


Abstract
The first step in gene expression is the transcription of DNA sequences into RNA. Regulation at the level of transcription leads to changes in steady-state concentrations of RNA transcripts, affecting the flux of downstream functions and ultimately cellular phenotypes. Changes in transcript levels are routinely followed in cellular contexts via genome-wide sequencing techniques. However, in vitro mechanistic studies of transcription have lagged with respect to throughput. Here, we describe the use of a real-time, fluorescent-aptamer-based method to quantitate steady-state transcription rates of the Mycobacterium tuberculosis RNA polymerase. We present clear controls to show that the assay specifically reports on promoter-dependent, full-length RNA transcription rates that are in good agreement with the kinetics determined by gel-resolved, α-32P NTP incorporation experiments. We illustrate how the time-dependent changes in fluorescence can be used to measure regulatory effects of nucleotide concentrations and identity, RNAP and DNA concentrations, transcription factors, and antibiotics. Our data showcase the ability to easily perform hundreds of parallel steady-state measurements across varying conditions with high precision and reproducibility to facilitate the study of the molecular mechanisms of bacterial transcription.
Graphical Abstract
Graphical Abstract.

INTRODUCTION
Cellular RNA abundance is dictated by the relative steady-state rates of RNA production and degradation. In particular, the rate of RNA production is ubiquitously the target of gene regulatory mechanisms and often represents a good proxy for protein synthesis flux (1). More specifically, the rate at which full-length RNA transcripts are generated is typically controlled by the rate of transcription initiation. This is because the overall initiation rate is often slower than subsequent elongation and termination steps, and because multiple RNA polymerases (RNAPs) may be elongating at the same locus at the same time. Bacterial transcription initiation progresses through several intermediates, where the rates and equilibrium constants that describe the initial binding of the RNAP to the promoter and the subsequent isomerization steps that culminate in opening of promoter DNA can vary greatly depending on the sequence of the promoter (2,3). For example, Escherichia coli promoters can differ in rates of promoter opening by factors of 103–104, resulting in initiation events ranging from once per second to once per generation (4,5). Following promoter opening, binding of the first two initiating nucleoside triphosphates (NTP) forms an initially transcribing complex that begins producing a nascent RNA transcript. After formation of the initiating dinucleotide, each step of RNA synthesis requires translocation, stressing RNAP–promoter contacts (6,7). If this stress disrupts these contacts, the RNAP escapes from the promoter and begins processive elongation. Otherwise, complexes stall near the promoter or perform cycles of abortive transcription (5,8,9). After decades of careful biophysical dissection, driven mainly by pre-steady-state kinetic and structural biology approaches, bacterial initiation pathways are well-characterized on a handful of different promoters for model bacteria (reviewed in (2,3,10–12)). Many kinetic/structural intermediates have been identified, including off-pathway states that, in some cases, do not lead to full-length RNA production (8,13–17). The resulting models from all these studies are complex and can vary depending on the system studied (18–20), bringing into question model generalizability across bacteria and different promoter sequences.
Ideally, rate constants for RNAP binding, DNA opening, initial transcription and promoter escape obtained from pre-steady-state kinetic measurements could be used to calculate an overall initiation rate or the average time between initiation events. A demonstration of this can be seen in approaches where both basal and regulated RNA flux is calculated based on simple models of transcription initiation (3,21). However, while these models may address theoretical links between initiation kinetics and steady-state rates of RNA production, they cannot account for the large number of variables that determine the rate of transcription. This overall initiation rate is most simply described using Michaelis-Menten enzyme kinetics, where the RNA polymerase is the enzyme, promoter DNA is the substrate, and the full-length RNA transcript is the product (22). This model assumes a constant concentration of RNAP–promoter (enzyme–substrate) complexes generated by a balance between promoter binding and escape rates and predicts a constant reaction velocity or rate of transcript production. Within this formalism, regulated promoters are activated or repressed via changes in the Michaelis-Menten parameters Km (the concentration at which the half maximal rate is observed) and/or Vmax (the maximal rate observed under saturating conditions) without changes in the free RNAP concentration. Alternatively, constitutively active promoters are affected by cell growth-rate-dependent changes in the free RNAP concentration, independent of changes in Km and/or Vmax (23–25). Classic examples of environmental adaptation in bacteria affecting transcription initiation processes include the up-regulation of beta-galactosidase in response to the presence of lactose and the absence of glucose (26), and the genome-wide response to starvation known as the stringent response (27,28).
In vitro measurements of steady-state transcription rates can be used to test and develop more complex models but have been limited by current methodologies. Historically, measurements of in vitro basal and regulated transcription initiation kinetics have been made possible by monitoring RNA production via the incorporation of radio-labelled NTPs, resolved via polyacrylamide gel electrophoresis (29). The approach was used in the initial discovery of RNAP holoenzyme activity more than fifty years ago (30) and is the most common assay for probing transcriptional mechanisms in vitro. The strengths of the assay include its high sensitivity and single-nucleotide resolution even with small quantities of RNA. This allows for the separation of RNA products by length and to investigate individual steps in nucleotide addition reactions that underlie the fundamental mechanisms of RNAP. However, steady-state 32P-detected transcription assays have several drawbacks. A practical limitation is that the use of radioactivity is expensive, both for the purchase of the reagent itself and for the requirement of specialized protocols for safe shipping and use in the lab. The reaction must also be taken through several steps that may include the incorporation of proteases or chelators to quench the reaction at set timepoints, phenol/chloroform extractions, and/or denaturation and loading the sample on an electrophoretic polyacrylamide gel. All these steps, as well as the quantification of resolved product bands via image analysis, can introduce non-biochemical variability in the measured RNA amounts. In addition, these assays require significant amounts of time and training, where single experiments often take multiple days before complete quantification and where technical practice is needed for generating reproducible data. All these factors combined result in relatively limited throughput. As a result, the time-dependent measurements needed to quantitate steady-state initiation rates are infrequently performed and single time-point measurements are often used to infer mechanisms of gene regulation.
In contrast to the in vitro approaches described above, in vivo transcription studies have undergone a remarkable transformation where genome-wide transcript levels in cells under varying environmental and genetic backgrounds are routinely queried (31). We were motivated by the desire to increase the throughput of in vitro transcriptional studies to facilitate the efficient testing of mechanistic hypotheses, the development of predictive transcription initiation models, and to be able to compare transcript flux from reconstituted systems to the genome-wide information accessible via RNAseq. To this end, we explored the use of a fluorescent-aptamer-based detection system to report on in vitro steady-state transcription rates.
Fluorescent light-up aptamers are RNA sequences that bind small molecule fluorophore dyes and generate a large fluorescence enhancement. They have been used in numerous applications (reviewed in (32)) including the detection of nascent RNA transcripts in cells (33–38), in synthetic biology transcription-translation coupled in vitro systems for T7 RNAP (39–41) and with cell lysates from diverse bacterial species (42). These approaches rely on the use of a DNA template that encodes for an aptamer sequence so that each transcription event results in the generation of an RNA aptamer which folds and binds the dye. For each transcript produced, there is an accompanying increase in fluorescence (Figure 1). Importantly, as the change in dye fluorescence requires the synthesis of a full-length RNA transcript containing the aptamer, the fluorescent readout is not complicated from short abortive products that may be generated during promoter escape. Using this approach, recent work has illustrated how the rates of in vitro reactions can be quantified in a fluorimeter cuvette with E. coli RNAP (43) and in a plate-reader format with T7 RNAP (44). Here, we follow up on these studies and provide a description of essential control experiments needed to clearly link the fluorescent signal with the transcription of a promoter-derived product. Additionally, using data acquired with RNAP from Mycobacterium tuberculosis (Mtb), we describe a workflow for the calibration and quantification of RNA concentration in multi-round initiation kinetics analyzed with a Michaelis-Menten approach. Most significantly, we illustrate the utility of this approach for the investigation of mechanisms of transcriptional regulation by NTP concentration, transcription factors, and antibiotic inhibitors, all derived from high-throughput measurements under steady-state conditions.
Figure 1.

Overview of the fluorescent-aptamer-based assay. This assay requires RNAP, a DNA template containing the sequence for the Spinach-mini RNA aptamer (33), NTPs and the fluorescent dye DFHBI. Upon initiating the reaction, full-length RNAs containing the Spinach-mini aptamer capable of binding DFHBI are synthesized. The fluorescent signal change, monitored in real-time, results from the unbound to aptamer-bound dye transition and is used as a reporter of full-length transcription rates. An initial lag time is observed, followed by a steady-state regime where the slope of the linear phase represents the steady-state rate of full-length RNA synthesis.
MATERIALS AND METHODS
Reagents
Preparation of DNA constructs
Circular plasmid templates, 2557 base-pair (bp) in length, were ordered from Twist Bioscience (San Francisco, CA), containing the sequences corresponding to the Mtb ribosomal RNA promoter (rrnAP3), the Spinach-mini aptamer (33) and the E. coli rrnBP1 T1 terminator (45) (illustrated in Figure 1). Plasmids were isolated via the Qiagen Midi Prep Kit (catalogue #: 27106) and were predominantly supercoiled (Supplementary Figure S1). For additional sequences and further descriptions, see Supplementary Table S1. Linear PCR templates 250 bp in length were amplified from 200 nucleotide (nt) PAGE purified DNA Ultramer oligos (Integrated DNA Technologies, Inc., Coralville, IA) following annealing and end-filling. For more details including primer and final construct sequences, as well as further preparation details, see Supplementary Table S2.
Preparation of recombinant proteins
The Mtb RNAP σA holoenzyme complex was co-expressed and purified using an N-terminal His-tagged σA subunit via nickel affinity chromatography, followed by size exclusion chromatography, in accordance with previous methods (18,46). Mtb CarD and RbpA, in pET-SUMO plasmid vectors, were expressed and purified, and the His-SUMO tag was subsequently cleaved in accordance with methods previously described (18,46). E. coli RNAP σ70 holoenzyme was purchased from New England BioLabs, Inc. (Ipswich, MA; catalogue # M0551S). Purified E. coli GreB was a generous gift from Irina Artsimovitch (The Ohio State University) (47). Protein concentrations were determined using the following extinction coefficients at 280 nm: Mtb RNAP σA holoenzyme (280,452 M−1 cm−1), Mtb CarD (16,900 M−1 cm−1), Mtb RbpA (13,980 M−1 cm−1), and E. coli GreB (36,900 M−1 cm−1).
Reagents for transcription assays
All transcription reactions were performed with NTPs (Thermo Scientific), 3,5-difluoro-4-hydroxybenzylidene imidazolinone (DFHBI) dye (Sigma Aldrich; catalogue # SML1627), and RiboLock RNase inhibitors (Thermo Scientific; catalogue # EO0381). DFHBI concentration was determined using an extinction coefficient at 420 nm of 31,611 M−1 cm−1. Rifampicin (Sigma-Aldrich; catalogue #R3501) and Fidaxomicin (Selleckchem; catalogue # S4227) solids were dissolved in DMSO and molar concentrations were determined by weight (Fidaxomicin) or by absorbance using an extinction coefficient at 470 nm of 15,300 M−1 cm−1 (48) (Rifampicin).
Plate-reader fluorescence measurements
To measure multi-round kinetics under steady-state conditions in real-time, we monitored the change in DFHBI fluorescence upon binding to a transcribed, full-length RNA sequence containing the Spinach-mini aptamer. Data was collected using a Synergy2 multi-detection microplate reader (BioTek Instruments, Inc., Winooski, VT) with the corresponding Gen5 analysis software. DFHBI fluorescence was measured with a tungsten light source equipped with a 505 nm long-pass dichroic mirror, excited with a 460 ± 40 nm bandpass filter, and the resulting emission signal was monitored with a 528 ± 20 nm bandpass filter. Data was acquired at a read height of 7.00 mm, typically in 20–30 s intervals, with varying total acquisitions times, not exceeding 75 min.
Transcription reaction master mixes containing 90% of the final volume included the RNAP holoenzyme, NTPs, DFHBI, and RNase inhibitors. Based on the volumes added for each corresponding buffer addition and concentrated stock component, the final solution conditions were 20 mM Tris (pH 8.0 at 25°C), 40 mM NaCl, 75 mM K-glutamate, 10 mM MgCl2, 5 μM ZnCl2, 20 μM EDTA, 5% (v/v) glycerol (defined as transcription buffer) with 1 mM DTT and 0.1 mg/mL BSA. From these master mixes, a volume of 9 μl for each technical replicate was transferred to an individual well in a 384 well, low volume, round-bottom, non-binding polystyrene assay plate (Corning; catalogue # 4514). For the negative controls, which represent the entirety of the reaction components except for DNA, 1 μl of transcription buffer was added to account for the remaining 10% of the final reaction volume (10 μl). Wells were covered with an optical adhesive to prevent sample evaporation (Applied Biosystems; catalogue # 4360954). The plate was then incubated for 10 min at 25°C, followed by a 30 s shake agitation in the microplate reader. An initial reading of the negative controls was used to scale the automatic gain adjustment to the background signal with an arbitrary value of 1000 RFUs and was applied to all the subsequent reads. The adhesive cover was removed, and the transcription reaction was initiated with 1 μl DNA unless otherwise indicated. Multichannel pipettors were used to reduce the initiation time difference across wells. Once the DNA was added, the plate was agitated for 15 sec before starting the kinetic measurements. Unless otherwise stated, the reaction master mixes always contained 20 μM DFHBI and 0.4 U/μl RNase inhibitors. For additional details regarding reaction specifics for individual titrations, including titrations measured under single-round conditions, see Supplementary Materials and Methods.
Radio-labelled NTP incorporation transcription experiments
Transcription experiments were performed with 500 μM each NTP under identical solution conditions and temperature as multi-round fluorescence experiments. Mtb σA RNAP holoenzyme and DFHBI concentrations were varied and always included unless otherwise indicated. 20 nM of α-32P UTP was added to label the nascent RNA transcripts via incorporation by RNAP. Reactions were initiated by addition of circular plasmid constructs containing either 5 nM Mtb rrnAP3 or a template lacking a promoter (promoterLESS, see Supplementary Table S1). 5 μl aliquots were removed from the reaction mixture at 1, 5, 10, 15, 20 and 30 min and combined with 5 μl of quench solution containing 95% formamide, 0.015 M EDTA, 0.05% (w/v) xylene cyanol, and 0.05% (w/v) bromophenol blue. 5 μl of each quenched timepoint was then loaded onto a 5% denaturing PAGE gel. Gels were run in TBE for 2 to 3 h at 1,500 V and then transferred to a phosporimaging cassette. After exposing for 18 h, phosphorimaging screens were imaged via a Typhoon 9000 phosphorimager. Bands of interest were quantified using the ImageQuant software and converted to reaction RNA concentrations as previously described (5).
Analyses of fluorescent time courses
Extraction of steady-state rates
As the fluorescent signal in the absence of aptamer formation does not significantly change across conditions where DNA or NTPs were left out of the reaction (Supplementary Figure S3), the same negative control could be used to correct all experimental conditions within a titration. This applies only if the time course measurements are made under the same solution conditions and instrumental detection scaling. However, to minimize experimental and/or instrumental variation, each time a new experiment was performed, a minimum of 2 to 3 technical replicates of the negative control (leaving out DNA) were collected and measured concurrently with the experimental data. Prior to data analysis, the experimental traces underwent two subtractions: first, the fluorescent value recorded at the initial timepoint was subtracted from all timepoints, bringing the starting fluorescent value to zero, and second, the fluorescence from the experimental traces was subtracted using the corresponding signal from each time-point of the negative control.
Linear regression of the corrected fluorescent traces was performed with a custom MATLAB fitting program (described further in Results). Using a statistical weighting from multiple technical replicates per condition and a user inputted R2 value to define the goodness of fit (Supplementary Figure S6), an unbiased determination of the linear regime can be obtained, where the slope of the fitted line reports on the steady-state rate in units of change in fluorescence/time. In the work presented here, R2-thresholds >0.998 were used.
Non-linear regression analyses of concentration dependencies
For analyses of titration data that yielded hyperbolic concentration dependencies in steady-state rates, a Michaelis-Menten equation was applied, fitting the data to Equation 1
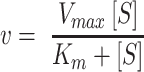 |
(1) |
where v represents the steady-state rate and S represents the titrated substrate, either DNA or individual NTPs. Steady-state rates determined in some NTP titrations were not well-described by Equation 1, displaying sigmoidal concentration dependences. These titrations were fit to Equation 2, a modified Michaelis-Menten equation with three parameters: Vmax, an apparent Km (Km,app), and the exponent n.
 |
(2) |
Here, we define Km,app as the NTP concentration that results in half maximal velocity, which is equivalent to the standard operational definition of Km when n = 1. It should be noted that while this equation is functionally identical to the Hill equation, if a non-unity n is the result of the kinetics from a single-site-binding system, then n is only effectively demonstrative of the type (positive/negative) and magnitude of cooperativity. In such a case, n has no specific physical meaning as it does in more traditional cases such as cooperative binding of multiple ligands (49). The kinetics of transcription inhibition as a function of antibiotic concentration at constant NTP concentrations were also found to be sigmoidal. For determination of the half maximal inhibitory concentration (IC50) of Rifampicin and Fidaxomicin, the concentration-dependent reduction in steady-state rates was fit to Equation 3.
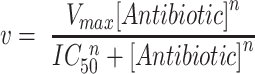 |
(3) |
Statistical analyses
For all fluorescent data presented unless otherwise indicated, between 2 and 4 independent experiments were collected for each condition tested. Within each independent experiment, standard deviations from 2 to 3 technical replicates were used as a statistical weight during the linear regression analyses. All non-linear regression analyses were performed using the standard deviations of the independent experiments as a statistical weight. Errors in Vmax, Km,app, n and IC50 values are those obtained from the fit of the averaged data set. For simplicity, only a single representative independent experiment is shown in the plots. For gel-based quantifications, average values and standard deviations from 2 to 3 independent experiments are reported.
Data availability
The MATLAB code used for automated linear regression analyses has been made publicly available and can be accessed with the following GitHub link (https://github.com/egalburt/aptamer-flux-fitting).
RESULTS
Promoter-dependent, aptamer-based measurements of full-length transcription
As similar assays have been described elsewhere, we briefly describe the specifics of the version we utilized for these studies. DNA templates were constructed such that an 80 nt Spinach-mini aptamer sequence (33) was inserted 30 bp downstream of the +1 transcription start site corresponding to the genomic Mtb ribosomal RNA promoter sequence (rrnAP3). Specifically, –60 to +31 of rrnAP3 was included to account for any upstream-promoter interactions as well as the initially transcribed sequences, both of which can regulate initiation kinetics. Inclusion of the genomic initially transcribed sequence, rather than just positioning the aptamer sequence to start at the +1 position, provides for unaltered promoter escape, which is known to become rate limiting under certain conditions (see (3) for examples of sequence effects on initiation kinetics). The E. coli intrinsic rrnBP1 T1 terminator sequence was included in circular plasmid DNA templates to dissociate the ternary polymerase, template, and transcript complex. Sequences for both circular plasmid and linear PCR templates are given in Supplementary Tables S1 and S2.
When transcribed downstream of the Mtb rrnAP3 promoter, the Spinach-mini aptamer sequence folds and binds the small molecule fluorophore DFHBI, resulting in a fluorescence enhancement. By mixing RNAP, DNA, all NTPs and DFHBI, the reaction is monitored in real-time where the increase in fluorescence reports on transcript production. All experiments were performed in a 384 well plate-reader format using 10 μl reaction volumes facilitating high-throughput measurements of steady-state initiation kinetics with minimal sample volume requirements. The slope of the fluorescent signal at early times reports on the steady-state rate of transcription initiation (Figure 1). In general, one expects initiation kinetics and not co-transcriptional aptamer folding (39,50,51) or dye binding (52) to be rate-limiting for functional aptamer production. However, even when the initiation rate is not limiting relative to the timescales of elongation or aptamer folding, the steady-state rate will still specifically report on the initiation rate (Supplementary Figure S2). This is because once one RNAP leaves the promoter, another RNAP may bind and begin the process of initiation irrespective of these downstream processes. This condition may be broken by pathological cases where downstream pausing creates a traffic jam that backs up onto the promoter, influencing the time needed for the next polymerase to bind the promoter.
Transcription reactions were initiated by adding DNA templates to Mtb RNAP preincubated with NTPs, DFHBI dye and RNase inhibitors. Fluorescence was monitored in real-time, where we observed three distinct kinetic phases to the trace using the circular plasmid template containing the rrnAP3 sequence: (i) a lag time, (ii) a linear increase and (iii) roll-off from the linear regime that begins to plateau over time (Figure 2A, black curve). We note that when starting from a well-controlled time zero, the lag time theoretically reports on the time required for pioneering RNAPs to complete the initiation process, transcribe the aptamer, and for dye to bind the folded RNA transcript (43) (Supplementary Figure S2). In our assay setup, since initiation of the reaction was done by hand, we do not attempt to quantitate the lag time, but focus on the linear regime. In the absence of either DNA or NTPs, only a slow decay in DFHBI fluorescence was observed as a function of time (Supplementary Figure S3), indicating that the increase in fluorescence when all reaction components are present is due to transcription-derived aptamer formation. For all plots shown, the signal in the absence of transcription is subtracted from the experimental traces so that the y-axis reports specifically on the fluorescence generated by transcription of the aptamer (Materials and Methods). Correcting for this time-dependent fluorescent change of the background signal is especially important when evaluating conditions of minimal transcript generation (Supplementary Figure S3).
Figure 2.

Real-time fluorescent signal is promoter dependent and due to multiple rounds of initiation. All experiments were performed using 100 nM Mtb RNAP, 5 nM DNA circular plasmid templates, and 500 μM NTPs. (A) Comparison of time courses of RNA synthesis from an Mtb plasmid template containing either both the rrnAP3 and aptamer sequences (black), the rrnAP3 sequence with no aptamer sequence (blue), or the ‘promoterLESS’ template, containing the aptamer, but lacking the rrnAP3 sequence (red). (B) Real-time traces comparing multi-round (black) and single-round conditions (gold) on the rrnAP3 circular plasmid template. Error shading indicates the standard deviation of 3 independent experiments.
To confirm that transcription is driven by the rrnAP3 promoter sequence, we performed the assay using a circular plasmid template lacking the promoter (i.e. promoterLESS; for sequence, see Supplementary Table S1) as a negative control. The signal generated in the presence of rrnAP3 is more than 20-fold higher than that obtained with the promoterLESS template (Figure 2A, red), confirming that the signal arises from promoter-driven transcription. However, the amount of signal generated by the promoterLESS template is not zero, as can be seen by comparing to a trace using a template that contains the promoter but lacks the aptamer sequence (Figure 2A, blue). This comparison suggests that even in the absence of a promoter sequence, some basal level of aptamer is transcribed (see below for further discussion).
Fluorescence can be monitored under both multi- and single-round conditions
Our specific goal was to obtain measurements of steady-state rates of transcription. We therefore needed to confirm that the observed signal is due to multiple rounds of transcription. We expect significantly higher signal amplitude under multi-round conditions compared to single-round conditions, where DNA traps are used to prevent RNAP rebinding. The preincubation of the reaction with salmon-sperm DNA, which does not actively dissociate RNAPs from the DNA like other competitors such as heparin (20), resulted in the absence of any signal upon the addition of NTPs (Supplementary Materials and Methods and Supplementary Figure S4A). This result shows that the competitor DNA acts as an effective trap for the Mtb RNAP and that its inclusion establishes single-round conditions. Upon initiating the transcription of pre-bound RNAP-DNA complexes with 500 μM NTPs and salmon-sperm DNA competitor, we observed no lag time, and the fluorescence rapidly reached saturation as RNAP re-binding was prevented (Figure 2B). These results are consistent with previous reports of single-round conditions for full-length transcript production (9,14,16). In the absence of competitor under otherwise identical conditions, we observed a much larger increase in the overall fluorescent signal (Figure 2B), confirming that in the absence of DNA trap, the assay is multi-round.
In theory, use of this assay under single-round conditions with pre-formed RNAP-DNA complexes should permit kinetic analyses of processes that are difficult to determine in multi-round conditions, such as promoter escape. Titrating all four NTPs in the presence of the DNA trap resulted in an NTP-concentration-dependent increase in signal amplitude, where the traces could be well-fit by a single-exponential function (Supplementary Figure S4B). This NTP-concentration-dependent change in amplitude suggests that the rate of RNAP dissociation from the promoter is on the same order as the rate of promoter escape under these conditions. This observation is consistent with our previous transient-state kinetic measurements of Mtb promoter escape kinetics, where we observed that increasing NTP concentration stabilizes the RNAP-DNA complexes, slowing the rate of dissociation and facilitating escape (20). These results illustrate that this fluorescent-aptamer-based assay can be used under single-round conditions to examine the kinetics of sub-steps in initiation (i.e. dissociation and promoter escape).
Extracting steady-state rates from real-time, multi-round fluorescent traces
Under multi-round conditions to examine the steady-state rates of transcription, we observed that the fluorescent time traces exhibited an apparent lag, followed by a linear increase in fluorescence over time. This rate of increase slows over longer timescales and eventually begins to plateau (Figure 3A, solid lines). We hypothesized that saturation of the fluorescence signal can be explained by the presence of paused and/or backtracked polymerases trapped on the template (reviewed in (53)), leading to a reduction in the RNAP molecules that can re-initiate at the promoter over time and an inability of active polymerases to complete transcription of a full length RNA. Consistent with this hypothesis, traces collected with E. coli RNAP in the presence and absence of the RNA cleavage factor GreB overlap at early timepoints and then diverged, where conditions containing GreB resulted in a higher fluorescent signal (Supplementary Figure S5). Based on these results, we suggest that GreB acts to increase the active RNAP concentration at longer times by facilitating recycling of long paused/backtracked elongation complexes (54,55). As GreB had no effect in the initial linear region, we suggest that this region reports on the true steady-state rate of initiation, without interference from inactive elongation states occurring downstream of the promoter. In addition, these results suggest that the fluorescent-aptamer-based assay can also be used to examine the regulation of rate-limiting elongation processes at long timescales.
Figure 3.

Quantification of the NTP-dependence of steady-state rates for full-length multi-round transcription. (A) Titration of the concentration of all NTPs with 100 nM Mtb RNAP and 5 nM rrnAP3 circular plasmid DNA results in an increase in the rate and amplitude of the fluorescent traces (solid, coloured lines). The unbiased linear fits of the early times are shown in grey dotted lines for each trace. (B) Quantification of steady-state rates on rrnAP3 (black) and promoterLESS (red) circular plasmid DNA templates plotted as a function of NTP concentration and fit to a modified Michaelis-Menten model (Equation 2) to account for the apparent sigmoidal behaviour.
Based on these results, we analyzed the initial fluorescence increase to quantitate the steady-state rate. Previous work manually determined a time regime of the trace to fit with either a line or an exponential. The slope was then determined directly or through differentiation, incorporating an initial time offset to account for the lag-phase (43). To define the steady-state (linear) regime, we designed an automated fitting protocol to reproducibly fit large amounts of data. With this strategy, there is no need to pre-determine either a time range to fit or a functional form representing the entire time course, both of which can change depending on the conditions of individual experiments. First, based on inspection of the traces to be fit, the user defines an initial time for the fitting procedure. In practice, this time should be past any lag or non-linear initial phase in the data. Then, each trace in the input data set are recursively fit with a line, using a statistical weight determined by the variation between technical replicates. The length of the time frame in each round of fitting is reduced by shortening the fit time frame (i.e. decreasing the time of the final data point considered in the fit), and the R2 value (goodness of fit) is compared to a threshold set by the user to evaluate whether the fit was adequately linear or whether a further reduction in the time frame should be attempted (Supplementary Figure S6). The code then plots the raw data and the fit for visual inspection by the user. Finally, the estimated rates are outputted for subsequent analyses.
Steady-state kinetic measurements are dependent on NTP concentration
We examined the NTP concentration dependence of reaction rates to ensure that the time-dependent signal change in fluorescence follows our expectations of steady-state behavior. In addition to promoter DNA, NTPs can also be considered as a substrate in the Michaelis-Menten analysis of the transcription reaction. In this case, concentrations below those required to reach Vmax can lead to a decrease in the rate of transcription, generally attributed to slower nucleotide incorporation rates and/or increases in the abortive fraction (14,20). Titrating all four NTPs together up to 500 μM on the rrnAP3 circular plasmid template, we observed a clear concentration dependence in the real-time traces (Figure 3A, solid lines). We fit the entire data set with our variable-time fitting algorithm described above, permitting us to extract the steady-state rates at each NTP concentration (Figure 3A, dotted lines). We plotted these rates as a function of NTP concentration and fit the data to a modified Michaelis-Menten equation, weighted by the exponential parameter n (Equation 2, Materials and Methods) (Figure 3B). The data did not fit the simpler form of the equation where n = 1 and an unconstrained fit results in an n = 2.1 ± 0.4. When n > 1, it signifies a steeper concentration dependence than expected from Michaelis-Menten equation for a single substrate binding site. Possible interpretations for this non-hyperbolic behavior can be found in the Discussion.
We performed analogous NTP titrations on the promoterLESS circular plasmid template. Compared to the kinetic parameters obtained with the rrnAP3 circular plasmid, we observed an ∼25-fold decrease in Vmax (6.5 ± 3.8 AU/min compared to 165 ± 7 AU/min) and an ∼5-fold increase in the apparent Km (Km,app defined in Methods; 230 ± 120 μM compared to 44 ± 5 μM) (Figure 3B, inset). As mentioned above, given that the signal from the promoterLESS circular plasmid is above that of the no-aptamer control obtained at saturating NTPs (Figure 2A), we interpret the NTP-dependent signal using the promoterLESS template as a measure of the non-rrnAP3 promoter derived background transcription. Being able to measure these NTP concentration-dependent trends in signal demonstrates that the assay possesses the detection sensitivity to measure low rates of full-length transcription, including those produced from sequences nominally devoid of promoters. As the promoterLESS control sets a lower bound for detecting rrnAP3 promoter-dependent transcription, it should always be included and, if need be, used as a correction. In this specific case, subtraction of the promoterLESS signal from that obtained with rrnAP3 resulted in no significant change in Michaelis-Menten parameters Vmax, Km,app or n (data not shown). These results indicate that we can confidently assign the fitted kinetic parameters to a rrnAP3-derived product under these experimental conditions.
Steady-state measurements show expected dependence on both RNAP and DNA concentration
In bacterial cells there exists a large molar excess of genomic DNA relative to the amount of free RNAP, making the in vivo global rate of transcription largely independent of gene concentration (56–59). Under these conditions, if the free RNAP concentration is well below the Km, the initial rate becomes independent of DNA concentration and proportional to the free RNAP concentration. As a result, the specific ratios of total RNAP concentration, free RNAP concentration (i.e. RNAPs that are not non-specifically bound to DNA or actively performing transcription (60,61)) and the number of possible interaction sites on the DNA will determine the reaction rate. To illustrate these concepts in vitro, we performed DNA titrations at multiple RNAP concentrations, using both circular plasmid (2557 bp) and linear PCR templates (250 bp) to provide different numbers of non-rrnAP3 specific interaction sites for the RNAP.
Previous measurements using an aptamer-based assay with T7 RNAP illustrated that the DNA concentration dependencies of steady-state rates followed a hyperbolic curve and could be described by Michaelis-Menten kinetics (39). We performed titrations of the rrnAP3 circular plasmid (0.1–50 nM) at two different concentrations of Mtb RNAP (20 and 100 nM) (Figure 4A, B) and observed significantly higher rates at the higher RNAP concentration (Figure 4C) as expected given the excess of RNAP over DNA. However, we did not observe a monotonic increase in the rates as a function of DNA concentration at either RNAP concentration. At low nM concentrations of the plasmid, we observed the expected increase in steady-state rate; however, as the plasmid concentration was increased, the rate exhibited a concentration-dependent decrease rather than a plateau (Figure 4C). Of note, the peak velocity at both concentrations of RNAP occurred at a similar DNA:RNAP ratio, roughly when RNAP was in 10-fold excess of plasmid DNA (Figure 4D). These observations are consistent with a mechanism where rrnAP3 promoter-specific DNA binding dominates in conditions of limiting DNA concentration, but as the concentration of total binding sites is increased, RNAP binds to sites other than rrnAP3 more often, resulting in a reduction in specific activity.
Figure 4.

Steady-state rates exhibit a biphasic DNA-concentration-dependence at multiple RNAP concentrations. Real-time data obtained at 500 μM all NTPs, titrating Mtb rrnAP3 circular plasmid DNA template (0.1–50 nM) at either (A) 20 nM or (B) 100 nM Mtb RNAP concentrations. The unbiased linear fits of the early times are shown in grey dotted lines for each trace. (C) Steady-state rates obtained from the linear fits in (A) and (B) for 20 nM (green) and 100 nM (black) RNAP, plotted as a function of rrnAP3 circular plasmid DNA concentration. (D) Steady-state rates, normalized from zero to one based on the lowest and highest rate obtained at each RNAP concentration, plotted as a function of the normalized [DNA]:[RNAP] concentration ratios. Included titrations of steady-state rate data are those obtained from the circular plasmids templates (open circles) and from linear PCR templates (Supplementary Figure S7, closed circles). Linear PCR DNA template concentration was normalized to that of the plasmid by dividing by a factor of ten to account for the template length (linear PCR template = 250 bp; circular plasmid template = 2557 bp).
Analogous titrations done on linear PCR templates containing the rrnAP3 promoter sequence (see Supplementary Table S2 for template preparation, description, and sequence), (Supplementary Figure S7A, B) displayed a less prevalent reduction in steady-state rates at higher DNA concentrations (Supplementary Figure S7C, D). Specifically, the reduction in steady-state rate did not occur until reaching a DNA:RNAP ratio of ∼2:1 for the 20 nM RNAP condition (Supplementary Figure S7D) and no reduction was observed in the data collected with 100 nM RNAP over the DNA concentration range tested (Supplementary Figure S7C,D). Thus, on these short linear PCR templates, the maximal transcriptional activity occurred at a higher DNA:RNAP ratio compared to circular plasmid templates. A shift to a higher ratio of the peak velocity may be caused by a decrease in non-rrnAP3 sites. When we normalize the DNA concentration by length, the maximal activities for both linear PCR and circular plasmid templates overlayed, suggesting that the rate decrease is due to non-rrnAP3 specific binding (Figure 4D). Combined, these experiments demonstrate quantification of the dependencies of the steady-state rates on both RNAP and DNA concentration, as well as highlight the advantages of using low DNA concentrations when quantifying promoter-specific initiation rates. This is especially important on plasmid templates which typically contain a higher number of possible interaction sites other than the promoter sequence under study.
Comparisons between fluorescence and gel-based approaches
Our fluorescence measurements clearly exhibit a promoter dependence (Figure 3B). As expected, performing analogous low throughput experiments involving incorporation of 32P labeled UTP followed by separation via polyacrylamide gel electrophoresis (Methods) revealed a specific band of the expected length in accordance with the position of the termination sequence on the circular plasmid containing the rrnAP3 promoter but not on the promoterLESS circular plasmid template (Figure 5A). In addition, bands larger than the full-length product were observed with both templates (Supplementary Figure S8A). As these bands were of equal intensity in both the rrnAP3 and promoterLESS data, we concluded that their generation was due to a component of the plasmid (possibly other cryptic promoters found on the plasmid) other than the promoter region being studied and they are not considered further.
Figure 5.

Comparison and calibration of gel- and fluorescence-based kinetics. Transcription gels showing the increase in amount of the specific, full-length 32P-labeled transcript with time (see Supplementary Figure S8 for full gel images) obtained with (A) 100 nM Mtb RNAP and 5 nM of the promoterLESS or rrnAP3 circular plasmid templates and (B) 20 nM and 100 nM Mtb RNAP with 5 nM rrnAP3 circular plasmid template. (C) Comparison of time-dependent signals from the fluorescent-aptamer-based assay (lines) and the gel assay (open circles) as a function of time for 20 nM and 100 nM RNAP. Both gel data sets were divided by the signal at 30 min obtained with the 20 nM RNAP concentration, thus normalizing that signal to a value of 1. The fluorescent data sets were then normalized using a factor determined by the ratio of the fluorescent and gel data at the same timepoint. The two gel data points that deviate from the fluorescent time course are indicated with red asterisks. The inset shows a comparison of normalized steady state rates from the gel (open bars) and fluorescence (filled bars) data with 20 nM (green) and 100 nM (black) RNAP concentrations. (D) Fluorescent signal plotted against RNA concentration from the data shown in (B) excluding the 20 and 30 minute time points obtained with 100 nM RNAP. A linear fit (solid red line) along with 95% confidence regions (dashed red lines) are indicated. The best fit line has a slope of 75 ± 17 AU/nM RNA and an y-intercept of –125 ± 70 AU. Error bars indicate the standard deviations from 3 to 4 independent experiments for fluorescence data and 2–3 independent experiments for gel-based data in all sub-plots.
We next asked whether the time evolution of the promoter-specific band yielded the same kinetics as those measured in real-time via the aptamer assay. For this comparison, gel-based experiments were performed at 20 and 100 nM Mtb RNAP with 5 nM rrnAP3 circular plasmid DNA (Figure 5B; Supplementary Figure S8B). We then normalized both sets of data using the 30-minute timepoint from the 20 nM RNAP condition and compared the time dependences of each signal (Figure 5C). With the exception of the 100 nM RNAP 20- and 30-minute timepoints, the normalized curves overlap within error. Additionally, a comparison of the two approaches shows that the normalized steady-state rates at each RNAP concentration are consistent (Figure 5C, inset). This analysis provides further evidence that the fluorescence signal reports on the promoter-specific activity of the system. Furthermore, it suggests that one may cautiously use the calculated RNA concentration of the specific, promoter-derived band from gel quantifications to convert the arbitrary fluorescence signal into an estimated RNA concentration. Fluorescence signal and RNA concentration were plotted against one another to construct a calibration curve omitting the long time points from the 100 nM RNAP condition (Figure 5D). The best fit line (Figure 5D, red) resulted in a negative y-intercept suggesting that the radioactive assay is more sensitive at low RNA concentrations. In particular, it suggests a lower limit of detection via the aptamer assay of approximately 1 nM, consistent with previous estimates (41). The slope of this calibration was 75 ± 17 AU/nM RNA. We note this conversion factor will not be universal and will depend highly on the experimental setup and conditions (see Supplementary Discussion). In summary, results from the gel-based and fluorescence-based assays are consistent and performing both assays provides a means of converting the arbitrary fluorescence signal into absolute RNA concentration.
The spinach aptamer and DFHBI have no effect on the measured steady-state rates
We have demonstrated that use of the aptamer assay allows for the quantification of steady-state rates of transcription. However, the two additional elements that could theoretically alter the measured rate have not been discussed: the aptamer sequence and the DFHBI dye. To examine the effect of the aptamer sequence or the presence of the dye on the rate of transcription, we performed 32P incorporation assay measurements on circular plasmid templates containing the rrnAP3 promoter in the presence and absence of the aptamer sequence and in the presence and absence of dye. We observed roughly the same steady-state rate ± aptamer sequence (Supplementary Figure S9A, B), suggesting that the fluorescence assay can be taken to report on the rates of transcript production without effects from the aptamer. Additionally, in titrating DFHBI concentration up to 20 μM (the concentration used in the fluorescence assay) in 32P incorporation experiments, we observed no change in the amount of RNA generated 30 min after initiating the transcription reaction (Supplementary Figure S9C, D), consistent with previous measurements made with T7 RNAP (39,62).
We note that in the aptamer assay, we observe a change in the magnitude of the fluorescent signal when titrating dye concentrations in the context of the same transcription reaction (Supplementary Figure S10A). This can be explained by the finite affinity of the dye for the aptamer. As dye concentration is increased, higher and higher fractions of aptamer are bound, effectively increasing the gain of the signal. However, if experiments are always performed at a fixed dye concentration, preferably higher than the Kd to maximize signal (Supplementary Figure S10B), comparing fold-changes in fluorescence is a valid approach under steady-state conditions (Supplementary Figure S10C, D). Care should be taken when using large concentrations of dye, as a correction for an inner filter effect may be needed (63).
High-throughput capabilities of the fluorescent assay permits concentration dependencies of individual NTPs to be measured in a single experiment
Higher concentrations of the initiating nucleotide (iNTP) than the subsequent NTPs are frequently required for promoter-specific initiation, especially on ribosomal RNA promoters where incorporation of the iNTP increases the population of open complexes at equilibrium, which can be a rate-limiting step (20,64). We hypothesized that when measured under steady-state conditions, titrations of the iNTP would thus yield the highest Km compared to the other NTPs on the Mtb ribosomal RNA promoter. To measure the dependence of steady-state rates on the concentration of individual NTPs on the rrnAP3 circular plasmid template, we performed titrations of each NTP from 2.5–500 μM in a background of saturating concentrations (500 μM) of the other three NTPs (Figure 6A). Each independent experiment incorporated three technical replicates across ten concentrations per individual NTP titration resulting in a total of 120 total kinetic measurements obtained in parallel, highlighting the high-throughput nature of this assay. The resulting traces were fit to extract steady-state rates as a function of individual NTP concentration (Figure 6B). When the iNTP (GTP) or the second incorporated NTP (UTP) were titrated, the Michaelis-Menten equation (Equation 1) was unable to account for the data. For these titrations Equation 2 was used to account for the sigmoidal dependence. The Vmax for each titration was equal within error (Figure 6A,B; Supplementary Table S3). As predicted, the data revealed that the transcription rate depends most strongly on the iNTP (GTP) concentration with a Km,app of 16 ± 2 μM, consistent with the results from single time-point experiments on E. coli ribosomal RNA promoters (64). As the first incorporation site of the titrated nucleotide is found further downstream within the sequence of the initially transcribed region, the measured Km,app shifted to lower concentrations (Figure 6B; Supplementary Table S3). Notably, these trends in Km,app and Vmax as a function of nucleotide identity were recapitulated using short linear PCR templates containing the rrnAP3 sequence (Figure 6C, D). Furthermore, the Km,app values were either within error or higher in all cases compared to those determined with the circular plasmid template (Figure 6D; Supplementary Table S3), consistent with previous linear and plasmid template comparisons made on ribosomal RNA promoters (64,65). Even though we observed a substantially lower Vmax when considering the scale of arbitrary fluorescent units on linear as opposed to plasmid templates, the fact that these nucleotide-identity-dependent trends could be easily measured on either template, including the sigmoidal concentration dependencies, demonstrates the utility of the assay regardless of the type of template used.
Figure 6.

Titrations of individual NTPs reveal incorporation of the initiating nucleotide is rate limiting. (A) Real-time fluorescent traces and linear fits obtained using 100 nM Mtb RNAP, 5 nM rrnAP3 circular plasmid DNA, and individually titrating GTP, UTP, CTP, or ATP in the presence of 500 μM of the other three, non-titrated NTPs. (B) Rate dependence on the concentration of individual NTPs from the data in (A) as well as the titration of all NTPs equally. Note that for clarity, only the titration data out to 200 μM NTPs is shown. The y-axis was converted to RNA concentrations using the calibration presented in Figure 5D. (C) Real-time fluorescent traces and fits as in (A), except using 25 nM of the rrnAP3 linear PCR DNA template. (D) Rate dependence on the concentration of individual NTPs from the data in (C) as well as the titration of all NTPs equally. Error bars in (C) and (D) represent standard deviations of 2–4 independent experiments each with 2–3 technical replicates. Rate dependencies in (C) and (D) were fit to Equation 1 or 2 depending on the identity of the titrated NTP, and fitted parameters are summarized in Supplementary Table S3.
Quantification of transcription factor activity under steady-state conditions
When evaluating the effect of a transcription factor in vitro, the most common approach is to use single timepoint measurements to compare transcription in the presence and absence of the factor. However, by only evaluating a single timepoint for each condition there is no guarantee or confirmation that the measurements are representative of changes occurring throughout a steady-state process. In contrast, the aptamer assay directly measures the effect of transcription factors in real-time and by focusing the analysis on the linear regime in an unbiased manner, ensures that factor-dependent changes are quantified under steady-state conditions. To demonstrate the use of the assay in analysis of transcription factor effects, we turned to two well-studied Mtb transcription factors, CarD and RbpA.
Previous studies have shown that both CarD and RbpA stabilize the open complex (18,19,66,67), slow promoter escape (20) and activate transcription from the rrnAP3 promoter (66,68). In addition, these factors are known to bind the initiation complex cooperatively and act synergistically (19,20,67). We measured transcription in the presence of CarD, RbpA, or both at saturating concentrations on the linear PCR rrnAP3 template. We monitored fluorescence over time and fit the traces to obtain steady-state rates (Figure 7A). Between 2 and 5 independent experiments were used to calculate and compare the average rates at each condition. Consistent with previous work, we observed a 2.8-fold, 4-fold and 9.9-fold increase in the rate of RNA production in the presence of RbpA, CarD and both factors together, respectively (Figure 7B).
Figure 7.

Factor-dependent effects on steady-state transcription rates. (A) Real-time fluorescent traces collected by pre-incubating 100 nM Mtb RNAP and 500 μM NTPs in the presence of saturating Mtb CarD (1 μM) and RbpA (2 μM) (solid curves) prior to addition of 25 nM rrnAP3 linear PCR template. Raw traces are shown with associated linear fits (dotted lines). (B) Comparison of steady-state rates (average of 2–5 independent experiments) reveals the extent of transcriptional activation by RbpA (red), CarD (blue), and the combination of RbpA and CarD (purple). Fold changes in the steady-state rate relative to Mtb RNAP alone are indicated above each condition.
Quantification of inhibitory concentrations of antibiotics
Bacterial RNAPs are the direct targets of many antibiotics (reviewed in (12,69)) and the search for new antibiotics to overcome drug resistance is never-ending. This is particularly important in the battle against Mtb, the causative agent of tuberculosis, as multi-drug resistant strains are becoming more prevalent (70). To this end, the development of new small molecule inhibitors that work at the level of transcription is of high interest. Here, we demonstrate that the aptamer assay can be used to quantify antibiotic-dependent inhibition of steady-state rates. We illustrate this with the well-characterized antibiotics Rifampicin and Fidaxomicin, currently used to treat Mtb and Clostridium difficile infections by directly targeting the bacterial RNAP (71–74).
Titrations of both Rifampicin and Fidaxomicin were performed using either 100 nM Mtb or E. coli RNAP with 5 nM Mtb rrnAP3 circular plasmid template (Supplementary Materials and Methods), and the kinetic traces were fit to extract the steady-state rates. Although E. coli RNAP exhibits a lower maximal rate of transcription on this template, as inferred from the fluorescence signal in the absence of antibiotic (Supplementary Figure S11), for both the RNAPs, we observed a concentration-dependent decrease in the measured steady-state rates upon increasing antibiotic concentrations (Figure 8). Fitting the data to an inhibition curve (Equation 3) permitted the calculation of half-maximal inhibitory concentrations. Consistent with previous work, the IC50 for Rifampicin on E. coli RNAP (15 ± 2 nM) and Mtb RNAP (17 ± 2 nM) were within error (Figure 8A) (66,75). In addition, the same analysis for Fidaxomicin yielded an approximately three-fold higher IC50 on E. coli RNAP (400 ± 110 nM) than on Mtb RNAP (138 ± 92 nM) (Figure 8B), consistent with previous reports of Mtb being more sensitive to the drug than E. coli (72,76).
Figure 8.

Quantification of antibiotic IC50 values based on changes in observed steady-state rates. Normalized steady-state rates plotted as a function of (A) Rifampicin and (B) Fidaxomicin concentrations for Mtb (black) and E. coli (red) RNAP. All experiments were performed using 1 mM of each NTP and 5 nM rrnAP3 plasmid DNA. See Supplementary Figure S11 for the corresponding real-time data, linear fits, and un-normalized rates. The normalized steady-state rates are based on the associated fits using Equation 3 (Supplementary Figure S11E, F).
DISCUSSION
High-throughput kinetic measurements of full-length transcription
The fluorescence experiments presented here are an attractive alternative technique to the standard radiolabeled-nucleotide incorporated gel-based approaches when used to quantitate steady-state transcription kinetics, especially when used in a plate-reader format as described. Hundreds of conditions can be measured in real-time with several technical replicates in a single experiment. We estimate that the throughput is on the order of a hundred times that of standard gel-based approaches, where the real-time measurements facilitate a more precise and accurate determination and quantification of the steady-state regime. As the steady-state rate of full-length transcription (along with the RNA degradation rate) is a crucial metric when considering the flux of biological processes that use RNA as a substrate (1), use of this assay will significantly facilitate the exploration of diverse mechanisms of gene regulation with biological relevance.
We emphasize that radioactive techniques are advantageous and complementary when looking to resolve individual RNA transcripts, as well as to determine the relative lengths and amounts of side and abortive products (77). In fact, the first steady-state assays that reported on bacterial transcription were made under conditions where NTP synthesis was restricted to abortive transcription (78,79). However, as RNAP does not escape the promoter under these circumstances, these assays did not report on the overall rate of initiation, but rather the rate-limiting step involved in binding and isomerization to the catalytically active open complex (79). Fluorescent-labeled nucleotides that are incorporated into the RNA transcripts have also been used to monitor the steady-state rates of abortive RNA generation with E. coli RNAP (80–83), representing an alternative to radioactivity-based approaches. However, this method cannot discriminate between abortive and full-length RNA products. In comparison to other techniques, the fluorescent aptamer-based assay described here is advantageous, as the observed fluorescence signal reports specifically on full-length RNA products. This is demonstrated in conditions where single NTPs are not included, resulting in the inability of RNAP to escape the promoter and a lack of fluorescence signal change (Figure 6). We note that the effects of abortive synthesis are still borne out in the kinetics, as the process of promoter escape can be rate-limiting on some promoters and affected by the initially transcribed sequence (84,85), which we have included in our DNA constructs. As a result, the assay permits a straightforward way to measure the rate of full-length transcript production dictated by the overall rate of initiation.
We have illustrated that the assay can easily quantitate differences in kinetics due to promoter sequence (Figure 3), NTP concentration (Figure 6), and RNAP and promoter DNA concentrations (Figure 4; Supplementary Figure S7). The non-monotonic rate dependencies observed when titrating DNA (Figure 4; Supplementary Figure S7) suggest one could use this assay to evaluate how the presence of other promoters or number of non-promoter sites compete for RNAP binding and affect the steady-states rates from a particular promoter of interest. Furthermore, in addition to having great utility in multi-round assays, the assay can be used in single-round conditions to evaluate rates of initiation processes such as promoter escape (Supplementary Figure S4). The single-round approach has previously been well-described and used for monitoring co-transcriptional RNA folding processes (43,51).
We stress that meaningful quantifications of relative changes in flux can be made using arbitrary fluorescence units as long as buffer conditions, temperature, or any other variable that may affect aptamer folding or DFHBI binding are not varied. However, we have also shown the approach and necessary controls to calibrate the fluorescent signal using an independent measure of RNA concentration from identical reactions (Figure 5). Further thoughts on calibration and interpretation of the fluorescent signal can be found in the Supplementary Discussion.
Nucleotide-dependent kinetics
By performing titrations of NTP substrates, we have illustrated that Km,app and Vmax parameters can be obtained with a high level of precision under steady-state conditions (Figure 6). Using these data, we observed that not all titrations could be fit to a hyperbolic Michaelis-Menten model. Rather, sigmoidal trends were observed for both circular plasmid and linear PCR DNA templates in the titration of all NTPs together and GTP in the background of saturating concentrations of ATP, UTP, and CTP (Figure 6B, D; Supplementary Table S3). Initiation requires the first two nucleotides to form the first RNA phosphodiester bond and, depending on the conformation of the open DNA, base-stacking with the +1/+2 DNA sequences on the template strand may represent a rate-limiting step (17). Our results are consistent with this hypothesis in that we observe that the concentration of the first two NTP substrates had a larger effect as compared to the other NTPs (Figure 6B, D; Supplementary Table S3). For monomeric enzymes containing a single active site, a sigmoidal dependence of velocity on substrate concentration has been referred to as kinetic cooperativity and was first measured with glucokinase (86,87). Additionally, other mechanisms have been suggested to lead to kinetic cooperativity without direct interactions between binding sites (reviewed in (49)), including the existence of a slow conformational change that precedes substrate incorporation, the existence of multiple enzyme classes capable of binding initiating nucleotides with different affinities, and/or a substrate-induced conformational change to the catalytically active enzyme (49,88). Mechanistic explanations may include effects of the first two nucleotides on the ratio of abortive and productive synthesis or the necessity to bind two NTP substrates to synthesize the first phosphodiester bond. These models may not be mutually exclusive and, in fact, are supported by numerous experimental studies evaluating the initial nucleotide incorporation process with E. coli RNAP (13,14,16,17). While we cannot directly comment on the specific mechanism(s) that results in this apparent positive cooperativity with Mtb RNAP, the detection of this sigmoidal behavior would not have been possible without the fine sampling of NTP concentrations and precise measurements of steady-state rates facilitated by the ease and throughput of the aptamer-based assay.
Future uses for high-throughput detection of basal and regulated transcription kinetics
Given these fluorescent experiments were all performed in a 384 well plate, the assay lends itself naturally to questions that require large data sets. We conclude by summarizing a few exciting possible directions.
Using sequence to predict transcriptional activity remains a challenge, as the context of the entire promoter must be considered when examining how sequence affects initiation kinetics (3). Using the aptamer-based assay, one could generate promoter libraries where sequence context is investigated by introducing sequence mutations in promoter regions of interest (44,84,89). Alternatively, one could design and measure basal and regulated transcription rates on large numbers of genomic promoter sequences. This would allow for a side-by-side comparison with genome-wide, RNA-seq data (90). Use of the aptamer-based assay will greatly facilitate the ability to quantitatively probe the kinetics of larger genomic promoter libraries in vitro to expand our knowledge of gene regulatory mechanisms.
As we illustrated the utility of the fluorescence assay in measuring antibiotic inhibition (Figure 8; Supplementary Figure S11), the high-throughput screening of novel inhibitors against the bacterial transcription machinery may also benefit from this assay. Inhibitors that act at the level of initiation could be identified by a reduction of the initial steady-state rate, as observed for Rifampicin and Fidaxomicin (Figure 8; Supplementary Figure S11). In addition, inhibitors of elongation or termination that lead to stalled, template bound RNAPs may result in changes in the kinetics of the signal over longer-timescales as they eventually reduce the number of actively transcribing polymerases and may become roadblocks to RNAPs that are still active. As a result, use of this assay under multi-round conditions may permit the identification of compounds that inhibit transcription via different mechanisms. Furthermore, because one tracks the time-dependent changes in fluorescence and not single-timepoints, compounds that exhibit fluorescent spectral properties are less likely to produce artifactual results. Compounds identified as potential hits can then easily be analyzed via titrations to measure the IC50s as described here. To develop structural hypotheses for the mechanism of transcription initiation inhibitors (or classes of inhibitors), titrations of NTPs or promoter DNA could be performed to determine the type of inhibition (e.g. competitive, non-competitive, or uncompetitive) via quantification of changes in Km,app and/or Vmax (91).
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Drs Dylan Plaskon (University of Wisconsin at Madison) for helpful advice during the infancy of this work, Wayne Barnes (Washington University) for providing advice and reagents for the design of DNA templates, Irina Artsimovitch (The Ohio State University) for providing purified E. coli GreB, and Nicole Fazio for critical reading of the manuscript. Additionally, thanks to the Washington University High-Throughput Screening Centre (HTSC) staff for technical assistance with plate-reader measurements.
Authors’ contributions: Drake Jensen: Conceptualization, Formal analysis, Investigation, Methodology, Validation, and Writing—original draft. Ana Ruiz Manzano: Formal analysis, Investigation, Methodology, and Validation. Maxwell Rector: Formal analysis, Investigation and Validation. Eric J. Tomko: Resources and Methodology. M. Thomas Record: Funding acquisition and Supervision. Eric A. Galburt: Conceptualization, Data curation, Formal analysis, Funding acquisition, Methodology, Software, Supervision, Validation, Visualization and Writing—original draft. All authors reviewed and edited the final draft.
Contributor Information
Drake Jensen, Department of Biochemistry and Molecular Biophysics, Washington University School of Medicine, Saint Louis, MO 63108, USA.
Ana Ruiz Manzano, Department of Biochemistry and Molecular Biophysics, Washington University School of Medicine, Saint Louis, MO 63108, USA.
Maxwell Rector, Department of Biochemistry, University of Wisconsin, Madison, WI 53706, USA.
Eric J Tomko, Department of Biochemistry and Molecular Biophysics, Washington University School of Medicine, Saint Louis, MO 63108, USA.
M Thomas Record, Department of Biochemistry, University of Wisconsin, Madison, WI 53706, USA.
Eric A Galburt, Department of Biochemistry and Molecular Biophysics, Washington University School of Medicine, Saint Louis, MO 63108, USA.
DATA AVAILABILITY
The MATLAB code used for automated linear regression analyses has been made publicly available and can be accessed via the following link: https://doi.org/10.5281/zenodo.8319359.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R35GM144282 to E.A.G., R35GM118100 to M.T.R.]. Funding for open access charge: National Institutes of Health [R35GM144282].
Conflict of interest statement. None declared.
REFERENCES
- 1. Balakrishnan R., Mori M., Segota I., Zhang Z., Aebersold R., Ludwig C., Hwa T.. Principles of gene regulation quantitatively connect DNA to RNA and proteins in bacteria. Science. 2022; 378:eabk2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ruff E.F., Record M.T., Artsimovitch I.. Initial events in bacterial transcription initiation. Biomol. 2015; 5:1035–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jensen D., Galburt E.A.. The context-dependent influence of promoter sequence motifs on transcription initiation kinetics and regulation. J. Bacteriol. 2021; 203:e00512-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McClure W.R. Mechanism and control of transcription initiation in prokaryotes. Annu. Rev. Biochem. 1985; 54:171–204. [DOI] [PubMed] [Google Scholar]
- 5. Henderson K.L., Felth L.C., Molzahn C.M., Shkel I., Wang S., Chhabra M., Ruff E.F., Bieter L., Kraft J.E., Record M.T.. Mechanism of transcription initiation and promoter escape by E. coli RNA polymerase. Proc Natl. Acad. Sci. U.S.A. 2017; 114:E3032–E3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kapanidis A.N., Margeat E., Ho S.O., Kortkhonjia E., Weiss S., Ebright R.H.. Initial transcription by RNA polymerase proceeds through a DNA-scrunching mechanism. Science. 2006; 314:1144–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Belogurov G.A., Artsimovitch I.. The mechanisms of substrate selection, catalysis and translocation by the elongating RNA polymerase. J. Mol. Biol. 2019; 431:3975–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lerner E., Chung S., Allen B.L., Wang S., Lee J., Lu S.W., Grimaud L.W., Ingargiola A., Michalet X., Alhadid Y.et al.. Backtracked and paused transcription initiation intermediate of Escherichia coli RNA polymerase. Proc Natl. Acad. Sci. U.S.A. 2016; 113:E6562–E6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Henderson K.L., Evensen C.E., Molzahn C.M., Felth L.C., Dyke S., Liao G., Shkel I.A., Record M.T.. RNA polymerase: step-by-step kinetics and mechanism of transcription initiation. Biochemistry-US. 2019; 58:2339–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Saecker R.M., Record M.T., deHaseth P.L.. Mechanism of bacterial transcription initiation: RNA polymerase - promoter binding, isomerization to initiation-competent open complexes, and initiation of RNA synthesis. J. Mol. Biol. 2011; 412:754–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boyaci H., Saecker R.M., Campbell E.A.. Transcription initiation in mycobacteria: a biophysical perspective. Transcription. 2019; 11:53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen J., Boyaci H., Campbell E.A.. Diverse and unified mechanisms of transcription initiation in bacteria. Nat. Rev. Microbiol. 2020; 19:95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Susa M., Sen R., Shimamoto N.. Generality of the branched pathway in transcription initiation by Escherichia coli RNA polymerase. J. Biol. Chem. 2002; 277:15407–15412. [DOI] [PubMed] [Google Scholar]
- 14. Vo N.V., Hsu L.M., Kane C.M., Chamberlin M.J.. In vitro studies of transcript initiation by Escherichia coli RNA polymerase. 2. Formation and characterization of two distinct classes of initial transcribing complexes. Biochemistry-US. 2003; 42:3787–3797. [DOI] [PubMed] [Google Scholar]
- 15. Dulin D., Bauer D.L.V., Malinen A.M., Bakermans J.J.W., Kaller M., Morichaud Z., Petushkov I., Depken M., Brodolin K., Kulbachinskiy A.et al.. Pausing controls branching between productive and non-productive pathways during initial transcription in bacteria. Nat. Commun. 2018; 9:1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Plaskon D.M., Henderson K.L., Felth L.C., Molzahn C.M., Evensen C., Dyke S., Shkel I.A., Record M.T.. Temperature effects on RNA polymerase initiation kinetics reveal which open complex initiates and that bubble collapse is stepwise. Proc Natl. Acad. Sci. U.S.A. 2021; 118:e2021941118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Saecker R.M., Chen J., Chiu C.E., Malone B., Sotiris J., Ebrahim M., Yen L.Y., Eng E.T., Darst S.A.. Structural origins of Escherichia coli RNA polymerase open promoter complex stability. Proc Natl. Acad. Sci. U.S.A. 2021; 118:e2112877118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rammohan J., Ruiz Manzano A., Garner A.L., Stallings C.L., Galburt E.A.. CarD stabilizes mycobacterial open complexes via a two-tiered kinetic mechanism. Nucleic Acids Res. 2015; 43:3272–3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hubin E.A., Fay A., Xu C., Bean J.M., Saecker R.M., Glickman M.S., Darst S.A., Campbell E.A.. Structure and function of the mycobacterial transcription initiation complex with the essential regulator RbpA. Elife. 2017; 6:e22520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jensen D., Manzano A.R., Rammohan J., Stallings C.L., Galburt E.A.. CarD and RbpA modify the kinetics of initial transcription and slow promoter escape of the mycobacterium tuberculosis RNA polymerase. Nucleic Acids Res. 2019; 47:6685–6698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Galburt E.A. The calculation of transcript flux ratios reveals single regulatory mechanisms capable of activation and repression. Proc National Acad Sci. 2018; 115:201809454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Record M.T., Reznikoff W.S., Craig M.L., McQuade K.I., Schlax P.J.. Neidhardt F.C. Escherichia coli RNA polymerase (Eσ70), promoters, and the kinetics of the steps of transcription initiation. Escherichia coli and Salmonella: Cellular and Molecular Biology. 1996; Washington, DC: American Society for Microbiology. [Google Scholar]
- 23. Liang S.-T., Bipatnath M., Xu Y.-C., Chen S.-L., Dennis P., Ehrenberg M., Bremer H.. Activities of constitutive promoters in Escherichia coli. J. Mol. Biol. 1999; 292:19–37. [DOI] [PubMed] [Google Scholar]
- 24. Zhang X., Dennis P., Ehrenberg M., Bremer H.. Kinetic properties of rrn promoters in Escherichia coli. Biochimie. 2002; 84:981–996. [DOI] [PubMed] [Google Scholar]
- 25. Gerosa L., Kochanowski K., Heinemann M., Sauer U.. Dissecting specific and global transcriptional regulation of bacterial gene expression. Mol. Syst. Biol. 2013; 9:658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jacob F., Monod J.. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 1961; 3:318–356. [DOI] [PubMed] [Google Scholar]
- 27. Gourse R.L., Chen A.Y., Gopalkrishnan S., Sanchez-Vazquez P., Myers A., Ross W.. Transcriptional responses to ppGpp and DksA. Annu. Rev. Microbiol. 2018; 72:163–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bergkessel M. Bacterial transcription during growth arrest. Transcription. 2021; 12:232–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ross W., Gourse R.L.. Analysis of RNA polymerase-promoter complex formation. Methods. 2009; 47:13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Burgess R.R., Travers A.A., Dunn J.J., Bautz E.K.F.. Factor stimulating transcription by RNA polymerase. Nature. 1969; 221:43–46. [DOI] [PubMed] [Google Scholar]
- 31. Stark R., Grzelak M., Hadfield J.. RNA sequencing: the teenage years. Nat. Rev. Genet. 2019; 20:631–656. [DOI] [PubMed] [Google Scholar]
- 32. Neubacher S., Hennig S.. RNA structure and cellular applications of fluorescent light-up aptamers. Angew. Chem. Int. Ed. 2019; 58:1266–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Paige J.S., Wu K.Y., Jaffrey S.R.. RNA mimics of green fluorescent protein. Science. 2011; 333:642–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Strack R.L., Disney M.D., Jaffrey S.R.. A superfolding Spinach2 reveals the dynamic nature of trinucleotide repeat–containing RNA. Nat. Methods. 2013; 10:1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang J., Fei J., Leslie B.J., Han K.Y., Kuhlman T.E., Ha T.. Tandem spinach array for mRNA imaging in living bacterial cells. Sci Rep-uk. 2015; 5:17295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen X., Zhang D., Su N., Bao B., Xie X., Zuo F., Yang L., Wang H., Jiang L., Lin Q.et al.. Visualizing RNA dynamics in live cells with bright and stable fluorescent RNAs. Nat. Biotechnol. 2019; 37:1287–1293. [DOI] [PubMed] [Google Scholar]
- 37. Bouhedda F., Fam K.T., Collot M., Autour A., Marzi S., Klymchenko A., Ryckelynck M.. A dimerization-based fluorogenic dye-aptamer module for RNA imaging in live cells. Nat. Chem. Biol. 2019; 16:69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cawte A.D., Unrau P.J., Rueda D.S.. Live cell imaging of single RNA molecules with fluorogenic Mango II arrays. Nat. Commun. 2020; 11:1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nies P.v., Nourian Z., Kok M., Wijk R.v., Moeskops J., Westerlaken I., Poolman J.M., Eelkema R., Esch J.H.v., Kuruma Y.et al.. Unbiased tracking of the progression of mRNA and protein synthesis in bulk and in liposome-confined reactions. ChemBioChem. 2013; 14:1963–1966. [DOI] [PubMed] [Google Scholar]
- 40. Chizzolini F., Forlin M., Cecchi D., Mansy S.S.. Gene position more strongly influences cell-free protein expression from operons than T7 transcriptional promoter strength. Acs Synth Biol. 2014; 3:363–371. [DOI] [PubMed] [Google Scholar]
- 41. Nies P.v., Canton A.S., Nourian Z., Danelon C. Chapter ten monitoring mRNA and protein levels in bulk and in model vesicle-based artificial cells. Methods Enzymol. 2015; 550:187–214. [DOI] [PubMed] [Google Scholar]
- 42. Yim S.S., Johns N.I., Park J., Gomes A.L., McBee R.M., Richardson M., Ronda C., Chen S.P., Garenne D., Noireaux V.et al.. Multiplex transcriptional characterizations across diverse bacterial species using cell-free systems. Mol. Syst. Biol. 2019; 15:e8875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huang Y.-H., Trapp V., Puro O., Mäkinen J.J., Metsä-Ketelä M., Wahl M.C., Belogurov G.A.. Fluorogenic RNA aptamers to probe transcription initiation and co-transcriptional RNA folding by multi-subunit RNA polymerases. Methods Enzymol. 2022; 675:207–233. [DOI] [PubMed] [Google Scholar]
- 44. Qin W., Li L., Yang F., Wang S., Yang G.-Y.. High-throughput iSpinach fluorescent aptamer-based real-time monitoring of in vitro transcription. Bioresour Bioprocess. 2022; 9:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Orosz A., Boros I., Venetianer P.. Analysis of the complex transcription termination region of the Escherichia coli rrn B gene. Eur. J. Biochem. 1991; 201:653–659. [DOI] [PubMed] [Google Scholar]
- 46. Prusa J., Jensen D., Santiago-Collazo G., Pope S.S., Garner A.L., Miller J.J., Manzano A.R., Galburt E.A., Stallings C.L.. Domains within RbpA serve specific functional roles that regulate the expression of distinct mycobacterial gene subsets. J. Bacteriol. 2018; 200:e00690-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Perederina A.A., Vassylyeva M.N., Berezin I.A., Svetlov V., Artsimovitch I., Vassylyev D.G.. Cloning, expression, purification, crystallization and initial crystallographic analysis of transcription elongation factors GreB from Escherichia coli and Gfh1 from Thermus thermophilus. Acta Crystallogr Sect F. 2006; 62:44–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Reisbig R.R., Woody A.Y.M., Woody R.W.. Rifampicin as a spectroscopic probe of the mechanism of RNA polymerase from E. coli. Biochemistry-US. 1982; 21:196–200. [Google Scholar]
- 49. Cornish-Bowden A., Cárdenas M.L.. Co-operativity in monomeric enzymes. J. Theor. Biol. 1987; 124:1–23. [DOI] [PubMed] [Google Scholar]
- 50. Mitra J., Ha T.. Nanomechanics and co-transcriptional folding of spinach and Mango. Nat. Commun. 2019; 10:4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Huang Y.-H., Hilal T., Loll B., Bürger J., Mielke T., Böttcher C., Said N., Wahl M.C.. Structure-based mechanisms of a molecular RNA polymerase/chaperone machine required for ribosome biosynthesis. Mol. Cell. 2020; 79:1024–1036. [DOI] [PubMed] [Google Scholar]
- 52. Han K.Y., Leslie B.J., Fei J., Zhang J., Ha T.. Understanding the photophysics of the spinach–DFHBI RNA aptamer–Fluorogen complex to improve live-cell RNA imaging. J. Am. Chem. Soc. 2013; 135:19033–19038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kang J.Y., Mishanina T.V., Landick R., Darst S.A.. Mechanisms of transcriptional pausing in bacteria. J. Mol. Biol. 2019; 431:4007–4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Abdelkareem M., Saint-André C., Takacs M., Papai G., Crucifix C., Guo X., Ortiz J., Weixlbaumer A.. Structural basis of transcription: RNA polymerase backtracking and its reactivation. Mol. Cell. 2019; 75:298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Janissen R., Eslami-Mossallam B., Artsimovitch I., Depken M., Dekker N.H.. High-throughput single-molecule experiments reveal heterogeneity, state switching, and three interconnected pause states in transcription. Cell Rep. 2022; 39:110749. [DOI] [PubMed] [Google Scholar]
- 56. Churchward G., Bremer H., Young R.. Transcription in bacteria at different DNA concentrations. J. Bacteriol. 1982; 150:572–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jinks-Robertson S., Gourse R.L., Nomura M.. Expression of rRNA and tRNA genes in Escherichia coli: evidence for feedback regulation by products of rRNA operons. Cell. 1983; 33:865–876. [DOI] [PubMed] [Google Scholar]
- 58. Baracchini E., Bremer H.. Control of rRNA synthesis in Escherichia coli at increased rrn gene dosage. Role of guanosine tetraphosphate and ribosome feedback. J. Biol. Chem. 1991; 266:11753–11760. [PubMed] [Google Scholar]
- 59. Patrick M., Dennis P.P., Ehrenberg M., Bremer H.. Free RNA polymerase in Escherichia coli. Biochimie. 2015; 119:80–91. [DOI] [PubMed] [Google Scholar]
- 60. DeHaseth P.L., Lohman T.M., Burgess R.R., Record M.T.. Nonspecific interactions of Escherichia coli RNA polymerase with native and denatured DNA: differences in the binding behavior of core and holoenzyme. Biochemistry-US. 1978; 17:1612–1622. [DOI] [PubMed] [Google Scholar]
- 61. Hippel P.H., Berg O.G.. On the specificity of DNA-protein interactions. Proc. Natl. Acad. Sci. 1986; 83:1608–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Höfer K., Langejürgen L.V., Jäschke A.. Universal aptamer-based real-time monitoring of enzymatic RNA synthesis. J. Am. Chem. Soc. 2013; 135:13692–13694. [DOI] [PubMed] [Google Scholar]
- 63. Birdsall B., King R.W., Wheeler M.R., Lewis C.A., Goode S.R., Dunlap R.B., Roberts G.C.K.. Correction for light absorption in fluorescence studies of protein-ligand interactions. Anal. Biochem. 1983; 132:353–361. [DOI] [PubMed] [Google Scholar]
- 64. Gaal T., Bartlett M.S., Ross W., Turnbough C.L., Gourse R.L.. Transcription regulation by initiating NTP concentration: rRNA synthesis in bacteria. Science. 1997; 278:2092–2097. [DOI] [PubMed] [Google Scholar]
- 65. Sudzinová P., Kambová M., Ramaniuk O., Benda M., Šanderová H., Krásný L.. Effects of DNA topology on transcription from rRNA promoters in Bacillus subtilis. Microorg. 2021; 9:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hu Y., Morichaud Z., Chen S., Leonetti J.-P., Brodolin K.. Mycobacterium tuberculosis RbpA protein is a new type of transcriptional activator that stabilizes the σ A -containing RNA polymerase holoenzyme. Nucleic Acids Res. 2012; 40:6547–6557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rammohan J., Ruiz Manzano A., Garner A.L., Prusa J., Stallings C.L., Galburt E.A.. Cooperative stabilization of mycobacterium tuberculosis rrnAP3 promoter open complexes by RbpA and CarD. Nucleic Acids Res. 2016; 44:7304–7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Srivastava D.B., Leon K., Osmundson J., Garner A.L., Weiss L.A., Westblade L.F., Glickman M.S., Landick R., Darst S.A., Stallings C.L.et al.. Structure and function of CarD, an essential mycobacterial transcription factor. Proc Natl. Acad. Sci. 2013; 110:12619–12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ma C., Yang X., Lewis P.J.. Bacterial transcription as a target for antibacterial drug development. Microbiol. Mol. Biol. R. 2016; 80:139–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Allué-Guardia A., García J.I., Torrelles J.B.. Evolution of drug-resistant mycobacterium tuberculosis strains and their adaptation to the Human lung environment. Front Microbiol. 2021; 12:612675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mosaei H., Zenkin N.. Inhibition of RNA polymerase by rifampicin and rifamycin-like molecules. Ecosal Plus. 2020; 9: 10.1128/ecosalplus.ESP-0017-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Boyaci H., Chen J., Lilic M., Palka M., Mooney R.A., Landick R., Darst S.A., Campbell E.A.. Fidaxomicin jams mycobacterium tuberculosis RNA polymerase motions needed for initiation via RbpA contacts. Elife. 2018; 7:e34823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lin W., Das K., Degen D., Mazumder A., Duchi D., Wang D., Ebright Y.W., Ebright R.Y., Sineva E., Gigliotti M.et al.. Structural basis of transcription inhibition by Fidaxomicin (Lipiarmycin A3). Mol. Cell. 2018; 70:60–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cao X., Boyaci H., Chen J., Bao Y., Landick R., Campbell E.A.. Basis of narrow-spectrum activity of fidaxomicin on Clostridioides difficile. Nature. 2022; 604:541–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Campbell E.A., Korzheva N., Mustaev A., Murakami K., Nair S., Goldfarb A., Darst S.A.. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell. 2001; 104:901–912. [DOI] [PubMed] [Google Scholar]
- 76. Prusa J., Zhu D.X., Flynn A.J., Jensen D., Manzano A.R., Galburt E.A., Stallings C.L.. Molecular dissection of RbpA-mediated regulation of fidaxomicin sensitivity in mycobacteria. J. Biol. Chem. 2022; 298:101752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hsu L.M. Monitoring abortive initiation. Methods. 2009; 47:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. McClure W.R., Cech C.L., Johnston D.E.. A steady state assay for the RNA polymerase initiation reaction. J Biological Chem. 1978; 253:8941–8948. [PubMed] [Google Scholar]
- 79. McClure W.R. Rate-limiting steps in RNA chain initiation. Proc Natl. Acad. Sci. U.S.A. 1980; 77:5634–5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Schlageck J.G., Baughman M., Yarbrough L.R.. Spectroscopic techniques for study of phosphodiester bond formation by Escherichia coli RNA polymerase. J Biol. Chem. 1979; 254:12074–12077. [PubMed] [Google Scholar]
- 81. Bertrand-Burggraf E., Lefèvre J.F., Daune M.. A new experimental approach for studying the association between RNA polymerase and the tet promoter of pBR322. Nucleic Acids Res. 1984; 12:1697–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hoggett J.G., Brierley I.. Kinetics of activation of the P4 promoter of pBR322 by the Escherichia coli cyclic AMP receptor protein. Biochem. J. 1992; 287:937–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Suh W.C., Leirmo S., Record M.T.. Roles of magnesium in the mechanism of formation and dissociation of open complexes between Escherichia coli RNA polymerase and the .Lambda.PR promoter: kinetic evidence for a second open complex requiring magnesium. Biochemistry-US. 1992; 31:7815–7825. [DOI] [PubMed] [Google Scholar]
- 84. Heyduk E., Heyduk T.. DNA template sequence control of bacterial RNA polymerase escape from the promoter. Nucleic Acids Res. 2018; 46:4469–4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hsu L.M., Cobb I.M., Ozmore J.R., Khoo M., Nahm G., Xia L., Bao Y., Ahn C.. Initial transcribed sequence mutations specifically affect promoter escape properties. Biochemistry-US. 2006; 45:8841–8854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Niemeyer H., Cárdenas M., de la L., Rabajille E., Ureta T., Clark-Turri L., Peñaranda J.. Sigmoidal kinetics of glucokinase. Enzyme. 1975; 20:321–333. [DOI] [PubMed] [Google Scholar]
- 87. Cárdenas M.L. Michaelis and Menten and the long road to the discovery of cooperativity. FEBS Lett. 2013; 587:2767–2771. [DOI] [PubMed] [Google Scholar]
- 88. Rabin B.R. Co-operative effects in enzyme catalysis: a possible kinetic model based on substrate-induced conformation isomerization. Biochem. J. 1967; 102:22C–23C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Heyduk E., Heyduk T.. Next generation sequencing-based parallel analysis of melting kinetics of 4096 variants of a bacterial promoter. Biochemistry-US. 2014; 53:282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Sanchez-Vazquez P., Dewey C.N., Kitten N., Ross W., Gourse R.L.. Genome-wide effects on Escherichia coli transcription from ppGpp binding to its two sites on RNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:8310–8319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. McClure W.R., Cech C.L.. On the mechanism of rifampicin inhibition of RNA synthesis. J. Biol. Chem. 1978; 253:8949–8956. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MATLAB code used for automated linear regression analyses has been made publicly available and can be accessed with the following GitHub link (https://github.com/egalburt/aptamer-flux-fitting).
The MATLAB code used for automated linear regression analyses has been made publicly available and can be accessed via the following link: https://doi.org/10.5281/zenodo.8319359.