

Abstract
We engineered an ultrasensitive reporter of p16INK4a, a biomarker of cellular senescence. Our reporter detected p16INK4a-expressing fibroblasts with certain senescent characteristics that appeared shortly after birth in the basement membrane adjacent to epithelial stem cells in the lung. Furthermore, these p16INK4a+ fibroblasts had enhanced capacity to sense tissue inflammation and respond through increased secretory capacity to promote epithelial regeneration. Finally, p16INK4a expression was required in fibroblasts to enhance epithelial regeneration. This study highlights a role for p16INK4a+ fibroblasts as tissue-resident sentinels in the stem cell niche that monitor barrier integrity and rapidly respond to inflammation to promote tissue regeneration.
p16INK4a is a tumor suppressor encoded in the cyclin dependent kinase 2a (Cdkn2a) locus that is increased in cultured cells in vitro undergoing cellular senescence(1), defined as a form of irreversible cell cycle arrest often induced by stress and associated with a secretory profile(2). Mouse reporters using p16INK4a promoter to drive luciferase expression have demonstrated increased transcription of p16INK4a associated with aging and wound repair(3–5). On the basis of these and other studies quantifying p16INK4a transcripts in whole tissues, cells expressing p16INK4a (p16INK4a+) are thought to be rare or absent in young and healthy tissues. However, the use of luciferase combined with whole-body bioluminescence imaging precludes studying p16INK4a+ cells at the cellular resolution. Genetic models have demonstrated beneficial effects of killing p16INK4a+ cells in models of aging-related pathologies(6–12), but the identity and behavior of living p16INK4a+ cells in their cellular ecosystem within tissues remain largely undefined(13). Recent advances have been made using Cre to identify and delete p16INK4a+ cells, but this approach only identifies cells with high p16INK4a expression, which was rare in young adult tissue(14, 15). Although p16INK4a+ senescent cells have been described as having a rigid and non-responsive cell state(16), presence of p16INK4a+ cells in young tissue might indicate they have some normal physiological function. We examined the role and function of p16INK4a+ cells in non-aged tissue with a reporter that has improved sensitivity for p16INK4a.
High-sensitivity fluorescent reporter of p16INK4a in vivo
To build a sensitive reporter for p16INK4a+ in tissues, we constructed a bacterial artificial chromosome (BAC) in which tandem cassettes of fused Histone H2B-Green Fluorescent Protein (H2B-GFP) is expressed in frame with the p16INK4a gene product in the murine Cdkn2a locus, thus utilizing p16INK4a promoter to drive the expression of multiple copies of a stable fluorescent protein that would be incorporated into the nucleosome. The BAC was injected into mouse embryos to create a transgenic model named the INK4A H2B-GFP Reporter-In-Tandem (hereafter referred to as INKBRITE) mouse (Fig. 1A). Flow cytometry analysis of cells from lungs of healthy, young (postnatal day, PND60) INKBRITE mice showed highly fluorescent cells not observed in wild-type lungs, and sorted GFP+ cells were significantly enriched for the p16INK4a transcript detected by quantitative reverse transcription polymerase chain reaction (qPCR) (Fig. 1B). Flow cytometry with lineage markers showed that the majority of GFP+ cells were immune cells (protein tyrosine phosphatase receptor type C+, or CD45+) and fibroblasts (platelet-derived growth factor receptor alpha+, or PDGFRα+) (Fig. S1A). We failed to detect p16INK4a+ cells in the lung during embryogenesis, but p16INK4a+ cells started appearing in the basement membrane shortly after birth (Fig. S1B to E) when the oxygen environment dramatically changes in the lung prior to the onset of alveologenesis starting at PND3 through 5. INKBRITE pups (PND4) subjected to hyperoxia demonstrated a small but not statistically significant increase in GFP+ cells in the lung (Fig. S1F). Thick section images of postnatal day 60 (PND60) lungs showed nuclear GFP staining surrounded by laminin+ basement membrane beneath the airway epithelium (Fig. 1C, secretoglobin 1A1, or SCGB1A1, marks airway stem cells). We observed these p16INK4a+ cells in similar positions in other barrier organs such as small intestine, colon, and skin (Fig. S1G to I).
Figure 1. INKBRITE identifies p16INK4A+ cells with senescent characteristics in vivo.
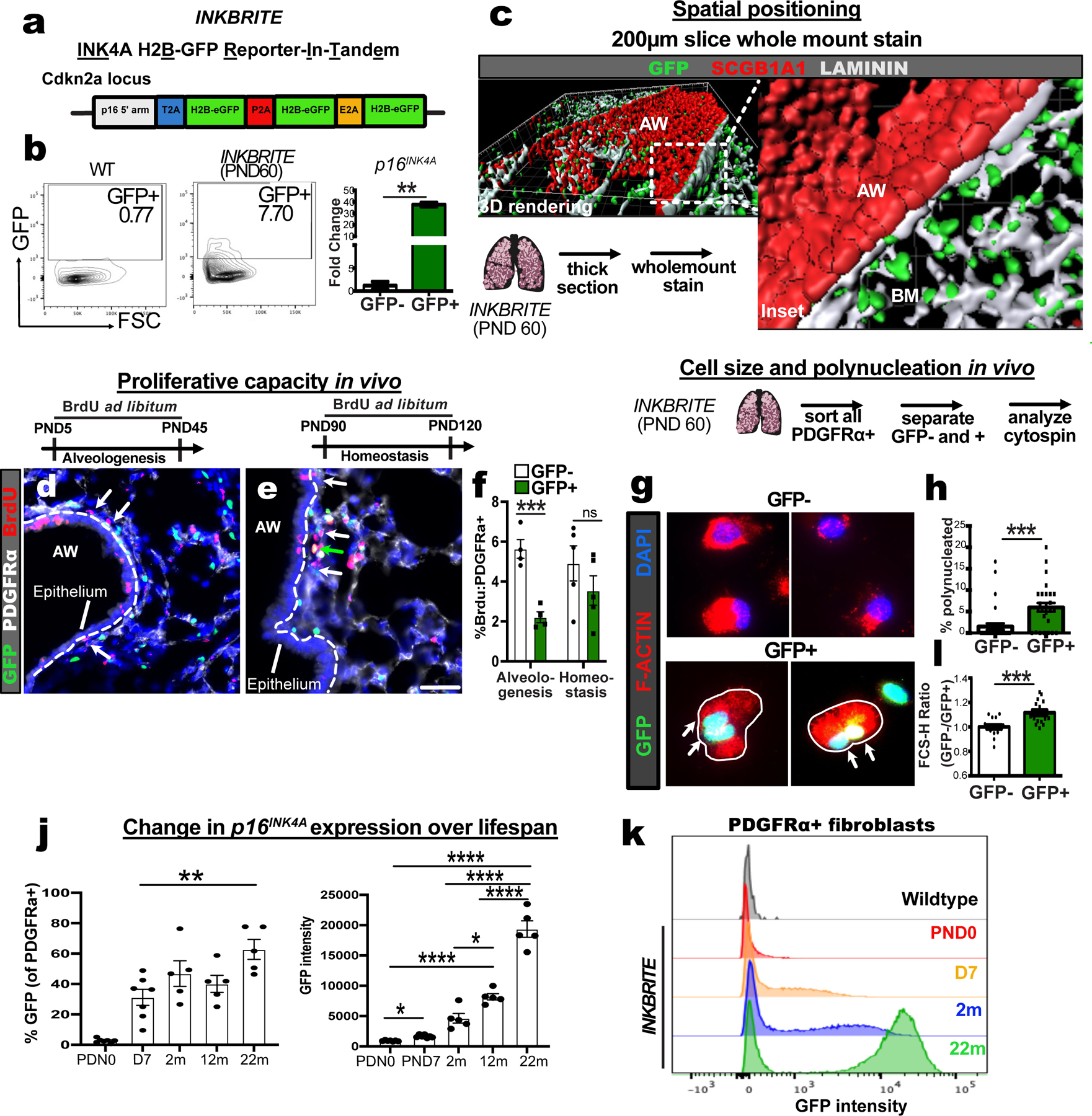
(A) Target construct design for INKBRITE. (B) FACS analysis of GFP+ cells from INKBRITE lungs and qPCR of sorted GFP+ and GFP−neg cells (n=2). (C) Wholemount image of the airway from thick-sectioned INKBRITE lung rendered on Imaris. (D to F) Immunohistochemistry (IHC) and quantification of BrdU incorporation into PDGFRα+ and GFP– (white arrows) or PDGFRα+ and GFP+ (green arrows) cells during alveologenesis or homeostasis (n=4 alveologenesis, n=5 homeostasis). (G and H) IHC and quantification of freshly sorted of GFP+ and GFP– fibroblasts for polynucleation (2 experiments, n=29–31 images per cell type). (I) FACS data with quantification of % GFP+ cells and mean GFP+ intensity of PDGFRα+ fibroblasts over the lifespan of INKBRITE animals (n; PND0=7, PND7=7, 2m=5, 22m=5). (J) Histogram display the of GFP intensity in PND0, PND7, 2m, and 22m PDGFRα+ lung fibroblasts (n= 5 per timepoint, 2 experiments). AW=airway, BM=basement membrane, PND=Postnatal day. Scale bars 100υm. Each point in graph represents one animal or one distinct image for in vitro studies with mean ± s.e.m. All p values determined by one-tailed t-test and two-way ANOVA when applicable. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
To determine whether p16INK4a+ fibroblasts divide more slowly than p16INK4a-negative fibroblasts, we administered bromodeoxyuridine (BrdU) continuously to separate cohorts of INKBRITE animals during the alveologenesis (PND5-45) or adult homeostasis (PND60-90) phases of postnatal lung development and maintenance respectively. Examining PDGFRα+ fibroblasts in the sub-airway epithelial compartment, we found a significant reduction of BrdU incorporation in the p16INK4a+ fibroblasts during alveologenesis but no statistically significant reduction during adult homeostasis compared to p16INK4a-negative fibroblasts (Fig. 1D to F). To trace the fate of p16INK4a+ cells over a longer period during homeostasis, we administered tamoxifen to p16creERT:RosatdTomato animals to permanently activate tdTomato expression in p16INK4a+ cells, followed by a chase period off tamoxifen to study the behavior of tdTomato+ cells (15). Histology analysis of tdTomato+ cells confirmed their localization in the subepithelial compartment in the airway, and tdTomato+ fibroblasts demonstrated a significant reduction in 5-ethynyl-2’-deoxyuridine (EdU) incorporation over a 60 day chase period (PND60-120) relative to tdTomato-neg fibroblasts (Fig. S2A and B). Long term trace of tdTomato+ fibroblasts demonstrated their persistence in the lung 7 months after animals were treated with tamoxifen to activate tdTomato expression (Fig. S2C). These studies demonstrate that p16INK4a+ fibroblasts constitute a stable, tissue-resident population with slower replication within the basement membrane during postnatal development and homeostasis.
Poly-nucleation is a feature of cellular senescence in cultured fibroblasts(1, 17), but has not been observed in intact tissues. Using nuclear and actin filament (F-actin) staining to define the morphology of freshly isolated fibroblasts from INKBRITE lungs (>PND60), we detected bi- and tri-nucleated fibroblasts that more prevalent in the p16INK4a+ fibroblasts (Fig. 1G and H). p16INK4a+ fibroblasts also demonstrated other features of senescent cells when cultured ex vivo, including an increase in size, presence of γHistone2AX (γH2AX) and senescence-associated β-galactosidase (Fig. S2D to F).
To examine the proportion of lung fibroblasts that express p16INKa over a mouse’s lifespan, we collected INKBRITE lungs at postnatal day 0, day 7, 2 months, 12 months, and 22 months for FACS. The proportion of fibroblasts expressing p16INK4a increased in the first week of life, followed by stability between the day 7 and 12 months, with the highest proportion of p16INK4a+ fibroblasts present at 22 months (Fig. 1I). While the proportion of fibroblasts expressing GFP increased about 2-fold with age, we observed a larger age-dependent increase in GFP intensity in GFP+ fibroblasts (Fig. 1I and J), indicated by the INKBRITE reporter, and confirmed by qPCR analysis of p16INK4a from GFP+ cells collected from young and aged animals (Fig. S2G). Thus p16INK4a+ fibroblasts in vivo exhibit features previously ascribed to senescent cells in vitro. This indicates that p16INK4a+ cells are not absent or particularly rare in healthy and young tissue, but rather present in relative abundance at the epithelial-mesenchymal interface where p16INK4a expression is increased throughout the lifespan.
Variable p16INK4a expression correlates with proliferative cell cycle arrest
We observed a broad range of GFP intensity in the GFP+ fibroblasts across lifespan, and further sorting of GFP+ fibroblasts into GFPhi (top 50 percentile fluorescent intensity) and GFPlo (bottom 50 percentile) confirmed correlation of GFP intensity with p16INK4a expression (Fig. 2A and B). p16INK4a expression was distributed across fibroblast subsets in the lung(18), with an enrichment of GFPhi fibroblasts in the adventitial fibroblast lineage that are associated with the airway and vasculature (Fig. S3A and B). To test whether high p16INK4a expression correlates with cell cycle arrest in response to proliferative stimuli, which is the canonical definition of senescence(19), we apply a chemical cell trace to p16INK4a+ cells that allows quantification of proliferative history using flow cytometry. We treated lung fibroblasts isolated from INKBRITE lungs with CellTrace Far Red (CTFR), a fluorescent cell dye that is diluted with each cell division, and grew them in culture. Serum-deprived fibroblasts retained CTFR as demonstrated by a narrow, bright CTFR peak on FACS indicative of cell cycle arrest (Fig. 2C, red histogram). In serum-enriched growth medium, we observe a broad range of CTFR intensity in cultured fibroblasts, with a significant enrichment of CTFR-retaining cells (% of cells with CTFR intensity similar to that of serum-deprived cells) in GFP+ fibroblasts. Further segregation based on GFP intensity showed GFPhi fibroblasts had increased CTFR retention relative to GFPlo (Fig. 2C and D). Analysis of CFTR retaining fibroblasts also confirmed that they were enriched for other senescence characteristics such as polyploidy, increased cell size, and DNA damage (Fig. S3C to E). We saw similar results when we isolate just adventitial fibroblasts (Fig. S3F). To confirm this behavior in vivo, we treated freshly isolated INKBRITE lung fibroblasts and transplanted them into injured host lungs without endogenous fluorescence to induce fibroblasts proliferation(18). 4 days after CTFR treatment and transplantation, we observed a broad range of CTFR retention in adoptive transferred fibroblasts (CTFR+). Segregation of GFP intensity showed highest CTFR retention in GFPhi fibroblasts, followed by GFPlo and GFPneg (Fig. 2E to I). We applied this technique to adult human lung fibroblasts (HLFs) freshly isolated from deceased donors without prior history of lung disease(20), and confirmed the presence of CTFR retaining HLFs that were enriched for p16INK4a expression along with other characteristics of senescent cells (Fig. S3G to J), highlighting a technique to isolate senescent cells from human tissues.
Figure 2. Range of p16INK4a expression correlates with proliferative cell cycle arrest.
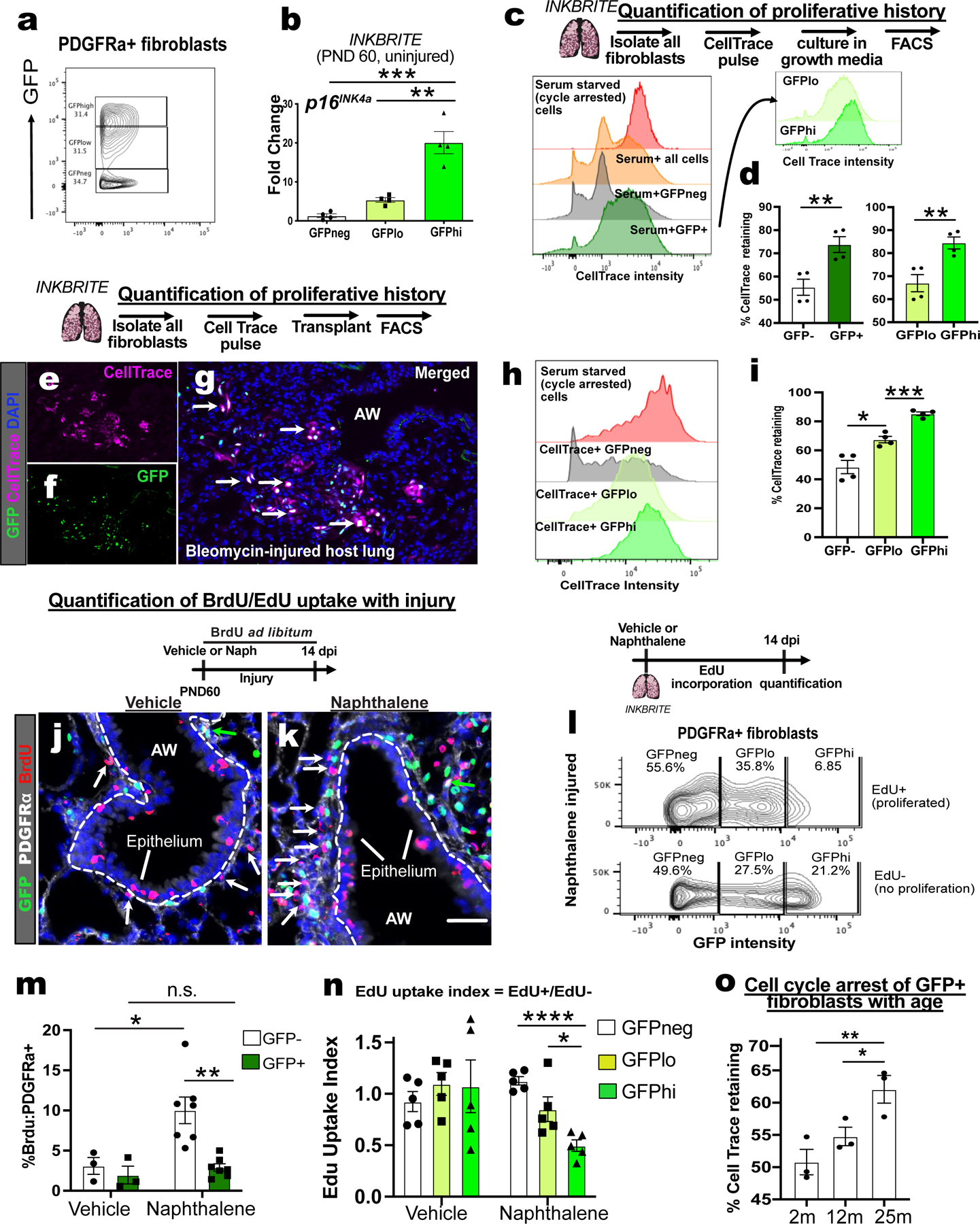
(A) Sorting strategy for GFP−high, low, and negative (hi/lo/neg) fibroblasts cells from uninjured INKBRITE lungs using GFP fluorescent intensity. (B) qPCR of p16INK4A transcript in GFPhi/lo/neg populations (n=4, >2 experiments). (C) Histogram of CellTrace Far Red (CTFR) intensity in INKBRITE lung fibroblasts. (D) quantification of % cell cycle arrest based on percentage of cells with CTFR intensity of serum-deprived cells (n=4, >2 experiments). (E to I) Histology, FACS and quantification of CTFR+ transplanted INKBRITE fibroblasts into Bleomycin injured NGS™ mice (2 experiments, n=4). (J to L) IHC and quantification of BrdU incorporation into PDGFRα+ and GFP–(white arrows) or PDGFRα+ and GFP+ (green arrows) cells in vehicle or naphthalene-injured (14 dpi) lungs (n = 3 for vehicle, 6 for naphthalene). (M) GFP intensity distribution of EdU+ and EdU− fibroblast isolated from naphthalene injured lungs (14 dpi). (N) EdU uptake Index (%EdU+/%EdU−) of GFPhi/lo/neg fibroblast populations in vehicle and naphthalene treated (14 dpi) fibroblasts (n = 5 per condition, 2 experiments). (O) CTFR retention in GFP+ fibroblasts of 2m, 12m, and 25m old INKBRITE lungs (n = 3 per timepoint, 2 experiments). AW = airway, dpi = days post injury. Each point in graph represents one animal with mean ± s.e.m. All p values determined by one-tailed t-test or two-way ANOVA when applicable. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
In a quiescent organ, cell cycle arrest can be attributed to either senescence or quiescence. Such cells differ in their response to a proliferative stimulus. Therefore, evaluating senescence requires the introduction of a proliferative stimulus to cells in vivo. To add a mitogenic stimulus to the lung, which normally exhibit very low cell turnover(21), we administered naphthalene to injure the lung of INKBRITE animals followed by continuous BrdU administration. Naphthalene induces damage to the airway epithelium accompanied by increase in proliferation of airway stem cells and fibroblasts during repair within 2 weeks of injury(22, 23). Naphthalene injury increased GFP+ cells in the lung (Fig. S3K), but this appeared to result from an increase in GFP+ immune cells, as the fraction of GFPneg, GFPlo, and GFPhi fibroblasts remained constant (Fig. S3B) and the percentage of GFP+ immune cells increased with injury (Fig. S3L), with a skewing towards myeloid lineages in GFPhi immune cells (Fig. S3M). There was minimal BrdU incorporation of PDGFRα+ fibroblasts that were GFP+ or negative over two weeks without injury. However, naphthalene injury was accompanied by an increase in BrdU incoporation that was almost exclusively in the p16INK4A-neg fibroblasts, with little to no BrdU incorporation in the p16INK4a+ fibroblasts (Fig. 2J to L). To compare proliferative changes based on GFP intensity, we repeated the BrdU experiment with EdU, which allows for FACS quantification concurrent with GFP measurement. There was no difference in EdU uptake between groups after two weeks of EdU administration in vehicle-treated controls, but a significant difference appeared when the lung was injured with naphthalene, with the least EdU incorporation in GFPhi fibroblasts (Fig. 2M and N).
To evaluate the propensity of p16INK4a+ fibroblasts to undergo cell cycle arrest as p16INK4a expression increases with age, we isolated GFP+ lung fibroblasts of animals at 2 months, 12 months, and 25 months of age for CTFR treatment and cell culture. We observed an age-dependent increase in the fraction of arrested (CTFR-retaining) fibroblasts in proliferative conditions (Fig. 2O). These experiments suggest that a physiologic increase in p16INK4a expression correlates with increased senescent characteristics in vivo during organ aging.
p16INK4a+ fibroblasts develop a secretory phenotype after epithelial injury
To profile the transcriptomic signature of p16INK4a+ fibroblasts, we performed bulk RNA sequencing on sorted GFP+ or GFP−neg and PDGFRα+ fibroblasts from uninjured (0 days post injury, or 0 dpi) and naphthalene injured (7, 14, and 28 dpi) INKBRITE lungs (Fig. 3A). Differentially expressed gene (DEG) analysis (p16INK4a+ vs. p16INK4a-neg) showed that Cdkn2a, the gene which encodes p16INK4a, was highly upregulated at all timepoints in the p16INK4a+ fibroblasts (Fig. 3B, fig. S4A). A key feature of senescent cells is the ability to upregulate a senescent-associated secretory program (SASP) to modify the extracellular environment(24). To identify potential SASP factors in p16INK4a+ fibroblasts, we compared DEGs overexpressed in p16INK4a+ and p16INK4a-neg fibroblasts (p <0.05) with a list of SASP factors previously identified in vitro(24). Gene overlap analysis showed a significant time-dependent increase in SASP expression in p16INK4a+ but not p16INK4a-neg fibroblasts after injury. (Fig. 3C, fig. S4B and C, Table S1)
Figure 3. Single cell and bulk RNA sequencing analysis of p16INK4A+ cells in the lung.
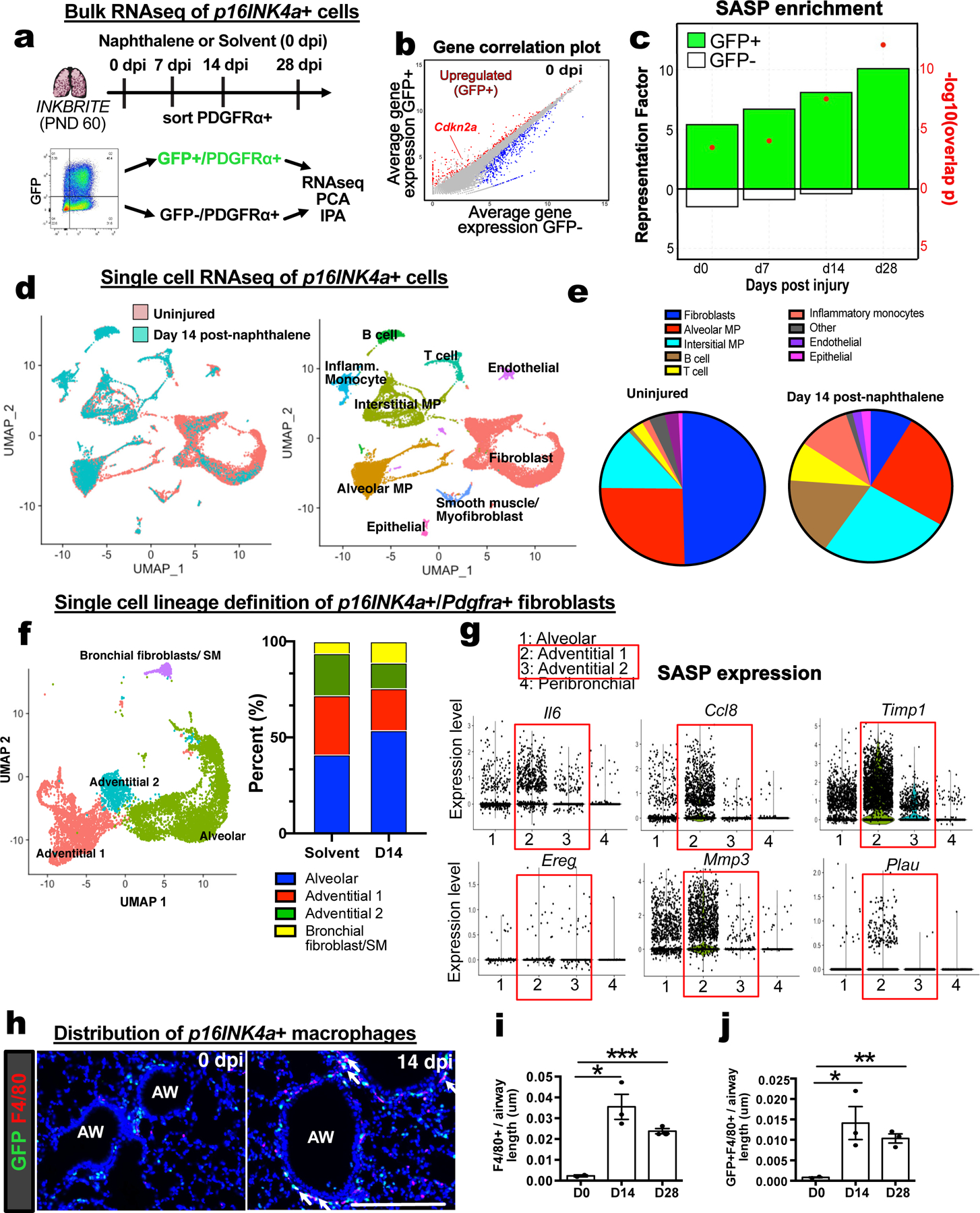
(A) Bulk RNAseq of p16INK4A+ and – fibroblasts during homeostasis (0 dpi) and injury (7,14,28 dpi) (n=3 animals per timepoint). (B) Gene correlation plot showing Cdkn2a expression. (C) Hypergeometric probability test for enrichment of differentially expressed genes (DEGs) between GFP+ and GFP−neg fibroblasts at each timepoint with SASP genes as quantified by Representation Factor and p-value. (D and E) Single cell analysis of all p16INK4A+ cells in 0 dpi and 14 dpi INKBRITE lungs. (F) Clustering of p16INK4A+ fibroblasts into distinct subsets in 0 dpi and 14 dpi INKBRITE lungs. (G) SASP gene expression at the single cell level in the 4 fibroblast subsets. (H to J) Histologic analysis of F4/80+ and GFP+ and F4/80+ macrophage/monocyte population in 0 dpi and 14 dpi INKBRITE lungs (0, 14, and 28 dpi; n=3). AW = airway, dpi = day post injury. Each point in graph represents one animal with mean ± s.e.m. All p values determined by one-tailed t-test. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
To assess the heterogeneity of p16INK4a+ cells in the lung, we performed single cell RNA sequencing (scRNAseq) on sorted GFP+ cells from healthy and naphthalene-injured (14 dpi) adult (2 months old) INKBRITE lungs by droplet-capture (10X Chromium Single Cell 3’v2). Cluster analysis confirmed our flow cytometry and immunohistochemistry (IHC) analysis, showing that the overwhelming majority of GFP+ cells are Pdgfra+ fibroblasts and Ptprc+ (CD45+) immune cells (Fig. 3D and E). ScRNAseq of GFP−neg cells in age-matched lungs (2 months old) showed fewer fibroblasts, and more T-cells in the immune population compared to GFP+ cells (Fig. S5A). Airway injury with naphthalene increased the fraction of immune p16INK4a+ cells relative to p16INK4a+ fibroblasts. Notably increased were infiltrating immune subsets associated with tissue injury such as inflammatory monocytes and monocyte-derived interstitial macrophages(25) (Fig. 3D and E). Further clustering of the fibroblasts according to previously annotated markers (18) confirmed our FACS analysis, showing that the majority of p16INK4a+ fibroblasts cluster within fibroblasts with adventitial markers (Pi16 and Adh7) predominantly found in connective tissue around the blood vessel, and the cluster distribution did not change significantly with injury or age (Fig. 3F, fig. S5B and C). SASP factors, which were upregulated in p16INK4a+ fibroblasts with injury as shown by bulk RNAseq (Fig. 3C and fig. S4B), were expressed in the adventitial fibroblasts (Fig 3G), which are found in various barrier organs(26). This suggests that p16INK4a+ adventitial fibroblasts respond to airway injury by increasing SASP. IHC of the airway confirmed the scRNAseq finding that infiltrating immune cells such as p16INK4a+ macrophages with interstitial subtype marker (Adgre1 encoding F4/80 antigen) are increased in the adventitial space adjacent to the airway and blood vessels after injury (Fig. 3H to J). Finally, scRNAseq analysis of p16INK4a+ cells in aged INKBRITE lungs (30 months old) showed increased number of cells from myeloid lineages (e.g. monocytes and interstitial macrophages) similar to those seen in young, injured lungs (Fig. S5C). This suggests that an increase in the number of p16INK4a+ myeloid cells is a feature of both tissue injury and aging in the lung.
p16INK4a+ fibroblasts promote epithelial stem cell regeneration
p16INK4a+ fibroblasts had increased SASP after epithelial injury that might alter epithelial stem cell behavior. To determine the effect of p16INK4a+ fibroblasts on airway stem cells responding to injury, we co-cultured airway stem cells with GFPhi, GFPlo, or GFPneg fibroblasts (PDGFRα+) isolated from uninjured (0 dpi) or naphthalene injured (14 dpi) INKBRITE lungs in a 3D cell culture. When cultured with Scgb1a1+ airway stem cells (isolated from Scgb1a1 reporter mouse), both uninjured GFPhi and GFPlo fibroblasts increased the number of Scgb1a1+ organoids more than did GFPneg fibroblasts (Fig. 4A and B). Naphthalene injury further increased the capacity of both GFPhi and lo fibroblasts to increase the number of Scgb1a1+ organoids relative to GFPneg fibroblasts (Fig. 4A and B). Naphthalene injured GFPhi and lo fibroblasts also increased the size of the organoids relative to GFPneg fibroblasts (Fig. 4C). To determine if p16INK4a expression is associated with enhanced ability to promote epithelial growth in human lung, we isolated p16INK4a-hi and p16INK4a-lo HLFs based on CTFR retention on FACS (Fig. S3G), and cocultured with airway stem cells isolated from donor lungs (27). p16INK4a-hi HLFs enhanced airway stem cell organoid growth (Fig. S6A and B), demonstrating that the capacity for p16INK4a+ fibroblasts to promote epithelial stem cell growth is conserved in mouse and human lungs.
Figure 4. Injured p16INK4A+ fibroblast enhances epithelial progenitor proliferation ex vivo.
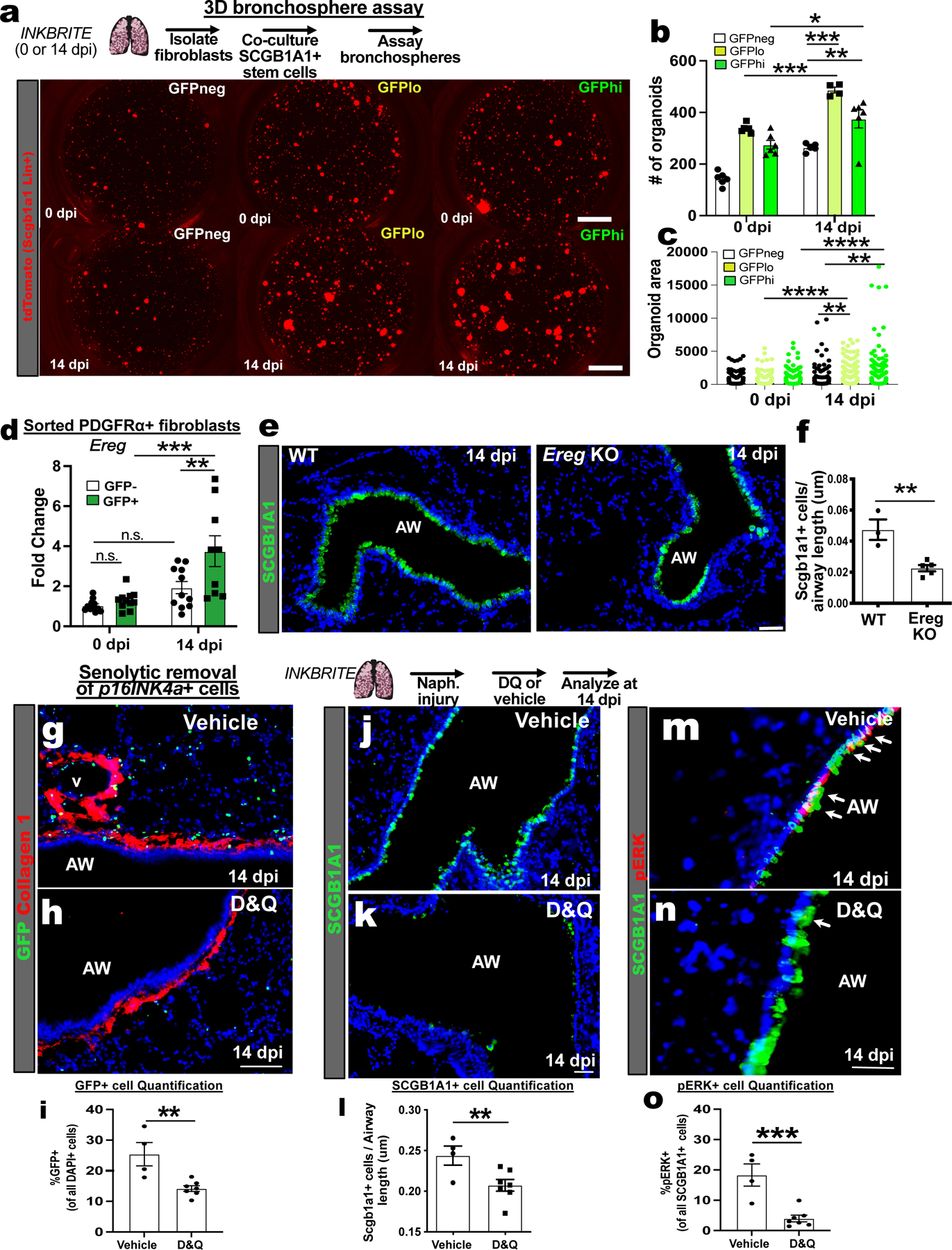
(A) 3D organoid assay combining uninjured Scgb1a1+ airway stem cells (tdTomato+) with p16INK4Ahi/lo/neg fibroblasts isolated from INKBRITE lungs at 0 or 14 dpi. (B) Quantification of Scgb1a1+ organoid numbers and (C) size (n=3 triplicate wells per condition, >2 experiments). (D) qPCR of Ereg on sorted GFP+ or – fibroblasts from INKBRITE lungs at 0 or 14 dpi (n = 9 per timepoint, >2 experiments). (E and F) Histologic quantification of airway stem cell regeneration in Ereg knockout (KO) vs. WT lungs at 14 dpi (n = 4 per genotype, 2 experiments) (G to I) IHC quantification of GFP+ cells, (J to L) SCGB1A1+ cells, and (M to O) pERK+SCGB1A1+ cells in vehicle vs. D&Q treated INKBRITE lungs after naphthalene injury (14 dpi, vehicle n=4, D&Q n=7; 2 experiments). AW = airway, D&Q = dasatnib/quercetin. Each point in graph represents one technical replicate (bronchosphere) or one animal with mean ± s.e.m. All p values determined by one-tailed t-test and two-way ANOVA when applicable. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
One of the genes found to be upregulated in p16INK4a+ fibroblasts following injury is Ereg that encodes epiregulin (Fig. S4A), which signals through epidermal growth factor receptor (EGFR) (28–30). qPCR analysis of sorted lung fibroblasts at 0 and 14 dpi confirmed that Ereg transcript is more abundant in p16INK4a+ fibroblasts after injury than in p16INK4a-neg fibroblasts at 14 dpi and p16INK4a+ fibroblasts at 0 dpi (Fig. 4D). ScRNAseq analysis of Ereg expression demonstrated preferential expression in the adventitial fibroblasts (Fig. 3G) and alveolar macrophages (Fig. S6C). To determine the effect of fibroblast-derived EREG on club cell growth, we isolated fibroblasts from naphthalene-injured lungs (14 dpi) of Ereg null (Ereg KO) and wildtype (WT) animals for 3D organoid assay, which showed a reduced capacity of the Ereg KO fibroblasts to promote airway stem cell growth (Fig. S6D and E). Furthermore, organoids cultured with Ereg KO fibroblasts exhibited reduced phosphorylated extracellular signal-related kinase (pERK), a readout EGFR activation (31) (Fig. S6F). While Ereg KO animals had normal quantity of SCGB1A1+ cells in the airway prior to injury (Fig. S6G), they exhibited a significant deficit in SCGB1A1+ cell regeneration in response to naphthalene in vivo (Fig. 4E and F).
To determine whether p16INK4a+ cells are required for airway stem cell regeneration in vivo, we treated INKBRITE animals with naphthalene and then the senolytics, dasatnib and quercetin (DQ), a combination that kills senescent cells (32). DQ treatment reduced the number of GFP+ cells in the lung after naphthalene injury (Fig. 4G to I). DQ-treated lungs had a significant deficit in SCGB1A1+ cell regeneration (Fig. 4J to L). Furthermore, pERK was less abundant in SCGB1A1+ airway stem cells in DQ treated lungs (Fig. 4M to O). These experiments demonstrated that p16INK4a+ fibroblasts adjacent to airway stem cells promote epithelial regeneration after injury by secreting a growth factor that enhance airway barrier repair.
p16INK4a+ fibroblasts sense inflammatory stimulus to augment epithelial repair
Ingenuity Pathway Analysis (IPA) of the DEGs in 16INK4a+ fibroblasts demonstrated a time-dependent activation of upstream regulators associated with NF-κB signaling (Fig. S7a). NF-κB responds to inflammatory stimuli to regulate the SASP (33, 34). To test whether p16INK4a+ fibroblasts can directly sense inflammatory stimuli, we examined NF-κB activation in p16INK4a+ and negative fibroblasts in response to lipopolysaccharide (LPS), a bacterial wall component. There was an increase in baseline nuclear localization of p65/RelA, a component of the NF-κB transciptional complex, in live sorted and unstimulated p16INK4a+ fibroblasts compared to p16INK4a-neg fibroblasts from uninjured INKBRITE lungs (Fig. 5A and B). Upon LPS induction in vitro, there was a further increase in nuclear localization of p65/RelA in the p16INK4a+ fibroblasts (Fig. 5A and B). Examining the transcriptional response of GFPhi,lo, and negative fibroblasts to LPS stimulation in vitro, we found that many of the SASP factors in p16INK4a+ fibroblasts were more abundantly expressed in the GFPhi subset than in GFPlo and negative (Fig. 5C). Similar to murine p16INK4a-hi fibroblasts, p16INK4a-hi HLFs also demonstrated increased expression of SASP factors such as EREG and IL6 in response to LPS (Fig. S7B). To test the ability of p16INK4a+ fibroblasts to respond to LPS in vivo, we administered LPS by intranasal inhalation into INKBRITE animals, which promotes an inflammatory response that mimics gram-negative bacterial infection(35), and isolated GFP+ and negative fibroblasts on 3 dpi for organoid assay. 3D airway stem cell organoid growth was enhanced when cultured with p16INK4a+ fibroblasts from LPS treated lungs, which was attenuated with the NF-κB inhibitor, BAY11-7082 (Fig. 5D and E). qPCR analysis of p16INK4a+ fibroblasts isolated from LPS treated lungs demonstrated a significant increase in Ereg expression (Fig. S7C).
Figure 5. p16INK4A+ fibroblast senses inflammatory stimuli to augment epithelial regeneration.
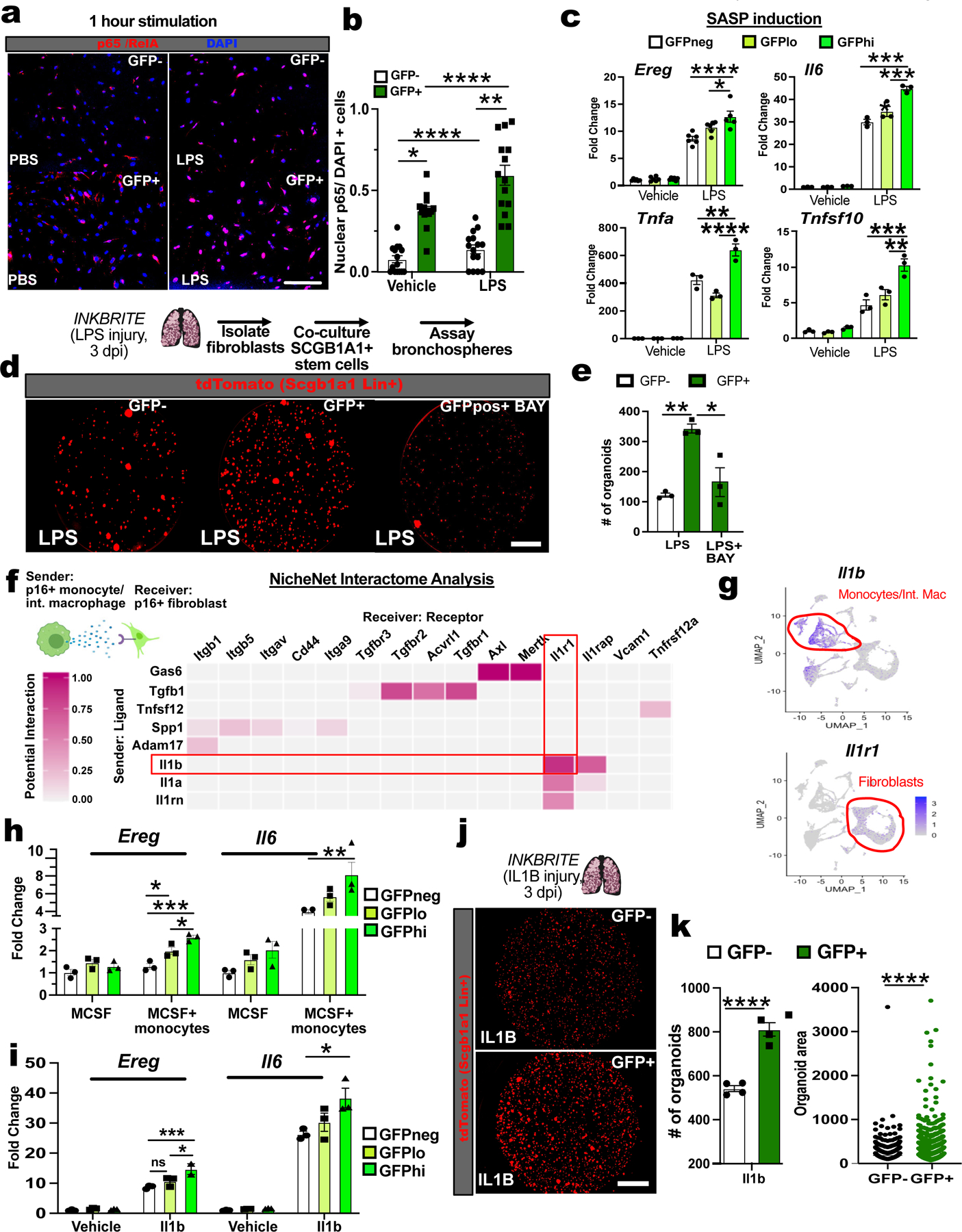
(A and B) Quantification of nuclear p65/RelA in p16INK4A+ or – fibroblasts from uninjured INKBRITE lungs (PND60) after 1 hour of vehicle or LPS treatment (n=3 wells, 14–15 images per condition). (C) SASP gene expression in p16INK4Ahi/lo/neg fibroblasts from uninjured INKBRITE lungs after 6 hours of PBS or LPS incubation (n=3 wells per condition, 2 experiments). (D and E) Scgb1a1+ 3D organoid assay with p16INK4A+/- fibroblasts isolated from LPS injured INKBRITE lungs and treated with vehicle or BAY11-7082 (n=3 wells per conditions, 2 experiments). (F) NicheNet interactome analysis of p16INK4A+ fibroblasts and monocyte/interstitial monocytes from scRNAseq of injured INKBRITE lungs (14 dpi). (G) Expression of Il1b and Il1r1 in p16INK4A+ cells in the lung. (H and I) Quantification of Scgb1a1+ organoids co-cultured with p16INK4A+ or – fibroblasts from IL-1B-treated INKBRITE lungs (n=4 wells per conditions). Scale bars 100υm. Each point in graph represents one triplicate well or distinct image with mean ± s.e.m. All p values determined by one-tailed t-test or two-way ANOVA when applicable. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001. Scale bars 100υm.
To define the potential source and identity of NF-κB activators during tissue repair, we performed an interactome analysis (NicheNet) of our scRNAseq of p16INK4a+ to evaluate potential ligand-receptor interactions(36). We found that the IL-1 family cytokines were particularly enriched in the monocyte and monocyte-derived interstitial macrophages (Fig 5F and G) found to be increased in the adventitial space after injury (Fig. 3H to I), corresponding with an enrichment of Il1r1 (receptor for IL-1A and B) in p16INK4a+ fibroblasts (Fig. 5F and G). To test the ability of monocytes to directly activate p16INK4a+ fibroblasts, we cultured inflammatory monocytes with lung fibroblasts isolated from INKBRITE lungs (+macrophage colony-stimulating factor, or M-CSF, to maintain monocyte survival). After 24 hours in co-culture, the monocytes increased the abundance of SASP factors including Ereg, and the expression was highest in GFPhi, fibroblasts (Fig. S7D). Induction with recombinant IL-1B produced an almost identical phenotype in the INKBRITE lung fibroblasts (Fig. S7E). Finally, to test the ability of p16INK4a+ fibroblasts to sense IL-1B in vivo, we administered recombinant IL-1B to the INKBRITE lung for 3D organoid assay. Similar to LPS injury, exposure to IL-1B in vivo primed the isolated p16INK4a+ fibroblasts to enhance airway stem cell organoid formation compared to p16INK4a-neg fibroblasts (Fig. 5H,I). These data highlight a role for p16INK4a+ in sensing and responding to inflammatory signals during injury through NF-κB, which regulates a secretory program that enhances stem cell repair.
Modulation of p16INK4a in fibroblasts alters epithelial regeneration
To determine whether modulation of p16INK4a expression alters SASP factors that induce epithelial proliferation, we designed a lentivirus expressing shRNA targeting p16INK4a (Lenti-shp16), and infected GFP+ fibroblasts isolated from INKBRITE lungs. Infection of Lenti-shp16 suppressed the expression of Ereg and Il6 in GFP+ fibroblasts after LPS stimulation (Fig. 6A), and increased fibroblast proliferation (Fig. S8A) along with reduction in p16INK4a transcript (Fig. S8B). We also knocked down p16INK4a by treating lung fibroblasts isolated from p16flox/flox animals with adenovirus expressing Cre recombinase (adeno-cre) followed by LPS stimulation, which demonstrated a similar attenuation of Ereg and Il6 expression following LPS (Fig. 6B). Conversely, we designed a dual lentiviral system (Lenti-tTS/rTTA+Lenti-TRE-p16-2A-tdTomato) to overexpress p16INK4a in GFP−neg fibroblast in a doxycycline (dox) dependent fashion, which did not change baseline SASP expression, but increased the expression of Ereg and other NF-κB-responsive genes in response to LPS (Fig. S8C). To determine whether fibroblast-specific p16INK4a expression is necessary for epithelial regeneration in vivo, we deleted p16INK4a with a mesenchymal-specific Cre-driver (Dermo1Cre/+)(37). Mesenchymal-specific deletion of p16INKa (Dermo1Cre/+:p16flox/flox, referred to as Dermo1p16CKO) did not alter the gross morphology of the uninjured adult lung (Fig. 6C and D). However, after injury with naphthalene, Dermo1p16CKO airways demonstrated reduced epithelial repair as quantified by the number of SCGB1A1+ club cells present as well as qPCR of Scgb1a1 transcripts in the whole lung (Fig. 6E to H). Airway injury in Dermo1p16CKO also resulted in increased airway fibrosis as demonstrated by increased fibrotic markers and collagen deposition along the airway (Fig. S8D to F). To confirm the epithelial regenerative defect, we also deleted p16INK4a with Gli1CreERT2 to specifically target Gli1+ adventitial fibroblasts that are enriched for GFPhi fibroblasts surrounding the airway (Fig. S8G to I). Tamoxifen treatment of Gli1CreERT2/+:p16flox/flox:R26RYFP/+ (referred to as Gli1p16CKO) animals followed by naphthalene injury significantly attenuated SCGB1A1+ airway stem cell recovery compared to that in controls (Gli1CreERT2/+:R26RYFP/+) (Fig. 6I to L). Analysis of Gli1 lineage-traced (Gli1 Lin+) fibroblasts in vivo demonstrated an increase in BrdU incorporation after inducible p16INK4a deletion and injury (Fig. S8J to M), and a reduction in Ereg and Il6 expression (Fig. 6M) similar to that observed with in vitro knockdown of p16INKa. The regenerating airway epithelium of Gli1p16CKO animals demonstrated a significant reduction in pERK+ airway stem cells (Fig. 6N to P). These data show that p16INKa is required to maintain cell-cycle arrest and promote the SASP in response to inflammation in senescent fibroblasts in vivo, the loss of which disrupts mesenchymal signals to progenitors required for the repair of the barrier epithelia.
Figure 6. Mesenchymal p16INK4A expression is required for epithelial regeneration in vivo.
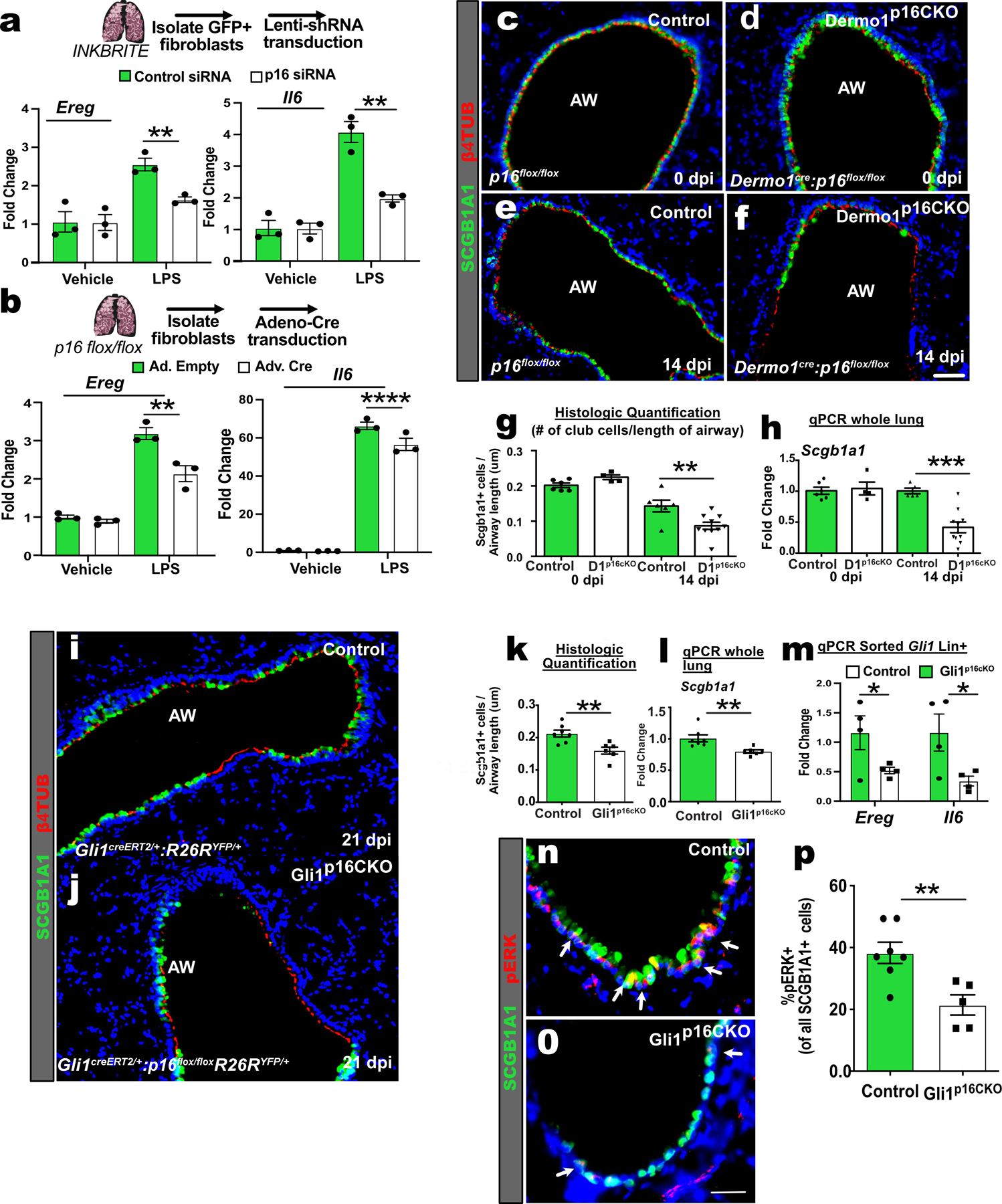
(A) qPCR of Ereg and Il6 in control or p16INK4A shRNA treated GFP+ fibroblasts after stimulation with vehicle or LPS for 6 hours (n=3 wells per conditions, 2 experiments). (B) qPCR of Ereg and Il6 in adeno-control or adeno-Cre treated p16flox/flox fibroblasts after stimulation with vehicle or LPS (n=3 wells per conditions, >2 experiments). (C to G) Images and quantification of the airways of control and Dermo1p16CKO lungs with and without naphthalene injury. (H) qPCR of Scgb1a1 of whole lung RNA (uninjured: n=6 Control, n=5 Dermo1p16CKO; injured: n=6 Control, n=10 Dermo1p16CKO,2 experiments ). (I to L) Images, histological quantification of SCGB1A1 club cells, and qPCR of Scgb1a1 on control and Gli1p16CKO lungs after naphthalene injury (n=7 Control, n=6 Gli1p16CKO). (M) qPCR of Ereg and Il6 of sorted Gli1 Lin+ cells after naphthalene (n=4 Control; n=4 Gli1p16CKO). (N to P) Images and quantification of pERK+ and SCGB1A1+ cells in control and Gli1p16CKO lungs after naphthalene injury (n=7 Control, n=6 Gli1p16CKO). AW=airway. Scale bars 100υm. Each point in graph represents one animal with mean ± s.e.m. All p values determined by one-tailed t-test and two-way ANOVA when applicable. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
Discussion
We generated an ultrasensitive reporter mouse to isolate and localize p16INK4a+ cells in tissues. We focused on p16INK4a because of the reported benefits of eliminating p16INK4a+ cells from aging tissues. Proposals that p16INK4a+ cells are in a rigid cellular state may be inaccurate, as we describe some p16INK4a+ cells that remain responsive to physiological cues and dynamically increase their secretory capacity in the reparative niche to enhance stem cell repair. We also show that physiological heterogeneity exists within a given p16INK4a+ cellular population in vivo, as not all p16INK4a+ cells exhibit uniform senescent characteristics. Thus, not all p16INK4a+ cells may be senescent, and expression of other biomarkers (e.g. Cdkn1a and Cdkn1b) may contribute to senescent characteristics in vivo. However, we demonstrated that cells within a range of p16INK4a expression levels can account for this heterogeneity in vivo, with a direct correlation of p16INK4a level with cell cycle arrest, response to inflammation, and SASP. Furthermore, we showed that p16INK4a expression increased throughout the lifespan of p16INK4a+ fibroblasts.
Our data show that p16INK4a+ fibroblasts, some with senescent characteristics, arise normally during postnatal tissue maturation in the basement membrane, persist within the stem cell niche and are activated upon barrier injury to promote epithelial repair (Fig. S9). p16INK4a+ fibroblast can sense the presence of infiltrating immune cells that appear during tissue injuries and rapidly integrates inflammatory stimuli through NF-κB activation to induce SASP after injury to restore barrier integrity. We uncovered EREG as a SASP component, the induction of which is regulated by p16INK4a to promote regional repair in the airway stem cell niche, but there are likely to be many SASP factors that function in other regional stem cell niches that are specific to the requirement of the resident stem cell. The enrichment of p16INK4a expression in the adventitial fibroblast subset suggests a cross-tissue role, as p16INK4a+ adventitial fibroblasts could serve as tissue sentinels at the barrier interface across multiple organs.
Supplementary Material
Acknowledgements
We thank the Parnassus Flow Cytometry Core for assistance with cell sorting for bulk and single cell RNA analysis (P30DK063720); Biological Imaging Development Core members (P30 DK063720); Eunice Wan and the Institute for Human Genetics Core for processing of single cell RNA samples and high-throughput sequencing.
Funding:
This work is supported by NIH grants DP2AG056034, R01HL142552, R01HL155622 to T.P., along with Tobacco Related Disease Research Program New Investigator Award and Pulmonary Hypertension Association award to T.P. and F32HL14226 to N.R.
Footnotes
Competing interests: authors do not have any competing interests.
Data and materials availability: The sequencing data reported in this paper is deposited in NCBI Gene Expression Omnibus (GEO) under the accession number GSE140654.
References and Notes
- 1.Zindy F, Quelle DE, Roussel MF, C. J. Oncogene 15, 203–211 (1997). [DOI] [PubMed] [Google Scholar]
- 2.Gorgoulis V et al. Cell 179, 813–827 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Takeuchi S et al. Cancer Res 70, 9381–9390 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Burd CE et al. Cell 152, 340–351 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Demaria M et al. Dev Cell 31, 722–733 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baker DJ et al. Nature 479, 232–236 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baker DJ et al. Nature 530, 184–189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang J et al. Nat Med 22, 78–83 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Childs BG et al. Science 354, 472–477 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baar MP et al. Cell 169, 132–147 e116 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jeon OH et al. Nat Med 23, 775–781 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bussian TJ et al. Nature 562, 578–582 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Deursen JM, Science 364, 636–637 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grosse L et al. Cell Metab 32, 87–99 e86 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Omori S et al. Cell Metab, (2020).
- 16.Scudellari M, Nature 550, 448–450 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Benn PA, Am J Hum Genet 28, 465–473 (1976). [PMC free article] [PubMed] [Google Scholar]
- 18.Tsukui T et al. Nat Commun 11, 1920 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hayflick L, Moorhead PS, Exp Cell Res 25, 585–621 (1961). [DOI] [PubMed] [Google Scholar]
- 20.Wang C et al. J Clin Invest 128, 4343–4358 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Basil MC et al. Cell Stem Cell 26, 482–502 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hong KU, Reynolds SD, Giangreco A, Hurley CM, Stripp BR, Am J Respir Cell Mol Biol 24, 671–681 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Peng T et al. Nature 526, 578–582 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coppe JP, Desprez PY, Krtolica A, Campisi J, Annu Rev Pathol 5, 99–118 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chakarov S et al. Science 363, (2019). [Google Scholar]
- 26.Buechler MB et al. Nature 593, 575–579 (2021). [DOI] [PubMed] [Google Scholar]
- 27.Bonser LR et al. Am J Respir Cell Mol Biol 64, 308–317 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Toyoda H et al. J Biol Chem 270, 7495–7500 (1995). [DOI] [PubMed] [Google Scholar]
- 29.Draper BK, Komurasaki T, Davidson MK, Nanney LB, J Cell Biochem 89, 1126–1137 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Li S et al. Proc Natl Acad Sci U S A 105, 3539–3544 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iijima M, Anai M, Kodama T, Shibasaki Y, Biochem Biophys Res Commun 489, 83–88 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Zhu Y et al. Aging Cell 14, 644–658 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laberge RM et al. Nat Cell Biol 17, 1049–1061 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chien Y et al. Genes Dev 25, 2125–2136 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bannerman DD, Goldblum SE, Am J Physiol Lung Cell Mol Physiol 284, L899–914 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Browaeys R, Saelens W, Saeys Y, Nat Methods 17, 159–162 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Sosic D, Richardson JA, Yu K, Ornitz DM, Olson EN, T Cell 112, 169–180 (2003). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.