

Significance
Adenovirus E1B-55K proteins can promote cell transformation, likely by operating as functional inhibitors of cellular p53. Through our comprehensive analysis of genomic localizations of chromatin-bound E1B-55K in transformed cells, we confirmed that this oncoprotein represses gene expression by indirectly binding to p53-dependent promoters via the tumor suppressor. Notably, our research has exposed undescribed interactions between E1B-55K and multiple p53-independent promoters and enhancers, resulting in transcriptional repression. These interactions involve host transcription factors that are well-known contributors to cancer and stress response signaling, including members of the TEAD (TEA domain) family, which play crucial roles as regulators of the Hippo pathway. Our findings revealed the remarkable versatility of E1B-55K oncoproteins as transcriptional deregulators of a wide variety of integral cellular pathways.
Keywords: AP-1, ChIP-seq, Hippo signaling pathway, p53, TPA response element (TRE)
Abstract
The multifunctional adenovirus E1B-55K oncoprotein can induce cell transformation in conjunction with adenovirus E1A gene products. Previous data from transient expression studies and in vitro experiments suggest that these growth-promoting activities correlate with E1B-55K-mediated transcriptional repression of p53-targeted genes. Here, we analyzed genome-wide occupancies and transcriptional consequences of species C5 and A12 E1B-55Ks in transformed mammalian cells by combinatory ChIP and RNA-seq analyses. E1B-55K-mediated repression correlates with tethering of the viral oncoprotein to p53-dependent promoters via DNA-bound p53. Moreover, we found that E1B-55K also interacts with and represses transcription of numerous p53-independent genes through interactions with transcription factors that play central roles in cancer and stress signaling. Our results demonstrate that E1B-55K oncoproteins function as promiscuous transcriptional repressors of both p53-dependent and -independent genes and further support the model that manipulation of cellular transcription is central to adenovirus-induced cell transformation and oncogenesis.
The first human virus that has been shown to induce cancer in various animal models belongs to the Adenoviridae family, a member of the small DNA tumor viruses (1–4). They are efficient in transforming cells in vitro and in vivo, similar to polyomaviruses and papillomaviruses (5, 6). The transformation of nonpermissive mammalian cells is a multistep process involving the inhibition of major tumor suppressor proteins, such as p53 (7) and the retinoblastoma protein (pRb) (8). Continuous expression of viral oncogenes ultimately results in the deregulation of cellular pathways and cell cycle checkpoints (9). It is well established that the 55-kDa protein, encoded in the early region 1B (E1B-55K) of human adenovirus species C type 5 (HAdV-C5, henceforth referred to as C5), contributes to this virus-induced complete transformation of cells by specifically antagonizing programmed cell death and growth arrest, which results from HAdV E1A–induced activation of p53 (10–12). During this process, the growth-promoting activities of the E1A protein cooperate efficiently with the ability of E1B-55K to act as a putative transcriptional repressor as well as through its role in the relocalization of p53 to perinuclear bodies (13–15). As a result of this interaction, p53 is unable to activate the transcription of genes involved in cell cycle control and apoptosis induction, such as MDM2, CCNG1, BAX, and CDKN1A, an observation that has been demonstrated in various in vitro transfection and overexpression experiments (13, 16). Although the exact mechanism by which E1B-55K blocks transcription is not well understood, previous findings from in vitro studies suggest that E1B-55K acts on transcription initiation and its inhibiting effect requires an undescribed cellular corepressor that copurifies with the RNA polymerase II (13, 17).
In the case of C5 E1-transformed cells, most of the E1B-55K and p53 proteins reside outside the nucleus and colocalize within perinuclear aggregates (18). This cytoplasmic restriction of the tumor suppressor was found to be accomplished by nucleocytoplasmic shuttling initiated by C5 E1B-55K (19), which is mediated by a leucine-rich nuclear export signal (NES) located in the amino-terminal region of the polypeptide (20). In addition to the impediment of p53-dependent tumor-suppressive functions, previous observations propose p53-independent mechanisms during virus-induced cell transformation (21). These include the sequestration of Mre11, which is part of the MRN complex and plays a role in double-strand break repair (21), or the proteasome-dependent degradation of the transcriptional regulator Daxx (22). Importantly, E1B-55K activities are regulated by post-translational modifications such as SUMOylation (23, 24) and phosphorylation (25, 26) that influence the localization as well as the molecular function of the protein (27, 28). SUMO-conjugation to E1B-55K mainly occurs at lysine 104 (K104) within the small ubiquitin-like modifier (SUMO) conjugation motif Ψ-K-x-E/D (23, 28) and phosphorylation by casein kinase II within the carboxy-terminal phosphorylation region of E1B-55K at serines 490, 491, and threonine 495 (29). It has been demonstrated that mutation of K104 to an arginine (R) completely abolishes SUMOylation of the oncoprotein, resulting in a strictly cytoplasmic phenotype, and renders the protein unable to fully cooperate with E1A to induce transformation (23). SUMOylation is likewise influenced by subcellular localization, or vice versa, since the mutation of leucines 83, 87, and 91 to alanines within the protein’s NES results in nuclear retention of E1B-55K, higher SUMOylation as well as greater transforming potential (20). Recently, we have shown that the lysine at position 101 (K101) of C5 E1B-55K potentially regulates SUMOylation at K104 as mutational inactivation results in higher SUMOylation of E1B-55K at K104 and decreased p53-dependent transcription when tested in dual luciferase assays (28).
While C5 belongs to the nononcogenic HAdVs, species A type 12 (HAdV-A12, hereafter referred to as A12) is the prototype of the highly oncogenic HAdVs in animal models, able to produce tumors within weeks of infection (4, 30–32). Furthermore, it has been shown that A12 E1B-55K also inhibits the transcriptional activity of p53 and induces its cellular relocalization, akin but not identical to C5 E1B-55K (15). Of note, A12 E1B-55K is neither SUMOylated, nor does it possess a canonical NES (28).
Multiple synergistic strategies have evolved for E1B-55K to interact with and inhibit p53 upon expression, but its postulated role as a transcriptional repressor of p53-targeted genes has been controversially discussed and the underlying mechanisms remained largely unknown. For that reason, we performed dual fixation chromatin immunoprecipitation, followed by high-throughput sequencing (ChIP-seq) experiments to reveal expansive and indirect in vivo E1B-55K occupancy across the genome of primary baby rat kidney (BRK) cells primarily via p53 but importantly also through various other host transcription factors (TFs). The integration of global RNA-seq data allowed us to precisely evaluate the gene expression levels of binding-associated genes and to thereby analyze the repressive consequences of this indirect E1B-55K–host genome interaction. Notably, repression of gene expression is highly conserved among the tested E1B-55K proteins of C5 and A12. This correlates with the oncogenic traits exhibited by these HAdVs in animal models, with more robust and widespread patterns of gene expression changes associated with A12 E1B-55K. The association between either C5- or A12 E1B-55K and cellular chromatin is closely tied to both the level of repression observed and the extent of its transforming potential. Our results conclusively determine E1B-55K as a viral transcriptional repressor interacting with DNA-bound p53 and additional previously unreported TFs in the context of adenovirus-induced cell transformation. By inhibiting their activity, E1B-55K can modulate downstream cellular responses irrespective of its SUMOylation status as long as it localizes to the nuclei of transformed cells.
In summary, we provide mechanistic insights into viral transformation by means of transcriptional repression through binding of the HAdV oncoprotein E1B-55K to p53 and various other cellular TFs that, inter alia, regulate tumorigenesis and cellular stress response pathways. The findings of this study will contribute to a better understanding of small DNA tumor virus oncogenes as well as virus-induced cell transformation.
Results
HAdV-C5 E1B-55K Predominantly Binds to Promoter Regions of Various Host Genes.
C5 E1B-55K binds to p53 and inhibits its role as a transcriptional activator of gene expression (33, 34). It has been discussed that this process defines E1B-55K’s role during the transformation of rodent cells, but the underlying mechanisms are not fully elucidated to date. The major goal of this research was to provide additional evidence that expands the knowledge of the putative functional role of E1B-55K as a transcriptional regulator by directly interacting with p53 and other TFs on the host chromatin.
To first map E1B-55K genomic binding sites and investigate the influence of intracellular localization, we performed dual fixation ChIP-seq assays (35) using E1-transformed BRK cell lines constitutively expressing either HA-tagged C5 wildtype (wt) E1B-55K, or different mutants with modified NES and/or SUMOylation-sites (Fig. 1A). These cell lines were generated via lentiviral transduction, fluorescence-activated cell sorting and polyclonal expansion. Expression of specific viral and cellular proteins was verified (SI Appendix, Fig. S1A) and localization patterns were quantified (SI Appendix, Fig. S2). Similar to the C5 wt E1B-55K, which almost exclusively localized in cytoplasmic perinuclear aggregates—along with cellular p53, the E1B-55K mutants also followed previously reported intracellular localization patterns: a strong nucleocytoplasmic localization of K101R; nuclear and nucleocytoplasmic localization patterns in cells transduced with the NES mutants; and a dominant cytoplasmic location of K104R. Importantly, all mutants retained their ability to interact and colocalize with p53 (SI Appendix, Fig. S2). Thus, our data confirmed earlier observations that intracellular E1B-55K localization is mediated by the presence of a functional NES as well as the SUMO modification at K104 (28, 36). The K104R cell line was used as a negative control as this E1B-55K mutant strictly localizes cytoplasmatically. The K101R, NES, and NES/K104R-expressing cell lines were originally chosen to potentially enhance the ChIP sensitivity as these E1B-55K mutants predominantly localize in the nucleus.
Fig. 1.
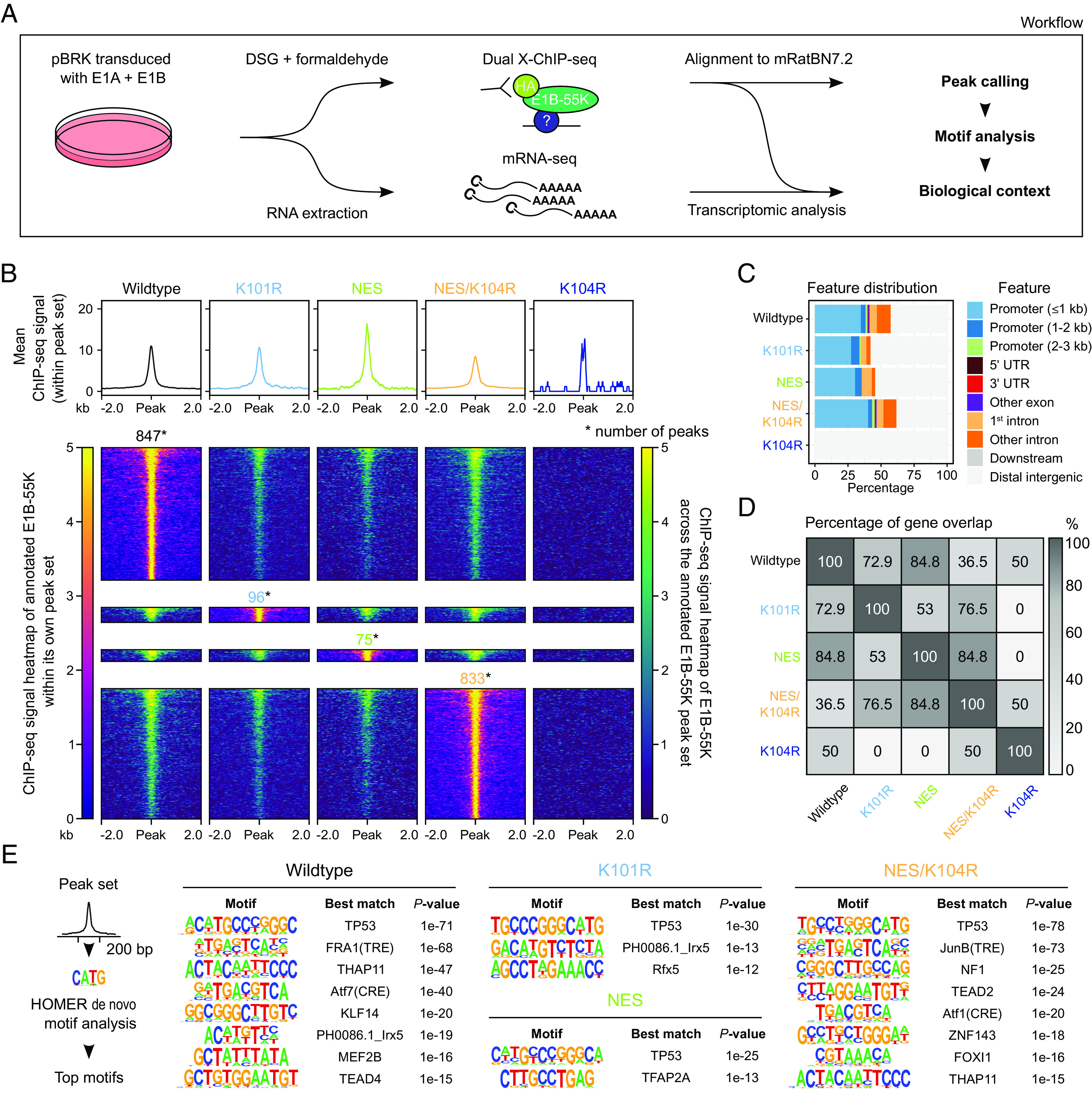
Characterization of the host genome interaction profiles of different HAdV-C5 E1B-55K proteins in BRK cells. (A) Utilized experimental and analysis workflow. BRK cells were transformed via lentiviral transduction and subsequent stable expression of the HAdV-C5 E1A and E1B gene regions. ChIP-seq binding profiles of E1B-55K and its mutants were obtained via alignment to the Ensembl 105 Rattus norvegicus (mRatBN7.2) genome assembly. Subsequent de novo motif analysis was integrated to identify potential underlying TFs. RNA-seq was utilized to trail potential transcriptomic alterations as a consequence of E1B-55K presence on the host chromatin. (B) Illustration and comparison of E1B-55K binding regions (q-value ≤ 0.05) with profile plots (upper part) and heatmaps (lower part), centered around a 4 kb region and normalized to RPGC (reads per genomic content). The profile plot displays the mean signal of E1B-55K within its respective peak set. The heatmaps either visualize this individual signal (left-side annotation) or display the binding profile within the peak sets of other E1B-55Ks (right-side annotation). The normalized ChIP signal intensity is displayed in both color gradients from dark blue (0, weak) to yellow (5, strong). Only one of the two replicates is shown here. The total number of peaks is annotated above the heatmap. (C) Feature classification of E1B-55K peaks in relation to their nearest gene. (D) Visualization of peak-to-gene overlap between the E1B-55Ks. Here, peaks were annotated based on their nearest up- or downstream located gene. The heatmap plot displays this overlap in percentages. Numerical values describe the ratio of the smaller group divided by the larger group, multiplied by 100. (E) De novo motif analysis of E1B-55K-associated peak sets. We used a 200 bp region around the peak summits, allowing for 2 mismatches to identify TF motifs, which were sorted by P-values of each called motif. Only the most significant motifs are shown with each individual best match annotated, with a P-value cutoff of 10–12. The original data are provided in Dataset S1A and S1B.
The ChIP-seq analyses were performed in biological duplicates (SI Appendix, Fig. S1A), and our workflow to analyze the sequencing data is outlined in SI Appendix, Fig. S3. Briefly, the ChIP-seq FASTQ sequence files were aligned to the rat reference genome, and binding sites (peaks) were identified via MACS2 (37), verified by the multiple sample peak calling (MSPC) software (38) and annotated based on their genomic regions using the R package ChIPseeker (39) (Fig. 1 B and C). We observed numerous E1B-55K binding events on the host genome (Fig. 1B). While the absolute number of significant peaks differed drastically between the mutants, we observed widespread overlaps between the binding patterns of E1B-55Ks in our ChIP experiments (Fig. 1B). In total, we identified 847 peaks (associated with 714 unique genes) in wt E1B-55K-transduced BRK cells, 96 peaks (85 genes) with K101R, 75 peaks (60 genes) with NES, and 833 peaks (702 genes) with NES/K104R, while the SUMO-null K104R mutant almost entirely lost its interaction capacity with the host genome (Fig. 1B), even though steady-state levels were equivalent to the other mutants (SI Appendix, Fig. S1A, K104R; replicate 3) (38, 40). In general, significant peak regions of each mutant were partly occupied by at least one of the other E1B-55Ks. To our surprise, the highly SUMOylated K101R and NES were associated with a markedly diminished presence on the host genome. Fig. 1C represents the relative position of interaction events based on the nearest up- or downstream gene feature. We found that the vast majority of peaks were located directly in promoter regions (<1 to 3 kb) or in the first intron, indicating a key role of E1B-55K in influencing transcriptional regulation. A further 10 to 20% of all peaks were located in other introns and exons, or the 5′ and 3′ untranslated regions of genes. The complete loss-of-function (LOF) phenotype observed in the K104R mutant may be due to its entrapment in the cytoplasm, where it is unable to interact with and deregulate nuclear p53 as the functional NES leads to immediate export of the protein from the nucleus upon entry. While presenting a comparatively similar localization pattern with the wt protein in immunofluorescence images (SI Appendix, Fig. S2), we can still observe trace amounts of wt protein in the nucleus. Additional inactivation of the NES (NES/K104R) rescues this LOF, permitting the protein to remain in the nucleus. Our results, therefore, confirm that intracellular localization of C5 E1B-55K is fine-tuned by its SUMOylation status together with its NES, retaining its interaction potential with cellular factors bound to the host genome.
Previous experiments have indicated that E1B-55K is unable to bind to DNA by itself (13, 14) and therefore requires interaction with DNA-bound host proteins to act as a transcriptional repressor. Consequently, we set out to identify E1B-55K interaction with diverse TFs through de novo motif analysis (Fig. 1E). An analysis of DNA enriched by ChIP-seq of E1B-55K-bound TF complexes revealed motifs that indicate interaction with various host TFs. As expected, except for K104R, the most significant interaction partner for all E1B-55Ks was p53 (Fig. 1E). Additional shared significant motifs included both TPA (12-O-tetradecanoylphorbol 13-acetate)-response element (TRE) and cyclic adenosine monophosphate (cAMP)-response element (CRE) motifs bound by activator protein 1 (AP-1) proteins (41), formed by a dimeric complex of activating transcription factor (ATF), Jun, or FOS protein family members as well as TEA domain (TEAD) protein binding motifs (Fig. 1E). We, therefore, conclude that E1B-55K interacts with various cellular TFs, including p53, to potentially modulate the host transcription machinery.
HAdV-C5 E1B-55K Interaction with DNA-Bound TFs Reveals Potential Influence on Cellular Pathways.
The most strongly binding C5 E1B-55Ks on the host chromatin were wt and NES/K104R (Fig. 1C), yet functional gene annotation was quite divergent with an overlap of only 36.5% (Fig. 1D), suggesting an influence of SUMOylation abrogation in combination with enhanced nuclear retention. By annotating the nearest genes to E1B-55K binding events, we identified well-established p53-targeted genes CDKN1A, MDM2, AEN, and TP53INP1 (42–45) among the most prominent genes in the Rattus norvegicus system (Dataset S1A) as predicted by previous motif analyses (Fig. 1E). Furthermore, through biological network analyses of all tested E1B-55K’s peak-associated genes, we detected pathways that are regulated by the p53 tumor suppressor (SI Appendix, Fig. S4, highlighted in red), with the exception being the LOF K104R mutant. Closely interconnected with p53-pathways are several cancer-associated clusters, e.g., “hepatocellular carcinoma” and “breast cancer” as well as stress-associated clusters, like “cellular responses to stress” and “DNA damage/telomere stress-induced senescence” (SI Appendix, Fig. S4, complete data are provided in Dataset S2A) (46, 47). Our findings confirm that the adenovirus oncoprotein interacts and thereby cooperates with p53 on the host genome. Intriguingly, E1B-55K was also identified to interact with TFs implicated in the highly conserved cell proliferation and growth-regulating developmental Hippo signaling pathway through regulation of genes like MYC, SERPINE1, WNT11, and WNT7B (48–53). With that, our work provides evidence for putative DNA-interacting partners that have not yet been described in the context of adenovirus-mediated cell transformation.
HAdV-C5 E1B-55K Expression Has a Strong Repressive Impact on the Host Transcriptome.
Using differential transcriptome analyses, we determined whether E1B-55K influences the expression of p53-signaling networks as predicted by our previous ChIP-seq results. Additionally, by investigating global up- and downregulated networks, we aimed to consolidate and attribute different pathways to the expression of E1B-55K in the context of host cell transformation. All C5 E1A and E1B-55K-expressing, transformed BRK cell lines were analyzed using poly(A) RNA-seq in comparison to newly transduced E1B-negative cell lines as a control (full data are provided in Dataset S3A). Through visualization of the first two principal components of our gene expression data, we observed that both the E1B-negative and K104R sets showed high intragroup variability (Fig. 2A). These findings can be explained by deviating E1A and E1B protein steady-state levels in the K104R replicates two and three (SI Appendix, Fig. S1). Of note, control BRK cells that expressed E1A without repression of pro-apoptotic processes due to the absence of E1B proteins seemingly underwent initial immortalization to different stages post-transduction, which presumably is captured by the first principal component (Fig. 2A).
Fig. 2.
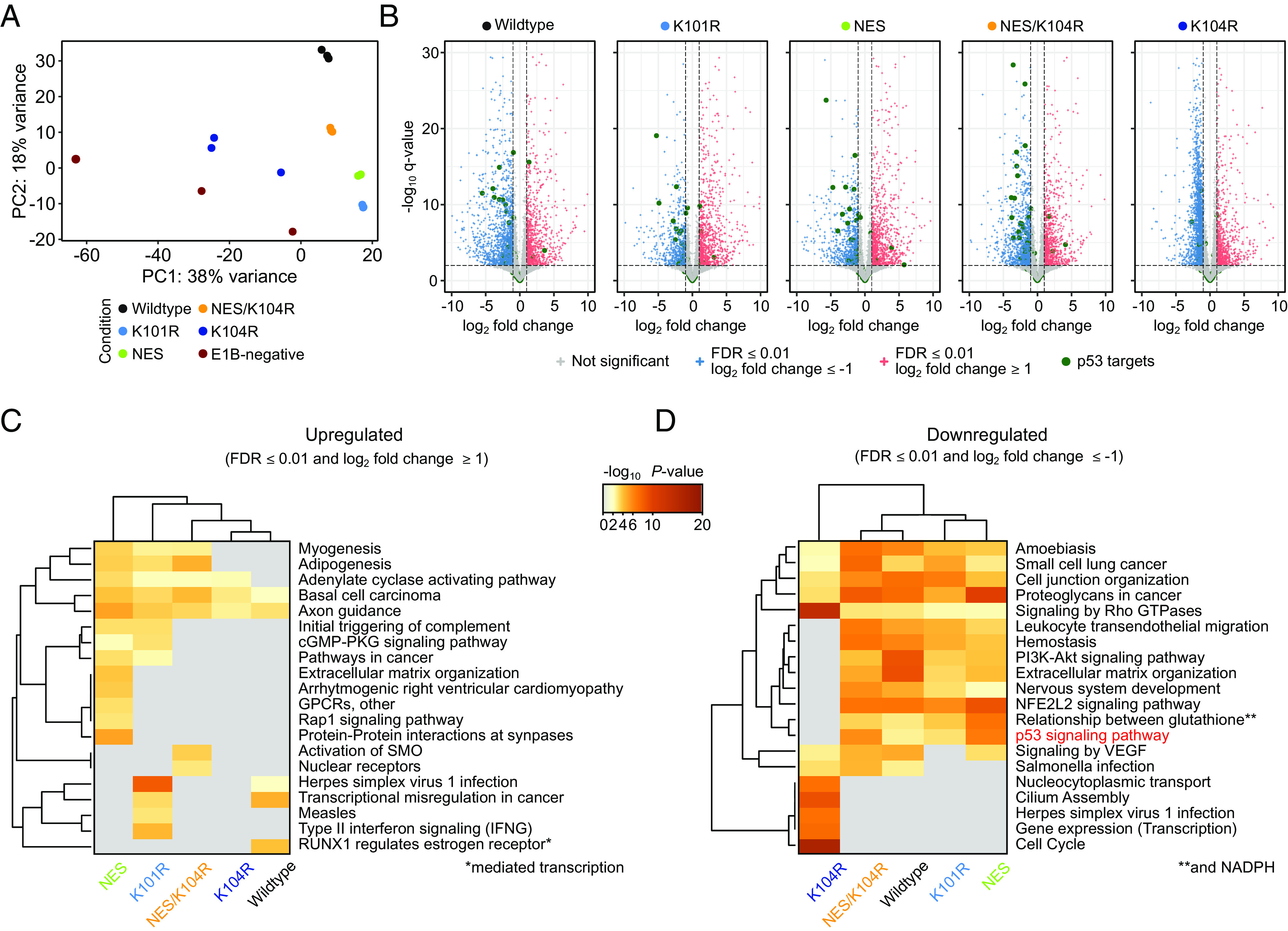
Effect of HAdV-C5 E1B-55K protein expression on the BRK cell transcriptome. (A) Principal component analysis (PCA) of transduced BRK cell RNA-seq data. “Condition” describes the individual E1B-55K protein expression cassette embedded in the lentiviral construct for the differentially expressed gene (DEG) analysis, compared to the E1B-negative control cell line. (B) Volcano plots highlighting the distribution of DEGs, induced by expression of different E1B-55Ks. Significantly upregulated (FDR ≤ 0.01 and log2 fold change ≥ 1) are colored red, while significantly downregulated (FDR ≤ 0.01 and log2 fold change ≤ –1) are colored blue. Green dots represent prominent p53 target genes curated by Fischer et al. (54) while non-significant genes are colored gray. The –log10 q-values for each gene are plotted on the y axis. The vertical and horizontal dashed lines represent the aforementioned significance cutoffs specified in the analysis. The complete list of underlying genes is provided in Dataset S4A. (C and D) Pathway analysis of the significant up- (C) or downregulated (D) genes. The heatmap plots are colored by P-values and grouped by hierarchical clustering. Here, pathways with a P-value of ≤ 0.01, a minimum count of three, and an enrichment factor of ≥ 1 are collected and grouped into clusters based on their membership similarities. The cluster term “p53 signaling pathway” is highlighted in red. Underlying data are summarized in Datasets S3A and S3B.
Among the statistically significant up- and downregulated genes presented via volcano plots, we highlighted p53 target genes that were described in previous high-throughput ChIP-seq datasets (54) (Fig. 2B). Focusing on this specifically curated set of p53-targeted genes only, we determined that all E1B-55K mutants that were previously found to interact with the tumor suppressor on the host chromatin predominantly downregulated p53 target genes, a repression that was absent in the LOF K104R mutant (Fig. 2B, highlighted in green).
Through contextualization via Metascape pathway and process enrichment analyses, we captured the potential influences of E1B-55K on biological pathways (Fig. 2 C and D, complete data are provided in Dataset S3B). Besides potent downregulation of the p53 signaling pathway, these analyses emphasize multiple additional pathways that are involved in cell differentiation. Remarkably, and in contrast to the pathway analysis for downregulated gene sets (Fig. 2D), we observed that the associated pathway list for upregulated gene sets (Fig. 2C) was generally less specific and more diverse. Our data suggest that the different E1B-55K SUMOylation levels contribute to these differences as hyper-SUMOylated mutants were more closely connected (K101R and NES in Fig. 2 C and D), indicating a yet-undescribed phenotype associated with divergent SUMOylation patterns of the oncogene. Many of the enriched pathways we found were related to various differentiation processes (e.g., “myogenesis”, “adipogenesis”, “axon guidance”) or included general transformation and cancer-associated pathways (“transcriptional misregulation in cancer”, “basal cell carcinoma”), similar to the general scheme of the ChIP-seq enriched pathways. Interestingly, only the K101R mutant induced immune response and virus-infection-related pathways (“type II interferon signaling”, “measles”), while NES and K101R both enhanced an innate immune system pathway (“initial triggering of complement”). Conversely, the K104R mutant shared only few downregulated pathways with the other E1B-55K proteins (Fig. 2D). Downregulated pathways in all other conditions included, for example, the “p53 signaling pathway”, the “PI3K-Akt signaling pathway”, and “leukocyte transendothelial migration”. The K104R mutant displayed a unique arrangement of infection and cell cycle-related set of ontologies (e.g., “cell cycle”) compared to the E1B-negative control, a phenotype we believe to be associated with differing E1A expression levels (SI Appendix, Fig. S1A, K104R, replicates 2+3). Several cancer-associated pathways were downregulated by all E1B-55K-expressing cell lines, regardless of the E1B-55K SUMOylation status (Fig. 2D). It is important to note that this transcriptome analysis provides an initial framework for further exploration of the mechanisms involved in E1B-55K-mediated cell transformation, which is fundamentally initiated by uncontrolled cell proliferation via the expression of E1A. We hereby show the different directions this transformation process can take in the presence of different functional variants of E1B-55K.
Interaction of E1B-55K with DNA-Bound p53 is a Likely Prerequisite for the Transcriptional Inhibition of p53-Targeted Genes.
To delve deeper into the effects of DNA-binding within predominant p53 target genes and provide evidence for a functional consequence of interaction with DNA-bound p53, we directly compared the transcriptomes of BRKs expressing wt, K101R, NES, and NES/K104R E1B-55K proteins, which were present on the DNA (Fig. 1B) with BRK cell lines that were expressing either K104R or were E1B-55K negative. We subsequently divided the aforementioned p53 target genes into three groups based on their expression patterns and significance and classified these sets of genes into clusters that were significantly upregulated (false discovery rate (FDR) < 0.1 and log2 fold change > 0), non-significant (FDR > 0.1) or significantly downregulated (FDR < 0.1 and log2 fold change < 0; Fig. 3A). The gene bodies from genes within these three clusters were visualized with the ChIP-seq data to compare whether the indirect occupancy of the E1B-55Ks on the host DNA resulted in altered transcription (Fig. 3B). While the upregulated genes showed no presence of any tested E1B-55K, the non-significantly changed genes directly presented with a moderate, primarily transcription start site (TSS)-associated E1B-55K ChIP-seq signal. It is critical to note that the non-significant gene set encompassed several genes that were downregulated by either one of the E1B-55K proteins, potentially explaining the ChIP-seq signal. This phenotype was markedly increased within the significantly downregulated cluster, where E1B-55Ks were located to both TSS and a secondary binding site, representing an alternative TSS of the CDKN1A and RPS27L genes (Fig. 3B). These results demonstrate that the presence of E1B-55K mutants at the TSS of p53-targeted genes correlates with transcriptional repression of these genes.
Fig. 3.
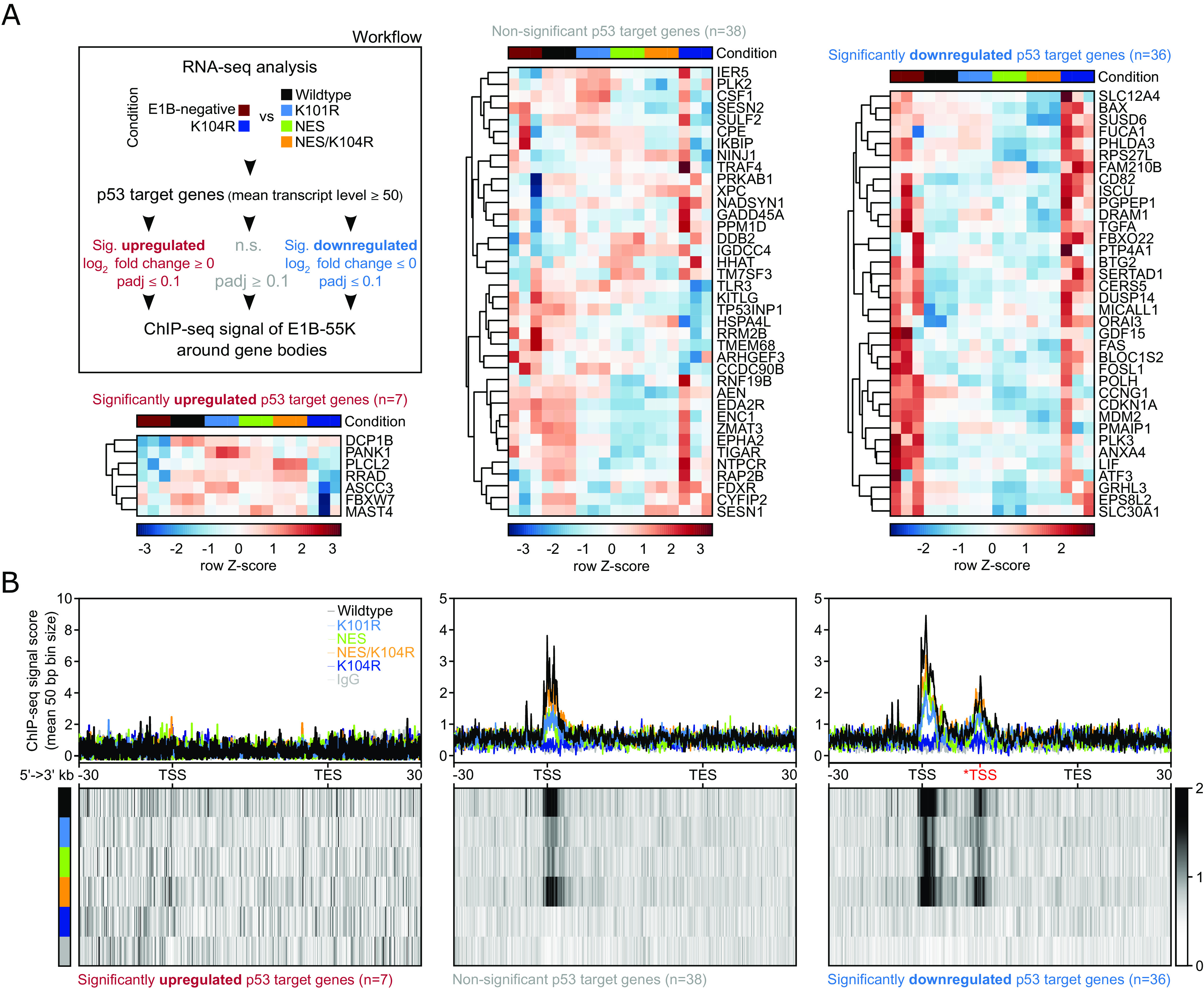
HAdV-C5 E1B-55K locates to regulatory regions of p53 target genes to directly repress their expression. (A) Workflow and heatmap of the subselection of p53 target genes, which were divided based on their respective expression level into significantly upregulated (FDR < 0.1 and log2 fold change > 0), non-significant (FDR > 0.1) or significantly downregulated (FDR < 0.1 and log2 fold change < 0) comparing the two E1B-55K conditions (indicated in the legend). Shown are the row Z-scores of normalized reads of indicated genes. We used a mean normalized transcript count of 50 reads as a cutoff to dismiss lowly expressed genes. Conditions are compared to the mean with negative (blue scores) and positive values (dark red scores) indicating fewer or more transcripts, respectively. The DEG analysis of this condition can be found in Dataset S3A (last sheet). (B) Visualized here are the gene bodies of p53 target genes from TSS to TES. The signal around the *TSS (in red) is attributed to interaction with an alternative transcriptional starting site of the CDKN1A and RPS27L genes. All gene bodies are normalized to a size of 50 kb (bin size of 50 bp) and visualized from 5′ to 3′ direction including a 30 kb up- and downstream region, showing the mean ChIP-seq signal score of E1B-55Ks via profile plots (Upper row) or heatmap plots (Lower row). The respective track is annotated in the top right corner of the first profile plot. The heatmaps visualize the profile below their respective profile plot, with the intensity of the respective signal represented by a grayscale, ranging from white (no signal) to black (strong signal). The absolute number of genes within each set is depicted below the respective heatmap plots. The genomic location of p53-targeted genes can be found in Dataset S4.
The HAdV-A12 E1B-55K Protein Exhibits a Global Transcriptional Repression Phenotype and Interferes with Numerous Different Host Pathways.
After these initial observations in the context of the nononcogenic HAdV-C5, we were particularly interested in an E1B-55K protein belonging to the highly oncogenic A12. This protein is remarkably unique compared to E1B-55Ks from other species as it lacks both SUMOylation- and NES-sites, mimicked by the NES/K104R mutant from C5. We, therefore, performed similar BRK cell transduction and transformation experiments with a combination of C5 E1A and A12 E1B-55K. By using this strategy, we kept the number of potential variables influencing our system to a minimum, allowing us to directly compare C5 and A12 E1B-55K. The protein steady-state levels and the localization of A12 E1B-55K are presented in SI Appendix, Fig. S1 B and C, respectively. To our surprise, we observed that the protein could be detected at considerably more sites compared to the C5 wt E1B-55K and any of the tested mutants (Fig. 4A). In total, we identified 18,792 unique binding sites associated with 8,728 genes by combining MACS2-called peaks from three transformation experiments, verified by MSPC as described above (SI Appendix, Figs. S1B and S6). The complete dataset we obtained by targeting A12 E1B-55K encompassed 87% (614 out of 708) of all genes associated with the C5 wt E1B-55K and 91% of genes (640 out of 702) associated with the C5 NES/K104R mutant (Fig. 4C). Intriguingly, the number of genes associated uniquely with either of these C5 proteins exceeds the number of shared genes. We found that the majority of these distinct genes are captured by A12 E1B-55K (378 and 404, respectively). This phenomenon indicates a conserved host TF-binding spectrum of the E1B-55K species compared here. In line with this, A12-enriched motifs were, in general, similar to the ones that were identified by C5 E1B-55K (Fig. 1E). Captivatingly, we observed a marked difference regarding the TEAD TF, which represents the most significantly enriched A12-interacting host factor on the genome (Fig. 4D). We also observed significant enrichment of a consensus motif recognized by the CTCF and BORIS (CTCFL) paralogs (55–57), the former of which represents a core architectural protein, suggesting a unique, or at least stronger interaction of A12 with the genome organization machinery not detectable within the C5 E1B-55K ChIP-seq data. Due to the sheer number of different A12-associated motifs identified by HOMER in this dataset, we chose to focus on the three most significant, namely, TEAD, AP-1, and p53, in a subsequent stringent biological pathway analysis. The genes linked to these motifs were associated with cancer- and apoptosis-related pathways, highlighting yet again the fundamental function ascribed here to E1B-55K (Fig. 4E). As expected, the respective motif-associated genes were also clustering in pathways related to their individual primary functional roles, e.g., TRE-associated motifs were linked to the stress response (“PI3K-Akt signaling pathway”), infection (“Salmonella infection”) and growth signaling (“cell junction organization”), while TEAD-associated motifs were connected to the Hippo signaling pathway. Similar to C5, A12 E1B-55K was also able to antagonize p53 (Fig. 4 B, E, and F) as we detected pronounced interactions within the regulatory regions of key p53-targeted genes, such as CDKN1A and MDM2 (Fig. 4B). Due to the availability of three replicates, we quantified the ChIP-seq signal from TSS to transcription end site (TES) of these genes, followed by quantile normalization using the EaSeq software with standard settings. By comparing A12 E1B-55K ChIP-seq signals within p53 target genes that were subdivided as previously described (Fig. 3A), we identified a significant difference between the differently regulated sets (Fig. 4 F, Left side and Inset). Here, indirect binding (as illustrated by ChIP-seq signal scores) was strongly enriched at genes that were found to be transcriptionally downregulated, in comparison to non-significant and particularly to upregulated genes. Intriguingly, a similar pattern was identified when investigating all genes that were associated with A12 E1B-55K binding events in their gene bodies (Fig. 4 F, Right side, Inset). A comparable trend could be identified when further decreasing the scope (SI Appendix, Figs. S8 and S9). These data provide clear and significant evidence for a conserved functionality of E1B-55K with apparent varying levels of deregulation of key underlying host regulatory networks, thereby potentially interfering with a multitude of biological pathways at the transcriptional level.
Fig. 4.
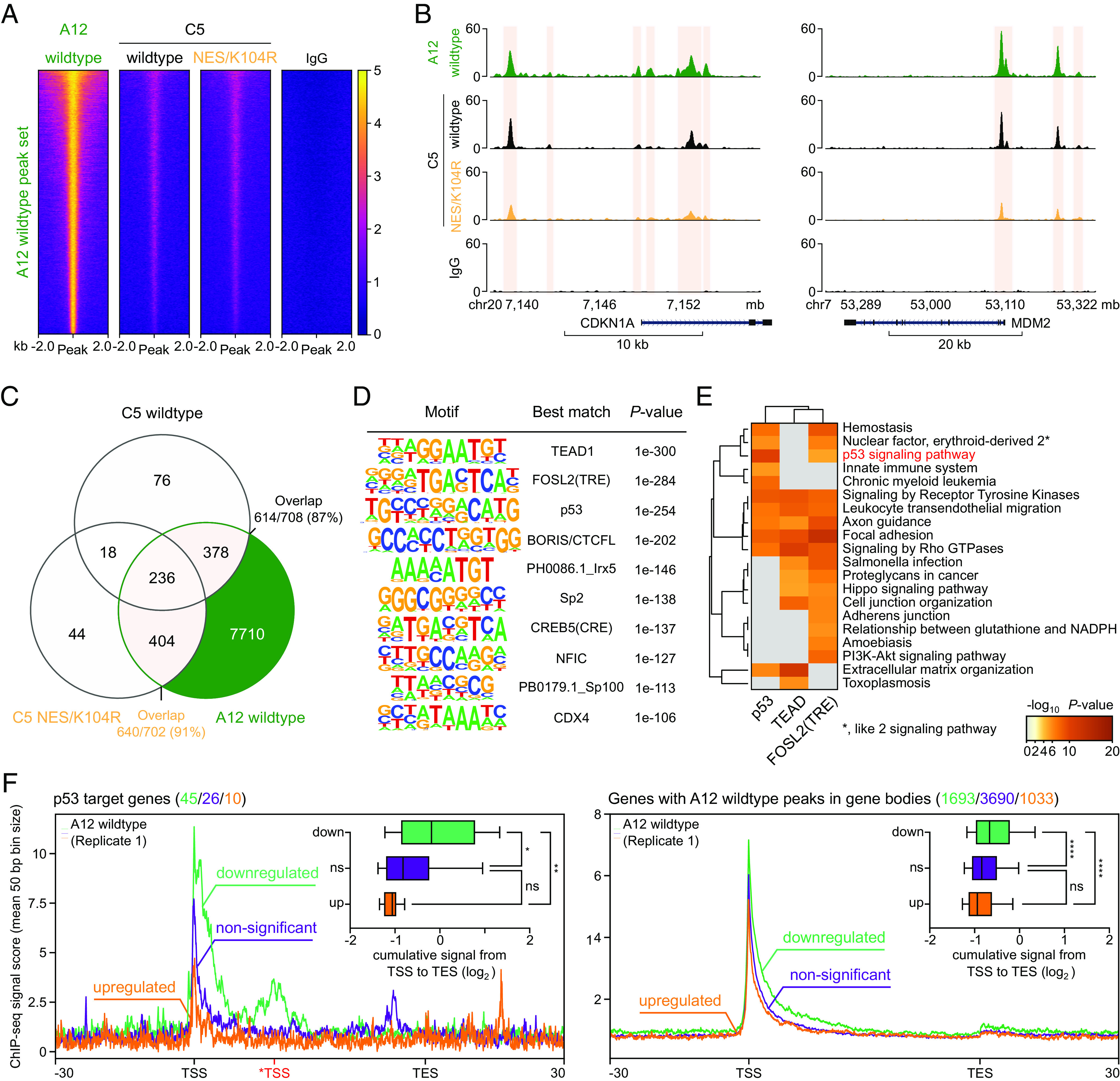
A12 E1B-55K extends its repressive binding-associated phenotype across the host genome. (A) Illustration of the A12 E1B-55K peak set with heatmaps, centered around a 4 kb region, normalized to RPGC (reads per genomic content). The normalized ChIP signal intensity of E1B-55K from species A12, C5 wt, and NES/K104R is displayed in a color gradient from dark blue (0, weak) to yellow (5, strong). (B) Visualization of identified peaks (light ochre) in two representative regulatory elements of p53-targeted gene regions (MDM2 and CDKN1A) of A12, as well as C5 wt and NES/K104R E1B-55K. The y axes indicate the respective ChIP-seq signal (normalized to reads per million per 1 kb). (C) Venn diagram representing ChIP-seq peak-associated gene overlaps comparing A12, C5 wt E1B-55K, and -NES/K104R. (D) De novo motif analysis of A12 E1B-55K peak sets around a 200 bp region, restricted to 2 mismatches, sorted by P-values of each called motif. Only the most significant motifs are shown with each individual best match annotated. (E) Pathway analysis of significantly downregulated (FDR < 0.1 and log2 fold change < 0) pathways compared to the E1B-negative control cell line for genes associated with the top three A12 E1B-55K identified motifs TEAD1, FOSL2(TRE), and p53 (see panel D). The heatmap plots are colored by P-values and grouped by hierarchical clustering. Pathways with a P-value of < 0.001, a minimum count of three, and an enrichment factor of > 1.5 are collected and grouped into clusters based on their membership similarities. The cluster term “p53 signaling pathway” is highlighted in red. (F) A12 E1B-55K ChIP-seq profile plots around p53 target genes (left plot) or genes with verified peaks within their gene body (right plot) that were further subselected into significantly upregulated (orange, FDR < 0.1 and log2 fold change > 0), non-significant (purple, FDR > 0.1), or significantly downregulated (green, FDR < 0.1 and log2 fold change < 0), based on their expression compared to the E1B-negative control cell line. Visualized here are the respective gene bodies from TSS to TES. The signal around the *TSS (in red) is attributed to enhanced interaction with an alternative transcriptional starting site of the CDKN1A and RPS27L genes, instead of the canonical TSS of these genes. All gene bodies were normalized to a size of 50 (bin size of 50 bp) and visualized from 5′ to 3′ direction, including a 30 kb up- and downstream region, showing the overlaying mean ChIP-seq signal score of A12 E1B-55K via profile plots. The respective track is annotated directly on the respective profile plot. The absolute number of genes within each set is depicted above the respective heatmap plots with corresponding colors. For each region, we quantified the ChIP-seq signal from TSS to TES, normalizing to reads per million and to a gene size of 1,000 bp (Upper Right Insets), assuming a fragment size of 150 bp. These data are presented as box-whisker-plots with the 10th to 90th percentiles indicated by whiskers. Areas above 1 Gbp or below 100 bp were set to those sizes. All quantified signals obtained from these genes across the aforementioned subselected groups were subsequently quantile normalized. We tested for statistical differences between the calculated signals of these groups via a nonparametric Kruskal–Wallis one-way ANOVA, followed by Dunn’s multiple comparisons test (Left side) or a two-way ANOVA test, followed by Tukey’s multiple comparisons test (Right side). ****adjusted P-value < 0.0001, **adjusted P-value < 0.01, *adjusted P-value < 0.05, ns; P-value > 0.5. All original data are provided in Datasets S1A and S1B, S2B, and S5.
Integration of ChIP and RNA-seq Analyses Indicates a Direct E1B-55K Interference with Specific TFs.
Significant enrichment of E1B-55Ks in repressed genes posed the question of whether the strength of interaction (indicated by the peak score) was related to transcriptional repression. Consequently, we combined significantly regulated genes (FDR ≤ 0.1) with motifs that were enriched in their close vicinity of our two largest peak sets C5 wt and NES/K104R as well as A12 E1B-55K (Fig. 5 A–C). Linear correlation analyses, along with a Pearson coefficient (ρ) calculation, indicated an inverse and significant correlation between the ChIP-seq peak scores and the mRNA fold changes of the respective nearest genes, suggesting a link between E1B-55K occupancy strength and the magnitude of the transcriptional outcome of associated genes. Here, we observed that the C5 NES/K104R mutant (ρ: –0.298) exhibited stronger repressive associations between binding events and transcriptional regulation than the C5 wt E1B-55K (ρ: –0.134), while the A12 protein ranked between the two (ρ: –0.169), although it occupied roughly 13 times more host genes compared to the C5 variants—including many genes with rather weak associations between binding and regulation, which may lead to an underestimation of this correlation. Taken together, all tested E1B-55Ks, nevertheless, showed a clear significant correlation with transcriptional repression, with largely comparable average log2 fold changes of the associated genes (Fig. 5 A–C).
Fig. 5.
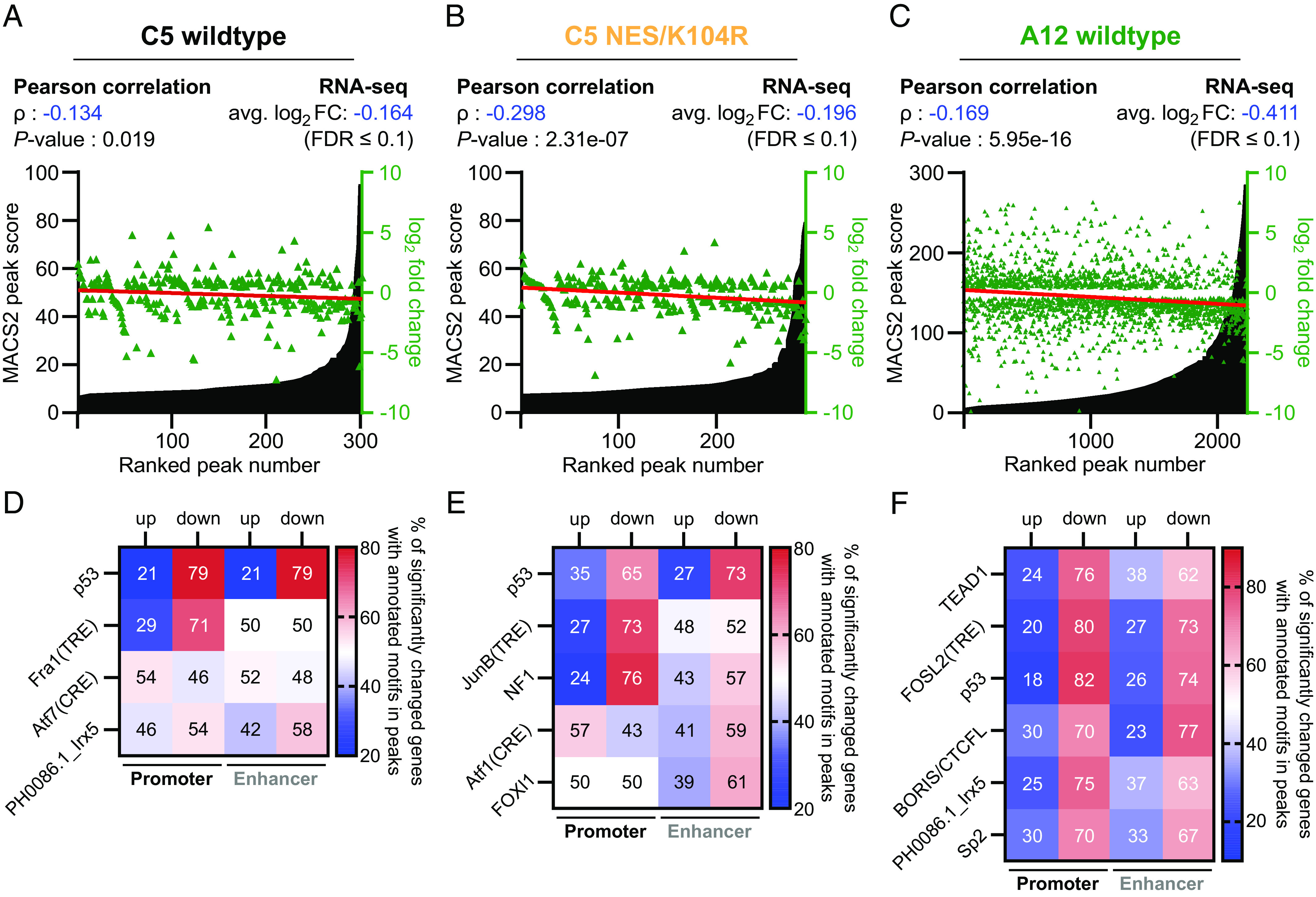
E1B-55K interactions with DNA-bound TFs are conserved and associated with transcriptional repression. (A–C) C5 wt (A), NES/K104R (B), and A12 E1B-55K (C) peaks, ranked by their score from least to most significant and annotated with the log2 fold change of its nearest gene (green triangles). As a cutoff, only genes with an FDR < 0.1 were considered in these analyses. The Pearson correlation coefficient (ρ) was calculated to quantify the relationship between E1B-55K binding events and transcriptional consequences. A negative ρ-value corresponds to an inverse relationship between peak strength and log2 fold change, while the P-value represents the calculated significance of ρ with a two-tailed t test. A linear regression analysis (red line) of log2 fold changes is included to visualize correlation and trend. The average log2 fold change of all associated genes is also indicated above the plots. (D–F) Motifs obtained from de novo motif calling from Figs. 1E and 4D were separated according to their respective gene positions into either promoter or enhancer-associated. Only motifs with ≥ 25 individual peaks were chosen for this analysis to dismiss motifs that were associated with too few genes to provide a reliable interpretative result, individually matched with the RNA-seq data of their respective nearest genes and separated into either up- or downregulated. The color gradient indicates the proportion of genes that occur in either set, ranging from blue (low relative amount) over white (equal amount) to red (high relative amount). The numbers in the squares represent the percentages of all motifs in either promoter or enhancer peaks that are associated with either up- or downregulated genes. The individual motifs were sorted by their significance (represented by P-values). The original data are provided in Datasets S6A and S6B.
As E1B-55K-binding to both p53-targeted genes via p53 (Figs. 2 D, 3, and 4 F, left side) and a generalized gene set (Fig. 4 F, Right side) had a clear negative transcriptional outcome of affected genes, we set out to determine whether we could attribute this repressive effect to TFs besides p53. In the final step, we therefore sought to determine whether we could attribute a specific pattern of downregulation of genes that had individual TF motifs occupied by E1B-55K (Fig. 5 D–F). To achieve that, we selected significant motifs that occurred in at least 25 combined peaks in promoters and enhancers. In agreement with our previous observations, oncogene presence on p53-associated motifs was accompanied by strong transcriptional downregulation, an effect slightly more pronounced in the C5 wt E1B-55K (79% in both promoter and enhancer regions) than in the NES/K104R mutant (65% in promoter and 73% in enhancer regions). Hence, both the C5 wt E1B-55K and the NES/K104R mutant comparably repress p53-mediated transcription. Intriguingly, our data also show transcriptional repression of the majority of promoter-associated TRE motifs (71% in C5 wt E1B-55K and 73% in NES/K104R). This observation cannot be attributed to a mere co-occurrence of both motifs as we also observed strong repression of genes that had TRE, but not p53 motifs present in their promoters (SI Appendix, Fig. S5 A and B). We identified a general enrichment of TRE compared to CRE-associated motifs, both of which were preferentially targeted by different compositions of the AP-1 dimer complex, with only the former being transcriptionally repressed (Fig. 5 D and E). Per this analysis, most significant motifs enriched with A12 E1B-55K were clearly repressed, with a slightly enhanced effect based on their presence in promoter over enhancer sites (Fig. 5F). While we observed an enrichment of genes with peaks containing TEAD motifs by both C5 E1B-55K variants, only A12 was associated with repression of these genes, especially when located in promoter regions (76%). In a more stringent analysis of isolated and overlapping motifs, we found that in the case of co-occurrence of either TEAD or TRE with p53 motifs in promoters, associated genes were transcriptionally repressed to overwhelmingly high levels by A12 E1B-55K (89.77% and 89.74%, respectively). Regarding co-occurrence, this was closely followed by the presence of all three motifs (80%), while TEAD1 and TRE motifs were associated with a smaller proportion of repression (74.07%; SI Appendix, Fig. S6 B and C, Left side). Generally, isolated motifs were less associated with transcriptional repression of adjacent genes compared to co-occurring motifs (SI Appendix, Fig. S6C, Left side). A muted effect was visible when motifs were found within enhancers of associated genes (SI Appendix, Fig. S6 B and C, Right side). Here, the presence of a p53 motif in an enhancer region remained the strongest predictor of transcriptional repression, the extent of which depended on the presence or absence of other motifs.
Together, these data provide additional in-depth evidence for a role of E1B-55K as a transcriptional repressor of p53-associated genes. Our study also expands the scope of deregulation to additional mammalian host TFs that may be selectively targeted by the adenovirus oncoprotein during cell transformation.
Discussion
Human DNA tumor viruses have common features that allow them to deregulate antiviral cellular processes to ensure efficient production of progeny (58, 59). In lytic infection, viral oncoproteins support efficient replication and are eradicated after cell death. However, abortive or long-term persistent infections, accompanied by constitutive expression of viral oncoproteins, can continuously inhibit cellular tumor suppressors, leading to virus-mediated tumorigenesis. pRb and p53 present important cellular targets and are preferentially targeted by viral oncogenes such as HAdV E1 proteins, SV40 large T antigen, and human papillomavirus E6/E7 to promote uncontrolled cellular replication and circumvent premature cell death (60). While E1B-55K is known to have transcriptional repression activity (13, 61), its specific role in the regulation of host gene transcription at a genome-wide level remained elusive.
In this study, we present a comprehensive and correlated analysis of ChIP-and mRNA-seq data of E1B-55K in E1-transformed BRK cells. Our results demonstrate that the interaction between E1B-55K and DNA-bound p53 leads to the repression of its transcriptional activity, addressing a long-standing debate in the field. In addition to p53-regulated sites, we identified numerous other indirect binding sites by the different tested E1B-55Ks (Fig. 1 B–D and Dataset S1). Mutations in the SUMOylation and/or NES motif sites of E1B-55K displayed only slight differences regarding these interactions as long as the protein remained in the nucleus, providing further insights into the mechanisms involved. Loss of SUMOylation (K104R) completely abrogated genome coverage, a phenotype that could be rescued by additional inactivation of the NES (NES/K104R), while the hyper-SUMOylated K101R and NES mutants displayed muted presence but generally retained their functionality. Earlier experiments indicated that enhanced E1B-55K SUMOylation positively correlates with its interaction with PML nuclear bodies, thereby likely reducing the steady-state levels on the host chromatin (23, 27, 62, 63). Our current findings are consistent with previous publications, confirming the strong repression of p53-associated pathways through traceable interactions (13, 61) (Figs. 1 and 2). Interestingly, our data differ from the results of transcriptome studies published by Miller et al. who performed microarray analyses and did not observe E1B-55K-mediated repression of p53-dependent transcription in adenovirus C5 wt-infected human foreskin fibroblasts (64). This indicates that E1B-55K expressed in productively infected human cells is likely to be functionally different from E1B-55K in transformed nonpermissive rodent cells. It is therefore tempting to speculate that the multifunctional properties of E1B-55K are host species-specific and thus are dependent on the genetic background of the cell (here: human vs. rodent). Such a model could also account, at least in part, for the long-standing observation that primary rodent but not primary human cells are highly susceptible to adenovirus E1-mediated transformation (5). Additionally, the functions of E1B-55K are differentially regulated by other viral proteins expressed during the course of a lytic infection in permissive human cells. Among these are the E1B-55K-binding proteins E4orf3 and E4orf6 (34). The latter regulates SUMOylation of E1B-55K, controls its intracellular localization (24), and forms an E3 ubiquitin ligase complex with E1B-55K that targets p53 and other key regulators of cell growth for proteolytic degradation during lytic infection (33). Moreover, it has been suggested that a factor specific to primate cells facilitates the functional interplay between E1B-55K and E4orf6 in human but not rodent cells (65). Finally, E4orf6 and E4orf3 can also inactivate p53 independently of E1B-55K in transfected and infected human cells (66, 67).
However, it is noteworthy that Hobom and Dobbelstein had previously reported similar results, stating that in addition to E1B-55K, E1B-19K also plays a role in antagonizing the activity of p53 during HAdV infection (68). These findings highlight the complexity of the interplay between viral proteins and host cellular machinery in the context of viral infection and transformation and emphasize the need for further investigation. Our transduction and transformation model offers a unique perspective, allowing for a comprehensive examination of isolated E1B-55K and its functions. With reduced background noise, we can more easily identify and understand the processes involved in the transcriptional repression of p53. This phenotype could be verified through analysis of A12 E1B-55K in transformed BRK cells when co-expressed with C5 E1A, which emerged as a remarkably more potent interaction partner with TFs bound to the host genome when compared to C5. Expressed in numbers, we could demonstrate around 22 times more interaction events across replicates, associated with 13 times more individual genes (Dataset S1A), which were equally—or even more so correlated with effective transcriptional repression of p53-targeted genes (Fig. 5 D–F). The potent ability to induce malignant tumors via infection of neonate rodents that is ascribed to group A of HAdVs (1, 69, 70) is, to some extent, attributed to a unique spacer region within their E1A protein (71). The findings presented here suggest a relationship between the E1 region proteins of A12 as both E1A and E1B gene products share an enhanced transforming activity when compared to group C HAdVs.
A key finding of our combined genome-wide exploration was a notable association of E1B-55Ks with the Hippo signaling pathway via the TEAD TF family (Figs. 1E, SI Appendix, Fig. S4, and 4 D and E). These networks have not previously been associated with E1B-55K, adding additional dimensions to our understanding of its role in cellular pathways. The highly conserved Hippo pathway regulates cell proliferation and differentiation (72). Additionally, it responds through mechanical cues sent via the extracellular matrix, neighboring cells, or the cellular geometry, and therefore represents an essential network that reacts to the cellular microenvironment (73, 74). Zemke et al. recently observed indirect repression of TEAD by E1A-dependent cytoplasmic sequestration of the YAP and TAZ oncoproteins, which are the terminal Hippo pathway effectors (75). This, in turn, led to the inhibition of genes associated with the extracellular matrix, a significant cluster of the pathway that we similarly detected to be repressed by all C5 E1B-55Ks present on the host genome (Fig. 2D). Although the TEAD TF family was only weakly associated with C5 E1B-55K, we observed an intriguing association with the A12 protein, for which the host TF was the most significant interaction partner based on our motif analysis (Fig. 4D). As a consequence, BRK cells that were transduced with E1B-55K of this highly oncogenic species displayed transcriptional inhibition of the Hippo signaling pathway (Fig. 4E and SI Appendix, Fig. S7B, in blue). In addition, the most prominently repressed pathways, “focal adhesion” and “extracellular matrix organization” (SI Appendix, Fig. S7B), were also the most significant pathways suppressed in 293 cells when E1A-dependent indirect repression of TEAD-mediated transcriptional activation via sequestration of YAP and TAZ remained active. Deregulation of these pathways plays an integral role in focal adhesion, cancer progression, and metastasis, and their control is influenced by the transcriptional activity of YAP/TAZ via TEAD (76–78). Additional evidence suggests that YAP/TAZ are regulators of the cellular differentiation state upon specific extracellular signals as their action is required in the maintenance of the actin cytoskeleton (75). Our work, therefore, provides data suggesting a synergistic role in the dedifferentiation of cells and the sensing of the cellular environment between the E1-proteins of HAdVs.
Although variations in the association of C5 and A12 E1B-55K with the TEAD TF family were evident, our analysis revealed shared characteristics of both species in terms of the binding and consequent repression of genes containing AP-1 family motifs within their regulatory regions (Fig. 5 D–F). Deregulation of these complex TFs by other oncogenic viruses in the context of cell transformation has also been reported (79, 80). Our global transcriptomic repression analysis of C5 E1B-55K-expressing BRK unveiled repression of genes highly associated with “PI3K-Akt signaling” and “small cell lung cancer”-pathways known to be frequently associated with the AP-1 TF family (81–83). AP-1 complexes are composed of homo or heterodimers of basic leucine zipper proteins belonging to the FOS, Jun, ATF, and Maf subfamilies. These protein complexes regulate multiple tumor and cancer-related pathways but are implicated in many more and diverse biological activities (84–86). Earlier publications reported that AP-1 complexes preferentially bind to either TRE (TGA(G/C)TCA) or CRE motifs (TGACGTCA), depending on their dimer composition: Heterodimers of Jun-FOS favor binding to TRE, with homodimers of Jun-Jun showing significantly lower enrichment, while Jun-ATF heterodimers prefer CRE motifs (41, 87). In our analysis, E1B-55Ks displayed a pronounced association with TRE rather than CRE motifs, thus suggesting an inclination to interact with either (or both) Jun or FOS to repress the AP-1-mediated cellular stress response or different cancer and infection-associated pathways (Fig. 5 D–F). Aberrantly increased FOS expression is associated with metastasis and epithelial-to-mesenchymal transition, especially in breast cancer (88, 89). By specifically targeting this function of AP-1, it is tempting to hypothesize that E1B-55K is additionally able to interfere via a yet undescribed mode of action in the differentiation and dedifferentiation programs of the cell.
The substantial amount of A12 E1B-55K-peak-associated genes enabled us to inquire the co-occurrence of the most prominent motifs we detected. We observed an enhanced propensity of transcriptional downregulation of genes when motifs co-occurred within their regulatory region (SI Appendix, Fig. S6 B and C), compared to an isolated occurrence of the respective motif. These genes were likely more prone to be transcriptionally active when E1B-55K is absent as synergistic effects between TFs have been described in the literature (90, 91). Notably, the presence of both TEAD and AP-1 at cellular enhancers is of particular interest (90). Future studies will focus on gaining a more comprehensive understanding of the intricate interplay between E1B-55K oncoproteins and these essential and multifaceted TF families.
This work presents the first genome-wide experimental setup to establish a direct link between the presence of E1B-55K in the regulatory regions of multiple p53-regulated target genes and transcriptional repression, strongly supporting the notion that E1B-55K indeed acts as a viral transcriptional regulator. Previous data indicated that E1B-55K can act as a general transcriptional repressor as it inhibits expression from several cellular and viral promoters (13, 14, 92). With this report, we provide unambiguous evidence that the enigmatic E1B-55K protein represses p53-mediated transcription by direct interaction on the host genome, independent of its SUMOylation state—as long as it is localized in the cell nucleus. Similarly, host cell TF SUMOylation does also not have a binary effect on their transcriptional activity; rather, it modulates transcriptional output to maintain appropriate gene expression levels (93). SUMOylation of target proteins thus helps prevent the excessive activation or repression of genes, thereby fine-tuning gene expression levels. In addition, we identified conserved E1B-55K-mediated transcriptional deregulation through non-p53 TFs. We thereby provide important data that expand the functional repertoire of E1B-55K and how it represses transcription, opening avenues for further analyses of E1B-55K functions. It is important to note that recent evidence from our lab suggests that p53 binding and inhibition alone may not necessarily be required for transformation (21). Consequently, our findings pave the way for further insights into the adenovirus oncogene E1B-55K and its synergy with E1A, irrespective of p53-mediated apoptosis.
Materials and Methods
Cells.
BRK cells were obtained from the kidneys of 3- to 5-d-old Sprague-Dawley rats as previously described (94). Briefly, kidneys were incubated with 1 mg/mL collagenase-dispase (Roche) at 37 °C for 3 h and single cells were seeded onto 150 mm culture plates and cultivated in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% fetal calf serum (FCS, Sigma-Aldrich) and 100 U/mL penicillin (PAN Biotech) and 100 µg/mL streptomycin (PAN Biotech). All generated BRK cell lines (please refer to SI Appendix for a detailed description of the BRK cell transduction protocol) and HEK 293T [ATCC CRL-1573 (95)] cells used for lentiviral pseudo-particle production were also grown in DMEM supplemented with 10% FCS and 1% penicillin/streptomycin in a 5% CO2 atmosphere at 37 °C. BRK cells were regularly tested for mycoplasma contaminations via the PCR Mycoplasma Test Kit I/C (PromoKine).
Antibodies.
The antibodies used in this study included commercially available antibodies as well as previously published ones (96–98). For detailed descriptions, please refer to SI Appendix.
ChIP.
ChIP assays were performed according to previously described methods (99). Please refer to SI Appendix for detailed descriptions.
RNA Extraction.
Approximately 1 × 106 cells harvested for subsequent RNA isolation were stored in 1 mL TRI Reagent (Sigma-Aldrich) at –80 °C. Total RNA was isolated by phenol-chloroform extraction according to the manufacturer’s instructions. RNA was resuspended in nuclease-free water and stored at –80 °C. Prior to sequencing, RNA quality was assessed with the Agilent 2100 Bioanalyzer System combined with an RNA 6000 Nano Chip (Agilent, USA).
Library Preparation and Sequencing.
Please refer to SI Appendix for comprehensive descriptions of our library preparations, sequencing, and data analyses, including the statistical analyses.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Dataset S08 (XLSX)
Dataset S09 (XLSX)
Dataset S10 (XLSX)
Dataset S11 (XLSX)
Dataset S12 (XLSX)
Dataset S13 (XLSX)
Acknowledgments
We extend our sincere gratitude to Dr. Sanamjeet Virdi (research unit Virus Genomics at the LIV, Hamburg) for his assistance and expert guidance during the initial stages of our RNA-seq analyses. K.v.S. received financial support from the EPILOG initiative (epilog-infect.org), which is funded by the Ministry of Science, Research, and Equalities, Hamburg. The LIV is supported by the Freie und Hansestadt Hamburg and the German Bundesministerium für Gesundheit.
Author contributions
K.v.S. and T.D. designed research; K.v.S., L.S., B.G., S.-C.W., and M.H. performed research; K.v.S. and T.G. analyzed data; and K.v.S., L.S., W.-H.I., L.D.B., and T.D. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
Konstantin von Stromberg, Email: konstantin.stromberg@leibniz-liv.de.
Thomas Dobner, Email: thomas.dobner@leibniz-liv.de.
Data, Materials, and Software Availability
All raw sequencing datasets used in this study are available via the European Nucleotide Archive (https://www.ebi.ac.uk/ena) under accession PRJEB63131 (100) and Zenodo: 10.5281/zenodo.8047240 (101), 10.5281/zenodo.8048294 (102), 10.5281/zenodo.8048302 (103), 10.5281/zenodo.8048509 (104), and 10.5281/zenodo.8048320 (105). All study data are included in the article and/or supporting information.
Supporting Information
References
- 1.Huebner R. J., Rowe W. P., Lane W. T., Oncogenic effects in hamsters of human adenovirus types 12 and 18. Proc. Natl. Acad. Sci. U.S.A. 48, 2051–2058 (1962). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yabe Y., Samper L., Bryan E., Taylor G., Trentin J. J., Oncogenic effect of human adenovirus type 12 mice. Science 143, 46–47 (1964). [DOI] [PubMed] [Google Scholar]
- 3.Javier R., Raska K. Jr., Macdonald G. J., Shenk T., Human adenovirus type 9-induced rat mammary tumors. J. Virol. 65, 3192–3202 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bertzbach L. D., Ip W. H., Dobner T., Animal models in human adenovirus research. Biology 10, 1253 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tessier T. M., et al. , Almost famous: Human adenoviruses (and what they have taught us about cancer). Tumour Virus Res. 12, 200225 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spurgeon M. E., Small DNA tumor viruses and human cancer: Preclinical models of virus infection and disease. Tumour Virus Res. 14, 200239 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Debbas M., White E., Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev. 7, 546–554 (1993). [DOI] [PubMed] [Google Scholar]
- 8.Dyson N., Guida P., McCall C., Harlow E., Adenovirus E1A makes two distinct contacts with the retinoblastoma protein. J. Virol. 66, 4606–4611 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berk A. J., Recent lessons in gene expression, cell cycle control, and cell biology from adenovirus. Oncogene 24, 7673–7685 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Querido E., et al. , Regulation of p53 levels by the E1B 55-kilodalton protein and E4orf6 in adenovirus-infected cells. J. Virol. 71, 3788–3798 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Querido E., Teodoro J. G., Branton P. E., Accumulation of p53 induced by the adenovirus E1A protein requires regions involved in the stimulation of DNA synthesis. J. Virol. 71, 3526–3533 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shenk T., Flint J., Transcriptional and transforming activities of the adenovirus E1A proteins. Adv. Cancer Res. 57, 47–85 (1991). [DOI] [PubMed] [Google Scholar]
- 13.Martin M. E., Berk A. J., Adenovirus E1B 55K represses p53 activation in vitro. J. Virol. 72, 3146–3154 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yew P. R., Liu X., Berk A. J., Adenovirus E1B oncoprotein tethers a transcriptional repression domain to p53. Genes Dev. 8, 190–202 (1994). [DOI] [PubMed] [Google Scholar]
- 15.Wienzek S., Roth J., Dobbelstein M., E1B 55-kilodalton oncoproteins of adenovirus types 5 and 12 inactivate and relocalize p53, but not p51 or p73, and cooperate with E4orf6 proteins to destabilize p53. J. Virol. 74, 193–202 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steegenga W. T., Shvarts A., Riteco N., Bos J. L., Jochemsen A. G., Distinct regulation of p53 and p73 activity by adenovirus E1A, E1B, and E4orf6 proteins. Mol. Cell Biol. 19, 3885–3894 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin M. E., Berk A. J., Corepressor required for adenovirus E1B 55,000-molecular-weight protein repression of basal transcription. Mol. Cell Biol. 19, 3403–3414 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y., Shevchenko A., Shevchenko A., Berk A. J., Adenovirus exploits the cellular aggresome response to accelerate inactivation of the MRN complex. J. Virol. 79, 14004–14016 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krätzer F., et al. , The adenovirus type 5 E1B–55K oncoprotein is a highly active shuttle protein and shuttling is independent of E4orf6, p53 and Mdm2. Oncogene 19, 850–857 (2000). [DOI] [PubMed] [Google Scholar]
- 20.Endter C., Härtl B., Spruss T., Hauber J., Dobner T., Blockage of CRM1-dependent nuclear export of the adenovirus type 5 early region 1B 55-kDa protein augments oncogenic transformation of primary rat cells. Oncogene 24, 55–64 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Härtl B., Zeller T., Blanchette P., Kremmer E., Dobner T., Adenovirus type 5 early region 1B 55-kDa oncoprotein can promote cell transformation by a mechanism independent from blocking p53-activated transcription. Oncogene 27, 3673–3684 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Schreiner S., et al. , Adenovirus type 5 early region 1B 55K oncoprotein-dependent degradation of cellular factor Daxx is required for efficient transformation of primary rodent cells. J. Virol. 85, 8752–8765 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Endter C., Kzhyshkowska J., Stauber R., Dobner T., SUMO-1 modification required for transformation by adenovirus type 5 early region 1B 55-kDa oncoprotein. Proc. Natl. Acad. Sci. U.S.A. 98, 11312–11317 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fiedler M., et al. , Protein-protein interactions facilitate E4orf6-dependent regulation of E1B–55K SUMOylation in HAdV-C5 infection. Viruses 14, 463 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Teodoro J. G., et al. , Phosphorylation at the carboxy terminus of the 55-kilodalton adenovirus type 5 E1B protein regulates transforming activity. J. Virol. 68, 776–786 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Teodoro J. G., Branton P. E., Regulation of p53-dependent apoptosis, transcriptional repression, and cell transformation by phosphorylation of the 55-kilodalton E1B protein of human adenovirus type 5. J. Virol. 71, 3620–3627 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wimmer P., et al. , Cross-talk between phosphorylation and SUMOylation regulates transforming activities of an adenoviral oncoprotein. Oncogene 32, 1626–1637 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Kolbe V., et al. , Conserved E1B–55K SUMOylation in different human adenovirus species is a potent regulator of intracellular localization. J. Virol. 96, e0083821 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ching W., Dobner T., Koyuncu E., The human adenovirus type 5 E1B 55-kilodalton protein is phosphorylated by protein kinase CK2. J. Virol. 86, 2400–2415 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones M. S., Chodosh J., Seto D. M., Adenoviruses. Ref. Module Life Sci., 10.1016/b978-0-12-809633-8.06017-9 (2017). [DOI] [Google Scholar]
- 31.Doerfler W., “Human adenovirus type 12” in Adenovirus Methods and Protocols, W. S. Wold, A. E. Tollefson, Eds. (Humana Press, Totowa, NJ, USA, 2007), 10.1007/978-1-59745-277-9_14, chap. 14, pp. 197–211. [DOI] [Google Scholar]
- 32.Endter C., Dobner T., Cell transformation by human adenoviruses. Curr. Top Microbiol. Immunol. 273, 163–214 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Blackford A. N., Grand R. J., Adenovirus E1B 55-kilodalton protein: Multiple roles in viral infection and cell transformation. J. Virol. 83, 4000–4012 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hidalgo P., Ip W. H., Dobner T., Gonzalez R. A., The biology of the adenovirus E1B 55K protein. FEBS Lett. 593, 3504–3517 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Tian B., Yang J., Brasier A. R., Two-step cross-linking for analysis of protein-chromatin interactions. Methods Mol. Biol. 809, 105–120 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kindsmüller K., et al. , Intranuclear targeting and nuclear export of the adenovirus E1B–55K protein are regulated by SUMO1 conjugation. Proc. Natl. Acad. Sci. U.S.A. 104, 6684–6689 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feng J., Liu T., Qin B., Zhang Y., Liu X. S., Identifying ChIP-seq enrichment using MACS. Nat. Protocols 7, 1728–1740 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jalili V., Matteucci M., Masseroli M., Morelli M. J., Using combined evidence from replicates to evaluate ChIP-seq peaks. Bioinformatics 31, 2761–2769 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Yu G., Wang L. G., He Q. Y., ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Jalili V., Matteucci M., Morelli M. J., Masseroli M., MuSERA: Multiple sample enriched region assessment. Brief Bioinform. 18, 367–381 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Isakova A., et al. , SMiLE-seq identifies binding motifs of single and dimeric transcription factors. Nat. Methods 14, 316–322 (2017). [DOI] [PubMed] [Google Scholar]
- 42.el-Deiry W. S., et al. , WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817–825 (1993). [DOI] [PubMed] [Google Scholar]
- 43.Juven T., Barak Y., Zauberman A., George D., Oren M., Wild type p53 can mediate sequence-specific transactivation of an internal promoter within the mdm2 gene. Oncogene 8, 3411–3416 (1993). [PubMed] [Google Scholar]
- 44.Kawase T., et al. , p53 target gene AEN is a nuclear exonuclease required for p53-dependent apoptosis. Oncogene 27, 3797–3810 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Okamura S., et al. , p53DINP1, a p53-inducible gene, regulates p53-dependent apoptosis. Mol. Cell 8, 85–94 (2001). [DOI] [PubMed] [Google Scholar]
- 46.Brown C. J., Lain S., Verma C. S., Fersht A. R., Lane D. P., Awakening guardian angels: Drugging the p53 pathway. Nat. Rev. Cancer 9, 862–873 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Olivares-Illana V., Fahraeus R., p53 isoforms gain functions. Oncogene 29, 5113–5119 (2010). [DOI] [PubMed] [Google Scholar]
- 48.Pantalacci S., Tapon N., Leopold P., The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nat. Cell Biol. 5, 921–927 (2003). [DOI] [PubMed] [Google Scholar]
- 49.Harvey K. F., Pfleger C. M., Hariharan I. K., The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell 114, 457–467 (2003). [DOI] [PubMed] [Google Scholar]
- 50.Huang J., Wu S., Barrera J., Matthews K., Pan D., The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 122, 421–434 (2005). [DOI] [PubMed] [Google Scholar]
- 51.Rajbhandari P., et al. , Cross-cohort analysis identifies a TEAD4-MYCN positive feedback loop as the core regulatory element of high-risk neuroblastoma. Cancer Discov. 8, 582–599 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marquard S., et al. , Yes-associated protein (YAP) induces a secretome phenotype and transcriptionally regulates plasminogen activator Inhibitor-1 (PAI-1) expression in hepatocarcinogenesis. Cell Commun. Signal 18, 166 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu S., et al. , Yap promotes noncanonical wnt signals from cardiomyocytes for heart regeneration. Circ. Res. 129, 782–797 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fischer M., Census and evaluation of p53 target genes. Oncogene 36, 3943–3956 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pugacheva E. M., et al. , The structural complexity of the human BORIS gene in gametogenesis and cancer. PLoS One 5, e13872 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sleutels F., et al. , The male germ cell gene regulator CTCFL is functionally different from CTCF and binds CTCF-like consensus sites in a nucleosome composition-dependent manner. Epigenet. Chrom. 5, 8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hore T. A., Deakin J. E., Marshall Graves J. A., The evolution of epigenetic regulators CTCF and BORIS/CTCFL in amniotes. PLoS Genet. 4, e1000169 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Murata T., et al. , Molecular basis of epstein-barr virus latency establishment and lytic reactivation. Viruses 13, 2344 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Broussard G., Damania B., Regulation of KSHV latency and lytic reactivation. Viruses 12, 1034 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu X., et al. , Human virus transcriptional regulators. Cell 182, 24–37 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao L. Y., Santiago A., Liu J., Liao D., Repression of p53-mediated transcription by adenovirus E1B 55-kDa does not require corepressor mSin3A and histone deacetylases. J. Biol. Chem. 282, 7001–7010 (2007). [DOI] [PubMed] [Google Scholar]
- 62.Pennella M. A., Liu Y., Woo J. L., Kim C. A., Berk A. J., Adenovirus E1B 55-kilodalton protein is a p53-SUMO1 E3 ligase that represses p53 and stimulates its nuclear export through interactions with promyelocytic leukemia nuclear bodies. J. Virol. 84, 12210–12225 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Günther T., Schreiner S., Dobner T., Tessmer U., Grundhoff A., Influence of ND10 components on epigenetic determinants of early KSHV latency establishment. PLoS Pathog. 10, e1004274 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miller D. L., Rickards B., Mashiba M., Huang W., Flint S. J., The adenoviral E1B 55-kilodalton protein controls expression of immune response genes but not p53-dependent transcription. J. Virol. 83, 3591–3603 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goodrum F. D., Shenk T., Ornelles D. A., Adenovirus early region 4 34-kilodalton protein directs the nuclear localization of the early region 1B 55-kilodalton protein in primate cells. J. Virol. 70, 6323–6335 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dobner T., Horikoshi N., Rubenwolf S., Shenk T., Blockage by adenovirus E4orf6 of transcriptional activation by the p53 tumor suppressor. Science 272, 1470–1473 (1996). [DOI] [PubMed] [Google Scholar]
- 67.Ou H. D., et al. , A structural basis for the assembly and functions of a viral polymer that inactivates multiple tumor suppressors. Cell 151, 304–319 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hobom U., Dobbelstein M., E1B–55-kilodalton protein is not required to block p53-induced transcription during adenovirus infection. J. Virol. 78, 7685–7697 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Trentin J. J., Yabe Y., Taylor G., The quest for human cancer viruses: A new approach to an old problem reveals cancer induction in hamsters by human adenovirus. Science 137, 835–841 (1962). [DOI] [PubMed] [Google Scholar]
- 70.Yabe Y., Trentin J. J., Taylor G., Cancer induction in hamsters by human type 12 adenovirus. Effect of age and of virus dose. Proc. Soc. Exp. Biol. Med. 111, 343–344 (1962). [DOI] [PubMed] [Google Scholar]
- 71.Jelinek T., Pereira D. S., Graham F. L., Tumorigenicity of adenovirus-transformed rodent cells is influenced by at least two regions of adenovirus type 12 early region 1A. J. Virol. 68, 888–896 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pan D., The hippo signaling pathway in development and cancer. Dev. Cell 19, 491–505 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dupont S., et al. , Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183 (2011). [DOI] [PubMed] [Google Scholar]
- 74.Meng Z., et al. , RAP2 mediates mechanoresponses of the Hippo pathway. Nature 560, 655–660 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zemke N. R., Gou D., Berk A. J., Dedifferentiation by adenovirus E1A due to inactivation of Hippo pathway effectors YAP and TAZ. Genes Dev. 33, 828–843 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Battilana G., Zanconato F., Piccolo S., Mechanisms of YAP/TAZ transcriptional control. Cell Stress 5, 167–172 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Calses P. C., Crawford J. J., Lill J. R., Dey A., Hippo pathway in cancer: Aberrant regulation and therapeutic opportunities. Trends Cancer 5, 297–307 (2019). [DOI] [PubMed] [Google Scholar]
- 78.Nardone G., et al. , YAP regulates cell mechanics by controlling focal adhesion assembly. Nat. Commun. 8, 15321 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gazon H., Barbeau B., Mesnard J. M., Peloponese J. M. Jr., Hijacking of the AP-1 signaling pathway during development of ATL. Front. Microbiol. 8, 2686 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mirzaei H., Khodadad N., Karami C., Pirmoradi R., Khanizadeh S., The AP-1 pathway; A key regulator of cellular transformation modulated by oncogenic viruses. Rev. Med. Virol. 30, e2088 (2020). [DOI] [PubMed] [Google Scholar]
- 81.Feng Y., Pan L., Zhang B., Huang H., Ma H., BATF acts as an oncogene in non-small cell lung cancer. Oncol Lett. 19, 205–210 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kikuchi J., et al. , Simultaneous blockade of AP-1 and phosphatidylinositol 3-kinase pathway in non-small cell lung cancer cells. Br. J. Cancer 99, 2013–2019 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ruiz E. J., et al. , JunD, not c-Jun, is the AP-1 transcription factor required for Ras-induced lung cancer. JCI Insight 6, e124985 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Casalino L., Talotta F., Cimmino A., Verde P., The Fra-1/AP-1 oncoprotein: From the “undruggable” transcription factor to therapeutic targeting. Cancers (Basel) 14, 1480 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Eferl R., Wagner E. F., AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 3, 859–868 (2003). [DOI] [PubMed] [Google Scholar]
- 86.Wu Z., Nicoll M., Ingham R. J., AP-1 family transcription factors: A diverse family of proteins that regulate varied cellular activities in classical hodgkin lymphoma and ALK+ ALCL Exp. Hematol. Oncol. 10, 4 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hai T., Curran T., Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc. Natl. Acad. Sci. U.S.A. 88, 3720–3724 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bakiri L., et al. , Fra-1/AP-1 induces EMT in mammary epithelial cells by modulating Zeb1/2 and TGFbeta expression. Cell Death Differ. 22, 336–350 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tam W. L., et al. , Protein kinase C alpha is a central signaling node and therapeutic target for breast cancer stem cells. Cancer Cell 24, 347–364 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zanconato F., et al. , Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 17, 1218–1227 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Goldstein I., Paakinaho V., Baek S., Sung M. H., Hager G. L., Synergistic gene expression during the acute phase response is characterized by transcription factor assisted loading. Nat. Commun. 8, 1849 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hidalgo P., et al. , E1B–55K is a phosphorylation-dependent transcriptional and post-transcriptional regulator of viral gene expression in HAdV-C5 infection. J. Virol. 96, JVI0206221 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Boulanger M., Chakraborty M., Tempe D., Piechaczyk M., Bossis G., SUMO and transcriptional regulation: The lessons of large-scale proteomic, modifomic and genomic studies. Molecules 26, 828 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Speiseder T., Nevels M., Dobner T., Determination of the transforming activities of adenovirus oncogenes. Methods Mol. Biol. 1089, 105–115 (2014). [DOI] [PubMed] [Google Scholar]
- 95.Graham F. L., Smiley J., Russell W. C., Nairn R., Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 36, 59–74 (1977). [DOI] [PubMed] [Google Scholar]
- 96.Harlow E., Franza B. R. Jr., Schley C., Monoclonal antibodies specific for adenovirus early region 1A proteins: Extensive heterogeneity in early region 1A products. J. Virol. 55, 533–546 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sarnow P., Sullivan C. A., Levine A. J., A monoclonal antibody detecting the adenovirus type 5 E 1 b-58Kd tumor antigen: Characterization of the E 1 b-58Kd tumor antigen in adenovirus-infected and -transformed cells. Virology 120, 510–517 (1982). [DOI] [PubMed] [Google Scholar]
- 98.Lomonosova E., Subramanian T., Chinnadurai G., Mitochondrial localization of p53 during adenovirus infection and regulation of its activity by E1B–19K. Oncogene 24, 6796–6808 (2005). [DOI] [PubMed] [Google Scholar]
- 99.Günther T., Theiss J. M., Fischer N., Grundhoff A., Investigation of viral and host chromatin by ChIP-PCR or ChIP-Seq analysis. Curr. Protoc. Microbiol. 40, 1E.10.1–1E.10.21 (2016). [DOI] [PubMed] [Google Scholar]
- 100.von Stromberg K., et al. , The human adenovirus E1B-55K oncoprotein coordinates cell transformation through regulation of DNA-bound host transcription factors. European Nucleotide Archive (ENA). https://www.ebi.ac.uk/ena/browser/view/PRJEB63131. Accessed 1 September 2023. [DOI] [PMC free article] [PubMed]
- 101.von Stromberg K., et al. , BRK mRNA-seq quantifying bash script. Zenodo. 10.5281/zenodo.8047240. Accessed 16 June 2023. [DOI]
- 102.von Stromberg K., et al. , BRK mRNA-seq DEG analysis R script. Zenodo. 10.5281/zenodo.8048294. Accessed 16 June 2023. [DOI]
- 103.von Stromberg K., et al. , BRK ChIP-seq data alignment, peak calling and motif enrichment bash script. Zenodo. 10.5281/zenodo.8048302. Accessed 16 June 2023. [DOI]
- 104.von Stromberg K., et al. , BRK ChIP-seq peak-to-gene annotation analysis R script. Zenodo. 10.5281/zenodo.8048509. Accessed 16 June 2023. [DOI]
- 105.von Stromberg K., et al. , EnhancerAtlas 2.0-based Enhancer consensus list creation bash script. Zenodo. 10.5281/zenodo.8048320. Accessed 16 June 2023. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Dataset S08 (XLSX)
Dataset S09 (XLSX)
Dataset S10 (XLSX)
Dataset S11 (XLSX)
Dataset S12 (XLSX)
Dataset S13 (XLSX)
Data Availability Statement
All raw sequencing datasets used in this study are available via the European Nucleotide Archive (https://www.ebi.ac.uk/ena) under accession PRJEB63131 (100) and Zenodo: 10.5281/zenodo.8047240 (101), 10.5281/zenodo.8048294 (102), 10.5281/zenodo.8048302 (103), 10.5281/zenodo.8048509 (104), and 10.5281/zenodo.8048320 (105). All study data are included in the article and/or supporting information.