

Abstract
The pseudokinase-endoribonuclease RNase L plays important roles in antiviral innate immunity and is also implicated in many other cellular activities. Inhibition of RNase L showed therapeutic potential for Aicardi-Goutières syndrome (AGS). Thus, RNase L is a promising drug target. In this study, using an enzyme assay and NMR screening, we discovered 13 inhibitory fragments against RNase L. Co-crystal structures of RNase L in complex with two fragments were determined, and both fragments bind to the ATP-binding pocket of the pseudokinase domain. Myricetin, vitexin and hyperoside, three natural products sharing similar scaffolds with the fragment AC40357, demonstrated potent inhibitory activity in vitro. In addition, myricetin has promising cellular inhibitory activity. A co-crystal structure of RNase L with myricetin provided a structural basis for inhibitor design by allosterically modulating its rnase activity. Our findings demonstrate that fragment screening can lead to the discovery of natural product inhibitors of RNase L.
INTRODUCTION
RNase L exerts its important function in interferon (IFN)-induced antiviral innate immunity through cleaving viral and cellular single stranded RNA (ssRNA).1–5 RNase L is a pseudokinase-endoribonuclease comprised of three domains6, 7: an N-terminal ankyrin-repeat (ANK) domain involved in 2-5A (2’-5’ linked oligoadenylate, a second messenger) binding, a pseudokinase (PK) domain participating RNase L dimerization and a C-terminal ribonuclease (RNase) domain that cleaves substrate RNA (Figure 1). Once a viral infection is detected, cells produce type I IFNs that induce expression of oligoadenylate synthetases (OASs) through the JAK-STAT signaling cascade.8 OASs 1-3 are then activated by viral double-stranded RNA (dsRNA) to synthesize 2-5A from ATP.9 2-5A and ATP bind to inactive monomeric RNase L and promote the formation of an intertwined dimeric conformation with active enzyme activity. 6, 7, 10, 11 Finally, active RNase L exerts antiviral activity by cleaving viral and cellular ssRNAs to eliminate viral components and inhibit cellular and viral protein synthesis.
Figure 1.
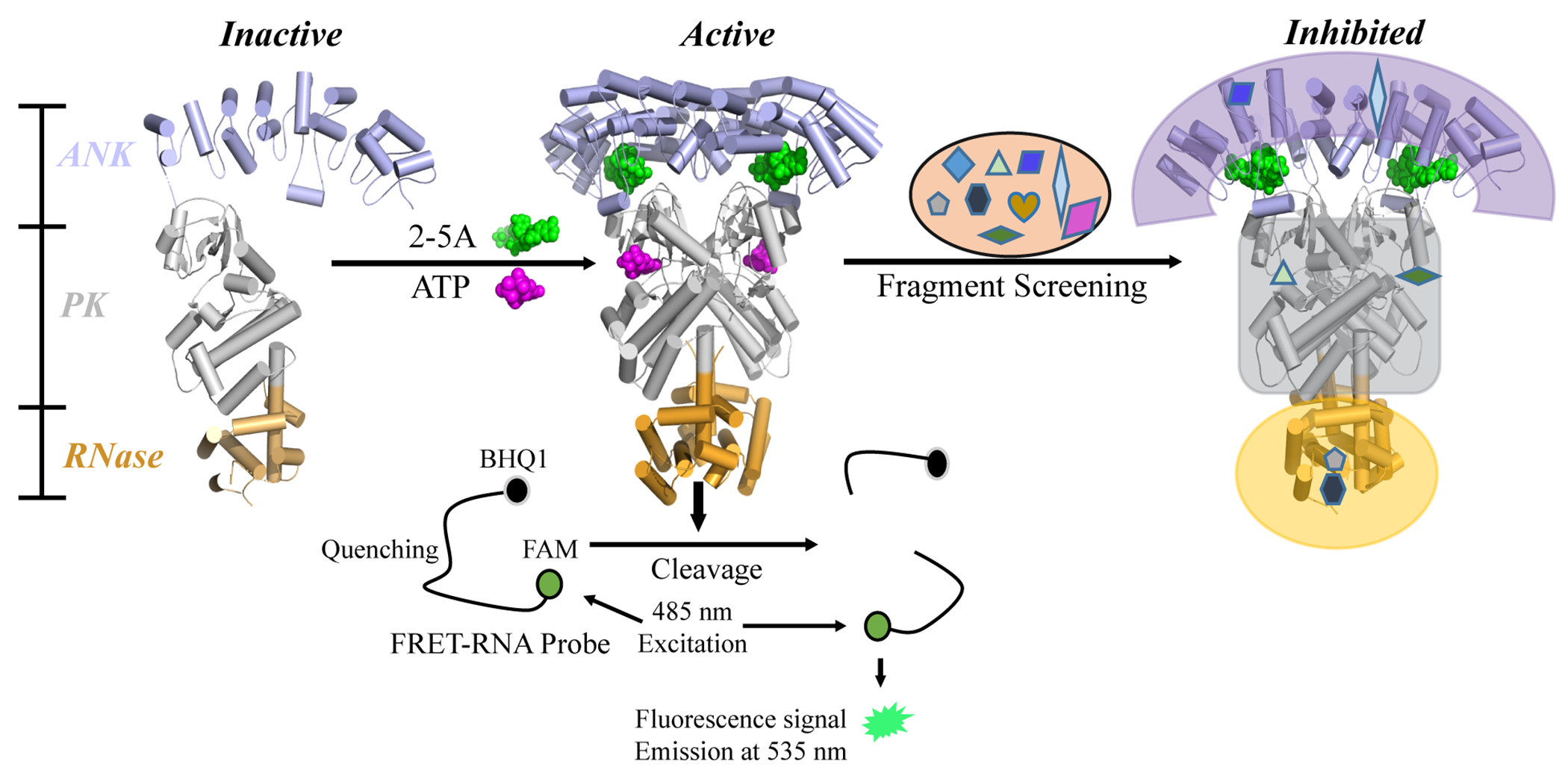
Diagram of RNase L activation and the FRET screening assay. The binding of 2-5A to the ANK domain and the binding of ATP to the PK domain co-operatively induce an active dimer conformation from inactive monomers. Activated RNase L cleaves viral and cellular ssRNA. In the FRET assay, an RNA substrate was labeled with a fluorophore (FAM) and a quencher (BHQ1) at its 5’ and 3’ termini, respectively. Upon substrate cleavage by RNase L, the fluorescence signal of FAM is detected, with excitation at 485 nm and emission at 535 nm. Active fragments were selected for next-stage validation.
In addition to its antiviral roles, RNase L has also been reported to be involved in apoptosis 12, IFN-β production13, autophagy14 and antibacterial effects.15, 16 A recent study also indicated that reduced adenosine deaminase (ADAR1) activity causes the activation of the OAS-RNase L pathway, which induced cell death.17 ADAR1 destabilizes dsRNA through deamination of adenosines, producing inosines in double stranded (dsRNA), which in turn decreases the activation of OAS.17, 18 Aicardi-Goutiéres syndrome (AGS) is a childhood neurodevelopmental and autoimmune inflammatory disease that lacks an effective treatment.19 Knockout of RNase L can rescue cells with ADAR1 mutations,17 suggesting that targeting RNase L with small molecule inhibitors may be a potential therapy for AGS cases caused by ADAR1 mutations. In fact, an RNase L inhibitor, the compound valoneic acid dilactone (VAL), has been shown to attenuate apoptosis caused by the loss of ADAR1 activity.20
Oncolytic viruses (OVs) are being used as anticancer therapy.21 Many factors hinder the efficacy of OVs, one of which is the inhibition of viral replication by the OAS signaling pathway. A study showed that inhibition of RNase L activity with sunitinib improves the anti-tumor effect of the Vesicular Stomatitis Virus (VSV).22 Therefore, inhibition of RNase L can potentially enhance oncolytic viral therapy.22 Furthermore, RNase L has a pro-inflammatory function23 and was suggested to be involved in cardiac acute ischemic injury.24 Therefore, RNase L inhibitors may also be useful as an anti-inflammatory or cardioprotective agents.
As described above, small molecule inhibitors of RNase L possess high therapeutic potential. However, the development of effective inhibitors for RNase L is still at an early stage. It has been reported that RNase L can be inhibited by sunitinib with an IC50 in the micromolar range in vitro.25 However, the relatively weak potency of sunitinib against RNase L compared with its primary targets VEGFR and PDFGR, limits its usefulness as a drug candidate for RNase L inhibition. In a recent study, VAL showed nanomolar inhibition activity on RNase L in vitro.20 Nevertheless, the EC50 of VAL for RNase L in intact cells was in the micromolar range. Therefore, there is an urgent and unmet need to develop effective novel inhibitors against RNase L to explore its functions in cellular activities and potentially to treat diseases in which RNase L activity is excessive or detrimental.
Our recent co-crystal structure showed that sunitinib allosterically inhibits RNase L activity by binding to the ATP pocket of the PK domain,26 which inspired us to design potent inhibitory compounds of RNase L starting from promising fragments.
In this study, we carried out a Fragment Based Drug Discovery (FBDD) campaign against RNase L and identified inhibitors using enzymatic and Saturation Transfer Difference-Nuclear Magnetic Resonance (STD-NMR) assays. From our screen, fragments of several novel chemotypes were identified. Co-crystal structures of RNase L with fragments show that two fragments bind to the ATP pocket of the PK domain. Because myricetin, vitexin and hyperoside share a similar scaffold with the fragment AC40357, the in vitro and in cyto inhibitory activities of these three compounds were further investigated. A co-crystal structure of RNase L with myricetin was determined and provided insights for future inhibitor optimization against RNase L.
RESULTS
Identification of active fragments against RNase L.
To discover small molecular inhibitors for RNase L, a commercial fragment library consisting of 840 core fragments was screened against human RNase L (H-RNase L) using a well-established FRET based enzyme assay (Figure 1 and 2).20, 26, 27 This sensitive assay has been used to characterize compounds which are able to modulate the RNA substrate cleavage activity of RNase L.20, 28 In the initial screen against H-RNase L, 31 fragments at 3 mM displayed inhibition rates over 50% and were selected for further validation (3.7% hit rate) (Figure 2B). Subsequently, we attempted to solve the co-crystal structures of H-RNase L with fragment hits. However, the expression and crystallization of H-RNase L requires considerable time, effort and expense.7 Because of the high sequence and structural similarities between human and porcine RNase L,6, 7 porcine RNase L (P-RNase L) has been previously used to explore the inhibition mechanism by sunitinib in a recent study.26 Therefore, these 31 hits were further evaluated for their inhibitory effects and binding abilities on P-RNase L using enzyme assays and STD-NMR spectroscopy, respectively (Figure 2A).
Figure 2.
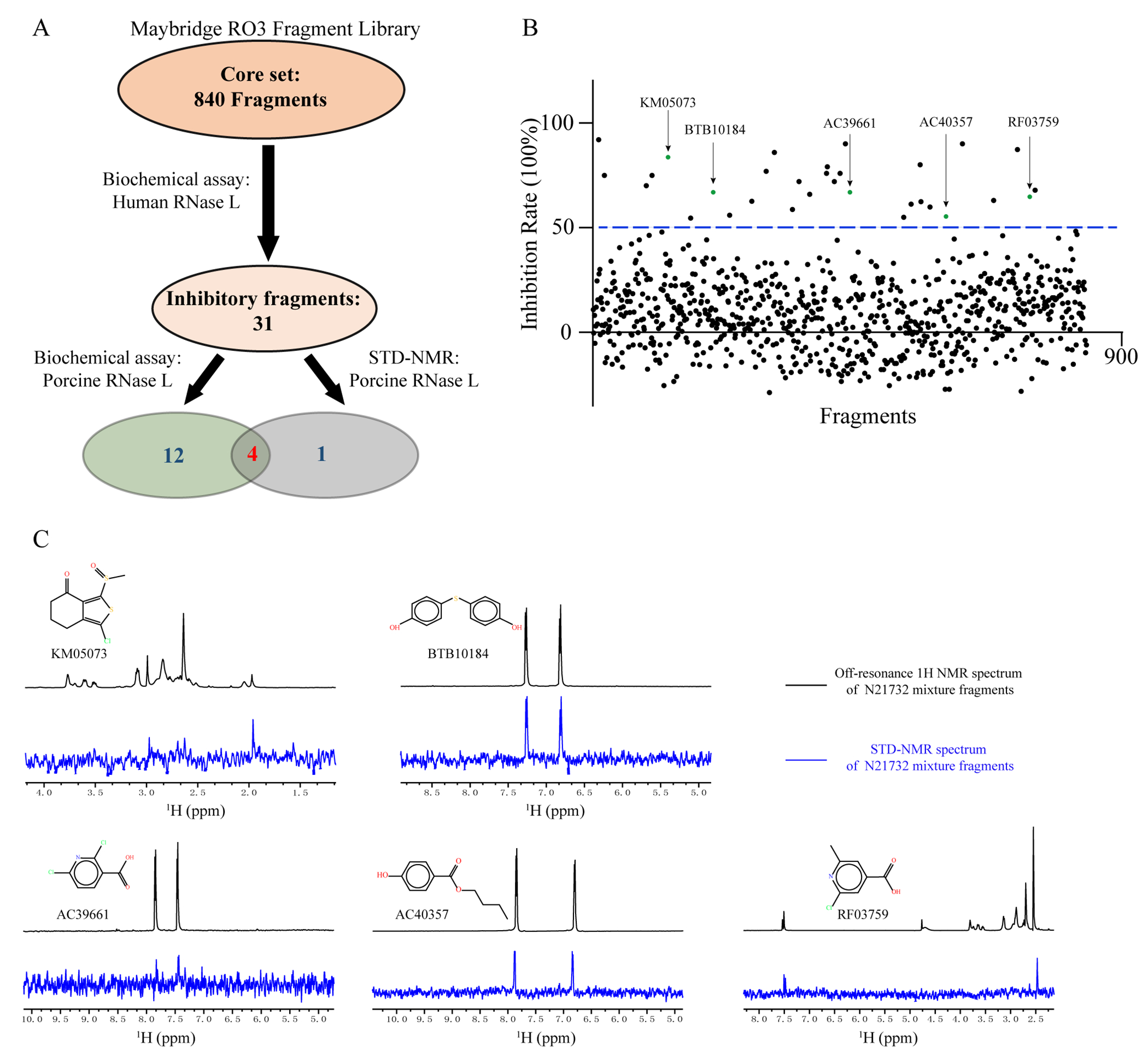
Screening cascade against RNase L using biochemical and STD-NMR assays. (A) A fragment library was first screened on H-RNase L. Thirty-one positive hits were further tested for potency and binding against the P-RNase L, because it is prone to crystallization. Venn diagram showed the numbers of fragment hits identified at each stage. (B) The screening of a fragment library of 840 compounds against H-RNase L using an in vitro FRET-based assay. Five green dots represent the STD-positive fragments identified in (C). (C) STD-NMR binding analysis of P-RNase L21-732 (N21732) and fragments. The top spectrum is the 1H-NMR of P-RNase L (10 μM) in mixture with each fragment hit (400 μM), while the bottom spectrum is the STD-NMR of the same sample. The chemical structures of fragments are shown.
Twelve of 31 fragments showed inhibitory effect on P-RNase L at 3 mM (Supporting Information Figure S1). Sequence and structural differences between H- and P-RNase L may explain why the remaining 19 fragments did not inhibit P-RNase L. Ligand-based STD-NMR, a simple and reliable method for ligand screening and characterization of protein binding with no limits on the size of the protein,29, 30 has been previously employed to confirm direct binding of VAL to RNase L.20 We observed that five fragments produced positive signals in STD-NMR (Figure 2C). Among these five fragments, four showed inhibitory activity against both H- and P-RNase L, and one fragment, AC40357, inhibited only H-RNase L (Figure 2 and Supporting Information Figure S1). However, AC40357 is able to bind to P-RNase L with a positive STD-NMR peak (Figure 2C and Supporting Information Table S1). Thus, these five fragment hits were selected for subsequent structural studies: KM05073, BTB10184, AC39661, AC40357 and RF03759 (Figure 2C and Supporting Information Figure S1).
In parallel, the half maximal inhibitory concentration (IC50) of these fragments was determined for both H- and P-RNase L (Supporting Information Table S1). The ligand efficiencies (LE) of seven fragments are greater than 0.3 on both H- and P-RNase L. All 13 fragments displayed IC50 values in the high micromolar to low millimolar range (100 - 3000 μM).
Structures of P-RNase L in complex with fragments.
To elucidate the mechanistic details of the inhibition of RNase L by fragments, the P-RNase L apo crystals were soaked with each of the above 5 fragments. The co-crystal structures of RNase L with fragment (AC40357 or KM05073) were solved at a resolution of 2.6 Å in the P212121 space group with a homodimer in each asymmetric unit (Figure 3, Figure S2 and Table 1). The fragment omit maps were shown in Supporting Information Figure S3A and S3B). In each structure, the fragment resides in the ATP-binding pocket of the PK domain (Figure 3 and Figure S2), as was previously shown for sunitinib.26 Although AC40357 and KM05073 occupy the same ATP pocket, these two fragments have different interaction modes with RNase L. AC40357 forms direct hydrogen bonds with the side chain of D500 and the backbone NH of C435 (Figure 3B and Supporting Information Figure S3D). The hydroxyl group of AC40357 also forms water-bridged hydrogen bonds with residues E402 and D500. In addition, AC40357 forms hydrophobic interactions with surrounding amino acids I369, A370, V416, L432, A433, L434, and L489. As shown in Supporting Information Figure S3B, the chlorine (Cl) atom of KM05073 forms a halogen bond with the backbone carbonyl oxygen of C435 in the hinge region of PK. The carbonyl and methyl-sulfinyl oxygens of KM07053 form three water-mediated hydrogen bonds with residues E402, A433, and D500, respectively. KM05073 additionally forms an H-π stacking interaction with L489 (Supporting Information Figure S3C). Nonspecific van der Waals interactions are formed between KM05073 and I369, A370, L432, A433, L434 and L489. Residues C435, E402 and D500, which form polar interactions with these two fragments, are conserved in H- and P-RNase L sequences (Supporting Information Figure S4). Similar to our previous study,26 these two fragments targeting the PK domain of RNase L could allosterically modulate the ribonuclease activity of RNase L.
Figure 3.
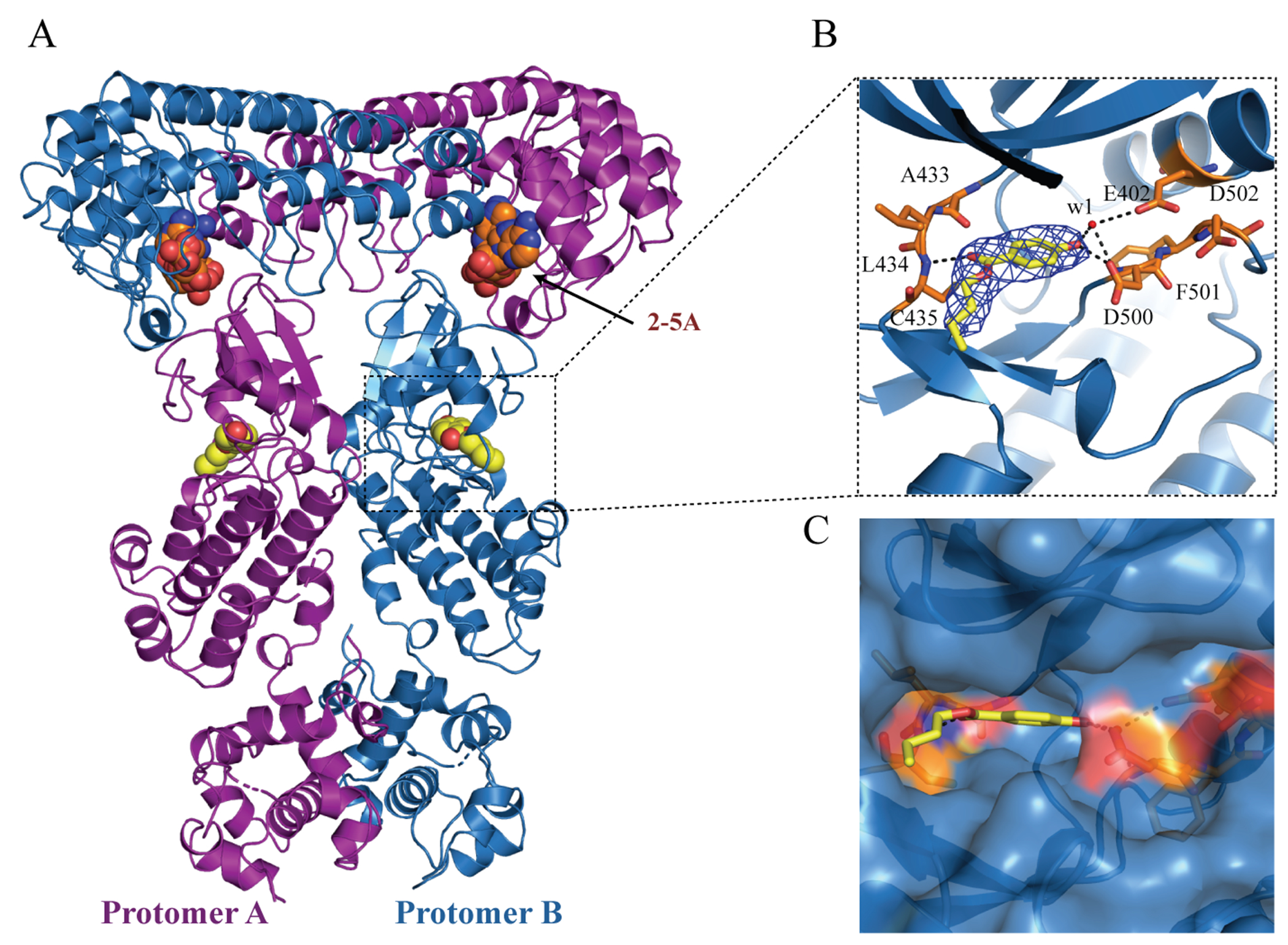
Co-crystal structure of the dimeric P-RNase L in complex with the fragment AC40357. (A) The dimer structure of P-RNase L in complex with 2-5A and AC40357. AC40357 bind to the PK domain in both protomers. (B) Stick model of AC40357 in P-RNase L, where the 2Fo-Fc density map of AC40357 is shown as blue mesh and contoured at 1 σ. Hydrogen bonds are shown as dotted black lines, and water molecules as red spheres. (C) The surface model AC40357 in P-RNase L.
Table 1.
Crystallographic data collection and refinement statistics for the complex structures of porcine-RNase L and compounds
| P-RNase L-AC40357 | P-RNase L-KM05073 | P-RNase L-Myricetin | |
|---|---|---|---|
| PDB ID | 7DTS | 7DSY | 7ELW |
|
| |||
| Data collection | |||
|
| |||
| Space group | P212121 | P212121 | P212121 |
| Cell dimensions | |||
| a, b, c (Å) | 59.01 111.28 264.09 | 59.32 111.05 262.83 | 59.61 111.93 264.93 |
| α, β, γ (°) | 90 90 90 | 90 90 90 | 90 90 90 |
| Wavelength (Å) | 0.9792 | 0.9792 | 1.54 |
| Resolution range (Å) | 30.36 - 2.63 (2.724 - 2.63) | 31.5 - 2.65 (2.746 - 2.651) | 22.92 - 3.55 (3.676 - 3.55) |
| Rsym or Rmerge | 0.08199 (1.297) | 0.06505 (0.9465) | 0.1096 (0.249) |
| I / s (I) | 15.02 (1.45) | 16.18 (1.27) | 13.03 (5.84) |
| Completeness (%) | 99.52 (99.94) | 99.55 (98.60) | 99.30 (99.86) |
|
| |||
| Refinement | |||
|
| |||
| Resolution (Å) | 2.63 | 2.65 | 3.55 |
| No. reflections | 52595 (5182) | 51275 (4987) | 22127 (2166) |
| Rwork / Rfree | 0.192 / 0.271 | 0.199/0.270 | 0.241/0.280 |
| No. atoms | 10992 | 10986 | 10909 |
| Protein residues | 1352 | 1348 | 1351 |
| Water | 98 | 113 | 0 |
| Favored (%) | 95.13 | 95.79 | 96.17 |
| Allowed (%) | 4.65 | 3.98 | 3.75 |
| Outlier (%) | 0.22 | 0.23 | 0.08 |
| Average B-factors | 99.39 | 103.09 | 72.17 |
| Bond lengths (Å) | 0.011 | 0.010 | 0.002 |
| Bond angles (°) | 1.63 | 1.56 | 0.44 |
Myricetin and hyperoside, derivatives of AC40357, showed potent inhibitory effects on RNase L.
AC40357 showed a low millimolar IC50 against H-RNase L and it has a similar para-hydroxyl benzoyl scaffold with flavonoid compounds (Figure 4A). In a previous study, it has been reported that some flavonoid compounds can inhibit H-RNase L.25 Thus, we measured the RNase L inhibitory activity of four previously unreported flavonoids which contained the major part of the scaffold of fragment AC40357 (shown in blue) (Figure 4A–C and Supporting Information Table S2). Two of these four compounds (myricetin and hyperoside) inhibited both H- and P-RNase L in vitro. Hyperoside is 200-fold more potent than myricetin against H-RNase L (Figure 4B and 4C). In particular, hyperoside showed an IC50 value of 1.63 μM, which resulted in a 1500-fold increase of potency compared with the parent fragment AC40357 against H-RNase L (Supporting Information Table S1 and Figure 4C). Of the other two flavonoids, phloretin inhibited H-RNase L, while liquiritin did not (Supporting Information Table 2).
Figure 4.
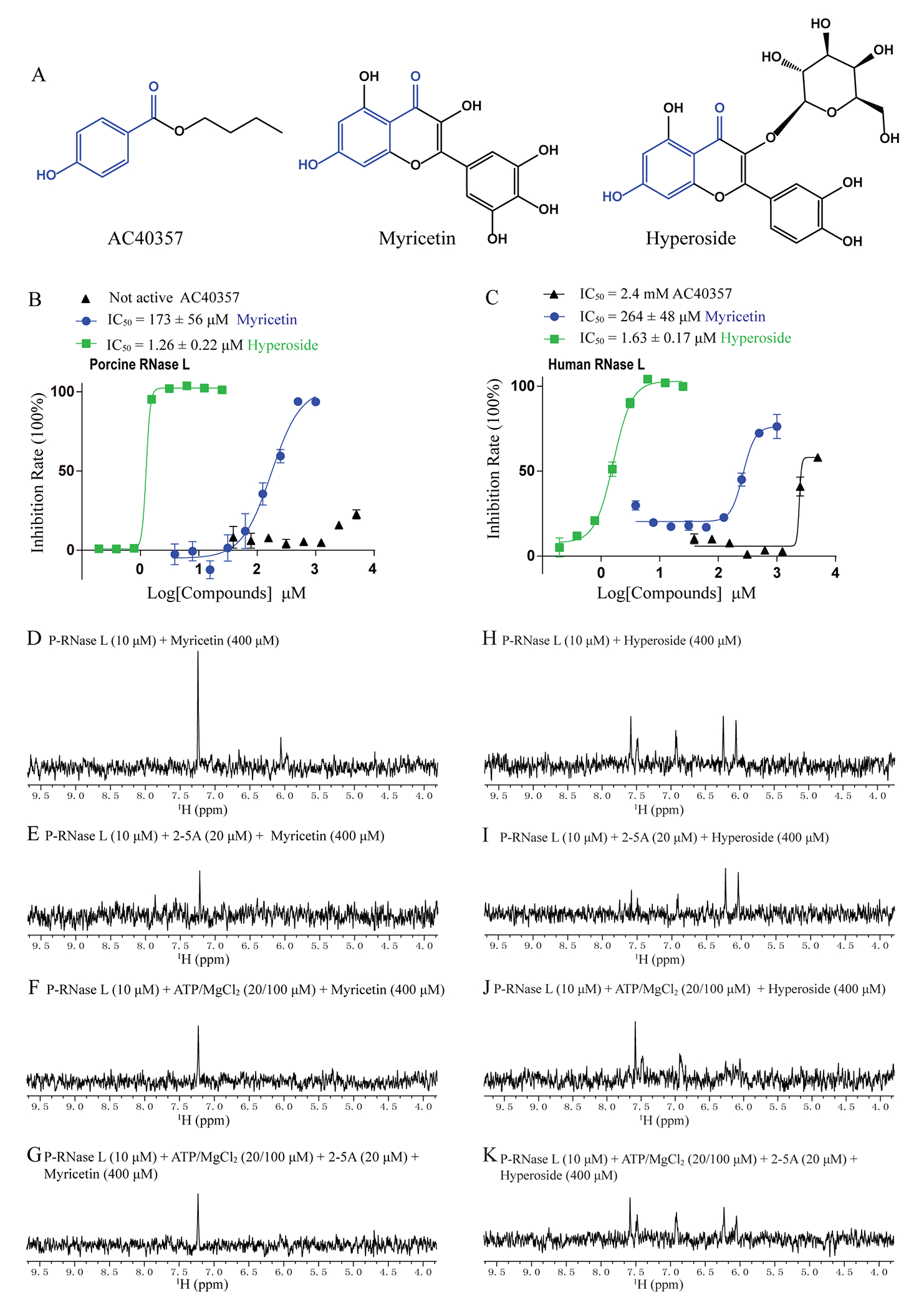
Myricetin and hyperoside exhibited potent inhibitory effects on both H- and P-RNase L. (A) Chemical structures of AC40357, myricetin and hyperoside. The blue scaffold is the same in these three compounds. (B, C) The potency of compounds against both H- and P-RNase L. (D-K) The STD-NMR showed the binding of myricetin and hyperoside to P-RNase L, in the presence of the indicated concentrations of protein, 2-5A and ATP/MgCl2.
Furthermore, six myricetin natural product derivatives were tested for their potency against both H- and P-RNase L (supporting information Figure S5). Among these compounds, vitexin showed in vitro inhibitory activities of 109 μM and 190 μM against P- and H-RNase L, respectively. This potency is similar to myricetin but other compounds were inactive. Although the chemical structure of morin is highly similar to myricetin, it is not active against RNase L highlighting the importance of positions of the hydroxy groups on C2-phenyl ring.
Three different concentrations of RNA substrate (50 nM, 100 nM and 200 nM) were used to confirm that the inhibition by hyperoside is not an artifact (Supporting Information Figure S6).
Ligand based STD-NMR was used to establish that the binding of myricetin or hyperoside to RNase L occurred in either the presence or absence of 2-5A and/or ATP/MgCl2. The STD-NMR spectra showed that myricetin or hyperoside directly interacts with monomeric RNase L in the absence of 2-5A (Figure 4D and 4H). It is known that 2-5A binds to the ANK domain with picomolar affinity,31 and induces the dimerization of RNase L. In the samples with 2-5A in molar excess to RNase L, myricetin and hyperoside also showed positive STD signal, which indicates that they can bind to the RNase L dimer (Figure 4E and 4I). Upon the addition of ATP/MgCl2 or the mixture of 2-5A and ATP/MgCl2, in molar excess to RNase L, the STD experiments remained positive indicating the binding of compounds to RNase L (Figure 4F, 4G, 4J and 4K). The STD-NMR tests also showed that vitexin directly binds to P-RNase L (supporting information Figure S7).
Crystal Structure of P-RNase L with myricetin.
To understand the mechanism by which myricetin inhibits RNase L, and to aid future inhibitor design efforts, we obtained crystal structure of P-RNase L with myricetin. The structure is a dimeric RNase L, and the statistics are shown in Table 1. The omit Fo - Fc density map of myricetin before refinement are shown in Supporting Information Figure S8A. Myricetin, like AC40357, bound to the ATP-binding pocket of each PK domain with a similar binding pose to AC40357 (Figure 5A and 5C). Figure 5B showed polar interactions of myricetin with RNase L and the 2Fo - Fc electron density map of myricetin contoured at 1.5 σ. Myricetin formed one hydrogen bond with the side chain of D500 in the DFD (Asp-Phe-Asp) motif and two hydrogen bonds with the side chain of E441 in the C-lobe of the PK domain. In addition, myricetin also formed three hydrogen bonds with the main-chain of A433 and C435 in the hinge region of the PK domain. By comparing the RNase L/myricetin structure with the RNase L/AMP-PNP structure (PDB 4O1P), the chromen-4-one group of myricetin occupies the position of the adenine group of AMP-PNP, which is similar to sunitinib (PDB 6M11)26 (Supporting Information Figure S8B). Same as AC40357, the binding of myricetin also caused a slight swing of the side chain of D500 in comparation with the structure of AMP-PNP bound RNase L (PDB 4O1P)6 (Supporting Information Figure S8C).
Figure 5.
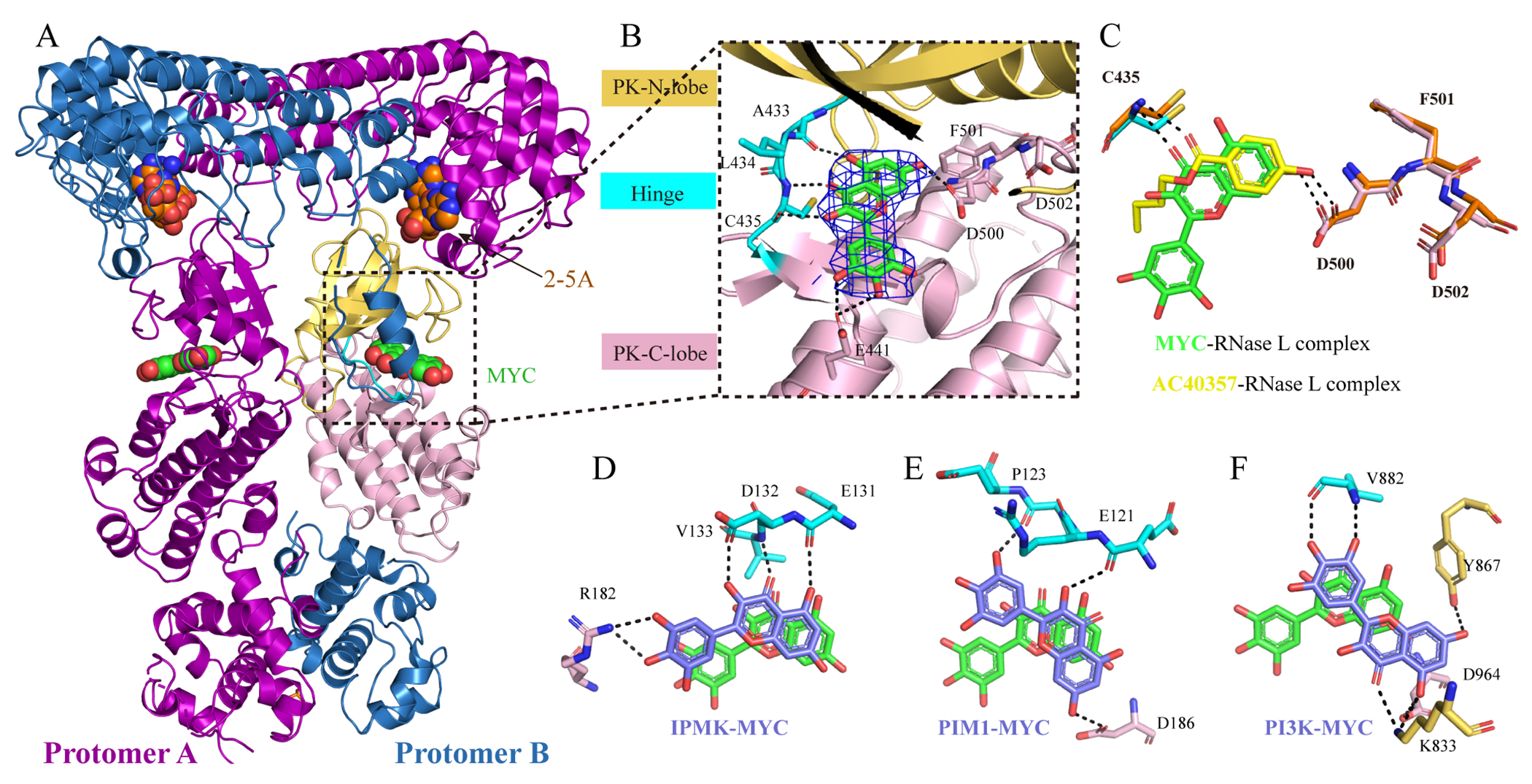
The co-crystal structure of P-RNase L with myricetin (MYC). (A) The three-dimensional structure of 2-5A and myricetin bound to the P-RNase L dimer. (B) The stick model of myricetin in the ATP-binding pocket in P-RNase L. The electron density map (2Fo-Fc) of myricetin in blue mesh and contoured at 1.5 σ. Hydrogen bonds were shown in dotted black lines. The C- and N-lobes of PK domain of P-RNase L are in yellow and pink, respectively; the hinge region is in cyan and myricetin is shown as a green stick model with oxygen atoms in red. (C) The same pose was taken by AC40357 and myricetin in the ATP-binding pocket of P-RNase L. (D-F) Comparison of the relative orientations of myricetin in different kinases. Myricetin (green) in the PK domain of P-RNase L was superimposed onto the following kinases: IPMK ((D), PDB 6M88), PIM1 ((E), PDB 2O63), and PI3K ((F), PDB 1E90), where myricetin is shown in slate blue. Black dotted lines represent hydrogen bond interactions. Residues in the C-lobe, N-lobe and hinge region are in yellow, pink and cyan, respectively. MYC: myricetin.
In comparison with available co-structures of myricetin in other kinases, such as inositol polyphosphate multikinase (IPMK),32 serine/threonine-protein kinase pim-1 (PIM1)33 and phosphoinositide 3-kinase (PI3K),34 the orientation of myricetin in RNase L is the same as in IPMK (Figure 5D). In both of these two structures, the 3,5,7-trihydroxy-4H-chromen-4-one moiety of myricetin makes hydrogen bonds with respective hinge regions and the benzene-1,2,3-triol group form polar interactions with the C-lobe of PK. Different from the above two kinases, the position of myricetin in PIM1 rotated approximately 40 degrees compared with in RNase L (Figure 5E). Compared with the orientation of myricetin in RNase L, myricetin in PI3K showed a flipped orientation such that its keto group is now pointing away from the hinge region (Figure 5F).
Hyperoside is a potent inhibitor of H- and P-RNase L
Ligand based STD-NMR confirmed the binding of hyperoside to P-RNase L (Figure 4H and 4I). However, our attempts at determining the co-crystal structure of RNase L with hyperoside were not successful. In supporting information Figure S5, it was shown that hyperoside binding did not compete with RNA substrate. Then we investigated if hyperoside would compete with 2-5A binding in the ANK domain. The IC50 of hyperoside against H- and P-RNase L were determined under three different concentrations of 2-5A (1, 5 and 20 nM) (Figure 6A and 6B). The results showed that 2-5A does not seem to have an effect on the binding of hyperoside to either H- or P-RNase L (Figure 6A and 6B). Next, the effect of hyperoside on 2-5A induced RNase L dimerization and oligomerization was tested using in vitro cross-linking assay. In Figure 6C, it was shown that 40 μM hyperoside did not suppress the dimerization induced by 2-5A at concentrations up to 2 μM; supporting information Figure S9A, shows a lack of suppression of dimerization in a hyperoside concentration gradient. Our previous study showed mutants K164E and R353E reduced the binding of 2-5A to P-RNase L and partially impaired its enzyme activity 6. Thus, we compared the inhibitory activity of hyperoside on wild type (wt-) P-RNase L to its mutants K164E and R353E. Hyperoside displayed similar potency against wt-RNase L to these two mutants (supporting information Figure S9B, wt-RNase L IC50 = 0.90 μM, K164E IC50 = 1.02 μM, R353E IC50 = 0.65 μM). Together, these studies clearly showed that hyperoside inhibits RNase L independently on 2-5A binding.
Figure 6.
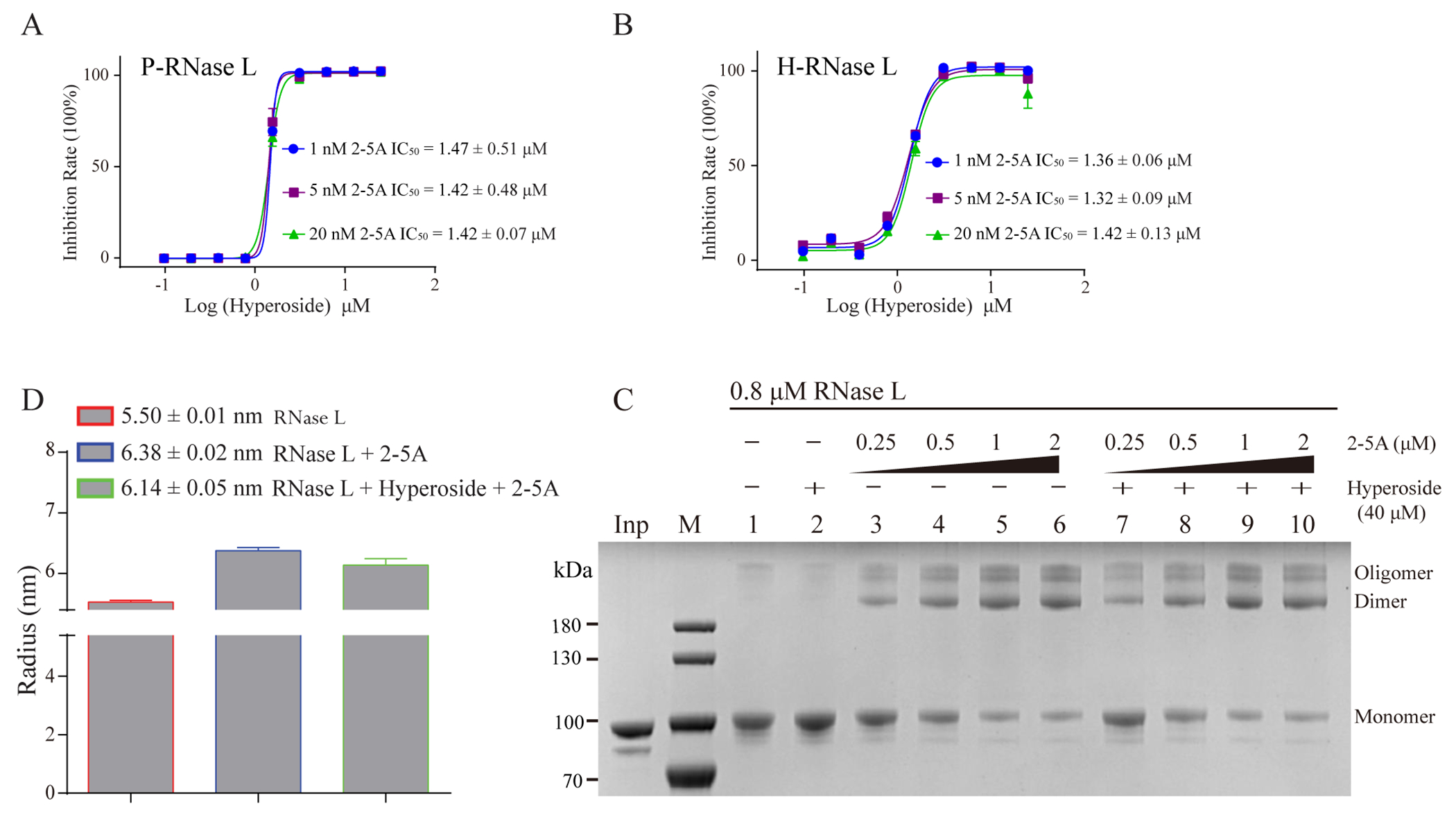
Hyperoside is an inhibitor of H- and P-RNase L. (A and B) 2-5A does not affect the potency of hyperoside against P- or H-RNase L. IC50 values represent the mean ± SEM of three inhibition profiles. (C) Hyperoside did not affect 2-5A induced RNase L oligomerization. (D) Hyperoside had a minimal effect on 2-5A-induced P-RNase L dimerization as determined by DLS.
Because sunitinib can destabilize the RNase L dimer in solution state,26 we performed Dynamic Light Scattering (DLS) experiments for P-RNase L in presence of hyperoside. Even though a slight decrease of the radius of RNase L dimer was observed for hyperoside (Figure 6D), there was no effect of hyperoside on dimerization in response to 2-5A as determined by protein-protein crosslinking with Dimethyl Suberimidate (DMS) (Figure 6C and Supporting Information Figure S9A). Therefore, we speculate that the inhibitory mechanism of hyperoside may be different from sunitinib.
The inhibition effect of compounds against H-RNase L in intact cells
To evaluate and compare the potency of valoneic acid dilactone (VAL), hyperoside, myricetin and vitexin against RNase L at the cellular level, we determined effects of these inhibitors on RNase L mediated rRNA cleavage in the human lung cancer cell line A549. Cells were pre-incubated with inhibitors for four hours, and then transfected for two hours with purified trimeric 2-5A (p35’A2’p5’A2’p5’A) to activate RNase L in the cells. RNase L activity was detected by monitoring highly specific and characteristic cleavages of rRNA in intact ribosomes.35 The RNase L inhibitor, VAL, inhibited RNase L activity at 10 to 30 μM (Figure 7A, compare lane 2 to lanes 7&11). Inhibition was complete at 30 μM of VAL. Myricetin showed identical inhibitory activity to that of VAL (Figure 7A, compare lanes 7&9, and 11&13). However, hyperoside and vitexin did not show any inhibitory activity against RNase L in intact cells (Figure 7, see lanes 16&18). Possible factors which may hinder hyperoside and vitexin inhibition function at the cellular level include protein binding specificity, metabolic stability, membrane permeability and efflux characteristics.
Figure 7.
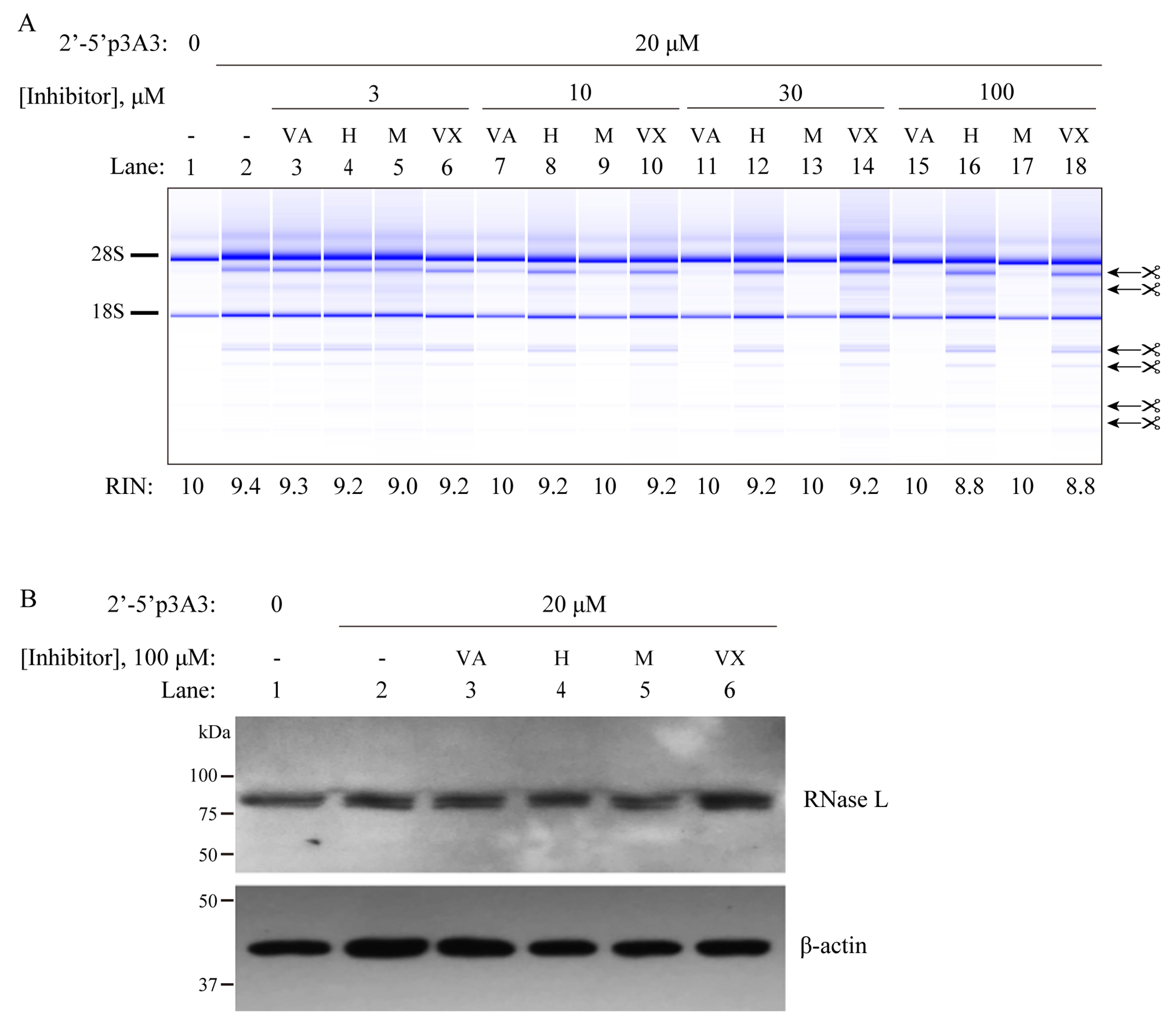
VAL and myricetin, but not hyperoside or vitexin, inhibit RNase L when added to intact A549 cells. (A) The cells were cultured in the presence of inhibitors for four hours as indicated. Subsequently, cells were transfected with 20 μM of trimeric 2-5A for two hours. Total RNA was isolated and separated on RNA chips (Agilent) to monitor RNase L dependent rRNA cleavage products (arrows). Positions of 28S and 18S rRNA are indicated. (−) untreated cells, (VA) valoneic acid dilactone; H (hyperoside), M (myricetin); VX (vitexin). (B) Western blot for RNase L and beta-actin in identically treated cells (with 100 μM of inhibitors). The experiment was done twice with identical results.
To rule out effects of the inhibitors on RNase L stability, western blotting was done (Figure 7B). Cells incubated in presence or absence of 100 μM of the inhibitors for four hours were then transfected with trimeric 2-5A (20 μM) for an additional two hours. No apparent degradation of RNase L in response to the inhibitors was observed. These finding show that VAL and myricetin are equally potent inhibitors of RNase L when added to intact cells, and that the inhibition is not caused by proteolysis of RNase L.
DISCUSSION AND CONCLUSIONS
RNase L participates in the regulation of various cellular activities in human cells.5 Studies have shown that targeted inhibition of RNase L activity has therapeutic potential for some diseases, such as AGS,17, 20 and for the promotion of oncolytic viral therapy.22 To date, only a few small molecule inhibitors have been reported to inhibit RNase L. Therefore, there is a need to develop novel inhibitors of RNase L to further evaluate their therapeutic potential in certain pathological conditions.
Myricetin, vitexin and hyperoside are flavonoids which belong to polyphenolic compounds. Myricetin is commonly found in different plant-based dietary agents, such as berries, fruits, vegetables, walnuts, red wine and teas.36 Vitexin is an active component in traditional Chinese medicine and usually found in many medicinal plants.37 Hyperoside is often isolated from various medicinal plants, for example Hypericum perforatum38 and Crataegus davisii.39 Many pharmacological studies reported that myricetin, vitexin and hyperoside can be used in anti-inflammatory responses, cardioprotective, antiviral and anti-tumor treatments.36, 37, 40 Myricetin has also been reported as an inhibitor against multiple kinases implicated in tumorigenesis.41 Furthermore, hyperoside was identified as an effective inhibitor of human Cytochrome P450 2D6 (CYP2D6) with an IC50 of 1.2 μM.42
Our co-crystal structure showed myricetin allosterically inhibited RNase L activity by binding to the ATP pocket, which is similar to sunitinib as characterized in our previous study.26 In the protein data bank (PDB), no co-crystal structures of hyperoside with any proteins was available up to June, 2021 and thus far we have been unable to determine the co-crystal structure of RNase L with hyperoside. The mechanism of action for hyperoside remains mysterious. The STD-NMR results showed that hyperoside could directly bind to both monomeric and dimeric RNase L with or without the presence of 2-5A. Furthermore, DLS experiments indicated that hyperoside did not affect the radius of particle of 2-5A induced RNase L dimer in solution state. Taken together, these results suggest that hyperoside may exert an allosteric regulation by a new mechanism distinct and different from that of sunitinib or myricetin. The detailed mechanism of how hyperoside inhibits RNase L activity will need to be elucidated in depth through crystallographic structure studies in the future, which may help develop potent inhibitors of RNase L.
Although myricetin only inhibits RNase L with sub-micromolar activity in the in vitro assay while hyperoside has a single-digit micromolar IC50 against RNase L, myricetin shows stronger inhibitory activity than hyperoside in intact cells. This may be explained by the differences in their molecular size and cell permeabilities. Also, myricetin was about 20-fold more active against H-RNase L when added to intact cells than it was in vitro (compare Figures 4C & 7A). These results suggest that a metabolite of myricetin may have a greater inhibitory activity against RNase L than myricetin itself. Future studies aimed at identifying the active metabolite of myricetin could lead to a better understanding of the mode of RNase L inhibition in cells, while also informing SAR efforts aimed at generating more potent inhibitors.
Several co-crystal structures of RNase L in complex with sunitinib and its analogs (PDB: 6M11, 6M12, 6M13)26 have been reported and a mechanism of destabilization of active RNase L dimer has been previously proposed. Together with the above-mentioned co-crystal structures, our new co-crystal structures of RNase L with myricetin and two fragments provided structural basis for further structure-activity relationship studies for these allosteric inhibitors targeting the ATP-binding pocket of RNase L.
Two recent studies showed that myricetin and its derivatives could effectively inhibit the SARS-CoV-2 3CL protease in vitro43, 44 and with micromolar in cyto potency.44 It has been suggested that myricetin derivatives could possess better selectivity for their target proteins as compared with myricetin.44 Therefore, the co-crystal structure of RNase L in complex with myricetin, or with myricetin metabolites, could help with the design of more potent and selective inhibitors for RNase L.
In this study, a few low affinity compounds show differential inhibitory activities against H- or P-RNase L, such as AC40357 and phloretin, this may be caused by protein instability and compound solubility. However, the corresponding high affinity compounds, e.g. myricetin and hyperoside, have shown consistent inhibition against both H- and P-RNase L.
The similar chemical scaffold between myricetin (and hyperoside) and the fragment AC40357 led us to identify three novel flavonoid inhibitors for RNase L. Similarly, natural product-derived fragments have been successfully applied to the development of p38a MAP kinase and phosphatase inhibitors.45 Therefore, our study provides another example that small molecule fragments derived from natural products can serve in FBDD as starting points for further drug optimization.
EXPERIMENTAL SECTION
Expression plasmid and preparation of proteins
The vector pGEX-2T-H-RNase L was used for the expression of full-length H-RNase L (residues 1-741) as previously reported.26 The plasmids containing full-length and truncated P-RNase L protein constructs (residues 1-743 for biochemical assay, residues 21-732 for structural and NMR experiments) were previously described.6, 26 The P-RNase L mutations, K164E and R353E, were constructed using a QuikChange site-directed mutagenesis kit and all plasmids are verified by sequencing.
Proteins including H-RNase L, P-RNase L and truncated P-RNase L21-732 were expressed and purified as described.26 Briefly, the plasmids were transformed into E. coli BL21 (DE3) (CAT#: EC1003, Weidi Biotech) and then the cell colonies were picked and grown in TB medium at 37 °C. When the bacterial density OD600 reached 0.8, 0.4 mM of isopropyl-β-D-1-thiogalactopyranoside (IPTG, CAT# IB0168, Sangon Biotech) was added to induce protein expression. After induction for 16-18 hours at 18 °C, the bacterial cells were centrifuged. The collected cell pellets were re-suspended in lysis buffer and lysed by sonication and the recombinant proteins were subsequently purified with affinity chromatography using Glutathione Sepharose™ 4B. The eluted proteins were further purified by size exclusion chromatography using a HiLoad 16/600 Superdex 200 column (CAT# 28-9893-35, GE Healthcare). The proteins were analyzed by SDS-PAGE and Coomassie brilliant blue staining. H-RNase L in buffer A (30 mM HEPES pH 7.8, 100 mM NaCl, 5 mM DTT, 2 mM MgCl2, and 10% glycerol) was used for biochemical enzyme assays. P-RNase L and P-RNase L21-732 in buffer B (30 mM HEPES pH 7.8, 100 mM NaCl, 5 mM DTT, and 2 mM MgCl2) were used for biochemical assay, NMR, and X-ray crystallography experiments.
Trimeric 2-5A (p35’A2’p5’A2’p5’A) was enzymatically synthesized, HPLC purified, lyophilized and dissolved in water as described previously.46
Biochemical activity assays
A convenient and sensitive fluorescence resonance energy transfer (FRET) based assay was used to determine RNase L activity (Figure 1),27 which is briefly described as follows. The FRET RNA probe is a 5’-FAM and 3’-BHQ-1 -labeled 36-nucleotide substrate (5’ FAM-UUA-UCA-AAU-UCU-UAU-UUG-CCC-CAU-UUU-UUU-GGU-UUA-BHQ-1 3’, Takara Inc.) derived from respiratory syncytial virus genomic RNA. This probe containing multiplicity of cleavage sites (UU or UA)47, 48 for RNase L which was highly susceptible to cleavage by RNase L. RNase L activator 2-5A (2’-5’pA3; > 95% purity) was synthesized by ChemGenes (MA, USA). Recombinant RNase L was incubated with 2-5A (0.5 nM) and the FRET probe (100 nM) in cleavage buffer (25 mM Tris-HCl, pH 7.4, 100 mM KCl, 10 mM MgCl2, and 2.5 mM DTT). Samples were then incubated for 60 min at 22 °C. The fluorescence signal was measured by excitation at 480 nm and emission at 535 nm with a 2104 EnVision Multilabel Plate Reader (PerkinElmer) at three-minute intervals. Each experiment was performed in triplicate and graphs were plotted using GraphPad Prism.
Screening of inhibitory fragments against RNase L
A Maybridge Ro3 Diversity Fragment Library, consisting of 840 compounds, was screened for inhibitory fragments against H-RNase L using a FRET-based RNase L activity assay (see above method). The screening was performed on 384-well black plates. Reaction mixtures contained 10 nM H-RNase L, 100 nM FRET RNA probe, 0.5 nM 2-5A, and 3.0 mM fragments in a buffer with 25 mM Tris pH 7.4, 100 mM KCl, 10 mM MgCl2 and 2.5 mM DTT in a final volume of 40 μL per well. Then, each fragment from a 200 mM stock in d6-DMSO was added, mixed, and incubated at room temperature for 30 min. Sunitinib (100 μM), which was reported as a H-RNase L inhibitor,25, 26 was used as a positive control in every screening plate. The fluorescence signal was measured using a 2104 EnVision Multilabel Plate Reader (PerkinElmer).
The half maximal inhibitory concentration (IC50) determination for compounds
The IC50 of fragments and flavonoid derivatives was measured by incubating at different concentrations of compounds with proteins in 25 mM Tris pH 7.4, 100 mM KCl, 10 mM MgCl2, 2.5 mM DTT and 0.5 nM 2-5A for 10 min on ice. FRET RNA probe was then added into the above mixture with a final concentration of 10 nM H-RNase L or 2.5 nM P-RNase L for 1 h at room temperature. Each experiment was performed in triplicate and graphs were plotted using GraphPad Prism. All compounds in this study are from commercial sources and analyzed by HPLC, their purities were > 95%.
Compounds binding validation using Nuclear Magnetic Resonance (NMR)
The positive hits against H-RNase L were prepared in cocktails containing three or four fragments (400 μM each) without overlapping chemical shifts, which were then incubated with 10 μM of protein (P-RNase L21-732) in solution containing 20 mM sodium phosphate, 100 mM NaCl, 2 mM DTT, pH 7.5 and 5% d6-DMSO. Positive hits in the cocktail were tested again using STD. STD-NMR spectra were acquired on a Bruker Avance III-600 MHz spectrometer equipped with a cryogenically cooled probe (Bruker Biospin, Germany) at 25 °C. The saturation time was set at 2 s.
Soaking of P-RNase L crystals with compounds
Purified P-RNase L21-732 was concentrated to 10-12 mg/mL with buffer (30 mM HEPES, 100 mM NaCl, 5 mM DTT, 2 mM MgCl2, pH 7.8). Apo crystals were grown in 24-well hanging-drop plates in the reservoir solution: 100 mM Tris-HCl, pH 7.0 - 8.0, 150 mM (NH4)2SO4, and 16-20% PEG4000. The crystal was grown at 20 °C in a 2 μL protein sample with an equal volume of reservoir solution. Then the crystals were soaked in drops with compounds at 20-50 mM final concentration with ~10% DMSO overnight at 20 °C. Crystals were cryo-protected using the corresponding reservoir solution plus 25% ethylene glycol and were flash-frozen in liquid N2.
X-ray data collection and structure solution and refinement
The diffraction data from crystals soaked with compounds were collected at the BL17U1 beamline of the Shanghai Synchrotron Radiation Facility or an in-house light source Rigaku MicroMax-007 HF and indexed, integrated, and scaled using XDS.49 The structure was solved by molecular replacement using the PHASER program,50 with a dimeric structure of RNase L (PDB ID: 6M11) in which sunitinib and 2-5A were removed as the search model. Structure refinement was performed using the CCP4 suite refmac551 or PHENIX.52 The fragment, 2-5A and water molecules were manually fitted into the initial Fo-Fc maps using the Coot graphics program.53 The MolProbity was used to evaluated structural models.54 All structure figures were prepared using PyMOL.55 Detailed statistics of the crystal diffraction data and refinement are shown in Table 1. Co-crystal structures of P-RNase L/AC40357 (7DTS), P-RNase L/KM05073 (7DSY) and P-RNase L/myricetin (7ELW) have been deposited in PDB.
RNase L oligomerization assay
Dimethyl Suberimidate (DMS) based cross-linking was used to detect 2-5A-induced RNase L dimerization and oligomerization.11, 28 Purified full-length P-RNase L (30 μL, 0.8 μM) was supplemented with 5 mM MgCl2 and incubated with or without hyperoside for 15 min on ice. Next, 2-5A was added to the mixture, and the mixture was further incubated for 15 min on ice. One microliter of 120 mg/mL DMS in 0.4 M triethanolamine hydrochloride (pH 8.5) was added to the mixture, and the mixture was then incubated at room temperature for 1 h and denatured by adding 5× loading buffer (250 mM Tris-HCl, pH 6.8, 0.2% bromophenol blue, 1% β-mercaptoethanol, 10% SDS, 50% glycerol) and heated at 95°C for 5 min. Samples were then separated by SDS-PAGE with a gradient gel (4-12%), which were subsequently stained with Coomassie blue G-250. The gels were finally photographed by ChemiDoc MP Imaging System (Biorad).
The particle size of dimeric RNase L was measured by Dynamic light scattering
A DynaPro NanoStar instrument (Wyatt Technology Corporation) was used to perform light scattering experiments at 25 °C. P-RNase L21-732 (5 μM in SEC buffer) was incubated with or without hyperoside (200 μM) for 10 min on ice. The mixture was further incubated with 10 μM 2-5A for 10 min on ice. All samples were centrifuged (15,000 × g for 10 min, 4 °C) and were tested with five replicates (20 scans/replicate). GraphPad Prism was used to plot the figure.
Determination of RNase L inhibition by compounds in intact cells
A549 cells were seeded in 24-well plates and cultured in RPMI media with 10% FBS for 16 hours. Cell were then incubated without or with 3, 10, 30 and 100 μM inhibitors in fresh media for four hours. The cells were next transfected with 20 μM of 2’-5’p3A3 (trimeric 2-5A) with lipofectamine 2000, in the continued presence of the inhibitors, and cultured for an additional two hours before isolation of total RNA. The RNA was isolated with EZ-10 Spin Columns Total RNA minipreps Super kit (BIO BASIC). Total RNA was separated on RNA chips with an Agilent Bioanalyzer 2000. Protein extracts were made from identically treated cells with non-idet P40 lysis buffer.56 Western blots were probed with monoclonal antibody against H-RNase L11 and antibody against beta-actin (Sigma-Aldrich).
Supplementary Material
ACKNOWLEDGMENTS
All NMR experiments were performed at the Beijing NMR Center and the NMR facility of National Center for Protein Sciences at Peking University. We thank the Shanghai Synchrotron Radiation Facility (SSRF) for data collection at the beamline BL-17U1. This research paper was supported by the National Natural Science Foundation of China (No. 21778008 to H.H. and 22107009 to J.T.), Shenzhen Science and Technology Program (KQTD20190929174023858 and JCYJ2020210940401752 to H.H.) and National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Awards (No. R01AI135922) to R.H.S.
ABBREVIATIONS
- AGS
Aicardi-Goutières syndrome
- IFN
interferon
- ANK
ankyrin-repeat domain
- PK
pseudokinase
- JAK-STAT
Janus kinase (JAK)-signal transducer of activators of transcription (STAT) pathway
- ADAR1
adenosine deaminase 1
- ssRNA
single-stranded RNA
- dsRNA
double-stranded RNA
- OAS
oligoadenylate synthetase
- OVs
oncolytic viruses
- VSV
vesicular stomatitis virus
- FBDD
fragment-based drug discovery
- VEGFR
vascular endothelial growth factor receptor
- PDFGR
platelet-derived growth factor receptor
- STD-NMR
saturation transfer difference-nuclear magnetic resonance
- FRET
fluorescence resonance energy transfer
- VAL
valoneic acid dilactone
- LE
ligand efficiencies
- IPMK
inositol polyphosphate multikinase
- PIM1
Serine/threonine-protein kinase pim-1
- PI3K
phosphoinositide 3-kinase
- DLS
dynamic light scattering
- DMS
Dimethyl Suberimidate
- PDB
Protein Data Bank
- CYP2D6
Cytochrome P450 2D6
Footnotes
Supporting Information
Inhibition constants (IC50) and ligand efficiencies (LE) of fragments identified in activity-based screening against H- and P-RNase L proteins; The potency of AC40357 derivatives against H- and P-RNase L; The inhibition rates of several fragments against P-RNase L; Co-crystal structure of the dimeric P-RNase L in complex with the fragment KM05073; The Fo-Fc electron density omit map of fragments and the 2D Ligand interaction diagrams; Critical residues in the fragments binding pocket are conserved in both H- and P-RNase L; The potency of six myricetin derivatives against H- and P-RNase L; RNA substrate concentration does not affect the inhibition of hyperoside against P-RNase L; STD-NMR showed vitexin directly binds to P-RNase L; The density map of myricetin and structural comparison; The potency of hyperoside was not affected by 2-5A binding
Molecular formula strings (CSV)
The authors declare no competing financial interest.
Contributor Information
Jinle Tang, State Key Laboratory of Chemical Oncogenomics, Laboratory of Structural Biology and Drug Discovery, School of Chemical Biology and Biotechnology, Peking University Shenzhen Graduate School, Shenzhen 518055, China.
Beihua Dong, Department of Cancer Biology, Lerner Research Institute, Cleveland Clinic, Cleveland, OH 44195, USA.
Ming Liu, State Key Laboratory of Chemical Oncogenomics, Laboratory of Structural Biology and Drug Discovery, School of Chemical Biology and Biotechnology, Peking University Shenzhen Graduate School, Shenzhen 518055, China.
Shuyan Liu, National Clinical Research Center for Infectious Diseases, Shenzhen Third People’s Hospital, Southern University of Science and Technology, Shenzhen 518112, China.
Xiaogang Niu, College of Chemistry and Molecular Engineering, Beijing Nuclear Magnetic Resonance Center, Peking University, Beijing 100871, China.
Christina Gaughan, Department of Cancer Biology, Lerner Research Institute, Cleveland Clinic, Cleveland, OH 44195, USA.
Abhishek Asthana, Department of Cancer Biology, Lerner Research Institute, Cleveland Clinic, Cleveland, OH 44195, USA.
Huan Zhou, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai 201204, China.
Zhengshuang Xu, State Key Laboratory of Chemical Oncogenomics, Laboratory of Structural Biology and Drug Discovery, School of Chemical Biology and Biotechnology, Peking University Shenzhen Graduate School, Shenzhen 518055, China.
Guoliang Zhang, National Clinical Research Center for Infectious Diseases, Shenzhen Third People’s Hospital, Southern University of Science and Technology, Shenzhen 518112, China.
Robert H. Silverman, Department of Cancer Biology, Lerner Research Institute, Cleveland Clinic, Cleveland, OH 44195, USA.
Hao Huang, State Key Laboratory of Chemical Oncogenomics, Laboratory of Structural Biology and Drug Discovery, School of Chemical Biology and Biotechnology, Peking University Shenzhen Graduate School, Shenzhen 518055, China.
References
- 1.Silverman RH A scientific journey through the 2-5A/RNase L system. Cytokine & growth factor reviews 2007, 18, 381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Silverman RH Viral encounters with 2′, 5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. Journal of virology 2007, 81, 12720–12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drappier M; Michiels T Inhibition of the OAS/RNase L pathway by viruses. Curr Opin Virol 2015, 15, 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chakrabarti A; Jha BK; Silverman RH New insights into the role of RNase L in innate immunity. Journal of Interferon & Cytokine Research 2011, 31, 49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gusho E; Baskar D; Banerjee S New advances in our understanding of the “unique” RNase L in host pathogen interaction and immune signaling. Cytokine 2020, 133, 153847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang H; Zeqiraj E; Dong B; Jha BK; Duffy NM; Orlicky S; Thevakumaran N; Talukdar M; Pillon MC; Ceccarelli DF Dimeric structure of pseudokinase RNase L bound to 2-5A reveals a basis for interferon-induced antiviral activity. Molecular cell 2014, 53, 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han Y; Donovan J; Rath S; Whitney G; Chitrakar A; Korennykh A Structure of human RNase L reveals the basis for regulated RNA decay in the IFN response. Science 2014, 343, 1244–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stark GR; Kerr IM; Williams BR; Silverman RH; Schreiber RD How cells respond to interferons. Annual review of biochemistry 1998, 67, 227–264. [DOI] [PubMed] [Google Scholar]
- 9.Kerr IM; Brown RE pppA2’p5’A2’p5’A: an inhibitor of protein synthesis synthesized with an enzyme fraction from interferon-treated cells. Proceedings of the National Academy of Sciences 1978, 75, 256–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dong B; Xu L; Zhou A; Hassel BA; Lee X; Torrence PF; Silverman RH Intrinsic molecular activities of the interferon-induced 2-5A-dependent RNase. Journal of Biological Chemistry 1994, 269, 14153–14158. [PubMed] [Google Scholar]
- 11.Dong B; Silverman RH 2-5A-dependent RNase molecules dimerize during Activation by 2-5A (*). Journal of Biological Chemistry 1995, 270, 4133–4137. [DOI] [PubMed] [Google Scholar]
- 12.Castelli JC; Hassel BA; Wood KA; Li X-L; Amemiya K; Dalakas MC; Torrence PF; Youle RJ A study of the interferon antiviral mechanism: apoptosis activation by the 2–5A system. The Journal of experimental medicine 1997, 186, 967–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malathi K; Dong B; Gale M; Silverman RH Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 2007, 448, 816–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chakrabarti A; Ghosh PK; Banerjee S; Gaughan C; Silverman RH RNase L triggers autophagy in response to viral infections. Journal of virology 2012, 86, 11311–11321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X-L; Ezelle HJ; Kang T-J; Zhang L; Shirey KA; Harro J; Hasday JD; Mohapatra SK; Crasta OR; Vogel SN An essential role for the antiviral endoribonuclease, RNase-L, in antibacterial immunity. Proceedings of the National Academy of Sciences 2008, 105, 20816–20821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Long TM; Chakrabarti A; Ezelle HJ; Brennan-Laun SE; Raufman J-P; Polyakova I; Silverman RH; Hassel BA RNase-L deficiency exacerbates experimental colitis and colitis-associated cancer. Inflammatory bowel diseases 2013, 19, 1295–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y; Banerjee S; Goldstein SA; Dong B; Gaughan C; Rath S; Donovan J; Korennykh A; Silverman RH; Weiss SR Ribonuclease L mediates the cell-lethal phenotype of double-stranded RNA editing enzyme ADAR1 deficiency in a human cell line. Elife 2017, 6, e25687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tomaselli S; Galeano F; Locatelli F; Gallo A ADARs and the balance game between virus infection and innate immune cell response. Current Issues in Molecular Biology 2015, 17, 37–52. [PubMed] [Google Scholar]
- 19.Crow YJ; Shetty J; Livingston JH Treatments in Aicardi–Goutières syndrome. Developmental Medicine & Child Neurology 2020, 62, 42–47. [DOI] [PubMed] [Google Scholar]
- 20.Daou S; Talukdar M; Tang J; Dong B; Banerjee S; Li Y; Duffy NM; Ogunjimi AA; Gaughan C; Jha BK; Gish G; Tavernier N; Mao D; Weiss SR; Huang H; Silverman RH; Sicheri F A phenolic small molecule inhibitor of RNase L prevents cell death from ADAR1 deficiency. Proc Natl Acad Sci U S A 2020, 117, 24802–24812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fukuhara H; Ino Y; Todo T Oncolytic virus therapy: a new era of cancer treatment at dawn. Cancer science 2016, 107, 1373–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jha BK; Dong B; Nguyen CT; Polyakova I; Silverman RH Suppression of antiviral innate immunity by sunitinib enhances oncolytic virotherapy. Molecular Therapy 2013, 21, 1749–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chakrabarti A; Banerjee S; Franchi L; Loo Y-M; Gale M Jr; Núñez G; Silverman RH RNase L activates the NLRP3 inflammasome during viral infections. Cell host & microbe 2015, 17, 466–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma Q; Li J; Zhou H; Tong W; Chen Y The function of RNase L and its degradation mechanism in cardiac acute ischemic injury. Apoptosis 2020, 25, 400–411. [DOI] [PubMed] [Google Scholar]
- 25.Jha BK; Polyakova I; Kessler P; Dong B; Dickerman B; Sen GC; Silverman RH Inhibition of RNase L and RNA-dependent protein kinase (PKR) by sunitinib impairs antiviral innate immunity. Journal of Biological Chemistry 2011, 286, 26319–26326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang J; Wang Y; Zhou H; Ye Y; Talukdar M; Fu Z; Liu Z; Li J; Neculai D; Gao J; Huang H Sunitinib inhibits RNase L by destabilizing its active dimer conformation. Biochem J 2020, 477, 3387–3399. [DOI] [PubMed] [Google Scholar]
- 27.Thakur CS; Xu Z; Wang Z; Novince Z; Silverman RH A convenient and sensitive fluorescence resonance energy transfer assay for RNase L and 2′, 5′ oligoadenylates. In Interferon Methods and Protocols, Springer: 2005; pp 103–113. [DOI] [PubMed] [Google Scholar]
- 28.Thakur CS; Jha BK; Dong B; Gupta JD; Silverman KM; Mao H; Sawai H; Nakamura AO; Banerjee AK; Gudkov A Small-molecule activators of RNase L with broad-spectrum antiviral activity. Proceedings of the National Academy of Sciences 2007, 104, 9585–9590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Viegas A; Manso J; Nobrega FL; Cabrita EJ Saturation-transfer difference (STD) NMR: a simple and fast method for ligand screening and characterization of protein binding. Journal of chemical Education 2011, 88, 990–994. [Google Scholar]
- 30.Angulo J; Nieto PM STD-NMR: application to transient interactions between biomolecules—a quantitative approach. European biophysics journal 2011, 40, 1357–1369. [DOI] [PubMed] [Google Scholar]
- 31.Silverman RH; Jung DD; Nolan-Sorden NL; Dieffenbach CW; Kedar VP; SenGupta DN Purification and analysis of murine 2-5A-dependent RNase. Journal of Biological Chemistry 1988, 263, 7336–7341. [PubMed] [Google Scholar]
- 32.Gu C; Stashko MA; Puhl-Rubio AC; Chakraborty M; Chakraborty A; Frye SV; Pearce KH; Wang X; Shears SB; Wang H Inhibition of inositol polyphosphate kinases by quercetin and related flavonoids: a structure-activity analysis. J Med Chem 2019, 62, 1443–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holder S; Zemskova M; Zhang C; Tabrizizad M; Bremer R; Neidigh JW; Lilly MB Characterization of a potent and selective small-molecule inhibitor of the PIM1 kinase. Mol Cancer Ther 2007, 6, 163–172. [DOI] [PubMed] [Google Scholar]
- 34.Walker EH; Pacold ME; Perisic O; Stephens L; Hawkins PT; Wymann MP; Williams RL Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Molecular Cell 2000, 6, 909–919. [DOI] [PubMed] [Google Scholar]
- 35.Silverman RH; Skehel J; James T; Wreschner D; Kerr I rRNA cleavage as an index of ppp (A2’p) nA activity in interferon-treated encephalomyocarditis virus-infected cells. Journal of virology 1983, 46, 1051–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song X; Tan L; Wang M; Ren C; Guo C; Yang B; Ren Y; Cao Z; Li Y; Pei J Myricetin: A review of the most recent research. Biomedicine & Pharmacotherapy 2021, 134, 111017. [DOI] [PubMed] [Google Scholar]
- 37.He M; Min JW; Kong WL; He XH; Li JX; Peng BW A review on the pharmacological effects of vitexin and isovitexin. Fitoterapia 2016, 115, 74–85. [DOI] [PubMed] [Google Scholar]
- 38.Zhou TT; Chen B; Fan GR; Chai YF; Wu YT Application of high-speed counter-current chromatography coupled with high-performance liquid chromatography-diode array detection for the preparative isolation and purification of hyperoside from Hypericum perforatum with online purity monitoring. J Chromatogr A 2006, 1116, 97–101. [DOI] [PubMed] [Google Scholar]
- 39.Sözer U; Dönmez AA; Meriçli AH Constituents from the leaves of Crataegus davisii Browicz. Scientia pharmaceutica 2006, 74, 189–201. [Google Scholar]
- 40.Raza A; Xu X; Sun H; Tang J; Ouyang Z Pharmacological activities and pharmacokinetic study of hyperoside: A short review. Tropical Journal of Pharmaceutical Research 2017, 16, 483–489. [Google Scholar]
- 41.Stoll S; Bitencourt S; Laufer S; Ines Goettert M Myricetin inhibits panel of kinases implicated in tumorigenesis. Basic Clin Pharmacol Toxicol 2019, 125, 3–7. [DOI] [PubMed] [Google Scholar]
- 42.Song M; Hong M; Lee MY; Jee J-G; Lee YM; Bae J-S; Jeong TC; Lee S Selective inhibition of the cytochrome P450 isoform by hyperoside and its potent inhibition of CYP2D6. Food and chemical toxicology 2013, 59, 549–553. [DOI] [PubMed] [Google Scholar]
- 43.Gossen J; Albani S; Hanke A; Joseph BP; Bergh C; Kuzikov M; Costanzi E; Manelfi C; Storici P; Gribbon P; Beccari AR; Talarico C; Spyrakis F; Lindahl E; Zaliani A; Carloni P; Wade RC; Musiani F; Kokh DB; Rossetti G A Blueprint for High Affinity SARS-CoV-2 Mpro Inhibitors from Activity-Based Compound Library Screening Guided by Analysis of Protein Dynamics. ACS Pharmacol Transl Sci 2021, 4, 1079–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Su H; Yao S; Zhao W; Zhang Y; Liu J; Shao Q; Wang Q; Li M; Xie H; Shang W Identification of pyrogallol as a warhead in design of covalent inhibitors for the SARS-CoV-2 3CL protease. Nature Communications 2021, 12, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Over B; Wetzel S; Grütter C; Nakai Y; Renner S; Rauh D; Waldmann H Natural-product-derived fragments for fragment-based ligand discovery. Nature chemistry 2013, 5, 21–28. [DOI] [PubMed] [Google Scholar]
- 46.Asthana A; Gaughan C; Dong B; Weiss SR; Silverman RH Specificity and mechanism of coronavirus, rotavirus, and mammalian two-histidine phosphoesterases that antagonize antiviral innate immunity. Mbio 2021, 12, e01781–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wreschner DH; McCauley JW; Skehel JJ; Kerr IM Interferon action—sequence specificity of the ppp (A2′ p) n A-dependent ribonuclease. Nature 1981, 289, 414–417. [DOI] [PubMed] [Google Scholar]
- 48.Floyd-Smith G; Slattery E; Lengyel P Interferon action: RNA cleavage pattern of a (2’-5’) oligoadenylate--dependent endonuclease. Science 1981, 212, 1030–1032. [DOI] [PubMed] [Google Scholar]
- 49.Kabsch W Xds. Acta Crystallographica Section D: Biological Crystallography 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser crystallographic software. J Appl Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murshudov GN; Skubák P; Lebedev AA; Pannu NS; Steiner RA; Nicholls RA; Winn MD; Long F; Vagin AA REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallographica Section D: Biological Crystallography 2011, 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adams PD; Afonine PV; Bunkóczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung L-W; Kapral GJ; Grosse-Kunstleve RW PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica Section D: Biological Crystallography 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Emsley P; Cowtan K Coot: model-building tools for molecular graphics. Acta crystallographica section D: biological crystallography 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 54.Chen VB; Arendall WB 3rd; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.DeLano WL Pymol: An open-source molecular graphics tool. CCP4 Newsletter on protein crystallography 2002, 40, 82–92. [Google Scholar]
- 56.SILVERMAN RH; CAYLEY PJ; KNIGHT M; GILBERT CS; KERR IM Control of the ppp (A2′ p) nA system in HeLa cells: effects of interferon and virus infection. European journal of biochemistry 1982, 124, 131–138. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.