

ABSTRACT
To eradicate bacterial pathogens, neutrophils are recruited to the sites of infection, where they engulf and kill microbes through the production of reactive oxygen and chlorine species (ROS/RCS). The most prominent RCS is the antimicrobial oxidant hypochlorous acid (HOCl), which rapidly reacts with various amino acid side chains, including those containing sulfur and primary/tertiary amines, causing significant macromolecular damage. Pathogens like uropathogenic Escherichia coli (UPEC), the primary causative agent of urinary tract infections, have developed sophisticated defense systems to protect themselves from HOCl. We recently identified the RcrR regulon as a novel HOCl defense strategy in UPEC. Expression of the rcrARB operon is controlled by the HOCl-sensing transcriptional repressor RcrR, which is oxidatively inactivated by HOCl resulting in the expression of its target genes, including rcrB. The rcrB gene encodes a hypothetical membrane protein, deletion of which substantially increases UPEC’s susceptibility to HOCl. However, the mechanism behind protection by RcrB is unclear. In this study, we investigated whether (i) its mode of action requires additional help, (ii) rcrARB expression is induced by physiologically relevant oxidants other than HOCl, and (iii) expression of this defense system is limited to specific media and/or cultivation conditions. We provide evidence that RcrB expression is sufficient to protect E. coli from HOCl. Furthermore, RcrB expression is induced by and protects from several RCS but not from ROS. RcrB plays a protective role for RCS-stressed planktonic cells under various growth and cultivation conditions but appears to be irrelevant for UPEC’s biofilm formation.
IMPORTANCE
Bacterial infections pose an increasing threat to human health, exacerbating the demand for alternative treatments. Uropathogenic Escherichia coli (UPEC), the most common etiological agent of urinary tract infections (UTIs), are confronted by neutrophilic attacks in the bladder, and must therefore be equipped with powerful defense systems to fend off the toxic effects of reactive chlorine species. How UPEC deal with the negative consequences of the oxidative burst in the neutrophil phagosome remains unclear. Our study sheds light on the requirements for the expression and protective effects of RcrB, which we recently identified as UPEC’s most potent defense system toward hypochlorous acid (HOCl) stress and phagocytosis. Thus, this novel HOCl stress defense system could potentially serve as an attractive drug target to increase the body’s own capacity to fight UTIs.
KEYWORDS: oxidative stress, transcriptional regulation, reactive chlorine species, bacterial defense systems, hypochlorous acid
INTRODUCTION
The human host uses oxidative stress as a strategy to limit bacterial infections (1, 2). For instance, during phagocytosis, neutrophils and macrophages produce high levels of reactive oxygen species (ROS) and reactive chlorine species (RCS) to kill ingested microbes (3, 4). To initiate the powerful oxidative burst, activated neutrophils release antimicrobial enzymes from granules into the phagosome, such as NADPH oxidase 2 (NOX2) and myeloperoxidase (MPO) (5). The process starts with the assembly of NOX2 into the phagosomal membrane, where the enzyme reduces molecular oxygen into superoxide by oxidizing NADPH. Superoxide is then dismutated into hydrogen peroxide (H2O2) (6), a ROS that is fairly well tolerated by bacteria. However, MPO uses H2O2 as a substrate to catalyze the oxidation of halides (i.e., Cl- and Br-) and pseudohalides (i.e., SCN-) into highly antimicrobial hypohalous acids (HOX). These include hypochlorous acid (HOCl), hypobromous acid (HOBr), and hypothiocyanous acid (HOSCN), respectively (3, 5, 7). The outcome of HOX production is determined by the differences in availability of the three anions as well as the distinct selectivity of MPO for each (pseudo-)halide. Approximately 70%–80% of all H2O2 consumed by MPO yields HOCl and the remainder is accounted for by HOSCN, as the formation of HOBr is negligibly small due to the low bromide concentration available (8).
HOCl is characterized by its high oxidizing capacity with virtually any cellular macromolecule, including lipids, nucleic acid, and proteins (9 – 11). These oxidative modifications can cause protein aggregation, DNA strand cleavage, mis-metalation, ATP depletion, and a substantial reduction in free thiols, ultimately causing significant macromolecular damage and cell death. A common target of all HOX is the amino acid cysteine, which is subject to reversible (i.e., sulfenic acids, disulfide bonds) or irreversible thiol modifications (i.e., sulfinic and sulfonic acid) (3, 12 – 14). While irreversible thiol modifications can lead to protein aggregation and degradation, reversible thiol modifications often come along with significant structural and functional consequences that may play important roles for the cellular redox homeostasis (13 – 15). Notably, many bacteria utilize reversible cysteine modifications as a tool to activate appropriate response and defense systems against oxidative stress (14, 16). Moreover, HOCl reacts with primary and secondary amines (i.e., in arginine and lysine) resulting in the production of chloramines, which are also considered antimicrobial despite their four to five magnitudes lower reactivity compared to HOCl (17). Regardless, formation of these additional RCS further extends the oxidizing capabilities of HOCl on cellular macromolecules and illustrates the difficulty of dissecting their individual contributions to antimicrobial killing in vivo (11). In addition to cysteine, HOCl also targets methionine residues, resulting in the formation of methionine sulfoxides, which can result in damage or gain of function in proteins (18). However, many bacteria have evolved methionine sulfoxide reductases to reduce methionine sulfoxides (19).
To mitigate the toxic effects of HOX, bacteria have evolved defense systems on both transcriptional and post-translational levels. Redox-sensing transcriptional regulators use conserved cysteine and/or methionine residues to modulate their activity (14, 16, 20, 21). Their HOX-mediated (in-)activation results in elevated transcript level of their target genes, many of which have been shown to protect the organism from ROS/RCS stress (17). Four HOCl-responsive transcriptional regulators have been identified in Escherichia coli. NemR is a transcriptional repressor that is inactivated by HOCl, resulting in the expression of nemA, an N-Ethylmaleimide reductase, and gloA, a glyoxylase, both of which have been shown to contribute to HOCl resistance in vitro (22, 23). The HOCl-sensing transcriptional activator RclR induces the transcription of rclABC (24). While the functions of RclB and RclC are still unknown, recent mechanistic studies shed light into RclA’s protective role for HOCl- and HOSCN-stressed E. coli (25, 26). While NemR and RclR sense HOCl through conserved cysteine residues, the transcriptional regulator HypT is activated through methionine oxidation and induces the transcription of genes involved in methionine and cysteine biosynthesis (27, 28). We recently reported that in comparison to E. coli K-12 and enteropathogenic E. coli (EPEC) strains, uropathogenic E. coli (UPEC) are substantially more resistant to HOCl exposure and killing by neutrophils due to the activation of the RcrR regulon, an additional HOCl defense system that many of the intestinal E. coli pathotypes lack (29). The UPEC RcrR regulon is regulated by the TetR-family transcriptional repressor RcrR, which becomes inactivated through reversible cysteine oxidation by HOCl resulting in the de-repression of the three structural genes rcrARB. UPEC’s increased HOCl resistance appears to be mediated by RcrB, a putative inner membrane protein of unknown function, as rcrB-deficient UPEC strains were as susceptible to HOCl as the HOCl-sensitive intestinal E. coli strains tested (29). However, the most intriguing mystery yet to decipher concerns RcrB’s precise mode of action. In a first attempt to understand the role of this protein during HOCl stress, we were wondering whether RcrB requires additional help for its protective effects. Recombinant expression of RcrB in E. coli strains that lack the operon suggests that RcrB is exclusively responsible for the increased HOCl resistance. We further provide evidence that RcrB expression is induced by and protects from several RCS, but not from ROS stress. RcrB plays a highly protective role for RCS-stressed planktonic cells under various growth and cultivation conditions but appears to be irrelevant for UPEC biofilm formation. Lastly, expression studies suggest a major role for RcrB, which cannot be complemented by its homolog RclC.
RESULTS
Recombinant expression of RcrB significantly improves HOCl resistance in RcrB-deficient E. coli strains
We recently reported that UPEC strains, including CFT073, an isolate from the blood of a patient with acute pyelonephritis (30), tolerate higher levels of HOCl compared to other E. coli pathotypes (29). We identified the protective gene cluster responsible for this phenotype, which is predominantly present in UPEC as well as in various invasive E. coli strains and consists of the three uncharacterized genes rcrA, rcrR, and rcrB. While we identified RcrR as an HOCl-sensing transcriptional repressor, the function of rcrA and rcrB remained unknown (29). Deletion of rcrB completely abolished CFT073’s increased resistance to HOCl, suggesting that the putative membrane protein RcrB is responsible for UPEC’s elevated HOCl resistance. To determine whether RcrB alone is indeed sufficient for the bacterial protection against HOCl and to examine whether its protective role extends beyond CFT073, we compared the growth behavior of different E. coli strains in the presence and absence of RcrB during sublethal HOCl stress. To reduce the possibility that media components react with and potentially quench HOCl, we performed our established lag phase extension (LPE)-based growth assay in MOPS-glucose (MOPSg) minimal media. We tested the impact of the presence and absence of RcrB in different strain backgrounds, including strains that naturally lack rcrB, by expressing plasmid-encoded RcrB in the presence of various HOCl concentrations. Empty vector (EV) strains served as controls. Transformation of a ∆rcrB strain in the CFT073 background resulted in full complementation of the UPEC’s increased HOCl resistance (Fig. S1A). Likewise, the same plasmid restored the HOCl resistance of the probiotic strain E. coli Nissle 1917 (EcN), a close relative of UPEC strain CFT073 (31), with an in-frame deletion in the gene homologous to rcrB (i.e., ∆rcrB-H) (Fig. S1B). Taken together, these data suggest that leaky RcrB expression from the pET28a plasmid was sufficient to restore full HOCl resistance.
In our previous study, we showed that K-12 strain MG1655, which does not contain rcrB, is significantly more sensitive to HOCl than UPEC strain CFT073 (29). This is significant given that MG1655 is considered much more HOCl resistant than most other non-pathogenic laboratory E. coli strains. We transformed the rcrB-encoding pET28a plasmid into MG1655 and performed our LPE assay in the presence and absence of increasing HOCl concentrations. As expected, MG1655 transformed with the empty vector control pET28a was substantially more sensitive to HOCl than the pET28a-containing UPEC strain CFT073 (Fig. 1A). However, leaky expression of pET28a-encoded RcrB resulted in LPE that was almost identical to CFT073, suggesting that RcrB alone and in the amount produced is sufficient to render MG1655 as resistant to HOCl as CFT073 (Fig. 1A). CFT073 cells carrying the rcrB-pET28a plasmid also showed a slight but significant increase in HOCl resistance. Like MG1655, the cystitis isolate UTI89, another UPEC model strain (32), also lacks rcrB, and its LPE under sublethal HOCl stress resembled that of MG1655 (Fig. 1B). Expression of pET28a-encoded RcrB in UTI89 resulted in substantially reduced LPE, which was even shorter than what we observed in pET28a-containing CFT073 (Fig. 1B). To confirm that the observed effect is indeed due to sufficient production of RcrB, we have cloned rcrB into the pBAD18 vector, transformed the plasmid into the ∆rcrB strain, and induced its expression by the presence of different concentrations of arabinose. ∆rcrB cells carrying the rcrB-pBAD18 plasmid were characterized by significantly shorter LPE than cells containing the empty vector even in the absence of the inducer arabinose, suggesting that leaky RcrB expression is sufficient to increase CFT073’s HOCl resistance (Fig. 1C). The presence of arabinose further stimulated the HOCl resistance, which is evidenced by even shorter LPE, likely due to the increased production of RcrB. Addition of arabinose had no effect on the HOCl resistance of empty vector control. To test whether the arabinose-induced overproduction of RcrB results in a decrease in cytoplasmic HOCl stress, we compared the transcript level of rcrA (first gene of the rcrARB operon) and ibpA (encodes the molecular chaperone IbpA, which is upregulated during HOCl stress) between the two strains in the presence of 0.2% arabinose. Our data show that increased expression of RcrB reduces the cellular demand for RcrA and IbpA, suggesting that the production of RcrB protects the cytoplasm from HOCl influx (Fig. 1D). All in all, our experiments confirm the prominent role that RcrB has for protecting various E. coli strains from the deleterious effects of HOCl and indicate that the protein likely does so without any additional help.
Fig 1.

. Recombinant expression of RcrB significantly improves HOCl resistance in different E. coli strains. (A, B) Growth phenotype analyses of the K-12 E. coli strain MG1655 and UPEC strains CFT073 and UTI89 carrying pET28a or rcrB-pET28a were performed in MOPSg media in the presence of the indicated HOCl concentrations. HOCl-mediated LPE was calculated for each strain (see Materials and Methods for a detailed protocol). Squares, MG1655; circles, CFT073; triangles, UTI89; orange filling, +RcrB. Expression of RcrB in (A) MG1655 and (B) UTI89 resulted in an increased HOCl resistance and is comparable to what we observed in the CFT073 strain, (n = 4–6, ± SD). The statistics for (A) MG1655 + pET28a versus MG1655 + rcrB-pET28a and CFT073 + pET28a versus CFT073 + rcrB-pET28a and (B) UTI89 + pET28 and UTI89 + rcrB-pET28 were calculated using Student’s paired t-test in prism. (C) Growth phenotype analyses of the UPEC strain CFT073 ∆rcrB carrying pBAD18a or rcrB-pBAD18a were performed in MOPSg media in the presence of the indicated HOCl concentrations. HOCl-mediated LPE was calculated for each strain (see Materials and Methods for a detailed protocol). The statistics for ∆rcrB + pBAD18a versus ∆rcrB + rcrB-pBAD18a in the presence and absence of 0.2% arabinose were calculated using Student’s paired t-test in prism (n = 4, ± SD). (D) The induction of ibpA and rcrA transcript levels was determined by quantitative real-time PCR. Gene expression was normalized to the housekeeping gene rrsD and calculated as fold changes based on expression levels in the untreated control. Symbols are * = P < 0.05; ** = P < 0.01; *** = P < 0.001; no *, P > 0.05.
The HOCl resistance of UPEC clinical isolates depends on the presence of RcrB
While both UTI89 and CFT073 were originally isolated from cystitis and pyelonephritis patients, respectively, they have been in the lab environment for decades and therefore may no longer serve as reliable representatives of UPEC infections. To further investigate the impact of RcrB on UPEC’s defense to HOCl stress, we used whole genome-sequenced clinical UPEC isolates from asymptomatic patients, as well as those suffering from cystitis and pyelonephritis, respectively (33). We used quantitative real-time PCR (qRT-PCR) as a first test to determine whether these UPEC clinical isolates upregulate rcrA and rcrB upon exposure to sublethal HOCl stress. Both genes are members of the RcrR regulon, the expression of which was induced in HOCl-stressed CFT073 (29). We confirmed the transcriptional upregulation of both genes in VUTI229, VUTI288, and VUTI313, which are representative clinical isolates from pyelonephritis, cystitis, and asymptomatic patients, respectively (Fig. 2A). Next, we compared the growth of 13 sequenced clinical UPEC isolates at sublethal HOCl stress to the HOCl-sensitive K-12 strain MG1655 and the HOCl-resistant UPEC strain CFT073. Our LPE assays revealed a strong correlation between increased HOCl resistance and the presence of rcrB, independent of whether the strain causes cystitis, pyelonephritis, or an asymptomatic infection (Fig. 2B through D). The HOCl resistance profiles of strains carrying the rcrB gene in their chromosome (i.e., VUTI150/188/229/288/308/313/317/319/332) were similar to those observed in CFT073 (Fig. 2B through D, compare blue and black circles). Similarly, strains that lack rcrB (i.e., VUTI207/334/369/409) resemble HOCl-sensitive E. coli strains such as MG1655 (Fig. 2B through D, compare red diamonds and white squares).
Fig 2.

The HOCl resistance of UPEC clinical isolates depends on the presence of RcrB. (A) UPEC clinical isolates VUTI229, VUTI288, and VUTI313 were grown to mid-log phase in MOPSg and changes in the expression of rcrA (black bars) and rcrB (white bars) upon 15-min exposure to 1 mM HOCl were determined by qRT-PCR. rcrA and rcrB mRNA levels were elevated in all three strains in the presence of HOCl. The statistics for the HOCl-induced rcrA and rcrB expressions were calculated using Student’s paired t-test in prism (n = 3–4, ± SD). * = P < 0.05. (B–D) Growth phenotype analyses of the indicated UPEC clinical isolates causing (B) an asymptomatic infection, (C) cystitis, and (D) pyelonephritis were performed in MOPSg media in the presence of the indicated HOCl concentrations and compared to the K-12 E. coli strain MG1655 and UPEC strain CFT073. HOCl-mediated LPE was calculated for each strain (see Materials and Methods for a detailed protocol). The presence (+) and absence (−) of the rcrB gene in the genome of each strain is indicated in the legend. Diamonds, clinical isolates lacking rcrB; circles, UPEC carrying rcrB; squares, MG1655. The increased HOCl resistance of UPEC clinical isolates depends on the presence of rcrB (n ≤ 4, ± SD).
RcrB plays an important role for UPEC’s HOCl resistance under various growth conditions
In nature, bacteria often experience nutrient-limiting conditions, and their growth more likely resembles stationary phase cells in batch culture. Stationary phase and non-growing, dormant bacteria are typically more resistant to stress compared to exponentially growing cells, which is attributed to differences in their gene regulation and metabolic activities (34 – 36). Thus, we were wondering whether the cellular impact of RcrB during HOCl stress differs under various growth conditions. We performed LPE-based growth analyses of exponentially growing CFT073 and ΔrcrB cells in the presence of increasing HOCl concentrations and compared the outcome to cells that were in the late logarithmic (log)/early stationary phase of growth. We found that independent of the growth phase, ΔrcrB cells exhibited significantly higher sensitivity to HOCl as evidenced by their longer LPE in comparison to the wild type (Fig. 3A and B). We made very similar observations in experiments with different cell densities upon which HOCl was added (Fig. S2). Interestingly, we observed a declining difference in HOCl sensitivity between the wild-type and mutant strains at higher starting optical densities (ODs) (Fig. S2). We further performed survival analyses for both the exponential and stationary cultures of each strain under growing and non-growing conditions to test whether metabolically active and inactive bacteria rely on RcrB differently. We cultivated both strains in MOPSg, washed the cells, and shifted one part into fresh MOPSg media (containing glucose) to allow for continuous growth prior to the exposure to HOCl. We transferred the other part into fresh MOPS media without glucose (MOPS), which resulted in an immediate growth arrest. We reasoned that this approach should allow us to keep the media composition constant while only varying the growth status of the bacteria. Under all the conditions tested, cells that lack RcrB (i.e., ∆rcrB) showed between two- and five-log higher sensitivity to HOCl (Fig. 3C and D). The least difference in survival between the wild-type and ΔrcrB mutant was observed upon shift of late log/stationary cells to MOPS resulting in only one-log difference in survival (Fig. 3D). In summary, our data indicate that RcrB is an important player in UPEC’s HOCl response independent of the bacterial growth status.
Fig 3.

RcrB plays an important role for UPEC’s HOCl resistance under various growth conditions. (A, B) Growth phenotype analyses of UPEC strains CFT073 and ∆rcrB were performed in MOPSg media in the presence of the indicated HOCl concentrations. HOCl-mediated LPE was calculated for each strain (see Materials and Methods for a detailed protocol). (A) Exponential cultures (left): overnight cultures were diluted into fresh MOPSg to an OD600 = 0.01 and grown until they reached an OD600 = 0.1 before they were split up and cultivated in the presence of the indicated HOCl concentrations. (B) Stationary cultures (right): overnight cultures were diluted 25-fold into fresh MOPSg and grown until they reached late log/early stationary phase (OD600 ~2) before they were diluted again to OD600 = 0.1 and cultivated in the presence of the indicated HOCl concentrations; (n ≤ 3, ± SD). (C) Effects of HOCl on the survival of growing and non-growing UPEC strains CFT073 and ∆rcrB. CFT073 and ∆rcrB were grown in MOPSg media to early exponential phase (OD600 = 0.1) or early stationary phase (OD600 ~2) before they were shifted to MOPS media in the presence or absence of glucose. The indicated concentrations of HOCl were added. After 145 min of incubation, excess HOCl was quenched by the addition of fivefold excess thiosulfate, cells were serially diluted with phosphate-buffered saline (PBS), spot-titered onto luria broth agar plates, and incubated overnight at 37°C. The statistics for the HOCl-mediated killing was calculated using Student’s unpaired t-test in prism (n = 3–4, ± SD). * = P < 0.05; ** = P < 0.01.
RcrB does not protect from HOCl stress when UPEC is grown as biofilms
The formation of biofilms is a robust response of bacteria to switch from a planktonic and motile to a sessile lifestyle, which provides UPEC isolates with increased bladder fitness and antimicrobial resistance (37). UPEC biofilms are also formed on abiotic surfaces such as catheters, increasing the risk for the development of catheter-associated urinary tract infections (UTIs). Recent studies provided evidence for an increased tendency of bacteria to switch from planktonic to the biofilm lifestyle when they encounter sublethal HOCl stress, likely as an adaptive stress response (14, 38 – 40). To further investigate RcrB’s role for UPEC’s increased HOCl resistance, we compared biofilm formation of UPEC wild-type and ΔrcrB cells in the presence of HOCl. The two strains were grown in luria broth (LB) without salt for 6 h to facilitate initial attachment to the well before the incubation was resumed for another 16 h in the presence of different concentrations of HOCl. We quantified the biofilm formed on the liquid-solid interface using crystal violet (CV) staining. While we found ~30% increased biofilm formation upon exposure of both strains to 4 mM HOCl, neither the presence nor absence of HOCl resulted in significant differences in CV staining between both strains (Fig. 4), suggesting that RcrB has no influence on HOCl-mediated biofilm responses or biofilm formation in general.
Fig 4.
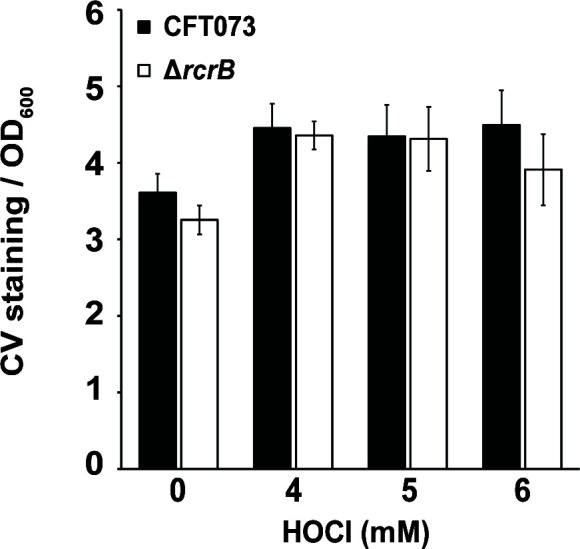
RcrB does not protect from HOCl stress when UPEC is grown as biofilms. Overnight cultures of CFT073 (black bars) and ∆rcrB (white bars) were diluted into LB without NaCl and cultivated under static conditions in 96-well plates in the absence and presence of the indicated HOCl concentrations. After 16 h, OD600 was determined, and planktonic cells were removed. The biofilms were washed twice with PBS, stained with 0.1% CV for 15 min, washed again twice with PBS to remove unbound CV dye. CV bound to biofilm cells was extracted with 30% (vol/vol) glacial acetic acid and OD595 was determined. CV staining (OD595) was normalized to the OD600 of planktonic cells. The statistics for the HOCl-mediated change in biofilm formation was calculated using Student’s unpaired t-test in prism [n = 2 (with eight technical replicates each), ± SD]. no *, P > 0.05.
RcrB protects UPEC from HOCl stress when cultivated in artificial urine media (AUM)
We have shown that RcrB provides protection from HOCl stress when the cells were cultivated in MOPSg, a minimal media used to reduce the possibility that media components react with and potentially quench HOCl. However, this environment may not necessarily resemble the physiological conditions in the urinary tract although UPEC is considered extremely versatile with regard to the different environments it can inhabit. UPEC strains grow as harmless commensals in the nutrient- and carbon-rich human gastrointestinal tract but transition rapidly into a pathogenic lifestyle upon entry into the urinary tract, an environment that is characterized by its high osmolarity and nitrogen abundance but also lower nutrient content and bacterial competition (41). Thus, we sought to confirm our findings in AUM, which resembles more closely the conditions UPEC typically encounter in the urinary tract (42). MG1655, CFT073, and ∆rcrB grown in AUM were treated with 380 µM HOCl, a concentration that resulted in an LPE of ~6 h for the CFT073 wild type (Fig. 5A). Consistent with data in MOPSg, LPE was significantly increased by HOCl in the ∆rcrB and MG1655 strains, indicating that the presence of RcrB in CFT073 wild type contributes to a better recovery from the HOCl exposure (Fig. 5A). To verify that an extended lag phase in HOCl-treated MG1655 and ∆rcrB cells correlates with decreased survival, we performed time-killing assays, in which we exposed the three strains to 668 µM HOCl, a concentration that resulted in ~25% reduction in CFT073 wild-type survival (Fig. 5B). The absence of rcrB, either in CFT073 (i.e., ∆rcrB) or in MG1655, caused a much more drastic effect and resulted in a ~ 75% survival decline (Fig. 5B). To confirm an analogous effect in clinical UPEC isolates, we performed overnight growth assays in AUM focusing on strains that either contain rcrB (i.e., VUTI308) or lack the gene (i.e., VUTI207). Cultures of each clinical isolate were allowed to grow in a 96-well plate overnight in the presence and absence of increasing concentrations of HOCl, and the optical density at 600 nm was recorded after 18 h. Upon exposure to 350 µM HOCl, growth of the RcrB-containing strain VUTI308 was unaffected, whereas VUTI207 showed a drastic reduction in optical density (Fig. 5C). Therefore, we conclude that RcrB’s protective role from HOCl stress is also prominent in AUM.
Fig 5.

RcrB protects UPEC from HOCl stress when cultivated in AUM. (A) Growth phenotype analyses of the K-12 E. coli strain MG1655 and UPEC strains CFT073 and ∆rcrB were performed in AUM media in the presence of 380 µM HOCl. HOCl-mediated LPE was calculated for each strain (see Materials and Methods for a detailed protocol). HOCl exposure in MG1655 and ∆rcrB caused ~6 h higher LPE compared to CFT073. The statistics for the HOCl-mediated growth inhibition was calculated using one-way analysis of variance (ANOVA) in prism, (n = 5, ± SD). (B) Survival of MG1655, CFT073, and ∆rcrB upon HOCl exposure was examined in a time-killing assay. Overnight strains grown in LB were diluted ~25-fold into AUM (OD600 = 0.1) and exposed to 668 µM HOCl. After 60 min of incubation, cells were serially diluted in PBS, spot-titered onto LB agar, and incubated overnight for colony forming unit counts. Survival of MG1655 and ∆rcrB was significantly reduced compared to CFT073. The statistics for the HOCl-mediated killing was calculated using one-way ANOVA in prism, (n = 6, ± SD). (C) UPEC clinical isolates VUTI308 (possesses chromosomal rcrB) and VUTI207 (chromosomal rcrB absent) were grown in AUM in the presence of the indicated HOCl concentrations. After 16 h of growth at 37°C, OD600 was measured and plotted against the HOCl concentrations tested. VUTI308 showed growth between 300 μM and 400 μM HOCl exposure while VUTI207 did not (n = 4, ± SD). * = P < 0.05; ** = P < 0.01.
Transcription of the RcrR regulon is induced over a longer time following exposure to HOCl
Our previous RNA-seq transcriptomic analyses identified the members of the RcrR regulon among the most upregulated genes in HOCl-stressed CFT073 cells (29). To better understand the factors that result in the induction of the RcrR regulon, we aimed to (i) examine the range of HOCl concentrations that result in transcriptional induction of rcrARB, (ii) determine the kinetics of rcrARB upregulation, and (iii) compare their expression to members of the NemR and RclR regulons, two additional HOCl defense systems that contribute to increased survival in HOCl-stressed E. coli (22, 24, 26). We cultivated CFT073 in MOPSg to mid-log phase and exposed the cells to increasing concentrations of HOCl for 15 min. We used qRT-PCR to analyze the transcript levels of each member of the RcrR regulon. The addition of HOCl concentrations between 0.1 mM and 2.5 mM resulted in similar mRNA level for all three genes, which were between 20- and 75-fold higher than in untreated CFT073 cells, suggesting that very little HOCl is already sufficient to mount a response (Fig. 6A). Treatment with 5 mM HOCl did not result in a significant upregulation of gene expression for any of the three genes, likely because the concentration was no longer sublethal and caused substantial killing (Fig. S3A). A time course analysis upon exposure of CFT073 to 1 mM HOCl, a sublethal concentration that does not cause killing, revealed that rcrARB induction remains elevated over at least 45 min (Fig. 6B, Fig. S3b). Likewise, the expressions of nemR and nemA, a member of the NemR regulon, as well as rclR and rclA, a gene under the control of the HOCl-sensing transcriptional activator RclR, were also rapidly induced by HOCl (Fig. 6C and D). However, their transcript level declined rapidly over 45 min, which is consistent with previous studies in the non-pathogenic E. coli strain MG1655 (22, 43). These results further indicate that the presence of the RcrR regulon, particularly RcrB, is needed for the cellular protection for longer time periods in comparison to the other two defense systems.
Fig 6.

Transcription of the RcrR regulon is induced over a longer time following exposure to HOCl. UPEC strain CFT073 was grown in MOPSg to mid-log phase (OD600 = 0.5–0.55) before it was incubated with the indicated HOCl concentrations for the indicated time. Transcription was stopped by the addition of ice-cold methanol. RNA was extracted, residual DNA removed, and mRNA reverse transcribed into cDNA. Transcript levels of the indicated genes were determined by qRT-PCR. Gene expression was normalized to the housekeeping gene rrsD and calculated as fold changes based on expression levels in the untreated control. (A) Fold change in transcript level of select members of the RcrR regulon following treatment with increasing concentrations of HOCl for 15 min (n = 3, ± SD). (B–D) Fold changes in transcript level of select members of the (B) RcrR, (C) NemR, and (D) RclR regulons following treatment with 1 mM HOCl for the indicated time points, (n = 3, ± SD). The statistics for the HOCl-induced gene expression was calculated using Student’s paired t-test in prism. ns, P > 0.05; * = P < 0.05.
Transcription of the RcrR regulon is induced in the presence of HOCl and its byproducts but not by H2O2 or HOSCN
Next, we examined whether the induction of rcrARB transcription is specific to HOCl or can also be induced by any of the other ROS/RCS that are either produced in the neutrophil phagosome during oxidative burst (i.e., HOBr, HOSCN, H2O2) or are byproducts from the reaction of HOCl with different biological compounds (NCT, NH2Cl, Ca(OCl)2, Gly-Cl). Using qRT-PCR, we analyzed the fold change in rcrR/rcrB gene expression following exposure to sublethal concentrations of each oxidant, which we had determined in survival assays before. Except for H2O2 and HOSCN, which resulted in insignificant transcriptional changes or a fivefold downregulation, respectively, exposure to all other stressors resulted in an upregulation of both rcrR and rcrB that was comparable to what we observed with HOCl (Fig. 7). These results indicate that expression of RcrR regulon is stimulated by RCS in general but not by ROS.
Fig 7.
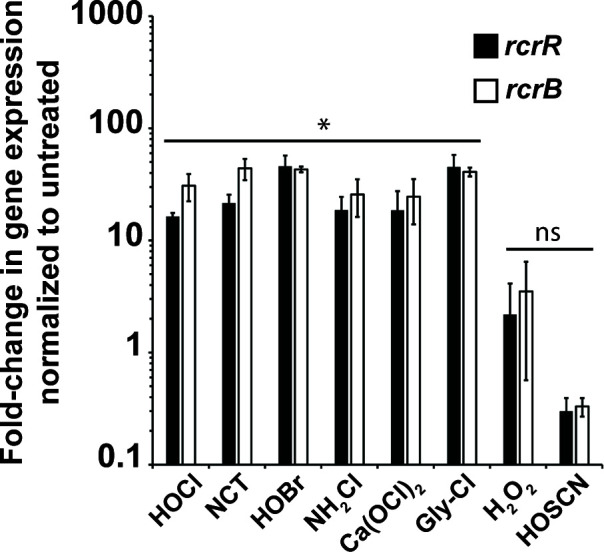
Transcription of the RcrR regulon is induced in the presence of HOCl and its byproducts but not H2O2 or HOSCN. UPEC strain CFT073 was grown in MOPSg to mid-log phase (OD600 = 0.5–0.55) prior to incubation with sublethal concentrations of the indicated stressors for 15 min (HOCl, 1 mM; NCT, 0.3 mM; NH2Cl, 0.3 mM; HOSCN, 0.35 mM; HOBr, 0.5 mM; Ca(OCl)2, 0.2 mM; Gly-Cl, 0.5 mM; H2O2, 10 mM). Transcription was stopped by the addition of ice-cold methanol. RNA was extracted, residual DNA removed, and mRNA reverse transcribed into cDNA. The induction of transcript levels of the indicated genes was determined by qRT-PCR. Gene expression was normalized to the housekeeping gene rrsD and calculated as fold changes based on expression levels in the untreated control. rcrB and rcrR transcript levels were increased following treatment with HOCl, NCT, HOBr, NH2Cl, Ca(OCl)2, and Gly-Cl, respectively. No change in transcript level was seen in the presence of H2O2, while exposure to HOSCN resulted in downregulation of rcrB/rcrR (n = 3, ± SD). The statistics for the oxidant-induced gene expression were calculated using Student’s paired t-test in prism. ns, P > 0.05; * = P < 0.05.
The RcrR regulon protects from HOCl/HOBr stress and its byproducts but not from H2O2 and HOSCN
Given that the expression of the RcrR regulon is induced by several RCS, we next examined whether these findings correlate with improved survival. Using our growth curve-based assays, we exposed CFT073, ∆rcrB (lacks RcrB), and ∆rcrR (constitutively expresses RcrB) to increasing concentrations of the indicated oxidants and recorded their growth. Likewise, we followed the same set-up using MG1655 (lacks RcrB), which was transformed with the RcrB-expressing plasmid (i.e., MG1655 + rcrB-pET28a), and compared its growth to MG1655 cells containing the empty vector control (i.e., MG1655 + pET28a). Consistent with our qRT-PCR data (Fig. 7), we neither observed significant differences in growth and/or LPE between the three strains in the CFT073 background nor between those in the MG1655 backgrounds when cells were treated with HOSCN and H2O2, respectively (Fig. 8, Fig. S4 and 5). In contrast, exposure of NCT and Gly-Cl revealed a higher susceptibility of strains that lack RcrB (i.e., ∆rcrB and MG1655 + pET28a) compared to those that express the membrane protein (i.e., CFT073 wild type and MG1655 + rcrB-pET28a) (Fig. 8, Fig. S4 and 5). However, the differences in growth deficit between these strains were smaller than what we observed upon HOCl exposure. Constitutive expression of RcrB, as it occurs in the ∆rcrR strain, provides additional protection from HOCl, Gly-Cl, and NCT, respectively. In summary, our results indicate that the RCS-induced expression of RcrR regulon provides substantial protection from RCS in general but not from ROS.
Fig 8.

The RcrR regulon protects from HOCl stress and its byproducts but not from H2O2 and HOSCN. Growth phenotype analyses of the K-12 E. coli strain MG1655 carrying pET28a or rcrB-pET28a, respectively, as well as UPEC strains CFT073, ∆rcrB, and ∆rcrR were performed in MOPSg media in the presence of the indicated stressor concentrations. LPE was calculated for each strain (see Materials and Methods for a detailed protocol). Endogenous and recombinant expression of RcrB in CFT073 and MG1655, respectively, resulted in an increase in resistance to all stressors tested except to HOSCN and H2O2 (n ≤ 4, ± SD).
RcrB and RclC do not complement each other during HOCl exposure
RcrB encodes a hypothetical protein, which according to our bioinformatic analysis shares partial homology with the membrane protein RclC, a member of the HOCl-sensing RclR operon with unknown function (24). Therefore, we examined the extent to which recombinant expressions of RcrB and RclC affect CFT073 strains defective in either rcrC (i.e., ΔrcrC), rclB (i.e., ΔrclB), or both genes (i.e., ΔrcrBΔrclC) during HOCl stress (Fig. 9). EV strains served as controls. We compared the growth behavior of the different strains after exposing them to sublethal HOCl concentrations. In contrast to ΔrcrB, which as expected showed substantially higher LPE than the wild-type cells (Fig. 9, compare black and gray circles), no such difference was observed for ΔrclC (Fig. 9, compare black circles and white diamonds). Moreover, LPE was not significantly increased in ∆rcrB∆rclC compared to ΔrcrB (Fig. 9, compare white and gray circles), suggesting that in contrast to MG1655, RclC may not play an important role for HOCl resistance in UPEC. Consistent with these data, recombinant expression of RclC had no positive effect on the growth of ΔrcrB (Fig. 9, compare gray and gray-striped circles), while expression of RcrB in the ΔrclC mutant resulted in substantial resistance to HOCl, as evident by very little to no LPE of this strain (Fig. 9, compare white and gray diamonds). We therefore conclude that the function of RcrB is independent of RclC.
Fig 9.

RcrB and RclC do not complement each other during HOCl exposure. Complementation analyses of the UPEC strains CFT073, ∆rcrB, ∆rclC, and ∆rcrB∆rclC in the presence of rcrB-pET28a, rclC-pET28a, and the empty vector (pET28a) control were performed in MOPSg media in the presence of the indicated HOCl concentrations. HOCl-mediated LPE was calculated for each strain (see Materials and Methods for a detailed protocol). The absence of CFT073 rclC does not cause increased HOCl sensitivity nor does recombinant RclC expression complement the ∆rcrB strain (n = 3, ± SD). The statistics for each sample pair described in Fig. 9 were calculated using Student’s paired t-test in prism.
DISCUSSION
In this study, summarized in Fig. 10, we characterized the significance of the RcrR regulon, comprised of the three genes rcrA, rcrR, and rcrB, for UPEC’s survival during RCS stress. We provide evidence that the elevated expression of rcrARB provides protection against HOCl, the major RCS, but also to HOBr and chlorinated byproducts that HOCl forms with taurine, glycine, ammonium, and calcium, respectively (44 – 47). In contrast, no rcrARB upregulation was observed in the presence of thiol-specific ROS (i.e.,H2O2) and the hypohalous acid HOSCN. The protective effect of the RcrR regulon is exclusively mediated by RcrB given that expression of plasmid-encoded RcrB is sufficient to significantly increase the HOCl resistance of E. coli strains that lack the RcrR regulon. RCS induces RcrB expression in both exponentially growing and stationary phase UPEC cells cultivated in minimal and artificial urine media, respectively. However, no such RcrB-mediated protective effect was observed when UPEC was present as a biofilm. In contrast to members of the NemR and RclR regulons, which provide protection against HOCl in E. coli K-12 strains, expression of the RcrR regulon appears to be the main RCS defense system in UPEC given its prolonged expression during RCS exposure and the lack of complementation between RcrB and its homolog RclC (Fig. 10).
Fig 10.

Summary of the findings of this study. The RcrR regulon plays an important role for UPEC’s survival during RCS stress, including their exposure to HOCl, HOBr, and the byproducts of HOCl, such as NCT, Gly-Cl, NH2Cl, and Ca(OCl)2, respectively. The protective effect of the RcrR regulon is exclusively mediated by RcrB given that expression of plasmid-encoded RcrB is sufficient for an increased HOCl resistance in E. coli strains that lack the RcrR regulon. In contrast, RcrR responds neither to HOSCN nor to H2O2. RCS induces RcrB expression in exponentially growing and stationary phase UPEC in minimal and artificial urine media. No such protective effect was observed in biofilm-forming UPEC cells. In contrast to the NemR and RclR regulons which play significant roles for protection of E. coli K-12 strains from HOCl, expression of the RcrR regulon appears to be the main RCS defense system in UPEC given its prolonged expression during RCS exposure.
rcrARB is controlled by the HOCl-sensing transcriptional repressor RcrR, which represses rcrARB transcription under non-stress conditions. However, HOCl negatively affects RcrR’s transcriptional repressor activity by reversibly oxidizing two conserved cysteine residues (29). As a result of conformational changes, RcrR dissociates from the promoter and rcrARB transcript levels increase. RcrB appears to be the major driver for UPEC’s increased resistance to HOCl in vitro and phagocytosis by activated neutrophils (29). The rcrARB operon is primarily found in members of invasive E. coli pathotypes, including adherent-invasive E. coli, avian pathogenic E. coli, and UPEC (29). In contrast, non-invasive E. coli, such as the enteropathogenic strain O127:H6 and the K-12 strain MG1655, lack the presence of the gene cluster and were shown to be significantly more sensitive to HOCl. However, recombinant expression of RcrB rendered MG1655 significantly more resistant to HOCl, similar to what we observed for the RcrB-containing UPEC strain CFT073 (Fig. 1). Likewise, the HOCl resistance of UPEC strain UTI89, a cystitis isolate that lacks the RcrR regulon (32), was significantly higher when the cells were transformed with the RcrB expression plasmid. When the rcrB homolog was deleted from the probiotic E. coli strain Nissle 1917, the sensitivity to HOCl became more profound, resembling what we observed in MG1655, rcrB-deficient CFT073 cells, or clinical isolates that lack the gene cluster (Fig. 1 and 2). The absence of rcrB resulted in an increased expression of the rcrARB operon and the molecular chaperone ibpA, which suggests that these cells experience higher levels of HOCl stress in the cytoplasm and that RcrB, as a putative membrane protein (Fig. S6), may control the HOCl influx into the cell. E. coli is an important member of the mammalian intestine and characterized by its great diversity. While most E. coli strains colonize the gut as harmless commensals, several E. coli pathotypes have evolved that cause a wide variety of intestinal and extraintestinal diseases (48). Among them are extraintestinal E. coli, such as UPEC, which are the primary causative agent of UTIs (48, 49). UTIs are considered one of the most common bacterial infections worldwide and affect ~150 million people, primarily women at all ages, each year (50). Different forms of UTIs exist depending on the location of the infection, such as cystitis, where UPEC infection occurs in the bladder, or pyelonephritis in the kidney (48). However, prior to successful colonization in the bladder, activated neutrophils infiltrate the bladder to clear bacterial intruders such as UPEC through a powerful oxidative burst (49). Oxidative burst occurs in the phagosome of activated neutrophils and involves the production of highly antimicrobial RCS/ROS to damage and kill the engulfed pathogen. Our work in clinical isolates revealed a strong correlation between increased HOCl resistance and the presence of rcrB, independent of whether the strain was isolated from patients with asymptomatic or symptomatic infections in either the lower (i.e., cystitis) or upper (i.e., pyelonephritis) urinary tract (Fig. 2). Whether UPEC’s ability to express RcrB in response to HOCl, an oxidant prominent during inflammation, provides a survival benefit in vivo is an interesting question that we would like to answer in the future. Deciphering the strategies that bacterial pathogens utilize to defend themselves against the deleterious effects of ROS/RCS is therefore important to better understand their pathogenesis. Moreover, it may provide new perspectives on potential novel drug targets, such as RcrB, which could impair pathogen survival in this highly oxidizing environment and thus increase the efficacy of the immune system. This becomes increasingly pressing in times of increasing antibiotic resistance mechanisms reported for UPEC. Furthermore, we showed that protection by RcrB is provided in actively growing as well as dormant cells and occurs independent of the growth phase (Fig. 2). Limitation appears to be that cells must be in their planktonic state, as the presence of RcrB did not affect UPEC’s biofilm-forming abilities (Fig. 4). While UPEC form biofilms, these occur mostly intracellularly after invasion of uroepithelial cells, which presents a more protective environment and shields them from extracellular stress (49). Although the precise biological functions of rcrA and rcrB are yet to be elucidated, it is now evident that at least RcrB is a major player for bacterial HOCl resistance in vitro and that its function likely is conserved across several E. coli pathotypes and possibly different bacterial species.
Besides methionine and thiol groups, HOCl shows high reactivity toward amino groups, resulting in the formation of chloramines (4). However, it is likely that protein chloramines decompose into smaller chloramines and/or aldehydes, which are more stable and readily penetrate the bacterial cell. Moreover, chloramines formation is also a result of the reaction between HOCl and ammonium, glycine, and/or the non-proteinogenic amino acid taurine (44 – 47). Despite having lower reactivities than HOCl, chloramines still show prolonged bactericidal properties and account for the majority of the added HOCl (51). Indeed, it has been shown that tyrosine chlorination was more pronounced in phagosomal proteins compared to proteins of ingested bacteria (52), suggesting that phagosomal proteins are exposed to a higher HOCl concentration and that chloramines may be the more physiological species that pathogens experience. Whether RCS other than HOCl have similar effects on the expression of the RcrR regulon had not yet been studied. In our current study, we show that rcrARB upregulation is induced by several members of the RCS family, including Ca(OCl)2, NH2Cl, Gly-Cl, and NCT (Fig. 7). Moreover, rcrARB expression is also induced by HOBr, an equally powerful yet physiologically less relevant oxidant (53). Our data therefore suggest that each of these RCS can effectively oxidize RcrR and cause the necessary conformational changes in the repressor that result in elevated RcrB expression and UPEC survival (Fig. 7 and 8). While we did not observe any significant differences in the extent to which the rcrARB operon was induced by the individual RCS, RcrB was most protective during HOCl stress, while its effect was less pronounced in the presence of NH2Cl, Gly-Cl, and NCT (Fig. 7 and 8). We observed upregulation of rcrARB and protection by RcrB in the presence of NCT, a product of the chlorination of taurine after HOCl exposure, which is unable to penetrate through the plasma membrane. However, we have performed our studies in MOPS-glucose minimal media, which contains ammonia in sufficiently high concentrations (54), to allow the conversion of NCT into NH2Cl (44), which readily enters the cytoplasm. However, neither did we observe any significant upregulation of rcrARB nor any impact of RcrB on survival when UPEC was exposed to H2O2 and HOSCN (Fig. 7 and 8). Both oxidants are characterized by their high thiol specificity, which makes them distinctly different from RCS (55). A study in Pseudomonas aeruginosa revealed overlapping outcomes for treatments with HOCl and HOBr as both oxidants target non-growing cells more efficiently than growing cells and elicit similar bacterial responses (53). In contrast, treatment with HOSCN has been found to affect primarily actively growing cells and evoking different defense mechanisms, likely due to its highly thiol-specific nature (53). Recent progress in the field of bacterial oxidative stress defense systems was made by the discovery of HOSCN reductases, which efficiently reduce HOSCN. The presence of these enzymatic systems potentially provide an explanation why RcrR does not sense thiol-specific oxidants such as HOSCN or H2O2 (25, 56 – 59).
Proteins constitute for >50% of the cellular macromolecules and are known to rapidly react with HOCl (60). Numerous studies in different HOCl-treated bacterial species revealed the strong upregulation of the heat shock regulon, indicating an accumulation of misfolded proteins and supporting the idea that proteins are the major targets of HOCl (23, 29, 53 – 62–63). Similarly, H2O2 can cause substantial protein aggregation as the result of methionine and cysteine oxidation (64). It is therefore not surprising that bacteria have evolved a number of molecular chaperones as defense mechanisms to limit and repair RCS-mediated damage, such as Hsp33 (65), CnoX (66, 67), RidA (68, 69), and polyP (70). Intriguingly, inhibitors of polyP synthesis have been shown to be effective against UPEC and reduce the bacterial load during acute UTIs (71, 72). Moreover, microbial responses to ROS/RCS often involve redox-sensitive transcriptional regulators, which use conserved cysteine and/or methionine residues to modulate their activity (18). This, in turn, upregulates the transcription of their target genes, many of which have been shown to protect the organism from ROS/RCS. Three RCS-responsive transcriptional regulators have been identified in E. coli, all of them in the K-12 strain MG1655: (i) HypT (27), (ii) the TetR-family transcriptional repressor NemR (23), and (iii) the AraC-family transcriptional activator RclR (25, 44). NemR is a transcriptional repressor that is inactivated by HOCl, resulting in the expression of nemA, an N-Ethylmaleimide reductase, and gloA, a glyoxylase, both of which have been shown to contribute to HOCl resistance in E. coli (22). The transcriptional activator RclR is specifically sensitive to RCS and HOSCN and regulates the rclABC gene cluster (25, 26). While the functions of RclB and RclC are still unknown, RclA was identified as a Cu(II) reductase that protects E. coli from HOCl stress and, more recently, an HOSCN reductase (25). With the RcrR regulon, we have added another regulatory system to the list of RCS-sensing transcriptional regulators (29). Questions remained as to why UPEC requires the presence of three regulons (i.e., RcrR, NemR, and RclR) to defend against HOCl stress. We found that all three systems were upregulated after just 5 min of exposure to HOCl (Fig. 6). While nemR/nemA and rclR/rclA expression started to decline after 10 min, rcrARB expression remained unaffected throughout the 45 min tested. Furthermore, our LPE assays indicate that ΔrcrB mutants are much more susceptible to HOCl than cells that are defective in a member of the two other regulons (Fig. 9) (29). Likewise, RcrB expression significantly increased the HOCl resistance of a ∆rclC mutant, while the expression of RclC had no additional protective effect on ΔrcrB, suggesting that both membrane proteins are functionally different despite their sequence homology. While expression of rclABC was induced following phagocytosis by neutrophils, the ΔrclABC mutant did not show any differences in neutrophil killing compared to the wild type (43), which is in stark contrast to a ΔrcrB mutant, which was significantly more sensitive to neutrophil killing compared to the corresponding wild-type CFT073 (29). This shows the substantial role of RcrB in resistance to both HOCl and neutrophils compared to the other stress defense systems. The bacterial response and defense strategies are expected to be critical for their ability to survive the immune cell attack, as reported in a recent review of several studies (16). Moreover, independent studies confirmed that the presence of functional oxidative stress defense systems positively affects pathogen colonization in the host, emphasizing their importance for pathogenesis (71 – 74).
MATERIALS AND METHODS
Strains, plasmids, oligonucleotides, and growth conditions
All strains, plasmids, and oligonucleotides used in this study are listed in Table S1. Unless otherwise mentioned, bacteria were cultivated at 37°C and 300 rpm in LB (Millipore Sigma) or in 3-(N-morpholino)propanesulfonic acid minimal media containing 0.2% glucose, 1.32 mM K2HPO4, and 10 µM thiamine (MOPSg) (54). Kanamycin (100 µg/mL), ampicillin (200 µg/mL), or chloramphenicol (34 µg/mL) were added when required.
Construction of in-frame gene deletions in CFT073 and E. coli Nissle 1917
In-frame deletion mutants were constructed using the lamda red-mediated site-specific recombination (75). CFT073 genes rclC, rcrR, and rcrB as well as the rcrB homologous gene in E. coli Nissle 1917 were replaced with a chloramphenicol resistance cassette, which was resolved using pCB20 to yield the nonpolar in-frame deletion strains CFT073∆rcrB∆rclC, CFT073∆rclC , CFT073∆rcrR, CFT073∆rcrB, and EcN∆rcrB-H, respectively (75). All chromosomal mutations were confirmed by PCR.
Plasmid construction
The rcrB and rclC genes were amplified from UPEC strain CFT073 genomic DNA with primers listed in Table S1 and cloned into the NdeI and BamHI sites of plasmid pET28a to generate the N-terminally His6-tagged RcrB and RclC expression plasmids pJUD29 and pJUD46, respectively. Likewise, rcrB was cloned into XbaI and HindIII sites of plasmid pBAD18 to generate the untagged RcrB expression plasmid pJUD61. All constructs were verified by DNA sequencing (Eurofins).
Preparation of oxidants
Sodium hypochlorite (Millipore-Sigma) stock solution was utilized in the preparation of many oxidants. The molar concentration of the stock solution was determined by measuring the A 292 nm after diluting in 10 mM NaOH and using ε292 = 350 M−1 cm−1. As previously described (43), chloramines were prepared freshly by mixing HOCl with excess ammonium sulfate, glycine, and taurine, respectively. The concentration of chloramines was determined by measuring absorbance after diluting in 10 mM NaOH, using ε250 nm= 429 M−1 cm−1 (76). The molar concentration of hydrogen peroxide was determined by measuring the A 240 nm after diluting the stock solution in 50 mM KPi buffer and using ε240 = 43.6 M−1 cm−1. HOSCN was prepared in an enzyme-catalyzed reaction using lactoperoxidase as previously described in references (53, 77).
Studies in AUM
AUM was prepared as described in reference (42) with the following components in their final concentrations: 78.5 mM sodium chloride, 9 mM sodium sulfate, 2.2 mM sodium citrate dihydrate, 0.1 mM sodium oxalate, 3.6 mM potassium dihydrogen phosphate, 21.5 mM potassium chloride, 3 g/L tryptic soy broth, 3 mM calcium chloride, 2 mM magnesium chloride, 15 mM ammonium chloride, 6 mM creatinine, and 200 mM urea (pH 6.3). AUM was prepared fresh every 7–10 days.
Determining oxidant susceptibility through LPE analyses
Overnight cultures of the indicated strains were diluted 25-fold into the indicated media (i.e., MOPSg or AUM) and cultivated until late-exponential/early stationary phase (OD600 = ~2). Cultures were back diluted into fresh MOPSg/AUM to an OD600 = 0.03 and cultivated in a Tecan Infinite 200 plate reader in the presence or absence of increasing concentrations of the indicated oxidant. To test the role of RcrB in exponentially growing cultures (Fig. 3A, Fig. S2B), overnight cultures were diluted into fresh MOPSg to an OD600 = 0.01 and grown to the indicated OD600 before they were split up and cultivated in the presence of the indicated HOCl concentrations. To test the role of RcrB in late log cultures (Fig. 3B, Fig. S2A), overnight cultures were diluted 25-fold into fresh MOPSg and grown until they reached late log/early stationary phase (OD600 ~2) before they were diluted again to the indicated OD600 and cultivated in the presence of the indicated HOCl concentrations. A600 nm measurements were recorded every 10 min for 16 h. Oxidant sensitivities of the strains tested were examined by quantifying their oxidant-mediated LPE. LPE was calculated by determining the differences in time for oxidant-treated samples to reach A 600 nm >0.3 compared to the untreated controls as described in references (26, 29).
Gene expression analyses using qRT-PCR
Overnight LB cultures of the indicated strains were diluted into MOPSg to an A 600 nm= 0.1 and cultivated until OD600 = 0.5–0.55 was reached before they were either left untreated or treated with the indicated concentrations of oxidant. At the indicated time points, 1 mL cells were harvested onto 1 mL of ice-cold methanol to stop transcription. After centrifugation, total RNA was prepared from the cell pellet of three biological replicates of the untreated and oxidant-treated strains using a commercially available RNA extraction kit (Macherey-Nagel). Remaining DNA was removed using the TURBO DNA-free kit (Thermo-Scientific) and cDNA generated using the PrimeScript cDNA synthesis kit (Takara). qRT-PCR reactions were set up according to the manufacturer’s instructions (Alkali Scientific). Transcript levels of the indicated genes were normalized against transcript level of the 16S rRNA-encoding rrsD gene, and relative fold changes in gene expression were calculated using the 2-ΔΔCT method (78).
Bacterial survival assay after exposure to HOCl
Unless stated otherwise, pre-cultures of the UPEC strains CFT073 and ΔrcrB were grown aerobically at 37°C for 16–18 h in LB. For the survival analyses of actively growing cells, the overnight cultures were diluted into MOPSg media to an OD600 = 0.01 and cultivated until OD600 nm reached 0.1. One milliliter of cells was pelleted at 3,000 × g for 5 min, washed twice, and then resuspended in pre-warmed MOPS (no glucose, non-growing) and MOPSg (actively growing). One hundred ninety microliters of the resuspended cultures was immediately transferred into a pre-warmed 96-well plate containing the indicated concentrations of HOCl and incubated aerobically at 37°C. For stationary cultures, the indicated strains were grown in MOPSg media for ~22 h before they were pelleted and resuspended in MOPS (no glucose, non-growing) and MOPSg (actively growing) to an OD600 nm = 0.1. Cultures were then incubated in 96-well plates in the presence/absence of HOCl stress for 145 min, serially diluted in phosphate-buffered saline (PBS), and spotted onto LB agar plates. Colony forming unit (CFU) counts were examined after 16 h of incubation.
Biofilm formation in the presence of HOCl
The effect of HOCl on UPEC biofilm formation was examined for CFT073 and ∆rcrB following a previously published protocol (40) with minor modifications. Overnight LB cultures were diluted into LB without salt to an OD600 = 0.05. One hundred fifty microliters of cultures was then transferred into polystyrene 96-well plates and incubated statically at 37°C to allow for attachment. After 6 h, HOCl was added at the indicated concentrations, the plate was gently rotated to mix HOCl into the culture and incubated for additional 16 h under the same static conditions. Next, OD600 was measured, and the planktonic cells were removed. The biofilms were washed twice with PBS, stained with 0.1% CV for 15 min, washed again twice with PBS to remove unbound CV dye. CV bound to biofilm cells was extracted with 30% (vol/vol) glacial acetic acid and A595 was determined.
Time-killing assays
Overnight MG1655, CFT073, and ∆rcrB cultures were diluted ~25-fold into AUM by normalizing cultures to an OD600 = 0.1 in a six-well sterile cell culture plate. HOCl was added at a final concentration of 664 µM and the plates were incubated at 37°C at 150 rpm under aerobic conditions. After 60 min, OD600 of cultures were recorded, and sample volumes were normalized to the lowest optical density measured, serially diluted in PBS (pH = 7.4), and plated on LB agar to quantify CFU after 16 h. Percent survival was calculated as the ratio of surviving colonies from treated to untreated samples.
Oxidant susceptibility determined by an overnight growth assay and end point OD600 measurement
The assay was performed as described for the LPE assay with the only exception that the 96-well plate was incubated in a shaking incubator at 37°C and 300 rpm. One final OD600 measurement was recorded after 16–18 h.
ACKNOWLEDGMENTS
This work was supported by the NIAID grants R15AI164585 and 1R03AI174033-01A1 and the Illinois State University Pre-Tenure Faculty Initiative Grant (to J.-U.D.). M.E.C. was supported by a Weigel fellowship by the Phi-Sigma Biological Sciences Honors Society and the Jill Hutchison endowed scholarship. L.F.G. and L.P. were supported by a DAAD RISE fellowship from the German Academic Exchange Service. M.B. and B.R. received FireBIRD grants by the Illinois State University Undergraduate Research Support Program. We thank Dr. Maria Hadjifrangiskou (Vanderbilt University) for providing the UPEC clinical isolates as gifts and Dr. Michael J. Gray (University of Alabama) for the E. coli Nissle 1917 strain. Members of the Dahl lab are acknowledged for feedback and proofreading of the manuscript. Figure 10 was generated using Biorender.
Contributor Information
Jan-Ulrik Dahl, Email: jdahl1@ilstu.edu.
Patricia A. Champion, University of Notre Dame, Notre Dame, Indiana, USA
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/jb.00064-23.
Fig. S1: Recombinant expression of CFT073 RcrB complements the HOCl sensitivity phenotypes of E. coli strains deficient in rcrB or its homolog. Fig. S2: RcrB plays an important role for UPEC's HOCl resistance under various growth conditions. Fig. S3: Survival assays performed in conjunction with gene expression analyses. Fig. S4: The RcrR regulon protects from HOCl stress and its byproducts but not from H2O2 and HOSCN. Fig. S5: The RcrR regulon protects from HOCl stress and its byproducts but not from H2O2 and HOSCN. Fig. S6: The predicted alphafold structure of RcrB.
Strains, plasmids, and oligonucleotides used in this study.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Imlay JA. 2019. Where in the world do bacteria experience oxidative stress? Environ Microbiol 21:521–530. doi: 10.1111/1462-2920.14445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Winterbourn CC. 2018. Biological production, detection, and fate of hydrogen peroxide. Antioxid Redox Signal 29:541–551. doi: 10.1089/ars.2017.7425 [DOI] [PubMed] [Google Scholar]
- 3. Winterbourn CC, Kettle AJ, Hampton MB. 2016. Reactive oxygen species and neutrophil function. Annu Rev Biochem 85:765–792. doi: 10.1146/annurev-biochem-060815-014442 [DOI] [PubMed] [Google Scholar]
- 4. Klebanoff SJ, Kettle AJ, Rosen H, Winterbourn CC, Nauseef WM. 2013. Myeloperoxidase: a front-line defender against phagocytosed microorganisms. J Leukoc Biol 93:185–198. doi: 10.1189/jlb.0712349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hurst JK. 2012. What really happens in the neutrophil phagosome? Free Radic Biol Med 53:508–520. doi: 10.1016/j.freeradbiomed.2012.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Winterbourn C.C, Hampton MB, Livesey JH, Kettle AJ. 2006. Modeling the reactions of superoxide and myeloperoxidase in the neutrophil phagosome: implications for microbial killing. J Biol Chem 281:39860–39869. doi: 10.1074/jbc.M605898200 [DOI] [PubMed] [Google Scholar]
- 7. Davies MJ. 2011. Myeloperoxidase-derived oxidation: mechanisms of biological damage and its prevention. J Clin Biochem Nutr 48:8–19. doi: 10.3164/jcbn.11-006FR [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Meotti FC, Jameson GNL, Turner R, Harwood DT, Stockwell S, Rees MD, Thomas SR, Kettle AJ. 2011. Urate as a physiological substrate for myeloperoxidase: implications for hyperuricemia and inflammation. J Biol Chem 286:12901–12911. doi: 10.1074/jbc.M110.172460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Winterbourn CC, van den Berg JJM, Roitman E, Kuypers FA. 1992. Chlorohydrin formation from unsaturated fatty acids reacted with hypochlorous acid. Arch Biochem Biophys 296:547–555. doi: 10.1016/0003-9861(92)90609-z [DOI] [PubMed] [Google Scholar]
- 10. Hawkins CL. 2020. Hypochlorous acid-mediated modification of proteins and its consequences. Essays Biochem 64:75–86. doi: 10.1042/EBC20190045 [DOI] [PubMed] [Google Scholar]
- 11. Prütz WA. 1996. Hypochlorous acid interactions with thiols, nucleotides, DNA, and other biological substrates. Arch Biochem Biophys 332:110–120. doi: 10.1006/abbi.1996.0322 [DOI] [PubMed] [Google Scholar]
- 12. Winterbourn CC, Kettle AJ. 2013. Redox reactions and microbial killing in the neutrophil phagosome. Antioxid Redox Signal 18:642–660. doi: 10.1089/ars.2012.4827 [DOI] [PubMed] [Google Scholar]
- 13. Dahl J-U, Gray MJ, Jakob U. 2015. Protein quality control under oxidative stress conditions. J Mol Biol 427:1549–1563. doi: 10.1016/j.jmb.2015.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sultana S, Foti A, Dahl J-U, Richardson AR. 2020. Bacterial defense systems against the neutrophilic oxidant hypochlorous acid. Infect Immun 88:e00964-19. doi: 10.1128/IAI.00964-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cremers CM, Jakob U. 2013. Oxidant sensing by reversible disulfide bond formation. J Biol Chem 288:26489–26496. doi: 10.1074/jbc.R113.462929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sultana S, Dahl J-U. 2023. A year at the forefront of bacterial defense systems against neutrophilic oxidants. Biol Open 12:bio059809. doi: 10.1242/bio.059809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gray MJ, Wholey W-Y, Jakob U. 2013. Bacterial responses to reactive chlorine species. Annu Rev Microbiol 67:141–160. doi: 10.1146/annurev-micro-102912-142520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vincent MS, Ezraty B. n.d. Methionine oxidation in bacteria: a reversible post-translational modification. Mol Microbiol [DOI] [PubMed] [Google Scholar]
- 19. Ezraty B, Gennaris A, Barras F, Collet J-F. 2017. Oxidative stress, protein damage and repair in bacteria. Nat Rev Microbiol 15:385–396. doi: 10.1038/nrmicro.2017.26 [DOI] [PubMed] [Google Scholar]
- 20. Varatnitskaya M, Degrossoli A, Leichert LI. 2021. Redox regulation in host-pathogen interactions: thiol switches and beyond. Biol Chem 402:299–316. doi: 10.1515/hsz-2020-0264 [DOI] [PubMed] [Google Scholar]
- 21. Linzner N, Loi VV, Fritsch VN, Antelmann H. 2021. Thiol-based redox switches in the major pathogen Staphylococcus aureus. Biol Chem 402:333–361. doi: 10.1515/hsz-2020-0272 [DOI] [PubMed] [Google Scholar]
- 22. Gray MJ, Wholey W-Y, Parker BW, Kim M, Jakob U. 2013. NemR is a bleach-sensing transcription factor. J Biol Chem 288:13789–13798. doi: 10.1074/jbc.M113.454421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gray MJ, Li Y, Leichert L-O, Xu Z, Jakob U. 2015. Does the transcription factor NemR use a regulatory sulfenamide bond to sense bleach Antioxid Redox Signal 23:747–754. doi: 10.1089/ars.2015.6346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Parker BW, Schwessinger EA, Jakob U, Gray MJ. 2013. The RclR protein is a reactive chlorine-specific transcription factor in Escherichia coli. J Biol Chem 288:32574–32584. doi: 10.1074/jbc.M113.503516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Meredith JD, Chapman I, Ulrich K, Sebastian C, Stull F, Gray MJ. 2022. Escherichia Coli RclA is a highly active hypothiocyanite reductase. Proc Natl Acad Sci U S A 119:e2119368119. doi: 10.1073/pnas.2119368119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Derke RM, Barron AJ, Billiot CE, Chaple IF, Lapi SE, Broderick NA, Gray MJ. 2020. The Cu(II) reductase RclA protects Escherichia Coli against the combination of hypochlorous acid and intracellular copper. mBio 11:e01905-20. doi: 10.1128/mBio.01905-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gebendorfer KM, Drazic A, Le Y, Gundlach J, Bepperling A, Kastenmüller A, Ganzinger KA, Braun N, Franzmann TM, Winter J. 2012. Identification of a hypochlorite-specific transcription factor from Escherichia coli. J Biol Chem 287:6892–6903. doi: 10.1074/jbc.M111.287219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Drazic A, Miura H, Peschek J, Le Y, Bach NC, Kriehuber T, Winter J. 2013. Methionine oxidation activates a transcription factor in response to oxidative stress. Proc Natl Acad Sci U S A 110:9493–9498. doi: 10.1073/pnas.1300578110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sultana S, Crompton ME, Meurer K, Jankiewicz O, Morales GH, Johnson C, Horbach E, Hoffmann KP, Kr P, Shah R, Anderson GM, Mortimer NT, Schmitz JE, Hadjifrangiskou M, Foti A, Dahl J-U. 2022. Redox-mediated inactivation of the transcriptional Repressor RcrR is responsible for uropathogenic Escherichia coli's increased resistance to reactive chlorine species. mBio 13:e0192622. doi: 10.1128/mbio.01926-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mobley HL, Green DM, Trifillis AL, Johnson DE, Chippendale GR, Lockatell CV, Jones BD, Warren JW. 1990. Pyelonephritogenic Escherichia coli and killing of cultured human renal proximal tubular epithelial cells: role of hemolysin in some strains. Infect Immun 58:1281–1289. doi: 10.1128/iai.58.5.1281-1289.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sonnenborn U. 2016. Escherichia coli strain Nissle 1917-from bench to bedside and back: history of a special Escherichia coli strain with probiotic properties. FEMS Microbiol Lett 363:fnw212. doi: 10.1093/femsle/fnw212 [DOI] [PubMed] [Google Scholar]
- 32. Chen SL, Hung C-S, Xu J, Reigstad CS, Magrini V, Sabo A, Blasiar D, Bieri T, Meyer RR, Ozersky P, Armstrong JR, Fulton RS, Latreille JP, Spieth J, Hooton TM, Mardis ER, Hultgren SJ, Gordon JI. 2006. Identification of genes subject to positive selection in uropathogenic strains of Escherichia coli: a comparative genomics approach. Proc Natl Acad Sci U S A 103:5977–5982. doi: 10.1073/pnas.0600938103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eberly AR, Beebout CJ, Tong CMC, Van Horn GT, Green HD, Fitzgerald MJ, De S, Apple EK, Schrimpe-Rutledge AC, Codreanu SG, Sherrod SD, McLean JA, Clayton DB, Stratton CW, Schmitz JE, Hadjifrangiskou M. 2020. Data highlighting phenotypic diversity of urine-associated Escherichia Coli isolates. Data Brief 31:105811. doi: 10.1016/j.dib.2020.105811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mascio CTM, Alder JD, Silverman JA. 2007. Bactericidal action of daptomycin against stationary-phase and nondividing Staphylococcus aureus cells. Antimicrob Agents Chemother 51:4255–4260. doi: 10.1128/AAC.00824-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Agrawal A, Rangarajan N, Weisshaar JC. 2019. Resistance of early stationary phase E. coli to membrane permeabilization by the antimicrobial peptide cecropin A. Biochim Biophys Acta Biomembr 1861:182990. doi: 10.1016/j.bbamem.2019.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hengge R. 2011. Stationary-phase gene regulation in Escherichia coli §. EcoSal Plus 4. doi: 10.1128/ecosalplus.5.6.3 [DOI] [PubMed] [Google Scholar]
- 37. Klein RD, Hultgren SJ. 2020. Urinary tract infections: microbial pathogenesis, host-pathogen interactions and new treatment strategies. Nat Rev Microbiol 18:211–226. doi: 10.1038/s41579-020-0324-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Perkins A, Tudorica DA, Teixeira RD, Schirmer T, Zumwalt L, Ogba OM, Cassidy CK, Stansfeld PJ, Guillemin K, Cooper VS. 2021. A bacterial inflammation sensor regulates C-di-GMP signaling, adhesion, and biofilm formation. mBio 12:e0017321. doi: 10.1128/mBio.00173-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Strempel N, Nusser M, Neidig A, Brenner-Weiss G, Overhage J. 2017. The oxidative stress agent hypochlorite stimulates C-di-GMP synthesis and biofilm formation in Pseudomonas Aeruginosa. Front Microbiol 8:2311. doi: 10.3389/fmicb.2017.02311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang X-S, García-Contreras R, Wood TK. 2007. YcfR (BhsA) influences Escherichia coli biofilm formation through stress response and surface hydrophobicity . J Bacteriol 189:3051–3062. doi: 10.1128/JB.01832-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Alteri CJ, Mobley HLT. 2012. Escherichia coli physiology and metabolism dictates adaptation to diverse host microenvironments. Curr Opin Microbiol 15:3–9. doi: 10.1016/j.mib.2011.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brooks T, Keevil CW. 1997. A simple artificial urine for the growth of urinary pathogens. Lett Appl Microbiol 24:203–206. doi: 10.1046/j.1472-765x.1997.00378.x [DOI] [PubMed] [Google Scholar]
- 43. Königstorfer A, Ashby LV, Bollar GE, Billiot CE, Gray MJ, Jakob U, Hampton MB, Winterbourn CC. 2021. Induction of the reactive chlorine-responsive transcription factor RclR in Escherichia coli following ingestion by neutrophils. Pathog Dis 79:ftaa079. doi: 10.1093/femspd/ftaa079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ashby LV, Springer R, Hampton MB, Kettle AJ, Winterbourn CC. 2020. Evaluating the bactericidal action of hypochlorous acid in culture media. Free Radic Biol Med 159:119–124. doi: 10.1016/j.freeradbiomed.2020.07.033 [DOI] [PubMed] [Google Scholar]
- 45. Peskin AV, Winterbourn CC. 2001. Kinetics of the reactions of hypochlorous acid and amino acid chloramines with thiols, methionine, and ascorbate. Free Radic Biol Med 30:572–579. doi: 10.1016/s0891-5849(00)00506-2 [DOI] [PubMed] [Google Scholar]
- 46. Peskin AV, Midwinter RG, Harwood DT, Winterbourn CC. 2004. Chlorine transfer between glycine, taurine, and histamine: reaction rates and impact on cellular reactivity. Free Radic Biol Med 37:1622–1630. doi: 10.1016/j.freeradbiomed.2004.08.010 [DOI] [PubMed] [Google Scholar]
- 47. Pattison DI, Davies MJ. 2006. Reactions of myeloperoxidase-derived oxidants with biological substrates: gaining chemical insight into human inflammatory diseases. Curr Med Chem 13:3271–3290. doi: 10.2174/092986706778773095 [DOI] [PubMed] [Google Scholar]
- 48. Kaper JB, Nataro JP, Mobley HL. 2004. Pathogenic Escherichia coli. Nat Rev Microbiol 2:123–140. doi: 10.1038/nrmicro818 [DOI] [PubMed] [Google Scholar]
- 49. Flores-Mireles AL, Walker JN, Caparon M, Hultgren SJ. 2015. Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat Rev Microbiol 13:269–284. doi: 10.1038/nrmicro3432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McLellan LK, Hunstad DA. 2016. Urinary tract infection: pathogenesis and outlook. Trends Mol Med 22:946–957. doi: 10.1016/j.molmed.2016.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hawkins CL, Davies MJ. 2002. Hypochlorite-induced damage to DNA, RNA, and polynucleotides: formation of chloramines and nitrogen-centered radicals. Chem Res Toxicol 15:83–92. doi: 10.1021/tx015548d [DOI] [PubMed] [Google Scholar]
- 52. Green JN, Kettle AJ, Winterbourn CC. 2014. Protein chlorination in neutrophil phagosomes and correlation with bacterial killing. Free Radic Biol Med 77:49–56. doi: 10.1016/j.freeradbiomed.2014.08.013 [DOI] [PubMed] [Google Scholar]
- 53. Groitl B, Dahl J-U, Schroeder JW, Jakob U. 2017. Pseudomonas aeruginosa defense systems against microbicidal oxidants: defense systems against microbicidal oxidants. Mol Microbiol 106:335–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Neidhardt FC, Bloch PL, Smith DF. 1974. Culture medium for enterobacteria. J Bacteriol 119:736–747. doi: 10.1128/jb.119.3.736-747.1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Skaff O, Pattison DI, Davies MJ. 2009. Hypothiocyanous acid reactivity with low-molecular-mass and protein thiols: absolute rate constants and assessment of biological relevance. Biochem J 422:111–117. doi: 10.1042/BJ20090276 [DOI] [PubMed] [Google Scholar]
- 56. Shearer HL, Paton JC, Hampton MB, Dickerhof N. 2022. Glutathione utilization protects Streptococcus pneumoniae against lactoperoxidase-derived hypothiocyanous acid. Free Radic Biol Med 179:24–33. doi: 10.1016/j.freeradbiomed.2021.12.261 [DOI] [PubMed] [Google Scholar]
- 57. Shearer HL, Kaldor CD, Hua H, Kettle AJ, Parker HA, Hampton MB. 2022. Resistance of Streptococcus pneumoniae to hypothiocyanous acid generated by host peroxidases. Infect Immun 90:e0053021. doi: 10.1128/IAI.00530-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shearer HL, Pace PE, Paton JC, Hampton MB, Dickerhof N. 2022. A newly identified flavoprotein disulfide reductase Har protects Streptococcus pneumoniae against hypothiocyanous acid. J Biol Chem 298:102359. doi: 10.1016/j.jbc.2022.102359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Meredith JD, Gray MJ. 2023. Hypothiocyanite and host-microbe interactions. Mol Microbiol 119:302–311. doi: 10.1111/mmi.15025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hawkins CL, Davies MJ. 2019. Detection, identification and quantification of oxidative protein modifications. J Biol Chem 294:19683–19708. doi: 10.1074/jbc.REV119.006217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Thakur PB, Long AR, Nelson BJ, Kumar R, Rosenberg AF, Gray MJ. 2019. Complex responses to inflammatory oxidants by the probiotic bacterium Lactobacillus reuteri . Microbiology. Microbiology. doi: 10.1101/605881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tung QN, Busche T, Van Loi V, Kalinowski J, Antelmann H. 2020. The redox-sensing MarR-type repressor HypS controls hypochlorite and antimicrobial resistance in Mycobacterium smegmatis. Free Radic Biol Med 147:252–261. doi: 10.1016/j.freeradbiomed.2019.12.032 [DOI] [PubMed] [Google Scholar]
- 63. Hillion M, Bernhardt J, Busche T, Rossius M, Maaß S, Becher D, Rawat M, Wirtz M, Hell R, Rückert C, Kalinowski J, Antelmann H. 2017. Monitoring global protein thiol-oxidation and protein S-mycothiolation in Mycobacterium smegmatis under hypochlorite stress. Sci Rep 7:1195. doi: 10.1038/s41598-017-01179-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Imlay JA. 2008. Cellular defenses against superoxide and hydrogen peroxide. Annu Rev Biochem 77:755–776. doi: 10.1146/annurev.biochem.77.061606.161055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Winter J, Ilbert M, Graf PCF, Ozcelik D, Jakob U. 2008. Bleach activates a redox-regulated chaperone by oxidative protein unfolding. Cell 135:691–701. doi: 10.1016/j.cell.2008.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Goemans CV, Beaufay F, Arts IS, Agrebi R, Vertommen D, Collet J-F. 2018. The chaperone and redox properties of CnoX chaperedoxins are tailored to the proteostatic needs of bacterial species. mBio 9:mBio doi: 10.1128/mBio.01541-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Goemans CV, Vertommen D, Agrebi R, Collet J-F. 2018. CnoX is a chaperedoxin: a holdase that protects its substrates from irreversible oxidation. Mol Cell 70:614–627. doi: 10.1016/j.molcel.2018.04.002 [DOI] [PubMed] [Google Scholar]
- 68. Müller A, Langklotz S, Lupilova N, Kuhlmann K, Bandow JE, Leichert LIO. 2014. Activation of RidA chaperone function by N-chlorination. Nat Commun 5:5804. doi: 10.1038/ncomms6804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Varatnitskaya M, Fasel J, Müller A, Lupilov N, Shi Y, Fuchs K, Krewing M, Jung C, Jacob T, Sitek B, Bandow JE, Carroll KS, Hofmann E, Leichert LI. 2022. An increase in surface hydrophobicity mediates chaperone activity in N-chlorinated RidA. Redox Biol 53:102332. doi: 10.1016/j.redox.2022.102332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gray MJ, Wholey W-Y, Wagner NO, Cremers CM, Mueller-Schickert A, Hock NT, Krieger AG, Smith EM, Bender RA, Bardwell JCA, Jakob U. 2014. Polyphosphate is a primordial chaperone. Mol Cell 53:689–699. doi: 10.1016/j.molcel.2014.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dahl J-U, Gray MJ, Bazopoulou D, Beaufay F, Lempart J, Koenigsknecht MJ, Wang Y, Baker JR, Hasler WL, Young VB, Sun D, Jakob U. 2017. The anti-inflammatory drug mesalamine targets bacterial polyphosphate accumulation. Nat Microbiol 2:16267. doi: 10.1038/nmicrobiol.2016.267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Peng L, Zeng L, Jin H, Yang L, Xiao Y, Lan Z, Yu Z, Ouyang S, Zhang L, Sun N. 2020. Discovery and antibacterial study of potential PPK1 inhibitors against uropathogenic E. Coli. J Enzyme Inhib Med Chem 35:1224–1232. doi: 10.1080/14756366.2020.1766453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hryckowian AJ, Welch RA. 2013. RpoS contributes to phagocyte oxidase-mediated stress resistance during urinary tract infection by Escherichia coli CFT073. mBio 4:e00023-13. doi: 10.1128/mBio.00023-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bessaiah H, Pokharel P, Habouria H, Houle S, Dozois CM. 2019. yqhG contributes to oxidative stress resistance and virulence of uropathogenic Escherichia Coli and identification of other genes altering expression of type 1 fimbriae. Front Cell Infect Microbiol 9:312. doi: 10.3389/fcimb.2019.00312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Thomas EL, Grisham MB, Jefferson MM. 1986. Preparation and characterization of chloramines. Methods Enzymol 132:569–585. doi: 10.1016/s0076-6879(86)32042-1 [DOI] [PubMed] [Google Scholar]
- 77. Chandler JD, Nichols DP, Nick JA, Hondal RJ, Day BJ. 2013. Selective metabolism of hypothiocyanous acid by mammalian thioredoxin reductase promotes lung innate immunity and antioxidant defense. J Biol Chem 288:18421–18428. doi: 10.1074/jbc.M113.468090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:45e–445. doi: 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1: Recombinant expression of CFT073 RcrB complements the HOCl sensitivity phenotypes of E. coli strains deficient in rcrB or its homolog. Fig. S2: RcrB plays an important role for UPEC's HOCl resistance under various growth conditions. Fig. S3: Survival assays performed in conjunction with gene expression analyses. Fig. S4: The RcrR regulon protects from HOCl stress and its byproducts but not from H2O2 and HOSCN. Fig. S5: The RcrR regulon protects from HOCl stress and its byproducts but not from H2O2 and HOSCN. Fig. S6: The predicted alphafold structure of RcrB.
Strains, plasmids, and oligonucleotides used in this study.