

Abstract
The nucleosome remodeling and histone deacetylase (NuRD) complex physically associates with BCL11B to regulate murine T‐cell development. However, the function of NuRD complex in mature T cells remains unclear. Here, we characterize the fate and metabolism of human T cells in which key subunits of the NuRD complex or BCL11B are ablated. BCL11B and the NuRD complex bind to each other and repress natural killer (NK)‐cell fate in T cells. In addition, T cells upregulate the NK cell‐associated receptors and transcription factors, lyse NK‐cell targets, and are reprogrammed into NK‐like cells (ITNKs) upon deletion of MTA2, MBD2, CHD4, or BCL11B. ITNKs increase OPA1 expression and exhibit characteristically elongated mitochondria with augmented oxidative phosphorylation (OXPHOS) activity. OPA1‐mediated elevated OXPHOS enhances cellular acetyl‐CoA levels, thereby promoting the reprogramming efficiency and antitumor effects of ITNKs via regulating H3K27 acetylation at specific targets. In conclusion, our findings demonstrate that the NuRD complex and BCL11B cooperatively maintain T‐cell fate directly by repressing NK cell‐associated transcription and indirectly through a metabolic‐epigenetic axis, providing strategies to improve the reprogramming efficiency and antitumor effects of ITNKs.
Keywords: CHD4, MBD2, MTA2, OPA1, T‐cell fate
Subject Categories: Cancer, Immunology, Metabolism
Mature T cell reprogramming into natural killer cells is actively inhibited by NuRD complex and BCL11B‐mediated regulation of H3K27 acetylation.

Introduction
Bcl11b is a transcription factor for murine T‐cell commitment (Ikawa et al, 2010; Li et al, 2010a, 2010b; Isoda et al, 2017; Hosokawa et al, 2018; Hu et al, 2018) and directly represses the transcription of Id2 and Zbtb16, which are essential for NK cell development (Constantinides et al, 2014; Delconte et al, 2016; Hosokawa et al, 2018, 2020). Other groups and we reported that inactivating BCL11B in human T cells induces their reprogramming into NK‐like cells (induced T‐to‐NK cells, ITNKs) (Maluski et al, 2019; Sottile et al, 2021; Wu et al, 2021; Jiang et al, 2022). ITNKs retain the expression of a diverse TCR repertoire and upregulate NK‐cell‐associated markers and transcription factors. In addition, ITNKs recognize and lyse tumor cells efficiently in vitro and in vivo. Preliminary clinical results (NCT:03882840) show that autologous ITNKs provide modest benefits in patients with refractory and advanced solid tumors without causing severe adverse effects (Jiang et al, 2022).
BCL11B physically associates with the nucleosome remodeling and histone deacetylase (NuRD) complex, a major transcriptional corepressor, via interaction with MTA1 and MTA2 to repress the expression of downstream targets, including HIV‐1 LTR, Id2, and Cdkn1c in T cells (Cismasiu et al, 2005, 2008; Topark‐Ngarm et al, 2006; Dubuissez et al, 2016; Hosokawa et al, 2018). The NuRD complex also plays essential roles in T‐cell fate and immune response (Lu et al, 2008; Rothenberg, 2012; Dege & Hagman, 2014; Loughran et al, 2017; Shen et al, 2018). It is a critical cofactor binding to transcription factors to control T‐cell differentiation (Gao et al, 2022b). Ablation of Mta2 derepresses the expression of genes that are directly bound to and repressed by Bcl11b in murine T cells (Hosokawa et al, 2018). However, it remains to assess the function of the NuRD complex and BCL11B in maintaining T cell identity.
Mitochondria are highly dynamic organelles that undergo fission and fusion and provide energy for cellular activities, including T cell development and function (Ghesquière et al, 2014; Dimeloe et al, 2017). T cells exhibit unique metabolic profiles in different states (MacIver et al, 2013; Kishton et al, 2017). Naïve T (Tn) cells remain in a quiescent state through oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO), while effector T (Te) cells upregulate aerobic glycolysis to support differentiation and rapid proliferation with activation (Chang et al, 2013). When antigens are cleared, long‐lived memory T (Tm) cells facilitate OXPHOS and FAO to meet energy requirements (van der Windt et al, 2012; Sukumar et al, 2013; O'Sullivan et al, 2014). Memory T cells rely on FAO to fuel OXPHOS, in which mitochondria fuse to form linear or tubular networks. At the same time, mitochondria can also undergo fission, generating discrete and fragmented mitochondria in activated T cells (West et al, 2011; Buck et al, 2016; Kishton et al, 2017). Mitochondrial fusion is mediated by outer membrane fusion proteins (mitofusin1 and mitofusin2) and inner membrane fusion proteins (optic atrophy 1, OPA1) (Chen et al, 2003; Song et al, 2009; Kishton et al, 2017). OPA1‐mediated mitochondrial fusion enhances FAO and OXPHOS metabolism in Tm cells (Buck et al, 2016; Kishton et al, 2017). Alternative splicing of OPA1 gives rise to long forms that can be cleaved into short forms by peptidases (Ishihara et al, 2006; Del Dotto et al, 2018). The presence of long‐(L) and short‐form (S)‐OPA1 is necessary for mitochondrial dynamics (Song et al, 2007). L‐OPA1 is indispensable for mitochondrial fusion, while S‐OPA1 promotes mitochondrial fragmentation (Anand et al, 2014; Baker et al, 2014). In contrast, dynamin‐related protein 1 (Drp1), a mitochondrial fission protein, regulates mitochondrial fragmentation to decrease OXPHOS metabolism through phosphorylation (Losón et al, 2013; Sukumar et al, 2013).
To understand the NuRD complex function in mature T cells, we ablated MTA2, MBD2, and CHD4, three critical subunits of the NuRD complex, individually in human primary T cells using CRISPR/Cas9 and characterized the transcription profiles and cytotoxicity of these genetically modified T cells. We also studied how BCL11B and the NuRD complex control the morphology and function of mitochondria in T cells.
Results
Ablation of subunits of the NuRD complex reprograms human T cells into ITNKs
To investigate whether BCL11B associates with the NuRD complex to maintain T‐cell identity, we performed coimmunoprecipitation (co‐IP) assays in human T cells and found that NuRD subunits, including MTA2, MBD2, and CHD4, physically interact with BCL11B (Fig 1A). We next ablated MTA2, MBD2, or CHD4 individually with the CRISPR/Cas9 technology in primary human T cells and cultured them with standard T‐cell culture conditions (Appendix Fig S1A). Like sgBCL11B‐transduced T cells, cultured sgMTA2‐transduced T cells showed CD8+NKp46+ and CD4+NKp30+ populations, although the percentages of CD8+NKp46+ and CD4+NKp30+ cells were lower in sgMTA2‐transduced T cells than in sgBCL11B‐transduced T cells (Fig 1B and C). In cultures of sgMBD2‐transduced or sgCHD4‐transduced T cells, NKp30 expression was significantly upregulated in both CD4+ and CD8+ T cells as compared to sgCtrl T cells, although fewer CD8+NKp46+ cells were detected (Fig 1B and C). In contrast, the ablation of MTA1, another component of the NuRD complex, did not alter the expression of NKp30 or NKp46 in T cells (Appendix Fig S1B). In addition, sgBCL11B‐, sgMTA2‐, sgMBD2‐, and sgCHD4‐transduced T cells efficiently lysed K562 cells in vitro (Fig 1D), suggesting that these cells recognized NK‐cell targets. Collectively, these results show that human primary T cells can be reprogrammed into ITNKs when lacking subunits of the NuRD complex.
Figure 1. Deletion of subunits of the NuRD complex reprograms human T cells into ITNKs.
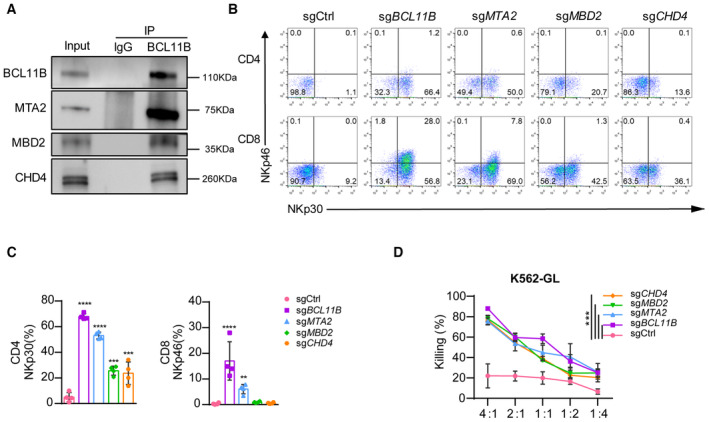
- Immunoprecipitation of the nuclear extract of peripheral blood mononuclear cells (PBMC)‐derived T cells with anti‐BCL11B antibodies and western blot analysis with antibodies against the NuRD components MBD2, MTA2, and CHD4.
- Human T cells from PBMC were electroporated with sgCtrl, sgBCL11B, sgMBD2, sgMTA2, or sgCHD4 and Cas9 protein after activation with CD3/CD28 antibodies for 36 h. Those T cells were cultured in T cell culture medium containing rh‐IL2 (300 U/ml) for 10 days. Representative flow cytometric detection of NKp30 and NKp46 expression in PBMC‐derived T cells transduced with sgCtrl, sgBCL11B, sgMBD2, sgMTA2, or sgCHD4. The data shown is representative of four individual healthy donors.
- Graph summarizing the percentages of NKp30+ in CD4 T cells and the percentages of NKp46+ cells in CD8 T cells that were transduced with sgCtrl, sgBCL11B, sgMBD2, sgMTA2, or sgCHD4.
- After culture for 10 days, T cells in (B) were incubated with the K562‐GL target cells at various E:T ratios for 24 h. Killing assays showing the percent cytotoxicity of T cells transduced with sgCtrl, sgBCL11B, sgMBD2, sgMTA2, or sgCHD4 against K562‐GL cells. The data represents killing percentage of cells from a donor.
Data information: In (C and D), data are presented as mean ± SD (N = 4 individual healthy donors). **P ≤ 0.01, ***P ≤ 0.001 and ****P ≤ 0.001, one‐way ANOVA (C) or two‐way ANOVA (D) with Tukey's multiple comparisons test.
Source data are available online for this figure.
BCL11B associates with the NuRD complex to repress the expression of NK‐cell‐associated genes in human T cells
To further understand the characteristics of NuRD complex‐deficient ITNKs, CD8+NKp30+ and CD4+NKp30+ populations from BCL11B‐, MTA2‐, MBD2‐, and CHD4‐deficient ITNKs were purified and subjected to RNA‐sequencing (RNA‐seq) analysis. The transcriptional profiles of ITNKs derived from sgMTA2‐, sgMBD2‐, and sgCHD4‐transduced T cells were like those of sgBCL11B‐transduced T cells but not to those of sgCtrl‐transduced T cells (Figs 2A and EV1A and B). Gene Ontology (G.O.) enrichment analysis showed that derepressed genes in both CD4+ and CD8+ populations in sgMTA2‐, sgMBD2‐, sgCHD4‐, and sgBCL11B‐transduced T cells as compared to sgCtrl‐transduced T cells were enriched in genes related to T‐cell activation and NK‐cell‐mediated immunity (Fig EV1C and D). In addition, there were 450 and 172 Differentially Expressed Genes (DEGs), including ID2, ZNF683, and ITGAX encoding CD11C, FCER1G, and TNFRSF18, that were derepressed in sgBCL11B‐, sgMTA2‐, sgMBD2‐, and sgCHD4‐transduced CD8+ and CD4+ T cells, respectively, compared to control T cells (Fig 2B and C). These analyses demonstrate that ITNKs generated from MTA2‐, MBD2‐, and CHD4‐deficient T cells share similar gene expression profiles with BCL11B‐deficient ITNKs in T‐cell activation and NK‐cell‐mediated immunity.
Figure 2. BCL11B associates with the NuRD complex to repress the expression of NK‐cell‐associated genes in human T cells.
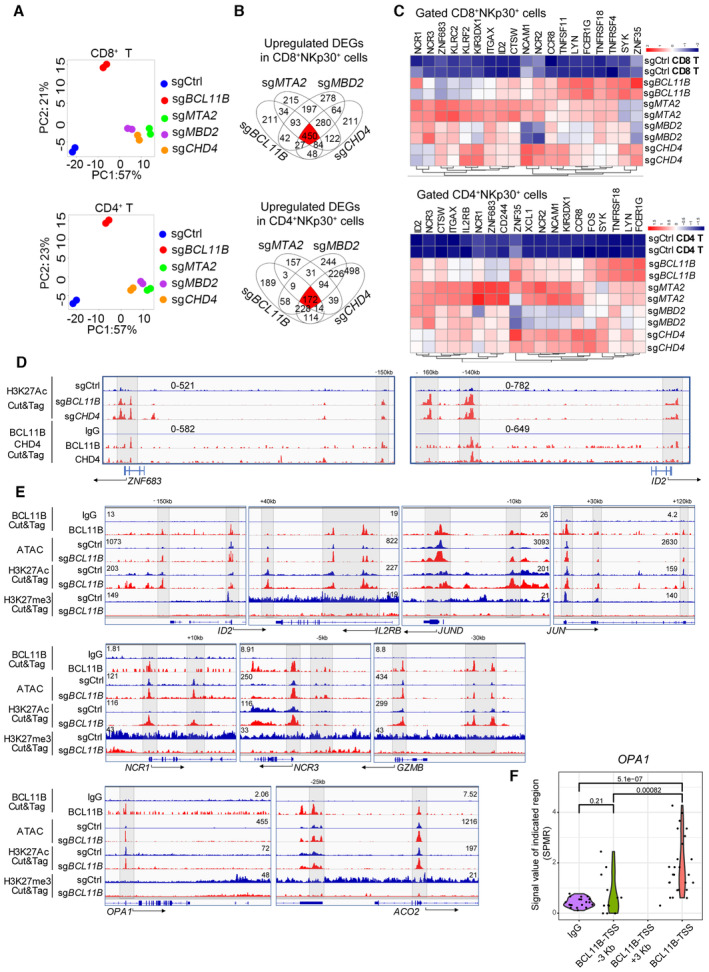
- PBMC‐derived T cells from two healthy donors were transduced with sgCtrl, sgBCL11B, sgMTA2, sgMBD2, or sgCHD4 and cultured for 10 days. CD8+NKp30+ and CD4+NKp30+ cells (purity > 90%) were enriched from PBMC‐derived T cells transduced with sgBCL11B‐, sgMTA2‐, sgMBD2‐, and sgCHD4. CD4 and CD8 T cell subsets were enriched from T cells transduced with sgCtrl. Principal component analysis (PCA) was used to evaluate the similarities in the global gene expression profiles of the listed populations.
- Venn diagrams for the number of upregulated differentially expressed genes (DEGs) in (A) overlapping among CD8+NKp30+ (450 genes) and CD4+NKp30+ (172 genes) subsets from sgBCL11B‐, sgMTA2‐, sgMBD2‐, and sgCHD4‐transduced T cells, compared to CD8+ and CD4+ sgCtrl‐transduced T cells.
- Heatmap of the overlapping upregulated DEGs from (B).
- PBMC‐derived CD8+ T cells transduced with sgCtrl, sgBCL11B‐, and sgCHD4 were cultured for 10 days in vitro before CUT&Tag experiments. Selected genome views for the H3K27Ac CUT&Tag data for sgBCL11B‐ and sgCHD4‐transduced CD8+NKp46+ cells. H3K27Ac peak data for listed NK‐cell‐associated genes in the indicated T cells. The data are representative of two individual healthy donors. CUT&Tag analysis of the binding of CHD4 to the loci of these genes in CD8+ T cells. IgG antibodies were used as a blank control.
- PBMC‐derived T cells transduced with sgCtrl and sgBCL11B were cultured for 10 days in vitro. T cells were harvested and extracted DNA fragments marked by BCL11B antibody; sgCtrl‐transduced T cells and sgBCL11B‐transduced T cells (CD3+NKp46+) were harvested and extracted DNA fragments marked by H3K27Ac antibody and H3K27me3 antibody for CUT&Tag sequencing, respectively. These sgCtrl‐transduced T cells and sgBCL11B‐transduced T cells (CD3+NKp46+) were also lysed to extract nuclei for ATAC‐seq profile. Selected genome views for the BCL11B, H3K27Ac CUT&Tag, and ATAC‐seq data of T cells. CUT&Tag analysis of the binding of BCL11B to the loci of NK‐related genes, AP‐1, and metabolism‐related genes in T cells. IgG antibodies were used as a blank control. ATAC‐seq data of sgCtrl T cells and corresponding sgBCL11B T cells are shown in views of the indicated gene loci. H3K27Ac and H3K27me3 peak data of sgCtrl T cells and corresponding sgBCL11B T cells are shown in views of the indicated gene loci. The data are representative of two individual healthy donors.
- Violin plot of the distribution of BCL11B binding peaks spanning 3 kb upstream to downstream of the TSS region of the OPA1 gene (BCL11B CUT&Tag data from (E)). SPMR: signal per million reads.
Data information: In (F), the points represent the windows with signal values in the CUT&Tag data (N = 10–25 signal values per region). We extracted the signal values from the bedgraph file generated by MACS2, and used two‐tailed unpaired Student's t‐test to evaluate the significant differences between the data.
Source data are available online for this figure.
Figure EV1. ITNKs derived from NuRD‐subunit‐deficient cells have similar characteristics to BCL11B‐deleted ITNKs as compared with those of T cells.
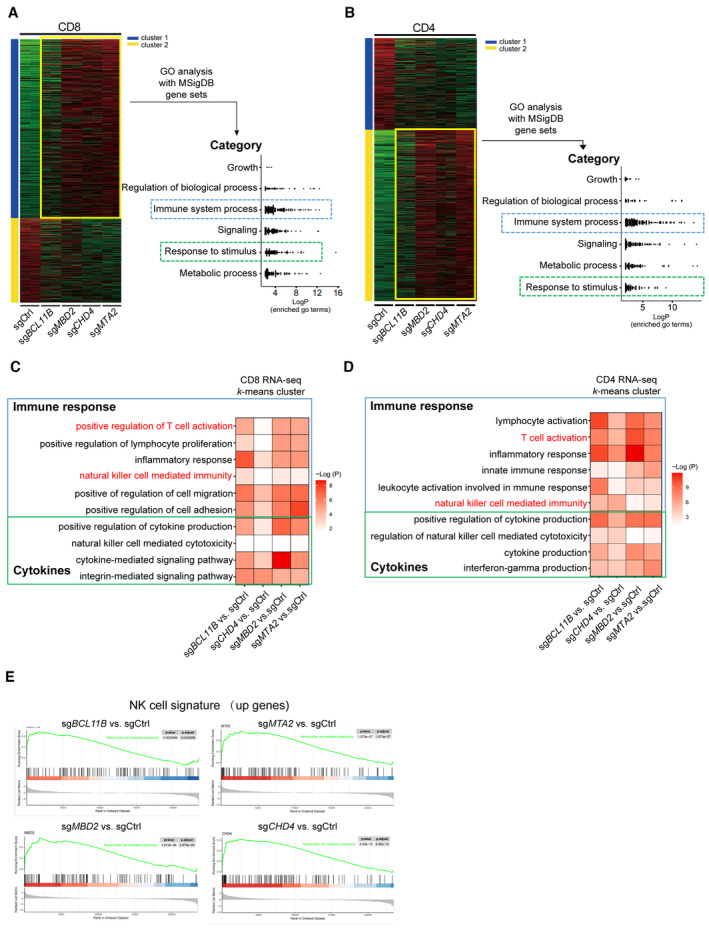
- k‐means clustering analysis of differentially expressed genes in RNA‐seq transcriptomes revealed two distinct clusters of genes in purified CD8+NKp30+ subsets of T cells that were transduced with sgBCL11B, sgMTA2, sgMBD2, or sgCHD4 and CD8+ sgCtrl‐transduced T cells. Relative enrichment of gene sets in the k‐means cluster by hierarchical clustering heatmap analysis. Pathway enrichment (gene set and GO) was performed on cluster 1. Gene sets enriched in cluster 1 were binned into different categories and plotted.
- k‐means clustering of differentially expressed genes in purified CD4+NKp30+ subsets of T cells that were transduced with sgBCL11B, sgMTA2, sgMBD2, or sgCHD4 and CD4+ sgCtrl‐transduced T cells. Gene sets enriched in cluster 2 were binned into different categories and plotted.
- Representative gene sets enriched by Gene Ontology (GO) analysis of the k‐means clusters 1 from (A). GO enrichment analysis was performed by R package clusterprofile (version 4.6.2). The was visualized as heatmaps generated by customed R scripts using ggplot2 (version 3.4.0).
- Representative gene sets enriched by GO term k‐means clusters 2 from (B). GO enrichment analysis was performed by R package clusterprofile (version 4.6.2). The was visualized as heatmaps generated by customed R scripts using ggplot2 (version 3.4.0).
- GSEA enrichment plots for the indicated gene sets in the transcriptome of CD4+ CTL (enrichment plot: NATURAL_KILLER_CELL_MEDIATED_CYTOTOXICITY; Table EV1). The top portion of the plot shows the running enrichment score (RES) for the gene set as the analysis walks down the ranked list of genes and reflects the degree to which the gene set is overrepresented at the top or bottom of the ranked list of genes. The middle portion of the plot shows where the members of the gene set (indicated as black lines) appear in the ranked list of genes. The bottom portion of the plot shows the value of the ranking metric.
Source data are available online for this figure.
CD4+ cytotoxic T lymphocytes (CD4‐CTLs), which are essential for immune responses to various viral infections (Juno et al, 2017; Takeuchi & Saito, 2017), share similar transcription profiles with TEMRA (defined as CD3+CD4+CD45RA+CCR7−cells), which exhibit the cytotoxic function of CD8+ T lymphocytes and NK cells (Patil et al, 2018). Gene set enrichment analysis (GSEA) of CD4+ ITNKs shows that the “natural killer cell‐mediated cytotoxicity” gene set was enriched in BCL11B‐, MTA2‐, MBD2‐, and CHD4‐deficient CD4+ ITNKs (Fig EV1E).
Of note, the PCA analysis shows that the expression profiles of MTA2‐, MBD2‐, and CHD4‐deficient ITNKs were similar but different from those of BCL11B‐deficient ITNKs (Fig 2A and Appendix Fig S2A and B). GO analysis suggests that upregulated genes in MTA2‐, MBD2‐, and CHD4‐deficient ITNKs were mainly involved in cellular morphogenesis pathways, while downregulated genes were enriched in immune responses and biosynthetic processes, compared with BCL11B‐deficient ITNKs (Appendix Fig S2C and D).
Bcl11b‐repressed genes harbor high H3K27Ac peak levels when Bcl11b is absent in murine T cells (Hosokawa et al, 2018). We thus performed cleavage under targets and tagmentation (CUT&Tag) analysis to identify H3K27Ac‐enriched regions and associated genes (Fig EV2A and B, Dataset EV1). We found that global H3K27 acetylation (scaled to 3 kb upstream and downstream of transcriptional start sites (TSSs)) was markedly increased in sgBCL11B‐, sgMBD2‐, and sgCHD4‐transduced CD8+ T cells as compared to CD8+ sgCtrl‐transduced T cells (Fig EV2C). In particular, we found that H3K27Ac binding peaks were enriched at TSSs and putative enhancer elements sites of T‐cell‐activation and NK‐cell‐mediated immunity gene sets, including ZNF683 and ID2 in sgBCL11B‐ and sgCHD4‐transduced CD8+ T cells, as compared to CD8+ sgCtrl‐transduced T cells (Figs 2D and EV2D and E), which is consistent with GO enrichment of RNA‐seq (Fig EV1C and D). To identify binding sites of BCL11B and CHD4 in human primary CD8+ T cells, we also performed CUT&Tag analysis. We found that their binding sites overlapped with H3K27Ac binding sites at TSSs and putative enhancer element sites of ZNF683 and ID2 in primary human CD8+ T cells (Figs 2D and EV2A and B). These results were in line with their derepressed expression (Fig 2B and C). In addition, BCL11B binding peaks were enriched at the loci of identified genes, including NK‐cell‐associated genes (ID2, IL2RB, NCR1, NCR3, and GZMB), AP‐1 family member genes (JUN and JUND), and metabolism‐related genes (OPA1 and ACO2) (Fig 2E). To investigate the roles of these BCL11B‐repressed genes during reprogramming of T cells into ITNKs, we ablated ID2 or JUND in combination with BCL11B in T cells and found that loss of ID2 or JUND significantly reduced the percentages of CD8+NKp46+ and CD4+NKp30+ cells in BCL11B‐deficient T cells (Appendix Fig S2E).
Figure EV2. ITNKs increase the H3K27Ac at TSS region of pluripotency gene sets.
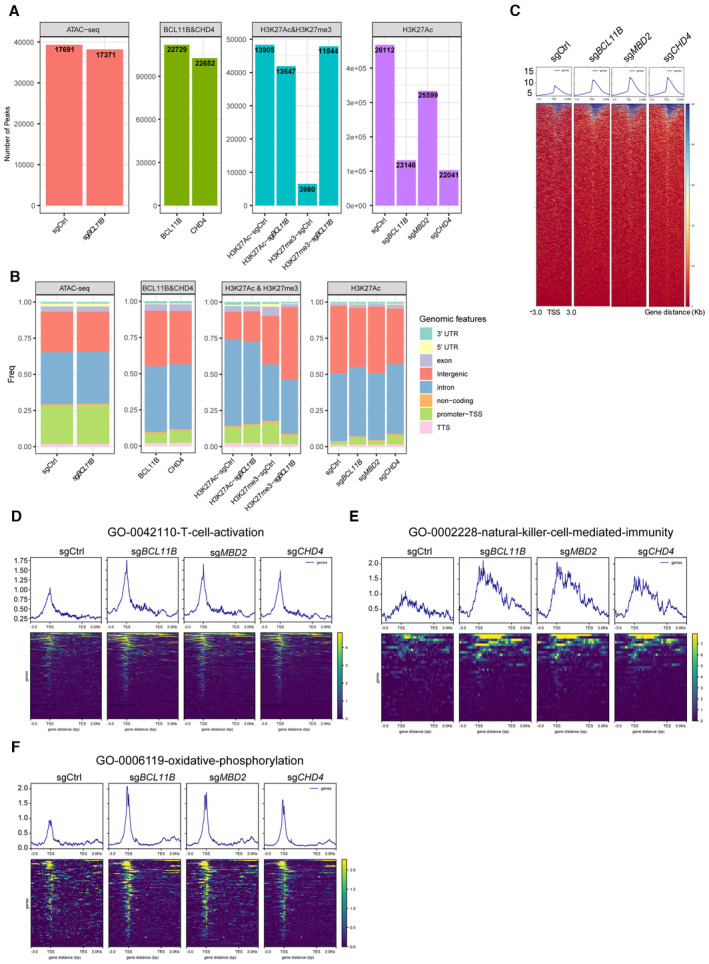
- Bar chart indicating the number of peaks and the number of genes at the peak position in these omics‐seq.
- Bar chart demonstrating the percentage of peaking with a TSS, TSS‐promoter, transcription end site (TES), and other sites in these omics‐seq.
- Tag density pileups of H3K27Ac peaks in CD8+ T cells transduced with sgBCL11B, sgMBD2, or sgCHD4 and CD8+ sgCtrl‐transduced T cells. Two individual healthy donors were used for the CUT&Tag assay.
- Tag density pileups of H3K27Ac peaks at the T‐cell‐activation gene set (see Table EV2) in CD8+ T cells transduced with sgBCL11B, sgMBD2, or sgCHD4 and CD8+ sgCtrl‐transduced T cells.
- Tag density pileups of H3K27Ac peaks at the natural‐killer‐cell‐mediated‐immunity gene set (see Table EV3) in CD8+ T cells transduced with sgBCL11B, sgMBD2, or sgCHD4 and CD8+ sgCtrl‐transduced T cells.
- Tag density pileups of H3K27Ac peaks at the oxidative‐phosphorylation gene set (see Table EV4) in CD8+ T cells transduced with sgBCL11B, sgMBD2, or sgCHD4 and CD8+ sgCtrl‐transduced T cells.
Source data are available online for this figure.
To further reveal the state of chromatin, we performed transposase‐accessible chromatin with sequencing (ATAC‐seq) (Fig EV2A and B, Dataset EV1). The results showed that the loci of these genes overlapped with the binding sites of BCL11B and were more accessible in sgBCL11B‐transduced T cells than in sgCtrl‐transduced T cells (Fig 2E). Moreover, these loci exhibited an increase in active H3K27Ac modification and a decrease in H3K27me3 modification in sgBCL11B‐transduced T cells as compared with sgCtrl‐transduced T cells (Fig 2E).
Thus, these results were consistent with the RNA‐seq data and show that BCL11B directly represses NK‐cell‐associated transcriptional signatures in cooperation with the NuRD complex in human T cells.
ITNKs contain fused mitochondria with enhanced metabolic function
We noted an elevated enrichment of metabolic gene signatures in ITNKs derived from both BCL11B‐ and NuRD‐subunit‐deficient T cells (Fig EV1A and B). We also found that H3K27Ac binding peaks increased at gene set associated with oxidative phosphorylation (Fig EV2F). In addition to NK‐cell‐associated genes, BCL11B also bound to the promoter region of OPA1 (Fig 2E). To identify more potential BCL11B binding sites at nearby regions of the OPA1 locus, we analyzed the peak from 3 kb upstream to downstream of the TSS region and found the BCL11B peaks were only significantly enriched at the TSS of OPA1 but not at other regions (Fig 2F, Dataset EV1). We also performed a BCL11B CUT&Tag‐qPCR assay and demonstrated that BCL11B indeed binds to the TSS region of OPA1 but not the nearby control regions (Figs 3A and EV3A). Moreover, the binding site of BCL11B in the OPA1 locus became more accessible with increased levels of active H3K27Ac modification in human T cells upon BCL11B ablation (Fig 2E). Consistently, we found that the OPA1 transcription was upregulated in ITNKs, compared to T cells from the re‐analysis of the bulk RNA‐seq data from our previous study (Jiang et al, 2022; Fig EV3B). Quantitative real‐time polymerase chain reaction (qPCR) also showed that the expression of OPA1 was derepressed in ITNKs upon BCL11B ablation, as compared with T cells (Figs 3B and EV3C). These results suggest that the expression of OPA1, which promotes mitochondrial fusion (Youle & van der Bliek, 2012; Buck et al, 2016), is directly repressed by BCL11B.
Figure 3. ITNKs contain fused mitochondria with enhanced metabolic function.
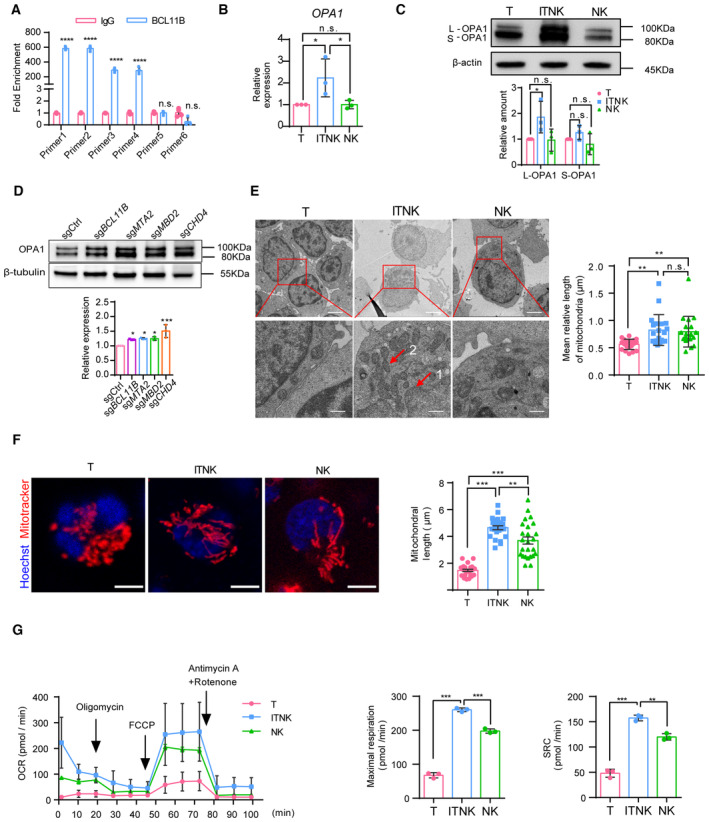
- Quantifying the fold enrichment of OPA1 at different binding sites of BCL11B using CUT&Tag‐qPCR.
- Relative mRNA levels of OPA1 in samples of human T cells, ITNKs (CD3+NKp46+), and NK cells (CD3−CD56+) based on quantitative RT‐PCR.
- Western blot analysis of mitochondrial fusion protein OPA1 levels in samples of human T cells, ITNKs (CD3+NKp46+), and NK cells (CD3−CD56+). Graph summarizing the relative protein levels of OPA1, including the long‐form OPA1 (L‐OPA1) and the short‐form OPA1 (S‐OPA1), in ITNKs compared to T cells.
- Western blot analysis of OPA1 protein levels in ITNKs that were derived from T cells transduced with sgBCL11B, sgMTA2, sgMBD2, or sgCHD4 and sgCtrl‐transduced T cells.
- Transmission electron micrograph of purified T, ITNK, and NK cells from Appendix Fig S3C. ITNKs had a low nucleocytoplasmic ratio compared to T cells. 1, nucleus; 2, mitochondria. Scale bar, 2 μm (top) and 500 nm (bottom).
- Confocal microscopy images showing purified T cells, ITNKs, and NK cells in which the mitochondria (MitoTracker; red) and nuclei (Hoechst; blue) are stained. Scale bars: 5 μm. Each dot represents the mean relative length of the mitochondria in a sample.
- ITNKs, T cells, and NK cells derived from the same donors who provided PBMCs were cultured for 10 days without activation or priming before assaying, and cell metabolism was analyzed. OXPHOS (OCR: O2 consumption rate) assays were performed in real‐time after injection of oligomycin (2 μM), FCCP (1 μM), and antimycin A (1 μM) plus rotenone (1 μM) as indicated.
Data information: In (A–G), data are presented as mean ± SD, N = 3 (B–G) or N = 4 (A) individual healthy donors. N = 20 (E) and N = 25 (F) mitochondria per replicate. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 and ****P ≤ 0.0001, two‐tailed paired Student's t‐test (B–D), one‐way ANOVA with Tukey's multiple comparisons test (E–G) or two‐way ANOVA with Sidak's multiple comparisons test (A).
Source data are available online for this figure.
Figure EV3. Mitochondrial dynamics‐related protein expression in ITNKs.
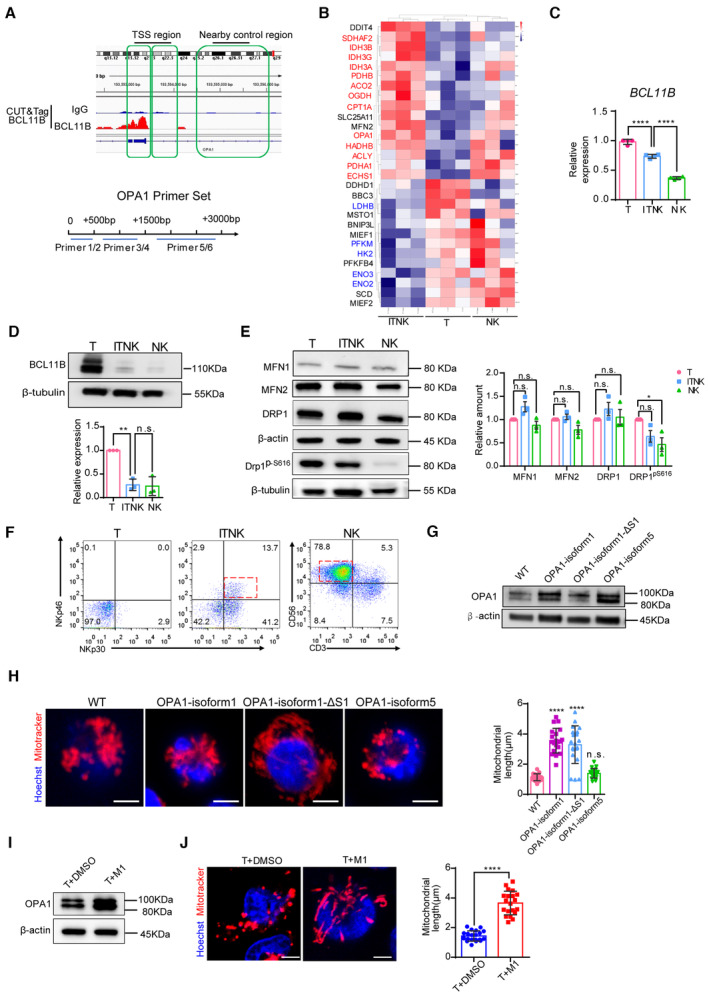
- Schematic diagram of BCL11B binding sits in the TSS region of OPA1 and OPA1 qPCR primer design strategies. OPA1 qPCR Primers 1 and 2 were designed in the range of 500 bp downstream of the OPA1 TSS; primers 3 and 4 were designed in the range of 500–1,500 bp; and primers 5 and 6 were designed in the nearby control region from 1,500 to 3,000 bp.
- The heatmap shows the upregulation of OPA1 and transcripts associated with TCA (tricarboxylic acid) and FAO (fatty acid oxidation) processes in ITNKs (N = 3 individual donors). Cutoff: absolute log2 (fold change) ≥ 1; adjusted P value ≤ 0.05.
- Relative mRNA levels of BCL11B in samples of human T cells, ITNKs (CD3+NKp46+), and NK cells (CD3−CD56+) based on quantitative RT‐PCR.
- Western blot analysis of BCL11B levels in samples of human T cells, ITNKs (CD3+NKp46+), and NK cells (CD3−CD56+). Graph summarizing the relative protein levels of BCL11B is shown in the right panel.
- Western blot analysis of mitochondrial fusion (MFN1, MFN2) and fission (DRP1, DRP1pS616) protein levels in samples of PBMC‐derived T cells, ITNKs (CD3+NKp46+), and NK cells (CD3−CD56+). A graph summarizing the relative protein levels of MFN1, MFN2, DRP1, and DRP1pS616 in ITNKs compared to T cells is shown in the right panel.
- T cells transduced with sgCtrl and sgBCL11B and NK cells enriched from PBMC from the same donor were cultured for 10 days in vitro and collected for FACS sorting. Representative flow cytometry analysis of purified T cells (CD3+NKp30−), ITNKs (CD3+NKp46+), and NK cells (CD3−CD56+).
- Representative western blot of OPA1 in T cells with different OPA1 isoforms overexpression. β‐actin was used as a control. Isoform 1 is the long form and its mutant S1 only produces a single long form OPA1. Isoform 5 is the short form of OPA1.
- Mitochondrial morphology of T cells with different OPA1 isoform overexpression in which the mitochondria (MitoTracker; red) and nuclei (Hoechst; blue) are stained. Scale bars: 5 μm. Each dot represents the mean relative length of the 20 mitochondria per replicate.
- Representative western blot of OPA1 in T cells with M1 treatment. β‐actin was used as a control.
- Mitochondrial morphology of T cells with M1 treatment in which the mitochondria (MitoTracker; red) and nuclei (Hoechst; blue) are stained. Each dot represents the mean relative length of the 20 mitochondria per replicate. Scale bars: 5 μm.
Data information: In (C–E, H, and J), data are presented as mean ± SD, N = 3 individual healthy donors. *P ≤ 0.05, **P ≤ 0.01 and ****P ≤ 0.0001, two‐tailed paired Student's t‐test (C–E and J) or one‐way ANOVA with Tukey's multiple comparisons test (E).
Source data are available online for this figure.
We next measured the levels of mitochondrial dynamics‐related proteins and found that both the long‐ and short‐form of the mitochondrial fusion protein OPA1 were significantly upregulated in BCL11B‐deficient T cells as compared to normal T cells, in line with BCL11B ablation (Figs 3C and EV3D). In particular, the ratio of the long‐form OPA1 to its short‐form was increased, which promotes mitochondrial inner‐membrane fusion (Fig 3C; Del Dotto et al, 2017; Ge et al, 2020). The expression of other mitochondrial dynamics‐related proteins, including MFN1, MFN2, Drp1, and Drp1ps616, was unchanged between T cells and ITNKs (Fig EV3E). However, the Drp1ps616 protein was significantly decreased in NK cells (Fig EV3E). Like BCL11B‐deficient ITNKs, MTA2‐, MBD2‐, and CHD4‐deficient ITNKs also showed upregulation of OPA1 protein levels as compared to sgCtrl‐transduced T cells (Fig 3D).
As OPA1 regulates mitochondrial dynamics (Buck et al, 2016), we purified ITNKs (CD3+NKp46+) that were derived from BCL11B‐deficient T cells and compared their mitochondrial morphology to that of T cells (CD3+NKp30−) and NK cells (CD3−CD56+) from the same normal donors by transmission electron microscopy (TEM) (Fig EV3F). ITNK and NK cells mostly harbor large and tubular mitochondria, while T cells had small and dispersed mitochondria (Fig 3E). In addition, we observed that ITNKs exhibited elongated and fused tubular mitochondria using confocal laser scanning microscopy (CLSM) (Fig 3F). To investigate the effects of different splice isoforms of OPA1 on mitochondrial fusion in T cells, we overexpressed two long OPA1 isoforms (isoform 1, isoform 1 with mutation at the S1 cleavage site (∆S1 mutation)) and a short isoform of OPA1 (isoform 5) in primary human T cells. Isoform 1 functions as the fusion‐active species, giving rise to long forms of OPA1 that can be cleaved into short forms. In contrast, isoform 1 ∆S1 produces an uncleavable isoform 1 (Ishihara et al, 2006; Song et al, 2007; Del Dotto et al, 2017). Conversely, isoform 5 produces short forms of OPA1 with little fusion activity (Ishihara et al, 2006; Song et al, 2007; Fig EV3G). In line with previous studies (Ehses et al, 2009), the expression of both long isoforms resulted in the tubulation of mitochondria, while the OPA1 isoform 5 overexpression did not promote mitochondrial elongation (Fig EV3H). Furthermore, we induced an upregulation of OPA1 protein in human T cells with M1 treatment that promotes mitochondrial fusion (Wang et al, 2012; Ding et al, 2020) and found that M1‐treated T cells exhibited elongated mitochondria (Fig EV3I and J).
We then evaluated the mitochondrial morphology in different subpopulations of T cells and ITNKs. CD8+ T cells exhibited predominantly punctate mitochondria compared to CD4+ T cells (Fig EV4A and B). However, both CD4+ ITNKs and CD8+ ITNKs possessed similar elongated tubular mitochondria (Fig EV4A and B). Moreover, ITNKs derived from T cells by ablating NuRD subunits also contained elongated tubular mitochondria (Fig EV4C). These data suggest that OPA1 expression could induce mitochondrial elongation in human T cells as seen in BCL11B‐deficient and NuRD‐subunit‐deficient ITNKs.
Figure EV4. Mitochondrial morphology and functions of ITNKs.
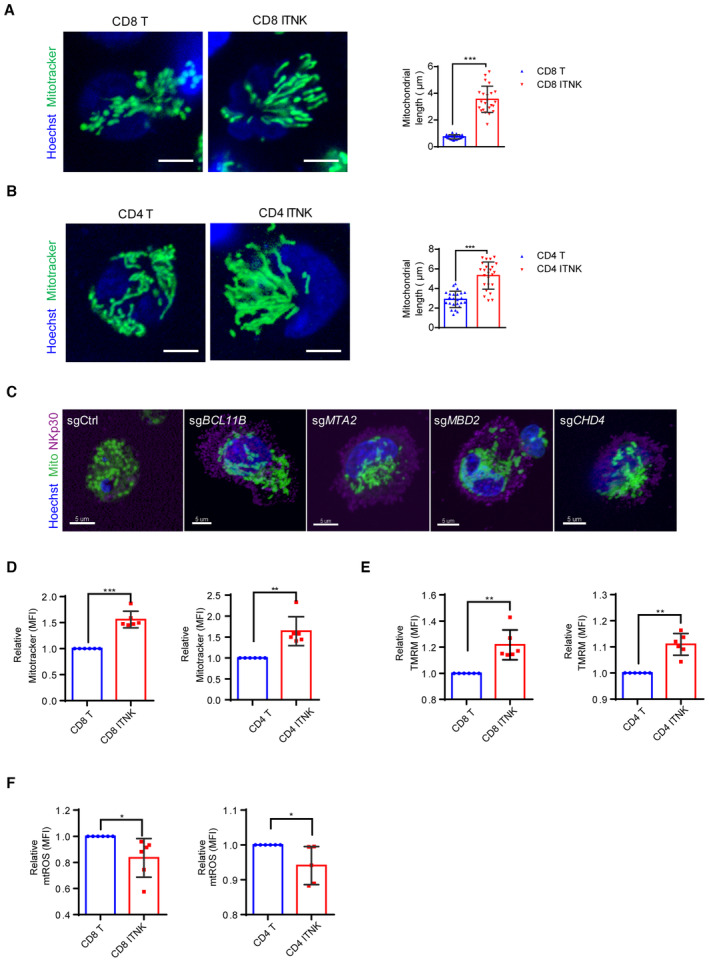
- Confocal microscopy images showing purified CD8+ T cells and CD8+NKp30+ ITNKs in which the mitochondria (MitoTracker; green) and nuclei (Hoechst; blue) are stained. Each dot represents the mean relative length of the 24 mitochondria per replicate. Scale bars: 5 μm.
- Confocal microscopy images showing purified CD4+ T cells and CD4+NKp30+ ITNKs in which the mitochondria (MitoTracker; green) and nuclei (Hoechst; blue) are stained. Each dot represents the mean relative length of the 24 mitochondria per replicate. Scale bars: 5 μm.
- ITNKs that were derived from T cells transduced with sgBCL11B, sgMTA2, sgMBD2, or sgCHD4 or sgCtrl‐transduced T cells were cultured for 10 days for observation of mitochondrial morphology. Confocal microscopy images are shown; mitochondria (MitoTracker; green); nuclei (Hoechst; blue), and NKp30 (purple) are stained. Scale bars: 5 μm.
- CD4+NKp30+ and CD8+NKp30+ ITNKs derived from sgBCL11B‐transduced T cells were purified and assessed the mitochondrial functions by FACS. CD8+ T cells and CD8+NKp30+ ITNKs (left) and CD4+ T cells and CD4+NKp30+ ITNKs (right) were stained with MitoTracker green and analyzed by flow cytometry.
- CD8+ T cells and CD8+NKp30+ ITNKs (left) and CD4+ T cells and CD4+NKp30+ ITNKs (right) were stained with TMRM and analyzed by flow cytometry.
- CD8+ T cells and CD8+NKp30+ ITNKs (left) and CD4+ T cells and CD4+NKp30+ ITNKs (right) were stained with MitoSOX and analyzed by flow cytometry.
Data information: In (A, B, and D–F), data are presented as mean ± SD, N = 3 (A and B) or 6 (D–F) individual healthy donors. *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001, two‐tailed paired Student's t‐test.
Source data are available online for this figure.
As tubular mitochondria exhibit increased OXPHOS (Zheng et al, 2019), we examined the metabolic output of BCL11B‐deficient ITNKs and found that the oxygen consumption rate (OCR), which is an indicator of OXPHOS (Zheng et al, 2019), was higher in ITNKs than in T or NK cells (Fig 3G). In addition, the maximal respiration and mitochondrial spare respiratory capacity (SRC) measured after the addition of carbonyl cyanide‐4‐(trifluoromethoxy) phenylhydrazone (FCCP) were significantly increased in ITNKs as compared with T and NK cells (Fig 3G). These results suggest that BCL11B ablation upregulates the oxidative metabolism in ITNKs. CD4+ and CD8+ subsets of BCL11B‐deficient ITNKs had significantly increased mitochondrial mass (as measured by MitoTracker green staining) (Fig EV4D) and mitochondrial membrane potential, as evaluated by staining with tetramethylrhodamine methyl ester (TMRM), as compared to CD4+ and CD8+ T cells, respectively (Fig EV4E). Moreover, the levels of reactive oxygen species (ROS) in these CD4+ and CD8+ ITNKs were lower than those in CD4+ and CD8+ T cells, respectively (Fig EV4F). Taken together, these results suggest that ITNKs exhibit fused mitochondria with augmented metabolic function.
OPA1‐mediated mitochondrial fusion facilitates the reprogramming of ITNKs and their cytotoxicity
To determine whether OPA1‐mediated mitochondrial fusion was essential for reprogramming T cells into ITNKs, we ablated OPA1 and BCL11B in T cells with the CRISPR/Cas9 technology (Appendix Fig S3A). In line with previous reports (Buck et al, 2016), mitochondria in sgOPA1‐transduced T cells and those in BCL11B and OPA1 double‐knockout T cells were fragmented (Fig 4A). In addition, the percentages of CD4+ ITNKs (CD4+NKp30+) and CD8+ ITNKs (CD8+NKp46+) with BCL11B and OPA1 double knockout in T cells were significantly decreased as compared to those in BCL11B‐deficient T cells (Fig 4B). These results suggest that mitochondrial fusion facilitates the reprogramming of T cells into ITNKs upon BCL11B loss.
Figure 4. OPA1‐mediated mitochondrial fusion facilitates the reprogramming of ITNKs and their cytotoxicity.
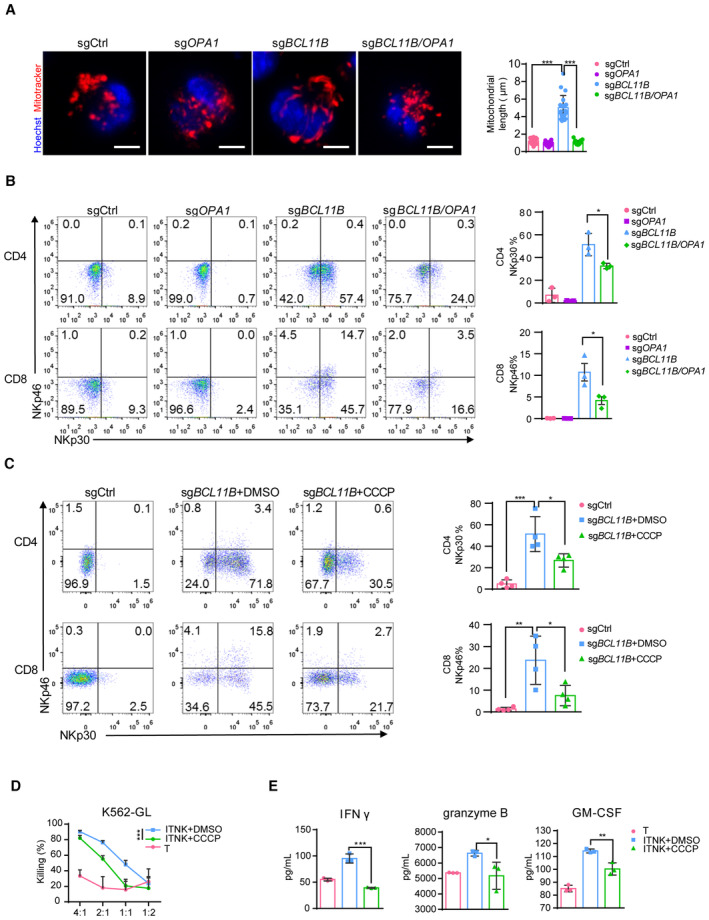
- Confocal microscopy images showing T cells transduced with sgCtrl, sgOPA1, sgBCL11B, or sgBCL11B/OPA1 in which the mitochondria (MitoTracker, red) and nuclei (Hoechst; blue) are stained. Scale bars: 5 μm. Mean lengths of the 20 mitochondria per replicate, as analyzed by confocal microscopy, are shown.
- Representative flow cytometric analysis of NKp30 and NKp46 expression in T cells transduced with sgCtrl, sgOPA1, sgBCL11B, or the combination of sgBCL11B and sgOPA1. A graph summarizing the percentages of NKp30+ cells in CD4+ T cells and percentages of NKp46+ cells in CD8+ T cells transduced with sgCtrl, sgOPA1, sgBCL11B, or the combination of sgBCL11B and sgOPA1 is in the right panel.
- CCCP (5 μM, mitochondrial fission inducer) was added to a culture of sgBCL11B‐transduced human T cells derived from PBMCs 24 h after electroporation. Ten days later, the T cells were subjected to FACS analysis. A graph summarizing the percentages of NKp30+ and NKp46+ cells in CD4+ and CD8+ T cells transduced with sgCtrl, sgBCL11B, or sgBCL11B and treated with CCCP is shown in the right panel.
- Killing assays showing the percent cytotoxicity of T cells, ITNKs, and ITNKs treated with CCCP from (C) against K562 cells. The data represents killing percentage of cells from a donor.
- Cytokine secretion profiles of T cells, ITNKs, and ITNKs treated with CCCP and cocultured with K562 cells. T cells, ITNKs, and ITNKs treated with CCCP from (C) were incubated with K562 cells at an E:T ratio of 1:1 for 24 h. The supernatants were then harvested, and the concentrations of the indicated cytokines were measured by a multiplex immunoassay. The data represents the concentrations of the indicated cytokines from (D).
Data information: In (A, B, D and E), data are presented as mean ± SD, N = 3 or N = 4 (C) individual healthy donors. *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001, two‐tailed paired Student's t‐test (B–D), one‐way ANOVA (A–C) or two‐way ANOVA (D and E) with Tukey's multiple comparisons test.
Source data are available online for this figure.
Carbonyl cyanide m‐chlorophenylhydrazone (CCCP), a mitochondrial electron transport chain uncoupler, can affect OPA1 stability and induce mitochondrial fission (Griparic et al, 2007). Indeed, we found that CCCP treatment resulted in OPA1 cleavage of the long form into the short form in ITNKs and induced mitochondrial fission (Appendix Fig S3B and C). In addition, the OCR was significantly decreased in the CCCP‐treated ITNKs, in line with mitochondrial spare respiratory capacity (Appendix Fig S3D). Like OPA1 ablation, the CCCP treatment also decreased NKp30 and NKp46 expression in BCL11B‐deficient T cells (Fig 4C). These results suggest that ablating OPA1 to disrupt mitochondrial fusion impairs the reprogramming of T cells into ITNKs.
Since mitochondrial fusion enhances the antitumor activity of NK cells (Zheng et al, 2019), we investigated whether it was the same case for ITNKs by inhibiting mitochondrial fusion in ITNKs via CCCP treatment. The cytotoxicity of ITNKs against K562 cells was compromised upon CCCP treatment (Fig 4D). In addition, CCCP‐treated ITNKs secreted less IFNγ, granzyme B, and GM‐CSF in coculture with K562 cells than untreated ITNKs (Fig 4E). These results demonstrate that OPA1 inhibition reduces the antitumor activity of ITNKs.
Acetyl‐CoA augments the expression of NK‐cell‐associated genes and antitumor effects in ITNKs by regulating histone acetylation
Mitochondrial fusion favors OXPHOS metabolism in T cells (Buck et al, 2016). Our previous data showed that the tricarboxylic acid (TCA) cycle and FAO process‐related gene were upregulated in ITNKs (Jiang et al, 2022; Fig EV3B). Consistently, a recent publication demonstrates that Th17 cells have fused mitochondria and require OPA1 for its control of the TCA cycle to regulate effector programs (Baixauli et al, 2022), which implies that the increase in OPA1 may elevate TCA metabolism through mitochondrial fusion in ITNKs. Metabolites act as a source for biomacromolecule synthesis in cells and provide key substrates for epigenetic modification (Matilainen et al, 2017; Wu et al, 2022). Acetyl‐CoA and α‐KG are key metabolites in the TCA cycle and participate in the regulation of histone modification in mammalian cells (Sutendra et al, 2014; Pietrocola et al, 2015). We measured the levels of acetyl‐CoA and α‐KG in ITNKs and found that Acetyl‐CoA but not α‐KG were increased in ITNKs, compared to T cells (Fig EV5A and B). In addition, the loss of OPA1 decreased Acetyl‐CoA levels in ITNKs (Fig 5A). Moreover, acetate, a precursor of acetyl‐CoA (Zhou et al, 2019), increased the acetyl‐CoA level in BCL11B and OPA1 double‐knockout ITNKs (Fig 5A). Strikingly, acetate treatment elevated the expression of NKp30 and NKp46 in CD4+ and CD8+ BCL11B and OPA1 double‐knockout ITNKs, respectively (Figs 5B and EV5C). Furthermore, the loss of OPA1 compromising the cytotoxicity of BCL11B‐deficient ITNKs could be rescued by acetate supplementation (Fig 5C). In addition, the decreased secretion of IFNγ, granzyme B, and GM‐CSF in BCL11B‐deficient ITNKs in coculture with HepG2 cells observed when OPA1 is depleted, could be reversed by acetate treatment (Fig EV5D). These results suggest that acetyl‐CoA facilitates the reprogramming of ITNKs and promotes their antitumor effects.
Figure EV5. Acetate restored the reprogramming efficacy and function of ITNKs.
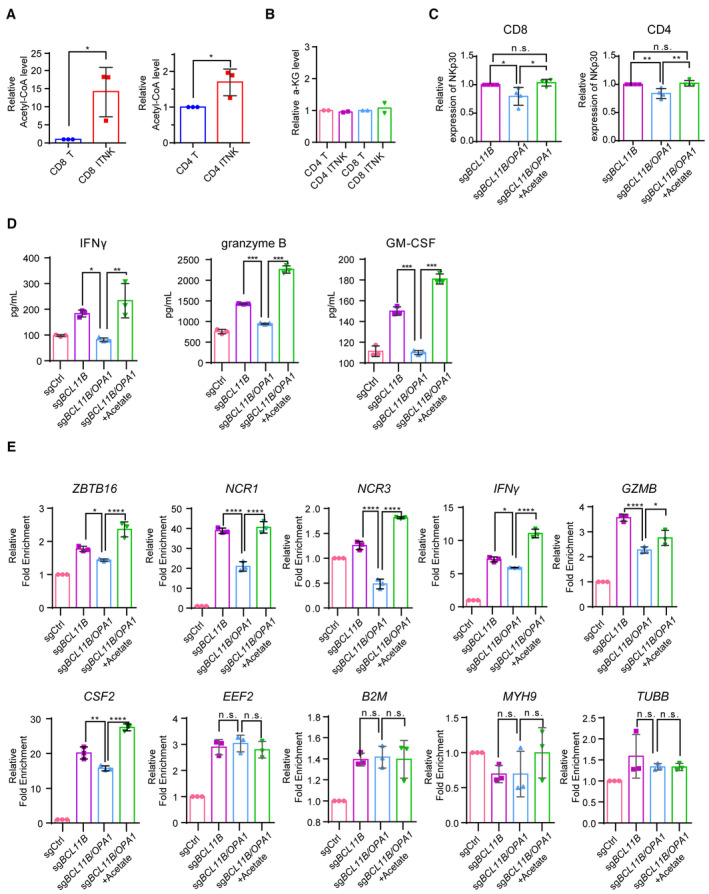
- Relative acetyl‐CoA levels in CD8+ (left) and CD4+ (right) ITNKs on Day 10 that were reprogrammed from T cells transduced with sgBCL11B and sgCtrl‐transduced T cells.
- Relative α‐KG levels in CD4+ (left) and CD8+ (right) ITNKs on Day 10 that were reprogrammed from T cells transduced with sgBCL11B and sgCtrl‐transduced T cells.
- Graph summarizing the percentages of NKp30+ in CD4 T cells and CD8 T cells transduced with sgBC11B or the combination of sgBCL11B and sgOPA1 and treated with or without acetate (from Fig 5B).
- Cytokine secretion profiles of T cells and ITNKs from Fig 5C that were incubated with HepG2 cells at an E:T ratio of 1:1 for 24 h. The supernatants were then harvested, and the concentrations of the indicated cytokines were measured by a multiplex immunoassay.
- Relative mRNA levels of ZBTB16, NCR1, NCR2, NCR3, IFNγ, CSF2, and GZMB in T cells and ITNKs from Fig 5A, housekeeping genes (EEF2, B2M, MYH9, and TUBB) as negative controls, measured by quantitative RT‐PCR.
Data information: In (A–E), data are presented as mean ± SD, N = 2 (B) or 3 (A and C–E) individual healthy donors. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 and ****P ≤ 0.0001, two‐tailed paired Student's t‐test (A) or one‐way ANOVA with Tukey's multiple comparisons test (C–E).
Source data are available online for this figure.
Figure 5. Acetyl‐CoA augments the expression of NK‐cell‐associated genes and antitumor effects in ITNKs by regulating histone acetylation.
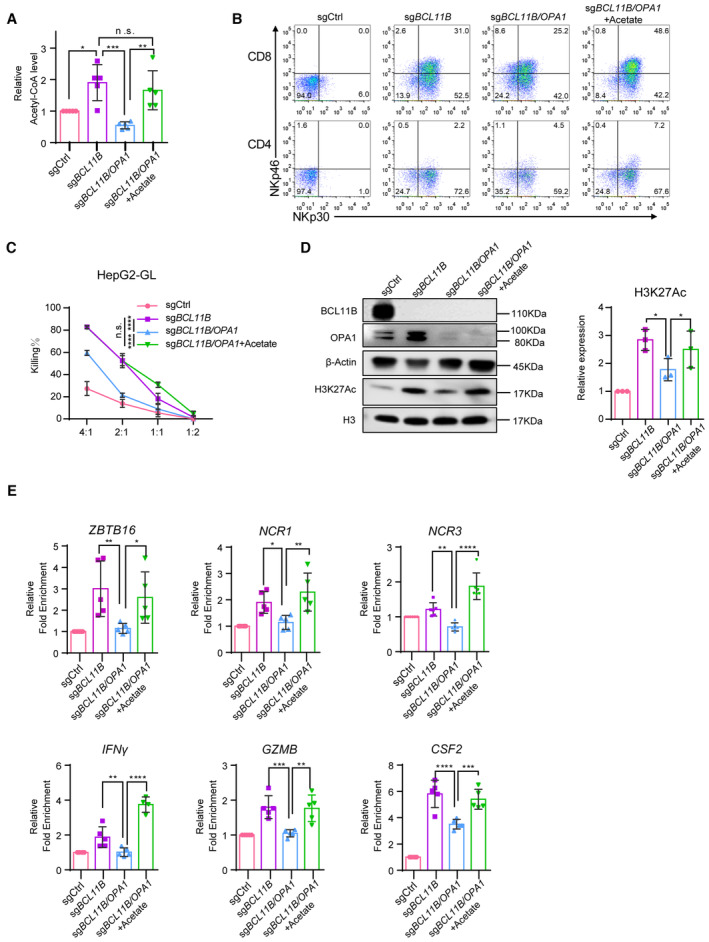
- T cells transduced with sgCtrl and ITNKs that were reprogrammed from T cells transduced with sgBC11B or the combination of sgBCL11B and sgOPA1 and were treated with or without acetate (20 mM) 24 h after electroporation. Relative acetyl‐CoA levels of those T cells were measured on Day 10 of the cell culture.
- Representative flow cytometry detection of NKp30 and NKp46 expression in T cells and ITNKs from (A).
- Killing assays showing the percent cytotoxicity of T cells and ITNKs from (A) against HepG2 cells. The data represents killing percentage of cells from a donor.
- Western blot analysis of H3K27Ac in samples of T cells and ITNKs from (A).
- ChIP‐qPCR analysis of H3K27Ac in the loci of indicated genes in T cells and ITNKs from (A).
Data information: In (A and C–E), data are presented as mean ± SD, N = 3 (C and D) or 5 (A and E) individual healthy donors. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 and ****P ≤ 0.0001, one‐way ANOVA (A) or two‐way ANOVA (C) with Tukey's multiple comparisons test, two‐tailed paired Student's t‐test (D), or one‐way ANOVA with Sidak's multiple comparisons test (E).
Source data are available online for this figure.
Since levels of acetyl‐CoA were increased in BCL11B‐deficient T cells (Fig 5A), we measured the acetylation levels of H3K27Ac in total histone proteins and found that the overall level of H3K27Ac was increased in BCL11B‐deficient ITNKs compared to sgCtrl‐transduced T cells (Fig 5D). Consistent to the acetyl‐CoA level, the H3K27Ac level in BCL11B and OPA1 double‐knockout ITNKs was lower than that in BCL11B‐deficient ITNKs but still higher than that in sgCtrl‐transduced T cells (Fig 5D). Of interest, acetate treatment augmented the level of H3K27Ac in BCL11B and OPA1 double‐knockout ITNKs (Fig 5D).
In particular, chromatin immunoprecipitation coupled with quantitative PCR (ChIP–qPCR) assessment of H3K27Ac showed that H3K27Ac was enriched at the loci of NK‐cell related genes, including ZBTB16, NCR1, NCR3, IFNG, GZMB, and CSF2 (Louis et al, 2020), in sgBCL11B‐transduced T cells compared to sgCtrl‐transduced T cells (Fig 5E). These results were consistent with the analyses of H3K27Ac CUT&Tag and ATAC‐seq in BCL11B‐deficient ITNKs and sgCtrl‐transduced T cells (Fig 2E). There was a decreased H3K27Ac level at the loci of these of NK‐cell‐associated genes in BCL11B and OPA1 double‐knockout ITNKs (Fig 5E). However, the decrease in H3K27Ac at these loci was restored by acetate treatment in BCL11B and OPA1 double‐knockout ITNKs (Fig 5E). Consistently, the mRNA levels of the NK‐cell‐related genes varied and were correlated to the H3K27Ac levels at their loci in different types of ITNKs with or without acetate treatment, while the expression of housekeeping genes (EEF2, B2M, MYH9, and TUBB) did not (Fig EV5E). Taken together, these results suggest that acetyl‐CoA promotes the expression of NK‐cell‐associated genes, cytotoxic cytokine secretion, and ITNK cytotoxicity through regulating H3K27Ac modifications.
To further confirm whether T cells acquire the NK‐cell potential through a metabolic‐epigenetic axis, we measured the levels of OPA1 and observed the morphology of mitochondria in T cells at different time‐points post‐BCL11B ablation to determine the time‐point when OPA1‐mediated fusion occurred. We found a gradual increase of OPA1 expression following BCL11B ablation, with a significant elevation of OPA1 expression observed on Day 4 upon BCL11B loss, along with the occurrence of mitochondrial fusion (Fig 6A and B). Subsequently, we performed a mitochondrial‐stress test by Seahorse on Days 2 and 4 post‐BCL11B ablation at the early stage of the reprogramming and found that OCR of T cells with BCL11B ablation was significantly improved on Day 4 (Fig 6C). These results suggest that the metabolic changes occur in T cells on Day 4 upon BCL11B ablation. We then confirmed whether epigenetic changes and gaining of NK cell potential in these T cells occur on Day 4 post‐BCL11B loss, by measuring the levels of H3K27Ac modification at the loci of OPA1, ID2, ZBTB16, and ZNF683 and their expression. Indeed, H3K27Ac CUT&Tag‐qPCR showed that H3K27Ac modification at the loci of OPA1, ID2, ZBTB16, and ZNF683 was also significantly increased on Day 4 upon BCL11B ablation but not earlier (Fig 6D). In addition, RT‐qPCR analysis confirmed that the expression of ID2, ZBTB16, and ZNF683 was upregulated on Day 4 but not earlier (Fig 6E and Appendix Fig S4A and B). Taken together, these results show that the metabolic and epigenetic changes alongside the upregulation of mRNA and protein levels of NK‐cell markers all initiate on Day 4 upon BCL11B ablation, suggesting that metabolic‐epigenetic changes at least partially contribute to the acquisition of NK cell identity in BCL11B‐deficient T cells.
Figure 6. ITNKs acquire the NK‐cell identity through a metabolic‐epigenetic axis.
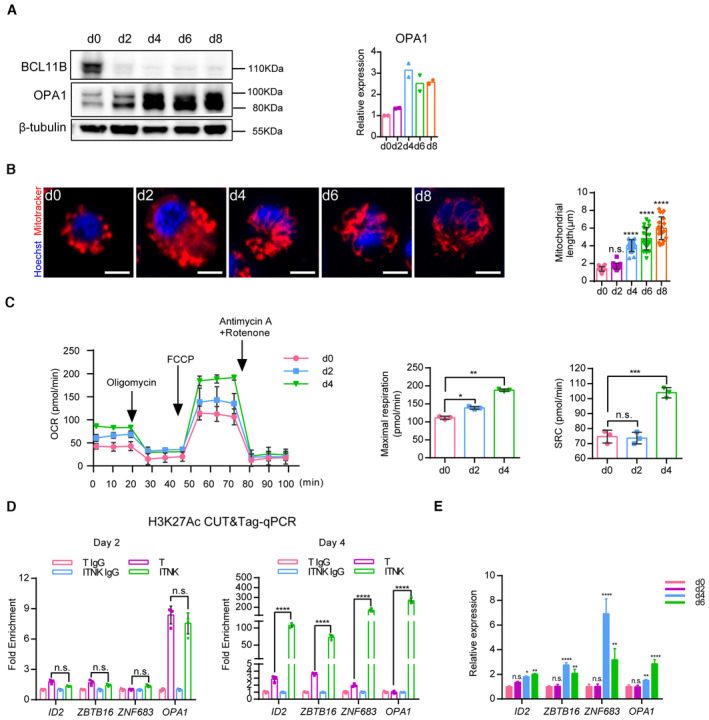
- Representative western blot analysis of BCL11B and OPA1 protein levels in ITNKs from Days 0 to 8.
- Representative confocal microscopy images showing ITNKs from Days 0 to 8 in the reprogramming process in which the mitochondria (MitoTracker; Red) and nuclei (Hoechst; blue) are stained. Scale bars: 5 μm. Each dot represents the mean length of the 20 mitochondria per replicate.
- Mitochondrial‐stress test on Days 2 and 4 of ITNKs.
- CUT&Tag‐qPCR analysis of H3K27Ac enrichment in the promoter regions of indicated genes on Days 2 and 4 of ITNKs.
- Time‐course analysis of NK‐related transcription factors and OPA1 expression by RT‐qPCR.
Data information: In (A–E), data are presented as mean ± SD, N = 2 (A and B) or 3 (C) or 4 (D and E) individual healthy donors. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 and ****P ≤ 0.0001, one‐way (C) or two‐way (E) ANOVA with Dunnett's multiple comparisons test, or two‐way ANOVA with Bonferroni's multiple comparisons test (D).
Source data are available online for this figure.
Discussion
BCL11B maintains T‐cell identity, as its loss induces T cells to reprogram into NK‐like cells (Li et al, 2010b; Jiang et al, 2022). In murine T cells and human T cells, BCL11B physically associates with subunits of the NuRD complex (Cismasiu et al, 2005; Hosokawa et al, 2018). Hosokawa et al (2018) reported that ablation of Mta1 and Mta2 blocks T‐cell differentiation and affects the transcription of Bcl11b‐regulated genes in murine T‐cell progenitors. However, whether the NuRD complex guards T‐cell fate remains unknown. Here, for the first time, we demonstrate that human T cells were reprogrammed into NK‐like cells by deletion of MTA2, MBD2, or CHD4, suggesting that BCL11B associates with the NuRD complex to impede the NK‐cell program in an evolutionarily conserved manner (Fig 1B). Previous studies show that BCL11B interacts with the NuRD complex via its subunits MTA1/2/3 (Cismasiu et al, 2005; Dubuissez et al, 2016). However, our results showed that MTA1‐deficient T cells did not reprogram into NK‐like cells (Appendix Fig S1B). This is probably due to the convergence of genomic occupancy sites of Mta2 and Bcl11b in T cells (Hosokawa et al, 2018). These results suggest that deletion of the MTA2 is more likely to weaken the transcriptional repression of BCL11B targeting genes, including NK‐related genes.
Recent studies demonstrate that mitochondrial dynamics dictate T‐cell fates via regulating metabolism in mature T cells, including T cell activation and exhaustion (Buck et al, 2016; Ron‐Harel et al, 2016; Yu et al, 2020). The roles of Bcl11b during the formation of effector and memory T cells have been investigated extensively showing complexity (Zhang et al, 2010; Califano et al, 2014; Lorentsen et al, 2018; Sidwell & Rothenberg, 2021). Nevertheless, whether Bcl11b regulates metabolism through mediating mitochondrial dynamics in T cells has not yet been elucidated. Our findings showed that BCL11B directly repressed OPA1 transcription and OPA1‐mediated mitochondrial fusion facilitating ITNK reprogramming and cytotoxicity, thus revealing a potential role of BCL11B in mediating metabolic activities in T cells, besides guarding T‐cell fate. Further studies are warranted to investigate whether BCL11B suppresses mitochondrial fusion via OPA1 during T‐cell development and differentiation.
It is interesting to notice that NK cells expressed lower amount of the short OPA1 protein than T cells, though they had similar levels of OPA1 mRNA. This discrepancy is possibly due to the lower translation efficiency of the short form OPA1 in NK cells, or accelerated protein degradation following translation (Ehses et al, 2009). As short OPA1 promotes mitochondrial fragmentation, a lower amount of short OPA1 may result in elongated mitochondria in NK cells (Ishihara et al, 2006; Ehses et al, 2009). In addition, we found that levels of Drp1pS616, which promotes Drp1‐mediated mitochondrial fission (Zheng et al, 2019; Gao et al, 2022a), were lower in NK cells than in T cells (Fig EV3E). This could also contribute to increase the length of mitochondria in NK cells.
Metabolic pathways provide not only energy but also necessary substrates for epigenetic modification to regulate gene expression (Yu et al, 2020; Fanucchi et al, 2021). We confirmed that OXPHOS was increased in T cells upon BCL11B ablation (Figs 3G and EV2F). To reveal how OPA1 impacts acetyl‐CoA levels, we re‐analyzed the bulk RNA‐seq data of ITNKs and T cells from our previous study (Jiang et al, 2022) and found that the tricarboxylic acid (TCA) and fatty acid oxidation (FAO) processes‐related genes were upregulated in ITNKs, as compared to T cells (Fig EV3B). Previous studies demonstrate that mitochondrial fusion favors TCA and FAO (Buck et al, 2016; Baixauli et al, 2022). In addition, Acetyl‐CoA and α‐KG are key metabolites in the TCA cycle and participate in the regulation of histone modification (Shen et al, 2015; Liu et al, 2017; Morris et al, 2019). We found that the levels of Acetyl‐CoA but not α‐KG were increased in ITNKs, compared to T cells (Fig EV5A and B). Consistently, ITNKs expressed OPA1 at higher levels than T cells (Fig 3C). Taken together, it is likely that OPA1 increases Acetyl‐CoA levels through promoting mitochondrial fusion and TCA during ITNK reprogramming. Therefore, the loss of OPA1 decreased Acetyl‐CoA levels in ITNKs (Fig 5A). Consistently, a recent publication demonstrates that Th17 cells have fused mitochondria and require OPA1 for its control of the TCA cycle to regulate effector programs (Baixauli et al, 2022). However, the specific source of acetyl‐CoA needs to be determined by isotope‐based metabolic flux analysis.
Our data showed that acetyl‐CoA level was positively correlated to the expression of OPA1 in ITNKs (Fig 5A). In addition, the enrichment of H3K27Ac at the loci of NK‐cell‐associated genes was positively correlated to the acetyl‐CoA level in ITNKs. Together, these results suggest that OPA1 facilitates the ITNK reprogramming by raising acetyl‐CoA levels for acetylation of H3K27 modifications at the loci of NK‐cell related genes. Current efforts are devoted to understand how acetylation of H3K27 modifications mediated by acetyl‐CoA occurred specifically at the loci of NK cell‐associated genes. The histone acetyltransferases (HAT), such as EP300 and GCN5, promote the expression of specific genes through regulating dynamic hyper‐acetylated chromatin states and acetylation modifications at enhancers (Sen et al, 2019; Wang et al, 2020; Durbin et al, 2022). Possibly, HATs participate the upregulation of NK cell‐related gene expression via inducing H3K27 acetylation modifications (Fig 7).
Figure 7. The BCL11B‐NuRD complex mediates mitochondrial dynamics to promote the reprogramming of T cells to ITNKs via epigenetic remodeling.
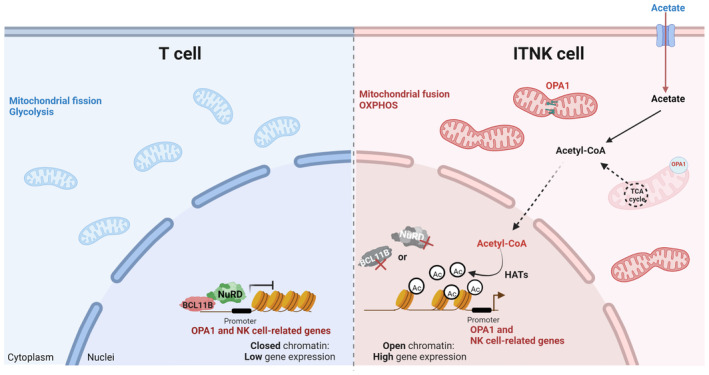
Schematic representation of the mechanism by which BCL11B and the NuRD complex inhibit the expression of OPA1 and NK‐cell‐associated genes in human T cells (left). OPA1‐mediated mitochondrial fusion regulated by the BCL11B‐NuRD complex promotes the reprogramming from T cells into ITNKs by regulating H3K27Ac modification (right). The graphics were created with BioRender.com.
In summary, we investigated the functions of the NuRD complex and BCL11B in primary human T cells with loss of function studies. Our findings show that BCL11B and the NuRD complex not only directly suppress the expression of NK‐cell‐associated genes but also may suppress their transcription indirectly through regulating OPA1‐mediated mitochondrial metabolism. Our results extend current knowledge on how mitochondrial dynamics regulate T cell fates. In addition, our work reports alternative methods to derive ITNKs, potent tumor killer cells, from T cells and provides strategies to improve the reprogramming efficiency and antitumor effects of BCL11B‐deficient ITNKs.
It is important to uncover whether BCL11B suppresses mitochondrial fusion via OPA1 during T‐cell development and differentiation. However, we did not address this question in this work. Further studies are warranted to investigate potential mechanisms on how BCL11B regulates mitochondrial dynamics during T cell development.
Although BCL11B protein level in NK cells was lower than that in T cells, the levels of OPA1 mRNA and protein in NK cells were similar to that in T cells, suggesting that the transcription of OPA1 may be regulated by other factors, rather than BCL11B in NK cells. Further investigation is warranted to reveal how OPA1 transcription is regulated in NK cells.
Materials and Methods
Isolation, transduction, and expansion of primary human T cells and NK cells
Healthy PBMC donors provided informed consent for the use of their samples for research purposes, and all procedures were approved by the Research Ethics Board of the Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences (GIBH, CAS). For all preclinical experiments in this study, human primary T cells were negatively selected using human T‐cell negative enrichment kits (STEMCELL Technologies, Vancouver, BC, Canada) from peripheral blood mononuclear cells (PBMCs), which were isolated from healthy donors using Lymphoprep (STEMCELL Technologies, Vancouver, Canada). Subsequently, T cells were activated with 5 μl MACS T Cell TransAct™ (130‐111‐160, Miltenyi Biotec, Bergisch Gladbach, Germany) at a bead:cell ratio of 1:2 and a density of 1 × 106 cells/ml for 24 h in T551‐H3 (Takara, Japan) medium supplemented with 5% heat‐inactivated fetal bovine serum (FBS), 500 U/ml recombinant human IL‐2, 10 mM HEPES, 2 mM glutamine, and 1% penicillin/streptomycin. NK cells were activated with the NK Cell Activation/Expansion Kit (130‐094‐483, Miltenyi Biotec, Germany), which contains microbeads loaded with anti‐NKp46 and anti‐CD2 antibodies, and were cultured in T551‐H3 medium supplemented with IL‐2 (500 U/ml) and 5% FBS at an initial density of 1 × 106 cells/ml.
Induction and expansion of ITNKs
For electroporation, on post‐activation Day 1, Cas9 RNPs were prepared by incubating 20 μM Cas9 with 20 μM sgRNA at a 1:1 ratio in Human T‐Cell Nucleofector buffer to a final concentration of 10 μM at 37°C for 10 min (Stadtmauer et al, 2020). T cells were electroporated immediately with sgBCL11B RNP (5 × 106 T cells per reaction) using Amaxa Nucleofector 2b (Amaxa® Human T Cell Nucleofector® Kit, Lonza, Germany) with electroporation program T‐023. Twelve hours after electroporation, T cells were cultured in T‐cell culture medium containing rh‐IL2 (500 U/ml). Subsequently, fresh medium was added every 2 days to maintain the cell density within the range of 0.5–1 × 106 cells/ml. CD3+NKp46+/CD3+NKp30+ T cells were defined as ITNKs and used in this study.
Cell lines
Human HCC cell lines (HepG2/HepG2‐GL) were maintained in Dulbecco's modified Eagle's medium (DMEM) (Gibco, Grand Island, NY, USA). Human leukemia cell lines (K562/K562‐GL) were maintained in RPMI‐1640 medium supplemented with 10% heat‐inactivated FBS (Gibco, Grand Island, NY, USA), 10 mM HEPES, 2 mM glutamine (Gibco, Grand Island, NY, USA) and 1% penicillin/streptomycin (Gibco, Grand Island, NY, USA). All cells were cultured at 37°C in an atmosphere of 5% carbon dioxide. Cell line identity was confirmed by STR sequencing.
Protein isolation and immunoblotting
Cells were lysed with RIPA buffer (Pierce, Rockford, Illinois, USA), and proteins were quantified using a BCA Protein Assay Kit (Pierce, Rockford, IL, USA). Samples were loaded onto a 4–20% SDS–PAGE gel, blotted onto a PVDF membrane, and sequentially probed with primary antibodies. A species‐matched HRP‐conjugated secondary antibody was purchased from Cell Signaling Technology (Boston, USA) and then added to detect proteins by autoradiography using an enhanced chemiluminescence kit (ECL Plus, General Electric Healthcare, Little Charfont, UK).
Coimmunoprecipitation (Co‐IP) assay
A total of 5 × 107 T cells were lysed in 1 ml of IP lysis buffer (150 mM KCl, 1% Triton X‐100, 50 mM Tris–HCl pH 7.6, 1 mM EDTA, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride (PMSF), 10 mg/l aprotinin, and 10 mg/l leupeptin), and cleared cell lysates were incubated with 10 μl Protein A/G beads (88802) (Thermo Scientific, USA) and the appropriate antibody (5–10 μg) overnight at 4°C. Following incubation, the resin was washed three times with IP wash buffer (150 mM KCl, 0.1% Triton X‐100, 50 mM Tris–HCl pH 7.6, 1 mM EDTA, and 1 mM PMSF), and protein samples were eluted by boiling in 1 × SDS sample buffer (30 μl) for western blotting analysis.
Flow cytometry and cell sorting
Flow cytometric analysis was performed on a FACSCanto or FACSFortessa (BD, USA). Fluorescence‐activated cell sorting (FACS) was performed on a FACS AriaII platform (BD, USA). Surface staining for flow cytometry and cell sorting was performed by pelleting cells and resuspending them in 50 μl of FACS buffer (2% FBS in PBS) with antibodies for 30 min at 4°C in the dark. Cells were washed once in FACS buffer before resuspension. Antibodies for cell‐surface markers are shown in Table 1 and were purchased from BioLegend (San Diego, USA). The gating strategy for flow cytometry is shown in Fig EV3F.
Table 1.
Antibodies and sgRNAs used in this study.
| Targets | Clone | Tag | Vendor | Catalog number | Application |
|---|---|---|---|---|---|
| CD3 | OKT3 | PE‐Cy7 | Biolegend | 317334 | FACS |
| CD4 | OKT4 | APC‐Cy7 | Biolegend | 317428 | FACS |
| CD8 | RPA‐T8 | FITC | Biolegend | 301006 | FACS |
| CD56 | 5.1H11 | PE | Biolegend | 306722 | FACS |
| NKp30 | P30‐15 | PE | Biolegend | 325208 | FACS |
| NKp30 | P30‐15 | APC | Biolegend | 325210 | FACS |
| NKp46 | 9E2 | PE | Biolegend | 331908 | FACS |
| NKp46 | 9E2 | APC | Biolegend | 331918 | FACS |
| mIgG1 | MOPC‐21 | Biolegend | 400101 | Control | |
| GAPDH | mAbcam9484 | Abcam | ab9484 | Western blot | |
| BCL11B | 25B6 | Abcam | ab18465 | Western blot | |
| OPA1 | D7C1A | Cell Signaling | 67589 | Western blot | |
| DRP1 | D6C7 | Cell Signaling | 8570 | Western blot | |
| MFN1 | D6E2S | Cell Signaling | 14739 | Western blot | |
| MFN2 | D2D10 | Cell Signaling | 9482 | Western blot | |
| Tom20 | D8T4N | Cell Signaling | 42406 | Western blot | |
| MTA2 | EPR8537(2) | Abcam | Ab171073 | Western blot | |
| MBD2 | EPR18361 | Abcam | Ab188474 | Western blot | |
| CHD4 | D4B7 | Cell Signaling | 12011 | Western blot | |
| β‐actin | 8H10D10 | Cell Signaling | 3700 | Western blot | |
| β‐tubulin | D65A4 | Cell Signaling | 5666 | Western blot | |
| H3K27Ac | D5E4 | Cell Signaling | 15485 | Western blot | |
| H3 | D1H2 | Cell Signaling | 60932SF | Western blot | |
| MBD2 | 106B691 | Abcam | Ab45027 | Cut&Tag | |
| CHD4 | D4B7 | Cell Signaling | 12011 | Cut&Tag | |
| BCL11B | D6F1 | Cell Signaling | 12120s | Cut&tag | |
| H3K27Ac | Polyclonal | Activemotif | 39133 | Cut&tag | |
| sg‐BCL11B | GAAGCAGTGTGGCGGCAGCT | GGTCAGACGGAGGCTCCCTT | Function | ||
| sg‐OPA1 | GCTGGCCAAAAATTCCTGCG | Function | |||
| sg‐MTA2 | AGGAGCATAAGGCCGTCGGG | GCAAAGGAACGGCTACGACC | Function | ||
| sg‐MTA1 | AAGTGGTGTGCTTCTACCGG | TGACGTTGTTGACGCTGATT | Function | ||
| sg‐MBD2 | AGCCGGTCCCTTTCCCGTCG | AGGGCAAGTCTCCGCTGCTG | Function | ||
| sg‐CHD4 | TGTGGAGGTGCCTATCCGCA | ATGGCTCTGTCCCCGTTGTA | Function | ||
| sg‐Ctrl | CCGGGTCTTCGAGAAGACCT | Function | |||
Transmission electron microscopy (TEM)
Cells were washed twice with phosphate‐buffered saline (PBS) and then fixed with 3% glutaraldehyde. The ultrastructure of mitochondria was examined by TEM.
Mitochondrial dynamic imaging
For mitochondrial imaging, cultured T, ITNK, and NK cells were stained with MitoTracker® Green (Yeasen, 40742ES50, China) or Deep Red FM (40743ES50, Yeasen, China) for 30 min and then stained with Hoechst 33342 (C1029, Beyotime, China) for nuclear staining for 30 min at 37°C. After washing twice with preheated culture medium, the cells were surface stained and processed for flow cytometry or confocal microscopy. Confocal laser scanning microscopy (CLSM) was carried‐out on an LSM 710 NLO scanning confocal imaging workstation (Oberkochen, Germany). The length of stained mitochondria was measured using ImageJ. The mean relative length of mitochondria was obtained by measuring the length of mitochondria in each cell and statistically analyzing 20–30 cells.
ATAC sequencing
ATAC‐seq was performed as previously described (Li et al, 2017). In brief, a total of 50,000 cells were washed once with 50 μl of cold PBS and suspended in 50 μl of lysis buffer (10 mM Tris–HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, and 0.2% (v/v) IGEPAL CA‐630). Suspended nuclei were then centrifuged for 10 min at 500 g at 4°C, followed by the addition of 50 ml of transposition reaction mix (25 μl TD buffer, 2.5 μl Tn5 transposase, and 22.5 μl nuclease‐free H2O) from the Nextera DNA Library Preparation Kit (96 samples) (FC‐121‐1031, Illumina, USA). Samples were then PCR amplified and incubated at 37°C for 30 min. DNA was isolated using a MinElute Kit (QIAGEN, Germany). ATAC‐seq libraries were first subjected to 5 cycles of preamplification. To determine the suitable number of cycles required for the second round of PCR, the library was assessed by quantitative PCR as described, and the library was then PCR amplified for the appropriate number of cycles. Libraries were purified with a QIAquick PCR (QIAGEN, Germany) column. The library concentration was measured using the KAPA Library Quantification Kit (KK4824) according to the manufacturer's instructions. The library's integrity was checked by gel electrophoresis. Finally, the ATAC library was sequenced on a NextSeq 500 using the NextSeq 500 High Output Kit v2 (150 cycles) (FC‐404‐2002, Illumina) according to the manufacturer's instructions.
CUT&Tag sequencing
CUT&Tag experiments were performed using Hyperactive In‐Situ ChIP LibraryPrep kit for Illumina (TD901‐02, Vazyme, China) as described previously with modifications (Dan et al, 2020). Briefly, 100,000 cells were incubated with activated concanavalin A‐coated magnetic beads. The bead‐bound cells were permeabilized and incubated first with primary antibody, followed by secondary antibody. Diluted pG‐Tn5 adaptor complex was then added followed by the tagmentation reaction. Extracted DNA fragments were used for library preparation. Finally, the CUT&Tag library was sequenced on the Illumina NovaSeq platform at Novogene Co., LTD.
Acetyl‐CoA and α‐KG measurement
Acetyl‐CoA levels were measured using PicoProbe™ Acetyl CoA Fluorometric Assay Kit (#K317‐100, BioVision, Milpitas, CA, USA). Briefly, 1,000,000 cells were washed with PBS and lysed with acetyl‐CoA assay buffer on ice. Then the supernatant was collected by centrifugation (10,000 g × 10 min, 4°C). Ten microliters of CoA Quencher were added to each sample to correct for background followed by 2 μl of quencher remover. Fifty microliters of acetyl‐CoA reaction mixture containing the Substrate Mix, Conversion Enzyme, Enzyme mix, and PicoProbe were subsequently added and incubated at 37°C for 30 min. Fluorescence of Excitation/Emission = 535/587 nm was measured. After correction for the matched background well (acetyl‐CoA reaction mixture added without conversion enzyme) of all readings, the values for each sample were normalized to the protein concentration in each sample determined using BCA protein assay kit (# E‐BC‐K318‐M, Elabscience).
α‐KG levels were measured using α‐KG Colorimetric/Fluorometric Assay Kit (#K677‐100, BioVision, Milpitas, CA, USA). Briefly, 2 × 106 cells are rapidly homogenized with 100 μl of ice cold α‐KG assay buffer. Add 1–50 μl samples of a 96‐well plate and bring volume to 50 μl with Assay buffer. Then 50 μl the Reaction Mix to each well containing samples and background control. Incubate for 30 min at 37°C and Fluorescence of Excitation/Emission = 535/587 nm was measured.
In vitro killing assays
The K562‐GL and HepG2‐GL target cells were incubated with the indicated killing cells at the indicated ratio in triplicate wells of U‐bottomed 96‐well plates. Target cell viability was monitored 24 h later by adding 100 μl of the substrate D‐luciferin (potassium salt) (Cayman Chemical, Michigan, USA) at 150 μg/ml to each well. Background luminescence was negligible (< 1% of the signal from wells containing only target cells). The percent cytotoxicity (killing %) values were calculated as (blank signal − experimental signal)/blank signal × 100%.
Cytokine release assays
Cytokine secretion profiles of indicated killing cells were incubated with cancer cells at an E:T ratio of 1:1 for 24 h. Hundred microliters of supernatants were then collected and stored at −20°C for further measurement. The concentration of cytokines was quantified by enzyme‐linked immunosorbent assay (ELISA) according to manufacturers' protocols. ELISA kits for human IFN‐γ, Granzyme B, and GM‐CSF were purchased from Thermo Fisher Scientific Inc, USA.
Extracellular metabolic flux analysis
For the mitochondria stress test, OCR was measured using a 24‐well XFe Analyzer system (Seahorse Bioscience). Indicated cells (2 × 105 cells/well) were plated on pretreated Seahorse plates (BD Biosciences) in XF media (10 mM glucose, 2 mM glutamine and 1 mM pyruvate). Basal OCR was measured for 30 min. Cells were cocultured with 2 μM oligomycin, 1 μM FCCP, and 1 μM each of rotenone and antimycin A (all drugs were from Agilent Technologies), to measure maximal respiration and spare respiratory capacity (SRC = maximal respiration‐basal respiration).
Metabolic assays
For analysis of mitochondrial mass, fresh cells were stained with 200 nM MitoTracker Green in culture medium for 30 min at 37°C. For measurement of the mitochondrial membrane potential, fresh cells were stained with 100 nM TMRM in culture medium for 10 min at 37°C. For measurement of mitochondrial superoxide production, fresh cells were incubated with 10 μM MitoSOX in culture medium for 20 min at 37°C. After washing twice with preheated PBS, cells were surface‐stained and measured by flow cytometry.
Real‐time PCR
Total RNA was prepared with FastPure Cell/Tissue Total RNA Isolation Kit V2 (RC112‐01, Vazyme, China) according to the manufacturer's instructions. cDNA was reverse transcribed from mRNA with TransScript Uni All‐in One First‐Strand Cdna Synthesis SuperMix for qPCR kit (AU341, TransGene, China). qPCR was performed with TransStart Tip Green qPCR SuperMix (AQ141, TransGene, China) on a Bio‐Rad CFX96 real‐time PCR machine (Bio‐Rad, Hercules, CA). The primers are listed in Table 2.
Table 2.
Primers for ChIP‐qPCR and RT‐qPCR.
| Genes | Forward primer 5′→3′ | Reverse primer 5′→3′ |
|---|---|---|
| OPA1 | ATGTGGCGACTACGTCGGG | TTTCTCCTGATGAAGAGCTT |
| ID2 | GCTATACAACATGAACGACTGCT | AATAGTGGGATGCGAGTCCAG |
| ZBTB16 | GAGATCCTCTTCCACCGCAAT | CCGCATACAGCAGGTCATC |
| NCR1 | CTAGGCCGGCAGAATCTGAG | TTTGGGAGAGTCTGCTGCTG |
| NCR2 | TTCCCTGTGCCCACAGAATC | CCTGGAGAAGACTGGGGAGA |
| NCR3 | TACAGTCCTCCTCCTTCGGG | TTCCAGGTCAGACATTTGCC |
| GZMB | ACCAATCCTGCTTCTGCTGG | GTGGAGAAAGGGCAGGAGAC |
| IFNγ | AGAAACCTGTACCATTGGGGG | AGCTCAACAAAGCTGATACTCCA |
| CSF2 | GACACTGCTGCTGAGATGGT | TATCAAGCTGACAGGCGTGG |
| EEF2 | TGAACAAGATGGACCGCG | GGATCGATCATGATGTTGCC |
| B2M | GATGAGTATGCCTGCCGTGT | TGCGGCATCTTCAAACCTCC |
| MYH9 | AAGAACAAGCTCAGGCGC | GCTGTGGTGTCTGTCTGTCC |
| TUBB | CCACCGGCACCTACCAC | CTGCCCCAGACTGACCAAAT |
| TCF7 | TTGATGCTAGGTTCTGGTGTACC | CCTTGGACTCTGCTTGTGTC |
| LEF1 | TTCTTGGCAGAAGGTGGCAT | AGGCAGCTGTCATTCTTGGA |
| CCR7 | ACAGCCTTCCTGTGTGGTTT | CTTGACACAGGCATACCTGGAA |
| BCL11B | TCCAGCTACATTTGCACAACA | GCTCCAGGTAGATGCGGAAG |
The sequences of primers used in this study.
ChIP‐qPCR
Cell lysis, sonication, and immunoprecipitation were performed using the SimpleChIP Enzymatic Chromatin IP Kit (Magnetic Beads) (Cell Signaling Technology, 9003S) according to the manufacturer's instructions. The antibodies for immunoprecipitation were acetyl‐histone H3 (Lys27) (Cell Signaling Technology, no. 8173, 1:100) and IgG (Cell Signaling Technology, no. 2729, 1:100). All reactions were performed with TransStart Tip Green qPCR SuperMix (AQ141, TransGene, China) on a Bio‐Rad CFX96 real‐time PCR machine (Bio‐Rad, Hercules, CA). The ChIP–qPCR primers are listed in Table 2.
CUT&Tag qPCR
CUT&Tag experiments were performed using Hyperactive In‐Situ ChIP LibraryPrep kit for Illumina (TD901‐02, Vazyme, China) as described above. The extracted DNA fragments were performed by PCR amplification. The PCR products were purified and then performed with TransStart Tip Green qPCR SuperMix (AQ141, TransGene, China) on a Bio‐Rad CFX96 real‐time PCR machine (Bio‐Rad, Hercules, CA). Twenty nanograms of DNA library were used for each PCR reaction. The CUT&Tag‐qPCR primers are listed in Table 3.
Table 3.
Primers for CUT&Tag‐qPCR.
| Genes | Forward primer 5′→3′ | Reverse primer 5′→3′ |
|---|---|---|
| OPA1 Primer 1 | GGCTCTTGCGGAAGTCCAT | GGAATGACCCAGGAAGTGGC |
| OPA1 Primer 2 | GCCACTTCCTGGGTCATTCC | GACGTAGTCGCCACATCCC |
| OPA1 Primer 3 | GCTCTGTCCATGAGTCACCT | GGCTAGGGAAAGAGACGGAC |
| OPA1 Primer 4 | GCTCATTGTTGCAGGCATTTGA | ATGCATTCCATTCCTTATAAACACG |
| OPA1 Primer 5 | AGCATTCTCTGTTGGAGCAAT | TGCCATCACCAGGAGACATTT |
| OPA1 Primer 6 | CCTCCCACTCCCTTTGGATAC | AGGAAATTCCCCAGGACATAAAAT |
| ID2 | GCTATACAACATGAACGACTGCT | AATAGTGGGATGCGAGTCCAG |
| ZBTB16 | GAGATCCTCTTCCACCGCAAT | CCGCATACAGCAGGTCATC |
| ZNF683 | CACCCCACCTGTTCACCTATG | CCCCAGCTCATTGACCATCA |
The sequences of primers used in this study.
Bulk RNA sequencing
mRNA extracted from purified T cells (purity > 90%) transduced with sgCtrl, sgBCL11B, sgMTA2, sgMBD2, or sgCHD4 from CD4 and CD8 T cell populations was performed on a BGISEQ‐500 (Mak et al, 2017) (BGI, Wuhan, China). Sequenced reads were trimmed for adaptor sequences and masked for low‐complexity or low‐quality sequences. The number of raw reads mapped to genes was calculated by RSEM (rsem‐1.2.4), and the sample results were combined and normalized by EDAseq (1.99.1). Gene expression fold changes were calculated using normalized raw reads. The downstream analysis used glbase scripts. Gene differential expression analysis was performed by R package DESeq2 (version 1.38.1). The number of differentially expressed genes was counted as visualized by customed R scripts using ggplot2 (version 3.4.0). Gene Ontology (G.O.) enrichment analysis was performed by R package clusterprofile (version 4.6.2). The was visualized as heatmaps generated by customed R scripts using ggplot2 (version 3.4.0).
CUT&Tag‐seq and ATAC‐seq data analysis
The sequencing Raw reads were processed by fastp (version 0.20.1) to filter out bad reads and cut adapters. Clean reads were aligned to the human genome defined by NCBI (GCF_000001405.40) with bowtie2 (version 2.2.5). Aligned reads were indexed and sorted with samtools (version 1.3.1). PCR duplicates were removed using the “markdup” function of sambamba (version 4.2.0.0). MACS2 (version 2.2.7.1) was used for peakcalling, blacklist region of the human genome was excluded from peakcalling results. Homer (version 4.11.1) was used to annotate peaks to genomic regions. Deeptools (version 3.5.1) was used to calculate scores per genome regions of interesting and create heatmaps for scores associated with genomic regions. The bedGraphToBigWig (version 2.9) was used to convert bedGraph file to bigwig format. BigWig tracks were visualized in the Integrative Genomic Viewer genome (IGV) browser (version 2.8.12).
Statistics
Statistical significance was determined using Student's t‐test (two groups) or ANOVA with Tukey, Sidak, and Dunnett's multiple comparison test (three or more groups). All statistical analyses were performed using Prism version 7.0 (GraphPad, Inc., San Diego, CA, USA). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001 were considered statistically significant.
Author contributions
Peng Li: Conceptualization; resources; data curation; supervision; funding acquisition; investigation; writing – original draft; project administration; writing – review and editing. Rui Liao: Conceptualization; data curation; formal analysis; validation; investigation; visualization; methodology; writing – original draft; writing – review and editing. Yi Wu: Data curation; formal analysis; funding acquisition; validation; investigation; visualization; methodology; writing – review and editing. Le Qin: Funding acquisition; methodology. Zhiwu Jiang: Funding acquisition; investigation; methodology. Shixue Gou: Formal analysis; validation; methodology. Linfu Zhou: Formal analysis; validation; methodology. Qilan Hong: Formal analysis. Yao Li: Validation; methodology. Jingxuan Shi: Formal analysis; validation. Yao Yao: Funding acquisition. Liangxue Lai: Resources. Yangqiu Li: Resources; funding acquisition. Pentao Liu: Resources. Jean Paul Thiery: Resources; writing – review and editing. Dajiang Qin: Resources. Thomas Graf: Resources; writing – review and editing. Xingguo Liu: Conceptualization; resources; data curation; supervision; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Dataset EV1
Source Data for Expanded View and Appendix
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank Diwei Zheng, Yuanbin Cui, Qiting Wu, Youguo Long, Suna Wang, Shouheng Lin, and Duo Hu for their help and contributions to this study. We also thank the teams at the Analysis and Test Center of GIBH for their invaluable support in providing core facilities for our research. This study was supported by the National Key Research and Development Plan, No. 2021YFE0202800 (PL), 2017YFE0131600 (YL); National Natural Science Foundation of China, No. 81972672 (PL), 32170946 (ZJ), 82003054 (SL), 82273377 (SL), 82202031 (LQ); The Youth Innovation Promotion Association of the Chinese Academy of Sciences (2020351, ZJ; 2021355, YW); Guangdong Natural Science Foundation, No. 2022A1515012569 (ZJ), 2022A1515012484 (SL), 2021A1515220077 (SL), 2021A1515110005 (LQ), 2022A1515012360 (LQ), 2022A1515010604 (YY), 2022A1515110349 (DZ); 2020B1212060052; Science and Technology Program of Guangzhou, No. 202102080470 (YY), 2023A04J0103(RL). Partially supported by a grant from the University Grants Committee/Research Grants Council of the Hong Kong Special Administrative Region, China (Project No. AoE/M‐401/20), Innovation and Technology Fund (ITF).
The EMBO Journal (2023) 42: e113448
Contributor Information
Xingguo Liu, Email: liu_xingguo@gibh.ac.cn.
Peng Li, Email: li_peng@gibh.ac.cn.
Data availability
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive (Chen et al, 2021) in National Genomics Data Center (CNCB‐NGDC Members and Partners, 2022), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA‐Human: HRA004801) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa‐human.
References
- Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, Langer T (2014) The i‐AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol 204: 919–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baixauli F, Piletic K, Puleston DJ, Villa M, Field CS, Flachsmann LJ, Quintana A, Rana N, Edwards‐Hicks J, Matsushita M et al (2022) An LKB1‐mitochondria axis controls TH17 effector function. Nature 610: 555–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker MJ, Lampe PA, Stojanovski D, Korwitz A, Anand R, Tatsuta T, Langer T (2014) Stress‐induced OMA1 activation and autocatalytic turnover regulate OPA1‐dependent mitochondrial dynamics. EMBO J 33: 578–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck MD, O'Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, Qiu J, Kretz O, Braas D, van der Windt GJ et al (2016) Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 166: 63–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Califano D, Sweeney KJ, Le H, VanValkenburgh J, Yager E, O'Connor W Jr, Kennedy JS, Jones DM, Avram D (2014) Diverting T helper cell trafficking through increased plasticity attenuates autoimmune encephalomyelitis. J Clin Invest 124: 174–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Curtis JD, Maggi LB Jr, Faubert B, Villarino AV, O'Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J et al (2013) Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153: 1239–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160: 189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Chen X, Zhang S, Zhu J, Tang B, Wang A, Dong L, Zhang Z, Yu C, Sun Y et al (2021) The genome sequence archive family: toward explosive data growth and diverse data types. Genomics Proteomics Bioinformatics 19: 578–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cismasiu VB, Adamo K, Gecewicz J, Duque J, Lin Q, Avram D (2005) BCL11B functionally associates with the NuRD complex in T lymphocytes to repress targeted promoter. Oncogene 24: 6753–6764 [DOI] [PubMed] [Google Scholar]
- Cismasiu VB, Paskaleva E, Suman Daya S, Canki M, Duus K, Avram D (2008) BCL11B is a general transcriptional repressor of the HIV‐1 long terminal repeat in T lymphocytes through recruitment of the NuRD complex. Virology 380: 173–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- CNCB‐NGDC Members and Partners (2022) Database resources of the national genomics data center, China National Center for bioinformation in 2022. Nucleic Acids Res 50: D27–D38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinides MG, McDonald BD, Verhoef PA, Bendelac A (2014) A committed precursor to innate lymphoid cells. Nature 508: 397–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan L, Liu L, Sun Y, Song J, Yin Q, Zhang G, Qi F, Hu Z, Yang Z, Zhou Z et al (2020) The phosphatase PAC1 acts as a T cell suppressor and attenuates host antitumor immunity. Nat Immunol 21: 287–297 [DOI] [PubMed] [Google Scholar]
- Dege C, Hagman J (2014) Mi‐2/NuRD chromatin remodeling complexes regulate B and T‐lymphocyte development and function. Immunol Rev 261: 126–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Dotto V, Mishra P, Vidoni S, Fogazza M, Maresca A, Caporali L, McCaffery JM, Cappelletti M, Baruffini E, Lenaers G et al (2017) OPA1 isoforms in the hierarchical organization of mitochondrial functions. Cell Rep 19: 2557–2571 [DOI] [PubMed] [Google Scholar]
- Del Dotto V, Fogazza M, Carelli V, Rugolo M, Zanna C (2018) Eight human OPA1 isoforms, long and short: what are they for? Biochim Biophys Acta Bioenerg 1859: 263–269 [DOI] [PubMed] [Google Scholar]
- Delconte RB, Shi W, Sathe P, Ushiki T, Seillet C, Minnich M, Kolesnik TB, Rankin LC, Mielke LA, Zhang JG et al (2016) The helix‐loop‐helix protein ID2 governs NK cell fate by tuning their sensitivity to interleukin‐15. Immunity 44: 103–115 [DOI] [PubMed] [Google Scholar]
- Dimeloe S, Burgener AV, Grahlert J, Hess C (2017) T‐cell metabolism governing activation, proliferation and differentiation; a modular view. Immunology 150: 35–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding M, Liu C, Shi R, Yu M, Zeng K, Kang J, Fu F, Mi M (2020) Mitochondrial fusion promoter restores mitochondrial dynamics balance and ameliorates diabetic cardiomyopathy in an optic atrophy 1‐dependent way. Acta Physiol 229: e13428 [DOI] [PubMed] [Google Scholar]
- Dubuissez M, Loison I, Paget S, Vorng H, Ait‐Yahia S, Rohr O, Tsicopoulos A, Leprince D (2016) Protein kinase C‐mediated phosphorylation of BCL11B at serine 2 negatively regulates its interaction with NuRD complexes during CD4+ T‐cell activation. Mol Cell Biol 36: 1881–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin AD, Wang T, Wimalasena VK, Zimmerman MW, Li D, Dharia NV, Mariani L, Shendy NAM, Nance S, Patel AG et al (2022) EP300 selectively controls the enhancer landscape of MYCN‐amplified neuroblastoma. Cancer Discov 12: 730–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehses S, Raschke I, Mancuso G, Bernacchia A, Geimer S, Tondera D, Martinou JC, Westermann B, Rugarli EI, Langer T (2009) Regulation of OPA1 processing and mitochondrial fusion by m‐AAA protease isoenzymes and OMA1. J Cell Biol 187: 1023–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanucchi S, Domínguez‐Andrés J, Joosten LAB, Netea MG, Mhlanga MM (2021) The intersection of epigenetics and metabolism in trained immunity. Immunity 54: 32–43 [DOI] [PubMed] [Google Scholar]
- Gao Q, Tian R, Han H, Slone J, Wang C, Ke X, Zhang T, Li X, He Y, Liao P et al (2022a) PINK1‐mediated Drp1S616 phosphorylation modulates synaptic development and plasticity via promoting mitochondrial fission. Signal Transduct Target Ther 7: 103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Zamisch M, Vacchio M, Chopp L, Ciucci T, Paine EL, Lyons GC, Nie J, Xiao Q, Zvezdova E et al (2022b) NuRD complex recruitment to Thpok mediates CD4+ T cell lineage differentiation. Sci Immunol 7: eabn5917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Shi X, Boopathy S, McDonald J, Smith AW, Chao LH (2020) Two forms of Opa1 cooperate to complete fusion of the mitochondrial inner‐membrane. Elife 9: e50973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghesquière B, Wong BW, Kuchnio A, Carmeliet P (2014) Metabolism of stromal and immune cells in health and disease. Nature 511: 167–176 [DOI] [PubMed] [Google Scholar]
- Griparic L, Kanazawa T, van der Bliek AM (2007) Regulation of the mitochondrial dynamin‐like protein Opa1 by proteolytic cleavage. J Cell Biol 178: 757–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa H, Romero‐Wolf M, Yui MA, Ungerbäck J, Quiloan MLG, Matsumoto M, Nakayama KI, Tanaka T, Rothenberg EV (2018) Bcl11b sets pro‐T cell fate by site‐specific cofactor recruitment and by repressing Id2 and Zbtb16. Nat Immunol 19: 1427–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa H, Romero‐Wolf M, Yang Q, Motomura Y, Levanon D, Groner Y, Moro K, Tanaka T, Rothenberg EV (2020) Cell type‐specific actions of Bcl11b in early T‐lineage and group 2 innate lymphoid cells. J Exp Med 217: e20190972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G, Cui K, Fang D, Hirose S, Wang X, Wangsa D, Jin W, Ried T, Liu P, Zhu J et al (2018) Transformation of accessible chromatin and 3D nucleome underlies lineage commitment of early T cells. Immunity 48: 227–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikawa T, Hirose S, Masuda K, Kakugawa K, Satoh R, Shibano‐Satoh A, Kominami R, Katsura Y, Kawamoto H (2010) An essential developmental checkpoint for production of the T cell lineage. Science 329: 93–96 [DOI] [PubMed] [Google Scholar]
- Ishihara N, Fujita Y, Oka T, Mihara K (2006) Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J 25: 2966–2977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isoda T, Moore AJ, He Z, Chandra V, Aida M, Denholtz M, Piet van Hamburg J, Fisch KM, Chang AN, Fahl SP et al (2017) Non‐coding transcription instructs chromatin folding and compartmentalization to dictate enhancer‐promoter communication and T cell fate. Cell 171: 103–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Qin L, Tang Y, Liao R, Shi J, He B, Li S, Zheng D, Cui Y, Wu Q et al (2022) Human induced‐T‐to‐natural killer cells have potent anti‐tumour activities. Biomark Res 10: 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juno JA, van Bockel D, Kent SJ, Kelleher AD, Zaunders JJ, Munier CM (2017) Cytotoxic CD4 T cells‐friend or foe during viral infection? Front Immunol 8: 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishton RJ, Sukumar M, Restifo NP (2017) Metabolic regulation of T cell longevity and function in tumor immunotherapy. Cell Metab 26: 94–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Leid M, Rothenberg EV (2010a) An early T cell lineage commitment checkpoint dependent on the transcription factor Bcl11b. Science 329: 89–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Burke S, Wang J, Chen X, Ortiz M, Lee SC, Lu D, Campos L, Goulding D, Ng BL et al (2010b) Reprogramming of T cells to natural killer‐like cells upon Bcl11b deletion. Science 329: 85–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Liu J, Yang X, Zhou C, Guo J, Wu C, Qin Y, Guo L, He J, Yu S et al (2017) Chromatin accessibility dynamics during iPSC reprogramming. Cell Stem Cell 21: 819–833 [DOI] [PubMed] [Google Scholar]
- Liu PS, Wang H, Li X, Chao T, Teav T, Christen S, Di Conza G, Cheng WC, Chou CH, Vavakova M et al (2017) α‐ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol 18: 985–994 [DOI] [PubMed] [Google Scholar]
- Lorentsen KJ, Cho JJ, Luo X, Zuniga AN, Urban JF Jr, Zhou L, Gharaibeh R, Jobin C, Kladde MP, Avram D (2018) Bcl11b is essential for licensing Th2 differentiation during helminth infection and allergic asthma. Nat Commun 9: 1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losón OC, Song Z, Chen H, Chan DC (2013) Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell 24: 659–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughran SJ, Comoglio F, Hamey FK, Giustacchini A, Errami Y, Earp E, Göttgens B, Jacobsen SEW, Mead AJ, Hendrich B et al (2017) Mbd3/NuRD controls lymphoid cell fate and inhibits tumorigenesis by repressing a B cell transcriptional program. J Exp Med 214: 3085–3104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis C, Souza‐Fonseca‐Guimaraes F, Yang Y, D'Silva D, Kratina T, Dagley L, Hediyeh‐Zadeh S, Rautela J, Masters SL, Davis MJ et al (2020) NK cell‐derived GM‐CSF potentiates inflammatory arthritis and is negatively regulated by CIS. J Exp Med 217: e20191421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Kovalev GI, Chang H, Kallin E, Knudsen G, Xia L, Mishra N, Ruiz P, Li E, Su L et al (2008) Inactivation of NuRD component Mta2 causes abnormal T cell activation and lupus‐like autoimmune disease in mice. J Biol Chem 283: 13825–13833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIver NJ, Michalek RD, Rathmell JC (2013) Metabolic regulation of T lymphocytes. Annu Rev Immunol 31: 259–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak SST, Gopalakrishnan S, Caroe C, Geng C, Liu S, Sinding MS, Kuderna LFK, Zhang W, Fu S, Vieira FG et al (2017) Comparative performance of the BGISEQ‐500 vs Illumina HiSeq2500 sequencing platforms for palaeogenomic sequencing. Gigascience 6: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maluski M, Ghosh A, Herbst J, Scholl V, Baumann R, Huehn J, Geffers R, Meyer J, Maul H, Eiz‐Vesper B et al (2019) Chimeric antigen receptor‐induced BCL11B suppression propagates NK‐like cell development. J Clin Invest 129: 5108–5122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matilainen O, Quirós PM, Auwerx J (2017) Mitochondria and epigenetics ‐ crosstalk in homeostasis and stress. Trends Cell Biol 27: 453–463 [DOI] [PubMed] [Google Scholar]
- Morris JP 4th, Yashinskie JJ, Koche R, Chandwani R, Tian S, Chen CC, Baslan T, Marinkovic ZS, Sánchez‐Rivera FJ, Leach SD et al (2019) α‐Ketoglutarate links p53 to cell fate during tumour suppression. Nature 573: 595–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan D, van der Windt GJ, Huang SC, Curtis JD, Chang CH, Buck MD, Qiu J, Smith AM, Lam WY, DiPlato LM et al (2014) Memory CD8+ T cells use cell‐intrinsic lipolysis to support the metabolic programming necessary for development. Immunity 41: 75–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil VS, Madrigal A, Schmiedel BJ, Clarke J, O'Rourke P, de Silva AD, Harris E, Peters B, Seumois G, Weiskopf D et al (2018) Precursors of human CD4+ cytotoxic T lymphocytes identified by single‐cell transcriptome analysis. Sci Immunol 3: eaan8664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrocola F, Galluzzi L, Bravo‐San Pedro JM, Madeo F, Kroemer G (2015) Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab 21: 805–821 [DOI] [PubMed] [Google Scholar]
- Ron‐Harel N, Santos D, Ghergurovich JM, Sage PT, Reddy A, Lovitch SB, Dephoure N, Satterstrom FK, Sheffer M, Spinelli JB et al (2016) Mitochondrial biogenesis and proteome remodeling promote one‐carbon metabolism for T cell activation. Cell Metab 24: 104–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberg EV (2012) Transcriptional drivers of the T‐cell lineage program. Curr Opin Immunol 24: 132–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen P, Lan Y, Li CY, Sidoli S, Donahue G, Dou Z, Frederick B, Chen Q, Luense LJ, Garcia BA et al (2019) Histone acetyltransferase p300 induces de novo super‐enhancers to drive cellular senescence. Mol Cell 73: 684–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Wei W, Zhou DX (2015) Histone acetylation enzymes coordinate metabolism and gene expression. Trends Plant Sci 20: 614–621 [DOI] [PubMed] [Google Scholar]
- Shen E, Wang Q, Rabe H, Liu W, Cantor H, Leavenworth JW (2018) Chromatin remodeling by the NuRD complex regulates development of follicular helper and regulatory T cells. Proc Natl Acad Sci USA 115: 6780–6785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidwell T, Rothenberg EV (2021) Epigenetic dynamics in the function of T‐lineage regulatory factor Bcl11b. Front Immunol 12: 669498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Chen H, Fiket M, Alexander C, Chan DC (2007) OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol 178: 749–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Ghochani M, McCaffery JM, Frey TG, Chan DC (2009) Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol Biol Cell 20: 3525–3532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottile R, Panjwani MK, Lau CM, Daniyan AF, Tanaka K, Barker JN, Brentjens RJ, Sun JC, Le Luduec JB, Hsu KC (2021) Human cytomegalovirus expands a CD8+ T cell population with loss of BCL11B expression and gain of NK cell identity. Sci Immunol 6: eabe6968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, Mangan PA, Kulikovskaya I, Gupta M, Chen F et al (2020) CRISPR‐engineered T cells in patients with refractory cancer. Science 367: eaba7365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, Roychoudhuri R, Palmer DC, Muranski P, Karoly ED et al (2013) Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest 123: 4479–4488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutendra G, Kinnaird A, Dromparis P, Paulin R, Stenson TH, Haromy A, Hashimoto K, Zhang N, Flaim E, Michelakis ED (2014) A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl‐CoA and histone acetylation. Cell 158: 84–97 [DOI] [PubMed] [Google Scholar]
- Takeuchi A, Saito T (2017) CD4 CTL, a cytotoxic subset of CD4+ T Cells, their differentiation and function. Front Immunol 8: 194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topark‐Ngarm A, Golonzhka O, Peterson VJ, Barrett B Jr, Martinez B, Crofoot K, Filtz TM, Leid M (2006) CTIP2 associates with the NuRD complex on the promoter of p57KIP2, a newly identified CTIP2 target gene. J Biol Chem 281: 32272–32283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, Pearce EJ, Pearce EL (2012) Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 36: 68–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Wang J, Bonamy GM, Meeusen S, Brusch RG, Turk C, Yang P, Schultz PG (2012) A small molecule promotes mitochondrial fusion in mammalian cells. Angew Chem Int Ed Engl 51: 9302–9305 [DOI] [PubMed] [Google Scholar]
- Wang Y, Huang Y, Liu J, Zhang J, Xu M, You Z, Peng C, Gong Z, Liu W (2020) Acetyltransferase GCN5 regulates autophagy and lysosome biogenesis by targeting TFEB. EMBO Rep 21: e48335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Shadel GS, Ghosh S (2011) Mitochondria in innate immune responses. Nat Rev Immunol 11: 389–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Lau CM, Sottile R, Le Luduec JB, Panjwani MK, Conaty PM, Srpan K, Laib Sampaio K, Mertens T, Adler SP et al (2021) Human cytomegalovirus infection promotes expansion of a functionally superior cytoplasmic CD3+ NK cell subset with a Bcl11b‐regulated T cell signature. J Immunol 207: 2534–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Chen K, Li L, Hao Z, Wang T, Liu Y, Xing G, Liu Z, Li H, Yuan H et al (2022) Plin2‐mediated lipid droplet mobilization accelerates exit from pluripotency by lipidomic remodeling and histone acetylation. Cell Death Differ 29: 2316–2331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM (2012) Mitochondrial fission, fusion, and stress. Science 337: 1062–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YR, Imrichova H, Wang H, Chao T, Xiao Z, Gao M, Rincon‐Restrepo M, Franco F, Genolet R, Cheng WC et al (2020) Disturbed mitochondrial dynamics in CD8+ TILs reinforce T cell exhaustion. Nat Immunol 21: 1540–1551 [DOI] [PubMed] [Google Scholar]
- Zhang S, Rozell M, Verma RK, Albu DI, Califano D, VanValkenburgh J, Merchant A, Rangel‐Moreno J, Randall TD, Jenkins NA et al (2010) Antigen‐specific clonal expansion and cytolytic effector function of CD8+ T lymphocytes depend on the transcription factor Bcl11b. J Exp Med 207: 1687–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Qian Y, Fu B, Jiao D, Jiang Y, Chen P, Shen Y, Zhang H, Sun R, Tian Z et al (2019) Mitochondrial fragmentation limits NK cell‐based tumor immunosurveillance. Nat Immunol 20: 1656–1667 [DOI] [PubMed] [Google Scholar]
- Zhou W, Zhao T, Du J, Ji G, Li X, Ji S, Tian W, Wang X, Hao A (2019) TIGAR promotes neural stem cell differentiation through acetyl‐CoA‐mediated histone acetylation. Cell Death Dis 10: 198 [DOI] [PMC free article] [PubMed] [Google Scholar]