

Abstract
Fanconi anaemia (FA), ataxia telangiectasia (A-T), Nijmegen breakage syndrome (NBS) and Bloom syndrome (BS), are clinically distinct, chromosome instability (or breakage) disorders. Each disorder has its own pattern of chromosome damage, with cells sensitive to particular drugs that indicate a likely different underlying defect in each case. In addition, each syndrome shows a predisposition to cancer. Understanding the molecular and genetic basis of these disorders has revealed mechanisms of recognition and repair of DNA double strand breaks, DNA interstrand crosslinks and DNA repair during DNA replication. Specialist clinics for each disorder have provided the concentration of expertise needed to tackle their characteristic clinical problems and improve outcomes. While some treatments of the consequences of a disorder may be possible, for example hematopoietic stem cell transplantation in FA and NBS, future early intervention to prevent complications of disease will depend on a greater understanding of the roles of the affected DNA repair pathways in development. An important realisation has been the predisposition to cancer in carriers of some of these gene mutations.
Introduction
The chromosome instability syndromes Fanconi anaemia (FA), ataxia telangiectasia (A-T), Nijmegen breakage syndrome (NBS) and Bloom syndrome (BS) are a group of predominantly recessively inherited conditions associated with defects in DNA repair mechanisms, leading to chromosomal instability, chromosomal breakage and an array of phenotypic consequences, including an increased tendency to develop malignancies. Each condition has distinct molecular features. In FA, mutations in any of the 22 FANC genes (but most commonly FANCA, FANCC and FANCG1–5 affect the repair of DNA interstrand crosslinks (ICLs) — a component of which is homology directed repair (Figure 1). In A-T and NBS, mutations in ATM and NBN, respectively, affect the resolution of DNA double strand breaks (DSBs) (Figure 2). In BS, mutations in BLM (encoding Bloom syndrome protein)6 affects several aspects of homologous recombination pathways, including DNA stability of replication forks during unperturbed and perturbed DNA replication, DNA end resection (Figure 3) and the dissolution of double Holliday junctions (Figure 4), leading to the presence of chromatid interchanges (quadriradials) and highly elevated levels of sister chromatid exchanges (SCEs). Mutations in TOP3A (encoding topoisomerase 3α), RMI1 and RMI2 (encoding the RecQ-mediated genome instability proteins) have been reported recently as conferring a BS-like disorder (BSLD), featuring small body size and, in those with TOP3A and RMI2 mutations, some dermal abnormalities. A-T-like disorder (ATLD) is caused by mutations of MRE11 (encoding DSB repair protein MRE11). A single case of NBS-like disorder (NBSLD) caused by mutation of RAD50 (encoding DNA repair protein RAD50) has been reported, with increased radiosensitivity. The mutations in these ‘related’ disorders affect components of the same protein complexes as in the respective disorders. We might expect the number of these related disorders to increase in the coming years as new genetic disorders are recognized.
Figure 1. Repair of DNA interstrand crosslinks.
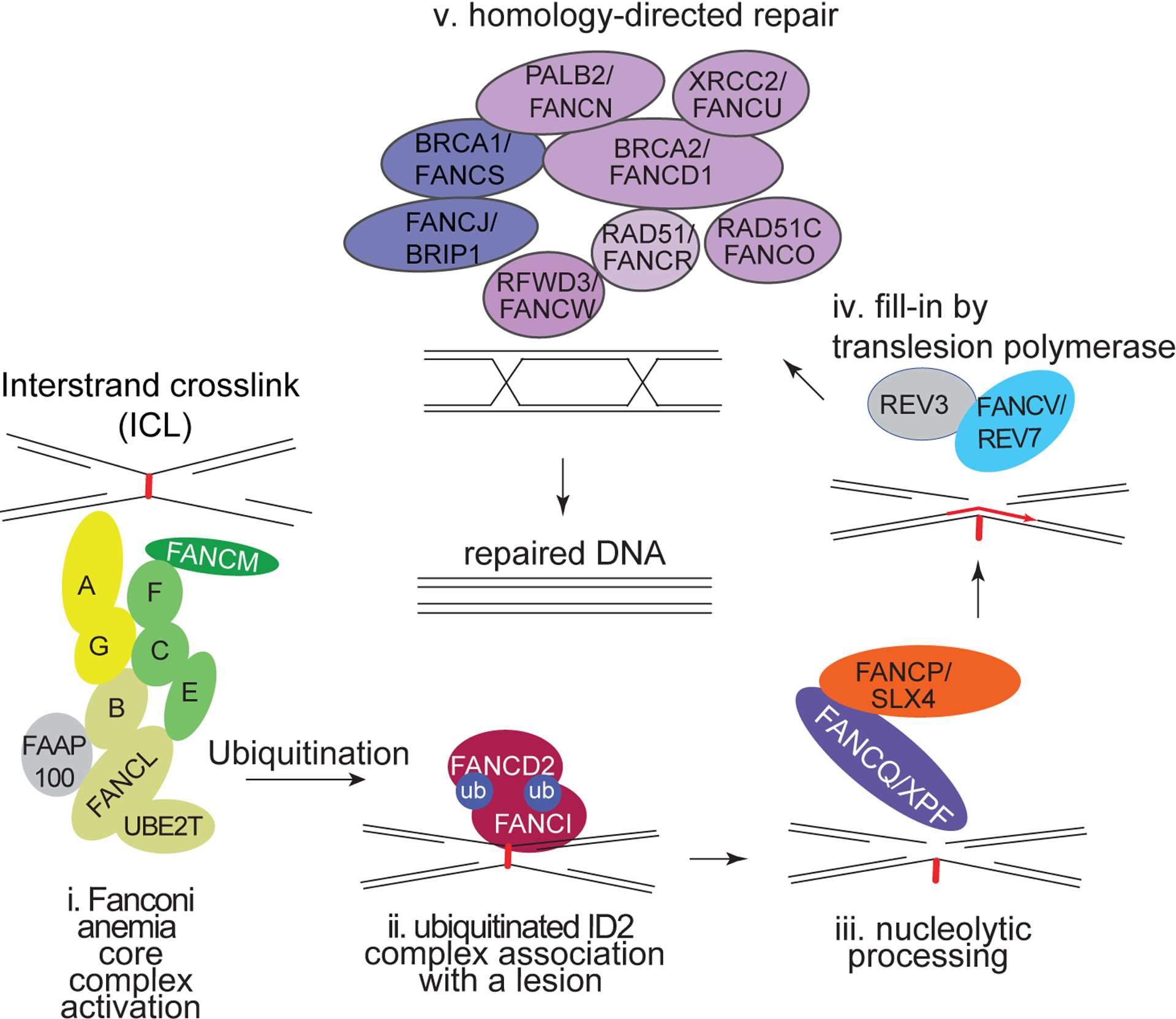
A series of steps, allows removal of the crosslink beginning with its recognition by the core complex (i) of Fanconi proteins (FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, FANCM and FANCT). This in turn activates the ID2 complex (ii) that allows the structure specific nuclease FANQ (also known as XPF) bound to scaffold protein FANCP (also known as SLX4) to cut the DNA on one strand (iii). This is followed by a DNA synthesis step (iv) performed by the translesion polymerase FANCV (also known as REV7). The final step is involves homology directed repair (v) involving the homologous recombination repair proteins (FANCS/BRCA1, FANJ/BRIP1, FANCR/RAD51, FANCO/RAD51C, FANCN/PALB2, FANCD1/BRCA2, FANCU/XRCC2 and FANW/RFWD3).
Figure 2. DNA double strand break (DSB) repair – the role of NBN and ATM in the recognition and signalling of the breaks.
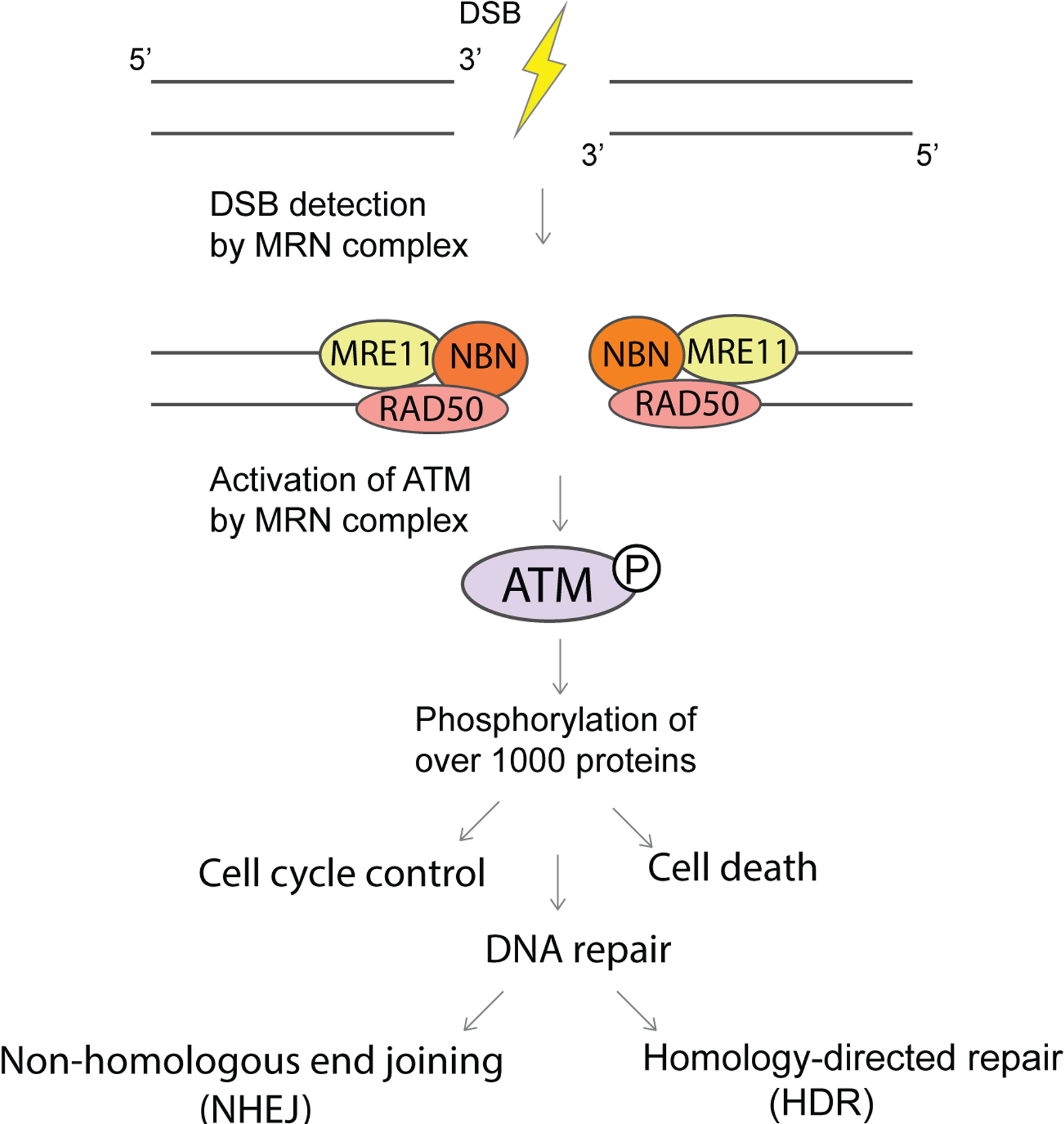
DNA double-strand break repair. Nibrin (NBN) recognizes DNA double-strand breaks (DSBs) via its involvement in the MRN complex, which is composed of DSB repair protein MRE11, DNA repair protein RAD50 and NBN. This recognition is required to activate serine-protein kinase ATM (ATM). ATM phosphorylates many downstream proteins to regulate DNA damage response pathways that include DNA repair, which in the case of DSBs can proceed through non-homologous end-joining or homology-directed repair. Mutations in NBN are pathognomonic of Nijmegen breakage syndrome and mutations in ATM are pathognomonic of ataxia telangiectasia. ATLD, ataxia telangiectasia-like disorder; NBSLD, Nijmegen breakage syndrome-like disorder.
Figure 3. DNA end resection of the 5’ end.
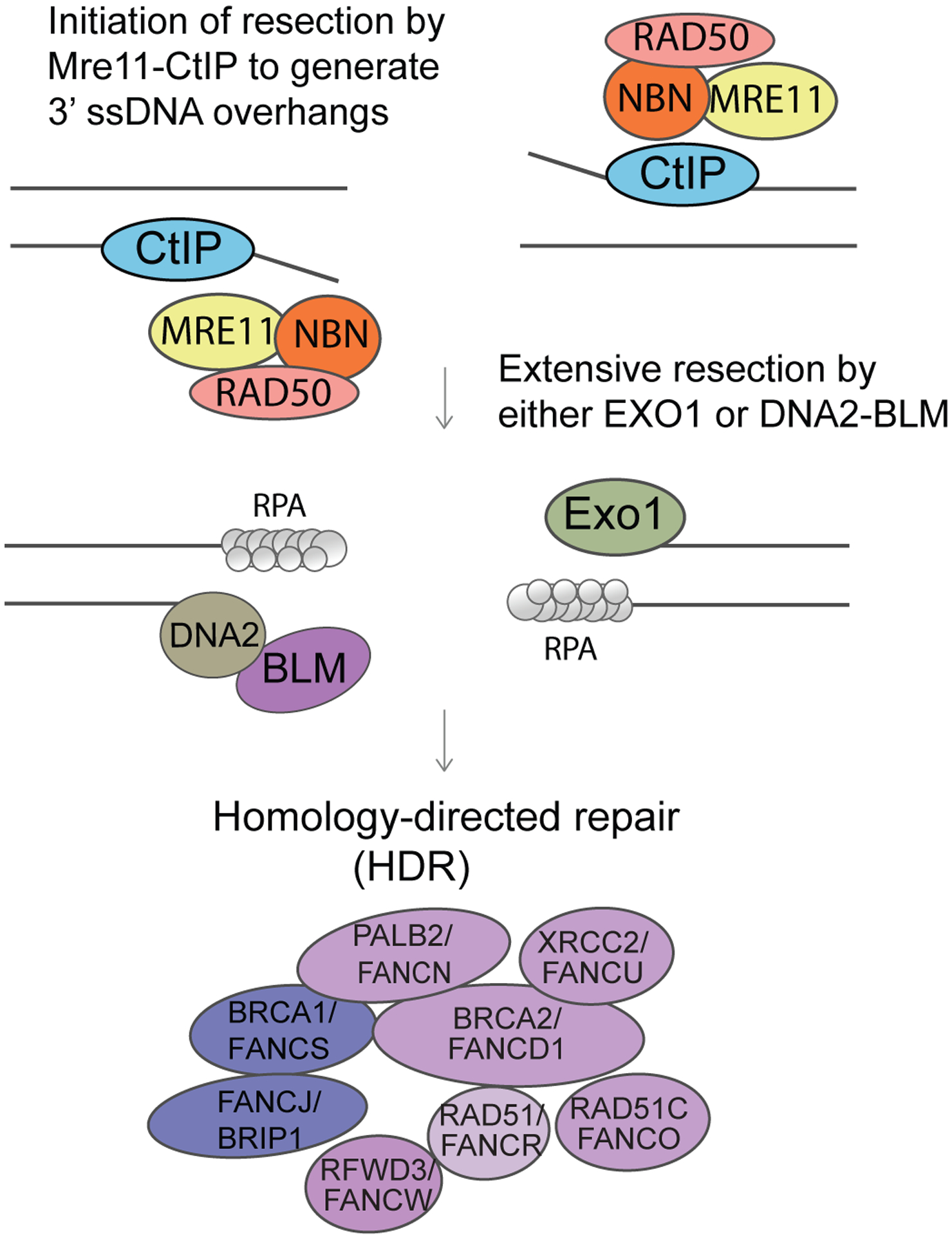
Undertaken by the MRN complex together with CtIP that results in RPA-3’ coated ssDNA overhangs. Long-range resection is carried by the BLM/DNA2 (helicase/nuclease) complex. 53BP1 and Rif1 suppress any excessive resection. Here BLM is promoting HR. The BRCA1/FANCS, BRCA2/FANCD1, PALB2/FANCN complex allows removal of RPA and the formation of RAD51 coated ssDNA nucleofilaments that catalyse strand invasion of the unbroken homologous template. The BLM helicase can suppress HR by destabilising the RAD51 coated nucleofilaments. Recombination repair is facilitated by Rad51 and co-factors
Figure 4. Dissolution of double Holliday (dHJs) junctions.
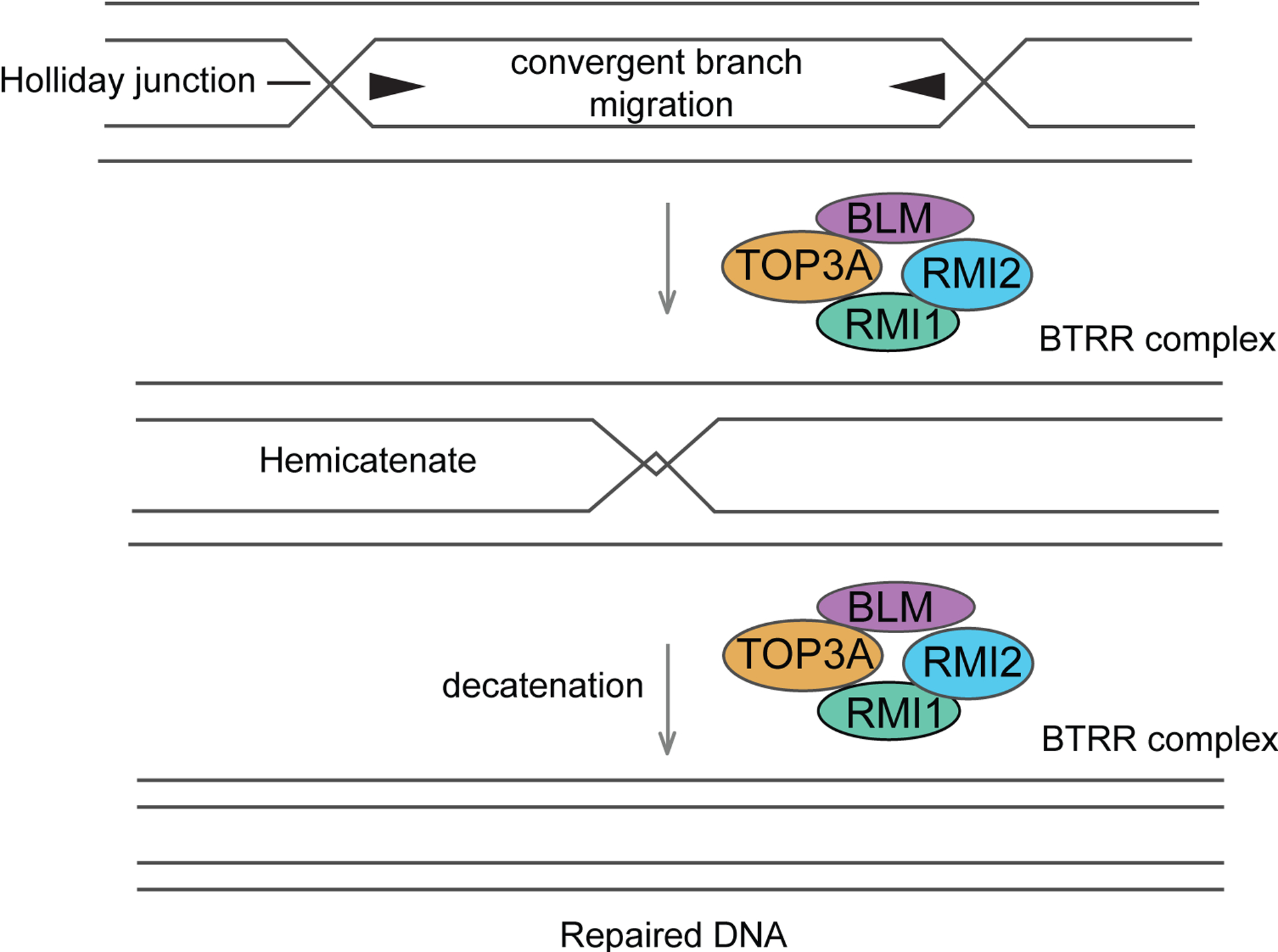
Mechanism of double Holliday junction (dHJ) dissolution by the combined action of the BTRR complex. BLM catalyzes convergent branch migation of the HJs to convert the dHJ into a hemicatenane, which is then decatenated by Topo IIIα.
At the clinical level, all these syndromes share a predisposition to cancer but with each having its own spectrum of tumours. The greatest phenotypic heterogeneity is observed in FA and A-T, with the populations of patients with NBS and BS each being more homogeneous. FA is commonly diagnosed in childhood in individuals with variable but distinct patterns of congenital or developmental abnormalities including short stature, microcephaly and café au lait spots and malformations affecting the thumbs or radial ray. Bone marrow failure (BMF) and predisposition in particular to acute myeloid leukaemia (AML) and squamous cell carcinoma (SCC) of the aero-digestive system are characteristic features. Cells from patients with FA display chromosomal breakage and cellular hypersensitivity to ICL-inducing agents (such as diepoxybutane, mitomycin C and cisplatin, a characteristic that is used as a diagnostic test for FA7; importantly some FA patients also show increased radiosensitivity.
A-T is a progressive neurodegenerative disease, with onset in early childhood. Characterized by increased radiosensitivity at the cellular level (in which cultured cells are unusually sensitive to the effects of ionizing radiation (for example, by reduced cell survival or increased chromosome damage) and at the clinical level (whereby careful consideration is required before exposing patients to radiotherapy or radiomimetic cytotoxic drugs), A-T also shows typical chromosome translocations in T cells (mainly involving chromosomes 7 and 14) and predisposition to lymphoid tumours in childhood. Patients with A-T show an increased risk of carcinoma including GI tract tumours, endocrine tumours and female breast cancer in adulthood8,9 (including one male breast cancer9). Ataxia telangiectasia-like disorder (ATLD) is clinically very similar to A-T and is caused by specific mutations of MRE11.
NBS is another radiosensitivity disorder that was first described in two Dutch brothers from a consanguineous family10 with microcephaly, growth deficiency, learning difficulty, immunodeficiency and chromosomal rearrangements (resembling those in A-T). Patients with NBS have craniofacial features that include receding forehead, prominent mid-face with long nose and philtrum, receding mandible, epicanthic folds, sparse hair, large ears and subtle sclera telangiectasia. Some have learning difficulty, and congenital malformations include brain malformation, clinodactyly, syndactyly, anal atresia, hydronephrosis, hip dysplasia and ovarian failure. Skin abnormalities common in NBS include café au lait spots, vitiligo spots, sun sensitivity of eyelids, pigment deposits in eye fundus, cutaneous telangiectasias and skin and organ granulomas. NBS predisposes mainly to lymphoid malignancies. A single case of Nijmegen breakage syndrome-like disorder (NBSLD) caused by mutation of RAD50 has been reported11, with increased radiosensitivity.
Patients with BS display proportional small body size, including microcephaly as the most characteristic clinical feature. Small size is frequently accompanied by various other features including a telangiectatic sun-sensitive facial erythema, café au lait spots and other dermal pigmentation abnormalities, a characteristic facial appearance with a high-pitched, squeaky voice, immune system deficiencies with increased infections, reduced fertility, gastrointestinal upsets and feeding problems and endocrine abnormalities. The most common complication of BS is the development of cancer. Cancer develops earlier than normal and many persons with BS have developed multiple cancers. Almost all cancer types are reported to occur in BS, the most common ones being leukemias, lymphomas, colorectal cancers, and breast cancers. An early onset of type II diabetes and chronic obstructive lung disease are also common complications in BS. Mutations in TOP3A, RMI1, and RMI2 have been recently reported as conferring a Bloom syndrome-like disorder, featuring small size and in persons with TOP3A and RMI2 mutations some dermal abnormalities. As in BS, these conditions exhibit an increased frequency of SCEs.
In this Primer, we describe our understanding of the genetic and molecular bases of these disorders, including the relationship of the defects to the predisposition to different cancers. We point out examples in which the pathogenesis of some of the presenting clinical features remain unclear and describe improvements in patient care that have an impact on survival and quality of life.
Epidemiology
Fanconi anaemia
Causative mutations with an estimated average global carrier frequency of 1/180 have been found, so far, in 22 FANC genes. Of these, >80% of mutations occur in FANCA, FANCG and FANCC1–5 with no sex preference; mutations in FANCE and FANCF comprise ~8% and FANCD1 ~3% of FANC mutations. FA caused by mutations in the other 16 FANC genes is rare, comprising small numbers of cases of each. Although FA is uncommon, the incidence varies owing to founder mutations in specific ethnic groups, such as Askenazi Jews and Spanish Gitanos (gypsies)12,13. With improved management the prevalence of FA is rising; data from the northwest of England suggest a current prevalence of 5 per million population14, which seems to have doubled in the past two decades.
Ataxia telangiectasia
The prevalence of A-T in the UK is ~3 per million population15, with an estimated ~200 cases (ascertainment close to 100%); similar proportions are expected in Germany, France and Italy. The estimate for the number of affected individuals in the United States is ~1,000 in a population of ~325 million. Median survival in A-T is 25y with a wide range16 Individuals of both sexes, all races and ethnicities are equally affected by A-T; however, the prevalence of A-T may be higher in consanguineous populations or those populations with a founder effect. ATLD accounts for ~20 published cases worldwide so far.
Nijmegen breakage syndrome
Although single patients with NBS are reported from all over the world, the majority of patients with NBS have a restricted geographical origin (Slavic and, in particular, Polish or Czech descent) and carry a common founder mutation, 657del5 in exon 6 of the NBN (formerly NBS1) gene. The prevalence of the founder mutation in the Czech Republic (1:154), Ukraine (1:182) and Poland (1:190) is high17. It was estimated that the founder mutation occurred less than 300 generations ago18, thus supporting the view that the original mutation predated the historic split and subsequent spread of the “Slavic people”19. The founder mutation and other mutations have been found in many countries in Western Europe, North and South America and New Zealand20,21. By contrast NBSLD is extremely rare with ~5 patients identified in Europe ever.
Bloom syndrome
Based on an estimate from the ExAc database, which is a sample of convenience consisting largely of Western European whites, the frequency of disease-causing alleles in BLM is 0.00138. Consequently, the expected incidence of BS would be 2 cases for every million live births in this population. As expected from this calculation, BS is a remarkably rare disorder with <300 reported cases worldwide. However, this is likely to be a significant underestimate due to inconsistent recording in many countries. For example, based on the frequency of a founder allele in Slavic peoples, there should be several thousand cases in the Russian Federation yet only a few cases have been reported in the literature. Females are just as likely to be affected as males. However, there is a slight under-diagnosis of females because expression of the facial erythema is not quite as severe18. As with many autosomal recessive disorders, the frequency of consanguinity is higher than expected; the parents of affected individuals are known to be related in approximately one-third of families. In another third of families, the parents are Ashkenazi Jewish, which is discussed below (Diagnosis, screening and prevention). The major cause of death in BS is from cancer. The average lifespan has been reported as 27 years, but this estimate is low because it is weighted by deaths from earlier cohorts and does not yet take into account improvements in cancer treatments22.
Since the chromosome instability syndromes are associated with large increased risks of cancer it might be predicted that heterozygous normal carriers of these gene mutations might also be at an increased risk of cancer through mechanisms such as loss of heterozygosity (Box1) and hapoinsufficincy.
BOX1. Increased cancer risk in heterozygous carriers of ‘Chromosome Instability Disorder’ gene mutations.
Being a carrier of a gene mutation associated with a chromosome instability disorder may increase the risk of cancer. Several studies have investigated the cancer risk of FANC mutation carriers. While individual cases of cancer in FANC mutation carriers have been described209 there is no evidence of a statistically significant increased cancer risk for mutation carriers (i.e. first degree relatives) of the commonly mutated FANC genes210 211. An increased risk of cancer is presumed in FANC family members that are carriers of the hereditary breast and ovarian cancer associated FANC mutations, which include FANCN (otherwise known as PALB2), FANCO (otherwise known as RAD51C), FANCS (otherwise known as BRCA1) and FANCD1 (otherwise known as BRCA2), which is the most commonly mutated gene of this subgroup of FA genes14. Also for these FA cases, which typically have a severe phenotype, the family history is not always positive, but the number of cases is small. Germline FANCD1 variants were also identified in a small but significant subgroup of non-FA childhood malignancies212 .The role of other FA gene variants in susceptibility to sporadic cancer, in particular SCC continues to be investigated, but any clinical risk contribution of these variants for non-FA cancer is not fully understood213.
Studies on families with A-T have established that female carriers in these families have a doubling of the relative risk for breast cancer compared with the general population in the UK (and a ~5 fold increase in those <50 years of age)214; indeed, carrying an ATM mutation is considered a moderate risk factor for breast cancer. Some data suggested that there is a further increased risk of breast cancer in carriers of specific ATM mutations, in particular the c.7271T>G;p.Gly2424Val ATM missense mutation215,216. Some evidence also suggests that these carriers have excess risks of colorectal cancer (RR = 2.54, 95% CI = 1.06 to 6.09) and stomach cancer (RR = 3.39, 95% CI = 0.86 to 13.4). Additional long-term studies on A-T families will further clarify these risks. Recent publications on >10,000 tumours in the general population with germline variants and cancer driver genes using The Cancer Genome Atlas data have highlighted the occurrence of biallelic ATM mutations across multiple cancer types217,218 with a strong association with both prostate and gastric carcinoma and a suggestive association with breast cancer, lung adenocarcinoma and pancreatic adenocarcinoma, in accordance with previous work219,220.
The NBN657del5 mutation is associated with an elevated risk of cancer in heterozygotes221. About a 3-fold increase in breast cancer risk for female NBS heterozygotes has been demonstrated222. There is an increase in prostate cancer in men and a predisposition to medulloblastoma in paediatric patients223 224. More recently, however, MRE11, RAD50 and NBN were also reported as intermediate risk breast cancer susceptibility alleles225.
Heterozygous Bloom syndrome mutation carriers are apparently asymptomatic, although the incidence of some cancers might be mildly elevated226. Confirmation of there being any association between BLM polymorphisms and an increased incidence of cancer will require large genome-wide association or sequencing studies.
Mechanisms/pathophysiology
Fanconi anaemia
Most of the mutations of any of 22 different FANC genes implicated in FA are recessively inherited, with the exception of FANCB (which is X-linked) and RAD51/FANCR (which is dominant). A much more severe clinical phenotype is attributed to mutations in genes such as FANCS and FANCD1. The proteins encoded by FANC genes are implicated in a common pathway necessary for the repair of DNA ICLs, lesions that covalently link two strands of DNA and block both replication and transcription. Unrepaired or misrepaired ICLs lead to stem cell failure (and in turn to developmental abnormalities and bone marrow failure) and genomic instability (leading to cancer).
The Fanconi repair pathway is activated during DNA replication whereby Fanconi proteins (Box 2) are recruited to the ICL-stalled replisome. FANCL23, an E3 ubiquitin ligase in a multisubunit protein complex known as the core complex (consisting of FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, FANCM and FANCT (also known as UBE2T)), mono-ubiquitylates FANCI and FANCD2, which stably localize to the lesion24,25. Once ubiquitylated, FANCI and FANCD2 recruit effectors that are responsible for cleaving the DNA, a step performed by the nuclease FANCQ (also known as XPF) in association with FANCP (also known as SLX4)26,27, and a DNA synthesis step that is performed by a translesion polymerase FANCV (also known as REV7)1. Once the lesion is excised and partially repaired, proteins necessary for homology-directed repair (FANCD1, FANCR (also known as RAD51), FANCS, FANCJ (also known as BRIP1), FANCN (also known as PALB2), FANCO (also known as RAD51C), FANCU (also known as XRCC2)) complete the repair28,29. This last step is regulated by another E3 ubiquitin ligase in the pathway, FANCW (also known as RFWD3)3,30,31.
Box 2. The affected proteins.
FANC
FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, FANCM, FANCT/UBE2T, together, form the Fanconi anaemia core complex. FANCL is the E3 ubiquitin ligase, and FANCT/UBE2T is an E2 Ubiquitin ligase. If the core complex is inactive, FANCI and FANCD2 are not monoubiquitinated resulting in Fanconi anemia.
FANCI and FANCD Form a heterodimer (ID2 complex) which is monoubiquitinated by FANCL in the core complex. ID2 localizes to DNA interstrand crosslinks and is thought to recruit pathway effectors.
FANCP/SLX4 is the protein scaffold for FANCQ/XPF, MUS81, SLX1. It is necessary for ICL repair but also for Holliday junction resolution.
FANCQ/XPF is a structure-specific nuclease necessary for DNA ICL repair when complexed with FANCP/SLX4 and Nucleotide Excision Repair (NER) independent of FANCP/SLX4 interaction.
FANCV/REV7 is a component of a translesion synthesis (TLS) polymerases Polζ
FANCD1/BRCA2, FANCJ/BRIP1, FANCN/PALB2 FANCO/RAD51C, FANCR/RAD51, FANCS/BRCA1, FANCU/XRCC2 and FANCW/RFWD3 are proteins that participate in or regulate homology-directed repair during DNA ICL repair but also in response to many other lesions including DSBs. Multiple components have enzymatic activities including 5’−3’ helicase activity of FANCJ, ATPase activity of RAD51, and E3 ubiquitin ligase of FANCS and FANCWa
ATM
Member of the PI-3-kinase-like family of Ser/Thr kinases (PIKKs) also containing ATR and DNA-PKCS
When mutated gives rise to the neurodegenerative, chromosome instability disorder, Ataxia Telangiectasia
Predominatly localises to the nucleus but is also found in cytoplasmic organelles/vesicles e.g. mitochondria, peroxisomes
Exists as a inactive dimer that is activated in response to DNA double strand breaks by trans autophosphorylation, which is stimulated by binding to the MRN complex
Can be activated in response to reactive oxygen species via an MRN-independent mechanism involving the oxidation of Cys-2991
Phosphorylates >700 different nuclear/cytoplasmic protein substrates involved in regulating DNA repair, replication, cell cycle checkpoint activation, apoptosis, telomere maintenance, transcription, chromatin structure, metabolism, growth factor signaling, RNA splicing, protein synthesis, autophagy and vesicular trafficking.
Somatically mutated in a number of sporadic lymphoid and epithelial tumours e.g. B-CLL, T-PLL, mantle cell lymphoma
NBN
Non-catalytic subunit of the MRE11/RAD50/NBN complex
When mutated gives rise to the developmental, chromosome instability disorder, Nijmegen Breakage Syndrome (NBS)
Contains an N-terminal FHA domain and two BRCT domains that mediate the MRN complex binding to MDC1, TCOF1 and CtIP in a phospho-dependent manner
Contains a C-terminal motif that is important for binding and activating ATM at the sites of DNA double strand breaks
Implicated in regulating the nuclear localisation of the MRN complex
Functions to regulate ATM-dependent DNA damage signaling, DNA double strand break end-resection, DNA damage-induced cell cycle checkpoint activation, DNA damage-induced apoptosis, the replication stress response, ATR activation
Somatically mutated in some lymphoid/epithelial tumours
BLM227–229
Member of the RecQ helicase family
Translocates along ssDNA in a 3’−5’ direction and mediates dissolution of recombination and late-replication intermediates in conjunction with Topoisomerase IIIα and RMI1/2. Dissolution of double Holliday junctions into non-crossover products are proposed to be the main reason why SCEs arise in the absence of BLM
Catalytic domain contains the helicase active site and a RecQ C-terminal (RQC) region, which comprises both winged-helix domain (for DNA binding) and a Zn2+-binding subdomain (for structural integrity)
Helicase and RNaseD C-terminal (HRDC) domain are implicated in binding to complex and branched DNA structures
N-terminal domain mediates protein-protein interactions, and is a target for several post-translational modifications
BS-associated mutations in BLM lead either to protein truncation or a catalytically inactive protein
aThe phenotypes of patients with mutations in genes coding for the HR proteins are variable with mutations in FANCO, FANCR, FANCS, FANCU leading to no spontaneous bone marrow failure (Fanconi anemia-like phenotype) and mutations in FANCD1 and PALB2 leading to a very severe cancer predisposition phenotype.
Although responses to DNA ICLs have been best studied, the Fanconi proteins are also activated as a result of a plethora of other problems that occur during DNA replication. Their function has been identified at common fragile sites, which represent difficult to replicate regions32 and at sites of collisions between replication and transcription machinery, in which they are implicated in the clearance of R-loops (a three-stranded nucleic acid structure, composed of a DNA:RNA hybrid, that forms during transcription)33,34. Finally, Fanconi proteins including the BRCA proteins, are also involved in protection of stalled replication forks against degradation by DNA nucleases35–37. It remains to be determined if the non-ICL repair functions of the majority of Fanconi proteins contribute to the phenotypes of patients with FA. It is likely, however; that the defect in global homology directed repair in patients with biallelic mutations in BRCA2/FANCD1 and PALB2/FANCN explains the very severe disease characterized by early onset AML and embryonal tumours including medullobastoma in these patients28,38–40.
The pathophysiology of haemopoietic stem cell (HSC) failure and acceleration of tumorigenesis in FA continues to be under investigation. It is clear that FA-deficient HSCs have an autonomous DNA repair defect. Damaged HSCs die due to the activation of p53 and p21-dependent apoptosis41 resulting in a progressive decrease of bone marrow cellularity necessitating HSC transplantation in FA patients. The key question in the field is what the source is of DNA damage. Mere re-entry of HSCs from quiescence into the cell cycle results in DNA damage that precipitates bone marrow failure if the Fanconi pathway is deficient in the mouse42. This finding would imply that lesions encountered during replication are to be blamed. A strong case is being built that such lesions would come from endogenous metabolites in the form of reactive aldehydes, including acetaldehyde and formaldehyde43–45. Consistent with data from mouse studies patients with FA with concomitant inherited deficiency of ALDH2, the enzyme that metabolizes acetaldehyde and is responsible for preventing alcohol-induced flushing, have increased number of developmental abnormalities, earlier onset of bone marrow failure and leukaemia43.
Ataxia telangiectasia and Nijmegen breakage syndrome
Despite similarities between the cellular defects displayed by cells derived from patients with underlying DNA repair deficiencies, the impact that this has on the development and maintenance of specific tissues/organs can be strikingly different, particularly with respect to the nervous system. Broadly speaking, DNA repair deficiencies either give rise to microcephaly or progressive cerebellar degeneration. The underlying reason for this stark contrast in disease-associated neuropathology and how it is related to specific repair deficiencies is not well understood. This is exemplified by the related chromosome instability disorders A-T and NBS, which exhibit overlapping clinical and cellular phenotypes but one is associated with neurodegeneration and the other abnormal neurodevelopment. For this reason, how particular DNA repair deficiencies contribute to the different neuropathologies exhibited by patients with A-T, NBS and other related genome instability disorders, will be discussed together to allow specific comparisons to be made. However, it should be noted that whilst both A-T and NBS are considered as related disorders, often the overlapping clinical and cellular phenotypes e.g. immunodeficiency or radiosensitivity, do differ in severity.
The protein kinase ATM (Box 2), in conjunction with the related protein kinases DNA-PK and ATR, are master regulators of the phosphorylation-dependent cellular response to DNA damage. Over 700 potential substrates of ATM46 have been identified, which has advanced our understanding of the role of ATM in regulating DNA DSB repair, cell cycle checkpoint activation and DNA damage-induced apoptotic pathways47. Furthermore, patients with A-T, ATLD, NBS, NBSLD (and RIDDLE syndrome caused by mutations in RNF168) all display a cellular hypersensitivity to ionising radiation (with A-T being the most radiosensitive by cell survival), and in A-T and NBS a clinical radiosensitivity. They also often display immunodeficiency, problems with fertility and an increased predisposition to lymphoid tumours. Given the role of physiologically-induced DSBs in promoting meiotic recombination and somatic recombination in the adaptive immune system, and that a failure to repair DSBs generated during the latter process is known to facilitate lymphoid tumourigenesis, some aspects of the clinical phenotype of these diseases align well with the underlying cellular DNA repair defect.
How ATM loss contributes to the major neurological features of A-T, that is progressive cerebellar degeneration, remains unclear. The prevailing dogma in the field is that specific neuronal cells within the cerebellum (primarily Purkinje and granule cells) are particularly sensitive to the loss of ATM. One hypothesis is that accumulated unrepaired DNA double strand breaks over time contribute to characteristic cerebellar pathology affecting these cells. However, from the study of other human disorders caused by inherited mutations in DSB repair genes, a DSB repair defect does not commonly give rise to cerebellar degeneration (Figure 2). The exception to this trend is ATLD48, whose gene product makes up the enzymatic component of the highly conserved MRE11-RAD50-NBN (MRN) DSB repair complex. The involvement of the MRN complex required to efficiently activate ATM following the induction of DNA breaks and that participates in many ATM-regulated DNA damage response (DDR) pathways47 has strengthened the idea that defective DSB repair may represent the underlying cause of the progressive neuronal decline in patients lacking ATM.
In contrast, hypomorphic mutations in NBN and RAD50 give rise NBS (Box 2) and NBSLD, respectively, which are characterised by the presence of microcephaly but not cerebellar degeneration11,49–51, therefore, arguing against the hypothesis that an underlying DSB repair defect per se is responsible for the neurodegeneration associated with A-T. Notably, a few patients with MRE11 mutations have also been identified that exhibit microcephaly and not cerebellar ataxia, indicating that hypomorphic loss of MRE11 and destabilisation of the entire MRN complex does not necessarily predispose to neurodegeneration52. Whilst the mechanism governing whether MRE11 mutations give rise to cerebellar ataxia or microcephaly is unclear, it is possible that a certain threshold level of cellular NBN and RAD50 is required to protect against the development of microcephaly but is not sufficient to prevent cerebellar atrophy. Intriguingly, in contrast to the embryonic lethality associated with a complete loss of Nbn, a mouse model in which Nbn is only disrupted in the central nervous system displayed both microcephaly and cerebellar ataxia, which could be reversed by the inactivation of TP5353. This suggests that the ability of unrepaired DNA damage to activate the p53-dependent apoptotic response may play a role in determining the pathological outcome of a DNA repair deficiency in the developing versus mature brain54.
Given that the MRN complex and ATM have been implicated in regulating HR-dependent DSB repair, protecting telomeres from inappropriate repair, processing and repairing programmed DSBs, activating the G1-, intra-S and G2/M-phase DNA damage checkpoints and inducing DNA damage-dependent apoptosis55, it is difficult to ascribe loss of a particular DDR function of these proteins to a specific clinical deficit. Furthermore, it is known that ATM and the MRN complex also have roles within the cellular response to DNA damage that are independent of each other, for example, facilitating non-homologous DNA end-joining, degrading stalled, unprotected replication forks, the regulation of transcription, mRNA splicing and translation47,55. Thus it is conceivable that it is a combination of specific DDR defects including DSB repair that dictates the development of the different neuropathologies observed in A-T, ATLD, NBS and NBSLD. To confuse matters further, mutations in the DNA damage responsive E3 ubiquitin ligase RNF168, which coordinates the ubiquitin-dependent DDR downstream of ATM and the MRN complex56, have also been identified in a human syndrome exhibiting cerebellar ataxia57. However, interestingly, mutations in RNF168 were originally identified in RIDDLE syndrome, an immunodeficiency syndrome lacking both microcephaly and overt cerebellar degeneration58.
Defects in other DNA repair pathways not directly linked to DSBs more consistently give rise to cerebellar degeneration rather than microcephaly. Mutations in five factors known to be involved in regulating DNA end processing have been identified in patients who exhibit progressive cerebellar ataxia: APTX (mutated in ataxia with oculomotor apraxia, type 1; AOA1)59, PNKP (mutated in AOA4)60, XRCC1 (mutated in AOA5)61, TDP1 (mutated in spinocerebellar ataxia with axonal neuropathy, type 1; SCAN1)62 and TDP2 (mutated in spinocerebellar ataxia autosomal recessive type 23; SCAR23)63. Functionally, these factors have been implicated in mediating the repair of reaction intermediates arising from failed topoisomerase 1- (TOP1) and topoisomerase 2 (TOP2)-dependent processes64–66. Given the neurological similarities between A-T and these ataxias, and the physiological relevance of abortive topoisomerase lesions, which are likely to arise at relatively high frequency in transcriptionally active cells such as those in the cerebellum, defective signalling and/or repair of these lesions could contribute to the progressive neurodegeneration in these disorders67–69. Consistent with this, cells devoid of ATM are hypersensitive to genotoxic agents that inhibit both TOP1 and TOP2. Moreover, ATM has been demonstrated to phosphorylate both TDP1 and PNKP; in the case of TDP1, phosphorylation stabilises the protein and facilitates its binding to XRCC1, whereas phosphorylation of PNKP enhances both its DNA kinase and phosphatase activities and its localisation to DNA breaks70–72.
In addition to responding to abortive Top1-associated DNA lesions, TDP1, XRCC1 and PNKP in conjunction with APTX have also been implicated in responding to and repairing oxidative DNA damage induced by reactive oxygen species (ROS). Since mitochondria are one of the main intracellular sources of ROS, it is perhaps not surprising that these four proteins in combination with a specific isoform of Top1, Top1-mt, and DNA ligase III, which is thought to play a role in ligating repair intermediates occurring during the repair of oxidative damage, are all localised to the mitochondria73. Intriguingly, unlike ATR and DNA-PK, the ATM protein is not just localised within the nucleus but is also present, to varying degrees, in a number of cytoplasmic organelles, for example, peroxisomes and the mitochondria74. Moreover, it has been demonstrated that ROS can directly activate the kinase activity of ATM, independently of the MRN complex, involving oxidation of the Cys-2991 residue, located just C-terminal to its kinase domain75. Whilst the functional relevance of ROS-dependent activation of ATM and its relationship to the neurodegeneration characteristic of A-T are not fully understood, it likely that this allows ATM to react to oxidative DNA damage that lacks a DNA end (i.e. a DSB) in both the nucleus and mitochondria and to trigger an appropriate anti-oxidative stress response, potentially in part by phosphorylating the repair proteins APTX and PNKP. In keeping with a ROS-dependent function of ATM being important for maintaining cellular homeostasis, it has been known for a long time that A-T cells exhibit elevated levels of oxidative stress, structural and functional mitochondrial abnormalities, dysfunctional mitophagy and an inability to properly repair mitochondrial DNA damage76 77.
Cerebellar degeneration is also caused by mutations in the SETX gene (mutated in AOA2), which encodes an RNA/DNA-directed DNA helicase78. Several studies have demonstrated that SETX is involved in resolving R-loops that occur at sites of transcriptional pausing or termination, collisions between the transcription and replication machinery and DNA DSBs localised in transcriptionally active genes79–81. However, loss of the function of SETX is unlikely to contribute significantly to the cerebellar pathology in AOA2 via collisions between transcription and replication machinery, because all neurons in the developed brain are post-mitotic. This is consistent with an inability to detect increased R-loop formation and chromosome breakage in the brains of Setx knockout mice, contrasting the situation in the testes of these mice82,83. Although, it should be noted that, in a manner similar to mouse knockout models of other human syndromes associated with cerebellar ataxia, Setx deficient mice did not exhibit any cerebellar abnormalities or ataxia82,83. However, its role in removing R-loops links well with the functions of APTX, PNKP, XRCC1, TDP1 and TDP2, in terms of repairing DNA breaks from oxidative DNA damage or the failed removal of torsional stress by TOP1 and TOP2 during active transcription. Based on this, it is tempting to speculate that any pathogenetic process that interferes with transcription or increases oxidative stress leads to the progressive degeneration of cells within the brain and nervous system. However, in reality it is unlikely to be this simple. Furthermore, if transcriptional abnormalities are a common link underlying all neurodegenerative processes, then it would seem more likely that specific types of neurodegeneration are caused by specific types of transcriptional abnormalities in specific neuronally-associated genes e.g. the presence/absence of R-loops, whether gene transcription initiation/elongation requires TOP2-dependent DSB induction, the chromatin context of the affected gene i.e. presence/absence of cohesin/CTCF84–87. In this respect, it has been suggested that the cerebellar degeneration-associated with loss of SETX may arise due to aberrant termination and splicing of specific genes important for neuronal maintenance88.
The question of why Purkinje cells in A-T are particularly sensitive to the loss of ATM remains a mystery. The high metabolic and transcriptional activity of neurons coupled with their inability to proliferate means that they are highly dependent on intrinsic protective mechanisms such as anti-oxidants and DDR pathways to maintain the integrity of both their nuclear and mitochondrial genomes. However, whilst this helps to explain why cells within the nervous system are more severely affected when DDR is compromised, this does not explain why Purkinje cells are targeted over other neurons. ATM has been linked to many of the cytoplasmic cellular processes and pathways89,90 described in ≥40 hereditary spinocerebellar ataxias (SCAs), which superficially share aspects of their neuropathology with A-T (reviewed in91). There are also examples of SCA gene products having direct roles in regulating homologous recombination-dependent DNA repair92. Taken together, whilst the cytoplasmic functions of ATM are likely to be important for neuroprotection, it is difficult to completely separate these from the role of ATM in regulating the nuclear DDR following either enzymatically-induced DNA breaks or those occurring through indirect mechanisms (for example, the production of highly reactive metabolic intermediates).
Bloom syndrome
BLM (Box 2) associates with TOPIIIα, RMI1 and RMI2 in the BTRR complex93. Acting in concert, these proteins promote the dissolution of a key intermediate in homologous recombination, the double Holliday junction94. This function ensures that certain recombination intermediates are processed without crossing over between the recombining molecules, which is one hypothesis for how BLM serves to limit the frequency of SCEs, which are increased as the hallmark cellular feature of BS95. Consistent with these proteins engaging in functional interactions, hypomorphic mutations in TOP3A and RMI1 have been shown to give rise to a BS-like disorder associated with microcephalic dwarfism96. As well as processing recombination intermediates, BLM also acts as a general anti-recombinase through its ability to dissociate recombination intermediates, a function that would similarly serve to suppress SCEs97. BLM also has a role in promoting the initiation of recombination through an ability to catalyze exonucleolytic resection of the ends of DSBs in association with the DNA2 nuclease98; this process creates single stranded DNA onto which the key activator of recombination, RAD51, is loaded. This function might also explain why BLM binds directly to RAD51, which it does in a SUMOylation-dependent manner99. In cells lacking telomerase, BLM has also been implicated in promoting the recombination-dependent alternative lengthening of telomeres (ALT) telomerase mechanism, which functions to maintain telomere integrity100. The binding of BLM to telomeric repeat-binding factor 2 (TRF2) at the telomere might be relevant to this function. Several connections also exist between the BTRR complex and the Fanconi pathway, including interactions with FANCM, FANCJ and the Fanconi core complex101–103.
Although cells derived from patients with BS have been used to study BLM function, BLM has now been inactivated in numerous human cell lines and in several model organisms. BLM-deficient cells universally show chromosomal instability with excessive chromosome breaks, and exchanges between sister chromatids and homologs, as well as a reduced ability to accurately segregate sister chromatids during mitosis104. Over 80 different mutations in BLM have been shown to give rise to BS, and these mutations either cause premature protein translation termination or affect highly conserved amino acids in the helicase and associated protein domains6. The gene is essential for embryonic development in the mouse, but hypomorphic alleles of mouse BLM have been generated, which confer some BS-like features104. DNA replication abnormalities are a consistent feature of BLM-deficient cells, including a reduced rate of maturation of replication intermediates. Following replication fork blockade using inhibitory drugs, BLM is required for replication fork stability, protecting against irreversible fork collapse. Excessive fork collapse in BS cells is associated with an increased rate of initiation of new replication forks (new origin firing), which increases the density of replication forks in both unperturbed cells and cells exposed to DNA damaging agents or replication inhibitors105. BLM might also be important for the disruption of certain DNA secondary structures, such as G-quadruplexes, that can impede fork progression. This activity might have a direct role in telomere maintenance through facilitating leading strand DNA synthesis106,107.
BLM also has a role in mitotic chromosome segregation. During anaphase, most human cells display threads of DNA called ultra-fine DNA bridges (UFBs) that cannot be stained with DNA dyes. BTRR binds to UFBs alongside the PICH translocase108. The hierarchical binding of these proteins to UFB DNA was modeled using optical tweezers. This showed how PICH recruits BTRR to bridge DNA, exemplifying how partner proteins can influence the properties of the BTRR complex109. It is likely that BLM’s association with UFBs is for the purpose of decatenating inter-linked replication or recombination intermediates that have persisted into mitosis. The small size in BS is most likely the result of the increased incidence of replication stress and the overall higher frequency of chromosomal mutations. This leads to a slower proliferation rate of cells and a higher rate of apoptosis, especially during embryonic and fetal development and in tissues where rapid cellular division is required, such as in the development of the immune system. The high frequency of cancer in BS, being its main complication, is most likely the result a 4-fold higher rate of mutation coupled with a 50-fold higher rate of loss of heterozygosity from inter-homolog recombination110.
Diagnosis, screening and prevention
Given the rarity of these disorders and their inherent clinical complexity, diagnosis and management can be challenging in underdeveloped or developing parts of the world. The clinical features of all the chromosome instability disorders are quite distinct often enabling a highly probable diagnosis based on clinical signs, symptoms and routine laboratory testing. Desired diagnostic certainty can be achieved with genetic testing. Prevention is not possible, although prenatal diagnosis is available for families with an affected child.
Fanconi anaemia
Presentation.
The clinical suspicion of FA arises in childhood in individuals with variable but distinct patterns of congenital or developmental abnormalities111 (Figure 5). The most common presenting symptoms are haematological abnormalities in children and young people, including cytopenias (which can affect any lineage), bone marrow hypoplasia or failure, myelodysplasia or AML with complex cytogenetic changes (characteristically involving gains of the chromosomal segment 3q)112 (Figure 6). Severe phenotypes of FA can also present in the neonatal period with a combination of vertebral anomalies, anal atresia, cardiac malformations, tracheo-oesophageal fistula with oesophageal atresia, structural renal and limb (VACTER-L) spectrum of abnormalities4,14. An uncommon but important group of patients can present with early childhood tumours, and FA should be considered if affected children have congenital abnormalities with severe toxicity from cytotoxic treatment. These patients can be affected by mutations in FANC genes associated with familial cancer, such as FANCD1 and FANCN14,40. At the other end of the spectrum, FA should also be considered in the differential diagnosis of aplastic anaemia, myelodysplasia, AML or early squamous cell carcinoma (SCC) in younger individuals and also when physical findings are not obvious, as the phenotype can be variable. Manifestations of FA can be very subtle, but may still be associated with severe side effects when treated with cytotoxics113. Most males with FA are infertile, and although several women with FA have had children, most are sub-fertile and go through menopause early114.
Figure 5. Characteristic features of the chromosome instability syndromes.
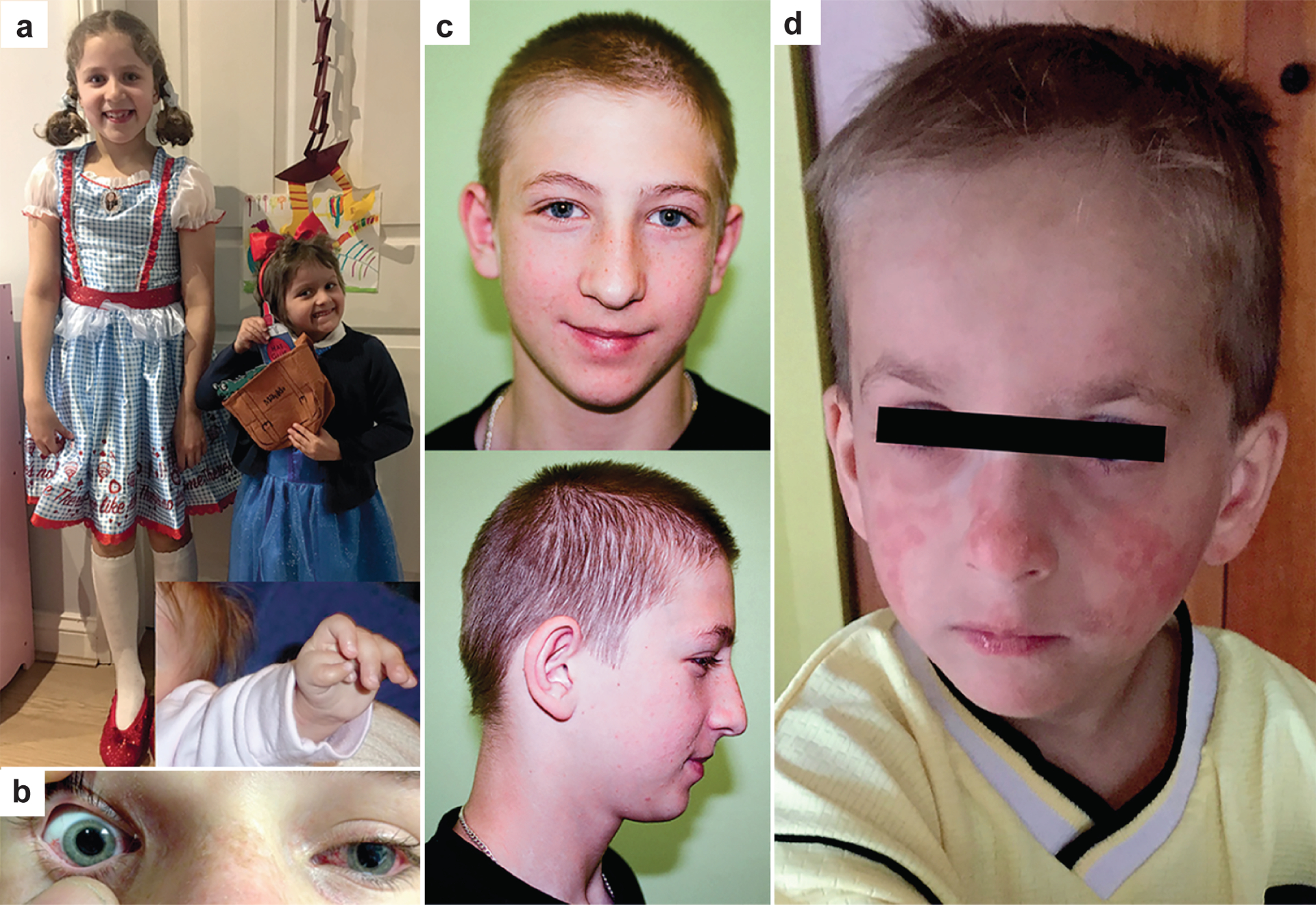
Characteristic features of the chromosome instability syndromes. a. The characteristic clinical features of children with Fanconi anaemia include extreme short stature, microcephaly and mid-facial hypoplasia, as illustrated in a 5-year-old girl (right) next to her unaffected 8-year-old sister. Inset shows the duplex thumb of the affected girl before surgical correction. b. Characteristic ocular telangiectasis of the exposed, but not the unexposed, bulbar conjunctiva in ataxia telangiectasia. c. The craniofacial features of those with Nijmegen breakage syndrome include receding forehead, receding mandible and prominent mid face with long nose. d. Characteristic sun-sensitive facial erythema in a young boy with Bloom syndrome.
Figure 6. The natural history of the chromosome breakage disorders.
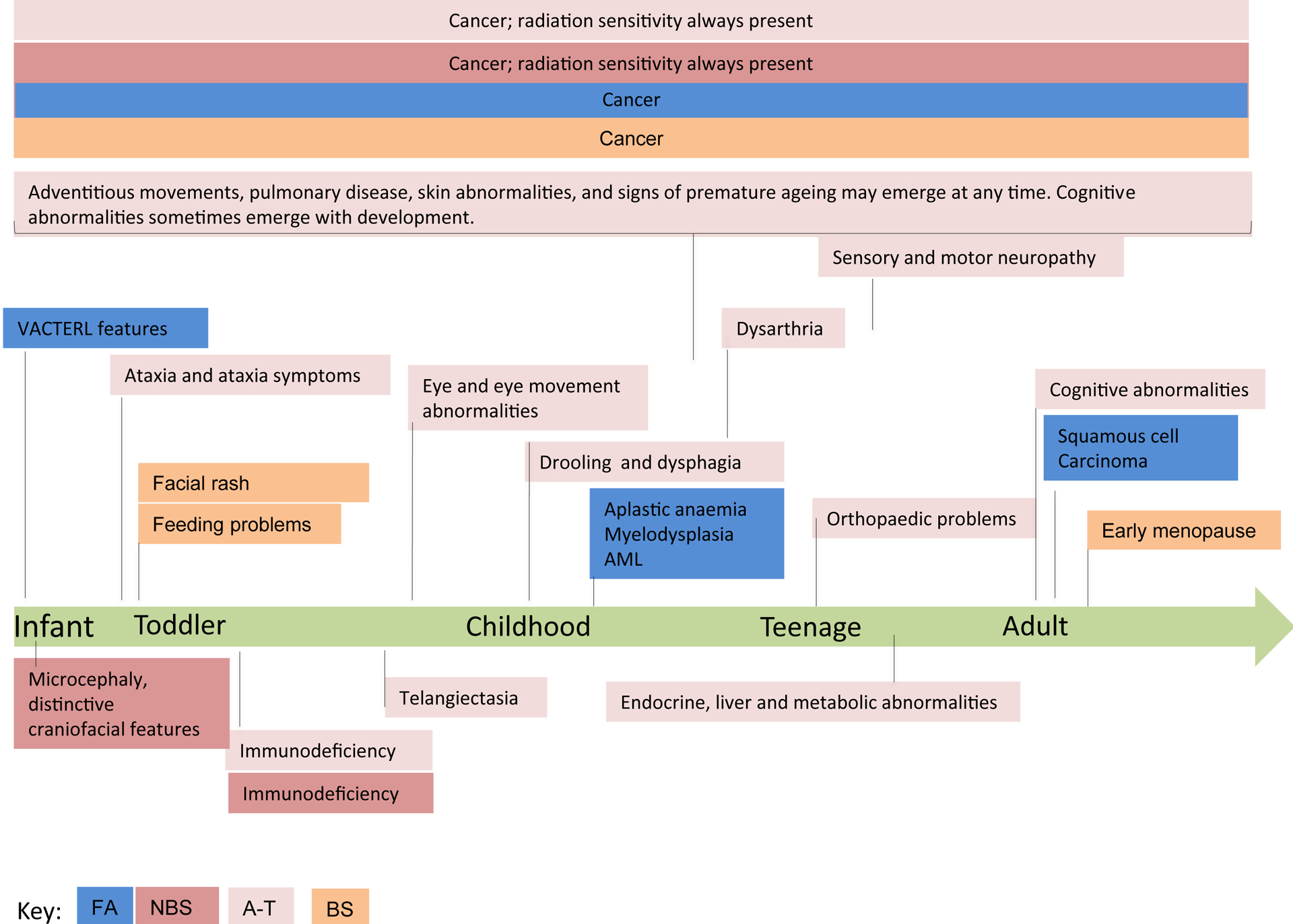
Each disorder has its own spectrum of age-related clinical features; in the case of most individuals with Fanconi anaemia this is focused on the consequences of bone marrow failure; in those with ataxia telangiectasia the progressive neurodegeneration and requirement for wheelchair use; in NBS the immunodeficiency and learning difficulties and in Bloom Syndrome small size and slowness with certainty about the diagnosis. All the disorders have a greatly increased likelihood of developing cancer.
Diagnosis.
FA is often diagnosed on the basis of bone marrow failure, even when other clinical findings have received prior medical attention. The diagnosis is confirmed by demonstration of the characteristic cellular cross linker sensitivity upon exposure to mitomycin C (MMC) or diepoxybutane (DEB). Ambiguous results obtained with peripheral blood lymphocytes can be caused by genetic mosaicism, and need confirmation using fibroblasts115. The detailed genetic diagnosis is determined using exon and panel approaches on next-generation sequencing platforms116. Once confirmed, the individual phenotypic manifestations should be assessed in detail, and include a bone marrow aspirate with cytogenetic analysis, including FISH for chromosomal gains of 3q and loss of chromosome 7112. For other clinical manifestations, which can involve every organ system, functional assessment and imaging of the central nervous system (CNS), kidneys, heart, ears and hearing, eyes and vision, is carried out, and includes detailed endocrine investigations, as hypothyroidism, in particular, is common in those with FA114,117–119. Therefore, a workup of suspected FA should include an abdominal ultrasound and a hearing test and routine biochemistry workup including thyroid function test.
The detailed genetic information can be used for antenatal diagnosis and family screening for mutations in familial cancer-associated FANC genes. Siblings should always be screened, even in the absence of clinical findings, as the clinical manifestations can be variable even between those with the same mutations4.
Ataxia telangiectasia
Presentation.
A-T is complex and substantial variability exists in the severity and appearance of different features120. We use the designations of “classic” and “mild” A-T to distinguish ends of the clinical spectrum. In the classic, or more severe, form of the disease (also known as typical, early onset or childhood onset A-T), ataxia first becomes apparent as children start to sit and walk, and an initial wobbly gait fails to improve. Children also have problems standing or sitting still and may sway slowly side-to-side or backwards. In childhood, ataxia progresses with requirement for wheelchair mobility typically beginning in the second decade of life. Eye movement abnormalities emerge in early school years. Dysarthria of speech can occur at any time and may or may not progress. Swallowing difficulties typically worsen in late school and early teen years. Involuntary movements can occur at any age. An important early manifestation may be the increased tendency for sino-pulmonary infections due to variable immunodeficiency and increasing difficulty with swallowing. Cancer and pulmonary disease are the two major causes of death by early adulthood121. An increasing number of individuals manifest a less severe form of A-T (also known as mild, variant, atypical, late onset or adult onset A-T). Those with mild A-T present with less severe features or later onset manifestations and generally have longer survival.122–125 .
Early in life, patients with A-T often manifest features of variable immunodeficiency with associated laboratory features126. They may also experience poor growth, delayed pubertal development with gonadal dysgenesis and early menopause. As the patients age, they may experience neuropathy, glucose intolerance and insulin-resistant diabetes, elevated cholesterol and triglycerides, non-alcoholic steatosis and cirrhosis, elevated serum transaminases, low vitamin D levels and osteopenia/osteoporosis127,128. Indeed, signs of premature ageing such as graying hair and skin changes may also occur in those with A-T127.
The paradigmatic ocular telangiectasia often appear after onset of neurological symptoms; their absence is a common cause for delayed diagnosis129 (Figure 3). Other disorders have features that partially overlap with the A-T phenotype, including cerebral palsy, congenital oculo-motor apraxia, Friedreich’s ataxia, AOA1, AOA2, ATLD, NBS and SCAN1. These disorders can be distinguished from A-T by the whole of the clinical course, neurological examination and selected laboratory tests. In some cases, genetic or protein assessment is necessary. Genetic analysis, and the absence of ATM protein or function, generally correlates with the A-T phenotype130,131 . Detection of more cases of mild A-T can be expected with increasing use of whole-exome sequencing.
Diagnosis and cascade screening.
A clinical diagnosis of A-T is suggested by combination of characteristic neurological and non-neurological clinical symptoms and laboratory findings. Although, no single laboratory abnormality is invariantly present, individuals with A-T can show an elevated alpha-fetoprotein (AFP) level after 1 year of age, spontaneous and X-ray-induced chromosomal breaks and/or rearrangements in cultured lymphoblastoid cell lines (LCLs), reduced cell survival following irradiation132, and cerebellar atrophy on imaging that progresses and does not necessarily correlate with clinical phenotype. Immune abnormalities may include low low serum IgA, IgE, IgG and IgG subclasses; lymphopenia (especially affecting T cells) and decreased immune repertoire diversity126,133,134. Confirmatory evidence becomes important to those without the full constellation of symptoms. A definitive diagnosis is secured by confirming the absence or deficiency of ATM kinase activity, measured in either a lymphoblastoid cell line made from the patient’s blood or in fibroblasts derived from a skin biopsy, the identification of pathological mutations in ATM, or a combination of these findings. Elevated serum AFP is evident in ≥95% of patients with A-T, and should be evaluated in any child with unexplained ataxia of stance or gait >1 year of age20,135.
Pre-natal genetic diagnosis is possible when prospective parents each have identifiable pathogenetic mutations in ATM136,137. A recent advance has led to frequent pre-symptomatic diagnosis. In combination with exome sequencing, the newborn screening test for severe combined immunodeficiency (SCID) can identify infants born with other disorders, including A-T, that involve a deficiency or absence of T and B lymphocytes138,139. Despite the lack of a disease modifying therapy, early diagnosis permits timely genetic counselling and family education as well as aggressive supportive care. Furthermore, cost-effective carrier testing can be performed in families in whom the ATM mutations have been identified in an affected child. In situations where the pathogenetic mutations are not known, but a definitive diagnosis of AT has been made, ATM-region haplotype analysis can be used to determine carrier status amongst related family members. Carrier testing in the general population is costly and challenging because of frequent variants of unknown significance in the very large ATM gene.
Nijmegen breakage syndrome
Presentation.
A hallmark symptom of NBS is a progressive microcephaly, which is observed from birth onwards, and typical distinctive craniofacial features (Figure 5)20.The dysmorphic facial features are very similar among all patients and become more obvious with age21. Somatic development is delayed, birth weight, length and head circumference (OFC) are typically below normal. Infants show a growth deficit until the age of 2 or 3 years, when some gain in weight and height is observed. The growth spurt in boys is poor, in girls absent21. Girls show no pubertal spurt and poor development of secondary sex characteristics, due to ovarian insufficiency140. Puberty in boys is initiated spontaneously and progresses normally. Congenital genito-urinary tract anomalies occur. Both the immunodeficiency and the chromosome instability may predispose patients with NBS to tumour development at an early age. By the age of 20 years >40% of patients with NBS develop cancer141. The great majority of malignancies are of lymphoid origin; the most frequent is non-Hodgkin lymphoma. Several patients are known to have developed a second malignancy. Solid tumours including rhabdomyosarcoma have less frequently been noted.
Respiratory infections are present in most children. Recurrent pneumonia and bronchitis may result in bronchiectasis, respiratory insufficiency and premature death from respiratory failure. Meningitis, sinusitis and otitis media with draining ears are observed in some children, as are gastrointestinal infections with diarrhoea and urinary tract infections. Opportunistic infections are very rare20. Disturbed antibody responses to tetanus, Haemophilus influenzae type B, diphtheria, polio and hepatitis B have been reported.
Diagnosis and cascade screening.
A clinical diagnosis is suggested by a microcephaly observed from birth onwards. Dysmorphic features become more obvious with age. Low serum levels of IgA, IgG and/or IgG2, lymphopenia, spontaneous and X ray induced chromosomal breaks and/or rearrangements in cultured cells from patients confirm the diagnosis. The characteristic immunodeficiency includes deficits of serum immunoglobulins, the most frequent of which is IgG (62%), followed by low or undetectable levels of IgA (57%). In contrast, IgM concentrations are normal in 61% and elevated in 14% of patients21. Deficiency of IgG subclasses (especially IgG2) can be masked in patients with normal concentrations of total IgG142. Lymphocyte subpopulations show reduction in absolute numbers of total CD3+ T cells and of CD4+ T cells in most patients. CD4+CD45RA+ T cells are almost lacking, there is a profound decrease in αβ CD8+ T cells but up to threefold increase in γδ CD8+ T cells. Natural killer cell counts are normal in most patients143. The absolute number of CD19+CD20+ B cells is reduced in most patients144. In 2003 two case reports were published. Both boys suffered from medulloblastoma and were treated with craniospinal irradiation. This resulted in severe toxicity and both boys died145,146.
Cytogenetic aberrations are present in 10–45% of metaphases of phytohaemagglutinin (PHA)-cultured T cells from NBS patients. Most of the rearrangements occur preferentially between chromosomes 7 and 14 and are typically inversions and translocations, with breakpoints at the site of immunoglobulin or T cell receptor genes.20. In colony-forming assays, NBS cells are 3–5 times more sensitive to ionising radiation or radiomimetic drugs than normal cells. NBS cells also display radioresistant DNA synthesis147.
Neonatal screening for severe primary immunodeficiencies began in 2008. Patients with severe combined immunodeficiency (SCID) have absent or reduced T cell numbers and reduced or non-functional B cells, similar to NBS, which can be detected using dried blood spot testing138. A patient with NBS detected by newborn screening has been described148.
Bloom syndrome
Presentation.
Suspicion of a diagnosis of BS is generally based on failure to thrive combined with the observation of other features, including microcephaly, a facial rash, non-facial skin pigmentation abnormalities, repeated chest and ear infections, as well as a lack of normal growth and weight gain (Figure 5). Males show infertility and there is subfertility in females149. With a few notable exceptions (prostate cancer and melanoma), virtually all cancer types are reported to occur150, which distinguishes BS from other chromosome instability disorders. Many of the reported cases are amongst persons of Ashkenazi Jewish ancestry, reflecting a founder mutation present in approximately 1% of that population. The other significant founder mutation occurs in Slavic populations with an allele frequency of approximately 0.4%.
Diagnosis.
Small size and a rash on the face are fairly non-specific and frequently lead to misdiagnosis. A path to the correct diagnosis usually requires the expertise of a clinical geneticist. Even in well-resourced settings, the diagnosis can take 3–5 years from birth. Many cases of BS were identified from general categories such as idiopathic intrauterine growth deficiency, primordial dwarfism, and failure to thrive. Some cases have been misdiagnosed as other rare syndromes; for example, a misdiagnosis of Russell Silver dwarfism is not infrequent. Historically, suspected cases were tested with a cytogenetic assay to determine the frequency of SCEs in peripheral lymphocytes, because until recently this test was pathognomonic for BS. However, elevated SCEs have been identified in cells from individuals with several BS-like disorders caused by hypomorphic mutations in TOP3A or RMI1. Consequently, direct DNA sequencing of the BLM gene is a more definitive test, although in some cases the results can give ambiguous data if the identified variant is not obviously disease-causing. In the case of persons of Ashkenazi Jewish ancestry, the prevalence of the founder BLMAsh mutation makes this analysis more definitive. The rarity of the disorder has largely precluded the development of widespread screening programmes, although BLMAsh mutation analysis within the Ashkenazi Jewish population is now more common. Prevention of conception is practised in a very limited sense, via the pre-nuptual identification of carriers of the BLMAsh mutation in certain orthodox Jewish communities151. Prenatal diagnosis is possible with the SCE assay or by BLM mutation analysis.
Management
A common requirement for all these disorders is the need for surveillance for cancer development. Cancer diagnosis can be at any age in FA, A-T and NBS and most frequently in early adulthood in BS. It is important, therefore, that consideration is given, at any age, to the possibility that a tumour is the cause of any unexplained symptoms and that appropriate tests are carried out.
Fanconi anaemia
Historically, bone marrow failure is the most common and significant manifestation of FA. Platelet counts above 30 ×109/L can often be tolerated for years without substantial complications and managed conservatively with watch and wait. The need for intervention arises if bleeding, transfusion dependency or infectious complications evolve. As with other bone marrow failure syndromes, FA-associated hypoplastic haematopoiesis can respond to low dose androgens, which seems to be safe and reasonably tolerated, with many patients maintaining satisfactory blood counts for up to several years152. Haematopoietic manifestations of FA and importantly the risk of leukaemic transformation are corrected with haematopoietic stem cell transplantation (HSCT); with the use of T-cell depleted bone marrow grafts and fludarabine-based conditioning, patients undergoing matched family or unrelated transplants have excellent outcomes153. HSCT outcome in adults and later stages of disease progression with pre-leukaemic changes and overt leukaemia is also improving154,155. When a matched sibling or unrelated donor is available, transplantation can be considered early and elective.
With a growing number of teenagers, young and middle-aged adults with FA, many of whom have had HSCT, non-haematological problems evolve and can become life-limiting117. Chronic organ dysfunction as a result of FA itself or HSCT for FA-associated BMF, such as endocrine dysfunction (hypothyroidisms, growth failure, early menopause and infertility) or impaired heart, lung or kidney function, need assessment, monitoring and appropriate management. The most concerning problem for adults with FA is the development of SCCs in the aero-digestive and ano-genital regions156 (Figure 2), which are difficult to manage as they are often multifocal; due to the patients’ inherited cross-linker sensitivity, the treatment can be very toxic157,158. Enrolment in a dedicated screening programme with regular detailed inspection of the head and neck and ano-genital region and upper GI endoscopy is important for early effective management, and many centres provide a dedicated service for those patients.
Ataxia telangiectasia
A-T is a multisystem disease, in which management is symptomatic and supportive. Regarding the neurological symptoms, no therapy can slow degeneration, but in some patients intervention may partially ameliorate symptoms. Drugs that may be prescribed for neurological symptoms include trihexphenidyl (an antimuscarinic), amantadine (an antiparkinsonian), baclofen (an antispastic) and botulinum toxin (a paralytic) and less commonly gabapentin (an anticonvulsant), clonazepam (a tranquilizer and antiseizure medication) and pregabalin (a calcium channel blocker typically used to treat epilepsy)159. Vision is typically normal, although reading and other saccade-based visual tasks are difficult. Large print or visual targeting techniques may be helpful160. Bracing or surgical correction (e.g. tendon transfer) may improve ankle stability to enable walking or weight bearing. Severe scoliosis requiring surgical intervention is relatively uncommon161.
All people with A-T should have at least one comprehensive immunologic evaluation to assess the number and type of B and T cells (which should be reassessed if the patient undergoes chemotherapy or is treated for longer than a few weeks with a corticosteroid), to assess levels of serum immunoglobulins (especially IgG, IgM and IgA) and to assess antibody responses to T cell- dependent and T cell-independent vaccines161. If antibody function is normal, all routine childhood immunizations should be given, except the measles, mumps, rubella (MMR) vaccine (see below)162. The risk:benefit ratio of the MMR vaccine may need to be reassessed if any of those diseases become locally endemic; if that occurs, another strategy would be to use prophylactic gamma globulin until the outbreak is under control. Individuals with normal ability to make antibody should receive an annual influenza vaccine, and additional pneumococcal vaccines at intervals to maintain high levels of anti-pneumococcal antibodies. All household members should also receive the influenza vaccine. People with impaired antibody function should receive standard immunoglobulin replacement therapy. Despite having low T-cell numbers, prophylactic antibiotics to prevent opportunistic infections are generally not necessary unless people are treated with chronic corticosteroids, other T-cell immunosuppressive drugs, or chemotherapy. Immunological tests should be repeated if problems with infections occur or worsen161,162.
Chronic cutaneous granulomas can be associated with A-T163,164. These have been associated with replication incompetent vaccine strain rubella virus detected by PCR165–167. Smaller or superficial granulomas can be treated with high-potency topical corticosteroids and/or cyclosporine A whereas more extensive lesions may respond to TNF inhibitors168, direct injection of steroids into the lesion(s)169 or combination therapy (for example, topical steroids and IV gamma globulin)170. No anti-viral drug has yet been found to be effective.
Chronic lung disease is responsible for approximately one-third of the deaths in A-T and early intervention is crucial for preventing or slowing its development. Pulmonary function tests (PFTs) should be performed in all children with A-T starting at 6 years of age and continued annually162,171. Management may include the liberal use of antibiotics and corticosteroids (BOX 3). Recurrent lung infections may involve dysfunctional swallow with aspiration162. Some people with A-T can be taught to drink, chew and swallow more safely reducing the risk of aspiration. As the nutritional deficit in some people with A-T may be more severe than previously appreciated172 173, early nutritional intervention and ongoing nutritional support and education for patients, families and caregivers are crucial. Dieticians can recommend ways to improve nutrition (e.g. use of high calorie foods or food supplements). A gastrostomy tube (G-tube or feeding tube) may be recommended174 175 if a child cannot eat enough to grow or weight at any age cannot be maintained; if dysphagia with aspiration results in respiratory compromise and/or mealtimes are too long or stressful176
BOX 3. Management of pulmonary symptoms in A-T162,230.
- Liberal use of antibiotics for
- persistent and/or prolonged upper and lower respiratory symptoms including those that follow a respiratory illness
- chronic cough with mucus or cough that does not respond to pulmonary clearance techniques
- individuals with muco-purulent secretions from the chest or sinuses
Examination of respiratory secretions (from bronchoscopy or induced sputum) may direct antibody therapy for lung infections and help prevent bronchiectasis
Prophylactic macrolides, inhaled aminoglycoside and/or fluoroquinolones may help reduce exacerbations in people with low lung function, recurrent pneumonias, or bronchiectasis
Corticosteroids may be beneficial for people with AT and ILD
Bronchodilators may be useful for treating restrictive (with a component of obstructive) lung disease in A-T
- Clearance of oral and bronchial secretions (using the manual method or with an acapella device or chest physiotherapy vest) can help limit injury from acute and chronic pulmonary infections
- Evaluation by a pulmonary specialist is necessary
- Use of chest physiotherapy requires an adequate cough to remove secretions
- An acapella device is useful for those with a weak cough or decreased lung reserve
Inspiratory muscle training (IMT) may improve respiratory strength and quality of life231. Low dose chest and sinus CT scans should be performed if symptoms are unresponsive to therapy to rule out bronchiectasis, fibrosis, ILD and tumors
Notes:
A pulmonary evaluation should be performed prior to surgical procedures requiring anesthesia.
All people with A-T should avoid secondhand smoke exposure and have minimal exposure to other environmental pollutants.
Adequate nutrition to maintain normal body mass index may help maintain respiratory muscle strength and minimize progression of lung disease.
Gender differences with regard to lung disease may exist.
Cancer treatment should take place only at specialist oncology centers and after consultation with a clinician who has specific expertise in A-T. Standard cancer therapy regimens need to be modified to minimize or avoid cytotoxicity from radiomimetic drugs. Radiation therapy should be used rarely and only at reduced doses. Cyclophosphamide use must be monitored as it has been associated with a later onset of severe haemorrhage from bladder telangiectasia177. Even with therapy modifications, some people with A-T who have late stage cancers will develop chemotherapy toxicities178. Bone marrow transplants have been successfully performed for haematopoietic cancers in A-T179,180 but routine use is not currently recommended.
During the school years, children with A-T will need special attention to the barriers faced in school. Recommended modifications for education are described in BOX 4.
BOX 4. Recommendations for school in patients with AT.
As individuals with A-T have neurological problems from an early age (e.g.resulting in abnormal eye movements for reading, hand movements for writing/typing etc) in the absence of any learning difficulty, considerable practical help with schooling is the standard.
Speech-language pathologists may aid communication skills and help educate others about the need for longer response times for people with A-T; however, traditional speech therapy is rarely helpful
Early use of computers with word completion software and other technologies are helpful
As hearing is normal and does not deteriorate, an emphasis should be placed on oral learning (e.g. audiobooks)
Classroom aides can help with writing, mealtimes, toileting and with transportation throughout the school
Fatigue is a significant part of life with A-T, therefore the need for rest time, shortened school days, a reduced class schedule, reduced homework and modified tests should be revisited as often as circumstances warrant
As with all children, social interactions with peers are important and should always be taken into consideration
Children with A-T often have excellent insight into how best to solve functional problems and their involvement should be encouraged
Nijmegen breakage syndrome
Monitoring of the immune system is important throughout the whole life of a patient with NBS as even patients with normal absolute B lymphocyte counts experience significant humoral deficiencies requiring IVIG therapy, which is used in ~68% of patients144. HSCT can correct the hematopoietic defect and underlying immunodeficiency in NBS181. Survival is superior when reduced-intensity conditioning (RIC) is used, with patients not experiencing relapse of malignancy (median follow up 6 years) in one retrospective analysi144. Umbilical cord blood transplantation is less common but in one study rapid and substantial progress in the development of psychomotor and physical skills occurred in the post-transplant period182.
The prognosis for patients with NBS and malignancies is still poor. Chemotherapy has to be adapted and radiotherapy omitted. In haematological malignancies, curative treatment is possible, adjusting the intensity of therapy to individual risk factors183,184. For example, reducing chemotherapy up to 50% especially when using anthracyclines, methotrexate and alkylating agents, is possible. Epipophyllotoxins (etoposide, teniposide), bleomycin and radiotherapy should be omitted141. Dosage-reduction of chemotherapeutic drugs seemed to have no disadvantages and reduced toxic adverse effects but does not prevent second malignancies185.
Bloom syndrome
Cancer is the main cause of early death in those with BS, and the predisposition includes the development of multiple cancers and cancer types, including leukaemias, lymphomas and carcinomas150. An early onset of the disease is also a prominent feature, with a mean age at cancer diagnosis of ~25 years. The main approach to cancer management is heightened surveillance, supported by lifestyle interventions that can help lessen cancer incidence (including minimization of tobacco use, sun exposure (which can also help address facial rash in BS), and irradiation from medical devices or naturally occurring sources such as radon). Awareness of symptoms of cancer and seeking prompt medical attention is considered to be the first defence.
For lymphomas and carcinomas, surgical resection of early lesions most frequently results in cure. Recommendations for cancer surveillance have been developed based on experience in other cancer-prone syndromes150. Although clinical trials on the efficacy of the surveillance recommendations have yet to be conducted, the successful increase in long-term survival of persons with Li-Fraumeni syndrome through frequent imaging studies offers hope that a similar success can be achieved in BS186. The recommended imaging studies in BS include abdominal ultrasonography every 3 months beginning at diagnosis and ending at age 8 years for Wilms tumour; whole body MRI every 1–2 years beginning at age 12–13 for lymphoma; annual colonoscopy and biannual fecal immunochemical test beginning at age 10–12 years for colorectal cancer; annual breast MRI beginning at age 18 years; annual skin examination for skin cancer; HPV vaccine for both boys and girls and annual Pap smears for females after reaching adolescence. When individuals with BS have developed cancer, medical providers should be aware of the risk of therapy-related, secondary malignancies. Standard weight-based chemotherapy regimens have resulted in life-threatening toxicities150. Dose reduction of the genotoxic chemotherapeutic agents by at least 50% is essential and usually well tolerated. Radiotherapy should be minimized unless it is the only realistic option for cure, and alkylating agents, such as busulfan and cyclophosphamide, should be avoided. Some chemotherapeutic could be tolerated at full weight-based doses, including kinase inhibitors and steroids149.
At present no remedy can address growth restriction in BS. Growth hormone therapy has had varying effects on growth; however, the question of whether this increases cancer risk is unresolved187. Feeding problems are common in children and infants, and there is a marked reduction in adipose tissue. Feeding intervention has been tried at some centres, but no systematic studies have been conducted. Use of high-calorie diets and anti-reflux medication should be considered. Approximately 20% of individuals with BS have developed type II diabetes. Fasting blood sugar measurements and screening for impaired glucose tolerance with haemoglobin A1c are recommended annually beginning at age 10 years to identify pre-diabetes and initiate standard preventive measures. An annual lipid profile and testing thyroid function should begin at age 10 years.
Individuals with BS often have deficiencies in immunoglobulins and are subject to recurrent infections150. Those individuals with recurrent sinusitis, more than one incidence of pneumonia in a 10-year period, multiple episodes of bronchitis, or other opportunistic infections, should consult an immunologist. Defects in humoral immunity can be managed with a preferred weekly subcutaneous injection of immunoglobulin or monthly Intravenous immunoglobulin. Finally, women with BS may have early menopause and may benefit from assisted reproductive technology. No remedy for infertility has been found for men with BS, although there is a single case report of confirmed paternity in a man with BS.
Quality of Life
Various voluntary patient organisations and support groups in different countries collaborate closely with scientific and medical experts to find effective life improving therapies and provide education and support to families affected by Fanconi anaemia, Ataxia telangiectasia and Nijmegen Breakage Syndrome; the rarest of these groups is supported by the Bloom Syndrome Association.
Fanconi anaemia
The impact of FA on the quality of life depends on the severity of the phenotype with organ dysfunction, timing and consequences of bone marrow failure and the need for HSCT, and cancer development. As with other chronic and life limiting diseases, the effect on the family can be profound188. Individuals affected with FA and individuals with a very mild phenotype can have a nearly normal life until their fourth decade; then sometimes the diagnosis is made when subtle clinical patterns are recognised113. Classical cases with bone marrow failure in childhood, radial ray abnormalities and short stature normally improve for a long period following successful haematopoietic reconstitution after HSCT, but in many cases this period can be affected by extreme short stature and disability from limb abnormalities. The dramatically increasing incidence of SCC affecting individuals with FA in the third and fourth decade is having a detrimental effect on quality of life in adult patients with FA, sometimes requiring repeated major and sometimes disfiguring surgery158, and is often life limiting.
Ataxia telangiectasia
Children with A-T will experience varying degrees of difficulty with school performance due to impaired fine and gross motor coordination (limiting ability to write and use a computer); dysarthria, delay in speech initiation, lack of facial expression, and delayed response times to visual and verbal cues (limiting ability to communicate); and oculomotor apraxia (limiting ability to read). Mental and physical fatigue are common. Individuals with A-T may be further burdened by the appearance of cognitive impairment, even when the impairment itself is mild or does not exist; however, social awareness is typically normal. This disparity can lead to social isolation and depression. Even with these difficulties, many people with A-T have found ways to overcome these difficulties; especially in the presence of a supportive environment in the school. As survival and quality of life have improved, a small but increasing number of people with A-T have been able to transition to higher education and independent living with support.
Nijmegen breakage syndrome
Developmental milestones are reached at expected times during the first years of life. Patients with normal intelligence or learning difficulty of variable degree have been reported. Longitudinal follow up studies indicate that small children (pre-school) are mostly within the normal range, but go on to develop IQ deficiency, which ranges from mild to moderate. Most children have a striking psychomotor hyperactivity. At older ages, all patients tested were mildly or moderately delayed. All are capable of good social interactions21. So far, there have not been any reports of NBS patients having offspring.
Bloom syndrome
BS impacts both the affected person and the parents or guardians entrusted with that person’s upbringing and education. For the guardians, there can be an emotional struggle of medical uncertainty until a definitive diagnosis is made, made more difficult by having to deal with the feeding problems and sun sensitivity, as well as a search for the explanation for the small size. After the diagnosis, there is the struggle to understand its impact, to learn what is known and not known about the syndrome, and to identify and organize the personal and societal resources needed to cope with that diagnosis. For the persons with BS, aside from for the accommodations that need to be made for the sun sensitivity and small size, day-to-day life might not be that different compared to anyone else, except for the extra attention received due to their small size. For most persons with BS, intellectual development is normal, although some have difficulty with subjects that require a high level of abstract thought. As children become adults, they come to know the risks that are attached to their diagnosis, and the likelihood that their lifespan might be foreshortened189.
Outlook
Fanconi anaemia
The past two decades have seen dramatic progress in the understanding of the genetics, molecular biology and disease mechanism in FA, and placed FA research firmly in the context of cancer and aging. Clinically, with wide availability of donors and improved conditioning regimens, HSCT has become a routine treatment modality for haematological manifestations of FA, which is likely to further improve as even higher risk transplantions in adults and using unmatched or haplo-identical donors are now successfully carried out154,190. While there is still a lot to learn about the role of the FANC pathway in haematopoietic maintenance, clinically for many patients the haematopoietic defect of FA can be successfully corrected. Also for patients without any suitable donor, results of current gene therapy trials aiming to restore impaired haematopoiesis by correction of patient-derived haematopoietic progenitor cells are promising191. This progress has transformed FA to a chronic condition of variable severity affecting an increasing number of adults who are now in their fourth and fifth decades. While efforts in identifying compounds that might affect the cellular FA defect show some promise192,193, the most pressing clinical problem is the high incidence of epithelial cancers and their management. Detailed understanding of the pathogenesis of SCC in FA will be important for effective prevention and monitoring, and targeted strategies for treatment are urgently needed.
Ataxia telangiectasia
Whole genome sequencing and epigenetic analyses may help identify modifiers of A-T disease severity and reveal additional genotype-phenotype correlations. Analyses of data from growing patient registries194,195will inform natural history, improve disease management and aid therapy development.
Difficulty with coordination, to varying degrees, is experienced by all patients with A-T. However, it is not yet known why the cerebellum is so severely affected in A-T, while other areas of the brain are unaffected. Although small animal models of A-T have failed to accurately recapitulate the human neurological phenotype the neurophenotype of the larger and longer lived porcine model for A-T is currently being studied. Researchers are investigating ways to apply recent breakthroughs in the fields of gene and mutation targeted therapies to A-T.
Risk factors for pulmonary decline need to be identified, and the contribution of inflammation to pulmonary disease in A-T needs further investigation. Optimal protocols for preventing decline in lung function and treating lung disease do not yet exist. MRI lung imaging techniques will help advance the field. Additionally, biomarkers and risk factors for the development of cancer in A-T need to be identified. Less toxic treatment regimens are critically needed. Present attention to symptomatic disease modifying therapies include, low-dose corticosteroids (e.g. dexamethasone196–198 and betamethasone199,200, 4-aminopyridine201 202, cannabinoids (e.g. Cannabidiol oil and Marinol), nicotinamide riboside203 as well as mutation-targeted204,205 and non-viral gene therapy approaches.
Nijmegen breakage syndrome
More knowledge of the immunodeficiency in NBS might provide a better understanding of the development of malignancy, especially lymphomas. NBS patients showed much lower numbers of αβ T cells (both CD4+ as well as CD8+) but normal numbers of γδ T cells. Circulating T cells show signs of a senescent phenotype present from young age, which might explain the T cell immune deficiency206. Patients with NBS have a high risk to develop a malignancy. Improvements of survival is possible with haematopoietic stem cell transplantation. Reduced conditioning regimens, similar to those used for patients with Fanconi anemia, are well tolerated. Due to the substantial risk of mixed chimerism patients with NBS may tolerate more intensive conditioning regimens than patients with Fanconi anemia, although this requires further observation (Wolska-Kusnierz 2015). It was demonstrated recently that antisense oligonucleotides could enforce alternative splicing in NBS patient cells, generating a p80-nibrin protein. Injecting the same antisense sequences as morpholinos in humanized NBS mice led to efficient alternative splicing in vivo207.
Bloom syndrome
Cancer risk is the most pressing issue for persons living with BS; the development of novel cancer therapies that target the particular cellular vulnerabilities of BS cancers holds the best hope for extending life expectancy. At present, however, there are no therapies that exploit this so-called ‘synthetic lethality’ approach to treatment in the way that PARP inhibitors have been used in BRCA1-deficient and BRCA2-deficient tumours208. Identifying treatments without toxicity in BS are needed. Furthermore, the efficacy of new immunotherapy approaches to treat cancer needs to be evaluated in BS. Although BS mouse models exist that could be used to test new therapeutics and biological questions relating to body size, there is an urgent need to develop better human cancer models (such as cultured tumour cell lines and patient-derived xenograft models) in BS. Other novel animal models are also needed to address biological questions where mouse models are less valuable, such as the use of porcine epidermis as a model for human skin.
The idea of gene therapy or correction in BS is often considered by the families affected189, especially in the age of CRISPR–Cas9 genome editing, but the practical issues surrounding reagent delivery make BS a poor first choice to test new advances in this area. There are also many questions regarding the somatic chimerism of corrected, uncorrected, and genetically damaged cells that need to be addressed.
Acknowledgements
Fanconi anaemia studies in the AS laboratory are supported by RO1HL120922 and R01CA204127. AS is a HHMI faculty scholar.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Bluteau D et al. Biallelic inactivation of REV7 is associated with Fanconi anemia. The Journal of clinical investigation 126, 3580–3584, doi: 10.1172/jci88010 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ceccaldi R, Sarangi P & D’Andrea AD The Fanconi anaemia pathway: new players and new functions. Nature reviews. Molecular cell biology 17, 337–349, doi: 10.1038/nrm.2016.48 (2016). [DOI] [PubMed] [Google Scholar]; Biochemistry and cell biology of the FANC pathway proteins
- 3.Knies K et al. Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anemia. The Journal of clinical investigation 127, 3013–3027, doi: 10.1172/jci92069 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neveling K, Endt D, Hoehn H & Schindler D Genotype-phenotype correlations in Fanconi anemia. Mutation research 668, 73–91, doi: 10.1016/j.mrfmmm.2009.05.006 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Rosenberg PS, Tamary H & Alter BP How high are carrier frequencies of rare recessive syndromes? Contemporary estimates for Fanconi Anemia in the United States and Israel. American journal of medical genetics. Part A 155a, 1877–1883, doi: 10.1002/ajmg.a.34087 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.German J, Sanz MM, Ciocci S, Ye TZ & Ellis NA Syndrome-causing mutations of the BLM gene in persons in the Bloom’s Syndrome Registry. Human mutation 28, 743–753, doi: 10.1002/humu.20501 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Auerbach AD Fanconi anemia and its diagnosis. Mutation research 668, 4–10, doi: 10.1016/j.mrfmmm.2009.01.013 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reiman A et al. Lymphoid tumours and breast cancer in ataxia telangiectasia; substantial protective effect of residual ATM kinase activity against childhood tumours. British journal of cancer 105, 586–591, doi: 10.1038/bjc.2011.266 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suarez F et al. Incidence, presentation, and prognosis of malignancies in ataxia-telangiectasia: a report from the French national registry of primary immune deficiencies. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 33, 202–208, doi: 10.1200/jco.2014.56.5101 (2015). [DOI] [PubMed] [Google Scholar]; Incidence, presentation and prognosis of malignant disease in ataxia telangiactasia
- 10.Weemaes CM et al. A new chromosomal instability disorder: the Nijmegen breakage syndrome. Acta paediatrica Scandinavica 70, 557–564 (1981). [DOI] [PubMed] [Google Scholar]
- 11.Waltes R et al. Human RAD50 deficiency in a Nijmegen breakage syndrome-like disorder. American journal of human genetics 84, 605–616, doi: 10.1016/j.ajhg.2009.04.010 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Callen E et al. A common founder mutation in FANCA underlies the world’s highest prevalence of Fanconi anemia in Gypsy families from Spain. Blood 105, 1946–1949, doi: 10.1182/blood-2004-07-2588 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Kutler DI & Auerbach AD Fanconi anemia in Ashkenazi Jews. Familial cancer 3, 241–248, doi: 10.1007/s10689-004-9565-8 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Meyer S et al. Fanconi anaemia, BRCA2 mutations and childhood cancer: a developmental perspective from clinical and epidemiological observations with implications for genetic counselling. Journal of medical genetics 51, 71–75, doi: 10.1136/jmedgenet-2013-101642 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Woods CG, Bundey SE & Taylor AM Unusual features in the inheritance of ataxia telangiectasia. Human genetics 84, 555–562 (1990). [DOI] [PubMed] [Google Scholar]
- 16.Crawford TO, Skolasky RL, Fernandez R, Rosquist KJ & Lederman HM Survival probability in ataxia telangiectasia. Archives of disease in childhood 91, 610–611, doi: 10.1136/adc.2006.094268 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Varon R et al. Clinical ascertainment of Nijmegen breakage syndrome (NBS) and prevalence of the major mutation, 657del5, in three Slav populations. European journal of human genetics : EJHG 8, 900–902, doi: 10.1038/sj.ejhg.5200554 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Bouman A, van Koningsbruggen S, Karakullukcu MB, Schreuder WH & Lakeman P Bloom syndrome does not always present with sun-sensitive facial erythema. European journal of medical genetics 61, 94–97, doi: 10.1016/j.ejmg.2017.10.010 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Seemanova E et al. The Slavic NBN Founder Mutation: A Role for Reproductive Fitness? PloS one 11, e0167984, doi: 10.1371/journal.pone.0167984 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nijmegen breakage syndrome. The International Nijmegen Breakage Syndrome Study Group. Archives of disease in childhood 82, 400–406 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chrzanowska KH, Gregorek H, Dembowska-Baginska B, Kalina MA & Digweed M Nijmegen breakage syndrome (NBS). Orphanet journal of rare diseases 7, 13, doi: 10.1186/1750-1172-7-13 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cunniff C, Bassetti JA & Ellis NA Bloom’s Syndrome: Clinical Spectrum, Molecular Pathogenesis, and Cancer Predisposition. Molecular syndromology 8, 4–23, doi: 10.1159/000452082 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; Comprehensive coverage of the features of Bloom Syndrome and underlying molecular pathology
- 23.Meetei AR et al. A novel ubiquitin ligase is deficient in Fanconi anemia. Nature genetics 35, 165–170, doi: 10.1038/ng1241 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Garcia-Higuera I et al. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Molecular cell 7, 249–262 (2001). [DOI] [PubMed] [Google Scholar]
- 25.Smogorzewska A et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 129, 289–301, doi: 10.1016/j.cell.2007.03.009 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim Y et al. Regulation of multiple DNA repair pathways by the Fanconi anemia protein SLX4. Blood 121, 54–63, doi: 10.1182/blood-2012-07-441212 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klein Douwel D et al. XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Molecular cell 54, 460–471, doi: 10.1016/j.molcel.2014.03.015 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Howlett NG et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science (New York, N.Y.) 297, 606–609, doi: 10.1126/science.1073834 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Long DT, Raschle M, Joukov V & Walter JC Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science (New York, N.Y.) 333, 84–87, doi: 10.1126/science.1204258 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elia AE et al. RFWD3-Dependent Ubiquitination of RPA Regulates Repair at Stalled Replication Forks. Molecular cell 60, 280–293, doi: 10.1016/j.molcel.2015.09.011 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Inano S et al. RFWD3-Mediated Ubiquitination Promotes Timely Removal of Both RPA and RAD51 from DNA Damage Sites to Facilitate Homologous Recombination. Molecular cell 66, 622–634.e628, doi: 10.1016/j.molcel.2017.04.022 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Howlett NG, Taniguchi T, Durkin SG, D’Andrea AD & Glover TW The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Human molecular genetics 14, 693–701, doi: 10.1093/hmg/ddi065 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Garcia-Rubio ML et al. The Fanconi Anemia Pathway Protects Genome Integrity from R-loops. PLoS genetics 11, e1005674, doi: 10.1371/journal.pgen.1005674 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwab RA et al. The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Molecular cell 60, 351–361, doi: 10.1016/j.molcel.2015.09.012 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rickman K & Smogorzewska A Advances in understanding DNA processing and protection at stalled replication forks. The Journal of cell biology 218, 1096–1107, doi: 10.1083/jcb.201809012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schlacher K et al. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542, doi: 10.1016/j.cell.2011.03.041 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schlacher K, Wu H & Jasin M A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer cell 22, 106–116, doi: 10.1016/j.ccr.2012.05.015 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alter BP, Rosenberg PS & Brody LC Clinical and molecular features associated with biallelic mutations in FANCD1/BRCA2. Journal of medical genetics 44, 1–9, doi: 10.1136/jmg.2006.043257 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Offit K et al. Shared genetic susceptibility to breast cancer, brain tumors, and Fanconi anemia. Journal of the National Cancer Institute 95, 1548–1551 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Reid S et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nature genetics 39, 162–164, doi: 10.1038/ng1947 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Ceccaldi R et al. Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell stem cell 11, 36–49, doi: 10.1016/j.stem.2012.05.013 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walter D et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature 520, 549–552, doi: 10.1038/nature14131 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Hira A et al. Variant ALDH2 is associated with accelerated progression of bone marrow failure in Japanese Fanconi anemia patients. Blood 122, 3206–3209, doi: 10.1182/blood-2013-06-507962 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Langevin F, Crossan GP, Rosado IV, Arends MJ & Patel KJ Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 475, 53–58, doi: 10.1038/nature10192 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Pontel LB et al. Endogenous Formaldehyde Is a Hematopoietic Stem Cell Genotoxin and Metabolic Carcinogen. Molecular cell 60, 177–188, doi: 10.1016/j.molcel.2015.08.020 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matsuoka S et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science (New York, N.Y.) 316, 1160–1166, doi: 10.1126/science.1140321 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Shiloh Y & Ziv Y The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nature reviews. Molecular cell biology 14, 197–210 (2013). [PubMed] [Google Scholar]; Biochemistry and cell biology of the ATM protein.
- 48.Stewart GS et al. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 99, 577–587 (1999). [DOI] [PubMed] [Google Scholar]
- 49.Carney JP et al. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell 93, 477–486 (1998). [DOI] [PubMed] [Google Scholar]
- 50.Matsuura S et al. Positional cloning of the gene for Nijmegen breakage syndrome. Nature genetics 19, 179–181, doi: 10.1038/549 (1998). [DOI] [PubMed] [Google Scholar]
- 51.Varon R et al. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell 93, 467–476 (1998). [DOI] [PubMed] [Google Scholar]
- 52.Matsumoto Y et al. Two unrelated patients with MRE11A mutations and Nijmegen breakage syndrome-like severe microcephaly. DNA repair 10, 314–321, doi: 10.1016/j.dnarep.2010.12.002 (2011). [DOI] [PubMed] [Google Scholar]
- 53.Frappart PO et al. An essential function for NBS1 in the prevention of ataxia and cerebellar defects. Nature medicine 11, 538–544, doi: 10.1038/nm1228 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Shull ER et al. Differential DNA damage signaling accounts for distinct neural apoptotic responses in ATLD and NBS. Genes & development 23, 171–180, doi: 10.1101/gad.1746609 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Syed A & Tainer JA The MRE11-RAD50-NBS1 Complex Conducts the Orchestration of Damage Signaling and Outcomes to Stress in DNA Replication and Repair. Annual review of biochemistry 87, 263–294, doi: 10.1146/annurev-biochem-062917-012415 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stewart GS et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell 136, 420–434, doi: 10.1016/j.cell.2008.12.042 (2009). [DOI] [PubMed] [Google Scholar]
- 57.Devgan SS et al. Homozygous deficiency of ubiquitin-ligase ring-finger protein RNF168 mimics the radiosensitivity syndrome of ataxia-telangiectasia. Cell death and differentiation 18, 1500–1506, doi: 10.1038/cdd.2011.18 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stewart GS et al. RIDDLE immunodeficiency syndrome is linked to defects in 53BP1-mediated DNA damage signaling. Proceedings of the National Academy of Sciences of the United States of America 104, 16910–16915, doi: 10.1073/pnas.0708408104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moreira MC et al. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nature genetics 29, 189–193, doi: 10.1038/ng1001-189 (2001). [DOI] [PubMed] [Google Scholar]
- 60.Bras J et al. Mutations in PNKP cause recessive ataxia with oculomotor apraxia type 4. American journal of human genetics 96, 474–479, doi: 10.1016/j.ajhg.2015.01.005 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hoch NC et al. XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia. Nature 541, 87–91, doi: 10.1038/nature20790 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takashima H et al. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nature genetics 32, 267–272, doi: 10.1038/ng987 (2002). [DOI] [PubMed] [Google Scholar]
- 63.Gomez-Herreros F et al. TDP2 protects transcription from abortive topoisomerase activity and is required for normal neural function. Nature genetics 46, 516–521, doi: 10.1038/ng.2929 (2014). [DOI] [PubMed] [Google Scholar]
- 64.Kawale AS & Povirk LF Tyrosyl-DNA phosphodiesterases: rescuing the genome from the risks of relaxation. Nucleic acids research 46, 520–537, doi: 10.1093/nar/gkx1219 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pommier Y et al. Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2). DNA repair 19, 114–129, doi: 10.1016/j.dnarep.2014.03.020 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sakasai R & Iwabuchi K The distinctive cellular responses to DNA strand breaks caused by a DNA topoisomerase I poison in conjunction with DNA replication and RNA transcription. Genes & genetic systems 90, 187–194, doi: 10.1266/ggs.15-00023 (2016). [DOI] [PubMed] [Google Scholar]
- 67.Alvarez-Quilon A et al. ATM specifically mediates repair of double-strand breaks with blocked DNA ends. Nature communications 5, 3347, doi: 10.1038/ncomms4347 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Katyal S et al. Aberrant topoisomerase-1 DNA lesions are pathogenic in neurodegenerative genome instability syndromes. Nature neuroscience 17, 813–821, doi: 10.1038/nn.3715 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sordet O et al. Ataxia telangiectasia mutated activation by transcription- and topoisomerase I-induced DNA double-strand breaks. EMBO reports 10, 887–893, doi: 10.1038/embor.2009.97 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Das BB et al. Optimal function of the DNA repair enzyme TDP1 requires its phosphorylation by ATM and/or DNA-PK. The EMBO journal 28, 3667–3680, doi: 10.1038/emboj.2009.302 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Segal-Raz H et al. ATM-mediated phosphorylation of polynucleotide kinase/phosphatase is required for effective DNA double-strand break repair. EMBO reports 12, 713–719, doi: 10.1038/embor.2011.96 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zolner AE et al. Phosphorylation of polynucleotide kinase/ phosphatase by DNA-dependent protein kinase and ataxia-telangiectasia mutated regulates its association with sites of DNA damage. Nucleic acids research 39, 9224–9237, doi: 10.1093/nar/gkr647 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sykora P, Wilson DM 3rd & Bohr VA Repair of persistent strand breaks in the mitochondrial genome. Mechanisms of ageing and development 133, 169–175, doi: 10.1016/j.mad.2011.11.003 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Watters D et al. Localization of a portion of extranuclear ATM to peroxisomes. The Journal of biological chemistry 274, 34277–34282 (1999). [DOI] [PubMed] [Google Scholar]
- 75.Guo Z, Kozlov S, Lavin MF, Person MD & Paull TT ATM activation by oxidative stress. Science (New York, N.Y.) 330, 517–521, doi: 10.1126/science.1192912 (2010). [DOI] [PubMed] [Google Scholar]
- 76.Ambrose M, Goldstine JV & Gatti RA Intrinsic mitochondrial dysfunction in ATM-deficient lymphoblastoid cells. Human molecular genetics 16, 2154–2164, doi: 10.1093/hmg/ddm166 (2007). [DOI] [PubMed] [Google Scholar]
- 77.Valentin-Vega YA et al. Mitochondrial dysfunction in ataxia-telangiectasia. Blood 119, 1490–1500, doi: 10.1182/blood-2011-08-373639 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moreira MC et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nature genetics 36, 225–227, doi: 10.1038/ng1303 (2004). [DOI] [PubMed] [Google Scholar]
- 79.Alzu A et al. Senataxin associates with replication forks to protect fork integrity across RNA-polymerase-II-transcribed genes. Cell 151, 835–846, doi: 10.1016/j.cell.2012.09.041 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cohen S et al. Senataxin resolves RNA:DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nature communications 9, 533, doi: 10.1038/s41467-018-02894-w (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Skourti-Stathaki K, Proudfoot NJ & Gromak N Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Molecular cell 42, 794–805, doi: 10.1016/j.molcel.2011.04.026 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Becherel OJ et al. Senataxin plays an essential role with DNA damage response proteins in meiotic recombination and gene silencing. PLoS genetics 9, e1003435, doi: 10.1371/journal.pgen.1003435 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yeo AJ et al. R-loops in proliferating cells but not in the brain: implications for AOA2 and other autosomal recessive ataxias. PloS one 9, e90219, doi: 10.1371/journal.pone.0090219 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bunch H et al. Transcriptional elongation requires DNA break-induced signalling. Nature communications 6, 10191, doi: 10.1038/ncomms10191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Canela A et al. Genome Organization Drives Chromosome Fragility. Cell 170, 507–521.e518, doi: 10.1016/j.cell.2017.06.034 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gothe HJ et al. Spatial Chromosome Folding and Active Transcription Drive DNA Fragility and Formation of Oncogenic MLL Translocations. Molecular cell, doi: 10.1016/j.molcel.2019.05.015 (2019). [DOI] [PubMed] [Google Scholar]
- 87.Tresini M et al. The core spliceosome as target and effector of non-canonical ATM signalling. Nature 523, 53–58, doi: 10.1038/nature14512 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Suraweera A et al. Functional role for senataxin, defective in ataxia oculomotor apraxia type 2, in transcriptional regulation. Human molecular genetics 18, 3384–3396, doi: 10.1093/hmg/ddp278 (2009). [DOI] [PubMed] [Google Scholar]
- 89.Huang M & Verbeek DS Why do so many genetic insults lead to Purkinje Cell degeneration and spinocerebellar ataxia? Neuroscience letters, doi: 10.1016/j.neulet.2018.02.004 (2018). [DOI] [PubMed] [Google Scholar]
- 90.Soong BW & Morrison PJ Spinocerebellar ataxias. Handbook of clinical neurology 155, 143–174, doi: 10.1016/b978-0-444-64189-2.00010-x (2018). [DOI] [PubMed] [Google Scholar]
- 91.Choy KR & Watters DJ Neurodegeneration in ataxia-telangiectasia: Multiple roles of ATM kinase in cellular homeostasis. Developmental dynamics : an official publication of the American Association of Anatomists 247, 33–46, doi: 10.1002/dvdy.24522 (2018). [DOI] [PubMed] [Google Scholar]
- 92.Pfeiffer A et al. Ataxin-3 consolidates the MDC1-dependent DNA double-strand break response by counteracting the SUMO-targeted ubiquitin ligase RNF4. The EMBO journal 36, 1066–1083, doi: 10.15252/embj.201695151 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu Y & West SC More complexity to the Bloom’s syndrome complex. Genes & development 22, 2737–2742, doi: 10.1101/gad.1732808 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bizard AH & Hickson ID The dissolution of double Holliday junctions. Cold Spring Harbor perspectives in biology 6, a016477, doi: 10.1101/cshperspect.a016477 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chaganti RS, Schonberg S & German J A manyfold increase in sister chromatid exchanges in Bloom’s syndrome lymphocytes. Proceedings of the National Academy of Sciences of the United States of America 71, 4508–4512 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Martin CA et al. Mutations in TOP3A Cause a Bloom Syndrome-like Disorder. American journal of human genetics 103, 221–231, doi: 10.1016/j.ajhg.2018.07.001 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chu WK & Hickson ID RecQ helicases: multifunctional genome caretakers. Nature reviews. Cancer 9, 644–654, doi: 10.1038/nrc2682 (2009). [DOI] [PubMed] [Google Scholar]
- 98.Nimonkar AV, Ozsoy AZ, Genschel J, Modrich P & Kowalczykowski SC Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proceedings of the National Academy of Sciences of the United States of America 105, 16906–16911, doi: 10.1073/pnas.0809380105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ouyang KJ et al. SUMO modification regulates BLM and RAD51 interaction at damaged replication forks. PLoS biology 7, e1000252, doi: 10.1371/journal.pbio.1000252 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bhattacharyya S, Sandy A & Groden J Unwinding protein complexes in ALTernative telomere maintenance. Journal of cellular biochemistry 109, 7–15, doi: 10.1002/jcb.22388 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Deans AJ & West SC FANCM connects the genome instability disorders Bloom’s Syndrome and Fanconi Anemia. Molecular cell 36, 943–953, doi: 10.1016/j.molcel.2009.12.006 (2009). [DOI] [PubMed] [Google Scholar]
- 102.Dhar S & Brosh RM BLM’s balancing act and the involvement of FANCJ in DNA repair. Cell cycle (Georgetown, Tex.) 17, 2207–2220, doi: 10.1080/15384101.2018.1520567 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Meetei AR et al. A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Molecular and cellular biology 23, 3417–3426 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.de Renty C & Ellis NA Bloom’s syndrome: Why not premature aging?: A comparison of the BLM and WRN helicases. Ageing research reviews 33, 36–51, doi: 10.1016/j.arr.2016.05.010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Davies SL, North PS & Hickson ID Role for BLM in replication-fork restart and suppression of origin firing after replicative stress. Nature structural & molecular biology 14, 677–679, doi: 10.1038/nsmb1267 (2007). [DOI] [PubMed] [Google Scholar]
- 106.Drosopoulos WC, Kosiyatrakul ST & Schildkraut CL BLM helicase facilitates telomere replication during leading strand synthesis of telomeres. The Journal of cell biology 210, 191–208, doi: 10.1083/jcb.201410061 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tippana R, Hwang H, Opresko PL, Bohr VA & Myong S Single-molecule imaging reveals a common mechanism shared by G-quadruplex-resolving helicases. Proceedings of the National Academy of Sciences of the United States of America 113, 8448–8453, doi: 10.1073/pnas.1603724113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Biebricher A et al. PICH: a DNA translocase specially adapted for processing anaphase bridge DNA. Molecular cell 51, 691–701, doi: 10.1016/j.molcel.2013.07.016 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sarlos K et al. Reconstitution of anaphase DNA bridge recognition and disjunction. Nature structural & molecular biology 25, 868–876, doi: 10.1038/s41594-018-0123-8 (2018). [DOI] [PubMed] [Google Scholar]
- 110.Langlois RG, Bigbee WL, Jensen RH & German J Evidence for increased in vivo mutation and somatic recombination in Bloom’s syndrome. Proceedings of the National Academy of Sciences of the United States of America 86, 670–674, doi: 10.1073/pnas.86.2.670 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kutler DI et al. A 20-year perspective on the International Fanconi Anemia Registry (IFAR). Blood 101, 1249–1256, doi: 10.1182/blood-2002-07-2170 (2003). [DOI] [PubMed] [Google Scholar]
- 112.Quentin S et al. Myelodysplasia and leukemia of Fanconi anemia are associated with a specific pattern of genomic abnormalities that includes cryptic RUNX1/AML1 lesions. Blood 117, e161–170, doi: 10.1182/blood-2010-09-308726 (2011). [DOI] [PubMed] [Google Scholar]
- 113.Huck K et al. Delayed diagnosis and complications of Fanconi anaemia at advanced age--a paradigm. British journal of haematology 133, 188–197, doi: 10.1111/j.1365-2141.2006.05998.x (2006). [DOI] [PubMed] [Google Scholar]
- 114.Petryk A et al. Endocrine disorders in Fanconi anemia: recommendations for screening and treatment. The Journal of clinical endocrinology and metabolism 100, 803–811, doi: 10.1210/jc.2014-4357 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Oostra AB, Nieuwint AW, Joenje H & de Winter JP Diagnosis of fanconi anemia: chromosomal breakage analysis. Anemia 2012, 238731, doi: 10.1155/2012/238731 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chandrasekharappa SC et al. Massively parallel sequencing, aCGH, and RNA-Seq technologies provide a comprehensive molecular diagnosis of Fanconi anemia. Blood 121, e138–148, doi: 10.1182/blood-2012-12-474585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schneider M, Chandler K, Tischkowitz M & Meyer S Fanconi anaemia: genetics, molecular biology, and cancer - implications for clinical management in children and adults. Clinical genetics 88, 13–24, doi: 10.1111/cge.12517 (2015). [DOI] [PubMed] [Google Scholar]; Clinical management of Fanconi anaemia
- 118.Sathyanarayana V et al. Patterns and frequency of renal abnormalities in Fanconi anaemia: implications for long-term management. Pediatric nephrology (Berlin, Germany) 33, 1547–1551, doi: 10.1007/s00467-018-3952-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Stivaros SM et al. Central nervous system abnormalities in Fanconi anaemia: patterns and frequency on magnetic resonance imaging. The British journal of radiology 88, 20150088, doi: 10.1259/bjr.20150088 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Crawford TO et al. Quantitative neurologic assessment of ataxia-telangiectasia. Neurology 54, 1505–1509, doi: 10.1212/wnl.54.7.1505 (2000). [DOI] [PubMed] [Google Scholar]
- 121.RP S & E B Handbook of Clinical Neurology. (North Holland Publishing, 1972). [Google Scholar]
- 122.Gilad S et al. Genotype-phenotype relationships in ataxia-telangiectasia and variants. American journal of human genetics 62, 551–561, doi: 10.1086/301755 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Micol R et al. Morbidity and mortality from ataxia-telangiectasia are associated with ATM genotype. The Journal of allergy and clinical immunology 128, 382–389.e381, doi: 10.1016/j.jaci.2011.03.052 (2011). [DOI] [PubMed] [Google Scholar]
- 124.Schon K et al. Genotype, extrapyramidal features, and severity of variant ataxia-telangiectasia. Annals of neurology 85, 170–180, doi: 10.1002/ana.25394 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Verhagen MM et al. Presence of ATM protein and residual kinase activity correlates with the phenotype in ataxia-telangiectasia: a genotype-phenotype study. Human mutation 33, 561–571, doi: 10.1002/humu.22016 (2012). [DOI] [PubMed] [Google Scholar]
- 126.Nowak-Wegrzyn A, Crawford TO, Winkelstein JA, Carson KA & Lederman HM Immunodeficiency and infections in ataxia-telangiectasia. The Journal of pediatrics 144, 505–511, doi: 10.1016/j.jpeds.2003.12.046 (2004). [DOI] [PubMed] [Google Scholar]
- 127.Crawford TO Ataxia telangiectasia. Seminars in pediatric neurology 5, 287–294 (1998). [DOI] [PubMed] [Google Scholar]
- 128.Nissenkorn A et al. Endocrine abnormalities in ataxia telangiectasia: findings from a national cohort. Pediatric research 79, 889–894, doi: 10.1038/pr.2016.19 (2016). [DOI] [PubMed] [Google Scholar]
- 129.Cabana MD, Crawford TO, Winkelstein JA, Christensen JR & Lederman HM Consequences of the delayed diagnosis of ataxia-telangiectasia. Pediatrics 102, 98–100 (1998). [DOI] [PubMed] [Google Scholar]
- 130.Alterman N et al. Ataxia-telangiectasia: mild neurological presentation despite null ATM mutation and severe cellular phenotype. American journal of medical genetics. Part A 143a, 1827–1834, doi: 10.1002/ajmg.a.31853 (2007). [DOI] [PubMed] [Google Scholar]
- 131.Worth PF et al. Very mild presentation in adult with classical cellular phenotype of ataxia telangiectasia. Movement disorders : official journal of the Movement Disorder Society 28, 524–528, doi: 10.1002/mds.25236 (2013). [DOI] [PubMed] [Google Scholar]
- 132.Sun X et al. Early diagnosis of ataxia-telangiectasia using radiosensitivity testing. The Journal of pediatrics 140, 724–731, doi: 10.1067/mpd.2002.123879 (2002). [DOI] [PubMed] [Google Scholar]
- 133.Driessen GJ et al. Antibody deficiency in patients with ataxia telangiectasia is caused by disturbed B- and T-cell homeostasis and reduced immune repertoire diversity. The Journal of allergy and clinical immunology 131, 1367–1375.e1369, doi: 10.1016/j.jaci.2013.01.053 (2013). [DOI] [PubMed] [Google Scholar]
- 134.Sadighi Akha AA, Humphrey RL, Winkelstein JA, Loeb DM & Lederman HM Oligo-/monoclonal gammopathy and hypergammaglobulinemia in ataxia-telangiectasia. A study of 90 patients. Medicine 78, 370–381, doi: 10.1097/00005792-199911000-00002 (1999). [DOI] [PubMed] [Google Scholar]
- 135.Stray-Pedersen A et al. Alpha fetoprotein is increasing with age in ataxia-telangiectasia. European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology Society 11, 375–380, doi: 10.1016/j.ejpn.2007.04.001 (2007). [DOI] [PubMed] [Google Scholar]
- 136.Hellani A, Lauge A, Ozand P, Jaroudi K & Coskun S Pregnancy after preimplantation genetic diagnosis for Ataxia Telangiectasia. Molecular human reproduction 8, 785–788 (2002). [DOI] [PubMed] [Google Scholar]
- 137.Verlinsky Y et al. Preimplantation diagnosis for immunodeficiencies. Reproductive biomedicine online 14, 214–223 (2007). [DOI] [PubMed] [Google Scholar]
- 138.Borte S et al. Neonatal screening for severe primary immunodeficiency diseases using high-throughput triplex real-time PCR. Blood 119, 2552–2555, doi: 10.1182/blood-2011-08-371021 (2012). [DOI] [PubMed] [Google Scholar]
- 139.Mallott J et al. Newborn screening for SCID identifies patients with ataxia telangiectasia. Journal of clinical immunology 33, 540–549, doi: 10.1007/s10875-012-9846-1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chrzanowska KH et al. High prevalence of primary ovarian insufficiency in girls and young women with Nijmegen breakage syndrome: evidence from a longitudinal study. The Journal of clinical endocrinology and metabolism 95, 3133–3140, doi: 10.1210/jc.2009-2628 (2010). [DOI] [PubMed] [Google Scholar]
- 141.Pastorczak A, Szczepanski T & Mlynarski W Clinical course and therapeutic implications for lymphoid malignancies in Nijmegen breakage syndrome. European journal of medical genetics 59, 126–132, doi: 10.1016/j.ejmg.2016.01.007 (2016). [DOI] [PubMed] [Google Scholar]
- 142.Gregorek H, Chrzanowska KH, Michalkiewicz J, Syczewska M & Madalinski K Heterogeneity of humoral immune abnormalities in children with Nijmegen breakage syndrome: an 8-year follow-up study in a single centre. Clinical and experimental immunology 130, 319–324 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Michalkiewicz J et al. Abnormalities in the T and NK lymphocyte phenotype in patients with Nijmegen breakage syndrome. Clinical and experimental immunology 134, 482–490 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wolska-Kusnierz B et al. Nijmegen Breakage Syndrome: Clinical and Immunological Features, Long-Term Outcome and Treatment Options - a Retrospective Analysis. Journal of clinical immunology 35, 538–549, doi: 10.1007/s10875-015-0186-9 (2015). [DOI] [PubMed] [Google Scholar]; Coverage of the immunodeficiency that is an important part of the presentation of NBS
- 145.Bakhshi S et al. Medulloblastoma with adverse reaction to radiation therapy in nijmegen breakage syndrome. Journal of pediatric hematology/oncology 25, 248–251 (2003). [DOI] [PubMed] [Google Scholar]
- 146.Distel L, Neubauer S, Varon R, Holter W & Grabenbauer G Fatal toxicity following radio- and chemotherapy of medulloblastoma in a child with unrecognized Nijmegen breakage syndrome. Medical and pediatric oncology 41, 44–48, doi: 10.1002/mpo.10275 (2003). [DOI] [PubMed] [Google Scholar]
- 147.Taalman RD, Jaspers NG, Scheres JM, de Wit J & Hustinx TW Hypersensitivity to ionizing radiation, in vitro, in a new chromosomal breakage disorder, the Nijmegen Breakage Syndrome. Mutation research 112, 23–32 (1983). [DOI] [PubMed] [Google Scholar]
- 148.Patel JP et al. Nijmegen breakage syndrome detected by newborn screening for T cell receptor excision circles (TRECs). Journal of clinical immunology 35, 227–233, doi: 10.1007/s10875-015-0136-6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Flanagan M & Cunniff CM in GeneReviews((R)) (eds Adam MP et al. ) (University of Washington, Seattle University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved., 1993). [Google Scholar]
- 150.Cunniff C et al. Health supervision for people with Bloom syndrome. American journal of medical genetics. Part A 176, 1872–1881, doi: 10.1002/ajmg.a.40374 (2018). [DOI] [PubMed] [Google Scholar]; Recommendations for diagnosis, screening and symptom treatment in Bloom Syndrome
- 151.Kornreich R, Ekstein J, Edelmann L & Desnick RJ Premarital and prenatal screening for cystic fibrosis: experience in the Ashkenazi Jewish population. Genetics in medicine : official journal of the American College of Medical Genetics 6, 415–420, doi:10.109701.Gim.0000139510.00644.F7 (2004). [DOI] [PubMed] [Google Scholar]
- 152.Scheckenbach K et al. Treatment of the bone marrow failure in Fanconi anemia patients with danazol. Blood cells, molecules & diseases 48, 128–131, doi: 10.1016/j.bcmd.2011.11.006 (2012). [DOI] [PubMed] [Google Scholar]
- 153.Peffault de Latour R et al. Allogeneic hematopoietic stem cell transplantation in Fanconi anemia: the European Group for Blood and Marrow Transplantation experience. Blood 122, 4279–4286, doi: 10.1182/blood-2013-01-479733 (2013). [DOI] [PubMed] [Google Scholar]
- 154.Bierings M et al. Transplant results in adults with Fanconi anaemia. British journal of haematology 180, 100–109, doi: 10.1111/bjh.15006 (2018). [DOI] [PubMed] [Google Scholar]
- 155.Mehta PA et al. Radiation-free, alternative-donor HCT for Fanconi anemia patients: results from a prospective multi-institutional study. Blood 129, 2308–2315, doi: 10.1182/blood-2016-09-743112 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Alter BP, Giri N, Savage SA & Rosenberg PS Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica 103, 30–39, doi: 10.3324/haematol.2017.178111 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; Cancer occurrence in four bone marrow failure disorders, including Fanconi anaemia
- 157.Rosenberg PS, Alter BP & Ebell W Cancer risks in Fanconi anemia: findings from the German Fanconi Anemia Registry. Haematologica 93, 511–517, doi: 10.3324/haematol.12234 (2008). [DOI] [PubMed] [Google Scholar]
- 158.Kutler DI et al. Natural history and management of Fanconi anemia patients with head and neck cancer: A 10-year follow-up. The Laryngoscope 126, 870–879, doi: 10.1002/lary.25726 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hoche F et al. Neurodegeneration in ataxia telangiectasia: what is new? What is evident? Neuropediatrics 43, 119–129, doi: 10.1055/s-0032-1313915 (2012). [DOI] [PubMed] [Google Scholar]
- 160.Farr AK et al. Ocular manifestations of ataxia-telangiectasia. American journal of ophthalmology 134, 891–896, doi: 10.1016/s0002-9394(02)01796-8 (2002). [DOI] [PubMed] [Google Scholar]
- 161.Rothblum-Oviatt C et al. Ataxia telangiectasia: a review. Orphanet journal of rare diseases 11, 159, doi: 10.1186/s13023-016-0543-7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.McGrath-Morrow SA et al. Evaluation and management of pulmonary disease in ataxia-telangiectasia. Pediatric pulmonology 45, 847–859, doi: 10.1002/ppul.21277 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chiam LY et al. Cutaneous granulomas in ataxia telangiectasia and other primary immunodeficiencies: reflection of inappropriate immune regulation? Dermatology (Basel, Switzerland) 223, 13–19, doi: 10.1159/000330335 (2011). [DOI] [PubMed] [Google Scholar]
- 164.Shoimer I, Wright N & Haber RM Noninfectious Granulomas: A Sign of an Underlying Immunodeficiency? Journal of cutaneous medicine and surgery 20, 259–262, doi: 10.1177/1203475415626085 (2016). [DOI] [PubMed] [Google Scholar]
- 165.Bodemer C et al. Live rubella virus vaccine long-term persistence as an antigenic trigger of cutaneous granulomas in patients with primary immunodeficiency. Clinical microbiology and infection : the official publication of the European Society of Clinical Microbiology and Infectious Diseases 20, O656–663, doi: 10.1111/1469-0691.12573 (2014). [DOI] [PubMed] [Google Scholar]
- 166.Neven B et al. Cutaneous and Visceral Chronic Granulomatous Disease Triggered by a Rubella Virus Vaccine Strain in Children With Primary Immunodeficiencies. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 64, 83–86, doi: 10.1093/cid/ciw675 (2017). [DOI] [PubMed] [Google Scholar]
- 167.Perelygina L et al. Rubella persistence in epidermal keratinocytes and granuloma M2 macrophages in patients with primary immunodeficiencies. The Journal of allergy and clinical immunology 138, 1436–1439.e1411, doi: 10.1016/j.jaci.2016.06.030 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Mitra A, Gooi J, Darling J & Newton-Bishop JA Infliximab in the treatment of a child with cutaneous granulomas associated with ataxia telangiectasia. Journal of the American Academy of Dermatology 65, 676–677, doi: 10.1016/j.jaad.2010.06.060 (2011). [DOI] [PubMed] [Google Scholar]
- 169.Pinzon-Charry A, Kimble R & Peake J Intralesional steroids for the treatment of cutaneous granulomas in ataxia telangiectasia Intern Med J 43, 25 (2013). [Google Scholar]
- 170.Privette ED, Ram G, Treat JR, Yan AC & Heimall JR Healing of granulomatous skin changes in ataxia-telangiectasia after treatment with intravenous immunoglobulin and topical mometasone 0.1% ointment. Pediatric dermatology 31, 703–707, doi: 10.1111/pde.12411 (2014). [DOI] [PubMed] [Google Scholar]
- 171.McGrath-Morrow SA et al. Pulmonary function in children and young adults with ataxia telangiectasia. Pediatric pulmonology 49, 84–90, doi: 10.1002/ppul.22760 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; Incidence of an important clinical feature of children and young adults with classic ataxia telangiectasia
- 172.Ross LJ et al. Nutritional status of patients with ataxia-telangiectasia: A case for early and ongoing nutrition support and intervention. Journal of paediatrics and child health 51, 802–807, doi: 10.1111/jpc.12828 (2015). [DOI] [PubMed] [Google Scholar]
- 173.Pommerening H et al. Body composition, muscle strength and hormonal status in patients with ataxia telangiectasia: a cohort study. Orphanet journal of rare diseases 10, 155, doi: 10.1186/s13023-015-0373-z (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Stewart E et al. Growth and nutrition in children with ataxia telangiectasia. Archives of disease in childhood 101, 1137–1141, doi: 10.1136/archdischild-2015-310373 (2016). [DOI] [PubMed] [Google Scholar]
- 175.Krauthammer A et al. Long-term nutritional and gastrointestinal aspects in patients with ataxia telangiectasia. Nutrition (Burbank, Los Angeles County, Calif.) 46, 48–52, doi: 10.1016/j.nut.2017.08.008 (2018). [DOI] [PubMed] [Google Scholar]
- 176.Lefton-Greif MA, Crawford TO, McGrath-Morrow S, Carson KA & Lederman HM Safety and caregiver satisfaction with gastrostomy in patients with Ataxia Telangiectasia. Orphanet journal of rare diseases 6, 23, doi: 10.1186/1750-1172-6-23 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lavin MF, Gueven N, Bottle S & Gatti RA Current and potential therapeutic strategies for the treatment of ataxia-telangiectasia. British medical bulletin 81–82, 129–147, doi: 10.1093/bmb/ldm012 (2007). [DOI] [PubMed] [Google Scholar]
- 178.Sandlund JT, Hudson MM, Kennedy W, Onciu M & Kastan MB Pilot study of modified LMB-based therapy for children with ataxia-telangiectasia and advanced stage high grade mature B-cell malignancies. Pediatric blood & cancer 61, 360–362, doi: 10.1002/pbc.24696 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Beier R et al. Allogeneic-matched sibling stem cell transplantation in a 13-year-old boy with ataxia telangiectasia and EBV-positive non-Hodgkin lymphoma. Bone marrow transplantation 51, 1271–1274, doi: 10.1038/bmt.2016.93 (2016). [DOI] [PubMed] [Google Scholar]
- 180.Ussowicz M, Musial J, Duszenko E, Haus O & Kalwak K Long-term survival after allogeneic-matched sibling PBSC transplantation with conditioning consisting of low-dose busilvex and fludarabine in a 3-year-old boy with ataxia-telangiectasia syndrome and ALL. Bone marrow transplantation 48, 740–741, doi: 10.1038/bmt.2012.207 (2013). [DOI] [PubMed] [Google Scholar]
- 181.Slack J et al. Outcome of hematopoietic cell transplantation for DNA double-strand break repair disorders. The Journal of allergy and clinical immunology 141, 322–328.e310, doi: 10.1016/j.jaci.2017.02.036 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; Outcomes on 26 NBS patients who underwent bone marrow transplantation.
- 182.Wozniak M, Krzywon M, Holda MK & Gozdzik J Reduced-intensity conditioning umbilical cord blood transplantation in Nijmegen breakage syndrome. Pediatric transplantation 19, E51–55, doi: 10.1111/petr.12420 (2015). [DOI] [PubMed] [Google Scholar]
- 183.Dembowska-Baginska B et al. Non-Hodgkin lymphoma (NHL) in children with Nijmegen Breakage syndrome (NBS). Pediatric blood & cancer 52, 186–190, doi: 10.1002/pbc.21789 (2009). [DOI] [PubMed] [Google Scholar]
- 184.Seidemann K et al. Non-Hodgkin’s lymphoma in pediatric patients with chromosomal breakage syndromes (AT and NBS): experience from the BFM trials. Annals of oncology : official journal of the European Society for Medical Oncology 11 Suppl 1, 141–145 (2000). [PubMed] [Google Scholar]
- 185.Bienemann K et al. Promising therapy results for lymphoid malignancies in children with chromosomal breakage syndromes (Ataxia teleangiectasia or Nijmegen-breakage syndrome): a retrospective survey. British journal of haematology 155, 468–476, doi: 10.1111/j.1365-2141.2011.08863.x (2011). [DOI] [PubMed] [Google Scholar]
- 186.Villani A et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: 11 year follow-up of a prospective observational study. The Lancet. Oncology 17, 1295–1305, doi: 10.1016/s1470-2045(16)30249-2 (2016). [DOI] [PubMed] [Google Scholar]
- 187.Brock PR, de Zegher F, Casteels-Van Daele M & Vanderschueren-Lodeweyckx M Malignant disease in Bloom’s syndrome children treated with growth hormone. Lancet (London, England) 337, 1345–1346 (1991). [DOI] [PubMed] [Google Scholar]
- 188.Zierhut HA & Bartels DM Waiting for the next shoe to drop: the experience of parents of children with fanconi anemia. Journal of genetic counseling 21, 45–58, doi: 10.1007/s10897-011-9394-5 (2012). [DOI] [PubMed] [Google Scholar]
- 189. www.bloomsyndromeassociation.org.
- 190.Ayas M et al. Allogeneic hematopoietic cell transplantation for fanconi anemia in patients with pretransplantation cytogenetic abnormalities, myelodysplastic syndrome, or acute leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 31, 1669–1676, doi: 10.1200/jco.2012.45.9719 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Rio P, Navarro S & Bueren JA Advances in Gene Therapy for Fanconi Anemia. Human gene therapy 29, 1114–1123, doi: 10.1089/hum.2018.124 (2018). [DOI] [PubMed] [Google Scholar]
- 192.Zhang H et al. TGF-beta Inhibition Rescues Hematopoietic Stem Cell Defects and Bone Marrow Failure in Fanconi Anemia. Cell stem cell 18, 668–681, doi: 10.1016/j.stem.2016.03.002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Zhang QS et al. Metformin improves defective hematopoiesis and delays tumor formation in Fanconi anemia mice. Blood 128, 2774–2784, doi: 10.1182/blood-2015-11-683490 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.--
- 195.--
- 196.Chessa L et al. Intra-erythrocyte infusion of dexamethasone reduces neurological symptoms in ataxia teleangiectasia patients: results of a phase 2 trial. Orphanet journal of rare diseases 9, 5, doi: 10.1186/1750-1172-9-5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Leuzzi V et al. Positive effect of erythrocyte-delivered dexamethasone in ataxia-telangiectasia. Neurology(R) neuroimmunology & neuroinflammation 2, e98, doi: 10.1212/nxi.0000000000000098 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Medicine., U. N. L. o. Clinicaltrials.gov. . (2016).
- 199.Zannolli R et al. A randomized trial of oral betamethasone to reduce ataxia symptoms in ataxia telangiectasia. Movement disorders : official journal of the Movement Disorder Society 27, 1312–1316, doi: 10.1002/mds.25126 (2012). [DOI] [PubMed] [Google Scholar]
- 200.Cirillo E et al. Minimum effective betamethasone dosage on the neurological phenotype in patients with ataxia-telangiectasia: a multicenter observer-blind study. European journal of neurology 25, 833–840, doi: 10.1111/ene.13606 (2018). [DOI] [PubMed] [Google Scholar]
- 201.Strupp M, Zwergal A & Brandt T Episodic ataxia type 2. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics 4, 267–273, doi: 10.1016/j.nurt.2007.01.014 (2007). [DOI] [PubMed] [Google Scholar]
- 202.Shaikh AG et al. Effects of 4-aminopyridine on nystagmus and vestibulo-ocular reflex in ataxia-telangiectasia. Journal of neurology 260, 2728–2735, doi: 10.1007/s00415-013-7046-4 (2013). [DOI] [PubMed] [Google Scholar]
- 203.Fang EF et al. NAD(+) Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell metabolism 24, 566–581, doi: 10.1016/j.cmet.2016.09.004 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Du L, Pollard JM & Gatti RA Correction of prototypic ATM splicing mutations and aberrant ATM function with antisense morpholino oligonucleotides. Proceedings of the National Academy of Sciences of the United States of America 104, 6007–6012, doi: 10.1073/pnas.0608616104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Lee P et al. SMRT compounds abrogate cellular phenotypes of ataxia telangiectasia in neural derivatives of patient-specific hiPSCs. Nature communications 4, 1824, doi: 10.1038/ncomms2824 (2013). [DOI] [PubMed] [Google Scholar]
- 206.Meijers RWJ et al. Circulating T Cells of Patients with Nijmegen Breakage Syndrome Show Signs of Senescence. Journal of clinical immunology 37, 133–142, doi: 10.1007/s10875-016-0363-5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Salewsky B et al. Directed Alternative Splicing in Nijmegen Breakage Syndrome: Proof of Principle Concerning Its Therapeutical Application. Molecular therapy : the journal of the American Society of Gene Therapy 24, 117–124, doi: 10.1038/mt.2015.144 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Farmer H et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921, doi: 10.1038/nature03445 (2005). [DOI] [PubMed] [Google Scholar]
- 209.Smetsers S et al. Heterozygote FANCD2 mutations associated with childhood T Cell ALL and testicular seminoma. Familial cancer 11, 661–665, doi: 10.1007/s10689-012-9553-3 (2012). [DOI] [PubMed] [Google Scholar]
- 210.Berwick M et al. Genetic heterogeneity among Fanconi anemia heterozygotes and risk of cancer. Cancer research 67, 9591–9596, doi: 10.1158/0008-5472.Can-07-1501 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Tischkowitz M, Easton DF, Ball J, Hodgson SV & Mathew CG Cancer incidence in relatives of British Fanconi Anaemia patients. BMC cancer 8, 257, doi: 10.1186/1471-2407-8-257 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Grobner SN et al. The landscape of genomic alterations across childhood cancers. Nature 555, 321–327, doi: 10.1038/nature25480 (2018). [DOI] [PubMed] [Google Scholar]
- 213.Chandrasekharappa SC et al. Assessing the spectrum of germline variation in Fanconi anemia genes among patients with head and neck carcinoma before age 50. Cancer 123, 3943–3954, doi: 10.1002/cncr.30802 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Thompson D et al. Cancer risks and mortality in heterozygous ATM mutation carriers. Journal of the National Cancer Institute 97, 813–822, doi: 10.1093/jnci/dji141 (2005). [DOI] [PubMed] [Google Scholar]
- 215.Goldgar DE et al. Rare variants in the ATM gene and risk of breast cancer. Breast cancer research : BCR 13, R73, doi: 10.1186/bcr2919 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Southey MC et al. PALB2, CHEK2 and ATM rare variants and cancer risk: data from COGS. Journal of medical genetics 53, 800–811, doi: 10.1136/jmedgenet-2016-103839 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Bailey MH et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 173, 371–385.e318, doi: 10.1016/j.cell.2018.02.060 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Huang KL et al. Pathogenic Germline Variants in 10,389 Adult Cancers. Cell 173, 355–370.e314, doi: 10.1016/j.cell.2018.03.039 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Helgason H et al. Loss-of-function variants in ATM confer risk of gastric cancer. Nature genetics 47, 906–910, doi: 10.1038/ng.3342 (2015). [DOI] [PubMed] [Google Scholar]
- 220.Roberts NJ et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer discovery 2, 41–46, doi: 10.1158/2159-8290.Cd-11-0194 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Seemanova E et al. Cancer risk of heterozygotes with the NBN founder mutation. Journal of the National Cancer Institute 99, 1875–1880, doi: 10.1093/jnci/djm251 (2007). [DOI] [PubMed] [Google Scholar]
- 222.Bogdanova N et al. Nijmegen Breakage Syndrome mutations and risk of breast cancer. International journal of cancer 122, 802–806, doi: 10.1002/ijc.23168 (2008). [DOI] [PubMed] [Google Scholar]
- 223.Cybulski C et al. NBS1 is a prostate cancer susceptibility gene. Cancer research 64, 1215–1219 (2004). [DOI] [PubMed] [Google Scholar]
- 224.Ciara E et al. Heterozygous germ-line mutations in the NBN gene predispose to medulloblastoma in pediatric patients. Acta neuropathologica 119, 325–334, doi: 10.1007/s00401-009-0608-y (2010). [DOI] [PubMed] [Google Scholar]
- 225.Damiola F et al. Rare key functional domain missense substitutions in MRE11A, RAD50, and NBN contribute to breast cancer susceptibility: results from a Breast Cancer Family Registry case-control mutation-screening study. Breast cancer research : BCR 16, R58, doi: 10.1186/bcr3669 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Gruber SB et al. BLM heterozygosity and the risk of colorectal cancer. Science (New York, N.Y.) 297, 2013, doi: 10.1126/science.1074399 (2002). [DOI] [PubMed] [Google Scholar]
- 227.Bohm S & Bernstein KA The role of post-translational modifications in fine-tuning BLM helicase function during DNA repair. DNA repair 22, 123–132, doi: 10.1016/j.dnarep.2014.07.007 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Ellis NA et al. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell 83, 655–666 (1995). [DOI] [PubMed] [Google Scholar]
- 229.Karow JK, Chakraverty RK & Hickson ID The Bloom’s syndrome gene product is a 3’−5’ DNA helicase. The Journal of biological chemistry 272, 30611–30614 (1997). [DOI] [PubMed] [Google Scholar]
- 230.Bhatt JM et al. ERS statement on the multidisciplinary respiratory management of ataxia telangiectasia. European respiratory review : an official journal of the European Respiratory Society 24, 565–581, doi: 10.1183/16000617.0066-2015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Felix E, Gimenes AC & Costa-Carvalho BT Effects of inspiratory muscle training on lung volumes, respiratory muscle strength, and quality of life in patients with ataxia telangiectasia. Pediatric pulmonology 49, 238–244, doi: 10.1002/ppul.22828 (2014). [DOI] [PubMed] [Google Scholar]