry points for therapeutic interventions. For example, re-expression of βIV-spectrin in a mouse model that lacks full-length βIV-spectrin-ΣVI and βIV-spectrin-ΣI54 partially restored PNS NoR organization independently of the intervention time180. CNS NoR restoration was slower and less efficient if the rescue started at a later timepoint, highlighting differences in the mechanisms and recovery window between CNS and PNS myelinated axons. Motor performance and axon functional parameters were only partially improved in these mice, probably because only 50% of PNS and 25% of CNS NoR were restored180.
Lastly, additional knock-in animal models of variants linked to spectrinopathies will expand the toolbox to unambiguously discern pathophysiological mechanisms. However, because animal models often fail to recapitulate pathogenic effects observed in humans, harnessing the unique features of human induced pluripotent stem cells and CRISPR–Cas9 editing technology to establish cell-based 2D and 3D brain organoid models relevant to spectrinopathies is an attractive complementary approach181. Combined with ‘omics’ and other unbiased high-throughput molecular technologies, this full arsenal will open avenues towards the identification of targets through traditional therapeutic discovery, an effort presently lacking for spectrinopathies. To this end, they will help to enable promising technologies that may offer alternative viable paths towards treatment, such as gene replacement and editing, and antisense oligonucleotide-based strategies182.
Supplementary Material
Acknowledgements
The authors apologize to those colleagues whose work could not be cited because of space limitations. This work was supported by National Institute of Mental Health (NIMH) grant R01MH127848 and National Institute of Neurological Disorders and Stroke (NINDS) grant R01NS110810.
Glossary
- Axon initial segment
(AIS). The domain 20-60μm long at the proximal axon–soma interface that has a high density of voltage-gated ion channels and other membrane proteins responsible for the initiation of the action potential.
- De novo variants
Changes in the sequence of a gene that are seen for the first time in an individual but are not present in the parents.
- Gain-of-function
A missense mutation (altered amino acid sequence) that results in enhanced or abnormal protein function.
- Haploinsufficient
A gene for which 50% of normal protein expression is insufficient for normal function and may result in disease.
- Juxtaparanode
A region adjacent to each side of a paranode in myelinated axons.
- Knock-in mouse
A mouse in which an endogenous gene sequence of interest is altered by a one-for-one substitution with a transgene or by adding gene sequences that are not found within the locus.
- Knockout mouse
A mouse in which expression of a gene of interest is inactivated.
- Loss-of-function
A mutation that abolishes protein function, often by partial or complete loss of protein expression.
- Nodes of Ranvier
(NoR). Ion channel-rich gaps along a myelinated axon that expose the neuronal membrane to the extracellular space and speed up the propagation of the action potential along the axon.
- Paranode
A region adjacent to each edge of nodes of Ranvier (NoR) in myelinated axons.
- Pinceaux terminals
The terminals of the Pinceau, a paintbrush-like network of cerebellar basket cell axon branchlets embracing the axon initial segment (AIS) of Purkinje neurons.
- Postsynaptic density
(PSD). The protein-dense molecular network located beneath the membrane of dendritic spines of excitatory neurons.
- Probands
The first individuals in a family who are suspected to be at risk of or affected by a genetic condition.
- Somatodendritic
A neuronal region that includes the cell body and dendrites but excludes the axon.
- Spectrinopathies
Diseases associated with loss or aberrant function of spectrins.
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41583-022-00674-6.
Peer review information Nature Reviews Neuroscience thanks M. Rasband, who co-reviewed with O. Sert; M. Sousa; and the other, anonymous reviewer for their contribution to the peer review of this work.
References
- 1.Bennett V & Lorenzo DN An adaptable spectrin/ankyrin-based mechanism for long-range organization of plasma membranes in vertebrate tissues. Curr. Top. Membr 77, 143–184 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Lorenzo DN Cargo hold and delivery: ankyrins, spectrins, and their functional patterning of neurons. Cytoskeleton 77, 129–148 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bennett V & Lorenzo DN Spectrin- and ankyrin-based membrane domains and the evolution of vertebrates. Curr. Top. Membr 72, 1–37 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Zhou R, Han B, Xia C & Zhuang X Membrane-associated periodic skeleton is a signaling platform for RTK transactivation in neurons. Science 365, 929–934 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marchesi VT & Steers EJ Selective solubilization of a protein component of the red cell membrane. Science 159, 203–204 (1968). [DOI] [PubMed] [Google Scholar]
- 6.Winkelmann JC et al. Full-length sequence of the cDNA for human erythroid β-spectrin. J. Biol. Chem 265, 11827–11832 (1990). [PubMed] [Google Scholar]
- 7.Bennett V, Davis J & Fowler WE Brain spectrin, a membrane-associated protein related in structure and function to erythrocyte spectrin. Nature 299, 126–131 (1982). [DOI] [PubMed] [Google Scholar]
- 8.Hu RJ, Watanabe M & Bennett V Characterization of human brain cDNA encoding the general isoform of β-spectrin. J. Biol. Chem 267, 18715–18722 (1992). [PubMed] [Google Scholar]
- 9.Berghs S et al. βIV spectrin, a new spectrin localized at axon initial segments and nodes of Ranvier in the central and peripheral nervous system. J. Cell Bio 151, 985–1002 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hund TJ et al. A β(IV)-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J. Clin. Invest 120, 3508–3519 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stabach PR & Morrow JS Identification and characterization of βV spectrin, a mammalian ortholog of Drosophila βH spectrin. J. Biol. Chem 275, 21385–21395 (2000). [DOI] [PubMed] [Google Scholar]
- 12.Papal S et al. The giant spectrin βV couples the molecular motors to phototransduction and Usher syndrome type I proteins along their trafficking route. Hum. Mol. Genet 22, 3773–3788 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Ohara O, Ohara R, Yamakawa H, Nakajima D & Nakayama M Characterization of a new β-spectrin gene which is predominantly expressed in brain. Brain Res. Mol. Brain Res 57, 181–192 (1998). [DOI] [PubMed] [Google Scholar]
- 14.Stankewich MC et al. A widely expressed βIII spectrin associated with Golgi and cytoplasmic vesicles. Proc. Natl Acad. Sci. USA 95, 14158–14163 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wasenius VM et al. Primary structure of the brain α-spectrin. J. Cell Bio 108, 79–93 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sahr KE et al. The complete cDNA and polypeptide sequences of human erythroid α-spectrin. J. Biol. Chem 265, 4434–4443 (1990). [PubMed] [Google Scholar]
- 17.Hayes NV et al. Identification of a novel C-terminal variant of βII spectrin: two isoforms of βII spectrin have distinct intracellular locations and activities. J. Cell Sci 113, 2023–2034 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Uemoto Y et al. Specific role of the truncated βIV-spectrin Sigma6 in sodium channel clustering at axon initial segments and nodes of Ranvier. J. Biol. Chem 282, 6548–6555 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Grum VL, MacDonald RI & Mondragón A Structures of two repeats of spectrin suggest models of flexibility. Cell 98, 523–535 (1999). [DOI] [PubMed] [Google Scholar]
- 20.Ipsaro JJ et al. Crystal structure and functional interpretation of the erythrocyte spectrin tetramerization domain complex. Blood 115, 4843–4852 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Speicher DW, Weglarz L & DeSilva TM Properties of human red cell spectrin heterodimer (side-to-side) assembly and identification of an essential nucleation site. J. Biol. Chem 267, 14775–14782 (1992). [PubMed] [Google Scholar]
- 22.Rief M, Pascual J, Saraste M & Gaub HE Single molecule force spectroscopy of spectrin repeats: low unfolding forces in helix bundles. J. Mol. Biol 286, 553–561 (1999). [DOI] [PubMed] [Google Scholar]
- 23.Krieger CC et al. Cysteine shotgun–mass spectrometry (CS-MS) reveals dynamic sequence of protein structure changes within mutant and stressed cells. Proc. Natl Acad. Sci. USA 108, 8269–8274 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heidemann SR & Bray D Tension-driven axon assembly: a possible mechanism. Front. Cell. Neurosci 9, 316 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Šmít D, Fouquet C, Pincet F, Zapotocky M & Trembleau A Axon tension regulates fasciculation/defasciculation through the control of axon shaft zippering. eLife 6, e19907 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leterrier C & Pullarkat PA Mechanical role of the submembrane spectrin scaffold in red blood cells and neurons. J. Cell Sci 135, jcs259356 (2022). [DOI] [PubMed] [Google Scholar]
- 27.Byers TJ & Branton D Visualization of the protein associations in the erythrocyte membrane skeleton. Proc. Natl Acad. Sci. USA 82, 6153–6157 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gardner K & Bennett V Modulation of spectrin–actin assembly by erythrocyte adducin. Nature 328, 359–362 (1987). [DOI] [PubMed] [Google Scholar]
- 29.Kuhlman PA, Hughes CA, Bennett V & Fowler VM A new function for adducin. Calcium/calmodulin-regulated capping of the barbed ends of actin filaments. J. Biol. Chem 271, 7986–7991 (1996). [DOI] [PubMed] [Google Scholar]
- 30.Weber A, Pennise CR, Babcock GG & Fowler VM Tropomodulin caps the pointed ends of actin filaments. J. Cell Biol 127, 1627–1635 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ursitti JA & Fowler VM Immunolocalization of tropomodulin, tropomyosin and actin in spread human erythrocyte skeletons. J. Cell Sci 107, 1633–1639 (1994). [DOI] [PubMed] [Google Scholar]
- 32.Pan L, Yan R, Li W & Xu K Super-resolution microscopy reveals the native ultrastructure of the erythrocyte cytoskeleton. Cell Rep 22, 1151–1158 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Han B, Zhou R, Xia C & Zhuang X Structural organization of the actin–spectrin-based membrane skeleton in dendrites and soma of neurons. Proc. Natl Acad. Sci. USA 114, E6678–E6685 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu K, Zhong G & Zhuang X Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science 339, 452–456 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leterrier C Putting the axonal periodic scaffold in order. Curr. Opin. Neurobiol 69, 33–40 (2021). [DOI] [PubMed] [Google Scholar]
- 36.D’Este E et al. Subcortical cytoskeleton periodicity throughout the nervous system. Sci. Rep 6, 22741 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He J et al. Prevalent presence of periodic actin–spectrin-based membrane skeleton in a broad range of neuronal cell types and animal species. Proc. Natl Acad. Sci. USA 113, 6029–6034 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhong G et al. Developmental mechanism of the periodic membrane skeleton in axons. eLife 3, e04581 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hofmann M et al. Cytoskeletal assembly in axonal outgrowth and regeneration analyzed on the nanoscale. Sci. Rep 12, 14387 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leite SC et al. The actin-binding protein α-adducin is required for maintaining axon diameter. Cell Rep 15, 490–498 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lorenzo DN et al. βII-Spectrin promotes mouse brain connectivity through stabilizing axonal plasma membranes and enabling axonal organelle transport. Proc. Natl Acad. Sci. USA 116, 15686–15695 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cousin MA et al. Pathogenic SPTBN1 variants cause an autosomal dominant neurodevelopmental syndrome. Nat. Genet 53, 1006–1021 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hammarlund M, Jorgensen EM & Bastiani MJ Axons break in animals lacking β-spectrin. J. Cell Biol 176, 269–275 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Law R et al. Cooperativity in forced unfolding of tandem spectrin repeats. Biophys. J 84, 533–544 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dubey S et al. The axonal actin–spectrin lattice acts as a tension buffering shock absorber. eLife 9, e51772 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang T et al. Radial contractility of actomyosin rings facilitates axonal trafficking and structural stability. J. Cell Biol 219, e201902001 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Costa AR et al. The membrane periodic skeleton is an actomyosin network that regulates axonal diameter and conduction. eLife 9, e55471 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Galiano MR et al. A distal axonal cytoskeleton forms an intra-axonal boundary that controls axon initial segment assembly. Cell 149, 1125–1139 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Albrecht D et al. Nanoscopic compartmentalization of membrane protein motion at the axon initial segment. J. Cell Biol 215, 37–46 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qu Y, Hahn I, Webb SE, Pearce SP & Prokop A Periodic actin structures in neuronal axons are required to maintain microtubules. Mol. Biol. Cell 28, 296–308 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lacas-Gervais S et al. βIVΣ1 spectrin stabilizes the nodes of Ranvier and axon initial segments. J. Cell Biol 166, 983–990 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang CY et al. αII spectrin forms a periodic cytoskeleton at the axon initial segment and is required for nervous system function. J. Neurosci 37, 11311–11322 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leterrier C et al. Nanoscale architecture of the axon initial segment reveals an organized and robust scaffold. Cell Rep 13, 2781–2793 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Komada M & Soriano P βIV-Spectrin regulates sodium channel clustering through ankyrin-G at axon initial segments and nodes of Ranvier. J. Cell Biol 156, 337–348 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang Y, Ogawa Y, Hedstrom KL & Rasband MN βIV spectrin is recruited to axon initial segments and nodes of Ranvier by ankyrinG. J. Cell Biol 176, 509–519 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang R et al. Neurodevelopmental mutation of giant ankyrin-G disrupts a core mechanism for axon initial segment assembly. Proc. Natl Acad. Sci. USA 116, 19717–19726 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jenkins PM et al. Giant ankyrin-G: a critical innovation in vertebrate evolution of fast and integrated neuronal signaling. Proc. Natl Acad. Sci. USA 112, 957–964 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu CH et al. β spectrin-dependent and domain specific mechanisms for Na+ channel clustering. eLife 9, e56629 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ho TS-Y et al. A hierarchy of ankyrin–spectrin complexes clusters sodium channels at nodes of Ranvier. Nat. Neurosc 17, 1664–1672 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rasband MN & Peles E Mechanisms of node of Ranvier assembly. Nat. Rev. Neurosci 22, 7–20 (2021). [DOI] [PubMed] [Google Scholar]
- 61.D’Este E, Kamin D, Gottfert F, El-Hady A & Hell SW STED nanoscopy reveals the ubiquity of subcortical cytoskeleton periodicity in living neurons. Cell Rep 10, 1246–1251 (2015). [DOI] [PubMed] [Google Scholar]
- 62.D’Este E, Kamin D, Balzarotti F & Hell SW Ultrastructural anatomy of nodes of Ranvier in the peripheral nervous system as revealed by STED microscopy. Proc. Natl Acad. Sci. USA 114, E191–E199 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yoshimura T, Stevens SR, Leterrier C, Stankewich MC & Rasband MN Developmental changes in expression of βIV spectrin splice variants at axon initial segments and nodes of Ranvier. Front. Cell. Neurosci 10, 304 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu CH et al. Nodal β spectrins are required to maintain Na+ channel clustering and axon integrity. eLife 9, e52378 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ogawa Y et al. Spectrins and ankyrinB constitute a specialized paranodal cytoskeleton. J. Neurosci 26, 5230–5239 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Amor V et al. The paranodal cytoskeleton clusters Na+ channels at nodes of Ranvier. eLife 6, e21392 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang C, Susuki K, Zollinger DR, Dupree JL & Rasband MN Membrane domain organization of myelinated axons requires βII spectrin. J. Cell Biol 203, 437–443 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huang CY-M, Zhang C, Zollinger DR, Leterrier C & Rasband MN An αII spectrin-based cytoskeleton protects large-diameter myelinated axons from degeneration. J. Neurosci 37, 11323–11334 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bär J, Kobler O, van Bommel B & Mikhaylova M Periodic F-actin structures shape the neck of dendritic spines. Sci. Rep 6, 37136 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sidenstein SC et al. Multicolour multilevel STED nanoscopy of actin/spectrin organization at synapses. Sci. Rep 6, 26725 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lorenzo DN et al. A PIK3C3–ankyrin-B–dynactin pathway promotes axonal growth and multiorganelle transport. J. Cell Biol 207, 735–752 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stankewich MC et al. Targeted deletion of βIII spectrin impairs synaptogenesis and generates ataxic and seizure phenotypes. Proc. Natl Acad. Sci. USA 107, 6022–6027 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Efimova N et al. βIII spectrin is necessary for formation of the constricted neck of dendritic spines and regulation of synaptic activity in neurons. J. Neurosci 37, 6442–6459 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Armbrust KR et al. Mutant β-III spectrin causes mGluR1α mislocalization and functional deficits in a mouse model of spinocerebellar ataxia type 5. J. Neurosci 34, 9891–9904 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ikeda Y et al. Spectrin mutations cause spinocerebellar ataxia type 5. Nat. Genet 38, 184–190 (2006). [DOI] [PubMed] [Google Scholar]
- 76.Wechsler A & Teichberg VI Brain spectrin binding to the NMDA receptor is regulated by phosphorylation, calcium and calmodulin. EMBO J 17, 3931–3939 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lambert S & Bennett V Postmitotic expression of ankyrinR and β R-spectrin in discrete neuronal populations of the rat brain. J. Neurosci 13, 3725–3735 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Malchiodi-Albedi F, Ceccarini M, Winkelmann JC, Morrow JS & Petrucci TC The 270 kDa splice variant of erythrocyte β-spectrin (βI Σ2) segregates in vivo and in vitro to specific domains of cerebellar neurons. J. Cell Sci 106, 67–78 (1993). [DOI] [PubMed] [Google Scholar]
- 79.Fifková E & Morales M Actin matrix of dendritic spines, synaptic plasticity, and long-term potentiation. Int. Rev. Cytol 139, 267–307 (1992). [DOI] [PubMed] [Google Scholar]
- 80.Sytnyk V, Leshchyns’ka I, Nikonenko AG & Schachner M NCAM promotes assembly and activity-dependent remodeling of the postsynaptic signaling complex. J. Cell Biol 174, 1071–1085 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ursitti JA et al. Spectrins in developing rat hippocampal cells. Brain Res. Dev. Brain Res 129, 81–93 (2001). [DOI] [PubMed] [Google Scholar]
- 82.Nestor MW, Cai X, Stone MR, Bloch RJ & Thompson SM The actin binding domain of βI-spectrin regulates the morphological and functional dynamics of dendritic spines. PLoS One 6, e16197 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Smith KR et al. Psychiatric risk factor ANK3/ankyrin-G nanodomains regulate the structure and function of glutamatergic synapses. Neuron 84, 399–415 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lorenzo DN et al. Spectrin mutations that cause spinocerebellar ataxia type 5 impair axonal transport and induce neurodegeneration in Drosophila. J. Cell Biol 189, 143–158 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cheney R, Hirokawa N, Levine J & Willard M Intracellular movement of fodrin. Cell Motil 3, 649–655 (1983). [DOI] [PubMed] [Google Scholar]
- 86.Takeda S et al. Kinesin superfamily protein 3 (KIF3) motor transports fodrin-associating vesicles important for neurite building. J. Cell Biol 148, 1255–1265 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Holleran EA et al. βIII spectrin binds to the Arp1 subunit of dynactin. J. Biol. Chem 276, 36598–36605 (2001). [DOI] [PubMed] [Google Scholar]
- 88.Muresan V et al. Dynactin-dependent, dynein-driven vesicle transport in the absence of membrane proteins: a role for spectrin and acidic phospholipids. Mol. Cell 7, 173–183 (2001). [DOI] [PubMed] [Google Scholar]
- 89.He M, Abdi KM & Bennett V Ankyrin-G palmitoylation and βIIspectrin binding to phosphoinositide lipids drive lateral membrane assembly. J. Cell Biol 206, 273e288 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hyvönen M et al. Structure of the binding site for inositol phosphates in a PH domain. EMBO J 14, 4676–4685 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sikorski AF, Terlecki G, Zagon IS & Goodman SR Synapsin I-mediated interaction of brain spectrin with synaptic vesicles. J. Cell Biol 114, 313–318 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stankewich MC et al. Cell organization, growth, and neural and cardiac development require αII-spectrin. J. Cell Sci 124, 3956–3966 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang Y et al. Critical roles of αII spectrin in brain development and epileptic encephalopathy. J. Clin. Invest 128, 760–773 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Beijer D et al. Nonsense mutations in α-II spectrin in three families with juvenile onset hereditary motor neuropathy. Brain 142, 2605–2616 (2019). [DOI] [PubMed] [Google Scholar]
- 95.Dong HL, Chen L & Wu ZY A novel de novo SPTAN1 nonsense variant causes hereditary motor neuropathy in a Chinese family. Brain 144, e11 (2021). [DOI] [PubMed] [Google Scholar]
- 96.Ylikallio E et al. De novo SPTAN1 mutation in axonal sensorimotor neuropathy and developmental disorder. Brain 143, 6–8 (2020). [DOI] [PubMed] [Google Scholar]
- 97.Miazek A et al. Age-dependent ataxia and neurodegeneration caused by an αII spectrin mutation with impaired regulation of its calpain sensitivity. Sci. Rep 11, 7312 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Van de Vondel L et al. De novo and dominantly inherited SPTAN1 mutations cause spastic paraplegia and cerebellar ataxia. Mov. Disord 10.1002/mds.28959 (2022). [DOI] [PMC free article] [PubMed]
- 99.Tang Y et al. Disruption of transforming growth factor-β signaling in ELF β-spectrin-deficient mice. Science 299, 574–577 (2003). [DOI] [PubMed] [Google Scholar]
- 100.Liu Y et al. Critical role of spectrin in hearing development and deafness. Sci. Adv 5, eaav7803 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rosenfeld JA et al. Heterozygous variants in SPTBN1 cause intellectual disability and autism. Am. J. Med. Genet. A 185, 2037–2045 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jackson M et al. Modulation of the neuronal glutamate transporter EAAT4 by two interacting proteins. Nature 410, 89–93 (2001). [DOI] [PubMed] [Google Scholar]
- 103.Clarkson YL et al. β-III spectrin underpins ankyrin R function in Purkinje cell dendritic trees: protein complex critical for sodium channel activity is impaired by SCA5-associated mutations. Hum. Mol. Genet 23, 3875–3882 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Perkins EM et al. Loss of β-III spectrin leads to Purkinje cell dysfunction recapitulating the behavior and neuropathology of spinocerebellar ataxia type 5 in humans. J. Neurosci 30, 4857–4867 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lise S et al. Recessive mutations in SPTBN2 implicate β-III spectrin in both cognitive and motor development. PLoS Genet 8, e1003074 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Clarkson YL, Gillespie T, Perkins EM, Lyndon AR & Jackson M β-III spectrin mutation L253P associated with spinocerebellar ataxia type 5 interferes with binding to Arp1 and protein trafficking from the Golgi. Hum. Mol. Genet 19, 3634–3641 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Parolin Schnekenberg R et al. De novo point mutations in patients diagnosed with ataxic cerebral palsy. Brain 138, 1817–1832 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jacob FD, Ho ES, Martinez-Ojeda M, Darras BT & Khwaja OS Case of infantile onset spinocerebellar ataxia type 5. J. Child. Neurol 28, 1292–1295 (2013). [DOI] [PubMed] [Google Scholar]
- 109.Nuovo S et al. Between SCA5 and SCAR14: delineation of the SPTBN2 p.R480W-associated phenotype. Eur. J. Hum. Genet 26, 928–929 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nicita F et al. Heterozygous missense variants of SPTBN2 are a frequent cause of congenital cerebellar ataxia. Clin. Genet 96, 169–175 (2019). [DOI] [PubMed] [Google Scholar]
- 111.Mizuno T et al. Infantile-onset spinocerebellar ataxia type 5 associated with a novel SPTBN2 mutation: a case report. Brain Dev 41, 630–633 (2019). [DOI] [PubMed] [Google Scholar]
- 112.Romaniello R et al. Novel SPTBN2 gene mutation and first intragenic deletion in early onset spinocerebellar ataxia type 5. Ann. Clin. Transl. Neurol 8, 956–963 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sancho P et al. Expanding the β-III spectrin-associated phenotypes toward non-progressive congenital ataxias with neurodegeneration. Int. J. Mol. Sci 22, 2505 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Parkinson NJ et al. Mutant β-spectrin 4 causes auditory and motor neuropathies in quivering mice. Nat. Genet 29, 61–65 (2001). [DOI] [PubMed] [Google Scholar]
- 115.Wang CC et al. βIV spectrinopathies cause profound intellectual disability, congenital hypotonia, and motor axonal neuropathy. Am. J. Hum. Genet 102, 1158–1168 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yang Y et al. βIV spectrins are essential for membrane stability and the molecular organization of nodes of Ranvier. J. Neurosci 24, 7230–7240 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Devaux JJ The C-terminal domain of βIV-spectrin is crucial for KCNQ2 aggregation and excitability at nodes of Ranvier. J. Physiol 588, 4719–4730 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Stevens SR et al. Ankyrin-R regulates fast-spiking interneuron excitability through perineuronal nets and Kv3.1b K+ channels. eLife 10, e66491 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Stankewich MC et al. Outer hair cell function is normal in βV spectrin knockout mice. Hear. Res 423, 108564 (2022). [DOI] [PubMed] [Google Scholar]
- 120.Saitsu H et al. Dominant-negative mutations in α-II spectrin cause West syndrome with severe cerebral hypomyelination, spastic quadriplegia, and developmental delay. Am. J. Hum. Genet 86, 881–891 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Writzl K et al. Early onset West syndrome with severe hypomyelination and coloboma-like optic discs in a girl with SPTAN1 mutation. Epilepsia 53, e106–e110 (2012). [DOI] [PubMed] [Google Scholar]
- 122.Hamdan FF et al. Identification of a novel in-frame de novo mutation in SPTAN1 in intellectual disability and pontocerebellar atrophy. Eur. J. Med. Genet 20, 796–800 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nonoda Y et al. Progressive diffuse brain atrophy in West syndrome with marked hypomyalination due to SPTAN1 gene mutation. Brain Dev 35, 280–283 (2013). [DOI] [PubMed] [Google Scholar]
- 124.Gilissen C et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 511, 344–347 (2014). [DOI] [PubMed] [Google Scholar]
- 125.Ream MA & Mikati MA Clinical utility of genetic testing in pediatric drug-resistant epilepsy: a pilot study. Epilepsy Behav 37, 241–248 (2014). [DOI] [PubMed] [Google Scholar]
- 126.Yavarna T et al. High diagnostic yield of clinical exome sequencing in Middle Eastern patients with Mendelian disorders. Hum. Genet 134, 967–980 (2015). [DOI] [PubMed] [Google Scholar]
- 127.Tohyama J et al. SPTAN1 encephalopathy: distinct phenotypes and genotypes. J. Hum. Genet 60, 167–173 (2015). [DOI] [PubMed] [Google Scholar]
- 128.Retterer K et al. Clinical application of whole-exome sequencing across clinical indications. Genet. Med 18, 696–704 (2016). [DOI] [PubMed] [Google Scholar]
- 129.Stavropoulos DJ et al. Whole genome sequencing expands diagnostic utility and improves clinical management in pediatric medicine. NPJ Genom. Med 1, 15012 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Syrbe S et al. Delineating SPTAN1 associated phenotypes: from isolated epilepsy to encephalopathy with progressive brain atrophy. Brain 140, 2322–2336 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rapaccini V et al. A child with a c.6923_6928dup (p.Arg2308_Met2309dup) SPTAN1 mutation associated with a severe early infantile epileptic encephalopathy. Int. J. Mol. Sci 19, 1976 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Terrone G et al. Intrafamilial variability in SPTAN1-related disorder: from benign convulsions with mild gastroenteritis to developmental encephalopathy. Eur. J. Paediatr. Neurol 28, 237–239 (2020). [DOI] [PubMed] [Google Scholar]
- 133.Leveille E et al. SPTAN1 variants as a potential cause for autosomal recessive hereditary spastic paraplegia. J. Hum. Genet 64, 1145–1151 (2019). [DOI] [PubMed] [Google Scholar]
- 134.Xie F, Chen S, Liu P, Chen X & Luo W SPTAN1 variants likely cause autosomal recessive complicated hereditary spastic paraplegia. J. Hum. Genet 67, 165–168 (2021). [DOI] [PubMed] [Google Scholar]
- 135.Gartner V et al. Novel variants in SPTAN1 without epilepsy: an expansion of the phenotype. Am. J. Med. Genet 176, 2768–2776 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Marco Hernández AV et al. Extending the clinical phenotype of SPTAN1: from DEE5 to migraine, epilepsy, and subependymal heterotopias without intellectual disability. Am. J. Med. Genet. A 188, 147–159 (2022). [DOI] [PubMed] [Google Scholar]
- 137.Luongo-Zink C et al. Longitudinal neurodevelopmental profile of a pediatric patient with de novo SPTAN1, epilepsy, and left hippocampal sclerosis. Epilepsy Behav. Rep 19, 100550 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Satterstrom FK et al. Autism spectrum disorder and attention deficit hyperactivity disorder have a similar burden of rare protein-truncating variants. Nat. Neurosci 22, 1961–1965 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lek M et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cho E & Fogel BL A family with spinocerebellar ataxia type 5 found to have a novel missense mutation within a SPTBN2 spectrin repeat. Cerebellum 12, 162–164 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bian X et al. Two novel missense variants in SPTBN2 likely associated with spinocerebellar ataxia type 5. Neurol. Sci 42, 5195–5203 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wang Y et al. A Japanese SCA5 family with a novel three-nucleotide in-frame deletion mutation in the SPTBN2 gene: a clinical and genetic study. J. Hum. Genet 59, 569–573 (2014). [DOI] [PubMed] [Google Scholar]
- 143.Zonta A, Brussino A, Dentelli P & Brusco A A novel case of congenital spinocerebellar ataxia 5: further support for a specific phenotype associated with the p.(Arg480Trp) variant in SPTBN2. BMJ Case Rep 13, e238108 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Valentino F et al. Exome sequencing in 200 intellectual disability/autistic patients: new candidates and atypical presentations. Brain Sci 11, 936 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Accogli A et al. Heterozygous missense pathogenic variants within the second spectrin repeat of SPTBN2 lead to infantile-onset cerebellar ataxia. J. Child. Neurol 35, 106–110 (2019). [DOI] [PubMed] [Google Scholar]
- 146.Yıldız Bölükbaşı E et al. Progressive SCAR14 with unclear speech, developmental delay, tremor, and behavioral problems caused by a homozygous deletion of the SPTBN2 pleckstrin homology domain. Am. J. Med. Genet. A 173, 2494–2499 (2017). [DOI] [PubMed] [Google Scholar]
- 147.Al-Muhaizea MA et al. A novel homozygous mutation in SPTBN2 leads to spinocerebellar ataxia in a consanguineous family: report of a new infantile-onset case and brief review of the literature. Cerebellum 17, 276–285 (2018). [DOI] [PubMed] [Google Scholar]
- 148.Fogel BL et al. Exome sequencing in the clinical diagnosis of sporadic or familial cerebellar ataxia. JAMA Neurol 71, 1237–1246 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Elsayed SM et al. Autosomal dominant SCA5 and autosomal recessive infantile SCA are allelic conditions resulting from SPTBN2 mutations. Eur. J. Hum. Genet 22, 286–288 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Liu LZ et al. A novel missense mutation in the spectrin β nonerythrocytic 2 gene likely associated with spinocerebellar ataxia type 5. Chin. Med. J 129, 2516–2517 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Rea G, Tirupathi S, Williams J, Clouston P & Morrison PJ Infantile onset of spinocerebellar ataxia type 5 (SCA-5) in a 6 month old with ataxic cerebral palsy. Cerebellum 19, 161–163 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Spagnoli C et al. Infantile-onset spinocerebellar ataxia type 5 (SCA5) with optic atrophy and peripheral neuropathy. Cerebellum 20, 481–483 (2021). [DOI] [PubMed] [Google Scholar]
- 153.Ranum LPW, Schut LJ, Lundgren JK, Orr HT & Livingston DM Spinocerebellar ataxia gene type 5 in a family descended from the paternal grandparents of President Lincoln maps to chromosome 11. Nat. Genet 8, 280–284 (1994). [DOI] [PubMed] [Google Scholar]
- 154.Stevanin G, Herman A, Brice A & Durr A Clinical and MRI findings in spinocerebellar ataxia type 5. Neurology 53, 1355–1357 (1999). [DOI] [PubMed] [Google Scholar]
- 155.Burk K et al. Spinocerebellar ataxia type 5: clinical and molecular genetic features of a German kindred. Neurology 62, 327–329 (2004). [DOI] [PubMed] [Google Scholar]
- 156.Sun M et al. Targeted exome analysis identifies the genetic basis of disease in over 50% of patients with a wide range of ataxia-related phenotypes. Genet. Med 21, 195–206 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Avery AW, Crain J, Thomas DD & Hays TS A human β-III-spectrin spinocerebellar ataxia type 5 mutation causes high-affinity F-actin binding. Sci. Rep 6, 21375 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Knierim E et al. A recessive mutation in β-IV-spectrin (SPTBN4) associates with congenital myopathy, neuropathy, and central deafness. Hum. Genet 136, 903–910 (2017). [DOI] [PubMed] [Google Scholar]
- 159.Häusler MG et al. A novel homozygous splice-site mutation in the SPTBN4 gene causes axonal neuropathy without intellectual disability. Eur. J. Med. Genet 63, 103826 (2020). [DOI] [PubMed] [Google Scholar]
- 160.Buelow M et al. Novel bi-allelic variants expand the SPTBN4-related genetic and phenotypic spectrum. Eur. J. Hum. Genet 29, 1121–1128 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Belkheir AM et al. Severe form of βIV-spectrin deficiency with mitochondrial dysfunction and cardiomyopathy—a case report. Front. Neurol 12, 643805 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Anazi S et al. Expanding the genetic heterogeneity of intellectual disability. Hum. Genet 136, 1419–1429 (2017). [DOI] [PubMed] [Google Scholar]
- 163.Pehlivan D et al. The genomics of arthrogryposis, a complex trait: candidate genes and further evidence for oligogenic inheritance. Am. J. Hum. Genet 105, 132–150 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Avery AW et al. Structural basis for high-affinity actin binding revealed by a β-III-spectrin SCA5 missense mutation. Nat. Commun 8, 1350 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Creighton BA et al. Giant ankyrin-B mediates transduction of axon guidance and collateral branch pruning factor sema 3A. eLife 10, e69815 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Khan A et al. SPTBN5, encoding the βV-spectrin protein, leads to a syndrome of intellectual disability, developmental delay, and seizures. Front. Mol. Neurosci 15, 877258 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Berghs S et al. Autoimmunity to βIV spectrin in paraneoplastic lower motor neuron syndrome. Proc. Natl Acad. Sci. USA 98, 6945–6950 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bartley CM et al. βIV-Spectrin autoantibodies in 2 individuals with neuropathy of possible paraneoplastic origin: a case series. Neurol. Neuroimmunol. Neuroinflamm 9, e1188 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Sanchez-Mut JV et al. DNA methylation map of mouse and human brain identifies target genes in Alzheimer’s disease. Brain 136, 3018–3027 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hüls A et al. Newborn differential DNA methylation and subcortical brain volumes as early signs of severe neurodevelopmental delay in a South African Birth Cohort Study. World J. Biol. Psychiatry 1–12 (2022). [DOI] [PMC free article] [PubMed]
- 171.Sihag RK & Cataldo AM Brain β-spectrin is a component of senile plaques in Alzheimer’s disease. Brain Res 743, 249–257 (1996). [DOI] [PubMed] [Google Scholar]
- 172.Czogalla A & Sikorski AF Spectrin and calpain: a ‘target’ and a ‘sniper’ in the pathology of neuronal cells. Cell Mol. Life Sci 62, 1913–1924 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Leverenz JB et al. Proteomic identification of novel proteins in cortical Lewy bodies. Brain Pathol 17, 139–145 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Peuralinna T et al. Genome-wide association study of neocortical Lewy-related pathology. Ann. Clin. Transl. Neurol 2, 920–931 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ordonez DG, Lee MK & Feany MB α-Synuclein induces mitochondrial dysfunction through spectrin and the actin cytoskeleton. Neuron 97, 108–124 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Susuki K et al. Glial βII spectrin contributes to paranode formation and maintenance. J. Neurosci 38, 6063–6075 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Neniskyte U & Gross CT Errant gardeners: glial-cell-dependent synaptic pruning and neurodevelopmental disorders. Nat. Rev. Neurosci 18, 658–670 (2017). [DOI] [PubMed] [Google Scholar]
- 178.Patel DC, Tewari BP, Chaunsali L & Sontheimer H Neuron–glia interactions in the pathophysiology of epilepsy. Nat. Rev. Neurosci 20, 282–297 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lukens JR & Eyo UB Microglia and neurodevelopmental disorders. Annu. Rev. Neurosci 45, 225–245 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Saifetiarova J, Shi Q, Paukert M, Komada M & Bhat MA Reorganization of destabilized nodes of Ranvier in βIV spectrin mutants uncovers critical timelines for nodal restoration and prevention of motor paresis. J. Neurosci 38, 6267–6282 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Whiteley JT et al. Reaching into the toolbox: stem cell models to study neuropsychiatric disorders. Stem Cell Rep 17, 187–210 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Rinaldi C & Wood MJA Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat. Rev. Neurol 14, 9–21 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.