

Abstract
MicroRNAs (miRNAs) together with Argonaute (AGO) proteins form the core of the RNA‐induced silencing complex (RISC) to regulate gene expression of their target RNAs post‐transcriptionally. Argonaute proteins are subjected to intensive regulation via various post‐translational modifications that can affect their stability, silencing efficacy and specificity for targeted gene regulation. We report here that in Caenorhabditis elegans, two conserved serine/threonine kinases – casein kinase 1 alpha 1 (CK1A1) and casein kinase 2 (CK2) – regulate a highly conserved phosphorylation cluster of 4 Serine residues (S988:S998) on the miRNA‐specific AGO protein ALG‐1. We show that CK1A1 phosphorylates ALG‐1 at sites S992 and S995, while CK2 phosphorylates ALG‐1 at sites S988 and S998. Furthermore, we demonstrate that phospho‐mimicking mutants of the entire S988:S998 cluster rescue the various developmental defects observed upon depleting CK1A1 and CK2. In humans, we show that CK1A1 also acts as a priming kinase of this cluster on AGO2. Altogether, our data suggest that phosphorylation of AGO within the cluster by CK1A1 and CK2 is required for efficient miRISC‐target RNA binding and silencing.
Keywords: Argonaute, CK1A1, CK2, microRNA, phosphorylation
Subject Categories: Post-translational Modifications & Proteolysis, RNA Biology, Signal Transduction
Two conserved casein kinases, casein kinase 1 alpha 1 and casein kinase 2, phosphorylate a highly conserved serine cluster on microRNA‐specific Argonaute proteins. Argonaute phosphorylation by these kinases is essential to regulate microRNA‐mediated gene silencing in Caenorhabditis elegans.
Introduction
MicroRNAs (miRNAs) belong to a highly conserved class of short non‐coding RNAs that are ~22 nucleotides (nts) in length and play a critical role in regulating diverse molecular pathways essential for cell growth, division, differentiation, and development in organisms (Bartel, 2018). They associate with Argonaute (AGO) proteins to form the core effector complex, known as the miRNA‐Induced Silencing Complex (miRISC), and guide the miRISC to complementary sites on the target mRNAs to initiate a series of gene silencing mechanisms such as translational repression and mRNA target destabilization including deadenylation, decapping, and decay (Jonas & Izaurralde, 2015; Frédérick & Simard, 2022). Both target destabilization and translation repression mechanisms can function either mutually or exclusively, however, the choice of favored mechanism can be context‐specific and can differ between cell types and developmental stages (e.g., Bazzini et al, 2012; Djuranovic et al, 2012; Jannot et al, 2016; Dallaire et al, 2018).
Argonautes are highly conserved proteins, which are found in almost all organisms from archaea to eukaryotes. The Argonaute superfamily contains numerous proteins that are mainly divided into three clades/branches according to sequence homology – the Ago clade, the Piwi clade, and the worm‐specific (Wago) clade (Almeida et al, 2019). In Caenorhabditis elegans, the two miRNA‐specific AGOs are ALG‐1 and ALG‐2, which are orthologous to the human AGO1‐4 proteins sharing a high sequence similarity (Grishok et al, 2001; Vasquez‐Rifo et al, 2012; Nakanishi, 2022). ALG‐5, which is mostly expressed in the germline, is another miRNA‐specific AGO required for proper developmental timing in C. elegans (Brown et al, 2017).
A hallmark of AGO proteins is their bilobed structure composed of four distinct domains: the N terminal, PAZ, MID and PIWI domains and two linker domains L1 and L2. The L1 linker between the N and PAZ domains and the L2 linker between the PAZ and MID domains makes the RISC structurally stable (Nakanishi, 2022). While numerous studies have helped us to understand how AGO can function in various small‐RNA‐mediated gene regulatory pathways, it is still unclear how these proteins can themselves be modulated to have such different effects on gene regulation. One of the mechanisms used by cells to modulate a protein's function and stability is by adding or removing diverse post‐translational modifications (PTMs). For instance, hydroxylation of AGO2 at proline 700 showed an increase in the stability of AGO2 in mouse and human cells and increased siRNA‐mediated target RNA cleavage (Qi et al, 2008). Under cellular stress, poly ADP‐ribosylation (PARylation) of AGO1‐4 inhibited miRNA activity by decreasing target accessibility (Leung et al, 2011). The most common protein modification is phosphorylation, which is also the best characterized AGO PTM. Phosphorylation of Serine 387 in the L2 region of AGO2 was found to be stimulated by both the p38 MAP kinase and AKT3 pathways facilitating the localization of AGO2 to processing bodies and upregulating translational repression (Zeng et al, 2008; Horman et al, 2013). This specific modification was also linked with the sorting of AGO2 into exosomes and is necessary for the association between AGO2 and LIMD1, a protein suggested to bridge AGO2 and GW182 (McKenzie et al, 2016; Bridge et al, 2017). Stimulated upon hypoxic stress, the epidermal growth factor receptor (EGFR) phosphorylates AGO2 at tyrosine 393 in the PAZ domain leading to the reduction of specific tumor‐suppressor‐like miRNAs (Shen et al, 2013). Tyrosine 393 residues of AGO2 can be dephosphorylated by PTPB1 phosphatase and its inhibition causes premature senescence of primary cells by affecting the function of H‐RASv12‐induced oncogenic miRNAs (Yang et al, 2014). The phosphorylation of specific residues on AGO can also affect its binding to miRNA and mRNA targets. For example, the phosphorylation of tyrosine 529 residue near the miRNA 5′‐end binding pocket located in the MID domain can prevent miRNA loading (Rüdel et al, 2011). Our recent study showed that phosphorylation of serine 642 residue by Protein kinase A (PKA) on AGO ALG‐1 decreases its ability to bind miRNAs during C. elegans development (Huberdeau et al, 2022). These studies suggest that miRNA silencing efficacy can be regulated by different AGO PTMs both under stress and normal cellular conditions.
We and another group previously discovered and characterized a highly conserved serine/threonine phosphorylation cluster at the C‐terminus of AGO proteins (Golden et al, 2017; Quévillon Huberdeau et al, 2017). The cluster is located at a surface‐exposed loop in the PIWI domain of AGO and consists of 4 phosphorylatable serine residues and a threonine residue‐ S824, S828, T830, S831, and S834 on human AGO2 (equivalent to S988, S992, S995, T997, and S998 on ALG‐1 in C. elegans). Our studies in C. elegans and human cells revealed that phosphorylation of the cluster is essential for miRNA‐mediated gene silencing in vivo and showed that its hyperphosphorylation affects binding to miRNA targets (Quévillon Huberdeau et al, 2017).
In this study, using a combination of biochemical, biophysical, and molecular analyses, we show that two conserved serine/threonine kinases casein kinase 1 alpha 1 (CK1A1) and casein kinase 2 (CK2) are involved in the regulation of the S988:S998 (S824:S834 in humans) phosphorylation cluster on the miRNA‐specific AGO proteins. We provide evidence that CK1A1 and CK2 phosphorylate C. elegans ALG‐1 and human AGO2. Through a series of genetic experiments, we demonstrate that phospho‐mimicking mutants of the entire S988:S998 phosphorylation cluster rescue the various developmental phenotypes associated with reduced miRNA pathway function observed upon depleting CK1A1 and CK2. In humans, we show that CK1A1 phosphorylates the S824:S834 cluster on AGO2 and primes for phosphorylation by another kinase(s). We propose a model where CK1A1 acts as the priming kinase and together with additional secondary kinases such as CK2, hyper‐phosphorylate AGO at the S988:S998 (S824:S834) cluster and regulates miRISC‐target mRNA binding and silencing.
Results
CK1A1 and CK2 are involved in the phosphorylation of the S988:S998 cluster on ALG‐1
The phosphorylation status of the conserved S988:S998 cluster in the PIWI domain of ALG‐1 is highly important for its function in vivo (Quévillon Huberdeau et al, 2017). Mutation of a single Serine residue at position 992 (S992) into an Alanine (S992A) within this cluster on ALG‐1 reiterated the complete loss of phosphorylation of the cluster in animals and lead to developmental phenotypes usually associated with the complete loss of function of ALG‐1 (Quévillon Huberdeau et al, 2017). Supporting our observations, it has been reported that the ANKRD52‐PPP6C phosphatase complex and CK1A1 regulate the phosphorylation of this cluster and miRNA targets binding in human cells (Golden et al, 2017). Interestingly, using in vitro kinase assays they showed that only a pre‐phosphorylated peptide at S828 of human AGO2 (equivalent to ALG‐1 S992) promotes robust phosphorylation of this cluster, and hence S828 (or ALG‐1 S992) may or may not be a direct substrate for CK1A1, as CK1A1 usually prefers pre‐phosphorylated substrates conforming to the consensus sequence of ([pS/pT]‐X1‐2‐[S/T], where X can be any amino acid) (Golden et al, 2017). Therefore, we can envision the possibility of other kinases being involved in phosphorylating the AGO2 S824:S834 (corresponding to S988:S998 on ALG‐1) cluster. Golden et al (2017) propose a model in which initial phosphorylation of S828, and potentially S824 by CK1A1 or some other kinase, stimulates efficient hierarchical phosphorylation of S831 followed by S834, leaving AGO2 incapable for target binding until ANKRD52–PPP6C phosphatase complex returns it to an active state.
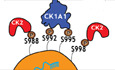
To fully understand the implications of this conserved and functionally relevant phosphorylation cluster in the miRNA‐mediated gene silencing process in vivo, it is important to uncover the kinases specifically targeting the S992 and other biologically conserved serine residues to regulate AGO function in C. elegans. We first used an in silico approach utilizing available online databases such as NetPhos3.1 to produce a list of putative kinases that can phosphorylate the S988:S998 cluster on ALG‐1 in C. elegans and the S824:S834 cluster on AGO2 in humans. The NetPhos3.1 program utilizes an artificial neural network to predict Serine, Threonine, and Tyrosine phosphorylation sites from an inputted amino acid sequence (Blom et al, 1999). CK1A1 and CK2 were both predicted to phosphorylate the cluster on both human and C. elegans (Fig EV1A and B). Casein kinase 1α (CK1α), a member of the CK1 family, is a serine/threonine protein kinase encoded by CSNK1A1 in humans and by KIN‐19 in C. elegans (Banerjee et al, 2010; Jiang et al, 2018). CK2 is a serine/threonine protein kinase composed of two catalytic α subunits and two regulatory β subunits in a tetrameric α2β2 configuration (encoded by KIN‐3 and KIN‐10 respectively in C. elegans) (Hu & Rubin, 1990; Litchfield, 2003). Interestingly, CK2 was previously reported to promote miRISC function in C. elegans by, potentially, phosphorylating the DEAD‐box RNA helicase CGH‐1 (Alessi et al, 2015).
Figure EV1. CK1A1 and CK2 were both predicted to be phosphorylating the S988:S998 cluster on ALG‐1 and S824:S834 cluster on AGO2 by Netphos3.1 online database.
- Netphos3.1 scores of CKI and CK2 phosphorylating the different phosphorylatable Serine residues (S988, S992, S995, and S998) and a Threonine residue (T997) within the S988:S998 cluster on ALG‐1 in Caenorhabditis elegans.
- Netphos3.1 scores of CKI and CK2 phosphorylating the different phosphorylatable Serine residues (S824, S828, S831, and S834) and a Threonine residue (T830) within the S824:S834 cluster on AGO2 in humans.
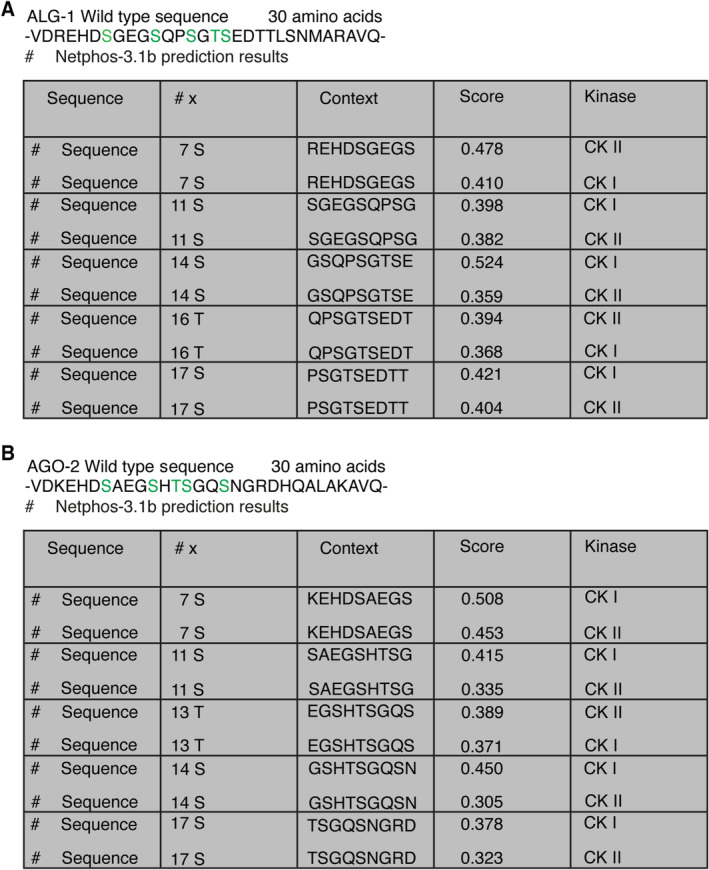
Casein kinase 1 alpha 1 mainly targets substrates containing acidic or phosphorylated amino acid residues (Knippschild et al, 2014). The canonical consensus motif for CK1A1 is [pS/pT]‐X1‐2‐[S/T], where pS/pT is a prior phosphorylated‐serine or threonine residue, X is any amino acid, and S/T are the residues to be phosphorylated (Kennelly & Krebs, 1991). CK1A1 also prefers motifs where the phospho‐serine or phospho‐threonine can be replaced by a cluster of negatively charged acidic amino acids (Knippschild et al, 2014). Thus, another consensus sequence for CK1A1 lies in the motif of [D/E]‐X1‐2‐[S/T], where D/E indicates an aspartate or a glutamate residue (Kennelly & Krebs, 1991). ALG‐1 contains two sites matching the CK1A1 consensus phosphorylation sequences‐ ([D/E]‐X1‐2‐[S/T]) at position S992 and ([pS/pT]‐X1‐2‐[S/T]) at position S995 within the cluster (Fig 1A). CK2 is an acidophilic kinase which requires the presence of one or more acidic residues like glutamate or an aspartate immediately C‐terminal to the phospho‐acceptor serine/threonine site (St‐Denis et al, 2015). The canonical consensus sequence for CK2 is represented by the motif‐ S/T‐X1‐2‐D/E (Meggio et al, 1994). ALG‐1 also contains two sites matching the CK2 consensus phosphorylation sequences‐ ([S/T]‐X‐X‐[D/E]) at position S988 and ([S/T]‐X‐[D/E]) at position S998 within the cluster (Fig 1B).
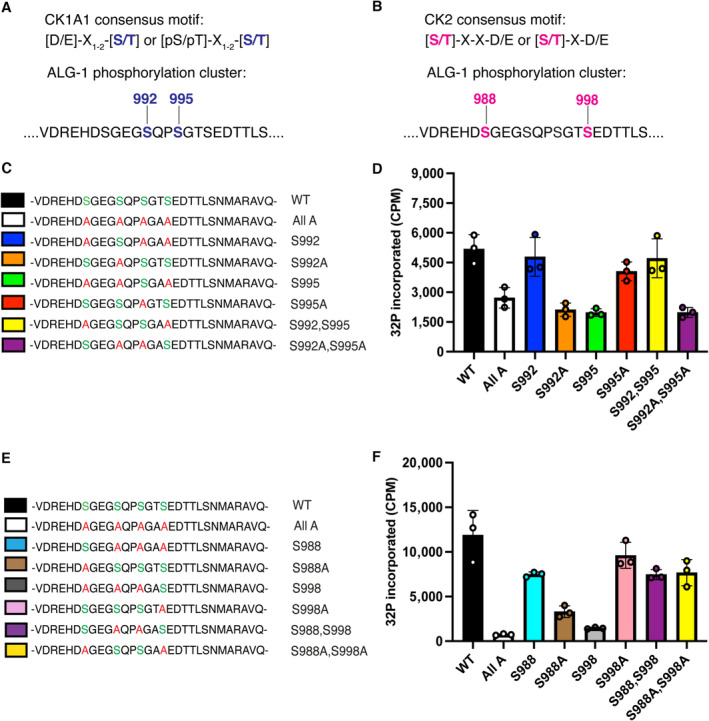
Figure 1. CK1A1 and CK2 phosphorylate the highly conserved S988:S998 cluster on ALG‐1.
- ALG‐1 harbors two CK1A1 recognition motifs within the cluster.
- ALG‐1 harbors two CK2 recognition motifs within the cluster.
- List of different 30 amino acid biotinylated peptides of ALG‐1 encompassing the S988:S998 phosphorylation cluster that were used for the in vitro CK1A1 kinase assay.
- ALG‐1 is phosphorylated in vitro by CK1A1 at S992 and S995 sites. Data are presented as the mean and the error bars represent the standard deviation (SD) from three technical replicates (n = 3).
- List of different 30 amino acid biotinylated peptides of ALG‐1 encompassing the S988:S998 phosphorylation cluster that were used for the in vitro CK2 kinase assay.
- ALG‐1 is phosphorylated in vitro by CK2 at S988 and S998 sites. Data are presented as the mean and the error bars represent the SD from three technical replicates (n = 3).
Source data are available online for this figure.
To determine whether ALG‐1 could be phosphorylated by CK1A1 and CK2, we first performed a series of in vitro kinase assays using different synthetic biotinylated ALG‐1 peptides (Fig 1C and E) corresponding to the S988:S998 cluster and incubated them in a kinase buffer with either recombinant CK1A1 (Fig 1D) or recombinant CK2 (Fig 1F) and 32P γ‐ATP. CK1A1 and CK2 phosphorylation of a wild‐type peptide where the entire S988:S998 cluster is retained was robustly detected (black; Fig 1D and F). We also used a peptide where all the phosphorylatable residues (S988, S992, S995, T997, and S998) were replaced with alanine residues to show the specificity of the in vitro phosphorylation of the cluster by CK1A1 and CK2. The incorporation of phosphate with this all‐alanine peptide was not measurable above background levels (white; Fig 1D and F). To evaluate whether CK1A1 specifically targets the S992 and S995 sites, we tested with different combinations of peptides where we either retained or mutated the serine residues at positions 992 and 995. Interestingly, retaining a single serine residue at position 992 within the cluster promoted robust phosphorylation by CK1A1 close to wild‐type levels (blue; Fig 1D). Replacing S992 with an alanine reiterated the complete loss of phosphorylation of the cluster and phosphorylation by CK1A1 was not measurable above background levels (orange; Fig 1D). As outlined above, CK1A1 prefers previously phosphorylated substrates conforming to the consensus sequence of [pS/pT]‐X1‐2‐[S/T] (Kennelly & Krebs, 1991). We failed to re‐capture wild‐type levels of phosphorylation with a peptide where only a single serine residue at position 995 was retained (green; Fig 1D) arguing that its phosphorylation likely needs the initial phosphorylation of S992. Replacing S995 with an alanine or retaining both S992 and S995 within the cluster was sufficient to re‐establish the phosphorylation of the cluster back to wild‐type levels due to the presence of serine residue at position 992 in these peptides (red and yellow; Fig 1D). These findings are in line with previous observations where initial CK1A1 phosphorylation of S992 (S828 in humans) might act as the priming site for robust hierarchical phosphorylation of the cluster (Golden et al, 2017). Replacing both S992 and S995 with alanine residues displayed background levels of CK1A1‐dependent phosphorylation suggesting that both sites are targeted by CK1A1 and are important for the regulation of the cluster (purple; Fig 1D).
To test whether CK2 targets the S988 and S998 sites, we carried out in vitro CK2 kinase assays with different combinations of peptides where we either retained or mutated the serine residues at positions 988 and 998. Retaining a single serine residue at position 988 promoted efficient phosphorylation by CK2 and replacing S988 with an alanine displayed background levels of phosphorylation (blue and brown; Fig 1F). This suggests that S988 is a strong substrate for CK2‐dependent phosphorylation. Unexpectedly, S998 turned out to be a weaker substrate for CK2 as retaining the serine residue at this position displayed background levels of phosphorylation (gray; Fig 1F). Replacing S998 with an alanine or retaining both S988 and S998 within the cluster was sufficient to re‐establish the phosphorylation of the cluster back to wild‐type levels as in these peptides, S988 residue is available for CK2 dependent phosphorylation (pink and purple; Fig 1F). Strikingly, in the absence of serine residues at both 988 and 998 positions, CK2 still phosphorylate the peptide at a wild‐type level of phosphorylation suggesting that S992 and S995 sites can also be targeted by CK2 (yellow; Fig 1F), although they are predicted to be strong substrates for CK1A1 (Fig EV1A). Taken together, our results suggest that S992/S995 and S988/S998 sites can be targeted by CK1A1 and CK2, respectively.
The phosphorylation of the S988:S998 cluster on ALG‐1 by CK1A1 and CK2 is essential for miRNA function in animals
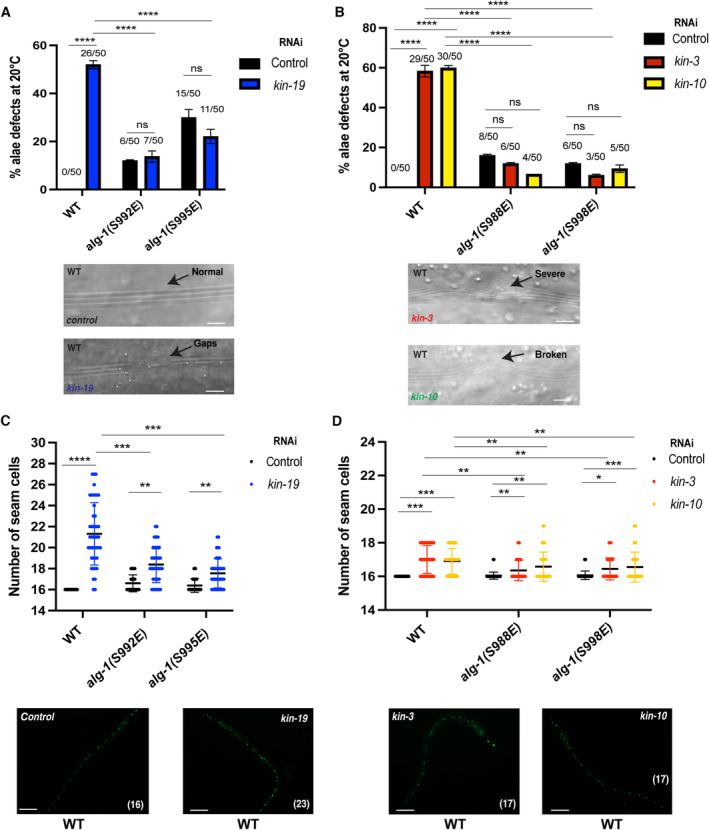
In C. elegans, the miRNA pathway plays an important role in different aspects of animal development. To determine whether CK1A1 and CK2 are involved in the miRNA pathway, we first monitored seam cell division patterning and adult alae formation. Seam cells are hypodermal cells, which are generated by asymmetric division during each larval stage (L1–L4) in a stem‐cell fashion, followed by a switch at the L4‐to‐adult transition, in which seam cells exit from the cell cycle and terminally differentiate to form the cuticular structure known as an alae. A single round of symmetric cell division during the L2 stage leads to a total of 16 lateral seam cells distributed on each side of the adult animal (Sulston & Horvitz, 1977). The developmental patterning of seam cells throughout development is tightly regulated by the heterochronic miRNAs lin‐4 (Ambros & Horvitz, 1984) and the let‐7 family (let‐7, miR‐48, miR‐84, and miR‐241) of miRNAs (Reinhart et al, 2000; Abbott et al, 2005). Disrupting genes associated with the miRNA pathway, like alg‐1 and the GW182 protein ain‐1 during development induces extra divisions of seam cells leading to defective cell fusion producing discontinuous, incomplete alae (gapped) or breaks along the cuticular structure (Grishok et al, 2001; Ding et al, 2005; Vasquez‐Rifo et al, 2013). We therefore monitored seam cell division patterning using a seam‐cell‐specific GFP reporter and adult alae formation in animals depleted of CK1A1 and CK2. Since CK1A1 and CK2 are both essential for the viability of the animal (arrested development at L4 molt) (Banerjee et al, 2010; Wang et al, 2014), we depleted these kinases using RNA interference (RNAi). KIN‐19 is the ortholog of CK1A1 in C. elegans (Banerjee et al, 2010). CK2 is composed of two catalytic α subunits and two regulatory β subunits encoded by KIN‐3 and KIN‐10 respectively in C. elegans (Hu & Rubin, 1990; Litchfield, 2003). In wild‐type animals, RNAi knockdown of kin‐19, kin‐3, and kin‐10 resulted in severe alae defects ranging from broken and gapped to incomplete alae structures (Figs 2A and B, and EV2A and B). Wild‐type animals have an invariable number of 16 seam cells at adulthood and animals depleted of CK1A1 displayed an abnormal number of seam cells (range 18–25, Figs 2C and EV3A). While animals depleted of CK2 also showed an increase in the number of seam cells (mean ~17, Figs 2D and EV3B), the range of increase was more severe upon CK1A1 depletion. These results indicate that CK1A1 and CK2 are essential for proper seam cell differentiation during development, as previously reported in (Banerjee et al, 2010) and (Alessi et al, 2015) respectively.
Figure 2. CK1A1 and CK2‐mediated phosphorylation of the S988:S998 cluster on ALG‐1 is essential for miRNA function in animals.
-
A, B(A) RNAi of kin‐19 on alg‐1(S992E) and alg‐1(S995E) animals significantly rescues defective alae formation as seen in wild‐type animals upon CK1A1 depletion. (B) RNAi of kin‐3 and kin‐10 on alg‐1(S988E) and alg‐1(S998E) animals significantly rescues defective alae formation as seen in wild‐type animals upon CK2 depletion. Animals were fed with bacteria expressing RNAi against kin‐19 (blue), kin‐3 (red), kin‐10 (yellow), or control RNAi (black) for 56 h and observed as young adults. KIN‐19 is an ortholog of CK1A1 in worms. KIN‐3, KIN‐10 are subunits of CK2 in worms. The P‐values indicated were measured by two‐tailed Fisher's exact t‐test (not significant [ns] P > 0.05, ****P < 0.0001). The number of animals with alae defects over the total number of animals scored (n = 50 for each condition) is indicated. Data are presented as the mean and the error bars represent the SD. Representative images with alae structure (normal and defective) are shown for wild‐type animals on kin‐19, kin‐3, kin‐10, or control RNAi. The scale bar represents 10 μm.
-
C, D(C) RNAi of kin‐19 on alg‐1(S992E) and alg‐1(S995E) animals expressing a seam cell gfp reporter (Pscm::gfp) significantly rescues seam cells hyperplasia post‐L4 as seen in wild‐type animals upon CK1A1 depletion. (D) RNAi of kin‐3 and kin‐10 on alg‐1(S988E) and alg‐1(S998E) animals expressing a seam cell gfp reporter (Pscm::gfp) significantly rescues seam cells hyperplasia post‐L4 as seen in wild‐type animals upon CK2 depletion. Mean and SD are plotted on the graph showing the distribution of seam cell number of wild‐type, alg‐1(S992E), and alg‐1(S995E) animals on kin‐19 RNAi (blue) versus control (black) (n = 45 for each condition). Mean and SD are plotted on the graph showing the distribution of seam cell number of wild‐type, alg‐1(S988E), and alg‐1(S998E) animals on kin‐3 (red), kin‐10 (yellow), or control RNAi (black) (n = 45 for each condition). Wild‐type animals have an invariable 16 seam cells at the adult stage. The P‐values indicated were measured by two‐tailed Student's t‐test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). Representative pictures of seam cell gfp reporter expression (normal and abnormal) are shown for wild‐type animals on kin‐19, kin‐3, kin‐10, or control RNAi. The number in brackets indicates the number of seam cells. The scale bar represents 100 μm.
Source data are available online for this figure.
Figure EV2. CK1A1 depletion in alg‐1(S992E) and alg‐1(S995E) animals and CK2 depletion in alg‐1(S988E) and alg‐1(S998E) animals rescue alae defects (a phenotype associated with reduced miRNA pathway function).

-
A, BRepresentative images with alae structure (normal and defective) are shown for all conditions. The scale bar represents 10 μm.
Figure EV3. CK1A1 depletion in alg‐1(S992E) and alg‐1(S995E) animals and CK2 depletion in alg‐1(S988E) and alg‐1(S998E) animals rescue seam cell defects (a phenotype associated with reduced miRNA pathway function).

-
A, BRepresentative pictures of seam cell gfp reporter expression (normal and abnormal) are shown for all conditions. The number in brackets indicates the number of seam cells. The scale bar represents 100 μm.
Recent detailed characterization of the ALG‐1 S988:S998 phosphorylation cluster revealed that phosphorylation of the phospho‐acceptor residue S992 is essential for the miRNA‐mediated gene silencing process during animal development (Quévillon Huberdeau et al, 2017). Loss of phosphorylation at this residue by mutating the specific serine residue to alanine (S992A, phospho‐lacking mutant) fully abolished the miRNA‐mediated gene silencing process in animals and led to complete lethality at higher temperatures by bursting from the vulva at adult stage. The drastic developmental phenotypes seen in the S992A mutant strongly suggested that the absence of phosphorylation at this residue is deleterious in animals. We hypothesized that if CK1A1 was targeting the S992 and S995 sites, then alg‐1(S992E) and alg‐1(S995E) phospho‐mimicking mutants would rescue the alae and seam cells defects seen in wild‐type animals upon kin‐19 RNAi depletion. Similarly, if CK2 was targeting the S988 and S998 sites, then alg‐1(S988E) and alg‐1(S988E) phospho‐mimicking mutants would rescue the alae and seam cells defects seen in wild‐type animals upon RNAi depletion of kin‐3 and kin‐10. When we performed those experiments, we observed that S992E and S995E animals led to a rescue of both alae defects and abnormal number of seam cells (although not complete) upon depletion of CK1A1 by kin‐19 RNAi knockdown (Figs 2A and C, and EV2A and EV3A). S988E and S998E animals led to a complete rescue of alae defects (Figs 2B and EV2B) and a partial but significant rescue of seam cells (Figs 2D and EV3B) upon depletion of CK2 by RNAi knockdown of kin‐3 and kin‐10.
Together with the defects observed in the seam cells differentiation pattern and the alae structure, our data suggest that both CK1A1 and CK2 are implicated in the developmental regulation controlled by the let‐7 family of miRNAs. These results suggest that CK1A1 and CK2 genetically interact with the let‐7 miRNA pathway and have an important biological function in the process of miRNA‐mediated gene silencing during animal development as evidenced by severe developmental phenotypes upon their depletion. Different phospho‐mimicking mutants of the S988:S998 cluster on ALG‐1 rescues these developmental phenotypes associated with reduced miRNA pathway function upon CK1A1 and CK2 depletion, further suggesting that CK1A1 and CK2 are the kinases phosphorylating the S988:S998 cluster in animals.
KIN‐19, the ortholog of CK1A1 in C. elegans phosphorylates ALG‐1 at the S992 site in vivo
Our in vitro and genetic data suggests that CK1A1 and CK2 are involved in the regulation of the S988:S998 cluster. We next wanted to investigate whether CK1A1 and CK2 phosphorylate ALG‐1 in vivo. We first produced a phospho‐specific antibody detecting the S992 site (Fig 3A) and tested the implication of CK1A1 and CK2 in ALG‐1 (S992) phosphorylation in vivo. No signal in the alg‐1(S992A) animals demonstrated the specificity of the pS992 Ab. We found a significant decrease of phosphorylation at the ALG‐1 (S992) site within the S988:S998 cluster in kin‐19 RNAi animals when compared to control RNAi animals (Fig 3A). However, the RNAi knockdown of kin‐3 and kin‐10 did not lead to any decrease in the ALG‐1 phosphorylation status of the S992 site (Fig 3A). To strengthen the evidence that CK1A1 phosphorylates ALG‐1 at the S992 site in vivo, we next tested whether kin‐19 deletion mutants would also exhibit a decrease in the phosphorylation status of the ALG‐1 S992 site. Since CK1A1 is essential for the development of the animal, kin‐19 (−/−) null mutants are maintained in a genetically balanced background. Consistent with our RNAi experiments, kin‐19 (ok602) homozygous mutant animals showed a strong loss of phosphorylation at the ALG‐1 (S992) site (Fig 3B). Taken altogether, our data strongly indicate that KIN‐19, the ortholog of CK1A1 in worms specifically phosphorylates ALG‐1 at the S992 site in vivo.
Figure 3. The ALG‐1 (S992) site within the cluster is phosphorylated in vivo by KIN‐19, the ortholog of CK1A1 in Caenorhabditis elegans .
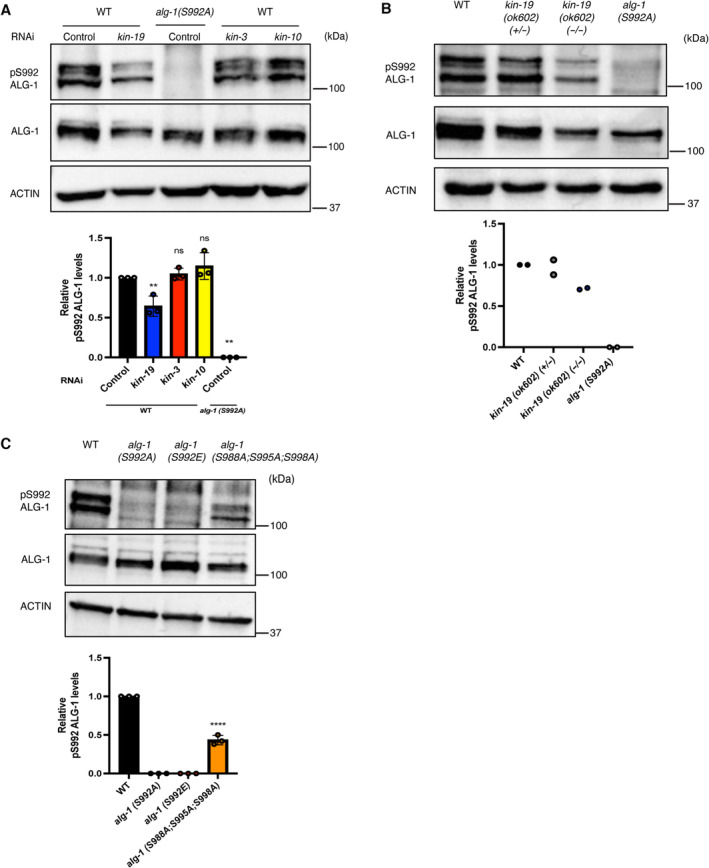
- RNAi of kin‐19 leads to a drastic loss of phosphorylation at ALG‐1(S992) site within the S988:S998 cluster. Representative Western blot of pALG‐1 (S992) and ALG‐1 in wild‐type or phospho‐lacking alg‐1(S992A) young adult worm extracts fed with bacteria expressing RNAi against kin‐19, kin‐3, kin‐10, or control RNAi. Western blot quantification from three biological replicates is shown (n = 3). pALG‐1 level is normalized to ALG‐1 levels. ACTIN is used as a loading control. Data are presented as the mean and the error bars represent the SD. P‐values were calculated with a two‐tailed Student's t‐test; **P < 0.01. A significant decrease (kin‐19 RNAi) and no change (kin‐3 and kin‐10 RNAi) in the phosphorylation status of ALG‐1 at S992 site is observed in wild‐type animals.
- kin‐19 (ok602) homozygous mutant animals show a strong loss of phosphorylation at ALG‐1(S992) site within the S988:998 cluster. Representative Western blot of pALG‐1 (S992) and ALG‐1 in wild‐type, kin‐19 (ok602) homozygotes, kin‐19 (ok602)/+ heterozygotes, and phospho‐lacking alg‐1 (S992A) young adult worm extracts. Western blot quantification from two biological replicates is shown (n = 2). pALG‐1 level is normalized to ALG‐1 levels. ACTIN is used as a loading control.
- The phospho‐specific antibody is specific to the S992 site as it does not recognize the phospho‐lacking alg‐1(S992A) or the phospho‐mimicking alg‐1(S992E) strains. The triple mutant of alg‐1(S988A;S995A;S998A) which still contains the S992 site is still recognized by the antibody. Representative Western blot of pALG‐1 (S992) and ALG‐1 in wild‐type, alg‐1(S992A), alg‐1(S992E), and alg‐1(S988A;S995A;S998A) young adult worm extracts. Western blot quantification from three biological replicates is shown (n = 3). pALG‐1 level is normalized to ALG‐1 levels. ACTIN is used as a loading control. Data are presented as the mean and the error bars represent the SD. P‐values were calculated with a two‐tailed Student's t‐test; ****P < 0.0001.
Source data are available online for this figure.
Our in vitro CK1A1 assays suggest that initial CK1A1 phosphorylation of S992 (S828 in humans) might act as the priming site for robust hierarchical phosphorylation of the cluster. We wanted to test with the phospho‐specific antibody whether S992 is at the top of the phosphorylation cascade in vivo. The phospho‐specific antibody does not recognize the phospho‐mimicking S992E mutation on ALG‐1 (Figs 3C and EV4). We next used CRISPR/Cas9 to generate a triple mutant of alg‐1(S988A;S995A;S998A). This triple mutant only retains the serine at the 992 position and has the other 3 serines mutated to alanines at positions 988, 995, and 998. Intriguingly, the antibody does recognize this triple mutant; however, the signal is significantly reduced compared to wild‐type animals (Figs 3C and EV4). We can envision that S992‐PO4 being at the top of the cascade, the antibody would also recognize the other residues within the cluster. Altogether, these findings are in line with our in vitro data supporting the notion that the S992 site initiates hierarchical phosphorylation of the other residues within the cluster on ALG‐1.
Figure EV4. Specificity of the phospho‐specific antibody to the S992 site within the S988:S998 cluster on ALG‐1.
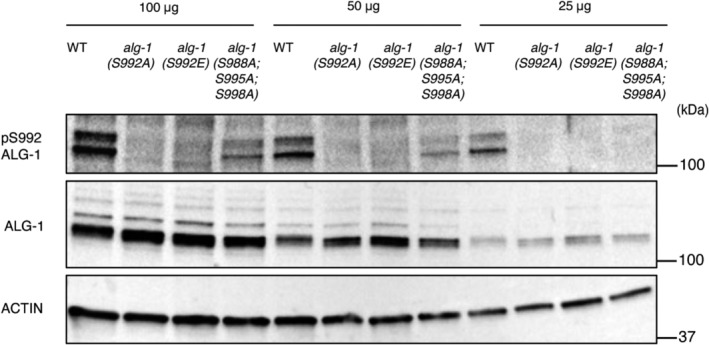
Representative Western blot of pALG‐1 (S992) and ALG‐1 in wild‐type, alg‐1(S992A), alg‐1(S992E), and alg‐1(S988A;S995A;S998A) young adult worm extracts at different protein concentrations of 100, 50, and 25 μg to demonstrate the specificity of the phospho‐specific antibody. ACTIN is used as a loading control.Source data are available online for this figure.
CK1A1 and CK2‐mediated phosphorylation of the S988:S998 cluster on ALG‐1 is important for proper miRNA‐mediated target silencing
To uncover by which mechanism CK1A1‐ and CK2‐mediated phosphorylation of S988:S998 cluster affects ALG‐1 function, we first assessed whether the knockdown of these kinases could affect the level of miRNAs globally as the alteration of ALG‐1 function leads often to a decrease of miRNAs in animals. We performed high‐throughput sequencing of small RNAs in wild‐type animals treated with control RNAi, kin‐19, kin‐3, and kin‐10 RNAi and observed no significant differences in the global levels of mature miRNAs upon CK1A1 and CK2 depletion (Fig 4A–C). The developmental timing phenotypes we observed previously upon CK1A1 and CK2 depletion are mainly associated with reduced miRNA function of the let‐7 family (let‐7, miR‐48, miR‐84, and miR‐241). CK1A1 and CK2 depletion does not change the levels of let‐7 family members as significantly compared to control RNAi (Fig 4A–C). These results indicate that the defective miRNA function caused by the alteration of CK1A1 and CK2‐dependent phosphorylation of the ALG‐1 S988:S998 cluster is not a consequence of a decrease of miRNA levels.
Figure 4. CK1A1 and CK2‐dependent phosphorylation of the S988:S998 cluster on ALG‐1 does not affect the miRNA levels.

-
A–CGlobal miRNA abundance after CK1A1 and CK2 depletion in wild‐type animals. Global miRNA abundance in wild‐type animals after kin‐19 (A), kin‐3 (B), and kin‐10 (C) RNAi compared to control RNAi. Mature miRNA levels are quantified by small RNA sequencing and represented as number of reads mapping to each mature miRNA normalized to total mapped library size in millions (reads per million—RPM). Each point represents the value for a specific miRNA averaged on four biological replicates (n = 4). Members of the let‐7 family are indicated in blue for (A), red for (B), and orange for (C).
Source data are available online for this figure.
Next, we tested if the heterochronic developmental timing defects seen in the wild‐type animals upon CK1A1 and CK2 RNAi depletion could be attributed to defective mRNA target silencing. Lin‐41 is a well‐established target of let‐7 miRNA that plays an important role in regulating the L4‐to‐adult developmental transition in C. elegans (Slack et al, 2000; Vella et al, 2004). During the L4 stage, lin‐41 mRNA levels are repressed by the let‐7 miRNA allowing developmental progression in C. elegans (Reinhart et al, 2000; Slack et al, 2000; Ecsedi et al, 2015; Aeschimann et al, 2019). To assess whether the CK1A1 and CK2‐dependent phosphorylation of the S988:S998 cluster affects miRISC‐mRNA target interaction, we looked at lin‐41 and two other endogenous mRNA targets of the let‐7 miRNA family‐ daf‐12 and hbl‐1 (Abbott et al, 2005; Hammell et al, 2009). We measured the levels of these endogenous mRNA targets in wild‐type animals treated with control RNAi, kin‐19, kin‐3, and kin‐10 RNAi (Fig 5A–C and E–G). In wild‐type animals, depletion of CK1A1 by kin‐19 RNAi leads to significant accumulation of lin‐41, daf‐12, and hbl‐1 mRNA levels (by 1.5‐ to 2‐fold) compared to control RNAi (Fig 5A–C). CK2 depletion by kin‐3 and kin‐10 RNAi in wild‐type animals also resulted in a significant increase of lin‐41 and daf‐12 (by 1.5‐ to 2‐fold) but not hbl‐1 mRNA levels compared to control RNAi (Fig 5E–G).
Figure 5. CK1A1 and CK2‐mediated phosphorylation of the S988:S998 cluster on ALG‐1 is important for proper miRNA‐mediated target silencing.
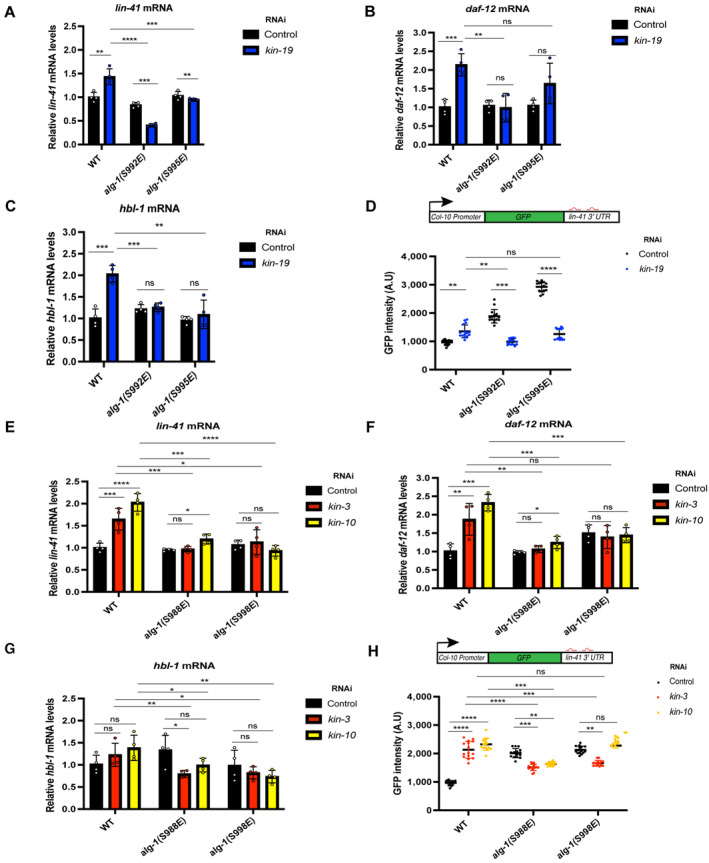
-
A–H(A–C) RNAi of kin‐19 on alg‐1(S992E) and alg‐1(S995E) animals rescues de‐repression of let‐7 targets as observed in wild‐type animals upon CK1A1 depletion. Wild‐type, alg‐1(S992E), and alg‐1(S995E) animals were fed with bacteria expressing RNAi against kin‐19 (blue) or control RNAi (black) for 56 h and collected as young adults. The mRNA levels of three let‐7 miRNA targets lin‐41 (A), daf‐12 (B), and hbl‐1 (C) were measured by RT–qPCR and normalized to the levels of control RNAi. tba‐1 mRNA was used as reference. Data are presented as the mean and the error bars represent the SD from four biological replicates (n = 4), and P‐values were calculated with a two‐tailed Student's t‐test; (not significant [ns] P > 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). (E–G) RNAi of kin‐3 and kin‐10 on alg‐1(S988E) and alg‐1(S998E) animals rescues de‐repression of let‐7 targets as observed in wild‐type animals upon CK2 depletion. Wild‐type, alg‐1(S988E) and alg‐1(S998E) animals were fed with bacteria expressing RNAi against kin‐3 (red), kin‐10 (yellow), or control RNAi (black). The mRNA levels of three let‐7 miRNA targets lin‐41 (E), daf‐12 (F), and hbl‐1 (G) were measured by RT–qPCR and normalized to the levels of control RNAi. tba‐1 mRNA was used as reference. Data are presented as the mean and the error bars represent the SD from four biological replicates (n = 4), and P‐values were calculated with a two‐tailed Student's t‐test; (not significant [ns] P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). (D and H) Functional analysis using a let‐7 miRNA reporter. Animals carrying the GFP reporter transgene under the control of a hypodermis‐specific col‐10 promoter and the lin‐41 3′ UTR containing the let‐7 miRNA binding sites (red: diagram) were crossed into wild‐type, alg‐1(S992E), alg‐1(S995E), alg‐1(S988E), and alg‐1(S998E) animals. Animals were fed with bacteria expressing RNAi against kin‐19 (blue), kin‐3 (red), kin‐10 (yellow), or control RNAi (black) for 48 h and observed at late L4 stage. Quantification of GFP in adult animals was performed by measuring the mean of the GFP detected in four different hypodermal cells for each animal. Data are plotted as the mean and the error bars represent the SD. The P‐values were calculated with a two‐tailed Student's t‐test (not significant [ns] P > 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). Fifteen animals were scored under each condition (n = 15).
Source data are available online for this figure.
To explain the rescue of the developmental defects, we hypothesized that different phospho‐mimicking mutants of the S988:S998 cluster would restore the mRNA target repression back to wild‐type levels upon CK1A1 and CK2 depletion. To test this, we measured the levels of these mRNA targets in S992E, S995E animals upon CK1A1 depletion and in S988E, S998E animals upon CK2 depletion and found significant downregulation in the levels of these mRNA targets compared to wild‐type animals (Fig 5A–C and E–G). Strikingly, when compared to wild‐type animals, S992E mutants have significantly lower lin‐41, daf‐12, and hbl‐1 mRNA levels (Fig 5A–C) and S995E have significantly lower lin‐41 and hbl‐1 mRNA levels (Fig 5A and C) upon CK1A1 depletion. When CK2 is depleted, both S988E and S998E mutants have significantly lower lin‐41, daf‐12, and hbl‐1 mRNA levels when compared to wild‐type animals (Fig 5E–G).
We further evaluated whether the mis‐regulation by let‐7 miRNA due to CK1A1 and CK2 RNAi depletion would also alter the protein levels of LIN‐41. We used transgenic animals expressing a GFP reporter under the control of lin‐41 3′ UTR and measured the repression by let‐7 in L4‐staged animals, as let‐7 is only expressed during late larval stages (Reinhart et al, 2000; Slack et al, 2000). We noticed a significant de‐repression of GFP reporter upon kin‐19, kin‐3, and kin‐10 RNAi at the young‐adult stage in wild‐type animals (Figs 5D and H, and EV5A and B). The depletion of CK1A1 by RNAi knockdown of kin‐19 in S992E and S995E animals re‐established the correct let‐7 repression of a GFP reporter gene under the control of the lin‐41 3′ UTR (Figs 5D and EV5A). Likewise, CK2 depletion by RNAi knockdown of kin‐3 and kin‐10 in S988E and S998E animals re‐established the correct let‐7 repression of the GFP reporter gene under the control of lin‐41 3′ UTR (Figs 5H and EV5B). Taken together, these results indicate that an orchestrated CK1A1 and CK2 phosphorylation activity on the S988:S998 cluster of ALG‐1 is required for proper miRNA‐guided target silencing.
Figure EV5. Functional analysis using a let‐7 miRNA reporter.

-
A, BCK1A1 depletion in alg‐1(S992E) and alg‐1(S995E) animals and CK2 depletion in alg‐1(S988E) and alg‐1(S998E) animals rescue let‐7 miRNA reporter de‐repression. Representative pictures of let‐7 miRNA reporter expression are shown for all conditions. The scale bar represents 20 μm.
CK1A1 primes C‐terminal cluster phosphorylation also in human AGO2
In human AGO2, the phosphorylation cluster is located between S824:S834 and it has also been reported that it is phosphorylated by CK1A1 in vitro with S828 being crucial for efficient phosphorylation of the full cluster (Golden et al, 2017; Bibel et al, 2022). In these assays, however, only recombinant proteins, fragments, or peptides have been studied. To assess the impact of different cluster residues on the phosphorylation levels by CK1A1 also in a more natural AGO2 full‐length context, we performed in vitro phosphorylation assays using single alanine mutants of transiently overexpressed Flag/HA‐tagged AGO2 (FH‐AGO2) (Fig 6A). Since our previous quantifications revealed that about 10–15% of cellular AGO2 is phosphorylated (Quévillon Huberdeau et al, 2017) a substantial amount of unphosphorylated AGO2 should remain as potential substrate for CK1A1. FH‐AGO2 constructs were transfected into HEK 293T cells, immunoprecipitated and in vitro phosphorylated using recombinant CK1A1 and 32P γ‐ATP. Phosphorylation of wild‐type protein was readily detected (Fig 6B and C). A clear reduction of phosphorylation was observed when S828 (corresponding to S992 in C. elegans) was mutated to Alanine (S828A). Substituting S824 or T830 by Alanine did not lead to prominent changes in phosphorylation, whereas S831A or S834A resulted in a decrease of phosphorylation. Our data, therefore, shows that phosphorylation of S828 by CK1A1 is important for full cluster phosphorylation also in the context of full‐length human AGO2.
Figure 6. S828 is important for CK1A1 phosphorylation of the S824:S834 cluster in human AGO2.
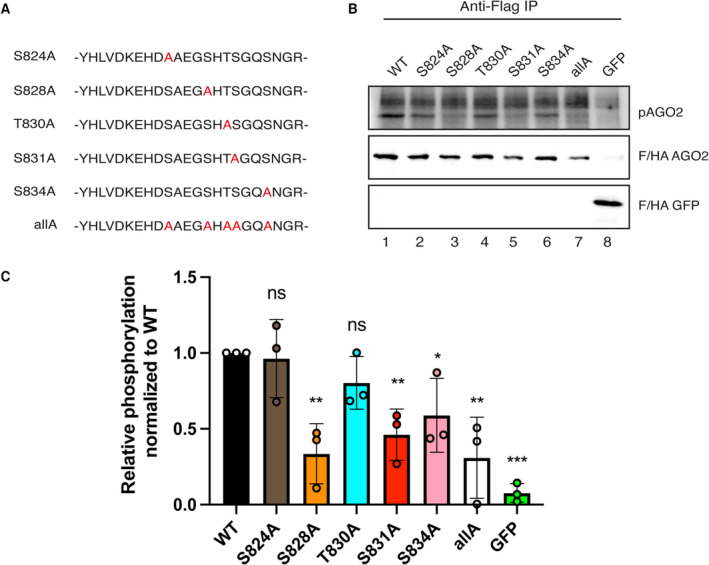
-
A, BIn vitro phosphorylation assay by CK1A1 (A) AGO2 alanine mutants that were used for the in vitro phosphorylation assay. The mutated residues are shown in red. (B) HEK 293T cells were transfected with Flag/HA‐tagged Ago2 WT and mutants. Flag‐tagged proteins were immunoprecipitated, phosphorylated by recombinant CK1A1 and separated on a 10% SDS gel (upper panel). Immunoprecipitated proteins were separated on a 10% SDS gel and analyzed by Western blotting using anti‐HA antibody (middle and lower panel).
-
CQuantification of in vitro phosphorylation assay. Signal of phosphorylated AGO2 was normalized to the AGO2 or EGFP Western blot signal and is shown relative to the WT phosphorylation level. Analysis was performed with three biological replicates (n = 3) and calculated as mean. Error bars represent the SD. P‐values were calculated with a two‐tailed Student's t‐test (not significant [ns] P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001).
Source data are available online for this figure.
Since not all residues are phosphorylated equally by CK1A1 in vitro, we further studied the hierarchy of phosphorylation of residues within the cluster. A panel of FH‐AGO2 mutants (Fig 7A) was transfected into HEK 293T cells, immunoprecipitated, and phosphorylation sites were identified by mass spectrometry. Phosphorylation levels were quantified by relative peptide intensities (Fig 7B). Mutating S824 still allows phosphorylation of all four remaining cluster residues, albeit with reduced efficiency. According to the CK1A1 consensus sequence ([D/E]‐X1‐2‐[S/T]) and ([pS/pT]‐X1‐2‐[S/T]), mutating S828 or S831 abolishes C‐terminal phosphorylation starting from both sites suggesting priming phosphorylation events that trigger downstream phosphorylation by so far unidentified kinases. S828 seems to also influence phosphorylation at S824, as its intensity decreased. Mutating T830 or S834 had no influence on the phosphorylation pattern. Interestingly, only when mutating T830, a peptide carrying four phosphorylated residues was found in our mass spectrometry analyses (Fig 7C). These findings further underline the importance of S828 for an initial phosphorylation event. Moreover, the phosphorylation of some residues within the cluster depends on the prior phosphorylation of other residues.
Figure 7. CK1A1 primes the phosphorylation of the cluster on human AGO2 for other kinases.
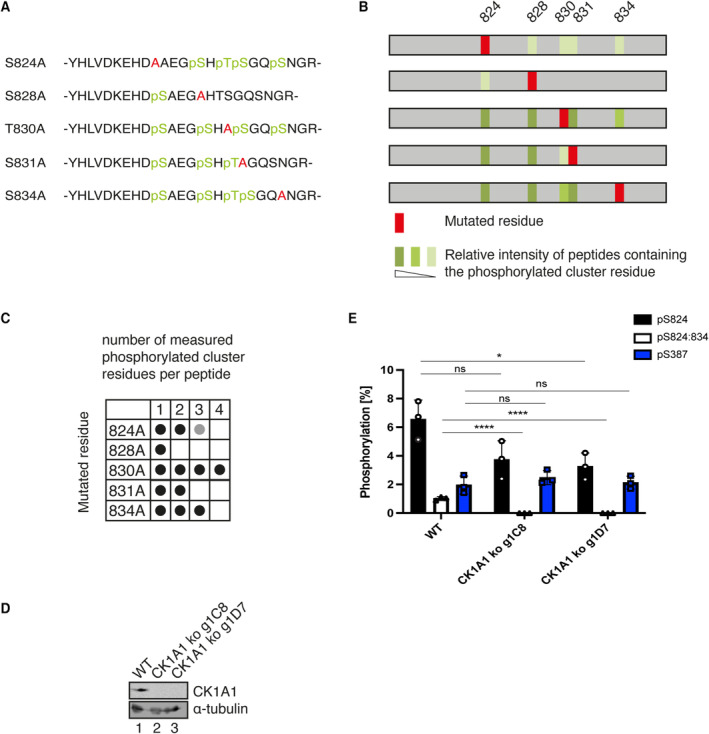
-
AAGO2 single alanine mutants. The mutated residue is shown in red, and the phosphorylated residues are shown in green.
-
BPhosphorylation was quantified by peptide intensities. Percentage of phosphorylation for each residue is shown as the ratio between the intensity of all peptides and the intensity of the phosphorylated peptide. In case of residues that were found phosphorylated in several peptides, the sum of these ratios is shown using the mean of two biological replicates. light green ratio < 2.0, green: ratio < 5, dark green: ratio > 5.
-
COverview of number of phosphorylated residues per peptide for each mutant. Black: in both replicates, Gray: in only one replicate.
-
D, EEndogenous AGO2 was immunoprecipitated from HEK 293T CK1A1 knockout cells and AGO2 cluster phosphorylation was quantified. (D) CK1A1 knockout was confirmed by Western blotting. (E) Targeted quantification of relative abundance for single phosphorylation at S824 and four times phosphorylation at S824, 828, 831 and 834. For quantification via Selected reaction monitoring, stable isotope‐labeled peptides were spiked into the tryptic digest. Percentual phosphorylation was calculated as fraction of the individual phosphopeptide species assuming the sum of all measured singly, multiply, and non‐phosphorylated peptides to be 100%. Analysis was performed with three biological replicates (n = 3) and calculated as mean. Error bars represent the standard deviation. P‐values were calculated with a two‐tailed Student's t‐test (not significant [ns] P > 0.05, *P < 0.05, ****P < 0.0001).
We next asked, which additional kinase could be involved and tested CK2 since it is important for cluster phosphorylation in C. elegans. Consistently, S824 is embedded in a potential consensus sequence of CK2 ([S/T]‐X‐X‐[D/E]) and ([S/T]‐X‐[D/E]). Unfortunately, we could not detect a clear signal with in vitro phosphorylation assays using CK2. To nevertheless confirm the action of additional kinases on this cluster in tissue culture cells, we generated HEK 293T CK1A1 knockout cells (Fig 7D). We assumed that the potential remaining phosphorylation of the cluster is performed by an additional kinase. For targeted quantification, we spiked in a non‐phosphorylated peptide, a peptide with a single phosphorylation at either S824 or S828, and a four‐times phosphorylated peptide (S824, S828, S831, and S834) and performed Selected reaction monitoring (SRM). In this way, we were able to quantify the amount of the respective phosphorylated peptide species as a percentage of the sum of unphosphorylated and phosphorylated peptides (100%) in wild‐type and two different CK1A1 knockout cell lines. Even after CK1A1 knockout, single‐phosphorylated peptides were still detectable, and the mass spectra point toward S824 as the phosphorylated residue (Fig 7E). These results are consistent with our in vitro assays and mutational studies and show that, besides CK1A1, additional kinases contribute to AGO2 cluster phosphorylation in human cells.
Discussion
In this study, we explored the kinases phosphorylating the highly conserved serine/threonine phosphorylation cluster in the C‐terminal PIWI domain of AGO proteins. We report that CK1A1 and CK2 are involved in the phosphorylation of the S988:S998 cluster on the miRNA‐specific ALG‐1 and that CK1A1 acts as a priming kinase for S824:S834 cluster on human AGO2 in cellulo. The cluster was previously shown in vivo to be essential for miRNA‐mediated gene silencing in humans and C. elegans (Quévillon Huberdeau et al, 2017). Functional characterization revealed that although phosphorylation of the S824:S834 cluster on human AGO2 did not affect miRNA loading and AGO localization, hyper‐phosphorylation of the cluster impaired mRNA binding (Quévillon Huberdeau et al, 2017). A mechanistic model involving a phosphorylation/dephosphorylation cycle of the AGO cluster was proposed to promote AGO‐target RNA association and dissociation. However, the identities of the priming kinase, additional secondary kinases, or the phosphatases regulating this cycle particularly in an animal model organism were elusive.
The ANKRD52‐PPP6c phosphatase complex and CK1A1 were reported to regulate the dephosphorylation/phosphorylation cycle of the S824:S834 cluster in human cells (Golden et al, 2017). Experiments with CK1A1 to date were mostly in vitro with pre‐phosphorylate peptides and it was still unclear if CK1A1 contributes to the phosphorylation of this cluster in vivo. Also, in vitro phosphorylation assay with CK1A1 showed that only a pre‐phosphorylated peptide at S828 could promote efficient hierarchical phosphorylation of S831 followed by S834 within the cluster. Since CK1A1 usually prefers pre‐phosphorylated substrates, it is possible that it either primes the S828 site for efficient hierarchical phosphorylation of the cluster by itself, or some other kinases are also involved in regulating the cluster. Here, we demonstrate that in human cells, CK1A1 phosphorylates the S824:S834 cluster on AGO2 and primes the S828 site for phosphorylation by another kinase. Our studies in C. elegans show that CK1A1 phosphorylates ALG‐1 at sites S992 and S995, whereas CK2 phosphorylates ALG‐1 at sites S988 and S998 both in vitro and in vivo.
Casein kinase 2 was previously shown to promote miRISC function in C. elegans by, potentially, phosphorylating the DEAD‐box RNA helicase CGH‐1 in vitro (Alessi et al, 2015). CGH‐1 is a miRISC cofactor which promotes miRISC activity by interacting with the deadenylation and degradation enzymes. Consistent with their findings, we also showed that CK2 is required for the effective functioning of the miRNA pathway as its inactivation led to severe developmental defects that phenocopy the loss of miRISC cofactors such as ALG‐1. Through genetic analyses, they suggest that CK2 phosphorylation of CGH‐1 is important to promote miRISC activity (Alessi et al, 2015). However, the cgh‐1 phosphorylation mutants were multicopy transgenic lines, which can lead to either overexpression or silencing in the germline beyond a critical threshold, causing a mutant phenotype. Also, all their genetic rescue experiments with cgh‐1 phospho‐mimicking mutants were done in a genetically sensitized background and only led to partially significant rescues.
Intriguingly, they proposed that mechanistically, CK2 functions specifically at the step of miRISC association with mRNA targets which is downstream of miRNA biogenesis and its loading onto the miRISC (Alessi et al, 2015). Here, we show that most of the developmental phenotypes caused due to CK2 depletion are due to the decreased phosphorylation status of the S988:S998 cluster on ALG‐1. Our data show that phospho‐mimicking mutants of the S988:S998 cluster on ALG‐1 were sufficient to significantly rescue all the developmental defects exhibited upon CK1A1 and CK2 depletion. Furthermore, we demonstrate that this rescue is due to the mRNA target repression being restored back to wild‐type levels. Since kinases can have multiple different substrates to regulate a variety of processes, it is possible that CK2 is also targeting CGH‐1 in vivo. Our data indicate that the CK2‐dependent phosphorylation of the S988 and S998 sites is the major contributor of the involvement of this kinase in the miRNA pathway.
The biochemical, molecular, and structural events triggering the phosphorylation of the cluster by the kinases to modulate miRISC‐mRNA target interactions are still unclear. A recent study suggested that hierarchical phosphorylation of the cluster by CK1A1 is triggered by target binding to the miRISC (Bibel et al, 2022). The binding of the miRNA‐loaded human AGO2 to target mRNA with complementarity to the seed and 3′ supplementary regions of the miRNA primes the S828 site for hierarchical phosphorylation by CK1A1 (Bibel et al, 2022). They suggested that the need for seed plus supplementary pairing for triggering the CK1A1‐mediated phosphorylation upon target binding supports a role for a conformational change. They argue that target binding with the miRISC would position the RNA molecules in an ideal conformation to interact with the kinase(s) properly and the addition of negative charges onto Argonaute will promote its release from the mRNA target (Bibel et al, 2022). Interestingly, a recent study highlights the idea that Csnk1a1 and Csnk2a1 may hierarchically cooperate in their phosphorylation of AGO2 within the S824:S834 cluster. Expression of these two kinases is suppressed upon oncogenic K‐Ras expression leading to an alteration of global miRNA activity (Shui et al, 2023). In this report, they identified CK2 as the candidate kinase that could potentially target S825 of AGO2 in mouse (corresponding to S988 in C. elegans and S824 in humans) (Shui et al, 2023).
With this study, we propose a model in which CK1A1 acts as the priming kinase and together with additional secondary kinases such as CK2, hyper‐phosphorylate AGO at the S988:S998 (S824:S834) cluster to regulate miRISC‐target mRNA binding and silencing. Target binding would trigger the CK1A1 and CK2‐dependent phosphorylation of the cluster. As proposed by Bibel et al (2022), this could lead to the negatively charged phosphates electrostatically repelling mRNA from AGOs, culminating with the release of mRNA target. After the dephosphorylation of AGOs by a phosphatase complex, it might be recycled and guided to a new target mRNA for another round of binding and disassociation. This way a phosphorylation/dephosphorylation cycle promotes efficient miRNA‐mediated gene silencing by regulating the release of target mRNAs. The phosphatase complex, which would carry out the dephosphorylation event, remains to be explored in C. elegans and other model organisms.
Since the cluster is highly conserved in mice, flies, rats, humans, and worms, a tightly controlled regulatory strategy like a phosphorylation/dephosphorylation cycle could help to maintain consistent and efficient miRNA‐mediated silencing of a wide variety of targets in different cell types. Our approach with this study contributed to revealing how AGO activity in the miRNA pathway is regulated by PTMs such as phosphorylation, the different kinases regulating the process, and helped in defining their precise implication during animal development.
Materials and Methods
C. elegans strains and methods
The C. elegans strains used in this study are listed in Table EV1. All strains were cultured under standard conditions (Brenner, 1974). Strains were grown at 20°C on Nematode Media Growth (NGM) plates and fed with Escherichia coli strain OP50 unless specified.
RNA interference
The kin‐10 RNAi clone was obtained from the Ahringer RNAi library (Fraser et al, 2000). The oligonucleotides used to generate the plasmids as well as the different plasmids for creating the RNAi vector targeting kin‐19 and kin‐3 are listed in Table EV2. RNAi knockdown of kin‐19, kin‐3, and kin‐10 were carried out by feeding. cDNA fragments of kin‐19, kin‐3, and kin‐10 were cloned into the RNAi feeding vector L4440 and transformed in the inducible IPTG HT115 (DE3) bacterial strain as described in Fire et al (1998). Around 200 L1 staged worms in triplicates were plated and grown at 20°C on IPTG Agar plates. Animals were exposed to bacteria expressing gene specific dsRNA or L4440 control plasmid from the L1 stage and scored at either L4 stage (for miRNA activity reporter) or at young adult stage (for alae and seam cells).
Imaging and microscopy
DIC Nomarski images of alae structure, GFP fluorescence of hypodermal let‐7 miRNA reporter and seam cells were collected in animals using a Zeiss AxioCam HRm digital camera mounted on a Zeiss Axio Imager M1 microscope using the same settings and exposition time for each animal. Fluorescence intensity was measured with the Zen software (Zeiss).
Genome editing using CRISPR‐Cas9
Genome editing of C. elegans with CRISPR‐Cas9 methods was carried out as described in Paix et al (2017). Injection mix contained reconstituted Cas9 RNP mix (IDT Cas9 protein [10 μg/μl], tracrRNA [0.4 μg/μl], CRISPR guide RNA [crRNA] [0.4 μg/μl], repair templates with short homology arms [ssODN; 25 μM]), 1 M KCl, 200 mM Hepes pH 7.4. The sequences of all the CRISPR RNAs and the repair templates to generate alg‐1(S992E), alg‐1(S988E), alg‐1(S998E) and alg‐1(S988A;S995A;S998A) animals are listed in Table EV2. Around 20 young‐adult wild‐type (N2) animals were injected, and the edits were determined by PCR genotyping and Sanger sequencing.
In vitro phosphorylation assay
In vitro CK1A1 and CK2 kinase assays were performed using assay conditions adapted from the manufacturer's recommendations (Recombinant CK1A1, SRP5013, Sigma Aldrich and Recombinant CK2, P6010S, NEB). All reactions were performed in a 25 μl volume for 60 min at 30°C. For CK1A1, assay buffer was composed of 25 mM MOPS (pH 7.2), 12.5 mM β‐glycerophosphate, 25 mM MgCl2, 5 mM EGTA, 2 mM EDTA, 0.25 mM DTT, 1.25 mM peptide, 200 ng of recombinant CK1A1, and 200 μM [γ‐32P]ATP [6000 Ci/mmol; Perkin Elmer]. For CK2, assay buffer was composed of 50 mM Tris–HCl (pH 7.5), 10 mM MgCl2, 5 mM EGTA, 0.1 mM EDTA, 2 mM DTT, 0.01% Brij 35, 2 units of recombinant CK2, and 200 μM [γ‐32P]ATP [6000 Ci/mmol; Perkin Elmer]. Reactions were terminated on ice using 7.5 mM Guanidine hydrochloride and the biotinylated peptides were spotted on streptavidin coated membranes. Samples were washed three times in 2 M NaCl followed by four times in 2 M NaCl with 1% H3PO4. Incorporated 32P was measured using a liquid scintillation counter in CPM (counts per minute).
Preparation of protein extracts from adult worms and Western blotting
Synchronized worm populations were obtained by alkaline hypochlorite solution treatment and overnight hatching in M9 buffer and plated onto NGM agar plates seeded with E. coli OP50 bacteria or with bacteria expressing RNAi against kin‐19, kin‐3, kin‐10, or control RNAi. Animals were grown at 20°C until young adult stage and boiled directly in SDS Sample loading buffer (1 mM Tris–HCl [pH 6.8], 2% [w/v] SDS, 100 mM DDT and 10% [v/v] glycerol) for Western blot analysis. Protein extracts were heated at 95°C for 10 min and resolved onto an 8% polyacrylamide gel and transferred to 0.45 μm nitrocellulose blotting membranes (GE Healthcare). Membranes were incubated overnight at 4°C with the primary antibodies‐ rabbit polyclonal anti‐ALG‐1‐pS992 (custom made for this study; Catalog # HAB2014, Thermo Scientific Pierce) diluted at 1:5,000 in TBS supplemented with 1% [w/v] BSA, rabbit polyclonal anti‐ALG‐1 (custom made, Vasquez‐Rifo et al, 2012) diluted at 1:1,000 in PBST supplemented with 1% [w/v] BSA, and mouse anti‐beta‐ACTIN (Catalog #AB49900, Abcam) diluted at 1:10,000 in PBST supplemented with 5% [w/v] dried milk. Membranes were incubated with secondary antibodies against the species of the primary antibody used: Sheep Anti‐Mouse IgG (Catalog #515‐035‐062, Jackson ImmunoResearch Labs) or Goat Anti‐Rabbit IgG HRP (Catalog #111‐035‐144, Jackson ImmunoResearch Labs).
RNA isolation, reverse transcription, and quantitative real‐time PCR
Worms were harvested from RNAi plates using M9 buffer, washed three times, resuspended in 80 μl M9, and frozen at −80°C. Total RNA was extracted using TRI Reagent (Sigma) and resuspended in 25 μl sterile, RNase‐free water. For mRNA analysis, cDNA was synthesized from 800 ng of total RNA using the Multiscribe Reverse Transcriptase Kit (Applied Biosystems) following manufacturer procedures. The lin‐41, daf‐12, hbl‐1, and lin‐14 mRNAs were analyzed by RT‐qPCR using SsoAdvanced Universal SYBR Green Supermix (Bio‐Rad). The relative mRNA levels were normalized based on the ΔΔ2Ct method using tba‐1 as an internal control. The primer sequences for RT‐qPCR are listed in Table EV2.
Small RNA cloning, sequencing, and miRNA analysis
Wild‐type animals were grown on bacteria expressing RNAi against kin‐19, kin‐3, kin‐10, or control RNAi at 20°C for 56 h and collected as young adults. Worms were harvested from RNAi plates using M9 buffer, washed three times, resuspended in 80 μl M9, and frozen at −80°C. Total RNA was extracted using TRI Reagent (Sigma) and resuspended in 25 μl sterile, RNase‐free water. Small RNA cloning was performed with the NEBNext multiplex small RNA library kit and according to the manufacturer's protocol. The small RNA sequencing data are available through the Gene Expression Omnibus (GEO) with the accession number: GSE239453. Hiseq 4000 SR50 sequencing reads were mapped to the genome and cDNA using custom PERL (5.10.1) scripts and Bowtie 0.12.7 (Langmead et al, 2009). Databases used include C. elegans genome (WormBase release WS215), Repbase 15.10 (Jurka et al, 2005), and miRBase 16 (Kozomara & Griffiths‐Jones, 2011). The samples were normalized to the total small RNAs including miRNAs, 22G‐RNAs, and 21U‐RNAs. Mature miRNA abundance is reported as reads per million (RPM) mapped reads (Dataset EV1).
Phosphosite measurements by mass spectrometry
Phosphosite measurements of AGO2 proteins by mass spectrometry were performed as described earlier in Huberdeau et al (2022). For database searching the following search parameters were applied: database Swiss‐Prot Homo sapiens with mutant AGO2 protein sequences added, enzyme specificity trypsin with 2 missed cleavages allowed precursor tolerance 0.02 Da, MS/MS tolerance 0.04 Da, variable modifications: carbamidomethylation and propionamide modification of cysteine, oxidation of methionine, deamidation of asparagine and glutamine, phosphorylation of serine, threonine and tyrosine. Mascot peptide ion‐score cut‐off was set 15. Phosphopeptide fragment spectra were evaluated manually. Calculation of relative abundances of phosphopeptide species at the S824‐S834 phospho‐cluster was based on peptide intensities. The sum of peptide intensities of all peptide species detected at the cluster position was considered as 100%. Peptide intensities of individual phosphorylated or non‐phosphorylated species were then used to determine the relative abundance of each peptide as a percentage. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD043913.
SRM‐quantification of phosphopeptides
Stable isotope‐labeled phosphopeptides as well as their non‐phosphorylated counterparts were used as internal standards for phosphorylation stoichiometry determination of selected phosphopeptides from human AGO2. The following heavy peptides were synthesized as quantified SpikeTides‐TQL (JPT Innovative Peptide solutions) with a 13C15N‐labeled C‐terminal lysine or arginine:
(i) SASFNTDPYVR, (ii) SA pS FNTDPYVR, (iii) YHLVDKEHDSAEGSHTSGQSNGR, (iv) YHLVDKEHD pS AEGSHTSGQSNGR, (v) YHLVDKEHDSAEG pS HTSGQSNGR, (vi) YHLVDKEHD pS AEG pS HT pS GQ pS NGR.
One hundred fmol of each heavy peptide were spiked into the overnight in‐gel tryptic digests which were otherwise processed as described above. To create a SRM (Selected Reaction Monitoring) method, the open source software Skyline (MacCoss Lab Software, Seattle, USA) was used. First, a spectral library was built from several LC–MS/MS discovery runs (DDA, data dependent analysis) on the hybrid triple quadrupole/linear ion trap instrument QTRAP4500 (SCIEX). According to their occurrence in the DDA runs, precursor charge states +2, +3, +4 with either 3 or 4 transitions were included in the targeted method and the resulting transition list was imported into the instrument software (Analyst 1.6.1). In addition, the following parameters were set for the SRM‐method: Q1 and Q3 set to unit resolution (0.7 m/z half‐maximum peak width), dwell time 20 ms, cycle time < 3 s. Second, a scheduled SRM method was created in Skyline by annotating peptide retention times from the initial SRM run and setting the following parameters: cycle time: 2 s, retention time window: 5 min.
The LC–MS/MS system consisted of an UltiMate 3000 RSLCnano System (Thermo Scientific, Dreieich) coupled via a NanoSprayII source (SCIEX) to a QTRAP4500. Peptides were separated on an Acclaim Pepmap100 C18 nano column (75 μm i.d. × 150 mm, Thermo Fisher) with a C18 Acclaim Pepmap100 preconcentration column (100 μm i.d. × 20 mm, Thermo Fisher) in front. At a flow rate of 300 nl/min a 60 min linear gradient of 4–40% acetonitrile in 0.1% formic acid was run. The resulting .wiff files of the SRM‐measurements were imported into Skyline, which facilitated the quantification of endogenous phosphorylated or non‐phosphorylated peptides by calculating the heavy‐to‐light ratios of the peak areas of the respective transitions. Relative quantification of phosphorylated peptides was performed in Excel by first calculating the absolute amount of either peptide species followed by adding up the amounts of the nonmodified peptide species and the related phosphorylated peptide species. Assuming this sum to represent 100%, it was possible to calculate the percentage of the individual phosphopeptide species. Raw data from the SRM measurements have been deposited to PeptideAtlas with the submission number PASS05836.
Plasmids
The vectors for Flag/HA‐tagged human AGO2 as well as Flag/HA‐tagged EGFP (Meister et al, 2004) and the AGO2 824:834A mutant (Quévillon Huberdeau et al, 2017) have been described before. Single alanine mutations of AGO2 were introduced by site‐directed mutagenesis strategy using the primers listed in Table EV3.
Cell culture and transfection
HEK 293T cells were cultivated under standard conditions (37°C, 5% CO2) using Dulbecco's modified Eagle Medium (DMEM, Gibco) supplemented with 10% FBS (Sigma‐Aldrich) and 1% penicillin–streptomycin (Sigma‐Aldrich). For in vitro phosphorylation assays, cells were grown on 15 cm dishes and calcium‐phosphate transfected using 10 μg of plasmid DNA per dish. For transfection of EGFP, 5 μg of DNA were used. For mass spectrometric analysis, cells were grown on 15 cm dishes and transfected using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer's protocol.
Flp‐In TREx293 CK1A1 knockout cell lines were generated by CRISPR/Cas9‐directed genome editing using the CK1A1 complementary sgRNA (5′‐ATGGCGAGTAGCAGCGGCTCC‐3′) to induce a frameshift mutation. The sgRNA sequence, which was inserted into the pX459 vector, was transfected using Lipofectamine 2000 (Thermo Fisher Scientific). Twenty‐four hours after transfection, transfected cells were selected by puromycin (6 μg/ml) for 24 h. After 2 days of recovery, cells were transfected again and selected by puromycin as described above. Cells were separated into single clones. Flp‐In TREx293 CK1A1 knockout clones were analyzed by Western blotting.
For preparation of cell extracts, cells were harvested 48 h after transfection, washed with PBS and lysed in 1 ml NET buffer (50 mM Tris/HCl pH 7.5, 150 mM NaCl, 5 mM EDTA, 0.5% [v/v] NP‐40 alternative, 10% glycerol, 1 mM NaF, 1 mM DTT and 1 mM AEBSF) for 20 min on ice. For mass spectrometric analysis, cells were lysed in NET buffer supplemented with additional 4 mM NaF, PhosSTOP™ phosphatase inhibitor (Roche) and 1 μg/ml RNase A (Thermo Fisher Scientific). Lysates were cleared by centrifugation at 15,000 g for 20 min at 4°C, and input samples were taken before performing immunoprecipitation.
Immunoprecipitation (IP) of Flag‐tagged human AGO2, SDS PAGE and Western blot analysis
For IP of Flag‐tagged proteins, 50 μl packed volume of ANTI‐FLAG® M2 agarose beads (Sigma‐Aldrich) was used. Prior to use, beads were washed twice with cold PBS (1 min, 1,000 g, 4°C). For IP of endogenous AGO2 protein, 100 μl packed volume of protein G beads (GE Healthcare) was coupled over night with anti‐AGO2 11A9 antibody. Before use, the antibody‐coupled beads were washed with PBS. The beads were incubated with lysate for 2.5 h at 4°C while rotating. Afterward, the samples were washed three times with NET wash buffer (50 mM Tris/HCl pH 7.5, 300 mM NaCl, 5 mM EDTA, 0.5% NP‐40 alternative, 10% glycerol, 1 mM NaF, 1 mM DTT, 1 mM AEBSF) followed by one washing step with PBS. For in vitro phosphorylation assays, the samples were split during the last washing step into phosphorylation reaction (75%) and Western blot (25%) samples. After adding 50 μl of 2.5 × Laemmli sample buffer to the samples for Western Blot or Coomassie staining, samples were incubated for 5 min at 95°C. The denatured proteins were separated on a 10% SDS gel and semidry blotted or Coomassie stained. Flag/HA‐tagged AGO2 proteins were detected with anti‐HA antibody (16B12, Covance, 1:1,000) in combination with IRDye® 800CW goat anti‐mouse IgG secondary antibody (Li‐Cor Biosciences, 1:10,000). Signals were detected and quantified with the Odyssey® Imaging System (Li‐Cor Biosciences).
In vitro phosphorylation assay of immunoprecipitated human AGO2
Flag/HA‐tagged proteins were immunoprecipitated as described above. After one additional washing step with phosphorylation buffer (25 mM Mops pH 7.2, 12.5 mM β‐glycerophosphate, 25 mM MgCl2, 5 mM EGTA, 2 mM EDTA, 0.25 mM DTT, 200 μM ATP), 45 μl of the in vitro phosphorylation reaction (25 mM Mops pH 7.2, 12.5 mM β‐glycerophosphate, 25 mM MgCl2, 5 mM EGTA, 2 mM EDTA, 0.25 mM DTT, 200 μM ATP, 100 ng recombinant CK1α (Sigma‐Aldrich), 5 μCi [γ‐32P] ATP) was added to the beads. The reaction was incubated for 1 h at 30°C while shaking with 300 rpm. 5× Laemmli sample buffer was added to a final concentration of 1× and the samples were loaded onto a 10% SDS gel. After separation, the gel was transferred onto a Whatman paper and dried for 2 h at 80°C. Signals were detected by exposure to a screen and scanning with the PMI imaging system (Bio‐Rad).
Author contributions
Vivek Nilesh Shah: Conceptualization; data curation; formal analysis; investigation; methodology; writing – original draft; writing – review and editing. Julia Neumeier: Data curation; formal analysis; investigation; methodology; writing – original draft; writing – review and editing. Miguel Quévillon Huberdeau: Investigation; methodology. Daniela M Zeitler: Investigation. Astrid Bruckmann: Data curation; formal analysis; investigation; methodology. Gunter Meister: Formal analysis; supervision; funding acquisition; methodology; writing – original draft; writing – review and editing. Martin J Simard: Conceptualization; formal analysis; supervision; funding acquisition; methodology; writing – original draft; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Dataset EV1
Source Data for Expanded View
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We would like to thank François Houle and Eduard Hochmuth for technical help as well as members of our laboratory for helpful comments. We would also like to acknowledge, Dr Éric Biron from the Service de Synthèse Peptidique from the CHU de Québec‐Université Laval Research Center for synthesizing all the peptides for the kinase assays. Some nematode strains used in this study were kindly provided by the Caenorhabditis Genetic Center (which is funded by the National Institutes of Health [NIH] Office of Research Infrastructure Programs [P40 OD010440]). The Canadian Institutes of Health Research (CIHR) supported this work (PJT‐173445). VNS is a recipient of scholarship from the Fonds de Recherche du Québec‐Santé (FRQ‐S). GM and AB received funding from the Deutsche Forschungsgemeinschaft (SFB 960).
EMBO reports (2023) 24: e57250
Data availability
The datasets and computer code produced in this study are available in the following databases: (i) microRNA‐Seq data: Gene Expression Omnibus GSE239453 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE239453), (ii) MS proteomics data: PRIDE PXD044139 (https://www.ebi.ac.uk/pride/archive/projects/PXD044139), (iii) SRM‐measurements of phosphopeptides data: PeptideAtlas PASS05836 (http://www.peptideatlas.org/PASS/PASS05836).
References
- Abbott AL, Alvarez‐Saavedra E, Miska EA, Lau NC, Bartel DP, Horvitz HR, Ambros V (2005) The let‐7 MicroRNA family members mir‐48, mir‐84, and mir‐241 function together to regulate developmental timing in Caenorhabditis elegans . Dev Cell 9: 403–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aeschimann F, Neagu A, Rausch M, Großhans H (2019) let‐7 coordinates the transition to adulthood through a single primary and four secondary targets. Life Sci Alliance 2: e201900335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi AF, Khivansara V, Han T, Freeberg MA, Moresco JJ, Tu PG, Montoye E, Yates JR, Karp X, Kim JK (2015) Casein kinase II promotes target silencing by miRISC through direct phosphorylation of the DEAD‐box RNA helicase CGH‐1. Proc Natl Acad Sci USA 112: E7213–E7222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida MV, Andrade‐Navarro MA, Ketting RF (2019) Function and evolution of nematode RNAi pathways. Noncoding RNA 5: 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambros V, Horvitz HR (1984) Heterochronic mutants of the nematode Caenorhabditis elegans . Science 226: 409–416 [DOI] [PubMed] [Google Scholar]
- Banerjee D, Chen X, Lin SY, Slack FJ (2010) kin‐19/casein kinase Iα has dual functions in regulating asymmetric division and terminal differentiation in C. elegans epidermal stem cells. Cell Cycle 9: 4748–4765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP (2018) Metazoan microRNAs. Cell 173: 20–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzini AA, Lee MT, Giraldez AJ (2012) Ribosome profiling shows that miR‐430 reduces translation before causing mRNA decay in zebrafish. Science 336: 233–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibel B, Elkayam E, Silletti S, Komives EA, Joshua‐Tor L (2022) Target binding triggers hierarchical phosphorylation of human Argonaute‐2 to promote target release. Elife 11: e76908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom N, Gammeltoft S, Brunak S (1999) Sequence and structure‐based prediction of eukaryotic protein phosphorylation sites. J Mol Biol 294: 1351–1362 [DOI] [PubMed] [Google Scholar]
- Brenner S (1974) The genetics of Caenorhabditis elegans . Genetics 77: 71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridge KS, Shah KM, Li Y, Foxler DE, Wong SCK, Miller DC, Davidson KM, Foster JG, Rose R, Hodgkinson MR et al (2017) Argonaute utilization for miRNA silencing is determined by phosphorylation‐dependent recruitment of LIM‐domain‐containing proteins. Cell Rep 20: 173–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KC, Svendsen JM, Tucci RM, Montgomery BE, Montgomery TA (2017) ALG‐5 is a miRNA‐associated Argonaute required for proper developmental timing in the Caenorhabditis elegans germline. Nucleic Acids Res 45: 9093–9107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallaire A, Frédérick P‐M, Simard MJ (2018) Somatic and germline microRNAs form distinct silencing complexes to regulate their target mRNAs differently. Dev Cell 47: 239–247 [DOI] [PubMed] [Google Scholar]
- Ding L, Spencer A, Morita K, Han M (2005) The developmental timing regulator AIN‐1 interacts with miRISCs and may target the Argonaute protein ALG‐1 to cytoplasmic P bodies in C. elegans . Mol Cell 19: 437–447 [DOI] [PubMed] [Google Scholar]
- Djuranovic S, Nahvi A, Green R (2012) miRNA‐mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science 336: 237–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecsedi M, Rausch M, Großhans H (2015) The let‐7 microRNA directs vulval development through a single target. Dev Cell 32: 335–344 [DOI] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC (1998) Potent and specific genetic interference by double‐stranded RNA in Caenorhabditis elegans . Nature 391: 806–811 [DOI] [PubMed] [Google Scholar]
- Fraser AG, Kamath RS, Zipperlen P, Martinez‐Campos M, Sohrmann M, Ahringer J (2000) Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature 408: 325–330 [DOI] [PubMed] [Google Scholar]
- Frédérick P‐M, Simard MJ (2022) Regulation and different functions of the animal microRNA‐induced silencing complex. Wiley Interdiscip Rev RNA 13: e1701 [DOI] [PubMed] [Google Scholar]
- Golden RJ, Chen B, Li T, Braun J, Manjunath H, Chen X, Wu J, Schmid V, Chang T‐C, Kopp F et al (2017) An Argonaute phosphorylation cycle promotes microRNA‐mediated silencing. Nature 542: 197–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishok A, Pasquinelli AE, Conte D, Li N, Parrish S, Ha I, Baillie DL, Fire A, Ruvkun G, Mello CC (2001) Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell 106: 23–34 [DOI] [PubMed] [Google Scholar]
- Hammell CM, Karp X, Ambros V (2009) A feedback circuit involving let‐7‐family miRNAs and DAF‐12 integrates environmental signals and developmental timing in Caenorhabditis elegans . Proc Natl Acad Sci USA 106: 18668–18673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horman SR, Janas MM, Litterst C, Wang B, MacRae IJ, Sever MJ, Morrissey DV, Graves P, Luo B, Umesalma S et al (2013) Akt‐mediated phosphorylation of Argonaute 2 downregulates cleavage and upregulates translational repression of MicroRNA targets. Mol Cell 50: 356–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu E, Rubin CS (1990) Casein kinase II from Caenorhabditis elegans. Properties and developmental regulation of the enzyme; cloning and sequence analyses of cDNA and the gene for the catalytic subunit. J Biol Chem 265: 5072–5080 [PubMed] [Google Scholar]
- Huberdeau MQ, Shah VN, Nahar S, Neumeier J, Houle F, Bruckmann A, Gypas F, Nakanishi K, Großhans H, Meister G et al (2022) A specific type of Argonaute phosphorylation regulates binding to microRNAs during C. elegans development. Cell Rep 41: 111822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jannot G, Michaud P, Quévillon Huberdeau M, Morel‐Berryman L, Brackbill JA, Piquet S, McJunkin K, Nakanishi K, Simard MJ (2016) GW182‐free microRNA silencing complex controls post‐transcriptional gene expression during Caenorhabditis elegans embryogenesis. PLoS Genet 12: e1006484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S, Zhang M, Sun J, Yang X (2018) Casein kinase 1α: biological mechanisms and theranostic potential. Cell Commun Signal 16: 23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas S, Izaurralde E (2015) Towards a molecular understanding of microRNA‐mediated gene silencing. Nat Rev Genet 16: 421–433 [DOI] [PubMed] [Google Scholar]
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J (2005) Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110: 462–467 [DOI] [PubMed] [Google Scholar]
- Kennelly PJ, Krebs EG (1991) Consensus sequences as substrate specificity determinants for protein kinases and protein phosphatases. J Biol Chem 266: 15555–15558 [PubMed] [Google Scholar]
- Knippschild U, Krüger M, Richter J, Xu P, García‐Reyes B, Peifer C, Halekotte J, Bakulev V, Bischof J (2014) The CK1 family: contribution to cellular stress response and its role in carcinogenesis. Front Oncol 4: 96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A, Griffiths‐Jones S (2011) miRBase: integrating microRNA annotation and deep‐sequencing data. Nucleic Acids Res 39: D152–D157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AKL, Vyas S, Rood JE, Bhutkar A, Sharp PA, Chang P (2011) Poly(ADP‐ribose) regulates stress responses and microRNA activity in the cytoplasm. Mol Cell 42: 489–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litchfield DW (2003) Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J 369: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie AJ, Hoshino D, Hong NH, Cha DJ, Franklin JL, Coffey RJ, Patton JG, Weaver AM (2016) KRAS‐MEK signaling controls Ago2 sorting into exosomes. Cell Rep 15: 978–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meggio F, Marin O, Pinna LA (1994) Substrate specificity of protein kinase CK2. Cell Mol Biol Res 40: 401–409 [PubMed] [Google Scholar]
- Meister G, Landthaler M, Patkaniowska A, Dorsett Y, Teng G, Tuschl T (2004) Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol Cell 15: 185–197 [DOI] [PubMed] [Google Scholar]
- Nakanishi K (2022) Anatomy of four human Argonaute proteins. Nucleic Acids Res 50: 6618–6638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paix A, Folkmann A, Seydoux G (2017) Precision genome editing using CRISPR‐Cas9 and linear repair templates in C. elegans . Methods 121–122: 86–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi HH, Ongusaha PP, Myllyharju J, Cheng D, Pakkanen O, Shi Y, Lee SW, Peng J, Shi Y (2008) Prolyl 4‐hydroxylation regulates Argonaute 2 stability. Nature 455: 421–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quévillon Huberdeau M, Zeitler DM, Hauptmann J, Bruckmann A, Fressigné L, Danner J, Piquet S, Strieder N, Engelmann JC, Jannot G et al (2017) Phosphorylation of Argonaute proteins affects mRNA binding and is essential for microRNA‐guided gene silencing in vivo . EMBO J 36: 2088–2106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G (2000) The 21‐nucleotide let‐7 RNA regulates developmental timing in Caenorhabditis elegans . Nature 403: 901–906 [DOI] [PubMed] [Google Scholar]
- Rüdel S, Wang Y, Lenobel R, Körner R, Hsiao H‐H, Urlaub H, Patel D, Meister G (2011) Phosphorylation of human Argonaute proteins affects small RNA binding. Nucleic Acids Res 39: 2330–2343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Xia W, Khotskaya YB, Huo L, Nakanishi K, Lim S‐O, Du Y, Wang Y, Chang W‐C, Chen C‐H et al (2013) EGFR modulates microRNA maturation in response to hypoxia through phosphorylation of AGO2. Nature 497: 383–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shui B, Beyett TS, Chen Z, Li X, La Rocca G, Gazlay WM, Eck MJ, Lau KS, Ventura A, Haigis KM (2023) Oncogenic K‐Ras suppresses global miRNA function. Mol Cell 83: 2509–2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack FJ, Basson M, Liu Z, Ambros V, Horvitz HR, Ruvkun G (2000) The lin‐41 RBCC gene acts in the C. elegans heterochronic pathway between the let‐7 regulatory RNA and the LIN‐29 transcription factor. Mol Cell 5: 659–669 [DOI] [PubMed] [Google Scholar]
- St‐Denis N, Gabriel M, Turowec JP, Gloor GB, Li SS‐C, Gingras A‐C, Litchfield DW (2015) Systematic investigation of hierarchical phosphorylation by protein kinase CK2. J Proteomics 118: 49–62 [DOI] [PubMed] [Google Scholar]
- Sulston JE, Horvitz HR (1977) Post‐embryonic cell lineages of the nematode, Caenorhabditis elegans . Dev Biol 56: 110–156 [DOI] [PubMed] [Google Scholar]
- Vasquez‐Rifo A, Jannot G, Armisen J, Labouesse M, Bukhari SIA, Rondeau EL, Miska EA, Simard MJ (2012) Developmental characterization of the microRNA‐specific C. elegans Argonautes alg‐1 and alg‐2. PLoS One 7: e33750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez‐Rifo A, Bossé GD, Rondeau EL, Jannot G, Dallaire A, Simard MJ (2013) A new role for the GARP complex in microRNA‐mediated gene regulation. PLoS Genet 9: e1003961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vella MC, Choi E‐Y, Lin S‐Y, Reinert K, Slack FJ (2004) The C. elegans microRNA let‐7 binds to imperfect let‐7 complementary sites from the lin‐41 3′UTR. Genes Dev 18: 132–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Gupta P, Fairbanks J, Hansen D (2014) Protein kinase CK2 both promotes robust proliferation and inhibits the proliferative fate in the C. elegans germ line. Dev Biol 392: 26–41 [DOI] [PubMed] [Google Scholar]
- Yang M, Haase AD, Huang F‐K, Coulis G, Rivera KD, Dickinson BC, Chang CJ, Pappin DJ, Neubert TA, Hannon GJ et al (2014) Dephosphorylation of tyrosine 393 in Argonaute 2 by protein tyrosine phosphatase 1B regulates gene silencing in oncogenic RAS‐induced senescence. Mol Cell 55: 782–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Sankala H, Zhang X, Graves PR (2008) Phosphorylation of Argonaute 2 at serine‐387 facilitates its localization to processing bodies. Biochem J 413: 429–436 [DOI] [PubMed] [Google Scholar]