

ABSTRACT
Advances in single-cell RNA sequencing provide an unprecedented window into cellular identity. The abundance of data requires new theoretical and computational frameworks to analyze the dynamics of differentiation and integrate knowledge from cell atlases. We present ‘single-cell Type Order Parameters’ (scTOP): a statistical, physics-inspired approach for quantifying cell identity given a reference basis of cell types. scTOP can accurately classify cells, visualize developmental trajectories and assess the fidelity of engineered cells. Importantly, scTOP does this without feature selection, statistical fitting or dimensional reduction (e.g. uniform manifold approximation and projection, principle components analysis, etc.). We illustrate the power of scTOP using human and mouse datasets. By reanalyzing mouse lung data, we characterize a transient hybrid alveolar type 1/alveolar type 2 cell population. Visualizations of lineage tracing hematopoiesis data using scTOP confirm that a single clone can give rise to multiple mature cell types. We assess the transcriptional similarity between endogenous and donor-derived cells in the context of murine pulmonary cell transplantation. Our results suggest that physics-inspired order parameters can be an important tool for understanding differentiation and characterizing engineered cells. scTOP is available as an easy-to-use Python package.
Keywords: Cell types, Differentiation, Developmental trajectory, Single-cell RNA sequencing, Computational method
Summary: Computational method to quantify cell identity and visualize the dynamics of differentiation using concepts from statistical physics. The method provides meaningful coordinates for comparing single-cell RNA-sequencing data across experiments.
INTRODUCTION
Humans and other animals have many specialized cells that arise through complex developmental processes (Zeng, 2022). At the blastocyst/epiblast stages of embryonic development, cells are pluripotent and possess the flexibility to develop into fates from all three germ layers (Wolpert, 2019). However, as embryos mature, cellular identity becomes less plastic and more specified through a complex spatiotemporal process involving signaling and other environmental cues. The questions of how and why cells end up in one fate or another are fundamental to developmental biology (Stephenson et al., 2012; Zhu and Zernicka-Goetz, 2020; Rand et al., 2021). More practically, understanding the process of cell-fate specification is crucial for developing directed differentiation protocols for engineering cells in vitro, with important clinical applications for human health and disease (Rossant, 2011; Rowe and Daley, 2019; Basil et al., 2020).
One powerful set of techniques for characterizing cellular phenotype is transcriptome-wide measurement using single-cell RNA sequencing (scRNA-seq). Typical scRNA-seq experiments involve thousands of genes and dozens to millions of cells. For this reason, data from such experiments are intrinsically high-dimensional and require specialized computational tools for analysis and interpretation (Zhu and Zernicka-Goetz, 2020; Setty et al., 2019; Schiebinger et al., 2019). This task is especially difficult because the technical challenges of sequencing RNA from individual cells result in data that is noisy and sparse (Hicks et al., 2018; Lähnemann et al., 2020; Argelaguet et al., 2021). Genes with relatively low expression are often recorded as not being expressed at all, resulting in zero counts (called dropouts). Experiment-to-experiment differences in sequencing methods, platforms and other difficult-to-standardize conditions result in technical variations in scRNA-seq measurements called batch effects. These challenges merit careful consideration of how to normalize and analyze the data in a way that permits the study of biological variation without conflation by technical variation.
scRNA-seq is often used for cellular identification. The most frequently used workflow involves applying dimensional reduction, such as uniform manifold approximation and projection (UMAP) (McInnes et al., 2018 preprint) or SPRING (Weinreb et al., 2018), to visualize the data in two dimensions, then using unsupervised clustering to group individual cells. These groups are then often manually annotated according to expression levels of known marker genes. Machine-learning classifiers, such as support vector machines, are also commonly used to identify cells after training with labeled data (Abdelaal et al., 2019). Another common analysis task is trajectory inference, in which the goal is to infer developmental trajectories from static time shots (reviewed by Saelens et al., 2019; Wagner and Klein, 2020). Trajectory inference arranges cells on a low-dimensional manifold and assigns pseudo-times according to transcriptomic similarity. There are also methods, such as partition-based graph abstraction (Wolf et al., 2019), that purport to combine clustering and trajectory inference to simultaneously calculate pseudo-times and provide discrete labels at varying resolutions. RNA velocity-based methods, such as Dynamo (Qiu et al., 2022), use splicing information or time-resolved data to predict paths of differentiation from RNA data.
Despite the power and utility of these computational tools, most commonly employed methods for analyzing scRNA-seq data share certain limitations. Dimensional reduction is often one of the first steps in visualizing scRNA-seq data, but current methods highly distort cell-to-cell and cluster-to-cluster quantitative relationships (Chari et al., 2023). Distorted embeddings inconsistently represent distances between individual data points and between cell types, making it difficult to interpret cell-fate transitions. Similar issues arise in trajectory inference and RNA-velocity methods because local neighborhoods can also vary substantially with the choice of embedding. Another limitation of these methods is the lack of reproducibility inherent to methods such as t-distributed stochastic neighbor embedding (t-SNE) and UMAP, which output different results on separate executions even when hyperparameters remain the same (Wattenberg et al., 2016). Many algorithms also perform feature selection (e.g. limiting their analysis to the most variable genes or highly expressed genes) or require parameters, such as perplexity, to be chosen, making results even more sensitive to the inclusion or exclusion of different datasets in the analysis.
An ideal analysis algorithm for scRNA-seq data should possess certain desirable properties. First, the algorithm should incorporate explicit assumptions that can be kept consistent between analyses. Second, the results of the algorithm should be reproducible, so that direct comparison between data sets is possible. Third, the axes in visualization should be interpretable, intuitive, and remain fixed across analysis and datasets. Fourth, the algorithm should assign a continuous value to cell identity instead of assigning discrete identity labels, thus enabling a more subtle and fine-grained analysis of cellular identity and developmental trajectories that reflect the continuous nature of biology. Finally, it should be possible to easily incorporate datasets from multiple data sources, including the wealth of new single-cell atlases now being generated (Quake, 2022).
Here, we present a new physics-inspired algorithm, single-cell Type Order Parameters (scTOP) (see Fig. 1), that has all of these desired properties. The algorithm is based on the statistical physics concept of an order parameter – a coarse-grained quantity that can distinguish between phases of matter. An order parameter takes on different values depending on the phase of the system. Typically, in a disordered phase, the corresponding order parameter is zero, and in an ordered phase it is nonzero. Arguably, the most famous example of an order parameter is magnetization in a ferromagnetic system, which is a collection of spins (in the simplest case, spins can be ‘up’ or ‘down’), which align with one another. In a ferromagnet, the magnetization measures the alignment of spins and can be used to distinguish magnetic and paramagnetic phases (Landau and Lifshitz, 2013; Sethna, 2021). Below a particular temperature (known as the ‘critical temperature’), the spins in the ferromagnet align with one another. In a simple model, the order parameter (magnetization, which measures the average spin) is approximately one if all the spins are up and approximately negative one if all the spins are down. When the temperature exceeds the critical temperature, the system changes from the magnetic phase to the paramagnetic phase, and the spins do not align; instead, they are randomly up or down, and the order parameter is approximately zero.
Fig. 1.

Steps involved in the scTOP algorithm. (A) Each cell in the query data is preprocessed independently of the other cells in the dataset. (B) scRNA-seq atlases, such as the Mouse Cell Atlas, are used to define the reference basis in the algorithm. (C) scTOP scores are the projections of query data onto the non-orthogonal subspace of cell types. Because there is no statistical fitting, no tuning parameters are involved. (D) The process of finding scTOP scores is shown in the gray section, and the blue section illustrates cell-type space. This space has as many dimensions as there are cell types in the reference basis. We can visualize this concept in 1, 2 or 3 dimensions, depending on which cell types are most relevant. In the three-dimensional case, the shadows on the bounds of the scatter plot are included to better illustrate the position of points in three-dimensional space.
It is straightforward to generalize the idea of magnetization to more complex settings, such as spin glasses and attractor neural networks (Amit et al., 1985). In epigenetic landscape models of cell identity based on attractor networks, this takes the form of generalized magnetizations that measure how aligned a gene expression state is with each of the attractor basins for different cell fates (Lang et al., 2014; Pusuluri et al., 2017). Whereas magnetization for the ferromagnetic system measures whether the system is in a magnetic or paramagnetic phase, these generalized magnetizations measure which phase a gene regulatory network is in (where the different phases correspond to different cell types). These generalized magnetizations serve as natural order parameters for cellular identity and can be calculated directly from data (Lang et al., 2014; Pusuluri et al., 2017; Dame et al., 2017; Ikonomou et al., 2020). The scTOP algorithm builds on previous work by developing methods for integrating scRNA-seq data, greatly expanding the power and utility of this approach. See supplementary information, section A for a further discussion on interpreting score values.
The inputs to scTOP are the scRNA-seq measurements of query data as well as a reference basis containing the archetypal gene expression profiles for cell types of interest. The outputs of scTOP are the coordinates of the query data in cell-type order parameter space (i.e. generalized magnetizations), with each output dimension serving as a measure of similarity between the query data and a distinct cell type in the reference basis. In this way, scTOP naturally projects gene expression deterministically onto a space with dimension equal to the number of cell types in the reference basis. These generalized coordinates can be used for both cell-type identification and visualization. One appealing feature of scTOP is that the assumptions of the algorithm are explicit and are contained entirely in the choice of reference basis. This allows direct comparison across data sets and settings because the coordinates of and distances between data points do not change as long as one uses the same reference basis. One important limitation of scTOP worth noting is that it cannot be easily used to identify novel cell fates. The underlying reason for this is that scTOP assumes one already knows what cell types are of interest in the form of a reference basis and, for this reason, is less suited for exploratory work with undocumented cell types.
In this article, we show that scTOP can be used to assess cell identity reliably and to classify cell type with high accuracy at the resolution of individual cells. We demonstrate this using mouse and human samples from lungs, brains and other organs, including transplanted cells that were generated through the directed differentiation of pluripotent stem cells (PSCs). By analyzing lineage-tracing data and embryonic development data, we also establish the capacity of scTOP to quantify transitions between cell types, including bifurcations between closely related cell fates. We also show that scTOP can be used to assess quantitatively the transcriptional similarity of engineered and natural cells by analyzing murine pulmonary transplantation experiments.
We have implemented scTOP in an easy-to-use Python package, available on the Python Package Index and GitHub. The code for the analyses presented here is available in a separate GitHub repository.
RESULTS
Benchmarking and validation: classification of cell fate
scTOP scores can be used to predict cell identity reliably and accurately. To show this, we applied the algorithm to several scRNA-seq datasets across species and laboratories. scTOP is organism agnostic as long as an appropriate reference basis is used. Mice and humans are among the most common subjects of scRNA-seq measurements, and we restricted our analysis in this article to data from these organisms. We made use of single-cell atlases, such as the Mouse Cell Atlas (Han et al., 2018) and the Atlas of Lung Development (Negretti et al., 2021).
Table 1 lists each of the datasets examined here with their respective scTOP score accuracies. For cases in which the training (or ‘reference’) data source was the same as the test data source, we set aside a randomly selected subset of cells for the training set and did not use them in the test data. TopN refers to the percentage of cells for which their true cell types were in the top N scTOP scores, with a score greater than 0.1. Unspecified refers to the percentage of cells for which the highest scTOP score was less than 0.1; this represents a failure of the algorithm to identify the cell confidently. The accuracies across species and tissues were high, and the unspecified percentages were very low, even in datasets exceeding a million cells.
Table 1.
Accuracy scores of included sample types for each of the described data sets

scTOP performed comparably to state-of-the-art methods for cell identification. Abdelaal et al. (2019) compared the performance of automatic cell identification algorithms for various datasets, including the CellBench dataset and an older version of the Allen Mouse Brain atlas. They found that the median F1-score for the CellBench dataset ranged from 0.9 to 1; scTOP had a median F1-score of 1 for the same data. The version of the Allen Mouse Brain atlas they analyzed contained three levels of cell population annotations, with 3, 16 and 92 populations for each annotation level. The median F1-score ranged from 0.64 to 1 for the 16-population case and 0 to 0.98 for the 92-population case. The version of the brain atlas we analyzed contained 40 populations, and scTOP had a median F1-score of 0.88 for this dataset.
Example 1: lung lineages
To demonstrate the efficacy of scTOP in the case in which the reference basis and query data come from different sources, we projected mouse lung data from Herriges et al. (2023) onto the mouse cell atlas. The Herriges et al. dataset measures gene expression levels of six different specialized lung types from healthy mice: AT1, AT2, basal, ciliated, secretory and neuroendocrine (see Fig. 2A). The reference basis used to find the individual scTOP scores for this dataset was derived from the Mouse Cell Atlas. Although the Herriges et al. dataset and the Mouse Cell Atlas were created in different laboratories using different sequencing methods, the resulting scTOP scores distinctly separated the Herriges et al. dataset into the expected cell types. Furthermore, the individual scTOP scores strongly correlated with the expression of lineage marker sets (Fig. 2B). Altogether, this suggests that scTOP provides a reliable method to annotate scRNA-seq datasets without the need for the expertise required to generate lineage marker gene sets.
Fig. 2.

scTOP identifies mouse lung cell types. (A-D) Using the Mouse Cell Atlas as the reference basis, we show that mouse lung types separate clearly on the scTOP score axes. The data points correspond to individual cells, and in A the marker shape and color indicate the true cell type as determined by annotations from Herriges et al. (2023). The axes used are scTOP scores for similarity with lung ciliated cells and alveolar types 1 and 2 (AT1, AT2). In B-D, individual cells are colored by the marker genes for the type indicated by the color bars at the end of each row (see supplementary information, section H for details on the marker genes used).
Example 2: mouse brain atlas
The mouse brain atlas (Yao et al., 2021) sequenced over 1,000,000 cells and clustered them into 42 subclasses and 101 supertypes. For a reference basis, we reserved a subset of 200 cells from each of the identified subclasses to use as training data. The test data, from which the accuracy score was calculated, was the remaining cells of each type not included in the training data. Ten different cell types are visualized in a grid (Fig. 3A). The color of each grid square corresponds to the average scTOP score of cells annotated as the type on the y-axis, with the cell type corresponding to the scTOP dimension on the x-axis. As shown by the distinct blue diagonal, the average scTOP score correctly matched the corresponding annotated cell type. The distributions of correctly-matched scores for a few cell types are shown in Fig. 3B. Each of the correctly-matched scTOP score distributions has a relatively high mean and low variance. This demonstrates that scTOP is able to distinguish cell fates even from closely related neural lineages.
Fig. 3.
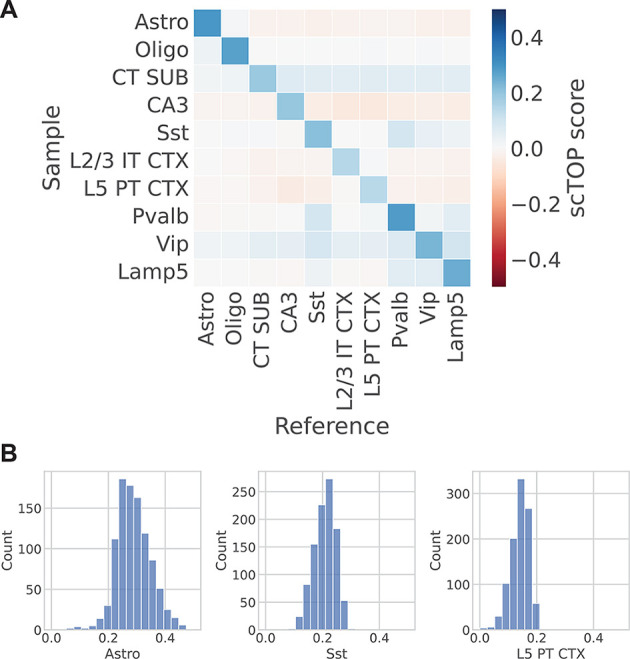
scTOP identifies mouse brain cell types. (A) Heatmap showing the average scTOP score for individual cells of the type indicated on the y-axis, compared with the reference types on the x-axis. A subset of cells from the Mouse Brain Atlas is used as the reference basis to query other cells from the same data set. The diagonal indicates that scTOP accurately matches query cells to the true reference type. (B) Histograms showing the distribution of scTOP scores for individual cells of the type labeled on the x-axis. The scores shown are of the same type as the sample data. The left-hand histogram shows the distribution of Astro scores for Astro cells, and the average of this distribution is indicated by the color of the upper left corner box in the heatmap shown in A. The cell types shown here are astrocytes (Astro), oligodendrocytes (Oligo), L6 corticothalamic in the subiculum (CT SUB), hippocampal field 3 (CA3), isocortex intratelencephalic layers 2/3 (L2/3 IT CTX), isocortex pyramidal tract layer 5 (L5 PT CTX) and GABAergic inhibitory neuronal subclasses (labelled by their corresponding marker genes Sst, Pvalb, Vip and Lamp5).
Example 3: matching cell types to tissues
scTOP can also be used to match cells with tissues/organs. We illustrate this by comparing cells from the Mouse Cell Atlas with a reference basis consisting of Tabula Muris organs (Schaum et al., 2018). To do so, we averaged RNA counts across cell populations from the Mouse Cell Atlas, then preprocessed the averaged data and calculated scTOP scores. These aggregate pseudo-bulk scores tended to be higher than individual cell scores because averaging over many cells compensates for the dropout effect. As shown in Fig. 4A, we found the aggregate scTOP scores for cells from the Mouse Cell Atlas were correctly and strongly identified with the annotated tissue of origin in the Tabula Muris atlas. Parenchymal cell types were the most associated with the correct organ, whereas stromal cells were sometimes misidentified – for example, the mammary gland endothelial cells. This suggests that stromal cells are less specified to each organ. A discussion of the accuracy of scTOP, when applied to stromal types versus parenchymal types, may be found in supplementary information, section G1.
Fig. 4.

scTOP correctly classifies tissues across biological contexts. The shade of each bin indicates the average scTOP score for individually queried cells projected on the reference cell type indicated on the x-axis. The y-axis lists the true types of the queried cells. The dark diagonals demonstrate that scTOP scores correctly match the queried data to the corresponding true types. (A) Aggregate pseudo-bulk samples from the Mouse Cell Atlas are compared with a reference basis consisting of organs from Tabula Muris. The scTOP scores are significantly higher when comparing a cell type with the organ of origin. (B) The CellBench dataset contains human cell cancer lines specifically processed to benchmark scRNA-seq algorithms. We take a subset of cells to use as the reference basis in analyzing the rest of the CellBench data. (C) Cells from the Human Lung Atlas are compared with a reference basis constructed from cells from the Human Lung Atlas.
Example 4: human data
The CellBench dataset (Tian et al., 2019) was designed specifically to benchmark scRNA-seq analysis methods. We used the subset of data pertaining to five cancer lines. Two-hundred cells from each line were used as training data to create the reference basis, then the rest of the cells were input as query data. As shown in Fig. 4B and Table 1, scTOP classified the cell line identity of the test data with 100% accuracy.
To illustrate the power of scTOP for classifying human samples, we analyzed data from a human lung atlas dataset (Travaglini et al., 2020). This dataset sequenced thousands of human lung cells and identified 58 phenotypic populations. For our analysis, we restricted the training and test data to epithelial and stromal types that had at least 200 cells in each population. The reference basis was created using 80% of the cells from each population, and the query data consisted of the remaining 20% of the data. Fig. 4C shows that the scTOP scores were consistently high in cases in which the score type matched the true type of the query.
As demonstrated by the diverse data sources examined here, as long as a well-defined reference basis is available, scTOP works extremely well for cell-type identification across different measurement conditions, species, organs and resolutions. This performance is especially impressive given that, as discussed earlier, the algorithm does not use any statistical fitting procedures or dimensional reduction methods such as principle components analysis, tSNE, UMAP or SPRING.
Visualizing cell-fate dynamics using scTOP
Having demonstrated the robust ability of scTOP to identify stable cell types, we next demonstrate how scTOP can be used in combination with scRNA-seq time series to visualize transient cell states in dynamic processes, such as development and differentiation.
Lung development
The alveolar epithelium of the lung undergoes dramatic maturation and morphological changes during embryonic and perinatal development, ultimately giving rise to AT1 cells and AT2 cells. To investigate this process with scTOP, we re-analyzed alveolar epithelial cells from a recent dataset by Zepp et al. (2021), which includes seven time points between embryonic day (E) 12.5 and postnatal day (P) 42. These cells were then compared against a reference basis developed from adult cell types in the Mouse Cell Atlas and an E12.5 epithelial progenitor cell type from the Atlas of Lung Development (Negretti et al., 2021).
The resulting AT1 and AT2 scTOP scores were plotted to visualize maturation into these two lineages (Fig. 5). As expected, early embryonic progenitor cells (E12.5, E15.5), which express few of the mature lineage markers, aligned poorly with both lineages. In contrast, the E17.5 alveolar epithelial cells were distributed across a wide spectrum with cells aligning to AT1 cells, AT2 cells, or both lineages. In the postnatal time points, this continuous spectrum gave way to more distinct clusters, eventually resolving into AT2 and AT1 cell clusters at P42. Similar to our analysis of adult mouse lungs (Fig. 2), the scTOP scores changed over the course of development, correlating well with our lineage marker gene sets.
Fig. 5.

Visualizing the developing mouse lung with scTOP. (A-C) scTOP illustrates the specification of alveolar type 1 (AT1; A) and type 2 (AT2; B) cells in murine embryonic development. Alveolar cells from Zepp et al. (2021) are compared with a reference basis constructed from adult lung cells from Herriges et al. (2023) and early epithelial cells from the Atlas of Lung Development. Each subplot corresponds to the age of the mouse from which the sample was extracted, from post-conception day 12.5 (E12.5) to 42 days post-birth (P42). Each cell is colored by the average of that cell's expression of AT1 marker genes (A) and AT2 marker genes (B). The scTOP scores of AT1 (or AT2) cells are high when the average AT1 (or AT2) marker gene expression is high.
In addition to the expected postnatal AT2 and AT1 cell clusters, this scTOP analysis illustrated a transient AT2/AT1 hybrid population that persisted through P7. The existence of these hybrid AT2/AT1 cells had already been detected by Zepp et al. using a combination of graph-based clustering and gene enrichment profiling (see figure 1C,I in Zepp et al., 2021). The authors conjectured that these AT2/AT1 cells were a transitional state of AT2 similar to the Spock2+/Axin2+ AT2 cells previously identified in adult lungs (Frank et al., 2016). To assess whether this was the case, we displayed Axin2 expression on our scTOP plots (Fig. S5A). These cells did not exhibit significant upregulation of Axin2 relative to perinatal or adult AT2 cells, suggesting that the postnatal hybrid AT2/AT1 cells are distinct from adult Spock2+/Axin2+ AT2 cells (Frank et al., 2016).
We then performed a similar analysis of markers for other published postnatal hybrid AT1/AT2 or transitional alveolar cell states, including alveolar differentiation intermediate cells (ADI cells; markers Krt8 and Trp53), pre-alveolar type-1 transitional cell state (PATS; markers Cldn4 and Krt19) and damage-associated transient progenitors (DATPs; markers Il1r1 and Hif1a) (Verheyden and Sun, 2020; Strunz et al., 2020; Kobayashi et al., 2020; Choi et al., 2020). In each case, markers were not uniquely upregulated in the perinatal hybrid AT1/AT2 cells relative to either the perinatal or adult committed lineages (Fig. S5B-D). Furthermore, differential gene expression analysis failed to identify any genes that were uniquely differentially expressed in the hybrid AT1/AT2 cells relative to either timepoint-matched AT1 or AT2 cells. These results suggest that this population is most likely transcriptionally distinct from previously identified adult AT2-transitional cell types.
scTOP for hematopoiesis using lineage-tracing data
In the previous section, we used scTOP to visualize differentiation into two closely related lung epithelial cell fates. In this section, we show how scTOP can also be used to gain insights into more complicated developmental processes, such as hematopoiesis, in which hematopoietic stem cells (HSCs) give rise to multiple differentiated blood cell types. To do so, we make use of the unique dataset by Weinreb et al. (2020) on hematopoiesis in mice using a new technique, lineage and RNA recovery (LARRY), that combines scRNA-seq and lineage-tracing data. LARRY uses heritable barcodes that can be detected using scRNA-seq, allowing for the tracking of clone families during differentiation. Here, we focus on visualizing the ex vivo experiments from Weinreb et al., in which cells were sequenced at a sufficient depth to infer lineage relationships. In these experiments, HSCs were extracted from mice, barcoded, and allowed to expand in primary culture. scRNA-seq measurements were performed on days 2, 4 and 6 post-barcoding. The cells were then annotated using marker genes into nine differentiated blood cell fates.
We restricted our analysis to a subset of these annotated cell types with at least 150 cells. We also excluded cells labeled as erythroids from our analysis because we found that this population was highly heterogeneous and gave rise to poor projections [likely owing to the fact it was annotated using a single marker gene, Hbb-bs (Weinreb et al., 2020), which did not sufficiently define a distinct population]. Furthermore, we treated all day 2 undifferentiated cells as a single population that we called progenitors. This resulted in a reference basis consisting of seven cell types: undifferentiated progenitors, neutrophils, monocytes, megakaryocytes, mast cells, eosinophils and basophils. To construct the basis, we averaged the RNA counts for 150 random cells of each of these types, only including cells if they had no sister clones. Then, we preprocessed the averaged gene expression profiles and inserted them into the reference basis.
Fig. 6 shows the differentiation dynamics of three different clonal families using scTOP. The histograms show the distribution of scTOP projection scores on each cell type for days 2, 4 and 6 post-barcoding, and the scatter plots show two-dimensional cross-sections of scTOP projection scores as a function of time for the indicated cell types. In all the plots, the scTOP projections for the undifferentiated hematopoietic progenitor stem cell decreased over time, with a high projection on day 2 and a low projection on day 6, whereas the scores for the mature types increased over time.
Fig. 6.

Visualizing hematopoietic lineage tracing with scTOP. (A-C) Scatter plots and distributions of scTOP scores for individual in vitro clone families from Weinreb et al. (2020). The scatter plots show the x-axis scTOP score compared with the y-axis scTOP score for individual cells, and the distributions show the scTOP scores of the type indicated on the x-axis of the corresponding column. The color and shape of each marker (and the color of each distribution) indicate the days in culture on which the cells were extracted. (A) A clone family in which all of the cells end up becoming neutrophils. (B) A clone family in which some clones end up becoming neutrophils and some become monocytes. At day 2, the three sister cells have low neutrophil and monocyte scores and high progenitor scores. At days 4 and 6, the progenitor scores decrease, and a portion of the cells increase in monocyte scores whereas others increase in neutrophil scores. (C) A clone family in which the descendant cells become neutrophils, eosinophils and basophils.
Interestingly, the three clones showed very different behaviors over the course of maturation. Fig. 6A shows a clone for which a single barcode detected at day 2 gave rise to multiple cells in the neutrophil lineage. This can be seen in the plots by noting that the day 6 cells have near zero scores on both progenitor cells and other differentiated lineages, such as monocytes, but project significantly on the neutrophils. Fig. 6B shows a barcode that was detected in three cells on day 2 that gave rise to a mix of neutrophils and monocytes on day 6. Interestingly, the day 4 cells moved in the direction of both of these lineages before bifurcating at day 6. These data are consistent with those of Weinreb et al., who previously highlighted the existence of such a bipotent neutrophil-monocyte progenitor population. We also found evidence that progenitor cells can even give rise to three distinct cellular populations. Fig. 6C shows a single barcode found on day 2 that resulted in day 6 cells with three distinct populations with non-zero scores on basophils, eosinophils and neutrophils, suggesting that a single clone can bifurcate into three different types of differentiated cell types.
Fig. 6 focuses on scTOP scores for individual clonal families, whereas Fig. 7 shows the scTOP projection scores across all 1816 multi-generational clonal families in the dataset. In each subplot, the horizontal axes show progenitor scores, and the vertical axes correspond to the six differentiated cell types included in the reference basis. Each bin of the two-dimensional histogram is colored according to the average day of all the cells that fall within that bin. For this reason, although the cells were only sampled on days 2, 4 and 6, the color bar is continuous, with a range between 2 and 6 days. The histograms show that, as expected, the day 2 cells generally had high projections on progenitor cells, but as time passed, progenitor scores decreased whereas the scores on differentiated cell types either increased (indicating cells that differentiated into the cell type on the y-axis) or remained close to zero (indicating cells that differentiated into a different cell type than that on the y-axis). Cell types predicted by the scores match the expected marker gene expression, as shown in Fig. S4. Somewhat surprisingly, the plots for different cell types look qualitatively different from each other (for example, compare the plots for megakaryocytes and monocytes).
Fig. 7.
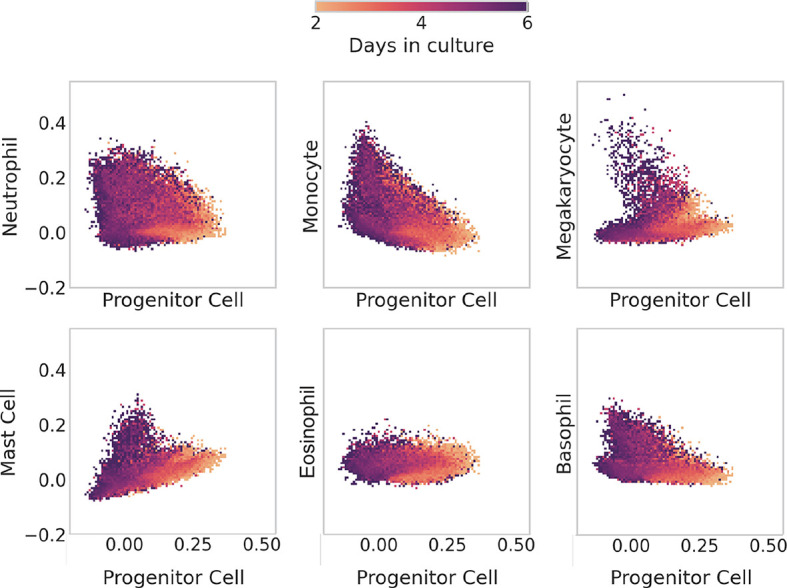
Tracing differentiation of hematopoietic cells from progenitor to mature types. Two-dimensional histograms of all of the in vitro clone families from Weinreb et al. (2020), with the x-axis of each one indicating the scTOP score for hematopoietic progenitor type and the y-axis showing the score for a mature type. Each bin of the histogram is colored by the average day in culture of the data points that fall within the bin. Early cells have high progenitor scores and low mature-type scores. As time passes, cells end up with high scores in mature types.
To try to understand this better, we made additional two-dimensional histograms comparing scTOP scores between all differentiated cell types in our basis (see Fig. 8). What is striking about these plots is that they show two qualitatively different behaviors depending on which pairs of cells are being compared: the differentiation pathways for some pairs are mutually exclusive, whereas other pairs show a more graded behavior. For example, the histogram for megakaryocytes and monocytes exhibits a distinct L-shape, indicating that these developmental programs are mutually exclusive because cells never have significant projection scores of both cell types simultaneously. This is in stark contrast with the histogram for monocytes and neutrophils, where there exists a continuous spectrum of cells that are monocyte-like, neutrophil-like, and every step in between.
Fig. 8.

Comparing differentiation of hematopoietic cells between mature types. As in Fig. 7, the plots shown here are two-dimensional histograms of all of the in vitro clone families from Weinreb et al. (2020). The x-axis and y-axis of each histogram indicate the scTOP score of the labeled mature type. Each bin of the histogram is colored by the average day in culture of the data points that fall within the bin. Some pairs of types have cells with high scores of both types. This is apparent in the neutrophil-monocyte histogram, which presents a continuous spectrum of scores from the horizontal axis to the vertical. Other pairs of types only have cells expressing one type or the other, as in the L-shaped neutrophil-megakaryocyte histogram.
Our scTOP-based visualizations of developmental decisions during hematopoiesis complement the analysis of Weinreb et al. (2020) using pseudo-time. Like in the original paper, we found strong evidence for the existence of progenitor cells that can give rise to multiple lineages (Fig. 6), including a neutrophil/monocyte bivalent progenitor and possibly a basophil/eosinophil/neutrophils trivalent progenitor. The existence of these multi-potent progenitors seems to give rise to graded developmental dynamics whereby intermediate cells have significant projections on multiple cell types. This is in stark contrast with developmental decisions between other cell lineages (megakaryocytes versus monocytes, basophils versus megakaryocytes, monocytes versus basophils), which seem to be mutually exclusive. One compelling feature of these visualizations using scTOP is that they require no statistical fitting, dimensional reduction, or ordering of cells. Once the relevant reference basis (in this case, progenitors and six differentiated lineages) has been chosen, the resulting plots give an interpretable way of assessing complex developmental dynamics.
Using scTOP to compare endogenous and transplanted cells
Recent advancements in cell culture and cell transplantation techniques have allowed researchers to generate cells that were maintained or differentiated in vitro to approximate and eventually replace endogenous cells generated during in vivo development. However, even with scRNA-seq, it is difficult to consistently quantify the transcriptomic similarity between donor-derived and endogenous lineages from transplant recipients. As it provides a quantitative measure of cell type similarity, scTOP can be useful for comparing cell populations. A relevant example can be found in pulmonary cell replacement therapy, where one goal is to differentiate or maintain alveolar cells in vitro and then transplant them into injured lungs.
To compare two distinct pulmonary cell transplant protocols, we used scTOP to analyze the results of scRNA-seq from two recent publications featuring murine pulmonary cell transplantation (Herriges et al., 2023; Louie et al., 2022). In the Louie et al. study, adult mouse AT2 cells were maintained in culture with support cells for 3 weeks to generate a donor cell population. In contrast, in the Herriges et al. study the authors developed a protocol for directed differentiation of mouse PSCs into lung distal tip-like cells, which mimic an embryonic progenitor of AT2 cells. In both papers, the donor cells were transplanted into injured mouse lungs, where they survived and gave rise to multiple cell lineages, including AT2-like cells.
To highlight the usefulness of scTOP, we first considered another dimensional reduction method. Dimensional reduction methods like UMAP and t-SNE are often used to compare similar cells from different sources, such as transplanted and endogenous cells. Fig. 9 shows the AT1 and AT2 cell populations from the Herriges et al. and Louie et al. studies, both of which were donor derived and endogenous. In Fig. 9A, three possible UMAP plots are shown with varying algorithmic parameters. The qualitative relations between the clusters are generally consistent, with AT1 cells clustering together apart from the AT2 cells. However, the distances between cells and the relative positions of clusters are not consistent when the UMAP parameter is varied. This illustrates how UMAP is useful for visualization and gaining qualitative intuition for a particular dataset but cannot be used to make a rigorous quantitative comparison between cells.
Fig. 9.

Comparing donor-derived and endogenous alveolar cells from Herriges et al. (2023) and Louie et al. (2022). The cell-type annotations come from a separate Louvain clustering and are the same for all plots in this figure. (A) UMAP visualizations of the data, colored by cell-type annotations. The three have different values for the UMAP parameter n_neighbors, which changes whether the embedding preserves more local or global structure. (B) scTOP plot showing the AT1 score on the x-axis and the AT2 score on the y-axis. (C) Box-and-whisker plot showing the distributions of scTOP AT2 scores for the various cell populations. For each population, the boundaries of the box indicate the first and third quartiles. The line inside the box is the median. Each whisker ends at the farthest point within 1.5x the interquartile range. Fig. S6 shows that the scores for other types are consistently low.
By contrast, Fig. 9B shows the scTOP scores for AT1 and AT2 for the same populations. The distances between cells will only change if the reference basis is changed, and even then will not change significantly (see supplementary information, section G for discussion). The AT1 and AT2 populations were separated along the relevant axes, and the highly similar cell populations were largely overlapping. Because scTOP provides many relevant dimensions to choose from instead of trying to capture high-dimensional distances in two dimensions, it is possible to compare different populations directly to the same cell type. Fig. 9C shows the distributions of AT2 scores for each of the populations, providing a clear quantitative way to compare the various engineered cells. As expected, the endogenous populations from both data sources were similar in distribution. The means and overall distributions of the AT2-derived (Louie et al.) cells were higher than those for the PSC-derived (Herriges et al.) cells, suggesting that the primary cell transplantation better emulates the endogenous AT2 cells.
Fig. S7 provides a comparison between cell populations using aggregate scTOP scores. Similar to Fig. 9C, Fig. S7 provides a quantitative comparison between the populations. This figure confirms that although both AT2-derived and PSC-derived transplants generate cells that have high AT2 scores, the AT2-derived cells are quantitatively more similar to the reference AT2 profile. Altogether, this analysis demonstrates how scTOP can be used to compare two distinct cell populations against a primary control. scTOP satisfies the need for quantitative assessment of transplantation protocols and can be used in combination with functional tests to identify protocols that most effectively replace endogenous cell line ages.
DISCUSSION
Here, we have introduced a new physics-inspired method, scTOP, for analyzing scRNA-seq data. scTOP projection scores provide clear and meaningful axes to visualize differentiation and classify cells. Because scTOP requires no statistical fitting, clustering, or dimensional reduction techniques, plots can be made quickly and easily, even for an individual cell. The input to scTOP is a reference basis of cell fates of interest, allowing us to take advantage of the wealth of new scRNA-seq expression atlases now being generated. Importantly, scTOP scores are robust to the choice of basis and reproducible across experiments, labs and datasets, allowing integration of disparate data sources.
scTOP is especially suited for tasks for which the cell types of interests are well characterized. For example, we have found in our own work that scTOP is a sensitive measure for assessing the fidelity of engineered cells from directed differentiation protocols and reprogramming (Herriges et al., 2023). Because scTOP does not require data harmonization, joint embeddings, or make use of cellular neighborhood information, scTOP is able to distinguish between technical noise and biologically meaningful differences. scTOP also can be used to classify cells with minimal computational costs and thus represents a potentially simple, scalable and reproducible way of dealing with the proliferation of scRNA-seq datasets. As scTOP yields an alternative coordinate system for representing developmental dynamics in cell-fate space, scTOP-based visualizations are a powerful way of thinking about complex developmental processes.
As an illustration, we analyzed the development of the lung epithelial AT1 and AT2 cell fates (Zepp et al., 2021) and developmental dynamics during hematopoiesis (Weinreb et al., 2020). Using scTOP, we showed that it was easy to identify a transient hybrid AT2/AT1 population 3 to 5 days post-birth in which the gene expression profile is distinct from previously investigated transient AT2-to-AT1 states. By plotting scTOP scores as a function of time for AT1 and AT2, hybrid AT2/AT1 cells were easily identified because they formed a distinct cluster that was well separated from mature AT1 and AT2 cells. This example illustrates how given an accurate reference basis, scTOP is able to pick up on small but biologically meaningful differences in global gene expression profiles.
In the context of hematopoietic development, during which HSCs give rise to multiple downstream cell fates, we took advantage of the fact that scTOP maps each cell to a seven-dimensional coordinate (each direction in the coordinate systems corresponds to one of the seven cell types in the reference basis: undifferentiated progenitors, neutrophils, monocytes, megakaryocytes, mast cells, eosinophils and basophils) to generate multiple two-dimensional visualizations using scTOP. We found that a single clonal family can often give rise to up to three different mature cell fates. Our visualizations also suggest that developmental decisions between different cell fates can be classified into two broad categories: mutually exclusive and graded. For cell fates such as megakaryocytes and monocytes, cells never have significant projection scores on both lineages simultaneously, suggesting that the underlying developmental decisions are mutually exclusive. In contrast, for other pairs of cell fates, such as monocytes and neutrophils, cells often exhibit significant projections on both members of the pair, suggesting that these developmental switches function in a more graded manner. It will be interesting to see if this general distinction is also present in other developmental systems or is specific to hematopoiesis.
In the analysis presented here, we have limited ourselves to considering scRNA-seq data. However, in principle, our approach could be easily extended to include other data modalities, such as chromatic accessibility and histone modifications. Recently, several interesting works using techniques such as Dynamo have used RNA velocity to learn cellular dynamics directly (Xing, 2022; Qiu et al., 2022). It will be interesting to see whether these methods can be combined with scTOP to learn vector flows directly in cell-type space. Similarly, methods based on optimal transport, such as Waddington-OT (Schiebinger et al., 2019), or developmental directory reconstruction based on graph embedding, such as Monocle (Qiu et al., 2017), can also be performed in the cellular identity space instead of gene expression space. These represent promising directions for future research because the scTOP cell identity coordinates are often better suited for analyzing developmental dynamics than gene expression coordinates.
We have demonstrated that scTOP can be used to quantitatively assess the fidelity of cultured cells, as in the case of donor-derived lung alveolar cells. However, we currently have no systematic manner within the scTOP framework for providing guidance on adjusting gene expression or designing differentiation protocols to steer cells toward desired fates. Developing such methods would provide powerful new computational tools for engineering cells via directed differentiation.
We are also interested in extending scTOP to improve our understanding of the signaling pathways and genetic signatures underlying developmental dynamics. Although scTOP provides a way to translate between gene expression space and cell type space, there is currently no direct interface with the biologically essential signaling space. Further work in uniting the three relevant spaces, combined with studying the possible bifurcations in these spaces, may provide insight into the complex process of differentiation.
MATERIALS AND METHODS
Mathematical details
To calculate cell type similarity scores, scTOP uses a linear algebra projection method inspired by the concept of cell types as attractors in a dynamical system (Huang et al., 2005). This projection method has been applied previously to bulk RNA-seq samples, and we will summarize the theoretical background here (Lang et al., 2014). A cell type is a state of the gene regulatory network. A natural way to represent attractors in this system is with an attractor neural network, in which attractors correspond to different cell types. If we represent each gene as a node in a network with an associated value representing the gene expression level (positive value representing high gene expression, negative value indicating low gene expression), then a cell type may be denoted by a vector of gene expression values Si, where i=1, …, G spans the G genes being measured. The gene expression profiles corresponding to the C cell type attractors are also G dimensional vectors 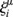 , where μ=1, …, C spans the C cell types being stored in the network. In what follows, we assume that G>C; in other words, the number of genes being measured is greater than the number of cell types.
, where μ=1, …, C spans the C cell types being stored in the network. In what follows, we assume that G>C; in other words, the number of genes being measured is greater than the number of cell types.
Cell types are often highly similar in their patterns of gene regulation. Kanter and Sompolinsky (1987) defined a pattern storage method for spin-glass-like neural networks in which even correlated patterns robustly act as attractors. For the same system, they also defined order parameters: generalized magnetizations aμ, where μ iterates through each of the stored cell-type attractors. The aμ can be understood as de-correlated versions of the conventional spin glass magnetization order parameter that measures the similarity between a given network state and the network state ξμ. We refer to ξ as the reference basis. Explicitly, the C generalized magnetizations aμ can be calculated for a gene expression state Si via the expression:
| (1) |
where
| (2) |
is the overlap matrix of gene expression profiles of different cell types. Previous works have shown the order parameters aμ provided an excellent similarity score for analyzing bulk RNA-seq data (Lang et al., 2014; Pusuluri et al., 2017; Dame et al., 2017; Ikonomou et al., 2020). However, bulk RNA-seq measurements average gene expression over many cells, rendering it impossible to measure cell-fate transitions beyond an average tissue state.
scTOP uses the same order parameters, aμ, to track cell type at the resolution of individual cells, making it possible to directly observe cells in different stages of differentiation. In both attractor neural networks and previous applications to bulk RNA-seq data, the expression vectors Si were binary variables. However, we have found that for working with scRNA-seq data, it is helpful to treat Si as continuous.
Preprocessing
As shown in Fig. 1A, the first stage of the algorithm is preprocessing and normalization of the input data. In our algorithm, measurements from each cell are normalized independently and do not depend on the expression profiles of any other cells in the dataset. We assume that the scRNA-seq data has been processed, resulting in a gene-wise count matrix: for each cell, each gene has an integer value that counts the number of assigned RNA reads. Genes are then rank ordered. The rank order is subsequently converted to a z-score representing the percentile rank of the expression of a gene within the cell relative to all other genes being measured (assuming a normal distribution with mean zero and standard deviation one, for the sake of simplicity). For example, a gene that has higher expression than 97.8% of genes being measured is assigned a score of z=2, whereas a gene that has higher expression than half of the genes is assigned a score of z=0. The details of the process and its effects on data distributions are shown in Fig. S1 and described in supplementary information, section C. The preprocessing step is a key component of the algorithm. By processing each cell independently, the output for one cell is independent of other cells included in the analysis. This is in contrast with other algorithms which normalize gene expression across cells by, for example, selecting the most variable genes. This preprocessing method reduces differences between results caused by batch effects, as shown in Figs S2 and S3.
Construction of reference basis
Constructing a representative, accurate reference basis for the data being analyzed is vital to the scTOP algorithm. This process is shown in Fig. 1B. First, relevant scRNA-seq atlases are gathered to be processed. For example, for analysis of mouse lung cells, we used the Mouse Cell Atlas (Han et al., 2018), which contains single-cell samples of mouse tissues across all major organs. Once the relevant atlases are identified, we take an average across each cell population that corresponds to a distinct cell type. To mitigate the potent effects of noise and avoid training data imbalances, a sufficient number of cells must be included in each population. We have empirically found that the minimum sample size to achieve reasonable results is 100-200 cells. Noise affects the algorithm most strongly when it appears in the basis because the decorrelation operation involved in the projection separates cells according to the canonical expression levels included in the reference basis. After each cell population is averaged to create the gene expression profiles for the desired cell types, each cell type profile is preprocessed separately using the same procedure as that used for processing single cells: normalization followed by z-scoring. This creates a modular reference basis whereby cell types can easily be replaced, removed or added at will.
Calculation of scTOP scores
Given a reference basis, we can calculate the order parameters aμ using Eqn 1. Although these order parameters were originally inspired by attractor neural networks and spin-glass physics, the aμ can also be understood as non-orthogonal projections onto the subspace spanned by the reference basis (i.e. cell type space). This is illustrated in Fig. 1C. An scRNA-seq measurement of a sample can be thought of as a vector in G-dimensional gene expression space, where G is the number of genes. The C-cell types in the reference basis are also each represented by a G-dimensional vector and form a non-orthogonal basis for the C-dimensional cell type space (we assume C<G). To find the coordinates aμ in cell type space, we project the sample vector onto the reference basis using Eqn 1.
Mathematically, we can write the sample vector as a sum of the projected components and a component 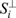 perpendicular to the cell type subspace:
perpendicular to the cell type subspace:
| (3) |
Biologically, S⊥ contains information about processes that affect gene expression but are not associated with cell identity, such as the cell cycle. We refer to the aμ as the scTOP scores. Each component of this vector measures the projection of the cell with gene expression profile Si onto the μ – the cell type in the reference basis. These scores can be used to classify cell identity accurately and provide a natural visualization of gene expression in the space of possible cell fates (see Fig. 1E). Gene regulatory dynamics that are unrelated to transitions between cell types in the reference basis would not be captured by aμ; instead, these would contribute to S⊥. As a result, these fate-unrelated dynamics cannot be visualized using scTOP.
In general, we are interested in calculating scTOP scores for individual cells. However, it is also sometimes useful to compute ‘aggregate’ scTOP scores for cellular populations by averaging the mRNA counts first, then preprocessing the data. This produces a gene expression profile and a set of scTOP aggregate scores for each population. Aggregate scores tend to be higher because averaging over the RNA counts mitigates the well-documented effects of scRNA-seq noise (Hicks et al., 2018; Lähnemann et al., 2020); the averaged gene expression profile for the populations are not as sparse as individual profiles and thus are more similar to the averaged profiles that form the reference basis.
In the supplementary information, we give a detailed discussion of scTOP, including the effects of basis choice, score distributions for correct and incorrect cell types, and other technical details.
Assumptions and computational complexity
scTOP does not require tuning of hyper-parameters or statistical fitting procedures. The assumptions of the algorithm are explicit: that the gene expression profiles included in the reference basis are truly representative of the relevant cell types and that the reference cell types correspond to distinct cellular populations. We have found that constructing a reference basis for a cell type typically requires at least 100 cells. Additionally, if the cell types are not sufficiently different from one another, as in early embryonic development or certain stromal cells where the same cell type is compared between organs, the resulting scTOP scores may be unreliable and exhibit extreme sensitivity to small changes in the choice of reference basis. More information on the limitations of scTOP may be found in the supplementary information. Generally, outside these very limited edge cases, we have found that scTOP can reliably distinguish even extremely closely related cells [as shown in the example of pulmonary alveolar epithelial type 1 (AT1) and type 2 (AT2) cell lineages below].
Finally, we note that scTOP is extremely computationally efficient. The core of the scTOP is Eqn 1. This requires a single matrix inversion involving the reference basis that can be precomputed. For this reason, the computational complexity of scTOP scales linearly with the number of cells. The end result is that scTOP takes milliseconds to run, even for very large datasets.
Supplementary Material
Acknowledgements
We acknowledge useful discussions with Jason Rocks, Robert Marsland and Alex Lang. We would like to thank the Mehta group and Kotton lab for their comments on the manuscript. We also acknowledge programming support from Alena Yampolskaya and Sumner Hearth. This work has benefited from access to the Boston University Shared Computing Cluster, located at the Massachusetts Green High Performance Computing Center.
Footnotes
Author contributions
Conceptualization: M.Y., P.M.; Methodology: M.Y., P.M.; Software: M.Y.; Validation: M.Y., M.J.H., L.I., P.M.; Investigation: M.Y., P.M.; Data curation: M.Y., M.J.H., L.I., D.N.K., P.M.; Writing - original draft: M.Y., P.M.; Writing - review & editing: M.Y., M.J.H., L.I., D.N.K., P.M.; Visualization: M.Y., P.M.; Supervision: D.N.K.; Project administration: P.M.; Funding acquisition: D.N.K., P.M.
Funding
The work was funded by a grant to the Boston University Kilachand Multicellular Design Program (to D.N.K. and P.M.) and the National Institute of General Medical Sciences (1R35GM119461 to P.M.). L.I. was supported by the National Institutes of Health (R01 HL158965-01). Deposited in PMC for release after 12 months.
Data availability
scTOP is available as a package on the Python Package Index (https://pypi.org/project/scTOP/) and the code is accessible via GitHub (https://github.com/Emergent-Behaviors-in-Biology/scTOP). The scripts with the analyses for this article are also available on GitHub (https://github.com/Emergent-Behaviors-in-Biology/scTOP-manuscript).
The scRNA-seq data analyzed can be downloaded from a range of sources. The endogenous and transplanted mouse lung data from the Kotton lab are available on NCBI's Gene Expression Omnibus (GEO) under accession codes GSE200886, GSE200883, GSE200884 and GSE200885, as well as the Kotton Lab's Bioinformatics Portal (https://www.bumc.bu.edu/kottonlab/bioinformatics-portal-kotton-lab/) (Herriges et al., 2023). The Mouse Cell Atlas data are available on FigShare (https://figshare.com/articles/dataset/MCA_DGE_Data/5435866) (Han et al., 2018). The mouse brain atlas 10x data are available on the Neuroscience Multi-omic Data Archive (https://assets.nemoarchive.org/dat-jb2f34y) (Yao et al., 2021). The CellBench data are available in GEO under accession number GSE118767 and on GitHub (https://github.com/LuyiTian/sc_mixology/blob/master/data/) (Tian et al., 2019). The human lung atlas data are available on Synapse (www.synapse.org/#!Synapse:syn21041850) as well as the European Genome-Phenome Archive under accession number EGAS00001004344 (Travaglini et al., 2020). Tabula Muris data are available through FigShare (https://figshare.com/projects/Tabula_Muris_Transcriptomic_characterization_of_20_organs_and_tissues_from_Mus_musculus_at_single_cell_resolution/27733) and in GEO under accession number GSE109774 (Schaum et al., 2018). The developing mouse lung data are available under accession number GSE149563 (Zepp et al., 2021). The hematopoietic lineage tracing data are available in GEO under accession number GSE140802, and the metadata can be found on GitHub (https://github.com/AllonKleinLab/paper-data/tree/master/Lineage_tracing_on_transcriptional_landscapes_links_state_to_fate_during_differentiation) (Weinreb et al., 2020). The endogenous and engineered mouse lung data from the Kim lab are available in GEO under accession number GSE190565 (Louie et al., 2022). The human pancreatic data used for the batch effects figure in the supplementary information was downloaded from Docker (https://hub.docker.com/repository/docker/jinmiaochenlab/batch-effect-removal-benchmarking) (Tran et al., 2020), and is available in ArrayExpress under accession code E-MTAB-5061 (Muraro et al., 2016), or in GEO under accession numbers GSE85241 (Baron et al., 2016), GSE84133 (Segerstolpe et al., 2016), GSE83139 (Wang et al., 2016) and GSE81608 (Xin et al., 2016).
Contributor Information
Maria Yampolskaya, Email: mariay@bu.edu.
Pankaj Mehta, Email: pankajm@bu.edu.
Peer review history
The peer review history is available online at https://journals.biologists.com/dev/lookup/doi/10.1242/dev.201873.reviewer-comments.pdf
References
- Abdelaal, T., Michielsen, L., Cats, D., Hoogduin, D., Mei, H., Reinders, M. J. T. and Mahfouz, A. (2019). A comparison of automatic cell identification methods for single-cell RNA sequencing data. Genome Biol. 20, 194. 10.1186/s13059-019-1795-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit, D. J., Gutfreund, H. and Sompolinsky, H. (1985). Spin-glass models of neural networks. Phys. Rev. A 32, 1007. 10.1103/PhysRevA.32.1007 [DOI] [PubMed] [Google Scholar]
- Argelaguet, R., Cuomo, A. S., Stegle, O. and Marioni, J. C. (2021). Computational principles and challenges in single-cell data integration. Nat. Biotechnol. 39, 1202-1215. 10.1038/s41587-021-00895-7 [DOI] [PubMed] [Google Scholar]
- Baron, M., Veres, A., Wolock, S. L., Faust, A. L., Gaujoux, R., Vetere, A., Ryu, J. H., Wagner, B. K., Shen-Orr, S. S., Klein, A. M.et al. (2016). A single-cell transcriptomic map of the human and mouse pancreas reveals inter-and intra-cell population structure. Cell Syst. 3, 346-360.e4. 10.1016/j.cels.2016.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basil, M. C., Katzen, J., Engler, A. E., Guo, M., Herriges, M. J., Kathiriya, J. J., Windmueller, R., Ysasi, A. B., Zacharias, W. J., Chapman, H. A.et al. (2020). The cellular and physiological basis for lung repair and regeneration: past, present, and future. Cell Stem Cell 26, 482-502. 10.1016/j.stem.2020.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chari, T. and Pachter, L. (2023). The specious art of single-cell genomics. PLoS Comput. Biol. 19, e1011288. 10.1371/journal.pcbi.1011288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, J., Park, J.-E., Tsagkogeorga, G., Yanagita, M., Koo, B.-K., Han, N. and Lee, J.-H. (2020). Inflammatory signals induce AT2 cell-derived damage-associated transient progenitors that mediate alveolar regeneration. Cell Stem Cell 27, 366-382.e7. 10.1016/j.stem.2020.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dame, K., Cincotta, S., Lang, A. H., Sanghrajka, R. M., Zhang, L., Choi, J., Kwok, L., Wilson, T., Kańduła, M. M., Monti, S.et al. (2017). Thyroid progenitors are robustly derived from embryonic stem cells through transient, developmental stage-specific overexpression of Nkx2-1. Stem Cell Rep. 8, 216-225. 10.1016/j.stemcr.2016.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank, D. B., Peng, T., Zepp, J. A., Snitow, M., Vincent, T. L., Penkala, I. J., Cui, Z., Herriges, M. J., Morley, M. P., Zhou, S.et al. (2016). Emergence of a wave of Wnt signaling that regulates lung alveologenesis by controlling epithelial self-renewal and differentiation. Cell Rep. 17, 2312-2325. 10.1016/j.celrep.2016.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, X., Wang, R., Zhou, Y., Fei, L., Sun, H., Lai, S., Saadatpour, A., Zhou, Z., Chen, H., Ye, F.et al. (2018). Mapping the mouse cell atlas by microwell-seq. Cell 172, 1091-1107.e17. 10.1016/j.cell.2018.02.001 [DOI] [PubMed] [Google Scholar]
- Herriges, M. J., Yampolskaya, M., Thapa, B. R., Lindstrom-Vautrin, J., Wang, F., Na, C.-L., Ma, L., Montminy, M. M., Huang, J., Villacorta-Martin, C.et al. (2023). Durable alveolar engraftment of PSC-derived lung epithelial cells into immunocompetent mice. Cell Stem Cell 30, 1217-1234.e7. 10.1016/j.stem.2023.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks, S. C., Townes, F. W., Teng, M. and Irizarry, R. A. (2018). Missing data and technical variability in single-cell RNA-sequencing experiments. Biostatistics 19, 562-578. 10.1093/biostatistics/kxx053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, S., Eichler, G., Bar-Yam, Y. and Ingber, D. E. (2005). Cell fates as high-dimensional attractor states of a complex gene regulatory network. Phys. Rev. Lett. 94, 128701. 10.1103/PhysRevLett.94.128701 [DOI] [PubMed] [Google Scholar]
- Ikonomou, L., Herriges, M. J., Lewandowski, S. L., Marsland, R., Villacorta-Martin, C., Caballero, I. S., Frank, D. B., Sanghrajka, R. M., Dame, K., Kańduła, M. M.et al. (2020). The in vivo genetic program of murine primordial lung epithelial progenitors. Nat. Commun. 11, 635. 10.1038/s41467-020-14348-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanter, I. and Sompolinsky, H. (1987). Associative recall of memory without errors. Phys. Rev. A 35, 380-392. 10.1103/PhysRevA.35.380 [DOI] [PubMed] [Google Scholar]
- Kobayashi, Y., Tata, A., Konkimalla, A., Katsura, H., Lee, R. F., Ou, J., Banovich, N. E., Kropski, J. A. and Tata, P. R. (2020). Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat. Cell Biol. 22, 934-946. 10.1038/s41556-020-0542-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lähnemann, D., Köster, J., Szczurek, E., Mccarthy, D. J., Hicks, S. C., Robinson, M. D., Vallejos, C. A., Campbell, K. R., Beerenwinkel, N., Mahfouz, A.et al. (2020). Eleven grand challenges in single-cell data science. Genome Biol. 21, 31. 10.1186/s13059-020-1926-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau, L. D. and Lifshitz, E. M. (2013). Statistical Physics, vol. 5. Elsevier. [Google Scholar]
- Lang, A. H., Li, H., Collins, J. J. and Mehta, P. (2014). Epigenetic landscapes explain partially reprogrammed cells and identify key reprogramming genes. PLoS Comput. Biol. 10, e1003734. 10.1371/journal.pcbi.1003734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louie, S. M., Moye, A. L., Wong, I. G., Lu, E., Shehaj, A., Garcia-De Alba, C., Ararat, E., Raby, B. A., Lu, B., Paschini, M.et al. (2022). Progenitor potential of lung epithelial organoid cells in a transplantation model. Cell Rep. 39, 110662. 10.1016/j.celrep.2022.110662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes, L., Healy, J., Saul, N. and Großberger, L. (2018). J. Open Source Softw. 3, 861. 10.21105/joss.00861 [DOI] [Google Scholar]
- Muraro, M. J., Dharmadhikari, G., Grün, D., Groen, N., Dielen, T., Jansen, E., Van Gurp, L., Engelse, M. A., Carlotti, F., De Koning, E. J.et al. (2016). A single-cell transcriptome atlas of the human pancreas. Cell Syst. 3, 385-394. 10.1016/j.cels.2016.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negretti, N. M., Plosa, E. J., Benjamin, J. T., Schuler, B. A., Habermann, A. C., Jetter, C. S., Gulleman, P., Bunn, C., Hackett, A. N., Ransom, M.et al. (2021). A single-cell atlas of mouse lung development. Development 148, dev199512. 10.1242/dev.199512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusuluri, S. T., Lang, A. H., Mehta, P. and Castillo, H. E. (2017). Cellular reprogramming dynamics follow a simple 1D reaction coordinate. Phys. Biol. 15, 016001. 10.1088/1478-3975/aa90e0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu, X., Mao, Q., Tang, Y., Wang, L., Chawla, R., Pliner, H. A. and Trapnell, C. (2017). Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979-982. 10.1038/nmeth.4402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu, X., Zhang, Y., Martin-Rufino, J. D., Weng, C., Hosseinzadeh, S., Yang, D., Pogson, A. N., Hein, M. Y., Min, K. H. J., Wang, L.et al. (2022). Mapping transcriptomic vector fields of single cells. Cell 185, 690-711.e45. 10.1016/j.cell.2021.12.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quake, S. R. (2022). A decade of molecular cell atlases. Trends Genet. 38, 805-810. 10.1016/j.tig.2022.01.004 [DOI] [PubMed] [Google Scholar]
- Rand, D. A., Raju, A., Sáez, M., Corson, F. and Siggia, E. D. (2021). Geometry of gene regulatory dynamics. Proc. Natl. Acad. Sci. USA 118, e2109729118. 10.1073/pnas.2109729118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossant, J. (2011). The impact of developmental biology on pluripotent stem cell research: successes and challenges. Dev. Cell 21, 20-23. 10.1016/j.devcel.2011.06.010 [DOI] [PubMed] [Google Scholar]
- Rowe, R. G. and Daley, G. Q. (2019). Induced pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Genet. 20, 377-388. 10.1038/s41576-019-0100-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saelens, W., Cannoodt, R., Todorov, H. and Saeys, Y. (2019). A comparison of single-cell trajectory inference methods. Nat. Biotechnol. 37, 547-554. 10.1038/s41587-019-0071-9 [DOI] [PubMed] [Google Scholar]
- Schaum, N., Karkanias, J., Neff, N. F., May, A. P., Quake, S. R., Wyss-Coray, T., Darmanis, S., Batson, J., Botvinnik, O. and Chen, M. B.et al. ; The Tabula Muris Consortium, Overall coordination, Logistical coordination, Organ collection and processing, Library preparation and sequencing, Computational data analysis, Cell type annotation, Writing group, Supplemental text writing group, and Principal investigators. (2018). Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367-372. 10.1038/s41586-018-0590-4 https://www.nature.com/articles/s41586-018-0590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiebinger, G., Shu, J., Tabaka, M., Cleary, B., Subramanian, V., Solomon, A., Gould, J., Liu, S., Lin, S., Berube, P.et al. (2019). Optimal-transport analysis of single-cell gene expression identifies developmental trajectories in reprogramming. Cell 176, 928-943.e22. 10.1016/j.cell.2019.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segerstolpe, Å̊., Palasantza, A., Eliasson, P., Andersson, E.-M., Andréasson, A.-C., Sun, X., Picelli, S., Sabirsh, A., Clausen, M., Bjursell, M. K. eet al. (2016). Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab. 24, 593-607. 10.1016/j.cmet.2016.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethna, J. P. (2021). Statistical Mechanics: Entropy, Order Parameters, and Complexity. Vol. 14. USA: Oxford University Press. [Google Scholar]
- Setty, M., Kiseliovas, V., Levine, J., Gayoso, A., Mazutis, L. and Pe'er, D. (2019). Characterization of cell fate probabilities in single-cell data with palantir. Nat. Biotechnol. 37, 451-460. 10.1038/s41587-019-0068-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson, R. O., Rossant, J. and Tam, P. P. L. (2012). Intercellular interactions, position, and polarity in establishing blastocyst cell lineages and embryonic axes. Cold Spring Harb. Perspect. Biol. 4, a008235. 10.1101/cshperspect.a008235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strunz, M., Simon, L. M., Ansari, M., Kathiriya, J. J., Angelidis, I., Mayr, C. H., Tsidiridis, G., Lange, M., Mattner, L. F., Yee, M.et al. (2020). Alveolar regeneration through a krt8+ transitional stem cell state that persists in human lung fibrosis. Nat. Commun. 11, 3559. 10.1038/s41467-020-17358-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, L., Dong, X., Freytag, S., Lê Cao, K.-A., Su, S., Jalalabadi, A., Amann-Zalcenstein, D., Weber, T. S., Seidi, A., Jabbari, J. S.et al. (2019). Benchmarking single cell RNA-sequencing analysis pipelines using mixture control experiments. Nat. Methods 16, 479-487. 10.1038/s41592-019-0425-8 [DOI] [PubMed] [Google Scholar]
- Tran, H. T. N., Ang, K. S., Chevrier, M., Zhang, X., Lee, N. Y. S., Goh, M. and Chen, J. (2020). A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol. 21, 12. 10.1186/s13059-019-1906-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travaglini, K. J., Nabhan, A. N., Penland, L., Sinha, R., Gillich, A., Sit, R. V., Chang, S., Conley, S. D., Mori, Y., Seita, J.et al. (2020). A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 587, 619-625. 10.1038/s41586-020-2922-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheyden, J. M. and Sun, X. (2020). A transitional stem cell state in the lung. Nat. Cell Biol. 22, 1025-1026. 10.1038/s41556-020-0561-5 [DOI] [PubMed] [Google Scholar]
- Wagner, D. E. and Klein, A. M. (2020). Lineage tracing meets single-cell omics: opportunities and challenges. Nat. Rev. Genet. 21, 410-427. 10.1038/s41576-020-0223-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. J., Schug, J., Won, K.-J., Liu, C., Naji, A., Avrahami, D., Golson, M. L. and Kaestner, K. H. (2016). Single-cell transcriptomics of the human endocrine pancreas. Diabetes 65, 3028-3038. 10.2337/db16-0405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattenberg, M., Viégas, F. and Johnson, I. (2016). How to use t-SNE effectively. Distill 1, e2. 10.23915/distill.00002 [DOI] [Google Scholar]
- Weinreb, C., Wolock, S. and Klein, A. M. (2018). SPRING: a kinetic interface for visualizing high dimensional single-cell expression data. Bioinformatics 34, 1246-1248. 10.1093/bioinformatics/btx792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb, C., Rodriguez-Fraticelli, A., Camargo, F. D. and Klein, A. M. (2020). Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science 367, eaaw3381. 10.1126/science.aaw3381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf, F. A., Hamey, F. K., Plass, M., Solana, J., Dahlin, J. S., Göttgens, B., Rajewsky, N., Simon, L. and Theis, F. J. (2019). PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 20, 59. 10.1186/s13059-018-1612-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolpert, L. (2019). Principles of Development. 6th edn. Oxford University Press. [Google Scholar]
- Xin, Y., Kim, J., Okamoto, H., Ni, M., Wei, Y., Adler, C., Murphy, A. J., Yancopoulos, G. D., Lin, C. and Gromada, J. (2016). Rna sequencing of single human islet cells reveals type 2 diabetes genes. Cell Metab. 24, 608-615. 10.1016/j.cmet.2016.08.018 [DOI] [PubMed] [Google Scholar]
- Xing, J. (2022). Reconstructing data-driven governing equations for cell phenotypic transitions: integration of data science and systems biology. Phys. Biol. 19, 061001. 10.1088/1478-3975/ac8c16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, Z., Van Velthoven, C. T., Nguyen, T. N., Goldy, J., Sedeno-Cortes, A. E., Baftizadeh, F., Bertagnolli, D., Casper, T., Chiang, M., Crichton, K.et al. (2021). A taxonomy of transcriptomic cell types across the isocortex and hippocampal formation. Cell 184, 3222-3241.e26. 10.1016/j.cell.2021.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, H. (2022). What is a cell type and how to define it? Cell 185, 2739-2755. 10.1016/j.cell.2022.06.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zepp, J. A., Morley, M. P., Loebel, C., Kremp, M. M., Chaudhry, F. N., Basil, M. C., Leach, J. P., Liberti, D. C., Niethamer, T. K., Ying, Y.et al. (2021). Genomic, epigenomic, and biophysical cues controlling the emergence of the lung alveolus. Science 371, eabc3172. 10.1126/science.abc3172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, M. and Zernicka-Goetz, M. (2020). Principles of self-organization of the mammalian embryo. Cell 183, 1467-1478. 10.1016/j.cell.2020.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.