

Abstract
RNA‐based therapeutics have the potential to revolutionize the treatment and prevention of human diseases. While early research faced setbacks, it established the basis for breakthroughs in RNA‐based drug design that culminated in the extraordinarily fast development of mRNA vaccines to combat the COVID‐19 pandemic. We have now reached a pivotal moment where RNA medicines are poised to make a broad impact in the clinic. In this review, we present an overview of different RNA‐based strategies to generate novel therapeutics, including antisense and RNAi‐based mechanisms, mRNA‐based approaches, and CRISPR‐Cas‐mediated genome editing. Using three rare genetic diseases as examples, we highlight the opportunities, but also the challenges to wide‐ranging applications of this class of drugs.
Keywords: antisense oligonucleotide drug, CRISPR‐Cas therapy, mRNA therapeutics, rare genetic disease, RNA interference
Subject Categories: Genetics, Gene Therapy & Genetic Disease; Pharmacology & Drug Discovery; RNA Biology
This review discusses the latest research on RNA‐based therapeutics, from design to the clinic.

Introduction
The rapid development of mRNA vaccines in response to the COVID‐19 pandemic has led to a resurgence in interest in RNA as a molecule class in diagnostics, prevention, and treatment of diseases. Whether for infectious diseases, cancer, neurodegeneration, metabolic disorders, or rare genetic diseases, RNA holds great promise for combatting human diseases previously intractable to therapy. Much of this promise builds on the fundamental cellular role of RNA – as a template, catalyst, scaffold, or regulator. The unique structural and biochemical properties of RNA allow it to be targeted or applied in a programmable manner since the rules that govern RNA binding affinity and specificity – complementary base pairing – are well understood. While conventional drug development often involves labor‐ and time‐intensive screening to identify lead compounds, RNA drugs can be rationally designed as long as the target is known. As a result, RNA therapies are emerging as platform technologies that can be applied to a variety of diseases. Moreover, delivery strategies can be developed and optimized independently from the RNA component of the drug, which not only speeds up therapeutic development but also opens the possibility to target ultra‐rare diseases.
Despite its potential, the use of RNA as a therapeutic faces obstacles, including its poor pharmacological properties, difficulty with intracellular delivery, and immune‐related toxicity. Technological advances in medicinal chemistry and a better understanding of natural antisense RNA phenomena were necessary to make RNA drugs a reality. The scientific breakthroughs that revolutionized basic biological research such as the discovery of microRNAs, RNA interference, and CRISPR‐Cas systems (Box 1) are now finding their applications in the clinic.
Box 1. Relevant discoveries in antisense RNA biology.
The notion of RNA as a regulatory molecule in eukaryotic cells took hold in 1993, when Victor Ambros, Gary Ruvkun and colleagues identified lin‐4 as a regulator of Caenorhabditis elegans development (Lee et al, 1993; Wightman et al, 1993). Curiously, lin‐4 did not encode a protein, but expressed a precursor RNA that is processed into a short, double‐stranded RNA. Lin‐4 post‐transcriptionally suppresses Lin‐14, a protein crucial for C. elegans larval progression, by recognizing a partially complementary sequence in the 3′ UTR of lin‐14 mRNA. lin‐4 is now recognized as the founding member of a family of small regulatory RNAs, the miRNAs. Although it was not until the early 2000s that miRNA‐mediated gene regulation was shown to be evolutionarily conserved and widespread throughout metazoans (Pasquinelli et al, 2000), the landmark discovery of lin‐4 can be seen as a first hint that the cellular functions of RNA are much more complex than simply coding proteins.
Meanwhile, scientists had been observing unexpected RNA‐mediated gene‐silencing phenomena in plants and other experimental systems, including C. elegans. Initially, the molecular basis was thought to be an antisense mechanism that depends on hybridization between the regulatory RNA and cellular mRNA transcripts. Then, in 1998, Andrew Fire and Craig Mello showed that, in nematodes, the administration of double‐stranded RNA triggers sequence‐specific mRNA silencing, in a process they coined “RNA interference” (RNAi). Their results argued against stochiometric interference with endogenous mRNA and suggested a catalytic component that amplified the process (Fire et al, 1998). This work, and the demonstration that 21‐ and 22‐nt double‐stranded RNAs induce post‐transcriptional gene silencing in plants (Hamilton & Baulcombe, 1999), set the stage for subsequent studies that characterized the molecular mechanism underlying the RNAi pathway. In 2000, two papers reported that the key to RNAi is the conversion of double‐stranded RNAs into small interfering RNAs (siRNAs), which guide an “RNA‐induced silencing complex” (RISC) to enzymatically cleave complementary mRNAs (Hammond et al, 2000; Zamore et al, 2000; Fig 4). This observation was pivotal to the development of siRNAs that could silence genes in mammalian cells without eliciting innate immune responses (Caplen et al, 2001; Elbashir et al, 2001). Soon, siRNAs became ubiquitous tools for the targeted inhibition of any gene of interest, based on sequence alone.
The next game changer in RNA biology came with the discovery of an RNA‐guided immune defense system in bacteria and archaea. CRISPR‐Cas systems are now widely known due to their fame as programmable genome‐editing tools. After the initial experimental demonstration that CRISPR‐Cas systems provide adaptive immunity against foreign mobile genetic elements (Barrangou et al, 2007) and the identification of the defense mechanism (Garneau et al, 2010; Deltcheva et al, 2011; Gasiunas et al, 2012; Jinek et al, 2012), the potential of RNA‐guided Cas nucleases for genome engineering was soon realized. This concept was established in a set of studies published in early 2013, less than 6 months after the demonstration of programmable DNA cleavage by Cas9 (Gasiunas et al, 2012; Jinek et al, 2012), when in vivo proof that RNA‐guided Cas9 could be used to edit genes in both mouse and human cell lines was provided (Cong et al, 2013; Jinek et al, 2013; Mali et al, 2013). This new, easy‐to‐use genome‐editing tool captured the attention of scientists across a wide range of disciplines and the technology rapidly took a key position in biological and biomedical research.
RNA‐based therapeutic strategies
Conceptually, RNA therapeutics can be divided into three major categories: compounds that target cellular RNA, which are typically heavily modified nucleic acid‐based antisense oligomers; treatments in which RNA itself is the therapeutic agent that is delivered into cells to mediate the expression of a protein; and therapeutic genome editing, in which RNA serves as the guide for an effector protein that modifies the cellular DNA sequence (Fig 1). In this review, we will provide a broad overview of these different strategies with a focus on their molecular modes of action. For more details, we refer the interested reader to recent comprehensive reviews that center on each approach in turn, specifically antisense technologies (Crooke et al, 2021a, 2021b; Egli & Manoharan, 2023), RNAi‐based therapeutics (Setten et al, 2019), mRNA‐based approaches (Chaudhary et al, 2021; Rohner et al, 2022) and CRISPR‐Cas‐mediated genome editing (Anzalone et al, 2020; Doudna, 2020; Raguram et al, 2022; Wang & Doudna, 2023).
Figure 1. Strategies for RNA‐based medicine.
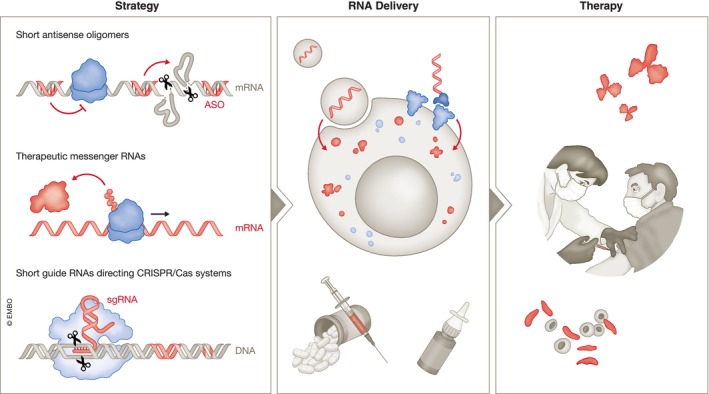
Strategies for RNA‐based medicines include antisense oligomers, mRNA‐based approaches and CRISPR‐Cas‐mediated genome editing. Most of these therapeutic modalities require efficient delivery vehicles such as lipid nanoparticles and targeting ligands for therapeutic development.
Antisense therapeutics
The quest for antisense‐based therapeutics dates back almost half a century to 1978, when Mary Stephenson and Paul Zamecnik designed a synthetic antisense oligonucleotide (ASO) to inhibit Rous sarcoma virus replication in tissue culture (Stephenson & Zamecnik, 1978; Zamecnik & Stephenson, 1978). These studies pioneered the notion of harnessing the unique chemical properties of nucleic acids for drug design. It took another 20 years until the United States Food and Drug Administration (FDA) approved the first ASO drug for clinical use. This drug – fomivirsen – is a synthetic 21 nucleotide (nt) ASO that binds to a complementary sequence of cytomegalovirus (CMV) mRNA and blocks the translation of proteins essential for CMV replication. It was indicated for the treatment of CMV retinitis, a serious infection of the retina that can lead to blindness, in patients with acquired immune deficiency syndrome (AIDS; Vitravene Study Group, 2002a, 2002b). Despite therapeutic benefits, the drug was withdrawn from the market due to the success of anti‐retroviral therapy. Still, fomivirsen provided the first proof‐of‐concept of the clinical value of ASOs.
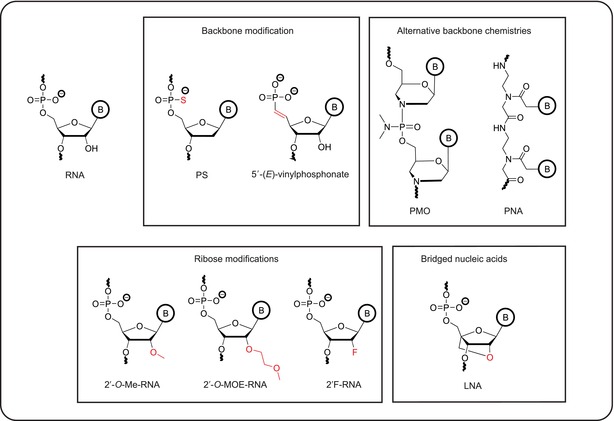
Principally, ASOs are short synthetic nucleic acids that bind to cellular RNA through complementary base pairing. RNA itself, in its unmodified form, is rapidly degraded by nucleases, making it unsuitable for use as an ASO drug. Chemical modifications of the nucleosides, nucleobases, and the ASO backbone are therefore essential for drug development. Several classes of nucleic acid analogs with improved stability and resistance to nucleases are currently in use and the field is developing rapidly (Box 2).
Box 2. Selected backbone, sugar or nucleobase modifications of ASO and siRNA therapeutics.
The goals of oligonucleotide medicinal chemistry are to increase resistance to nucleases, to enhance affinity to the target, to improve pharmacokinetics and to reduce pro‐inflammatory responses. An initial focus lay on modifications of the phosphodiester backbone. In unmodified DNA or RNA oligonucleotides, this backbone is highly susceptible to degradation by nucleases. A major breakthrough was the introduction of phosphorothioate (PS) linkages, in which one of the non‐bridging oxygen in the phosphodiester is replaced by a sulfur atom. This modification increases nuclease resistance, decreases hydrophilicity and promotes the binding of serum proteins, which, in turn, improves circulation life‐time. It also increases binding to cell surface proteins, thereby facilitating ASO uptake into cells (Roberts et al, 2020; Crooke et al, 2021a). Yet, ASOs with a phosphorothioate backbone retain immunostimulatory activity (Kulkarni et al, 2021) and show lower target binding affinity than unmodified ASOs (Freier & Altmann, 1997). 5′‐vinylphosphonate, a metabolically stable phosphate mimic, protects siRNA drugs from phosphatases and improves their silencing activity by enhanced binding to human Argonaute 2 (Ago2; Prakash et al, 2015; Parmar et al, 2016; Elkayam et al, 2017). The modification also increases siRNA accumulation and retention in multiple tissues and extends the duration of silencing in vivo (Haraszti et al, 2017). PMO morpholinos, which have a backbone of methylenemorpholine rings linked through phosphorodiamidate groups, have improved target affinity and stability, but are uncharged, which decreases serum protein binding and circulation lifetime. They are rapidly eliminated from the body following systemic injection and poorly taken up by cells (Crooke et al, 2021a). They also do not activate RNase H1, and therefore rely on regulatory mechanism that are based on steric hindrance (Summerton, 1999). Peptide nucleic acids (PNAs) harbor a pseudo‐peptide backbone that links the four natural nucleobases. This chemical structure also confers resistance toward nucleases and proteases. Like PMOs, PNAs are uncharged, leading to strong binding to complementary sequences, but they face a cellular delivery barrier (Pradeep et al, 2023). Other chemical modifications that increase the affinity of oligonucleotides for their targets and thereby improve potency and selectivity center on 2′ ribose modifications, such as 2′‐O‐methyl (2′‐O‐Me), 2′‐O‐methoxyethyl (2′ MOE), and 2′‐fluoro (2′‐F; Kulkarni et al, 2021). Full modification is often incompatible with specific mechanisms of actions such as RNase H1 recruitment. Nevertheless, partial modification, especially in flanking sequences, is generally well tolerated. Increased affinity can also be gained by using oligonucleotides modified with locked nucleic acids (LNA), which contain a methylene bridge between the 2′ and 4′ position of the ribose. This bridge secures the ribose ring in a conformation that is ideal for binding to complementary sequences (Braasch & Corey, 2001). Nucleobase modifications are less common, although replacing cytosine with 5‐methylcytosine (5mC) reduces the immunostimulatory effects of ASOs (Henry et al, 2000). Introducing these modifications into oligonucleotides is compatible with DNA and RNA synthesis. Therefore, ASOs can be designed to incorporate multiple modifications to combine their advantageous properties.
ASOs alter mRNA expression through a variety of mechanisms that either trigger the decay of target RNAs or prevent processing steps such as RNA splicing or translation (Fig 2; Crooke et al, 2021a). One important mode of action is the induction of Ribonuclease H1 (RNase H1)‐mediated cleavage and subsequent degradation (Fig 2A). RNase H1 is an endonuclease that cuts RNA in double‐stranded RNA:DNA hybrids. The resulting RNA fragments are degraded by 5′‐ and 3′‐exonucleases. RNase H1 requires 8–10 contiguous ribonucleotide‐containing base pairs (bp) in a substrate for optimal activity. This has led to the conception of “gapmers” — ASOs that have a central core of deoxyribonucleotides to support RNase H1‐mediated cleavage, which is flanked by 2′ modified nucleotides at both the 5′ and 3′ ends. This design enhances the affinity of the ASO for its target RNA and increases its resistance to nucleases (Crooke et al, 2021a).
Figure 2. Molecular mechanisms of ASOs.
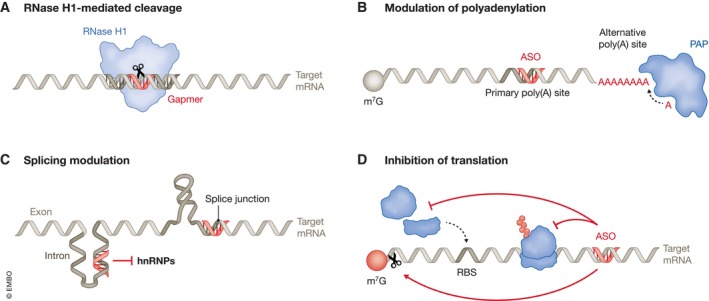
ASOs are extensively modified RNA analogs that bind to complementary sequences in target RNAs. They act via various molecular mechanisms such as (A) RNase H1‐mediated RNA cleavage induced by gapmers, which have a central core of deoxyribonucleotides (pink) flanked by 2′ modified nucleotides at both the 5′ and 3′ ends (gray), (B) modulation of polyadenylation, (C) modulation of splicing either by directly blocking splice junctions or by interfering with the binding of splice modulators, e.g heterogeneous nuclear ribonucleoproteins (hnRNPs) or (D) inhibition of translation by blocking ribosome scanning, interfering with translation initiation factors or causing the cleavage of the mRNA 5′ cap structure (m7G).
In addition to RNase H1‐induced degradation, there are other mechanisms by which ASOs can alter mRNA levels. For example, they can redirect mRNA polyadenylation and thereby modify RNA stability (Vickers et al, 2001; Fig 2B). ASOs can also act on precursor mRNA to modulate splicing, either by directly blocking splice junctions or by interfering with the binding of splice modulators, proteins that promote or inhibit splicing (Hodges & Crooke, 1995; Hua et al, 2008; Rigo et al, 2012a; Fig 2C). These effects can be the goal itself, as in diseases caused by aberrant splicing (see sections on spinal muscular atrophy (SMA) or Duchenne muscular dystrophy (DMD) below). Alternatively, changes in splicing patterns may lead to mRNAs containing premature termination codons, which are selectively degraded through nonsense‐mediated mRNA decay (Ward et al, 2014). Moreover, ASOs can directly interfere with mRNA translation (Fig 2D). This can trigger no‐go decay (Liang et al, 2019), an mRNA quality control mechanism in which mRNAs with stacks of stalled ribosomes are recognized and degraded. Alternatively, ASOs can be designed to inhibit translation initiation by blocking ribosome scanning or by interfering with the interaction between mRNA and translation initiation factors (Melton, 1985; Baker et al, 1997). Additionally, ASOs can trigger the cleavage of 5′ cap structures, which inhibits translation and leads to mRNA decay (Baker et al, 1999; Fig 2D).
ASO delivery in a clinical setting faces challenges, including their degradation by serum nucleases, renal clearance, poor tissue penetration, and inefficient cellular uptake. The chemical modifications of ASOs discussed above and in Box 2 help alleviate some of these problems (Roberts et al, 2020). For example, the commonly used phosphorothioate backbone promotes interactions with serum proteins, which slows excretion by the kidneys (Sands et al, 1994). Chemical modifications also promote cellular uptake by cell surface receptors that facilitate oligonucleotide endocytosis (Koller et al, 2011; Roberts et al, 2020). Once inside the cell, ASOs must escape the endosome in order to be active, posing an additional challenge (Dowdy, 2023).
Another obstacle for ASO therapeutics is their immunogenicity. The human immune system recognizes both single‐stranded and double‐stranded RNA via pattern recognition receptors (PRRs; Takeuchi & Akira, 2010; Okude et al, 2020; Fig 3). Extracellular recognition is mediated by endosomal Toll‐like receptors (TLRs), especially TLR‐3, −7 and −8 (Lind et al, 2022). Intracellular recognition occurs via cytoplasmic defense mechanisms such as RIG‐I‐like receptors, protein kinase R (PKR), and oligoadenylate synthases (OASes; Hur, 2019). Activation of these pathways can trigger inflammatory responses, arrest of cellular translation, and RNA degradation, respectively. Therefore, 2′‐ribose modifications are commonly applied to synthetic RNA therapeutics to reduce their immunogenicity (Box 2; Crooke et al, 2021a).
Figure 3. Cellular response to exogenous RNA.
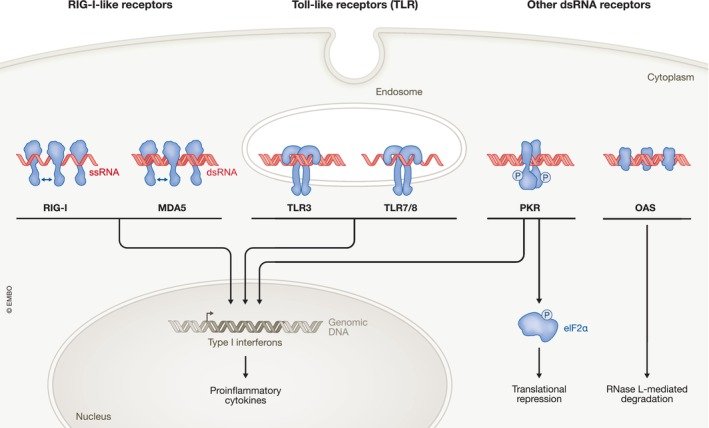
Exogenous RNA is recognized by various innate immune receptors such as retinoic‐acid‐inducible gene I (RIG‐I), melanoma differentiation‐associated protein 5 (MDA5), endosome associated Toll‐like receptors (TLRs), protein kinase RNA‐activated (PKR) and 2′–5′ oligoadenylate synthase (OAS). Upon detection of their nucleic acid activator, these sensors initiate innate immune responses that often result in the production of Type I interferons, pro‐inflammatory cytokines, translational repression via phosphorylation of eukaryotic translation initiation factor 2 α (eIF2α) and RNA degradation by ribonuclease L (RNase L).
Progress in understanding the molecular mechanisms of activity, distribution, cellular uptake, and toxicity of ASOs provides a framework for adapting this versatile technology to many diseases. While all currently approved ASO drugs are for use in patients with rare diseases (Table 1), many ASOs in clinical development are intended to treat common diseases such as cardiovascular and metabolic diseases and cancer. Although widespread implementation still faces challenges such as cell‐type‐specific delivery and potential adverse effects upon chronic treatment, ASO therapies are expected to have a substantial impact on many diseases that currently have limited or no treatment options.
Table 1.
FDA‐approved RNA therapeutics.
| Product | Target | Mechanism of action | Indication | Route of delivery | Company | Approval year |
|---|---|---|---|---|---|---|
| ASOs | ||||||
| Formivirsen | CMV mRNA | Downregulation | CMV retinitis | IVT | Ionis Pharmaceuticals, Novartis | 1998 (withdrawn 2002) |
| Mipomersen | Apolipoprotein B‐100 mRNA | Downregulation | Familial Hypercholesterolemia | SC | Ionis Pharmaceuticals | 2013 |
| Nusinersen | SMN2 pre‐mRNA | Splicing modulation | Spinal muscular atrophy | ITH | Ionis Pharmaceuticals, Biogen | 2016 |
| Eteplirsen | Exon 51 of dystrophin pre‐mRNA | Splicing modulation | DMD | IV | Sarepta Therapeutics | 2016 |
| Inotersen | TTR mRNA | Downregulation | Transthyretin‐mediated amyloidosis | SC | Ionis Pharmaceuticals | 2018 |
| Golodirsen | Exon 53 of DMD | Splicing modulation | DMD | IV | Sarepta Therapeutics | 2019 |
| Volanesoren | Apolipoprotein CIII mRNA | Downregulation | Familial chylomicronemia syndrome | SC | Ionis Pharmaceuticals, Akcea | 2019 |
| Viltolarsen | Exon 53 of dystrophin pre‐mRNA | Splicing modulation | DMD | IV | NS Pharma, Inc | 2020 |
| Casimersen | Exon 45 of dystrophin pre‐mRNA | Splicing modulation | DMD | IV | Sarepta Therapeutics | 2021 |
| RNAi‐based therapeutics | ||||||
| Patisiran | TTR mRNA | Downregulation | Transthyretin‐mediated amyloidosis | IV | Alnylam | 2018 |
| Givosiran | ALS1 mRNA | Downregulation | Acute hepatic porphyria | SC | Alnylam | 2020 |
| Lumasiran | HAO1 mRNA | Downregulation | Primary hyperoxaluria type 1 | SC | Alynlam | 2020 |
| Inclisiran | PCSK9 | Downregulation | Atherosclerotic cardiovascular disease | SC | Novartis | 2021 |
| Vutrisiran | TTR mRNA | Downregulation | Transthyretin‐mediated amyloidosis | SC | Alynlam | 2022 |
| mRNA therapeutics | ||||||
| BNT162b2 | SARS‐CoV‐2 Spike mRNA | Expression of SARS‐CoV‐2 Spike protein | COVID‐19 | IM | BioNTech, Pfizer | 2020 |
| mRNA‐1273 | SARS‐CoV‐2 Spike mRNA | Expression of SARS‐CoV‐2 Spike protein | COVID‐19 | IM | Moderna | 2020 |
IM, intramuscular; ITH, intrathecal; IV, intravenous; IVT, intravitreal; SC, subcutaneous.
RNAi‐based therapeutics
In 1998, Andrew Fire and Craig Mello identified double‐stranded RNA as the trigger for RNA interference in Caenorhabditis elegans (Fire et al, 1998; Box 1). Their observations challenged the prevailing view that antisense RNAs acted by directly binding and sterically interfering with target mRNA expression, and led to the discovery of an enzymatic silencing mechanism. Double‐stranded RNAs, either supplied exogenously or expressed within cells as precursor RNAs with stem loops or short hairpin structures, are converted by the cytoplasmic RNase III enzyme Dicer to small interfering RNAs (siRNAs) or microRNAs (miRNAs), respectively. Target mRNA regulation is mediated by the RNA‐induced silencing complex (RISC), a ribonucleoprotein complex composed of the siRNA or miRNA, which acts as the specificity determinant, and an Argonaute protein, which, together with other complex components, acts as the effector molecule (Meister, 2013; Ipsaro & Joshua‐Tor, 2015). siRNAs interact with their targets with perfect or near‐perfect complementarity and induce sequence‐specific cleavage of mRNAs through the slicer activity of Argonaute 2 (Ago2; Zamore et al, 2000; Liu et al, 2004). In contrast, miRNAs tend to interact with imperfectly complementary targets and cause translational repression and transcript degradation (Pillai et al, 2005; Wu et al, 2006; Djuranovic et al, 2012; Fig 4). The RNAi pathway in human cells is remarkably efficient due to the ability of the activated RISC to direct multiple rounds of RNA cleavage (Hutvágner & Zamore, 2002). RNAi‐based therapies take advantage of this feature and of the versatility and programmability of the RNAi machinery, but the need to engage this machinery also creates constraints on the design of siRNA drugs.
Figure 4. RNA interference.
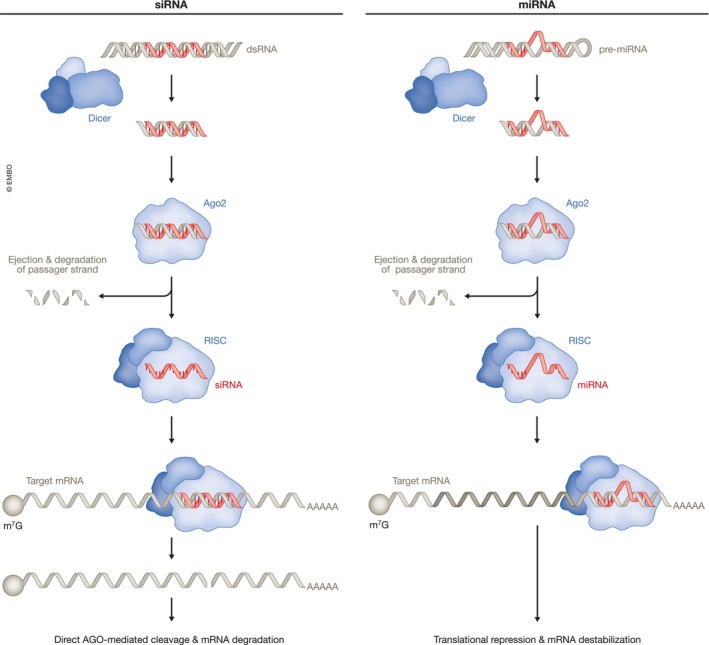
RNA interference (RNAi) pathways are guided by small interfering RNAs (siRNAs; left) or microRNAs (miRNAs; right). siRNAs typically derive from exogenous sources while canonical miRNA precursors are derived from transcripts with internal hairpins. After endonucleolytic processing by Dicer, the RNA is loaded into Ago2 forming the RNAi‐induced silencing complex (RISC). siRNAs recognize targets with perfect or near‐perfect sequence complementarity and direct mRNA cleavage by Ago2. miRNAs typically recognizes target sites with imperfect complementarity, usually in the 3′ untranslated region (3′ UTR) of mRNAs and induce silencing by translational repression and target deadenylation and destabilization. miRNAs with perfect complementarity can also induce endonucleolytic cleavage by Ago2.
Generally, synthetic RNAi triggers are perfectly base‐paired double‐stranded RNAs that must be unwound so that one of the strands can be selected as the “guide” strand to be loaded into RISC. Since only the antisense strand hybridizes to the target mRNA, RNAi drug design must ensure that the correct strand is chosen. For example, siRNAs with a blunt end on one side and a 2‐nt 3′ overhang on the other tend to bias guide strand selection to the strand with the 3′ overhang (Sano et al, 2008). In addition, chemical modifications can also promote antisense strand loading (Varley & Desaulniers, 2021). RNAi triggers that are longer than 21 bp require Dicer for cleavage and handoff to RISC. Notably, Dicer processing is linked to a more reliable selection of the antisense strand as the RISC guide (Snead et al, 2013). Shorter siRNAs bypass the early steps of the RNAi pathway and can be loaded into RISC directly. This has the advantage that they are less likely to interfere with gene regulation by endogenous miRNAs (Grimm et al, 2006).
siRNA drugs face similar challenges as ASOs in terms of delivery, stability, and immunogenicity. Fortunately, the insights into backbone, base, and sugar modifications initially established for ASO therapeutics (Box 2) largely apply to siRNA therapeutics, although the molecular requirements for effective recruitment of the RNAi machinery impose limitations on the chemical modification of siRNAs (Khvorova & Watts, 2017). Moreover, the delivery of duplex siRNAs is more challenging than the delivery of single‐stranded ASOs, partly due to their increased size and hydrophilicity. In siRNAs, the external‐facing phosphate groups create a hydrated surface that adheres poorly to cells leading to their rapid excretion from the body. As a result, researchers have developed delivery vehicles such as lipid nanoparticles and targeting ligands (Box 3) to improve siRNA delivery. Further, the use of chemical optimization strategies is leading to the design of modified siRNAs that will expand the reach of RNAi therapeutics (Davis et al, 2022).
Box 3. RNA delivery.
RNA is large and negatively charged, making it difficult to cross cell membranes. It is also rapidly degraded by nucleases that are active in all fluids of the body. Therefore, several approaches have been developed to increase the efficiency of in vivo delivery of RNA therapeutics, including encapsulation into nanoparticles. Lipid‐based nanoparticles (LNPs) are the most advanced for mRNA delivery and offer benefits such as ease of formulation, biocompatibility, and a large carrying capacity (Chaudhary et al, 2021; Hou et al, 2021). LNPs typically include ionizable lipids, cholesterol, a phospholipid, and a PEGylated lipid. Initially, cationic lipids were used due to their positive charge, which facilitated the encapsulation of negatively charged RNA (Kauffman et al, 2016). However, cationic lipids trigger toxic and proinflammatory responses (Cui et al, 2018). To overcome these drawbacks, ionizable lipids were developed (Cullis & Hope, 2017). These lipids are neutral at physiological pH, improving their safety and extending their circulation time. In acidic conditions, they are positively charged. This aids RNA packaging, but also promotes fusion with the endosomal membrane after cellular uptake to release the cargo into the cytoplasm. The other lipid components of LNPs play a role in their formation and function. Cholesterol enhances stability and aids membrane fusion during uptake, helper lipids modulate nanoparticle fluidity and enhance efficacy, and PEGylated lipids stabilize LNPs and regulate their size and half‐life (Chaudhary et al, 2021). Polymers, which offer similar advantages to lipids, and cell‐penetrating peptides can also deliver mRNA into cells. Chemical conjugation to trivalent N‐acetylgalactosamine (GalNAc; Nair et al, 2014; Matsuda et al, 2015; Rajeev et al, 2015), which is recognized by the hepatocyte‐restricted asialoglycoprotein receptor (ASGPR) represents an efficient way of increasing liver uptake, especially of short RNAs.
The first therapy based on RNAi‐mediated gene silencing received FDA approval in 2018. Patisiran is a double‐stranded siRNA indicated for the treatment of hereditary transthyretin‐mediated amyloidosis (hATTR; Adams et al, 2018). This is a progressive neurodegenerative disease caused by the deposition of amyloid fibrils formed by the misfolded protein transthyretin. Patisiran silences transthyretin mRNA in the liver and decreases serum levels of the protein, thus reducing the amyloid deposits. It is composed of two modified 21‐mer oligonucleotides and encapsulated in a lipid nanoparticle formulated for hepatocyte uptake. Vutrisiran, a successor to patisiran, came on the market in 2022. It uses the same RNAi mechanism but takes advantage of enhanced stabilization chemistry. This siRNA is coupled to N‐acetylgalactosamine (GalNAc; Box 3), which increases its uptake in liver cells and allows the administration of lower doses (Adams et al, 2023). While patisiran required intravenous injection every 3 weeks, treatment with vutrisiran involves only one subcutaneous injection every 3 months. Currently, five RNAi‐based drugs have been approved by the FDA (Table 1), and the many oligonucleotide drugs currently in pre‐clinical and clinical development indicate that RNAi therapeutics will soon be used in a broad range of applications. For example, several strategies to achieve extrahepatic delivery are being explored, including antibody conjugates (Cuellar et al, 2015; Dugal‐Tessier et al, 2021; Malecova et al, 2023), peptide conjugates (Klabenkova et al, 2021), hydrophobic (Biscans et al, 2019) or lipophilic conjugates (Brown et al, 2022) as well as multivalency (Alterman et al, 2019). In addition, programmable siRNA pro‐drugs that are activated in response to specific cellular RNA biomarkers show promise in selective targeting of diseased cells over healthy bystander tissue (Han et al, 2022).
miRNAs are important gene regulators that influence many physiological processes linked to disease, making them attractive therapeutic targets in their own right (Rupaimoole & Slack, 2017). miRNAs require only partial complementarity for target recognition. Therefore, a single miRNA can interact with multiple mRNAs with different affinities. Interfering with or mimicking miRNA function thus enables the simultaneous manipulation of complex gene expression networks. Targeting multiple, potentially compensatory pathways at once is appealing, but this approach also carries the risk of unforeseen side effects.
miRNA therapeutics come in two flavors: miRNA mimics and antimiRs. miRNA mimics are synthetic oligonucleotide duplexes that imitate the function of a naturally occurring miRNA akin to siRNA drugs. antimiRs are structurally similar to ASOs and are designed to bind directly to the mature strand of the targeted miRNA and to block its function. More details on this topic can be found in a comprehensive review on miRNA‐targeted therapeutics (Rupaimoole & Slack, 2017). For the purpose of this overview, we want to highlight the antimiR miravirsen as a notable example. Miravirsen targets miR‐122, an abundant liver miRNA that regulates lipid metabolism. miR‐122 also plays a critical role during infection with Hepatitis C virus (HCV), a major cause of chronic liver diseases such as cirrhosis and hepatocellular carcinoma. Binding of miR‐122 to the 5′‐untranslated region (UTR) of HCV RNA is essential for viral replication (Jopling et al, 2005), and miravirsen was designed to interfere with this process. It is composed of locked nucleic acid (LNA) ribonucleotides interspersed throughout a DNA phosphorothioate sequence that is complementary to miR‐122. Miravirsen hybridizes to mature miR‐122 and blocks its interaction with HCV RNA, inhibiting HCV replication (Lanford et al, 2010). It showed prolonged antiviral activity in initial clinical trials and was well‐tolerated in patients infected with HCV (Janssen et al, 2013), but the success of potent small‐molecule antiviral treatments for hepatitis C diminished the clinical need for miravirsen and its clinical development was recently discontinued.
mRNA‐based therapeutics
The RNA drugs discussed above act as effectors, but RNA, in the form of mRNA, is also a carrier of genetic information that can serve as a therapeutic agent by mediating protein expression. Applications for mRNA‐based therapeutics include vaccines against infectious diseases and cancer as well as protein replacement. Clinically applied synthetic mRNAs are usually in vitro transcribed (IVT) from a DNA plasmid using a bacteriophage RNA polymerase. They are structured similarly to cellular mRNA and include elements such as a 5′ cap, a 5′ UTR, an open reading frame (OTR), 3′ UTR, and poly(A) tail. These features are important for mRNA translation and stability and therefore affect efficacy of the mRNA drug. The synthesized mRNA is purified to remove contaminants, reactants, and incomplete transcripts, which is essential to reduce immune‐stimulatory effects (Karikó et al, 2011). To maximize translation, modified nucleosides like pseudouridine and N1‐methylpseudouridine are often incorporated into the mRNA molecule. The use of these modified nucleosides, particularly modified uridine, also prevents the recognition of the IVT mRNA by the innate immune system, thus allowing for higher dosing (Karikó et al, 2005).
Early efforts to use IVT mRNA for therapeutic applications laid the groundwork for the rapid development of highly efficient mRNA vaccines against SARS‐CoV‐2 (Chaudhary et al, 2021). By the end of 2019, several preclinical and clinical studies had established the potential of mRNA vaccines to protect against pathogens such as Zika virus, respiratory syncytial virus (RSV), Influenza A virus, and rabies virus, but it was expected that it would take another 5–6 years before an mRNA vaccine would be approved for clinical use (Chaudhary et al, 2021). The COVID‐19 pandemic accelerated this development, and the mRNA vaccines developed by BioNTech/Pfizer and Moderna received approval within a mere 10 months. Both vaccines are formulated with ionizable lipid nanoparticles (LNPs; Box 3) and deliver a nucleoside‐modified mRNA encoding the viral spike glycoprotein. They demonstrated more than 90% efficacy in clinical trials. Aside from transient local and systemic reactions, no safety concerns were identified (Polack et al, 2020; Baden et al, 2021), although potential long‐term effects must be further evaluated.
mRNA vaccines are usually administered as a single injection into the skin, muscle, or subcutaneous space, where they are taken up by immune or non‐immune cells and translated into antigens that stimulate an immune response. In contrast to plasmid DNA and viral DNA vectors, IVT mRNA does not need to enter the nucleus to be effective. The high sensitivity of the immune system enables the generation of strong immune responses even at low antigen levels, making high and sustained expression of the IVT mRNA unnecessary.
The mRNA platform has several benefits for pandemic vaccine production, notably a rapid development time and cost‐effective, scalable production, allowing a fast response in case a new pandemic virus emerges. Other advantages include flexibility in antigen design and the ability to deliver multiple antigens in a single formulation. These features can be exploited in the development of “universal” vaccines that provide broad protection against multiple viral strains. While regulatory and approval pathways for these vaccines still need to be fully established, we are likely to see the development of more mRNA vaccines for infectious diseases in the near future.
Another promising application of mRNA vaccines is the personalized treatment of cancer (Sahin & Türeci, 2018). In ongoing clinical studies, researchers are using mRNA vaccines to stimulate an immune response against specific cancer‐associated neoantigens (Kranz et al, 2016; Sahin et al, 2017; Palmer et al, 2022; Rojas et al, 2023), with early results suggesting that these therapies can yield clinical benefit (Dolgin, 2023). These vaccines are individualized for each patient based on RNA sequencing analysis of their tumor tissue, and are designed to instruct the patient's immune system to target and attack the cancer cells (Lang et al, 2022).
mRNA vaccines can also be used to treat autoimmune diseases by selectively dampening autoimmune responses without compromising normal immune function (Krienke et al, 2021). This is achieved through systemic delivery of mRNA encoding disease‐related autoantigens, designed to be taken up and presented by lymphoid antigen‐presenting cells with low‐level surface expression of co‐stimulatory molecules. This leads to peripheral tolerance through reduction of effector T cells and development of regulatory T cell populations that mediate bystander immunosuppression (Krienke et al, 2021).
Besides mRNA vaccines, an obvious use of therapeutic mRNA is the expression of proteins that are absent or not functional in the body. This approach has the potential to treat a wide range of diseases, but comes with additional challenges like the need for high and sustained expression of the therapeutic protein in specific cell types, as well as difficulties in delivering mRNA to solid organs other than the liver. Treatment of chronic diseases would require repeated dosing, which can activate the immune system and reduce the effectiveness of the therapeutic protein, even with the use of modified mRNA and advanced delivery vehicles. The therapeutic protein itself can also elicit an immune response if it is not expressed endogenously in the body. To date, only a small number of clinical studies have shown promising results for this approach in terms of safety and efficacy. One notable example is the use of VEGF mRNA to promote vasculogenesis during cardiac regeneration (Anttila et al, 2020).
Therapeutic genome editing
While mRNA‐based protein replacement therapies allow temporary expression of a functional copy of a transcript, therapeutic genome editing offers the promise of a permanent cure by correcting pathogenic mutations in genomic DNA. The clinical feasibility of this approach is the result of the discovery of ancient bacterial adaptive immune systems – the CRISPR‐Cas nucleases (Mojica et al, 2005; Barrangou et al, 2007; Brouns et al, 2008; Box 1) – and their development as genome editing tools.
The first described and still widely used genome editing tool is the type‐II CRISPR‐Cas9 system from Streptococcus pyogenes (SpCas9; Deltcheva et al, 2011; Jinek et al, 2012). SpCas9 is an RNA‐guided DNA endonuclease that generates targeted double‐strand breaks (DSBs) at specific genomic loci (Gasiunas et al, 2012; Jinek et al, 2012). The enzyme recognizes its target sites through a small guide RNA that hybridizes to complementary regions in DNA (Fig 5A). This programmable RNA can be designed to guide Cas9 to genomic regions of interest (Cong et al, 2013; Jinek et al, 2013; Mali et al, 2013). The only sequence requirement is that the target sequence must be flanked on the 3′ side by a short protospacer adjacent motif (PAM). This prerequisite limits the genomic sites that can be edited to locations of PAM sequences. Cas nucleases isolated from different bacterial species recognize different PAM sequences, which broadens the targeting space. In addition to natural Cas variants, there are now lab‐evolved mutants with alternative PAM recognition (Hu et al, 2018; Nishimasu et al, 2018; Miller et al, 2020; Walton et al, 2020).
Figure 5. CRISPR‐Cas‐based genome editing tools.
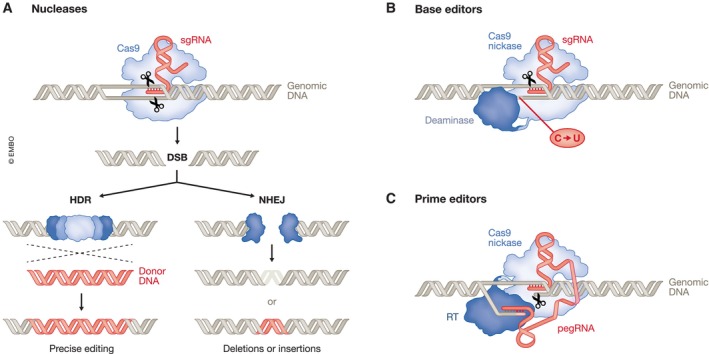
(A) Cas9 nucleases are guided to their target region by a small guide RNA (sgRNA). There they generate double‐strand breaks (DSBs) that are repaired by either homology‐directed repair (HDR) or non‐homologous end joining (NHEJ). HDR repairs DSBs by homologous recombination using a donor DNA template. This can be the sister chromatid when it is present during the late S or G2 phase of the cell cycle or an exogenously supplied donor that supports gene correction. In contrast, NHEJ, the dominant path for DSB repair, often leads to uncontrolled nucleotide deletions or insertions. (B) Base editors mediate targeted single‐nucleotide conversions using a fusion between a Cas9 nickase and a deaminase domain, which modifies single bases through deamination. Nicking of the nondeaminated strand biases cellular DNA repair to replace the unedited strand, thereby resolving the mismatch and leading to stable conversion. Base editors are able to mediate targeted C > U, A > G, C > G, A > I and C > T conversions. (C) Prime editors consists of a reverse transcriptase (RT) fused to a Cas9 nickase. The Cas9 nickase binds to and nicks the non‐target DNA strand and the reverse transcriptase subsequently uses the resulting free 3′ end to copy the sequence of the prime‐editing guide RNA (pegRNA).
Once the Cas nuclease has been guided to a target site in the genome, it generates a DSB, which is subsequently repaired by cellular DNA repair systems, either by homology‐directed repair (HDR) or, more commonly, by error‐prone non‐homologous end joining (NHEJ) or microhomology‐mediated end joining (Fig 5A; Hustedt & Durocher, 2016). These repair pathways pose challenges for Cas nuclease‐mediated therapeutic genome editing. Specifically, although HDR in principle allows the precise correction of a mutation through the use of a donor DNA template to repair the break, this pathway is restricted to the S and G2 phase of the cell cycle, making it unusable for treating diseases that involve non‐dividing cells. And while the NHEJ pathway is active in non‐dividing cells, it ligates the broken DNA ends in a template‐independent manner. This results in small insertions and deletions at the break point, excluding the generation of exact site‐specific mutations. DSBs in genomic DNA can also cause large deletions, chromosomal translocations, or other chromosomal abnormalities (Giannoukos et al, 2018; Kosicki et al, 2018; Turchiano et al, 2021). While rare, this poses safety risks.
Recently developed precision genome editing strategies that do not rely on DSBs overcome these limitations. Prominent among these are base editors (BEs), which enable single‐nucleotide conversions in genomic DNA (Rees & Liu, 2018; Fig 5B). BEs are fusions of a Cas9 nickase (nCas9), that is a Cas9 variant that produces a single‐stranded rather than a double‐stranded break, and an enzyme that catalyzes a nucleobase deamination (Komor et al, 2016; Nishida et al, 2016). Like Cas9 nucleases, the nCas9‐deaminase fusion is directed to a genomic target site by a small guide RNA. Base pairing between the guide RNA and the target DNA exposes a region of single‐stranded DNA that is then accessible to deamination. The resulting base mismatch is resolved through cellular repair mechanisms. While the current BE toolbox mediates a range of single nucleotide conversions, base editing does not yet extend to all possible exchanges, and it cannot perform targeted insertions or deletions. Nevertheless, BEs show promising results in pre‐clinical studies (Arbab et al, 2023; Mayuranathan et al, 2023) and are progressing towards the clinic, for example, in an early‐stage clinical trial for the treatment of familial hypercholesterolemia (Kingwell, 2022).
Prime editors (PEs) provide additional flexibility to engineer changes beyond single‐nucleotide conversions (Chen & Liu, 2023; Fig 5C). PEs consist of a Cas9 nickase fused to a reverse transcriptase (Anzalone et al, 2019). They use an engineered prime editing guide RNA (pegRNA) that directs the enzyme to a specific target locus, and also serves as a template for the edit of interest. PEs first nick the non‐target DNA strand and use the resulting free 3′ end to prime reverse transcription using the pegRNA as a template. While several examples of in vivo gene editing using PEs have been reported (Newby & Liu, 2021), their efficiency currently lags behind BEs (Wang & Doudna, 2023). Still, recent improvements bode well for their future (Chen et al, 2021; Nelson et al, 2022).
For therapeutic applications, delivery remains a major bottleneck. Current delivery strategies are divided into two types of approaches: ex vivo, where cells are removed from the patient, edited outside the body, and reintroduced into the patient; and in vivo, where cells are edited directly in the patient following delivery of CRISPR components. Ex vivo approaches are often used for editing hematopoietic stem and progenitor cells, but most other cell types are not amenable to ex vivo manipulation and transplantation. In vivo approaches offer treatment for a wider range of genetic diseases, but require efficient and safe delivery of the editing agents to specific cell types.
Common delivery vehicles include viral vectors, typically adeno‐associated virus (AAV), adenoviruses or lentiviruses, as well as nanoparticles (Raguram et al, 2022; Box 3). Viral delivery offers advantages in terms of efficiency and tissue selectivity. AAVs are especially attractive because of their inherent tissue tropism and clinically manageable immunogenicity, but they have a low packaging capacity. Adeno‐ and lentiviruses offer higher packaging efficiencies, but have immunogenicity concerns. Production at scale and a good manufacturing practice at affordable cost are other unresolved issues for viral‐based delivery. Nanoparticles, on the other hand, are easier to produce and are often considered safer. Yet, they have lower delivery efficiency compared to viral vectors, and when systemically delivered naturally accumulate in the liver. Recently, virus‐like particles (VLPs) have emerged as promising delivery platforms. These viral protein assemblies can package desired cargo and transduce cells, but mostly lack viral genetic material (Raguram et al, 2022). They offer the high delivery efficiencies of viral vectors without the associated safety concerns and have the potential to be targeted to specific cell types by exploiting the cellular tropism of different viral envelope glycoproteins (Banskota et al, 2022). Nevertheless, the feasibility of scaling up production of VLPs in quantities required for pre‐clinical or clinical studies still needs to be established.
Immunogenicity concerns associated with the in vivo delivery of gene editing agents also need to be considered. These include potential preexisting immunity to delivery vehicles (Weber, 2021) or cellular immunity to Cas9 and other components of the gene editing machinery (Charlesworth et al, 2019; Wagner et al, 2021a). In addition, their prolonged expression in edited cells might provoke adaptive immune responses and may increase the chances of off‐target activity. Therefore, transient delivery that minimizes exposure to gene editing agents is desirable.
Therapeutic approaches that use genome editing to treat hereditary diseases are currently mostly in the preclinical stage, although some have advanced to clinical trials. For example, Cas nuclease‐mediated gene therapy has achieved promising initial clinical results in the treatment of beta‐thalassemia and sickle cell disease, two inherited blood disorders caused by reduced production of hemoglobin. The approach taken in the most advanced trial uses ex vivo genome editing and aims to reactivate the synthesis of fetal hemoglobin, which is normally deactivated shortly after birth (Frangoul et al, 2021). This strategy offers treatment to both diseases and circumvents the need to precision edit a disease‐associated mutation. Instead, it seeks to disrupt an erythroid‐specific enhancer region of BCL11A, a transcription factor that represses expression of fetal hemoglobin (Canver et al, 2015; Wu et al, 2019).
Notable examples for successful in vivo delivery of CRISPR‐based therapeutics are clinical trials to treat the neurodegenerative disease hATTR (Gillmore et al, 2021) and the treatment of Leber congenital amaurosis 10 (LCA10), a type of congenital blindness (Maeder et al, 2019). The former constitutes the first systemic in vivo delivery of CRISPR components to the human liver using targeted LNPs, while the latter involves direct injection of AAV harboring Cas9 and two small guide RNAs into the eye.
Opportunities and challenges for RNA therapeutics exemplified by specific genetic diseases
Some RNA‐based therapies have shown remarkable clinical success while others have unexpectedly encountered limited efficacy. The recent suspension of the CRISPR‐editing approach to LCA10, the rare inherited blindness disorder mentioned above, is a case in point. The setting could be considered a best‐case‐scenario because the eye is easily accessible for direct injection of therapeutic modalities and it has an immune‐privileged status (Suh et al, 2022). The strategy – CRISPR‐Cas‐mediated correction of a splice defect in the CEP290 gene, which causes LCA10 – had shown promising preclinical results (Maeder et al, 2019). Nevertheless, although the treatment was well tolerated, only three out of 14 patients experienced clinically meaningful vision improvement, based on company announcements. These disappointing results underline that the road ahead is not without challenges. In this section, we will discuss three genetic disorders that have been targeted by RNA‐based therapeutics, with varying success. We will consider the specific challenges associated with each of these diseases, and highlight how RNA drugs could facilitate treatment.
Spinal muscular atrophy
One of the key success stories of ASO therapeutics is nusinersen, approved for the treatment of spinal muscular atrophy (SMA). SMA is an autosomal recessive neuromuscular disease caused by deletions or loss‐of‐function mutations in the gene survival motor neuron 1 (SMN1) (Lefebvre et al, 1995). Without functional SMN protein, the motor neurons in the spinal cord and brain stem degenerate, resulting in muscle weakness and atrophy (Fig 6A). Of the infants born with the most severe form of SMA, 60% show symptoms before 6 months of age and the median life expectancy is less than 2 years. Until nusinersen came on the market in 2016, there were no approved therapies for SMA, and medical care focused on supportive and palliative measures.
Figure 6. Disease mechanisms and RNA‐based therapeutic approaches for SMA, DMD and cystic fibrosis.
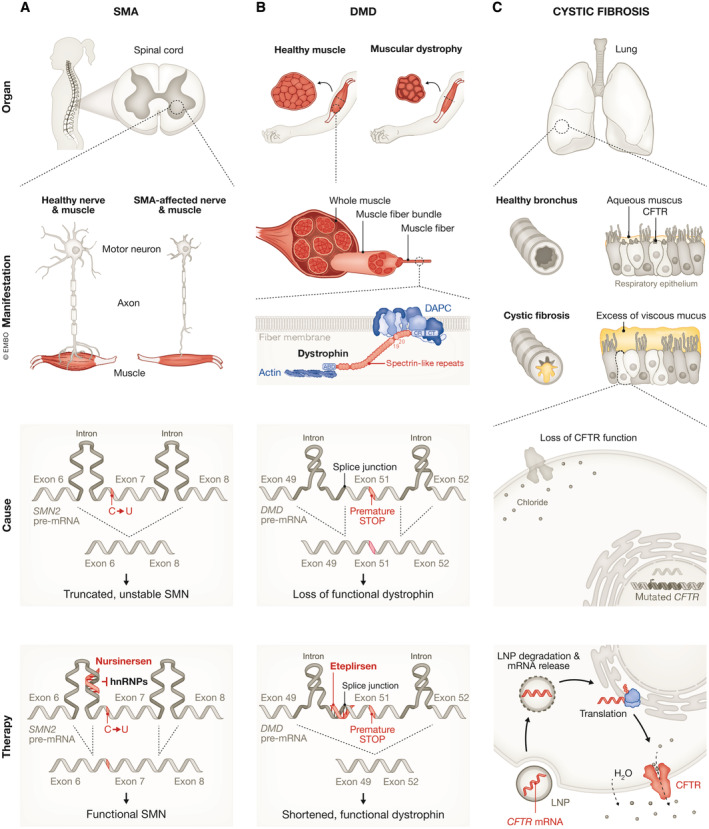
(A) SMA results in the loss of motor neurons in the spinal cord leading to muscular atrophy. The disease is caused by a lack of functional SMN protein due to mutations in the SMN1 gene and aberrant splicing of its paralog SMN2. The SMN2 splicing defect is due to a synonymous C‐to‐T substitution within exon 7, which causes the production of a truncated, unstable polypeptide. Binding of the ASO nusinersen to the SMN2 pre‐mRNA displaces the splice repressor hnRNP, resulting in the production of a mature mRNA that includes exon 7 and translation of the full‐length SMN protein. (B) DMD is characterized by progressive muscle degeneration due to mutations in the protein dystrophin. Dystrophin has a modular organization with an N‐terminal actin binding domain (ABD), a rod domain consisting of 24 spectrin‐like repeats and four interspersed hinges, a cysteine‐rich domain (CR) and a C‐terminal (CT). Dystrophin interacts with the actin cytoskeleton and the dystrophin‐associated protein complex (DAPC), a transmembrane scaffold, thereby supporting the maintenance of muscle cell integrity and contractility. One of the more common causes of the disease is a change in the DMD reading frame caused by deletion of exon 50, which leads to a nonsense mutation and the loss of functional dystrophin. Eteplirsen binds to exon 51 and favors exclusion of exon 51, thereby restoring the reading frame and enabling production of partially functional, internally deleted dystrophin. (C) Cystic fibrosis leads to the accumulation of excessively viscous mucus that obstruct passageways, e.g. in the lung. The disease is caused by dysfunction of the cystic fibrosis transmembrane conductance regulator (CFTR), an anion channel that maintains fluid‐ and electrolyte‐homeostasis. One of the RNA‐based therapeutic approaches currently in development is the delivery of CFTR mRNA packaged into lipid nanoparticles (LNPs) to drive cellular expression of functional CFTR.
Humans have a second gene, survival motor neuron 2 (SMN2) that encodes an identical SMN protein. But SMN2 contains a synonymous C‐to‐T substitution within exon 7 that weakens the binding of splice activators to the SMN2 pre‐mRNA (Hofmann et al, 2000; Cartegni & Krainer, 2002). This leads to aberrant splicing, with 90% of mature SMN2 transcripts lacking exon 7 and producing a truncated, unstable polypeptide (Fig 6A). Instead of targeting SMN1, the tactic chosen to restore functional levels of SMN protein was to use ASOs to promote SMN2 exon 7 inclusion (Rigo et al, 2012b). An early approach was to engineer bifunctional ASOs that operated as synthetic splice activators: a peptide mimicking a splice activator was covalently linked to an ASO that hybridized to exon 7 (Cartegni & Krainer, 2003). Over the years, the strategy to control exon 7 inclusion was optimized. It was shown that ASOs targeting a site near the 5′ splice site in SMN2 intron 7 could efficiently promote exon 7 inclusion without the need of an appended peptide moiety. They acted by preventing binding of the splice repressors hnRNP A1 and hnRNP A2 (Hua et al, 2008; Rigo et al, 2012a; Fig 6A). In addition, chemical modifications in the backbone and nucleotides of the ASOs improved their pharmacological properties.
Based on preclinical studies in mice and non‐human primates (Hua et al, 2011; Passini et al, 2011; Rigo et al, 2014), nusinersen advanced to clinical development and underwent highly successful clinical trials (Finkel et al, 2016, 2017). An interim analysis of a phase III clinical trial for patients with infantile‐onset SMA showed that 40% of children treated with nusinersen achieved improvement in motor functions, such as head control, sitting, rolling, crawling, standing and walking, whereas none of the control patients did. These results led to the early termination of the trial and the drug was approved for use in the US in 2016. It has since become available in over 40 countries. Nusinersen has revolutionized the treatment of SMA patients, and it is also the first antisense drug to achieve a substantial commercial success. This feat, along with the approval of seven other ASOs (Table 1) for the treatment of rare genetic diseases, suggests that the ASO technology has potential to fulfill the hopes placed in it.
Duchenne muscular dystrophy
Other monogenic diseases such as Duchenne muscular dystrophy (DMD) have proven more challenging to treat by RNA therapeutics. DMD is a progressive X‐linked muscle‐wasting disease caused by deletions, duplications, or point mutations in the DMD gene (Koenig et al, 1987). DMD is one of the largest genes in the human genome, spanning ~2.3 Mb of DNA, and exhibits a complex intron‐exon organization. Thousands of different DMD mutations have been found in patients with DMD. They cluster in hotspot regions and approximately 47% of patients carry mutations in exons 45–55 (Nakamura et al, 2017). DMD encodes the protein dystrophin (Hoffman et al, 1987), which protects muscle fibers from contraction‐induced injury (Fig 6B). Dystrophin has a modular organization with an N‐terminus that interacts with the actin cytoskeleton and a C‐terminus that anchors the protein to a transmembrane scaffolding complex, termed the dystrophin‐associated protein complex (DAPC; Gao & McNally, 2015). The central region of dystrophin, which is encoded by the genomic region that includes exons 45–55, is composed of a rod‐domain divided into 24 redundant spectrin‐like repeats and four interspersed hinges (Fig 6B). Dystrophin and the DAPC have both structural and signaling roles that support the maintenance of muscle cell integrity and contractility. DAPC disassembly caused by the absence of functional dystrophin has severe effects on muscle cell function. Patients experience increasing difficulties in movement and eventually need assisted ventilation; the disease inevitably culminates in premature death from respiratory and cardiac failure.
Therapeutic approaches attempting to maintain muscle cell function in DMD include treatment with agents that block inflammation, fibrosis, calcium overload, and oxidative stress (Duan et al, 2021). Yet none of these treatments address the primary cause of the disease – lack of functional dystrophin. Extensive efforts have been directed toward the development of ASOs that restore the reading frame of dystrophin transcripts. This strategy leads to the expression of a partially functional, internally deleted dystrophin with fewer spectrin‐like repeats, but both terminal interaction domains. This “exon‐skipping” approach is based on the observation that naturally occurring mutations that maintain the DMD reading frame cause Becker muscular dystrophy, a milder form of the disease with later onset and slower progression (Mercuri et al, 2019).
The first drug approved by the FDA as a specific DMD therapy was the ASO eteplirsen, which masks a splice acceptor sequence in exon 51 of DMD thereby promoting restoration of dystrophin expression in patients with deletions of exon 50 (Mendell et al, 2016; Alfano et al, 2019; Fig 6B). It was followed by ASOs designed to skip exon 53 (golodirsen (Frank et al, 2020) and viltolarsen (Roshmi & Yokota, 2019)) or exon 45 (casimersen (Wagner et al, 2021b)). FDA approval was based on low levels of dystrophin restoration in small cohorts of patients rather than on confirmation of functional efficacy and continued approval is contingent upon validation of a clinical benefit in on‐going confirmatory trials. Based on the initial clinical trial data, the European Medicines Agency (EMA) refused approval of eteplirsen, while approval of the other three ASOs is awaited. In the interim, efforts are underway to develop more effective ASO therapies. All four FDA‐approved ASOs are uncharged phosphorodiamidate morpholino oligomers (PMOs) and different chemical modifications and conjugation to arginine‐rich or muscle‐homing peptides are being explored to improve ASO efficacy and uptake by skeletal muscle and heart. Although the approved ASOs are generally well tolerated, the durability of the response is short. Because of protein and ASO turnover, the drugs need to be administered once weekly as a 35–60 min intravenous infusion.
CRISPR‐based gene editing of DMD would avoid life‐long treatment. Restoration of only a small amount of the normal level of dystrophin provides therapeutic benefits in mice (Long et al, 2014). Since skeletal muscles are multinucleated, this might be achieved if only a fraction of the myonuclei are corrected. On the other hand, muscle cells are postmitotic, so that precise DMD correction via HDR is not feasible. Instead, the strategy is reliant on error‐prone NHEJ and depends on the redundancy in dystrophin's central rod domain, which permits deletion of internal segments if the ORF is maintained. While proof of concept has been achieved confirming that genome editing can restore dystrophin levels in cells and animal models (Long et al, 2016; Min et al, 2019, 2020), challenges have to be overcome to apply this approach systematically in humans. These include optimal delivery of genome‐editing components, the risk of off‐target editing and a potential immune response against Cas9 (Olson, 2021).
“Classic” gene therapy approaches to restore dystrophin expression using viral vectors are in more advanced stages. While most viruses do not have a natural tropism for skeletal or heart muscle, AAVs can infect these tissues efficiently. However, due to AAV's limited carrying capacity, only micro‐dystrophin constructs that lack all but the most crucial domains can be used. Phase 1 clinical trials have shown micro‐dystrophin expression in muscle fibers but it is not yet established if treatment will ameliorate disease progression (Mendell et al, 2020). Despite this uncertainty, elevidys, a micro‐dystrophin AAV gene therapy has recently been granted accelerated approval by the FDA. Although it is unclear which therapeutic modality will ultimately prove to be effective, there is hope that continuous research efforts will result in the development of a successful DMD therapy.
Cystic fibrosis
In the competitive drug development market, RNA therapeutics and small molecule drugs vie with each other. The effectiveness of each modality depends on the molecular mechanism of the disease and the “druggability” of the target. An example of a disease where small molecule drugs have been highly successful is cystic fibrosis (CF). CF is an inherited disorder caused by mutations in the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR; Kerem et al, 1989; Riordan et al, 1989; Rommens et al, 1989). CFTR is an anion channel that helps maintain fluid‐ and electrolyte‐homeostasis in multiple organs (Knowles et al, 1983; Quinton, 1983; Rich et al, 1990; Kartner et al, 1991). Loss‐of‐function leads to the accumulation of excessively viscous mucus that obstruct passageways. In the lung, this leads to airway blockage and causes repeated cycles of bacterial infections and chronic inflammation. Lung disease and respiratory failure are currently the main causes of morbidity and mortality in patients (Shteinberg et al, 2021; Fig 6C).
The CFTR gene displays mutational heterogeneity; to date, around 2,000 CFTR variants have been identified in patients with CF and CFTR‐related disorders. These mutations are classed into six groups based on how they affect the production, trafficking, function or stability of CFTR (Welsh & Smith, 1993). Class II mutations, which interfere with CFTR trafficking to the cell surface, are the most common, with an in‐frame deletion of phenylalanine 508 (F508del) affecting more than 70% of CF patients.
For decades, therapies were palliative and focused on medications that reduce symptoms, such as mucolytics, anti‐infectives, and pancreatic enzyme replacement. The development of small molecule modulators of CFTR that promote channel trafficking or function revolutionized CF treatment (Cutting, 2015). Nearly 90% of people with CF stand to benefit from these drugs, which are typically used in combination. There are, however, subsets of patients who carry CFTR mutations that lead to a complete loss of the protein, precluding the effective use of CFTR modulator therapy.
RNA‐based therapies might fill this gap. ASOs with different mechanisms of action such as inhibition of nonsense‐mediated mRNA decay or CFTR splice modulation are currently in pre‐clinical development (Kim & Krainer, 2023). These ASOs are often mutation‐specific and may therefore be suitable for only a small subset of CF patients. Earlier efforts to commercially develop ASOs for CF therapy have met with limited success. Development of eluforsen, an ASO that aims to insert the three missing bases in the F508del CFTR mRNA through an unknown mechanism, has recently been suspended, despite showing potential to improve lung function in homozygous F508del CF patients (Sermet‐Gaudelus et al, 2019; Drevinek et al, 2020). This ASO targets a similar patient group as the highly successful triple‐drug combination of the small molecule modulators ivacaftor, elexacaftor, and tezacaftor.
Gene replacement therapy allows treatment of all CF patients, regardless of the underlying genetic mutation. This approach has been explored since shortly after the identification of CFTR as the CF gene (Zabner et al, 1993). While initial clinical trials using AAV‐mediated gene delivery did not produce major improvements in symptoms (Wagner et al, 2002), a new therapy based on advanced vector engineering is currently in phase 1/2 clinical trials (NCT05248230). Delivery of CFTR mRNA as an alternative to DNA‐mediated gene replacement is actively pursued as well, with mixed results so far (Fig 6C). After encouraging initial reports, MRT5005, a drug that delivers CFTR mRNA packaged into lipid nanoparticles as an inhalable aerosol, failed to show improved lung function during the second interim analysis of the phase1/2 clinical trial data (NCT03375047), according to company statements. Nonetheless, MRT5005 was well‐tolerated, and the trial will continue.
CF is also amenable to CRISPR‐based genome editing approaches. Experiments in patient‐derived organoids have shown that allele‐specific corrections of aberrant CFTR splicing (Maule et al, 2019) and base editing to correct specific mutations within the CFTR gene (Geurts et al, 2020) are feasible. Still, to achieve durable therapeutic effects, the disease‐causing mutations must be corrected within lung stem cells, and targeted delivery of the therapeutic modalities to these cells remains a major challenge.
Outlook
Advances in medicinal chemistry, a better understanding of the versatile cellular functions of RNA, and the experiences gained from decades of preclinical and clinical studies have established a strong foundation for the future of RNA therapeutics. Continuous optimization of RNA drug design, be it ASOs, siRNA drugs, mRNAs, and CRISPR‐Cas editing tools, will improve drug efficacies. An impressive illustration of this concept is the improvement in metabolic stability, durability, and potency of siRNA therapeutics achieved through advanced chemical modifications. For example, the prototype GalNAc–siRNA conjugate revusiran, which targeted transthyretin mRNA for the treatment of hATTR, was metabolically labile and therefore required high and frequent dosing. The drug was poorly tolerated in a phase 3 clinical trial and its development was discontinued. Optimized chemistry led to siRNA drugs with a satisfying safety profile and increased potency, facilitating dosing as infrequently as once every 6 months (Ray et al, 2023). Likewise, RNA drugs that exhibit limited efficacy in current clinical trials can serve as the developmental basis for next‐generation drug candidates.
Despite the spirit of optimism, RNA therapeutics face remaining challenges, including economic ones. High manufacturing and regulatory costs and the expense of clinical trials combined with the limited size of the potential market in cases of rare diseases might mean that the retail price charged for a treatment, if it is developed, may be unaffordable for most patients. Another major problem that currently stands in the way of achieving broad applications for RNA therapeutics is targeted in vivo delivery. For example, most ASO therapeutics to‐date use local delivery to specific sites, e.g. the eye or spinal cord, or delivery to the liver. It is expected that optimized combinations of RNA chemical modifications, conjugation with cell‐targeting ligands, and improved nanoparticle carrier systems will enhance the efficiency of RNA drug delivery and enable therapeutic molecules to reach previously inaccessible target tissues.
Apart from the RNA therapeutic modalities discussed in this review, there are others in preclinical and clinical development. These include ASOs or siRNA drugs that activate gene expression (Li et al, 2006; Janowski et al, 2007; Liang et al, 2016, 2017; Johnson & Corey, 2023), tRNAs that have been re‐coded to facilitate read‐through of nonsense mutations (Porter et al, 2021), and CRISPR interference and activation systems. The latter are comprised of a catalytically inactive Cas9 mutant fused to either a transcriptional repressor or a transcriptional activator, respectively (Gilbert et al, 2013, 2014; Maeder et al, 2013; Perez‐Pinera et al, 2013). Aptamers – short structured RNAs with high specificity and affinity for target molecules – can be used to modulate these targets or employed as carriers for delivering other therapeutic agents to specific cells or tissues (Zhou & Rossi, 2017).
While current clinical RNA therapeutics all target eukaryotic cells, RNA drugs can also be applied to prokaryotes. Proof of principle of the efficacy of ASOs in eliminating diverse bacterial species by targeting essential bacterial genes at the mRNA level has been established, including in animal models (Good et al, 2001; Daly et al, 2018). This highlights opportunities for the development of programmable RNA antibiotics to combat multidrug‐resistant pathogens (Sully & Geller, 2016; Vogel, 2020).
Looking ahead, we expect to see developments in all areas of RNA medicine in the near future – the possible benefits for patients are too great to ignore.
Author contributions
Jörg Vogel: Conceptualization; writing – review and editing. Anke Sparmann: Writing – original draft; writing – review and editing.
Disclosure and competing interests statement
JV is a member of the Advisory Editorial Board of The EMBO Journal. This has no bearing on the editorial consideration of this article for publication.
Acknowledgements
This work was supported by the RNAmed International Doctorate Program (Elite Network of Bavaria/Elitenetzwerk Bayern). We thank Helen Pickersgill for helpful comments on the manuscript, Sandy Westermann (www.scigraphix.com) for illustrator support and Chandradhish Ghosh for help in rendering the chemical formulas in Box 2.
The EMBO Journal (2023) 42: e114760
References
- Adams D, Gonzalez‐Duarte A, O'Riordan WD, Yang CC, Ueda M, Kristen AV, Tournev I, Schmidt HH, Coelho T, Berk JL et al (2018) Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med 379: 11–21 [DOI] [PubMed] [Google Scholar]
- Adams D, Tournev IL, Taylor MS, Coelho T, Planté‐Bordeneuve V, Berk JL, González‐Duarte A, Gillmore JD, Low SC, Sekijima Y et al (2023) Efficacy and safety of vutrisiran for patients with hereditary transthyretin‐mediated amyloidosis with polyneuropathy: a randomized clinical trial. Amyloid 30: 1–9 [DOI] [PubMed] [Google Scholar]
- Alfano LN, Charleston JS, Connolly AM, Cripe L, Donoghue C, Dracker R, Dworzak J, Eliopoulos H, Frank DE, Lewis S et al (2019) Long‐term treatment with eteplirsen in nonambulatory patients with Duchenne muscular dystrophy. Medicine (Baltimore) 98: e15858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alterman JF, Godinho B, Hassler MR, Ferguson CM, Echeverria D, Sapp E, Haraszti RA, Coles AH, Conroy F, Miller R et al (2019) A divalent siRNA chemical scaffold for potent and sustained modulation of gene expression throughout the central nervous system. Nat Biotechnol 37: 884–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anttila V, Saraste A, Knuuti J, Jaakkola P, Hedman M, Svedlund S, Lagerström‐Fermér M, Kjaer M, Jeppsson A, Gan LM (2020) Synthetic mRNA encoding VEGF‐A in patients undergoing coronary artery bypass grafting: design of a phase 2a clinical trial. Mol Ther Methods Clin Dev 18: 464–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, Chen PJ, Wilson C, Newby GA, Raguram A et al (2019) Search‐and‐replace genome editing without double‐strand breaks or donor DNA. Nature 576: 149–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzalone AV, Koblan LW, Liu DR (2020) Genome editing with CRISPR‐Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol 38: 824–844 [DOI] [PubMed] [Google Scholar]
- Arbab M, Matuszek Z, Kray KM, Du A, Newby GA, Blatnik AJ, Raguram A, Richter MF, Zhao KT, Levy JM et al (2023) Base editing rescue of spinal muscular atrophy in cells and in mice. Science 380: eadg6518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baden LR, El Sahly HM, Essink B, Kotloff K, Frey S, Novak R, Diemert D, Spector SA, Rouphael N, Creech CB et al (2021) Efficacy and Safety of the mRNA‐1273 SARS‐CoV‐2 Vaccine. N Engl J Med 384: 403–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker BF, Lot SS, Condon TP, Cheng‐Flournoy S, Lesnik EA, Sasmor HM, Bennett CF (1997) 2'‐O‐(2‐Methoxy)ethyl‐modified anti‐intercellular adhesion molecule 1 (ICAM‐1) oligonucleotides selectively increase the ICAM‐1 mRNA level and inhibit formation of the ICAM‐1 translation initiation complex in human umbilical vein endothelial cells. J Biol Chem 272: 11994–12000 [DOI] [PubMed] [Google Scholar]
- Baker BF, Lot SS, Kringel J, Cheng‐Flournoy S, Villiet P, Sasmor HM, Siwkowski AM, Chappell LL, Morrow JR (1999) Oligonucleotide‐europium complex conjugate designed to cleave the 5′ cap structure of the ICAM‐1 transcript potentiates antisense activity in cells. Nucleic Acids Res 27: 1547–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banskota S, Raguram A, Suh S, Du SW, Davis JR, Choi EH, Wang X, Nielsen SC, Newby GA, Randolph PB et al (2022) Engineered virus‐like particles for efficient in vivo delivery of therapeutic proteins. Cell 185: 250–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science 315: 1709–1712 [DOI] [PubMed] [Google Scholar]
- Biscans A, Coles A, Haraszti R, Echeverria D, Hassler M, Osborn M, Khvorova A (2019) Diverse lipid conjugates for functional extra‐hepatic siRNA delivery in vivo. Nucleic Acids Res 47: 1082–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braasch DA, Corey DR (2001) Locked nucleic acid (LNA): fine‐tuning the recognition of DNA and RNA. Chem Biol 8: 1–7 [DOI] [PubMed] [Google Scholar]
- Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, van der Oost J (2008) Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321: 960–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KM, Nair JK, Janas MM, Anglero‐Rodriguez YI, Dang LTH, Peng H, Theile CS, Castellanos‐Rizaldos E, Brown C, Foster D et al (2022) Expanding RNAi therapeutics to extrahepatic tissues with lipophilic conjugates. Nat Biotechnol 40: 1500–1508 [DOI] [PubMed] [Google Scholar]
- Canver MC, Smith EC, Sher F, Pinello L, Sanjana NE, Shalem O, Chen DD, Schupp PG, Vinjamur DS, Garcia SP et al (2015) BCL11A enhancer dissection by Cas9‐mediated in situ saturating mutagenesis. Nature 527: 192–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplen NJ, Parrish S, Imani F, Fire A, Morgan RA (2001) Specific inhibition of gene expression by small double‐stranded RNAs in invertebrate and vertebrate systems. Proc Natl Acad Sci USA 98: 9742–9747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartegni L, Krainer AR (2002) Disruption of an SF2/ASF‐dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet 30: 377–384 [DOI] [PubMed] [Google Scholar]
- Cartegni L, Krainer AR (2003) Correction of disease‐associated exon skipping by synthetic exon‐specific activators. Nat Struct Biol 10: 120–125 [DOI] [PubMed] [Google Scholar]
- Charlesworth CT, Deshpande PS, Dever DP, Camarena J, Lemgart VT, Cromer MK, Vakulskas CA, Collingwood MA, Zhang L, Bode NM et al (2019) Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat Med 25: 249–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary N, Weissman D, Whitehead KA (2021) mRNA vaccines for infectious diseases: principles, delivery and clinical translation. Nat Rev Drug Discov 20: 817–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PJ, Liu DR (2023) Prime editing for precise and highly versatile genome manipulation. Nat Rev Genet 24: 161–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PJ, Hussmann JA, Yan J, Knipping F, Ravisankar P, Chen PF, Chen C, Nelson JW, Newby GA, Sahin M et al (2021) Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell 184: 5635–5652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooke ST, Baker BF, Crooke RM, Liang XH (2021a) Antisense technology: an overview and prospectus. Nat Rev Drug Discov 20: 427–453 [DOI] [PubMed] [Google Scholar]
- Crooke ST, Liang XH, Baker BF, Crooke RM (2021b) Antisense technology: A review. J Biol Chem 296: 100416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuellar TL, Barnes D, Nelson C, Tanguay J, Yu SF, Wen X, Scales SJ, Gesch J, Davis D, van Brabant SA et al (2015) Systematic evaluation of antibody‐mediated siRNA delivery using an industrial platform of THIOMAB‐siRNA conjugates. Nucleic Acids Res 43: 1189–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui S, Wang Y, Gong Y, Lin X, Zhao Y, Zhi D, Zhou Q, Zhang S (2018) Correlation of the cytotoxic effects of cationic lipids with their headgroups. Toxicol Res (Camb) 7: 473–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullis PR, Hope MJ (2017) Lipid nanoparticle systems for enabling gene therapies. Mol Ther 25: 1467–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutting GR (2015) Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet 16: 45–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly SM, Sturge CR, Marshall‐Batty KR, Felder‐Scott CF, Jain R, Geller BL, Greenberg DE (2018) Antisense inhibitors retain activity in pulmonary models of Burkholderia infection. ACS Infect Dis 4: 806–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis SM, Hariharan VN, Lo A, Turanov AA, Echeverria D, Sousa J, McHugh N, Biscans A, Alterman JF, Karumanchi SA et al (2022) Chemical optimization of siRNA for safe and efficient silencing of placental sFLT1. Mol Ther Nucleic Acids 29: 135–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E (2011) CRISPR RNA maturation by trans‐encoded small RNA and host factor RNase III. Nature 471: 602–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djuranovic S, Nahvi A, Green R (2012) miRNA‐mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science 336: 237–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolgin E (2023) Personalized cancer vaccines pass first major clinical test. Nat Rev Drug Discov 22: 607–609 [DOI] [PubMed] [Google Scholar]
- Doudna JA (2020) The promise and challenge of therapeutic genome editing. Nature 578: 229–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowdy SF (2023) Endosomal escape of RNA therapeutics: how do we solve this rate‐limiting problem? RNA 29: 396–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drevinek P, Pressler T, Cipolli M, De Boeck K, Schwarz C, Bouisset F, Boff M, Henig N, Paquette‐Lamontagne N, Montgomery S et al (2020) Antisense oligonucleotide eluforsen is safe and improves respiratory symptoms in F508DEL cystic fibrosis. J Cyst Fibros 19: 99–107 [DOI] [PubMed] [Google Scholar]
- Duan D, Goemans N, Takeda S, Mercuri E, Aartsma‐Rus A (2021) Duchenne muscular dystrophy. Nat Rev Dis Primers 7: 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugal‐Tessier J, Thirumalairajan S, Jain N (2021) Antibody‐oligonucleotide conjugates: a twist to antibody‐drug conjugates. J Clin Med 10: 838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egli M, Manoharan M (2023) Chemistry, structure and function of approved oligonucleotide therapeutics. Nucleic Acids Res 51: 2529–2573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T (2001) Duplexes of 21‐nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411: 494–498 [DOI] [PubMed] [Google Scholar]
- Elkayam E, Parmar R, Brown CR, Willoughby JL, Theile CS, Manoharan M, Joshua‐Tor L (2017) siRNA carrying an (E)‐vinylphosphonate moiety at the 5΄ end of the guide strand augments gene silencing by enhanced binding to human Argonaute‐2. Nucleic Acids Res 45: 3528–3536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel RS, Chiriboga CA, Vajsar J, Day JW, Montes J, De Vivo DC, Yamashita M, Rigo F, Hung G, Schneider E et al (2016) Treatment of infantile‐onset spinal muscular atrophy with nusinersen: a phase 2, open‐label, dose‐escalation study. Lancet 388: 3017–3026 [DOI] [PubMed] [Google Scholar]
- Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, Chiriboga CA, Saito K, Servais L, Tizzano E et al (2017) Nusinersen versus Sham control in infantile‐onset spinal muscular atrophy. N Engl J Med 377: 1723–1732 [DOI] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC (1998) Potent and specific genetic interference by double‐stranded RNA in Caenorhabditis elegans . Nature 391: 806–811 [DOI] [PubMed] [Google Scholar]
- Frangoul H, Altshuler D, Cappellini MD, Chen YS, Domm J, Eustace BK, Foell J, de la Fuente J, Grupp S, Handgretinger R et al (2021) CRISPR‐Cas9 gene editing for sickle cell disease and β‐thalassemia. N Engl J Med 384: 252–260 [DOI] [PubMed] [Google Scholar]
- Frank DE, Schnell FJ, Akana C, El‐Husayni SH, Desjardins CA, Morgan J, Charleston JS, Sardone V, Domingos J, Dickson G et al (2020) Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology 94: e2270–e2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freier SM, Altmann KH (1997) The ups and downs of nucleic acid duplex stability: structure‐stability studies on chemically‐modified DNA:RNA duplexes. Nucleic Acids Res 25: 4429–4443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao QQ, McNally EM (2015) The dystrophin complex: structure, function, and implications for therapy. Compr Physiol 5: 1223–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau JE, Dupuis M, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadán AH, Moineau S (2010) The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468: 67–71 [DOI] [PubMed] [Google Scholar]
- Gasiunas G, Barrangou R, Horvath P, Siksnys V (2012) Cas9‐crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci USA 109: E2579–E2586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geurts MH, de Poel E, Amatngalim GD, Oka R, Meijers FM, Kruisselbrink E, van Mourik P, Berkers G, de Winter‐de Groot KM, Michel S et al (2020) CRISPR‐based adenine editors correct nonsense mutations in a cystic fibrosis organoid biobank. Cell Stem Cell 26: 503–510 [DOI] [PubMed] [Google Scholar]
- Giannoukos G, Ciulla DM, Marco E, Abdulkerim HS, Barrera LA, Bothmer A, Dhanapal V, Gloskowski SW, Jayaram H, Maeder ML et al (2018) UDiTaS™, a genome editing detection method for indels and genome rearrangements. BMC Genomics 19: 212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern‐Ginossar N, Brandman O, Whitehead EH, Doudna JA et al (2013) CRISPR‐mediated modular RNA‐guided regulation of transcription in eukaryotes. Cell 154: 442–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC et al (2014) Genome‐scale CRISPR‐mediated control of gene repression and activation. Cell 159: 647–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillmore JD, Gane E, Taubel J, Kao J, Fontana M, Maitland ML, Seitzer J, O'Connell D, Walsh KR, Wood K et al (2021) CRISPR‐Cas9 in vivo gene editing for transthyretin amyloidosis. N Engl J Med 385: 493–502 [DOI] [PubMed] [Google Scholar]
- Good L, Awasthi SK, Dryselius R, Larsson O, Nielsen PE (2001) Bactericidal antisense effects of peptide‐PNA conjugates. Nat Biotechnol 19: 360–364 [DOI] [PubMed] [Google Scholar]
- Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR, Marion P, Salazar F, Kay MA (2006) Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 441: 537–541 [DOI] [PubMed] [Google Scholar]
- Hamilton AJ, Baulcombe DC (1999) A species of small antisense RNA in posttranscriptional gene silencing in plants. Science 286: 950–952 [DOI] [PubMed] [Google Scholar]
- Hammond SM, Bernstein E, Beach D, Hannon GJ (2000) An RNA‐directed nuclease mediates post‐transcriptional gene silencing in Drosophila cells. Nature 404: 293–296 [DOI] [PubMed] [Google Scholar]
- Han SP, Scherer L, Gethers M, Salvador AM, Salah MBH, Mancusi R, Sagar S, Hu R, DeRogatis J, Kuo YH et al (2022) Programmable siRNA pro‐drugs that activate RNAi activity in response to specific cellular RNA biomarkers. Mol Ther Nucleic Acids 27: 797–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraszti RA, Roux L, Coles AH, Turanov AA, Alterman JF, Echeverria D, Godinho B, Aronin N, Khvorova A (2017) 5΄‐Vinylphosphonate improves tissue accumulation and efficacy of conjugated siRNAs in vivo. Nucleic Acids Res 45: 7581–7592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry S, Stecker K, Brooks D, Monteith D, Conklin B, Bennett CF (2000) Chemically modified oligonucleotides exhibit decreased immune stimulation in mice. J Pharmacol Exp Ther 292: 468–479 [PubMed] [Google Scholar]
- Hodges D, Crooke ST (1995) Inhibition of splicing of wild‐type and mutated luciferase‐adenovirus pre‐mRNAs by antisense oligonucleotides. Mol Pharmacol 48: 905–918 [PubMed] [Google Scholar]
- Hoffman EP, Brown RH Jr, Kunkel LM (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51: 919–928 [DOI] [PubMed] [Google Scholar]
- Hofmann Y, Lorson CL, Stamm S, Androphy EJ, Wirth B (2000) Htra2‐beta 1 stimulates an exonic splicing enhancer and can restore full‐length SMN expression to survival motor neuron 2 (SMN2). Proc Natl Acad Sci USA 97: 9618–9623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou X, Zaks T, Langer R, Dong Y (2021) Lipid nanoparticles for mRNA delivery. Nat Rev Mater 6: 1078–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JH, Miller SM, Geurts MH, Tang W, Chen L, Sun N, Zeina CM, Gao X, Rees HA, Lin Z et al (2018) Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556: 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR (2008) Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet 82: 834–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, Sahashi K, Rigo F, Hung G, Horev G, Bennett CF, Krainer AR (2011) Peripheral SMN restoration is essential for long‐term rescue of a severe spinal muscular atrophy mouse model. Nature 478: 123–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur S (2019) Double‐stranded RNA sensors and modulators in innate immunity. Annu Rev Immunol 37: 349–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hustedt N, Durocher D (2016) The control of DNA repair by the cell cycle. Nat Cell Biol 19: 1–9 [DOI] [PubMed] [Google Scholar]
- Hutvágner G, Zamore PD (2002) A microRNA in a multiple‐turnover RNAi enzyme complex. Science 297: 2056–2060 [DOI] [PubMed] [Google Scholar]
- Ipsaro JJ, Joshua‐Tor L (2015) From guide to target: molecular insights into eukaryotic RNA‐interference machinery. Nat Struct Mol Biol 22: 20–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janowski BA, Younger ST, Hardy DB, Ram R, Huffman KE, Corey DR (2007) Activating gene expression in mammalian cells with promoter‐targeted duplex RNAs. Nat Chem Biol 3: 166–173 [DOI] [PubMed] [Google Scholar]
- Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez‐Torres M, Patel K, van der Meer AJ, Patick AK, Chen A, Zhou Y et al (2013) Treatment of HCV infection by targeting microRNA. N Engl J Med 368: 1685–1694 [DOI] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual‐RNA‐guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J (2013) RNA‐programmed genome editing in human cells. Elife 2: e00471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KC, Corey DR (2023) RNAi in Cell Nuclei: potential for a new layer of biological regulation and a new strategy for therapeutic discovery. RNA 29: 415–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P (2005) Modulation of hepatitis C virus RNA abundance by a liver‐specific MicroRNA. Science 309: 1577–1581 [DOI] [PubMed] [Google Scholar]
- Karikó K, Buckstein M, Ni H, Weissman D (2005) Suppression of RNA recognition by Toll‐like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23: 165–175 [DOI] [PubMed] [Google Scholar]
- Karikó K, Muramatsu H, Ludwig J, Weissman D (2011) Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside‐modified, protein‐encoding mRNA. Nucleic Acids Res 39: e142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kartner N, Hanrahan JW, Jensen TJ, Naismith AL, Sun SZ, Ackerley CA, Reyes EF, Tsui LC, Rommens JM, Bear CE et al (1991) Expression of the cystic fibrosis gene in non‐epithelial invertebrate cells produces a regulated anion conductance. Cell 64: 681–691 [DOI] [PubMed] [Google Scholar]
- Kauffman KJ, Webber MJ, Anderson DG (2016) Materials for non‐viral intracellular delivery of messenger RNA therapeutics. J Control Release 240: 227–234 [DOI] [PubMed] [Google Scholar]
- Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC (1989) Identification of the cystic fibrosis gene: genetic analysis. Science 245: 1073–1080 [DOI] [PubMed] [Google Scholar]
- Khvorova A, Watts JK (2017) The chemical evolution of oligonucleotide therapies of clinical utility. Nat Biotechnol 35: 238–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Krainer AR (2023) Antisense oligonucleotide therapeutics for cystic fibrosis: recent developments and perspectives. Mol Cells 46: 10–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingwell K (2022) Base editors hit the clinic. Nat Rev Drug Discov 21: 545–547 [DOI] [PubMed] [Google Scholar]
- Klabenkova K, Fokina A, Stetsenko D (2021) Chemistry of peptide‐oligonucleotide conjugates: a review. Molecules 26: 5420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles MR, Stutts MJ, Spock A, Fischer N, Gatzy JT, Boucher RC (1983) Abnormal ion permeation through cystic fibrosis respiratory epithelium. Science 221: 1067–1070 [DOI] [PubMed] [Google Scholar]
- Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM (1987) Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 50: 509–517 [DOI] [PubMed] [Google Scholar]
- Koller E, Vincent TM, Chappell A, De S, Manoharan M, Bennett CF (2011) Mechanisms of single‐stranded phosphorothioate modified antisense oligonucleotide accumulation in hepatocytes. Nucleic Acids Res 39: 4795–4807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR (2016) Programmable editing of a target base in genomic DNA without double‐stranded DNA cleavage. Nature 533: 420–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosicki M, Tomberg K, Bradley A (2018) Repair of double‐strand breaks induced by CRISPR‐Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol 36: 765–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, Meng M, Fritz D, Vascotto F, Hefesha H et al (2016) Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 534: 396–401 [DOI] [PubMed] [Google Scholar]
- Krienke C, Kolb L, Diken E, Streuber M, Kirchhoff S, Bukur T, Akilli‐Öztürk Ö, Kranz LM, Berger H, Petschenka J et al (2021) A noninflammatory mRNA vaccine for treatment of experimental autoimmune encephalomyelitis. Science 371: 145–153 [DOI] [PubMed] [Google Scholar]
- Kulkarni JA, Witzigmann D, Thomson SB, Chen S, Leavitt BR, Cullis PR, van der Meel R (2021) The current landscape of nucleic acid therapeutics. Nat Nanotechnol 16: 630–643 [DOI] [PubMed] [Google Scholar]
- Lanford RE, Hildebrandt‐Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, Kauppinen S, Ørum H (2010) Therapeutic silencing of microRNA‐122 in primates with chronic hepatitis C virus infection. Science 327: 198–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang F, Schrörs B, Löwer M, Türeci Ö, Sahin U (2022) Identification of neoantigens for individualized therapeutic cancer vaccines. Nat Rev Drug Discov 21: 261–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V (1993) The C. elegans heterochronic gene lin‐4 encodes small RNAs with antisense complementarity to lin‐14. Cell 75: 843–854 [DOI] [PubMed] [Google Scholar]
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M et al (1995) Identification and characterization of a spinal muscular atrophy‐determining gene. Cell 80: 155–165 [DOI] [PubMed] [Google Scholar]
- Li LC, Okino ST, Zhao H, Pookot D, Place RF, Urakami S, Enokida H, Dahiya R (2006) Small dsRNAs induce transcriptional activation in human cells. Proc Natl Acad Sci USA 103: 17337–17342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang XH, Shen W, Sun H, Migawa MT, Vickers TA, Crooke ST (2016) Translation efficiency of mRNAs is increased by antisense oligonucleotides targeting upstream open reading frames. Nat Biotechnol 34: 875–880 [DOI] [PubMed] [Google Scholar]
- Liang XH, Sun H, Shen W, Wang S, Yao J, Migawa MT, Bui HH, Damle SS, Riney S, Graham MJ et al (2017) Antisense oligonucleotides targeting translation inhibitory elements in 5' UTRs can selectively increase protein levels. Nucleic Acids Res 45: 9528–9546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang XH, Nichols JG, Hsu CW, Vickers TA, Crooke ST (2019) mRNA levels can be reduced by antisense oligonucleotides via no‐go decay pathway. Nucleic Acids Res 47: 6900–6916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind NA, Rael VE, Pestal K, Liu B, Barton GM (2022) Regulation of the nucleic acid‐sensing Toll‐like receptors. Nat Rev Immunol 22: 224–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song JJ, Hammond SM, Joshua‐Tor L, Hannon GJ (2004) Argonaute2 is the catalytic engine of mammalian RNAi. Science 305: 1437–1441 [DOI] [PubMed] [Google Scholar]
- Long C, McAnally JR, Shelton JM, Mireault AA, Bassel‐Duby R, Olson EN (2014) Prevention of muscular dystrophy in mice by CRISPR/Cas9‐mediated editing of germline DNA. Science 345: 1184–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long C, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez‐Ortiz E, Bhattacharyya S, Shelton JM, Bassel‐Duby R, Olson EN (2016) Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 351: 400–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder ML, Linder SJ, Cascio VM, Fu Y, Ho QH, Joung JK (2013) CRISPR RNA‐guided activation of endogenous human genes. Nat Methods 10: 977–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder ML, Stefanidakis M, Wilson CJ, Baral R, Barrera LA, Bounoutas GS, Bumcrot D, Chao H, Ciulla DM, DaSilva JA et al (2019) Development of a gene‐editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat Med 25: 229–233 [DOI] [PubMed] [Google Scholar]
- Malecova B, Burke RS, Cochran M, Hood MD, Johns R, Kovach PR, Doppalapudi VR, Erdogan G, Arias JD, Darimont B et al (2023) Targeted tissue delivery of RNA therapeutics using antibody‐oligonucleotide conjugates (AOCs). Nucleic Acids Res 51: 5901–5910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM (2013) RNA‐guided human genome engineering via Cas9. Science 339: 823–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda S, Keiser K, Nair JK, Charisse K, Manoharan RM, Kretschmer P, Peng CG, Kel'in AV, Kandasamy P, Willoughby JLS et al (2015) siRNA conjugates carrying sequentially assembled trivalent N‐acetylgalactosamine linked through nucleosides elicit robust gene silencing in vivo in hepatocytes. ACS Chem Biol 10: 1181–1187 [DOI] [PubMed] [Google Scholar]
- Maule G, Casini A, Montagna C, Ramalho AS, De Boeck K, Debyser Z, Carlon MS, Petris G, Cereseto A (2019) Allele specific repair of splicing mutations in cystic fibrosis through AsCas12a genome editing. Nat Commun 10: 3556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayuranathan T, Newby GA, Feng R, Yao Y, Mayberry KD, Lazzarotto CR, Li Y, Levine RM, Nimmagadda N, Dempsey E et al (2023) Potent and uniform fetal hemoglobin induction via base editing. Nat Genet 55: 1210–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister G (2013) Argonaute proteins: functional insights and emerging roles. Nat Rev Genet 14: 447–459 [DOI] [PubMed] [Google Scholar]
- Melton DA (1985) Injected anti‐sense RNAs specifically block messenger RNA translation in vivo. Proc Natl Acad Sci USA 82: 144–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, Goemans N, Lowes LP, Alfano LN, Berry K, Shao J, Kaye EM, Mercuri E (2016) Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol 79: 257–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, Sahenk Z, Lehman K, Nease C, Lowes LP, Miller NF, Iammarino MA, Alfano LN, Nicholl A, Al‐Zaidy S et al (2020) Assessment of systemic delivery of rAAVrh74.MHCK7.micro‐dystrophin in children with Duchenne muscular dystrophy: a nonrandomized controlled trial. JAMA Neurol 77: 1122–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercuri E, Bönnemann CG, Muntoni F (2019) Muscular dystrophies. Lancet 394: 2025–2038 [DOI] [PubMed] [Google Scholar]
- Miller SM, Wang T, Randolph PB, Arbab M, Shen MW, Huang TP, Matuszek Z, Newby GA, Rees HA, Liu DR (2020) Continuous evolution of SpCas9 variants compatible with non‐G PAMs. Nat Biotechnol 38: 471–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min YL, Li H, Rodriguez‐Caycedo C, Mireault AA, Huang J, Shelton JM, McAnally JR, Amoasii L, Mammen PPA, Bassel‐Duby R et al (2019) CRISPR‐Cas9 corrects Duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci Adv 5: eaav4324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min YL, Chemello F, Li H, Rodriguez‐Caycedo C, Sanchez‐Ortiz E, Mireault AA, McAnally JR, Shelton JM, Zhang Y, Bassel‐Duby R et al (2020) Correction of three prominent mutations in mouse and human models of Duchenne muscular dystrophy by single‐cut genome editing. Mol Ther 28: 2044–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojica FJ, Díez‐Villaseñor C, García‐Martínez J, Soria E (2005) Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol 60: 174–182 [DOI] [PubMed] [Google Scholar]
- Nair JK, Willoughby JL, Chan A, Charisse K, Alam MR, Wang Q, Hoekstra M, Kandasamy P, Kel'in AV, Milstein S et al (2014) Multivalent N‐acetylgalactosamine‐conjugated siRNA localizes in hepatocytes and elicits robust RNAi‐mediated gene silencing. J Am Chem Soc 136: 16958–16961 [DOI] [PubMed] [Google Scholar]
- Nakamura A, Shiba N, Miyazaki D, Nishizawa H, Inaba Y, Fueki N, Maruyama R, Echigoya Y, Yokota T (2017) Comparison of the phenotypes of patients harboring in‐frame deletions starting at exon 45 in the Duchenne muscular dystrophy gene indicates potential for the development of exon skipping therapy. J Hum Genet 62: 459–463 [DOI] [PubMed] [Google Scholar]
- Nelson JW, Randolph PB, Shen SP, Everette KA, Chen PJ, Anzalone AV, An M, Newby GA, Chen JC, Hsu A et al (2022) Engineered pegRNAs improve prime editing efficiency. Nat Biotechnol 40: 402–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newby GA, Liu DR (2021) In vivo somatic cell base editing and prime editing. Mol Ther 29: 3107–3124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida K, Arazoe T, Yachie N, Banno S, Kakimoto M, Tabata M, Mochizuki M, Miyabe A, Araki M, Hara KY et al (2016) Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 353: aaf8729 [DOI] [PubMed] [Google Scholar]
- Nishimasu H, Shi X, Ishiguro S, Gao L, Hirano S, Okazaki S, Noda T, Abudayyeh OO, Gootenberg JS, Mori H et al (2018) Engineered CRISPR‐Cas9 nuclease with expanded targeting space. Science 361: 1259–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okude H, Ori D, Kawai T (2020) Signaling through nucleic acid sensors and their roles in inflammatory diseases. Front Immunol 11: 625833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson EN (2021) Toward the correction of muscular dystrophy by gene editing. Proc Natl Acad Sci USA 118: e2004840117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CD, Rappaport AR, Davis MJ, Hart MG, Scallan CD, Hong SJ, Gitlin L, Kraemer LD, Kounlavouth S, Yang A et al (2022) Individualized, heterologous chimpanzee adenovirus and self‐amplifying mRNA neoantigen vaccine for advanced metastatic solid tumors: phase 1 trial interim results. Nat Med 28: 1619–1629 [DOI] [PubMed] [Google Scholar]
- Parmar R, Willoughby JL, Liu J, Foster DJ, Brigham B, Theile CS, Charisse K, Akinc A, Guidry E, Pei Y et al (2016) 5′‐(E)‐vinylphosphonate: a stable phosphate mimic can improve the RNAi activity of siRNA‐GalNAc conjugates. Chembiochem 17: 985–989 [DOI] [PubMed] [Google Scholar]
- Pasquinelli AE, Reinhart BJ, Slack F, Martindale MQ, Kuroda MI, Maller B, Hayward DC, Ball EE, Degnan B, Müller P et al (2000) Conservation of the sequence and temporal expression of let‐7 heterochronic regulatory RNA. Nature 408: 86–89 [DOI] [PubMed] [Google Scholar]
- Passini MA, Bu J, Richards AM, Kinnecom C, Sardi SP, Stanek LM, Hua Y, Rigo F, Matson J, Hung G et al (2011) Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med 3: 72ra18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Pinera P, Kocak DD, Vockley CM, Adler AF, Kabadi AM, Polstein LR, Thakore PI, Glass KA, Ousterout DG, Leong KW et al (2013) RNA‐guided gene activation by CRISPR‐Cas9‐based transcription factors. Nat Methods 10: 973–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai RS, Bhattacharyya SN, Artus CG, Zoller T, Cougot N, Basyuk E, Bertrand E, Filipowicz W (2005) Inhibition of translational initiation by Let‐7 MicroRNA in human cells. Science 309: 1573–1576 [DOI] [PubMed] [Google Scholar]
- Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, Perez JL, Pérez Marc G, Moreira ED, Zerbini C et al (2020) Safety and efficacy of the BNT162b2 mRNA Covid‐19 vaccine. N Engl J Med 383: 2603–2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JJ, Heil CS, Lueck JD (2021) Therapeutic promise of engineered nonsense suppressor tRNAs. Wiley Interdiscip Rev RNA 12: e1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradeep SP, Malik S, Slack FJ, Bahal R (2023) Unlocking the potential of chemically modified peptide nucleic acids for RNA‐based therapeutics. RNA 29: 434–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash TP, Lima WF, Murray HM, Li W, Kinberger GA, Chappell AE, Gaus H, Seth PP, Bhat B, Crooke ST et al (2015) Identification of metabolically stable 5′‐phosphate analogs that support single‐stranded siRNA activity. Nucleic Acids Res 43: 2993–3011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinton PM (1983) Chloride impermeability in cystic fibrosis. Nature 301: 421–422 [DOI] [PubMed] [Google Scholar]
- Raguram A, Banskota S, Liu DR (2022) Therapeutic in vivo delivery of gene editing agents. Cell 185: 2806–2827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajeev KG, Nair JK, Jayaraman M, Charisse K, Taneja N, O'Shea J, Willoughby JL, Yucius K, Nguyen T, Shulga‐Morskaya S et al (2015) Hepatocyte‐specific delivery of siRNAs conjugated to novel non‐nucleosidic trivalent N‐acetylgalactosamine elicits robust gene silencing in vivo. Chembiochem 16: 903–908 [DOI] [PubMed] [Google Scholar]
- Ray KK, Troquay RPT, Visseren FLJ, Leiter LA, Scott Wright R, Vikarunnessa S, Talloczy Z, Zang X, Maheux P, Lesogor A et al (2023) Long‐term efficacy and safety of inclisiran in patients with high cardiovascular risk and elevated LDL cholesterol (ORION‐3): results from the 4‐year open‐label extension of the ORION‐1 trial. Lancet Diabetes Endocrinol 11: 109–119 [DOI] [PubMed] [Google Scholar]
- Rees HA, Liu DR (2018) Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet 19: 770–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich DP, Anderson MP, Gregory RJ, Cheng SH, Paul S, Jefferson DM, McCann JD, Klinger KW, Smith AE, Welsh MJ (1990) Expression of cystic fibrosis transmembrane conductance regulator corrects defective chloride channel regulation in cystic fibrosis airway epithelial cells. Nature 347: 358–363 [DOI] [PubMed] [Google Scholar]
- Rigo F, Hua Y, Chun SJ, Prakash TP, Krainer AR, Bennett CF (2012a) Synthetic oligonucleotides recruit ILF2/3 to RNA transcripts to modulate splicing. Nat Chem Biol 8: 555–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigo F, Hua Y, Krainer AR, Bennett CF (2012b) Antisense‐based therapy for the treatment of spinal muscular atrophy. J Cell Biol 199: 21–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigo F, Chun SJ, Norris DA, Hung G, Lee S, Matson J, Fey RA, Gaus H, Hua Y, Grundy JS et al (2014) Pharmacology of a central nervous system delivered 2'‐O‐methoxyethyl‐modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates. J Pharmacol Exp Ther 350: 46–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL et al (1989) Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245: 1066–1073 [DOI] [PubMed] [Google Scholar]
- Roberts TC, Langer R, Wood MJA (2020) Advances in oligonucleotide drug delivery. Nat Rev Drug Discov 19: 673–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohner E, Yang R, Foo KS, Goedel A, Chien KR (2022) Unlocking the promise of mRNA therapeutics. Nat Biotechnol 40: 1586–1600 [DOI] [PubMed] [Google Scholar]
- Rojas LA, Sethna Z, Soares KC, Olcese C, Pang N, Patterson E, Lihm J, Ceglia N, Guasp P, Chu A et al (2023) Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 618: 144–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N et al (1989) Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 245: 1059–1065 [DOI] [PubMed] [Google Scholar]
- Roshmi RR, Yokota T (2019) Viltolarsen for the treatment of Duchenne muscular dystrophy. Drugs Today (Barc) 55: 627–639 [DOI] [PubMed] [Google Scholar]
- Rupaimoole R, Slack FJ (2017) MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov 16: 203–222 [DOI] [PubMed] [Google Scholar]
- Sahin U, Türeci Ö (2018) Personalized vaccines for cancer immunotherapy. Science 359: 1355–1360 [DOI] [PubMed] [Google Scholar]
- Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Löwer M, Bukur V, Tadmor AD, Luxemburger U, Schrörs B et al (2017) Personalized RNA mutanome vaccines mobilize poly‐specific therapeutic immunity against cancer. Nature 547: 222–226 [DOI] [PubMed] [Google Scholar]
- Sands H, Gorey‐Feret LJ, Cocuzza AJ, Hobbs FW, Chidester D, Trainor GL (1994) Biodistribution and metabolism of internally 3H‐labeled oligonucleotides. I. Comparison of a phosphodiester and a phosphorothioate. Mol Pharmacol 45: 932–943 [PubMed] [Google Scholar]
- Sano M, Sierant M, Miyagishi M, Nakanishi M, Takagi Y, Sutou S (2008) Effect of asymmetric terminal structures of short RNA duplexes on the RNA interference activity and strand selection. Nucleic Acids Res 36: 5812–5821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sermet‐Gaudelus I, Clancy JP, Nichols DP, Nick JA, De Boeck K, Solomon GM, Mall MA, Bolognese J, Bouisset F, den Hollander W et al (2019) Antisense oligonucleotide eluforsen improves CFTR function in F508del cystic fibrosis. J Cyst Fibros 18: 536–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setten RL, Rossi JJ, Han SP (2019) The current state and future directions of RNAi‐based therapeutics. Nat Rev Drug Discov 18: 421–446 [DOI] [PubMed] [Google Scholar]
- Shteinberg M, Haq IJ, Polineni D, Davies JC (2021) Cystic fibrosis. Lancet 397: 2195–2211 [DOI] [PubMed] [Google Scholar]
- Snead NM, Wu X, Li A, Cui Q, Sakurai K, Burnett JC, Rossi JJ (2013) Molecular basis for improved gene silencing by Dicer substrate interfering RNA compared with other siRNA variants. Nucleic Acids Res 41: 6209–6221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson ML, Zamecnik PC (1978) Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc Natl Acad Sci USA 75: 285–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh S, Choi EH, Raguram A, Liu DR, Palczewski K (2022) Precision genome editing in the eye. Proc Natl Acad Sci USA 119: e2210104119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sully EK, Geller BL (2016) Antisense antimicrobial therapeutics. Curr Opin Microbiol 33: 47–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summerton J (1999) Morpholino antisense oligomers: the case for an RNase H‐independent structural type. Biochim Biophys Acta 1489: 141–158 [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140: 805–820 [DOI] [PubMed] [Google Scholar]
- Turchiano G, Andrieux G, Klermund J, Blattner G, Pennucci V, El Gaz M, Monaco G, Poddar S, Mussolino C, Cornu TI et al (2021) Quantitative evaluation of chromosomal rearrangements in gene‐edited human stem cells by CAST‐Seq. Cell Stem Cell 28: 1136–1147 [DOI] [PubMed] [Google Scholar]
- Varley AJ, Desaulniers JP (2021) Chemical strategies for strand selection in short‐interfering RNAs. RSC Adv 11: 2415–2426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickers TA, Wyatt JR, Burckin T, Bennett CF, Freier SM (2001) Fully modified 2' MOE oligonucleotides redirect polyadenylation. Nucleic Acids Res 29: 1293–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitravene Study Group (2002a) A randomized controlled clinical trial of intravitreous fomivirsen for treatment of newly diagnosed peripheral cytomegalovirus retinitis in patients with AIDS. Am J Ophthalmol 133: 467–474 [DOI] [PubMed] [Google Scholar]
- Vitravene Study Group (2002b) Randomized dose‐comparison studies of intravitreous fomivirsen for treatment of cytomegalovirus retinitis that has reactivated or is persistently active despite other therapies in patients with AIDS. Am J Ophthalmol 133: 475–483 [DOI] [PubMed] [Google Scholar]
- Vogel J (2020) An RNA biology perspective on species‐specific programmable RNA antibiotics. Mol Microbiol 113: 550–559 [DOI] [PubMed] [Google Scholar]
- Wagner JA, Nepomuceno IB, Messner AH, Moran ML, Batson EP, Dimiceli S, Brown BW, Desch JK, Norbash AM, Conrad CK et al (2002) A phase II, double‐blind, randomized, placebo‐controlled clinical trial of tgAAVCF using maxillary sinus delivery in patients with cystic fibrosis with antrostomies. Hum Gene Ther 13: 1349–1359 [DOI] [PubMed] [Google Scholar]
- Wagner DL, Peter L, Schmueck‐Henneresse M (2021a) Cas9‐directed immune tolerance in humans‐a model to evaluate regulatory T cells in gene therapy? Gene Ther 28: 549–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner KR, Kuntz NL, Koenig E, East L, Upadhyay S, Han B, Shieh PB (2021b) Safety, tolerability, and pharmacokinetics of casimersen in patients with Duchenne muscular dystrophy amenable to exon 45 skipping: A randomized, double‐blind, placebo‐controlled, dose‐titration trial. Muscle Nerve 64: 285–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton RT, Christie KA, Whittaker MN, Kleinstiver BP (2020) Unconstrained genome targeting with near‐PAMless engineered CRISPR‐Cas9 variants. Science 368: 290–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JY, Doudna JA (2023) CRISPR technology: a decade of genome editing is only the beginning. Science 379: eadd8643 [DOI] [PubMed] [Google Scholar]
- Ward AJ, Norrbom M, Chun S, Bennett CF, Rigo F (2014) Nonsense‐mediated decay as a terminating mechanism for antisense oligonucleotides. Nucleic Acids Res 42: 5871–5879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber T (2021) Anti‐AAV antibodies in AAV gene therapy: current challenges and possible solutions. Front Immunol 12: 658399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh MJ, Smith AE (1993) Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 73: 1251–1254 [DOI] [PubMed] [Google Scholar]
- Wightman B, Ha I, Ruvkun G (1993) Posttranscriptional regulation of the heterochronic gene lin‐14 by lin‐4 mediates temporal pattern formation in C. elegans . Cell 75: 855–862 [DOI] [PubMed] [Google Scholar]
- Wu L, Fan J, Belasco JG (2006) MicroRNAs direct rapid deadenylation of mRNA. Proc Natl Acad Sci USA 103: 4034–4039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Zeng J, Roscoe BP, Liu P, Yao Q, Lazzarotto CR, Clement K, Cole MA, Luk K, Baricordi C et al (2019) Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat Med 25: 776–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabner J, Couture LA, Gregory RJ, Graham SM, Smith AE, Welsh MJ (1993) Adenovirus‐mediated gene transfer transiently corrects the chloride transport defect in nasal epithelia of patients with cystic fibrosis. Cell 75: 207–216 [DOI] [PubMed] [Google Scholar]
- Zamecnik PC, Stephenson ML (1978) Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc Natl Acad Sci USA 75: 280–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamore PD, Tuschl T, Sharp PA, Bartel DP (2000) RNAi: double‐stranded RNA directs the ATP‐dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 101: 25–33 [DOI] [PubMed] [Google Scholar]
- Zhou J, Rossi J (2017) Aptamers as targeted therapeutics: current potential and challenges. Nat Rev Drug Discov 16: 181–202 [DOI] [PMC free article] [PubMed] [Google Scholar]