

Abstract
Since the original description of amyloid-β plaques and tau tangles more than 100 years ago, these lesions are considered the neuropathological hallmarks of Alzheimer disease (AD). The prevalence of plaques, tangles and dementia increases with age, and the lesions are considered to be causally related to the cognitive symptoms of AD. Current schemes for assessing AD lesion burden examine the distribution, abundance and characteristics of plaques and tangles at post-mortem, yielding an estimate of the likelihood of cognitive impairment. Although this is highly predictive for most individuals, in some instances a striking mismatch between lesions and symptoms can be observed. A small subset of individuals harbour a high burden of plaques and tangles at autopsy, which would be expected to have had devastating clinical consequences, but remain at their cognitive baseline, indicating ‘resilience’. The study of these brains might provide the key to understanding the ‘black box’ between accumulation of plaques and tangles and cognitive impairment, and show the way towards disease-modifying treatments for AD. In this Review, we begin by considering the heterogeneity of clinical manifestations associated with the presence of plaques and tangles, and then focus on insights derived from the rare yet informative individuals who display high amounts of amyloid and tau deposition in their brains (observed directly at autopsy) without manifesting dementia during life. The resilient response of these individuals to the gradual accumulation of plaques and tangles has potential implications for assessing an individual’s risk of AD and for the development of interventions aimed at preserving cognition.
Introduction
A plethora of autopsy studies have convincingly shown that the frequency of both amyloid-β (Aβ) plaques and tau neurofibrillary tangles — the classic neuropathological hallmarks of Alzheimer disease (AD) — increases with age, as does the prevalence of dementia1. Currently, the dominant view is that these pathological changes silently accumulate in the brain over years or decades before the development of clinical symptoms of AD2–4. However, the definition of AD exclusively through plaques and tangles, regardless of the presence or absence of clinical symptoms of dementia, remains problematic. Plaques and tangles are objective and quantifiable pathological findings that can, but do not always, correlate with changes in cognitive function at individual level. Increasing evidence from cohort studies of ageing and dementia suggests that not everyone who harbors plaques and tangles in the brain at post-mortem had clinical symptoms of the disease during life.
For example, in the Nun Study cohort, 12% of participants with intact cognition at time of death had abundant Aβ plaques and neurofibrillary tangles at post-mortem examination5. Consistent with these observations, in the Religious Orders Study and the Memory and Aging Project, one-third of brains from people who were aged 80 years or older and without cognitive impairment contained enough AD lesions to meet pathological criteria for AD6. The 90+ Study was a population-based study designed specifically to study ageing and dementia, as well as their neuropathological correlates, in participants 90 years of age and older. In this study, 10% of individuals without ante-mortem dementia had a high probability of AD when assessed at autopsy using NIA-Reagan neuropathological criteria (the criteria used at the time of the study)7. Therefore, some individuals seem to tolerate the full burden of plaques and tangles without developing dementia during their lifetime.
This phenomenon of absence of overt cognitive deterioration in the face of neuropathological lesions that would be expected to have substantial clinical consequences has been termed ‘resilience’ to AD pathology (and we refer to the tissue from such individuals as ‘resilient brains’). Over the past several decades, this phenomenon has been recognized in various settings, and given a variety of other terms, including “high pathology controls”8, “AD-Resilient”9 and “Non-Demented with Alzheimer’s Neuropathology (NDAN)”10. We prefer the broader term “resilience” because it makes no assumptions about the mechanism(s) that are responsible for the cognitive impairment that is typically present in individuals with a high burden of neuropathological lesions.
When considering biological phenomena such as cognitive function, we might expect to observe a continuous (typically normal) distribution of scores. Resilient individuals with a high burden of plaques and tangles are outliers on the distribution of cognitive function when neuropathological lesions of all sorts are controlled for — that is, these individuals constitute are a distinct peak of healthy cognitive performance while their peers in terms of plaque and tangle burden fall in a separate distribution close to the other (more impaired) end of the scale11. This separation indicates that, either these resilient individuals have an intrinsic capacity to tolerate the presence of the lesions without developing the expected neuronal and synaptic derangement, or they respond in a distinct manner that protects cognition. Just as the relatively rare but genetically determined forms of AD highlighted important pathways in disease pathogenesis12studying these rare resilient brains might help us understand more fully the link between plaques, tangles and cognition as well as revealing clues for the development of interventions to halt neuronal and synaptic damage and subsequent cognitive impairment in older adults.
In this Review, we explore from a molecular perspective what is known about how resilient individuals, identified at autopsy, might have escaped the typical consequences of AD neuropathological lesions. By considering existing data on the pathways that normally result in neuronal dysfunction as the substrate for altered cognition in AD, we hope to highlight critical pathways that might differ in resilient brains, allowing preserved cognition.
How do lesions relate to cognition?
Extensive post-mortem analyses on individuals from 1 to 100 years of age have shown that classic neurofibrillary tangle pathology can appear in the entorhinal cortex as early as the third decade of life and is followed by the development of plaques in the neocortex in the fourth decade1. In agreement with these observations, brain amyloid can be detected by in vivo neuroimaging some 15 years before the mean age of symptom onset in members of Colombian families who carry the E280A mutation in PSEN1, which causes early onset familial AD3. Similarly, serial amyloid neuroimaging studies that used PiB PET and were conducted in cognitively healthy individuals estimated that amyloid plaque deposition takes about 15 years to build up before it reaches a plateau13, and found more than 20% of clinically unimpaired individuals over 65 to be amyloid positive14–17. These findings have helped to shape the concept of a ‘preclinical’ stage of AD; individuals are considered to have preclinical AD if they are clinically asymptomatic but have neuropathological or biomarker evidence of amyloid and tau lesions18. However, data from multiple clinical–pathological correlation studies (discussed in more detail below) spanning the clinical spectrum of AD have shown that relationships between plaques, tangles and cognition are not very strong and are not sufficient to predict future decline in function on an individual basis.
Plaques
In multiple studies, neither the regional distribution nor the overall burden of amyloid plaques correlated well with the severity of ante-mortem cognitive decline or the amount of neuronal cell death and synaptic loss quantified at autopsy in individuals with symptomatic AD19–22. The existence of autosomal-dominant forms of AD that result from mutations in the genes encoding amyloid precursor protein (APP), presenillin 1 and presenillin 2 highlight the crucial role of Aβ in the initiation of AD pathogenesis (presenillin 1 and 2 are involved in the cleavage processes that result in Aβ23). However, high levels of expression of mutant human amyloid precursor protein (APP) in mice result in cerebral amyloid deposits and cognitive impairment to some extent, but these deposits are not sufficient to trigger tangle formation — even though mouse tau is able to form paired helical filaments in vitro24 — or robust neuronal loss25–28.
Tangles
When compared with plaque and/or amyloid burden, the pattern of neurofibrillary tangle distribution in AD matches the pattern of brain regional atrophy more faithfully, and the number of neurofibrillary tangles tends to correlate better than the number of plaques with the severity and duration of clinical symptoms19,20. However, the results of quantitative studies suggest that some neuronal populations are vulnerable to cell loss in the presence of only limited tangle formation29. Even in areas that develop abundant tangles, the vast majority of the neuronal loss in individuals with clinically manifest AD cannot be explained by the number of tangles present at autopsy20,30,31 — importantly, an estimated 9 out of 10 neurons lost do not bear a tangle. In mice, altered tau is required to mimic AD neuronal loss but tangles on their own do not seem sufficient to permanently disrupt cognitive function and, similarly to human AD, the extent of neuronal loss far exceeds tangle number32. Together, the above observations suggest that, even though plaques and tangles are likely to be upstream instigators, other downstream mechanisms are directly responsible for the brain tissue responses (for example, neuronal and synaptic loss) that ultimately result in functional changes and impaired cognition, recognized as the dementing disorder in AD.
The results of autopsy studies have shown that, among individuals whose brains display the full burden of lesions as assessed by the current neuropathological scheme for ADNC (stage A3B3C3)33, a clear continuum of cognitive function is no longer observed. The vast majority of such individuals have already reached the advanced clinical stages of the disease34–37, and resilient individuals, with robust loads of amyloid and tau pathology but without clinical AD, represent outliers11.
In vivo biomarkers
Imaging and fluid biomarkers can now be used to assess brain amyloid burden, tangle presence, and neurodegeneration during life, although the measures of neurodegeneration (for example, volume loss and cortical thinning)38–42 are less specific and not as well-validated as those of amyloid and tau. The connection between plaques, tangles and neurodegeneration has been captured in the A/T/N biomarker framework, which was developed for clinical research purposes43. This framework enables the categorization of individuals on the basis of the biomarker changes present — A corresponds to evidence of amyloid deposition, T to evidence of tau accumulation and N to evidence of neurodegeneration. Older adults who have biomarker evidence of amyloid- and tau-containing lesions, but are asymptomatic, have been identified, raising several questions15,16,44,45. First, will these individuals all inevitably develop clinically manifest AD if they live long enough? If so, over what time frame will symptoms develop? Second, will a subset of these individuals be able to tolerate the presence of plaques and tangles without developing dementia? If so, do such individuals have an absolute resilience? Or do they just respond more slowly to pathology than most individuals, preventing them from becoming symptomatic before death? Ongoing longitudinal studies that involve measurement of these biomarkers alongside cognitive assessment could provide answers to these questions. However, in this Review we focus on the results of autopsy-based studies, which provide richer insights into the type and distribution of pathology present. Combining autopsy studies with longitudinal biomarker studies will begin to address some of the selection and temporal biases inherent in autopsy-based studies and guide the development of additional in vivo biomarkers.
Challenges and confounders
Trying to understand the relationship between cognition and neuropathological lesions — including plaques, tangles, synaptic alterations, neuronal loss and changes in connections among brain regions — can be complicated by a range of variables. For example, the anatomical distribution of neuropathological lesions can differ among individuals, as is mostly commonly observed with the clinical presentation of posterior cortical atrophy46 or the hippocampal-sparing pattern of AD47. Although a range of genetic factors have been found to influence the risk of developing AD, with the ε4 allele of the apolipoprotein E gene (APOEε4) representing the strongest susceptibility gene for clinically manifest AD48, a separate set of genetic loci might affect the degree of brain structural changes and clinical outcome resulting from formation of plaques and tangles49. In another example, evidence suggests that there are distinct tau aggregate strains with different biological properties, including ability to induce aggregation of soluble tau and the intensity of phosphorylation, resulting in differences in the overall brain burden of tau aggregates and the aggressiveness of clinical progression50. In addition to these biological differences, lifestyle factors such as educational attainment51 and cognitive activity across the life span52, could also contribute to inter-individual variation in the processes that link plaques and tangles with cognitive decline — consistent with the cognitive reserve hypothesis.
This large variability in the biological response, as well as the complex relationship between anatomic burden of injury and clinical manifestation of dementia, complicates the search for a simple link between classical brain AD neuropathological changes and dementia. Such complexity is also likely to decrease the robustness and consistency of treatment effects observed in AD clinical trials on symptomatic patients, and remains an obstacle for the rational design of prevention trials in asymptomatic individuals who harbour plaques and tangles in their brains. Nevertheless, the unrecognized mechanisms and biological factors that link plaques and tangles with clinical dementia represent potential targets for disease-altering interventions.
Influence of co-morbidities
One of the key challenges of understanding the link between plaques and tangles and functional changes in the brain is the complicating effect of co-morbidities that also increase in prevalence with age. These co-morbidities include other neurodegenerative processes, such as those marked by inclusions of α-synuclein and TAR DNA-binding protein 43 (TDP43), and vascular brain lesions, for example, stroke and small vessel diseases such as arteriolar sclerosis and cerebral amyloid angiopathy (CAA)53–55. The results of clinical–pathological correlation analyses indicate that the presence of these co-morbidities lowers the threshold at which the classic AD neuropathological changes result in a diagnosis of dementia56,57. Thus, the presence of co-morbid disease processes is not required for plaques and tangles to result in dementia, but would be expected to increase the likelihood of dementia.
Lewy bodies are another common co-morbidity among individuals with plaques and tangles in their brain at autopsy, particularly in individuals who are <60 years of age when they die55. The frequency and significance of TDP43-immunoreactive protein aggregates and hippocampal sclerosis have also been increasingly recognized, particularly in individuals over 8056,58. The presence of TDP43-containing inclusions in the brain, independent of the associated hippocampal sclerosis, has been found to be an independent contributor to cognitive impairment at death in individuals with AD neuropathological changes59. In another study, the presence of both AD neuropathological changes and TDP43-containing inclusions was associated with a faster rate of cognitive decline than either pattern of disease when alone60.
Vascular brain injury resulting from disease processes of both large and small cerebral arteries has also been recognized as an important contributor to neuronal damage and impaired cognition in individuals with clinically manifest AD61,62. Several longitudinal community-based studies have identified both macroinfarcts and microinfarcts as important independent predictors of cognitive decline63–65, despite the underlying pathophysiological mechanisms remaining unclear. In a study by Patricia Boyle and colleagues, described in more detail below, the presence of CAA was associated with an increased risk of cognitive impairment even after taking into account plaque and tangle burden and the presence of other common age-related neurodegenerative lesions61. This finding further highlights the important contribution of vascular brain injury to symptom burden in individuals with AD dementia.
In a large clinical–pathological study in 1,161 participants who came to autopsy at an average of 90 years of age, Boyle and colleagues examined the associations of eight common age-related neuropathological indices with clinically manifest AD and quantified the percentage of cases attributable to each of those indices61. In only 41% of participants was the diagnosis of AD dementia attributable to AD neuropathological changes. TDP43-containing lesions (11.7%), LBD (10.8%), macroinfarcts (8.9%), CAA (8.1%), atherosclerosis (6.0%), hippocampal sclerosis (5.2%), and arteriolosclerosis (5.2%) also significantly contributed to clinical disease expression. Importantly, after adjusting for other factors, in only 67.5% of participants was the dementia diagnosis determined to be attributable to all eight neuropathological indices combined, suggesting that other factors are also important in the heterogeneous clinical expression of the otherwise common neuropathological phenotype of plaques and tangles. Evidence from the 90+ study indicates that, in comparison with the participants in the study by Boyle and colleagues, the burden of chronic systemic conditions in individuals over 90 years of age increased, and the relationship between plaques, tangles and cognition is further attenuated, even after adjusting for the presence of other common neuropathological processes7.
Putative mechanisms of resilience
Data indicate that, even after excluding individuals with substantial brain co-morbid processes and those who die from other diseases before developing symptoms of AD, some people have high amounts of amyloid and tau deposition in their brains (observed directly at autopsy) without manifesting dementia5–8,66. Literature concerning these unique resilient brains is scarce, and several potential caveats need to be taken into consideration when studying these brains. The most parsimonious explanation for the absence of dementia in these apparently resilient individuals would be that their overall plaque and tangle burdens are substantially lower than in individuals with clinically manifest AD. Alternatively, or in addition, a subtle cognitive decline from premorbid baseline before death might have not been detected in some of these apparently resilient individuals — although standard cognitive testing is a useful screening tool when cognitive impairment is overt, it is imperfect and insensitive to more modest changes in cognitive function67,68. Nevertheless, meaningful impairment in brain function very rarely goes undetected either by the affected individuals, their families or cognitive testing, and the presence of a full burden of AD lesions (stage A3B3C3 in current criteria) is expected to be associated with robust cognitive deficits sufficient to meet clinical criteria for dementia. Therefore, brain function of resilient individuals who exhibit very high loads of plaques and tangles at autopsy without meeting ante-mortem criteria for dementia can be considered, being conservative, much better preserved than in individuals with typical AD dementia.
Neuronal and synaptic loss
In a study published in 1996, Lih-Fen Lue and colleagues evaluated synapse integrity in the entorhinal cortex and the superior frontal gyrus of elderly individuals without dementia, but with elevated amyloid and tau deposits measured at autopsy (referred to here as resilient participants, in keeping with the terminology used elsewhere in this Review). Also included in the study were age-matched and gender-matched participants with AD dementia and control participants with negligible AD neuropathological changes and no dementia8. Although the average number of plaques did not differ between the resilient and AD groups, the number of neurofibrillary tangles in the superior frontal gyrus was substantially lower in the former group. Consistent with their lack of dementia and in contrast to the AD group, participants in the resilient group had no evidence of synapse loss in the entorhinal cortex and the superior frontal gyrus, or brain weight reduction, when compared with the control participants. Indeed, participants in the resilient group tended to have higher levels of synaptophysin immunoreactivity, as detected with Western Blot, and had significantly higher brain weights than participants in the control group. Consistent with these observations, a significant nuclear hypertrophy in the anterior cingulate gyrus and CA1 hippocampal neurons was observed in participants with asymptomatic AD neuropathological changes from the Baltimore Longitudinal Study of Aging69. Similarly, a significant hypertrophy of neuronal cell bodies, nuclei and nucleoli was reported in the cortex and the CA1 region in clinically asymptomatic participants with AD pathology from the Nun Study, and it was suggested that this could represent an early brain reaction to the presence of plaques and tangles, or a compensatory mechanism that prevents clinical progression to dementia70,71.
In a study published in 2013, Beatriz Perez-Nievas and colleagues conducted detailed quantitative neuropathological and biochemical assessments on post-mortem brain samples containing the banks of the superior temporal sulcus66. The samples came from three groups of age-matched individuals: those who had neither dementia before death nor substantial AD lesion burden at post-mortem examination; those without dementia but with abundant AD lesions (resilient participants); and those with AD lesions and dementia. Importantly, the group of resilient participants and the group of participants with clinically manifest AD were also carefully matched for burden and regional distribution of plaques and tangles; however, the effects of amyloid and tau lesions on the brain were very different in the two groups. Brains from participants with clinically manifest AD displayed the expected signs of neurodegeneration, including profound loss of neurons and synaptic markers and consequent reduction in cortical thickness. In contrast, the number of neurons, levels of pre-synaptic and post-synaptic markers and the thickness of the cortex in brains from resilient individuals were indistinguishable from age-matched controls free of AD neuropathological changes66. Similarly, in another study, neuronal densities and pre-synaptic and post-synaptic elements in the midfrontal gyrus were preserved in cognitively healthy individuals with high burden of AD pathology at autopsy when compared with individuals with clinically manifest AD9.
Studies in the brains of individuals with AD and in transgenic mice have identified widespread alterations in the geometry of dendrites and axons, including disrupted trajectories and dystrophic swellings, that occur both near plaques and distant from them72–75. These changes, along with neuronal and synaptic loss, are likely to contribute to altered neural system function and cognitive impairment in AD. Perez-Nievas and colleagues observed that the trajectory and morphology of neurites were much better preserved in the brains of resilient individuals than in the brains of individuals with clinically manifest AD — axons close to plaques were significantly less distorted, axons far from plaques had normal straight trajectories indistinguishable from control participants, and the number of dystrophic neurites associated with amyloid plaques was substantially reduced66.
These data favour the idea that preservation of neurons and synapses in the setting of plaques and tangles can be used to identify resilient brains, and argue further against a direct causal relationship of plaques and tangles to loss of anatomical integrity of neurons and cognitive symptoms in AD. Unrecognized downstream mechanisms and biological factors, beyond plaques and tangles, are likely to be more directly involved in the brain tissue responses and the clinical expression and progression of the disease; and this is a factor that might have relevant implications for the future development of fluid and neuroimaging biomarkers predictive of clinical outcomes. When considering individuals who harbour amyloid and tau lesions in their brain, in vivo markers of neuronal cell death and synaptic loss might predict more accurately than just measures of plaques and tangles who (and also when, if ever during their lifetime), is most likely to develop cognitive impairment.
Soluble Aβ and tau
Investigating phenotypic divergences between the brains of resilient individuals and those with clinically manifest AD might provide information about the biological factors and mechanisms resulting in their opposite ante-mortem clinical outcomes. Therefore, we and others have studied in more detail the profiles of potentially neurotoxic soluble Aβ and tau species in these brains. Soluble Aβ, and specifically soluble oligomeric forms of Aβ, has been proposed to have a larger role in disrupting cognitive function in AD than Aβ plaques themselves76. Studies in wild-type rodents showed that Aβ oligomers, including dimers and trimers, were sufficient to disrupt synaptic function and integrity and to impair memory77–83. In another study, blocking de novo production of Aβ oligomers reversed synapse loss and memory impairment in APP transgenic mice in the absence of changes in plaque load84. In the brains of individuals with clinically symptomatic AD, Aβ oligomers (measured at autopsy) were present at higher levels than in brains from individuals without plaques and tangles, were associated with synapses, and correlated with antemortem measures of cognition85–87. Studies in tau transgenic mice also identified potentially neurotoxic soluble tau species, the levels of which correlated with memory loss and neuronal cell loss in the absence of tangles88,89. Injection of tau oligomers in the brain of wild-type mice was sufficient to impair memory and to induce synaptic and mitochondrial dysfunction90.
We and others have also investigated potential differences in the Aβ phenotype — that is, the combination of Aβ plaque subpopulations and/or soluble Aβ species — present in brains from individuals with clinically manifest AD and resilient individuals8,66. Neither the total number of plaques (diffuse and dense cored), nor the number of Thioflavin-S-positive dense cored plaques (a subset considered to be more deleterious than diffuse plaques)8,66 or the levels of different soluble Aβ assemblies (monomers, dimers and oligomeric species) could differentiate resilient brains from clinically manifest AD brains66. However, one study reported an absence of Aβ oligomers in post-synaptic terminals of the hippocampus in brains from individuals with NDAN87.
In addition to its well-described axonal localization, normal soluble tau is also present at both pre-synaptic and post-synaptic terminals in the brains of healthy individuals. In the brains of individuals with AD, this synaptic tau becomes hyperphosphorylated and ubiquitinated, and forms stable multimeric assemblies resistant to denaturation91. In agreement with those observations, Perez-Nievas and colleagues observed a robust aberrant accumulation of soluble monomeric and multimeric species of hyperphosphorylated tau within the synaptic compartment in the brains of individuals with clinically manifest AD66. Those species, however, were only present in very small quantities or absent in the synapses of brains from resilient individuals, despite the presence of similar tangle burdens66. This observation suggests that soluble pathological species of tau are more directly involved than tangles in the neuronal cell death and synaptic loss that results in clinically manifest AD. Similarly, in another study, the level of tau oligomers at synapses was lower in the brains of individuals with full burden of AD lesions and no dementia than in individuals with clinically manifest AD92. Electrophysiological recording of micro-transplanted synaptic membranes prepared from the postmortem samples indicated that, compared with individuals with clinically manifest AD, the absence of tau oligomers at synapses in resilient individuals was associated with greater synaptic functional integrity92.
Increasing evidence from in vitro and in vivo studies in mouse models of tauopathy supports a role for soluble pathological tau assemblies in several pathways that lead to synapse and neuron death93. These pathways include altered microtubule-based axonal transport with subsequent impaired trafficking of organelles, particularly mitochondria, and dysregulation of calcium homeostasis in dendrites93–95. Furthermore, the results of studies in mice suggest that these soluble species of tau can be transmitted between neurons and contribute to or are responsible for the pathological spread of disease through the brain96,97. Importantly, in vivo multiphoton imaging studies conducted in tau transgenic mice showed that caspase activation cleaves tau to initiate tangle formation but, surprisingly, that tangle-bearing neurons are long-lived98 and adequately respond to physiologically relevant stimuli without impairing local circuits99.
These unexpected results in mouse models, along with the observed lack of aberrant accrual of soluble hyperphosphorylated tau in the synapses of resilient individuals, further suggest that tangles themselves do not inevitably lead to gross impairment of brain function and are not likely to be part of the pathway to acute neuronal death and synaptic loss. Taken together, these insights favour the idea that accumulation of mistargeted hyperphosphorylated soluble tau assemblies within the synaptic compartment, rather than the formation of tangles, might be the critical toxic moiety leading to neurodegeneration (for example, loss of neurons and synapses) and clinical expression of AD (Fig. 1).
Figure 1 |. Putative role of tau and microglia in resilience.
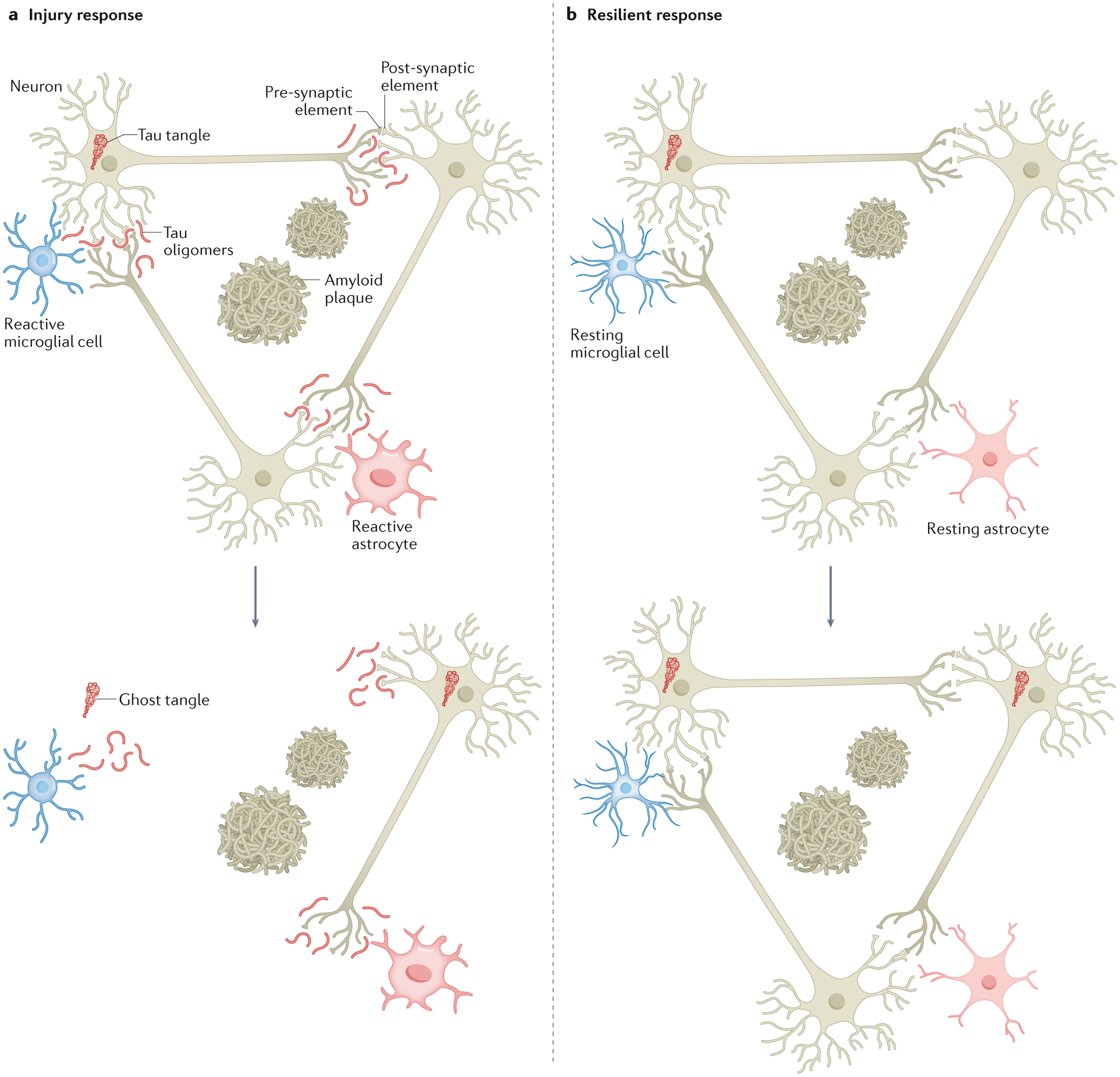
a | In individuals with Alzheimer disease (AD), tau, which is largely bound to axonal microtubules in normal conditions, detaches from microtubules, becomes hyperphosphorylated and misfolded, and coalesces into neurofibrillary tangles in the neuronal soma. In individuals with clinically manifest AD, soluble hyperphosphorylated tau species are mis-targeted and preferentially accumulate within the synaptic compartment in parallel with microglial cell activation. Over time, these changes are associated with the most frequently observed tissue injury response in AD brains, that is, neuronal cell death and synaptic loss. b | In the brains of ‘resilient’ individuals, a negligible accumulation of soluble hyperphosphorylated tau in synapses and a suppressed inflammatory brain response have been observed. These features are associated with the preservation of neurons and synapses amid a burden of plaques and tangles equal to that observed in the brains of individuals with clinically manifest AD. This differential tissue response to the progressive accumulation of plaques and tangles in individuals with clinically manifest AD and resilient individuals is likely to contribute to their widely divergent clinical phenotypes.
Glia and neuroinflammation
Activation of astrocytes and microglia — accompanied by release of cytokines — is consistently present in the brains of individuals with AD and transgenic mouse models of the disease, and is temporally and anatomically associated with plaques and tangles100,101. Brain glial responses are present in proximity to plaques and tangles, beginning early in AD and increasing as the lesion burden grows with disease progression102. However, whether the aberrant activation of glial elements in AD contributes to neuronal damage and loss of brain function or reflects a mere response to neuronal injury remains controversial.
Lue and colleagues reported a robust increase in the C5b–9 membrane attack complex of the complement cascade and activated LN3+ microglia in the brains of individuals with AD dementia compared with control participants, but a reduction of these markers of inflammation in resilient participants8. Of note, overall levels of C5b-9 immunoreactivity and activated LN3+ microglia correlated much better than plaques and tangles with measures of synapse loss. In agreement with those observations, stereologically-based assessments of glial cell responses in the brains of individuals with clinically manifest AD identified a robust and significant increase in the numbers of activated GFAP+ astrocytes and CD68+ microglial cells in comparison with control participants without AD lesions102,103. However, in the study by Perez-Nievas, this neuroinflammatory response was suppressed in the brains of resilient individuals66.
Interestingly, in one study, this reduction glial activation in brains of resilient individuals was associated with a distinct cytokine expression profile in the entorhinal cortex and the superior temporal sulcus that distinguished those brains not only from the brains of participants with clinically manifest AD but also from the brains of control participants free of AD neuropathological lesions103. The differential profile observed in resilient brains included up-regulated expression of IL-6, IL-13 and IL-10 — all of which have known roles in pathogen clearance and/or the resolution of inflammation — and neurotrophic factors such as basic FGF and PDGF-bb. This diversity of cytokine expression profiles, which are the primary instruments of immune communication, suggests a complex and heterogeneous relationship between astrocytes and microglia, and the possible existence of a differential ‘protective’ cytokine signaling program in resilient brains. Such a signalling program might be capable of maintaining homeostatic regulation of neuroinflammation and neuronal and synaptic integrity despite the presence of plaques and tangles, thus modulating the degree of cognitive impairment. Intriguingly, the peripheral expression of an innate pro-inflammatory cytokine profile in middle-aged individuals with a parental history of late-onset AD was identified as a risk factor for clinically manifest late-onset AD104, suggesting that crosstalk between the peripheral and the local brain immune system in early years can contribute to the development of AD pathology and its clinical expression in later life (reviewed in105).
Studies have now identified a diverse heterogeneity of populations of astrocytes and microglial cells with as yet incompletely understood roles in the clinical expression of brain responses to plaques and tangles and other common neurodegenerative lesions106–110. Renewed interest in a better understanding of the role of glia and immune-related mechanisms in AD has also been fuelled by the identification of AD risk loci in genes related to the innate immune system, including CLU, PICALM, CR1, TREM2 and CD33111–116. Novel insights into the involvement of microglia in synaptic pruning and modulation of synaptic activity have also highlighted the importance of understanding the contribution of these mechanisms to the pathophysiology of AD117,118.
Studies in mice showed that during development, the classical complement cascade components C1q and C3 localize to synapses and have a crucial role in the elimination of synapses by phagocytic microglia117,118. In a more recent study, the aberrant activation of this complement-dependent developmental synaptic pruning pathway in young APP transgenic mice was associated with microglial engulfment of synaptic elements in advance of Aβ plaque deposition118. Importantly, in young APP mice free of Aβ plaques, inhibition of complement receptor 3 (CR3) or C1q was associated with a reduction in the number of phagocytic microglia and the prevention of synapse loss118. Similarly, inhibition of C1q was associated with evidence of reduced microglial engulfment of synapses in Tau-P301S mice119. These findings suggest that microglia are involved in early synapse loss in AD (reviewed in120). Indeed, the elimination of microglia in 5xfAD mice was associated with a reversal of dendritic spine loss, prevention of neuronal loss and improvement in contextual memory function, whereas Aβ burden was unaffected121. In agreement with these findings, pharmacologically driven reduction of astrocyte and microglial activation in APP mice was associated with the rescue of alterations in neurite geometry and morphology, and memory deficits without modification of total Aβ plaque burden122.
These novel insights derived from human resilient brains, asymptomatic at risk individuals and AD transgenic mice all favour a model in which the responses of glial cells, in particular microglia, and other immune-related mechanisms mediate the loss of neuronal and synaptic integrity and impaired brain function in the setting of plaques and tangles (Fig. 1).
Genetic and external factors
Well-established genetic risk factors contribute to the likelihood of an individual developing AD (both at the clinical and neuropathological levels)48,111–116, and various aspects of lifestyle and prior experiences have an influence on the risk of dementia123–125. The proposed disease-modifying factors, including early-life experiences, educational attainment, level of physical activity and ongoing participation in cognitive activities, would be expected to result in a continuum of clinical outcomes, given that these factors exist on a continuum in the population. However, the resilient individuals that are the focus of this Review are outliers in terms of clinical outcome and are not part of a normal distribution. Shaping of the brain’s response to plaque and tangle accumulation either by genetic determinants or by life experiences is clearly possible but, to date, the relatively small proportion of resilient individuals and the fact that such individuals are identified only at autopsy has precluded the investigation of this possibility.
Conclusions and future directions
The range of biomarkers indicative of the presence of plaques and tangles in the brain is rapidly changing how we approach the diagnosis of AD and research into possible interventions. In 2018, a National Institute on Aging and Alzheimer’s Association (NIA-AA) workgroup proposed a research framework in which AD is defined by neuropathological or biomarker evidence of amyloid and tau lesions, regardless of the presence or absence of clinical symptoms126. The hypothetical benefit of this framework is to help the identification of asymptomatic individuals with plaques and tangles in their brains, with the aim of intervening before irreversible damage has occurred. However, the framework relies on two assumptions that, to date, have not been unequivocally proven. The first assumption is that plaques and tangles are causally related to the cognitive symptoms in AD. The second assumption is that all individuals who harbour amyloid plaques and neurofibrillary tangles in their brain will develop dementia given enough time. When faced with a similar difficulty of relating neuropathological findings to symptomatic disease, the neuropathology community opted for the terminology of “Alzheimer disease neuropathological changes, [ADNC]” because of the expected mismatch between lesions and symptoms, as is the case in many other diseases33,127.
In the absence of in vivo measures that can estimate how far an individual might be from entering the symptomatic phase of AD, the high prevalence of age-associated biomarker findings of amyloid and tau abnormalities among asymptomatic individuals128 can be confusing and troubling to patients, families and physicians. The observations discussed in this Review, suggest that additional pathological and biochemical brain changes — beyond plaques and tangles — might more accurately predict the presence of neuronal and synaptic derangement and the future outcome of an individual who is cognitively healthy but harbours amyloid and tau pathology in the brain. These additional changes include aberrant mis-targeting of soluble assemblies of hyperphosphorylated tau in synapses and activation of microglia with distinct cytokine expression profiles. The development of surrogate in vivo markers capable of detecting such changes has the potential to more precisely identify individuals who will develop clinical symptoms of dementia and over what time frame; this ability would greatly enhance the power of secondary prevention trials.
Over the last decade, the therapeutic interventions trialled in AD have focused mainly on amyloid-related pathological changes — either clearing deposits or interrupting pathways which generate Aβ. After many unsuccessful trials129–132, two newer agents — aducanumab and donanemab — were found to clear amyloid deposits, as reflected in PET imaging data from phase III trials133,134. However, these trials did not report a robust effect on cognition. Given the evidence from the studies of resilience discussed in this Review, it might be that better therapeutic targets will be found in the pathways that link the protein aggregation and deposition processes for tau and Aβ to cellular injury. Indeed, an optimal therapeutic approach to AD might be to induce the resilient pattern of response to the gradual accumulation of plaques and tangles.
Increasing evidence suggests that many other age-related pathological processes, beyond plaques and tangles, as well as heterogeneous individual profiles of risk and protective factors substantially contribute to both the current cognitive function of individuals as well as the trajectory of any future cognitive decline. Even after excluding individuals with significant brain concomitant neurodegenerative and vascular lesions, and carefully matching burdens and regional distribution of plaques and tangles, several studies have convincingly demonstrated that some individuals who exhibit robust amounts of amyloid and tau deposition in their brains at autopsy never manifested dementia during life5–9,66. The study of these resilient brains gives us the opportunity to understand the pathological and biochemical brain changes as well as individual genetic and epigenetic factors that ultimately drive neurodegeneration (for example, loss of neuronal and synaptic integrity) and change from baseline function in the face of plaques and tangles. Identifying such changes will likely facilitate development of novel therapies that can stop cognitive impairment, identification of in vivo biomarkers that can more accurately predict the future of asymptomatic individuals who harbour plaques and tangles in their brain, and guidance on the need and optimal timing for personalized preventive intervention. The string of disappointing results from amyloid-targeting drug trials indicate that it might be time to rethink our approach to AD and diversify the drug search. The existence of subset of individuals who seem to be able to successfully cope with plaques and tangles in their brain during their lifetime also reminds us that, at least in some cases, we may not need to fix what it is not broken.
Key points.
Plaques of amyloid-β (Aβ) and tangles containing hyperphosphorylated tau accumulate in the brain over time as part of the Alzheimer disease process; as the lesion burden increases, most people become cognitively impaired.
A subset of individuals are identified at autopsy as having a lesion burden that would be expected to have caused cognitive impairment during life, yet they remain clinically unaffected; this dissociation between lesions and symptoms is termed resilience.
Biomarker studies are identifying similar mismatch between lesions and cognition in some people, although more prospective longitudinal data will need to be collected to determine the clinical trajectory of such individuals.
Various mechanisms link the deposition of Aβ and tau to neuronal and synaptic loss, and it remains uncertain which of these is most associated with resilience; however, differences in lesion-associated immune response and properties of soluble tau aggregates are both likely to be contributors.
Understanding resilience could provide insights into key mechanisms of brain injury in Alzheimer disease, and identify new therapeutic opportunities.
Glossary
- C5b–9 membrane attack complex
terminal components of the complement cascade
- APP transgenic mice
transgenic mice that have been engineered to over-express disease-associated mutant forms of human amyloid precursor protein and develop elevated levels of amyloid-β in the brain
- Tau-P301S mice
transgenic mice that are engineered to over-express disease-associated mutant forms of human tau and develop abnormally phosphorylated tau and tau aggregates
- 5xfAD mice
transgenic mice harbouring mutant forms of human APP, PSEN1 and MAPT.
Footnotes
Competing interests
T. G.-I. has participated as speaker in an Eli Lilly sponsored educational symposium and serves as member of an Eli Lilly Data Monitoring Committee (DMC). M. P. F. declares no competing interests.
References
- 1.Braak H, Thal D, Ghebremedhin E & Tredici K. Del. Stages of the Pathologic Process in Alzheimer Disease: Age Categories From 1 to 100 Years. J. Neuropathol. Exp. Neurol 70, 960–969 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Landau S et al. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann. Neurol 72, 578–86 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bateman R et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med 367, 795–804 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McDade E et al. Longitudinal cognitive and biomarker changes in dominantly inherited Alzheimer disease. Neurology 91, e1295–e1306 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Riley KP, Snowdon D, Desrosiers M & Markesbery W Early life linguistic ability, late life cognitive function, and neuropathology: findings from the Nun Study. Neurobiol. Aging 26, 341–347 (2005). [DOI] [PubMed] [Google Scholar]; This was was one of the first studies to show a dissociation between Alzheimer disease neuropathological lesions and symptoms; up to 12% of participants with intact cognition at time of death had abundant amyloid-β plaques and neurofibrillary tangles at post-mortem examination.
- 6.Schneider J, Aggarwal N, Barnes L, Boyle P & Bennett D The neuropathology of older persons with and without dementia from community versus clinic cohorts. J. Alzheimers. Dis 18, 691–701 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; This autopsy study in participants from two community-based cohorts and one clinic-based cohort showed that one-third of brains from people aged 80 years or older without cognitive impairment contained enough AD lesions to meet pathological criteria for Alzheimer disease.
- 7.Corrada M, Berlau DJ & Kawas C A population-based clinicopathological study in the oldest-old: the 90+ study. Curr. Alzheimer Res 9, 709–717 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This population-based study in participants 90 years of age and older showed that 10% of those without ante-mortem dementia met neuropathological criteria for high probability of Alzheimer disease.
- 8.Lue L, Brachova L, Civin WH & Rogers J Inflammation, A beta deposition, and neurofibrillary tangle formation as correlates of Alzheimer’s disease neurodegeneration. J. Neuropathol. Exp. Neurol 55, 1083–1088 (1996). [PubMed] [Google Scholar]; This post-mortem study was the first to report lower levels of microglial activation in the brains of resilient individuals than individuals with AD dementia, and found that microglial activation correlated more closely with measures of synapse loss than did levels of plaques and tangles.
- 9.Arnold S, Louneva N, Cao K, Wang LS & Bennett D Cellular, synaptic, and biochemical features of resilient cognition in Alzheimer’s disease. Neurobiol. Aging 34, 157–168 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zolochevska O, Bjorklund N, Woltjer R, Wiktorowicz JE & Taglialatela G Postsynaptic Proteome of Non-Demented Individuals with Alzheimer’s Disease Neuropathology. J. Alzheimers. Dis 65, 659–682 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.SantaCruz KS et al. Alzheimer disease pathology in subjects without dementia in 2 studies of aging: the Nun Study and the Adult Changes in Thought Study. J. Neuropathol. Exp. Neurol 70, 832–840 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singleton A & Hardy J The Evolution of Genetics: Alzheimer’s and Parkinson’s Diseases. Neuron 90, 1154–1163 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jack CR et al. Brain β-amyloid load approaches a plateau. Neurology 80, 890–896 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lopresti B et al. Simplified quantification of Pittsburgh Compound B amyloid imaging PET studies: a comparative analysis. J. Nucl. Med 46, 1959–1972 (2005). [PubMed] [Google Scholar]
- 15.Mintun MA et al. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology 67, 446–452 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Rowe CC et al. Imaging beta-amyloid burden in aging and dementia. Neurology 68, 1718–1725 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Aizenstein H et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch. Neurol 65, 1509–1517 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dubois B, Hampel H, Feldman H, Scheltens P & of the Meeting of the International Wo, 2015 Washington D C U S A Proceedings. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimer’s Dement. 12, 292–323 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arriagada P, Marzloff K & Hyman B Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer’s disease. Neurology 42, 1681–8 (1992). [DOI] [PubMed] [Google Scholar]
- 20.Gómez-Isla T et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann. Neurol 41, 17–24 (1997). [DOI] [PubMed] [Google Scholar]; This milestone paper showed that, although the number of tangles correlates much better with loss of neurons in Alzheimer brains than does the number of plaques, the amount of neuronal loss exceeds tangle formation by an order of magnitude.
- 21.Terry R Cell death or synaptic loss in Alzheimer disease. J. Neuropathol. Exp. Neurol 59, 1118–1119 (2000). [DOI] [PubMed] [Google Scholar]
- 22.Scheff S & Price D Synapse loss in the temporal lobe in Alzheimer’s disease. Ann. Neurol 33, 190–9 (1993). [DOI] [PubMed] [Google Scholar]
- 23.Lane C, Hardy J & Schott J Alzheimer’s disease. Eur. J. Neurol 25, 59–70 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Kampers T, Pangalos M, Geerts H, Wiech H & Mandelkow E Assembly of paired helical filaments from mouse tau: implications for the neurofibrillary pathology in transgenic mouse models for Alzheimer’s disease. FEBS Lett. 451, 39–44 (1999). [DOI] [PubMed] [Google Scholar]
- 25.Irizarry M et al. Aβ Deposition Is Associated with Neuropil Changes, but not with Overt Neuronal Loss in the Human Amyloid Precursor Protein V717F (PDAPP) Transgenic Mouse. J. Neurosci 17, 7053–7059 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Irizarry MC, Fedorchak K, Hsiao K & Hyman B APPSW Transgenic Mice Develop Age-related Aβ Deposits and Neuropil Abnormalities, but no Neuronal Loss in CA1. J. Neuropathol. Exp. Neurol 56, 965–73 (1997). [DOI] [PubMed] [Google Scholar]
- 27.Calhoun M et al. Neuron loss in APP transgenic mice.Nature 395, 755–756 (1998). [DOI] [PubMed] [Google Scholar]
- 28.Urbanc B et al. Neurotoxic effects of thioflavin S-positive amyloid deposits in transgenic mice and Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A 99, 13990–13995 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bondareff W, Mountjoy C, Roth M & Hauser DL Neurofibrillary degeneration and neuronal loss in alzheimer’s disease. Neurobiol. Aging 10, 709–715 (1989). [DOI] [PubMed] [Google Scholar]
- 30.Vogt BA, Van Hoesen GW & Vogt LJ Laminar distribution of neuron degeneration in posterior cingulate cortex in Alzheimer’s disease. Acta Neuropathol. 80, 581–589 (1990). [DOI] [PubMed] [Google Scholar]
- 31.Gómez-Isla T et al. Profound Loss of Layer II Entorhinal Cortex Neurons Occurs in Very Mild Alzheimer’s Disease. J. Neurosci 16, 4491–4500 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.SantaCruz K et al. Tau Suppression in a Neurodegenerative Mouse Model Improves Memory Function. Science 309, 476–481 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hyman B, Phelps C, Beach T, Bigio E & Montine T National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s Dement. 8, 1–13 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nelson P et al. Correlation of Alzheimer Disease Neuropathologic Changes With Cognitive Status: A Review of the Literature. J. Neuropathol. Exp. Neurol 71, 362–81 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nelson PT et al. Modeling the association between 43 different clinical and pathological variables and the severity of cognitive impairment in a large autopsy cohort of elderly persons. Brain Pathol. 20, 66–79 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tiraboschi P, Hansen LA, Thal LJ & Corey-Bloom J The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology 62, 1984–1989 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Matthews FE et al. Epidemiological pathology of dementia: attributable-risks at death in the Medical Research Council Cognitive Function and Ageing Study. PLoS Med 6, e1000180 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Calvin CM, de Boer C, Raymont V, Gallacher J & Koychev I Prediction of Alzheimer’s disease biomarker status defined by the ‘ATN framework’ among cognitively healthy individuals: results from the EPAD longitudinal cohort study. Alzheimers. Res. Ther 12, 143 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rafii M et al. The AT(N) framework for Alzheimer’s disease in adults with Down syndrome. Alzheimer’s Dement. 12, e12062 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shen XN et al. Plasma amyloid, tau, and neurodegeneration biomarker profiles predict Alzheimer’s disease pathology and clinical progression in older adults without dementia. Alzheimer’s Dement. Diagnosis, Assess. Dis. Monit 12, e12104 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cousins K et al. ATN status in amnestic and non-amnestic Alzheimer’s disease and frontotemporal lobar degeneration. Brain 143, 2295–2311 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Harten AC et al. CSF biomarkers in Olmsted County: Evidence of 2 subclasses and associations with demographics. Neurology 95, e256–e267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jack C et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 87, 539–547 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lowe VJ et al. Widespread brain tau and its association with ageing, Braak stage and Alzheimer’s dementia. Brain 141, 271–287 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Biel D et al. Tau-PET and in vivo Braak-staging as prognostic markers of future cognitive decline in cognitively normal to demented individuals. Alzheimers. Res. Ther 13, 137 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Firth NC et al. Longitudinal neuroanatomical and cognitive progression of posterior cortical atrophy. Brain 142, 2082–2095 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murray M, Graff-Radford N, Ross OA, Petersen R & Dickson D Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. Lancet Neurol. 10, 785–796 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Naj AC & Schellenberg GD Genomic variants, genes, and pathways of Alzheimer’s disease: An overview. Am. J. Med. Genet. Part B, Neuropsychiatr. Genet. Off. Publ. Int. Soc. Psychiatr. Genet 174, 5–26 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dumitrescu L et al. Genetic variants and functional pathways associated with resilience to Alzheimer’s disease. Brain 143, 2561–2575 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dujardin S et al. Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease. Nat. Med 26, 1256–1263 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bennett DA et al. Education modifies the relation of AD pathology to level of cognitive function in older persons. Neurology 60, 1909–1915 (2003). [DOI] [PubMed] [Google Scholar]
- 52.Wilson RS et al. Life-span cognitive activity, neuropathologic burden, and cognitive aging. Neurology 81, 314–321 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kawas C et al. Multiple pathologies are common and related to dementia in the oldest-old. Neurology 85, 535–542 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Farfel J et al. Alzheimer’s disease frequency peaks in the tenth decade and is lower afterwards. Acta Neuropathol. Commun 7, 104 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beach T & Malek-Ahmadi M Alzheimer’s Disease Neuropathological Comorbidities are Common in the Younger-Old. J. Alzheimers. Dis 79, 389–400 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Robinson J et al. Non-Alzheimer’s contributions to dementia and cognitive resilience in The 90+ Study. Acta Neuropathol. 136, 377–388 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kapasi A, DeCarli C & Schneider J Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol. 134, 171–186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.James B et al. TDP-43 stage, mixed pathologies, and clinical Alzheimer’s-type dementia. Brain 139, 2983–2993 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Josephs K et al. TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol. 127, 811–824 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kapasi A et al. Limbic-predominant age-related TDP-43 encephalopathy, ADNC pathology, and cognitive decline in aging. Neurology 95, e1951–e1962 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boyle P et al. Attributable risk of Alzheimer’s dementia attributed to age-related neuropathologies. Ann. Neurol 85, 114–124 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This is one of the largest clinical-pathological correlation studies to show that just over two-thirds of clinically diagnosed cases of Alzheimer disease are attributable to classic Alzheimer neuropathological changes (e.g. plaques and tangles) and other common age-related neuropathologies, suggesting that other disease and resilience factors are important.
- 62.Sweeney MD, Montagne A, Sagare A, Nation D & Zlokovic B Vascular dysfunction-The disregarded partner of Alzheimer’s disease. Alzheimer’s Dement. 15, 158–167 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kapasi A & Schneider J Vascular contributions to cognitive impairment, clinical Alzheimer’s disease, and dementia in older persons. Biochim. Biophys. Acta 1862, 878–886 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Corrada M, Sonnen J, Kim R & Kawas C Microinfarcts are common and strongly related to dementia in the oldest-old: The 90+ study. Alzheimer’s Dement. 12, 900–908 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Iadecola C et al. Vascular Cognitive Impairment and Dementia: JACC Scientific Expert Panel. J. Am. Coll. Cardiol 73, 3326–3344 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Perez-Nievas BG et al. Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain 136, 2510–2526 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study confirmed that the robust glial activation that accompanies plaques and tangles in individuals with typical Alzheimer disease dementia was remarkably reduced in resilient individuals, and was the first study to show lower amounts of pathological species of oligomeric tau in synapses of resilient individuals than individuals with Alzheimer disease dementia.
- 67.Duara R et al. Diagnosis and staging of mild cognitive impairment, using a modification of the clinical dementia rating scale: the mCDR. Int. J. Geriatr. Psychiatry 25, 282–289 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ozer S, Young J, Champ C & Burke M A systematic review of the diagnostic test accuracy of brief cognitive tests to detect amnestic mild cognitive impairment. Int. J. Geriatr. Psychiatry 31, 1139–1150 (2016). [DOI] [PubMed] [Google Scholar]
- 69.Riudavets MA et al. Resistance to Alzheimer’s pathology is associated with nuclear hypertrophy in neurons. Neurobiol. Aging 28, 1484–1492 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Iacono D et al. Neuronal Hypertrophy in Asymptomatic Alzheimer Disease. J. Neuropathol. Exp. Neurol 67, 578–589 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Iacono D et al. The Nun study: clinically silent AD, neuronal hypertrophy, and linguistic skills in early life. Neurology 73, 665–673 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Knowles RB et al. Plaque-induced neurite abnormalities: implications for disruption of neural networks in Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A 96, 5274–5279 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Le R et al. Plaque-Induced Abnormalities in Neurite Geometry in Transgenic Models of Alzheimer Disease: Implications for Neural System Disruption. JNEN J. Neuropathol. Exp. Neurol 60, 753–8 (2001). [DOI] [PubMed] [Google Scholar]
- 74.D’Amore JD et al. In Vivo Multiphoton Imaging of a Transgenic Mouse Model of Alzheimer Disease Reveals Marked Thioflavin-S-Associated Alterations in Neurite Trajectories. JNEN J. Neuropathol. Exp. Neurol 62, 137–45 (2003). [DOI] [PubMed] [Google Scholar]
- 75.Spires T et al. Dendritic Spine Abnormalities in Amyloid Precursor Protein Transgenic Mice Demonstrated by Gene Transfer and Intravital Multiphoton Microscopy. J. Neurosci 25, 7278–7287 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Selkoe D The molecular pathology of Alzheimer’s disease. Neuron 6, 487–498 (1991). [DOI] [PubMed] [Google Scholar]
- 77.Cleary J et al. Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat. Neurosci 8, 79–84 (2005). [DOI] [PubMed] [Google Scholar]
- 78.Lesné S et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440, 352–357 (2006). [DOI] [PubMed] [Google Scholar]
- 79.Poling A et al. Oligomers of the amyloid-beta protein disrupt working memory: confirmation with two behavioral procedures. Behav. Brain Res. 193, 230–234 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shankar G et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med 14, 837–842 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Figueiredo CP et al. Memantine rescues transient cognitive impairment caused by high-molecular-weight aβ oligomers but not the persistent impairment induced by low-molecular-weight oligomers. J. Neurosci 33, 9626–9634 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ledo JH et al. Amyloid-β oligomers link depressive-like behavior and cognitive deficits in mice. Molecular psychiatry vol. 18 1053–4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lourenco MV et al. TNF-α mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s β-amyloid oligomers in mice and monkeys. Cell Metab. 18, 831–843 (2013). [DOI] [PubMed] [Google Scholar]
- 84.Fowler SW et al. Genetic modulation of soluble Aβ rescues cognitive and synaptic impairment in a mouse model of Alzheimer’s disease. J. Neurosci 34, 7871–7885 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tomic JL, Pensalfini A, Head E & Glabe C Soluble fibrillar oligomer levels are elevated in Alzheimer’s disease brain and correlate with cognitive dysfunction. Neurobiol. Dis 35, 352–358 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McDonald JM et al. The presence of sodium dodecyl sulphate-stable Abeta dimers is strongly associated with Alzheimer-type dementia. Brain 133, 1328–1341 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bjorklund NL et al. Absence of amyloid β oligomers at the postsynapse and regulated synaptic Zn2+ in cognitively intact aged individuals with Alzheimer’s disease neuropathology. Mol. Neurodegener 7, 23 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Berger Z et al. Accumulation of Pathological Tau Species and Memory Loss in a Conditional Model of Tauopathy. J. Neurosci 27, 3650–3662 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hampton D et al. Cell-Mediated Neuroprotection in a Mouse Model of Human Tauopathy. J. Neurosci 30, 9973–9983 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lasagna-Reeves CA et al. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener 6, 39 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tai H-C et al. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am. J. Pathol 181, 1426–1435 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Singh A et al. Functional Integrity of Synapses in the Central Nervous System of Cognitively Intact Individuals with High Alzheimer’s Disease Neuropathology Is Associated with Absence of Synaptic Tau Oligomers. J. Alzheimers. Dis 78, 1661–1678 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kopeikina KJ, Hyman B & Spires-Jones T Soluble forms of tau are toxic in Alzheimer’s disease. Transl. Neurosci 3, 223–233 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kopeikina KJ et al. Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer’s disease brain. Am. J. Pathol 179, 2071–2082 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.de Calignon A, Spires-Jones TL, Pitstick R, Carlson GA & Hyman BT Tangle-bearing neurons survive despite disruption of membrane integrity in a mouse model of tauopathy. J. Neuropathol. Exp. Neurol 68, 757–761 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.de Calignon A et al. Propagation of Tau Pathology in a Model of Early Alzheimer’s Disease. Neuron 73, 685–697 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu L et al. Trans-Synaptic Spread of Tau Pathology In Vivo. PLoS One 7, e31302 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.de Calignon A et al. Caspase activation precedes and leads to tangles. Nature 464, 1201–1204 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kuchibhotla K et al. Neurofibrillary tangle-bearing neurons are functionally integrated in cortical circuits in vivo. Proc. Natl. Acad. Sci 111, 510–514 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mcgeer E & Mcgeer P The importance of inflammatory mechanisms in alzheimer disease. Exp. Gerontol 33, 371–378 (1998). [DOI] [PubMed] [Google Scholar]
- 101.Wyss-Coray T Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat. Med 12, 1005–1015 (2006). [DOI] [PubMed] [Google Scholar]
- 102.Serrano-Pozo A et al. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. Am. J. Pathol 179, 1373–1384 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Barroeta-Espar I, Weinstock LD, Perez-Nievas B, Meltzer A & Gómez-Isla T Distinct cytokine profiles in human brains resilient to Alzheimer’s pathology. Neurobiol. Dis 121, 327–337 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper was the first to show the existence of a different cytokine expression profile in the enthorinal cortex of individuals resilient to Alzheimer pathology than in age-matched individuals with typical Alzheimer disease dementia and individuals free of Alzheimer disease neuropathological changes.
- 104.van Exel E et al. Vascular factors and markers of inflammation in offspring with a parental history of late-onset Alzheimer disease. Arch. Gen. Psychiatry 66, 1263–1270 (2009). [DOI] [PubMed] [Google Scholar]
- 105.Paouri E & Georgopoulos S Systemic and CNS inflammation crosstalk: implications for Alzheimer’s Disease. Curr. Alzheimer Res. 16, 559–574 (2019). [DOI] [PubMed] [Google Scholar]
- 106.Simpson JE et al. Microarray analysis of the astrocyte transcriptome in the aging brain: relationship to Alzheimer’s pathology and APOE genotype. Neurobiol. Aging 32, 1795–1807 (2011). [DOI] [PubMed] [Google Scholar]
- 107.Sekar S et al. Alzheimer’s disease is associated with altered expression of genes involved in immune response and mitochondrial processes in astrocytes. Neurobiol. Aging 36, 583–591 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bachstetter AD et al. Disease-related microglia heterogeneity in the hippocampus of Alzheimer’s disease, dementia with Lewy bodies, and hippocampal sclerosis of aging. Acta Neuropathol. Commun 3, 32 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Simpson JE et al. Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol. Aging 31, 578–590 (2010). [DOI] [PubMed] [Google Scholar]
- 110.Kamphuis W et al. Glial fibrillary acidic protein isoform expression in plaque related astrogliosis in Alzheimer’s disease. Neurobiol. Aging 35, 492–510 (2014). [DOI] [PubMed] [Google Scholar]
- 111.Harold D et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet 41, 1088–1093 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lambert J-C et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet 41, 1094–1099 (2009). [DOI] [PubMed] [Google Scholar]
- 113.Seshadri S et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 303, 1832–1840 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hollingworth P et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet 43, 429–435 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Guerreiro R et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med 368, 117–127 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jonsson T et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med 368, 107–116 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Stevens B et al. The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell 131, 1164–1178 (2007). [DOI] [PubMed] [Google Scholar]; This paper was the first one to show that complement-mediated synapse elimination by microglia becomes aberrantly activated in adult mice.
- 118.Hong S et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dejanovic B, Huntley M, Mazière A. De, Meilandt W & Sheng M Changes in the Synaptic Proteome in Tauopathy and Rescue of Tau-Induced Synapse Loss by C1q Antibodies. Neuron 100, 1322–1336.e7 (2018). [DOI] [PubMed] [Google Scholar]
- 120.Stephan AH, Barres B & Stevens B The complement system: an unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci 35, 369–389 (2012). [DOI] [PubMed] [Google Scholar]
- 121.Spangenberg EE et al. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-β pathology. Brain 139, 1265–1281 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Coma M, Serenó L, Rocha-Souto BD, Scotton TC & Gómez-Isla T Triflusal reduces dense-core plaque load, associated axonal alterations and inflammatory changes, and rescues cognition in a transgenic mouse model of Alzheimer’s disease. Neurobiol. Dis 38, 482–491 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Verghese J et al. Leisure activities and the risk of dementia in the elderly. N. Engl. J. Med 348, 2508–2516 (2003). [DOI] [PubMed] [Google Scholar]
- 124.Laurin D, Verreault R, Lindsay J, MacPherson K & Rockwood K Physical activity and risk of cognitive impairment and dementia in elderly persons. Arch. Neurol 58, 498–504 (2001). [DOI] [PubMed] [Google Scholar]
- 125.Sharp ES & Gatz M Relationship between education and dementia: an updated systematic review. Alzheimer Dis. Assoc. Disord 25, 289–304 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jack C et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers. Dement 14, 535–562 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Montine T et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheime’s disease: a practical approach. Acta Neuropathol. 123, 1–11 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jack C et al. Prevalence of Biologically vs Clinically Defined Alzheimer Spectrum Entities Using the National Institute on Aging-Alzheimer’s Association Research Framework. JAMA Neurol. 76, 1174–1183 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Doody RS et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med 369, 341–350 (2013). [DOI] [PubMed] [Google Scholar]
- 130.Salloway S et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med 370, 322–333 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Honig LS et al. Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N. Engl. J. Med 378, 321–330 (2018). [DOI] [PubMed] [Google Scholar]
- 132.Wessels AM et al. Efficacy and Safety of Lanabecestat for Treatment of Early and Mild Alzheimer Disease: The AMARANTH and DAYBREAK-ALZ Randomized Clinical Trials. JAMA Neurol. 77, 199–209 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sevigny J et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537, 50–56 (2016). [DOI] [PubMed] [Google Scholar]
- 134.Mintun MA et al. Donanemab in Early Alzheimer’s Disease. N. Engl. J. Med 384, 1691–1704 (2021). [DOI] [PubMed] [Google Scholar]