

Abstract
Multiple sclerosis (MS) is an autoimmune disease of the central nervous system (CNS) that results in significant neurodegeneration in the majority of those affected and is a common cause of chronic neurological disability in young adults1,2. To provide insight into the mechanisms determining progression, we conducted a genome-wide association study of the age-related MS severity score in 12,584 cases and replicated our findings in a further 9,805 cases. We identified a significant association with rs10191329 in the DYSF-ZNF638 locus (P=3.6×10−9), the risk allele shortening the median time to require a walking aid by up to 3.7 years and increasing brainstem and cortical pathology in brain tissue. We also identified suggestive association with rs149097173 in the DNM3-PIGC locus (P=2.3×10−7) and significant enrichment for expression in CNS tissues. Mendelian randomization analyses indicated a protective role for higher educational attainment. In contrast to immune-driven susceptibility3, these findings indicate a key role of CNS resilience and neurocognitive reserve in determining outcome in MS.
INTRODUCTION
Multiple sclerosis (MS) affects more than 2.8 million individuals worldwide and profoundly reducing quality of life for the majority of affected individuals1,2. Clinically, the disease is characterized by recurrent episodes of largely reversible neurological dysfunction, known as relapses, together with steady and unrelenting accumulation of chronic neurological disability, referred to as progression1. The relative impact of these largely independent features varies between patients and during the course of illness within individuals. Over the past few decades, the introduction of a range of immunological treatments has transformed the ability to control relapse activity in the disease, leaving therapy capable of controlling progression as the greatest currently unmet clinical need4.
Case-control genome-wide association studies (GWAS) have identified over 200 variants influencing susceptibility to the disease, with the strongest effects coming from the major histocompatibility complex (MHC) and the implicated genes being overwhelmingly enriched for immune relevance3. Although these risk variants have been found to reduce the age at onset5, it is notable that they do not appear to have any impact on disease severity6. These findings, together with the concordance for outcome within families7, suggest that an independent genetic architecture determines the clinical course of the disease, as has been seen in other autoimmune8 and neurological conditions9. However, published efforts to systematically interrogate severity have, thus far, only involved modest numbers of cases, and unanimously fall short of identifying any convincingly associated genetic variants5,10,11.
Through long-standing international collaborations, we have completed the largest in-depth effort to date aimed at characterizing the genetic architecture underlying MS severity. In this study, we combined cross-sectional and longitudinal analyses of MS-specific disability outcomes, and correlated findings with neuropathology and tissue-specific expression patterns. We contrasted the genetic determinants of susceptibility and severity, and examined potential modifiable risk factors for MS progression. Given the significantly increased potential for the development of rational therapies attached to drug targets with genetic support12, our work will likely help to advance patients’ priorities with regard to treatment and prognosis.
RESULTS
Cohort description
Here we describe a genetic analysis of disease severity performed in 12,584 people with MS of European ancestry. After imputation to the Haplotype Reference Consortium and rigorous quality control (Methods), a total of 7.8 million autosomal single nucleotide variants with a minor allele frequency (MAF) > 0.01 were analyzed. The discovery population consisted of 21 cohorts collected from centers across North America, Europe, and Australia (Supplementary Fig. 1 and Supplementary Table 1). In line with standard practice, neurological disability was measured using the Expanded Disability Status Scale (EDSS), an ordinal numerical scale that increases as neurodegeneration progresses. To control for the effects of aging, individual EDSS measures were converted to the age-related MS severity (ARMSS) score by ranking disability within age-specific strata13 (Methods). To reduce the influence of disability fluctuation related to relapses and lessen the imprecision of attempting to predict outcome in patients early in the disease, we focused recruitment on older individuals with longer duration of disease who had effectively declared their clinical outcome. Consequently, mean age at last follow-up and disease duration were 51.7 and 18.2 years, respectively (Extended Data Fig. 1 and Supplementary Table 2). Replication of variant associations was tested in existing data from nine independent cohorts totaling 9,805 cases (Extended Data Fig. 1, Supplementary Fig. 1, Supplementary Tables 1 and 2). The replication population was organized into four strata matched by genotyping platform and was subjected to equivalent quality control procedures (Supplementary Note, Extended Data Fig. 2, Supplementary Fig. 2 and 3, Supplementary Tables 3 and 4).
Heritability and tissue enrichment
The SNP-based heritability estimate (h2SNP) for variants with a MAF > 0.01 was 0.13 (s.e. 0.04) (Supplementary Table 5). Partitioned heritability analysis by functional annotation with 53 categories14 did not identify strong enrichment in any category after correction for multiple testing (Supplementary Table 6), likely due to insufficient power. To uncover disease-relevant tissues, we combined variant association statistics with specifically expressed gene sets from 205 tissues and cell types in a heritability enrichment analysis15. We observed a significant enrichment, adjusted for multiple testing, exclusively in CNS tissues across multiple brain regions and the C1 segment of the cervical spinal cord (Fig. 1 and Supplementary Table 7). In contrast, repeating the same analysis for MS susceptibility3 revealed strong enrichment in lymphoid organs, immune lymphoid and myeloid cells, as well as in tissues with recognized immunological functions and microbiota interactions (pharynx, lung, terminal ileum and endocervix; Fig. 1 and Supplementary Table 8). This pattern faithfully recapitulates the immune-related nature of susceptibility associations, further highlighting the striking difference from the heritability pattern observed for disease severity.
Fig. 1 |. Tissue and cell type heritability enrichment.
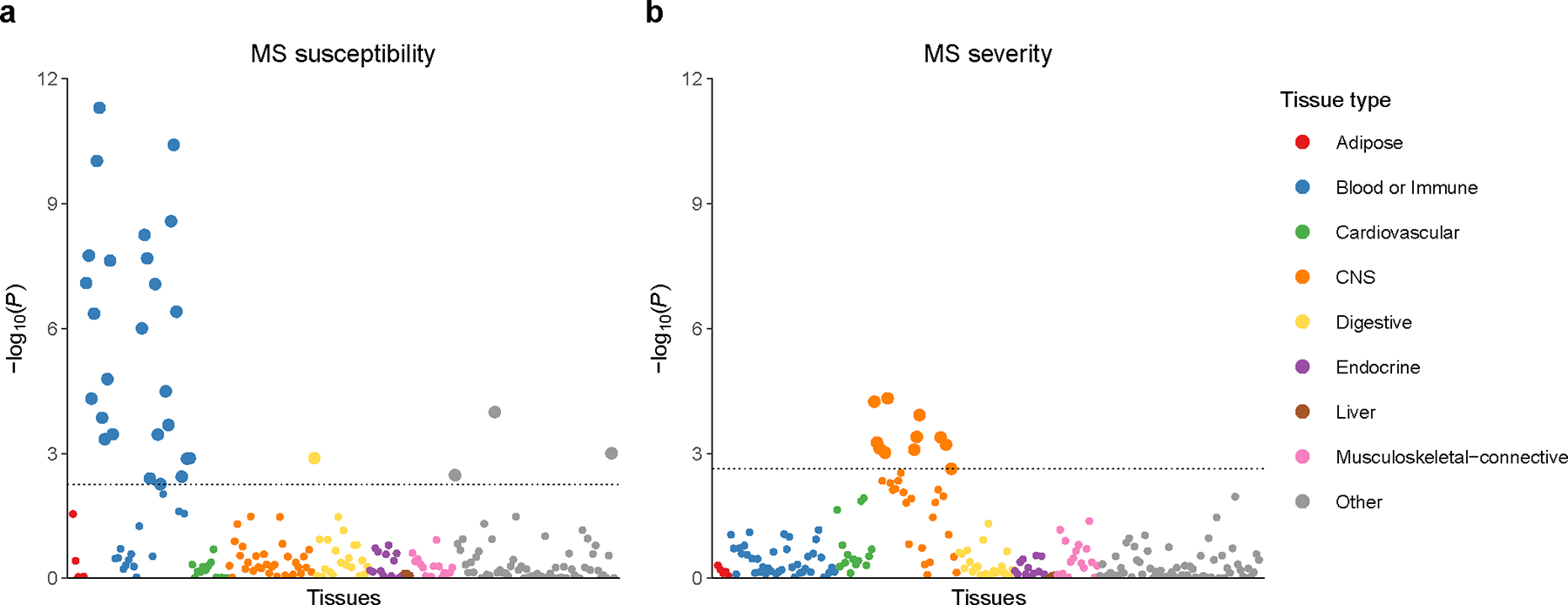
a, MS susceptibility from previous meta-analysis3. b, MS severity from this study. While susceptibility associations display strong immunological lymphoid and myeloid enrichment, our analysis of MS severity uncovered significant enrichment exclusively in CNS tissues. Each point represents one of 205 tissues and cell types, grouped by color into 9 categories. Large circles are significant at a false discovery rate cutoff of 0.05 (dotted line). Full results including tissue and cell type labels are provided in Supplementary Tables 7 and 8.
Discovery of an MS severity locus
To identify genetic variants associated with MS severity, we first performed a cross-sectional GWAS using ARMSS scores with the entire discovery cohort, adjusting for age, sex, date of birth, EDSS source, center, genotyping batch and the first ten principal components. Use of MS disease modifying therapy was not included as a covariate given the potential for collider bias (Methods). We observed only modest inflation of the test statistics (λGC = 1.016; Supplementary Fig. 4) and linkage disequilibrium (LD) score regression (LDSC) yielded an intercept not significantly different from 1 (1.006, 95% confidence interval [CI] 0.993 to 1.019), consistent with polygenicity driving inflation. An association signal in the DYSF–ZNF638 locus reached genome-wide significance (P = 9.7×10−9; Fig. 2 and Table 1). The lead variant rs10191329 (MAF = 0.17) was not close to (> 3 Mb) or in LD with (r2 ≤ 0.006) any of the lead MS susceptibility variants3. Eleven additional loci showed suggestive association with ARMSS score (P < 5×10−6; Fig. 2), thereby identifying 12 independent loci that were brought forward for replication (Supplementary Table 9). Conditional and joint analysis did not identify secondary signals.
Fig. 2 |. Within-cases GWAS identifies a novel locus associated with MS severity.
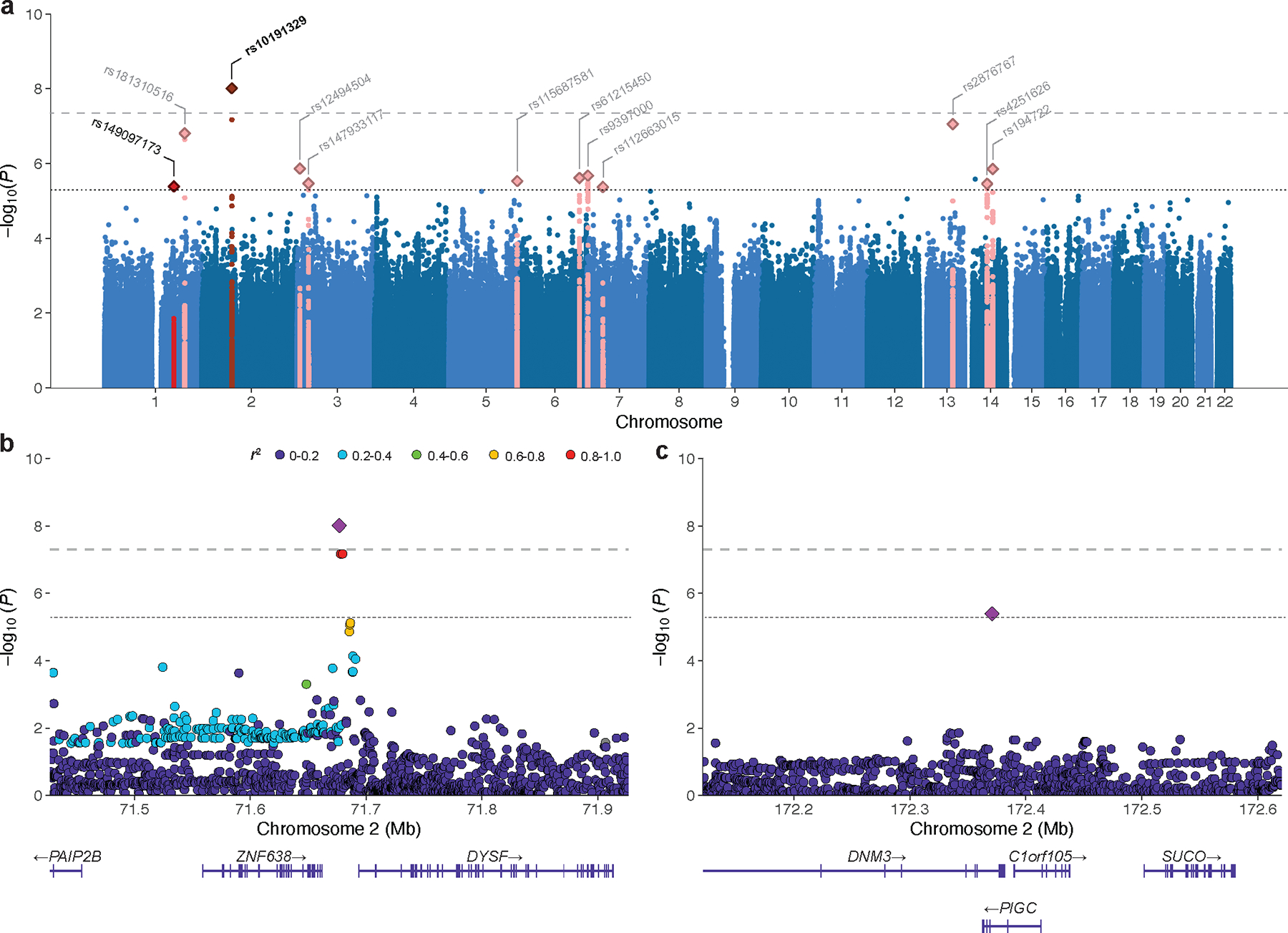
a, Genome-wide association statistics obtained by linear regression of ARMSS scores. The −log10(P) are plotted against chromosomal position. The horizontal dashed line corresponds to the genome-wide significant threshold (P < 5×10−8) and the horizontal dotted line reflects the threshold for suggestive association (P < 5×10−6). The bold label indicates the lead genome-wide significant and replicated variant. Variants labeled in gray were not replicated. b, Locus Zoom plot for rs10191329 (DYSF-ZNF638 locus). c, Locus Zoom plot for rs149097173 (DNM3-PIGC locus). Top, −log10(P) from the GWAS for variants at each locus (left y-axis) with the recombination rate indicated by the blue line (right y-axis); bottom, gene positions. Colors represent LD (r2 values) with the lead variant. LD, linkage disequilibrium.
Table 1 |.
Variants associated with MS severity.
| Chr. | Position (bp) | ID | EA | EAF | R2 | Effect (s.e.) | P discovery | P replication | P combined | Genes |
|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 71676999 | rs10191329 | A | 0.17 | 0.97 | 0.089 (0.015) | 9.7×10−9 | 0.021 | 3.6×10−9 | DYSF–ZNF638 |
| 1 | 172370873 | rs149097173 | T | 0.01 | 0.94 | 0.256 (0.056) | 4.1×10−6 | 0.010 | 2.3×10−7 | DNM3–PIGC |
Effect on ARMSS score in MS patients. Two variants were genome-wide significant (bold) or suggestive in the discovery GWAS and confirmed in the replication population; two-sided P values were calculated using regression models. Pcombined represents the fixed-effects meta-analysis P value of the discovery and replication data. Chr., chromosome; bp, base pair (GRCh37); EA, effect allele; EAF, risk allele frequency; R2, imputation quality score; s.e., standard error.
The DYSF–ZNF638 locus was confirmed in the replication population and retained genome-wide significance in fixed-effects meta-analysis (P = 3.6×10−9, Table 1). The direction of effect was consistent across all replication centers without evidence of heterogeneity (Q-statistic = 1.5, P = 0.99; I2 = 0%; Extended Data Fig. 3). A suggestive association signal in the DNM3-PIGC locus replicated (P = 0.010) but did not reach genome-wide significance in the combined analysis (P = 2.3×10−7, Table 1). The lead variant (rs149097173) did not overlap with known MS susceptibility loci3. The ten other suggestive loci were not replicated. Statistical fine-mapping16 supported the replicated lead variants to be causal at their respective loci (rs10191329 posterior inclusion probability (PIP) = 0.75, rs149097173 PIP = 0.95; Supplementary Fig. 5). In addition, we examined rs10191329 and rs149097173 for their association with severity in African-American (n = 1,407) and Hispanic (n = 1,718) cohorts. Results were not significant, but the analysis was limited by a lack of statistical power owing to small sample size (median 28%) compounded by substantial imprecision in outcome measures (high proportion of EDSS scores approximated from questionnaire, Supplementary Table 10).
Modifiers of longitudinal outcomes in MS
We next investigated whether the associations identified using the cross-sectional ARMSS score-based GWAS could be confirmed using additional disability outcomes from patients who had been assessed longitudinally. For this analysis, we identified 8,325 patients with EDSS documented at three or more timepoints, including 5,565 from the discovery cohort and 2,760 from the replication cohort. Cumulatively, these patients were evaluated over 54,113 visits spanning up to 13.9 years (Methods). A generalized linear mixed model (LMM) analysis of serial EDSS across all visits revealed that DYSF–ZNF638 risk allele carriers displayed faster disability progression (P = 0.002; Fig. 3a). Moreover, adjusted Cox proportional hazards analyses showed that the risk allele rs10191329A at the DYSF–ZNF638 locus was associated with faster 24-week confirmed disability worsening (hazard ratio [HR] = 1.1 per unit increase in allele dosage, 95% CI 1.02–1.18, P = 7.9×10−3; Fig. 3b), a metric used as the primary outcome in progressive MS therapeutic trials4. In homozygous carriers, the lead variant also conferred a 3.7-year shorter median time to using a walking aid (EDSS 6.0; HR = 1.22, 95% CI 1.09–1.38, P = 9.3×10−4; Fig. 3c), a clinically relevant MS disability milestone that typically tracks with the progressive phase of the disease and fixed neurological disability17.
Fig. 3 |. MS severity variant accelerates disability accumulation in longitudinal analysis.
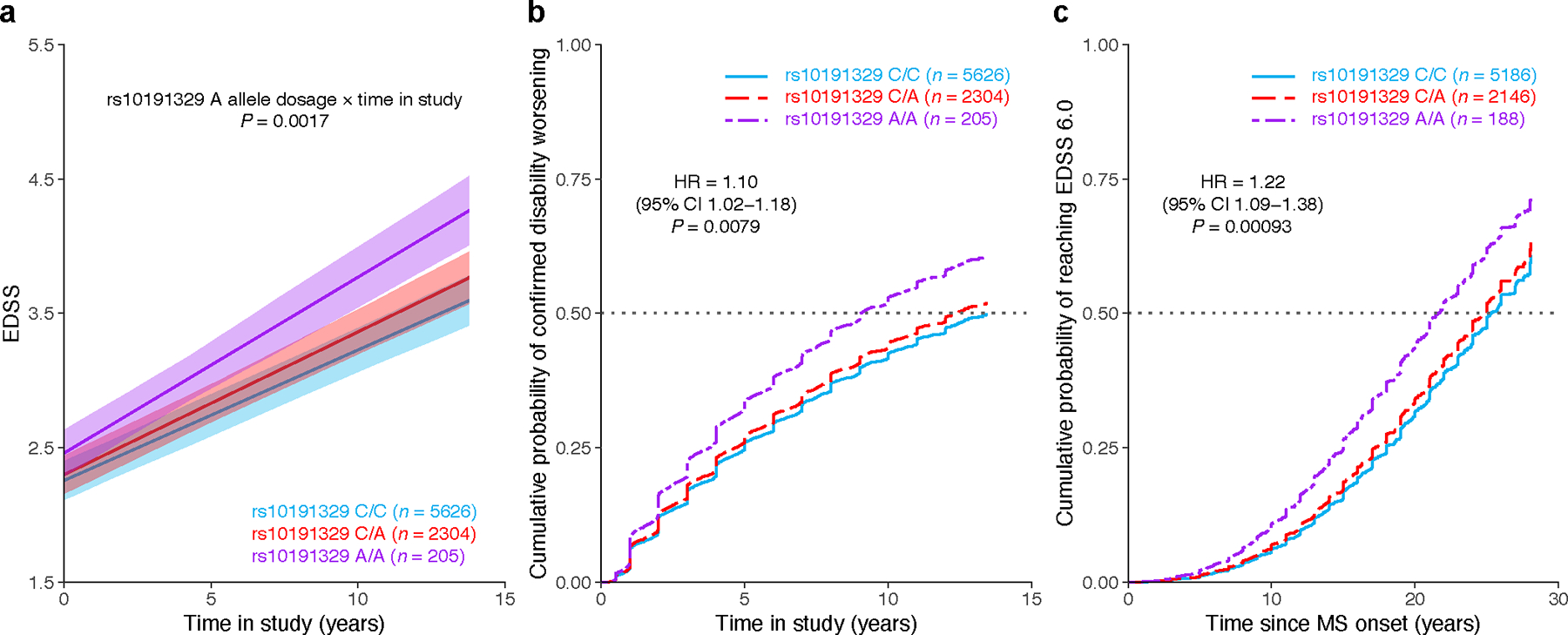
a, Adjusted mean EDSS scores over time predicted from LMM analysis showed faster disability worsening in rs10191329 risk allele carriers. Shaded ribbons indicate the standard error of the mean over time; P value from LMM. b, Covariate-adjusted cumulative incidence of 24-week confirmed disability worsening in MS patients based on rs10191329 genotype. Similar to MS clinical trials, worsening was defined as an increase in EDSS by 1.0 if the baseline score was < 5.5 and by 0.5 if the baseline was ≥ 5.5. c, Covariate-adjusted cumulative incidence of requiring a walking aid for the same lead variant. Homozygous carriers had a 3.7-year shorter median time to require a walking aid. HR and two-sided P values were obtained from Cox proportional hazards models using imputed allele dosage (b–c; Methods). Left-censoring of participants with EDSS ≥ 6.0 at study entry resulted in different sample sizes for genotype groups in the time to walking aid analysis. CI, confidence interval; HR, hazard ratio.
Carriage of the low frequency (MAF = 0.01) risk allele rs149097173T at the DNM3-PIGC locus was only nominally associated with accelerated disability accrual (P = 0.041), faster 24-week confirmed disability worsening (HR = 1.29, 95% CI 1.02–1.65, P = 0.037), and shorter time to EDSS 6.0 (HR = 1.56, 95% CI 1.05–2.34, P = 0.029; Extended Data Fig. 4). These results were not significant after correction for multiple testing (P > 0.05/6), although a sensitivity analysis revealed a statistically significant association that withstood correction for multiple testing with time to sustained EDSS 6.0 (HR = 1.85, 95% CI 1.23 to 2.76, P = 0.0029; Methods) and a 3.3-year shorter median time to require a walking aid for risk allele carriers (for rs10191329, HR = 1.25, 95% CI 1.10 to 1.41, P = 0.0006).
Severity locus impacts CNS tissue injury
To further explore the relationship between rs10191329 and MS severity, we examined the variant’s association with disease-relevant markers of tissue injury in an independent MS autopsy cohort comprising 4,652 tissue blocks in 290 individuals. Consistent with estimates from our longitudinal analysis, homozygous risk allele carriers had experienced a 4-year shorter median time to EDSS 6.0, although differences were not significant in this smaller cohort (Supplementary Table 11). Pathologically, homozygous carriers displayed a 1.83-fold higher number of lesions in the brainstem (95% CI 1.09 to 3.06, P = 0.023; Methods), as well as a 1.76-fold higher rate of cortical lesions across sampled supratentorial tissue (95% CI 1.15 to 2.69, P = 0.001; Fig. 4), confirming that the risk allele at the DYSF-ZNF638 locus is associated with worse injury at key brain locations. It is well established that focal lesions such as those in the brainstem result in axonal loss, and that cortical demyelination, which occurs independently of white matter lesions, is associated with selective neuronal loss18; both these degenerative features are prominent determinants of progression18,19. Our pathological cohort was too small to allow any meaningful analysis of the low frequency variant rs149097173.
Fig. 4 |. Cortical lesion rate and brainstem lesion count are elevated in homozygous rs10191329 risk allele carriers.
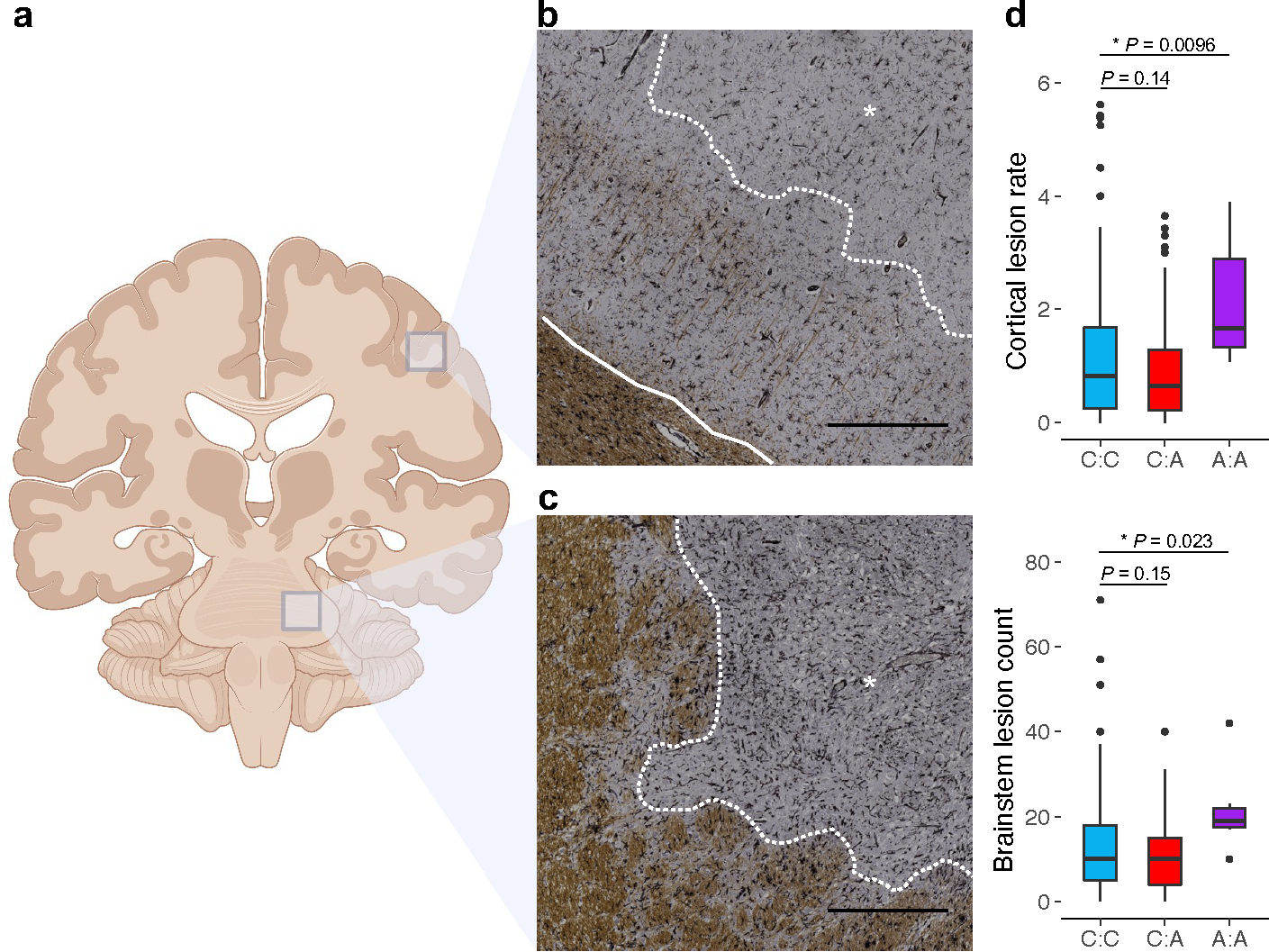
a, Schematic representation of tissue sampling locations (created with BioRender.com). Demyelinating lesions were quantified on a brainstem section dissected in a consistent manner across individuals. Cortical lesions were identified on supratentorial tissue blocks targeted to macroscopic or MRI-visible MS lesions. b, Brain tissue section immunostained for the proteolipid protein marker of myelin (brown). A subpial cortical lesion characterized by loss of myelin is marked by an asterisk and delineated by the dotted white line. The solid white line separates normal-appearing gray matter (sparse brown) from white matter (dense brown). c, A lesion spanning gray and white matter in the brainstem of the same donor, marked by an asterisk and delineated from normal-appearing tissue by the dotted white line. Black scale bars indicate 0.5 mm. The donor was an A allele homozygote for rs10191329. d, Box plots show median, first, and third quartiles; whiskers represent the smallest and largest values within 1.5-times the interquartile range; outliers are depicted as dots. Two-sided P values were obtained from generalized linear models comparing lesion count in the cortex (offset by the relevant number of tissue blocks; n = 174 donors) and brainstem (n = 181 donors) across genotype groups adjusting for covariates; significant differences are marked with an asterisk. The displayed cortical lesion rate was calculated by dividing the number of lesions by the number of tissue blocks containing cortex.
Gene prioritization and related traits
To identify possible biological mechanisms at the discovered loci, we applied several approaches to prioritize putative causal genes (Methods, Supplementary Table 12). The intergenic MS severity variant rs10191329 is nearest to DYSF (3,692 base pairs to the transcription start site), and this gene was prioritized by the combined SNP-to-gene (cS2G)20 strategy based on enhancer-gene linking. This variant also displayed a methylation quantitative trait locus (QTL) effect in the promoter region of DYSF (ENSR00001922663) in the dorsolateral prefrontal cerebral cortex21 (Supplementary Table 13). In addition, rs10191329 showed correlation (r2 > 0.6) with fine-mapped expression QTLs for the upstream gene ZNF638 (Supplementary Table 14) and weaker correlation with splicing QTLs for the same gene in brain (r2 0.3 to 0.4). Predicted expression of ZNF638 in the dorsolateral prefrontal cerebral cortex also associated with MS severity (Z = 3.1, P = 0.002; Methods). Both these genes are highly expressed in neuronal and glial cells in the CNS with shared specificity for oligodendrocytes (Extended Data Fig. 5 and 6) and are important in biological processes of potential relevance. DYSF is implicated in membrane repair22; ZNF638 mediates the silencing of unintegrated viral DNA23. The suggestive variant rs149097173 is intronic to DNM3 and PIGC, the latter also being nominated by cS2G. DNM3 participates in the morphogenesis of the postsynaptic density and excitatory synaptic transmission24 and is preferentially expressed in the CNS, specifically in neurons and oligodendrocyte lineage cells (Extended Data Fig. 5 and 6). PIGC initiates biosynthesis of the glycosylphosphatidylinositol anchor25 (Extended Data Fig. 7). These prioritized genes were differentially expressed in MS brain lesion types relative to control white matter26,27 (Supplementary Note, Supplementary Fig. 6). Integrated analysis of genetically regulated and compound-perturbed gene expression28 revealed significant enrichment for CNS-acting compounds and, along with an alternative locus-based approach, identified chromatin remodeling via histone deacetylase inhibitors as a potential therapeutic strategy for slowing progression (Supplementary Note, Supplementary Fig. 7), an approach with support in preclinical models including a potential for neuroprotection29,30.
Among other traits, rs10191329A has been inversely associated with intelligence, whereas for rs149097173 association is limited to height (Supplementary Tables 15 and 16). Genome-wide, we found no evidence of a shared genetic contribution between MS severity and a range of neurological, psychiatric, and autoimmune disorders. In contrast, cognitive phenotypes and aging traits displayed inverse genetic correlations with MS severity (Extended Data Fig. 8 and Supplementary Table 17). A polygenic score (PGS) for MS severity was not associated with other neurological diseases in the UK Biobank (Supplementary Note).
Education, smoking modulate MS severity
We investigated putative causal and potentially modifiable risk factors for MS severity using two-sample Mendelian randomization (MR). We focused our analyses on traits with prior evidence for association with MS outcomes and suitable genetic instruments, namely 25-hydroxyvitamin D (25OHD) levels, body mass index (BMI), lifetime smoking index, and educational attainment31,32 (Supplementary Table 18). The latter was further motivated by the implication of brain reserve in MS progression33 and our finding of CNS heritability enrichment. MR analyses did not indicate a causal role for either 25OHD levels or BMI in MS severity (Fig. 5a). In contrast, the main inverse-variance weighted MR estimate provided support for an association between higher years of education and milder MS severity (β = −0.15, PIVW = 0.005) and between heavier smoking and worse MS severity (β = 0.23, PIVW = 0.005; Fig. 5a). These results were substantiated by pleiotropy-robust MR sensitivity analyses at different P value thresholds for instrument selection, in the absence of heterogeneity or outliers (Supplementary Table 19). The association with education persisted in multivariable MR adjusting for smoking (β = −0.13, P = 0.04). Reverse analysis did not support an effect of genetic liability to MS severity on the traits considered (Supplementary Table 19). PGS analysis of lifetime smoking index (β = 0.022 per standard deviation score increase, P = 0.004) and education also indicated consistent and independent associations with MS severity (Fig. 5b–c, Supplementary Tables 20 and 21, Methods).
Fig. 5 |. Association of MS severity with educational attainment and smoking.
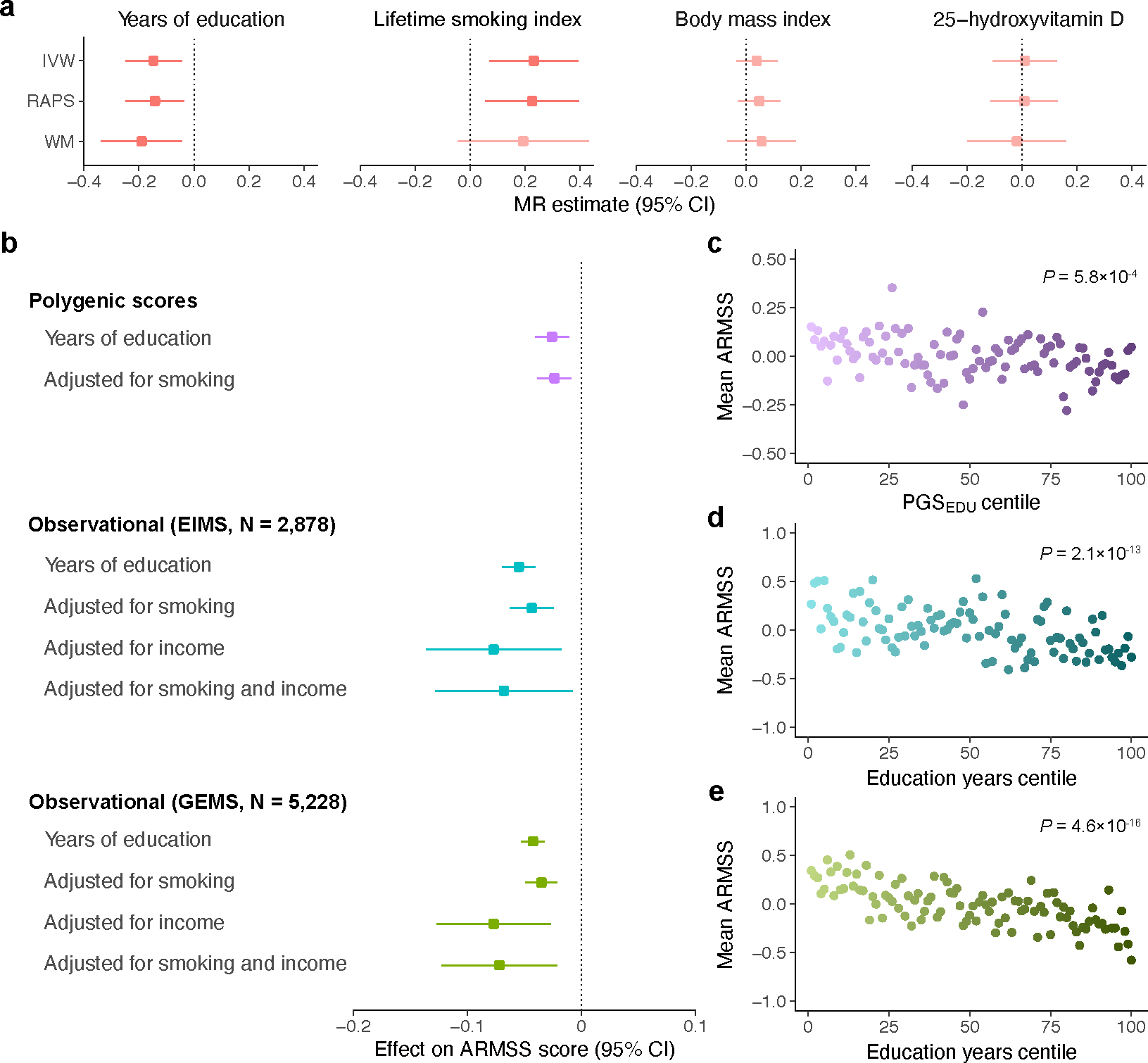
a, MR estimates for the effect of years of education (n = 765,283), lifetime smoking index (n = 462,690), body mass index (n = 681,275), and 25-hydroxyvitamin D (n = 441,291) on ARMSS scores; the lighter color represents nonsignificant results. b, Similarly, adjusted polygenic risk score (n = 12,584) and observational analyses of two MS cohorts (n = 2,878 and 5,228) demonstrated reduced MS severity with higher years of education in linear regression models. This effect persisted following adjustment for smoking and income. c, Mean ARMSS scores decreased with higher PGSEDU percentile. d, Similarly, higher percentile of recorded years of education associated with lower mean ARMSS scores in the EIMS cohort. e, Mean ARMSS scores decreased with higher percentile years of education in the GEMS cohort. P values were obtained from a regression of ARMSS scores on PGSEDU (c) or years of education (d-e), adjusted for baseline covariates. In the MR and observational analyses, point estimates (squares) reflect a 1-year increase in education, while polygenic score estimates are per standard deviation score increase. ARMSS, age-related multiple sclerosis severity score (rank-based inverse-normal transformed); IVW, inverse-variance weighted; PGSEDU, polygenic score for years of education; RAPS, robust adjusted profile score; WM, weighted median.
As genetic influences on educational attainment also capture indirect genetic effects from relatives and social factors34, we then assessed whether the observed association between education and MS severity persisted following adjustment for indicators of socioeconomic status. We extended our analysis to two independent population-based MS cohorts with recorded educational attainment, smoking status, and income. Even after adjusting for these indicators and their interactions, years of education remained associated with MS severity (Fig. 5b–e, Supplementary Tables 20 and 21, Methods). Together, these results demonstrate a detrimental effect of smoking in people with MS and implicate educational attainment as a potential protective factor.
Limited influence of MS risk variants
We undertook multiple approaches to determine whether previously described MS susceptibility variants3 also drive disease severity. First, we observed only weak non-significant genetic correlation between MS severity and susceptibility (rg = 0.17, p = 0.25). Next, the proportion of susceptibility variants showing concordant direction of effect in the severity GWAS was not different from that expected by chance (Pbinom = 0.097). We then aggregated the genome-wide significant MS susceptibility variants into a PGS (PGSMS) and evaluated the gain in coefficient of determinant (incremental R2) when adding PGSMS to a regression of the phenotype on a set of baseline covariates (Methods). We found a weak but statistically significant positive correlation with ARMSS score (incremental R2 = 0.001, P = 7.1×10−5) across MHC and non-MHC regions (Supplementary Fig. 8). However, higher genetic susceptibility for MS leads to earlier age at onset, which in turn is associated with increased MS severity (Supplementary Fig. 9). Therefore, we repeated this analysis adjusting for age at onset and observed that the effect of PGSMS on ARMSS score was substantially attenuated (incremental R2 = 3.9×10−4, P = 0.014; Supplementary Fig. 8). In addition, we interrogated the association of susceptibility variants with longitudinal disability. Individually, none of the variants influenced these outcomes after adjusting for the number tested (Extended Data Fig. 9a–c and Supplementary Table 22). Furthermore, none showed consistent nominal association (P < 0.05) across outcomes (Extended Data Fig. 9d). Comparing individuals in the highest PGSMS quartile to those in the lowest, we detected no differences in their longitudinal outcomes (Extended Data Fig. 10). In short, we found no evidence that susceptibility variants exert a meaningful effect on the outcome of the disease.
DISCUSSION
In this GWAS, which included over 22,000 people with MS, we have identified the first genome-wide significant modifier of long-term outcome in MS, and have thereby identified high-value targets for drug discovery12. The lead variant and an additional suggestive association replicated and showed concordant effects in a range of MS-specific longitudinal outcomes across tens of thousands of patient visits, likely reflecting progressive mechanisms (Supplementary Note). Both severity variants had a clinically meaningful impact on time to needing a walking aid, with the median interval from onset shortened by 3.7 years for homozygous risk allele carriers of the DYSF-ZNF638 variant (rs10191329) and 3.3 years for risk allele carriers of the DNM3-PIGC variant (rs149097173). Although not comparable in terms of likely mechanism, the magnitude of this effect matches that of treatment with a disease modifying agent such as beta-interferon35. Besides these clinical differences, homozygous rs10191329 risk allele carriers also demonstrated more severe MS-specific brainstem and cortical pathology, which result in axonal and neuronal degeneration respectively and drive progression18,19. Furthermore, we show that genetic susceptibility burden has little influence on cross-sectional and longitudinal outcomes outside of its effect on age at onset. MR analyses also provide evidence for smoking and educational attainment as potential modifiable risk factors for MS progression.
Our findings demonstrate that at least 13% of the variance in long-term MS severity can be attributed to common and low frequency single nucleotide variation, explaining some of the considerable variability in MS outcome. Notably, this heritability was enriched in the brain and spinal cord, in marked contrast to the immune signal seen for MS susceptibility. Although divergent genetic determinants of susceptibility and progression have been noted in other conditions8,9,36, the observation of distinct tissue enrichment is to our knowledge unique to MS. This result has potentially significant clinical implications. A persistent challenge in understanding MS progression has been determining the relative contributions of inflammatory activity (including CNS-compartmentalized immune responses) and neurodegeneration4. Here, we show that variation in genes preferentially expressed within the CNS modulate MS severity. Moreover, the prioritized MS severity genes displayed shared cell type specificity in oligodendrocyte lineage cells. This strongly implicates neuronal and glial mechanisms as key determinants of MS progression and, together with our exploratory genomics-driven drug discovery analyses, provides genetic evidence to support the search for new therapeutic targets focused on neuroprotection and brain repair. It may also partly explain why immunosuppressive therapies have thus far had little or no effect on disability accumulation in progressive MS trials4. Our observations are also in concordance with the proposed enhanced penetrance of monogenic causes of neurological disease reported to result from comorbidity with MS37,38.
Our gene prioritization analyses implicated four biologically plausible genes at the identified loci, including ZNF638 upstream of rs10191329. ZNF638 encodes the DNA-binding zinc finger protein 638, which mediates transcriptional repression of unintegrated retroviral DNA through recruitment of the human silencing hub (HUSH) complex and the histone methyltransferase SETDB123. The same chromatin repressors are involved in epigenetic silencing of endogenous retroviruses39. Several exogenous and endogenous viruses have been considered in MS pathogenesis, with the most compelling evidence implicating respectively Epstein-Barr virus (EBV)40 and human endogenous retrovirus type-W (HERV-W)41. The possibility of ZNF638 silencing EBV or HERV-W could have therapeutic implications in MS, as demonstrated by the ongoing development of EBV T-cell therapy (NCT03283826) and HERV-W envelope protein-binding monoclonal antibody42. Furthermore, convergent evidence supports a role, still to be determined, for ZNF638 in the CNS, including in the context of MS. The gene is highly expressed in the brain, particularly in oligodendrocytes and their precursor cells, and has been implicated in large-scale genetic studies of intelligence and general cognitive ability43. In single-nucleus RNA sequencing from brain white matter areas in MS patients and controls, ZNF638 was preferentially expressed in an oligodendrocyte cluster with a predicted actively myelinating phenotype26. Moreover, cell expression of ZNF638 was proportionally enriched in control brain tissue and chronic inactive MS lesions compared to other MS lesions26.
DYSF, the nearest gene to rs10191329, encodes dysferlin, a type II transmembrane protein. Although widely expressed, its functions are mainly characterized in skeletal muscle where it participates in calcium-mediated membrane repair and regeneration22. Recessive pathogenic variants lead to muscular dystrophies (OMIM 254130, 253601, 606768). DYSF is also specifically expressed in oligodendrocytes and excitatory neurons, and the protein has been found to accumulate in Aβ-containing extracellular neuritic plaques, in proportion to Alzheimer’s disease severity44. Although its role in the CNS has yet to be determined, participation in membrane maintenance of neurons or glia could influence neuronal and axonal survival (such as in response to axonal injury33) or subsequent remyelination.
The suggestive variant rs149097173 is located in intron 20 of DNM3, which encodes dynamin-3 and mediates synaptic vesicle endocytosis. As with other prioritized genes, expression is preferentially in oligodendrocytes lineage cells and neurons. Interestingly, the paralog dynamin-2 participates in membrane repair by wound-induced endocytosis in skeletal muscle45, which may point to a convergence of mechanisms with DYSF. Variant rs149097173 is also intronic to PIGC, mutations in which can lead to intellectual disability and epilepsy25.
Our MR results do not support a causal role for serum 25OHD levels or BMI on MS severity. This agrees with the inconclusive results of randomized trials of vitamin D supplementation in MS46 and a recent prospective study that found no association between BMI and clinical disability47. In contrast, our results provide evidence for a causal effect of smoking on worsening disability, in line with strong observational evidence of faster disability progression in smokers that reverses following cessation48. Furthermore, a few observational studies have documented an inverse association between educational attainment and subsequent MS disability31 as well as retinal neurodegeneration49. In accordance with these data, we have found genetic support for educational attainment having a causal effect on reducing long-term MS severity. The effect size was substantial, with 4 years of additional education (equivalent to an undergraduate degree) predicted to reduce disability rank by a quintile. Similar protective effects of education have been observed in Alzheimer’s disease and frontotemporal dementia50,51, indicating some commonality with other neurodegenerative conditions. Genetic determinants of education may partly operate through indirect familial influences and socioeconomic factors34. The persistence of this association following adjustment for smoking and income may suggest direct biological effects. These findings would be consistent with education promoting neurocognitive reserve51, increasing resilience to neuronal degeneration resulting from MS injury and aging. This is further supported by negative genetic correlations with cognitive traits and aging, a factor previously implicated in MS progression immunology and neurobiology33. We caution that neurocognitive reserve is a complex construct that is operationalized using proxies such as education, but it cannot be directly measured51. We have not tested the robustness of these findings to alternative measures of educational attainment (e.g., on a continuous scale) or additional proxies of neurocognitive reserve.
We acknowledge several limitations. Despite its widespread use and regulatory precedent, the EDSS has several shortcomings including its non-linear ordinal nature, variability between raters, overemphasis on ambulation, and inadequate capture of cognitive impairment52. In the survival analyses, events could only be observed at clinic visits and not in real-time. This may bias survival time estimates from clinical settings where follow-up intervals can vary. The MR analysis assumes linearity and may not be applicable to individuals at the extremes of trait distributions, including for vitamin D and BMI. Also, collider bias may occur when considering risk factors affecting both disease onset and progression. Although the MR sensitivity analyses did not find evidence of horizontal pleiotropy, it can only be tested indirectly and violation of this instrumental variable assumption cannot be entirely excluded. Educational attainment and smoking are complex traits influenced by both genetic and environmental factors, and genetic predisposition may not have the same biological consequences as environmental changes (through policy), such that the predicted effect may not be realized. To gain a deeper understanding of the pathways underlying the relationship between education and MS severity, future studies should consider a broader range of social determinants (e.g., neighborhood environments, work exposures, pollution, and patterns of healthcare utilization).
In conclusion, this study presents robust evidence for the role of genetic variation in influencing MS progression. MS has undergone a therapeutic revolution in the past few decades, with the emergence of ever more effective immune therapies that reduce and even halt relapses. Despite this, treatment of progression remains an unmet need. We have identified genetic drivers of disability in MS, providing new directions for functional characterization and drug development targeted on the neurodegenerative component of the disease. Successful unraveling of the genetic basis for disease susceptibility has implicated dysregulation across immune cells as a driver of MS onset. Our findings identify CNS resilience and reserve as likely determinants of MS progression, and may have broader implications for neurodegeneration.
METHODS
GWAS study participants and outcome.
The discovery population consisted of patients with MS recruited through 21 centers from North America, Europe, and Australia. A total of 15,072 patients were genotyped on a common platform (Illumina Global Screening Array) in five cohorts. Samples from patients with longer disease duration, older age, and availability of longitudinal outcome measures were preferentially submitted for genotyping. A primary progressive onset was reported in 8.6% of patients with a documented disease phenotype. Supplementary Tables 1 and 2 respectively describe the case counts per center and additional demographic characteristics. The replication population consisted of a combination of already genotyped MS patients and controls with available clinical information assembled through 9 European centers and genotyped on various Illumina arrays, resulting in 17 cohorts (Supplementary Table 3). Cases of European ancestry that passed sample quality control and had at least one disability measure were included in the analysis (Supplementary Fig. 1). All participants gave written informed consent in accordance with approval from the relevant local ethical committees or institutional review boards (Supplementary Note). Patients with MS were ascertained and diagnosed by a neurologist locally according to established criteria. Neurological disability was measured using the EDSS53, an ordinal scale which incorporates a range of neurological functions relevant to MS. EDSS was scored by neurologist assessment in all but 1,040 cases (4.6%), where it was approximated via questionnaire. For each individual, the last recorded EDSS was converted to an ARMSS score by ranking disability against participants with the same age (±2 years) from the same cohort and from an additional 26,058 patients with MS13.
Quality control and imputation.
For each cohort, we performed individual- and variant-level quality control, after which cohorts were merged into strata based on genotyping platform and submitted to additional stratum-level quality control (Supplementary Note). Sample overlap across strata and between the discovery and replication populations was assessed, and duplicates removed. Imputation to the Haplotype Reference Consortium panel (release 1.1)54 was performed using Minimac4 (v1.0.2)55. The resulting variant counts and imputation quality metrics are described in Supplementary Table 4 and Supplementary Fig. 2, respectively.
GWAS and replication.
To identify genetic variants associated with MS severity, we performed a linear regression model implemented in fastGWA56 using genotype dosages. We applied a rank-based inverse-normal transformation (RINT) to the ARMSS scores and fit as covariates in the model age, sex, date of birth, EDSS source (neurologist assessment vs. questionnaire), center, genotyping batch, and the first ten principal components. Results were unchanged when using a LMM or untransformed ARMSS scores (Supplementary Table 23). Disease modifying therapy was not included as it is not a confounder (i.e. does not influence genotype) and may instead introduce collider bias57. A variable is described as a collider when it is directly affected by both the exposure and the outcome of interest (genetic variants and disease severity in our case), or some unmeasured variables that also influence the outcome (e.g. comorbidities)58. Conditioning on a collider or its descendants can introduce bias in either direction (spurious associations or false negatives). To assess any residual confounding due to population stratification or cryptic relatedness, we calculated the genomic inflation factor and LDSC intercept using HapMap3 variants and LD scores from 1000 Genomes phase 359. Conditional and joint (COJO) analysis60 was performed to identify potential secondary association signals. Lead variants with association P ≤ 5×10−8 were considered genome-wide significant and were tested in the replication population, together with those with suggestive association P ≤ 5×10−6.
As above, linear regression of ARMSS scores was performed in the replication population using the same covariates. Individual-level imputed genotypes were merged across strata prior to joint analysis. Principal components were calculated on a set of hard-called high-quality (imputation R2 ≥ 0.9, genotype missingness < 0.01, MAF > 0.05) and LD-pruned genotypes. To examine for heterogeneity, we recalculated the association between lead variants and MS severity in the replication stratified by center (n=9) and computed Q-statistics and I2 tests. Association statistics from the discovery and replication were combined using fixed-effects meta-analysis. Finally, we examined the association of the two replicated severity variants (rs10191329 and rs149097173) in self-identified African-American (n = 1,407) and Latinx/Hispanic (n = 1,718; herein referred to as Hispanic) participants with MS with an available disability measure, recruited by the Alliance for Research in Hispanic Multiple Sclerosis61. Principal component projections for the retained African-American samples overlapped with those from 1000 Genomes African populations. LMM analysis was conducted using the same covariates as in the GWAS.
Heritability estimation.
To estimate SNP-based heritability, we constructed a genomic relationship matrix (GRM) from all variants and used it to remove individuals (n = 848) with a coefficient of relationship > 0.025. The resulting GRM was used to estimate SNP-heritability with restricted maximum likelihood (single-component GREML)62. As SNP-heritability can be sensitive to LD and allele frequency assumptions63, we also fitted a model with ten GRMs (GREML-LDMS) constructed from variants assigned to five MAF bins (0.01–0.1, 0.1–0.2, 0.2–0.3, 0.3–0.4 and 0.4–0.5) each divided into two by the median LD score in each bin. To calculate LD scores, variants were first hard-called (PLINK2 –hard-call-threshold 0.1) then filtered for missingness < 0.05, MAF > 0.01 and HWE P > 10−6. Heritability analyses were adjusted for the same set of covariates as the GWAS.
Heritability enrichment analyses.
We used stratified LDSC (version 1.0.1) to calculate SNP-based heritability enrichment for 53 functional categories (baseline model version 1.2)14. Next, we assessed the SNP-based heritability associated with different tissues by applying stratified LDSC to our GWAS summary statistics using a gene expression dataset consisting of 205 tissues and cell types (as provided in the LDSC software)15. Tissues and cell types were grouped into nine categories for visualization (Supplementary Tables 7 and 8). The same analysis was repeated with the summary statistics from the discovery phase of our previous GWAS meta-analysis of MS susceptibility3 to compare the enrichment patterns. We applied FDR correction for multiple testing within each enrichment analysis, and FDR-corrected P < 0.05 were considered statistically significant. Summary statistics were filtered for variants in HapMap3, with an imputation R2 ≥ 0.9, and outside of the MHC region prior to analysis. We also extended this framework to human CNS cell types from single-nucleus RNA sequencing studies64–66 (Supplementary Note, Supplementary Fig. 10).
Analysis of longitudinal outcomes.
We identified a subset of 8,325 MS patients from our study population with a minimum of 3 visits separated by at least 6 months (5,565 from the discovery cohort and 2,760 from the replication cohort). These patients contributed a total of 56,966 visits, of which 54,113 (95%) occurred within 13.9 years of follow-up from the first study visit (mean 5.2 years). To assess the influence of MS severity variants on the rate of disability progression, we constructed a generalized LMM with serial EDSS scores as the dependent variable. The primary predictor was the interaction term between genotype (dosage or carrier status) and time in the study (years), with individuals and centers as random terms. Subject-level fixed covariates were sex, age at onset and study entry, date of birth, and the first ten principal components. This analysis was performed using penalized quasi-likelihood estimation as implemented in the glmmPQL function from the ‘MASS’ package (version 7.3–54) in R to address the non-normal distribution of EDSS.
In addition, two key MS-specific disability outcomes were examined in survival analyses. First, we estimated the influence of MS severity variants on time to a clinically meaningful increase in neurological disability. Similar to MS clinical trials67, worsening was defined as an increase in EDSS by 1.0 if the baseline score was < 5.5 and by 0.5 if the baseline was ≥ 5.5. To increase specificity, the endpoint also required this EDSS increase to be maintained on a subsequent visit and for at least 24 weeks. Second, we examined the influence of genotype on time (from disease onset) to reaching EDSS 6.0 (defined as requiring unilateral assistance to walk more than 100 meters). A sensitivity analysis also evaluated the time to sustained EDSS 6.0, requiring subsequent scores to remain at or above 6.0 until censoring. Following left-censoring, 7,695 patients and 51,189 study visits remained, extending to 28.3 years from disease onset. Cox proportional hazards analyses were carried out using the coxph function in the ‘survival’ package (version 3.2–11) in R, with Efron approximation for tie handling. Sex, age at onset, date of birth, center, genotyping platform, and the first ten principal components were included as covariates. Adjustment for baseline EDSS was included in the 24-week confirmed disability worsening analysis to account for the non-linear nature of this scale; this was not applicable for the time to EDSS 6.0 analysis. The proportional hazards assumption was examined by inspection of scaled Schoenfeld residuals. Hazard ratios were calculated using dosages for rs10191329 and carrier status for rs149097173 given its low frequency. P values < 0.0083 were considered significant following Bonferroni correction for the number of variants and outcomes tested. Sensitivity analysis found no evidence of bias introduced by including participants who partially overlapped with the ARMSS score-based GWAS (Supplementary Note).
Fine-mapping.
For each lead variant, effect estimates on MS severity in a 250 kb region centered on the variant were extracted. A variant correlation matrix was computed with LDstore2 (version 2.0)68 from the same genotype dosage used to generate the GWAS summary statistics. Fine-mapping with shotgun stochastic search was performed using FINEMAP (version 1.4)16 with equal prior probabilities.
MS autopsy cohort and associations with neuropathology.
Following informed consent, brain donors with pathologically confirmed MS recruited to the Netherlands Brain Bank since 1990 were clinically and pathologically characterized (Supplementary Table 11). Autopsy procedures were approved by the Ethical Committee of the VU University Medical Center in Amsterdam, the Netherlands. As previously described69, blocks were dissected at standardized CNS locations (including the brainstem), with additional blocks targeted to MS lesions using macroscopic and post-mortem MRI assessment. Sections were double-immunostained for proteolipid protein and human leukocyte antigen. For each individual, a brainstem lesion count was quantified using one section per standardized block. Areas of cortical gray matter demyelination were identified and classified by location (subpial, intracortical, leukocortical, pancortical). These lesion locations were selected based on their recognized importance to MS pathophysiology69,70 and their count frequency. DNA was extracted from whole blood or frozen cerebellar tissue, or when neither were available from formalin-fixed paraffin-embedded cerebellar tissue. Genotyping for rs10191329 was performed using the KASP genotyping platform (LGC Genomics, Hoddesdon, UK). Pathological characterization was undertaken blind to genotype status. Differences in brainstem lesion load and rate of cortical lesions between genotype groups were examined using quasi-Poisson regression adjusted for sex, age at onset and initial disease course. To account for a variable number of supratentorial blocks sampled between individuals, cortical lesions were considered as a rate by adding the number of tissue blocks with visible cortex as an offset. Individuals with missing dependent variables or covariates were excluded. P values less than 0.025 were considered significant (adjusting for two pathological variables).
Gene prioritization.
To prioritize putative causal genes, we applied a combination of functional and non-functional strategies: (1) the closest gene(s), defined as genes with overlapping bodies or closest transcription start site; (2) genes that overlap with a genomic range of 200 kb centered around the variant; (3) genes with missense or loss of function coding variants in LD (r2 > 0.6) with the lead variant; (4) genes with fine-mapped (PIP > 0.1) cis-eQTL or splicing QTL in LD (r2 > 0.6) with the lead variant; (5) genes prioritized by Open Targets Genetics using a V2G71 threshold of 0.5; (6) genes prioritized by the combined SNP-to-gene (cS2G) strategy20; (7) genes whose imputed expression is associated with MS severity72. We retrieved fine-mapped QTLs from GTEx73 (version 8) and the eQTL catalogue74. The V2G aggregates weighted evidence from variant functional prediction, colocalization with molecular QTLs, chromatic interaction and gene distance. The cS2G strategy consists of seven components, with gene assignments most often driven by a single feature. The association between MS severity and predicted gene expression in a ±1 megabase window around each lead variant was assessed using FUSION72 and an expression reference panel of the dorsolateral prefrontal cortex from 452 CommonMind Consortium participants. A Bonferroni-corrected significance level was set using the number of local genes present in the reference panel (P < 0.05/6; ZNF638, MPHOSPH10, TGFA, CYP26B1, VAMP4, TNFSF18). Moreover, we evaluated the influence of MS severity variants on brain dorsolateral prefrontal cortex methylation based on 543 individuals from ROSMAP (Bonferroni-corrected P < 5×10−9)21.
Associations with other traits.
To investigate the effects of the MS severity variants on previously reported phenotypes, we retrieved phenome-wide associations in the Open Target Genetics portal75 obtained from the GWAS Catalog, UK Biobank and FinnGen. We also calculated genome-wide genetic correlations between MS severity and 17 neurological, psychiatric, autoimmune, cognitive, and aging phenotypes (Supplementary Table 17) using cross-trait LDSC76. Since we expect no sample overlap with our within-case GWAS, the LDSC intercept was constrained on the assumption of no shared population stratification. Benjamini-Hochberg-adjusted P values below 0.05 were considered significant.
Gene expression profiles.
Gene expression values in human tissues for the prioritized genes at the two MS severity loci were obtained from GTEx73 (version 8). Cell type expression profiles for the same genes were evaluated using single-cell RNA sequencing data in 76 cell types from the Human Protein Atlas77. We examined genes for cell type specificity, defined as expression that is at least fourfold higher in a cell type compared to the mean of all others (cell type enhanced)77. Since PIGC expression in brain neuronal and glial cell types was missing, we obtained it from a study of 4 progressive MS patients and 5 non-neurological controls with single nuclear RNA expression in white matter tissues26.
Mendelian randomization.
We applied MR analysis to investigate the effects of four exposures with robust genetic associations and strong prior evidence of association with MS severity. In the case of body mass index and 25-hydroxyvitamin D, previous MR studies additionally provided support for a causal role in the development of MS78. A description of the GWAS used to proxy the exposures is provided in Supplementary Table 18. For each of these, variants were selected at two different association thresholds (P < 5×10−8 and P < 5×10−5), as in previous studies36, and LD clumped (r2 < 0.001) to ensure independence. Palindromic variants were excluded. For variants absent from our MS severity GWAS, we selected a strong LD proxy (r2 > 0.8) when possible. The variants included were examined for instrument strength79 (mean F-statistic > 10; Supplementary Table 18).
The main analysis was performed using the inverse-variance weighted MR approach with a random-effects model. We also tested for heterogeneity across the genetic variants as a potential indicator of horizontal pleiotropy, using the Cochran’s Q-statistic and MR-pleiotropy residual sum and outlier (PRESSO) global test80,81. To further examine the assumption of no horizontal pleiotropy, we applied four additional MR methods: robust adjusted profile score, weighted median, MR-PRESSO, and MR-Egger regression (reviewed in ref81). Consistent results across these methods reduce the likelihood of bias. For the MR-Egger regression, we focused on the intercept as a test for unbalanced pleiotropy given that the association estimate is considerably underpowered82, although beta-coefficients are reported in Supplementary Table 19. Multivariable MR was also conducted to determine the effect of education adjusted for smoking on MS severity.
To determine the direction of effect, we also conducted a reverse analysis examining the effect of genetic liability to MS severity on each of the traits considered. Because there was only one genome-wide significant variant, the reverse analysis was only performed using the instrument selection threshold of P < 5×10−5. Finally, to provide an interpretable estimate of the effect size of education on MS severity, we repeated the educational attainment MR analysis using a GWAS of untransformed ARMSS scores. Analysis was conducted using the ‘MendelianRandomization’ and ‘TwoSampleMR’ R packages.
Education and smoking polygenic scores.
We constructed PGSs using LD clumped (r2 < 0.001) genome-wide significant variants (P < 5×10−8) associated with educational attainment34 and lifetime smoking index, a measure capturing smoking initiation (i.e. ever and never smokers) and, among ever smokers, also accounts for smoking intensity, duration and cessation83. For educational attainment, associations from the full meta-analysis including 23andMe samples (n = 3,037,499) were considered. Each PGS was regressed on ARMSS scores adjusting for age, sex, center, batch, date of birth, EDSS source, initial disease course, age at onset, and the first 10 principal components. To test for independence between education and smoking, we repeated the regression analysis including both PGSs and their interaction.
Observational analysis of educational attainment.
The association between educational attainment and long-term MS disability was assessed in two independent population-based Swedish cohorts, the Epidemiological Investigation of Multiple Sclerosis (EIMS) and Genes and Environment in MS (GEMS) studies. Cohort and variable descriptions are reported in the Supplementary Note. In each cohort, linear regression analyses assessed the association between recorded years of education (Supplementary Table 24) and MS severity adjusting for age, sex, date of birth, initial disease course, and age at onset. We also examined whether the observed association was dependent upon smoking status or income level by adding them to the model separately and then together, allowing for interaction with years of education.
MS susceptibility variants.
To compare the genetic architecture of MS susceptibility and severity, we calculated the genome-wide genetic correlation excluding the MHC region using bivariate LDSC with unconstrained intercept (version 1.0.1)59. A free intercept was modeled to allow for sample overlap. We then focused our analyses on the 232 autosomal MS susceptibility associations we previously reported3. For non-MHC variants, we included the association statistics from the joint analysis and labeled them using the discovery variant (‘SNP discovery’). We excluded variants that were palindromic (n=1), missing from the current study (n=1) or with a joint P > 5×10−8 (n=2). For MHC associations, we included those reported as non-palindromic single nucleotide variants (as opposed to HLA alleles) and added rs3135388 to tag HLA-DRB1*15:0184. In total, 209 variants (197 non-MHC and 12 MHC) were examined (Supplementary Table 22). A two-sided exact binomial test was used to assess concordance of direction of effect on MS susceptibility and severity. The same variants were tested for association with longitudinal outcomes using a Bonferroni-corrected significance threshold [P < 0.05/(209×3) or 8.0×10−5] and evaluated for concordance of nominal association (P < 0.05) across four disability outcomes (ARMSS score, 24-week confirmed disability worsening, time to EDSS 6.0 and rate of EDSS change).
To determine the aggregate effect of MS susceptibility on disability outcomes, we constructed a PGS (PGSMS) using 178 variants retained following LD clumping (r2 < 0.01) of the 209 susceptibility associations. Variants were weighted by the natural log of their joint odds ratio. We then regressed the ARMSS scores on PGSMS adjusting for the same covariates as in the GWAS. We also regressed the phenotype on the covariates alone and measured the difference in R2 with and without PGSMS, reported as the incremental R2. We performed similar analyses using age at onset, as well as ARMSS scores adjusted for age at onset. Next, we compared individuals in the highest and lower quartile of PGSMS based on the same survival and LMM analyses as previously described for the MS severity variants.
Extended Data
Extended Data Fig. 1 |. Demographic characteristics by population and center.

a, Discovery population (n = 12,584). b, Replication population (n = 9,805). Bars represent the proportion of patients in each category. Centers are ordered as in the box plot legend (bottom right subpanel). Box plots show median, first, and third quartiles; whiskers represent the smallest and largest values within 1.5-times the interquartile range; outliers are depicted as dots. The countries corresponding to the abbreviations in the box plot legend are shown in Supplementary Table 1. ARMSS, age-related multiple sclerosis severity; EDSS, expanded disability status scale; Primary prog., primary progressive; yrs, years.
Extended Data Fig. 2 |. Principal component analysis of the discovery and replication populations.

MS cases were recruited from 13 countries for the discovery (a) and 8 for the replication (b). After removing population outliers, all remaining cases were of European ancestry. The first two principal components respectively captured the north-to-south and east-to-west gradients of European genetic structure. US and Canadian participants overlapped with those from other countries. Based on self-reported ancestry, East European and Ashkenazi Jewish individuals constituted the majority of the predominantly US subcluster located at the bottom right of the discovery population (a). The scree plots for our principal component analysis in the discovery (c) and replication (d) populations confirm that the first few principal components capture most of the variance attributable to the minimal population structure remaining after quality control.
Extended Data Fig. 3 |. Replication of MS severity variants by center.

a, Genome-wide significant lead variant rs10191329. b, Suggestive lead variant rs149097173. Forest plots show successful replication of the two variants with minimal heterogeneity between centers as indicated by the Cochran’s Q and I2 statistics (n = 9,805 participants). ARMSS scores are rank-based inverse-normal transformed. Error bars represent 95% CIs. ARMSS, age-related multiple sclerosis severity; CI, confidence interval.
Extended Data Fig. 4 |. Association of rs149097173 with longitudinal disability outcomes.

a, Adjusted mean EDSS scores over time by carrier status for rs149097173 predicted from LMM analysis. Shaded ribbons indicate the standard error of the mean over time; P value from LMM. b, Covariate-adjusted cumulative incidence of 24-week confirmed disability worsening for the same groups of individuals. c, Covariate-adjusted cumulative incidence of requiring a walking aid; carriers had a 2.2-year shorter median time to require a walking aid. HR and two-sided P values were obtained from Cox proportional hazards models using imputed allele dosage (b–c; Methods). Results were not significant after adjusting for multiple testing across two variants (see Fig. 3 for rs10191329 associations) and three outcomes (P < 0.05/6), although the latter are not expected to be independent. CI, confidence interval; HR, hazard ratio.
Extended Data Fig. 5 |. Tissue expression for nominated MS severity genes.

Gene expression profiles were obtained from GTEx73 (version 8). Transcripts were collapsed to the gene level and expressed in natural log-transformed transcript per million (TPM) units. DYSF, ZNF638, DNM3 and PIGC are expressed in the brain. Box plots show median, first, and third quartiles; whiskers represent the smallest and largest values within 1.5-times the interquartile range; outliers are depicted as dots. Bold x-axis labels identify CNS tissues. Colors represent tissue types as defined in GTEx.
Extended Data Fig. 6 |. Cell type expression profiles for nominated MS severity genes.

Single-cell RNA sequencing data from 25 human tissues and peripheral blood mononuclear cells were obtained from the Human Protein Atlas77. Transcript expression levels were summarized per gene and reported as average normalized transcripts per million (nTPM) in 76 cell types. Asterisks mark cell type specificity for the gene, defined as at least fourfold higher expression in a cell type compared to the mean of others. We note that three of the genes show specificity for oligodendrocyte lineage cells. PIGC expression in brain neuronal and glial cells, missing here, is demonstrated in Extended Data Fig. 8. Colors represent cell type categories; bold x-axis labels identify neuronal and glial cell categories.
Extended Data Fig. 7 |. Cell type expression for PIGC in brain white matter tissue.

Single nuclear RNA expression from 4 progressive MS patients and 5 non-neurological controls26 confirms PIGC expression in neuronal and glial cells including oligodendrocyte lineage cells. COPs, committed oligodendrocyte precursors; ImOLGs, immune oligodendroglia; Oligo, oligodendrocyte; OPCs, oligodendrocyte precursor cells; Vasc, vascular.
Extended Data Fig. 8 |. Genetic correlations with MS severity.

Shared genetic contribution obtained from cross-trait LDSC. Colors correspond to genetic correlation (rg) estimates (blue, negative; red, positive). An asterisk indicates a correlation that is significantly different from zero, based on two-sided P values calculated using LDSC (*FDR < 0.05, **FDR < 0.01). Full results are in Supplementary Table 17. Aging-GIP1 was constructed using principal component analysis to capture GWASs of healthspan, father lifespan, mother lifespan, longevity, frailty, and self-rated health85.
Extended Data Fig. 9 |. Association of individual MS susceptibility variants (n = 209) with longitudinal disability outcomes.

a, Distribution of P values from adjusted LMM analysis of EDSS change across all study visits. Distribution of two-sided P values from adjusted Cox proportional hazards analyses of (b) time to 24-week confirmed disability worsening and (c) time to require a walking aid. The dashed orange line represents the Bonferroni-corrected significance threshold adjusted for the number of susceptibility variants. d, Venn diagram of nominal associations (Punadjusted < 0.05) between individual MS susceptibility variants and all disability outcomes considered; no variant showed consistent association across three or more outcomes. The labels in this panel correspond to the following outcomes: ARMSS, association with ARMSS scores following rank-based inverse normal transformation; Disability worsening, time to 24-week confirmed disability worsening; Walking aid, time to require a walking aid (EDSS 6.0); EDSS rate, rate of EDSS change across all study visits.
Extended Data Fig. 10 |. MS susceptibility PGS and longitudinal disability outcomes.

a, Adjusted mean EDSS scores over time by PGS quartile predicted from LMM analysis. Shaded ribbons indicate the standard error of the mean over time; P value from LMM. b, Covariate-adjusted cumulative incidence of 24-week confirmed disability worsening comparing individuals in the highest versus those in the lowest quartile of MS susceptibility PGS. c, Covariate-adjusted cumulative incidence of requiring a walking aid for the same groups of individuals. HR and two-sided P values were obtained from Cox proportional hazards models using imputed allele dosage (b–c; Methods). Across all analyses, the MS susceptibility PGS had no influence on longitudinal outcomes.
Supplementary Material
Acknowledgments.
We thank all study participants for their support and for making this work possible. This work was supported by funding from the NIH/NINDS (R01NS099240) to S.E.B. and S.J.S., and the European Union’s Horizon 2020 Research and Innovation Funding Programme (EU RIA 733161) to MultipleMS. We acknowledge support from the National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre. A.H. is supported by the NMSS-ABF Clinician Scientist Development Award (FAN-1808-32256) funded by the National Multiple Sclerosis Society (NMSS) and the Multiple Sclerosis Society of Canada (MSSC). We thank Dr. Ganqiang Liu (Sun Yat-Sen University) for help with the longitudinal analyses. P.S. is supported by the Magretha af Ugglas foundation and Horizon 2020 EU grant (MultipleMS, 733161). S.E.B holds the Professorship in Neurology I and the Heidrich Family and Friends Endowed Chair in Neurology. The UCSF DNA biorepository is supported by the NMSS (Si-2001-35701). J.L.M. acknowledges funding support from the NIH/NINDS (R01NS096212) and the Genentech Health Equity Innovation Fund (G-79758). L.A. has received academic grant support from the Swedish Research Council, the Swedish Research Council for Health, Working Life and Welfare and the Swedish Brain foundation. S.R.D. has received institutional research grant funding from the NMSS and the NIH/NINDS. T.O. has received academic grant support from the Swedish Research Council, the Swedish Brain foundation, Knut and Alice Wallenberg foundation and Margaretha af Ugglas foundation. M.J.F.-P. has received grant support from the Multiple Sclerosis Society of Western Australia (MSWA). M.V. is a PhD fellow (11ZZZ21N) and B.D. is a Clinical Investigator of the Research Foundation-Flanders (FWO-Vlaanderen). B.D. and A.G. have received academic grant support from the Research Fund KU Leuven (C24/16/045) and the Research Foundation Flanders (FWO G.07334.15). S.L. holds research support from the Spanish Government (PI21/010189, PI18/01030, PI15/00587), funded by the Instituto de Salud Carlos III-Subdirección General de Evaluación and co-funded by the European Union, and the Red Española de Esclerosis Múltiple (REEM: RD16/0015/0002, RD16/0015/0003). S.B. and F.Z. have received funding from the German Research Foundation (CRC-TR-128). F.Z. also acknowledges support from the Progressive MS Alliance (BRAVEinMS PA-1604-08492) and the Federal Ministry of Education and Research (VIP+ HaltMS-03VP07030). A.M. is supported by Margaretha af Ugglas foundation. B.H. is associated with DIFUTURE (Data Integration for Future Medicine) [BMBF 01ZZ1804[A-I]]. He received funding for the study by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy within the framework of the Munich Cluster for Systems Neurology [EXC 2145 SyNergy – ID 390857198]. The study was supported by the Italian Foundation of Multiple Sclerosis (FISM, 2011/R/14 2015/R/10, 2019/R-Multi/033, grants), Ricerca finalizzata, Italian Ministry of Health (RF-2016-02361294 grant), the AGING Project for Department of Excellence at the Department of Translational Medicine (DIMET), Università del Piemonte Orientale, Novara, Italy. N.B. is partly supported by the MultipleMS project (Horizon 2020 European, Grant N. 733161). N.A.P. was supported in part by the NMSS (grants JF-1808-32223 and RG-1707-28657). In.K. was partly supported by the MultipleMS project (Horizon 2020 European, Grant N. 733161), the Swedish Research Council (Grant N. 2020–01638), and the Swedish Brain foundation. L.F.B. is supported by the NIH (R01ES017080, R01AI076544, R01NS049510, and R01NR017431)
This manuscript is dedicated to the memory of Rogier Q. Hintzen, a member of the International Multiple Sclerosis Genetics Consortium, in recognition of his contributions to human genetic research.
The International Multiple Sclerosis Genetics Consortium
Adil Harroud1*, Pernilla Stridh2, Jacob L. McCauley3,4, Janna Saarela5,6, Aletta M. R. van den Bosch7, Hendrik J. Engelenburg7, Ashley H. Beecham3, Lars Alfredsson2, Katayoun Alikhani8, Lilyana Amezcua9, Till F. M. Andlauer10, Maria Ban11, Lisa F. Barcellos12, Nadia Barizzone13, Tone Berge14,15, Achim Berthele10, Stefan Bittner16, Steffan D. Bos17,18, Farren B. S. Briggs19, Stacy J. Caillier1, Peter A. Calabresi20, Domenico Caputo21, David X. Carmona-Burgos22, Paola Cavalla23, Elisabeth G. Celius17,18, Gabriel Cerono1, Angel R. Chinea22,24, Tanuja Chitnis25,26, Ferdinando Clarelli27, Manuel Comabella28, Giancarlo Comi29,30, Chris Cotsapas31,32, Bruce C. A. Cree1, Sandra D’Alfonso13, Efthimios Dardiotis33, Philip L. De Jager34, Silvia R. Delgado35, Bénédicte Dubois36,37, Sinah Engel16, Federica Esposito38, Marzena J. Fabis-Pedrini39,40, Massimo Filippi41,30, Kathryn C. Fitzgerald20, Christiane Gasperi10, Lissette Gomez3, Refujia Gomez1, Georgios Hadjigeorgiou42, Jörg Hamann43,7, Friederike Held10, Roland G. Henry1, Jan Hillert2, Jesse Huang2, Inge Huitinga7,44, Talat Islam45, Noriko Isobe46, Maja Jagodic2, Allan G. Kermode47,40, Michael Khalil48, Trevor J. Kilpatrick49,50,51, Ioanna Konidari3, Karim L. Kreft52, Jeannette Lechner-Scott53,54, Maurizio Leone55, Felix Luessi16, Sunny Malhotra28, Ali Manouchehrinia2, Clara P. Manrique3, Filippo Martinelli-Boneschi56,57, Andrea C. Martinez9, Viviana Martinez-Maldonado22, Elisabetta Mascia27, Luanne M. Metz8, Luciana Midaglia28, Xavier Montalban28, Jorge R. Oksenberg1, Tomas Olsson2, Annette Oturai58, Kimmo Pääkkönen6, Grant P. Parnell59,60, Nikolaos A. Patsopoulos61,62,63, Margaret A. Pericak-Vance3,4, Fredrik Piehl2, Justin P. Rubio50,51, Adam Santaniello1, Silvia Santoro27, Catherine Schaefer64, Finn Sellebjerg58,65, Hengameh Shams1, Klementy Shchetynsky2,66, Claudia Silva8, Vasileios Siokas33, Helle B. Søndergaard58, Melissa Sorosina27, Bruce Taylor67, Marijne Vandebergh37, Elena S. Vasileiou20, Domizia Vecchio68, Margarete M. Voortman48, Howard L. Weiner25,26, Dennis Wever7, V. Wee Yong8, David A. Hafler61,69, Graeme J. Stewart70,71, Alastair Compston11, Frauke Zipp16, Hanne F. Harbo17,18, Bernhard Hemmer10,72, An Goris37, Joost Smolders73,7, Stephen L. Hauser1, Ingrid Kockum2, Stephen J. Sawcer11,78, Sergio E. Baranzini1,78.
The MultipleMS Consortium
Adil Harroud1*, Pernilla Stridh2, Janna Saarela5,6, Ingileif Jónsdóttir74,75, Till F. M. Andlauer10, Maria Ban11, Tone Berge14,15, Yolanda Blanco76, Steffan D. Bos17,18, Elisabeth G. Celius17,18, Ferdinando Clarelli27, Chris Cotsapas31,32, Sandra D’Alfonso13, Bénédicte Dubois36,37, Federica Esposito38, Christiane Gasperi10, Roland G. Henry1, Jesse Huang2, Maja Jagodic2, Maurizio Leone55, Sara Llufriu76, Lohith Madireddy1, Ali Manouchehrinia2, Filippo Martinelli-Boneschi56,57, Tomas Olsson2, Annette Oturai58, Kimmo Pääkkönen6, Fredrik Piehl2, Albert Saiz76, Silvia Santoro27, Finn Sellebjerg58,65, Klementy Shchetynsky2,66, Helle B. Søndergaard58, Melissa Sorosina27, Pablo Villoslada76,77, Kári Stefánsson74,75, Frauke Zipp16, Hanne F. Harbo17,18, Bernhard Hemmer10,72, An Goris37, Ingrid Kockum2, Stephen J. Sawcer11,78, Sergio E. Baranzini1,78.
1UCSF Weill Institute for Neurosciences, Department of Neurology, University of California, San Francisco, San Francisco, CA, USA. 2Department of Clinical Neuroscience, Karolinska Institutet, Center for Molecular Medicine, Karolinska University Hospital, Stockholm, Sweden. 3John P Hussman Institute for Human Genomics, Miller School of Medicine, University of Miami, Miami, FL, USA. 4The Dr. John T. Macdonald Foundation Department of Human Genetics, Miller School of Medicine, University of Miami, Miami, FL, USA. 5Centre for Molecular Medicine Norway, University of Oslo, Oslo, Norway. 6Institute for Molecular Medicine Finland, Helsinki Institute for Life Sciences, University of Helsinki, Helsinki, Finland. 7Neuroimmunology Research Group, Netherlands Institute for Neuroscience, Amsterdam, Netherlands. 8Department of Clinical Neurosciences and the Hotchkiss Brain Institute, University of Calgary, Calgary, Canada. 9Multiple Sclerosis Division, Department of Neurology, Keck School of Medicine, Univeristy of Southern California, Los Angeles, CA, USA. 10Department of Neurology, School of Medicine, Technical University of Munich, Munich, Germany. 11Department of Clinical Neurosciences, University of Cambridge, Cambridge, UK. 12Genetic Epidemiology and Genomics Laboratory, Division of Epidemiology, School of Public Health, University of California, Berkeley, Berkeley, CA, USA. 13Department of Health Sciences and Center on Auto-immune and Allergic Diseases (CAAD), University of Eastern Piedmont, Novara, Italy. 14Department of Research, Innovation and Education, Oslo University Hospital, Oslo, Norway. 15Institute of Mechanical, Electronics and Chemical Engineering, Faculty of Technology, Art and Design, Oslo Metropolitan University, Oslo, Norway. 16Department of Neurology, Focus Program Translational Neuroscience (FTN) and Immunotherapy (FZI), University Medical Center of the Johannes Gutenberg University Mainz, Mainz, Germany. 17Department of Neurology, Oslo University Hospital, Oslo, Norway. 18Institute of Clinical Medicine, University of Oslo, Oslo, Norway. 19Department of Population and Quantitative Health Sciences, School of Medicine, Case Western Reserve University, Cleveland, OH, USA. 20Department of Neurology, Johns Hopkins University School of Medicine, Baltimore, MD, USA. 21IRCCS Fondazione Don Gnocchi ONLUS, Milano, Italy. 22Caribbean Center for Clinical Research, Guaynabo, PR, USA. 23Department Neuroscience and Mental Health, City of Health and Science University Hospital of Turín, Turín, Italy. 24San Juan MS Center, Guaynabo, PR, USA. 25Ann Romney Center for Neurologic Diseases, Brigham and Women’s Hospital, Boston, MA, USA. 26Brigham Multiple Sclerosis Center, Brigham and Women’s Hospital, Boston, MA, USA. 27Laboratory of Human Genetics of Neurological Disorders, IRCCS San Raffaele Scientific Institute, Milan, Italy. 28Servei de Neurologia-Neuroimmunologia, Centre d’Esclerosi Múltiple de Catalunya (Cemcat), Vall d’Hebron Institut de Recerca, Vall d’Hebron Hospital Universitari, Barcelona, Spain. 29Casa di Cura Privata del Policlinico, Milan, Italy. 30Vita-Salute San Raffaele University, Milan, Italy. 31Departments of Neurology and Genetics, Yale School of Medicine, New Haven, CT, USA. 32Program in Medical and Population Genetics, Broad Institute of MIT and Harvard, Cambridge, MA, USA. 33Department of Neurology, University General Hospital of Larissa, Faculty of Medicine, School of Health Sciences, University of Thessaly, Larissa, Greece. 34Center For Translational & Computational Neuroimmunology and the Multiple Sclerosis Center, Department of Neurology, Columbia University Irving Medical Center, New York, NY, USA. 35Multiple Sclerosis Division, Department of Neurology, Miller School of Medicine, University of Miami, Miami, FL, USA. 36Department of Neurology, University Hospitals Leuven, Leuven, Belgium. 37KU Leuven, Leuven Brain Institute, Department of Neurosciences, Leuven, Belgium. 38Neurology Unit and Laboratory of Human Genetics of Neurological Disorders, IRCCS San Raffaele Scientific Institute, Milan, Italy. 39Centre for Molecular Medicine and Innovative Therapeutics, Murdoch University, Perth, Australia. 40Perron Institute for Neurological and Translational Science, University of Western Australia, Perth, Australia. 41Neurology Unit, Neurorehabilitation Unit, Neurophysiology Service and Neuroimaging Research Unit, IRCCS San Raffaele Scientific Institute, Milan, Italy. 42Medical School, University of Cyprus, Nicosia, Cyprus. 43Department of Experimental Immunology, Amsterdam Institute for Infection and Immunity, Amsterdam University Medical Centers, Amsterdam, The Netherlands. 44Swammerdam Institute for Life Sciences, Center for Neuroscience, University of Amsterdam, Amsterdam, The Netherlands. 45Division of Environmental Health, Department of Population and Public Health Sciences, University of Southern California, Los Angeles, CA, USA. 46Department of Neurology, Graduate School of Medical Sciences, Kyushu University, Fukuoka, Japan. 47Institute for Immunology and Infectious Diseases, Murdoch University, Perth, Australia. 48Department of Neurology, Medical University of Graz, Graz, Austria. 49Department of Neurology, Royal Melbourne Hospital, Melbourne, Australia. 50Florey Department of Neuroscience and Mental Health, University of Melbourne, Melbourne, Australia. 51Florey institute of Neuroscience and Mental Health, Melbourne, Australia. 52Department of Neurology, MS center ErasMS, Erasmus University Medical Center, Rotterdam, Netherlands. 53Department of Neurology, John Hunter Hospital, Hunter New England Health District, Newcastle, Australia. 54Hunter Medical Research Institute, University of Newcastle, Newcastle, Australia. 55Fondazione IRCCS Casa Sollievo della Sofferenza, San Giovanni Rotondo, Italy. 56Dino Ferrari Center, Department of Pathophysiology and Transplantation, University of Milan, Milan, Italy. 57IRCCS Fondazione Ca’ Granda Ospedale Maggiore Policlinico, Neurology Unit, Milan, Italy. 58Danish Multiple Sclerosis Center, Department of Neurology, Copenhagen University Hospital - Rigshospitalet, Glostrup, Denmark. 59Centre for Immunology and Allergy Research, The Westmead Institute for Medical Research, Westmead, Australia. 60School of Medical Sciences, Faculty of Medicine and Health, The University of Sydney, Sydney, Australia. 61Broad Institute of MIT and Harvard University, Cambridge, MA, USA. 62Division of Genetics, Department of Medicine, Brigham & Women’s Hospital, Harvard Medical School, Boston, MA, USA. 63Systems Biology and Computer Science Program, Ann Romney Center for Neurological Diseases, Department of Neurology, Brigham & Women’s Hospital, Boston, MA, USA. 64Kaiser Permanente Division of Research, Oakland, CA, USA. 65Department of Clinical Medicine, Faculty of Healthy and Medical Sciences, University of Copenhagen, Copenhagen, Denmark. 66Department of Neurology, Yale School of Medicine, New Haven, CT, USA. 67Menzies Institute for Medical Research, University of Tasmania, Hobart, Australia. 68Department of Translational Medicine and Interdisciplinary Research Center of Autoimmune Diseases (IRCAD), University of Eastern Piedmont, Novara, Italy. 69Departments of Neurology and Immunobiology, Yale School of Medicine, New Haven, CT, USA. 70University of Sydney, Sydney, Australia. 71Westmead Institute for Medical Research, Sydney, Australia. 72Munich Cluster for Systems Neurology (SyNergy), Munich, Germany. 73Departments of Neurology and Immunology, MS center ErasMS, Erasmus University Medical Center, Rotterdam, Netherlands. 74deCODE Genetics/Amgen, Inc., Reykjavik, Iceland. 75Faculty of Medicine, School of Health Sciences, University of Iceland, Reykjavik, Iceland. 76Department of Neurology, Hospital Clinic Barcelona, Institut d’Investigacions Biomediques August Pi Sunyer (IDIBAPS) and Universitat de Barcelona, Barcelona, Spain. 77Stanford University, Stanford, CA, USA. 78These authors jointly supervised this work: Stephen J. Sawcer and Sergio E. Baranzini. *During revision, Adil Harroud moved to The Neuro (Montreal Neurological Institute and Hospital), Department of Neurology and Neurosurgery, McGill University, Montréal, Québec, Canada.
Footnotes
Competing interests. T.O. has received compensation for advisory boards/lectures from Biogen, Novartis, Merck and Sanofi, as well as unrestricted MS research grants from the same companies, none of which are related to the current article. A.B. and his institution have received compensation for consultancy, lectures, and participation in clinical trials from Alexion, Biogen, Celgene, Merck, Novartis, Sandoz/Hexal, Sanofi, and Roche, all outside the current work. S.R.D. has received compensation for serving on advisory boards from Novartis, and institutional research grant funding from EMD Serono and Novartis, all outside the current work. M.F. is Editor-in-Chief of the Journal of Neurology, Associate Editor of Human Brain Mapping, Associate Editor of Radiology, and Associate Editor of Neurological Sciences; received compensation for consulting services and/or speaking activities from Alexion, Almirall, Bayer, Biogen, Celgene, Eli Lilly, Genzyme, Merck-Serono, Neopharmed Gentili, Novartis, Roche, Sanofi, Takeda, and Teva Pharmaceutical Industries; and receives research support from Biogen Idec, Merck-Serono, Novartis, Roche, Teva Pharmaceutical Industries, Italian Ministry of Health, Fondazione Italiana Sclerosi Multipla, and ARiSLA (Fondazione Italiana di Ricerca per la SLA). J.L.-S. received travel compensation from Biogen, Merck, Novartis; has been involved in clinical trials with Biogen, Novartis, Roche; her institution has received honoraria for talks and advisory board service from Biogen, Merck, Novartis, Roche, all outside the current work. M.J.F.-P. has received travel compensation from Merck outside the current work. A.G.K. has received speaker honoraria and Scientific Advisory Board fees from Bayer, BioCSL, Biogen-Idec, Lgpharma, Merck, Novartis, Roche, Sanofi-Aventis, Sanofi-Genzyme, Teva, NeuroScientific Biopharmaceuticals, Innate Immunotherapeutics, and Mitsubishi Tanabe Pharma, all outside of the current work. F.Z. has recently received research grants and/or consultation funds from Biogen, Ministry of Education and Research (BMBF), Bristol-Meyers-Squibb, Celgene, German Research Foundation (DFG), Janssen, Max-Planck-Society (MPG), Merck Serono, Novartis, Progressive MS Alliance (PMSA), Roche, Sanofi Genzyme, and Sandoz, all outside of the current work. B.D. has received consulting fees and/or funding from Biogen Idec, BMS, Sanofi-Aventis, and Teva. B.D. and A.G. have received consulting/travel fees and/or research funding from Novartis, Roche, and Merck, all outside the current work. SL received compensation for consulting services and speaker honoraria from Biogen Idec, Novartis, TEVA, Genzyme, Sanofi, and Merck, all outside the current work. S.B. has received honoraria from Biogen Idec, Bristol Meyer Squibbs, Merck Healthcare, Novartis, Roche, Sanofi Genzyme, and TEVA; his research is funded by the German Research Foundation (DFG), Hertie Foundation, and the Hermann and Lilly-Schilling Foundation. F.E. received compensation for consulting services and speaker honoraria from Novartis, Sanofi Genzyme, Almirall, Teva, and Merck-Serono. Jo.S. received consultancy and/or lecture fee from Biogen, Merck, Novartis, and Sanofi Genzyme, his institution received research funding by Biogen, GSK, Idorsia, and Merck, all outside the current work. B.H. has served on scientific advisory boards for Novartis; he has served as DMSC member for AllergyCare, Polpharma, Sandoz, and TG therapeutics; his institution received research grants from Regeneron and Roche for multiple sclerosis research. He holds part of two patents; one for the detection of antibodies against KIR4.1 in a subpopulation of patients with multiple sclerosis and one for genetic determinants of neutralizing antibodies to interferon. Ja.S. received speaker honoraria and a research grant for rare diseases from Sanofi Genzyme, and is a founder and minority shareholder of the University of Helsinki spin-off company VEIL.AI. J.L.M. has participated in advisory board meetings for Sanofi-Genzyme and received research funding from Genentech, Biogen Idec, and the Bristol-Myers Squibb Foundation. N.A.P. is currently an employee of Novartis Institutes for BioMedical Research (NIBR). Ká.S. and I.J. are employees of the biotechnology company deCODE genetics/AMGEN. Li.A. reports personal compensation for consulting and serving on steering committees or advisory boards for Biogen Idec, Novartis, Genentech, EMD Serono, and research funding from the Bristol-Myers Squibb Foundation, NMSS, Race to Erase MS, and NIH NINDS. P.C. reports consulting fees from Biogen, Nervgen, Idorsia, Avidea (now Vaccitech), and Disarm Therapeutics (now Lilly); research grant support from Genentech. A.R.C. reports personal compensation for participating as active speaker, consulting and serving on steering committees or advisory boards for Biogen Idec, Novartis, Genentech, EMD Serono, Bristol Myers Squib, Sanofi Genzyme, Banner Life Sciences, Alexion and Horizon. The remaining authors declare no competing interests related to this work.
Code availability. The following software packages were used for data analyses: R version 4.0.5 (https://www.r-project.org/) with additional packages ms.sev version 1.0.4, aberrant version 1.0, survminer version 0.4.9, survival version 3.2–11, metafor version 3.0–2, MASS version 7.3–54, lme4 version 1.1–27.1, lmerTest version 3.1–3, bootpredictlme4 version 0.1, gwasglue version 0.0.0.9000, MendelianRandomization version 0.5.1, TwoSampleMR version 0.5.6, mr.raps version 0.4, MRPRESSO version 1.0, data.table version 1.14.0, tidyverse version 1.3.1, ggplot2 version 3.3.5, ggpubr version 0.4.0, ggvenn version 0.1.9, scattermore version 0.7; bcftools version 1.12 (https://samtools.github.io/bcftools/), EAGLE version 2.4.1 (https://alkesgroup.broadinstitute.org/Eagle/), EIGENSOFT version 6.1.4 (https://github.com/DreichLab/EIG), FINEMAP version 1.4 and LDstore version 2.0 (http://www.christianbenner.com/), FOCUS version 0.6.10 (https://github.com/bogdanlab/focus), FUSION (https://github.com/gusevlab/fusion_twas), GCTA version 1.93.2beta (https://yanglab.westlake.edu.cn/software/gcta/), GenomeStudio version 2.0 (https://support.illumina.com/downloads/genomestudio-2-0.html), GWAMA version 2.2.2 (https://manpages.ubuntu.com/manpages/xenial/man1/GWAMA.1.html), KING version 2.2.5 (https://www.kingrelatedness.com/), LDSC version 1.0.1 (https://github.com/bulik/ldsc), Minimac4 version 1.0.2 (https://genome.sph.umich.edu/wiki/Minimac4), PLINK version 1.90beta (https://www.cog-genomics.org/plink/1.9/) and version 2.00 (https://www.cog-genomics.org/plink/2.0/), PRSice-2 version 2.3.3 (https://github.com/choishingwan/PRSice), qctool version 2.0.6 (https://www.well.ox.ac.uk/~gav/qctool_v2/), Trans-Phar (https://github.com/konumat/Trans-Phar).
Supplementary Information is available for this paper.
Reprints and permissions information is available at www.nature.com/reprints.
Data availability.
The GWAS summary statistics generated in this study can be accessed through the International Multiple Sclerosis Genetics Consortium website (https://imsgc.net/). Individual-level genetic and phenotype data are deposited in the European Genome-phenome Archive for European centers (accession number EGAS00001007162), and in dbGAP (accession number phs002929.v1.p1) for other centers. Access restrictions are detailed in the Supplementary Note. Swedish participant metadata is available on Figshare (https://doi.org/10.6084/m9.figshare.22551355.v1) and access to genotype data can be requested by contacting the senior principal investigator at the Karolinska Institutet (currently ingrid.kockum@ki.se) and signing the required legal agreement regarding data sharing. Gene expression profiles of human tissues used in this study can be downloaded from the GTEx Portal v8 (https://gtexportal.org/home/datasets). The single-cell type expression profiles in human tissues can be downloaded from the Human Protein Atlas (https://www.proteinatlas.org/about/download). Additional CNS single-nucleus RNA expression and cell-type annotation data were obtained from the Gene Expression Omnibus under accession numbers GSE71585, GSE97942, GSE118257, and GSE180759. We used publicly available data from the eQTL Catalogue release 4 (https://www.ebi.ac.uk/eqtl/Data_access/), the LDSC GitHub repository (https://github.com/bulik/ldsc/), the Gonçalo Castelo-Branco Group (https://ki.se/en/mbb/oligointernode/). Detailed information on the GWAS summary statistics used in the Mendelian randomization analysis is provided in Supplementary Table 18. The GRCh37 reference genome used for mapping was obtained from the 1000 Genomes Project (http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/).
MAIN REFERENCES
- 1.Thompson AJ, Baranzini SE, Geurts J, Hemmer B & Ciccarelli O Multiple sclerosis. Lancet 391, 1622–1636 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Walton C et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult. Scler. 26, 1816–1821 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 365, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hauser SL & Cree BAC Treatment of Multiple Sclerosis: A Review. Am. J. Med. 133, 1380–1390.e2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.International Multiple Sclerosis Genetics Consortium et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476, 214–219 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.George MF et al. Multiple sclerosis risk loci and disease severity in 7,125 individuals from 10 studies. Neurol Genet 2, e87 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chataway J et al. Multiple sclerosis in sibling pairs: an analysis of 250 families. J. Neurol. Neurosurg. Psychiatry 71, 757–761 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee JC et al. Genome-wide association study identifies distinct genetic contributions to prognosis and susceptibility in Crohn’s disease. Nat. Genet. 49, 262–268 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu G et al. Genome-wide survival study identifies a novel synaptic locus and polygenic score for cognitive progression in Parkinson’s disease. Nat. Genet. 53, 787–793 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.International Multiple Sclerosis Genetics Consortium. Genome-wide association study of severity in multiple sclerosis. Genes Immun. 12, 615–625 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.International Multiple Sclerosis Genetics Consortium (IMSGC) et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 45, 1353–1360 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.King EA, Davis JW & Degner JF Are drug targets with genetic support twice as likely to be approved? Revised estimates of the impact of genetic support for drug mechanisms on the probability of drug approval. PLoS Genet. 15, e1008489 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manouchehrinia A et al. Age Related Multiple Sclerosis Severity Score: Disability ranked by age. Mult. Scler. 23, 1938–1946 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finucane HK et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Finucane HK et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet. 50, 621–629 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benner C et al. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics 32, 1493–1501 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tremlett H et al. Impact of multiple sclerosis relapses on progression diminishes with time. Neurology 73, 1616–1623 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schirmer L et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature 573, 75–82 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Absinta M, Lassmann H & Trapp BD Mechanisms underlying progression in multiple sclerosis. Curr. Opin. Neurol. 33, 277–285 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gazal S et al. Combining SNP-to-gene linking strategies to identify disease genes and assess disease omnigenicity. Nat. Genet. 54, 827–836 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ng B et al. An xQTL map integrates the genetic architecture of the human brain’s transcriptome and epigenome. Nat. Neurosci. 20, 1418–1426 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bansal D et al. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 423, 168–172 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Zhu Y, Wang GZ, Cingöz O & Goff SP NP220 mediates silencing of unintegrated retroviral DNA. Nature 564, 278–282 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu J et al. Postsynaptic positioning of endocytic zones and AMPA receptor cycling by physical coupling of dynamin-3 to Homer. Neuron 55, 874–889 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edvardson S et al. Mutations in the phosphatidylinositol glycan C (PIGC) gene are associated with epilepsy and intellectual disability. J. Med. Genet. 54, 196–201 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Jäkel S et al. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 566, 543–547 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Absinta M et al. A lymphocyte-microglia-astrocyte axis in chronic active multiple sclerosis. Nature 597, 709–714 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Konuma T, Ogawa K & Okada Y Integration of genetically regulated gene expression and pharmacological library provides therapeutic drug candidates. Hum. Mol. Genet. 30, 294–304 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ge Z et al. Vorinostat, a histone deacetylase inhibitor, suppresses dendritic cell function and ameliorates experimental autoimmune encephalomyelitis. Exp. Neurol. 241, 56–66 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Kazantsev AG & Thompson LM Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat. Rev. Drug Discov. 7, 854–868 (2008). [DOI] [PubMed] [Google Scholar]
- 31.D’hooghe MB, Haentjens P, Van Remoortel A, De Keyser J & Nagels G Self-reported levels of education and disability progression in multiple sclerosis. Acta Neurol. Scand. 134, 414–419 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Olsson T, Barcellos LF & Alfredsson L Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat. Rev. Neurol. 13, 25–36 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Kuhlmann T et al. Multiple sclerosis progression: time for a new mechanism-driven framework. Lancet Neurol. 22, 78–88 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okbay A et al. Polygenic prediction of educational attainment within and between families from genome-wide association analyses in 3 million individuals. Nat. Genet. 54, 437–449 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palace J et al. Assessing the long-term effectiveness of interferon-beta and glatiramer acetate in multiple sclerosis: final 10-year results from the UK multiple sclerosis risk-sharing scheme. J. Neurol. Neurosurg. Psychiatry 90, 251–260 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Rheenen W et al. Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat. Genet. 53, 1636–1648 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kellar-Wood H, Robertson N, Govan GG, Compston DA & Harding AE Leber’s hereditary optic neuropathy mitochondrial DNA mutations in multiple sclerosis. Ann. Neurol. 36, 109–112 (1994). [DOI] [PubMed] [Google Scholar]
- 38.Ismail A et al. Concurrence of multiple sclerosis and amyotrophic lateral sclerosis in patients with hexanucleotide repeat expansions of C9ORF72. J. Neurol. Neurosurg. Psychiatry 84, 79–87 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Robbez-Masson L et al. The HUSH complex cooperates with TRIM28 to repress young retrotransposons and new genes. Genome Res. 28, 836–845 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bjornevik K, Münz C, Cohen JI & Ascherio A Epstein-Barr virus as a leading cause of multiple sclerosis: mechanisms and implications. Nat. Rev. Neurol. 19, 160–171 (2023). [DOI] [PubMed] [Google Scholar]
- 41.Kremer D et al. Human endogenous retrovirus type W envelope protein inhibits oligodendroglial precursor cell differentiation. Ann. Neurol. 74, 721–732 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Hartung H-P et al. Efficacy and safety of temelimab in multiple sclerosis: Results of a randomized phase 2b and extension study. Mult. Scler. 28, 429–440 (2022). [DOI] [PubMed] [Google Scholar]
- 43.Savage JE et al. Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat. Genet. 50, 912–919 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Galvin JE, Palamand D, Strider J, Milone M & Pestronk A The muscle protein dysferlin accumulates in the Alzheimer brain. Acta Neuropathol. 112, 665–671 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McDade JR, Naylor MT & Michele DE Sarcolemma wounding activates dynamin-dependent endocytosis in striated muscle. FEBS J. 288, 160–174 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bhargava P et al. The vitamin D to ameliorate multiple sclerosis (VIDAMS) trial: study design for a multicenter, randomized, double-blind controlled trial of vitamin D in multiple sclerosis. Contemp. Clin. Trials 39, 288–293 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Manuel Escobar J et al. Body mass index as a predictor of MS activity and progression among participants in BENEFIT. Mult. Scler. 13524585211061861 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Manouchehrinia A et al. Tobacco smoking and disability progression in multiple sclerosis: United Kingdom cohort study. Brain 136, 2298–2304 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vasileiou ES et al. Socioeconomic disparity is associated with faster retinal neurodegeneration in multiple sclerosis. Brain 144, 3664–3673 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gazzina S et al. Education modulates brain maintenance in presymptomatic frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry 90, 1124–1130 (2019). [DOI] [PubMed] [Google Scholar]
- 51.Cabeza R et al. Maintenance, reserve and compensation: the cognitive neuroscience of healthy ageing. Nat. Rev. Neurosci. 19, 701–710 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cohen JA, Reingold SC, Polman CH, Wolinsky JS & International Advisory Committee on Clinical Trials in Multiple Sclerosis. Disability outcome measures in multiple sclerosis clinical trials: current status and future prospects. Lancet Neurol. 11, 467–476 (2012). [DOI] [PubMed] [Google Scholar]
METHODS REFERENCES
- 53.Kurtzke JF Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 33, 1444–1452 (1983). [DOI] [PubMed] [Google Scholar]
- 54.McCarthy S et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 48, 1279–1283 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Das S et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jiang L, Zheng Z, Fang H & Yang J A generalized linear mixed model association tool for biobank-scale data. Nat. Genet. 53, 1616–1621 (2021). [DOI] [PubMed] [Google Scholar]
- 57.Aschard H, Vilhjálmsson BJ, Joshi AD, Price AL & Kraft P Adjusting for heritable covariates can bias effect estimates in genome-wide association studies. Am. J. Hum. Genet. 96, 329–339 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Greenland S Quantifying biases in causal models: classical confounding vs collider-stratification bias. Epidemiology 14, 300–306 (2003). [PubMed] [Google Scholar]
- 59.Bulik-Sullivan BK et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang J et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 44, 369–75, S1–3 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Beecham AH et al. The genetic diversity of multiple sclerosis risk among Hispanic and African American populations living in the United States. Mult. Scler. 26, 1329–1339 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee SH, Yang J, Goddard ME, Visscher PM & Wray NR Estimation of pleiotropy between complex diseases using single-nucleotide polymorphism-derived genomic relationships and restricted maximum likelihood. Bioinformatics 28, 2540–2542 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Evans LM et al. Comparison of methods that use whole genome data to estimate the heritability and genetic architecture of complex traits. Nat. Genet. 50, 737–745 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bryois J et al. Genetic identification of cell types underlying brain complex traits yields insights into the etiology of Parkinson’s disease. Nat. Genet. 52, 482–493 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lake BB et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol. 36, 70–80 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Habib N et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 14, 955–958 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kappos L et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet 391, 1263–1273 (2018). [DOI] [PubMed] [Google Scholar]
- 68.Benner C et al. Prospects of Fine-Mapping Trait-Associated Genomic Regions by Using Summary Statistics from Genome-wide Association Studies. Am. J. Hum. Genet. 101, 539–551 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Luchetti S et al. Progressive multiple sclerosis patients show substantial lesion activity that correlates with clinical disease severity and sex: a retrospective autopsy cohort analysis. Acta Neuropathol. 135, 511–528 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Geurts JJG & Barkhof F Grey matter pathology in multiple sclerosis. Lancet Neurol. 7, 841–851 (2008). [DOI] [PubMed] [Google Scholar]
- 71.Mountjoy E et al. An open approach to systematically prioritize causal variants and genes at all published human GWAS trait-associated loci. Nat. Genet. 53, 1527–1533 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gusev A et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 48, 245–252 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Consortium GTEx. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kerimov N et al. A compendium of uniformly processed human gene expression and splicing quantitative trait loci. Nat. Genet. 53, 1290–1299 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ghoussaini M et al. Open Targets Genetics: systematic identification of trait-associated genes using large-scale genetics and functional genomics. Nucleic Acids Res. 49, D1311–D1320 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bulik-Sullivan B et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Karlsson M et al. A single-cell type transcriptomics map of human tissues. Sci Adv 7, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Harroud A et al. The relative contributions of obesity, vitamin D, leptin, and adiponectin to multiple sclerosis risk: A Mendelian randomization mediation analysis. Mult. Scler. 27, 1994–2000 (2021). [DOI] [PubMed] [Google Scholar]
- 79.Bowden J et al. Improving the accuracy of two-sample summary-data Mendelian randomization: moving beyond the NOME assumption. Int. J. Epidemiol. 48, 728–742 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Verbanck M, Chen C-Y, Neale B & Do R Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 50, 693–698 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hemani G, Bowden J & Davey Smith G Evaluating the potential role of pleiotropy in Mendelian randomization studies. Hum. Mol. Genet. 27, R195–R208 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Burgess S, Foley CN, Allara E, Staley JR & Howson JMM A robust and efficient method for Mendelian randomization with hundreds of genetic variants. Nat. Commun. 11, 376 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wootton RE et al. Evidence for causal effects of lifetime smoking on risk for depression and schizophrenia: a Mendelian randomisation study. Psychol. Med. 50, 2435–2443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.de Bakker PIW et al. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat. Genet. 38, 1166–1172 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Timmers PRHJ et al. Mendelian randomization of genetically independent aging phenotypes identifies LPA and VCAM1 as biological targets for human aging. Nature Aging 2, 19–30 (2022). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The GWAS summary statistics generated in this study can be accessed through the International Multiple Sclerosis Genetics Consortium website (https://imsgc.net/). Individual-level genetic and phenotype data are deposited in the European Genome-phenome Archive for European centers (accession number EGAS00001007162), and in dbGAP (accession number phs002929.v1.p1) for other centers. Access restrictions are detailed in the Supplementary Note. Swedish participant metadata is available on Figshare (https://doi.org/10.6084/m9.figshare.22551355.v1) and access to genotype data can be requested by contacting the senior principal investigator at the Karolinska Institutet (currently ingrid.kockum@ki.se) and signing the required legal agreement regarding data sharing. Gene expression profiles of human tissues used in this study can be downloaded from the GTEx Portal v8 (https://gtexportal.org/home/datasets). The single-cell type expression profiles in human tissues can be downloaded from the Human Protein Atlas (https://www.proteinatlas.org/about/download). Additional CNS single-nucleus RNA expression and cell-type annotation data were obtained from the Gene Expression Omnibus under accession numbers GSE71585, GSE97942, GSE118257, and GSE180759. We used publicly available data from the eQTL Catalogue release 4 (https://www.ebi.ac.uk/eqtl/Data_access/), the LDSC GitHub repository (https://github.com/bulik/ldsc/), the Gonçalo Castelo-Branco Group (https://ki.se/en/mbb/oligointernode/). Detailed information on the GWAS summary statistics used in the Mendelian randomization analysis is provided in Supplementary Table 18. The GRCh37 reference genome used for mapping was obtained from the 1000 Genomes Project (http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/).