

Abstract
The etiology of Tauopathies, a diverse class of neurodegenerative diseases associated with the Microtubule Associated Protein (MAP) Tau, is usually described by a common mechanism in which Tau dysfunction results in the loss of axonal microtubule stability. Here, we reexamine and build upon the canonical disease model to encompass other Tau functions. In addition to regulating microtubule dynamics, Tau acts as a modulator of motor proteins, a signaling hub, and a scaffolding protein. This diverse array of functions is related to the dynamic nature of Tau isoform expression, posttranslational modification (PTM), and conformational flexibility. Thus, there is no single mechanism that can describe Tau dysfunction. The effects of specific pathogenic mutations or aberrant PTMs need to be examined on all of the various functions of Tau in order to understand the unique etiology of each disease state.
Graphical Abstract
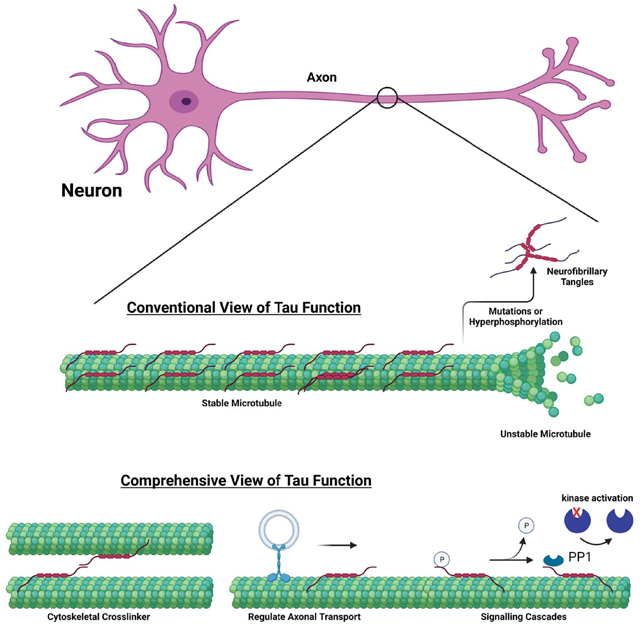
Tau is a microtubule associated protein involved in a number of neurodegenerative disorders. Conventionally, Tau is thought to stabilize microtubules with disease onset proposed to be due microtubule destabilization. We discuss the importance of other Tau physiological functions, modifications, as well as implications for mechanistic disease models.
Introduction
Tau, a microtubule associated protein (MAP) found abundantly in the axonal compartment of neurons [1], is associated with a class of heterogeneous neurodegenerative disorders, collectively known as Tauopathies. These include Progressive Supranuclear Palsy (PSP), Corticobasal Syndrome, Frontotemporal Dementia (FTD) and Alzheimer’s Disease (AD) [2]. These heterogeneous disorders present clinically in a variety ways, including as dementia, movement disorders, and/or motor neuron disease, affecting different areas of the nervous system [2].
Tau belongs to a family of MAPs that include neuronally expressed MAP2 and ubiquitous MAP4 [3]. The Tau/MAP2/MAP4 family is characterized by C-terminal microtubule binding repeats, containing a KXGS motif, a central proline-rich region, and an N-terminal projection domain. Tau also contains a pseudo-repeat C-terminally adjacent to the more conserved microtubule binding repeats. The expression of these related MAPs, including the MAPT gene on chromosome 17 that encodes Tau, are regulated through alternative splicing. Tau isoforms differ in the number of N-terminal acidic inserts and C-terminal microtubule binding repeats based on the inclusion of exons 2, 3 and 10. Inclusion of exons 2 and 3 determine the number of acidic inserts (0,1, or 2), resulting in short (S) or 0N, medium (M) or 1N, and long (L) or 2N Tau isoforms, respectively (Figure 1). The addition of exon 10 determines the number of microtubule binding repeats, either three or four, noted as 3R or 4R isoforms, respectively. Used in combination, this nomenclature can be used to define Tau isoforms. For example, the shortest Tau isoform, 3RS or 0N3R, without exons 2, 3 or 10 contains three microtubule binding repeats and no acidic inserts, while the longest Tau isoform, 4RL or 2N4R, contains four microtubule binding repeats (including exon 10) and two acidic inserts (including exons 2 and 3). Here, we will use S, M, and L to describe the number acidic inserts.
Figure 1.

Schematic of Tau protein domains. Primary amino acid sequence showing the domains of Tau. Alternatively spliced regions are shown in white text. The projection domain contains the phosphatase activating region (purple) and alternatively spliced acidic inserts (green). The proline rich region (blue) is combined from two parts. The microtubule binding region contains 3 or 4 repeats (red) and a pseudo repeat (maroon).
Tauopathy onset is proposed to occur through a loss of microtubule stability in the axon. In this model of Tau dysfunction, a disease-associated mutation or aberrant post-translational modifications (PTMs) decreases Tau-microtubule interactions. This is suggested to be through a reduction in the microtubule-binding affinity of Tau, an increase in Tau aggregation propensity, or both as these can be dependent on each other (Figure 2). For example, a reduction of microtubule affinity increases the fraction of Tau in solution and could increase aggregation propensity, as aggregation of Tau occurs off the microtubule surface. Similarly, an increase in aggregation could decrease the pool of soluble Tau able to bind with the microtubule. In accordance with this model, there are disease-associated missense mutations and PTMs that are known to alter Tau-microtubule interactions. Many of these alterations are found within the C-terminal microtubule binding region or the central proline rich region (Figure 3), known to directly and indirectly mediate Tau-tubulin interactions [4–8]. For example, in vitro studies have shown general Tau phosphorylation, through incubation with kinases, reduces microtubule affinity [9, 10]. Further studies went on to show that residue specific changes, such as phosphorylation at S214 or T231, decreases microtubule affinity [11, 12]. Disease-associated mutations, such as A152T, P301S, and R406W are also known to reduce microtubule affinity [13, 14]. Additionally, phosphorylation at S202/T205/S208, or disease-associated mutations (P301S, S352M), increase heparin or arachidonic acid induced Tau aggregation propensity in vitro compared to WT-Tau [15, 16]. Although these residues have been shown to alter Tau-microtubule interactions, the generally accepted model of Tauopathy onset indicates that Tau modifications reduce microtubule stability. In that regard, it is important to understand the role of Tau as a microtubule stabilizer.
Figure 2.

Canonical Disease Theory Mechanism. Tau disease onset is proposed to occur through a loss of Tau-microtubule interactions, either through decrease in microtubule affinity or increase in aggregation propensity. This is suggested to lead to a loss of microtubule stability. (Created with biorender.com)
Figure 3.

Schematic of Modifications to Tau protein. a). Disease associated mutations found in Tau protein. Figure based on [118]. b). Potential post-translational modifications of amino acids in Tau protein. Post-translational modifications include acetylation, glycation, methylation, nitration, N-glycosylation, O-GlcNAcylation, phosphorylation, SUMOylation, and ubiquitination. Figure based on [107].
Tau as a microtubule stabilizer
Tau was first discovered as a factor essential for in vitro microtubule polymerization [17]. Microtubules are formed by the polymerization of alpha-beta tubulin heterodimers, stabilized through longitudinal and lateral bonds. Generally, microtubules contain 13 linear protofilaments formed around a hollow core. Because tubulin subunits are added in a head to tail manner, microtubules are polar macromolecules with a more dynamic plus end and a less dynamic minus end. In most cell types, microtubules are highly dynamic, going through periods of polymerization and depolymerization, a process known as dynamic instability [18]. These dynamic microtubules are important for cell movement and division. However, in a mature axon, many microtubules are long lived, helping to maintain the axonal structure, and allowing for long range cargo transport [19]. Tau is predominantly expressed in neurons and although it is found in the soma and dendrites, expression is enriched in the axon of neurons [1, 20–23]. Based on this, it was proposed that Tau stabilizes axonal microtubules.
Since the initial discovery [17], research has been done to understand the role Tau plays in regulating microtubule dynamics. Early in vitro experiments showed the addition of Tau increases the rate and extent of tubulin polymerization measured by absorbance at 320 nm [24]. Using electron microscopy, it was also determined that Tau increases the number of microtubules by increasing microtubule nucleation [24]. Further work, using purified Tau protein, indicated Tau polymerizes microtubules below the critical concentration of tubulin [25]. Under non-polymerizing conditions, Tau has been shown to aid in the formation of tubulin rings [26]. Although in vitro experiments do not recapitulate the complexity of the physiological state, similar results have been also seen in cells. Tau is expressed in the growth cone where microtubule invasion forces the growth cone to move forward [27]. In RAT1 fibroblasts, Tau overexpression promotes microtubule assembly [28]. Although Tau is predominantly neuronal, similar results were seen in PC12 cells in which Tau expression increased microtubule assembly with less tubulin turnover [29]. These studies indicate that Tau is a potent microtubule nucleator, and additional work has also characterized Tau’s function in regulating microtubule dynamics.
Microtubules can undergo a process known as dynamic instability, alternating in between polymerization and depolymerization [18, 30, 31]. When tubulin dimers are added to the end of the microtubule, it polymerizes or grows. However, microtubules can switch from a polymerizing state to a rapidly depolymerizing state, a process known as a catastrophe. A rescue, or a switch back to polymerization occurs when tubulin is incorporated on the growing end [32]. Using video microscopy, it was shown that Tau increases the growth rate, slows the shrinkage rate, and reduce the catastrophe frequency of microtubules in vitro [33–36]. All of this work indicates that Tau is both a potent microtubule nucleator and alters, but does not completely stop, microtubule dynamics. Therefore, describing Tau’s primary function as a microtubule stabilizer can be misleading at best. Importantly, recent work has demonstrated that axonal microtubules contain both stable and labile domains, and that Tau is enriched in the labile microtubules [37]. Additionally, Tau depletion in either rat hippocampal or cortical neurons reduces tyrosinated tubulin, a marker of more dynamic microtubules [37], and decreases the total mass of microtubules in the axon [37, 38]. Furthermore, other factors, including specific posttranslational modifications [39] or other MAPs such as MAP6 [37, 40] are more likely to contribute to stabilizing microtubules in the axon than Tau. Of the many posttranslational modifications found on tubulin, acetylation [41–43], phosphorylation [44, 45] and polyamination [46, 47] are the most common types associated with microtubule stability [39], although no single factor has been shown to be both necessary and sufficient for stabilizing axonal microtubules. For example, there is also evidence that acetylation of tubulin at K40 does not stabilize microtubules but is merely a marker for long-lived microtubules that can accumulate this posttranslational modification slowly over time [48]. Phosphorylation can both promote [44, 45] and disrupt [49, 50] the incorporation of tubulin dimers into microtubules, depending on the site of phosphorylation, but the role of phosphorylation in stabilizing long-lived microtubules in axons is still unclear. Like acetylation, polyamination of tubulin is associated with stable microtubules in the axon [39, 46, 47]. Although speculative, one of the residues modified, Q15, is near the active site of β-tubulin and may block GTP hydrolysis thereby stabilizing the microtubule lattice [46, 47]. There are also numerous posttranslational modifications on the C-terminal tails of both α- and β-tubulin, collectively known as the “tubulin code” that may indirectly modulate microtubule dynamics in the neuron by regulating the interaction of different MAPs with the microtubule lattice [51–53].
Tau also functions to regulate microtubule bundle formation through its N-terminal projection domain [54, 55]. Quick-freeze, deep etch electron microscopy showed Tau projections towards adjacent microtubules [54]. By measuring microtubule bundle formation through small-angle X-ray scattering (SAXS), it has been proposed that the length and charge of the N-terminal projection domain alters the repulsive force between microtubules. This put forth the model that states high concentrations of Tau form a brush along the surface of the microtubule to sterically stabilize microtubule spacing [56]. Additionally, by measuring Tau interaction forces on coated mica, it was proposed the projection domain and proline rich region form an “electrostatic zipper” important for the spacing of microtubule bundles [57]. In cells, transfection of Tau into fibroblasts increased microtubule bundling and deletion of the projection domain caused a decrease in bundling [55]. Furthermore, Tau knockdown in hamster sensorimotor neurons reduced microtubule bundling, microtubule polarity, and the number of microtubules in the growth cone [58]. Thus, Tau appears to regulate both microtubule dynamics and organize microtubule architecture within the axon and growth cone of nerve cells.
It is clear from these studies that Tau increases the association of tubulin dimers into a microtubule and is therefore categorized as both a nucleating and a stabilizing MAP. This is in contrast to destabilizing MAPs, such as stathmin or katanin, which increase the dissociation of tubulin from the microtubule. Although this is convenient, this language does not encapsulate the entire function of Tau. As discussed above, microtubules are still dynamic in the presence of Tau, but those dynamics are altered relative to microtubules in the absence of Tau. Thus, Tau alone cannot account for the long-lived stable microtubules observed in neuronal axons as microtubules in the presence of Tau are dynamic. Furthermore, it is now recognized that there are populations of axonal microtubules that are both stable and more dynamic [59], and thus Tau, despite its long history as being thought of as a microtubule stabilizer, may be more important in regulating microtubule dynamics within the axon. Indeed, the relative stability of axonal microtubules is unlikely to be regulated by one specific mechanism but requires the balanced activity of many factors, including specific posttranslational modifications of tubulin and the interaction of different MAPs of which Tau is only one player. And Tau may also act indirectly in regulating microtubule dynamics by modulating other MAPs that directly affect microtubule stability. For example, acetylated microtubules have been shown to be more sensitive to severing by katanin, but this effect can be mitigated in axons by the presence of Tau [60]. Combinatorial effects such as these may explain why it has been challenging to define a single definitive stabilizing agent of axonal microtubules even though numerous factors including Tau have been implicated.
Other functional Roles of Tau
Tau has been proposed to have a myriad of different functions, other than microtubule stabilization, depending on its subcellular localization and specific binding partners (Figure 4). Tau interacts with a variety of intracellular targets, including membranes, other cytoskeletal proteins, and signaling molecules. A subset of these functions includes Tau’s ability to form structural cross-links between different intracellular structures. In addition to bundling and spacing microtubules [55], Tau can also cross-link microtubules with actin filaments [61] to help organize dynamic microtubules and actin network interactions in axonal growth cones [62]. Furthermore, Tau also binds to membrane associated proteins such as annexins via its N-terminal projection domain to cross-link the plasma membrane and microtubules and help define axonal morphology [63], or Golgi membranes to tether nascent intracellular cargo to microtubule tracks for subsequent transport [64]. Tau may also modulate local membrane morphology through interactions between its basic C-terminal domain and anionic phospholipids [65].
Figure 4.

Multi-faceted functions of Tau protein. Tau has many interrelated functions that go beyond its role as a microtubule stabilizer. These additional functions, which occur at different regions in the neuron, include acting as a signaling molecule, cross-linker, and modulator of transport. Each of these interrelated functions can be modified by the Tau isoform, Tau binding behavior, post-translational modifications, and disease mutations. (Created with biorender.com)
Tau is also an important regulator of microtubule-based cargo transport in the axon. Early studies demonstrated that Tau overexpression causes mislocalization of mitochondria, ER, and vesicles [66], altering the run length, but not the velocity, of cargos in both directions, preferentially inhibiting anterograde transport resulting in a shift in net movement of cargo towards the cell body [67]. Subsequent experiments using in vitro reconstituted systems have further elucidated specific details of Tau’s regulation of specific motor proteins, including various members of the kinesin family of anterograde motors, and the predominant retrograde motor, cytoplasmic dynein. Tau reduces the on-rate for microtubule binding of both kinesin-1 and the dynein-dynactin complex with a larger effect on kinesin-1 [68]. Tau also inhibits the overall run length of kinesin-1, but not the dynein-dynactin complex, in a concentration and isoform specific manner, with the shorter 3RS isoform having a stronger effect than the longer 4RL isoform [68]. As in the cellular studies [67], motor velocity was unchanged [68]. Work from our lab agrees with these studies, showing kinesin-1 run length is more strongly attenuated by 3RS-Tau compared to 4RL-Tau on microtubules stabilized with paclitaxel (Taxol-microtubules) [69]. However, this regulation is dependent on the microtubule lattice as Tau is more inhibitory to kinesin-1 motility on Taxol-microtubules, a model of the GDP-lattice, relative to microtubules stabilized with guanosine 5’-[(α,β)-methyleno] triphosphate (GMPCPP-microtubules), a model of the GTP-lattice [69]. Furthermore, not all kinesin motors are regulated by Tau. For example, our lab has shown that kinesin-2 is insensitive to Tau due to the ability of kinesin-2 to switch protofilaments in the presence of obstacles, such as Tau, on the microtubule surface [70, 71]. And kinesin-3 family members such as KIF1A are likely to be regulated by Tau in a different manner than kinesin-1 or kinesin-2. Both KIF1A and Tau have been shown to interact with tubulin C-terminal tails (CTTs). KIF1A has been shown to bind to tubulin C-terminal tails (CTT) via its K-Loop, increasing its run length 2–3 fold [72], while Tau interacts with CTTs diffusively [73] in an isoform specific manner, with the longer 4RL isoform binding more diffusively than the shorter 3RS isoform [74]. Thus, in contrast to kinesin-1, which is more strongly inhibited by 3RS-Tau [69], or kinesin-2, which is uninhibited by Tau [70, 71], KIF1A is likely to be more strongly inhibited by 4RL-Tau through competition for binding to CTTs, which contributes to the long processive run length of kinesin-3 motors [72].
Tau has been shown to regulate teams of motor proteins as well. A study was done using optical trapping of beads coated with varying numbers of kinesin-1 motor proteins and showed Tau effects the rate of motor reattachment to the microtubule, causing a reduction in cargo persistence and force [75]. Similar to other experiments using kinesin-1, it was shown that 3RS-Tau had a larger reduction in cargo persistence compared to 4RL-Tau [75]. Isolated phagosomes from ATCC cells, which contain motor complexes of kinesin-1, kinesin-2 and dynein, are also shown to be regulated by Tau, biasing cargo moved preferentially towards the minus ends of microtubules in the presence of 3RS-Tau [76].
Studies on Tau’s ability to differentially regulate motor proteins involved in axonal transport have also led to the discovery that Tau behavior on the microtubule surface is complex, depending on a number of different factors including the nucleotide state of the microtubule lattice, the isoform of Tau, and site-specific posttranslational modifications of Tau. Traditionally thought to bind microtubules statically in a stabilizing role, Tau has since been shown to interact with the microtubule in a dynamic equilibrium between static and diffusive states [73, 74]. Static Tau likely binds through its microtubule-binding repeats in extended conformation along individual protofilaments in the microtubule lattice [77], while diffusive Tau has been shown to interact through tubulin’s acidic, flexible CTTs [73]. However, Tau binding behavior is influenced by a number of factors. Tau’s behavior is shifted towards diffusive binding on GMPCPP-microtubules vs. Taxol-microtubules [74], in 4-Repeat isoforms vs. 3-Repeat isoforms [74], and in response to specific posttranslational modifications such phosphorylation at Y18 [78]. Furthermore, static Tau can also form larger order complexes on the microtubule surface known as patches [68], condensates [79], or cohesive islands [80], which have been shown to differentially regulate molecular motors [68, 69, 79] and other MAPs such as microtubule severing enzymes like katanin [60, 80].
Finally, Tau has been shown to activate Protein Phosphatase 1 (PP1) through its N-terminal Phosphatase Activating Domain (PAD), and this interaction can be regulated by phosphorylation of Tau at a specific tyrosine (Y18) adjacent to the PAD [81–83]. Specifically, phosphorylation of Y18 by kinases such as Fyn prevent activation of PP1 by the PAD of Tau [81, 82, 84]. In the axon it has been proposed that under normal physiological conditions Tau is phosphorylated at Y18 [82], preventing its interaction with PP1 and promoting a diffusive state on the microtubule surface that is less inhibitory for kinesin-1 mediated cargo transport [78]. Dephosphorylation of Y18 has been shown to be associated with activation of PP1 via the PAD of Tau [82]. Activation of PP1 has been shown to increase GSK3β activity in the axon, which can phosphorylate the kinesin-1 light chain resulting in decreased cargo binding [85]. Interestingly, dephosphorylation of Y18 in Tau not only activates PP1 [82] but also results in a shift to increased static binding of Tau [78], which inhibits kinesin-1 motility [69, 74]. Thus, dephosphorylation of Tau at Y18 may function to regulate kinesin-1 mediated cargo delivery at specific locations in the axon by both inhibiting the kinesin-1 motility [69, 74] and promoting cargo release [78, 81, 82, 84]. Interactions of Tau with PP1 or Fyn may also be involved in synaptic transmission. Tau has been shown to be recruited to excitatory postsynaptic compartments in an activity-dependent manner [86] where it interacts with Fyn and promotes NMDA-receptor activity [87]. On the presynaptic side, activation of PP1 and GSK3β by Tau has been linked to decreased synaptic vesicle release [88]. Finally, Tau activation of PP1 in the soma may promote the translocation of β-catenin to the nucleus where it acts as a transcription factor for antiapoptotic pro-survival genes [89], and Tau [90] and PP1 [91] have also been found in the nucleus suggesting roles for both in directly or indirectly regulating gene expression [83].
Tau Structure and Modification
An important question arises as to how a single protein like Tau can be so functionally diverse. The answer is that Tau is a highly dynamic and structurally heterogeneous protein that can undergo extensive posttranslational modifications. As previously discussed, Tau is produced as six alternatively spliced isoforms that vary in the number of N-terminal repeats (0, 1 or 2) and microtubule binding repeats (3 or 4) that are present in the expressed protein. The expression pattern of these isoforms is developmentally regulated, with the 3RS isoform expressed embryonically and all six isoforms eventually present in the adult human brain [92]. Recent evidence also suggests that Tau isoforms are differentially expressed in specific regions of the brain [93]. Together with the fact that isoform specific differences in Tau behavior [74] and function [68, 69] have been identified, it is likely that the inherent structural heterogeneity of Tau contributes to varied functions within different types of neurons at varied stages of development.
In addition to variability in its primary sequence, Tau is an intrinsically disordered protein (IDP) that lacks well defined secondary structure and adopts an ensemble of dynamic conformations [94–96], which likely contributes to its myriad of cellular functions. In solution Tau has been shown to adopt an ensemble of folded conformations in which the N- and C-terminal regions fold back to form dynamic interactions with the microtubule-binding domain [94, 97]. The structure of Tau on the microtubule surface is less clear. Cryo-electron microscopy (cryo-EM) studies suggest that Tau binds statically to the microtubule along individual protofilaments [98] in an extended conformation with individual microtubule-binding repeats stabilizing the interface between tubulin dimers [77]. The N-terminal projection has been shown to extend away from the microtubule surface [54], although recent nuclear magnetic resonance (NMR) evidence also suggests that the N-terminus of Tau may also interact weakly with the microtubule surface [99]. And the structure of diffusive Tau on the microtubule is completely unknown, but likely to be different than that of static Tau. Thus, Tau adopts multiple dynamic conformations both in solution and on the microtubule surface that contribute to its ability to interact with a large number of binding partners involved in a variety of different physiological processes within the neuron.
Further complicating the picture is the fact that Tau is heavily posttranslationally-modified, leading to alterations in Tau structure and function. As seen above, phosphorylation at Y18 alters both Tau’s behavior on the microtubule surface [78] and its ability to regulate axonal transport through a conformational change of the PAD [82]. Posttranslational modifications of Tau include acetylation [100], glycation [101], glycosylation [102], methylation [103], phosphorylation [104], SUMOylation [105], ubiquitination [106], and more [107] (Figure 3b). Of these, phosphorylation is by far the most prevalent posttranslational modification with more than 50 known, and at least another 30 putative, phosphorylation sites identified in Tau [104, 107]. Twenty of these sites are found to be phosphorylated in healthy brains, and at least 40 sites have been found to be hyperphosphorylated in tauopathy patients [107]. However, there is overlap as some sites phosphorylated under normal physiological conditions are also found in pathologic hyperphosphorylated Tau [107]. Like isoform expression, Tau phosphorylation is developmentally regulated [108], suggesting that site-specific phosphorylation is important for specific Tau functions. Furthermore, phosphorylation at physiologically relevant sites can be influenced by phosphorylation or other posttranslational modifications at other sites within the Tau molecule. For example, phosphorylation of Tau by GSK3β requires phosphorylation at other sites by kinases such as PKA [109], and glycation [110] and glycosylation [111] have also been shown to alter Tau phosphorylation patterns. The dynamic structural ensemble of Tau is therefore influenced by combinations of posttranslational modifications, which in turn influence Tau’s function both developmentally and spatiotemporally within the neuron.
Implications for Disease Mechanisms of Tau
The most widely accepted disease etiology of Tauopathies states that modifications to Tau, such as pathologic point mutations or hyperphosphorylation, decrease Tau-microtubule interactions and lead to loss of microtubule stability through decrease of microtubule affinity and/or increase in aggregation. However, given that Tau is, in fact, not a simple microtubule “stabilizer” and has a number of diverse physiological functions throughout the neuron at different developmental stages, it is evident that the current disease state model is at best oversimplistic. Furthermore, not all pathogenic mutations in Tau lead to decreased microtubule affinity and/or increased aggregation propensity. For example, the R5L mutation in the N-terminal projection domain does not alter microtubule affinity [99]. Some disease association missense mutations, such as N279K and S305N, do not alter microtubule affinity or assembly [112], and others, such as Q336H and E342V, are known to actually increase microtubule assembly [15, 113]. Each pathogenic mutation and/or aberrant posttranslational modification should be viewed as a unique pathology with its own set of molecular mechanisms. Therefore, while some pathogenic states may involve decreased microtubule affinity and/or increased aggregation, others may not and more directly affect some combination of alterations in microtubule dynamics, axonal transport, and/or intracellular signaling pathways (Figure 5). Thus, future research needs to be focused on not only Tau’s interaction with the microtubule lattice and/or itself during aggregation, but on the myriad of other binding partners that allow it to participate in a number of different physiological roles within the neuron. Proteomic approaches show promise for identifying key interacting partners with Tau in both well-established functions such as axonal transport and less well-studied roles like synaptic vesicle release [114, 115]. Interactomes for different Tau pathogenic mutations and posttranslational modifications can then be used to further define the complex physiological roles of Tau and identify novel therapeutic strategies for specific Tauopathies that exhibit distinct etiologies.
Figure 5.

Updated Tauopathy Disease Model. Tau disease onset is likely to occur through a number of different mechanisms including, decrease in microtubule affinity, increase in aggregation propensity, or altered binding behavior. This alters Tau functions including microtubule regulation, modulation of motor transport, participation of signaling cascades, etc. (created with biorender.com)
Conclusions and Perspectives
The microtubule-associated protein Tau plays a key role in a number of diverse neurodegenerative diseases collectively known as Tauopathies [116]. Despite the variability of the regions of the brain affected and of the symptoms presented, a common etiology of the different Tauopathies is often proposed [117]. In this model, the main function of Tau is to stabilize axonal microtubules, which are critical to neuronal function and viability. Tau dysfunction, caused by disease-associated mutations or aberrant post-translational modifications (PTMs), thus decreases Tau-microtubule interactions leading to microtubule instability, axonal dysfunction, and neuronal cell death. However, as discussed in this review, Tau is not simply a microtubule stabilizer, but a dynamic protein involved in a diverse set of neuronal processes. In other words, Tau is a regulator of microtubule dynamics and organization [37], axonal transport [68, 69], and intracellular signaling pathways [83]. Likewise, disruption of Tau function in pathogenic states disrupts a myriad of cellular functions which may include microtubule dynamics and organization, axonal transport, and/or intracellular signaling pathways. Thus, there is no singular mechanism that describes the initial onset of Tauopathies – each disease needs to be examined for the unique pathogenic state that it is. Only by understanding how each disease associated mutation or aberrant PTM affects the myriad of cellular functions that Tau participates in can the molecular mechanisms of different Tauopathies be properly defined. This is the first and foremost step needed to begin to develop therapeutics for a class of neurodegenerative diseases for which the prospects have been very challenging. The variability observed in the presentation of symptoms for different Tauopathies reflects heterogeneity in disease mechanism, rooted in the diversity of Tau structure, function, and interacting partners. Therefore, despite the simplistic model that disease onset arises from decreased Tau binding and microtubule stability, in reality there is not a single common etiology that describes the molecular mechanisms of all or even most Tauopathies. Ultimately, recognition that Tau is more than the “superglue” that stabilizes axonal microtubules reflects the dynamic nature of its structure and function and will continue to open new avenues of investigation into the complex nature of its role in both normal function and disease states of the neuron.
Acknowledgments
This work was supported by National Institutes of Health Grant R01 GM132646 to C.L.B.
References Cited:
- 1.Binder LI, Frankfurter A, Rebhun LI (1985). The distribution of tau in the mammalian central nervous system. J Cell Biol, 101, 1371–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Josephs KA (2017). Current Understanding of Neurodegenerative Diseases Associated With the Protein Tau. Mayo Clin Proc, 92, 1291–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dehmelt L, Halpain S (2005). The MAP2/Tau family of microtubule-associated proteins. Genome Biol, 6, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Butner KA, Kirschner MW (1991). Tau protein binds to microtubules through a flexible array of distributed weak sites. J Cell Biol, 115, 717–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gustke N, Trinczek B, Biernat J, Mandelkow EM, Mandelkow E. (1994). Domains of tau protein and interactions with microtubules. Biochemistry, 33, 9511–22. [DOI] [PubMed] [Google Scholar]
- 6.Goode BL, Feinstein SC (1994). Identification of a novel microtubule binding and assembly domain in the developmentally regulated inter-repeat region of tau. J Cell Biol, 124, 769–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Niewidok B, Igaev M, Sundermann F, Janning D, Bakota L, Brandt R. (2016). Presence of a carboxy-terminal pseudorepeat and disease-like pseudohyperphosphorylation critically influence tau’s interaction with microtubules in axon-like processes. Mol Biol Cell, 27, 3537–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goode BL, Denis PE, Panda D, Radeke MJ, Miller HP, Wilson L, . . . Feinstein SC (1997). Functional interactions between the proline-rich and repeat regions of tau enhance microtubule binding and assembly. Mol Biol Cell, 8, 353–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lindwall G, Cole RD (1984). Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem, 259, 5301–5. [PubMed] [Google Scholar]
- 10.Mandelkow EM, Biernat J, Drewes G, Gustke N, Trinczek B, Mandelkow E. (1995). Tau domains, phosphorylation, and interactions with microtubules. Neurobiol Aging, 16, 355–62; discussion 362–3. [DOI] [PubMed] [Google Scholar]
- 11.Illenberger S, Zheng-Fischhofer Q, Preuss U, Stamer K, Baumann K, Trinczek B, . . . Mandelkow E. (1998). The endogenous and cell cycle-dependent phosphorylation of tau protein in living cells: implications for Alzheimer’s disease. Mol Biol Cell, 9, 1495–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cho JH, Johnson GV (2004). Primed phosphorylation of tau at Thr231 by glycogen synthase kinase 3beta (GSK3beta) plays a critical role in regulating tau’s ability to bind and stabilize microtubules. J Neurochem, 88, 349–58. [DOI] [PubMed] [Google Scholar]
- 13.Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, . . . Lee VM (1998). Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science, 282, 1914–7. [DOI] [PubMed] [Google Scholar]
- 14.Coppola G, Chinnathambi S, Lee JJ, Dombroski BA, Baker MC, Soto-Ortolaza AI, . . . Geschwind DH (2012). Evidence for a role of the rare p.A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer’s diseases. Hum Mol Genet, 21, 3500–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Combs B, Gamblin TC (2012). FTDP-17 tau mutations induce distinct effects on aggregation and microtubule interactions. Biochemistry, 51, 8597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Despres C, Byrne C, Qi H, Cantrelle FX, Huvent I, Chambraud B, . . . Smet-Nocca C. (2017). Identification of the Tau phosphorylation pattern that drives its aggregation. Proc Natl Acad Sci U S A, 114, 9080–9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW (1975). A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A, 72, 1858–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitchison T, Kirschner M. (1984). Dynamic instability of microtubule growth. Nature, 312, 237–42. [DOI] [PubMed] [Google Scholar]
- 19.Baas PW, Slaughter T, Brown A, Black MM (1991). Microtubule dynamics in axons and dendrites. J Neurosci Res, 30, 134–53. [DOI] [PubMed] [Google Scholar]
- 20.Ahmad FJ, Pienkowski TP, Baas PW (1993). Regional differences in microtubule dynamics in the axon. J Neurosci, 13, 856–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baas PW (1997). Microtubules and axonal growth. Curr Opin Cell Biol, 9, 29–36. [DOI] [PubMed] [Google Scholar]
- 22.Heidemann SR, Landers JM, Hamborg MA (1981). Polarity orientation of axonal microtubules. J Cell Biol, 91, 661–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okabe S, Hirokawa N. (1990). Turnover of fluorescently labelled tubulin and actin in the axon. Nature, 343, 479–82. [DOI] [PubMed] [Google Scholar]
- 24.Murphy DB, Johnson KA, Borisy GG (1977). Role of tubulin-associated proteins in microtubule nucleation and elongation. J Mol Biol, 117, 33–52. [DOI] [PubMed] [Google Scholar]
- 25.Cleveland DW, Hwo SY, Kirschner MW (1977). Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J Mol Biol, 116, 227–47. [DOI] [PubMed] [Google Scholar]
- 26.Devred F, Barbier P, Douillard S, Monasterio O, Andreu JM, Peyrot V. (2004). Tau induces ring and microtubule formation from alphabeta-tubulin dimers under nonassembly conditions. Biochemistry, 43, 10520–31. [DOI] [PubMed] [Google Scholar]
- 27.Goldberg DJ, Burmeister DW (1986). Stages in axon formation: observations of growth of Aplysia axons in culture using video-enhanced contrast-differential interference contrast microscopy. J Cell Biol, 103, 1921–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Drubin DG, Kirschner MW (1986). Tau protein function in living cells. J Cell Biol, 103, 2739–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Janning D, Igaev M, Sundermann F, Bruhmann J, Beutel O, Heinisch JJ, . . . Brandt R. (2014). Single-molecule tracking of tau reveals fast kiss-and-hop interaction with microtubules in living neurons. Mol Biol Cell, 25, 3541–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Desai A, Mitchison TJ (1997). Microtubule polymerization dynamics. Annu Rev Cell Dev Biol, 13, 83–117. [DOI] [PubMed] [Google Scholar]
- 31.Walker RA, O’Brien ET, Pryer NK, Soboeiro MF, Voter WA, Erickson HP, . . . Salmon ED (1988). Dynamic instability of individual microtubules analyzed by video light microscopy: rate constants and transition frequencies. J Cell Biol, 107, 1437–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cassimeris L, Pryer NK, Salmon ED (1988). Real-time observations of microtubule dynamic instability in living cells. J Cell Biol, 107, 2223–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Drechsel DN, Hyman AA, Cobb MH, Kirschner MW (1992). Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell, 3, 1141–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Panda D, Goode BL, Feinstein SC, Wilson L. (1995). Kinetic stabilization of microtubule dynamics at steady state by tau and microtubule-binding domains of tau. Biochemistry, 34, 11117–27. [DOI] [PubMed] [Google Scholar]
- 35.Panda D, Miller HP, Wilson L. (1999). Rapid treadmilling of brain microtubules free of microtubule-associated proteins in vitro and its suppression by tau. Proc Natl Acad Sci U S A, 96, 12459–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trinczek B, Biernat J, Baumann K, Mandelkow EM, Mandelkow E. (1995). Domains of tau protein, differential phosphorylation, and dynamic instability of microtubules. Mol Biol Cell, 6, 1887–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qiang L, Sun X, Austin TO, Muralidharan H, Jean DC, Liu M, . . . Baas PW (2018). Tau Does Not Stabilize Axonal Microtubules but Rather Enables Them to Have Long Labile Domains. Curr Biol, 28, 2181–2189 e4. [DOI] [PubMed] [Google Scholar]
- 38.Qiang L, Yu W, Andreadis A, Luo M, Baas PW (2006). Tau protects microtubules in the axon from severing by katanin. J Neurosci, 26, 3120–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wloga D, Joachimiak E, Fabczak H. (2017). Tubulin Post-Translational Modifications and Microtubule Dynamics. Int J Mol Sci, 18, 2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Slaughter T, Black MM (2003). STOP (stable-tubule-only-polypeptide) is preferentially associated with the stable domain of axonal microtubules. J Neurocytol, 32, 399–413. [DOI] [PubMed] [Google Scholar]
- 41.LeDizet M, Piperno G. (1986). Cytoplasmic microtubules containing acetylated alpha-tubulin in Chlamydomonas reinhardtii: spatial arrangement and properties. J Cell Biol, 103, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Piperno G, LeDizet M, Chang XJ (1987). Microtubules containing acetylated alpha-tubulin in mammalian cells in culture. J Cell Biol, 104, 289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu Z, Schaedel L, Portran D, Aguilar A, Gaillard J, Marinkovich MP, . . . Nachury MV (2017). Microtubules acquire resistance from mechanical breakage through intralumenal acetylation. Science, 356, 328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abeyweera TP, Chen X, Rotenberg SA (2009). Phosphorylation of alpha6-tubulin by protein kinase Calpha activates motility of human breast cells. J Biol Chem, 284, 17648–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De S, Tsimounis A, Chen X, Rotenberg SA (2014). Phosphorylation of alpha-tubulin by protein kinase C stimulates microtubule dynamics in human breast cells. Cytoskeleton (Hoboken), 71, 257–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Song Y, Brady ST (2014). Stabilization of neuronal connections and the axonal cytoskeleton. Bioarchitecture, 4, 22–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Song Y, Kirkpatrick LL, Schilling AB, Helseth DL, Chabot N, Keillor JW, . . . Brady ST (2013). Transglutaminase and polyamination of tubulin: posttranslational modification for stabilizing axonal microtubules. Neuron, 78, 109–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szyk A, Deaconescu AM, Spector J, Goodman B, Valenstein ML, Ziolkowska NE, . . . Roll-Mecak A. (2014). Molecular basis for age-dependent microtubule acetylation by tubulin acetyltransferase. Cell, 157, 1405–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ley SC, Verbi W, Pappin DJ, Druker B, Davies AA, Crumpton MJ (1994). Tyrosine phosphorylation of alpha tubulin in human T lymphocytes. Eur J Immunol, 24, 99–106. [DOI] [PubMed] [Google Scholar]
- 50.Liu N, Xiong Y, Ren Y, Zhang L, He X, Wang X, . . . Zhou J. (2015). Proteomic Profiling and Functional Characterization of Multiple Post-Translational Modifications of Tubulin. J Proteome Res, 14, 3292–304. [DOI] [PubMed] [Google Scholar]
- 51.Garnham CP, Roll-Mecak A. (2012). The chemical complexity of cellular microtubules: tubulin post-translational modification enzymes and their roles in tuning microtubule functions. Cytoskeleton (Hoboken), 69, 442–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roll-Mecak A. (2014). Intrinsically disordered tubulin tails: complex tuners of microtubule functions? Semin Cell Dev Biol, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu I, Garnham CP, Roll-Mecak A. (2015). Writing and Reading the Tubulin Code. J Biol Chem, 290, 17163–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen J, Kanai Y, Cowan NJ, Hirokawa N. (1992). Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature, 360, 674–7. [DOI] [PubMed] [Google Scholar]
- 55.Kanai Y, Chen J, Hirokawa N. (1992). Microtubule bundling by tau proteins in vivo: analysis of functional domains. EMBO J, 11, 3953–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chung PJ, Choi MC, Miller HP, Feinstein HE, Raviv U, Li Y, . . . Safinya CR (2015). Direct force measurements reveal that protein Tau confers short-range attractions and isoform-dependent steric stabilization to microtubules. Proc Natl Acad Sci U S A, 112, E6416–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rosenberg KJ, Ross JL, Feinstein HE, Feinstein SC, Israelachvili J. (2008). Complementary dimerization of microtubule-associated tau protein: Implications for microtubule bundling and tau-mediated pathogenesis. Proc Natl Acad Sci U S A, 105, 7445–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Biswas S, Kalil K. (2018). The Microtubule-Associated Protein Tau Mediates the Organization of Microtubules and Their Dynamic Exploration of Actin-Rich Lamellipodia and Filopodia of Cortical Growth Cones. J Neurosci, 38, 291–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Baas PW, Rao AN, Matamoros AJ, Leo L. (2016). Stability properties of neuronal microtubules. Cytoskeleton (Hoboken), 73, 442–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sudo H, Baas PW (2010). Acetylation of microtubules influences their sensitivity to severing by katanin in neurons and fibroblasts. J Neurosci, 30, 7215–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.He HJ, Wang XS, Pan R, Wang DL, Liu MN, He RQ (2009). The proline-rich domain of tau plays a role in interactions with actin. BMC Cell Biology, 10, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Elie A, Prezel E, Guérin C, Denarier E, Ramirez-Rios S, Serre L, . . . Arnal I. (2015). Tau co-organizes dynamic microtubule and actin networks. Scientific Reports, 5, 9964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sallaberry CA, Voss BJ, Majewski J, Biernat J, Mandelkow E, Chi EY, . . . Vander Zanden CM. (2021). Tau and Membranes: Interactions That Promote Folding and Condensation. Front Cell Dev Biol, 9, 725241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Farah CA, Perreault S, Liazoghli D, Desjardins M, Anton A, Lauzon M, . . . Leclerc N. (2006). Tau interacts with Golgi membranes and mediates their association with microtubules. Cell Motil Cytoskeleton, 63, 710–24. [DOI] [PubMed] [Google Scholar]
- 65.Pooler AM, Hanger DP (2010). Functional implications of the association of tau with the plasma membrane. Biochem Soc Trans, 38, 1012–5. [DOI] [PubMed] [Google Scholar]
- 66.Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E. (1998). Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer’s disease. J Cell Biol, 143, 777–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Trinczek B, Ebneth A, Mandelkow EM, Mandelkow E. (1999). Tau regulates the attachment/detachment but not the speed of motors in microtubule-dependent transport of single vesicles and organelles. J Cell Sci, 112 ( Pt 14), 2355–67. [DOI] [PubMed] [Google Scholar]
- 68.Dixit R, Ross JL, Goldman YE, Holzbaur EL (2008). Differential regulation of dynein and kinesin motor proteins by tau. Science, 319, 1086–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McVicker DP, Chrin LR, Berger CL (2011). The nucleotide-binding state of microtubules modulates kinesin processivity and the ability of Tau to inhibit kinesin-mediated transport. J Biol Chem, 286, 42873–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hoeprich GJ, Mickolajczyk KJ, Nelson SR, Hancock WO, Berger CL (2017). The axonal transport motor kinesin-2 navigates microtubule obstacles via protofilament switching. Traffic, 18, 304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hoeprich GJ, Thompson AR, McVicker DP, Hancock WO, Berger CL (2014). Kinesin’s neck-linker determines its ability to navigate obstacles on the microtubule surface. Biophys J, 106, 1691–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lessard DV, Zinder OJ, Hotta T, Verhey KJ, Ohi R, Berger CL (2019). Polyglutamylation of tubulin’s C-terminal tail controls pausing and motility of kinesin-3 family member KIF1A. J Biol Chem, 294, 6353–6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hinrichs MH, Jalal A, Brenner B, Mandelkow E, Kumar S, Scholz T. (2012). Tau protein diffuses along the microtubule lattice. J Biol Chem, 287, 38559–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McVicker DP, Hoeprich GJ, Thompson AR, Berger CL (2014). Tau interconverts between diffusive and stable populations on the microtubule surface in an isoform and lattice specific manner. Cytoskeleton (Hoboken), 71, 184–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vershinin M, Carter BC, Razafsky DS, King SJ, Gross SP (2007). Multiple-motor based transport and its regulation by Tau. Proc Natl Acad Sci U S A, 104, 87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chaudhary AR, Berger F, Berger CL, Hendricks AG (2018). Tau directs intracellular trafficking by regulating the forces exerted by kinesin and dynein teams. Traffic, 19, 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kellogg EH, Hejab NMA, Poepsel S, Downing KH, DiMaio F, Nogales E. (2018). Near-atomic model of microtubule-tau interactions. Science, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stern JL, Lessard DV, Hoeprich GJ, Morfini GA, Berger CL (2017). Phosphoregulation of Tau modulates inhibition of kinesin-1 motility. Mol Biol Cell, 28, 1079–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tan R, Lam AJ, Tan T, Han J, Nowakowski DW, Vershinin M, . . . McKenney RJ (2019). Microtubules gate tau condensation to spatially regulate microtubule functions. Nat Cell Biol, 21, 1078–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Siahaan V, Krattenmacher J, Hyman AA, Diez S, Hernandez-Vega A, Lansky Z, . . . Braun M. (2019). Kinetically distinct phases of tau on microtubules regulate kinesin motors and severing enzymes. Nat Cell Biol, 21, 1086–1092. [DOI] [PubMed] [Google Scholar]
- 81.Kanaan NM, Morfini GA, LaPointe NE, Pigino GF, Patterson KR, Song Y, . . . Binder LI (2011). Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases. J Neurosci, 31, 9858–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kanaan NM, Morfini G, Pigino G, LaPointe NE, Andreadis A, Song Y, . . . Brady ST (2012). Phosphorylation in the amino terminus of tau prevents inhibition of anterograde axonal transport. Neurobiol Aging, 33, 826 e15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mueller RL, Combs B, Alhadidy MM, Brady ST, Morfini GA, Kanaan NM (2021). Tau: A Signaling Hub Protein. Front Mol Neurosci, 14, 647054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.LaPointe NE, Morfini G, Pigino G, Gaisina IN, Kozikowski AP, Binder LI, . . . Brady ST (2009). The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. J Neurosci Res, 87, 440–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Morfini G, Szebenyi G, Brown H, Pant HC, Pigino G, DeBoer S, . . . Brady ST (2004). A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. EMBO J, 23, 2235–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Frandemiche ML, De Seranno S, Rush T, Borel E, Elie A, Arnal I, . . . Buisson A. (2014). Activity-dependent tau protein translocation to excitatory synapse is disrupted by exposure to amyloid-beta oligomers. J Neurosci, 34, 6084–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, . . . Gotz J. (2010). Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell, 142, 387–97. [DOI] [PubMed] [Google Scholar]
- 88.Moreno H, Morfini G, Buitrago L, Ujlaki G, Choi S, Yu E, . . . Llinas RR (2016). Tau pathology-mediated presynaptic dysfunction. Neuroscience, 325, 30–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li HL, Wang HH, Liu SJ, Deng YQ, Zhang YJ, Tian Q, . . . Wang JZ (2007). Phosphorylation of tau antagonizes apoptosis by stabilizing beta-catenin, a mechanism involved in Alzheimer’s neurodegeneration. Proc Natl Acad Sci U S A, 104, 3591–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mansuroglu Z, Benhelli-Mokrani H, Marcato V, Sultan A, Violet M, Chauderlier A, . . . Bonnefoy E. (2016). Loss of Tau protein affects the structure, transcription and repair of neuronal pericentromeric heterochromatin. Sci Rep, 6, 33047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rebelo S, Santos M, Martins F, da Cruz e Silva EF, da Cruz e Silva OA (2015). Protein phosphatase 1 is a key player in nuclear events. Cell Signal, 27, 2589–98. [DOI] [PubMed] [Google Scholar]
- 92.Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA (1989). Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron, 3, 519–26. [DOI] [PubMed] [Google Scholar]
- 93.Trabzuni D, Wray S, Vandrovcova J, Ramasamy A, Walker R, Smith C, . . . Ryten M. (2012). MAPT expression and splicing is differentially regulated by brain region: relation to genotype and implication for tauopathies. Hum Mol Genet, 21, 4094–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Elbaum-Garfinkle S, Rhoades E. (2012). Identification of an aggregation-prone structure of tau. J Am Chem Soc, 134, 16607–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jeganathan S, von Bergen M, Brutlach H, Steinhoff HJ, Mandelkow E. (2006). Global hairpin folding of tau in solution. Biochemistry, 45, 2283–93. [DOI] [PubMed] [Google Scholar]
- 96.Mukrasch MD, Bibow S, Korukottu J, Jeganathan S, Biernat J, Griesinger C, . . . Zweckstetter M. (2009). Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol, 7, e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jeganathan S, Chinnathambi S, Mandelkow EM, Mandelkow E. (2012). Conformations of microtubule-associated protein Tau mapped by fluorescence resonance energy transfer. Methods Mol Biol, 849, 85–99. [DOI] [PubMed] [Google Scholar]
- 98.Al-Bassam J, Ozer RS, Safer D, Halpain S, Milligan RA (2002). MAP2 and tau bind longitudinally along the outer ridges of microtubule protofilaments. J Cell Biol, 157, 1187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cario A, Savastano A, Wood NB, Liu Z, Previs MJ, Hendricks AG, . . . Berger CL (2022). The pathogenic R5L mutation disrupts formation of Tau complexes on the microtubule by altering local N-terminal structure. Proc Natl Acad Sci U S A, 119, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cohen TJ, Guo JL, Hurtado DE, Kwong LK, Mills IP, Trojanowski JQ, . . . Lee VM (2011). The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun, 2, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yan SD, Chen X, Schmidt AM, Brett J, Godman G, Zou YS, . . . et al. (1994). Glycated tau protein in Alzheimer disease: a mechanism for induction of oxidant stress. Proc Natl Acad Sci U S A, 91, 7787–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liu F, Zaidi T, Iqbal K, Grundke-Iqbal I, Merkle RK, Gong CX (2002). Role of glycosylation in hyperphosphorylation of tau in Alzheimer’s disease. FEBS Lett, 512, 101–6. [DOI] [PubMed] [Google Scholar]
- 103.Balmik AA, Chinnathambi S. (2021). Methylation as a key regulator of Tau aggregation and neuronal health in Alzheimer’s disease. Cell Commun Signal, 19, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wegmann S, Biernat J, Mandelkow E. (2021). A current view on Tau protein phosphorylation in Alzheimer’s disease. Curr Opin Neurobiol, 69, 131–138. [DOI] [PubMed] [Google Scholar]
- 105.Luo HB, Xia YY, Shu XJ, Liu ZC, Feng Y, Liu XH, . . . Wang JZ (2014). SUMOylation at K340 inhibits tau degradation through deregulating its phosphorylation and ubiquitination. Proc Natl Acad Sci U S A, 111, 16586–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li L, Jiang Y, Wang JZ, Liu R, Wang X. (2021). Tau Ubiquitination in Alzheimer’s Disease. Front Neurol, 12, 786353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Alquezar C, Arya S, Kao AW (2020). Tau Post-translational Modifications: Dynamic Transformers of Tau Function, Degradation, and Aggregation. Front Neurol, 11, 595532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yu Y, Run X, Liang Z, Li Y, Liu F, Liu Y, . . . Gong CX (2009). Developmental regulation of tau phosphorylation, tau kinases, and tau phosphatases. J Neurochem, 108, 1480–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Leroy A, Landrieu I, Huvent I, Legrand D, Codeville B, Wieruszeski JM, . . . Lippens G. (2010). Spectroscopic studies of GSK3{beta} phosphorylation of the neuronal tau protein and its interaction with the N-terminal domain of apolipoprotein E. J Biol Chem, 285, 33435–33444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liu K, Liu Y, Li L, Qin P, Iqbal J, Deng Y, . . . Qing H. (2016). Glycation alter the process of Tau phosphorylation to change Tau isoforms aggregation property. Biochim Biophys Acta, 1862, 192–201. [DOI] [PubMed] [Google Scholar]
- 111.Liu F, Zaidi T, Iqbal K, Grundke-Iqbal I, Gong CX (2002). Aberrant glycosylation modulates phosphorylation of tau by protein kinase A and dephosphorylation of tau by protein phosphatase 2A and 5. Neuroscience, 115, 829–37. [DOI] [PubMed] [Google Scholar]
- 112.Hasegawa M, Smith MJ, Iijima M, Tabira T, Goedert M. (1999). FTDP-17 mutations N279K and S305N in tau produce increased splicing of exon 10. FEBS Lett, 443, 93–6. [DOI] [PubMed] [Google Scholar]
- 113.Pickering-Brown SM, Baker M, Nonaka T, Ikeda K, Sharma S, Mackenzie J, . . . Mann DM (2004). Frontotemporal dementia with Pick-type histology associated with Q336R mutation in the tau gene. Brain, 127, 1415–26. [DOI] [PubMed] [Google Scholar]
- 114.Sinsky J, Majerova P, Kovac A, Kotlyar M, Jurisica I, Hanes J. (2020). Physiological Tau Interactome in Brain and Its Link to Tauopathies. J Proteome Res, 19, 2429–2442. [DOI] [PubMed] [Google Scholar]
- 115.Tracy TE, Madero-Perez J, Swaney DL, Chang TS, Moritz M, Konrad C, . . . Gan L. (2022). Tau interactome maps synaptic and mitochondrial processes associated with neurodegeneration. Cell, 185, 712–728 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhang Y, Wu KM, Yang L, Dong Q, Yu JT (2022). Tauopathies: new perspectives and challenges. Mol Neurodegener, 17, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Barbier P, Zejneli O, Martinho M, Lasorsa A, Belle V, Smet-Nocca C, . . . Landrieu I. (2019). Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front Aging Neurosci, 11, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Goedert M, Jakes R. (2005). Mutations causing neurodegenerative tauopathies. Biochim Biophys Acta, 1739, 240–50. [DOI] [PubMed] [Google Scholar]