

The title compound exhibits I⋯π halogen bonding and π-stacking in its extended structure.
Keywords: crystal structure; 9,10-bis(iodoethynyl); anthracene; halogen bonding; π-stacking
Abstract
The title compound, C18H8I2, is an ethynyl-substituted anthracene. The C—C—I bond angles deviate from 180°, being affected by intermolecular I⋯π interactions. These interactions form a two-dimensional supramolecular structure further supported by offset π–π stacking of neighboring anthracene moieties.

Structure description
The flat, stable, and conjugated composition of anthracene derivatives makes them good candidates for two-dimensional molecular crystals. Two-dimensional crystals can have unique properties with applications in electronics, biomedicine, and sensors (Yan et al., 2023 ▸). The title compound is an iodoethynyl-substituted anthracene. The iodine functional groups provide the opportunity for halogen-bonding interactions. The synthesis and structure of the title compound are reported here.
The crystal stucture represents the first example of an ethynyl–anthracene halogenated with iodine (Fig. 1 ▸). The C—I bonds have an average length of 1.996 (4) Å, similar to that found in 1,4-bis(iodoethynyl)benzene [2.007 (7) Å; Barrès et al., 2008 ▸], 4-iodoethnynylanisole [1.990 (3) Å; Dumele et al., 2014 ▸], and other iodoethynyl derivatives (Lehnherr et al., 2015 ▸). The 180° bond angle expected from the alkynyl C atoms and iodine, C15—C16—I1 and C17—C18—I2, are slightly bent to 177.4 (3) and 178.0 (3)°, respectively. This may be attributed to halogen bonding between C(sp)—I moieties and the π-electrons of the adjacent anthracene rings (Fig. 2 ▸), where I1 maintains its shortest I⋯centroid contact to the centroid of the C2–C7 ring (Cg1) [I1⋯Cg1 = 3.528 (4) Å and C16—I1⋯Cg1 = 151.2 (3)°] and I2 has a short contact to the centroid of the C9–C14 ring (Cg2) [I2⋯Cg2 = 3.767 (4) Å and C18—I2⋯Cg2 = 150.1 (3)°]. The bent nature of the C—I⋯centroid interactions leads to short I⋯C contacts ranging from 3.352 (4) to 3.655 (4) Å. The shorter contact between I1 and Cg1 appears to influence more significantly the bending of the entire alkynyl substituent [C1—C15—I1 = 173.8 (3)° versus C8—C17—I2 = 178.7 (3)°], notably pulling the I1 atom away from the central ring of the neighboring anthracene molecule and toward its C2–C7 centroid.
Figure 1.
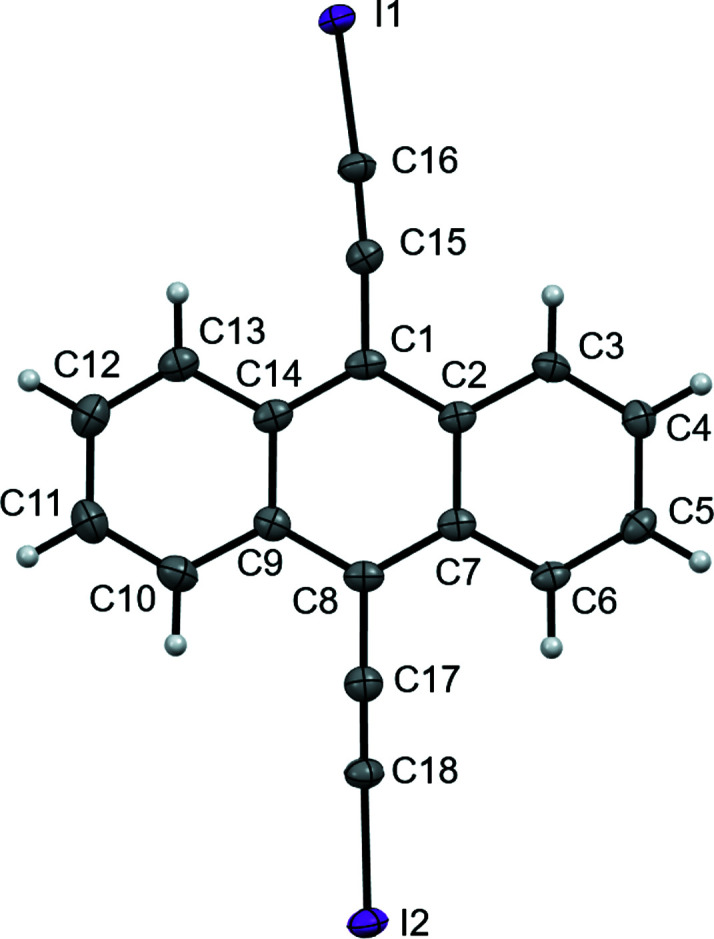
The title molecule, showing the atom-labeling scheme and with displacement ellipsoids drawn at the 50% probability level.
Figure 2.

I⋯π interactions (blue dashed lines) occurring to and from a central molecule of the title compound.
Propagation of the I⋯π interactions results in a two-dimensional supramolecular structure in the (101) plane (Fig. 3 ▸). Interestingly, in 1,4-bis(iodoethynyl)benzene, 4-iodoethnynylanisole, and 1-chloro-4-(iodoethynyl)benzene, the I⋯π interactions occur at a similar distance [for example, the shortest I⋯C contact is 3.427 (7) Å in 1,4-bis(iodoethynyl)benzene, 3.392 (3) Å in 4-iodoethnynylanisole, and 3.417 (4) Å in 1-chloro-4-(iodoethynyl)benzene], but occur to the alkynyl C atoms rather than to the aromatic rings as in the title compound (Barrès et al., 2008 ▸; Dumele et al., 2014 ▸; Lehnherr et al., 2015 ▸). The I atom in (tert-butyl)[4-(iodoethynyl)phenyl]carbamate (Kahlfuss et al., 2016 ▸) does appear to interact with the aromatic system of a neighboring molecule to form a one-dimensional I⋯π motif of similar C—I⋯centroid geometries to the title anthracene derivative. Anthracene portions of adjacent molecules are arranged in an offset stacking arrangement (Fig. 4 ▸), with an interplanar separation of 3.377 Å, a shortest C⋯C distance of 3.436 (5) Å, and a shortest centroid–centroid distance of 3.692 (5) Å. The interplanar spacing of the anthracene scaffold in the title compound is shorter than in the offset stacking in 9,10-diiodoanthracene (3.602 Å; Peters et al., 1996 ▸) and similar to that of the offset stacking in monoclinic 9,10-bis(phenylethynyl)anthracene (3.405 Å; Batsanov et al., 2013 ▸).
Figure 3.

The two-dimensional supramolecular motif formed via I⋯π interactions in the title compound.
Figure 4.

Top (left) and side (right) views of the offset stacking of neighboring molecules in the title compound.
Synthesis and crystallization
The procedure was modeled after an analogous functionalization of an alkynylsilane (Tse et al., 2021 ▸). 9,10-Bis(trimethylsilylethynyl)anthracene (0.0325 g, 0.0878 mmol), N-iodosuccinimide (0.0531 g, 0.236 mmol) and AgNO3 (0.0022 g, 0.0129 mmol) were added to dry dimethylformamide (5 ml), and the resulting mixture was stirred under nitrogen. After 5 h, the reaction mixture was diluted with EtOAc (30 ml) and washed with H2O (5 × 30 ml). The organic layer was dried in vacuo, resulting in an orange solid. The product was crystallized from the orange solid using vapor–vapor diffusion (CH2Cl2/hexanes).
1H NMR key spectroscopic features as determined from the crude product (400 MHz, chloroform-d): δ 8.33 (d, J = 9.2 Hz, 4H), 7.82 (m, 4H).
Refinement
Crystal data, data collection, and structural refinement details are summarized in Table 1 ▸.
Table 1. Experimental details.
| Crystal data | |
| Chemical formula | C18H8I2 |
| M r | 478.04 |
| Crystal system, space group | Monoclinic, P21/n |
| Temperature (K) | 100 |
| a, b, c (Å) | 8.0022 (3), 15.0735 (7), 12.2506 (5) |
| β (°) | 96.2749 (17) |
| V (Å3) | 1468.83 (11) |
| Z | 4 |
| Radiation type | Mo Kα |
| μ (mm−1) | 4.27 |
| Crystal size (mm) | 0.17 × 0.15 × 0.13 |
| Data collection | |
| Diffractometer | Bruker D8 Venture Photon 2 |
| Absorption correction | Multi-scan (SADABS; Krause et al., 2015 ▸) |
| T min, T max | 0.774, 1.000 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 37187, 3373, 3084 |
| R int | 0.037 |
| (sin θ/λ)max (Å−1) | 0.650 |
| Refinement | |
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.026, 0.063, 1.13 |
| No. of reflections | 3373 |
| No. of parameters | 181 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 1.41, −0.90 |
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S2414314623005539/hb4433sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2414314623005539/hb4433Isup2.hkl
Supporting information file. DOI: 10.1107/S2414314623005539/hb4433Isup3.cml
CCDC reference: 2191397
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
The authors thank John Lee for his advice on preparing this report. Funding for this research was provided by the UTC Irvine and Nita Grote Fund. Nehemiah Antoine was supported by the Tom Rybolt and Richard X. Zhang Endowed Undergraduate Research in Chemistry Scholarship. Marisa James was supported by the William H. Wheeler Center for Odor Research.
full crystallographic data
Crystal data
| C18H8I2 | F(000) = 888 |
| Mr = 478.04 | Dx = 2.162 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| a = 8.0022 (3) Å | Cell parameters from 9890 reflections |
| b = 15.0735 (7) Å | θ = 2.7–30.6° |
| c = 12.2506 (5) Å | µ = 4.27 mm−1 |
| β = 96.2749 (17)° | T = 100 K |
| V = 1468.83 (11) Å3 | Column, red |
| Z = 4 | 0.17 × 0.14 × 0.13 mm |
Data collection
| Bruker D8 Venture Photon 2 diffractometer | 3084 reflections with I > 2σ(I) |
| Radiation source: Incoatec IµS | Rint = 0.037 |
| φ and ω scans | θmax = 27.5°, θmin = 2.2° |
| Absorption correction: multi-scan (SADABS; Krause et al., 2015) | h = −10→10 |
| Tmin = 0.774, Tmax = 1.000 | k = −19→19 |
| 37187 measured reflections | l = −15→15 |
| 3373 independent reflections |
Refinement
| Refinement on F2 | Primary atom site location: dual |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.026 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.063 | H-atom parameters constrained |
| S = 1.13 | w = 1/[σ2(Fo2) + (0.0142P)2 + 7.1388P] where P = (Fo2 + 2Fc2)/3 |
| 3373 reflections | (Δ/σ)max = 0.001 |
| 181 parameters | Δρmax = 1.41 e Å−3 |
| 0 restraints | Δρmin = −0.90 e Å−3 |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| I1 | 0.82384 (3) | 0.70341 (2) | 0.18275 (2) | 0.02740 (8) | |
| I2 | 0.03465 (3) | 0.12055 (2) | 0.56404 (2) | 0.02483 (8) | |
| C1 | 0.5433 (5) | 0.4778 (2) | 0.3413 (3) | 0.0183 (7) | |
| C2 | 0.6141 (5) | 0.4243 (2) | 0.4292 (3) | 0.0178 (7) | |
| C3 | 0.7841 (5) | 0.4348 (3) | 0.4760 (3) | 0.0207 (7) | |
| H3 | 0.853257 | 0.477816 | 0.446422 | 0.025* | |
| C4 | 0.8486 (5) | 0.3842 (3) | 0.5624 (3) | 0.0223 (8) | |
| H4 | 0.961959 | 0.392364 | 0.592776 | 0.027* | |
| C5 | 0.7470 (5) | 0.3190 (3) | 0.6078 (3) | 0.0230 (8) | |
| H5 | 0.792490 | 0.284632 | 0.668972 | 0.028* | |
| C6 | 0.5867 (5) | 0.3059 (2) | 0.5643 (3) | 0.0196 (7) | |
| H6 | 0.520936 | 0.261846 | 0.594840 | 0.024* | |
| C7 | 0.5139 (5) | 0.3572 (2) | 0.4729 (3) | 0.0178 (7) | |
| C8 | 0.3466 (5) | 0.3430 (2) | 0.4245 (3) | 0.0190 (7) | |
| C9 | 0.2770 (5) | 0.3953 (2) | 0.3353 (3) | 0.0192 (7) | |
| C10 | 0.1094 (5) | 0.3826 (3) | 0.2846 (3) | 0.0266 (8) | |
| H10 | 0.042007 | 0.337285 | 0.311230 | 0.032* | |
| C11 | 0.0440 (5) | 0.4340 (3) | 0.1990 (4) | 0.0297 (9) | |
| H11 | −0.067695 | 0.424074 | 0.166417 | 0.036* | |
| C12 | 0.1413 (6) | 0.5018 (3) | 0.1588 (3) | 0.0282 (9) | |
| H12 | 0.094835 | 0.537029 | 0.098712 | 0.034* | |
| C13 | 0.3010 (5) | 0.5178 (3) | 0.2048 (3) | 0.0234 (8) | |
| H13 | 0.363688 | 0.564785 | 0.177326 | 0.028* | |
| C14 | 0.3761 (5) | 0.4645 (2) | 0.2943 (3) | 0.0190 (7) | |
| C15 | 0.6415 (5) | 0.5464 (2) | 0.2976 (3) | 0.0201 (7) | |
| C16 | 0.7133 (5) | 0.6047 (3) | 0.2564 (3) | 0.0226 (8) | |
| C17 | 0.2484 (5) | 0.2737 (3) | 0.4661 (3) | 0.0224 (8) | |
| C18 | 0.1684 (5) | 0.2157 (2) | 0.5008 (3) | 0.0227 (8) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| I1 | 0.02902 (14) | 0.02681 (14) | 0.02568 (14) | −0.00824 (10) | −0.00015 (10) | 0.01045 (10) |
| I2 | 0.03161 (14) | 0.01874 (12) | 0.02455 (13) | −0.00641 (10) | 0.00495 (10) | 0.00300 (9) |
| C1 | 0.0253 (18) | 0.0140 (16) | 0.0175 (16) | −0.0007 (14) | 0.0104 (14) | −0.0041 (13) |
| C2 | 0.0229 (18) | 0.0134 (16) | 0.0180 (16) | 0.0009 (13) | 0.0067 (14) | −0.0033 (13) |
| C3 | 0.0209 (18) | 0.0204 (18) | 0.0216 (18) | −0.0032 (14) | 0.0061 (14) | −0.0020 (14) |
| C4 | 0.0230 (18) | 0.0224 (19) | 0.0211 (18) | 0.0003 (15) | 0.0007 (15) | −0.0044 (15) |
| C5 | 0.030 (2) | 0.0184 (18) | 0.0208 (18) | 0.0028 (15) | 0.0031 (15) | 0.0004 (14) |
| C6 | 0.0267 (19) | 0.0137 (16) | 0.0193 (17) | −0.0010 (14) | 0.0066 (14) | −0.0018 (13) |
| C7 | 0.0238 (18) | 0.0136 (16) | 0.0171 (16) | 0.0001 (13) | 0.0066 (14) | −0.0057 (13) |
| C8 | 0.0231 (18) | 0.0151 (16) | 0.0203 (17) | −0.0009 (14) | 0.0094 (14) | −0.0038 (13) |
| C9 | 0.0209 (17) | 0.0172 (17) | 0.0200 (17) | 0.0009 (14) | 0.0052 (14) | −0.0049 (14) |
| C10 | 0.026 (2) | 0.028 (2) | 0.027 (2) | −0.0051 (16) | 0.0061 (16) | −0.0027 (16) |
| C11 | 0.0226 (19) | 0.034 (2) | 0.032 (2) | 0.0005 (17) | −0.0016 (16) | −0.0029 (18) |
| C12 | 0.033 (2) | 0.028 (2) | 0.0241 (19) | 0.0056 (17) | 0.0021 (16) | 0.0009 (16) |
| C13 | 0.0274 (19) | 0.0222 (19) | 0.0216 (18) | 0.0009 (15) | 0.0078 (15) | −0.0011 (15) |
| C14 | 0.0268 (18) | 0.0157 (16) | 0.0158 (16) | 0.0012 (14) | 0.0086 (14) | −0.0034 (13) |
| C15 | 0.0234 (18) | 0.0189 (18) | 0.0188 (17) | 0.0020 (14) | 0.0055 (14) | −0.0018 (14) |
| C16 | 0.0266 (19) | 0.0221 (18) | 0.0197 (17) | −0.0043 (15) | 0.0055 (15) | 0.0016 (14) |
| C17 | 0.0251 (19) | 0.0199 (18) | 0.0225 (18) | 0.0003 (15) | 0.0046 (15) | −0.0029 (14) |
| C18 | 0.0274 (19) | 0.0185 (18) | 0.0228 (18) | −0.0048 (15) | 0.0046 (15) | −0.0012 (14) |
Geometric parameters (Å, º)
| I1—C16 | 1.996 (4) | C7—C8 | 1.420 (5) |
| I2—C18 | 1.996 (4) | C8—C9 | 1.412 (5) |
| C1—C14 | 1.413 (5) | C8—C17 | 1.433 (5) |
| C1—C2 | 1.413 (5) | C9—C10 | 1.428 (6) |
| C1—C15 | 1.437 (5) | C9—C14 | 1.434 (5) |
| C2—C3 | 1.426 (5) | C10—C11 | 1.362 (6) |
| C2—C7 | 1.431 (5) | C10—H10 | 0.9500 |
| C3—C4 | 1.360 (5) | C11—C12 | 1.406 (6) |
| C3—H3 | 0.9500 | C11—H11 | 0.9500 |
| C4—C5 | 1.426 (5) | C12—C13 | 1.360 (6) |
| C4—H4 | 0.9500 | C12—H12 | 0.9500 |
| C5—C6 | 1.349 (6) | C13—C14 | 1.437 (5) |
| C5—H5 | 0.9500 | C13—H13 | 0.9500 |
| C6—C7 | 1.430 (5) | C15—C16 | 1.192 (5) |
| C6—H6 | 0.9500 | C17—C18 | 1.190 (5) |
| C14—C1—C2 | 120.9 (3) | C7—C8—C17 | 119.3 (3) |
| C14—C1—C15 | 118.8 (3) | C8—C9—C10 | 122.3 (3) |
| C2—C1—C15 | 120.3 (3) | C8—C9—C14 | 119.4 (3) |
| C1—C2—C3 | 121.9 (3) | C10—C9—C14 | 118.3 (3) |
| C1—C2—C7 | 119.6 (3) | C11—C10—C9 | 121.5 (4) |
| C3—C2—C7 | 118.5 (3) | C11—C10—H10 | 119.3 |
| C4—C3—C2 | 121.0 (3) | C9—C10—H10 | 119.3 |
| C4—C3—H3 | 119.5 | C10—C11—C12 | 120.1 (4) |
| C2—C3—H3 | 119.5 | C10—C11—H11 | 119.9 |
| C3—C4—C5 | 120.4 (4) | C12—C11—H11 | 119.9 |
| C3—C4—H4 | 119.8 | C13—C12—C11 | 121.0 (4) |
| C5—C4—H4 | 119.8 | C13—C12—H12 | 119.5 |
| C6—C5—C4 | 120.3 (4) | C11—C12—H12 | 119.5 |
| C6—C5—H5 | 119.9 | C12—C13—C14 | 120.8 (4) |
| C4—C5—H5 | 119.9 | C12—C13—H13 | 119.6 |
| C5—C6—C7 | 121.3 (3) | C14—C13—H13 | 119.6 |
| C5—C6—H6 | 119.4 | C1—C14—C9 | 119.7 (3) |
| C7—C6—H6 | 119.4 | C1—C14—C13 | 122.0 (3) |
| C8—C7—C6 | 122.1 (3) | C9—C14—C13 | 118.2 (3) |
| C8—C7—C2 | 119.4 (3) | C16—C15—C1 | 175.4 (4) |
| C6—C7—C2 | 118.5 (3) | C15—C16—I1 | 177.4 (4) |
| C9—C8—C7 | 120.9 (3) | C18—C17—C8 | 179.3 (4) |
| C9—C8—C17 | 119.8 (3) | C17—C18—I2 | 178.0 (3) |
| C14—C1—C2—C3 | 178.2 (3) | C7—C8—C9—C10 | 179.9 (3) |
| C15—C1—C2—C3 | −1.1 (5) | C17—C8—C9—C10 | 0.7 (5) |
| C14—C1—C2—C7 | −1.6 (5) | C7—C8—C9—C14 | −0.9 (5) |
| C15—C1—C2—C7 | 179.0 (3) | C17—C8—C9—C14 | 180.0 (3) |
| C1—C2—C3—C4 | 178.2 (3) | C8—C9—C10—C11 | 179.7 (4) |
| C7—C2—C3—C4 | −1.9 (5) | C14—C9—C10—C11 | 0.4 (6) |
| C2—C3—C4—C5 | 0.3 (6) | C9—C10—C11—C12 | −0.4 (6) |
| C3—C4—C5—C6 | 1.1 (6) | C10—C11—C12—C13 | −0.5 (6) |
| C4—C5—C6—C7 | −0.6 (5) | C11—C12—C13—C14 | 1.3 (6) |
| C5—C6—C7—C8 | 178.6 (3) | C2—C1—C14—C9 | −0.5 (5) |
| C5—C6—C7—C2 | −1.0 (5) | C15—C1—C14—C9 | 178.8 (3) |
| C1—C2—C7—C8 | 2.5 (5) | C2—C1—C14—C13 | −179.9 (3) |
| C3—C2—C7—C8 | −177.3 (3) | C15—C1—C14—C13 | −0.5 (5) |
| C1—C2—C7—C6 | −177.9 (3) | C8—C9—C14—C1 | 1.8 (5) |
| C3—C2—C7—C6 | 2.3 (5) | C10—C9—C14—C1 | −178.9 (3) |
| C6—C7—C8—C9 | 179.1 (3) | C8—C9—C14—C13 | −178.9 (3) |
| C2—C7—C8—C9 | −1.3 (5) | C10—C9—C14—C13 | 0.4 (5) |
| C6—C7—C8—C17 | −1.7 (5) | C12—C13—C14—C1 | 178.0 (4) |
| C2—C7—C8—C17 | 177.9 (3) | C12—C13—C14—C9 | −1.3 (5) |
References
- Barrès, A.-L., El-Ghayoury, A., Zorina, L. V., Canadell, E., Auban-Senzier, P. & Batail, P. (2008). Chem. Commun. pp. 2194–2196. [DOI] [PubMed]
- Batsanov, A. S., Collings, J. C. & Marder, T. B. (2013). Private communication (deposition number 855467). CCDC, Cambridge, England.
- Bruker (2016). SAINT. Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruker (2017). APEX3. Bruker AXS Inc., Madison, Wisconsin, USA.
- Dumele, O., Wu, D., Trapp, N., Goroff, N. & Diederich, F. (2014). Org. Lett. 16, 4722–4725. [DOI] [PubMed]
- Kahlfuss, C., Denis-Quanquin, S., Calin, N., Dumont, E., Garavelli, M., Royal, G., Cobo, S., Saint-Aman, E. & Bucher, C. (2016). J. Am. Chem. Soc. 138, 15234–15242. [DOI] [PubMed]
- Krause, L., Herbst-Irmer, R., Sheldrick, G. M. & Stalke, D. (2015). J. Appl. Cryst. 48, 3–10. [DOI] [PMC free article] [PubMed]
- Lehnherr, D., Alzola, J. M., Lobkovsky, E. B. & Dichtel, W. R. (2015). Chem. Eur. J. 21, 18122–18127. [DOI] [PubMed]
- Macrae, C. F., Sovago, I., Cottrell, S. J., Galek, P. T. A., McCabe, P., Pidcock, E., Platings, M., Shields, G. P., Stevens, J. S., Towler, M. & Wood, P. A. (2020). J. Appl. Cryst. 53, 226–235. [DOI] [PMC free article] [PubMed]
- Peters, K., Peters, E.-M. & Syassen, K. (1996). Z. Kristallogr. 211, 360.
- Sheldrick, G. M. (2015a). Acta Cryst. A71, 3–8.
- Sheldrick, G. M. (2015b). Acta Cryst. C71, 3–8.
- Tse, Y. C., Docker, A., Zhang, Z. & Beer, P. D. (2021). Chem. Commun. 57, 4950–4953. [DOI] [PubMed]
- Westrip, S. P. (2010). J. Appl. Cryst. 43, 920–925.
- Yan, X., Zhao, Y., Cao, G., Li, X., Gao, C., Liu, L., Ahmed, S., Altaf, F., Tan, H., Ma, X., Xie, Z. & Zhang, H. (2023). Adv. Sci. 10, 2203889. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S2414314623005539/hb4433sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2414314623005539/hb4433Isup2.hkl
Supporting information file. DOI: 10.1107/S2414314623005539/hb4433Isup3.cml
CCDC reference: 2191397
Additional supporting information: crystallographic information; 3D view; checkCIF report