

Abstract
ATPase family AAA domain‐containing protein 1 (ATAD1) maintains mitochondrial homeostasis by removing mislocalized tail‐anchored (TA) proteins from the mitochondrial outer membrane (MOM). Hepatitis C virus (HCV) infection induces mitochondrial fragmentation, and viral NS5B protein is a TA protein. Here, we investigate whether ATAD1 plays a role in regulating HCV infection. We find that HCV infection has no effect on ATAD1 expression, but knockout of ATAD1 significantly enhances HCV infection; this enhancement is suppressed by ATAD1 complementation. NS5B partially localizes to mitochondria, dependent on its transmembrane domain (TMD), and induces mitochondrial fragmentation, which is further enhanced by ATAD1 knockout. ATAD1 interacts with NS5B, dependent on its three internal domains (TMD, pore‐loop 1, and pore‐loop 2), and induces the proteasomal degradation of NS5B. In addition, we provide evidence that ATAD1 augments the antiviral function of MAVS upon HCV infection. Taken together, we show that the mitochondrial quality control exerted by ATAD1 can be extended to a novel antiviral function through the extraction of the viral TA‐protein NS5B from the mitochondrial outer membrane.
Keywords: hepatitis C virus infection, mitochondria, protein degradation, protein interaction, tail‐anchored protein
Subject Categories: Microbiology, Virology & Host Pathogen Interaction; Organelles; Post-translational Modifications & Proteolysis
ATAD1 removes the HCV tail‐anchored protein NS5B from mitochondria and mediates its proteasomal degradation, revealing an antiviral function associated with mitochondrial quality control.
Introduction
Hepatitis C virus (HCV) chronically infects 58 million people worldwide, and chronic hepatitis C (CHC) is a major cause of serious liver diseases such as potentially fatal hepatic cirrhosis and hepatocellular carcinoma (HCC). HCV infection accounts for one in four cases of liver cancer (Bartenschlager et al, 2018). According to the World Health Organization, there are approximately 1.5 million new infections and 290,000 HCV‐related deaths annually (WHO, 2022). Although the cure rate of CHC patients has significantly improved with the use of directly acting antiviral agents (DAAs), there is currently no prophylactic vaccine available. Additionally, cured patients may still be at risk for reinfection due to lack of protective immunity (Farci et al, 1992; Page et al, 2009). Moreover, the persistent risk of HCC development post‐treatment, emergence of drug resistance‐associated substitutions (RAS) and new subtypes, as well as limited access to DAA therapy in underdeveloped regions pose significant challenges to achieving the goal of eliminating viral hepatitis as a public health threat by 2030 (Bartenschlager et al, 2018; Shah et al, 2021). Therefore, HCV infection will continue to be a global public health concern.
Hepatitis C virus belongs to the Hepacivirus genus within the Flaviviridae family and possesses a positive‐strand RNA genome of ~9.6 kilobases, consisting of a single open reading frame (ORF) flanked by 5′ untranslated regions (5′UTR) and 3′UTR. The ORF is translated into a polyprotein that is cleaved by cellular and viral proteases, producing three structural proteins (Core, E1, and E2), p7 protein, and six non‐structural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (Moradpour & Penin, 2013). NS5B is an RNA‐dependent RNA polymerase (RdRp) essential for viral RNA replication (Behrens et al, 1996; Kolykhalov et al, 1997; Lohmann et al, 1997), and mutations enhancing its interaction with NS2 facilitate viral assembly (Zheng et al, 2021). The NS5B is featured as a member of the TA protein family (Schmidt‐Mende et al, 2001; Chu et al, 2011), which could target the organelle membrane by a single transmembrane domain (TMD) of hydrophobic amino acids (aa) close to the C‐terminus (Wattenberg & Lithgow, 2001; Horie et al, 2002; Borgese et al, 2003). The TMD at C‐terminus determines the subcellular membrane localization of NS5B (Yamashita et al, 1998; Ferrari et al, 1999; Schmidt‐Mende et al, 2001; Lee et al, 2004; Jonikas et al, 2009).
Mitochondria are highly dynamic organelles that constantly undergo fission (fragmentation), fusion, and mitophagy to maintain mitochondrial quality control (Chan, 2012). Viral infections can induce mitochondrial dysfunction and have been linked to various diseases including tumorigenesis. When the TRC40/GET pathway (transmembrane recognition complex of 40 kDa [TRC40] in mammals and guided entry of tail‐anchored proteins [GET] in yeast) is impaired or partially functional, TA proteins may be mislocalized to the mitochondrial outer membrane (MOM), thereby affecting mitochondrial function (Schuldiner et al, 2008; Jonikas et al, 2009; Favaloro et al, 2010; Aviram & Schuldiner, 2017). ATPase family AAA domain‐containing protein 1 (ATAD1), also known as Thorase, is a member of the AAA ATPase family that transduces energy and regulates synaptic plasticity, learning, and memory (Zhang et al, 2011). Additionally, ATAD1 plays a role in muscle plasticity and function through its interaction with PLAA‐UBXN4 complex (Aweida & Cohen, 2022). Furthermore, ATAD1 has been shown to function as a part of the mitochondrial protein quality control system by extracting the TA proteins that have been mislocalized to mitochondria (Chen et al, 2014; Okreglak & Walter, 2014). The functional domains and structure of ATAD1 are increasingly elucidated in recent years (Wang et al, 2020, 2022). As a TA protein, HCV NS5B has also been found to localize to endoplasmic reticulum (ER), mitochondrial matrix, and mitochondrial outer and inner membrane (Schmidt‐Mende et al, 2001; Ivashkina et al, 2002; Chu et al, 2011); however, the implications of its mitochondrial localization remain unexplored. Previous studies have also shown that HCV infection induces mitochondrial fission, which in turn promotes virus secretion, evasion of innate immunity, and attenuation of apoptosis, thereby contributing to persistent HCV infection (Kim et al, 2013, 2014). Mitochondria‐associated antiviral responses play crucial roles against HCV infection, and several HCV proteins show mitochondrial localization and alter mitochondrial function (Okuda et al, 2002; Brault et al, 2013). HCV is one of only seven viruses, and the only positive‐strand RNA virus known to be oncogenic in humans (Bartenschlager et al, 2018). A better understanding of the interplay between mitochondrial proteins and HCV infection would provide novel insights into the mechanism of mitochondria in antiviral responses and virus‐related oncogenesis. Additionally, we previously found that a 20‐nucleotide (nt) ATAD1 mRNA insertion rescued an HCV recombinant with deletion in the 5′UTR (Li et al, 2011). Therefore, we speculated that ATAD1 may be involved in regulating the HCV life cycle, yet the mechanism remains unknown.
In this study, we have demonstrated that ATAD1 negatively regulated HCV infection by removing the mislocalized viral NS5B protein from the mitochondria and mediated its degradation through proteasome pathway, thus suppressing HCV infection. This study expands our understanding of the functional role of ATAD1 in antiviral responses, beyond its established function in mitochondrial quality control.
Results
ATAD1 suppressed the infection of various HCV genotypes in cultured hepatoma cells
Recently, ATAD1 has emerged as an important player in diverse biological contexts (Wang & Walter, 2020; Dederer & Lemberg, 2021), and its function in removing the mislocalized TA proteins from the mitochondria is increasingly recognized (Chen et al, 2014; Okreglak & Walter, 2014; Dederer et al, 2019; Matsumoto et al, 2019; Wang et al, 2022). The NS5B of HCV is classified as a TA protein, and previous studies have shown its localization primarily in the mitochondrial matrix, outer and inner membrane, as well as the ER (Schmidt‐Mende et al, 2001; Ivashkina et al, 2002; Chu et al, 2011). However, the implications underlying its mitochondrial localization remain unknown. Additionally, we previously discovered that insertion of a 20‐nt ATAD1 mRNA rescued an HCV recombinant with a deletion in the 5′UTR stem‐loop I (Li et al, 2011). These observations suggest that ATAD1 may play a role in regulating HCV infection by interacting with NS5B protein.
To investigate the functional role of ATAD1 potentially involved in the HCV life cycle, we initially examined whether HCV infection disrupts ATAD1 expression. We infected Huh7.5 cells with full‐length infectious HCV clones of genotypes 1a (TNcc clone) (Li et al, 2012b) and 2a (JFH1 and J6cc) (Wakita et al, 2005; Li et al, 2012a), as well as intergenotypic recombinant 5a/2a (SA135′TUR‐NS5A/JFH1) (Li et al, 2014). The results showed that the total ATAD1 levels remained unchanged following infection with HCV genotypes 1a, 2a, and 5a/2a (Fig EV1A–D). We subsequently transfected Huh7.5 cells with a plasmid expressing ATAD1 and observed that the overexpression of ATAD1 specifically inhibited HCV infection (Fig EV1E). Therefore, the overall levels of ATAD1 were not significantly altered by HCV infection. However, these findings did not preclude the possibility that ATAD1 may exert regulatory effects on modulating HCV infection.
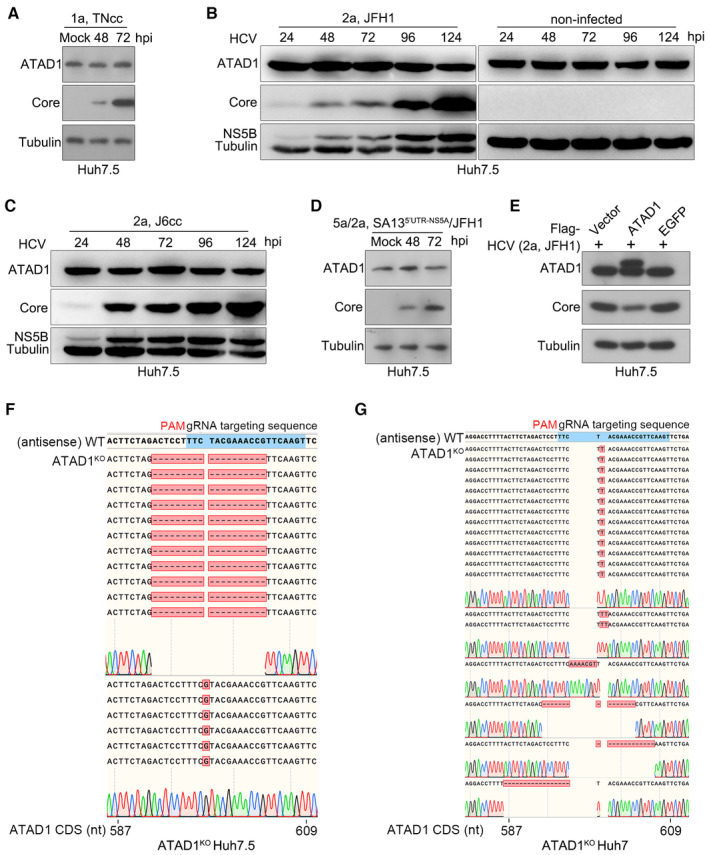
Figure EV1. HCV infection did not affect the expression level of total ATAD1 in Huh7.5 cells.
-
AHuh7.5 cells were infected with HCV genotype 1a TNcc for 48 and 72 h, and total ATAD1 and HCV Core proteins were detected by western blotting.
-
B, CHuh7.5 cells were infected with genotypes 2a clones, JFH1 (B) and J6cc (C), and the cells were harvested at 24, 48, 72, 96, and 124 h post‐infection (hpi). The expression levels of ATAD1, HCV Core, and NS5B were determined by western blotting. The cells without virus infection were treated in parallel as control.
-
DHuh7.5 cells were infected with HCV intergenotypic recombinant (5a/2a; SA135′UTR‐NS5A/JFH1) for 48 and 72 h, then the expression levels of ATAD1 and Core were determined by western blotting.
-
EHuh7.5 cells were transfected with plasmids expressing Flag‐ATAD1, Flag‐EGFP, and Flag‐vector for 24 h, then the cells were infected with HCV 2a JFH1 for 48 h. The cells were harvested and analyzed by western blotting using anti‐Core and anti‐ATAD1 antibodies. In panels (A–E), tubulin was detected as an internal control.
-
F, GSequencing analysis of the genome of ATAD1KO Huh7.5 cells (F) and ATAD1KO Huh7 cells (G). The genomic DNAs of ATAD1KO Huh7.5 and Huh7 cells were extracted, and a region spanning sgATAD1‐RNA‐targeting sequence was amplified by PCR. The PCR products were cloned and subjected to Sanger sequencing analysis. For ATAD1KO Huh7.5 cells (F), 17 clones were sequenced, of which 1‐nt insertion (n = 6) and 19‐nt deletion (n = 11) were identified in the clonal analysis. For ATAD1KO Huh7 cells (G), 19 PCR product clones were sequenced, of which 1‐nt insertion (n = 13), 2‐nt insertion (n = 2), 7‐nt insertion (n = 1), 15‐nt deletion (n = 1), 13‐nt deletion (n = 1), and 17‐nt deletion (n = 1) were identified in the clonal analysis.
To further explore the role of ATAD1 in HCV infection, we generated an ATAD1‐knockout (ATAD1KO) Huh7.5 cell line using CRISPR/Cas9 technology and found that knockout of ATAD1 did not significantly affect the cell growth kinetics (Figs 1A–C and EV1F). However, infection of HCV 2a strain JFH1 was significantly enhanced in ATAD1KO cells compared to both naive wild‐type (WT) Huh7.5 cells (Fig 1D) and CRISPR/Cas9‐knockout negative control Huh7.5 cells (NCKO) (Fig 1E). Knockout of ATAD1 also enhanced the infection of a 2a/2a chimera J6/JFH1‐EGFPΔ40 (referring to the reporter virus 2a(J6)‐EGFPΔ40 in Gottwein et al, 2011) and 1a TNcc at 48 and 72 h post‐infection (hpi) (Fig 1F and G). Additionally, knockout of ATAD1 slightly enhanced ZIKV and DENV infection in Huh7.5 cells (Appendix Fig S1). To verify these observations, we generated ATAD1KO Huh7 cells, which exhibited no impairment in cell growth (Figs 1H and EV1G). Huh7.5 cells are a derivative of Huh7 cells with high susceptibility to HCV replication (Blight et al, 2002). Similarly, the knockout of ATAD1 augmented HCV 2a JFH1 infection in Huh7 cells (Fig 1I and J).
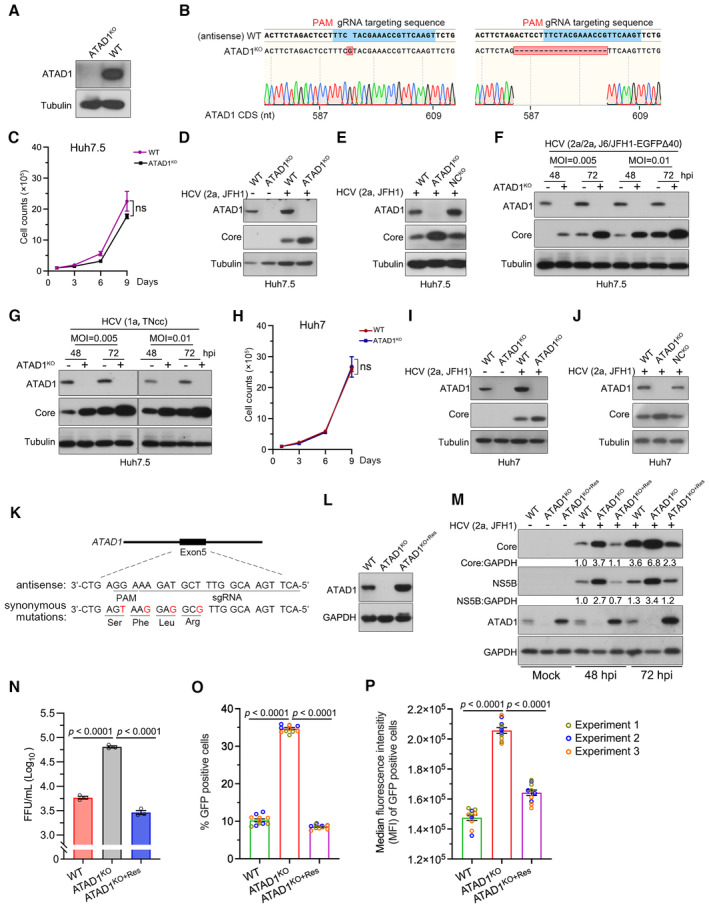
Figure 1. ATAD1 suppressed HCV infection in cultured hepatoma cells.
-
AGeneration of ATAD1KO cells. WT and ATAD1KO Huh7.5 cells were harvested and analyzed by western blotting with an anti‐ATAD1 antibody.
-
BSequencing analysis of ATAD1KO Huh7.5 cells. The genome of ATAD1KO Huh7.5 cells was extracted, and a region spanning sgATAD1 RNA‐targeting sequence was amplified by PCR. The PCR products were cloned for Sanger sequencing analysis. Seventeen clones of PCR products were sequenced, and 1‐nt insertion (n = 6) and 19‐nt deletion (n = 11) were identified in the clonal analysis.
-
CGrowth kinetics of WT and ATAD1KO Huh7.5 cells. The 1 × 105 cells were seeded and the cell counts were determined every 2 days. Data are from three independent experiments (n = 3).
-
DHCV infection in WT and ATAD1KO Huh7.5 cells. The cells were infected with HCV genotype 2a JFH1, and the cell lysates were collected for analysis by western blotting 48 h post‐infection (hpi). “+” and “−” indicate with HCV and without HCV infection, respectively.
-
EHCV infection in WT, ATAD1KO, and NCKO Huh7.5 cells. The cells were infected with JFH1 for 48 h. The NCKO was generated by transduction of lentivirus expressing irrelevant sgRNA‐targeting GFP.
-
F, GInfections of HCV genotypes 1a and 2a. WT and ATAD1KO Huh7.5 cells were infected with J6/JFH1‐EGFPΔ40 (F) and TNcc (G) at different multiplicities of infection (MOI) for 48 and 72 h.
-
HGrowth kinetics of WT and ATAD1KO Huh7 cells. The 1 × 105 cells were plated and the cell number was counted every 2 days (n = 3).
-
I, JHCV infection of WT, ATAD1KO, or NCKO Huh7 cells. The cells were infected with JFH1 for 48 h. In panels (D–G, I, and J), the cells were harvested and subjected to western blotting with anti‐ATAD1 and anti‐Core antibodies.
-
K, LThe synonymous mutations in ATAD1 complementation. The sequence of ATAD1 with four silent mutations (Mut4) was used to generate the ATAD1KO+Res Huh7.5 stable cell line (K), and the complement expression of ATAD1 in the ATAD1KO cells (ATAD1KO+Res cells) (L).
-
MHCV infection in WT, ATAD1KO, and ATAD1KO+Res Huh7.5 cells. The cells were infected with HCV 2a JFH1 (MOI = 0.015) and then the cells were collected at 48 and 72 hpi for analysis. The levels of Core and NS5B were determined by western blotting.
-
NThe infectivity titers of HCV. The FFU of supernatant virus at 48 hpi was determined from the infected culture supernatant shown in Fig EV2A (n = 3).
-
O, PThe percentage of HCV‐infected cells and the intensity of HCV protein in infected cells. WT, ATAD1KO, and ATAD1KO+Res Huh7.5 cells were infected with J6/JFH1‐EGFPΔ40 at same MOI for 72 h. Flow cytometry analysis was performed to determine the percentage of GFP‐positive cells (O) as well as the median fluorescence intensity (MFI) in HCV‐infected cells (P), which reflects the amount of HCV protein present. Three independent experiments are presented in different colors.
Data information: In panels (C, H, and N–P), data are presented as mean ± SEM from three independent experiments. The statistical significance was analyzed using unpaired two‐tailed Student's t‐test in panels (C and H). One‐way ANOVA with Dunnett's post‐hoc test was used in panels (N–P) (compared to ATAD1KO group). ns, not significant (P > 0.05).
Source data are available online for this figure.
Next, we complementally expressed ATAD1 in the ATAD1KO Huh7.5 cells (ATAD1KO+Res cells) (Fig 1K and L) and infected them with HCV 2a JFH1, in comparison with WT and ATAD1KO cells (Fig 1M). The results showed that ATAD1KO+Res cells showed a lower level of HCV infection than ATAD1KO cells at 48 and 72 hpi, as determined by the Core and NS5B levels (Figs 1M and EV2A), as well as the Core‐positive cells in the culture (Fig EV2B). Consistent with these observations, WT and ATAD1KO+Res cells released lower levels of progeny 2a JFH1 supernatant virus compared to ATAD1KO Huh7.5 cells, as determined by either the viral titers using focus‐forming unit (FFU) assay (Fig 1N) or the Core‐positive cells following infection of Huh7.5 cells with progeny virus (Fig EV2C). Moreover, we explored whether ATAD1 knockout led to a reduced number of infected cells or an increased HCV protein in infected cells. We infected WT, ATAD1KO, and ATAD1KO+Res cells with a reporter chimera virus 2a/2a J6/JFH1‐EGFPΔ40 (2a(J6)‐EGFPΔ40 in Gottwein et al, 2011) and quantified the percentage of infected cells as well as the median fluorescence intensity (MFI) of GFP‐positive infected cells (Fig 1O and P). The results showed that knockout of ATAD1 resulted in an increased number of HCV‐infected cells (Fig 1O) and a higher level of HCV in infected cells (Fig 1P).
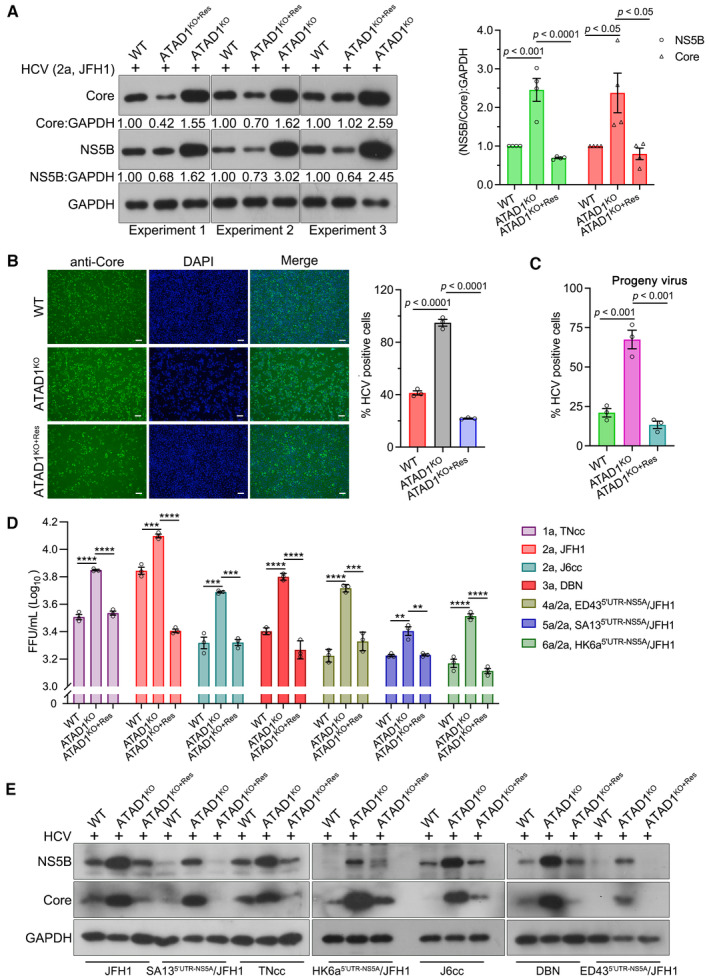
Figure EV2. HCV infection was suppressed in ATAD1KO+Res cells.
-
AWT, ATAD1KO+Res, and ATAD1KO Huh7.5 cells were infected with JFH1 (MOI = 0.015) for 48 h, and the levels of viral Core and NS5B were determined in three independent experiments (left panel). The ratios of NS5B:GAPDH and Core:GAPDH were analyzed by gel band intensities using ImageJ software. The values, together with that of Fig 1M, were normalized to the corresponding GAPDH and shown relative to the signal of WT group (right panel).
-
BWT, ATAD1KO, and ATAD1KO+Res Huh7.5 cells were infected with JFH1 for 72 h, and the cells were immunostained with anti‐Core, followed by goat anti‐mouse IgG (H+L) highly cross‐adsorbed secondary, Alexa Fluor® 488 (green). The nuclei were stained with Hoechst (blue). Typical pictures of HCV infection were captured (left panel), while the percentage of HCV‐positive cells from three independent experiments was shown (right panel). Scale bars, 20 μm.
-
CHuh7.5 cells in 96‐well plates were infected with 100 μl of virus for 48 h, which was collected from the experiment at 48 hpi time point (shown in panel A), and the percentage of HCV‐positive cells was calculated from three independent experiments.
-
D, EWT, ATAD1KO, and ATAD1KO+Res Huh7.5 cells were infected with HCV 1a TNcc, 2a JFH1, 2a J6cc, 3a DBN, 4a/2a ED435′UTR‐NS5A/JFH1, 5a/2a SA135′UTR‐NS5A/JFH1, or 6a/2a HK6a5′UTR‐NS5A/JFH1 for 48 h. The supernatant of three independent experiments was collected for FFU assay (D), and the cells from one of three experiments were collected for western blotting with anti‐NS5B and anti‐Core antibodies (E).
Data information: In panels (A–D), data are presented as mean ± SEM, as described in panel legends. The statistical significance was analyzed using one‐way ANOVA with Dunnett's post‐hoc test (comparing to ATAD1KO group). **P < 0.01, ***P < 0.001, ****P < 0.0001.
Subsequently, we investigated the effects of ATAD1KO and ATAD1KO+Res on the infection of various HCV genotypes, including full‐length infectious HCV clones, 1a TNcc, 2a J6cc, and 3a DBN, as well as chimera viruses 4a/2a ED435′UTR‐NS5A/JFH1, 5a/2a SA135′UTR‐NS5A/JFH1, and 6a/2a HK6a5′UTR‐NS5A/JFH1 (Figs EV2D and 2E). The results showed that knockout of ATAD1 enhanced the infections of tested HCV genotypes, whereas complementation of ATAD1 suppressed the virus infections to levels comparable to those observed in WT Huh7.5 cells, as evidenced by viral titers and intracellular levels of Core and NS5B proteins (Figs EV2D and 2E). Collectively, these results demonstrate that ATAD1 specifically negatively regulated HCV infection in cultured hepatoma cells.
Figure 2. HCV NS5B localized to mitochondria and induced mitochondrial fragmentation in Huh7.5 cells.
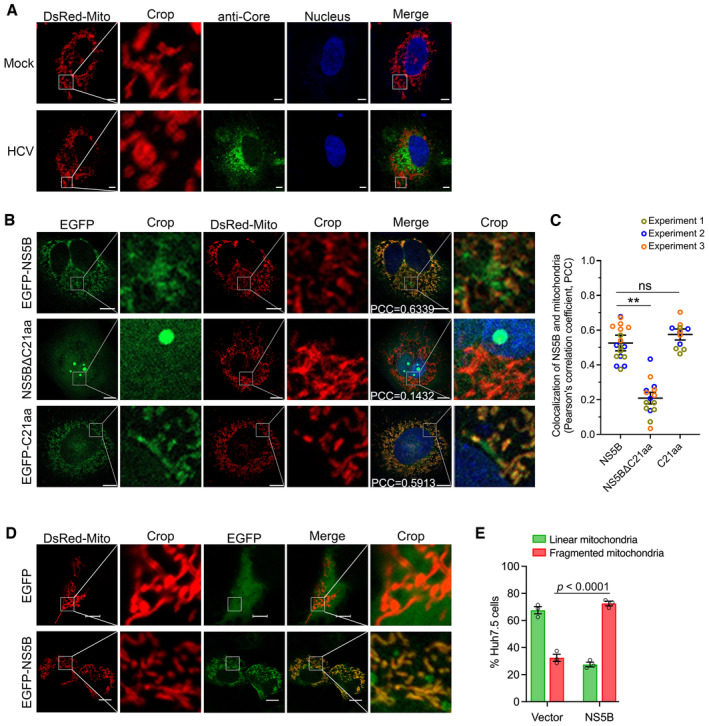
- HCV infection induced mitochondrial fragmentation. Huh7.5 cells were infected with HCV 2a JFH1 for 48 h, followed by transfection with DsRed‐Mito‐expressing plasmid. Twenty‐four hours post‐transfection, the cells were fixed and immunostained for HCV Core (green), using mouse primary antibody anti‐Core for 2 h at room temperature, followed by incubation of goat anti‐mouse‐conjugated IgG (H+L) highly cross‐adsorbed secondary antibody Alexa Fluor® 488 conjugate for 1 h at room temperature. Mitochondria were visualized by DsRed‐Mito (red) and nuclei were stained with Hoechst (blue). The images were captured by Zeiss LSM800, and the representative images are shown. Scale bars, 10 μm.
- HCV NS5B localization to mitochondria. Huh7.5 cells were transfected with EGFP‐NS5B, EGFP‐NS5BΔC21aa, or EGFP‐C21aa, alone with DsRed‐Mito plasmid for 48 h. The cells were fixed and images were taken by confocal microscopy. NS5B and mitochondria were visualized by EGFP (green) and DsRed‐Mito (red), respectively. The images were captured using Zeiss LSM800. Nuclei were stained with Hoechst (blue). The Pearson's correlation coefficient (PCC) between NS5B and mitochondria was indicated in the merged images. Scale bars, 10 μm.
- Co‐localization analysis of NS5B and mitochondria. The Huh7.5 cells were transfected with DsRed‐Mito and various version of NS5B. The images were taken by Zeiss LSM800, and the co‐localization of NS5B and mitochondria was analyzed for PCC. The experiment was conducted independently three times, and a total of 18 cell images from NS5B‐transfected cells, 15 cell images from NS5BΔC21aa‐transfected cells, and 12 cell images from C21aa‐transfected cells were analyzed using Imaris 8.4 software. The statistics analysis was conducted using the mean of the three independent experiments and shown as the mean ± SEM; meanwhile, the datapoints representing the cells derived from each experiment are indicated by different colors. One‐way ANOVA with Dunnett's post‐hoc test was used to analyze the difference (comparing to WT cells transfected with NS5B). **P < 0.01; ns, not significant (P > 0.05).
- NS5B induced mitochondrial fragmentation. Huh7.5 cells were transfected with EGFP‐NS5B or vector EGFP‐C1 plasmid, together with DsRed‐Mito, for 48 h. Then, the cells were fixed and processed for confocal microscopy analysis. NS5B was visualized by EGFP signal (green), and mitochondria were visualized by DsRed‐Mito (red). Scale bars, 10 μm.
- The ratio of Huh7.5 cells with linear and fragmented mitochondria was determined in the Huh7.5 cells following transfection of DsRed‐Mito and vector, or DsRed‐Mito and NS5B. Data are presented as mean ± SEM from three independent experiments. A total of 46 cell images from vector‐transfected cells, and 40 cell images from NS5B‐transfected cells were analyzed. Two‐way ANOVA with Tukey's post‐hoc test was used to analyze the difference.
Source data are available online for this figure.
HCV NS5B was localized to mitochondria and induced mitochondrial fragmentation in Huh7.5 cells
It has been reported that HCV infection induces mitochondrial fragmentation (Chu et al, 2011; Kim et al, 2013, 2014). However, the underlying mechanism has not been explored. Here, we infected Huh7.5 cells with HCV 2a JFH1 and observed mitochondrial fragmentation in HCV‐infected cells (Fig 2A), in consistent with previous findings (Chu et al, 2011; Kim et al, 2013, 2014). HCV NS5B is recognized as a TA protein and has been previously observed to localize to the mitochondrial and ER membrane systems (Schmidt‐Mende et al, 2001). The 21aa at the C‐terminus TMD (C21aa) is required for subcellular membrane localization of NS5B (Yamashita et al, 1998; Schmidt‐Mende et al, 2001; Lee et al, 2004). Thus, we proceeded to explore whether NS5B protein localized to mitochondria and contributed to mitochondrial fragmentation. We transfected Huh7.5 cells with plasmids expressing EGFP‐NS5B, EGFP‐NS5BΔC21aa (deletion of C21aa), and EGFP‐C21aa (fusion of the C21aa and EGFP), along with a plasmid expressing DsRed‐Mito for mitochondrial visualization, and examined their subcellular localization by confocal microscopy (Fig 2B). The results showed that both EGFP‐NS5B and EGFP‐C21aa exhibited a similar level of mitochondrial localization (Pearson's correlation coefficient [PCC], 0.6339 and 0.5913, respectively) (Fig 2B and C). In contrast, EGFP‐NS5BΔC21aa was condensed into globular structures within nucleoli and exhibited a lower degree of co‐localization with mitochondria (PCC = 0.1432) (Fig 2B and C). The NS5B with a deletion of C21aa (NS5BΔC21aa) was previously reported to exhibit a diffuse distribution pattern in both cytoplasm and nucleus (Schmidt‐Mende et al, 2001). These results demonstrate that a portion of NS5B was targeted to mitochondria and the C21aa region determined its subcellular membrane localization.
To investigate whether NS5B contributed to the mitochondrial fragmentation, we transfected Huh7.5 cells with DsRed‐Mito and EGFP‐NS5B, or DsRed‐Mito, and vector plasmid expressing EGFP alone. The results showed a higher percentage of cells underwent mitochondrial fragmentation in NS5B‐expressing cells (27% linear vs. 73% fragmented mitochondria), while fewer cells showed mitochondrial fragmentation when only EGFP was expressed (68% linear vs. 32% fragmented) (Fig 2D and E). Together, HCV NS5B was localized to mitochondria and induced mitochondrial fragmentation.
Knockout of ATAD1 resulted in NS5B accumulation to the mitochondria
ATAD1 functions as mitochondrial protein quality control by extracting TA proteins that are mislocalized to mitochondria, thus maintaining mitochondrial morphology and function (Chen et al, 2014; Okreglak & Walter, 2014). We proceeded to analyze the mitochondrial morphology in WT and ATAD1KO Huh7.5 cells by immunofluorescence confocal microscopy (Fig EV3A) and found that ATAD1KO Huh7.5 cells displayed more fragmented mitochondria (136 cells analyzed; linear vs. fragmented mitochondria, 10 vs. 90%), while WT cells had typically more tubular mitochondria (82 cells analyzed, linear vs. fragmented; 80 vs. 20%) (Fig EV3B). This observation is consistent with the previous observation in mouse embryonic fibroblast (MEFs) cells and Hela cells, in which ATAD1 deficiency induced fragmentation of mitochondria (Chen et al, 2014).
Figure EV3. Knockout of ATAD1 induced mitochondrial fragmentation.
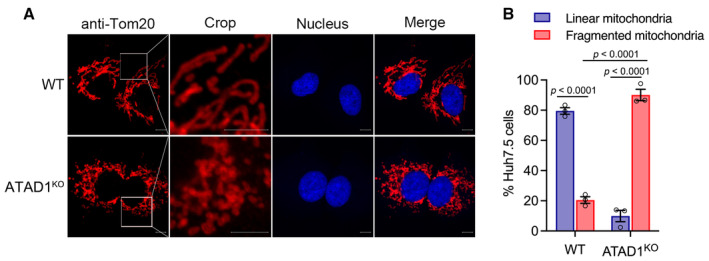
- WT and ATAD1KO cells were seeded in a 12 mm cell‐climbing sheet plated in a six‐well plate for 16 h. The cells were fixed and blotted with rabbit primary antibody anti‐Tom20 for immunostaining of mitochondria (red), followed by goat anti‐rabbit‐conjugated IgG (H+L) highly cross‐adsorbed secondary antibody Alexa Fluor® 555 for 1 h at room temperature. Nuclei were stained using Hoechst (blue). The typical images were captured by Zeiss LSM800. Scale bars, 10 μm.
- The images of a total of 136 ATAD1KO cells and 82 WT cells taken from three experiments were captured by Zeiss LSM800, and mitochondrial morphology was analyzed (fragmented or linear). The percentage of Huh7.5 cells with linear or fragmented mitochondria was calculated and presented. Data are presented as mean ± SEM from three independent experiments. Two‐way ANOVA with Tukey's post‐hoc test was used to analyze the difference.
Next, we examined whether ATAD1 affected NS5B localization to the mitochondria or ER. We transfected WT and ATAD1KO Huh7.5 cells with plasmids expressing EGFP‐NS5B and DsRed‐Mito (Fig 3A). The results revealed that ATAD1KO cells exhibited a higher degree of NS5B localization to mitochondria compared to WT Huh7.5 cells (WT vs. ATAD1KO cells, PCC, 0.6006 vs. 0.8137) (Fig 3A and B). Given that HCV NS5B is also localized to ER membrane system (Schmidt‐Mende et al, 2001), we examined the ER localization of NS5B by immunofluorescence using an anti‐Calnexin antibody. The results indicated that the ER localization of NS5B was comparable between WT (PCC, 0.5010) and ATAD1KO (PCC, 0.5163) Huh7.5 cells (Fig 3A and C). Notably, a fraction of EGFP‐NS5B did not co‐localize with DsRed‐Mito on mitochondria but instead localized to the ER in both WT and ATAD1KO Huh7.5 cells (solid box) (Fig 3A). In line with this observation, the ER isolation experiment demonstrated comparable amount of NS5B associated with the ER in both WT and ATAD1KO Huh7.5 cells (Appendix Fig S2). These findings suggest that NS5B was primarily targeted to the mitochondria and ER membrane, which is consistent with its characteristics as a TA protein. Overall, ATAD1 knockout promoted NS5B localization to the mitochondria without significantly affecting its localization to the ER.
Figure 3. Knockout of ATAD1 promoted NS5B accumulation to mitochondria.
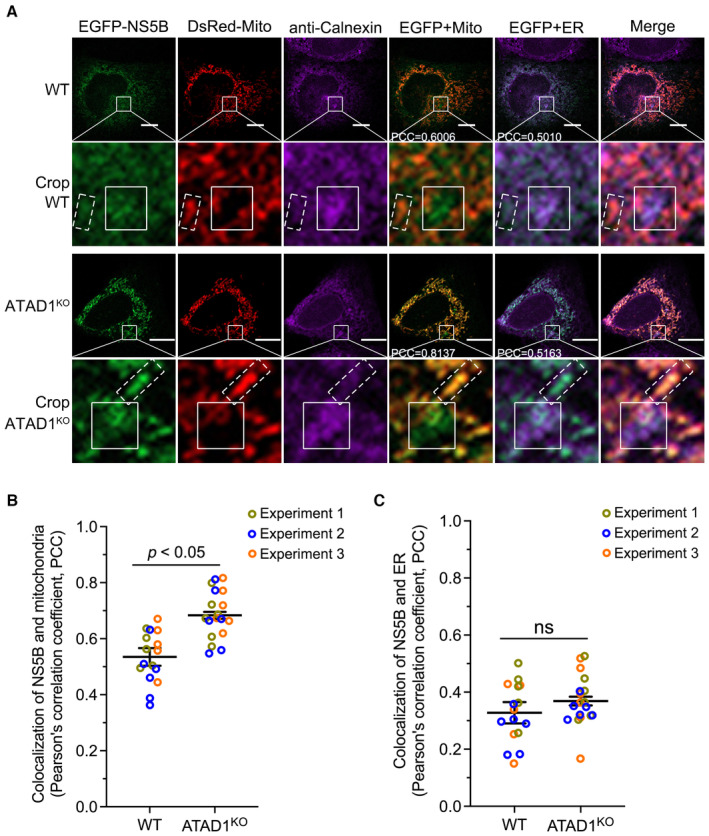
-
AWT and ATAD1KO Huh7.5 cells were co‐transfected with plasmids expressing EGFP‐NS5B and DsRed‐Mito for 48 h. The cells were fixed and labeled with rabbit primary antibody anti‐calnexin for 2 h at room temperature. Goat anti‐rabbit conjugated IgG (H+L) highly cross‐adsorbed secondary antibody Alexa Fluor® 647 conjugate was incubated for 1 h at room temperature for immunostaining of ER (purple). Nuclei were stained with Hoechst (blue). The images were captured using Zeiss LSM800. NS5B and mitochondria were visualized by EGFP (green) and DsRed‐Mito (red), respectively. A typical co‐localization between NS5B and mitochondria (in dashed box) or ER (in solid box) was shown in crop. The PCC between NS5B and mitochondria or ER was shown in the picture using white font. Scale bars, 10 μm.
-
B, CThe images were taken by Zeiss LSM800. The PCCs of NS5B and mitochondria (B) or ER (C) were analyzed by Imaris 8.4 software. The statistics analysis was conducted using the mean of the three independent experiments and shown as the mean ± SEM. A total of 18 cell images for ATAD1KO Huh7.5 and 16 cell images for WT Huh7.5 were analyzed, and the datapoints representing the cells derived from each experiment are indicated by different colors. The statistical significance was analyzed using unpaired two‐tailed Student's t‐test in panels (B and C). ns, not significant (P > 0.05).
Source data are available online for this figure.
ATAD1 interacted with NS5B protein containing transmembrane domain through its substrate‐binding domains
Given that ATAD1 enables to extract the TA proteins that are mislocalized to the mitochondrial membrane (Chen et al, 2014; Okreglak & Walter, 2014), and ATAD1 knockout enhanced NS5B localization to the mitochondria (Fig 3A and B), we speculated that ATAD1 may extract the mislocalized NS5B proteins from the mitochondria. To test this hypothesis, we transfected Huh7.5 cells with plasmids expressing EGFP‐NS5B and either HA‐ATAD1 (HA‐tag at the N‐terminus of ATAD1) or ATAD1‐HA (HA‐tag at the C‐terminus), respectively, and examined their co‐localization (Fig 4A). The results showed that both HA‐ATAD1 and ATAD1‐HA exhibited a similar degree of co‐localization with EGFP‐NS5B (PCCs, 0.5876 and 0.5607, respectively) (Fig 4A and B). Furthermore, the addition of HA‐tag at either the N‐ or C‐terminus did not affect the mitochondrial localization of ATAD1 (PCCs, 0.90 and 0.89, respectively) (Fig 4C and D). Moreover, we demonstrated a direct interaction between ATAD1 and NS5B through Co‐IP assay using 293T cells that co‐transfected with Flag‐NS5B and either HA‐ATAD1 or ATAD1‐HA (Fig 4E). Notably, deletion of C21aa membrane anchor eliminated the interaction between ATAD1 and NS5B (Fig 4E), thus indicating that ATAD1 selectively interacted with NS5B proteins capable of subcellular membrane localization. Additionally, the endogenous viral NS5B in 2a JFH1‐infected Huh7.5 cells also interacted with ATAD1 (Fig 4F), and JFH1 infection did not appear to alter the interaction between ATAD1 and Pex26ΔC30aa, a mitochondria‐targeting TA‐protein (Appendix Fig S4), indicating that HCV infection was unlikely to impact the functional association of ATAD1 with TA proteins. Collectively, these findings suggest that ATAD1 specifically interacted with TMD‐containing NS5B protein capable of membrane localization.
Figure 4. ATAD1 interacted with NS5B protein containing transmembrane domain through its substrate‐binding domains.
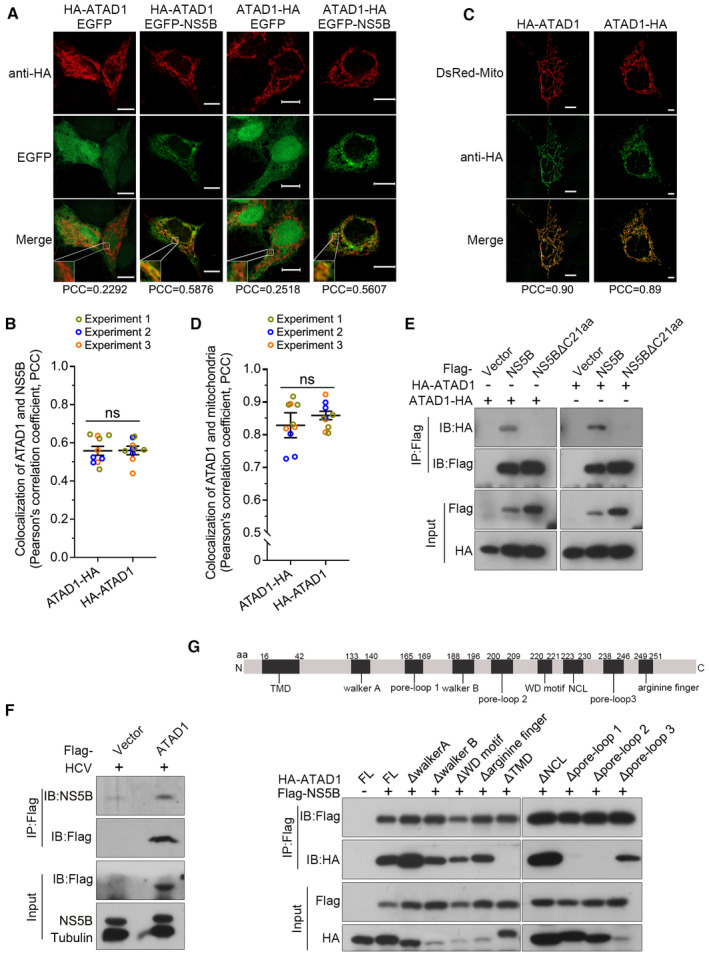
- Co‐localization of ATAD1 and NS5B. Huh7.5 cells were co‐transfected with plasmids expressing EGFP‐NS5B and HA‐ATAD1 (HA‐tag at the N‐terminus), EGFP and HA‐ATAD1, EGFP‐NS5B and ATAD1‐HA (HA‐tag at the C‐terminus), or EGFP and ATAD1‐HA, for 36 h. The cells were fixed and blotted with mouse primary antibody anti‐HA for 2 h at room temperature, followed by goat anti‐mouse‐conjugated IgG (H+L) highly cross‐adsorbed secondary antibody Alexa Fluor® 568 for 1 h at room temperature. The images were captured using Zeiss LSM800. Co‐localization of ATAD1 (red) and NS5B (green) was analyzed by analysis of PCC. Scale bars, 10 μm.
- The analysis of PCC of ATAD1 and NS5B. The PCC of NS5B and HA‐ATAD1 as well as NS5B and ATAD1‐HA were analyzed using Imaris 8.4 software. A total of 10 cell images were captured from three experiments. The statistics analysis was conducted using the mean of the three experiments and the results are presented as mean ± SEM; meanwhile, the datapoints representing the cells from each experiment are indicated by different colors. The statistical significance was analyzed using unpaired two‐tailed Student's t‐test. ns, not significant (P > 0.05).
- Localization of ATAD in mitochondria. Huh7.5 cells were co‐transfected with DsRed‐Mito and HA‐ATAD1, or DsRed‐Mito and ATAD1‐HA, for 36 h. The images were captured using Zeiss LSM800, and co‐localization of ATAD1 (green) and mitochondria (red) was analyzed. Scale bars, 10 μm.
- The PCC analysis of ATAD1 and mitochondria. The cells co‐transfected with DsRed‐Mito and HA‐ATAD1 and with DsRed‐Mito and ATAD1‐HA were analyzed using Imaris 8.4 software. A total of 10 cell images obtained from three experiments. The statistics analysis was conducted using the mean of the three experiments and the results are presented as mean ± SEM; meanwhile, the datapoints representing the cells from each experiment are indicated by different colors. The statistical significance was analyzed using unpaired two‐tailed Student's t‐test. ns, not significant (P > 0.05).
- Co‐IP analysis of ATAD1 and NS5B. 293T cells were co‐transfected with plasmids expressing HA‐ATAD1 and Flag‐NS5B, Flag‐NS5BΔC21aa, or the vector plasmid containing Flag‐tag (Flag‐vector), or expressing ATAD1‐HA and Flag‐NS5B, Flag‐NS5BΔC21aa, or Flag‐vector, for 24 h. The cells were harvested and subjected to IP analysis using an anti‐Flag antibody (1:100), followed by western blotting with anti‐HA and anti‐Flag antibodies.
- ATAD1 interacted with endogenous viral NS5B. Huh7.5 cells were infected JFH1 for 24 h, followed by transfection of Flag‐vector or Flag‐ATAD1. The cells were harvested at 48 hpi and subjected to IP analysis using an anti‐Flag antibody (1:100) and western blotting using anti‐Flag and anti‐NS5B antibodies.
- Interaction of NS5B with ATAD1 deletion mutants. Diagram of nine conserved domains in ATAD1 (upper panel) and Co‐IP analysis for ATAD1 deletion mutants with NS5B (lower panel). 293T cells were transfected with plasmids expressing HA‐ATAD1 or ATAD1 domain deletion mutants, together with plasmids expressing Flag‐NS5B or Flag‐vector for 24 h. The cells were harvested and subjected to Co‐IP analysis using an anti‐Flag antibody (1:100) and western blotting using anti‐HA and anti‐Flag antibodies.
Source data are available online for this figure.
ATAD1 comprises nine conserved elements, namely TMD, walker A, pore‐loop 1, walker B, pore‐loop 2, WD motif, nucleotide communication loop (NCL), pore‐loop 3, and arginine finger (Wohlever et al, 2017; Wang et al, 2020). Thus, we proceeded to investigate the specific region of ATAD1 responsible for its interaction with NS5B (Fig 4G). We transfected 293T cells with the plasmids expressing Flag‐NS5B and either full‐length (FL) or truncated mutants of HA‐ATAD1 and performed Co‐IP assay (Fig 4G). The results indicated that the deletion mutants ΔTMD, Δpore‐loop 1, and Δpore‐loop 2 did not interact with NS5B (Fig 4G), suggesting that the TMD, pore‐loop 1, and pore‐loop 2 domains were responsible for interaction with NS5B while other mutants retain interactions. As deletion mutants may be destabilized, resulting in undetectable interactions, we conducted cycloheximide (CHX) chase assay to examine their stability. The results indicated that the mutants ΔTMD, Δpore‐loop 1, and Δpore‐loop 2 maintained their stability comparable to the FL ATAD1 throughout the experiment, in contrast to the other mutants which became destabilized (Figs EV4A and 4B). In addition, structure prediction based on the parsed ATAD1 (Protein Data Bank: 7UPR) (Wang et al, 2022) and the TMD structure model predicted by Alpha Fold2 (Jumper et al, 2021) also supported the notion that these three mutants were most likely to maintain their stability (Appendix Fig S5). Of these three domains, TMD has been demonstrated to insert into the membrane and form hexamer to capture the substrate, while pore‐loop 1 and pore‐loop 2 have direct contact with the substrate (Fresenius & Wohlever, 2019; Matsumoto et al, 2019), thus corroborating their interactions with NS5B. In summary, we conclude that during HCV infection, ATAD1 interacted with membrane‐localized NS5B through its TMD domain as well as pore‐loops 1 and 2.
Figure EV4. CHX chase analysis of ATAD1 mutant and NS5B.
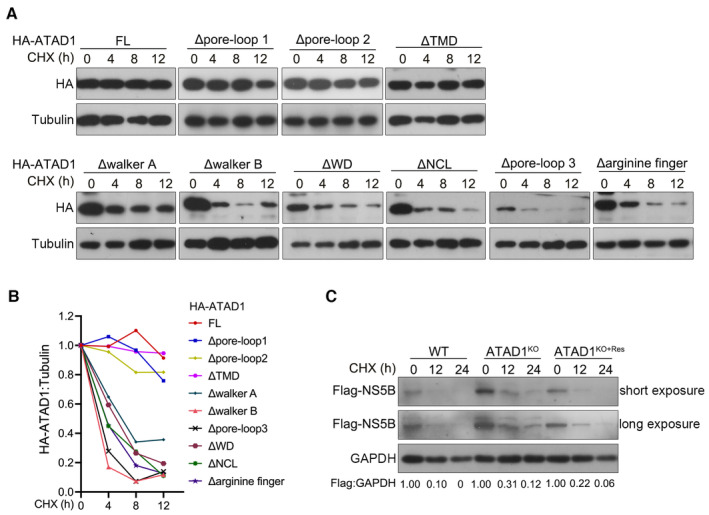
- 293T cells were transfected with HA‐ATAD1 or ATAD1 domain‐deletion mutants for 24 h, and then the cells were treated with CHX (100 μg/ml) for indicated time duration. The cells were harvested and analyzed by western blotting with anti‐HA antibody.
- The ratio of HA:tubulin was analyzed by gel band intensities using ImageJ software. The relative levels of HA‐ATAD1 or ATAD1 domain‐deletion mutant proteins at 0 h were normalized to 1.
- WT, ATAD1KO, and ATAD1KO+Res Huh7.5 cells were transfected with Flag‐NS5B for 24 h, and then the cells were treated with CHX (100 μg/ml) for indicated time. The cells were harvested and analyzed by western blotting with anti‐Flag antibody.
ATAD1 specifically induced degradation of mitochondria‐localized NS5B by proteasome pathway
A major function of ATAD1 is to extract the mislocalized TA proteins from mitochondria and subsequently facilitate their degradation through proteasome pathway (Chen et al, 2014; Okreglak & Walter, 2014; Wohlever et al, 2017). Here, we investigated whether ATAD1 mediated the degradation of NS5B. We co‐transfected 293T cells with plasmids expressing ATAD1‐HA and Flag‐NS5B and observed a gradual reduction in the level of NS5B as ATAD1 expression increased (Fig 5A). However, the level of NS5BΔC21aa remained unchanged (Fig 5A). The reduction in NS5B by ATAD1 occurred at the protein level (Fig 5B, left panel) rather than the mRNA level (Fig 5B, middle panel). The degradation of NS5B by ATAD1 was further validated by CHX chase assay in 293T cells and the WT, ATAD1KO, and ATAD1KO+Res Huh7.5 cells (Figs 5C and EV4C). The results demonstrated that the presence of ATAD1 facilitated the reduction in NS5B protein (Fig 5C and D), and faster degradation of NS5B was observed in WT and ATAD1KO+Res cells, compared to that in ATAD1KO cells (Fig EV4C). Therefore, ATAD1 specifically mediated the degradation of NS5B, which required the membrane anchor domain of NS5B.
Figure 5. ATAD1 specifically induced the degradation of mitochondria‐localized NS5B through proteasome pathway.
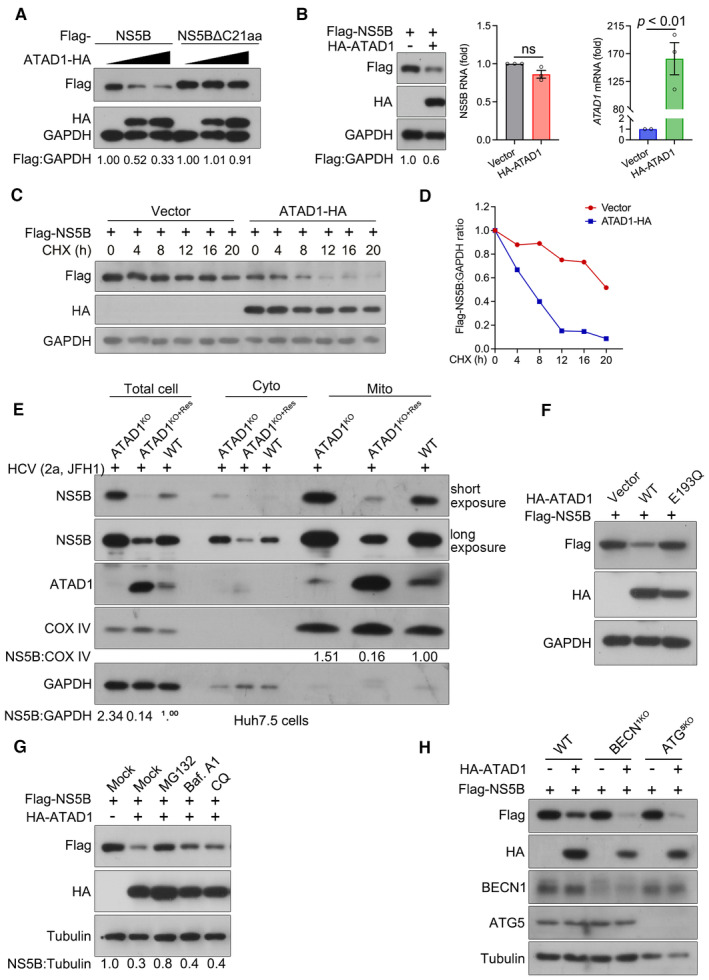
- ATAD1 mediated the degradation of NS5B. 293T cells were transfected with plasmids expressing either Flag‐NS5B or Flag‐NS5BΔC21aa, along with increasing amount of ATAD1‐HA plasmid (0, 500, and 1,500 ng). To ensure equal total plasmid amount in transfection, vector‐HA plasmid was added as a supplement. The cells were harvested at 24 h after transfection and analyzed by western blotting with anti‐Flag and anti‐HA antibodies. It is noteworthy that the mobility of full‐length Flag‐NS5B and Flag‐NS5BΔC21aa on SDS‐PAGE appears to be comparable, and was not affected by the Flag‐tag fused at either N‐ or C‐terminus (Appendix Fig S3). This observation may be attributed to the fact that the alteration in molecular weight upon removal of TMD is associated with changes in hydrophobicity, as transmembrane proteins typically possess a hydrophobic TMD while their extracellular structures are usually hydrophilic. Additionally, deletion of C21aa may have altered its interaction with SDS, resulting in changes in gel mobility.
- ATAD1 reduced the expression of NS5B protein. 293T cells were co‐transfected with Flag‐NS5B and HA‐ATAD1 or HA vector for 24 h. The cells were harvested, and then the protein and mRNA levels of Flag‐NS5B were analyzed by western blotting and qRT–PCR, respectively. The HA‐ATAD1 mRNA was also analyzed. Data are presented as mean ± SEM from three independent experiments. The statistical analysis was determined by unpaired two‐tailed Student's t‐test. ns, not significant (P > 0.05).
- CHX chase assay for NS5B protein. 293T cells were co‐transfected with Flag‐NS5B (500 ng) and ATAD1‐HA (900 ng) or vector‐HA (900 ng) for 24 h, and then the cells were treated with CHX (100 μg/ml) for indicated time duration. The cells were harvested and analyzed by western blotting with anti‐Flag and anti‐HA antibodies.
- The ratio of Flag:GAPDH in panel (C) was analyzed by gel band intensities using ImageJ software. The relative of Flag‐NS5B protein at 0 h was normalized to 1.
- Mitochondrial isolation assay. The same number of WT, ATAD1KO, and ATAD1KO+Res Huh7.5 cells were infected with JFH1 for 48 h. The cells were harvested and isolated for mitochondria using the Cell Mitochondria Isolation Kit. Equal amounts of protein (10 μg) were analyzed by western blotting with anti‐NS5B and anti‐ATAD1 antibodies. COX IV was detected as the mitochondria loading control, and GAPDH was detected as the total cell control.
- Hydrolysis activity of ATAD1 is required for the degradation of NS5B. 293T cells were transfected with plasmids expressing Flag‐NS5B and HA‐vector, Flag‐NS5B and HA‐ATAD1, or Flag‐NS5B and HA‐ATAD1‐E193Q for 24 h. The cells were harvested and lysed for western blotting with anti‐Flag and anti‐HA antibodies.
- Proteasome inhibitor prevents NS5B from degradation. 293T cells were transfected with the plasmids expressing Flag‐NS5B and HA‐ATAD1, or Flag‐NS5B and HA‐vector (mock) for 24 h, and then the cells were treated with inhibitors MG132 (10 μM), bafilomycin A1 (Baf. A1; 0.8 μM), or chloroquine (CQ; 50 μM) for 6 h and harvested for western blotting using anti‐Flag and anti‐HA antibodies.
- Degradation of NS5B independent of autophagic pathway. WT, BECN1KO, and ATG5KO 293T cells were transfected with the plasmids expressing Flag‐NS5B, together with HA‐ATAD1 or HA‐vector for 24 h. The cells were harvested and analyzed by western blotting with antibodies as indicated.
Source data are available online for this figure.
To further confirm the specific targeting of mitochondria‐localized NS5B by ATAD1 for degradation, we isolated mitochondria from cytoplasmic contents after HCV infection of WT, ATAD1KO, and ATAD1KO+Res Huh7.5 cells and quantified the amount of NS5B protein associated with mitochondria (Fig 5E). The results showed that NS5B was substantially enriched in the mitochondrial portion of ATAD1KO cells, while only moderate or lower levels of NS5B were observed in the mitochondria portion of both WT and ATAD1KO+Res cells (Fig 5E). Furthermore, we investigated whether the ATP hydrolysis activity of ATAD1 was required for NS5B degradation and found that the mutant form of ATAD1 (ATAD1‐E193Q) lacking hydrolysis activity failed to induce NS5B degradation (Fig 5F). The E193Q mutation abolished the ATP hydrolysis activity but retained the ATP binding activity, and thus ATAD1‐E193Q has been used as a substrate‐trap mutant (Hanson & Whiteheart, 2005; Chen et al, 2014; Okreglak & Walter, 2014; Wohlever et al, 2017). Taken together, our findings suggest that the degradation of NS5B was contingent upon ATAD1 hydrolysis activity and that ATAD1‐mediated NS5B degradation only occurred in instances where NS5B had mislocalized to mitochondria and directly interacted with ATAD1.
Proteasome and autophagy–lysosome pathways are the two major systems responsible for protein degradation (Pohl & Dikic, 2019). To determine whether ATAD1‐mediated degradation of NS5B occurs via proteasome pathway, as previously reported for other TA proteins that have been mislocalized to mitochondria (Chen et al, 2014; Okreglak & Walter, 2014; Matsumoto et al, 2019), we transfected 293T cells with plasmids expressing NS5B, either alone or with ATAD1, in the presence of proteasome inhibitor MG132 and autophagy inhibitors bafilomycin A1 (Baf. A1) and chloroquine (CQ). The results showed that MG132 effectively inhibited ATAD1‐mediated degradation of NS5B, whereas Baf. A1 and CQ were unable to impede the degradation (Fig 5G). In addition, the degradation of HCV NS5B was not hindered in 293T cells with knockout of either Beclin‐1 (BECN1) or ATG5 (Fig 5H), which are indispensable for autophagy activation (Wesselborg & Stork, 2015; Hao et al, 2022), thus indicating that autophagic degradation did not participate. Therefore, we conclude that ATAD1 promoted the degradation of HCV NS5B through the proteasome pathway.
ATAD1 also augmented the antiviral function of MAVS, independent of its action on NS5B
We have demonstrated that ATAD1 removed the mislocalized NS5B from mitochondria for proteasomal degradation, thus suppressing HCV infection. Given that ATAD1 functions as one of major players for mitochondrial homeostasis, we further investigated whether the knockout of ATAD1 interfered the antiviral effect of mitochondrial antiviral signaling protein (MAVS), which is a critical adaptor protein transducing antiviral signaling primarily through its interaction with activated RIG‐I and MDA5 receptors (Kawai et al, 2005; Li et al, 2005; Seth et al, 2005). Viral infection induces the formation of functional prion‐like aggregates of MAVS (Hou et al, 2011), while HCV evades antiviral immunity by cleaving MAVS through NS3/4A protease (Li et al, 2005). We initially examined whether the knockout of ATAD1 impacted MAVS expression and found that it did not affect the overall level of MAVS (Fig 6A, lanes without HCV infection). However, HCV 2a JFH1 infection resulted in a significant reduction in the total amount of MAVS, accompanied by extensive cleavage form of MAVS (ΔMAVS) (Fig 6A and B, lanes with HCV infection) as well as lower TBK1 activation (Fig 6B). In addition, the extent of MAVS aggregates was lower in ATAD1KO cells, compared to that in WT and ATAD1KO+Res Huh7.5 cells, as visualized by confocal microscopy (Fig 6C). Semi‐denaturing detergent agarose gel electrophoresis (SDD‐AGE) results also demonstrated that ATAD1 knockout impaired the formation of functional prion‐like aggregates following HCV infection (Fig 6D). Therefore, ATAD1 had an impact on the function of MAVS, and its knockout impaired the antiviral effect of MAVS.
Figure 6. ATAD1 also augmented the antiviral function of MAVS, independent of its action on NS5B.
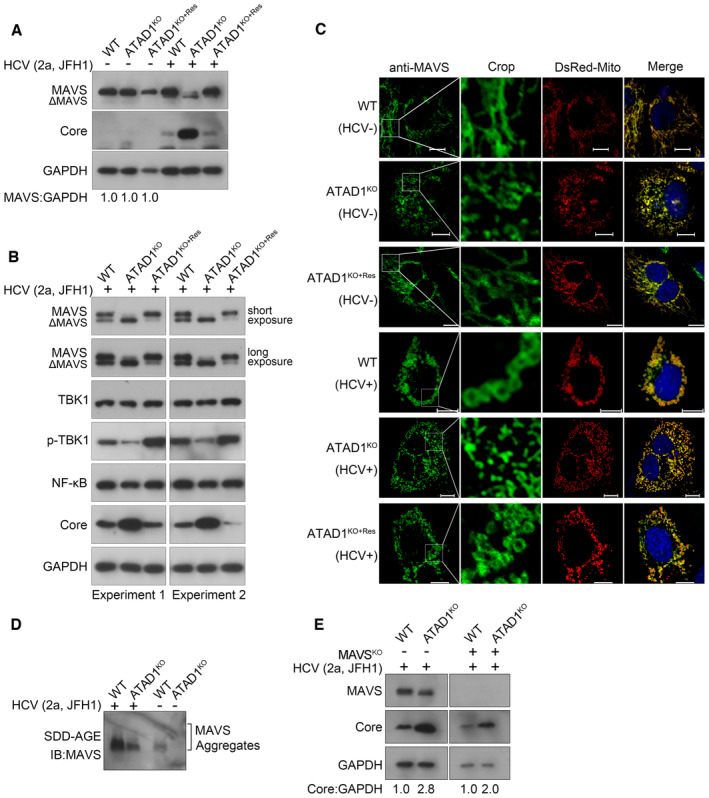
-
A, BExtensive cleavage of MAVS in ATADKO cells upon HCV infection. WT, ATAD1KO, and ATAD1KO+Res Huh7.5 cells were infected with HCV 2a JFH1 for 48 h. The cells were harvested and lysed with the IP lysis buffer containing phosphatase inhibitor cocktails, and analyzed by western blotting with antibodies anti‐MAVS, anti‐Core, anti‐TBK1, anti‐p‐TBK1, etc. Extensive cleavage of MAVS in ATAD1KO Huh7.5 cells was restored in ATAD1KO+Res cells, accompanied by increased phosphorylation level of TBK1.
-
CATAD1 promoted the aggregates of MAVS upon HCV infection. WT, ATAD1KO, and ATAD1KO+Res Huh7.5 cells were infected with J6/JFH1‐EGFPΔ40 for 72 h, followed by sorting the HCV‐positive cells using FACS. Then, HCV‐infected cells and control cells were transfected with DsRed‐Mito. The cells were fixed and blotted with mouse primary antibody anti‐MAVS for 2 h at room temperature, followed by anti‐mouse IgG (H+L), F(ab′)2 fragment (Alexa Fluor® 647 Conjugate) for 1 h at room temperature. The images were captured using Nikon Eclipse Ni‐E. The prion‐like aggregates of MAVS (green) and mitochondria (red) were analyzed. Scale bars, 10 μm.
-
DKnockout of ATAD1 impaired the aggregation of MAVS. WT and ATAD1KO Huh7.5 cells were infected with JFH1 for 72 h. The cells were harvested and analyzed using SDD‐AGE with anti‐MAVS.
-
EATAD1 remained negatively regulating HCV infection without MAVS. WT, ATAD1KO, MAVSKO, and MAVSKO/ATAD1KO Huh7.5 cells were infected with JFH1 for 48 h. The cells were harvested and analyzed by western blotting with anti‐MAVS and anti‐Core antibodies. An increased level of HCV infection was observed in MAVSKO/ATAD1KO cells compared to MAVSKO cells (right panel).
Source data are available online for this figure.
To rule out whether the removal and degradation of HCV NS5B from mitochondria by ATAD1 was independent of the functional association between ATAD1 and MAVS, we generated MAVS knockout cells in both WT and ATAD1KO Huh7.5 cells, designated as MAVSKO and MAVSKO/ATAD1KO Huh7.5 cells, respectively. Following infection with HCV 2a JFH1, we observed that MAVSKO/ATAD1KO cells exhibited a higher level of HCV infection than MAVSKO cells (Fig 6E). This result suggests that ATAD1 remained a negative regulator for HCV infection even in the absence of MAVS. In summary, we have demonstrated that ATAD1 inhibited HCV infection by removing mislocalized NS5B protein from mitochondria for degradation and also augmented the antiviral effect of MAVS. However, further investigation is needed to understand the mechanism underlying the interplay between ATAD1 and MAVS.
Discussion
The novel functional role of ATAD1 protein in regulating virus infection was discovered in this study. By using HCV infection model, we found that the TA protein NS5B of HCV was mislocalized to mitochondria and caused mitochondrial fragmentation in hepatoma cells. ATAD1 executed its quality control function for mitochondria by removing the mitochondria‐localized NS5B for downstream proteasomal degradation, thereby maintaining mitochondrial integrity. Therefore, ATAD1 exerted antiviral activity against HCV infection through at least two mechanisms: facilitating the degradation of HCV RNA polymerase NS5B after its extraction from mitochondria and maintaining mitochondria homeostasis (Fig 7). Furthermore, although the underlying mechanism required further investigation, we have also identified the involvement of ATAD1 in regulating the antiviral function of MAVS (Fig 7).
Figure 7. The working model of ATAD1 in HCV infection.
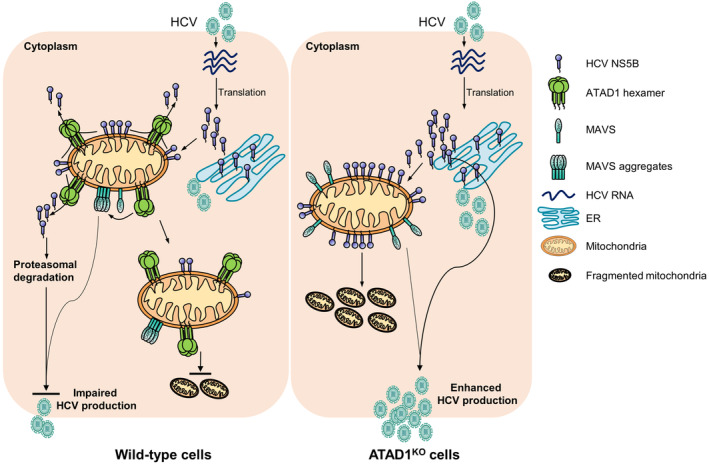
In HCV‐infected cells, the viral TA protein NS5B is partially mislocalized to mitochondria and induces mitochondrial fragmentation, thereby facilitating HCV infection. However, ATAD1, a mitochondrial membrane protein, extracts NS5B and mediates its proteasomal degradation, thus maintaining mitochondrial morphology. In addition to its action on NS5B, ATAD1 also augmented the antiviral function of MAVS.
Human ATAD1 and yeast mitochondrial sorting of proteins 1 (Msp1) share a 50% identity and 70% similarity. Both are located at mitochondria and peroxisome, possessing transmembrane domains and conserved AAA+ domains (Chen et al, 2014; Okreglak & Walter, 2014; Grimm et al, 2016). The mode of TA protein insertion into ER, and peroxisome is mainly through TRC40/GET pathway in mammalian and yeast (Borgese & Fasana, 2011). Our previous study found that a 20‐nt of ATAD1 mRNA was integrated into the HCV recombinant lacking the stem‐loop I structure in the 5′UTR, thereby rescuing the infectivity of this HCV mutant (Li et al, 2011). This initial observation suggests that ATAD1, whether in the form of mRNA or protein, may play a role in the life cycle of HCV. Here, we present evidence that the ATAD1 protein regulated HCV infection by removing viral TA protein NS5B from the mitochondrial outer membrane. Specifically, NS5B was found to be mislocalized to mitochondria and induced mitochondrial fragmentation leading to dysfunction. ATAD1 extracted NS5B from mitochondria and mediated its proteasome degradation, thereby protecting the dynamics of mitochondria. Moreover, the degradation of viral NS5B resulted in inhibition of HCV infection. Therefore, this study has expanded our understanding of the function of ATAD1 beyond its recognized role to encompass antiviral activity.
Mitochondria have strong association with HCV infection, and the latter could alter mitochondrial functions and signaling pathways, leading to significant consequences for viral infection and HCV‐related pathogenesis. These include at least mitochondrial dysfunction as well as mitochondria‐associated antiviral responses (Li et al, 2005; Meylan et al, 2005; Brault et al, 2013). It has been demonstrated that HCV infection induces mitochondrial fission, which facilitates virus secretion, immune evasion, and apoptosis suppression, thereby contributing to persistent HCV infection (Kim et al, 2013, 2014). Several HCV proteins exhibit localization to mitochondria, but the consequences of their effect on mitochondrial functions remain unclear or unexplored. Specifically, viral p7, NS4A, and NS5A have been found to localize to mitochondria and cause dynamic changes in mitochondria (Brault et al, 2013). HCV Core is associated with outer and inner membrane of mitochondria and induced reactive oxygen species (ROS) production and ER stress (Okuda et al, 2002). Partial NS5B was shown to localize to mitochondria in previous studies (Schmidt‐Mende et al, 2001; Chu et al, 2011), yet the mechanisms and consequences remain unknown. In this study, we have experimentally elucidated the implications of mitochondrial localization of NS5B and provided an interplay scenario between mitochondria and HCV. These findings may serve as a catalyst for further exploration into the association between other viral proteins and mitochondria.
Previous studies have shown that ATAD1 physically interacts with mislocalized TA proteins, such as Gos1 and Pex15 proteins (Chen et al, 2014; Okreglak & Walter, 2014; Matsumoto et al, 2019). ATAD1 forms hexametric spirals and extracts substrates through the central pore in mitochondria. The TMD domain facilitates the proper targeting and anchoring of ATAD1 to the MOM, while three pore‐loop structures (pore‐loops 1, 2, and 3) execute the translocation of substrates (Wohlever et al, 2017). Pore‐loop 3 domain is situated more distant from the pore center and does not make direct contact with the substrate (Wang et al, 2020). This mechanism of action for ATAD1 supports our findings that deletion of TMD, pore‐loop 1, and pore‐loop 2 individually resulted in the elimination of interaction between ATAD1 and NS5B (Fig 4). The 21 aa at C‐terminus (C21aa) is a partial of NS5B TMD and was required for the interaction between NS5B and ATAD1 (Fig 4). This suggests that only membrane‐localized NS5B had a higher chance of physically interacting with ATAD1. Furthermore, by separating mitochondrial from cytoplasmic contents, we further demonstrated that ATAD1 and NS5B largely localized to isolated mitochondria, and the degradation of NS5B was determined by the functional state of ATAD1 (Fig 5). In conjunction with other findings, our results provide a novel perspective on the involvement of ATAD1 in regulating the HCV life cycle (Fig 7).
In summary, we have identified a novel mechanism by which the mitochondrial protein ATAD1 negatively regulates HCV infection. This is the first time that ATAD1 has been shown to be involved in antiviral responses. ATAD1 eliminates the viral TA‐protein NS5B proteins that are mislocalized to mitochondria for downstream degradation, while also maintaining a normal mitochondrial function. These actions create an unfavorable cellular environment for virus infection.
Materials and Methods
Cell lines
Human hepatoma Huh7 and Huh7.5 cells were provided by Dr. Charles Rice (Apath, L.L.C and Rockefeller University, New York, USA) and Dr. Jin Zhong (Institute Pasteur of Shanghai, China). HEK293T cells were provided by Dr. Jun Cui (Sun Yat‐sen University, Guangzhou, China). The ATG5‐knockout (ATG5KO) and Beclin‐1 (BECN1)‐knockout (BECN1KO) 293T cells were constructed and used recently in one of our studies (Hao et al, 2022). The cells were cultured in Dulbecco's modified Eagle medium (DMEM; Gibco, 12800‐017, USA) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin (Hyclone, SV30010, USA), and 10% (vol/vol) fetal bovine serum (FBS; Excell, FSP500, China), which refers to complete medium, and incubated at 37°C with 5% CO2.
HCV recombinants
Hepatitis C virus genotype 2a infectious clone JFH1 was provided by Dr. Takaji Wakita (National Institute of Infectious Diseases, Tokyo, Japan) and Dr. Charles Rice (Apath, L.L.C and Rockefeller University, New York, USA) (Lindenbach et al, 2005; Wakita et al, 2005). HCV infectious clones of other genotypes were provided by Dr. Jens Bukh (CO‐HEP, Copenhagen University Hospital and University of Copenhagen, Denmark), including 1a (TNcc) (Li et al, 2012b) and 2a (J6cc) (Li et al, 2012a) developed in our previous studies, 3a (DBN) (Ramirez et al, 2016), together with intergenotypic recombinants 4a/2a (ED435′UTR‐NS5A/JFH1), 5a/2a (SA135′UTR‐NS5A/JFH1), and 6a/2a (HK6a5′UTR‐NS5A/JFH1) (Li et al, 2014), as well as a 2a reporter chimera J6/JFH1‐EGFPΔ40 (referring to 2a(J6)‐EGFPΔ40 in Gottwein et al, 2011). The J6/JFH1‐EGFPΔ40 is a J6‐JFH1 chimera expressing J6Core‐NS2 region and an EGFP fused in the NS5A protein, with a deletion of 40aa in the latter (Gottwein et al, 2011). All HCV full‐length clones and chimera recombinants were generated by transfection of Huh7.5 cells with in vitro‐transcribed RNA transcripts and passaged the supernatant virus to naive Huh7.5 cells to make virus stock for future use, as previously described (Jensen et al, 2008; Gottwein et al, 2011; Li et al, 2011, 2012a, 2012b, 2021; Chen et al, 2018; Duan et al, 2018).
Antibodies and reagents
The primary antibodies used in this study were as the followings: anti‐HCV Core C7‐50 mouse antibody (Santa Cruz Biotechnology, sc‐57800, USA), anti‐ATAD1 mouse antibody (Abcam, ab94583, UK), anti‐NS5B rabbit antibody (GeneTex, GTX131273, USA), anti‐HA mouse antibody (MBL, M130‐8, Japan), anti‐HA rabbit antibody (Cell Signaling Technology, C29F4, USA), anti‐Flag tag (MA4) mouse antibody (Ray Antibody Biotech, RM1002, China), anti‐Tom20 rabbit antibody (Proteintech, 11802‐1‐AP, China), anti‐COX IV rabbit antibody (Cell Signaling Technology, 3E11), anti‐MAVS mouse antibody (Santa Cruz Biotechnology, sc‐166583), anti‐Calnexin rabbit antibody (Proteintech, 66903‐1‐Ig), anti‐ATG5 rabbit antibody (Proteintech, 10181‐2‐AP), anti‐BECN1 rabbit antibody (Proteintech, 11306‐1‐AP), anti‐GAPDH antibody (Proteintech, HRP‐60004), and anti‐Alpha Tubulin antibody (Proteintech, HRP‐66031). The secondary antibodies used for western blotting analysis were goat anti‐mouse IgG (H+L)‐HRP (Ray Antibody Biotech, RM3001, China) and goat anti‐rabbit IgG (H+L)‐HRP (Ray Antibody Biotech, RM3002). The secondary antibodies used for immunofluorescence were goat anti‐mouse IgG (H+L) highly cross‐adsorbed secondary, Alexa Fluor® 488 (Thermo Fisher Scientific, A‐11029, USA), anti‐mouse IgG (H+L), F(ab′)2 fragment (Alexa Fluor® 647 Conjugate) (Cell Signaling Technology, 4410S), goat anti‐mouse IgG (H+L) highly cross‐adsorbed secondary, Alexa Fluor® 568 (Thermo Fisher Scientific, A‐11031), goat anti‐rabbit IgG (H+L) highly cross‐adsorbed secondary, Alexa Fluor® 555 (Thermo Fisher Scientific, A‐21429), and goat anti‐rabbit IgG (H+L) highly cross‐adsorbed secondary, Alexa Fluor® 647 (Thermo Fisher Scientific, A‐21245).
The chemical reagents proteasome inhibitor MG132 (MedChemExpress, HY‐13259, USA), autolysosome inhibitors bafilomycin A1 (Baf. A1; Selleck Chemicals, S1413, USA) and chloroquine phosphate (CQ; Sigma, PHR1258, USA), cycloheximide (CHX, ACMEC, A49960‐1 g, China), and phosphatase inhibitor cocktails (Roche, 4906845001, Switzerland) were purchased. Protein A+G Sepharose beads (7Sea Biotech, P001‐3, China) were used in co‐immunoprecipitation (Co‐IP) assay. Certified Megabase Agarose (Bio‐Rad, 1613109, USA), bromophenol blue (Biosharp, BS952‐5 g, China), and 10 × TBE buffer (Solarbio, T1051‐500 ml, China) were used in semi‐denaturing detergent agarose gel electrophoresis (SDD‐AGE) assay. Minute™ ER enrichment Kit (Invent Biotechnologies, ER‐036, USA) was used for ER isolation.
Construction of plasmids
To construct plasmid expressing ATAD1, total RNA was extracted from Huh7.5 cells and the cDNA was synthesized using HiScript II Q Select RT SuperMix. The ATAD1 coding sequence (CDS) was amplified by PCR with specific primers (ATAD1‐CDS‐F and ATAD1‐CDS‐R, Table EV1). The CDS was used as a template for a second round of PCR, wherein the desired peptide tags were introduced to facilitate western blotting analysis. The forward primer contained BamHI site and HA‐ or Flag‐tag sequences, and reverse primer contained XbaI site for cloning of PCR products into pcDNA3.1 vector to generate pcDNA3.1‐HA‐ATAD1 and pcDNA3.1‐Flag‐ATAD1 plasmids. Other plasmids pcDNA3.1‐ATAD1‐HA, pCDH‐CMV‐MCS‐EF1‐Puro‐ATAD1, or ATAD1 mutants with deletion of each domain or substitution E193Q were constructed based on the pcDNA3.1‐HA‐ATAD1 backbone. The NS5B sequence of HCV genotype 2a clone, J6cc (Li et al, 2012a), was amplified by PCR (Table EV1) and cloned into pEGFP‐C1 (Clonetech Laboratories, Japan) to obtain pEGFP‐NS5B, as well as into pcDNA3.1 to generate pcDNA3.1‐Flag‐NS5B. Other plasmids pEGFP‐NS5BΔC21aa and pEGFP‐C21aa were generated using pEGFP‐NS5B as a backbone plasmid, and pcDNA3.1‐Flag‐NS5BΔC21aa was generated using pcDNA3.1‐Flag‐NS5B as a backbone plasmid.
Quantitative real‐time PCR
Total RNA was extracted from 293T cells that were co‐transfected with NS5B and vector plasmid control, or plasmids expressing ATAD1, using MagZol regent (Magen, R4801‐2, China) according to the manufacturer's instructions. The cDNA was synthesized using HiScript II Q Select RT SuperMix for qPCR analysis (Vazyme, RR037A, China). Quantitative real‐time PCR (qRT–PCR) was performed with Magic SYBR Mixture reagent (CWBIO, CW3008M, China) using CFX96™ Real‐Time System (Bio‐Rad, USA) (Table EV1). The RNA levels were normalized to internal GAPDH mRNA as indicated.
Generation of ATAD1 knockout cell lines
To generate ATAD1‐knockout (ATAD1KO) Huh7 and Huh7.5 cells, small guide RNAs (sgRNAs) targeting ATAD1 sequence were cloned into lentiCRISPR v2 vector (Addgene plasmid # 52961) with BsmBI site to make pCRISPR‐sgATAD1 construct (Sanjana et al, 2014) (Table EV1). pCRISPR‐sgATAD1 (5 μg), psPAX2 (3 μg; Addgene plasmid # 12260), and pMD2.G (2 μg; Addgene plasmid # 12259) were co‐transfected into 293T cells using PEI reagent (30 μl; Polysciences, 24765, USA) according to the manufacturer's instructions. The culture supernatant containing lentivirus was harvested at 48 h post‐transfection (hpt) and filtered (0.45 μm; PALL, 4614, USA). Huh7 or Huh7.5 cells were infected with the sgATAD1‐expressing lentivirus and selected with puromycin (2 μg/ml) to obtain ATAD1KO cells. The knockout of ATAD1 was confirmed by western blotting using puromycin‐resistant cells and subsequent sequencing analysis of TA‐cloned PCR products spanning sgATAD1‐targeting and flanking sequences (pGEM‐T Easy Vector, Promega, A1360, USA), using genomic DNA extracted by EasyPure Genomic DAN Kit (TransGen, EE101, China). Huh7 or Huh7.5 cells expressing sgRNAs targeting green fluorescent protein (GFP) were used as irrelevant knockout negative control (NCKO) cells (Table EV1).
Generation of MAVS knockout Huh7.5 cells
MAVS was knocked out in both WT and ATAD1KO Huh7.5 cells using CRISPR/Cas9 technology. Briefly, lentiCRISPR v2 vector containing small guide RNA (sgRNA)‐targeting MAVS sequence was constructed (Table EV1). The WT and ATAD1KO Huh7.5 cells were infected with the sgMAVS‐expressing lentivirus and selected with puromycin (2 μg/ml) to obtain MAVSKO and MAVSKO/ATAD1KO cells. The knockout of MAVS was confirmed by western blotting demonstrating the absence of MAVS protein.
Complementation of ATAD1 in ATAD1 ‐knockout cells
To complementally express ATAD1 in the ATAD1KO Huh7.5 cells (ATAD1KO+Res), which must be resisted to the sgATAD1‐mediated knockout of wild‐type (WT) ATAD1, four silent mutations (Mut4) were introduced into the sgATAD1‐ and PAM‐binding sequences within the coding sequence of ATAD1 using ATAD1‐PAM‐F and ATAD1‐PAM‐R primers (Table EV1). The ATAD1‐Mut4 sequence was cloned into pCDH‐CMV‐MCS‐EF1‐Puro lentivirus vector by NheI‐BamHI sites. The resulting plasmid pCDH‐CMV‐MCS‐EF1‐Puro‐ATAD1 (5 μg), together with psPAX2 (3 μg) and pMD2.G (2 μg), were co‐transfected into 293T cells using PEI reagent (30 μl). The supernatant lentivirus‐harboring ATAD1‐expression cassette was harvested, filtered (0.45 μm), and used to infect ATAD1KO Huh7.5 cells. The cells complementally expressing ATAD1 were selected and confirmed the expression of ATAD1 by western blotting (designated ATAD1KO+Res).
Immunofluorescence and FFUs assay
Hepatitis C virus‐infected cells were determined by immunostaining of HCV Core using monoclonal antibody anti‐Core C7‐50 (Santa Cruz Biotechnology), as previously described (Li et al, 2012a, 2012b). Briefly, HCV‐infected cells were fixed with methanol (−20°C) and incubated with primary antibody (Core C7‐50) (1:400 dilutions) for 1 h at room temperature, followed by secondary antibody goat anti‐mouse IgG (H+L) highly cross‐adsorbed secondary, Alexa Fluor® 488 (Thermo Fisher Scientific), for 2 h at room temperature. The immunofluorescent images were captured with fluorescence microscope (Leica DMI4000B, Germany), and the percentage of HCV‐positive cells was counted by ImageJ software (National Institutes of Health, USA).
Hepatitis C virus infectivity titers were determined by FFUs assay as previously described (Li et al, 2011, 2021). Briefly, Huh7.5 cells (7 × 103 cells) were seeded in a 96‐well plate with 100 μl of complete DMEM and incubated for 16 h. Subsequently, the cells were infected with twofold serial dilutions of HCV in complete medium for 48 h, or as appropriate. The cells were fixed with methanol (−20°C) and immunostained with anti‐Core C7‐50 and goat anti‐mouse IgG (H+L) highly cross‐adsorbed secondary, Alexa Fluor® 488 (Thermo Fisher Scientific), to determine the presence of HCV‐positive cells. The number of FFUs was manually counted under the fluorescence microscope (Leica DMI4000B).
Confocal microscopy
To prepare cells for confocal microscopy analysis, the cell climbing sheet with 12 mm in diameter (WHB scientific, WHB‐12‐CS, China) was plated in a six‐well plate, and then 3.5 × 105 cells were seeded in each well and cultured for 16 h prior to plasmid transfection or virus infection. The cell climbing sheet was fixed with 4% paraformaldehyde (Sangon Biotech, E672002, China) for 10 min, permeabilized with 0.2% Triton X‐100 for 10 min, and blocked with 3% bovine serum albumin (BSA) for 1 h at room temperature. The fixed cells were incubated with primary antibodies (C7‐50, Tom20, Calnexin, MAVS, or HA antibodies) for 2 h, followed by secondary antibodies conjugated with Alexa Fluor® 488, 555, 568, or 647 for 1 h at room temperature. Nuclei were stained with Hoechst 33342 (Life technologies, 33258, USA). ProLong™ Glass Antifade Mountant (Invitrogen, P36982, USA) was applied to protect fluorescent signals from photobleaching across the entire visible and near‐infrared spectra. Cell images were captured by a confocal microscope (Zeiss, LSM800, Germany) with optimized laser parameters to visualize the co‐localization of red, green, and purple fluorescence. The Zen (Zeiss Imaging Software), Imaris 8.4 (Switzerland), and Adobe‐Photoshop (CS6, USA) were used for processing confocal images.
Western blotting
Western blotting was conducted as described in our previous study (Duan et al, 2018). Briefly, cells were harvested and lysed with immunoprecipitation (IP) lysis buffer (25 mM Tris–HCl, 150 mM NaCl, 1 mM EDTA, 1% NP‐40, and 5% glycerol, pH = 7.4) (Thermo Fisher Scientific, 87787) on ice for 15 min. Total protein was resolved by either a 10 or 12% sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS–PAGE), followed by transfer onto polyvinylidene fluoride (PVDF) membranes (Bio‐Rad, 1620177). The membrane was blocked with 5% skimmed milk for 1 h, followed by overnight incubation at 4°C with gentle rocking using primary antibodies as appropriate. The membrane was then washed three times (10 min per wash) with TBS plus 1‰ Tween 20 (TBST) (12.1 g Tris and 40 g NaCl in 1 l ddH2O, pH = 7.6), and subsequently incubated with horseradish peroxidase (HRP)‐conjugated anti‐mouse or anti‐rabbit secondary antibodies. Finally, the protein bands of interest were detected with ECL chemiluminescent substrate kit (Proteintech, B500013) and visualized with OPTIMAX X‐ray Film Processor (PROTEC GmbH, Germany).
Co‐immunoprecipitation (Co‐IP)
The cells were harvested and lysed with IP lysis buffer (Thermo Fisher Scientific, 87787) on ice for 15 min. The resulting whole‐cell extracts were incubated overnight at 4°C with gentle rocking in the presence of anti‐Flag antibody (1:100; Proteintech, 20543‐1‐AP), followed by a subsequent incubation step with protein A+G Sepharose beads (7Sea Biotech, P001‐4) at 4°C for 2 h. The beads were then washed five times with cold PBS (15 min per wash), and the protein pellets were eluted using a solution of 2 × LDS sample buffer that has been diluted with NuPAGE® 4 × LDS sample buffer (Thermo Fisher Scientific, NP0008) and analyzed by western blotting.
Isolation of mitochondria, ER, and cytoplasm fractions
The mitochondria and cytoplasmic contents were isolated from the WT, ATAD1KO, and ATAD1KO+Res Huh7.5 cells using the Cell Mitochondria Isolation Kit (Beyotime, C3601, China) according to the manufacturer's instructions. Briefly, 1.5 ml of mitochondrial separation reagent containing 1% phenylmethanesulfonyl fluoride (PMSF) was added to cell precipitation (2 × 107 cells) and incubated on ice for 15 min. The cells were homogenized 50–80 times using a homogenizer and centrifuged at 1,000 × g at 4°C for 10 min. The supernatant was collected and re‐centrifuged at 3,500 × g at 4°C for 10 min. The supernatant was collected as the cytoplasmic component free of mitochondria, and the precipitate was mitochondrial fraction. The precipitate was lysed with 100 μl of mitochondrial lysis buffer containing 1% PMSF on ice for 15 min and then centrifuged at 12,000 × g at 4°C for 10 min. The resulting supernatant was collected as the mitochondrial component. Equal amounts (10 μg) of protein from both mitochondrial and cytoplasmic fractions were analyzed by western blotting, respectively. Cox IV was used as a loading control for the mitochondrial fraction (Sun et al, 2021). The isolation of ER was performed according to the manufacture's protocol of Minute™ ER enrichment Kit (Invent Biotechnologies).
Semi‐denaturing detergent agarose gel electrophoresis (SDD‐AGE)
Semi‐denaturing detergent agarose gel electrophoresis experiments were performed according to previous reports with minor modifications (Hou et al, 2011; Liu et al, 2017). In brief, WT and ATAD1KO Huh7.5 cells were infected with HCV 2a JFH1 for 72 h, following which crude mitochondria were isolated from 1 × 107 cells using the Cell Mitochondria Isolation Kit (Beyotime, C3601) according to the manufacturer's instructions. The crude mitochondria were resuspended in 1 × sample buffer (0.5 × TBE containing 10% glycerol, 2% SDS, and 0.0025% bromophenol blue) and loaded onto a vertical 1.5% agarose gel (Bio‐Rad; 0.5 × TBE containing 0.1% SDS). After electrophoresis in the running buffer (1 × TBE containing 0.1% SDS) for 1 h with a constant voltage of 100 V at 4°C, the proteins were transferred to a PVDF membrane with a constant voltage of 20 V for 1 h in ice bath. The membrane was blotted with anti‐MAVS antibodies following western blotting procedures.
Statistical mean mitochondrial length
The Mitochondrial Network Analysis (MiNA) plugin of ImageJ software was employed to determine the average length of mitochondria, as previously reported (Valente et al, 2017). Linear and fragmented mitochondria were differentiated based on a mean length threshold of 2 μm.
Statistical analysis
The data are presented as mean and standard error of mean (mean ± SEM) from a minimum of three independent experiments. Statistical significance was determined using Student's unpaired t‐test, One‐way ANOVA, and Two‐way ANOVA with GraphPad Prism 8 software (California, USA), and P < 0.05 was considered significant.
Author contributions
Qing Zhou: Conceptualization; data curation; formal analysis; validation; investigation; visualization; methodology; writing – original draft. Yuhao Yang: Data curation; formal analysis; validation; investigation; visualization; methodology. Zhanxue Xu: Data curation; formal analysis; methodology. Kai Deng: Formal analysis; methodology. Zhenzhen Zhang: Software; formal analysis; methodology. Jiawei Hao: Methodology. Ni Li: Methodology. Yanling Wang: Methodology. Ziwen Wang: Software; formal analysis. Haihang Chen: Methodology. Yang Yang: Methodology. Fei Xiao: Resources; methodology. Xiaohong Zhang: Resources; methodology. Song Gao: Software; formal analysis. Yi‐Ping Li: Conceptualization; resources; data curation; formal analysis; supervision; funding acquisition; validation; investigation; visualization; methodology; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank Dr. Jens Bukh (Hvidovre Hospital and University of Copenhagen, Denmark), Dr. Charles Rice (Rockefeller University, USA), Dr. Takaji. Wakita (National Institute of Infectious Diseases, Japan) and Dr. Jin Zhong (Institut Pasteur of Shanghai, China) for providing HCV recombinants, Huh7, Huh7.5 cells, and research reagents. We thank Dr. Jun Cui (Sun Yat‐sen University) for constructive discussions. We thank Dr. Bingyu Liu and Dr. Chengjiang Gao (Shandong University School of Basic Medical Sciences, China) for constructive discussion and technical advice. This work was supported by the National Natural Science Foundation of China (81971938 for Y‐PL) and The Innovation Research Team for Basic and Clinical Studies on Chronic Liver Diseases of 2018 High‐Level Health Teams of Zhuhai (for Y‐PL and FX). The funding bodies were not involved in the design of the study, collection, analysis, and interpretation of data, and in writing the manuscript.
EMBO reports (2023) 24: e56614
Data availability
No data were deposited in the public database.
References
- Aviram N, Schuldiner M (2017) Targeting and translocation of proteins to the endoplasmic reticulum at a glance. J Cell Sci 130: 4079–4085 [DOI] [PubMed] [Google Scholar]
- Aweida D, Cohen S (2022) The AAA‐ATPase ATAD1 and its partners promote degradation of desmin intermediate filaments in muscle. EMBO Rep 23: e55175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartenschlager R, Baumert TF, Bukh J, Houghton M, Lemon SM, Lindenbach BD, Lohmann V, Moradpour D, Pietschmann T, Rice CM et al (2018) Critical challenges and emerging opportunities in hepatitis C virus research in an era of potent antiviral therapy: considerations for scientists and funding agencies. Virus Res 248: 53–62 [DOI] [PubMed] [Google Scholar]
- Behrens SE, Tomei L, De Francesco R (1996) Identification and properties of the RNA‐dependent RNA polymerase of hepatitis C virus. EMBO J 15: 12–22 [PMC free article] [PubMed] [Google Scholar]
- Blight KJ, McKeating JA, Rice CM (2002) Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol 76: 13001–13014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgese N, Fasana E (2011) Targeting pathways of C‐tail‐anchored proteins. Biochim Biophys Acta 1808: 937–946 [DOI] [PubMed] [Google Scholar]
- Borgese N, Colombo S, Pedrazzini E (2003) The tale of tail‐anchored proteins: coming from the cytosol and looking for a membrane. J Cell Biol 161: 1013–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brault C, Levy PL, Bartosch B (2013) Hepatitis C virus‐induced mitochondrial dysfunctions. Viruses 5: 954–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC (2012) Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet 46: 265–287 [DOI] [PubMed] [Google Scholar]
- Chen YC, Umanah GK, Dephoure N, Andrabi SA, Gygi SP, Dawson TM, Dawson VL, Rutter J (2014) Msp1/ATAD1 maintains mitochondrial function by facilitating the degradation of mislocalized tail‐anchored proteins. EMBO J 33: 1548–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Zheng F, Yuan G, Duan X, Rong L, Liu J, Feng S, Wang Z, Wang M, Feng Y et al (2018) Development of an infectious cell culture system for hepatitis C virus genotype 6a clinical isolate using a novel strategy and its sensitivity to direct‐acting antivirals. Front Microbiol 9: 2950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu VC, Bhattacharya S, Nomoto A, Lin J, Zaidi SK, Oberley TD, Weinman SA, Azhar S, Huang T‐T (2011) Persistent expression of hepatitis C virus non‐structural proteins leads to increased autophagy and mitochondrial injury in human hepatoma cells. PLoS One 6: e28551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dederer V, Lemberg MK (2021) Transmembrane dislocases: a second chance for protein targeting. Trends Cell Biol 31: 898–911 [DOI] [PubMed] [Google Scholar]
- Dederer V, Khmelinskii A, Huhn AG, Okreglak V, Knop M, Lemberg MK (2019) Cooperation of mitochondrial and ER factors in quality control of tail‐anchored proteins. Elife 8: e45506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan X, Anwar MI, Xu Z, Ma L, Yuan G, Chen Y, Liu X, Xia J, Zhou Y, Li YP (2018) Adaptive mutation F772S‐enhanced p7‐NS4A cooperation facilitates the assembly and release of hepatitis C virus and is associated with lipid droplet enlargement. Emerg Microbes Infect 7: 143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farci P, Alter HJ, Govindarajan S, Wong DC, Engle R, Lesniewski RR, Mushahwar IK, Desai SM, Miller RH, Ogata N et al (1992) Lack of protective immunity against reinfection with hepatitis C virus. Science 258: 135–140 [DOI] [PubMed] [Google Scholar]
- Favaloro V, Vilardi F, Schlecht R, Mayer MP, Dobberstein B (2010) Asna1/TRC40‐mediated membrane insertion of tail‐anchored proteins. J Cell Sci 123: 1522–1530 [DOI] [PubMed] [Google Scholar]
- Ferrari E, Wright‐Minogue J, Fang JW, Baroudy BM, Lau JY, Hong Z (1999) Characterization of soluble hepatitis C virus RNA‐dependent RNA polymerase expressed in Escherichia coli . J Virol 73: 1649–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fresenius HL, Wohlever ML (2019) Sorting out how Msp1 maintains mitochondrial membrane proteostasis. Mitochondrion 49: 128–134 [DOI] [PubMed] [Google Scholar]
- Gottwein JM, Jensen TB, Mathiesen CK, Meuleman P, Serre SBN, Lademann JB, Ghanem L, Scheel TKH, Leroux‐Roels G, Bukh J (2011) Development and application of hepatitis C reporter viruses with genotype 1 to 7 core‐nonstructural protein 2 (NS2) expressing fluorescent proteins or luciferase in modified JFH1 NS5A. J Virol 85: 8913–8928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm I, Erdmann R, Girzalsky W (2016) Role of AAA+‐proteins in peroxisome biogenesis and function. Biochim Biophys Acta 1863: 828–837 [DOI] [PubMed] [Google Scholar]
- Hanson PI, Whiteheart SW (2005) AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol 6: 519–529 [DOI] [PubMed] [Google Scholar]
- Hao J, Li J, Zhang Z, Yang Y, Zhou Q, Wu T, Chen T, Wu Z, Zhang P, Cui J et al (2022) NLRC5 restricts dengue virus infection by promoting the autophagic degradation of viral NS3 through E3 ligase CUL2 (cullin 2). Autophagy 19: 1332–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie C, Suzuki H, Sakaguchi M, Mihara K (2002) Characterization of signal that directs C‐tail‐anchored proteins to mammalian mitochondrial outer membrane. Mol Biol Cell 13: 1615–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ (2011) MAVS forms functional prion‐like aggregates to activate and propagate antiviral innate immune response. Cell 146: 448–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivashkina N, Wölk B, Lohmann V, Bartenschlager R, Blum HE, Penin F, Moradpour D (2002) The hepatitis C virus RNA‐dependent RNA polymerase membrane insertion sequence is a transmembrane segment. J Virol 76: 13088–13093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen TB, Gottwein JM, Scheel TK, Hoegh AM, Eugen‐Olsen J, Bukh J (2008) Highly efficient JFH1‐based cell‐culture system for hepatitis C virus genotype 5a: failure of homologous neutralizing‐antibody treatment to control infection. J Infect Dis 198: 1756–1765 [DOI] [PubMed] [Google Scholar]
- Jonikas MC, Collins SR, Denic V, Oh E, Quan EM, Schmid V, Weibezahn J, Schwappach B, Walter P, Weissman JS et al (2009) Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science 323: 1693–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A et al (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596: 583–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S (2005) IPS‐1, an adaptor triggering RIG‐I‐ and Mda5‐mediated type I interferon induction. Nat Immunol 6: 981–988 [DOI] [PubMed] [Google Scholar]
- Kim S‐J, Syed GH, Siddiqui A (2013) Hepatitis C virus induces the mitochondrial translocation of Parkin and subsequent mitophagy. PLoS Pathog 9: e1003285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S‐J, Syed GH, Khan M, Chiu W‐W, Sohail MA, Gish RG, Siddiqui A (2014) Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc Natl Acad Sci USA 111: 6413–6418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolykhalov AA, Agapov EV, Blight KJ, Mihalik K, Feinstone SM, Rice CM (1997) Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science 277: 570–574 [DOI] [PubMed] [Google Scholar]
- Lee KJ, Choi J, Ou JH, Lai MM (2004) The C‐terminal transmembrane domain of hepatitis C virus (HCV) RNA polymerase is essential for HCV replication in vivo . J Virol 78: 3797–3802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XD, Sun L, Seth RB, Pineda G, Chen ZJ (2005) Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci USA 102: 17717–17722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y‐P, Gottwein JM, Scheel TK, Jensen TB, Bukh J (2011) MicroRNA‐122 antagonism against hepatitis C virus genotypes 1‐6 and reduced efficacy by host RNA insertion or mutations in the HCV 5′ UTR. Proc Natl Acad Sci USA 108: 4991–4996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y‐P, Ramirez S, Gottwein JM, Scheel TKH, Mikkelsen L, Purcell RH, Bukh J (2012a) Robust full‐length hepatitis C virus genotype 2a and 2b infectious cultures using mutations identified by a systematic approach applicable to patient strains. Proc Natl Acad Sci USA 109: E1101–E1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y‐P, Ramirez S, Jensen SB, Purcell RH, Gottwein JM, Bukh J (2012b) Highly efficient full‐length hepatitis C virus genotype 1 (strain TN) infectious culture system. Proc Natl Acad Sci USA 109: 19757–19762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y‐P, Ramirez S, Humes D, Jensen SB, Gottwein JM, Bukh J (2014) Differential sensitivity of 5′UTR‐NS5A recombinants of hepatitis C virus genotypes 1‐6 to protease and NS5A inhibitors. Gastroenterology 146: 812–821 [DOI] [PubMed] [Google Scholar]
- Li J, Zhou Q, Rong L, Rong D, Yang Y, Hao J, Zhang Z, Ma L, Rao G, Zhou Y et al (2021) Development of cell culture infectious clones for hepatitis C virus genotype 1b and transcription analysis of 1b‐infected hepatoma cells. Antiviral Res 193: 105136 [DOI] [PubMed] [Google Scholar]
- Lindenbach BD, Evans MJ, Syder AJ, Wölk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA et al (2005) Complete replication of hepatitis C virus in cell culture. Science 309: 623–626 [DOI] [PubMed] [Google Scholar]
- Liu B, Zhang M, Chu H, Zhang H, Wu H, Song G, Wang P, Zhao K, Hou J, Wang X et al (2017) The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63‐linked polyubiquitination. Nat Immunol 18: 214–224 [DOI] [PubMed] [Google Scholar]
- Lohmann V, Körner F, Herian U, Bartenschlager R (1997) Biochemical properties of hepatitis C virus NS5B RNA‐dependent RNA polymerase and identification of amino acid sequence motifs essential for enzymatic activity. J Virol 71: 8416–8428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto S, Nakatsukasa K, Kakuta C, Tamura Y, Esaki M, Endo T (2019) Msp1 clears mistargeted proteins by facilitating their transfer from mitochondria to the ER. Mol Cell 76: 191–205 [DOI] [PubMed] [Google Scholar]
- Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J (2005) Cardif is an adaptor protein in the RIG‐I antiviral pathway and is targeted by hepatitis C virus. Nature 437: 1167–1172 [DOI] [PubMed] [Google Scholar]
- Moradpour D, Penin F (2013) Hepatitis C virus proteins: from structure to function. Curr Top Microbiol Immunol 369: 113–142 [DOI] [PubMed] [Google Scholar]
- Okreglak V, Walter P (2014) The conserved AAA‐ATPase Msp1 confers organelle specificity to tail‐anchored proteins. Proc Natl Acad Sci USA 111: 8019–8024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda M, Li K, Beard MR, Showalter LA, Scholle F, Lemon SM, Weinman SA (2002) Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology 122: 366–375 [DOI] [PubMed] [Google Scholar]
- Page K, Hahn JA, Evans J, Shiboski S, Lum P, Delwart E, Tobler L, Andrews W, Avanesyan L, Cooper S et al (2009) Acute hepatitis C virus infection in young adult injection drug users: a prospective study of incident infection, resolution, and reinfection. J Infect Dis 200: 1216–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl C, Dikic I (2019) Cellular quality control by the ubiquitin‐proteasome system and autophagy. Science 366: 818–822 [DOI] [PubMed] [Google Scholar]
- Ramirez S, Mikkelsen LS, Gottwein JM, Bukh J (2016) Robust HCV genotype 3a infectious cell culture system permits identification of escape variants with resistance to sofosbuvir. Gastroenterology 151: 973–985 [DOI] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F (2014) Improved vectors and genome‐wide libraries for CRISPR screening. Nat Methods 11: 783–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt‐Mende J, Bieck E, Hugle T, Penin F, Rice CM, Blum HE, Moradpour D (2001) Determinants for membrane association of the hepatitis C virus RNA‐dependent RNA polymerase. J Biol Chem 276: 44052–44063 [DOI] [PubMed] [Google Scholar]
- Schuldiner M, Metz J, Schmid V, Denic V, Rakwalska M, Schmitt HD, Schwappach B, Weissman JS (2008) The GET complex mediates insertion of tail‐anchored proteins into the ER membrane. Cell 134: 634–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth RB, Sun L, Ea CK, Chen ZJ (2005) Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF‐kappaB and IRF 3. Cell 122: 669–682 [DOI] [PubMed] [Google Scholar]
- Shah R, Ahovegbe L, Niebel M, Shepherd J, Thomson EC (2021) Non‐epidemic HCV genotypes in low‐ and middle‐income countries and the risk of resistance to current direct‐acting antiviral regimens. J Hepatol 75: 462–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Liu X, Ca S, Zhang W, Niu Y (2021) Adiponectin receptor agonist AdipoRon blocks skin inflamm‐ageing by regulating mitochondrial dynamics. Cell Prolif 54: e13155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente AJ, Maddalena LA, Robb EL, Moradi F, Stuart JA (2017) A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem 119: 315–326 [DOI] [PubMed] [Google Scholar]
- Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Kräusslich HG, Mizokami M et al (2005) Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11: 791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Walter P (2020) Msp1/ATAD1 in protein quality control and regulation of synaptic activities. Annu Rev Cell Dev Biol 36: 141–164 [DOI] [PubMed] [Google Scholar]
- Wang L, Myasnikov A, Pan X, Walter P (2020) Structure of the AAA protein Msp1 reveals mechanism of mislocalized membrane protein extraction. Elife 9: e54031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Toutkoushian H, Belyy V, Kokontis CY, Walter P (2022) Conserved structural elements specialize ATAD1 as a membrane protein extraction machine. Elife 11: e73941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattenberg B, Lithgow T (2001) Targeting of C‐terminal (tail)‐anchored proteins: understanding how cytoplasmic activities are anchored to intracellular membranes. Traffic 2: 66–71 [DOI] [PubMed] [Google Scholar]
- Wesselborg S, Stork B (2015) Autophagy signal transduction by ATG proteins: from hierarchies to networks. Cell Mol Life Sci 72: 4721–4757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2022) Hepatitis C. WHO page, accessed date November 1, 2022; https://www.who.int/news‐room/fact‐sheets/detail/hepatitis‐c
- Wohlever ML, Mateja A, McGilvray PT, Day KJ, Keenan RJ (2017) Msp1 is a membrane protein dislocase for tail‐anchored proteins. Mol Cell 67: 194–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Kaneko S, Shirota Y, Qin W, Nomura T, Kobayashi K, Murakami S (1998) RNA‐dependent RNA polymerase activity of the soluble recombinant hepatitis C virus NS5B protein truncated at the C‐terminal region. J Biol Chem 273: 15479–15486 [DOI] [PubMed] [Google Scholar]
- Zhang J, Wang Y, Chi Z, Keuss MJ, Pai YM, Kang HC, Shin JH, Bugayenko A, Wang H, Xiong Y et al (2011) The AAA+ ATPase thorase regulates AMPA receptor‐dependent synaptic plasticity and behavior. Cell 145: 284–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng F, Li N, Xu Y, Zhou Y, Li YP (2021) Adaptive mutations promote hepatitis C virus assembly by accelerating core translocation to the endoplasmic reticulum. J Biol Chem 296: 100018 [DOI] [PMC free article] [PubMed] [Google Scholar]