

ABSTRACT
Sporadic colorectal cancer (CRC) is a leading cause of worldwide cancer mortality. It arises from a complex milieu of host and environmental factors, including genetic and epigenetic changes in colon epithelial cells that undergo mutation, selection, clonal expansion, and transformation. The gut microbiota has recently gained increasing recognition as an additional important factor contributing to CRC. Several gut bacteria are known to initiate CRC in animal models and have been associated with human CRC. In this Review, we discuss the factors that contribute to CRC and the role of the gut microbiota, focusing on a recently described mechanism for cancer initiation, the so-called microbiota-induced bystander effect (MIBE). In this cancer mechanism, microbiota-driven parainflammation is believed to act as a source of endogenous mutation, epigenetic change and induced pluripotency, leading to the cancerous transformation of colon epithelial cells. This theory links the gut microbiota to key risk factors and common histologic features of sporadic CRC. MIBE is analogous to the well-characterized radiation-induced bystander effect. Both phenomena drive DNA damage, chromosomal instability, stress response signaling, altered gene expression, epigenetic modification and cellular proliferation in bystander cells. Myeloid-derived cells are important effectors in both phenomena. A better understanding of the interactions between the gut microbiota and mucosal immune effector cells that generate bystander effects can potentially identify triggers for parainflammation, and gain new insights into CRC prevention.
Keywords: Colorectal neoplasms, Gut microbiome, Radiation-induced bystander effect, Neoplastic cell transformation, Carcinogenesis, DNA damage, Cancer stem cells, Cell-of-origin, Chromosomal instability, Mutation, Paligenosis, Bacteria, Cell dedifferentiation, Doublecortin-like kinase 1
Summary: New mechanisms of sporadic colorectal cancer initiation link gut dysbiosis with recent concepts of parainflammation, paligenosis and bystander effects to drive chromosomal instability and malignant transformation in colon epithelial cells.
Introduction
The human microbiome has gained wide recognition as an important contributor to human health and disease, with recent advances highlighting its role in cardiovascular, respiratory, autoimmune, psychiatric, metabolic, gastrointestinal and neurologic diseases (Lynch and Pedersen, 2016). Of equal importance is the microbiome's potential to contribute to carcinogenesis and to the global cancer burden (Rajagopala et al., 2017). Although well-defined mechanisms have been described for the initiation of cancer following infection with oncogenic pathogens, such as Helicobacter pylori and human papilloma viruses in gastric and cervical cancers, respectively, it was recently concluded that there is still no “direct evidence that the human commensal microbiome is a key determinant in the aetiopathogenesis of cancer” (Scott et al., 2019). Unfortunately, many gaps still exist in our knowledge regarding the underlying mechanisms of cancer initiation potentially caused by commensal microorganisms.
Sporadic colorectal cancer (CRC) is a leading cause of cancer and death due to cancer worldwide (Bray et al., 2018). As such, it is important to clarify the mechanisms of commensal microbiome-driven malignant transformation, not least because understanding such mechanisms could lead to new strategies preventing this disease. Sporadic CRC progresses in a well-defined manner during which normal colonic epithelium forms precursor lesions in the form of adenomas, which then become overtly malignant (Vogelstein et al., 2013). Genomic and epigenetic changes in these precursor adenomas can be readily investigated to identify those that initiate cellular transformation. In addition, numerous animal models of intestinal cancer have been developed, which provide additional insights into the role of commensals in carcinogenesis.
Human CRC is multifactorial in origin. Both host and environmental factors play major roles in influencing the genetic mutations and the transcriptional and epigenetic changes associated with this cancer (Lichtenstein et al., 2000) and, therefore, in the development of cells-of-origin (see Box 1, Glossary) (Visvader, 2011). Cofactors that affect sporadic CRC development include age, genetic background and lifestyle factors, e.g. diet, obesity, alcohol and smoking, together with extrinsic modifiers, such as non-steroidal anti-inflammatory agents (Song et al., 2020). The complex intertwining of these variables and factors in the etiology of CRC has rendered the identification of commensal-driven initiation mechanisms particularly challenging.
Box 1. Glossary.
Azoxymethane (AOM): methyl-methylimino-oxidoazanium, a potent carcinogen and neurotoxin that induces colon cancer in rats and mice.
Cancer stem cells (CSCs): a small subpopulation of cells that can recapitulate a parent tumor through processes of self-renewal and differentiation.
Cell-of-origin: tumor-initiating cell that is a non-transformed precursor to cancer stem cells. They may arise from normal tissue-resident stem cells, such as those at the bottom of the intestinal crypt, or from fully differentiated post-mitotic cells. Cells-of-origin are non-malignant and cannot divide or reproduce indefinitely.
Colibactin: a non-protein bacterial compound synthesized by polyketide synthases and other enzymes, and encoded by a 54-kb genomic island designated pks. Colibactin is genotoxic and induces DNA double-strand breaks and chromosome aberrations in epithelial cells. It can induce colorectal carcinogenesis in animal models and has been associated with human CRC. The pks island is carried by selected strains of Enterobacteriaceae, including Escherichia coli.
Crypt stem cell: self-renewing cell at the base of crypts, which gives rise to differentiated cell types that maintain the integrity of the intestinal epithelium.
Culture-enriched molecular profiling: a combination of exhaustive bacterial culture techniques that use 16S rRNA gene sequencing to identify unique bacteria from complex ecological niches, such as the gastrointestinal tract.
Dextran sulfate sodium (DSS): a polysaccharide polymer that, when given orally, degrades the intestinal mucus barrier and induces severe colitis.
Dysbiosis: a perturbation in the healthy intestinal microbial population, which permits disease-associated pathobionts to emerge.
Gnotobiotic: an environment in which all present microorganisms are known; i.e. a gnotobiotic animal is an animal in which all present strains of bacteria and other microorganisms are known and defined.
Inflammaging: an inherent feature of the aging process characterized by the chronic, progressive and poorly controlled increase in proinflammatory status due to activation of innate immunity. Inflammaging is linked to numerous diseases of aging, including atherosclerosis, Alzheimer's disease, type II diabetes mellitus, osteoporosis and cancer among others.
Interleukin 10 (IL10): a potent anti-inflammatory cytokine that is produced by subsets of T cells and monocytes among many other cells that contributes to intestinal homeostasis and immune tolerance. Knockout of the Il10 gene results in an unremarkable phenotype in mice unless colonized by pathobionts that induce colonic inflammation and microbiota-driven CRC.
Lamina propria: a thin vascular layer of connective tissue beneath the epithelium of a mucous membrane.
Macrophage: myeloid-derived immune cell found in virtually all tissues and especially enriched in the colon, which function as professional phagocytes to provide defense against exogenous pathogens, and/or to serve as sentinels for tissue homeostasis and tolerance to commensals. Macrophages have remarkable cellular plasticity and are readily polarized from a resting state (M0) to M1 or M2 phenotypes, depending on cues in their tissue microenvironment.
Mucosal-associated invariant T (MAIT) cell: unique major histocompatibility complex class I-related protein 1 (MR1)-restricted innate-like T cell that bridges innate and adaptive immunity.
Mast cell: myeloid-derived immune cell that is rich in histamine and heparin, expresses high-affinity receptors for IgE, and plays a key role in inflammation by releasing multiple mediators from storage granules into the tissue microenvironment.
Mastocytosis: a rare medical condition caused by excess numbers of mast cells congregating in tissues.
Microbiota-induced bystander effect (MIBE): a theory regarding colorectal carcinogenesis whereby gut bacteria polarize tissue macrophages to induce stress signaling and DNA damage in epithelial bystander cells, leading to cellular transformation and malignancy.
Microbiota: ecological communities of commensal, symbiotic and pathogenic microorganisms and viruses.
Mono-association: colonization with microbes of a single species.
Neutrophils: the most-abundant granulocytes, making up 40-70% of all white blood cells in humans. They are a crucial part of the innate immune system.
Parainflammation: an inflammatory state characterized by chronic activation of genes involved in innate immunity through persistent DNA damage.
Pathobiont: a commensal microorganism that causes disease when specific genetic, environmental or ecological conditions are altered.
Prostaglandin-endoperoxide synthase 2 (PTGS2, also known as COX-2): an enzyme that converts arachidonic acid into prostaglandin H2, is expressed by macrophages during inflammation and plays a key role in colorectal carcinogenesis.
Polymicrobial: any condition involving multiple species of microorganisms.
Reactive oxygen species (ROS): chemically reactive molecules that, among others, include superoxide, hydroxyl radical and peroxides. ROS are by-products of aerobic metabolism and can play important roles in cell signaling. They can also lead to cellular damage of proteins, lipids and nucleic acids.
Radiation-induced bystander effect (RIBE): compilation of phenomena in which irradiated cells can induce stress signaling and DNA damage in bystander cells through diffusible mediators or gap-junction communication.
T cell: a type of lymphocyte; they are divided into several subsets, each of which has a crucial role in adaptive immune responses. Among others, there are TH1, TH2, TH9, TH17, cytotoxic, Treg, Tr1 and TCM cells.
Tr1 cell: T regulatory type 1 cell that is distinct from TH1, TH2 and TH17 cells. Tr1 cells express an anergic phenotype that induces high-level production of the anti-inflammatory cytokine IL10 but little to no IL2, IL4 or IFNG.
Treg: regulatory T cell; a subset of T helper cells (CD4+) that express the FOXP3 transcription factor and CD25, and help maintain tolerance by suppressing or downregulating the induction and proliferation of effector T cells.
This Review focuses on microbiota-driven mechanisms that initiate sporadic CRC and help elucidate the disease's known cofactors. The microbiota comprises living members of the microbiome – bacteria, fungi, protozoa and other eukaryotes, and is often also considered to include viruses and bacteriophages (Box 1). The preponderance of research on microbiota-driven mechanisms for cancer initiation in CRC involve bacteria, although a few studies have highlighted potential roles for viruses, bacteriophages and fungi (Coker et al., 2019; Goel et al., 2006; Hannigan et al., 2018). Here, we focus on bacterial studies and models that have helped to elucidate causal relationships among the gut microbiota, dysbiosis (Box 1) and CRC. Emphasis is placed on bystander effects that lead to mutations, epigenetic reprogramming and the cellular transformation of colon epithelial cells. We draw parallels between a theory for the microbiota-induced bystander effect (MIBE; Box 1) and the well-characterized radiation-induced bystander effect (RIBE; Boxes 1 and 2) (Burdak-Rothkamm and Rothkamm, 2018). Bacterial traits that promote the growth of malignant cells, facilitate epithelial-mesenchymal transition and metastasis, modulate cancer therapy or function as probiotics in CRC prevention are not discussed, as these topics have been recently reviewed elsewhere (Allen and Sears, 2019; Eslami et al., 2019; Janney et al., 2020; Tilg et al., 2018).
Box 2. Radiation-induced bystander effect (RIBE).
RIBE generates numerous bioreactive molecules that – with considerable implications for cancer initiation – chronically induce stress signaling and DNA damage in bystander cells. In addition to the direct effects on irradiated cells, ionizing radiation causes a range of cellular responses, particularly DNA damage and CIN (Bach et al., 2019; Hoevenaar et al., 2020) in unirradiated bystander cells exposed to irradiated cells (Mothersill and Seymour, 2004). RIBE was initially observed in individuals who had undergone whole-body irradiation and were found to have chromosome-breaking factors (clastogens) in their blood many years later (Goh and Sumner, 1968), indicating persistent clastogen production (Marozik et al., 2007; Pant and Kamada, 1977). This effect has been confirmed using irradiated tissue culture cells (Nagasawa and Little, 1992) and in bone marrow transplants form irradiated rodents (Watson et al., 2000). Hematopoietic and mesenchymal cells, including macrophages, are common effectors of RIBE (Burr et al., 2010; Dong et al., 2015; Lorimore et al., 2008; Pampfer and Streffer, 1989).
RIBE activates multiple signaling pathways in bystander cells, including NF-κB, MAPK and JNK (Azzam et al., 2002), leading to altered expression of stress response genes (e.g. PTGS2 and NOS2), activation of DNA damage repair (p53/p21 and ATM/ATR), epigenetic modification (through miRNA and/or CpG island methylator phenotypes), and proliferation, apoptosis and death of bystander cells (Azzam et al., 2002; Ilnytskyy et al., 2009; Najafi et al., 2014; Sokolov and Neumann, 2018; Zhou et al., 2005).
Bystander effects occur through diffusible mediators and gap-junction intercellular communication (Azzam et al., 2003). Mediators include ROS and mutagenic products of lipid peroxidation (Emerit et al., 1996; Lorimore et al., 2008; Wang et al., 2012), TNF-α, IL1A, and IL8 (Dong et al., 2015; Fu et al., 2016; Yang et al., 2012), extracellular vesicles (Al-Mayah et al., 2015; Ariyoshi et al., 2019; Mo et al., 2018; Rastogi et al., 2018; Szatmári et al., 2019), TGF-β1 (Gow et al., 2010); cell-free DNA (Konkova et al., 2019; Sergeeva et al., 2017), and cysteine protease cathepsin B and insulin-like growth factor receptor (Peng et al., 2017). Irradiation can also release chromatin particles that are aberrantly integrated into the DNA of bystander cells, leading to genomic damage (Kirolikar et al., 2018), and can induce signaling in bystander cells through pattern recognition receptors (Schaue et al., 2015).
Gut microbiota and dysbiosis in CRC
The gut microbiome is a complex ecosystem of living microorganisms that contains mixtures of metabolites, structural elements and other bioactive molecules (Berg et al., 2020). The colon contains a high density and diversity of microbiota (∼1011 microorganisms per ml) (Sender et al., 2016). By comparison, the human distal small intestine has a much lower density of bacteria than the colon (∼108 microorganisms per ml). Despite a 94% larger surface area (Helander and Fandriks, 2014), the incidence of adenocarcinomas in the small intestine is 99% less compared with that in the colorectum (Qubaiah et al., 2010). The magnitude of this difference, and the many animal models of intestinal cancer showing a requirement of microbiota for cancer initiation, provide compelling evidence that the colorectal microbiota play a key role in colon carcinogenesis. Several mechanisms have been described for how commensals might generate the genomic damage and epigenetic changes that precede malignant transformation in colon epithelial cells. These mechanisms are considered in this review, albeit with a focus on MIBE.
Numerous murine models have provided useful insights into microbiota-associated CRC initiation and progression. These models can be placed into several different categories: (1) carcinogen induced (see Box 3 carcinogen-based models of CRC); (2) engineered mutations in CRC driver genes or in genes that modulate driver genes; (3) initiated by agents or mutations that disrupt the intestinal barrier, leading to inflammation and; (4) gene knockouts that lead to intestinal inflammation (see Table 1). The role of the microbiota in these models is often investigated by using antibiotic treatment or is carried out under conditions that are either gnotobiotic, i.e. free of germs, mono-associated and defined as being polymicrobial, or free of specific pathogens (Box 1 and Table 1). As expected, each model has its advantages and disadvantages. For example, gnotobiotic models that investigate specific microbiota in intestinal carcinogenesis are inherently biased by systematic alterations in murine immune responses that arise from gnotobiosis and, potentially, affect inflammation-associated mechanisms (Kennedy et al., 2018). Chronic antibiotic therapy to induce intestinal dysbiosis might also not mimic the dysbiosis that precedes human sporadic CRC. Mutations in driver genes can oversimplify the complex mutational landscape and the epigenetic changes that occur in oncogenes and tumor suppressors in sporadic CRC. Engineered or spontaneous mutations in Apc, as exemplified in the mouse ApcMin/+ model of intestinal tumorigenesis (Su et al., 1992), do not accurately recapitulate the pathology of human sporadic CRC. Additionally, mutations in Apc primarily generate adenomas in the rodent small intestine, whereas precursor lesions in humans occur in the large intestine (McIntyre et al., 2015). Overall, although these models can help to refine and confirm microbiota-associated mechanisms in CRC, no model has as yet faithfully recapitulated the spectrum of genetic, epigenetic and histological findings observed in human sporadic CRC.
Box 3. Carcinogen-based models of CRC.
Azoxymethane (AOM) is commonly used to induce colon tumors in rodent models by intraperitoneal injection. AOM is a metabolite of 1,2-dimetylhydrazine and a metabolic precursor to methylazoxymethanol. The metabolic activation of these alkylating agents leads to the aberrant methylation of guanine at the O6- and N7-positions (Rogers and Pegg, 1977). When AOM treatment is followed by oral administration of DSS, an agent that disrupts the mucus and the intestinal barrier that protect against colonic inflammation, the incidence of colon tumors increases together with their size and frequency (Tanaka et al., 2003). The resulting adenomas show characteristics common to precursor lesions in human CRC, such as β-catenin activation and induction of PTGS2. An early study using gnotobiotic rats found that colon cancer initiation by 1,2-dimethylhydraine depended on the microbial conversion of 1,2-dimethylhydrazine to AOM (Reddy et al., 1975). This report was among the first to implicate the intestinal microbiome in CRC carcinogenesis. Although the AOM/DSS model is still widely used, it relies on an exogenous carcinogen and, thus, does not recapitulate the potential mechanisms for carcinogenesis involving endogenous mutagenesis.
Table 1.
Selected models of CRC initiation that show dependency on intestinal microbiota

In addition to animal models, culture-enriched molecular profiling provides further evidence for the involvement of microbiota in human CRC (Box 1) (Lau et al., 2016), as do metagenomic sequencing (Ranjan et al., 2016) and metabolomics (Brown et al., 2016). These technologies allow for the comprehensive investigation of the colon microbiota and its relationship to CRC. Metagenomic investigations in particular have identified associations between CRC and imbalances in the intestinal microbiota, such as those associated with dysbiosis (Wirbel et al., 2019). In a recent study of fecal samples and tumor biopsies, bacterial phylotypes were correlated with human colon adenomas and sporadic CRC (Wirbel et al., 2019). Data from large CRC cohorts have identified multiple bacterial species and phylotypes that are associated with CRC, such as Bacteroides fragilis, Fusobacterium spp., Porphyromonas spp., Parvimonas micra, Prevotella spp., Alistipes finegoldii, Gemella morbillorum and Thermanaerovibrio acidaminovorans among others (Dai et al., 2018; Wirbel et al., 2019). Additional findings suggest that other pathobionts (Box 1), such as Salmonella enterica (Lu et al., 2016; Mughini-Gras et al., 2018) and Streptococcus gallolyticus subsp. gallolyticus (Kumar et al., 2017), together with viruses (Hannigan et al., 2018) and fungi (Coker et al., 2019), are also linked to colorectal carcinogenesis. Unfortunately, these strategies are unable to identify or to address the specific mechanisms by which commensals or pathobionts generate the genomic damage and the epigenetic changes that lead to the malignant transformation of colon epithelial cells.
Several bacterial phenotypes have been shown to initiate CRC. The phenotypes and their underlying mechanisms include toxin-induced damage to colon epithelial cell DNA as caused by colibactin-producing Escherichia coli (Box 1), alterations in cell signaling pathways that regulate genomic stability as caused by enterotoxigenic B. fragilis and the polarization of tissue macrophages (Box 1) or the generation of lipid peroxidation and reactive oxygen species (ROS; Box 1) as caused by Enterococcus faecalis and Fusobacterium nucleatum (Allen and Sears, 2019; Faїs et al., 2018; Wang et al., 2012). A summary of proposed CRC-initiating mechanisms produced by these specific pathobionts is provided in Table 2. As discussed below, commensal-driven immune mechanisms represent a common theme for many of the proposed mechanisms for CRC initiation and provide an integrated perspective on the origins of cellular transformation (Irrazabal et al., 2014; Janney et al., 2020; Lasry et al., 2016). However, these mechanisms are best understood in the context of what is currently known about the genetics, cells-of-origin (Box 1), precursor lesions, histological morphology and inflammatory milieu of CRC. These topics are, therefore, reviewed below.
Table 2.
Proposed mechanisms for bacterium-mediated initiation of sporadic colorectal cancer

Colorectal cancer origins and genetics
Our current understanding of the origin of most cancers, including sporadic CRC, postulates that they arise as a consequence of genetic and/or epigenetic changes in somatic cells. These changes may be inherited or result from environmental insults to the genome, such as those discussed above. Over time, cells undergo selection and clonal expansion to culminate in cancer (Hanahan and Weinberg, 2011; Vogelstein et al., 2013). The transcriptomic profiling of CRCs has identified four consensus subtypes of tumor cells based on changes in gene expression: microsatellite instability immune, canonical, metabolic and mesenchymal (Guinney et al., 2015). However, this approach has been challenged because it does not define the cells-of-origin used to develop these transcriptional subtypes (Dunne et al., 2016). Recently, RNA sequencing (RNA-seq) analysis of single cells isolated from CRC tumors has identified transcriptional groups that correspond to canonical and metabolic consensus subtypes (Lee et al., 2020). Despite this progress, these approaches have yet to define the mechanisms of CRC initiation. By contrast, the analysis of somatic mutation signatures of human sporadic CRC may be better able to help clarify the potential variety of initiating mechanisms. For example, whole genome sequencing of 60 CRC biopsies identified 14 distinct single-base substitutions in >50% of tumors from 49 biologically relevant signatures that had been curated from 2650 human cancers (Alexandrov et al., 2020). Nine CRC signatures (64%) were associated with well-defined mutational or epigenetic processes, e.g. deamination of 5-methylcytosine, defective DNA-mismatch repair and ROS. Moreover, ROS-related mechanisms have been implicated in CRC initiation for enterotoxin-producing B. fragilis, F. nucleatum and superoxide-producing E. faecalis (Table 2). In an elegant study using colibactin-producing E. coli infection of clonal human colon organoids, a specific mutational signature was identified and, subsequently, found in 5–7.5% of CRC biopsies (Pleguezuelos-Manzano et al., 2020). Of particular interest was evidence for this signature in healthy human colon crypts, suggesting that E. coli-driven DNA damage is an initiating event in a small percentage of CRC cases. However, mechanisms underlying many of the other common mutation signatures have yet to be identified.
Precursor lesions in colorectal cancer
Colon polyps are precursor lesions for CRC and occur in 20-53% of adults in the USA aged >50 years (Strum, 2016). These lesions are classified as adenomas (75%) or serrated lesions (25%), with the latter consisting of hyperplastic polyps, sessile serrated lesions and traditional serrated lesions (Crockett and Nagtegaal, 2019; Strum, 2016). Unlike large (≥10 mm) polyps, diminutive (1-5 mm) and small (6-9 mm) polyps often regress (Strum, 2016). Aberrant crypt foci are an additional type of precursor lesion that are small and difficult to detect. These foci can be induced by colon-specific carcinogens in animal models, exhibit hyperplasia or dysplasia, and harbor the typical genetic and epigenetic changes found in adenomas and serrated lesions (Lopez-Ceron and Pellise, 2012). Azoxymethane (AOM; Box 1) is the prototypical carcinogen used in these models and requires activation by the gut microbiota to induce mutations (Table 1). Approximately 85% of CRCs arise from conventional adenomas and, following malignant transformation, show an average of 66 nonsynonymous mutations (Vogelstein et al., 2013). The source of these mutations is largely unknown, although enterotoxigenic B. fragilis, colibactin-producing E. coli, F. nucleatum and superoxide-producing E. faecalis have all been shown to damage epithelial cell DNA (Tables 1 and 2).
The most common genomic alternation in adenomas is chromosomal instability (CIN) (Shih et al., 2001b), which is characterized by extrachromosomal DNA, as well as insertions, deletions, amplifications and translocations in existing chromosomes. CIN can occur very early in precursor lesions and leads to the overexpression of oncogenes, and to the silencing of tumor suppressors to drive cellular proliferation and malignant transformation (Bach et al., 2019; Hoevenaar et al., 2020; Nowak et al., 2002). CIN and microsatellite instability are genomic hallmarks of most precursor lesions. Not surprisingly, a subset of CRCs express both microsatellite instability and CIN (Bolhaqueiro et al., 2019). These genomic changes are dynamic, accumulate over time and can lead to malignant transformation (Blokzijl et al., 2016; Tomasetti et al., 2013). Among the gut microbiota, E. faecalis in particular can generate CIN and aneuploidy in epithelial cells through ROS and MIBE (Table 2).
Sporadic CRCs arising from serrated lesions characteristically feature mutations in KRAS or BRAF (Crockett and Nagtegaal, 2019). BRAF mutations result in the widespread methylation of regions that comprise a high frequency of cytosine followed by guanine, i.e. CpG islands, causing the silencing of tumor suppressors. For example, hypermethylation of the MLH1 promoter region, which is involved in DNA mismatch repair, results in microsatellite instability, with typically >700 mutations in malignant tumor cells. Fusobacterium species and E. faecalis are associated with human CRC that expresses the CpG island methylator phenotype (Lennard et al., 2016; Tahara et al., 2014). It remains to be determined whether these associations are causal or, more simply, due to microsatellite instability tumors that exhibit enhanced colonization of bacteria.
Transforming somatic cells into cancer stem cells
CRC emerges from colon epithelial cells with mutations and epigenetic changes in genes that drive transformation, e.g. APC, TP53 and MLH1 as tumor suppressors, and KRAS and CTNNB1 as oncogenes (Vogelstein et al., 2013; Yaeger et al., 2018). Most cancer cells in a tumor mass contain the original genomic changes that led to malignancy, although tumor genomes also continuously adapt to changing tumor microenvironments. A minority of cells within a tumor mass (1-2%) are cancer (or tumor) stem cells (CSCs), which are characterized by self-renewal and lineage-production (Box 1). These cells can recapitulate the original tumor mass upon seeding to an appropriate niche (Zeuner et al., 2014). Lineage tracing in genetically engineered CRC models shows that CSCs can arise from self-renewing crypt stem cells (Box 1) that are exposed to transformation-promoting processes, e.g. chemical or radiation injury or chronic inflammation. Thus, these cells are candidates for the cells-of-origin for CRC (Barker et al., 2009). However, experimental evidence also suggests that constitutive activation of Wnt signaling or engineered mutations of Apc and Kras in fully differentiated colon epithelial cells can also induce transformation and form CSCs (Schwitalla et al., 2013; Tetteh et al., 2016).
Recent studies have shown that cells-of-origin for CRC can also arise from somatic cells that are fully (or terminally) differentiated and undergo reprogramming. This process is likely to occur following chronic mucosal injury or inflammation. The orchestration of tissue repair and regeneration involves a sequence of cellular autodegradation, metaplastic gene expression, reactivation of mammalian target of rapamycin complex 1 (mTORC1), and cell cycle re-entry through a process termed paligenosis (Boxes 1 and 4) (Burclaff and Mills, 2018; Willet et al., 2018). Somatic cell dedifferentiation and proliferation during paligenosis occurs via crosstalk among mTORC1 via Hippo-YAP, Notch1 and Wnt/β-catenin pathways (Kaur and Moreau, 2019). The process of paligenosis has been observed in gastric and pancreatic tissues (Willet et al., 2018). Ongoing injury, as might occur during chronic inflammation, and checkpoint failure following DNA damage could lead to cellular proliferation, repetitive cycles of paligenosis, and to the accumulation of mutations in a ‘cyclical hit’ model of carcinogenesis (Burclaff and Mills, 2018; Miao et al., 2020b). This way, cells may become fixed in a state of dedifferentiation or induced pluripotency and may propagate mutations as cells-of-origin for CSCs. Massively parallel transcriptome analyses of individual CRC tumor cells have shown that tumor cells co-segregate with normal stem-like and proliferative cell types (Lee et al., 2020), providing support for this concept, although direct evidence is still needed for these processes in colorectal carcinogenesis. To the extent that dysbiosis in the gut microbiota induces chronic mucosal injury and parainflammation (Box 1), bacteria might be expected to contribute to paligenosis and the eventual creation of cells-of-origin for CSCs.
Box 4. Paligenosis and a ‘cyclical hit’ model for carcinogenesis.
Paligenosis is the mechanism by which fully differentiated cells can re-enter the cell cycle in response to tissue injury and DNA damage (Willet et al., 2018). This is an evolutionarily conserved repair process that begins with the quenching of the mammalian target of rapamycin complex 1 (mTORC1), a master regulator of cellular energetics. Inhibition of mTORC1 occurs through injury-activated DNA damage-induced transcript 4 (DDIT4), which helps to ‘license’ post-mitotic cells to re-enter the cell cycle (Miao et al., 2020b). This is followed by increased autophagocytic activity to remove differentiated cellular features and damaged organelles. The reactivation of mTORC1 is associated with increased levels of injury-induced interferon-related developmental regulator 1 (IFRD1), thereby deactivating a key p53 checkpoint, and enabling cells to progress into and through the cell cycle (Miao et al., 2020a). This process occurs in response to tissue injury and does not occur in normally dividing stem cells, although a role for mTORC1 in stem cell activity has yet to be defined. Disruptions in paligenosis have been associated with tumorigenesis in the stomach and pancreas but not the colon (Miao et al., 2020b; Willet et al., 2018). Bypass or loss of function of the IFRD1/p53 checkpoint should allow for the ‘unlicensed’ division of cells, with the propagation of DNA mutations in a ‘cyclical hit’ model of carcinogenesis (Burclaff and Mills, 2018).
Researchers have modeled murine intestinal tumors that originate from fully differentiated colon epithelial cells that had undergone reprogramming and dedifferentiation. For example, fully differentiated intestinal epithelial cells in mice that are engineered to constitutively activate nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and Wnt/β-catenin undergo dedifferentiation and malignant transformation (Schwitalla et al., 2013). Enterotoxigenic B. fragilis, F. nucleatum and E. faecalis can each activate Wnt/βcatenin signaling in epithelial cells (Table 2). Similarly, tuft cells that express doublecortin-like kinase 1 (Dclk1) can serve as cells-of-origin for intestinal tumors in Apc knockout mice exposed to colitis-inducing agents (Westphalen et al., 2014). Colon tuft cells are fully differentiated, long-lived intestinal epithelial cells located in the crypt base and are scattered along the crypt axis. These cells perform chemosensory functions, modulate stromal immunity and regulate epithelial DNA damage responses through paracrine mechanisms (Middelhoff et al., 2017). Although Dclk1 expression was noticed in tumors initiated through MIBE by using polarized macrophages infected with E. faecalis (Wang et al., 2015), the role of this gene in paligenosis and cancer initiation remains unclear. Intestinal tumors can also be generated in mice by selectively engineering mutations in Apc and Kras in post-mitotic cells expressing carbonic anhydrase as a differentiation marker (Tetteh et al., 2016). In summary, it appears that – in murine models – almost any cell type within a colon crypt is capable of being transformed into cells-of-origin for CRC. How cells-of-origin divide to form early precursor lesions in sporadic CRC will provide significant additional insight regarding their source and is discussed in the following section.
Top-down versus bottom-up morphogenesis in colorectal adenomas
The careful morphological analysis of diminutive colon adenomas has helped clarify the location of epithelial cells-of-origin in sporadic CRC (Cole and McKalen, 1963; Maskens, 1979; Shih et al., 2001a). CSCs have commonly been thought to arise from crypt stem cells. If this were true, then transformed cells within small tumors, as noted by Shih et al., “…should give rise to new, completely dysplastic crypts that branch as lesions expand. Histopathological examination, however, has long shown that this expected pattern is not observed…” (Shih et al., 2001a). These investigators confirmed that adenomas in the human colon undergo a pattern of ‘top-down’ morphogenesis with cells located in the upper portion of colon crypts and in zones between crypt orifices rather than at the crypt base – as would be expected if crypt stem cells were the cells-of-origin (Fig. 1A). Nearly all small, sporadic human colon adenomas show top-down morphogenesis, with dysplastic cells at the crypt surface that show active Wnt/β-catenin signaling and loss of APC (Shih et al., 2001a). Epithelial cells at the crypt base that appear histologically normal do not show Wnt signaling and loss of APC, two features considered to be characteristic of CRC. A top-down pattern of morphogenesis had been difficult to reconcile with the concept of CRC arising from normal crypt stem cells. However, recent studies have helped with this by demonstrating that fully differentiated colon epithelial cells can be reprogrammed into cells that can serve as cells-of-origin for CSCs (Adachi and Scholer, 2012; Burclaff and Mills, 2018; Greten, 2017). These findings have reframed concepts about the origin of top-down morphogenesis.
Fig. 1.
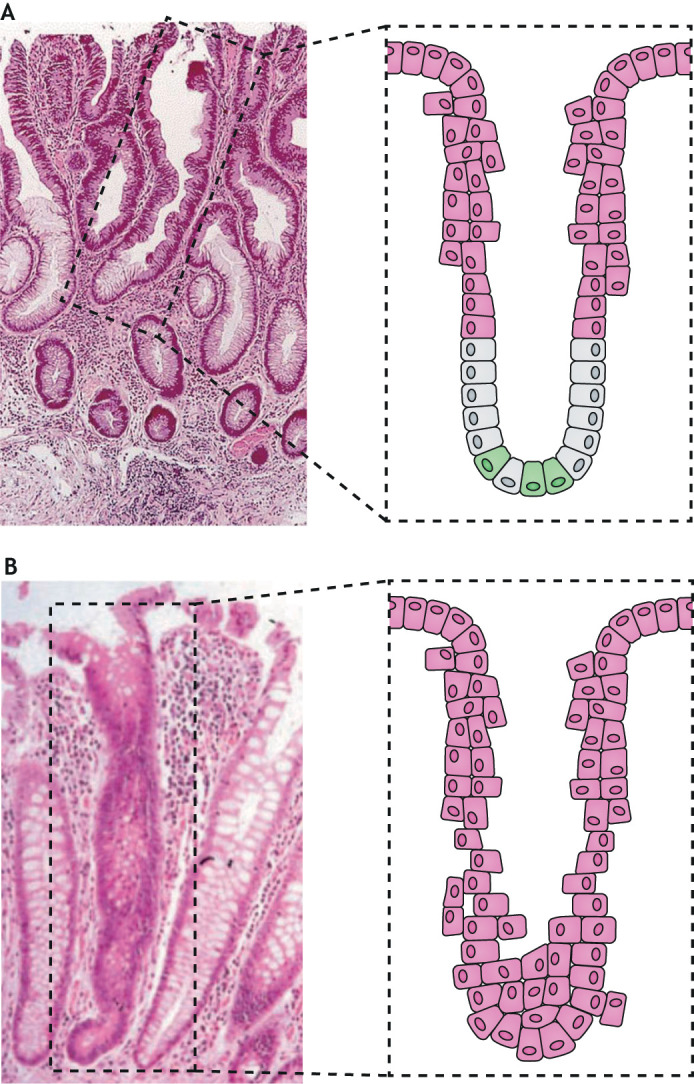
Early features of colon crypt morphogenesis in human adenomas. (A) Top-down morphogenesis. H&E-stained section of a typical small adenomatous polyp in the human colon. The highlighted structure shows dysplastic epithelium at the top of a crypt that is contiguous with the histologically normal epithelium at the base of the crypt. Notice the abrupt transition between dysplastic cells (pink) and the normal-appearing crypt stem cells and differentiating epithelial cells near the crypt mid-point (green and grey). The photomicrograph was reproduced with permission from Shih et al. (2001a). (B) Bottom-up morphogenesis. H&E-stained colonic section from an individual with familial adenomatous polyposis showing a monocryptal adenoma, in which all cells are dysplastic (pink). Dysplastic stem cells that are located at the crypt base always drive the dysplastic morphology for an entire crypt. The photomicrograph was reproduced with permission from (Preston et al., 2003).
Top-down morphogenesis may also partly explain why crypt stem cell markers have not proven useful for identifying the cells-of-origin in human sporadic CRC. A retrospective analysis of 11 putative markers in colon adenomas found that only four had higher levels of expression when compared to normal epithelial cells (Jang et al., 2016). However, no consistent pattern of marker expression was observed in this study.
Support for top-down morphogenesis was found in several murine models of intestinal cancer (Schwitalla et al., 2013; Tetteh et al., 2016; Westphalen et al., 2014). Tuft cells are fully differentiated somatic cells – making them prime candidates for cells-of-origin for CSCs. These cells constitutively express Dclk1, a protein kinase with tumor promoter activity and, as with crypt stem cells, persist in colon crypts for long periods (Chandrakesan et al., 2017; Ong et al., 2014; Westphalen et al., 2014). Such persistence can allow for the gradual accumulation of mutational and epigenetic burdens that drive transformation (Westphalen et al., 2014). In mice, expression of Dclk1 has been identified in cells-of-origin for CSCs (Chandrakesan et al., 2014, 2017; Nakanishi et al., 2013; Westphalen et al., 2014). In ApcMin/+ mice, Dclk1 correlates with enhanced pluripotency and increased self-renewal (Chandrakesan et al., 2017). Finally, inactivation of Dclk1 in Apc mutant mice markedly suppresses adenoma multiplicity (Chandrakesan et al., 2014).
Murine models of CRC also provide evidence for ‘bottom-up’ adenoma morphology. This particular histology has been observed in intestinal adenomas that arise after activation of the Wnt/β-catenin signaling pathway in rapidly dividing crypt stem cells (Barker et al., 2009; Zhu et al., 2009). Similarly, and depending on context, colon tumors have been shown to also originate from non-cycling reserve stem cells in the crypt base (Powell et al., 2012). Unicryptal microadenomas, i.e. dysplastic epithelia cells that occupy an entire crypt, represent bottom-up morphogenesis and are observed in polyps from patients with germline APC mutations (Fig. 1B) (Preston et al., 2003). Similarly, bottom-up morphogenesis is found in ApcMin/+ mice. Finally, activating mutations in Kras or NF-κB that drive dedifferentiation in cells with Apc mutations produce intestinal lesions in mice with both top-down and bottom-up morphology (Cammareri et al., 2017).
Overall, there is strong evidence from murine models that the cells-of-origin for CSCs in CRC can arise from fully differentiated somatic cells. Although migration of crypt stem cells from their normal location at the base to the upper crypt and to intrazonal regions between crypts cannot be entirely excluded as an explanation for top-down morphology, a more parsimonious explanation would be that post-mitotic epithelial cells dedifferentiate to become cells-of-origin. One potential driving force for such a process would be tissue injury from chronic parainflammation driven by dysbiosis and pathobionts. Multiple rodent models of intestinal cancer show that the gut microbiome can degrade or breach intestinal barriers to induce inflammation and initiate CRC (Tables 1 and 2). CSCs are inherent to CRC formation, although cells-of-origin for CSCs have yet to be identified in bacterium-driven models. Finally, it must be noticed that no murine model of CRC, particularly those due to germline mutations, has been shown to recapitulate the top-down morphogenesis seen in sporadic human adenomas. Therefore, new models or new analyses of established models are needed, which can generate cells-of-origin through commensal-driven mutations and reprogramming, and create top-down morphogenesis.
Immune cells and parainflammation in colorectal cancer initiation
Innate and adaptive immune systems, along with disruptions to intestinal barriers, such as mucus, tight junctions and basement membrane, play pivotal roles in CRC initiation (Lasry et al., 2016). The importance of inflammation to the development and progression of precursor lesions was highlighted in large human clinical trials showing that treatment with aspirin or non-steroidal anti-inflammatory drugs can reduce the risk of adenomas and CRC by 20-40% relative to placebo (Katona and Weiss, 2020). Colon adenomas are often infiltrated by polarized macrophages, neutrophils, mast cells and/or activated T cells, including regulatory T cells (Tregs) (see Box 1; McLean et al., 2011). Observations suggest that most colon adenomas exist in an intermediate state, somewhere between overt chronic inflammation, similar to that seen in colitis and the basal tolerance of the normal intestinal mucosa that shows no evidence of immune activation. This state has been termed parainflammation (Lasry et al., 2016; Pribluda et al., 2013).
There is scant evidence concerning the role of dysbiosis in parainflammation within human colon adenomas. Selected members of the gut microbiota have been shown to activate T cells that contribute colorectal carcinogenesis. This is best exemplified by enterotoxigenic B. fragilis-induced colitis and tumorigenesis in ApcMin/+ mice, which both require activation of T helper type 17 (TH17) cells via signal transducer and activator of transcription-3 (Stat3) (Wu et al., 2009). In this model, B. fragilis-induced expansion of Tregs limits the availability of interleukin 2 (IL2) and allows for TH17 responses to develop and drive carcinogenesis (Geis et al., 2015). Innate lymphoid cells and IL22 have also been shown to promote colorectal cancer in a H. hepaticus-driven model of carcinogenesis (Kirchberger et al., 2013). In ApcΔ468/+ mice, the dysregulation of Tregs induces mastocytosis (Box 1) and drives the adenoma-carcinoma sequence (Gounaris et al., 2009). Recent work has shown that mucosal associated invariant T (MAIT) cells (Box 1) are also linked to cancer initiation, growth and metastasis (Yan et al., 2020). MAIT cells are clusters of unconventional T cells primarily located in the lamina propria of the intestine (Box 1), lung and female genital tract. MAIT cell development requires the presence of both the intestinal microbiota and B cells (Treiner et al., 2003). The selection and expansion of MAIT cells is regulated by the major histocompatibility complex related protein 1 (MR1). MR1 recognizes a narrow range of unstable pyrimidine intermediates that are derived from bacteria and fungi (Corbett et al., 2014). MAIT cells occur in increased numbers in human CRC (Zabijak et al., 2015) but, as yet, there is no consensus concerning their role in anti- or pro-tumor responses. In a model of lung cancer, mice that lack the MR1 locus or in which MR1 is blocked by antibody show decreased tumor growth and reduced metastasis. These effects can be reversed by MAIT cell transplantation (Yan et al., 2020). The modulation of MAIT cells, other T-cell subsets and innate lymphoid cells by the gut microbiota requires further investigation to clarify their roles in colorectal carcinogenesis.
Histological analyses of human and murine intestinal adenomas, and of E. faecalis-triggered CRC, have shown mast cells infiltrating these lesions (Gounaris et al., 2009; Yang et al., 2013). In these lesions, mast cells and macrophages are associated with inorganic polyphosphates that activate neutrophils to release extracellular ‘traps’ composed of DNA. These traps have been implicated in tumor progression (Arelaki et al., 2018). Colon cancer cells can also attract mast cells that promote proliferation and tumor growth by releasing soluble mediators (Yu et al., 2018). The nature of these mediators and their overall contribution to cancer initiation, however, remains unclear.
Macrophages perform many niche-specific functions in the healthy colon (Davies et al., 2013). Many studies focus on their role in the cancer microenvironment as tumor-associated macrophages (TAMs) (Vitale et al., 2019). These cells, however, should be considered separately from the polarized macrophages that are involved in CRC initiation. TAMs are heterogeneous and express pro-inflammatory M1 macrophages that are positive for CD68, CD80, inducible nitric oxide synthase (iNOS) and TNF-α, or anti-inflammatory/pro-tumoral M2 macrophages that are positive for CD163, CD206 and Arg1 markers, or both – depending upon type, stage and immune composition of a tumor (Sainz et al., 2016). TAMs enhance the growth of existing tumors, promote metastasis, contribute to resistance to chemotherapy, and generally confer a poor prognosis. TAM plasticity is defined as the ability to polarize or depolarize, or adapt their phenotype, depending on signals in the microenvironment. For example, Toll-like receptor agonism, interferon-γ (IFNG) and TNF-α favor an M1 phenotype, whereas IL4, IL10 (Box 1), IL13, and TGF-β induce macrophage polarization from M1 to M2 (van Dalen et al., 2018). Perturbation of the gut microbiota can promote tumor growth and invasion through modulation of macrophage plasticity. The complexity of these interactions had been suggested for human sporadic CRCs, wherein increased Firmicutes and Bacteroides were observed in the gut microbiota of patients showing high infiltrations of M1 TAMs in their tumor masses (Kikuchi et al., 2020). In a xenograft model, intestinal antibiotic-induced dysbiosis activated (or polarized) macrophages and increased the growth of a grafted human CRC cell line (Wan et al., 2018).
Although there is extensive literature describing the functions of TAMs, much less is known about the role of macrophages and colonic parainflammation in tumor initiation. Histologic analyses have shown that an abundance of polarized macrophages is present in most colon adenomas (McLean et al., 2011). As the lesions they infiltrate are not malignant, these cells are not TAMs, usually express an M1 phenotype and show activation of NF-κB, p38 mitogen activated-protein kinase (MAPK) and c-Jun N-terminal kinase (JNK) pathways (Hardwick et al., 2001). In addition, prostaglandin-endoperoxide synthase 2 (PTGS2, also known as COX-2) (Box 1), TNF-α and iNOS are often induced (Ambs et al., 1998; Chapple et al., 2000). So far, mechanisms that maintain a state of chronic macrophage polarization and parainflammation have not been well defined. Among the numerous genes induced in M1-polarized macrophages, the one encoding the pro-carcinogenic enzyme PTGS2 (Box 1) plays an outsized role in cancer initiation. Inhibition of PTGS2 decreases tumor multiplicity in numerous murine models and, according to many randomized controlled clinical trials, prevents CRC (Fischer et al., 2011; Janakiram and Rao, 2014; Katona and Weiss, 2020). Among pathobionts, in vitro inhibition of PTGS2 by drugs or through gene knockdown in E. faecalis-infected macrophages diminished the development of CIN in bystander epithelial cells (Wang and Huycke, 2007). However, in ApcMin/+ mice, specific deletion of PTGS2 in myeloid cells but not in intestinal epithelial cells, showed no effect on intestinal tumor multiplicity (Cherukuri et al., 2014), yet again highlighting the difficulties that can occur when extrapolating findings from animal models to a human disease.
Agents that deplete colon macrophages and, thus, block or eliminate PTGS2 and other inflammatory cytokines, protect against CRC. In the E. faecalis-triggered IL10 knockout model of CRC, liposome-encapsulated clodronate selectively reduced colon macrophages, and prevented colitis and CRC (Watanabe et al., 2003; Yang et al., 2013). Clodronate is a bisphosphonate that depletes tissue macrophages by selectively inducing apoptosis. Zoledronate and alendronate are closely related bisphosphonates that were recently shown to suppress azoxymethane-induced CRC in rats (see Box 3) (Madka et al., 2020). Meta-analyses of bisphosphonate therapy in humans, which is usually given to treat osteoporosis, show that it is associated with marked reductions in CRC risk (Bonovas et al., 2013; Thosani et al., 2013). Overall, these data highlight the importance of parainflammation in CRC initiation and its possible associations with M1-polarized macrophages, Tregs, Tr1 (Box 1) and TH17 cells. A more-holistic theory for CRC initiation is discussed in the following section that links dysbiosis and the gut microbiota with parainflammation, paligenosis and cells-of-origin for CSCs.
Microbiota-induced bystander effect and initiation of colorectal cancer
MIBE is a term that describes the paracrine interaction of polarized macrophages with colon epithelial cells to generate DNA damage and to initiate malignant transformation (Wang et al., 2017). Among several mechanisms by which intestinal pathobionts initiate CRC (Table 2), MIBE uniquely links immune activation and parainflammation to genomic instability, epigenetic change, the development of cells-of-origin through induced pluripotency and top-down morphogenesis. Inflammation is a recognized hallmark of cancer (Hanahan and Weinberg, 2011) and colonic parainflammation is likely to play a prominent role in the initiation of sporadic CRC.
Our group has studied MIBE as a generalized mechanism for the transformation of somatic epithelial cells into CSCs (Wang et al., 2013; Wang and Huycke, 2007, 2015; Yang et al., 2013). We have investigated MIBE by using an E. faecalis colonized IL10 knockout mouse model for colitis-associated CRC. In this model, E. faecalis, as a human commensal and opportunistic pathobiont, chronically polarizes colon macrophages to a pro-inflammatory M1 phenotype that generates endogenous mutagens and inflammatory cytokines. Colon epithelial cells targeted by M1 macrophages sustain double-strand DNA damage, mutations, tetraploidy and aneuploidy (Wang et al., 2008; Wang and Huycke, 2007; Yang et al., 2012). Repetitive exposure of primary colon epithelial cells to macrophages polarized by E. faecalis leads to malignant transformation (Wang et al., 2015). As bystanders to these macrophages, intestinal epithelial cells develop Wnt/β-catenin signaling and induce transcription factors associated with induced pluripotency (Wang et al., 2017). Conceptually, MIBE is analogous to well-characterized observations in radiation biology, showing that irradiated cells can produce soluble factors that generate mutations and genomic instability in non-irradiated bystander cells (Mothersill and Seymour, 2004). A comparison of MIBE to radiation-induced bystander effect (RIBE) has provided important new insights into the mechanisms of CRC initiation (see Box 2 and Fig. 2).
Fig. 2.

Radiation-induced bystander effect (RIBE). The bystander effect can be generated by cells (orange) that are directly ‘activated’ by irradiation or can arise from the progeny of irradiated cells (not shown). Myeloid and mesenchymal cells are the cells that typically generate a bystander effect. Subsets of normal chromosomally stable bystander (or target) cells (gray) that are exposed to diffusible mediators generated by irradiated cells develop genomic instability (pink cells). DNA damage is usually observed in either the first generation of exposed cells (I) or in the descendants of exposed cells (II). Modified with permission from Springer Nature (Lorimore et al., 2003).
In the MIBE paradigm, the activation of immune effector cells by intestinal commensals creates endogenous mutational stress on colon epithelial cells, while simultaneously instigating epigenetic changes that drive malignant transformation (Fig. 3). MIBE shares many characteristics with RIBE. Macrophages in MIBE are chronically polarized and serve as effectors for DNA damage and CIN. This is analogous to the polarization of macrophages by irradiation to cause RIBE (Burr et al., 2010). In these scenarios, a bystander effect is induced by polarized macrophages, generating diffusible mediators and cytokines that damage DNA in targeted cells. Examples of these mediators include 4-hydroxy-2-nonenal (4-HNE), a breakdown product of ω-6 polyunsaturated acids and by-product of PTGS2, TNF-α and ROS (Burr et al., 2010; Yan et al., 2006; Yang et al., 2012; Zhou et al., 2005). In addition, both MIBE and RIBE depend on PTGS2 activity (Wang et al., 2013; Zhou et al., 2005). In one animal model, colon macrophages were implicated as the primary effector cells for MIBE in CRC (Yang et al., 2013), although neutrophils and Tregs are also likely to play important roles (Lakritz et al., 2015; Rao et al., 2006).
Fig. 3.

Proposed mechanism for sporadic colorectal cancer (CRC) initiation by a microbiota-induced bystander effect (MIBE). (A) Normal colon crypt: Normal homeostasis in a colon crypt involves healthy microbiota, adequate mucus and tight junctions to form an intact intestinal barrier, and efficient shedding and engulfment of apoptotic cells at the crypt apex. Tissue macrophages are maintained in a tolerogenic state (M0) through the regulatory influences of TGF-β expressed by phagocytic macrophages and anti-inflammatory actions of IL10 produced by Tr1 and Treg cells. MIBE and parainflammation: Aging leads to increased immune senescence and dysbiosis, which impair the homeostatic inhibition of mucosal macrophages. Disruption of the intestinal barrier, e.g. through the loss of mucus or tight junction integrity, contributes to aberrant macrophage polarization. This results in parainflammation and the polarization of tolerogenic M0 macrophages to pro-inflammatory M1 macrophages. Parainflammation is maintained through TH17 pathways. Top-down morphogenesis: Prolonged MIBE-mediated changes lead to abnormal, i.e. dysplastic, cells that collect in the upper part of the crypt. Although dysplastic colonocytes accumulate DNA and epigenetic changes, most small adenomas revert to normal histology upon restoration of the intestinal barrier or resolution of dysbiosis. Adenoma: However, some proliferative lesions continue to develop during chronic parainflammation. At this stage, precursor lesions acquire sufficient genomic instability to become cells-of-origin that can be transformed into cancer stem cells (CSCs). CRC: Chronic MIBE, therefore, may culminate in the emergence of CSCs, and tumors that feature colonizing pathobionts and tumor-associated immune cells, such as tumor-associated macrophages and myeloid-derived suppressor cells. (B) Close-up of a colon crypt affected by dysbiosis, loss of barrier integrity, MIBE and parainflammation. These processes activate NF-κB signaling in M1 macrophages, which induces PTGS2 and iNOS proteins, and the generation of genotoxic and pro-proliferative paracrine factors, such as reactive oxygen species (ROS), genotoxins (e.g. 4-HNE), cytokines (e.g. TNF-α), chemokines (e.g. CCL2) and prostaglandins. Additional mediators of the bystander effect may include extracellular DNA (ecDNA) and extracellular vesicles (EVs). These diffusible factors cause epithelial cell injury, leading to induced pluripotency and the expression of stem-like markers (dedifferentiation), as well as proliferation through paligenosis, mutations, genomic instability, epigenetic modifications, the activation of β-catenin and, ultimately to the creation of cells-of-origin for CSCs.
Mouse models of MIBE
MIBE has been primarily investigated using E. faecalis as a pathobiont in the colitis-associated Il10 knockout (Il10−/−) murine model of CRC (Kim et al., 2005; Wang et al., 2008, 2015, 2012; Wang and Huycke, 2007; Yang et al., 2013, 2012). Under germ-free or pathogen-free conditions, Il10−/− mice are healthy and show no evidence of colitis or cancer. However, when these mice are colonized with selected pathobionts, such as E. faecalis, E. coli or Helicobacter spp., colon macrophages become chronically polarized, followed by the subsequent formation of cancer (Balish and Warner, 2002; Kim et al., 2005; Sellon et al., 1998). These macrophages bear resemblance to the M1-polarized macrophages observed in human colon adenomas. These models, however, are complicated by observations that inflammation alone does not drive carcinogenesis. Colonization with E. faecalis causes colitis in Il10−/− mice but the extracellular production of superoxide appears necessary for cancer formation (Wang et al., 2012). Similarly, E. coli causes colitis in Il10−/− mice but colibactin is required for colon tumors to form (Arthur et al., 2012). Such findings highlight the importance of specific bacterial phenotypes, e.g. the production of colibactin or the formation of superoxide – and not just the global presence of species or genera, in the initiation of microbiota-driven carcinogenesis.
The Rag2−/−/ApcMin/+ model of intestinal carcinogenesis represents a variant of the bystander effect following intestinal colonization with H. hepaticus or Campylobacter jejuni (Rao et al., 2007). Instead of promoting adenoma formation, these bacteria initiate distant tumors in the mammary tissue of Rag2−/−/ApcMin/+ mice. Similar observations have been made using Tg(C3-1-TAg)cJeg/JegJ mice colonized with H. hepaticus (Lakritz et al., 2015). In these models, inflammatory cytokines (e.g. TNF-α and IFNG), together with ROS and diffusible mutagens, are implicated as mediators for the bystander effect.
MIBE and CRC risk factors
Any credible theory regarding initiation of CRC should conform to known risk factors for CRC. MIBE largely fits this requirement. Age is by far the greatest risk factor for CRC. In the USA, the age-adjusted incidence rate for CRC per 100,000 increases from ∼10 for individuals of 40-49 years of age to >140 for individuals of >80 years of age (Ansa et al., 2018). This increased risk is not solely due to the occurrence of spontaneous somatic mutations in adult crypt stem cells. For colon crypt stem cells, the average rate of spontaneous mutation was estimated to be 40 per year, with small intestinal crypt stem cells exhibiting a similar rate (Blokzijl et al., 2016). However, the overall age-adjusted incidence of CRC is 100-fold higher than it is for adenocarcinoma of the small intestine (Qubaiah et al., 2010). It would, therefore, seem that age-related mutations alone do not account for the higher incidence of CRC.
Lifestyle is an additional major risk factor for CRC and, as such, has been extensively investigated. Unfortunately, collinearity between dietary factors, such as sugar, fruit, vegetable and fiber intake, and other behaviors or traits, such as physical activity, alcohol consumption, smoking and body-mass index, limit the ability to isolate their independent effects on the overall risk of developing CRC (Chan and Giovannucci, 2010). In addition, the magnitude of risk due to these factors is considerably smaller than age, with average age-adjusted relative risks typically >80-95% less.
Dysbiosis in gut microbiota has become the latest risk consideration for CRC (Janney et al., 2020). Evidence for this largely derives from associations between intestinal dysbiosis − or loss of healthy intestinal commensals that limit the outgrowth of pathobionts – and CRC (Dai et al., 2018; Levy et al., 2017; Sobhani et al., 2011; Wirbel et al., 2019). Perturbations in the gut microbiota correlate with changes in diet, antibiotic use and systemic or localized inflammation, and become increasingly prevalent with advancing age (Buford, 2017). Dysbiosis allows CRC-associated pathobionts to emerge and, potentially, to initiate CRC; for example, through mechanisms as listed in Table 2. Furthermore, as people age, the immune system becomes increasingly subjected to immunosenescence and to chronic, low-grade inflammation or ‘inflammaging’ (Box 1) (Franceschi et al., 2000; Fulop et al., 2017). This effect has been demonstrated in mice, where age-associated dysbiosis of the gut microbiota promotes intestinal permeability and systemic inflammation mediated by TNF-α (Thevaranjan et al., 2017). The dual effects of dysbiosis and immunosenescence/inflammaging is likely to cause dysregulation of host tolerance to gut commensals and a predisposition to the aberrant activation of innate immune cells that generate MIBE. Immune activation is seen as parainflammation in the precursor lesions for sporadic CRC.
Macrophages as primary effectors for MIBE
Macrophages serve several key roles in gut homeostasis (Joeris et al., 2017). They clear apoptotic cells via efferocytosis and transiting bacteria via phagocytosis, perform tissue remodeling, and interact with T cells and innate lymphoid cells. They sample luminal antigens and transfer those antigens to neighboring dendritic cells. Under normal conditions, intestinal macrophages resist polarization from a resting M0 phenotype. This is primarily due to the anti-inflammatory effects of IL10 produced by Tregs and Tr1 cells (Bogdan et al., 1991; Chen et al., 2003; Vieira et al., 2004), TGF-β produced by phagocytic cells (Perruche et al., 2008), and lack of expression of innate response receptors (Smythies et al., 2005). Despite homeostatic tolerance, human intestinal macrophages retain robust phagocytic and bactericidal activity.
However, with advancing age, macrophages develop pro-inflammatory phenotypes, particularly in adipose and hepatic tissues (Jackaman et al., 2017). Peritoneal macrophages from aged mice are hyper-responsive to activation and show enhanced production of ROS (Smallwood et al., 2011). Other studies, however, found that aged peritoneal and splenic macrophages show decreased responsiveness to inflammatory stimuli (Yoon et al., 2004) but, a careful evaluation of age-related changes in human or rodent intestinal macrophages has yet to be carried out. Despite a lack of evidence, the importance of macrophage polarization to tumor initiation is supported by studies that used Il10−/− mice colonized with E. faecalis or E. coli, transgenic mice overexpressing the transcription inhibitor Zeb2 or the ApcMin/+ mouse model, as well as the treatment of mice with AOM and dextran sulfate sodium (DSS; Box 1) to induce colitis and tumor formation. In these systems, depletion of colon macrophages or blockage of macrophage migration in response to C-C motif chemokine ligand 2 (CCL2) effectively inhibits microbiota-driven intestinal inflammation and reduces or abolishes tumor formation (Gu et al., 2019; Popivanova et al., 2009; Slowicka et al., 2020; Watanabe et al., 2003; Yang et al., 2013). One study, in which mice were treated with DSS describes increased neutrophil infiltration and colitis when macrophages and dendritic cells were depleted (Qualls et al., 2006). However, this study did not assess cancer endpoints. Overall, these findings reinforce a role for polarized macrophages as primary effector cells in the initiation of CRC.
In humans, the importance of MIBE and intestinal macrophages in CRC initiation has only been investigated indirectly. Bisphosphonates are a class of drugs that inhibit macrophage function (Rogers et al., 2011). They are primarily prescribed to treat or prevent high-risk postmenopausal osteoporosis (Tella and Gallagher, 2014) and act by inhibiting osteoclasts, the specialized multinucleated macrophages that resorb bone. Large, retrospective cohort studies of subjects taking bisphosphonates report that these drugs significantly reduce CRC risk (Bonovas et al., 2013; Thosani et al., 2013). Another line of evidence involves the analysis of tissue macrophages in human colon adenomas. These studies found macrophages in human colon adenomas are often polarized to an M1 phenotype and express PTGS2, iNOS and TNF-α (Ambs et al., 1998; Chapple et al., 2000; Hardwick et al., 2001; McLean et al., 2011). In the normal healthy colon, none of these enzymes are expressed. iNOS can be induced by multiple cells types, including polarized macrophages, and forms the radical nitric oxide that has competing effects on carcinogenesis (Janakiram and Rao, 2014). PTGS2, in particular, is a pro-carcinogenic enzyme that catalyzes the synthesis of prostaglandins with anti-apoptotic, pro-angiogenic, and pro-proliferative properties (Janakiram and Rao, 2014; Wu et al., 2010). Other less well-known by-products of M1-polarized macrophages include 4-HNE, a diffusible aldehyde and potent mutagen (Gueraud, 2017; Wang et al., 2012). Finally, both TNF-α and IL1β are secreted by M1-polarized macrophages. These cytokines activate multiple signaling pathways in colon epithelial cells that inhibit apoptosis, generate mutations and promote inflammation (Lasry et al., 2016; Rao et al., 2006; Yan et al., 2006; Yang et al., 2012).
MIBE and induced pluripotency
MIBE represents a source of colon epithelial cell injury that can lead to reprogramming and induced pluripotency of fully differentiated post-mitotic cells. Under chronic mutagenic and proliferative pressures, these cells are primed to enter a pathway towards transformation (Wang et al., 2017). However, current evidence for MIBE remains limited. The repetitive in vitro exposure of primary murine colon epithelial cells to E. faecalis-triggered MIBE increased the expression of Dclk1 and other stem-like markers. The end result was malignant transformation (Wang et al., 2015, 2017). Cancerous clones from these experiments showed the activation of Myc and the induction Klf4, Oct4 and Sox2 (Wang et al., 2017). These transcription factors are considered key regulators of induced pluripotency (Takahashi and Yamanaka, 2006).
Colonic epithelial cells that express Dclk1 are attractive candidates for MIBE-induced pluripotency and as cells-of-origin for CSCs within CRCs (Chandrakesan et al., 2014; Nakanishi et al., 2013; Schwitalla et al., 2013; Westphalen et al., 2014). Intestinal epithelial cells that express this kinase participate in the repair of mucosal injury and DNA damage (Chandrakesan et al., 2016; May et al., 2014). In ApcMin/+ mice, Dclk1-positive colon epithelial cells show increased expression of pluripotency and self-renewal markers, and knockdown of Dclk1 in these mice attenuated intestinal adenoma formation (Chandrakesan et al., 2017). Finally, the induction of pluripotency in post-mitotic colon epithelial cells through MIBE could be consistent with the top-down morphogenesis as seen in colon adenomas (Fig. 3). MIBE arises from the polarization of – otherwise tolerogenic – colon macrophages, followed by reprogramming and damage to bystander (or target) epithelial cells. These effects include genomic instability, induced pluripotency and malignant transformation. However, despite evidence that MIBE can be generated in vitro and in vivo by selected members of the gut microbiome, direct evidence for this process in human disease has not – yet – been obtained. MIBE mediators, such as TNF-α, 4-HNE and ROS, have been detected in human colon adenomas, although such associations do not prove causation and many questions remain unanswered. For example, does intestinal dysbiosis in humans promote the polarization of colon macrophages? Does human aging sensitize intestinal macrophages to pathogenic polarization by the gut microbiome? What evidence is there for induced pluripotency in dysplastic cells that form small human adenomas with top-down morphology? Are biochemical signatures for paligenosis or epigenetic change evident in these adenomas? And, finally, does MIBE generate unique mutational signatures in CRC? Answers to these questions and any evidence thereof would support MIBE as an important mechanism of carcinogenesis in human disease and would further advance our understanding of how the gut microbiota initiates sporadic CRC.
Conclusions and future directions
Sporadic CRC arises within a complex milieu of overlapping risk factors that include age, host genetics, lifestyle and the gut microbiota. Understanding the mechanisms of microbiota-induced CRC initiation is essential for developing new prevention strategies for this disease. Recent investigations have identified several members of the human microbiota that express phenotypes that contribute to CRC initiation. The mechanisms by which human microbiota contribute to CRC initiation include the activation of β-catenin/Myc signaling, production of ROS, expression of DNA-damaging toxins and microbiota-induced polarization of colon macrophages to generate a bystander effect. Although there is limited evidence for MIBE contributing to CRC initiation in humans, the histology and immunology of sporadic human colon adenomas – together with findings from multiple animal models – link MIBE to parainflammation, paligenosis and top-down morphogenesis, as seen in precursor lesions to CRC. As such, MIBE has the potential to support multiple mechanisms by which gut bacteria initiate sporadic CRC. It provides a broad context for the major risk cofactors of CRC and helps to explain the preventive efficacy of anti-inflammatory drugs. Nevertheless, additional work is needed to better understand how intestinal barrier function, dysbiosis, and macrophage tolerance and responsiveness change with age. Finally, despite much recent progress, there are insufficient data on members of the gut microbiota that can chronically polarize colon macrophages and generate MIBE. Exploring these issues will help clarify how aging, intestinal dysbiosis and immunosenescence drive initiation of CRC.
In conclusion, a healthy and stable gut microbiota helps to maintain the mutually beneficial relationship between these microorganisms and the immune system. Perturbations of intestinal homeostasis due to aging and/or immunosenescence permits the emergence of bacterial pathobionts that can initiate DNA damage and reprogramming in colon epithelial cells through a bystander effect and, potentially, lead to malignant transformation over time. Unraveling the spectrum of mutations, epigenetic changes and induced pluripotency that occur in CRC is important in order to identify the cells-of-origin for CSCs within CRCs and to provide new strategies that can be used to prevent this disease.
Acknowledgements
We thank Drs C. V. Rao, Courtney W. Houchen, and Jason C. Mills for comments on the manuscript.
Footnotes
Funding
M.M.H. is supported by a R01 grant from the National Cancer Institute (CA230641) and the Stephenson Cancer Center. X.W. is supported by the National Natural Science Foundation of China (8197278) and the Jiangsu Commission of Health (H2018109 and Distinguished Medical Expert Program). N.A. is supported by the National Institutes of Health through an Oklahoma-IDeA Network of Biomedical Research Excellence grant (C3145817).
References
- Adachi, K. and Scholer, H. R. (2012). Directing reprogramming to pluripotency by transcription factors. Curr. Opin. Genet. Dev. 22, 416-422. 10.1016/j.gde.2012.07.001 [DOI] [PubMed] [Google Scholar]
- Al-Mayah, A., Bright, S., Chapman, K., Irons, S., Luo, P., Carter, D., Goodwin, E. and Kadhim, M. (2015). The non-targeted effects of radiation are perpetuated by exosomes. Mutat. Res. 772, 38-45. 10.1016/j.mrfmmm.2014.12.007 [DOI] [PubMed] [Google Scholar]
- Alexandrov, L. B., Kim, J., Haradhvala, N. J., Huang, M. N., Tian Ng, A. W., Wu, Y., Boot, A., Covington, K. R., Gordenin, D. A., Bergstrom, E. N.et al. (2020). The repertoire of mutational signatures in human cancer. Nature 578, 94-101. 10.1038/s41586-020-1943-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen, J. and Sears, C. L. (2019). Impact of the gut microbiome on the genome and epigenome of colon epithelial cells: contributions to colorectal cancer development. Genome Med. 11, 11. 10.1186/s13073-019-0621-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambs, S., Merriam, W. G., Bennett, W. P., Felley-Bosco, E., Ogunfusika, M. O., Oser, S. M., Klein, S., Shields, P. G., Billiar, T. R. and Harris, C. C. (1998). Frequent nitric oxide synthase-2 expression in human colon adenomas: implication for tumor angiogenesis and colon cancer progression. Cancer Res. 58, 334-341. [PubMed] [Google Scholar]
- Ansa, B. E., Coughlin, S. S., Alema-Mensah, E. and Smith, S. A. (2018). Evaluation of colorectal cancer incidence trends in the United States (2000-2014). J. Clin. Med. 7, 22. 10.3390/jcm7020022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arelaki, S., Arampatzioglou, A., Kambas, K., Sivridis, E., Giatromanolaki, A. and Ritis, K. (2018). Mast cells co-expressing CD68 and inorganic polyphosphate are linked with colorectal cancer. PLoS ONE 13, e0193089. 10.1371/journal.pone.0193089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariyoshi, K., Miura, T., Kasai, K., Fujishima, Y., Nakata, A. and Yoshida, M. (2019). Radiation-induced bystander effect is mediated by mitochondrial DNA in exosome-like vesicles. Sci. Rep. 9, 9103. 10.1038/s41598-019-45669-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur, J. C., Perez-Chanona, E., Muhlbauer, M., Tomkovich, S., Uronis, J. M., Fan, T. J., Campbell, B. J., Abujamel, T., Dogan, B., Rogers, A. B.et al. (2012). Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338, 120-123. 10.1126/science.1224820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur, J. C., Gharaibeh, R. Z., Mühlbauer, M., Perez-Chanona, E., Uronis, J. M., McCafferty, J., Fodor, A. A. and Jobin, C. (2014). Microbial genomic analysis reveals the essential role of inflammation in bacteria-induced colorectal cancer. Nat. Commun. 5, 4724. 10.1038/ncomms5724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzam, E. I., De Toledo, S. M., Spitz, D. R. and Little, J. B. (2002). Oxidative metabolism modulates signal transduction and micronucleus formation in bystander cells from alpha-particle-irradiated normal human fibroblast cultures. Cancer Res. 62, 5436-5442. [PubMed] [Google Scholar]
- Azzam, E. I., de Toledo, S. M. and Little, J. B. (2003). Oxidative metabolism, gap junctions and the ionizing radiation-induced bystander effect. Oncogene 22, 7050-7057. 10.1038/sj.onc.1206961 [DOI] [PubMed] [Google Scholar]
- Bach, D. H., Zhang, W. and Sood, A. K. (2019). Chromosomal instability in tumor initiation and development. Cancer Res. 79, 3995-4002. 10.1158/0008-5472.CAN-18-3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balish, E. and Warner, T. (2002). Enterococcus faecalis induces inflammatory bowel disease in interleukin-10 knockout mice. Am. J. Pathol. 160, 2253-2257. 10.1016/S0002-9440(10)61172-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker, N., Ridgway, R. A., van Es, J. H., van de Wetering, M., Begthel, H., van den Born, M., Danenberg, E., Clarke, A. R., Sansom, O. J. and Clevers, H. (2009). Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 457, 608-611. 10.1038/nature07602 [DOI] [PubMed] [Google Scholar]
- Belcheva, A., Irrazabal, T., Robertson, S. J., Streutker, C., Maughan, H., Rubino, S., Moriyama, E. H., Copeland, J. K., Surendra, A., Kumar, S.et al. (2014). Gut microbial metabolism drives transformation of MSH2-deficient colon epithelial cells. Cell 158, 288-299. 10.1016/j.cell.2014.04.051 [DOI] [PubMed] [Google Scholar]
- Berg, G., Rybakova, D., Fischer, D., Cernava, T., Verges, M. C., Charles, T., Chen, X., Cocolin, L., Eversole, K., Corral, G. H.et al. (2020). Microbiome definition re-visited: old concepts and new challenges. Microbiome 8, 103. 10.1186/s40168-020-00875-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstrom, K., Liu, X., Zhao, Y., Gao, N., Wu, Q., Song, K., Cui, Y., Li, Y., McDaniel, J. M., McGee, S.et al. (2016). Defective intestinal mucin-type O-glycosylation causes spontaneous colitis-associated cancer in mice. Gastroenterology 151, 152-164.e11. 10.1053/j.gastro.2016.03.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blokzijl, F., de Ligt, J., Jager, M., Sasselli, V., Roerink, S., Sasaki, N., Huch, M., Boymans, S., Kuijk, E., Prins, P.et al. (2016). Tissue-specific mutation accumulation in human adult stem cells during life. Nature 538, 260-264. 10.1038/nature19768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdan, C., Vodovotz, Y. and Nathan, C. (1991). Macrophage deactivation by interleukin 10. J. Exp. Med. 174, 1549-1555. 10.1084/jem.174.6.1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolhaqueiro, A. C. F., Ponsioen, B., Bakker, B., Klaasen, S. J., Kucukkose, E., van Jaarsveld, R. H., Vivié, J., Verlaan-Klink, I., Hami, N., Spierings, D. C. J.et al. (2019). Ongoing chromosomal instability and karyotype evolution in human colorectal cancer organoids. Nat. Genet. 51, 824-834. 10.1038/s41588-019-0399-6 [DOI] [PubMed] [Google Scholar]
- Bonovas, S., Nikolopoulos, G. and Bagos, P. (2013). Bisphosphonate use and risk of colorectal cancer: a systematic review and meta-analysis. Br. J. Clin. Pharmacol. 76, 329-337. 10.1111/bcp.12135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonanantanasarn, K., Gill, A. L., Yap, Y., Jayaprakash, V., Sullivan, M. A. and Gill, S. R. (2012). Enterococcus faecalis enhances cell proliferation through hydrogen peroxide-mediated epidermal growth factor receptor activation. Infect. Immun. 80, 3545-3558. 10.1128/IAI.00479-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A. and Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394-424. 10.3322/caac.21492 [DOI] [PubMed] [Google Scholar]
- Brown, D. G., Rao, S., Weir, T. L., O'Malia, J., Bazan, M., Brown, R. J. and Ryan, E. P. (2016). Metabolomics and metabolic pathway networks from human colorectal cancers, adjacent mucosa, and stool. Cancer Metab. 4, 11. 10.1186/s40170-016-0151-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buc, E., Dubois, D., Sauvanet, P., Raisch, J., Delmas, J., Darfeuille-Michaud, A., Pezet, D. and Bonnet, R. (2013). High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS ONE 8, e56964. 10.1371/journal.pone.0056964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buford, T. W. (2017). (Dis)Trust your gut: the gut microbiome in age-related inflammation, health, and disease. Microbiome 5, 80. 10.1186/s40168-017-0296-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burclaff, J. and Mills, J. C. (2018). Plasticity of differentiated cells in wound repair and tumorigenesis, part II: skin and intestine. Dis. Model. Mech. 11, dmm035071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdak-Rothkamm, S. and Rothkamm, K. (2018). Radiation-induced bystander and systemic effects serve as a unifying model system for genotoxic stress responses. Mutat. Res. 778, 13-22. 10.1016/j.mrrev.2018.08.001 [DOI] [PubMed] [Google Scholar]
- Burr, K. L., Robinson, J. I., Rastogi, S., Boylan, M. T., Coates, P. J., Lorimore, S. A. and Wright, E. G. (2010). Radiation-induced delayed bystander-type effects mediated by hemopoietic cells. Radiat. Res. 173, 760-768. 10.1667/RR1937.1 [DOI] [PubMed] [Google Scholar]
- Cammareri, P., Vincent, D. F., Hodder, M. C., Ridgway, R. A., Murgia, C., Nobis, M., Campbell, A. D., Varga, J., Huels, D. J., Subramani, C.et al. (2017). TGFβ pathway limits dedifferentiation following WNT and MAPK pathway activation to suppress intestinal tumourigenesis. Cell Death Differ. 24, 1681-1693. 10.1038/cdd.2017.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, A. T. and Giovannucci, E. L. (2010). Primary prevention of colorectal cancer. Gastroenterology 138, 2029-2043.e10. 10.1053/j.gastro.2010.01.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrakesan, P., Weygant, N., May, R., Qu, D., Chinthalapally, H. R., Sureban, S. M., Ali, N., Lightfoot, S. A., Umar, S. and Houchen, C. W. (2014). DCLK1 facilitates intestinal tumor growth via enhancing pluripotency and epithelial mesenchymal transition. Oncotarget 5, 9269-9280. 10.18632/oncotarget.2393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrakesan, P., May, R., Weygant, N., Qu, D., Berry, W. L., Sureban, S. M., Ali, N., Rao, C., Huycke, M., Bronze, M. S.et al. (2016). Intestinal tuft cells regulate the ATM mediated DNA damage response via Dclk1 dependent mechanism for crypt restitution following radiation injury. Sci. Rep. 6, 37667. 10.1038/srep37667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrakesan, P., Yao, J., Qu, D., May, R., Weygant, N., Ge, Y., Ali, N., Sureban, S. M., Gude, M., Vega, K.et al. (2017). Dclk1, a tumor stem cell marker, regulates pro-survival signaling and self-renewal of intestinal tumor cells. Mol. Cancer 16, 30. 10.1186/s12943-017-0594-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapple, K. S., Cartwright, E. J., Hawcroft, G., Tisbury, A., Bonifer, C., Scott, N., Windsor, A. C., Guillou, P. J., Markham, A. F., Coletta, P. L.et al. (2000). Localization of cyclooxygenase-2 in human sporadic colorectal adenomas. Am. J. Pathol. 156, 545-553. 10.1016/S0002-9440(10)64759-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, W., Jin, W., Hardegen, N., Lei, K. J., Li, L., Marinos, N., McGrady, G. and Wahl, S. M. (2003). Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J. Exp. Med. 198, 1875-1886. 10.1084/jem.20030152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, T., Li, Q., Wu, J., Wu, Y., Peng, W., Li, H., Wang, J., Tang, X., Peng, Y. and Fu, X. (2018). Fusobacterium nucleatum promotes M2 polarization of macrophages in the microenvironment of colorectal tumours via a TLR4-dependent mechanism. Cancer Immunol. Immunother. 67, 1635-1646. 10.1007/s00262-018-2233-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherukuri, D. P., Ishikawa, T. O., Chun, P., Catapang, A., Elashoff, D., Grogan, T. R., Bugni, J. and Herschman, H. R. (2014). Targeted Cox2 gene deletion in intestinal epithelial cells decreases tumorigenesis in female, but not male, ApcMin/+ mice. Mol. Oncol. 8, 169-177. 10.1016/j.molonc.2013.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, L., Thiele Orberg, E., Geis, A. L., Chan, J. L., Fu, K., DeStefano Shields, C. E., Dejea, C. M., Fathi, P., Chen, J., Finard, B. B.et al. (2018). Bacteroides fragilis toxin coordinates a pro-carcinogenic inflammatory cascade via targeting of colonic epithelial cells. Cell Host Microbe 23, 203-214.e5. 10.1016/j.chom.2018.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coker, O. O., Nakatsu, G., Dai, R. Z., Wu, W. K. K., Wong, S. H., Ng, S. C., Chan, F. K. L., Sung, J. J. Y. and Yu, J. (2019). Enteric fungal microbiota dysbiosis and ecological alterations in colorectal cancer. Gut 68, 654-662. 10.1136/gutjnl-2018-317178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole, J. W. and McKalen, A. (1963). Studies on the morphogenesis of adenomatous polyps in the human colon. Cancer 16, 998-1002. [DOI] [PubMed] [Google Scholar]
- Corbett, A. J., Eckle, S. B., Birkinshaw, R. W., Liu, L., Patel, O., Mahony, J., Chen, Z., Reantragoon, R., Meehan, B., Cao, H.et al. (2014). T-cell activation by transitory neo-antigens derived from distinct microbial pathways. Nature 509, 361-365. 10.1038/nature13160 [DOI] [PubMed] [Google Scholar]
- Cougnoux, A., Dalmasso, G., Martinez, R., Buc, E., Delmas, J., Gibold, L., Sauvanet, P., Darcha, C., Déchelotte, P., Bonnet, M.et al. (2014). Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut 63, 1932-1942. 10.1136/gutjnl-2013-305257 [DOI] [PubMed] [Google Scholar]
- Crockett, S. D. and Nagtegaal, I. D. (2019). Terminology, molecular features, epidemiology, and management of serrated colorectal neoplasia. Gastroenterology 157, 949-966.e4. 10.1053/j.gastro.2019.06.041 [DOI] [PubMed] [Google Scholar]
- Cuevas-Ramos, G., Petit, C. R., Marcq, I., Boury, M., Oswald, E. and Nougayrède, J.-P. (2010). Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc. Natl. Acad. Sci. USA 107, 11537-11542. 10.1073/pnas.1001261107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, Z., Coker, O. O., Nakatsu, G., Wu, W. K. K., Zhao, L., Chen, Z., Chan, F. K. L., Kristiansen, K., Sung, J. J. Y. and Wong, S. H. (2018). Multi-cohort analysis of colorectal cancer metagenome identified altered bacteria across populations and universal bacterial markers. Microbiome 6, 70. 10.1186/s40168-018-0451-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, L. C., Jenkins, S. J., Allen, J. E. and Taylor, P. R. (2013). Tissue-resident macrophages. Nat. Immunol. 14, 986-995. 10.1038/ni.2705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejea, C. M., Fathi, P., Craig, J. M., Boleij, A., Taddese, R., Geis, A. L., Wu, X., DeStefano Shields, C. E., Hechenbleikner, E. M., Huso, D. L.et al. (2018). Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 359, 592-597. 10.1126/science.aah3648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, C., He, M., Tu, W., Konishi, T., Liu, W., Xie, Y., Dang, B., Li, W., Uchihori, Y., Hei, T. K.et al. (2015). The differential role of human macrophage in triggering secondary bystander effects after either gamma-ray or carbon beam irradiation. Cancer Lett. 363, 92-100. 10.1016/j.canlet.2015.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunne, P. D., McArt, D. G., Bradley, C. A., O'Reilly, P. G., Barrett, H. L., Cummins, R., O'Grady, T., Arthur, K., Loughrey, M. B., Allen, W. L.et al. (2016). Challenging the cancer molecular stratification dogma: intratumoral heterogeneity undermines consensus molecular subtypes and potential diagnostic value in colorectal cancer. Clin. Cancer Res. 22, 4095-4104. 10.1158/1078-0432.CCR-16-0032 [DOI] [PubMed] [Google Scholar]
- Emerit, I., Garban, F., Vassy, J., Levy, A., Filipe, P. and Freitas, J. (1996). Superoxide-mediated clastogenesis and anticlastogenic effects of exogenous superoxide dismutase. Proc. Natl. Acad. Sci. USA 93, 12799-12804. 10.1073/pnas.93.23.12799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdman, S. E., Poutahidis, T., Tomczak, M., Rogers, A. B., Cormier, K., Plank, B., Horwitz, B. H. and Fox, J. G. (2003). CD4+ CD25+ regulatory T lymphocytes inhibit microbially induced colon cancer in Rag2-deficient mice. Am. J. Pathol. 162, 691-702. 10.1016/S0002-9440(10)63863-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eslami, M., Yousefi, B., Kokhaei, P., Hemati, M., Nejad, Z. R., Arabkari, V. and Namdar, A. (2019). Importance of probiotics in the prevention and treatment of colorectal cancer. J. Cell. Physiol. 234, 17127-17143. 10.1002/jcp.28473 [DOI] [PubMed] [Google Scholar]
- Faïs, T., Delmas, J., Barnich, N., Bonnet, R. and Dalmasso, G. (2018). Colibactin: more than a new bacterial toxin. Toxins (Basel) 10, 151. 10.3390/toxins10040151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer, S. M., Hawk, E. T. and Lubet, R. A. (2011). Coxibs and other nonsteroidal anti-inflammatory drugs in animal models of cancer chemoprevention. Cancer Prev. Res. (Phila) 4, 1728-1735. 10.1158/1940-6207.CAPR-11-0166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschi, C., Bonafè, M., Valensin, S., Olivieri, F., De Luca, M., Ottaviani, E. and De Benedictis, G. (2000). Inflamm-aging: an evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 908, 244-254. 10.1111/j.1749-6632.2000.tb06651.x [DOI] [PubMed] [Google Scholar]
- Fu, J., Wang, J., Wang, X., Wang, P., Xu, J., Zhou, C., Bai, Y. and Shao, C. (2016). Signaling factors and pathways of α-particle irradiation induced bilateral bystander responses between Beas-2B and U937 cells. Mutat. Res. 789, 1-8. 10.1016/j.mrfmmm.2016.04.004 [DOI] [PubMed] [Google Scholar]
- Fulop, T., Larbi, A., Dupuis, G., Le Page, A., Frost, E. H., Cohen, A. A., Witkowski, J. M. and Franceschi, C. (2017). Immunosenescence and inflamm-aging as two sides of the same coin: friends or foes? Front. Immunol. 8, 1960. 10.3389/fimmu.2017.01960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geis, A. L., Fan, H., Wu, X., Wu, S., Huso, D. L., Wolfe, J. L., Sears, C. L., Pardoll, D. M. and Housseau, F. (2015). Regulatory T-cell response to enterotoxigenic Bacteroides fragilis colonization triggers IL17-dependent colon carcinogenesis. Cancer Discov. 5, 1098-1109. 10.1158/2159-8290.CD-15-0447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel, A., Li, M. S., Nagasaka, T., Shin, S. K., Fuerst, F., Ricciardiello, L., Wasserman, L. and Boland, C. R. (2006). Association of JC virus T-antigen expression with the methylator phenotype in sporadic colorectal cancers. Gastroenterology 130, 1950-1961. 10.1053/j.gastro.2006.02.061 [DOI] [PubMed] [Google Scholar]
- Goh, K. and Sumner, H. (1968). Breaks in normal human chromosomes: are they induced by a transferable substance in the plasma of persons exposed to total-body irradiation? Radiat. Res. 35, 171-181. 10.2307/3572443 [DOI] [PubMed] [Google Scholar]
- Goodwin, A. C., Destefano Shields, C. E., Wu, S., Huso, D. L., Wu, X., Murray-Stewart, T. R., Hacker-Prietz, A., Rabizadeh, S., Woster, P. M., Sears, C. L.et al. (2011). Polyamine catabolism contributes to enterotoxigenic Bacteroides fragilis-induced colon tumorigenesis. Proc. Natl. Acad. Sci. USA 108, 15354-15359. 10.1073/pnas.1010203108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gounaris, E., Blatner, N. R., Dennis, K., Magnusson, F., Gurish, M. F., Strom, T. B., Beckhove, P., Gounari, F. and Khazaie, K. (2009). T-regulatory cells shift from a protective anti-inflammatory to a cancer-promoting proinflammatory phenotype in polyposis. Cancer Res. 69, 5490-5497. 10.1158/0008-5472.CAN-09-0304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gow, M. D., Seymour, C. B., Ryan, L. A. and Mothersill, C. E. (2010). Induction of bystander response in human glioma cells using high-energy electrons: a role for TGF-β1. Radiat. Res. 173, 769-778. 10.1667/RR1895.1 [DOI] [PubMed] [Google Scholar]
- Greten, F. R. (2017). Cancer: tumour stem-cell surprises. Nature 543, 626-627. 10.1038/543626a [DOI] [PubMed] [Google Scholar]
- Gu, T., Li, Q. and Egilmez, N. K. (2019). IFNb-producing CX3CR1+ macrophages promote T-regulatory cell expansion and tumor growth in the APCmin/+/Bacteroides fragilis colon cancer model. Oncoimmunology 8, e1665975. 10.1080/2162402X.2019.1665975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueraud, F. (2017). 4-Hydroxynonenal metabolites and adducts in pre-carcinogenic conditions and cancer. Free Radic. Biol. Med. 111, 196-208. 10.1016/j.freeradbiomed.2016.12.025 [DOI] [PubMed] [Google Scholar]
- Guinney, J., Dienstmann, R., Wang, X., de Reynies, A., Schlicker, A., Soneson, C., Marisa, L., Roepman, P., Nyamundanda, G., Angelino, P.et al. (2015). The consensus molecular subtypes of colorectal cancer. Nat. Med. 21, 1350-1356. 10.1038/nm.3967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale, V. L., Jeraldo, P., Mundy, M., Yao, J., Keeney, G., Scott, N., Cheek, E. H., Davidson, J., Greene, M., Martinez, C.et al. (2018). Synthesis of multi-omic data and community metabolic models reveals insights into the role of hydrogen sulfide in colon cancer. Methods 149, 59-68. 10.1016/j.ymeth.2018.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan, D. and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646-674. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- Hannigan, G. D., Duhaime, M. B., Ruffin, M. T. T., Koumpouras, C. C. and Schloss, P. D. (2018). Diagnostic potential and interactive dynamics of the colorectal cancer virome. mBio 9. 10.1128/mBio.02248-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick, J. C., van den Brink, G. R., Offerhaus, G. J., van Deventer, S. J. and Peppelenbosch, M. P. (2001). NF-kappaB, p38 MAPK and JNK are highly expressed and active in the stroma of human colonic adenomatous polyps. Oncogene 20, 819-827. 10.1038/sj.onc.1204162 [DOI] [PubMed] [Google Scholar]
- He, Z., Gharaibeh, R. Z., Newsome, R. C., Pope, J. L., Dougherty, M. W., Tomkovich, S., Pons, B., Mirey, G., Vignard, J., Hendrixson, D. R.et al. (2019). Campylobacter jejuni promotes colorectal tumorigenesis through the action of cytolethal distending toxin. Gut 68, 289-300. 10.1136/gutjnl-2018-317200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helander, H. F. and Fandriks, L. (2014). Surface area of the digestive tract - revisited. Scand. J. Gastroenterol. 49, 681-689. 10.3109/00365521.2014.898326 [DOI] [PubMed] [Google Scholar]
- Hoevenaar, W. H. M., Janssen, A., Quirindongo, A. I., Ma, H., Klaasen, S. J., Teixeira, A., van Gerwen, B., Lansu, N., Morsink, F. H. M., Offerhaus, G. J. A.et al. (2020). Degree and site of chromosomal instability define its oncogenic potential. Nat. Commun. 11, 1501. 10.1038/s41467-020-15279-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huycke, M. M. and Moore, D. R. (2002). In vivo production of hydroxyl radical by Enterococcus faecalis colonizing the intestinal tract using aromatic hydroxylation. Free Radic. Biol. Med. 33, 818-826. 10.1016/S0891-5849(02)00977-2 [DOI] [PubMed] [Google Scholar]
- Huycke, M. M., Abrams, V. and Moore, D. R. (2002). Enterococcus faecalis produces extracellular superoxide and hydrogen peroxide that damages colonic epithelial cell DNA. Carcinogenesis 23, 529-536. 10.1093/carcin/23.3.529 [DOI] [PubMed] [Google Scholar]
- Ilnytskyy, Y., Koturbash, I. and Kovalchuk, O. (2009). Radiation-induced bystander effects in vivo are epigenetically regulated in a tissue-specific manner. Environ. Mol. Mutagen. 50, 105-113. 10.1002/em.20440 [DOI] [PubMed] [Google Scholar]
- Irrazabal, T., Belcheva, A., Girardin, S. E., Martin, A. and Philpott, D. J. (2014). The multifaceted role of the intestinal microbiota in colon cancer. Mol. Cell 54, 309-320. 10.1016/j.molcel.2014.03.039 [DOI] [PubMed] [Google Scholar]
- Jackaman, C., Tomay, F., Duong, L., Abdol Razak, N. B., Pixley, F. J., Metharom, P. and Nelson, D. J. (2017). Aging and cancer: the role of macrophages and neutrophils. Ageing Res. Rev. 36, 105-116. 10.1016/j.arr.2017.03.008 [DOI] [PubMed] [Google Scholar]
- Janakiram, N. B. and Rao, C. V. (2014). The role of inflammation in colon cancer. Adv. Exp. Med. Biol. 816, 25-52. 10.1007/978-3-0348-0837-8_2 [DOI] [PubMed] [Google Scholar]
- Jang, B. G., Kim, H. S., Kim, K. J., Rhee, Y. Y., Kim, W. H. and Kang, G. H. (2016). Distribution of intestinal stem cell markers in colorectal precancerous lesions. Histopathology 68, 567-577. 10.1111/his.12787 [DOI] [PubMed] [Google Scholar]
- Janney, A., Powrie, F. and Mann, E. H. (2020). Host-microbiota maladaptation in colorectal cancer. Nature 585, 509-517. 10.1038/s41586-020-2729-3 [DOI] [PubMed] [Google Scholar]
- Joeris, T., Müller-Luda, K., Agace, W. W. and Mowat, A. M. (2017). Diversity and functions of intestinal mononuclear phagocytes. Mucosal Immunol. 10, 845-864. 10.1038/mi.2017.22 [DOI] [PubMed] [Google Scholar]
- Katona, B. W. and Weiss, J. M. (2020). Chemoprevention of colorectal cancer. Gastroenterology 158, 368-388. 10.1053/j.gastro.2019.06.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur, H. and Moreau, R. (2019). Role of mTORC1 in intestinal epithelial repair and tumorigenesis. Cell. Mol. Life Sci. 76, 2525-2546. 10.1007/s00018-019-03085-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy, E. A., King, K. Y. and Baldridge, M. T. (2018). Mouse microbiota models: comparing germ-free mice and antibiotics treatment as tools for modifying gut bacteria. Front. Physiol. 9, 1534. 10.3389/fphys.2018.01534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi, T., Mimura, K., Ashizawa, M., Okayama, H., Endo, E., Saito, K., Sakamoto, W., Fujita, S., Endo, H., Saito, M.et al. (2020). Characterization of tumor-infiltrating immune cells in relation to microbiota in colorectal cancers. Cancer Immunol. Immunother. 69, 23-32. 10.1007/s00262-019-02433-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S. C., Tonkonogy, S. L., Albright, C. A., Tsang, J., Balish, E. J., Braun, J., Huycke, M. M. and Sartor, R. B. (2005). Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology 128, 891-906. 10.1053/j.gastro.2005.02.009 [DOI] [PubMed] [Google Scholar]
- Kirchberger, S., Royston, D. J., Boulard, O., Thornton, E., Franchini, F., Szabady, R. L., Harrison, O. and Powrie, F. (2013). Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J. Exp. Med. 210, 917-931. 10.1084/jem.20122308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirolikar, S., Prasannan, P., Raghuram, G. V., Pancholi, N., Saha, T., Tidke, P., Chaudhari, P., Shaikh, A., Rane, B., Pandey, R.et al. (2018). Prevention of radiation-induced bystander effects by agents that inactivate cell-free chromatin released from irradiated dying cells. Cell Death Dis. 9, 1142. 10.1038/s41419-018-1181-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konkova, M. S., Kaliyanov, A. A. and Sergeeva, V. A. (2019). Oxidized cell-free DNA is a factor of stress signaling in radiation-induced bystander effects in different types of human cells. Int. J. Genomics 2019, 9467029. 10.1155/2019/9467029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic, A. D., Chun, E., Robertson, L., Glickman, J. N., Gallini, C. A., Michaud, M., Clancy, T. E., Chung, D. C., Lochhead, P., Hold, G. L.et al. (2013). Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 14, 207-215. 10.1016/j.chom.2013.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, R., Herold, J. L., Schady, D., Davis, J., Kopetz, S., Martinez-Moczygemba, M., Murray, B. E., Han, F., Li, Y., Callaway, E.et al. (2017). Streptococcus gallolyticus subsp. gallolyticus promotes colorectal tumor development. PLoS Pathog. 13, e1006440. 10.1371/journal.ppat.1006440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakritz, J. R., Poutahidis, T., Mirabal, S., Varian, B. J., Levkovich, T., Ibrahim, Y. M., Ward, J. M., Teng, E. C., Fisher, B., Parry, N.et al. (2015). Gut bacteria require neutrophils to promote mammary tumorigenesis. Oncotarget 6, 9387-9396. 10.18632/oncotarget.3328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasry, A., Zinger, A. and Ben-Neriah, Y. (2016). Inflammatory networks underlying colorectal cancer. Nat. Immunol. 17, 230-240. 10.1038/ni.3384 [DOI] [PubMed] [Google Scholar]
- Lau, J. T., Whelan, F. J., Herath, I., Lee, C. H., Collins, S. M., Bercik, P. and Surette, M. G. (2016). Capturing the diversity of the human gut microbiota through culture-enriched molecular profiling. Genome Med. 8, 72. 10.1186/s13073-016-0327-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, H. O., Hong, Y., Etlioglu, H. E., Cho, Y. B., Pomella, V., Van den Bosch, B., Vanhecke, J., Verbandt, S., Hong, H., Min, J.-W.et al. (2020). Lineage-dependent gene expression programs influence the immune landscape of colorectal cancer. Nat. Genet. 52, 594-603. 10.1038/s41588-020-0636-z [DOI] [PubMed] [Google Scholar]
- Lennard, K. S., Goosen, R. W. and Blackburn, J. M. (2016). Bacterially-associated transcriptional remodelling in a distinct genomic subtype of colorectal cancer provides a plausible molecular basis for disease development. PLoS ONE 11, e0166282. 10.1371/journal.pone.0166282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesiow, M. K., Pietrzyk, P., Kyzioł, A. and Komarnicka, U. K. (2019). Cu(II) complexes with FomA protein fragments of Fusobacterium nucleatum increase oxidative stress and malondialdehyde level. Chem. Res. Toxicol. 32, 2227-2237. 10.1021/acs.chemrestox.9b00269 [DOI] [PubMed] [Google Scholar]
- Levy, M., Kolodziejczyk, A. A., Thaiss, C. A. and Elinav, E. (2017). Dysbiosis and the immune system. Nat. Rev. Immunol. 17, 219-232. 10.1038/nri.2017.7 [DOI] [PubMed] [Google Scholar]
- Li, Q., Peng, W., Wu, J., Wang, X., Ren, Y., Li, H., Peng, Y., Tang, X. and Fu, X. (2019). Autoinducer-2 of gut microbiota, a potential novel marker for human colorectal cancer, is associated with the activation of TNFSF9 signaling in macrophages. Oncoimmunology 8, e1626192. 10.1080/2162402X.2019.1626192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenstein, P., Holm, N. V., Verkasalo, P. K., Iliadou, A., Kaprio, J., Koskenvuo, M., Pukkala, E., Skytthe, A. and Hemminki, K. (2000). Environmental and heritable factors in the causation of cancer - analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med. 343, 78-85. 10.1056/NEJM200007133430201 [DOI] [PubMed] [Google Scholar]
- Lopez-Ceron, M. and Pellise, M. (2012). Biology and diagnosis of aberrant crypt foci. Colorectal Dis. 14, e157-e164. 10.1111/j.1463-1318.2011.02925.x [DOI] [PubMed] [Google Scholar]
- Lorimore, S. A., Coates, P. J. and Wright, E. G. (2003). Radiation-induced genomic instability and bystander effects: inter-related nontargeted effects of exposure to ionizing radiation. Oncogene 22, 7058-7069. 10.1038/sj.onc.1207044 [DOI] [PubMed] [Google Scholar]
- Lorimore, S. A., Chrystal, J. A., Robinson, J. I., Coates, P. J. and Wright, E. G. (2008). Chromosomal instability in unirradiated hemaopoietic cells induced by macrophages exposed in vivo to ionizing radiation. Cancer Res. 68, 8122-8126. 10.1158/0008-5472.CAN-08-0698 [DOI] [PubMed] [Google Scholar]
- Lu, R., Wu, S., Zhang, Y. G., Xia, Y., Zhou, Z., Kato, I., Dong, H., Bissonnette, M. and Sun, J. (2016). Salmonella protein AvrA activates the STAT3 signaling pathway in colon cancer. Neoplasia 18, 307-316. 10.1016/j.neo.2016.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas, C., Salesse, L., Thi Hoang, M. H., Bonnet, M., Sauvanet, P., Larabi, A., Godfraind, C., Gagniere, J., Pezet, D., Rosenstiel, P.et al. (2020). Autophagy of intestinal epithelial cells inhibits colorectal carcinogenesis induced by colibactin-producing Escherichia coli in ApcMin/+ mice. Gastroenterology 158, 1373-1388. 10.1053/j.gastro.2019.12.026 [DOI] [PubMed] [Google Scholar]
- Lynch, S. V. and Pedersen, O. (2016). The human intestinal microbiome in health and disease. N. Engl. J. Med. 375, 2369-2379. 10.1056/NEJMra1600266 [DOI] [PubMed] [Google Scholar]
- Madka, V., Kumar, G., Pathuri, G., Zhang, Y., Lightfoot, S., Asch, A. S., Mohammed, A., Steele, V. E. and Rao, C. V. (2020). Bisphosphonates Zometa and Fosamax synergize with metformin to prevent AOM-induced colon cancer in F344 rat model. Cancer Prev. Res. (Phila) 13, 185-194. 10.1158/1940-6207.CAPR-19-0265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggio-Price, L., Treuting, P., Zeng, W., Tsang, M., Bielefeldt-Ohmann, H. and Iritani, B. M. (2006). Helicobacter infection is required for inflammation and colon cancer in SMAD3-deficient mice. Cancer Res. 66, 828-838. 10.1158/0008-5472.CAN-05-2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marozik, P., Mothersill, C., Seymour, C. B., Mosse, I. and Melnov, S. (2007). Bystander effects induced by serum from survivors of the Chernobyl accident. Exp. Hematol. 35, 55-63. 10.1016/j.exphem.2007.01.029 [DOI] [PubMed] [Google Scholar]
- Maskens, A. P. (1979). Histogenesis of adenomatous polyps in the human large intestine. Gastroenterology 77, 1245-1251. 10.1016/0016-5085(79)90164-1 [DOI] [PubMed] [Google Scholar]
- May, R., Qu, D., Weygant, N., Chandrakesan, P., Ali, N., Lightfoot, S. A., Li, L., Sureban, S. M. and Houchen, C. W. (2014). Brief report: Dclk1 deletion in tuft cells results in impaired epithelial repair after radiation injury. Stem Cells 32, 822-827. 10.1002/stem.1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre, R. E., Buczacki, S. J., Arends, M. J. and Adams, D. J. (2015). Mouse models of colorectal cancer as preclinical models. BioEssays 37, 909-920. 10.1002/bies.201500032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean, M. H., Murray, G. I., Stewart, K. N., Norrie, G., Mayer, C., Hold, G. L., Thomson, J., Fyfe, N., Hope, M., Mowat, N. A.et al. (2011). The inflammatory microenvironment in colorectal neoplasia. PLoS ONE 6, e15366. 10.1371/journal.pone.0015366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao, Z.-F., Lewis, M. A., Cho, C. J., Adkins-Threats, M., Park, D., Brown, J. W., Sun, J.-X., Burclaff, J. R., Kennedy, S., Lu, J.et al. (2020a). A dedicated evolutionarily conserved molecular network licenses differentiated cells to return to the cell cycle. Dev. Cell 55, 178-194.e7. 10.1016/j.devcel.2020.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao, Z.-F., Sun, J.-X., Adkins-Threats, M., Pang, M.-J., Zhao, J.-H., Wang, X., Tang, K.-W., Wang, Z.-N. and Mills, J. C. (2020b). DDIT4 licenses only healthy cells to proliferate during injury-induced metaplasia. Gastroenterology 160, 260-271.e10. 10.1053/j.gastro.2020.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middelhoff, M., Westphalen, C. B., Hayakawa, Y., Yan, K. S., Gershon, M. D., Wang, T. C. and Quante, M. (2017). Dclk1-expressing tuft cells: critical modulators of the intestinal niche? Am. J. Physiol. Gastrointest. Liver Physiol. 313, G285-G299. 10.1152/ajpgi.00073.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo, L. J., Song, M., Huang, Q. H., Guan, H., Liu, X. D., Xie, D. F., Huang, B., Huang, R. X. and Zhou, P. K. (2018). Exosome-packaged miR-1246 contributes to bystander DNA damage by targeting LIG4. Br. J. Cancer 119, 492-502. 10.1038/s41416-018-0192-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mothersill, C. and Seymour, C. B. (2004). Radiation-induced bystander effects—implications for cancer. Nat. Rev. Cancer 4, 158-164. 10.1038/nrc1277 [DOI] [PubMed] [Google Scholar]
- Mughini-Gras, L., Schaapveld, M., Kramers, J., Mooij, S., Neefjes-Borst, E. A., Pelt, W. V. and Neefjes, J. (2018). Increased colon cancer risk after severe Salmonella infection. PLoS ONE 13, e0189721. 10.1371/journal.pone.0189721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagamine, C. M., Rogers, A. B., Fox, J. G. and Schauer, D. B. (2008). Helicobacter hepaticus promotes azoxymethane-initiated colon tumorigenesis in BALB/c-IL10-deficient mice. Int. J. Cancer 122, 832-838. 10.1002/ijc.23175 [DOI] [PubMed] [Google Scholar]
- Nagasawa, H. and Little, J. B. (1992). Induction of sister chromatid exchanges by extremely low doses of alpha-particles. Cancer Res. 52, 6394-6396. [PubMed] [Google Scholar]
- Najafi, M., Fardid, R., Hadadi, G. and Fardid, M. (2014). The mechanisms of radiation-induced bystander effect. J. Biomed. Phys. Eng. 4, 163-172. [PMC free article] [PubMed] [Google Scholar]
- Nakanishi, Y., Seno, H., Fukuoka, A., Ueo, T., Yamaga, Y., Maruno, T., Nakanishi, N., Kanda, K., Komekado, H., Kawada, M.et al. (2013). Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nat. Genet. 45, 98-103. 10.1038/ng.2481 [DOI] [PubMed] [Google Scholar]
- Nougayrede, J. P., Homburg, S., Taieb, F., Boury, M., Brzuszkiewicz, E., Gottschalk, G., Buchrieser, C., Hacker, J., Dobrindt, U. and Oswald, E. (2006). Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 313, 848-851. 10.1126/science.1127059 [DOI] [PubMed] [Google Scholar]
- Nowak, M. A., Komarova, N. L., Sengupta, A., Jallepalli, P. V., Shih Ie, M., Vogelstein, B. and Lengauer, C. (2002). The role of chromosomal instability in tumor initiation. Proc. Natl. Acad. Sci. USA 99, 16226-16231. 10.1073/pnas.202617399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong, B. A., Vega, K. J. and Houchen, C. W. (2014). Intestinal stem cells and the colorectal cancer microenvironment. World J. Gastroenterol. 20, 1898-1909. 10.3748/wjg.v20.i8.1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pampfer, S. and Streffer, C. (1989). Increased chromosome aberration levels in cells from mouse fetuses after zygote X-irradiation. Int. J. Radiat. Biol. 55, 85-92. 10.1080/09553008914550091 [DOI] [PubMed] [Google Scholar]
- Pant, G. S. and Kamada, N. (1977). Chromosome aberrations in normal leukocytes induced by the plasma of exposed individuals. Hiroshima J. Med. 26, 149-154. [PubMed] [Google Scholar]
- Peng, Y., Zhang, M., Zheng, L., Liang, Q., Li, H., Chen, J. T., Guo, H., Yoshina, S., Chen, Y. Z., Zhao, X.et al. (2017). Cysteine protease cathepsin B mediates radiation-induced bystander effects. Nature 547, 458-462. 10.1038/nature23284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perruche, S., Zhang, P., Liu, Y., Saas, P., Bluestone, J. A. and Chen, W. (2008). CD3-specific antibody-induced immune tolerance involves transforming growth factor-β from phagocytes digesting apoptotic T cells. Nat. Med. 14, 528-535. 10.1038/nm1749 [DOI] [PubMed] [Google Scholar]
- Pleguezuelos-Manzano, C., Puschhof, J., Huber, A. R., van Hoeck, A., Wood, H. M., Nomburg, J., Gurjao, C., Manders, F., Dalmasso, G., Stege, P. B.et al. (2020). Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature 580, 269-273. 10.1038/s41586-020-2080-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popivanova, B. K., Kostadinova, F. I., Furuichi, K., Shamekh, M. M., Kondo, T., Wada, T., Egashira, K. and Mukaida, N. (2009). Blockade of a chemokine, CCL2, reduces chronic colitis-associated carcinogenesis in mice. Cancer Res. 69, 7884-7892. 10.1158/0008-5472.CAN-09-1451 [DOI] [PubMed] [Google Scholar]
- Powell, A. E., Wang, Y., Li, Y., Poulin, E. J., Means, A. L., Washington, M. K., Higginbotham, J. N., Juchheim, A., Prasad, N., Levy, S. E.et al. (2012). The pan-ErbB negative regulator Lrig1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell 149, 146-158. 10.1016/j.cell.2012.02.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston, S. L., Wong, W. M., Chan, A. O., Poulsom, R., Jeffery, R., Goodlad, R. A., Mandir, N., Elia, G., Novelli, M., Bodmer, W. F.et al. (2003). Bottom-up histogenesis of colorectal adenomas: origin in the monocryptal adenoma and initial expansion by crypt fission. Cancer Res. 63, 3819-3825. [PubMed] [Google Scholar]
- Pribluda, A., Elyada, E., Wiener, Z., Hamza, H., Goldstein, R. E., Biton, M., Burstain, I., Morgenstern, Y., Brachya, G., Billauer, H.et al. (2013). A senescence-inflammatory switch from cancer-inhibitory to cancer-promoting mechanism. Cancer Cell 24, 242-256. 10.1016/j.ccr.2013.06.005 [DOI] [PubMed] [Google Scholar]
- Proenca, M. A., Biselli, J. M., Succi, M., Severino, F. E., Berardinelli, G. N., Caetano, A., Reis, R. M., Hughes, D. J. and Silva, A. E. (2018). Relationship between Fusobacterium nucleatum, inflammatory mediators and microRNAs in colorectal carcinogenesis. World J. Gastroenterol. 24, 5351-5365. 10.3748/wjg.v24.i47.5351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qualls, J. E., Kaplan, A. M., van Rooijen, N. and Cohen, D. A. (2006). Suppression of experimental colitis by intestinal mononuclear phagocytes. J. Leukoc. Biol. 80, 802-815. 10.1189/jlb.1205734 [DOI] [PubMed] [Google Scholar]
- Qubaiah, O., Devesa, S. S., Platz, C. E., Huycke, M. M. and Dores, G. M. (2010). Small intestinal cancer: a population-based study of incidence and survival patterns in the United States, 1992 to 2006. Cancer Epidemiol. Biomarkers Prev. 19, 1908-1918. 10.1158/1055-9965.EPI-10-0328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopala, S. V., Vashee, S., Oldfield, L. M., Suzuki, Y., Venter, J. C., Telenti, A. and Nelson, K. E. (2017). The human microbiome and cancer. Cancer Prev. Res. (Phila) 10, 226-234. 10.1158/1940-6207.CAPR-16-0249 [DOI] [PubMed] [Google Scholar]
- Ranjan, R., Rani, A., Metwally, A., McGee, H. S. and Perkins, D. L. (2016). Analysis of the microbiome: advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem. Biophys. Res. Commun. 469, 967-977. 10.1016/j.bbrc.2015.12.083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao, V. P., Poutahidis, T., Ge, Z., Nambiar, P. R., Boussahmain, C., Wang, Y. Y., Horwitz, B. H., Fox, J. G. and Erdman, S. E. (2006). Innate immune inflammatory response against enteric bacteria Helicobacter hepaticus induces mammary adenocarcinoma in mice. Cancer Res. 66, 7395-7400. 10.1158/0008-5472.CAN-06-0558 [DOI] [PubMed] [Google Scholar]
- Rao, V. P., Poutahidis, T., Fox, J. G. and Erdman, S. E. (2007). Breast cancer: should gastrointestinal bacteria be on our radar screen? Cancer Res. 67, 847-850. 10.1158/0008-5472.CAN-06-3468 [DOI] [PubMed] [Google Scholar]
- Rastogi, S., Hwang, A., Chan, J. and Wang, J. Y. J. (2018). Extracellular vesicles transfer nuclear Abl-dependent and radiation-induced miR-34c into unirradiated cells to cause bystander effects. Mol. Biol. Cell 29, 2228-2242. 10.1091/mbc.E18-02-0130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy, B. S., Narisawa, T., Wright, P., Vukusich, D., Weisburger, J. H. and Wynder, E. L. (1975). Colon carcinogenesis with azoxymethane and dimethylhydrazine in germ-free rats. Cancer Res. 35, 287-290. [PubMed] [Google Scholar]
- Rogers, K. J. and Pegg, A. E. (1977). Formation of O6-methylguanine by alkylation of rat liver, colon, and kidney DNA following administration of 1,2-dimethylhydrazine. Cancer Res. 37, 4082-4087. [PubMed] [Google Scholar]
- Rogers, M. J., Crockett, J. C., Coxon, F. P. and Mönkkönen, J. (2011). Biochemical and molecular mechanisms of action of bisphosphonates. Bone 49, 34-41. 10.1016/j.bone.2010.11.008 [DOI] [PubMed] [Google Scholar]
- Rubinstein, M. R., Baik, J. E., Lagana, S. M., Han, R. P., Raab, W. J., Sahoo, D., Dalerba, P., Wang, T. C. and Han, Y. W. (2019). Fusobacterium nucleatum promotes colorectal cancer by inducing Wnt/β-catenin modulator Annexin A1. EMBO Rep. 20. 10.15252/embr.201847638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainz, B., Jr, Carron, E., Vallespinós, M. and Machado, H. L. (2016). Cancer stem cells and macrophages: implications in tumor biology and therapeutic strategies. Mediators Inflamm. 2016, 9012369. 10.1155/2016/9012369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaue, D., Micewicz, E. D., Ratikan, J. A., Xie, M. W., Cheng, G. and McBride, W. H. (2015). Radiation and inflammation. Semin. Radiat. Oncol. 25, 4-10. 10.1016/j.semradonc.2014.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwitalla, S., Fingerle, A. A., Cammareri, P., Nebelsiek, T., Goktuna, S. I., Ziegler, P. K., Canli, O., Heijmans, J., Huels, D. J., Moreaux, G.et al. (2013). Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 152, 25-38. 10.1016/j.cell.2012.12.012 [DOI] [PubMed] [Google Scholar]
- Scott, A. J., Alexander, J. L., Merrifield, C. A., Cunningham, D., Jobin, C., Brown, R., Alverdy, J., O'Keefe, S. J., Gaskins, H. R., Teare, J.et al. (2019). International Cancer Microbiome Consortium consensus statement on the role of the human microbiome in carcinogenesis. Gut 68, 1624-1632. 10.1136/gutjnl-2019-318556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellon, R. K., Tonkonogy, S., Schultz, M., Dieleman, L. A., Grenther, W., Balish, E., Rennick, D. M. and Sartor, R. B. (1998). Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect. Immun. 66, 5224-5231. 10.1128/IAI.66.11.5224-5231.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sender, R., Fuchs, S. and Milo, R. (2016). Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 14, e1002533. 10.1371/journal.pbio.1002533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeeva, V. A., Ershova, E. S., Veiko, N. N., Malinovskaya, E. M., Kalyanov, A. A., Kameneva, L. V., Stukalov, S. V., Dolgikh, O. A., Konkova, M. S., Ermakov, A. V.et al. (2017). Low-dose ionizing radiation affects mesenchymal stem cells via extracellular oxidized cell-free DNA: a possible mediator of bystander effect and adaptive response. Oxid. Med. Cell Longev. 2017, 9515809. 10.1155/2017/9515809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih, I. M., Wang, T. L., Traverso, G., Romans, K., Hamilton, S. R., Ben-Sasson, S., Kinzler, K. W. and Vogelstein, B. (2001a). Top-down morphogenesis of colorectal tumors. Proc. Natl. Acad. Sci. USA 98, 2640-2645. 10.1073/pnas.051629398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih, I. M., Zhou, W., Goodman, S. N., Lengauer, C., Kinzler, K. W. and Vogelstein, B. (2001b). Evidence that genetic instability occurs at an early stage of colorectal tumorigenesis. Cancer Res. 61, 818-822. [PubMed] [Google Scholar]
- Slowicka, K., Petta, I., Blancke, G., Hoste, E., Dumas, E., Sze, M., Vikkula, H., Radaelli, E., Haigh, J. J., Jonckheere, S.et al. (2020). Zeb2 drives invasive and microbiota-dependent colon carcinoma. Nat. Cancer 1, 620-634. 10.1038/s43018-020-0070-2 [DOI] [PubMed] [Google Scholar]
- Smallwood, H. S., López-Ferrer, D. and Squier, T. C. (2011). Aging enhances the production of reactive oxygen species and bactericidal activity in peritoneal macrophages by upregulating classical activation pathways. Biochemistry 50, 9911-9922. 10.1021/bi2011866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smythies, L. E., Sellers, M., Clements, R. H., Mosteller-Barnum, M., Meng, G., Benjamin, W. H., Orenstein, J. M. and Smith, P. D. (2005). Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J. Clin. Invest. 115, 66-75. 10.1172/JCI200519229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobhani, I., Tap, J., Roudot-Thoraval, F., Roperch, J. P., Letulle, S., Langella, P., Corthier, G., Tran Van Nhieu, J. and Furet, J. P. (2011). Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS ONE 6, e16393. 10.1371/journal.pone.0016393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolov, M. and Neumann, R. (2018). Changes in gene expression as one of the key mechanisms involved in radiation-induced bystander effect. Biomed. Rep. 9, 99-111. 10.3892/br.2018.1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, M., Chan, A. T. and Sun, J. (2020). Influence of the gut microbiome, diet, and environment on risk of colorectal cancer. Gastroenterology 158, 322-340. 10.1053/j.gastro.2019.06.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steck, N., Hoffmann, M., Sava, I. G., Kim, S. C., Hahne, H., Tonkonogy, S. L., Mair, K., Krueger, D., Pruteanu, M., Shanahan, F.et al. (2011). Enterococcus faecalis metalloprotease compromises epithelial barrier and contributes to intestinal inflammation. Gastroenterology 141, 959-971. 10.1053/j.gastro.2011.05.035 [DOI] [PubMed] [Google Scholar]
- Strum, W. B. (2016). Colorectal adenomas. N. Engl. J. Med. 375, 389-390. 10.1056/NEJMra1513581 [DOI] [PubMed] [Google Scholar]
- Su, L. K., Kinzler, K. W., Vogelstein, B., Preisinger, A. C., Moser, A. R., Luongo, C., Gould, K. A. and Dove, W. F. (1992). Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 256, 668-670. 10.1126/science.1350108 [DOI] [PubMed] [Google Scholar]
- Szatmári, T., Persa, E., Kis, E., Benedek, A., Hargitai, R., Sáfrány, G. and Lumniczky, K. (2019). Extracellular vesicles mediate low dose ionizing radiation-induced immune and inflammatory responses in the blood. Int. J. Radiat. Biol. 95, 12-22. 10.1080/09553002.2018.1450533 [DOI] [PubMed] [Google Scholar]
- Tahara, T., Yamamoto, E., Suzuki, H., Maruyama, R., Chung, W., Garriga, J., Jelinek, J., Yamano, H. O., Sugai, T., An, B.et al. (2014). Fusobacterium in colonic flora and molecular features of colorectal carcinoma. Cancer Res. 74, 1311-1318. 10.1158/0008-5472.CAN-13-1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, K. and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663-676. 10.1016/j.cell.2006.07.024 [DOI] [PubMed] [Google Scholar]
- Tanaka, T., Kohno, H., Suzuki, R., Yamada, Y., Sugie, S. and Mori, H. (2003). A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 94, 965-973. 10.1111/j.1349-7006.2003.tb01386.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tella, S. H. and Gallagher, J. C. (2014). Prevention and treatment of postmenopausal osteoporosis. J. Steroid Biochem. Mol. Biol. 142, 155-170. 10.1016/j.jsbmb.2013.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetteh, P. W., Kretzschmar, K., Begthel, H., van den Born, M., Korving, J., Morsink, F., Farin, H., van Es, J. H., Offerhaus, G. J. and Clevers, H. (2016). Generation of an inducible colon-specific Cre enzyme mouse line for colon cancer research. Proc. Natl. Acad. Sci. USA 113, 11859-11864. 10.1073/pnas.1614057113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevaranjan, N., Puchta, A., Schulz, C., Naidoo, A., Szamosi, J. C., Verschoor, C. P., Loukov, D., Schenck, L. P., Jury, J., Foley, K. P.et al. (2017). Age-associated microbial dysbiosis promotes intestinal permeability, Systemic inflammation, and macrophage dysfunction. Cell Host Microbe 21, 455-466.e4. 10.1016/j.chom.2017.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele Orberg, E., Fan, H., Tam, A. J., Dejea, C. M., Destefano Shields, C. E., Wu, S., Chung, L., Finard, B. B., Wu, X., Fathi, P.et al. (2017). The myeloid immune signature of enterotoxigenic Bacteroides fragilis-induced murine colon tumorigenesis. Mucosal Immunol. 10, 421-433. 10.1038/mi.2016.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thosani, N., Thosani, S. N., Kumar, S., Nugent, Z., Jimenez, C., Singh, H. and Guha, S. (2013). Reduced risk of colorectal cancer with use of oral bisphosphonates: a systematic review and meta-analysis. J. Clin. Oncol. 31, 623-630. 10.1200/JCO.2012.42.9530 [DOI] [PubMed] [Google Scholar]
- Tilg, H., Adolph, T. E., Gerner, R. R. and Moschen, A. R. (2018). The intestinal microbiota in colorectal cancer. Cancer Cell 33, 954-964. 10.1016/j.ccell.2018.03.004 [DOI] [PubMed] [Google Scholar]
- Tomasetti, C., Vogelstein, B. and Parmigiani, G. (2013). Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proc. Natl. Acad. Sci. USA 110, 1999-2004. 10.1073/pnas.1221068110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomkovich, S., Yang, Y., Winglee, K., Gauthier, J., Muhlbauer, M., Sun, X., Mohamadzadeh, M., Liu, X., Martin, P., Wang, G. P.et al. (2017). Locoregional effects of microbiota in a preclinical model of colon carcinogenesis. Cancer Res. 77, 2620-2632. 10.1158/0008-5472.CAN-16-3472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treiner, E., Duban, L., Bahram, S., Radosavljevic, M., Wanner, V., Tilloy, F., Affaticati, P., Gilfillan, S. and Lantz, O. (2003). Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature 422, 164-169. 10.1038/nature01433 [DOI] [PubMed] [Google Scholar]
- Tsoi, H., Chu, E. S. H., Zhang, X., Sheng, J., Nakatsu, G., Ng, S. C., Chan, A. W. H., Chan, F. K. L., Sung, J. J. Y. and Yu, J. (2017). Peptostreptococcus anaerobius Induces intracellular cholesterol biosynthesis in colon cells to induce proliferation and causes dysplasia in mice. Gastroenterology 152, 1419-1433.e5. 10.1053/j.gastro.2017.01.009 [DOI] [PubMed] [Google Scholar]
- van Dalen, F. J., van Stevendaal, M., Fennemann, F. L., Verdoes, M. and Ilina, O. (2018). Molecular repolarisation of tumour-associated macrophages. Molecules 24, 9. 10.3390/molecules24010009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira, P. L., Christensen, J. R., Minaee, S., O'Neill, E. J., Barrat, F. J., Boonstra, A., Barthlott, T., Stockinger, B., Wraith, D. C. and O'Garra, A. (2004). IL-10-secreting regulatory T cells do not express Foxp3 but have comparable regulatory function to naturally occurring CD4+CD25+ regulatory T cells. J. Immunol. 172, 5986-5993. 10.4049/jimmunol.172.10.5986 [DOI] [PubMed] [Google Scholar]
- Visvader, J. E. (2011). Cells of origin in cancer. Nature 469, 314-322. 10.1038/nature09781 [DOI] [PubMed] [Google Scholar]
- Vitale, I., Manic, G., Coussens, L. M., Kroemer, G. and Galluzzi, L. (2019). Macrophages and metabolism in the tumor microenvironment. Cell Metab. 30, 36-50. 10.1016/j.cmet.2019.06.001 [DOI] [PubMed] [Google Scholar]
- Vogelstein, B., Papadopoulos, N., Velculescu, V. E., Zhou, S., Diaz, L. A., Jr. and Kinzler, K. W. (2013). Cancer genome landscapes. Science 339, 1546-1558. 10.1126/science.1235122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan, G., Xie, M., Yu, H. and Chen, H. (2018). Intestinal dysbacteriosis activates tumor-associated macrophages to promote epithelial-mesenchymal transition of colorectal cancer. Innate Immun 24, 480-489. 10.1177/1753425918801496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. and Huycke, M. M. (2007). Extracellular superoxide production by Enterococcus faecalis promotes chromosomal instability in mammalian cells. Gastroenterology 132, 551-561. 10.1053/j.gastro.2006.11.040 [DOI] [PubMed] [Google Scholar]
- Wang, X. and Huycke, M. M. (2015). Colorectal cancer: role of commensal bacteria and bystander effects. Gut Microbes 6, 370-376. 10.1080/19490976.2015.1103426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X., Allen, T. D., May, R. J., Lightfoot, S., Houchen, C. W. and Huycke, M. M. (2008). Enterococcus faecalis induces aneuploidy and tetraploidy in colonic epithelial cells through a bystander effect. Cancer Res. 68, 9909-9917. 10.1158/0008-5472.CAN-08-1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X., Yang, Y., Moore, D. R., Nimmo, S. L., Lightfoot, S. A. and Huycke, M. M. (2012). 4-hydroxy-2-nonenal mediates genotoxicity and bystander effects caused by Enterococcus faecalis-infected macrophages. Gastroenterology 142, 543-551. 10.1053/j.gastro.2011.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X., Allen, T. D., Yang, Y., Moore, D. R. and Huycke, M. M. (2013). Cyclooxygenase-2 generates the endogenous mutagen trans-4-hydroxy-2-nonenal in Enterococcus faecalis-infected macrophages. Cancer Prev. Res. (Phila) 6, 206-216. 10.1158/1940-6207.CAPR-12-0350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X., Yang, Y. and Huycke, M. M. (2015). Commensal bacteria drive endogenous transformation and tumour stem cell marker expression through a bystander effect. Gut 64, 459-468. 10.1136/gutjnl-2014-307213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X., Yang, Y. and Huycke, M. M. (2017). Commensal-infected macrophages induce dedifferentiation and reprogramming of epithelial cells during colorectal carcinogenesis. Oncotarget 8, 102176-102190. 10.18632/oncotarget.22250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe, N., Ikuta, K., Okazaki, K., Nakase, H., Tabata, Y., Matsuura, M., Tamaki, H., Kawanami, C., Honjo, T. and Chiba, T. (2003). Elimination of local macrophages in intestine prevents chronic colitis in interleukin-10-deficient mice. Dig. Dis. Sci. 48, 408-414. 10.1023/A:1021960401290 [DOI] [PubMed] [Google Scholar]
- Watson, G. E., Lorimore, S. A., Macdonald, D. A. and Wright, E. G. (2000). Chromosomal instability in unirradiated cells induced in vivo by a bystander effect of ionizing radiation. Cancer Res. 60, 5608-5611. [PubMed] [Google Scholar]
- Westphalen, C. B., Asfaha, S., Hayakawa, Y., Takemoto, Y., Lukin, D. J., Nuber, A. H., Brandtner, A., Setlik, W., Remotti, H., Muley, A.et al. (2014). Long-lived intestinal tuft cells serve as colon cancer-initiating cells. J. Clin. Invest. 124, 1283-1295. 10.1172/JCI73434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willet, S. G., Lewis, M. A., Miao, Z. F., Liu, D., Radyk, M. D., Cunningham, R. L., Burclaff, J., Sibbel, G., Lo, H. G., Blanc, V.et al. (2018). Regenerative proliferation of differentiated cells by mTORC1-dependent paligenosis. EMBO J. 37, e98311. 10.15252/embj.201798311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirbel, J., Pyl, P. T., Kartal, E., Zych, K., Kashani, A., Milanese, A., Fleck, J. S., Voigt, A. Y., Palleja, A., Ponnudurai, R.et al. (2019). Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nat. Med. 25, 679-689. 10.1038/s41591-019-0406-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, S., Lim, K. C., Huang, J., Saidi, R. F. and Sears, C. L. (1998). Bacteroides fragilis enterotoxin cleaves the zonula adherens protein, E-cadherin. Proc. Natl. Acad. Sci. USA 95, 14979-14984. 10.1073/pnas.95.25.14979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, S., Morin, P. J., Maouyo, D. and Sears, C. L. (2003). Bacteroides fragilis enterotoxin induces c-Myc expression and cellular proliferation. Gastroenterology 124, 392-400. 10.1053/gast.2003.50047 [DOI] [PubMed] [Google Scholar]
- Wu, S., Rhee, K. J., Albesiano, E., Rabizadeh, S., Wu, X., Yen, H. R., Huso, D. L., Brancati, F. L., Wick, E., McAllister, F.et al. (2009). A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 15, 1016-1022. 10.1038/nm.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, W. K., Sung, J. J., Lee, C. W., Yu, J. and Cho, C. H. (2010). Cyclooxygenase-2 in tumorigenesis of gastrointestinal cancers: an update on the molecular mechanisms. Cancer Lett. 295, 7-16. 10.1016/j.canlet.2010.03.015 [DOI] [PubMed] [Google Scholar]
- Wu, J., Li, K., Peng, W., Li, H., Li, Q., Wang, X., Peng, Y., Tang, X. and Fu, X. (2019). Autoinducer-2 of Fusobacterium nucleatum promotes macrophage M1 polarization via TNFSF9/IL-1β signaling. Int. Immunopharmacol. 74, 105724. 10.1016/j.intimp.2019.105724 [DOI] [PubMed] [Google Scholar]
- Yaeger, R., Chatila, W. K., Lipsyc, M. D., Hechtman, J. F., Cercek, A., Sanchez-Vega, F., Jayakumaran, G., Middha, S., Zehir, A., Donoghue, M. T. A.et al. (2018). Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell 33, 125-136.e3. 10.1016/j.ccell.2017.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, B., Wang, H., Rabbani, Z. N., Zhao, Y., Li, W., Yuan, Y., Li, F., Dewhirst, M. W. and Li, C. Y. (2006). Tumor necrosis factor- α is a potent endogenous mutagen that promotes cellular transformation. Cancer Res. 66, 11565-11570. 10.1158/0008-5472.CAN-06-2540 [DOI] [PubMed] [Google Scholar]
- Yan, J., Allen, S., McDonald, E., Das, I., Mak, J. Y. W., Liu, L., Fairlie, D. P., Meehan, B. S., Chen, Z., Corbett, A. J.et al. (2020). MAIT cells promote tumor initiation, growth, and metastases via tumor MR1. Cancer Discov. 10, 124-141. 10.1158/2159-8290.CD-19-0569 [DOI] [PubMed] [Google Scholar]
- Yang, Y., Wang, X., Moore, D. R., Lightfoot, S. A. and Huycke, M. M. (2012). TNF- α mediates macrophage-induced bystander effects through netrin-1. Cancer Res. 72, 5219-5229. 10.1158/0008-5472.CAN-12-1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y., Wang, X., Huycke, T., Moore, D. R., Lightfoot, S. A. and Huycke, M. M. (2013). Colon macrophages polarized by commensal bacteria cause colitis and cancer through the bystander effect. Transl. Oncol. 6, 596-606. 10.1593/tlo.13412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y., Weng, W., Peng, J., Hong, L., Yang, L., Toiyama, Y., Gao, R., Liu, M., Yin, M., Pan, C.et al. (2017). Fusobacterium nucleatum increases proliferation of colorectal cancer cells and tumor development in mice by activating Toll-like receptor 4 signaling to nuclear factor-κB, and up-regulating expression of microRNA-21. Gastroenterology 152, 851-866. 10.1053/j.gastro.2016.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon, P., Keylock, K. T., Hartman, M. E., Freund, G. G. and Woods, J. A. (2004). Macrophage hypo-responsiveness to interferon-γ in aged mice is associated with impaired signaling through Jak-STAT. Mech. Ageing Dev. 125, 137-143. 10.1016/j.mad.2003.11.010 [DOI] [PubMed] [Google Scholar]
- Yoshida, Y., Ito, S., Kamo, M., Kezuka, Y., Tamura, H., Kunimatsu, K. and Kato, H. (2010). Production of hydrogen sulfide by two enzymes associated with biosynthesis of homocysteine and lanthionine in Fusobacterium nucleatum subsp. nucleatum ATCC 25586. Microbiology 156, 2260-2269. 10.1099/mic.0.039180-0 [DOI] [PubMed] [Google Scholar]
- Yu, Y., Blokhuis, B., Derks, Y., Kumari, S., Garssen, J. and Redegeld, F. (2018). Human mast cells promote colon cancer growth via bidirectional crosstalk: studies in 2D and 3D coculture models. Oncoimmunology 7, e1504729. 10.1080/2162402X.2018.1504729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabijak, L., Attencourt, C., Guignant, C., Chatelain, D., Marcelo, P., Marolleau, J. P. and Treiner, E. (2015). Increased tumor infiltration by mucosal-associated invariant T cells correlates with poor survival in colorectal cancer patients. Cancer Immunol. Immunother. 64, 1601-1608. 10.1007/s00262-015-1764-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeuner, A., Todaro, M., Stassi, G. and De Maria, R. (2014). Colorectal cancer stem cells: from the crypt to the clinic. Cell Stem Cell 15, 692-705. 10.1016/j.stem.2014.11.012 [DOI] [PubMed] [Google Scholar]
- Zhou, H., Ivanov, V. N., Gillespie, J., Geard, C. R., Amundson, S. A., Brenner, D. J., Yu, Z., Lieberman, H. B. and Hei, T. K. (2005). Mechanism of radiation-induced bystander effect: role of the cyclooxygenase-2 signaling pathway. Proc. Natl. Acad. Sci. USA 102, 14641-14646. 10.1073/pnas.0505473102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, L., Gibson, P., Currle, D. S., Tong, Y., Richardson, R. J., Bayazitov, I. T., Poppleton, H., Zakharenko, S., Ellison, D. W. and Gilbertson, R. J. (2009). Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature 457, 603-607. 10.1038/nature07589 [DOI] [PMC free article] [PubMed] [Google Scholar]