

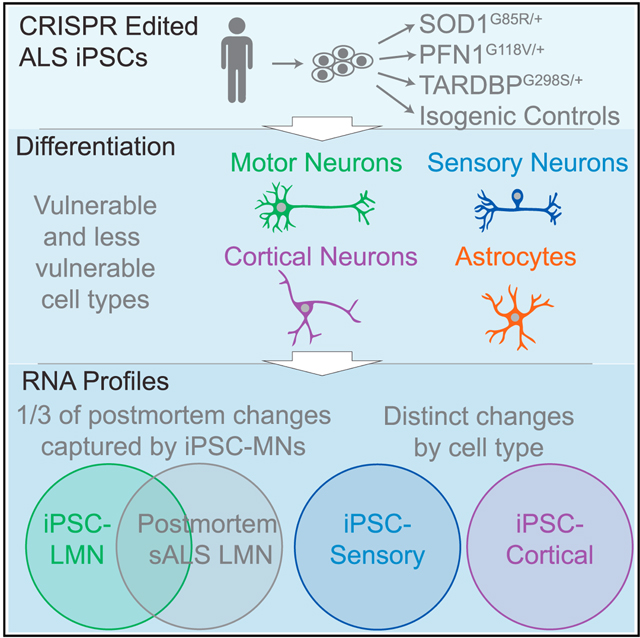
SUMMARY
Motor neuron degeneration, the defining feature of amyotrophic lateral sclerosis (ALS), is a primary example of cell-type specificity in neurodegenerative diseases. Using isogenic pairs of induced pluripotent stem cells (iPSCs) harboring different familial ALS mutations, we assess the capacity of iPSC-derived lower motor neurons, sensory neurons, astrocytes, and superficial cortical neurons to capture disease features including transcriptional and splicing dysregulation observed in human postmortem neurons. At early time points, differentially regulated genes in iPSC-derived lower motor neurons, but not other cell types, overlap with one-third of the differentially regulated genes in laser-dissected motor neurons from ALS compared with control postmortem spinal cords. For genes altered in both the iPSC model and bona fide human lower motor neurons, expression changes correlate between the two populations. In iPSC-derived lower motor neurons, but not other derived cell types, we detect the downregulation of genes affected by TDP-43-dependent splicing. This reduction takes place exclusively within genotypes known to involve TDP-43 pathology.
In brief
Amyotrophic lateral sclerosis causes motor neuron degeneration but leaves most other neurons intact. Held et al. find that familial ALS mutations cause distinct RNA profiles in iPSC-derived motor neurons compared with other cell types and that fALS iPSC motor neurons capture gene expression changes in postmortem sporadic ALS motor neurons.
Graphical Abstract
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by the degeneration of the motor nervous system.1 Approximately 90% of ALS cases are sporadic (sALS), while mutations in over twenty-five different genes can cause familial ALS (fALS). These fALS mutations sort broadly into functional groups implicated in the disruption of protein homeostasis, RNA processing, or cytoskeletal function1 and are exemplified by SOD1, TARDBP, and PFN1, respectively.2 Nearly all fALS mutations exhibit a dominant inheritance pattern1; these include the most common genetic cause, an intronic repeat expansion in the C9ORF72 gene.3,4 Two outstanding questions concern the mechanisms underlying the selective vulnerability of motor neurons in ALS and the convergence of a heterogeneous group of genes onto a homogeneous clinical presentation.
The selective vulnerability and degeneration of motor neurons in ALS contrasts with the relative sparing of other neuronal subtypes.1,5 First-order sensory neurons, similar to lower motor neurons (LMNs), extend long peripheral projections and have high energetic requirements to sustain axonal transport to and from distant compartments. Such similarities between LMNs and sensory neurons juxtaposed with their differential vulnerabilities in ALS provide an opportunity to identify cellular processes that are unique to LMNs and may contribute to their degeneration.
The main pathological hallmark of ALS, nuclear depletion and cytoplasmic aggregation of TDP-43 (encoded by the TARDBP gene),6–8 occurs in all sALS and fALS cases except for fALS due to SOD1 or FUS mutations.6,8 How diverse genes implicated in fALS converge on this molecular phenotype remains unclear. TDP-43 suppresses human genome-specific cryptic exons in key ALS-related genes,9–13 and this observation has led to an increased emphasis on human cell-based models of ALS, particularly using neurons derived from human induced pluripotent stem cells (iPSCs). While some reports indicate that iPSC-derived neurons across a range of fALS mutations exhibit components of TDP-43 pathology,14–20 interpretations may be confounded by the use of stressors or non-physiological expression modulation to elicit these features. Furthermore, understanding how sALS leads to TDP-43 pathology is even more challenging because of the lack of single strong genetic drivers and limitations of iPSC modeling to capture epigenetic changes.17,18,21,22
Given the complexity of sALS, one approach is to ask whether genes and mechanisms implicated in fALS can provide insight into sALS.23 Using sALS postmortem human tissue expression datasets, this possibility can be evaluated on a genome-wide scale to determine how accurately fALS iPSC-derived neurons model transcriptional changes found in postmortem sALS neurons. Unlike the neurons generated from iPSCs, the postmortem samples presumably incorporate important non-cell-autonomous influences of other cell types on neurons.
Here, to address the key questions of cell-type specificity and mechanistic convergence, we generated a matrix of different cell types and fALS mutations. Notably, the effects of fALS mutations varied largely across cell types. LMNs–but not other cell types–captured transcriptional features found in laser-captured LMNs from postmortem ALS spinal cords. Of the three investigated fALS genes, only those known to generate TDP-43 pathology yielded reduced transcripts of genes that undergo TDP-43-dependent aberrant splicing.9–13 Finally, in contrast with the detected downregulation of such genes, cryptic exon inclusion events were largely absent in the iPSC LMNs, at least in part reflecting RNA clearance mechanisms that were intact in the iPSC LMNs but may become compromised in disease.
RESULTS
Generation of different neuronal subtypes
Compared with small-molecule-based neuronal differentiation, direct expression of transcription factors in iPSCs can improve the purity of the target cell population.24 We therefore generated PiggyBac25 plasmids with tetracycline (tet)-inducible transcription factors to differentiate LMNs (NGN2, ISL1, LXH3),26 superficial cortical-like neurons (NGN2),27 or first-order sensory neurons (NGN2, BRN3A)28 (Figure 1A). iPSCs with successful integrations were differentiated for 3 days with doxycycline and then transferred to long-term media without doxycycline for maturation. After 10 days, we used cyclic immunofluorescence29 and automated imaging analysis (Figure 1B) to evaluate the LMN markers HB9, CHAT, NEUN, ISL1, and TUJ1. 96.8% ± 0.35% of cells were positive for all markers (Figures 1C and 1D). A similar analysis of NGN2 superficial cortical-like neurons showed staining for TUJ1 and the layer 2/3 cortical markers BRN2 and FOXG1 (Figures 1E and 1F). Finally, sensory neurons were positive for BRN3A, ISL1, and TUJ1 (Figures 1G and 1H). qRT-PCR validation confirmed an increase in cell-type-specific markers (Figures S1A–S1C).
Figure 1. Differentiation of LMNs, cortical neurons, and sensory neurons.
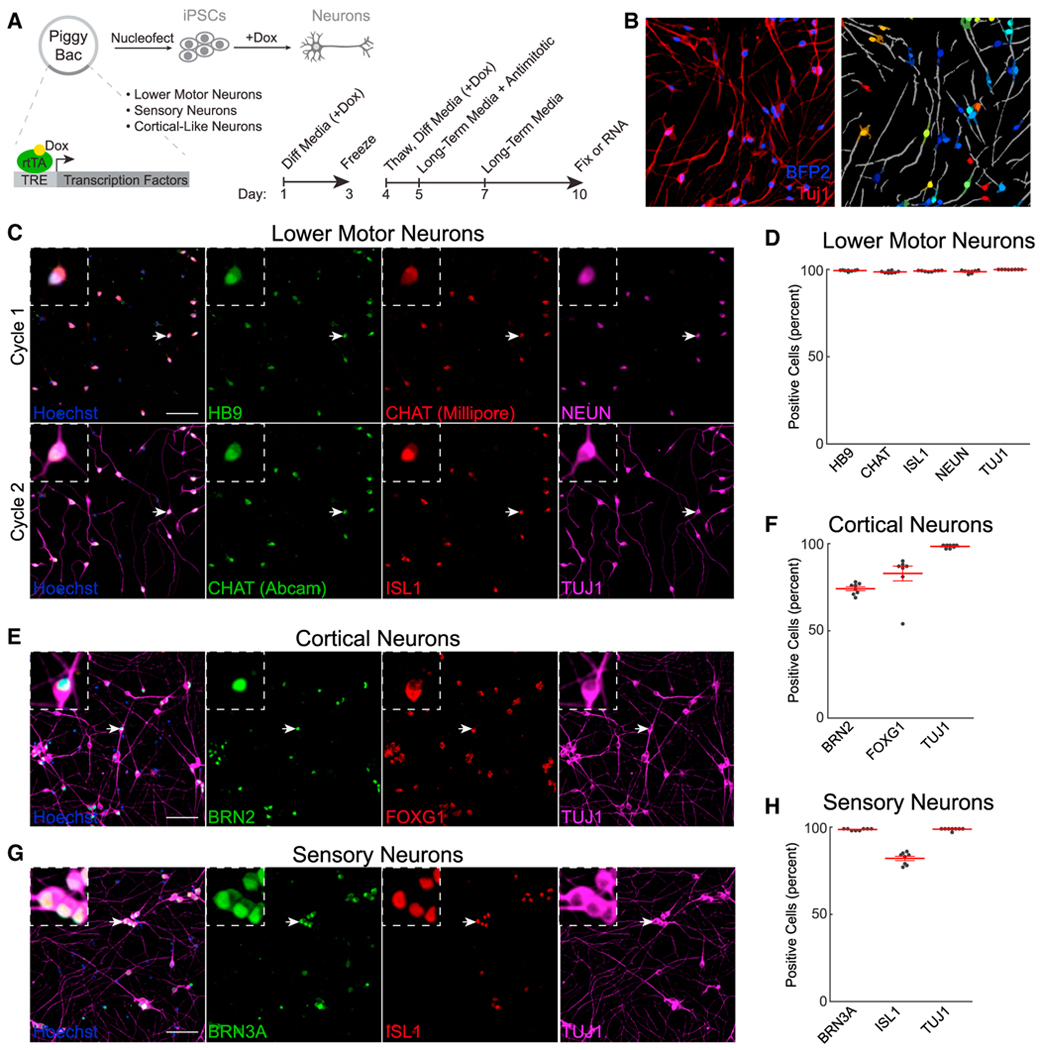
(A) Schematic for tet-inducible differentiation. Timeline summarizes the differentiation protocol.
(B) Masking of neurons using BFP2 and TUJ1. Lighter, darker, and gray colors indicate nuclei, cytoplasm, and neurites, respectively.
(C–H) Immunofluorescence and quantification of LMNs (C and D), cortical neurons (E and F), and sensory neurons (G and H). Scale bars: 100 μM; n = 8 wells for all groups. Arrows indicate magnified cells in insets.
RNA sequencing reveals cell-type-specific changes resulting from fALS mutations
We next sought to determine which disrupted cellular processes are shared among fALS mutations and whether such perturbations occur specifically in LMNs. We therefore differentiated LMNs, sensory neurons, and astrocytes30 (Figure S2A) from three isogenic pairs (SOD1G85R/+, TARDBPG298S/+, and PFN1G118V/+) we had previously generated through CRISPR editing2 (Figures 2A and 2B). We also differentiated NGN2 cortical-like neurons from iPSCs harboring the TARDBPG298S/+ mutation, as about 50% of frontotemporal dementia cases exhibit TDP-43 pathology.8,31 A principal-component analysis (PCA) showed a clear separation of astrocytes vs. neuronal cell types along principal component 1 (72% of the variance among libraries) as well as a separation of neuronal subtypes along principal component 2 (15% of the variance) (Figure S2B). To confirm the cellular identities of these libraries, we selected cellular markers and performed heatmap clustering (Figure 2C). Libraries of the same cell type clustered together and were positive for their respective cell type markers (e.g., LMNs with MNX1 expression, sensory neurons with SCN9A, and astrocytes with GFAP).
Figure 2. RNA sequencing across cell types and fALS mutations.

CRISPR editing strategy to generate heterozygous fALS mutations sharing the same genetic background.
(B) Differentiation of edited iPSCs.
(C) Heatmap for selected cell markers (n = 3 libraries per condition).
(D) Volcano plots comparing all control lines vs. all ALS lines.
(E) Differentially expressed genes due to fALS mutations across different cell types.
(F) Comparison of differentially expressed genes in fALS iPSC LMNs with laser-captured LMNs from postmortem sALS samples32 (n = 8 postmortem control and n = 13 postmortem sALS spinal cords).
We next performed a PCA within the LMN subgroup to identify differences between LMNs with fALS mutations and controls (Figure S2C). Two of the three LMN genotype isogenic pairs, TARDBP and PFN1, separated along PC1 (81% of variance), while SOD1 did not. We then compared iPSC LMNs with fALS mutations against all control libraries and found a substantial overlap in differentially expressed genes across fALS mutations (Figure S2D; Table S1). Because of the substantial intersection of differentially regulated genes among all three fALS mutations, we next compared all control LMNs with all fALS LMNs (Figure 2D). We observed a total of 3,804 upregulated genes and 3,700 downregulated genes between control and fALS LMNs, numbers that were substantially larger than the 354 upregulated and 591 downregulated genes observed when comparing between control and fALS sensory neurons (Figure 2E; Table S1). The TARDBPG298S mutation also led to fewer gene expression changes in NGN2 cortical-like neurons than LMNs (Figures 2E and S2E). Notably, we observed downregulation of STMN2, UNC13A, and KCNQ2, three genes with important contributions to ALS pathology,10–13,33–35 in LMNs but not in sensory neurons, astrocytes, or cortical neurons (Figure 2D). We then compared differentially expressed genes between cell types and observed a small overlap between LMNs and sensory neurons, with a more substantial overlap between LMNs and astrocytes (Figure 2E). Similarly, the dysregulated genes within TARDBPG298S NGN2 neurons were mostly non-overlapping with TARDBPG298S LMNs, despite mutations in the TDP-43 C-terminal domain being associated with both ALS and frontotemporal dementia (FTD)36 (Figure S2E; Table S1). Gene Ontology terms for upregulated genes specific to LMNs included “regulation of programmed cell death” (p = 1.7e–9), “cellular response to stress” (p = 4e–8), and “immune effector process” (p = 1.1e–6), while Gene Ontology terms from downregulated genes specific to LMNs included “modulation of chemical synaptic transmission” (p = 6.3e–19), “regulation of neuron projection development” (p = 2.6e–15), and “cell projection organization” (p = 2.2e–13) (Figure S2F; Table S2).
iPSC LMNs with fALS mutations capture gene expression changes in postmortem sALS LMNs
To validate the relevance of these expression changes, we compared dysregulated genes in the fALS iPSC LMNs with genes differentially regulated in microdissected LMNs from postmortem sALS vs. control spinal cords.32 In the original analysis of this postmortem dataset, the top 1,000 genes contributing to principal component 1 were excluded due to concerns regarding microglia contamination. However, 52% of these genes were also differentially expressed in the fALS iPSC LMN cultures, which do not contain microglia (Table S3). Thus, an alternative possibility is that some of these expression changes may genuinely occur within bona fide ALS LMNs, an interpretation consistent with recent findings of traditional inflammatory signaling intrinsic within neurons.37,38 We then compared our re-analysis of sALS postmortem LMNs (Figures S3A and S3B) with the fALS iPSC LMNs and found that the fALS iPSC LMNs captured 480/1,303 of the upregulated and 103/316 of the downregulated genes in postmortem sALS LMNs (Figure 2F; Table S3). Furthermore, the amplitudes of the gene expression changes were also significantly correlated (R = 0.26, p = 4.8e–27 for all genes differentially expressed in sALS postmortem LMNs; Table S4; R = 0.41, p = 3.3e–32 for genes differentially regulated in both datasets; Figure S3C). Significant GO terms for the overlap between sALS postmortem LMNs and fALS iPSC LMNs included “immune system process” (p = 1.7e–15) and “regulation of cell death” (p = 5.7e–14) for upregulated genes and “regulation of cell projection organization” (p = 2.2e–5) and “regulation of plasma membrane bounded cell projection” (p = 3.6e–5) for downregulated genes (Figure S3D). We then examined differentially expressed genes in postmortem ALS LMNs that were not captured by iPSC LMNs and found “cell surface receptor signaling pathway” (p = 7.2e–21), and “regulation of response to external stimulus” (p = 4e–11) in the upregulated genes and no significant Gene Ontology (GO) terms in the downregulated genes (Figure S3E; Table S3).
Genes with aberrant splicing in patients with ALS are downregulated in TARDBPG298S/+ and PFN1G118V/+ LMNs
Several genes that exhibit aberrant splicing due to loss of nuclear TDP-43,9–13 including STMN2, UNC13A, and KCNQ2, were spe cifically downregulated in LMNs. In fact, 34/61 genes in which TDP-43-dependent alternative splicing events were observed in FTD-ALS postmortem neurons13,39 were significantly reduced in the fALS iPSC LMNs (Table S5). When we broke down this analysis by isogenic pair, we observed a strong downregulation of these genes in TARDBPG298S/+ and PFN1G118V/+ LMNs (44/61 and 35/61 genes, respectively) but not in SOD1G85R/+ LMNs (2/61 genes) (Figure 3A). We considered whether this lack of differentially expressed genes in SOD1G85R/+ LMNs may be due to the SOD1WT (wild-type) control LMNs because they did not cluster as well with the other control LMNs in our PCA analysis; however, even when comparing SOD1G85R/+ LMNs to all control libraries, only 8/61 genes were differentially expressed. We also did not observe marked downregulation of these genes in sensory neurons or astrocytes for any genotypes (Figure 3A). Thus, the downregulation of these genes resulted from muta tions associated with TDP-43 pathology, TARDBPG298S/+ or PFN1G118V/+ but not SOD1G85R/+, and occurred specifically in LMNs.
Figure 3. Alternatively spliced genes in TDP-43-depleted FTD-ALS postmortem neurons are downregulated in iPSC LMNs.

(A) 61 genes are differentially spliced in postmortem neurons that are depleted for TDP-4312,13 and have sufficient read counts to analyze in iPSC LMNs. Those genes are plotted for each cell type and fALS mutation combination. p values were calculated using gene set enrichment analysis (GSEA).
(B) STMN2 splicing in the TARDBP LMN isogenic pair, TDP-43 knockdown,12 and sALS postmortem LMNs.32 Black arrows indicate exon 2a.
(C) Expression of splicing genes in C9ORF72 iPSC LMNs.40 Significant genes are colored in red; n = 6 libraries per condition.
(D) STMN2 splicing in C9ORF72 iPSC LMNs.
(E) Expression of splicing genes in SOD1A4V/+ iPSC LMNs.41 Significant genes are colored in red; n = 3 control libraries and n = 2 SOD1A4V/+ libraries.
We next considered whether these genes were downregulated because iPSC LMNs recapitulate the aberrant splicing observed in FTD-ALS postmortem neurons that lack nuclear TDP-43.13,39 As a control, we re-analyzed four published datasets in which cryptic splicing events have been reported11–13 and were able to replicate detection of cryptic exons (Figures 3B, S4A, and S4B; Table S5); however, we did not observe any cryptic exon inclusion in our fALS iPSC LMNs. We were able to replicate seven other alternative splicing events identified in postmortem FTD-ALS neurons in fALS iPSC LMNs (Table S5), but none of these events cause cryptic exon inclusion. The apparent dichotomy between the downregulation of these genes and the lack of cryptic exon detection led us to evaluate additional previously published datasets of C9ORF72 and SOD1A4V/+ iPSC LMNs41,40 and examine the same subset of genes.13,39 In C9ORF72 iPSC LMNs,40 we found that 26/61 genes were significantly downregulated (Figure 3C) without detectable cryptic exons (Figures 3D and S3). The expression level of only one gene was decreased in SOD1A4V/+ iPSC LMNs41 (Figure 3E), consistent with our own results in SOD1G85R/+ iPSC LMNs (Figure 3A).
We performed qRT-PCR for the STMN2 and UNC13A fulllength transcripts and their cryptic exons (Figures 4A and S4C–S4E). The expression of the full-length mRNAs mirrored the RNA sequencing results. We could now detect a small increase in the STMN2 cryptic exon in RNA from TARDBPG298S/+ LMNs but only when normalizing to the full-length transcript as has been done in two recent studies17,18 (Figures 4A, S4D, and S4E). No change was detected in the STMN2 cryptic exon in PFN1G118V/+ or the UNC13A cryptic exon in either genotype. We separately performed qRT-PCR with hydrolysis probes and obtained similar results (Figures S4D and S4E).
Figure 4. Transcripts containing cryptic exons are rapidly degraded.

qRT-PCR for full-length transcripts and cryptic exon transcripts in STMN2 and UNC13A; n = 3 wells per condition.
(B and C) Western blots for TDP-43 in the RIPA-soluble (S) fraction from LMNs.
(D) qRT-PCR for cryptic exon transcripts after inhibiting translation with CHX. Black circles and gray squares indicate separate experiments.
(E) Expression of NMD-sensitive transcripts in postmortem sALS LMNs and TARDBPG298S/+ iPSC LMNs. Two-sided t tests or GSEA were used to compare between groups. ***p < 0.001, **p < 0.01, and *p < 0.05.
On account of the reduced expression of TDP-43 splicing targets, we next investigated whether TDP-43 was dysregulated. We observed no change in TARDBP mRNA expression in TARDBPG298S/+ and PFN1G118V/+ LMNs relative to their isogenic controls (Table S1), and therefore we examined TDP-43 protein. We found no difference in total TDP-43 quantified by immunofluorescence and only a small increase in TDP-43 cytoplasmic localization in PFN1G118V/+ (Figures S5A and S5B). We then assessed TDP-43 solubility by separating LMNs into radioimmunoprecipitation assay (RIPA)-soluble and RIPA-insoluble fractions and performed western blotting (Figures 4B and S5C). We observed a small decrease in full-length TDP-43 in the RIPA-soluble fractions of TARDBPG298S/+ and PFN1G118V/+ LMNs relative to their isogenic controls (Figure 4C). We did not observe any TDP-43 cleavage in the soluble fraction, and we did not observe any TDP-43 or phosphorylated TDP-43 in the insoluble fractions, although we were able to detect both total TDP-43 and phosphorylated TDP-43 in a positive control of brain homogenates from hTDP-43-DNLS transgenic mice42 (Figure S5C).
Cryptic splicing in TARDBPG298S/+ LMNs is masked by rapid transcript degradation
The lack of strong cryptic exon expression in TARDBPG298S/+ and PFN1G118V/+ LMNs and previously published heterozygous C9ORF72 iPSC LMNs contrasts with the detection of cryptic exons in publications using homozygous TARDBP mutations or TARDBP knockdown.10–12,16 Notably, homozygous TARDBP mutations led to less frequent cryptic exon inclusion than TARDBP knockdown (Figures S4A and S4B), leading us to hypothesize that cryptic exon inclusion may occur at an even lower rate with heterozygous TARDBP mutations. In general, aberrant mRNA transcripts are degraded by translation-dependent RNA clearance mechanisms,43 including the nonsense-mediated decay (NMD) pathway but also no-go and non-stop mechanisms. To prevent the clearance of aberrant transcripts by these mechanisms, we treated TARDBPG298S/+ iPSC LMNs with cycloheximide (CHX)12 and observed increased expression of STMN2 and UNC13A cryptic exons (Figure 4D), indicating that these transcripts accumulate when translation-dependent RNA quality control measures are blocked. We did not observe an increase in STMN2 or UNC13A cryptic exons in TARDBPWT or PFN1G118V/+ iPSC LMNs after CHX treatment (Figure S5D). These results are consistent with preserved TDP-43 function in the TARDBPWT LMNs and a more modest extent of TDP-43 impairment in the PFN1G118V/+ LMNs (Figures 3A and S5D).
The rapid clearance of transcripts containing cryptic exons led us to hypothesize that RNA clearance mechanisms may be functional in iPSC LMNs but disrupted in sALS postmortem LMNs and thus that impairment of RNA clearance may contribute to cryptic exon accumulation in postmortem LMNs. To test this possibility, we used the upregulation of established NMD target genes as an index of blocked NMD.44–46 We observed an increase in the expression of NMD-sensitive transcripts in postmortem sALS LMNs (log2(fold change) = 1.6), a finding suggestive of disrupted NMD (Figure 4E). In contrast, we observed much smaller effects on NMD target genes in fALS iPSC LMNs (TARDBPG298S/+ = 0.07, PFN1G118V/+ = 0.49, SOD1G85R/+ = 0.08) (Figures 4E and S5E). Thus, intact RNA quality control mechanisms may reduce cryptic exon transcripts in fALS iPSC LMNs, whereas impairment of such mechanisms in postmortem neurons may contribute to cryptic exon persistence and consequent detection.
DISCUSSION
iPSC-based modeling studies of neurodegenerative disease have implicated individual candidate genes that have been validated in postmortem disease tissue.10–13,47,48 However, there have not been systemic comparisons to address, on a genome-wide level, how comprehensively iPSC models capture gene dysregulation in bona fide disease-relevant cell types. Here, we find that monocultures of LMNs harboring fALS mutations capture a third of the genes differentially regulated in sALS laser-captured LMNs from postmortem tissue. These transcriptional changes, which occur in only 10-day-old LMNs, broadly validate the utility of such iPSC models and the capacity of fALS gene mutations to yield insight into more complex and elusive sALS mechanisms. Furthermore, expression analysis within iPSC-derived neurons may provide reciprocal insight into the interpretation of the postmortem datasets. For example, in the iPSC LMNs, which do not contain immune cells, the alteration of genes typically associated with innate immune activation may support greater impact of such signaling pathways within neurons themselves37 and less influence of microglial contamination in laser-captured postmortem LMNs.32
By using a matrix of iPSCs harboring different fALS mutations and a range of differentiated cell types, we address both phenotypic convergence and cell-type vulnerability. Despite starting from the same iPSC lines, we observed almost completely different sets of differentially expressed genes in LMNs compared to sensory neurons or NGN2 superficial cortical neurons, illustrating the extent to which ALS mutations initiate broadly distinct transcriptional programs in vulnerable vs. non-vulnerable cell types. These results demonstrate how cell fate dramatically influences downstream responses. Within the LMNs, the substantial overlap among dysregulated genes across all three ALS genotypes supports a mechanism of phenotypic convergence in ALS and provides candidate genes that may be subsequently tested for such a role. Given the demonstrated value of disease modeling with specific cell types and multiple genotypes, the capacity of PiggyBac integrations to provide robust, rapid, and highly pure differentiations across cell types will be valuable for accurate modeling of selective vulnerability in neurodegenerative disease.
The example of transcript depletion of genes that require nuclear TDP-43 for proper splicing illustrates the specificity of iPSC modeling for both cell-type- and genotype-specific effects.9–13,39 While TDP-43 depletion or homozygous mutation of TDP-43 elicited aberrant splicing in multiple cell types,9–13,16,39 we found that heterozygous mutations–as occur in the disease–produced downregulation of such transcripts in LMNs but not in sensory neurons, NGN2 neurons, or astrocytes. This interaction between TDP-43 dysfunction and cellular identity is also consistent with previous reports of TARDBP knockdown yielding different cryptic splicing events in different cell types.49 In addition to cell-type effects, the transcript abundance of these genes shows genotype-specific patterns that parallel the presence or absence of TDP-43 pathology in human tissue: the decrease in transcript abundance occurred in fALS iPSC LMNs harboring TARDBP, PFN1, and C9ORF72 mutations, genes linked to TDP-43 pathology, but not in two different SOD1 mutations, which do not yield appreciable TDP-43 pathology in postmortem tissue.6,8
The reduction of transcripts that require nuclear TDP-43 for their correct splicing suggests a loss of TDP-43 function in TARDBPG298S/+ and PFN1G118V/+ iPSC LMNs. However, we observed no change in total TDP-43 protein and only minimal differences in TDP-43 protein localization by immunostaining.
We did observe a decrease in RIPA-soluble TDP-43 in both TARDBPG298S/+ and PFN1G118V/+ LMNs without an increase in RIPA-insoluble TDP-43. The expected low level of RIPA-insoluble TDP-43 may have been below the detection threshold of our assay. For TARDBPG298S/+ LMNs, we cannot distinguish whether the loss of TDP-43 function resulted primarily from a reduction in soluble protein or, as suggested by a previous study, due to the mutation, but without a loss of TDP-43 from the nucleus, change in TDP-43 protein levels, aggregation, or solubility.50 For PFN1G118V/+, our study is consistent with prior studies linking mutations in PFN1 with altered TDP-43 location and reduced solubility.51,52
While we observed reduced expression of transcripts that require nuclear TDP-43 for correct splicing, at this early time point, we did not detect cryptic exon transcripts directly in our RNA sequencing data or in datasets from similar studies.40,41 Low abundance of cryptic exons was supported by qRT-PCR in which small changes in cryptic exon levels depended on how the normalization was performed. Blocking translation-dependent RNA clearance pathways12,43 in TARDBPG298S/+ LMNs increased STMN2 and UNC13A cryptic exon transcripts, consistent with a rapid turnover of these transcripts that could limit their detection under native conditions. These results differ slightly from experiments performed in SH-SY5Y cells, in which blocking translation-dependent RNA clearance increased cryptic exon abundance for UNC13A, but not STMN2,12 but may potentially be explained by cell-type differences. Consistent with such cell-type variation in RNA clearance, analysis of consensus NMD decay target levels did not indicate NMD impairment in sALS brain tissue,53 but our similar analysis of postmortem sALS LMNs32 suggested that NMD may be compromised.
Limitations of the study
Defining the overlap between fALS iPSC LMNs and postmortem sALS LMNs may help delineate which gene expression changes in iPSC LMN monocultures are most disease relevant. However, some changes that are not captured in the iPSC models may reflect non-cell-autonomous, aging, or other effects, and determining what causes these other postmortem changes could provide greater insight into ALS pathogenesis. The effects we observed occurred at surprisingly early time points; further neuronal maturation may improve disease fidelity, including cryptic exon abundance. Emerging techniques and reagents for more precise quantitation of TDP-43 and cryptic exon transcripts and peptides,54–56 as well as modulation of RNA clearance mechanisms, may provide more sensitive detection of TDP-43 pathology and further molecular dissection of the turnover of aberrant splicing products.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Brian Wainger (brian.wainger@ mgh.harvard.edu )
Materials availability
Requests of material, including CRISPR edited iPSC lines, can be directed to the lead contact, Brian Wainger. Reagents and cell lines can be transferred after the completion of a materials transfer agreement. We have made all plasmids and their sequences available through Addgene (PB-tet-hNIL, #197089; PB-tet-NGN2, #197090; PB-tet-NB, #197092).
Data and code availability
RNA-sequencing data generated in this manuscript has been deposited in dbGaP (phs002440.v2.p1). These data are available under restricted access to comply with the conditions of the informed consent form agreed with the donors involved in the study, and access can be obtained by applying for controlled access through the dbGaP website. All other data reported in this paper will be shared by the lead contactupon request.
Custom scripts used to analyze immunofluorescence and RNA-seq data are available at https://github.com/waingerlab/iPSC-SMN.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
iPSC culture
iPSCs were plated on Vitronectin XF (Stemcell Technologies 07180) using mTeSR Plus (Stemcell Technologies 100–0275) supplemented with CET.66 After day 1, CET was removed, and cells were maintained in mTeSR Plus. Cells were passaged with Accutase (Thermofisher Scientific A1110501). All iPSC lines have been described previously.2 Cells were maintained at 37°C with 5% CO2.
PiggyBac integration
iPSCs were nucleofected using the Human Stem Cell Nucleofector Kit 1 (Lonza VPH-5012) and protocol A-23 with a Nucleofector I (Lonza). Both the transposase plasmid (2.75μg) and donor plasmid (2.75μg) were nucleofected into 800,000 iPSCs and then maintained in mTeSR Plus with CET for 24 h. Cells with successful nucleofection were then selected using puromycin (10μg/mL, InvivoGen ant-pr-1) for 48 h. Selection continued until all remaining cells had a nuclear BFP2 signal, indicating successful integration of the donor plasmid.
Neuron differentiation
iPSCs were differentiated using induction media (480mL DMEM/F12, Life Technlologies 11320082; 5mL N2 supplement, Gibco 17502–048; 5mL non-essential amino acids, Corning 25–025-CI; 5mL Glutamax, Thermo 35050061; 5mL Penicillin/Streptomycin, Life Technologies 15070–063) supplemented with doxycycline (2μg/mL, Sigma Aldrich D9891–1G) and CET. For LMN differentiations, Compound E (0.2uM, Calbiochem 565790–500UG) was also added. 10 million iPSCs with integrated donor plasmids were resuspended in 25mL supplemented induction media and plated in a T175 flask coated with Matrigel (Corning 354277). After 24 h, the media was exchanged for fresh supplemented induction media. The following day differentiated cells were either frozen in batches of 1 million cells/vial (250μL supplemented induction media, 200μL FBS (Hyclone SH30910.03HI), 50μL DMSO (Sigma D2650)) or replated for experiments.
For immunostaining experiments, 96-wells plates (Cellvis P96–1.5H-N) were coated with PEI (Sigma 03880) diluted 1:50 in borate buffer (100mM boric acid, 25mM sodium tetraborate, 75 mM sodium chloride, pH 8.4) for 24 h and then washed 5x in 1xPBS (Gibco 10010049). They were then coated for an additional 24 h in laminin (12μg/mL, Life Technologies 23017–015) and then washed an additional 3x in 1xPBS. Frozen, differentiated neurons were thawed in supplemented induction media and plated at either 40,000 cells/well (Figure 1) or 80,000 cells/well (Figure 3). The following day, neurons were transferred to long-term media supplemented with aphidicolin (5uM, Cell Signaling 32774). After two days, media was exchanged for fresh long-term media without aphidicolin. Neurons were cultured for an additional 3 days and then fixed for immunofluorescence. For qRT-PCR and western blotting experiments, frozen cells were thawed into 6-well plates (Corning 353046) coated in Matrigel at a density of 1 million cells/well in supplemented induction media. The next day, media was exchanged for long-term media with aphidicolin. Two days later, media was exchanged for long-term media without aphidicolin. Cells were harvested 3 days later by direct application of buffer RLT from the RNeasy mini kit (Qiagen 74104).
LMN long-term media consisted of Neurobasal (478mL, Life Technologies 21103049), N2 supplement (5mL, Gibco 17502–048), non-essential amino acids (5mL, Corning 25–025-CI), Glutamax (5mL, Thermo 35050061), Penicillin/Streptomycin (5mL, Life Technologies 15070–063), and beta mercaptoethanol (2mL, Sigma M6250–100ML). LMN media was supplemented with GDNF (10 ng/mL, Life Technologies PHC7044), BDNF (10 ng/mL, Life Technologies PHC7074), CTNF (10 ng/mL R&D Systems 257-NT-50UG), IGF-1 (10 ng/mL, R&D Systems 291-G1–200), and retinoic acid (1uM, Sigma R2625–50MG).
Long-term media for cortical-like neurons and sensory neurons consisted of Neurobasal (475mL, Life Technologies 21103049), B27 supplement (10mL, Gibco 17504–44), Glutamax (5mL, Thermo 35050061), Penicillin/Streptomycin (5mL, Life Technologies 15070–063), and sodium chloride (5mL of 5M solution, Sigma 71376–1KG). Cortical neuron media was supplemented with NT-3 (10 ng/mL, PeproTech, 450–03) and BDNF (10 ng/mL, Life Technologies PHC7074). Sensory neuron media was supplemented with NT-3 (10 ng/mL, PeproTech, 450–03), BDNF (10 ng/mL, Life Technologies PHC7074), GDNF (10 ng/mL, Life Technologies PHC7044), and NGF (10 ng/mL, R&D Systems 256-GF).
Astrocyte differentiation
1 million iPSCs were resuspended in 12mL N2B27 media (250mL Advanced DMEM/F12, Thermo 12634–028; 250mL Neurobasal, Life Technologies 21103049; 5mL Penicillin/Streptomycin, Life Technologies 15070–063; 5mL Glutamax, Life Technologies 35050061; 1mL beta mercaptoethanol, Sigma M6250–100ML; 10mL B27, Life Technologies 17504044; 5mL N2 Life Technologies 17502048) supplemented with LDN193189 (0.1uM, Stemgent 04–0074-02), SB431542 (20mM, DNSK DNSK-KI-12), Retinoic Acid (100nm, Sigma R2625–50MG), and Y27632 (10μM, abcam ab120129), and plated in a T75 flask coated with Matrigel. The following day, fresh supplemented N2B27 media was exchanged, except without Y27632. Media was exchanged again on day 4. On day 6, neural stem cells were lifted with accutase and 1 million cells were resuspended in 12mL astrocyte media with astrocyte supplement, FBS, and Penicillin/Streptomycin (all astrocyte media reagents included in Sciencell 1801) and then plated in a T75 flask coated with Matrigel. Media was exchanged every 2–3 days for 1 month. When cells reached 80% confluence, they were split 1:4 into a new T75 flask coated with Matrigel.
For immunofluorescence experiments, astrocytes were seeded into 96-wells plates (Cellvis P96–1.5H-N) coated with PEI and laminin (see neuron differentiation). 10,000 astrocytes per well were seeded and then grown for an additional week in supplemented astrocyte media before fixation with 4% formaldehyde (Life Technologies #28908).
SH-SY5Y culture
The neuroblastoma SH-SY5Y cells (ATCC) were cultured in DMEM/F12 (Gibco 1132–033) containing 1% of Penicillin/Streptomycin (Gibco 15140–122) and 10% of Fetal Bovine Serum (Sigma F0926) and kept at 37°C and 5% CO2. The knockdown was achieved by transfecting cells for 48h with siRNA against TDP-43 (ON-TARGETplus siRNA, Dharmacon L-012394) or a control sequence (ON-TARGETplus Non-targeting Control Pool, Dharmacon D001810–10) using Lipofectamine RNAiMAX Transfection Reagent (Thermofisher Scientific 13778075) in Opti-MEM (Gibco 31985070). We used a final siRNA amount of 2.5pmol per well.
METHOD DETAILS
Immunofluorescence
Cells were fixed using using 4% formaldehyde (Life Technologies #28908) for 15 min and then blocked and permeabilized for 1 h in PBT (1xPBS, Gibco #10010049; 0.5% Triton X-, Millipore Sigma #9400; 1% Bovine Serum Albumin, Millipore Sigma #A2153). Cells were then incubated overnight in PBT with primary antibodies. The following day, cells were washed 2x in 1xPBS and incubated for 1 h in PBT with secondary antibodies and Hoechst (1:1000, Thermofisher Scientific 62249). Cells were then washed 2x in 1xPBS and then imaged with an ImageXpress micro confocal (MetaXpress version 6 software; 10x objective) or a Zeiss LSM900 (Zen 3.3 software; 20x or 63x objective). Images were quantified using custom MATLAB (2018b) scripts (https://github.com/waingerlab/iPSC-LMN). Briefly, the nuclear signal was thresholded at 3 standard deviations above the mean of the background signal, and groups of pixels meeting this threshold were identified as nuclei. The same approach was used with TUJ1 to identify the cytoplasmic compartment. Thin segments of the TUJ1 signal extending away from the cytoplasm were classified as neurites. A cell was considered “positive” for a marker if the average of the cell’s signal for that marker was more than 2 standard deviations above the background noise.
The primary antibodies used in this study were: anti-Brn2 (1:1000, Cell Signaling 12137S), anti-FoxG1 (1:500, Millipore Sigma MABD79), anti-Tuj1 (1:500, Aves TUJ-0020), anti-Brn3a (1:200, Millipore Sigma MAB1585), anti-Isl1 (1:200, Abcam ab203406), anti-GFAP (1:250, Abcam ab194324), anti-S100β (1:200, Abcam ab52642), and anti-TDP-43 (1:500, Proteintech 10782–2-AP). Secondary antibodies used in this study were: donkey anti-mouse 488 (1:1000, Thermofisher Scientific A21202), donkey anti-mouse 568 (1:1000, Thermofisher Scientific A10037), donkey anti-rabbit 488 (1:1000, Thermofisher Scientific A21206), donkey anti-rabbit 568 (1:1000, Thermofisher Scientific A10042), and donkey anti-chicken 647 (1:1000, Jackson Immunoresearch 703–605-155).
Cyclic immunofluorescence
LMNs were first imaged before immunostaining to track the BFP2 signal. They were then blocked and permeabilized for 1 h in PBT and incubated overnight in a primary antibody solution of: PBT, anti-Hb9 (1:150, DSHB 81.5C10), anti-CHAT (1:100, EMD Millipore AB144P-200UL), and anti-NeuN (1:500, EMD Millipore ABN78). Cells were then washed 2x in 1xPBS and incubated for 1 h in a secondary solution of: PBT, Hoechst (1:1000, Thermofisher Scientific 62249), donkey anti-mouse 488 (1:1000, Thermofisher Scientific A21202), donkey anti-goat 555 (1:1000, Thermofisher Scientific A21432), and donkey anti-rabbit 647 (1:1000 Thermofisher Scientific A31573). Cells were then imaged on an ImageXpress micro confocal. Fluorophores were then bleached overnight using 25mM sodium hydroxide (Sigma-Aldrich 72068–100ML) in 1xPBS with 4.5% hydrogen peroxide (Thermofisher Scientific H325–500). Fluorophore bleaching was confirmed using an ImageXpress micro confocal before proceeding to additional staining. Cells were then stained overnight with a primary solution of: PBT, anti-CHAT 488 (1:100, Abcam ab192465), anti-Isl1 568 (1:200, Abcam ab203406), and anti-Tuj1 (1:500, Aves TUJ-0020). They were then washed 2x in 1xPBS and incubated for 1 h in a secondary solution of PBT with donkey anti-chicken 647 (Jackson Immunoresearch 703–605-155) and Hoechst (1:1000, Thermofisher Scientific 62249). Cells were washed an additional 2x in 1xPBS and imaged on an ImageXpress micro confocal. Images were then aligned between cycles using custom MATLAB scripts (https://github.com/waingerlab/iPSC-SMN).
RNA extraction and qRT-PCR
Cells were harvested using buffer RLT from the RNeasy mini kit (Qiagen 74104). The RNeasy mini kit was then used to extract RNA, with QIAshredder columns (Qiagen 79654) used to increase the yield. The High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems 4368814) was used to make cDNA. KiCqStart SYBR Green (Millipore Sigma KCQS00) was used with a BioRad CFX96 PCR system (running BioRad CFX Manager 3.1) to perform qRT-PCR. Primers are listed in Table S7. All gene expression was normalized to GAPDH, and fold change was calculated using ΔΔCt.
For qRT-PCR with hydrolysis probes, the GAPDH primers were: GAAGGTGAAGGTCGGAGTC (forward), GAAGATGGTGATG GGATTTC (reverse), and CAAGCTTCCCGTTCTCAGCC (probe). The STMN2 cryptic exon primers were CTTTCTCTAGCACGG TCCCAC (forward), ATGCTCACACAGAGAGCCAAATTC (reverse), and CTCTCGAAGGTCTTCTGCCG (probe). cDNA, primers, and iTaq (BioRad 1725131) were mixed together and analyzed using a BioRad CFX96 PCR system.
Cycloheximide treatment
iPSC LMNs were grown to day 10 as described in the neuron differentiation methods. iPSC LMNs were treated with DMSO or 100uM Cycloheximide (Millipore Sigma 01810) for 6 h and then harvested using buffer RLT from the RNeasy mini kit (Qiagen 74104).
Western blotting
Induced pluripotent stem cell (iPSC)-derived motor neurons were lysed with ice-cold 250 μL of radioimmunoprecipitation assay (RIPA) lysis and extraction buffer (Thermofisher Scientific 89901) supplemented with 1X Halt protease and phosphatase inhibitor cocktail (Roche 78440). Cell lysates were centrifuged at 21,000 rcf for 30 min at 4°C, and the resulting supernatant was collected as the RIPA-soluble fraction. The cell pellets were washed with the same RIPA buffer and centrifuged at 21,000 rcf for 5 min at 4°C before discarding the residual RIPA buffer. The remaining cell pellets were dissolved in 50 μL of urea buffer (7M urea, 2M thiourea, 4% 3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), 30 mM Tris, pH 8.5). The protein concentration was immediately measured using the Pierce BCA protein assay kit (Thermofisher Scientific 23252).
Brain homogenates from NEFH-hTDP-43ΔNLS transgenic mice42 were used as positive control (30 μg) for insoluble and hyperphosphorylated TDP-43 in the urea-soluble fraction. Samples were prepared in TE buffer (50 mM Tris, pH 7.4, 50 mM NaCl, 1 mM EDTA) with protease (Roche 1873580001) and phosphatase inhibitors (Sigma P5726–5mL). The resulting brain lysates (50 μL) were mixed with 100 μL of co-immunoprecipitation buffer (50 mM Tris, pH 7.4, 300 mM NaCl, 5 mM EDTA, 0.1% Triton X-100, 3.0% SDS).
For Western blot analyses of iPSC LMNs, 10 μg of protein from the RIPA-soluble fraction (denatured at 95°C for 5 min) and 10 μL of urea-soluble fraction were prepared in NuPAGE LDS sample buffer (Thermofisher Scientific NP0007) and electrophoresed in 4–20% Criterion TGX Precast Midi 18-well Protein Gel (Bio-Rad 5671094) at 120 V for 90 min. Proteins were transferred to a PVDF membrane at 15 V for 6 min using iBlot 2 transfer stacks, PVDF, regular size (Thermofisher Scientific IB24001), and the iBlot 2 Gel Transfer Device (ThermoFisher Scientific IB21001). Membranes were blocked for 1 h at room temperature in 1.0% skim milk in 1X Tris-buffered saline with 0.1% Tween 20, pH 7.4 (TBST) solution (Boston BioProducts IBB-180X) and incubated with primary antibodies against TDP-43 (ProteinTech 10782–2-A, 1:1,000), pTDP-43 (CosmoBio TIP-PTD-M01, 1:3,000), and GAPDH (Cell Signaling 2118S, 1:2,000) overnight at 4°C. Membranes were washed thrice with 1X TBST solution for 5 min before incubation with either the IRDye 800CW Donkey anti-Rabbit IgG Secondary Antibody (LI-COR Biosciences 926–32213, 1:5,000) or the IRDye 680RD Goat anti-Mouse IgG Secondary Antibody (LI-COR Biosciences 926–68070, 1:5,000) at room temperature for 2 h. Membranes were washed thrice with 1X TBST solution for 5 min before visualization using the Bio-Rad Chemidoc MP imaging system.
RNA sequencing
Separate differentiations were completed for each replicate. 1 million cells per differentiation were pelleted and sent to Genewiz for RNA extraction, library preparation (TruSeq RNA Library Prep Kit v2, mRNA selection), and RNA sequencing (Illumina HiSeq). An average of 26.5 million 2×150bp paired end reads were obtained per library. Reads were aligned to GRCh38 using STAR (2.7.3a)57 with refgene annotation and counted using STAR –quantmode GeneCounts. Differential expression was calculated using DESeq2 (1.36.0)58 in R (4.0.3). To identify general differences between ALS and control libraries, we used design = ~Gene+Disease, where “Gene” indicates the isogenic pair and “Disease” represents whether a mutation that causes ALS is present. PCA plots, heat-maps, and volcano plots were made in R (4.0.3) or MATLAB (2018b), Venn Diagrams in MATLAB (2018b), and Sashimi plots in IGV (2.11.9)59. For differential splicing analysis, reads were aligned to GRCh38 using STAR (2.7.3a) with Gencode v40 annotation. Aligned bams were indexed with SAMtools (1.13)60 and differential splicing identified with Majiq (2.3)61. Reads aligning to cryptic exons were counted using BEDTools (2.27.1)62. The previously published datasets reanalyzed in this study were obtained through the NCBI GEO database (GSE54409,41 GSE143743,40 GSE76220,32 GSE122069,11 GSE12654339) or the European Nucleotide Archive (PRJEB4276312). For the reanalysis of postmortem sporadic ALS LMNs,32 a model of design = ~Subject_Sex+Disease was used to normalize for effects by patient sex. For the reanalysis of TDP-43 sorted neurons,39 a model of design = ~Subject+TDP-43_status was used to pair libraries within the same patient. Genes with FDR<0.05 were considered significantly differentially expressed and used in downstream analyses. Differentially expressed genes were analyzed for gene ontology using GOrilla63 and summarized with Revigo.64 Only GO terms with corrected p values less than 0.001 were considered significant. For the analysis of differential splicing events described in Ma et al. 2022, 5 genes were excluded from our analysis because either there were not enough reads in our RNA-seq libraries to evaluate differential expression or the gene symbols did not match because of the use of different annotation files between studies.
NMD-sensitive transcripts
Previous studies identified differentially expressed genes after UPF1,44 UPF2,45 or UPF346 depletion. We compiled the upregulated genes (Table S7) as a readout NMD function and then assessed the expression of these transcripts in iPSC LMNs and postmortem LMNs. Significance was calculated using fgsea (1.22.0)65 in R (4.0.3), and the average log2(fold change) was determined by averaging the log2(fold change) of differentially regulated genes (adjusted p < 0.05).
QUANTIFICATION AND STATISTICAL ANALYSIS
For RNA sequencing experiments, each replicate was obtained from a separate batch differentiation. A negative binomial generalized linear model (DESeq2 1.36.0) was used to identify differences in gene expression. For immunofluorescence and qRT-PCR experiments, neurons were derived from the same batch differentiation, frozen into separate vials, and then plated in either 96-well or 6-well format. Individual plotted dots represent the average of technical replicates in a well (qRT-PCR) or the average of individual cells in a well (immunofluorescence). Red bars represent the average and standard error of the mean. Two sided t-tests (MATLAB 2018b) were used to compare between groups. Pearson correlations (MATLAB 2018b) compared differentially expressed genes in postmortem sALS LMN to differentially expressed genes in iPSC-neurons. Significance for downregulated alternative splicing genes was calculated with fgsea (1.22.0) in R (4.0.3). P-values are depicted in graphs as *, **, or ***, representing p < 0.05, p < 0.01, and p < 0.001, respectively.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-Brn2 | Cell Signaling | CAT# 12137S; RRID:AB_2797827 |
| Anti-FoxG1 | Millipore Sigma | CAT# MABD79 |
| Anti-Tuj1 | Aves | CAT# TUJ-0020 |
| Anti-Brn3a | Millipore Sigma | CAT# MAB1585; RRID:AB_94166 |
| Anti-GFAP | Abcam | CAT# ab194324 |
| Anti-S100β | Abcam | CAT# ab52642; RRID:AB_882426 |
| Anti-TDP-43 | Proteintech | CAT# 10782-2-AP; RRID:AB_615042 |
| Anti-Hb9 | DSHB | CAT# 81.5C10; RRID:AB_2145209 |
| Anti-CHAT | EMD millipore | CAT# AB144P-200UL; RRID:AB_2079751 |
| Anti-NeuN | EMD millipore | CAT# ABN78; RRID:AB_10807945 |
| Anti-CHAT 488 | Abcam | CAT# ab192465 |
| Anti-Isl1 568 | Abcam | CAT# ab203406 |
| Anti-pTDP-43 | CosmoBio | CAT# TIP-PTD-M01; RRID:AB_1961900 |
| Anti-GAPDH | Cell Signaling | CAT# 2118S; RRID:AB_561053 |
| Donkey anti-mouse 488 | Thermofisher Scientific | CAT# A21202; RRID:AB_141607 |
| Donkey anti-mouse 568 | Thermofisher Scientific | CAT# A10037; RRID:AB_253401 |
| Donkey anti-rabbit 488 | Thermofisher Scientific | CAT# A21206; RRID:AB_2535792 |
| Donkey anti-goat 555 | Thermofisher Scientific | CAT# A21432; RRID:AB_2535853 |
| Donkey anti-rabbit 568 | Thermofisher Scientific | CAT# A10042; RRID:AB_2534017 |
| Donkey anti-chicken 647 | Jackson Immunoresearch | CAT# 703-605-155; RRID:AB_2340379 |
| Donkey anti-rabbit 647 | Thermofisher Scientific | CAT# A31573; RRID:AB_2536183 |
| IRDye 680RD Goat anti-Mouse IgG | LI-COR | CAT# 926-68070; RRID:AB_10956588 |
| IRDye 800CW Donkey anti-Rabbit IgG | LI-COR | CAT# 926-32213; RRID:AB_621848 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| NEB 5-alpha Competent E. coli | New England Biolabs | CAT# C2987I |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| 16% Formaldehyde | Life Technologies | CAT# 28908 |
| PBS | Gibco | CAT# 10010049 |
| Triton X- | Millipore Sigma | CAT# 9400 |
| Bovine Serum Albumin | Millipore Sigma | CAT# A2153 |
| Hoechst | Thermofisher Scientific | CAT# 62249 |
| Sodium Hydroxide | Sigma-Aldrich | CAT# 72068-100ML |
| Hydrogen Peroxide | Thermofisher Scientific | CAT# H325-500 |
| Cycloheximide | Millipore Sigma | CAT# 01810 |
| Dimethyl Sulfoxide | Sigma | CAT# D2650 |
| Vitronectin XF | Stemcell Technologies | CAT# 07180 |
| mTeSR Plus | Stemcell Technologies | CAT# 100-0275 |
| Accutase | Thermofisher Scientific | CAT# A1110501 |
| Chroman1 | MedChemExpress | CAT# HY-15392 |
| Emricasan | Selleckchem | CAT# S7775 |
| Trans-ISRIB | Tocris | CAT# 5284 |
| Puromycin | InvivoGen | CAT# ant-pr-1 |
| DMEM/F12 | Life Technlologies | CAT# 11320082 |
| N2 supplement | Gibco | CAT# 17502-048 |
| Non-essential amino acids | Corning | CAT# 25-025-CI |
| Glutamax | Thermo | CAT# 35050061 |
| Penicillin/Streptomycin | Life Technlologies | CAT# 15070-063 |
| Doxycycline | Sigma Aldrich | CAT# D9891-1G |
| Compound E | Calbiochem | CAT# 565790-500UG |
| Matrigel | Corning | CAT# 354277 |
| Fetal Bovine Serum | Hyclone | CAT# SH30910.03HI |
| 96-wells plates | Cellvis | CAT# P96-1.5H-N |
| 6-well plates | Corning | CAT# 353046 |
| Polyethylenimene | Sigma | CAT# 03880 |
| Boric Acid | Millipore Sigma | CAT# B6768-500G |
| Sodium Tetraborate | Sigma-Aldrich | CAT# B9876-500G |
| Sodium Chloride | Sigma-Aldrich | CAT# 71376-1KG |
| Laminin | Life Technlologies | CAT# 23017-015 |
| Aphidicolin | Cell Signaling | CAT# 32774 |
| Neurobasal | Life Technologies | CAT# 21103049 |
| Beta mercaptoethanol | Sigma | CAT# M6250-100ML |
| GDNF | Life Technologies | CAT# PHC7044 |
| BDNF | Life Technologies | CAT# PHC7074 |
| CTNF | R&D Systems | CAT# 257-NT-50UG |
| IGF-1 | R&D Systems | CAT# 291-G1-200 |
| Retinoic acid | Sigma | CAT# R2625-50MG |
| B27 supplement | Gibco | CAT# 17504-44 |
| NT-3 | PeproTech | CAT# 450-03 |
| NGF | R&D Systems | CAT# 256-GF |
| Advanced DMEM/F12 | Thermo | CAT# 12634-028 |
| LDN193189 | Stemgent | CAT# 04-0074-02 |
| SB431542 | DNSK | CAT# DNSK-KI-12 |
| Y27632 | Abcam | CAT# ab120129 |
| Astrocyte Media | Sciencell | CAT# 1801 |
| DMEM/F12 | Gibco | CAT# 1132-033 |
| Penicillin/Streptomycin | Gibco | CAT# 15140-122 |
| Fetal Bovine Serum | Sigma | CAT# F0926 |
| Lipofectamine RNAiMAX | Thermofisher Scientific | CAT# 13778075 |
| Opti-MEM | Gibco | CAT# 31985070 |
| QIAshredder | Qiagen | CAT# 79654 |
| KiCqStart SYBR Green | Millipore Sigma | CAT# KCQS00 |
| iTaq | BioRad | CAT# 1725131 |
| RIPA Buffer | Thermofisher Scientific | CAT# 89901 |
| Halt protease and phosphatase inhibitor cocktail | Roche | CAT# 78440 |
| Urea | Millipore Sigma | CAT# U4883-6X25ML |
| Thiourea | Millipore Sigma | CAT# T8656-50G |
| CHAPS | Biotium | CAT# 22024-10g |
| Protease inhibitor cocktail | Roche | CAT# 1873580001 |
| Phosphatase inhibitor cocktail | Sigma | CAT# P5726-5mL |
| NuPAGE LDS sample buffer | Thermofisher Scientific | CAT# NP0007 |
| 4-20% CriterionTM TGXTM Precast Midi 18-well Protein Gel | Bio-Rad | CAT# 5671094 |
| iBlot 2 transfer stacks | Thermofisher Scientific | CAT# IB24001 |
| 1% blocking buffer | Boston BioProducts | CAT# IBB-180X |
|
| ||
| Critical commercial assays | ||
|
| ||
| Human Stem Cell Nucleofector Kit 1 | Lonza | CAT# VPH-5012 |
| RNeasy mini kit | Qiagen | CAT# 74104 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | CAT# 4368814 |
| Pierce BCA protein assay kit | Thermofisher Scientific | CAT# 23252 |
|
| ||
| Deposited data | ||
|
| ||
| Bulk RNA-seq from iPSC-derived cells | This paper | dbGAP: phs002440.v2.p1 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| CRISPR edited iPSCs | Pereira et al.2 | N/A |
| SH-SY5Y | ATTC | CAT# CRL-2266 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| NEFH-hTDP-43DNLS transgenic mice | Walker et al.42 | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| siRNA against TDP-43 | Dharmacon | CAT# L-012394 |
| siRNA control sequence | Dharmacon | CAT# D001810-10 |
| GAPDH forward: GAAGGTGAAGGTCGGAGTC | This paper | N/A |
| GAPDH reverse: GAAGATGGTGATGGGATTTC | This paper | N/A |
| GAPDH probe: CAAGCTTCCCGTTCTCAGCC | This paper | N/A |
| STMN2 CE forward: CTTTCTCTAGCACGGTCCCAC | This paper | N/A |
| STMN2 CE reverse: ATGCTCACACAGAGAGCCAAATTC | This paper | N/A |
| STMN2 CE probe: CTCTCGAAGGTCTTCTGCCG | This paper | N/A |
| Primers for qRT-PCR, see Table S7 | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| PB-tet-hNIL | This paper | Addgene 197089 |
| PB-tet-NGN2 | This paper | Addgene197090 |
| PB-tet-NB | This paper | Addgene197092 |
| pEIF1 a:Transposase | Michael Ward | Addgene172116 |
|
| ||
| Software and algorithms | ||
|
| ||
| MetaXpress version 6 software | MetaXpress | https://support.moleculardevices.com/ |
| Zen 3.3 software | Zeiss | https://www.zeiss.com/microscopy |
| MATLAB 2018b | MATLAB | https://www.mathworks.com/ |
| STAR (2.7.3a) | Dobin et al.57 | https://github.com/alexdobin/STAR |
| R (4.0.3) | The R Project for Statistical Computing | https://www.r-project.org/ |
| DESeq2 (1.36.0) | Love et al.58 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Integrated Genomics Viewer (2.11.9) | Robinson et al.59 | https://software.broadinstitute.org/software/igv/ |
| SAMtools (1.13) | Danecek et al.60 | http://www.htslib.org/ |
| MAJIQ (2.3) | Green et al.61 | https://majiq.biociphers.org/ |
| BEDtools (2.27.1) | Quinlan etal.62 | https://github.com/arq5x/bedtools2 |
| GOrilla | Eden et al.63 | https://cbl-gorilla.cs.technion.ac.il/ |
| REVIGO | Supek et al.64 | http://revigo.irb.hr/ |
| fgsea (1.22.0) | Subramanian et al.65 | https://github.com/ctlab/fgsea |
Highlights.
fALS iPSC LMNs capture one-third of gene expression changes in postmortem sALS LMNs
iPSC LMNs, but not other neurons, captured postmortem LMN gene expression changes
Dysregulated genes in fALS iPSC-LMNs include TDP-43 splicing targets
RNA clearance may be disrupted in postmortem LMNs but intact in iPSC LMNs
ACKNOWLEDGMENTS
This work was funded by NIH F32NS114319 and a Cullen Education and Research Fund Young Investigator Award (A.H.); the ALS Association Milton Safenowitz Fellowship (C.M.); the BrightFocus Foundation (A.R.A.A.Q.); ALS Finding A Cure (C.L.-T. and M.W.); NIH R01NS112503 and RF1NS124203 (C.L.-T.); and NIH DP2NS106664, NIH 1RF1NS127407, and the New York Stem Cell Foundation (B.J.W.). C.L.-T. is the recipient of the Araminta Broch-Healey Endowed Chair in ALS. B.J.W. is a New York Stem Cell Foundation - Robertson Investigator and is the recipient of the Alexander Healey Endowed Chair in ALS.
Footnotes
DECLARATION OF INTERESTS
B.J.W. is a consultant and member of the scientific advisory board of Quralis and is a coinventor on patent US9517223B2, issued for use of potassium channel openers in neurodegenerative diseases.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113046.
REFERENCES
- 1.Brown RH, and Al-Chalabi A. (2017). Amyotrophic Lateral Sclerosis. N. Engl. J. Med 377, 162–172. 10.1056/NEJMra1603471. [DOI] [PubMed] [Google Scholar]
- 2.Pereira JD, Dubreuil DM, Devlin AC, Held A, Sapir Y, Berezovski E, Hawrot J, Dorfman K, Chander V, and Wainger BJ (2021). Human sensorimotor organoids derived from healthy and amyotrophic lateral sclerosis stem cells form neuromuscular junctions. Nat. Commun 12, 4744–4817. 10.1038/s41467-021-24776-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch N. a., Adamson J, Flynn H, Adamson J, et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, et al. (2011). A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 72, 257–268. 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawamura Y, Dyck PJ, Shimono M, Okazaki H, Tateishi J, and Doi H. (1981). Morphometric Comparison of the Vulnerability of Peripheral Motor and Sensory Neurons in Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol 40, 667–675. 10.1097/00005072-198111000-00008. [DOI] [PubMed] [Google Scholar]
- 6.Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y, Beach TG, Buratti E, Baralle F, Morita M, et al. (2008). Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol 64, 60–70. 10.1002/ana.21425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, et al. (2006). Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 314, 130–133. 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 8.Mackenzie IRA, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, Kwong LK, Forman MS, Ravits J, Stewart H, et al. (2007). Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol 61, 427–434. 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- 9.Ling JP, Pletnikova O, Troncoso JC, and Wong PC (2015). TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 349, 650–655. 10.1126/science.aab0983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klim JR, Williams LA, Limone F, Guerra San Juan I, Davis-Dusenbery BN, Mordes DA, Burberry A, Steinbaugh MJ, Gamage KK, Kirchner R, et al. (2019). ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci 22, 167–179. 10.1038/s41593-018-0300-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Melamed Z, López-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y, Freyermuth F, McMahon MA, Beccari MS, Artates JW, et al. (2019). Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci 22, 180–190. 10.1038/s41593-018-0293-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown AL, Wilkins OG, Keuss MJ, Hill SE, Zanovello M, Lee WC, Bampton A, Lee FCY, Masino L, Qi YA, et al. (2022). TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature 603, 131–137. 10.1038/s41586-022-04436-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma XR, Prudencio M, Koike Y, Vatsavayai SC, Kim G, Harbinski F, Briner A, Rodriguez CM, Guo C, Akiyama T, et al. (2022). TDP-43 represses cryptic exon inclusion in the FTD–ALS gene UNC13A. Nature 603, 124–130. 10.1038/s41586-022-04424-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang S, Cooper-Knock J, Weimer AK, Shi M, Moll T, Marshall JNG, Harvey C, Nezhad HG, Franklin J, Souza CDS, Ning K, Wang C, Li J, Dilliott AA, Farhan S, Elhaik E, Pasniceanu I, Livesey MR, Eitan C, Hornstein E, Kenna KP, Project MinE ALS Sequencing Consortium; Veldink JH, Ferraiuolo L, Shaw PJ, and Snyder MP (2022). Genome-wide identification of the genetic basis of amyotrophic lateral sclerosis. Neuron 110, 992–1008.e11. 10.1016/j.neuron.2021.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hao J, Wells MF, Niu G, San Juan IG, Limone F, Fukuda A, Leyton-Jaimes MF, Joseph B, Qian M, Mordes DA, et al. (2021). Loss of TBK1 activity leads to TDP-43 proteinopathy through lysosomal dysfunction in human motor neurons. Preprint at bioRxiv, 1–22. 10.1101/2021.10.11.464011. [DOI] [Google Scholar]
- 16.Imaizumi K, Ideno H, Sato T, Morimoto S, and Okano H. (2022). Pathogenic mutation of TDP-43 impairs RNA processing in a cell type-specific manner : implications for the pathogenesis of ALS/FTLD. eNeuro 9, ENEURO.0061. 10.1523/ENEURO.0061-22.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Linares GR, Li Y, Chang WH, Rubin-Sigler J, Mendonca S, Hong S, Eoh Y, Guo W, Huang YH, Chang J, et al. (2023). SYF2 suppression mitigates neurodegeneration in models of diverse forms of ALS. Cell Stem Cell 30, 171–187.e14. 10.1016/j.stem.2023.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hung ST, Linares GR, Chang WH, Eoh Y, Krishnan G, Mendonca S, Hong S, Shi Y, Santana M, Kueth C, et al. (2023). PIKFYVE inhibition mitigates disease in models of diverse forms of ALS. Cell 186, 786–802.e28–17.. 10.1016/j.cell.2023.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi Y, Lin S, Staats KA, Li Y, Chang W-H, Hung S-T, Hendricks E, Linares GR, Wang Y, Son EY, et al. (2018). Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med 24, 313–325. 10.1038/nm.4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yuva-Aydemir Y, Almeida S, Krishnan G, Gendron TF, and Gao FB (2019). Transcription elongation factor AFF2/FMR2 regulates expression of expanded GGGGCC repeat-containing C9ORF72 allele in ALS/FTD. Nat. Commun 10, 5466. 10.1038/s41467-019-13477-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fujimori K, Ishikawa M, Otomo A, Atsuta N, Nakamura R, Akiyama T, Hadano S, Aoki M, Saya H, Sobue G, and Okano H. (2018). Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med 24, 1579–1589. 10.1016/0040-4020(75)85065-4. [DOI] [PubMed] [Google Scholar]
- 22.Workman MJ, Lim RG, Wu J, Frank A, Ornelas L, Panther L, Galvez E, Perez D, Meepe I, Lei S, et al. (2023). Large-scale differentiation of iPSC-derived motor neurons from ALS and control subjects. Neuron 111, 1191–1204.e5. 10.1016/j.neuron.2023.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Al-Chalabi A, Van Den Berg LH, and Veldink J. (2017). Gene discovery in amyotrophic lateral sclerosis: Implications for clinical management. Nat. Rev. Neurol 13, 96–104. 10.1038/nrneurol.2016.182. [DOI] [PubMed] [Google Scholar]
- 24.Briggs JA, Li VC, Lee S, Woolf CJ, Klein A, and Kirschner MW (2017). Mouse embryonic stem cells can differentiate via multiple paths to the same state. Elife 6, 269455–e27023. 10.7554/eLife.26945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yusa K, Zhou L, Li MA, Bradley A, and Craig NL (2011). A hyperactive piggyBac transposase for mammalian applications. Proc. Natl. Acad. Sci. USA 108, 1531–1536. 10.1073/pnas.1008322108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mazzoni EO, Mahony S, Closser M, Morrison CA, Nedelec S, Williams DJ, An D, Gifford DK, and Wichterle H. (2013). Synergistic binding of transcription factors to cell-specific enhancers programs motor neuron identity. Nat. Neurosci 16, 1219–1227. 10.1038/nn.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, Marro S, Patzke C, Acuna C, Covy J, et al. (2013). Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798. 10.1016/j.neuron.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beers J, Zou J, Nguyen MQ, Szczot M, Lam RM, Wang Q, Nickolls AR, Lee MM, Espinoza DF, Solinski HJ, et al. (2020). Transcriptional Programming of Human Mechanosensory Neuron Subtypes from Pluripotent Stem Cells. Cell Rep. 30, 932–946.e7. 10.1016/j.celrep.2019.12.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin JR, Fallahi-Sichani M, and Sorger PK (2015). Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method. Nat. Commun 6, 8390–8397. 10.1038/ncomms9390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.TCW J, Wang M, Pimenova AA, Bowles KR, Hartley BJ, Lacin E, Machlovi SI, Abdelaal R, Karch CM, Phatnani H, et al. (2017). An Efficient Platform for Astrocyte Differentiation from Human Induced Pluripotent Stem Cells. Stem Cell Rep. 9, 600–614. 10.1016/j.stemcr.2017.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ, Foong C, White CL, Schneider JA, Kretzschmar HA, et al. (2007). TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am. J. Pathol 171, 227–240. 10.2353/ajpath.2007.070182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krach F, Batra R, Wheeler EC, Vu AQ, Wang R, Hutt K, Rabin SJ, Baughn MW, Libby RT, Diaz-Garcia S, et al. (2018). Transcriptome–pathology correlation identifies interplay between TDP-43 and the expression of its kinase CK1E in sporadic ALS. Acta Neuropathol. 136, 405–423. 10.1007/s00401-018-1870-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wainger BJ, Macklin EA, Vucic S, McIlduff CE, Paganoni S, Maragakis NJ, Bedlack R, Goyal NA, Rutkove SB, Lange DJ, et al. (2021). Effect of Ezogabine on Cortical and Spinal Motor Neuron Excitability in Amyotrophic Lateral Sclerosis: A Randomized Clinical Trial. JAMA Neurol. 78, 186–196. 10.1001/jamaneurol.2020.4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tan HHG, Westeneng HJ, van der Burgh HK, van Es MA, van den Berg LH, Bakker LA, van Veenhuijzen K, van Eijk KR, van Eijk RPA, Veldink JH, and Berg LHVD (2020). The Distinct Traits of the UNC13A Polymorphism in Amyotrophic Lateral Sclerosis. Ann. Neurol 88, 796–806. 10.1002/ana.25841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park SB, Kiernan MC, and Vucic S. (2017). Axonal Excitability in Amyotrophic Lateral Sclerosis. Neurotherapeutics 14, 78–90. 10.1007/s13311-016-0492-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodriguez S, Sahin A, Schrank BR, Al-lawati H, Costantino I, Benz E, Fard D, Albers AD, Cao L, Gomez AC, et al. (2021). Genome-encoded cytoplasmic double-stranded RNAs , found in C9ORF72 ALS-FTD brain , propagate neuronal loss. Sci. Transl. Med 13, eaaz4699–14. 10.1126/scitranslmed.aaz4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y-J, Guo L, Gonzales PK, Gendron TF, Wu Y, Jansen-West K, O’Raw AD, Pickles SR, Prudencio M, Carlomagno Y, et al. (2019). Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science 363, eaav2606–9. 10.1126/science.aav2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu EY, Russ J, Cali CP, Phan JM, Amlie-wolf A, Lee EB, Liu EY, Russ J, Cali CP, Phan JM, et al. (2019). Loss of Nuclear TDP-43 Is Associated with Decondensation of LINE Retrotransposons. Cell Rep. 27, 1409–1421.e6. 10.1016/j.celrep.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abo-Rady M, Kalmbach N, Pal A, Schludi C, Janosch A, Richter T, Freitag P, Bickle M, Kahlert A-K, Petri S, et al. (2020). Knocking out C9ORF72 Exacerbates Axonal Trafficking Defects Associated with Hexanucleotide Repeat Expansion and Reduces Levels of Heat Shock Proteins. Stem Cell Rep. 14, 390–405. 10.1016/j.stemcr.2018.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kiskinis E, Sandoe J, Williams LA, Boulting GL, Moccia R, Wainger BJ, Han S, Peng T, Thams S, Mikkilineni S, et al. (2014). Pathways Disrupted in Human ALS Motor Neurons Identified through Genetic Correction of Mutant SOD1. Cell Stem Cell 14, 781–795. 10.1016/j.stem.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walker AK, Spiller KJ, Ge G, Zheng A, Xu Y, Zhou M, Tripathy K, Kwong LK, Trojanowski JQ, and Lee VMY (2015). Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol. 130, 643–660. 10.1007/s00401-015-1460-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Powers KT, Szeto JYA, and Schaffitzel C. (2020). New insights into no-go, non-stop and nonsense-mediated mRNA decay complexes. Curr. Opin. Struct. Biol 65, 110–118. 10.1016/j.sbi.2020.06.011. [DOI] [PubMed] [Google Scholar]
- 44.Mendell JT, Sharifi NA, Meyers JL, Martinez-Murillo F, and Dietz HC (2004). Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat. Genet 36, 1073–1078. 10.1038/ng1429. [DOI] [PubMed] [Google Scholar]
- 45.Wittmann J, Hol EM, and Jä ck HM (2006). hUPF2 Silencing Identifies Physiologic Substrates of Mammalian Nonsense-Mediated mRNA Decay. Mol. Cell Biol 26, 1272–1287. 10.1128/mcb.26.4.1272-1287.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nguyen LS, Jolly L, Shoubridge C, Chan WK, Huang L, Laumonnier F, Raynaud M, Hackett A, Field M, Rodriguez J, et al. (2012). Transcriptome profiling of UPF3B/NMD-deficient lymphoblastoid cells from patients with various forms of intellectual disability. Mol. Psychiatr 17, 1103–1115. 10.1038/mp.2011.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hawrot J, Imhof S, and Wainger BJ (2020). Neurobiology of Disease Modeling cell-autonomous motor neuron phenotypes in ALS using iPSCs. Neurobiol. Dis 134, 104680–104715. 10.1016/j.nbd.2019.104680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee S, and Huang EJ (2017). Modeling ALS and FTD with iPSC-derived neurons. Brain Res. 1656, 88–97. 10.1016/j.brainres.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jeong YH, Ling JP, Lin SZ, Donde AN, Braunstein KE, Majounie E, Traynor BJ, Laclair KD, Lloyd TE, and Wong PC (2017). Tdp-43 cryptic exons are highly variable between cell types. Mol. Neurodegener 12, 13–19. 10.1186/s13024-016-0144-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arnold ES, Ling S-C, Huelga SC, Lagier-Tourenne C, Polymenidou M, Ditsworth D, Kordasiewicz HB, McAlonis-Downes M, Platoshyn O, Parone PA, et al. (2013). ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc. Natl. Acad. Sci. USA 110, E736–E745. 10.1073/pnas.1222809110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu CH, Fallini C, Ticozzi N, Keagle PJ, Sapp PC, Piotrowska K, Lowe P, Koppers M, McKenna-Yasek D, Baron DM, et al. (2012). Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 488, 499–503. 10.1038/nature11280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Matsukawa K, Hashimoto T, Matsumoto T, Ihara R, Chihara T, Miura M, Wakabayashi T, and Iwatsubo T. (2016). Familial Amyotrophic lateral sclerosis-linked mutations in profilin 1 exacerbate TDP-43-induced degeneration in the retina of Drosophila melanogaster through an increase in the cytoplasmic localization of TDP-43. J. Biol. Chem 291, 23464–23476. 10.1074/jbc.M116.729152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun Y, Eshov A, Zhou J, Isiktas AU, and Guo JU (2020). C9orf72 arginine-rich dipeptide repeats inhibit UPF1-mediated RNA decay via translational repression. Nat. Commun 11, 3354–3359. 10.1038/s41467-020-17129-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zacco E, Kantelberg O, Milanetti E, Armaos A, Panei FP, Gregory J, Jeacock K, Clarke DJ, Chandran S, Ruocco G, et al. (2022). Probing TDP-43 condensation using an in silico designed aptamer. Nat. Commun 13, 3306. 10.1038/s41467-022-30944-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schnitzbauer J, Strauss MT, Schlichthaerle T, Schueder F, and Jungmann R. (2017). Super-resolution microscopy with DNA-PAINT. Nat. Protoc 12, 1198–1228. 10.1038/nprot.2017.024. [DOI] [PubMed] [Google Scholar]
- 56.Prasad A, Bharathi V, Sivalingam V, Girdhar A, and Patel BK (2019). Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front. Mol. Neurosci 12, 25–36. 10.3389/fnmol.2019.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550–621. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, and Li H. (2021). Twelve years of SAMtools and BCFtools. GigaScience 10, giab008–4. 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Green CJ, Gazzara MR, and Barash Y. (2018). MAJIQ-SPEL: WebTool to interrogate classical and complex splicing variations from RNA-Seq data. Bioinformatics 34, 300–302. 10.1093/bioinformatics/btx565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Quinlan AR, and Hall IM (2010). BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eden E, Navon R, Steinfeld I, Lipson D, and Yakhini Z. (2009). GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinf. 10, 48–57. 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Supek F, Bošnjak M, Škunca N, and Šmuc T. (2011). Revigo summarizes and visualizes long lists of gene ontology terms. PLoS One 6, 1–6. 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen Y, Tristan CA, Chen L, Jovanovic VM, Malley C, Chu P, Ryu S, Deng T, Ormanoglu P, Tao D, et al. (2021). A Versatile Polypharmacology Platform Promotes Cytoprotection and Viability of Human Pluripotent and Differentiated Cells (Springer US; ). 10.1038/s41592-021-01126-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-sequencing data generated in this manuscript has been deposited in dbGaP (phs002440.v2.p1). These data are available under restricted access to comply with the conditions of the informed consent form agreed with the donors involved in the study, and access can be obtained by applying for controlled access through the dbGaP website. All other data reported in this paper will be shared by the lead contactupon request.
Custom scripts used to analyze immunofluorescence and RNA-seq data are available at https://github.com/waingerlab/iPSC-SMN.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.