

Abstract
Azines, such as pyridines, quinolines, pyrimidines, and pyridazines, are widespread components of pharmaceuticals. Their occurrence derives from a suite of physiochemical properties that match key criteria in drug design and is tunable by varying their substituents. Developments in synthetic chemistry, therefore, directly impact these efforts, and methods that can install various groups from azine C–H bonds are particularly valuable. Furthermore, there is a growing interest in late-stage functionalization (LSF) reactions that focus on advanced candidate compounds that are often complex structures with multiple heterocycles, functional groups, and reactive sites. Because of factors such as their electron-deficient nature and the effects of the Lewis basic N atom, azine C–H functionalization reactions are often distinct from their arene counterparts, and the application of these reactions in LSF contexts is difficult. However, there have been many significant advances in azine LSF reactions, and this review will describe this progress, much of which has occurred over the past decade. It is possible to categorize these reactions as radical addition processes, metal-catalyzed C–H activation reactions, and transformations occurring via dearomatized intermediates. Substantial variation in reaction design within each category indicates both the rich reactivity of these heterocycles and the creativity of the approaches involved.
Graphical Abstract
1. INTRODUCTION
Late-stage functionalization (LSF), broadly defined as reactions that transform complex organic molecules into valuable derivatives via their C–H bonds, is a burgeoning area in synthetic organic chemistry.1–13 It is now common to see examples of LSF in reports of new chemical processes, and its prevalence is a result of an inevitable progression in the C–H functionalization field. LSF intersects strongly with the modus operandi of drug discovery where the logic of assembling molecules via their C–H bonds suits structure–activity relationship (SAR) studies on complex druglike molecules. Although discussed less frequently, similar principles apply to agrochemical development.14 This way, medicinal and agrochemists can obtain analogue compounds in one or two steps without recourse to extensive synthesis from a simpler precursor. Here, we will review recent applications of LSF to azines, which are key components of pharmaceuticals and now frequent targets in this regime.
Structural analysis of FDA-approved pharmaceuticals shows that azines are highly prevalent in drug compounds, and Figure 1 shows representative examples.15 The Njardarson et al. 2014 survey found pyridine in 62 drugs, and it ranked as the second most common nitrogen heterocycle; 16 pharmaceuticals contained pyrimidines, which ranked 10th at that time.16 The Paul and Kumar group’s study of drugs approved in the 2015–2020 period ranked pyridines first and pyrimidines fourth.17 This data indicates that azines have, and continue to be, a significant focus in medicinal chemistry programs, and their occurrence results from the effect of the heterocycle’s intrinsic properties combined with its adorning substituents.18,19 The heterocycle often forms the key drug–receptor binding interaction via hydrogen bonding to the N lone pair. Furthermore, these electron-deficient heterocycles resist oxidative metabolism, which enables drugs to be dosed lower with fewer side effects. The potential for π-stacking and increased aqueous solubility are other beneficial properties.20–22 The substituents allow precise tuning of the steric and electronic environment of the azine core and serve other roles, such as additional binding sites and bonds to other portions of the drug.
Figure 1.
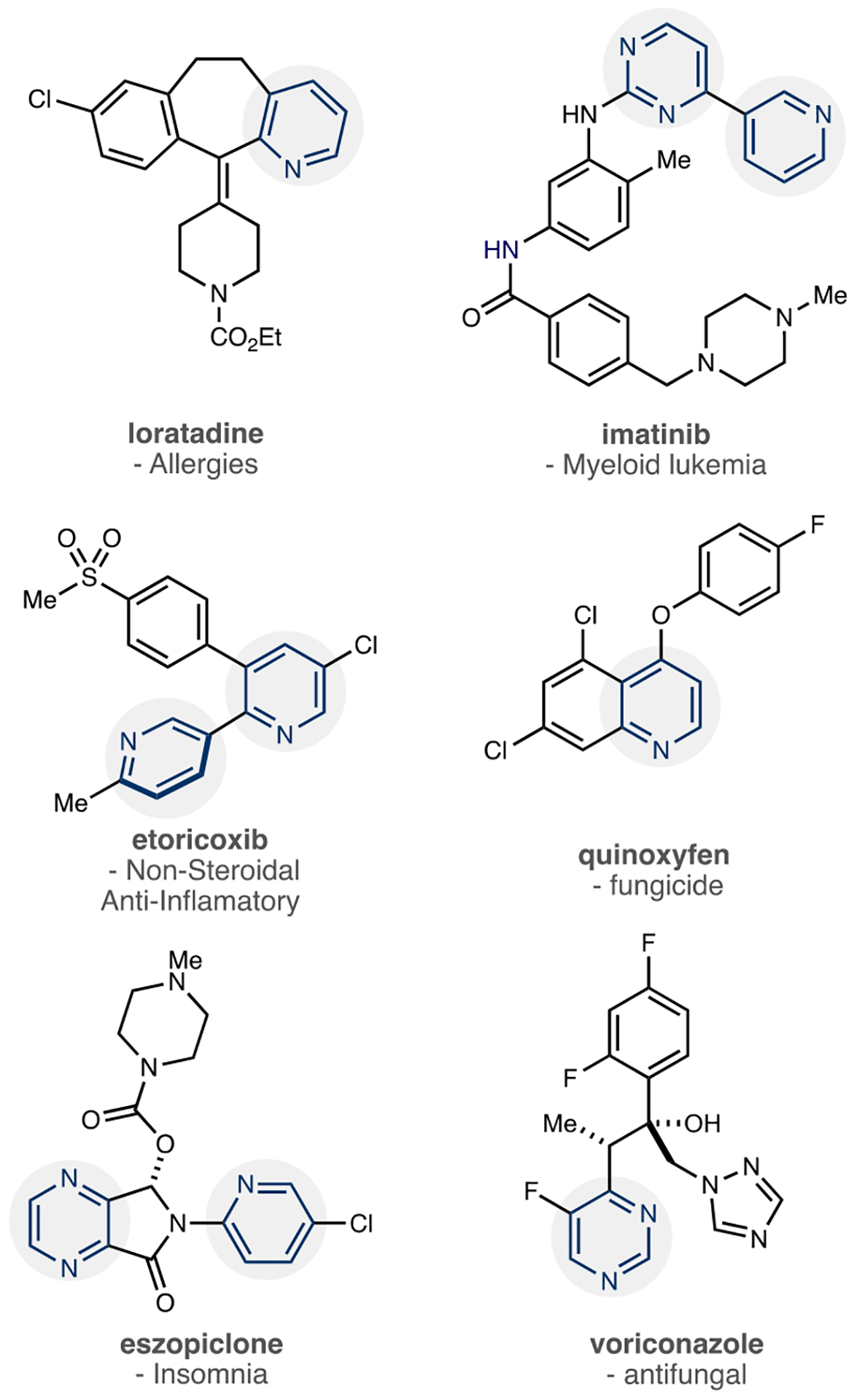
Examples of azines in pharmaceuticals and agrochemicals.
Given the multifaceted roles of azines in drug development, applying them to LSF applications is simultaneously an opportunity to rapidly optimize a candidate’s pharmacokinetic and pharmacodynamic (PK/PD) properties and is a formidable challenge for synthetic chemistry. The N lone pairs in these heterocycles, often exploited for drug–receptor interactions, can also bind to Lewis acids and form unwanted off-cycle binding in transition-metal-catalyzed reactions, for example. As azines are electron-deficient, they also perform poorly in electrophilic aromatic substitution (EAS) reactions.23,24 Controlling regioselectivity can be challenging and often requires directing groups, specific substrate patterns, or N atom prefunctionalization. Azine derivatives can have poor solubility in organic solvents. These challenges are inherent to azines, and LSF applications only amplify their difficulty.25 Furthermore, the central tenants of LSF processes are also at play. Reactions must tolerate a range of functional groups, select between multiple types of C–H bonds, differentiate between other Lewis basic heterocycles, and other criteria that others have extensively documented.
This review will show that the community has made significant progress in reactions for the LSF of azines. Synthetic chemists have adeptly translated reactions that functionalize the sp2 C–H bonds of arenes to azine cores. Also, several processes function on the premise that azines are electrophilic in conjunction with a rich diversity of chemistry that proceeds via pyridinium salts.26 It is instructive to classify the reactions by mechanism type, and we have grouped LSF reactions into three categories: radical processes, metal-catalyzed C–H functionalization reactions, and reactions proceeding via dearomatized intermediates. In terms of the usefulness of LSF reactions in this review, we agree with Ackermann and Johansson et al. in their 2021 review that LSF methods that install substituents, such as small alkyl and heteroatom-containing groups, halides, and other versatile functional groups, are most valuable.4 These modifications allow the subtle tuning of PK/PD properties required at late stages of drug development and serve as handles for compound diversification. Appending larger groups, such as arenes, can excessively distort a compound’s properties. However, there are scenarios before the late-stage frontier involving complex substrates where access to an arylated library, for example, would be valuable. As such, we believe broadening the scope of LSF applications is useful and allows the reader to view the full spectrum of transformation. Note that classic approaches to azine functionalization, such as pyridine N-oxide chemistry, occurred before the surge in LSF reactions and are rare in this review. However, given the developments in this area, these reagents are likely more useful for LSF than their representation in the recent literature suggests.27–29 We will not describe applications of hydrogen isotope exchange reactions that other reviews have summarized extensively.30
2. RADICAL-MEDIATED LSF REACTIONS
2.1. Introduction to Radical-Mediated Processes
In 1991, Sawada et al. reported an early example of azine LSF that led to the development of irenotecan, a treatment for colon cancer and small-cell lung cancer.31,32 Its precursor, camptothecin, is an alkaloid with inhibitory activity toward leukemia, but severe toxicity precluded any further development of the compound as a therapeutic. Scheme 1 shows a key LSF step toward irenotecan involving a radical alkylation using propionaldehyde as the alkylating reagent. While this process involves strongly acidic conditions that would not broadly apply to other azine-containing pharmaceutical candidates, it does show the major benefits of azine radical additions, many of which stem from the venerable Minisci reaction,33 which has enabled drug developers to obtain a valuable analogue in a single step from a C–H precursor on a complex structure. Eventually, the synthetic community recognized the power of these Minisci-type reactions for LSF, and they are now the most frequently reported methods in the chemical literature for this purpose.
Scheme 1.

Discovery of Irenotecan Using a Radical-Mediated Alkylation Reaction
In this section, we will outline several factors that explain this rapid growth of radical-based reactions for LSF. From a chemical standpoint, several important features make these processes inherently suitable. The energetic penalty when radicals add to azines and disrupt their aromatic system is evidently lower, in general, than two-electron processes. The venerable Minisci reaction, and subsequent reports, have indicated that numerous classes of heterocycles, including many azines, are amenable to these processes.33–35 Further advancements in synthetic method development (vide infra) have shown that the reactions could be performed under mild conditions and with more convenient radical precursors showing excellent functional group compatibility, thereby obviating the need to protect alcohols and amines, for example. Furthermore, these reactions lend themselves superbly to emerging technologies, such as high-throughput experimentation and flow reactors. With hindsight, it is unsurprising that the chemical community has gravitated toward these radical reactions, and further developments are continually being documented.
Between Minisci’s seminal report and Sawada’s discovery of irenotecan from camptothecin, there was relatively little activity in azine LSF via radical addition processes before a surge of papers in the preceding decade that we will outline below. In this hiatus, two examples are notable. In 2009, Lou et al. described a protocol for adding carbon-bearing groups to camptothecin from aldehyde precursor and found that obtaining alkylated and acylated products was possible (Scheme 2A, 1 and 2).36 In the same year, Duncton et al. reported a protocol for radical alkylation using FeSO4 and H2O2 with alkyl iodides as radical precursors (Scheme 2B).37 They showed that this method could couple valuable fragments, such as oxetanes, to gefitinib (3).
Scheme 2.
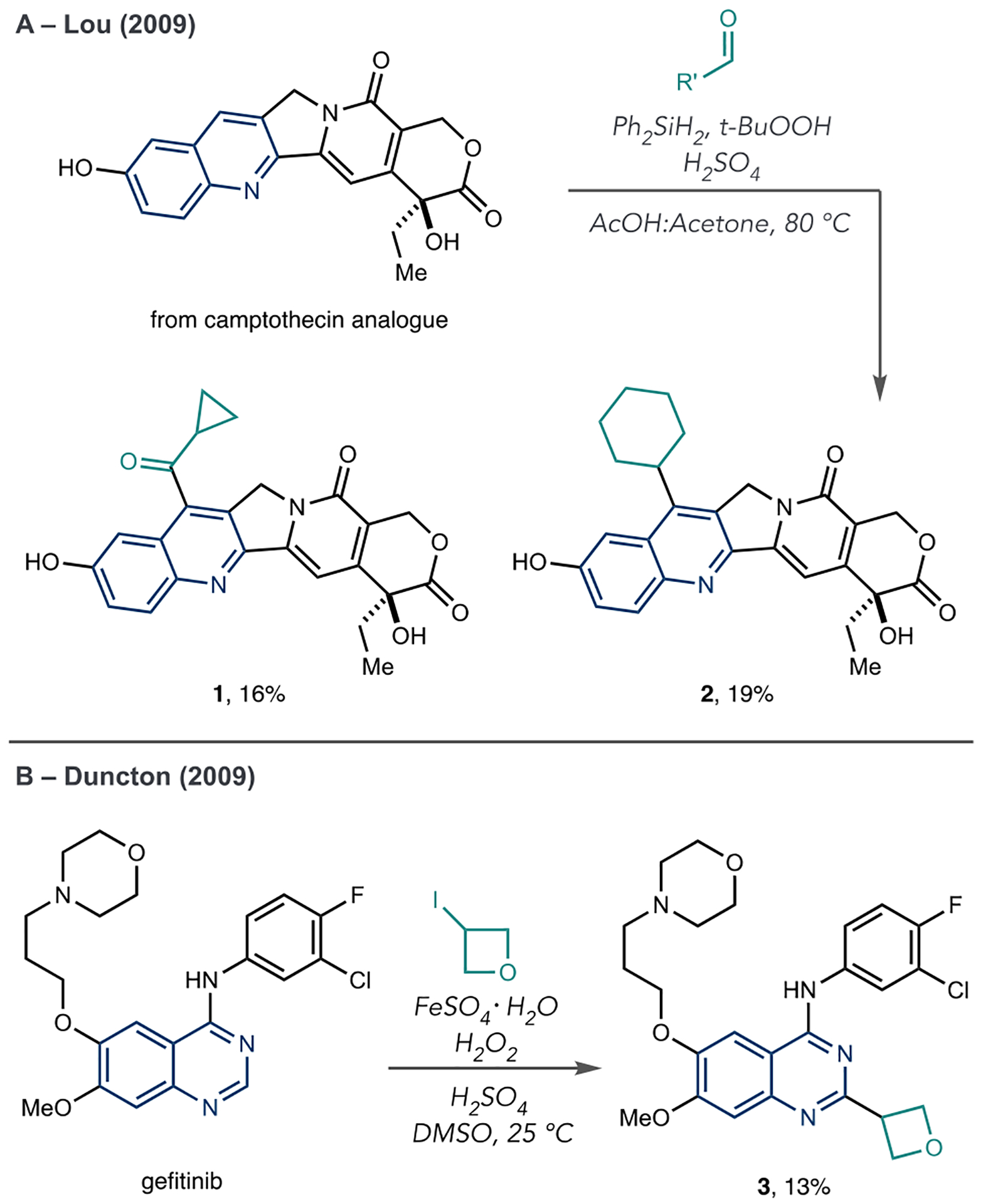
Lou et al. and Duncton et al. Examples of Azine LSF Using Minisci-Type Reactions
Baran et al. produced a seminal report in 2010, where his group showed that aryl boronic acids serve as radical precursors under oxidative conditions in a Minisci-type process (Scheme 3).38 This report is significant because it indicated that other classes of reagents, beyond those typically employed in Minisci reactions, could serve as coupling partners in these radical-mediated processes. Furthermore, mild oxidants and less reliance on strong acidic media make these processes more amenable to complex drugs and agrochemicals. The advent of photoredox catalysis and other visible-light-driven reactions was another significant impetus for the rapid development of azine radical addition reactions.
Scheme 3.

Aryl Boronic Acids as Radical Precursors for Azine Arylation
The following sections outline a range of methods for azine LSF reactions via open shell pathways. It is important to note that fundamental constraints exist in both the radical species and the azines typically employed. First, these reactions tend to feature more stabilized radicals, such as secondary or tertiary carbon-centered radicals or carbon-centered radicals stabilized by adjacent heteroatoms. Transformations that use methyl radicals are still rare, despite the importance of azine methylation reactions.39 Second, regiocontrol can be challenging and often depends on the azine’s structure, substitution pattern, and electronic disposition.40 Third, carbon–heteroatom bond formation via radical pathways is underdeveloped and suffers from a mismatch in electronics between azine cores and heteroatom-centered radicals. Despite these limitations, in little over a decade, radical addition processes are the biggest collection of reactions for azine LSF.
2.2. Reagent-Based Methods
The Baran lab has developed a family of metal sulfinates, based on Langlois’ reagent, for the radical alkylation and fluoroalkylation of azines (Scheme 4A).41–46 They propose that tert-butoxy radicals form in the reaction and oxidize the sulfinate salts, which subsequently lose SO2 and form the key carbon-centered radicals. These radicals readily add to azines, and reoxidation of the aromatic system, as described in Scheme 4B, completes the process. These reactions have a large substrate scope, and most azines undergo C–C bond formation under their reaction protocol. Scheme 4C shows examples of LSF reactions using these sulfinates. Fluoroalkylation is particularly valuable in drug and agrochemical development because these substituents can profoundly affect the biological properties of candidate molecules, particularly through improved binding and the modulation of lipophilicity. Examples from their study include trifluoromethylated derivatives of dihydroquinine and varenicline (5 and 8). Difluoromethyl groups act as lipophilic hydrogen bond donors that improve membrane permeability; 6 is an example of how this protocol can conveniently install this substituent on an azine framework. They can also add various alkyl groups, such as in 7. Baran’s group also showed that this method has potential for biorthogonal chemistry via the installation of an azide-containing alkyl chain to pioglitazone (9).
Scheme 4.

Baran’s Group Metal Sulfinate Reagents for Azine Functionalization
The regioselectivity of these reactions depends on several factors, including the nucleophilicity of the carbon-centered radical involved, the substituents appended to the azine, and the reaction solvent. A subsequent study details these effects and provides predictive guidelines. Nevertheless, the broad scope of these reaction will provide practitioners with a high chance of obtaining azine analogues in LSF applications.40
Since the Baran group’s initial report using metal sulfinates as radical sources for C–C bond formation, several other reagents have emerged as radical sources for Minisci reactions using chemical oxidants as radical initiators (Scheme 5).34 In general, radical generation follows a similar pathway to that described in Scheme 4B, where single-electron oxidation of the radical precursor results in a carbon-centered radical and performs the reoxidation step to reform the azine core. In 2017, the Molander group reported an alkylation of pyridines, quinolines, and isoquinolines with 1,4-dihydropyridines (1,4-DHP) as the radical source (Scheme 5A).47 Sodium persulfate initiates the radical formation by oxidizing 1,4-DHP. Then, a typical Minisci-type reaction occurs and forms the alkylated products. They showed that 1,4-dihydropyridines with a range of alkyl substituents apply to this protocol, including primary, secondary, and tertiary structures. Compound 10 also shows that radicals adjacent to heteroatoms are amenable (vide infra). They observed the typical regioselectivity associated with Minisci-type reactions, but they could, however, obtain single isomers when certain positions on the azine were blocked, such as in quinine derivative 11. In total, they reported eight LSF alkylation reactions using these reagents.
Scheme 5.
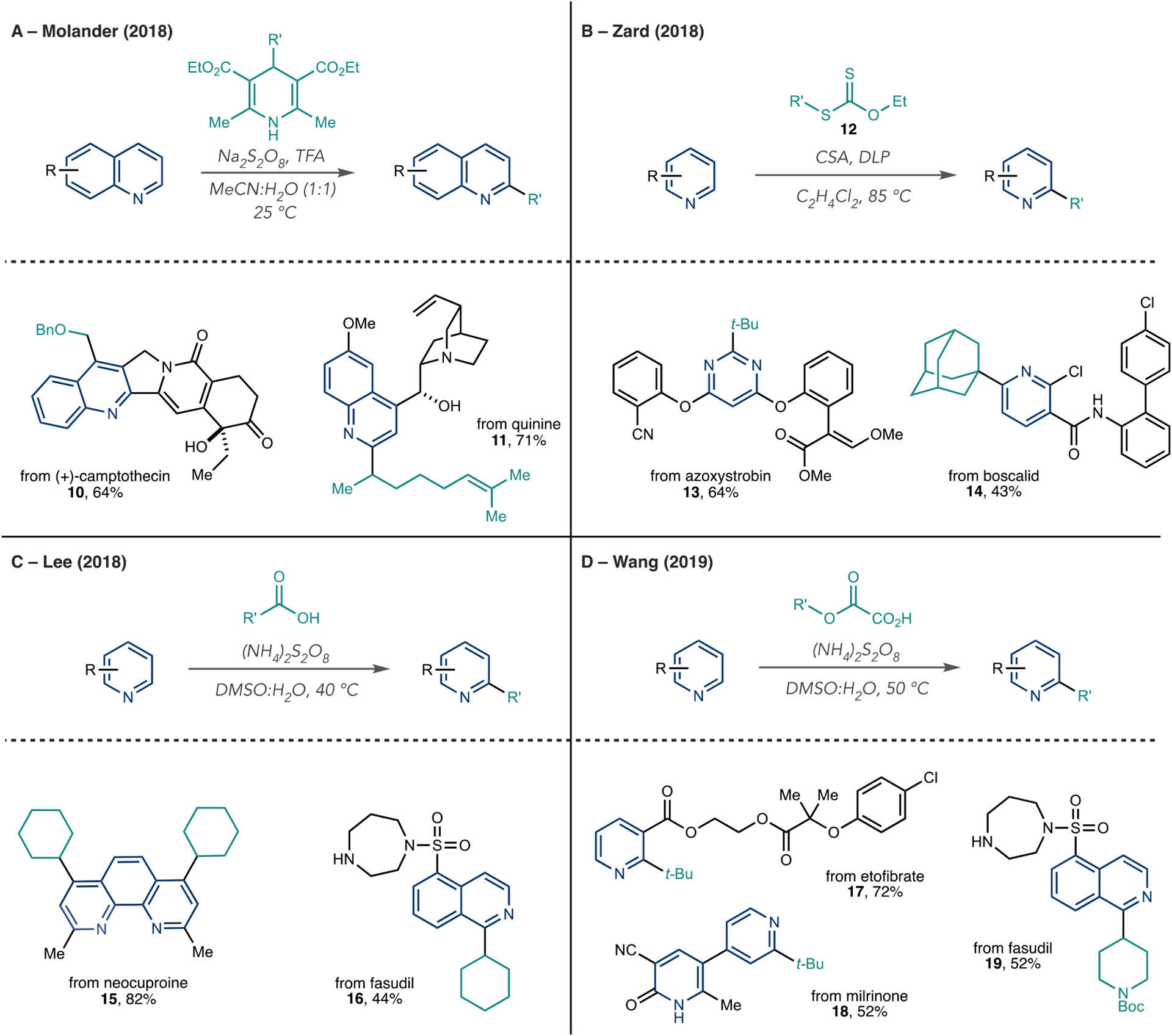
Other Reagents for Azine Alkylation Reactions under Oxidative Conditions
Zard et al. disclosed that xanthates serve as radical precursors for azine C–H alkylation reaction (Scheme 5B).48 In the reaction, dilauroyl peroxide (DLP) homolytically cleaves, and the resulting oxygen-centered radical adds to the xanthate 12 to ultimately form a carbon-centered radical, and a Minisci process ensues. Numerous azines are amenable, including pyridines, pyrazines, quinolines, and others. As in other cases, regioselectivity is challenging to control, and mono- and dialkylated products often form. The authors demonstrated the alkylation of nine LSF examples, including azoxystrobin and boscalid (13 and 14). In 2018, Lee et al. showed that carboxylic acids are viable as radical precursors under relatively mild conditions compared with previous reports, although the LSF scope in this report was more limited than when using other reagents (Scheme 5C, 15 and 16).49 Lastly, the Wang group reported oxalic acids as precursors for primary, secondary, and tertiary radicals using persulfates as the chemical oxidant (Scheme 5D).50,51 The oxalic acids protonate the azine, and the persulfate oxidizes the oxalate anion, which liberates two equivalents of CO2 when forming the radical intermediate. The authors showed several examples of azine LSF, including derivatives, such as etofibrate, milrinone, and fasudil (17–19).
2.3. Photoredox and Visible-Light-Mediated Processes
2.3.1. General Mechanisms for Photoredox Azine Alkylations.
Like the advances in reagent-based transformations described above, photoredox and photomediated processes have enabled many new azine radical addition processes.34,52,53 The ability of these methods to generate carbon-centered radicals under mild reaction conditions from convenient precursors also engenders these processes to LSF applications. There will be deviations in the precise ordering of events in the subsequent sections. However, many reactions operate within the catalytic cycles shown in Scheme 6, which are helpful as general representations of the order of steps in these radical addition reactions. Scheme 6A is an oxidative quenching cycle where the excited state photocatalyst reduces the radical precursor, which is a reagent designed with a functional group that becomes anionic as the carbon-centered radical forms. The radical then adds to a protonated azine to form a dearomatized radical cation; single-electron oxidation and subsequent deprotonation rearomatizes the ring, thereby completing the photoredox cycle and releasing the functionalized azine product. Scheme 6B shows a reductive photoredox cycle where the radical precursor is instead primed to lose an electron en route to forming the carbon-centered radical. These cycles usually involve a sacrificial oxidant to reform the azine ring system and regenerate the photocatalyst.
Scheme 6.
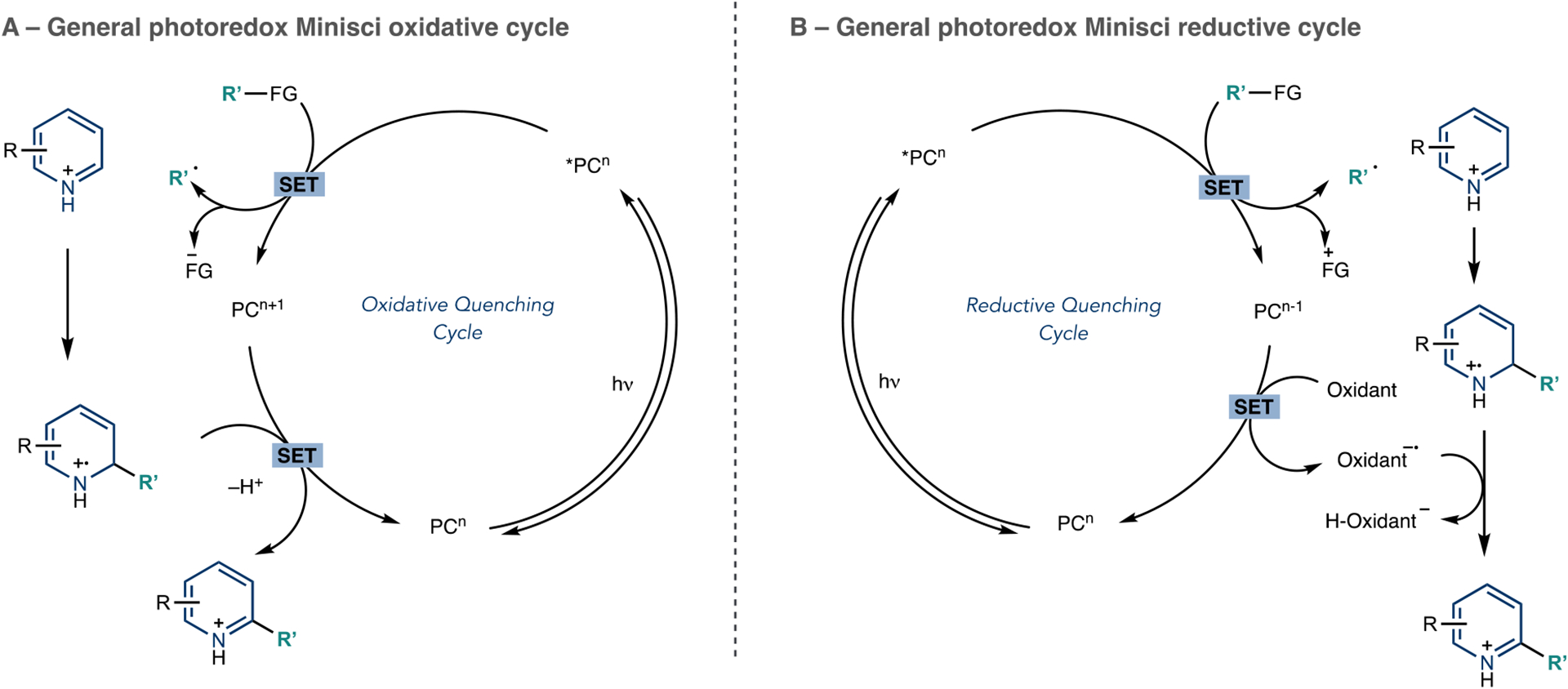
Photoredox Catalytic Cycles for Azine Radical Addition Reactions
2.3.2. Reactions That Append Alkyl and Acyl Fragments.
We will first examine reactions that append alkyl fragments to azines. DiRocco and co-workers used high-throughput experimentation to develop a photoredox-catalyzed alkylation of azine-containing pharmaceuticals (Scheme 7A).54 This process is particularly valuable for LSF applications because the authors showed several examples of azine C–H methylation. The so-called “magic methyl” effect derives from numerous studies where installing this group on drug candidates can significantly improve their PK/PD properties.39 The authors found that cyclometalated Ir photocatalysts, {Ir[dF(CF3)ppy]2(dtbpy)}PF6 or [Ir(ppy)2(dtpy)]PF6, were optimal, and peroxides were useful radical precursors. Their proposed mechanism follows an oxidative photoredox catalytic cycle. They propose that tert-butylperacetate undergoes homolytic cleavage of the O–O bond via proton-coupled electron transfer to form the tert-butoxy radical. Next, β-scission of the tert-butoxy radical generates the methyl radical, which adds to the protonated azine, and is followed by oxidation to produce the methylated product.
Scheme 7.
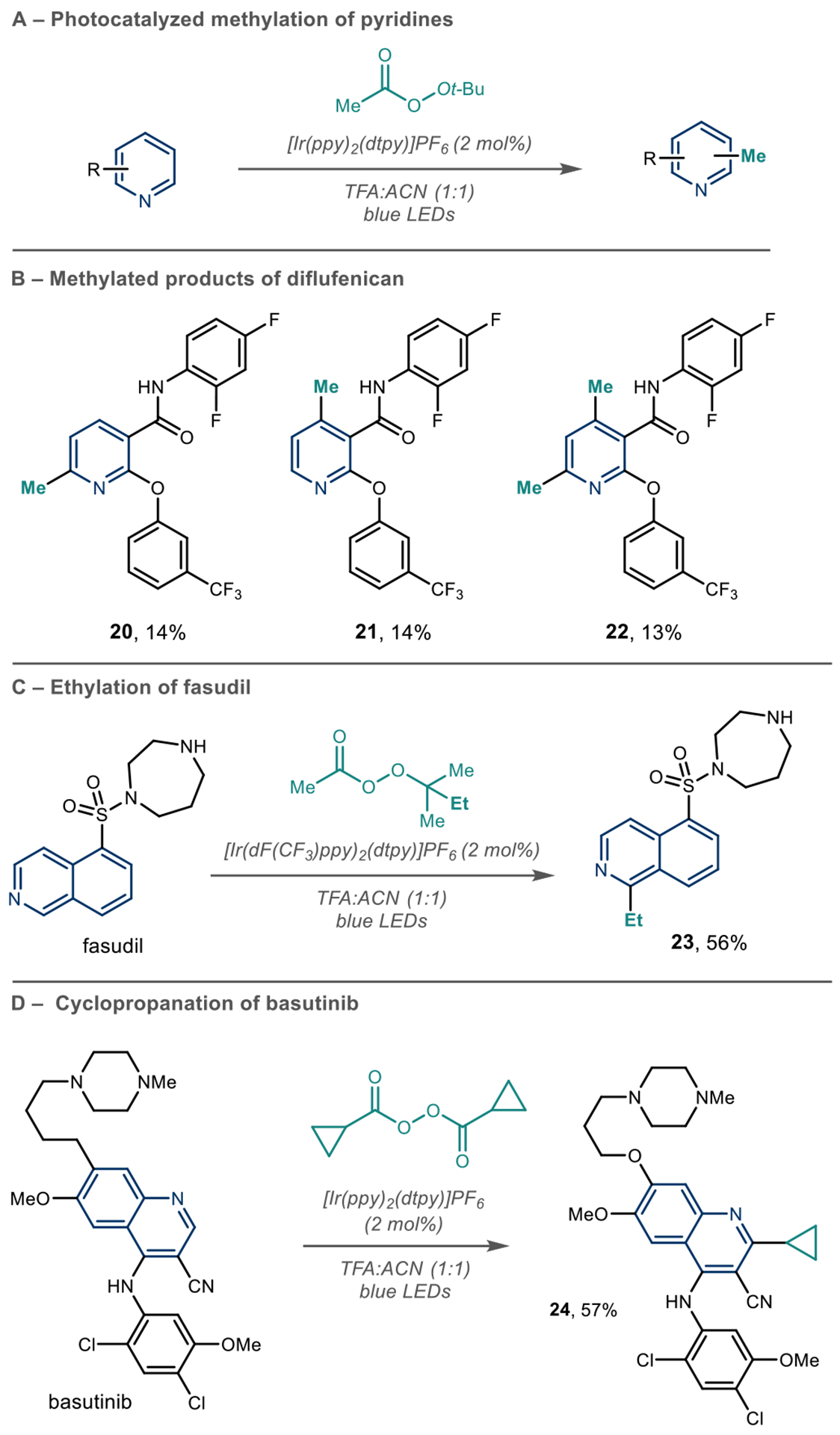
Peroxides as Alkyl Radical Precursors under Photoredox Conditions
This photoredox process can methylate pyridines, quinolines, isoquinolines, pyrimidine, and pyrazine containing free basic amines, alcohols, amides, and esters. The regioselectivity for this reaction follows the trends for nucleophilic addition into protonated azines, so it is common to observe regioselective mixtures and polymethylation in unbiased systems. Tuning of the light intensity avoids rapid heat generation, maintains low radical concentration to reduce possible side reactions, and therefore allows the desired pathways to dominate, which makes this method applicable to LSF as portrayed in their scope. When they subjected diflufenican to the reaction conditions, they observed a mixture of 2- and 4-mono- and 2,4-bis-methylated products 20, 21, and 22 (Scheme 7B). Although compound mixtures are often observed, the breadth of structures that are amenable to this process is striking. With modern separation techniques, it is arguably an advantage to produce multiple distinct analogues that can proceed into biological screening. The addition of other alkyl groups is possible by changing the structure of the peroxide reagent, such as the ethylation of fasudil (Scheme 7C) and cyclopropanation of bosutinib (Scheme 7D).
Scheme 8 shows five other examples of photoredox-catalyzed azine alkylation reactions using carboxylic acids and aldehydes as radical precursors. These reagents are appealing because of their high abundance and structural variability, as well as being relatively stable and inexpensive. There are several mechanistic pathways that can result in the carbon-centered radical in these processes. Most cases involve forming an activated carboxylate intermediate that enters an oxidative photoredox cycle (Scheme 6); the resulting carboxyl radical then loses CO2 to form the radical intermediate.
Scheme 8.
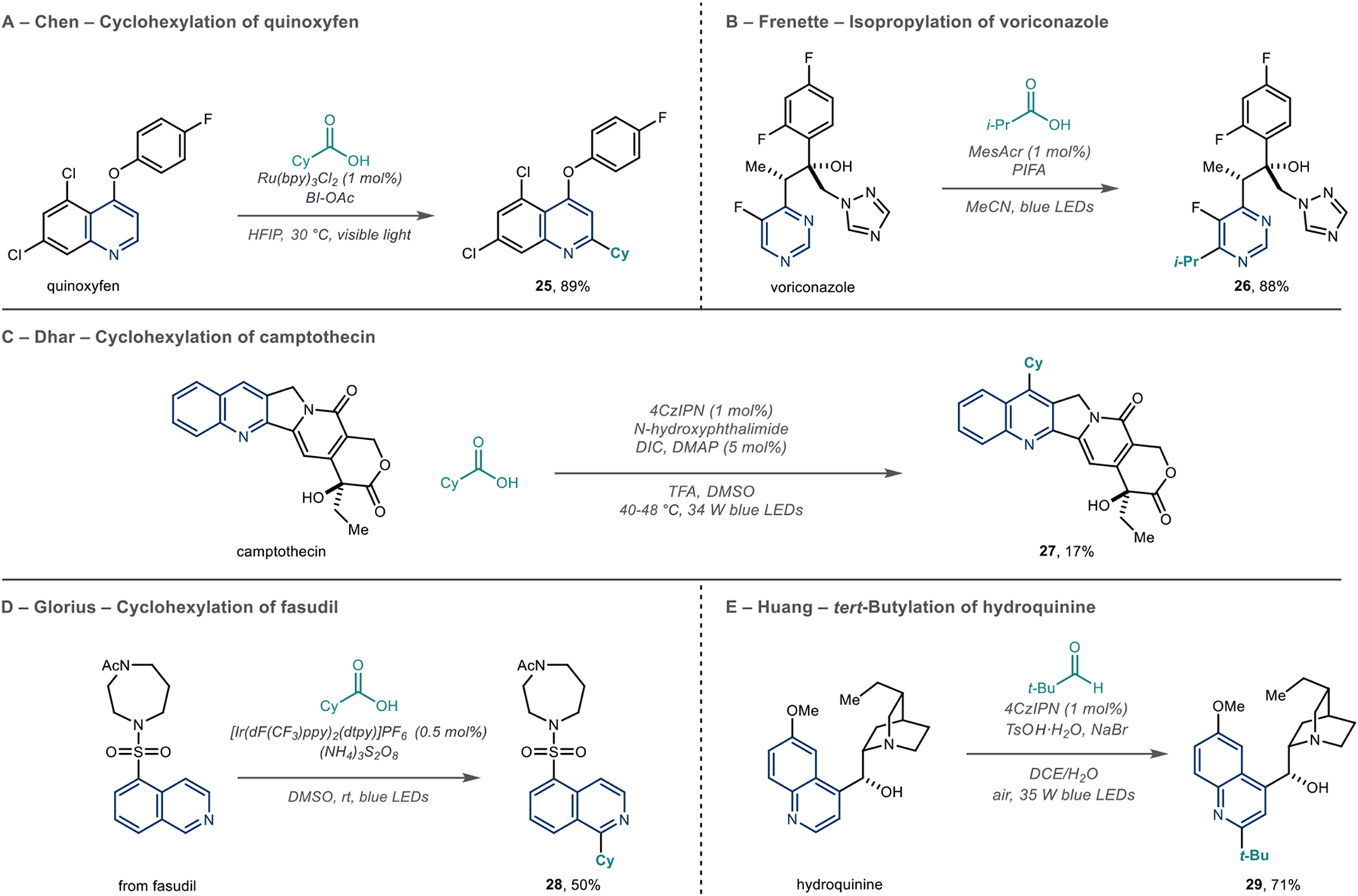
Carboxylic Acids and Aldehydes as Radical Precursors in Photoredox Reactions
Using this principle, Chen et al. reported a photoredox-catalyzed Minisci alkylation of azines with various aliphatic carboxylic acids (Scheme 8A).55 Here, the authors used Bl-OAc (acetoxybenziodoxole) to form an iodocarboxylate intermediate as the in situ radical precursor and used this method to alkylate the 2-position of the fungicide quinoxyfen (25). Frenette et al. used a hypervalent carboxylate as an intermediate derived from phenyliodine bis(trifluoroacetate) (PIFA) with an organic photocatalyst, 9-mesityl-10-methyl acridinium (MesAcr), to add an isopropyl group to voriconazole (Scheme 8B, 26).56 In this case, the authors proposed that a reduced form of the organic photocatlyst (MesAcr•) is the putative catalytic intermediate that reduces the iodocarboxylate. Dhar et al. formed an N-hydroxyphthalimide ester from cyclohexyl carboxylic acid in a photoredox reaction that alkylated camptothecin (Scheme 8C, 27).57 Glorius et al. proposed an alternative mechanism for radical generation in their 2017 report focusing on azine alkylation via photoredox catalysis.58 In this case, a sulfate radical anion performs a hydrogen atom abstraction (HAT) process on the carboxylic acid to generate the carboxyl radical that decarboxylates to form a carbon-centered radical. Using this method, the authors showed that fasudil could be alkylated in reasonable yield (Scheme 8D, 28). The Huang group described a C–C bond-forming reaction with aldehydes that was mediated by visible light photoredox with air as the sole oxidant and 4CzIPN as the photocatalyst (Scheme 8E).59 This reaction proceeds via a formyl radical intermediate that decarbonylates to form alkyl radicals. The authors found that TsOH·H2O was the optimal acid, and the NaBr additive resulted in fewer byproducts. Using this system, they successfully alkylated the quinoline ring in hydroquinine using pivalaldehyde (29).
Several examples show that organoboron reagents are effective radical precursors in photoredox-catalyzed alkylations and have potential for LSF processes (Scheme 9). The Molander group used alkyltrifluoroborate salts in conjunction with MesAcr and K2S2O8 for azine alkylation. In these cases, single-electron oxidation by the excited state of the MesAcr catalyst generates the carbon-centered radical, and the group demonstrated its use for more complex molecules by adding a tert-butyl group to quinine 30.60 Subsequently, the Chen group reported a photoredox-catalyzed azine C–H alkylation reaction with primary and secondary alkyl boronic acids using [Ru(bpy)3Cl2] as a photocatalyst.61 BI-OAc plays a key role in this process; this peroxide is first reduced, and its corresponding carboxyl radical adds to the boronic acid, thereby facilitating the carbon-centered radical formation (Scheme 9). In this way, the group impressively showed that they could add primary radicals to azines and used this method to form ethylated quinine derivative 31.
Scheme 9.
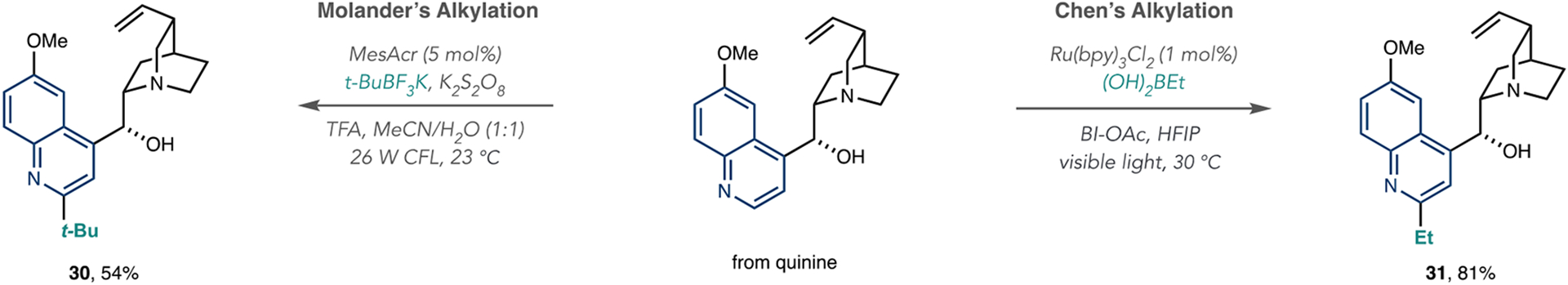
Organoboron Reagents as Radical Precursors
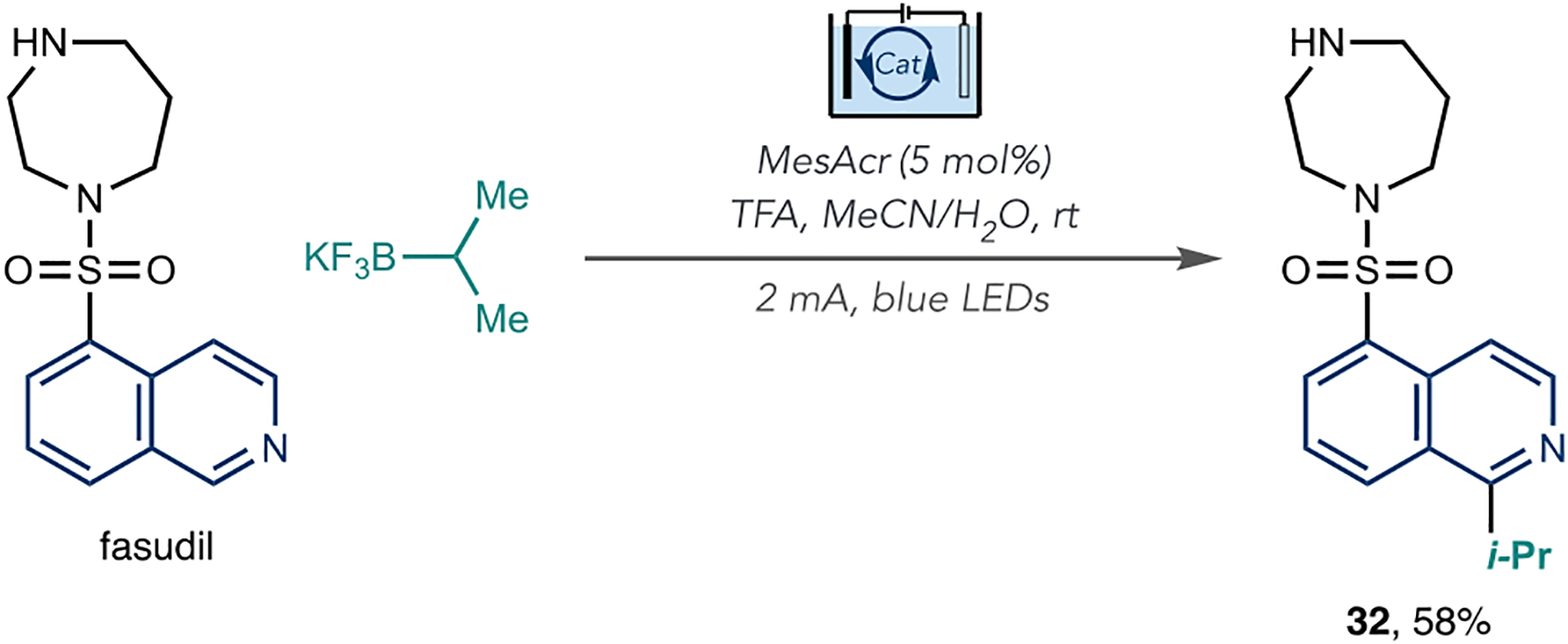
The Xu group reported a photoelectrochemical C–H alkylation of azines that overcame the challenge of preventing overoxidation of alkylation reactions under electrochemical conditions (Scheme 10).62 They conducted the electrolysis with a constant current in an undivided cell equipped with a reticulated vitreous carbon (RVC) anode, platinum plate cathode, and blue LEDs as the light source. The optimal reaction used MesAcr as the catalyst and TFA as an acid additive. Notably, this approach obviates the requirement for chemical oxidants, and by using organotrifluoroborate salts, the authors added primary, secondary and tertiary alkyl groups to a range of azines. They also showed four examples of azine LSF reactions, including fasudil, to form isopropylated derivative 32. This combined approach of photo- and electrochemistry could enable reactions on sensitive substrates that may not be viable using each approach individually.
Scheme 10.
Photoelectrochemical Approach to Azine Alkylation with Organotrifluoroborate Salts
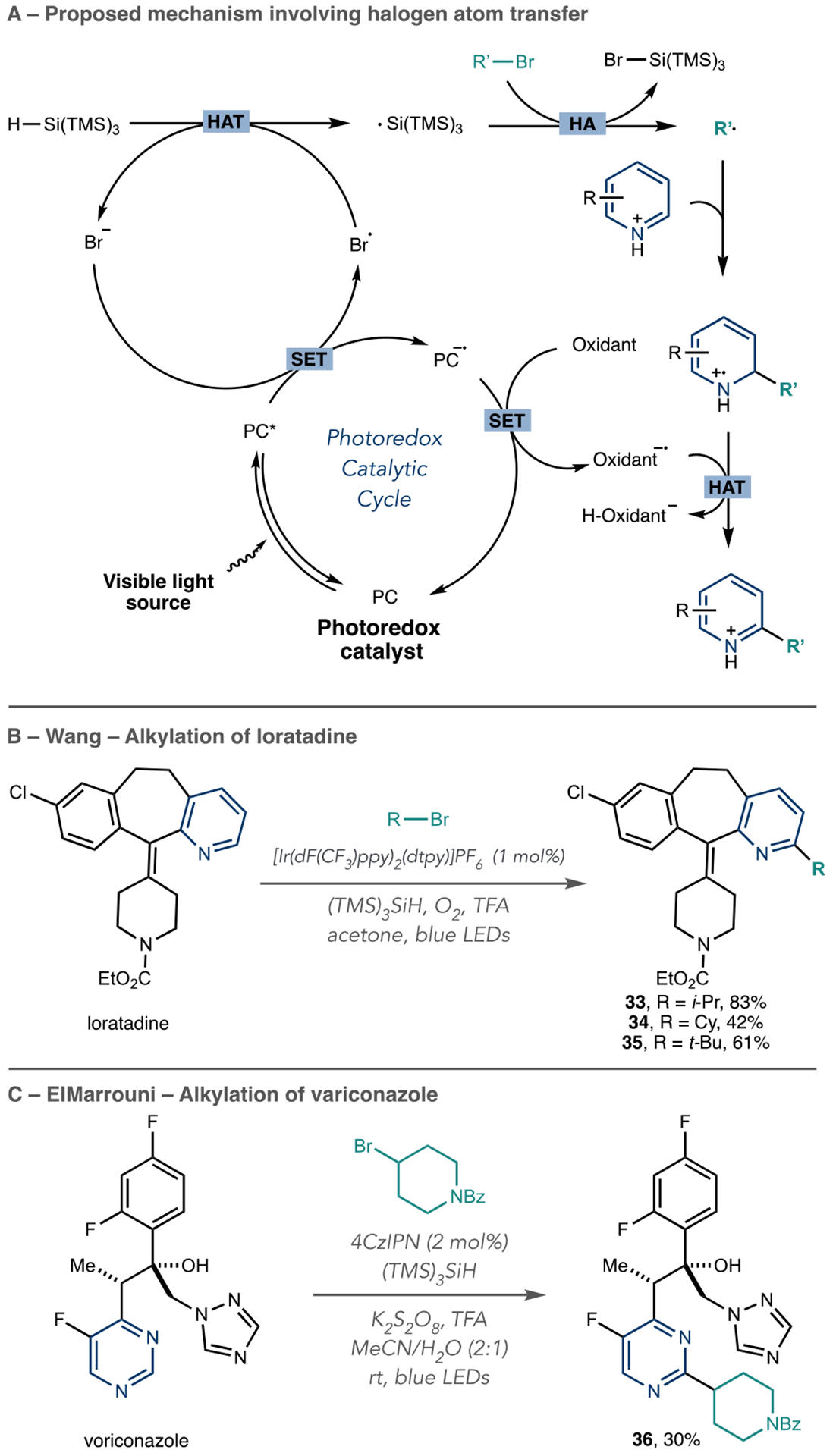
The use of alkyl halides is an advance in azine alkylation reactions because they are abundant, simple to prepare, and many variants are commercially available. These reactions required modifications of the typical photoredox process described above. In their Ni-catalyzed metallaphotoredox cross-electrophile coupling reactions, the MacMillan group employed silyl radical intermediates as halogen atom abstractors.63 Scheme 11A shows how these silicon radicals also enable Minisci-type radical addition process.64 To begin the cycle, the excited state photocatalyst oxidizes trace amounts of bromide ions that are postulated to form under the reaction conditions. The resulting bromine radical then abstracts a H atom from (TMS)3SiH, and the silyl radical then abstracts a Br atom from the alkyl halide. The typical radical addition–reoxidation sequence then ensues. The Wang group published a protocol for azine C–H alkylation using alkyl halides and molecular oxygen as the terminal oxidant (Scheme 11B). Alkyl iodides can be used in place of bromides, if necessary, but alkyl chlorides showed no reactivity. The authors showed this method’s amenability for LSF on a variety of bioactive azine-containing compounds and formed multiple alkyl derivatives of loratadine with selectivity for the 2-position of the pyridine ring (33–35). Similarly, ElMarrouni et al. used alkyl bromides, (TMS)3SiH, and CzIPN as a photocatalyst for azine alkylation and produced an alkylated voriconazole derivative using this protocol (Scheme 11C, 36).65
Scheme 11.
Alkyl Halides in Photoredox Azine Alkylation Reactions Using Silicon Radicals for Halogen Atom Transfer Reactions
A collaboration between the Frenette group and Pfizer used Mn2(CO)10 37 as an alternative to silanes (Scheme 12).66 They proposed that Mn(CO)5 radical 38 forms under visible light irritation, and this species abstracts the iodide atom from the alkyl halide to form the alkyl radical 39. They used high-throughput experimentation to evaluate the potential of this reaction for LSF. They reported 35 examples, including fasudil, varenicline, hydroquinine, and bosutinib with various alkyl iodides in moderate to good yields (40 and 41).
Scheme 12.

Mn-Catalyzed Photoredox Azine Alkylation Using Alkyl Halides
Fluorine-18 (18F) is one of the most commonly used isotopes for positron emission tomography (PET) experiments to measure biodistribution and receptor occupancy in the central nervous system.67 Despite this, there are limited methods to install 18F atoms late-stage. One report by the Luxen and Genicot groups used photoredox catalysis in flow to achieve radiolabeled difluoromethyled azines from the C–H bond (Scheme 13).68 A benzothiazolesulfone derivative acts as the difluoromethyl (CF2H) radical source in the reaction. The mechanism occurs like typical radical trifluoromethylation reactions on aromatics initiated by a photocatalyst, and the regioselectivity rationale is similar to other Minisci reports. The authors reported several LSF examples, including a moxonidine analogue and a SV2A PET tracer, thereby showing the LSF utility for isotopic labeling experiments (42–44). The process also extends to nucleosides, including uridine and cytidine (45 and 46), and it is noteworthy that the Li, Zhang, and He group showed a similar photoredox approach to fluoroalkylate nucleosides.69 It is important to note that several other groups have reported azine fluoroalkylation reactions, and while LSF did not feature in their reports, it would undoubtedly apply in those applications.34
Scheme 13.

Azine Difluoroalkylation for 18F Labeling
The examples shown thus far in this section have used preinstalled functional groups to form alkyl radicals at specific positions on alkyl groups. Scheme 14 shows two examples that involve forming radicals at unactivated positions on alkyl groups. In 2018, the Zhu group reported an azine alkylation reaction using alkyl alcohols and PIFA, which serve as the radical source and initiator, respectively (Scheme 14A).70 In this reaction, PIFA reacts with the alcohol 47 to form 48. Blue light then cleaves the O–I bond to generate reactive O-centered radical 49, which undergoes 1,5-HAT to form C-centered radical 50 (Scheme 14B). A typical Minisci-like process occurs to yield the alkylated products. As in other reports, numerous azines are amenable to this process. The authors demonstrated two LSF examples, including the alkylation of voriconazole and eszopiclone (51 and 52). Similarly, the Jin group reported an alkylation reaction of azines using Selectfluor 53 as a HAT reagent to generate alkyl radicals (Scheme 14C).71 They proposed that exposing Selectfluor to blue light forms radical cation 54. Compound 54 then performs HAT on cyclohexane and generates alkyl radical 55, which subsequently adds to the azine to form the C–C bond. The heteroarene scope is similar to the Zhu report, and Scheme 14D shows LSF examples involving quinoxyfen and fasudil and using cyclohexane as a reagent (56 and 57).
Scheme 14.

Azine Alkylation via Radical Generation at Unactivated C–H Bonds
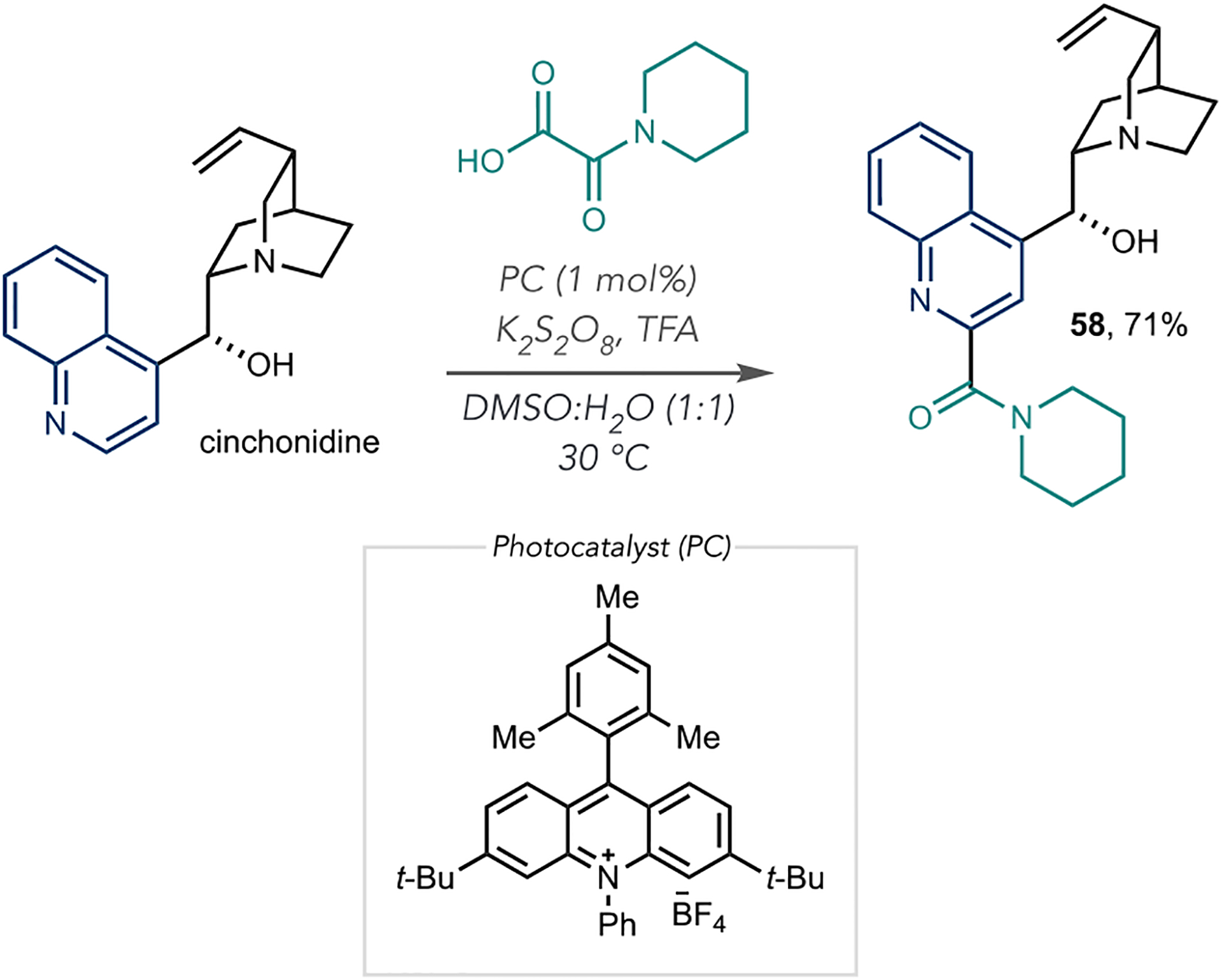
As well as alkylation reactions, these photoredox processes are useful for adding acyl groups to azines, and Jouffroy and Kong from Merck developed a visible-light-mediated amidation of heteroaromatics (Scheme 15).72 The reaction mechanism follows typical Minisci-type reactivity, where the carbamoyl radical generates from potassium oxamate salts or the corresponding oxamic acids in the presence of a photocatalyst and chemical oxidant. N-Alkyl and N,N-dialkyl carbamoyl groups are successfully installed on various heteroaromatics that are typical with Minisci-like reactivity, such as pyridines, pyrimidines, and quinolines. Cinchonidine undergoes amidation (58), as shown in Scheme 15. However, other LSF azines may be amenable, as reported in related Minisci-like transformations.
Scheme 15.
Photoredox-Catalyzed Amidation for Azine LSF
2.3.3. Reactions Proceeding via Radicals Adjacent to Heteroatoms.
There are a number of azine addition processes that exploit carbon-centered radicals adjacent to heteroatoms because of the inherent stability of these radicals, which allows selective radical formation among other C–H bonds.52,53 Furthermore, these products also have high value in drug development programs. The Wang group described a photoredox cross-dehydrogenative coupling (CDC) reaction for selective α-aminoalkylation of quinolines, isoquinolines, quinoxalines, and pyrimidines (Scheme 16).73 They found using {Ir[dF(CF3)ppy]2(dtbbpy)}PF6 as a photocatalyst, trifluoroacetic acid (TFA) as a proton source, and tert-butyl peracetate as an oxidant under blue light irradiation afforded α-aminoalkylated products. Scheme 16A shows a proposed mechanism where HAT between the N-protected pyrrolidine and the tert-butoxy radical generates α-amino radical 59, which adds to the protonated quinoline via a Minisci-type pathway to afford radical cation 60. Single-electron oxidation of this intermediate by the IrIV species and deprotonation gives the final α-aminoalkylated product 61 and closes the photoredox cycle. The Wang group showed one example of azine LSF using this protocol, where they coupled N-Boc pyrrolidine with fasudil to form 62 (Scheme 16B).
Scheme 16.

Photoredox-Catalyzed Azine Aminoalkylation Using a HAT Process
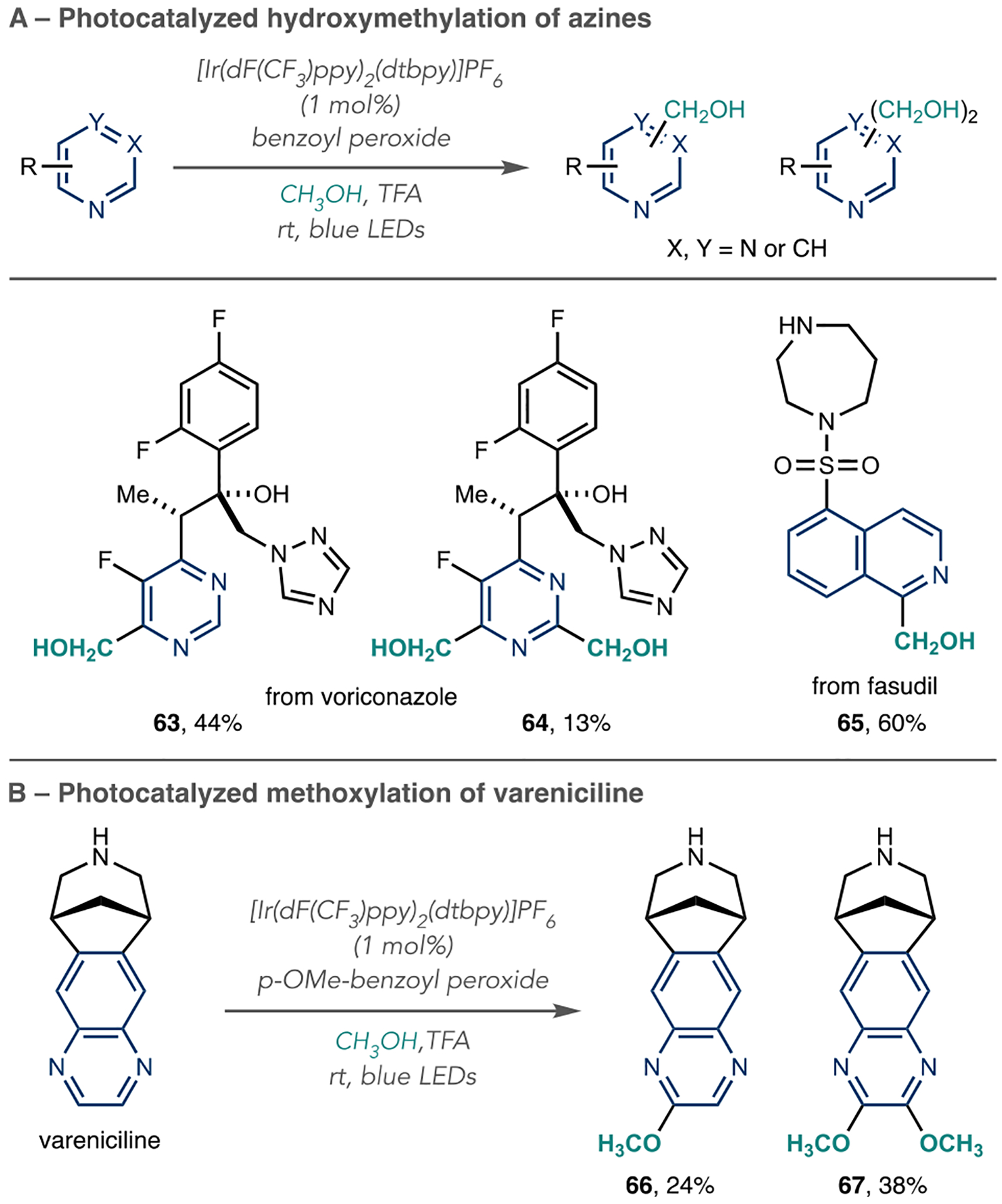
Huff and co-workers developed a hydroxymethylation on heteroarenes using methanol as a reagent (Scheme 17A).74 Their reaction protocol consists of {Ir[dF(CF3)ppy]2(dtbpy)}-PF6 as the photocatalyst, benzoyl peroxide (BPO) as the terminal oxidant, and blue LEDs as the light source and it provides an alternative to previous reactions of this type that use UV light. They also proposed an oxidative catalytic cycle where the excited state *IrIII reduces BPO, and the resulting phenyl radical abstracts a hydrogen atom from methanol. The nucleophilic hydroxymethyl radical then adds to the activated position of the protonated azine, and the hydroxymethylated products form after rearomatization in the usual fashion.
Scheme 17.
Photoredox Azine Hydroxymethylation and Alkoxylation Reactions
Scheme 17A shows that mono- and diaddition products form when this reaction is applied to the drug voriconazole (63 and 64). The Huff group also hydroxymethylated the isoquinoline ring in fasudil 65. Scheme 17B shows a remarkable change in chemoselectivity by changing the terminal oxidant to p-methoxy-benzoyl peroxide. This altered protocol results in mono- and bis-methoxylation of the quinoxaline ring in varenicline (66 and 67). We will further discuss other C–Heteroatom formations via radical addition in this review.
The MacMillan laboratory developed a visible-light-mediated dual photoredox organocatalytic platform for the direct C–H bond methylation of azines using methanol via a spin-center shift pathway with H2O as the only byproduct (Scheme 18A).75 Their proposed dual catalytic mechanism begins with visible light irradiation of IrIII to generate the long-lived *IrIII excited state (Scheme 18B). Next, *IrIII undergoes a single-electron transfer event with a sacrificial quantity of protonated heteroarene 68 (not shown) to initiate the first catalytic cycle and provide an oxidizing IrIV species. Then, single-electron transfer from the thiol catalyst 69 to IrIV occurs and, after deprotonation, generates the thiyl radical 72 while returning IrIII to the catalytic cycle. The thiyl radical 72 undergoes hydrogen atom transfer with methanol to provide the α-oxy radical 73 and regenerate the thiol catalyst 69. Polar effects in the transition state drive this process, which enables considerably endergonic C–H abstractions to be possible. The nucleophilic α-oxy radical 73 adds to the protonated electron-deficient heteroarene 68 in a Minisci-type pathway to afford the aminyl radical cation 74. Deprotonation of the α-C–H bond of 74 forms α-amino radical 75, which undergoes the subsequent spin-center shift (SCS) to eliminate H2O and generate benzylic radical 76. The resulting open-shell species undergoes protonation followed by a second single-electron transfer with the excited photocatalyst *IrIII to regenerate the active oxidant IrIV and provide the desired methylated product 77.
Scheme 18.
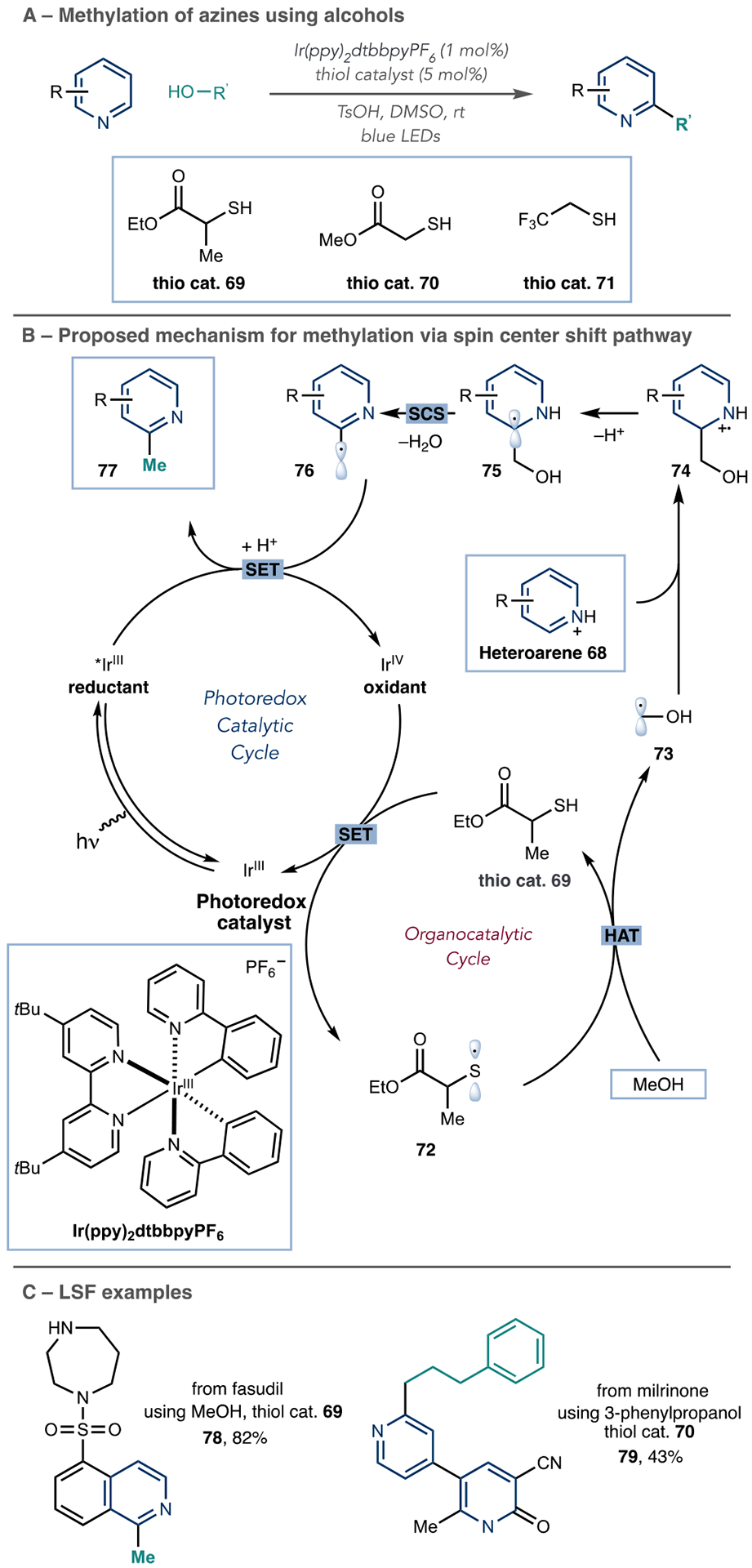
MacMillan et al. Azine Photoredox-Catalyzed Azine Alkylation Involving a Spin-Center Shift
For pyridines, methylation typically occurs at the 2- and 4-position when these sites are unsubstituted. The reaction also functions on quinolines, isoquinolines, phthalazines, and phenanthridines. Given the value of these methylated products and using methanol as a simple and abundant reagent, this reaction has great potential for LSF. The MacMillan laboratory formed the methylated derivative of fasudil 78 using this protocol and demonstrated that other alcohols are viable to give alkylated products, such as alkylated compound 79 (Scheme 18C).
The Melchiorre group reported an azine hydroxyalkylation reaction using 1,4-DHPs as the alkyl source and also invoked a SCS during the reaction mechanism (Scheme 19A).76 Unlike other reports using Minisci-like chemistry, this reaction proceeds without a photocatalyst or chemical oxidant to generate carbon-centered radicals. Instead, 1,4-DHP 80 undergoes visible light excitation to generate 80* (Scheme 19B). A small amount of aromatic Hantzsch ester is necessary to trigger SET to 80* and yield 81. Because 81 is unstable, it rapidly decomposes to an acyl-centered radical 82, which adds to the protonated azine of interest, and produces 83. Acidic conditions trigger the SCS and subsequent SET, and deprotonation liberates the hydroxyalkylated product 85. Alkyl and benzyl radicals are amenable for this reaction and follow typical Minisci regioselectivity. In addition, hydroxyalkylation occurs on various heterocycles commonly shown in numerous reports following Minisci-like pathways. The authors reported 17 LSF examples, including bosutinib, a roxadustat analogue, and a PKA inhibitor (Scheme 19C, 86–88). Furthermore, the resulting alcohol can serve as a functional handle for further derivatizations.
Scheme 19.
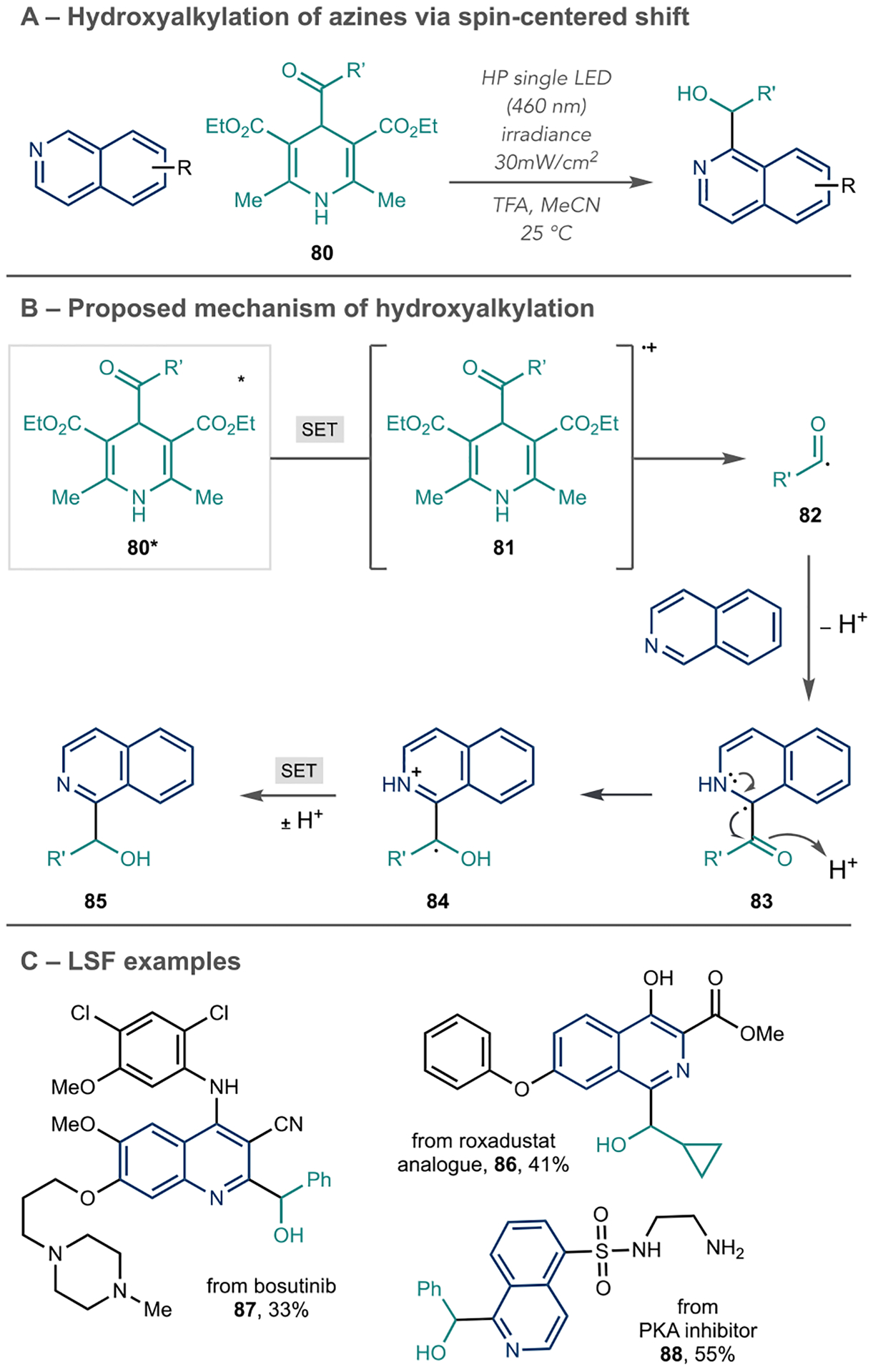
Azine Hydroxyalkylation Involving an SCS Process
The Fu group developed an azine α-amino alkylation reaction using photoredox and Brønsted acid catalysis (Scheme 20).77 They found that photocatalyst {Ir[dF(CF3)-ppy]2(dtbpy)}PF6 and BINOL-derived phosphoric acid (PA-1) couple amino-acid-derived redox-active esters to quinolines, isoquinoline, quinazoline, quinoxaline, and pyrimidines with blue light irradiation. They proposed that an IrII species forms in this reaction that reduces redox-active ester to generate the key α-amino radical (not shown). They showed 14 natural and unnatural –Boc or –Cbz-N-protected amino acids are suitable for this reaction. This decarboxylative α-amino alkylation reaction is applicable for LSF, as evidenced by examples 89 and 90.
Scheme 20.
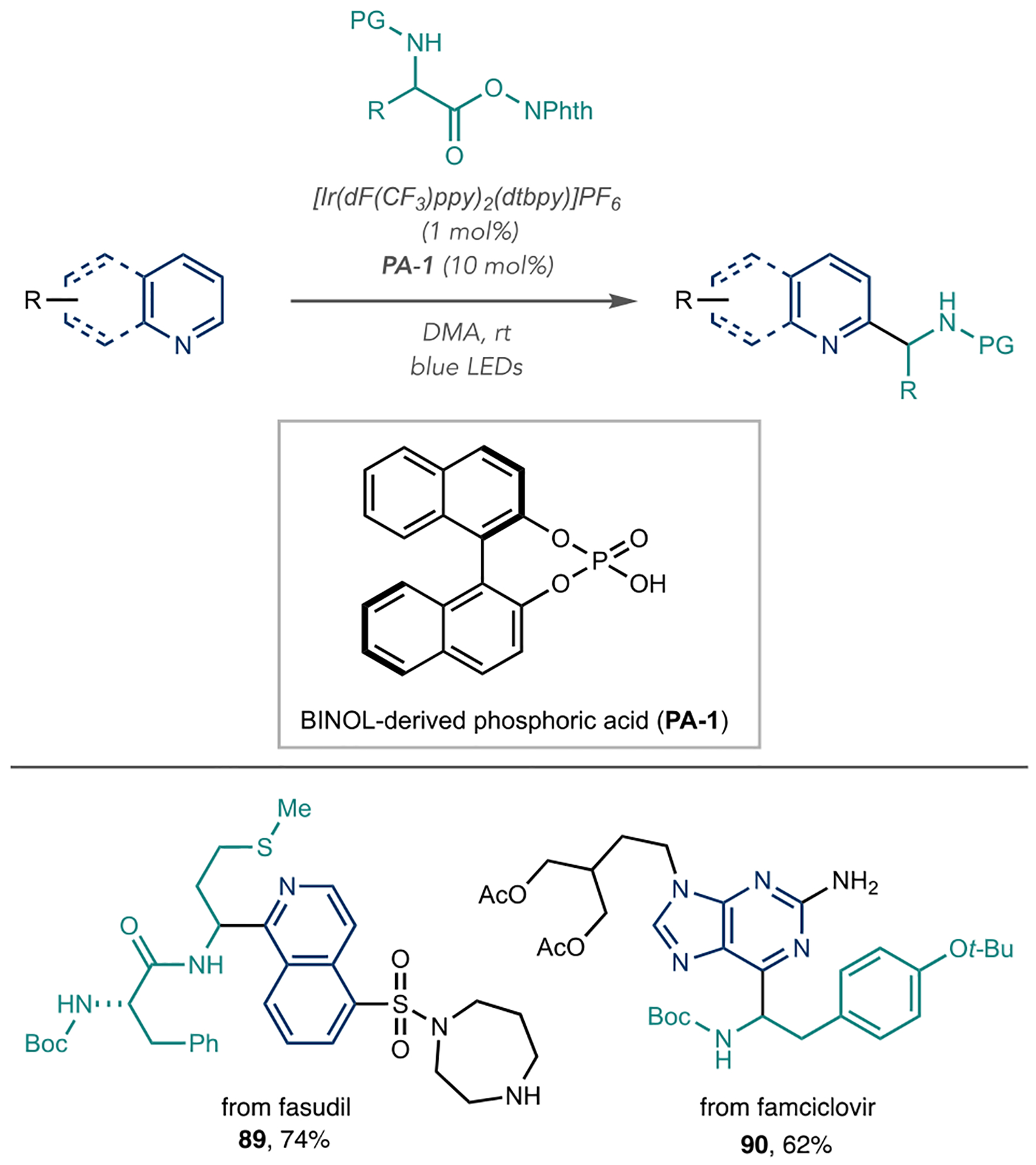
Photoredox and Brønsted Acid-Catalyzed Photoredox Aminoalkylation Using Redox Active Esters as Radical Precursors
The Phipps group made a significant advance in Minisci chemistry by showing that azine α-amino radical addition can be enantioselective using a chiral Brønsted acid catalyst (Scheme 21A).78 In a similar approach to Fu et al.’s report, they found that the amino acid protecting group was key for this transformation, and Scheme 21B shows the key C–C bond formation steps. The (R)-TRIP or (R)-TCYP catalysts both protonate the azine and engage in H-bonding with the N–H portion of the α-amino radical (Scheme 21B, Int 1). They propose that the radical addition step is fast and reversible, and the enantioselectivity results from deprotonation of TS-1 that reforms the aromatic ring.79 In addition to a series of building block azines, this enantioselective process is also viable for LSF applications. The authors showed that highly enantioenriched derivatives of metyrapone and etofibrate, 91 and 92, were obtainable in good yields (Scheme 21C).
Scheme 21.
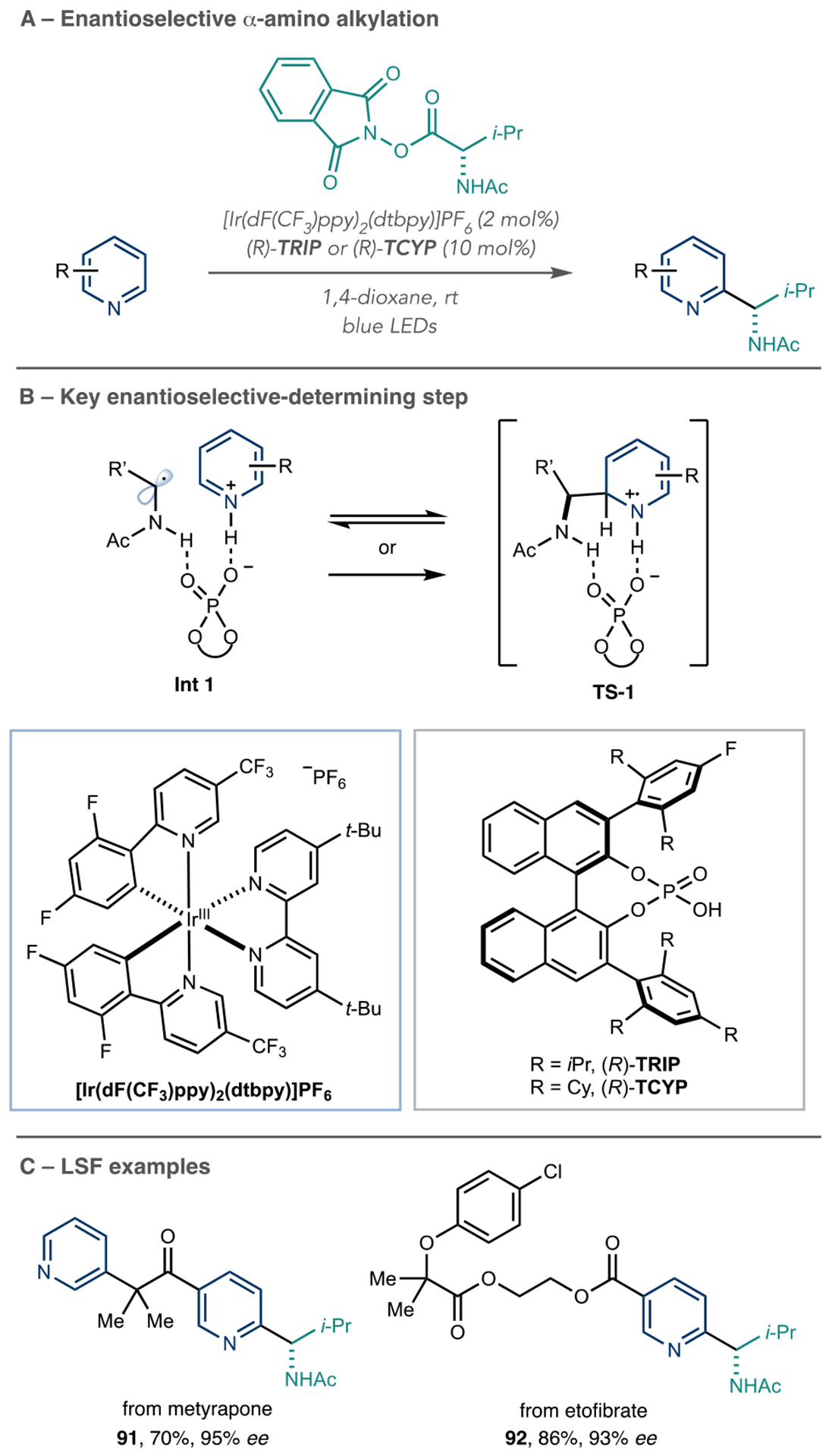
Enantioselective Azine Aminoalkylation Reactions Using Chiral Brønsted Acids
2.3.4. Radical Additions to N-Activated Pyridinium Salts.
N-Activating groups can significantly affect the reactivity and regioselectivity of azine radical addition processes. Hong’s group has developed a series of pyridine C–H functionalization reactions using N-amino- and N-alkoxy-activating groups for numerous bond transformations.80 Unlike typical Minisci reports in this review, these reactions are highly regioselective at either the 4- or 2-position depending on the activating group and nucleophile involved.
For N-amino pyridiniums, a 4-position addition is typically observed with high levels of regioselectivity, and Hong et al. has attributed two factors to explain this observation. First, the 4-position has a significantly lower-lying LUMO than at the 2-position. Second, the activating group sterically encumbers the 2-position selectivity and favors C4 addition of the incoming radical species. Alternatively, N-alkoxy pyridiniums favor the 2-position since the electronic distributions of the activated azine change depending on the identity of the activating group. In these cases, the 2-position LUMO is lower in energy than its 4-position counterpart. In addition, the N-alkoxy pyridinium is less sterically demanding than the N-amino pyridiniums, thus allowing more facile C2 addition. Most reactions outlined in this review follow these general regioselectivity guidelines, and exceptions to these trends will be explained in specific cases.
These reactions follow similar mechanistic pathways, and a general reaction mechanism is outlined in Scheme 22. For clarity, only the events of N-amino pyridinium functionalization are explained and are shown in Scheme 22A. However, N-alkoxy pyridiniums will typically follow a similar mechanism to the one outlined in Scheme 22B. First, the photocatalyst undergoes visible light excitation to produce PC*. Then, a single-electron transfer event to N-amino pyridinium species, 93, yields a sacrificial amount of aromatic pyridine and amino radical, 94. These radicals are highly reactive intermediates and combine with the coupling partner of interest to generate the nucleophilic radical species, 95. This radical species adds to another equivalent of N-amino pyridinium to generate a radical cation intermediate, 96. Deprotonation liberates the functionalized pyridine and another equivalent of the amino radical (94), which propagates the radical chain.
Scheme 22.
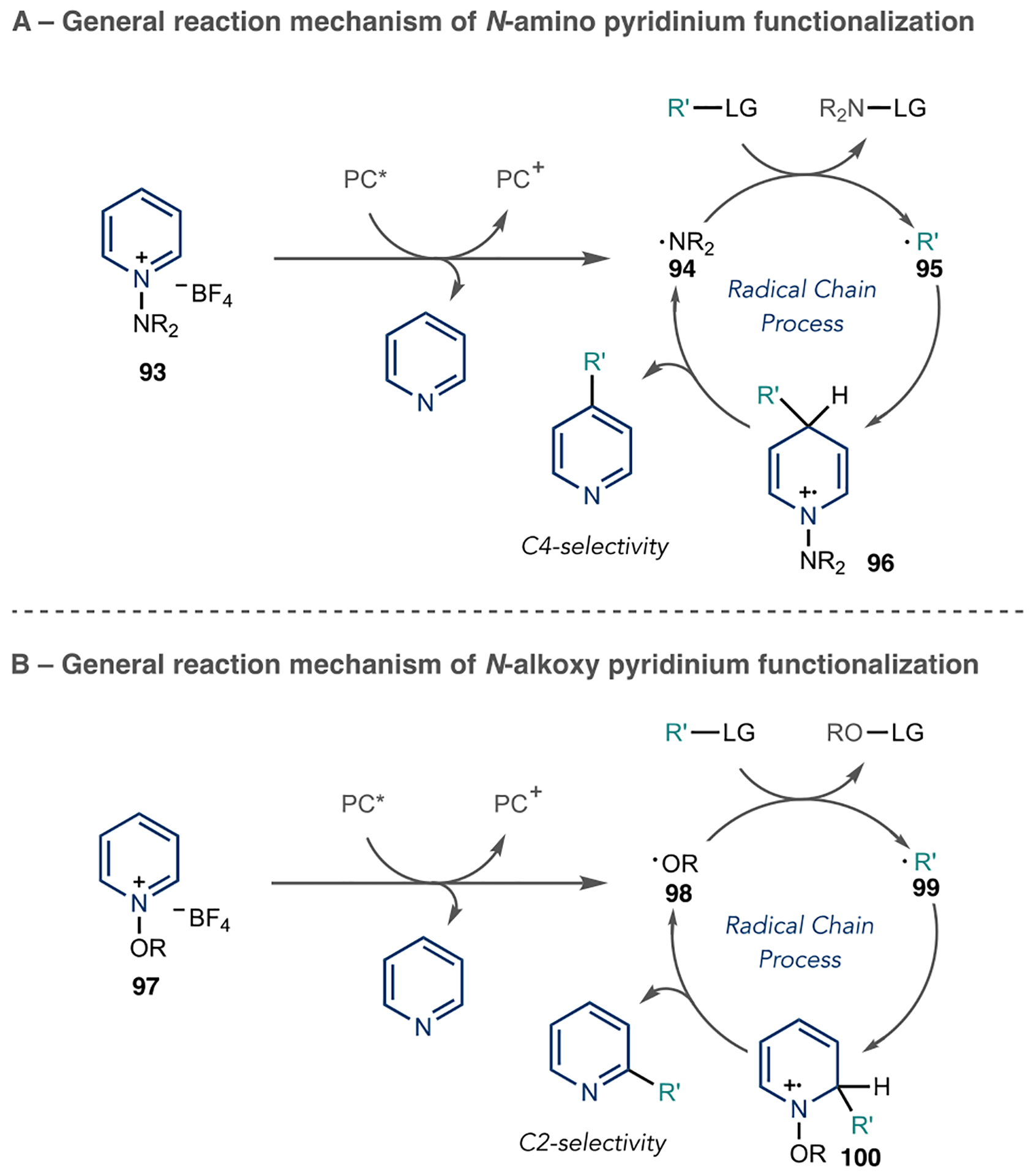
Regiodivergent Azine Radical Additions Using N-Activated Pyridinium Salts
Transformations from the N-amino pyridinium have a large substrate scope, and various C–C bond-forming reactions are reported, as pioneered by Hong et al. Scheme 23 represents various reaction manifolds that are accessed using this strategy. Bicyclo[1.1.1]pentanes (BCPs) are installed onto vismodegib, 101.81 This reaction is useful for LSF because BCPs serve as nonclassical bioisosteres for arenes and internal alkynes. In addition, the resulting BCP radical after addition to the azine allows for the installation of protected amines, which originates from the N-activating group present in the substrate. Radical additions to alkenes are also amenable using this platform (Scheme 23, 102).82 Again, the resulting alkyl radical formed after addition to the azine can be intercepted by an amino radical derived from the N-activating group, which allows for rapid installation of γ-amide groups on vismodegib. The amino radical also adds to strained ring systems, such as cyclopropanols, to install α-carbonyl groups (Scheme 23, 103).83 Coupling partners typically associated with Minisci reactions are amenable to these transformations, including aldehydes (Scheme 23, 104),84 1,4-dihydropyridines (105),85 and alkyl bromides (106).86 As N-amino pyridiniums are coupling partners for a series of versatile alkylation reactions, this reaction platform proves useful in LSF. In addition, their excellent regioselectivity can obviate challenging separation of isomers, thereby allowing for the facile introduction of these alkyl groups to the 4-position of pyridines.
Scheme 23.

Hong et al. Regioselective Radical Additions to N-Amino Pyridinium Salts
As described above, N-alkoxy pyridiniums switch regioselectivity to the 2-position, which allows for the rapid access of carbon-bearing groups. This controlled switch in regioselectivity is useful for LSF because regioisomers can have different binding effects in biological systems. The Hong group has also pioneered numerous reports for functionalizing N-alkoxy pyridiniums. In some cases, the radical forms on the O-activating group and reactions occur at the proximal 2-position (Scheme 24, 107 and 108).87,88 However, it is not necessary to form the radical on the activating group because bimolecular radical additions are viable. As shown in Scheme 24, an amidation reaction occurs at the 2-position of pyridines and is viable in an LSF context for 109.89
Scheme 24.
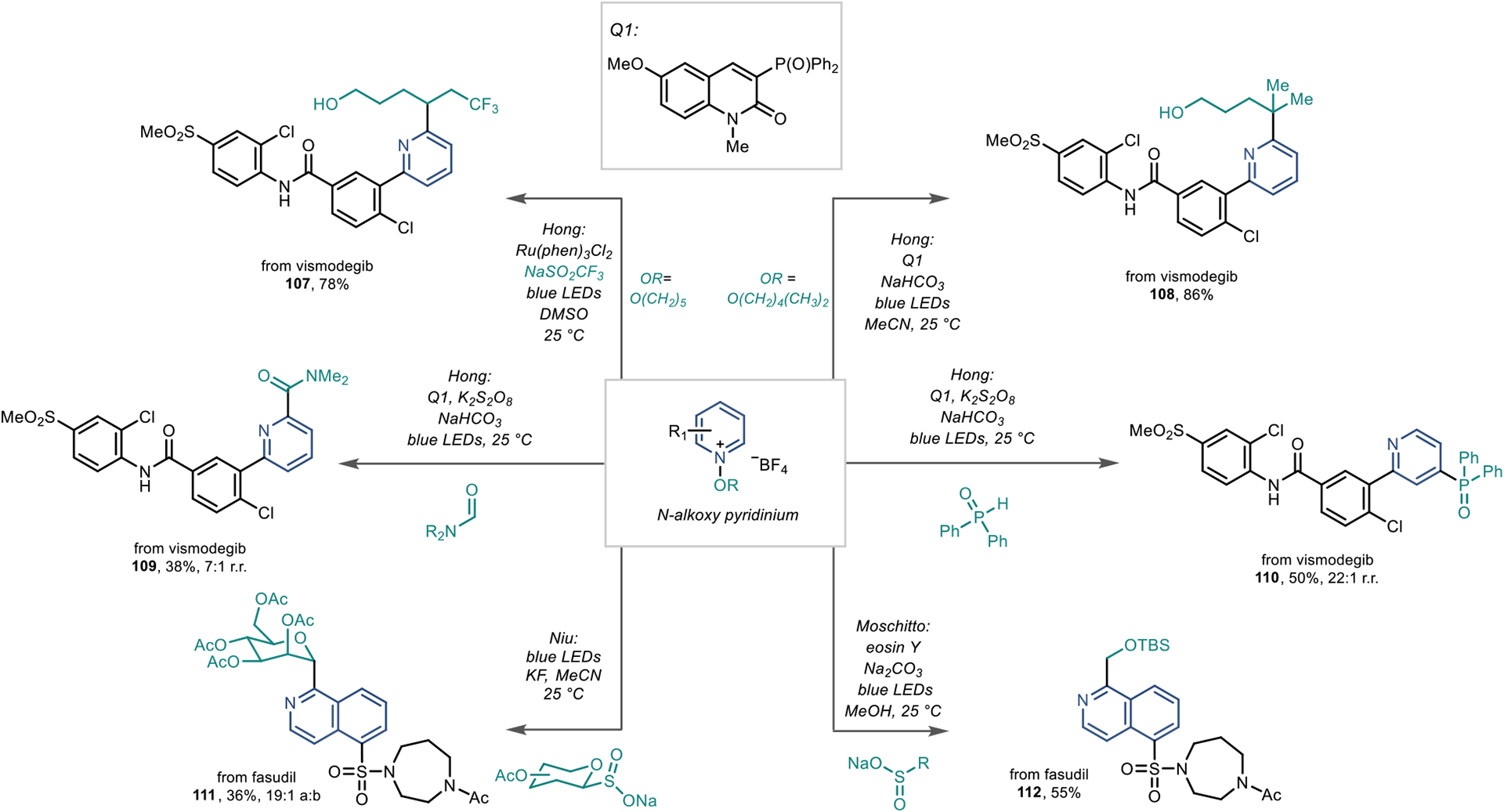
Regioselective Radical Addition Reaction to N-Alkoxy Pyridinium Salts
Interestingly, there are cases where certain radical species add to the 4-position of O-activated pyridinium salts; phosphinoyl radicals override the inherent regioselectivity of the N-alkoxypyridinium, and 4-position functionalization is preferred (Scheme 24, 110).89 The Hong group attributes the 4-position regioselectivity to the structural distortion the substrate must undergo in the transition state during the radical addition step, which favors C4 addition when the larger phosphinoyl radical adds to the pyridinium species by about 1.5 kcal/mol. This distortion is less pronounced for the much smaller carbamoyl radical; thus, typical 2-position selectivity is observed.
Other groups have also adopted this reaction platform to install α-heteroatom carbon-bearing groups to isoquinolines, such as fasudil (Scheme 24, bottom). The Niu group reported a glycosidation reaction of fasudil 111.90 In addition, the Moschitto group developed an α-heteroatom alkylation reaction shown in Scheme 24 (112).91 Because the synthesis of the N-alkoxy and N-amino activating groups occurs before the reaction, derivatization cannot arise directly from the C–H azine. However, the broad scope, impressive regioselectivity, and wide range of amenable coupling partners of these reactions will allow practitioners to rapidly access azine analogues for LSF applications.
2.3.5. C–B Bond Formation via Radical Addition Processes.
An azine radical addition protocol that installs a versatile functional group is useful for LSF because of the broad scope of these reactions and the potential to obtain a range of derivatives in a subsequent step. Leonori’s group has made a significant advance in this area via a C–H borylation reaction that proceeds via a Minisci-type mechanism and is amenable to azine substrates (Scheme 25A).92 This reaction uses Me3N–BH3 as the borylating reagent under oxidative photoredox conditions in the presence of TFA. Excited-state photocatalysts, either 4CzIPN or Ru(bpy)3(PF6)2, oxidize the persulfate anion (113), and then, radical 114 performs a B–H abstraction from Me3N–BH3 (Scheme 25B). The amine portion of the reagent facilitates this radical abstraction and results in nucleophilic σ-boryl radical 115. Radical addition to the protonated azine ensues, and then, the reoxidation of the dearomatized structure 116 forms borylated product 117 and concomitantly completes the catalytic cycle.
Scheme 25.
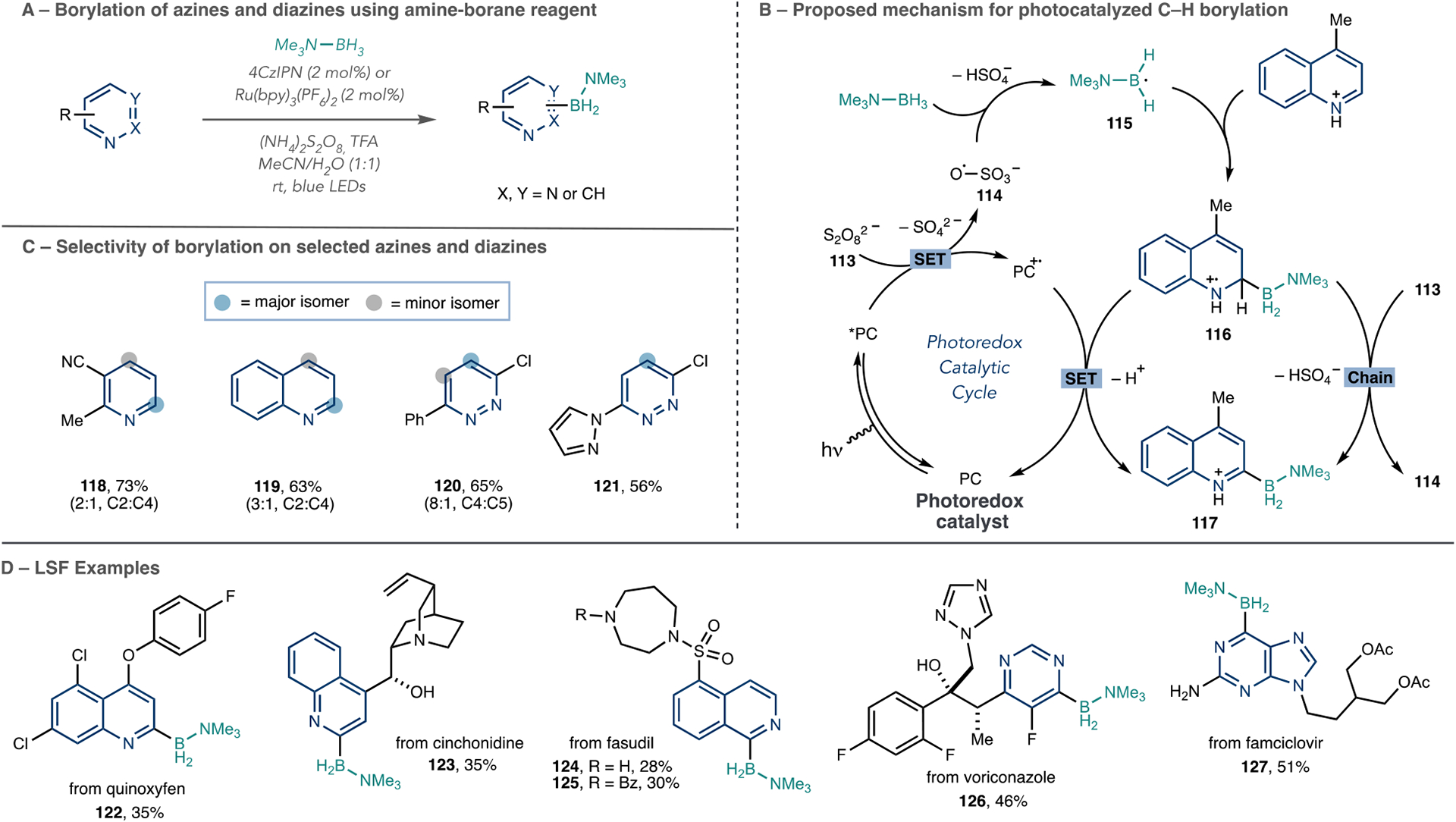
Leonori’s Group Azine Borylation Reaction via Boryl Radical Intermediates
The regioselectivity of the reaction follows the typical pattern observed in Minisci reactions, with biases toward the 2- and 4-positions of azines, such as pyridines and quinolines. Scheme 25C provides other examples of substrates and their regioselective outcomes (118–121). They also found that the C2-amine–borane pyridine products are stable and less sensitive toward protodeborylation than the corresponding boronic acids. They demonstrated that this borylation protocol has the potential for LSF on pharmaceuticals and agrochemicals (Scheme 25D). The purine core of famciclovir underwent C6-borylation with 4CzIPN as the photocatalyst. A switch to Ru(bpy)3(PF6)2 as the photocatalyst allowed for C2-borylation of quinoline in cinchonidine 123 containing benzylic alcohol, tertiary amine, and olefin functionalities (122, 123–127). Leonari and co-workers showed that these amine–borane products could undergo Suzuki–Miyaura crosscouplings, Chan–Lam amination and etherification reactions, and oxidation derivatizations to form products, such as pyridones, quinolones, and isoquinolones. Rapid access to borylated products across a range of azines has great promise for further LSF applications.
3. TRANSITION-METAL-CATALYZED C–H ACTIVATION REACTIONS
3.1. Introduction to Azine C–H Activation Reactions and Related Approaches
Direct metalation of azines to form reactive species, such as organolithium or organomagnesium intermediates, and trapping with electrophiles is an established means to obtain useful derivatives directly from azine C–H bonds.18,93–96 This approach typically requires strong bases, and the reactive nature of the metalated intermediates means that application of these protocols in LSF contexts is restricted. Metal-catalyzed C–H activation reactions offer a more applicable alternative. They typically use transition metals rather than alkali metals and operate into the azine C–H bonds. These reactions allow broader tolerance of functional groups and react more discriminately in complex organic molecules. This section will show examples of LSF reactions via these processes and other approaches that involve cleaving azine C–H bonds.
Metal-catalyzed C–H activation is an established field in synthetic organic chemistry, and sp2 C–H bonds are common targets for this approach. However, there are relatively few examples of LSF transformations involving the sp2 C–H bonds of azines since much of the development in this area occurred before LSF applications became common. There are mechanistic reasons that also make azine C–H activation challenging. These reactions often employ Lewis basic directing groups to facilitate the C–H bond cleavage step on other carbon frameworks, including the N atoms of azine heterocycles. Therefore, azines are often used as the directing group for other moieties and are not conducive to facilitating C–H functionalization on themselves. This section will show examples where azine-to-azine reactivity facilitates LSF, directing groups on the azines enable the C–H cleavage, and creative ligand design addresses azine C–H functionalization problems. It will also highlight nondirected C–H activation reactions, pyridine N-oxides in late-stage C–H arylation reactions, and other entirely different reaction designs to enable LSF.
3.2. Reactions Facilitated by Directing Groups
The Pilarski group developed an azine methylation reaction using Rh-catalyzed C–H activation under mechanochemical conditions. In this report, heteroaromatics are used as directing groups to form 5- or 6-membered rhodacycles, and Scheme 26 shows an example of how they applied this method to methylate etoricoxib.97 Here, the 2,3,5-substituted pyridine directs metalation to the 6-position of the other to form a 5-membered rhodacycle. Subsequent transmetalation and coupling with Me–BF3K forms methylated etoricoxib 128 in 60% yield. This approach allows for methylation under mechanochemical conditions, which offers some advantages compared with traditional transition metal catalysis. First, the reaction time is lower than typical transition-metal-catalyzed methylation reactions. Second, mono- to difunctionalized product ratios improve under mechanochemical conditions. As methylation can have dramatic effects on a compound’s PK/PD profile, this C–H activation strategy will be useful for candidate compounds that have the requisite directing groups proximal to azine C–H bonds.39
Scheme 26.
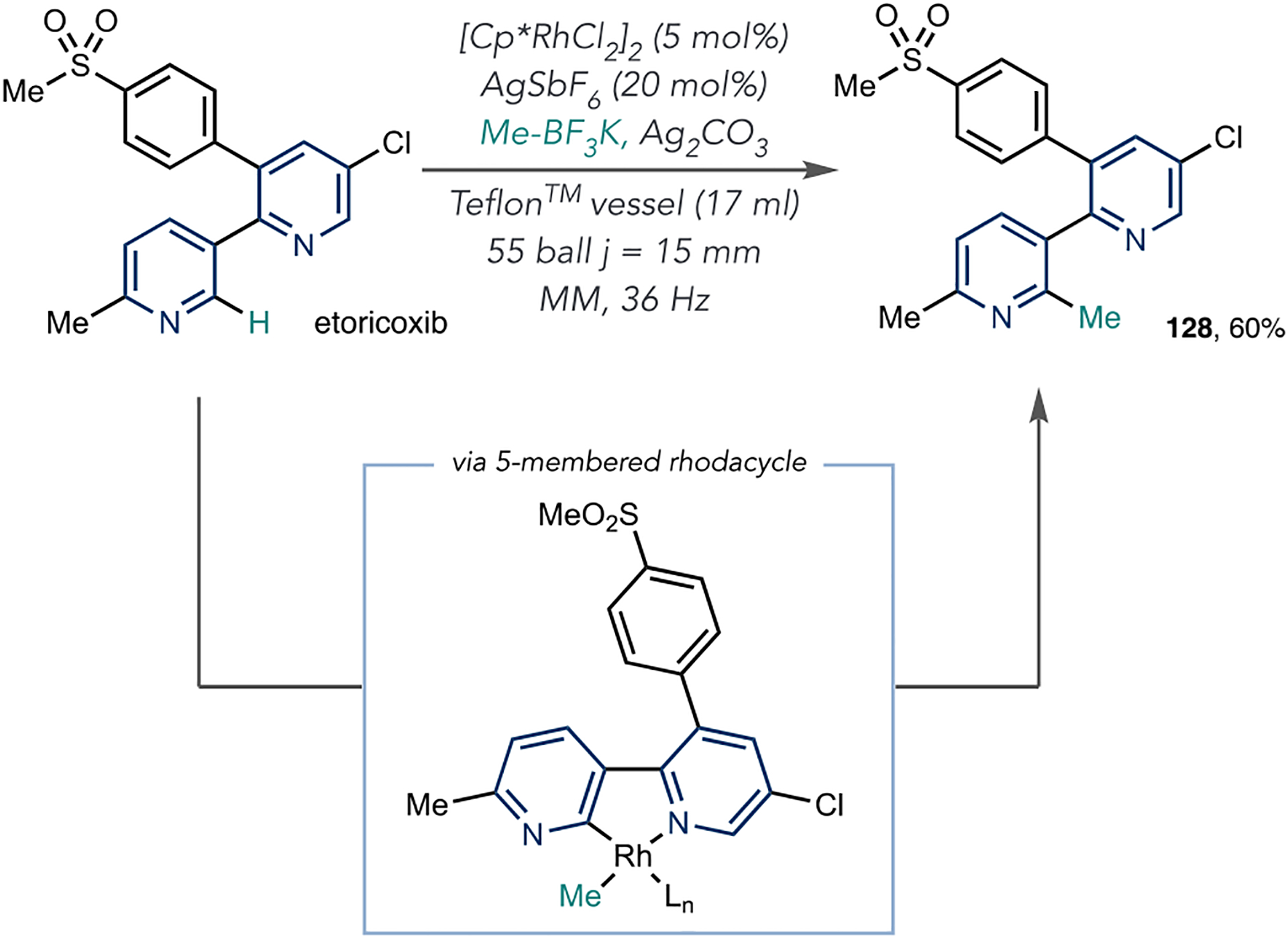
Rh-Catalyzed Azine Methylation of Etoricoxib under Mechanochemical Conditions
Yu’s group reported a (hetero)arene C–H hydroxylation in which a palladium catalyst and tautomeric ligand install the–OH group adjacent to carboxylic acids.98 This process functions on azines, such as pyridines, quinolines, and quinoxaline, and Scheme 27 exemplifies this reaction by the hydroxylation of clonixin, a nonsteroidal anti-inflammatory drug where the hydroxyl group derives from molecular oxygen (129). The ligand (L1) contains both pyridine and 2-pyridone groups; they assert that the capacity of the pyridone portion to adopt both pyridine and pyridone forms on the basis of tautomerization is key for the success of this reaction, and TS-2 proposes a six-membered palladacycle involving the pyridone form of the ligand. Further LSF application of this reaction will be restricted by the requirement for the carboxylic acid directing group. However, the scope of the reaction suggests that the process is robust provided that motif is present.
Scheme 27.

Pd-Catalyzed Hydroxylation of Azines Using Carboxylic Acid Directing Groups
3.3. Regioselective C–H Activation Reactions via Ligand Control
Ye and Huang’s group developed a bimetallic alkylation reaction of pyridines in which the Lewis basic nature of pyridine’s N atom guides the Al–Ni bimetallic species to the C2 position.99 The diamine phosphine oxide ligand, L2, shown in Scheme 28, is critical for the reactivity, regioselectivity, and diastereoselectivity of this reaction. Pyridines, quinolines, and quinoxalines undergo alkylation; however, the pyridazines, bipyridines, and terpyridines fail because of catalyst poisoning. Despite these limitations, six LSF examples undergo alkylation, including those shown in Scheme 28, and the resulting alkene could serve as a functional handle for further diversification.
Scheme 28.

Ni–Al Bimetal-Catalyzed Enantioselective Pyridine Allylation
Similarly, Yu’s group developed a pyridine alkenylation reaction using a bimetallic system with a ligand structure selective for the C3 position (Scheme 29A).100 To change regioselectivity from C2 to C3, they used an organic linker and an N-heterocyclic carbene (NHC) ligand to position the active Ni catalyst in proximity of the C3 position. Scheme 29B shows the proposed mechanism for this transformation that begins with pyridine binding to the aluminum portion of the catalyst. The linker then orientates the NHC-ligated Ni in the proximity of the 3-position C–H bond such that selective C–H insertion occurs to form 135. Migratory insertion into an alkyne generates intermediate 136, and reductive elimination installs the alkene and releases product 138. Catalyst turnover occurs via ligand exchange with another substrate. The Yu group’s report documents nine LSF examples that undergo alkenylation with high levels of regiocontrol, and Scheme 29C shows three cases from that collection. Their results on pyridine building blocks indicate amenability to nonsymmetrical alkynes.
Scheme 29.

C3-Alkenylation of Pyridines via an Al–Ni Tethered Catalyst
Selectively accessing remote positions on bicyclic azines is a significant challenge for C–H activation reactions, as electronic effects within the ring or directing effects from the N atom is insufficient for the level of control to switch functionalization between different positions. Accessing remote positions is a useful attribute for LSF because it allows access to distinct positional isomers that, for example, could project into different areas of a binding pocket. The design of ligand architectures has been used to address this problem, and Yu’s group showed that the C–H bonds at the C6, C7, and C8 positions of the quinoline in camptothecin could be selectively functionalized using Pd catalysis with various ligand scaffolds (Scheme 30).101 Using L3, C–H alkenylation preferentially occurs at C8 (142): accessing C7 is more difficult and requires ligand L4, which serves as a macrocyclic template to position palladium close to the C7 C–H bond, thereby forming alkenylated product 143.102 Modification of the ligand scaffold to L5 and L6 enables 6-arylated and alkylated products, respectively (144 and 145).103
Scheme 30.

Ligand Architecture Design for C–H Activation of Quinolines
3.4. Non-Directed Ir-Catalyzed Processes
Iridium-catalyzed borylation is one of the most widely applicable methods for (hetero)arene C–H functionalization.104 The reaction does not require directing groups, which makes a large collection of substrates applicable. The C–B bond can be transformed into various derivatives in a subsequent step, thereby making this strategy ideal for LSF applications. The Hartwig group studied the factors that influence regiocontrol on azines, and Scheme 31A shows 3-picoline as a model substrate in their proposed mechanism. First, the pyridine displaces the cyclooctene (COE) or 1,5-cyclooctadiene (COD) ligands bound to the iridium precatalyst to form Int 2, a resting state for the catalytic cycle. Second, the pyridine dissociates and releases the active catalyst 146 that undergoes a turnover-limiting C–H insertion reaction into the pyridine and forms 3-metalated Int 3. Third, the borylated product forms and regenerates active catalyst 146. The authors attributed regiocontrol to steric effects from the methyl substituent. They did not observe any 2- or 6-position borylated pyridines and stated that these products are unstable under the reaction conditions. They also found that C–H borylation does not generally occur next to the N atom in other azines, such as pyrimidines.
Scheme 31.

Mechanism and Applications of Ir-Catalyzed C–H Borylation for Azine LSF
Although the current literature contains a relatively small number of azine C–H borylation in LSF applications, the reaction has significant potential in drug and agrochemical discovery. Hartwig’s group applied a C–H borylation–halogenation sequence to nicotine and obtained 3-brominated derivative 147 using CuBr as the bromide source (Scheme 31B).105 They also showed that C–C bond formation is amenable in a LSF setting. After borylating loratadine, they formed methylated derivative 148 in a one-pot procedure using CuI and PO(OMe)3 as the methylating reagent (Scheme 31C).106 Sarpong et al. used a pyridine C–H borylation to prepare a cross-coupling precursor used in the total synthesis of complanadine A (Scheme 31D).107 This Lycopodium alkaloid contains a 2,3′-bipyridine motif, and the iridium-catalyzed borylation reaction delivered 149 in good yield, which is in line with the selectivity pattern observed in Scheme 31A. This pyridine then served as the nucleophilic partner in a subsequent Suzuki reaction–deprotection sequence with pyridine 150 to form complanadine A (151). Sarpong’s group took a similar approach in their synthesis of complanadine B, which also features an example of a pyridine N-oxide 2-chlorination in an advanced intermediate.108
Hartwig’s group also developed azine C–H silylation reactions capable of LSF (Scheme 32A).109 They found that [Ir(COD)OMe]2 as the catalyst, 2,4,7-trimethylphenanthroline as a ligand, and HSi(OSiMe3)2 as the silane functioned well for azine-containing compounds. Compounds 152 and 153, the latter containing a secondary aliphatic amine, are incompatible in Ir-catalyzed C–H borylation reactions (Scheme 32B). Like the C–B bond, the C–Si bond is also amenable to various derivatization reactions. Scheme 32C shows how they converted mirtazipine into brominated analogue 154 using NBS and AgF as reagents.
Scheme 32.

Applications of Ir-Catalyzed C–H Silylation for Azine LSF
3.5. LSF Reactions Using Pyridine N-Oxides
In 2005, Fagnou et al. reported a 2-position-selective Pd-catalyzed C–H arylation of pyridine N-oxides, and Tsukano et al. expertly used this protocol as a key step to synthesize complanadines A and B.110–112 Scheme 33 shows the step to construct the 2,3-bipyridine motif using pyridine N-oxide 156 and 3-bromopyridine 157. A subsequent reduction using Pd(OH)2 and ammonium formate reduces the N-oxide to the pyridine and cleaves the Cbz groups (151). Like Sarpong’s group, Tsukano et al. also used iridium-catalyzed C–H borylation as part of the synthesis; the C–Br bond in 157 derives from pyridine C–H borylation, followed by a CuBr2-mediated bromination step.
Scheme 33.

Pyridine N-Oxide C–H Arylation in Tsukano et al.’s Total Synthesis of Complanadine A
3.6. Base-Mediated Reactions
The Bandar lab exploited a base-catalyzed halogen transfer platform for azine C–H etherification (Scheme 34A).113 They proposed that KOt-Bu deprotonates the N-heteroarene (HetAr–H), then halogen transfer occurs with 2,3-diiodobenzothiophene, which is a halogen transfer reagent (Scheme 34B). The N-heteroaryl halide intermediate (HetAr–X) is then intercepted by an alkoxide present in the reaction mixture via a SNAr reaction to form the heteroarylether product.
Scheme 34.

C–H Etherification of Azine Using Base-Mediated Halogen Transfer Reactions
This C–H etherification is regioselective for the 4-position of pyridines and functions on diazines. In polyazine systems, the substitution occurs on the least basic and most sterically hindered azine, which complements the scope of (pseudo)-halogenation methods reliant on N-activation. The site-selectivity is exemplified in examples 157 and 158, where C–H etherification selectively occurs on the pyridazine over the other azines (Scheme 34C). The Bandar group concluded that the 2-halothiophene halogen transfer reagent design was key for this transformation. The regioselectivity and site-selectivity of this base-catalyzed halogen transfer protocol shows its capability for LSF.
4. REACTIONS VIA DEAROMATIZED INTERMEDIATES
4.1. Introduction to Azine Dearomatization Reactions
As azines are electrophilic, a collection of C–H functionalization reactions proceed by adding nucleophiles to these cores, which results in dearomatized intermediates.18 A classic approach to add carbon-bearing groups to azines and N-activated intermediates is through reactions with organolithium and organomagnesium reagents and subsequent oxidation to reform the aromatic ring. However, these reactive organometallics do not suit the criteria for LSF applications. The field has made significant progress since these early developments and has shown coupling reactions with a broad range of nucleophiles, and the careful design of pyridinium salts facilitates several examples. Furthermore, chemists have taken advantage of the inverted reactivity of the dearomatized intermediates to enable reactions with electrophiles. Via these distinct reaction mechanisms, different regioselective outcomes are also possible.
4.2. Direct Nucleophile Additions
As an alternative to organolithium and Grignard additions to azines, Baik and Cho et al. reported a C2-selective alkylation of quinolines using organozinc intermediates (Scheme 35A).114 The combination of dimethylzinc with LiOt-Bu forms Int 4, which subsequently reacts with 1,1-diborylalkanes, such as 160, and ultimately results in borylzincate Int 5 (Scheme 35B). Scheme 35C shows the proposed mechanism for the alkylations of quinolines. Int 5 selectively adds to the quinoline 2-position (not shown) to form a dearomatized adduct. Subsequently, two proposed pathways can result in the alkylated product. In path A, LiOt-Bu serves as a base to rearomatize the ring, whereas path B proceeds through intramolecular elimination via the Zn–Me bond. In either case, benzyl boronate ester 161 forms and undergoes protodeborylation to form the alkylated quinine 162. This report shows two LSF examples using O-TMS-protected quinoline, including methylated product 159. However, the application of this method to a broader set of azines is needed to evaluate its potential.
Scheme 35.
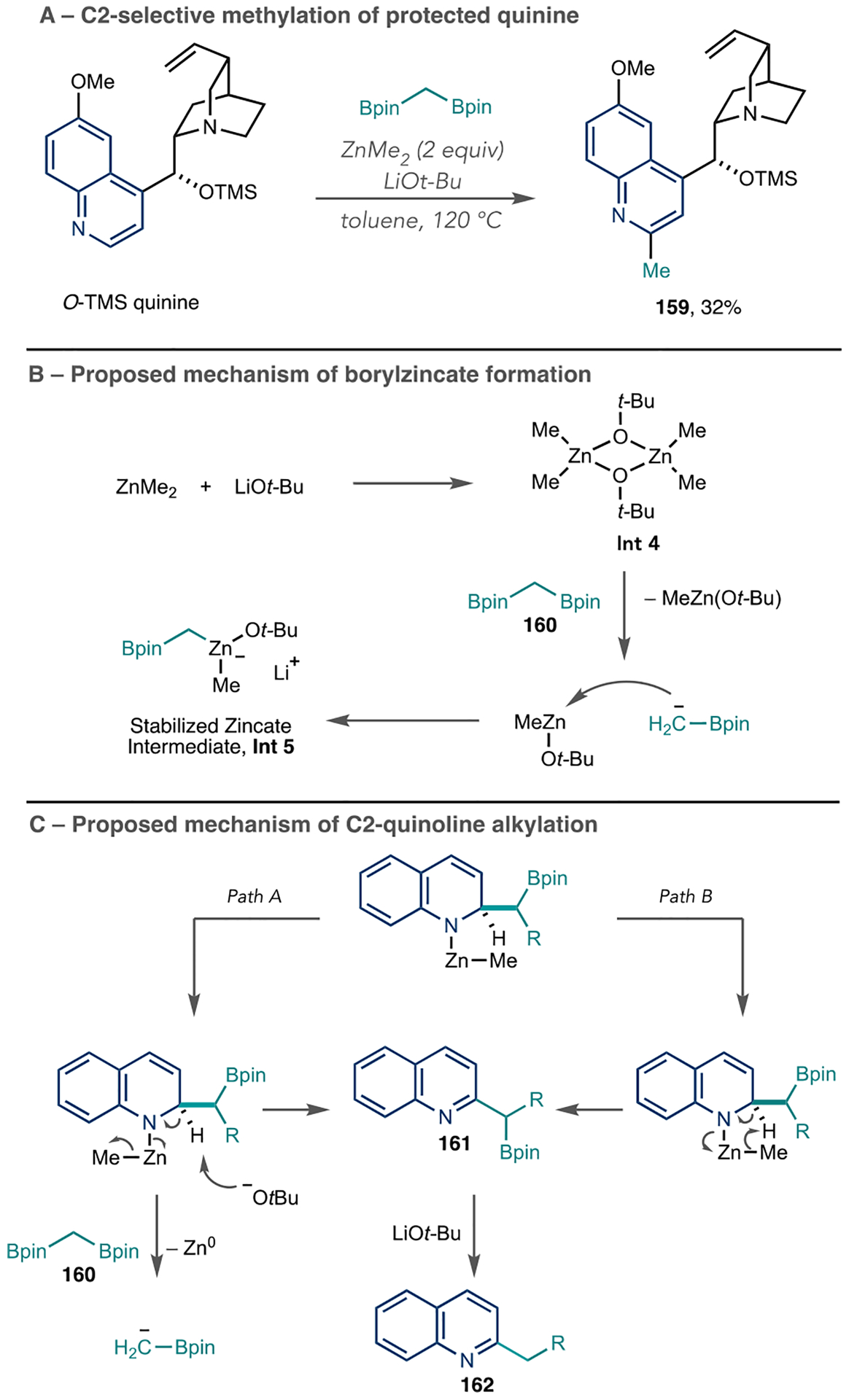
Azine Alkylation Using Borylzincate Intermediates
Although functionalization occurs from unactivated azines for organozinc additions, the additions of other nucleophiles often fail without preactivation of the azine with an electrophile. The Kanai group reported a C4-selective trifluoromethylation reaction of azines via a nucleophile addition–oxidation sequence using a Lewis acid activator (Scheme 36).115 First, they prepared azine–Lewis acid adducts using tris[2,3,5,6-tetrafluoro-4-(trifluoromethyl)phenyl]-borane, such as 163 derived from abiraterone acetate. Tetrabutylammonium difluorotriphenylsilicate (TBAT) then activated TMSCF3, and the resulting silicate transferred the trifluoromethyl group onto the pyridinium C4 position to form dearomatized Int 6. Finally, the addition of (diacetoxyiodo)-benzene (PIDA) reformed the aromatic system and formed trifluoromethylated derivative 164 in 39% yield, which is the only LSF example the authors reported that undergoes C4-trifuoromethylation.
Scheme 36.
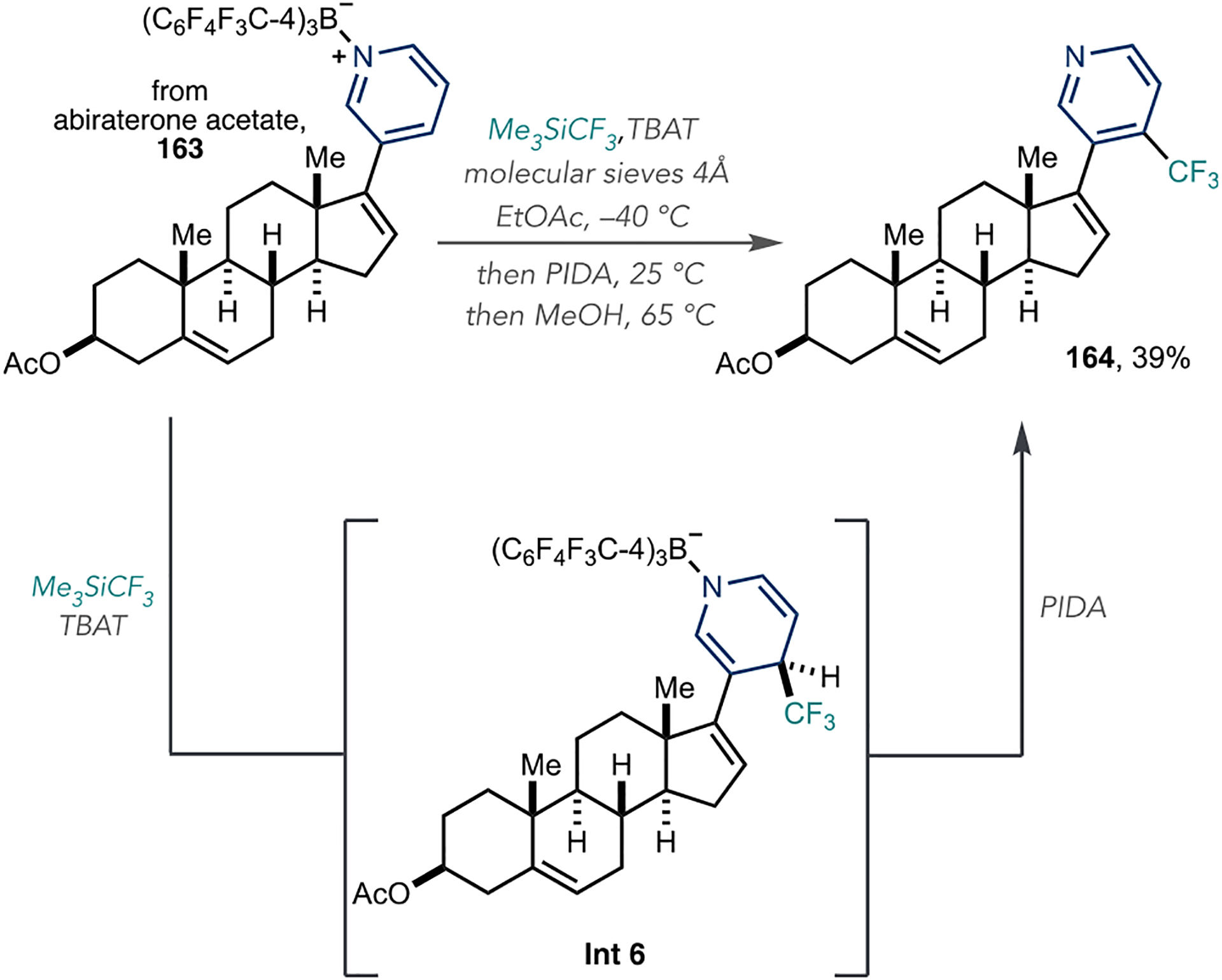
Direct C–H Trifluoromethylation of Azines Using a Boron Lewis Acid for N-Activation
4.3. Reactions with N-Tf and N-OTf Pyridinium Salts
The installation of nitrile groups onto azines is advantageous because they can serve as precursors to other functional groups, such as amides, amines, and carboxylic acids. An Oxford team added trimethylsilyl cyanide (TMSCN), a cyanide source, to N-Tf azines with the addition of N-methyl morpholine (NMM) as a base for a net C–H cyanation reaction (Scheme 37A).116 They observed the reaction is biased toward the 4-position of pyridines, but the 2- and 6-regioisomers form in several cases depending on the substitution pattern (Scheme 37B). However, 3-alkylated pyridines form 4-cyanated products with high regioselectivity. Other azines, such as quinolines, isoquinolines, and diazines, perform well in this C–H cyanation process.
Scheme 37.

Azine C–H Cyanation via N-Tf Pyridinium Salts
As shown in Scheme 37C, this reaction is capable of LSF to cyanate pyridine-, quinoline-, and quinazoline-containing drugs. The authors used computational studies to rationalize the regiocontrol and found that Fukui indices of the N-Tf pyridinium salts matched well with the regioisomeric outcomes. As shown in Scheme 37C, this process is capable of LSF to cyanate pyridine-, quinoline-, and quinazoline-containing drugs (165–168).
Manolikakes et al. reported a related approach for azine C–H sulfonation (Scheme 38).117 Again, they used Tf2O for N-Tf activation and added sulfinate salts and DABCO at –30 °C. Warming to room temperature rearomatized the heterocycle. While the reaction can be 4-selective for pyridines, 2- and 4-regioisomeric mixtures often resulted. The authors attributed the reactivity of this reaction to the formation of charge transfer complex 171, which prevents decomposition of the sulfinate nucleophile. In their 2022 report, they showed two examples of late-stage C–S bond formation using a protected quinine (169) and fasudil (170) as substrates. Given that sulfones are prevalent in pharmaceutical structures, this reaction could have relevance in medicinal chemistry programs.
Scheme 38.

Azine Sulfonation via N-Tf Pyridinium Salts
The Chang laboratory developed an interesting method to selectively aminate the C–H bond at the 2-position of pyrimidines (Scheme 39A).118 They converted pyrimidines into their corresponding N-oxides and then added Tf2O to form pyrimidinium N-OTf salts as activated Int 7. Pyridines or imidoyl chlorides can attack these reactive pyrimidinium salts via a nucleophilic substitution of hydrogen (SNH) pathway to form Int 8. Scheme 39B shows the full reaction details of this three-stage protocol, where MeReO3 catalyzes N-oxide formation, and 3-Bz- or 3-CF3-substituted pyridines form the pyridinium salts. Then, the addition of aqueous ammonia achieves the 2-amino azine 172. If imidoyl chlorides are used instead of pyridines to form an iminium salt intermediate 176, then a subsequent basic workup with NaHCO3 results in amidated pyrimidines 173. The iminium salt intermediates can undergo different derivatizations to achieve secondary and tertiary amine products by either adding NaOH or NaBH4.
Scheme 39.

Azine Amination and Amidation via N-OTf Pyridinium Salts
Scheme 39C shows how the pyridine’s electronics influence the regioselectivity of the amination reaction. If electron-rich pyridines, such as 4-dimethylaminopyridine and 4-OMepyridine, are used, then C2 and C4 mixtures of pyrimidine ammonium salts form (174). However, if the authors employed pyridines substituted with electron-withdrawing groups, such as 4-CF3, 4-CN, 3-CF3, and 3-Bz pyridines, then they observed high C2 selectivity (175). In the former, the electron-rich pyridines add nonselectively to the pyrimidinium N-OTf salt, and this process constitutes the selectivity-determining step. However, elimination of the triflate is the selectivity-determining step when electron-deficient pyridines are employed. The imidoyl chlorides follow the same selectivity pattern as electron-deficient pyridines and result in 2-iminium salts 176.
This method is particularly promising for the LSF of pyridimidine-containing pharmaceuticals, where Chang and co-workers showed three examples of LSF applications in their report (Scheme 39D). They transformed dabrafenib 177 into a 2-aminated pyrimidine using aqueous ammonia as the amine source. Using the imidoyl chloride protocol, they formed amidated derivatives of encorafenib analogue 178 and ruxolitinib 179. 2-Amination also occurred on other diazines, such as quinoxaline in varenicline 180. These examples allude to this method’s potential to expand to other complex pyrimidines and quinoxalines for LSF to achieve 2-aminated products.
4.4. Formation of Azine Phosphonium Salts and Subsequent Transformations
McNally et al. developed a program for azine C–H functionalization that involves the installation of phosphonium ions onto heterocycles and then the transformation of these salts to obtain a wide range of valuable azine derivatives (vide infra).119–126 Initial reports by Anders et al. showed that these salts could form on a limited set of azine examples; McNally’s group demonstrated that the C–P bond-forming process has a broad scope, particularly for pyridines, and is viable for LSF applications.127 Scheme 40A shows the mechanism for phosphonium salt formation using pyridines, where N-Tf pyridinium salt formation is key for reaction generality. Triphenylphosphine then adds selectively at the pyridinium 4-position to form dearomatized Int 9. Base-mediated elimination of the triflyl anion results in the pyridine phosphonium salt, and this 4-position selectivity is constant across a diverse array of pyridines with different substitution patterns and electronic variations.
Scheme 40.

Mechanism and Examples of Azine Phosphonium Salt Formation
In this approach, McNally’s group transformed a diverse collection of azine-containing pharmaceuticals into phosphonium salts, and Scheme 40B shows four representative examples (181–184). Furthermore, site-selective switching is possible in polyazines by relatively simple changes in the reaction conditions for phosphonium salt formation. For example, Scheme 40C shows that isomer 185 forms from an A-84543 analogue under standard conditions. Doubling the equivalents of Tf2O and phosphine and using NEt3 as a base changes the site of the C–P bond formation to the 2,5-disubstituted ring (186). As installation of a versatile functional handle at the 4-position of pyridine C–H precursors is rare, this approach offers a simple, reagent-based method to form C–C and C–Heteroatom bonds in a subsequent step, thereby producing a large number of potential derivatives at various points in drug development.
Azine phosphonium salts can react with anionic nucleophiles to form C–O, C–S, C–N, and C–halogen bonds (Scheme 41).122–124,128,129 Several classes of protic nucleophiles are viable in these C–Het bond formations, including alcohols; phenols; thiols; thiophenols; selenophenols; anilines; and nitrogen heterocycles, such as pyrroles, pyrazoles, and anilines, as shown in Scheme 40A. In the proposed mechanism, the anionic nucleophile adds to the phosphonium ion to form a P(V) intermediate; C–Het bond formation occurs via phosphorus ligand coupling, an asynchronous process where an apical ligand migrates to the ipso-position of the pyridine ring (vide infra). Scheme 41B shows examples of LSF, including protected versions of varenicline and cinchonidine, pyriproxyfen, and loratadine with four classes of heteroatom nucleophiles (187–191). Given the ability to make phosphonium salts on a wide variety of azine-containing structures, and the latitude of amenable nucleophile classes, this method is also useful for pharmaceutical library synthesis.
Scheme 41.
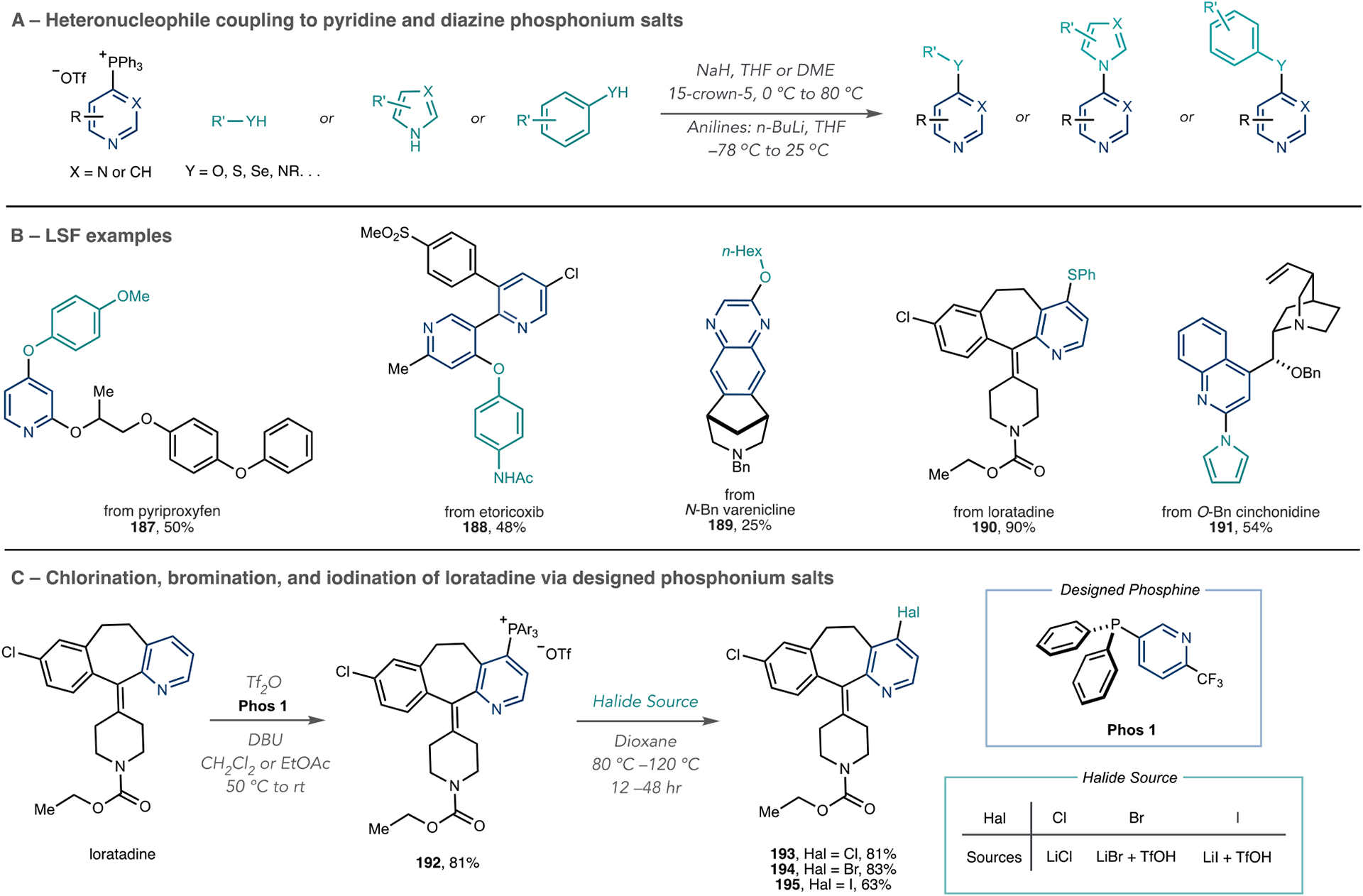
Reactions of Azine Phosphonium Salts with Heteroatom Nucleophiles
The installation of halides at the 4-position of pyridines is highly valuable for LSF applications given the extensive downstream chemistry that applies to the C–Halogen bond, and Scheme 41C shows how McNally’s group used designed phosphines for this purpose.129 However, when Phos 1 is used, three major features in salt 192 facilitate selective C–halogen bond formation, whereas PPh3 was ineffective to facilitate halogenation. First, the CF3-substituted pyridine does not interfere with phosphonium salt formation because of poor reactivity with Tf2O. Second, because two heterocycles are present in 192, they can both be activated by Brønsted or Lewis acids, thereby making the phosphonium ion more electrophilic. Third, reactivity occurs on the pyridine of interest because the C–P bond is in a para relationship to the N atom and, hence, more nucleophilic aromatic substitution (SNAr)-active than the meta relationship in the CF3-substituted pyridine. McNally’s report shows eight LSF examples, including halogenated versions of loratadine (193–195). LiCl is effective for chlorination, whereas LiBr and LiI must be combined with TfOH for effective bromination and iodination reactions. Computational studies indicated that this reaction proceeds via a SNAr mechanism as opposed to a phosphorus ligand-coupling reaction. The steric environment around the phosphonium ion also plays a significant role during the C–halogen bond-forming step.
McNally’s group exploited phosphorus ligand-coupling reactions in a strategy for bis-azine biaryl synthesis.125 This approach offers an alternative to metal-catalyzed cross-coupling reactions that can be challenging using two azines as coupling partners (Scheme 42). Furthermore, this phosphorus ligand-coupling approach commences from azine C–H precursors, thereby obviating the need to synthesize halogenated azines and nucleophilic partners, such as boronic acid or other azine organometallics.
Scheme 42.

Bis-Azine Biaryl Synthesis Using Phosphorus Ligand-Coupling Reactions
Scheme 42A shows the protocol for this three-step process that involves transforming the first azine into a heteroaryl phosphine, forming a phosphonium salt with the second azine, and finally subjecting this salt to acidic alcohol solutions at 80 °C. Scheme 42B describes the first stage that uses fragmentable phosphine (Phos 2) for a salt formation–base elimination sequence that results in pyridyl phosphine (Phos 3).
McNally and Paton et al. studied the mechanism of the C–C bond-forming step where both heterocycles are protonated, and ethanol addition to the phosphonium ion is the ratelimiting step of the reaction (Scheme 42C). The two heterocycles then undergo selective phosphorus-ligand-coupling via P(V) intermediate Int 10. This elementary step is an asynchronous process where the apical pyridine undergoes a 1,2-migration to the pyridinium ligand in the equatorial plane. A buildup of electron density at the apical positions in Int 10 explains why protonated pyridine groups preferentially occupy these sites over the phenyl ligands and it explains the role of Int 10 and explains why pyridines are preferential donors. After the formation of dearomatized Int 11, it irreversibly collapses to the bipyridine product. Scheme 42D shows examples of LSF using this phosphorus-mediated approach (196–207). While the addition of groups, such as pyridines, to pharmaceuticals at the final stages of drug development is less useful as an LSF application, it is conceivable that such bond constructions are more appropriate at intermediate stages to incorporate complex azine-containing structures.
Using the principles of phosphorus-ligand-coupling reactions described above, McNally’s group extended this platform to sp2–sp3 coupling reactions via azine C–H trifluoromethylation and difluoromethylation reactions (Scheme 43A).126 These substituents have numerous effects in modulating the bioactivity of candidate compounds, including increasing their lipophilicity, binding constants, and serving as hydrogen bond donors in the case of CF2H. Thus, these transformations are highly sought after during LSF campaigns. As shown in Scheme 43B, fluoroalkylphosphines Phos 4 and Phos 5 mediate these transformations. An advantage of this process over the bis-azine biaryl couplings in Scheme 42 is that the intermediate fluoroalkylphosphinium salts do not need to be isolated, and it is sufficient to add an alcoholic solvent, water, and an acid to the reaction to promote C–C bond formation in a one-pot reaction. Phosphorus ligand-coupling proceeds via P(V) Int 12; the apical fluoroalkyl group stabilizes the δ– charge via inductive effects and fulfills the criteria used to rationalize selective ligand-coupling in Scheme 43B. The McNally group report shows 19 examples of LSF from a diverse collection of pharmaceuticals and agrochemicals, including imatinib, pyriproxyfen, and triprolidine (208–212, Scheme 43C). Because this process has a distinct scope from other azine C–H fluoroalkylation reactions, it is practical for drug and agrochemical discovery programs. Because the reaction functions on complex substrates, it is likely to be a useful tool for LSF applications.
Scheme 43.

Fluoroalkylation of Azines via Phosphorus Ligand Coupling Reactions
Azine phosphonium salts can serve as inputs into metal-catalyzed cross-coupling reactions to make C–C bonds. McNally et al. reported a nickel-catalyzed coupling reaction with arylboronic acids that generates 4-arylated azines (Scheme 44A).130 The identity of the metal is critical for selective coupling with the azine group on the phosphonium ion rather than one of the three phenyl groups. Palladium catalysts typically result in mixtures of azine and phenyl coupling products. In contrast, the optimal nickel catalyst ligated with an N-heterocyclic carbene (NHC) exclusively couples arylboronic acid with the pyridine portion of the salt. Using this catalyst system, the authors reported arylated derivates of bisacodyl, and N-benzyl varenicline (213 and 214). To extend this method to sp2–sp3 couplings, McNally and Zhang reported a Co-catalyzed coupling of azine phosphonium salts with alkyl zinc reagents (Scheme 44B).131 Interestingly, both nickel and palladium catalysts resulted in mixtures of azine and phenyl couplings; a Co(III) catalyst with a bipyridine ligand, however, resulted in highly selective azine couplings. Examples 215–217 show that it is possible to append small alkyl groups, such as cyclobutyl, cyclopropyl, and methyl groups, to pharmaceuticals, which makes this method particularly useful for LSF applications.
Scheme 44.

Azine Phosphonium Salts As Inputs into Metal-Catalyzed Cross-Coupling Reactions
In a related report, Feng et al. reported a 4-selective arylation, alkenylation, and alkynylation of phosphonium salts using a palladium catalyst (Scheme 45).132 In this case, the azine phosphonium salts serve as the nucleophilic coupling partner, which enables coupling with aryl and vinyl iodides. Mechanistically, they exploit a carbonate-mediated fragmentation process where P(V) intermediate decomposes to form CO2, O=Phh3, and an incipient C4-pyridyl anion reacts with an electrophilic Ag salt. The authors proposed that the resultant organosilver species transmetallates with a Pd(II)-aryl species, which is formed via oxidative addition of the aryl iodide. Reductive elimination completes the catalytic cycle. Aryl iodides containing electron-withdrawing and electron-donating substituents are effective coupling partners in the reaction. Using this method, Feng et al. reported five LSF examples, including 218–220. Widely available aryl iodides make this a potentially useful method for creating diverse libraries of arylated and alkenylated azines.
Scheme 45.
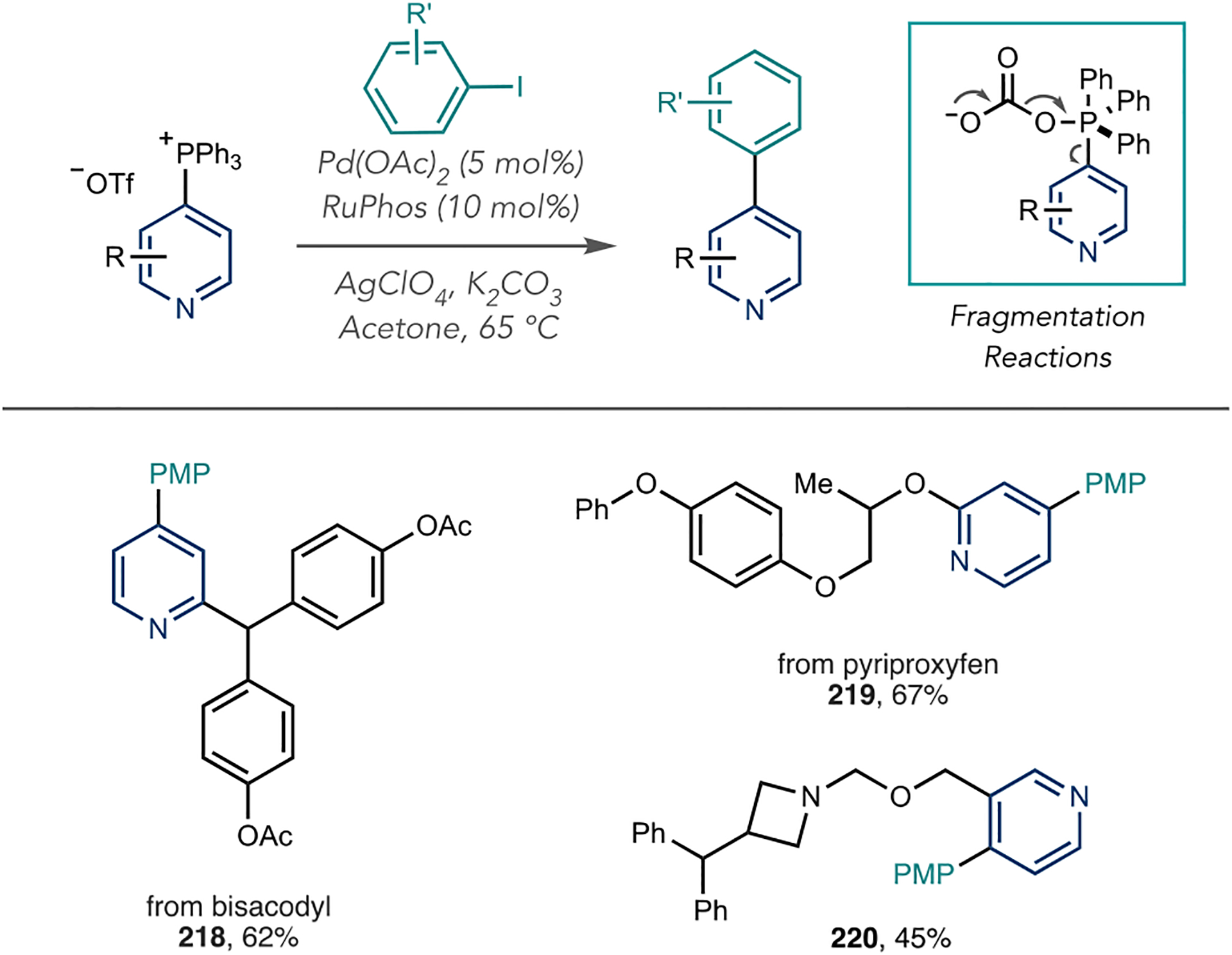
Pd-Catalyzed Phosphonium Salt Cross-Coupling Reactions via Organosilver Intermediates
McNally’s group reported two examples of Csp2–Csp3 and Csp2–Csp2 couplings with azine phosphonium salts that involve radical–radical couplings in the key C–C bond-forming step. In the first case, they found that the combination of azine phosphonium salts with cyanopyridines and B2pin2 regioselectively forms 2,4-bipyridines (Scheme 46A).133 In their proposed mechanism, a series of electron transfer steps promotes the radical–radical coupling between boryl cyanopyridine 2-position Int 13 and radical cation Int 14. Subsequent elimination of PPh3 and deborylation mediated by NEt3 forms a dearomatized adduct, where reoxidation occurs rapidly in air to form the bipyridine core. They showed that this reaction can function on bioactive molecules, although the method is more likely to be appropriate prior to LSF applications on advanced drug candidates (Scheme 46B, 221 and 222).
Scheme 46.

Azine Phosphonium Salt Coupling Reactions via Radical Intermediates
In their next approach, they developed a pyridine alkylation reaction using photoredox catalysis (Scheme 46C).134 This study showed that pyridine phosphonium salts mimic the reactivity of cyanopyridines in single-electron reduction processes. The radical zwitterion intermediate is analogous to radical anions derived from cyanopyridines that can undergo several subsequent coupling reactions with other stabilized radicals, such as stabilized benzylic radicals via single-electron oxidation of BF3K salts. An advantage of using pyridine phosphonium salts is that they are prepared from a wide array of pyridine C–H precursors and lend themselves to LSF applications. In this report, the authors disclosed five LSF examples, including 223 and 224 (Scheme 46D).
McNally’s group applied Wittig olefination chemistry in a nonconventional method for pyridine 4-alkylation (Scheme 47A).135 After forming N-triazinylpyridinium salts, they added tributylphosphine (PBu3) to form dearomatized phosphonium ion 225 and, then, MeLi at low temperature to obtain the key ylide Int 15 (Scheme 47B). A subsequent Wittig olefination–rearomatization sequence formed the desired 4-alkylated pyridine. Aromatic and aliphatic aldehydes are both competent in the olefination step, albeit with lower yields in the latter case. Using this protocol, the McNally team synthesized benzylated versions of abiraterone acetate and loratadine, thereby showing that this approach is viable for LSF applications (Scheme 47C, 226 and 227). Furthermore, modification of the reaction protocol enabled pyridine 4-methylation: benzotriazole 228 is a formaldehyde surrogate that, when treated with base and added to the ylide at low temperature, formed methylated derivatives 229 and 230 (Schemes 47D,E).136
Scheme 47.

Engaging Dearomatized Phosphonium Salts in Wittig-Type Reactions for Azine Alkylation
4.5. Reactions Mediated By AgF2 and Subsequent Transformations
Hartwig et al. developed a AgF2-mediated azine fluorination reaction that proceeds via a mechanism resembling the classic Chichibabin reaction (Scheme 48A).137 The mechanism begins with reversible coordination of the silver reagent to the azine N atom Int 16, and the Ag–F bond subsequently adds across the pyridinium π-system to form the amidosilver(II)-fluoride Int 17 (Scheme 48B). The authors propose that another equivalent of AgF2 promotes an H atom abstraction step to reform the azine core and release AgF and HF as byproducts.
Scheme 48.

AgF2-Mediated 2-Fluorination of Pyridines
Notably, this reaction is 2-selective for 2-, 3-, and 4-substituted pyridines. Some 6-fluoro regioisomer formation occurs when the 3-substitutent on the pyridine is an amide, ester, or alkyl (Scheme 48C). A series of competition and computation experiments determined the regioselectivity of these reactions and are outlined in Scheme 48C.138 In addition, more Lewis basic pyridines fluorinated in preference to less Lewis basic pyridines, presumably because of an increased rate of coordination of silver to the N lone pair.
This fluorination method tolerates pyridines containing halogens; electron-withdrawing groups such as ketones, esters, and amides; and electron-donating groups, including alkoxy substituents, on azines. However, free amines or alcohols, carboxylic acids, aldehydes, and electron-rich five-membered rings are incompatible under the reaction conditions. Fluorination occurred successfully in the cases of tropicamide (231), calcitonin gene-related peptide (CGRP) receptor antagonist core (232), and N-Me roflumilast (233), thereby supporting the use of this fluorination method for LSF on medicinally relevant molecules (Scheme 48D).
Subsequent transformation of the 2-fluoroazine products with a variety of heteroatom nucleophiles via SNAr reactions is a significant advantage of this strategy138. For example, in Scheme 49A, Hartwig et al. converted fluorinated derivatives of etoricoxib 234 into various derivatives using alcohols, amines, cyanides, and five-membered ring heteroaromatics under basic conditions (235–238). Isopropyl alcohol added to the 2-position of fluorinated N-Me roflumilast 239 using KOt-Bu as the base (240). 2-Amination occurred on N-Me roflumilast using an alkyl amine with Hunig’s base (241).
Scheme 49.
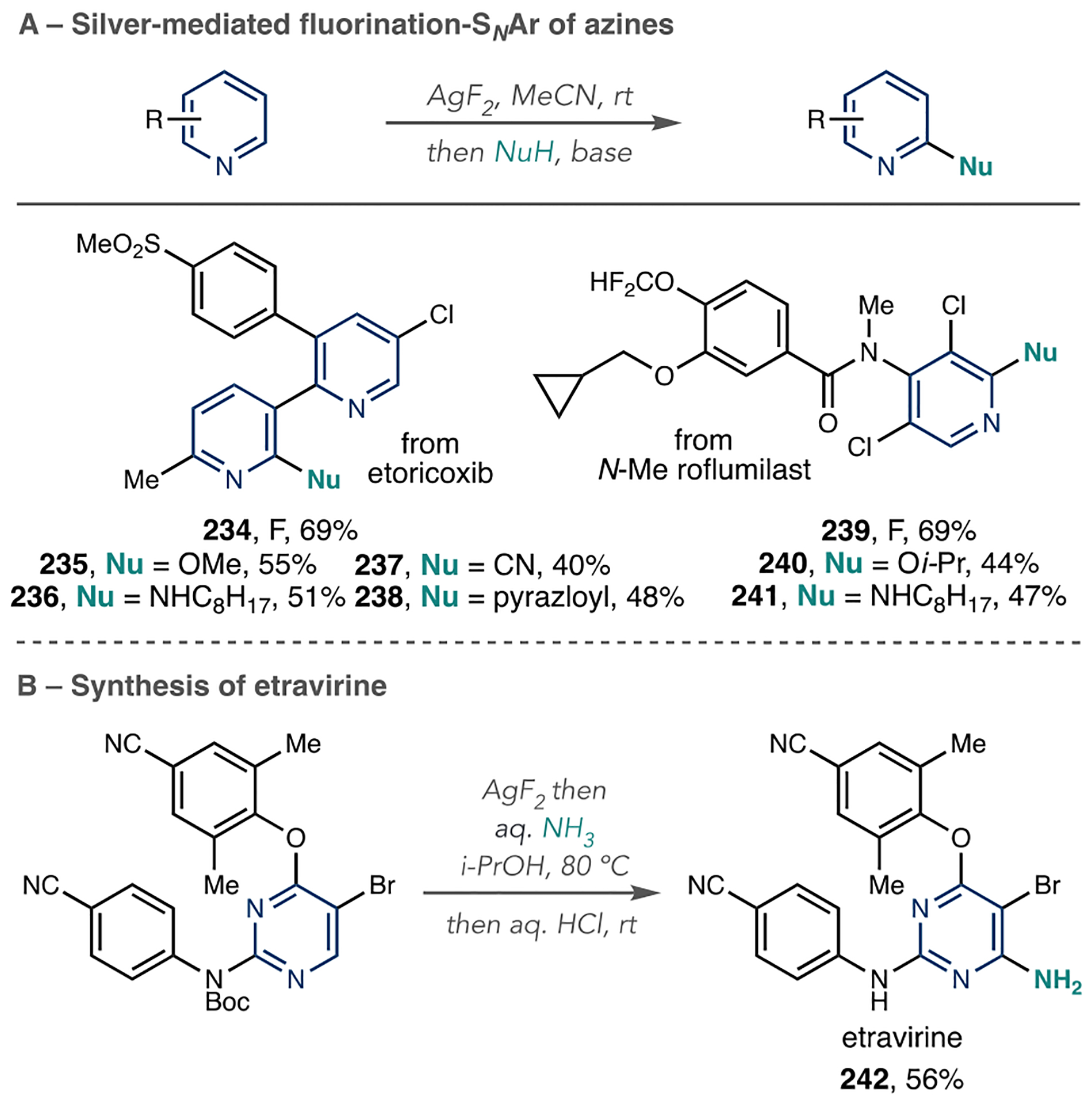
Transformations of Fluoroazines via SNAr Reactions
The fluorination–SNAr sequence is useful in discovery chemistry, as shown in the synthesis of the non-nucleoside reverse transcriptase inhibitor, etravirine, used to treat HIV (Scheme 49B). The last step in the synthesis installs the amino group via SNAr with aqueous ammonia (242). The synthesis required only three total isolations under six hours of reaction time compared with the medicinal chemist route that acquires 9% yield in five steps. This tandem C–H fluorination and SNAr reaction allows for the functionalization of medicinally important compounds, and the examples shown indicate that this method is useful for LSF applications.
Similarly, the Tang group developed a C–H trifluoromethoxylation reaction of azines that proceeds via a similar mechanism to the Hartwig et al. fluorination (Scheme 50).139 The use of silver additive in the reaction guides the nucleophile to the C2 position via coordination with the nitrogen lone pair. Notably, the reaction is regioselective for the C2 position for 2- and 4-substituted pyridines. Regioselectivity of 3-substituted pyridines is improved by blocking one or more reactive site. Despite this, the authors reported successful trifluoromethoxylation on seven LSF examples, including rosuvastatin (243) and etoricoxib (244), in encouraging yields.
Scheme 50.

Installation of Trifluoromethoxy Groups from 2-Fluoropyridines
4.6. Designed N-Activating Groups for 2-Selective Pyridine Functionalization Reactions
Fier developed a bifunctional reagent, α-chloro O-methanesulonyl aldoxime (245), for pyridine 2-cyanation (Scheme 51A).140 In the first stage of the reaction, pyridines react with 245 to form pyridinium salts (246); cyanide then adds at the 2-position to form dearomatized intermediate 247 and, then, a carbonate-mediated elimination result in the cyanated product (Scheme 51B). Again, the substituents on the azine ring dictate regioselectivity and are outlined in Scheme 51C. However, this cyanation method is amendable for LSF on medicinally relevant compounds such as 248. Cyanation occurred on progesterone agonist/antagonist 249 containing two electrophilic ketones with an epimerizable stereocenter in high yields with a 2.5:1 regioselectivity between the 2- and 6-positions (Scheme 50D). In addition, other nucleophiles, such as Grignards, organozincs, and malonates, added to the 4-position of pyridinium salts 246, which resulted in 4-functionalized products. Alkoxides, however, resulted in 2-functionalized pyridines. This bifunctional reagent 245, developed by Fier, enables LSF on azines for selective cyanation and potentially other nucleophiles beyond cyanides and organometallics.
Scheme 51.
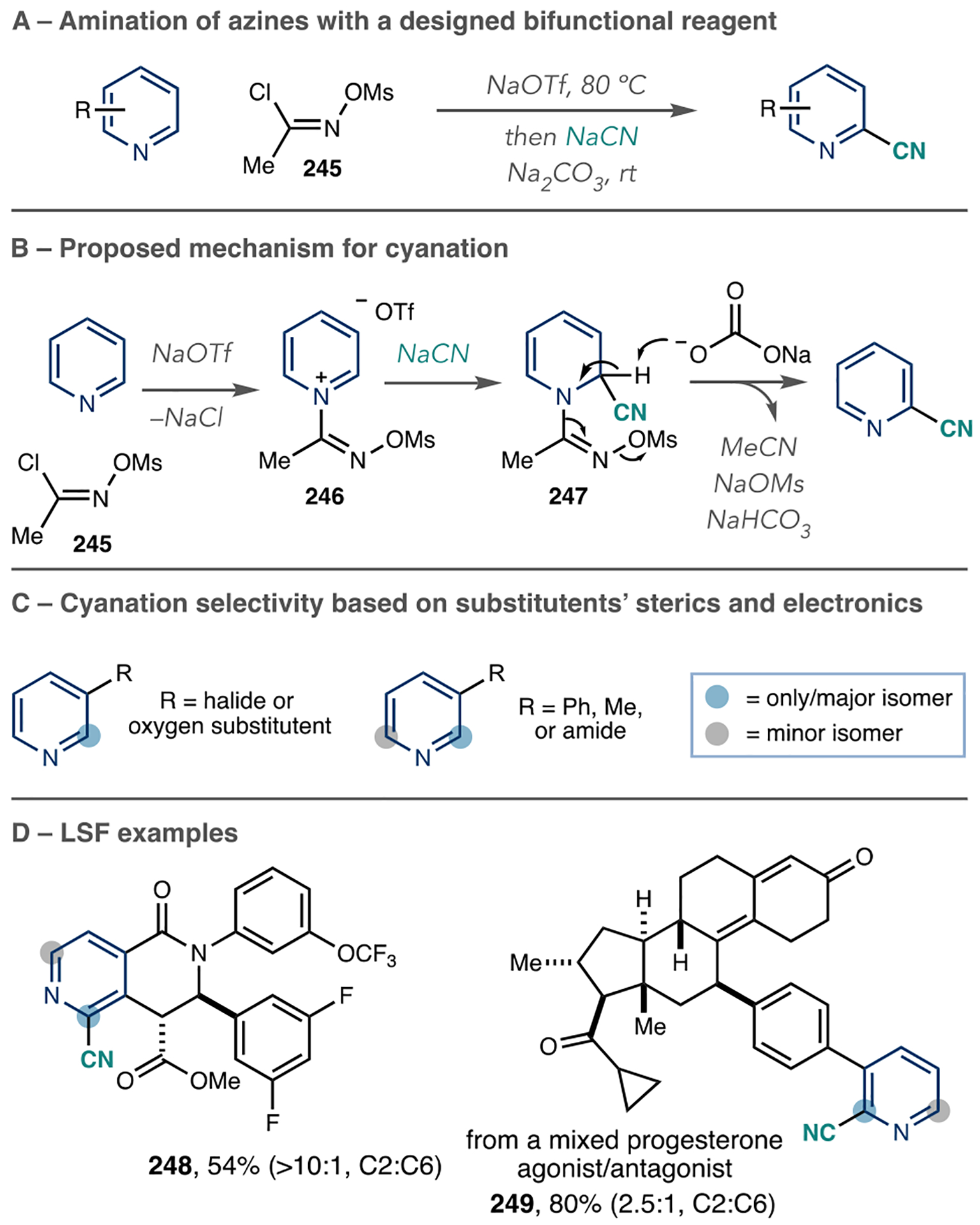
α-Chloro O-Methanesulonyl Aldoxime Reagent for Pyridine 2-Cyanation
Fier and co-workers subsequently reported a related strategy for pyridine 2-amination using a pyrazine-based multifunctional reagent 250 (Scheme 52A).141 In their proposed mechanism, pyridine first reacts with the chloropyrazine via SNAr reaction, and the pendent NHBoc group attacks the 2-position of the pyridinium salt to form dearomatized adduct 251 (Scheme 52B). The HCl formed from this step reacts with N,O-bis(trimethylsilyl)acetamide (BSA) to generate TMSCl and AcNHTMS as byproducts. Next, an elimination forms the imine and an oxygen anion that gets rapidly TMS-protected (252). At 80 °C, the adduct 252 isomerizes to the more stable intermediate 253. Then, Zn-mediated reduction of the electrophilic pyrazine extrudes the 2-NHBoc pyridine, followed by a further reduction to form the pyrazine byproduct.
Scheme 52.

2-Amination of Pyridines Using a Multifunctional Pyrazine Reagent
The selectivity for this reaction is similar to known 2-position pyridine functionalization methods such as pyridine N-oxide reactions, cyanation reactions, and fluorination reactions with AgF2. Fier and co-workers rationalized that 3-fluoro, 3-bromo, or 3-methoxy groups gave 2,3-disubstituted products with minor regioisomers at the 6-position because of increased reactivity of the C2-position relative to the C6-position from inductive or resonance effects and relief of eclipsing interactions during rehybridization in the nucleophilic addition step (Scheme 52C). The selectivity switches to form 2,5-disubstituted pyridines when larger substituents are present at the 3-position. This multifunctional reagent 250 developed by Fier et al. is amenable to LSF for the amination of complex biologically active molecules, such as examples 254 and 255 (Scheme 52D).
4.7. Regiocontrolled Pyridine Silylation via Ion Pair Effects
In 2019, Martin et al. disclosed a base-mediated regioselective azine C–H silylation reaction using Et3SiBpin as a reagent.142 Given the versatility of (hetero)aryl silanes for further transformations, this strategy offers access to a wide variety of azine derivatives. Notably, the regioselectivity of this reaction is highly dependent on solvent effects and is switchable between the 2- and 4-positions on pyridines. Specifically, the use of potassium hexamethyldisilazide (KHMDS) as a base and conducting the reaction in DME resulted in 10:1 C4/C2 selectivity on pyridine, whereas the use of 1,4-dioxane formed a 6:1 mixture with the C2-silylated isomer as the predominant outcome. The authors attributed this regioselective switch to the solvent’s chelation capacity and the aggregation state of the K+ counterion (Scheme 53). In DME, a solvent-separated ion pair forms Int 18 that sterically shields the pyridine 2-position, and the silyl anion species adds to C4. In dioxane, conversely, the authors assert that an inability to engage in bidentate binding resulted in a contacted ion pair Int 19, which directed the silyl anion equivalent to the proximal 2-position. In both cases, the oxidation of dearomatized intermediates in air reforms the azine cores.
Scheme 53.
Ion Pair Effect in Pyridine Silylation Reactions
The collection of bioactive compounds that successfully engage with this C–H silylation process demonstrates its applicability for LSF (Scheme 54). Examples include silylated derivatives of nevirapine 256, doxylamine 257, buspirone 258, loratadine 259, estrone 260, and piribedil 261 and indicate that both pyridine- and diazine-containing pharmaceuticals are viable. Functional groups, such as NH-amides, imides, ketones, and aryl chlorides, are well tolerated under the reaction conditions. This method complements transition-metal-catalyzed C–H azine silylation.
Scheme 54.
Examples of Azine LSF Reaction via C–H Silylation
4.8. Organocatalytic Azine Functionalization
The List group reported an azine alkylation reaction using silyl ketene acetals and silylium catalysis (Scheme 55A).143 The silylium catalyst forms in situ from designed Brønsted acid 264; the organocatalyst structure and acidity creates unstabilized and highly reactive silylium ions that subsequently transfer to azines and serve as the N-activating group (262). For pyridines, subsequent attack of the silyl ketene acetal is highly C4-regioselective, and 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) oxidation of the dihydropyridine derivative 263 forms the alkylated products (Scheme 55B). The report clearly shows that the acidity of the catalysts and reactivity of the silylium ion are important. Noncovalent interactions from the catalyst structure are also suggested to assist in this transformation. The reaction appears sensitive to sterically encumbering groups at the 2-position of azines. In the report, they alkylate epiandrosterone analogue 265 and a desloratadine analogue 266 as examples of LSF applications (Scheme 55C). While this protocol may not yet have the broad azine scope of other reactions in this review, it does raise the point that LSF reactions may need to be rendered scalable in certain scenarios in drug development, and organocatalysis will be valuable in those endeavors.
Scheme 55.
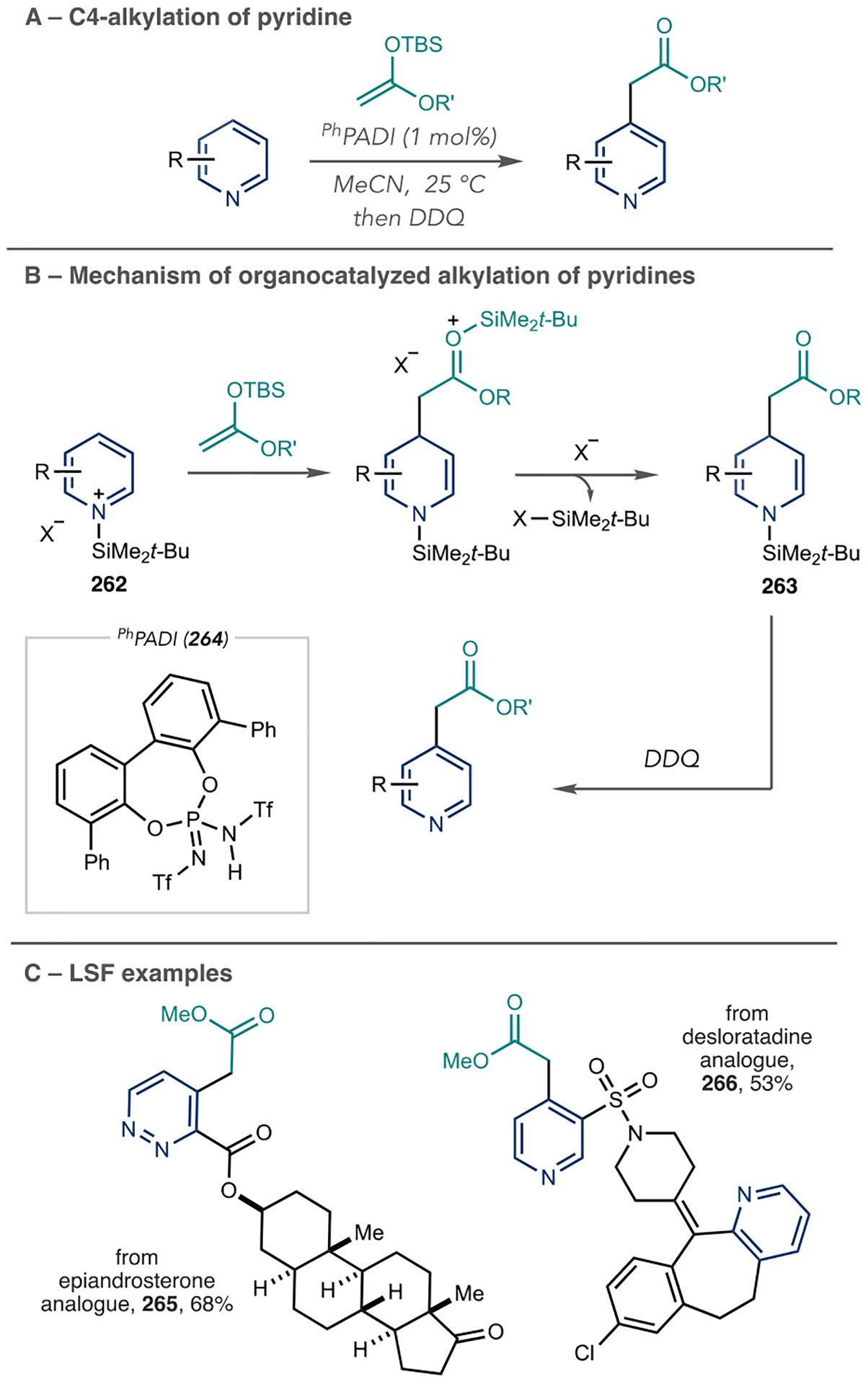
Brønsted Acid-Catalyzed Azine Alkylation Using Silylium Ion Intermediates
4.9. Exploiting Temporary Dearomatized Intermediates for Reactions with Electrophiles and Radicals
Recently, Wang et al. reported a strategy for pyridine C3-functionalization using a borane-catalyzed hydroboration and electrophile-trapping oxidation sequence (Scheme 56).144 Specifically, the 1,4-hydroboration step forms dihydropyridine Int 20, and the enamine-like bonds selectively react with electrophiles shown in Scheme 56 at the pyridine 3-position. Oxidation in air then reforms the aromatic system and results in 3-functionalized products. The authors’ first report showed that imines, aldehydes, and ketones were all competent electrophiles, and they described a single example of LSF using a vitamin E derivative. Then, they extended this platform to trifluorothiomethylation (SCF3) and difluorothiomethylation (SCF2H) reactions, including several derivatives of pharmaceuticals. (Scheme 56, 267–269).145 Installing SCF3 and SCF2H groups is valuable in drug development because of their capacity to modulate lipophilicity and promote hydrogen bonding interactions. LSF methods would further enhance the utility of these groups. For 2-subsituted pyridines, they found the control of mono- versus bis-addition challenging, such as for 269, and difluorothiomethylation was not successful on this substrate class. Nevertheless, they demonstrated a broad pyridine scope and reported 18 LSF examples.
Scheme 56.
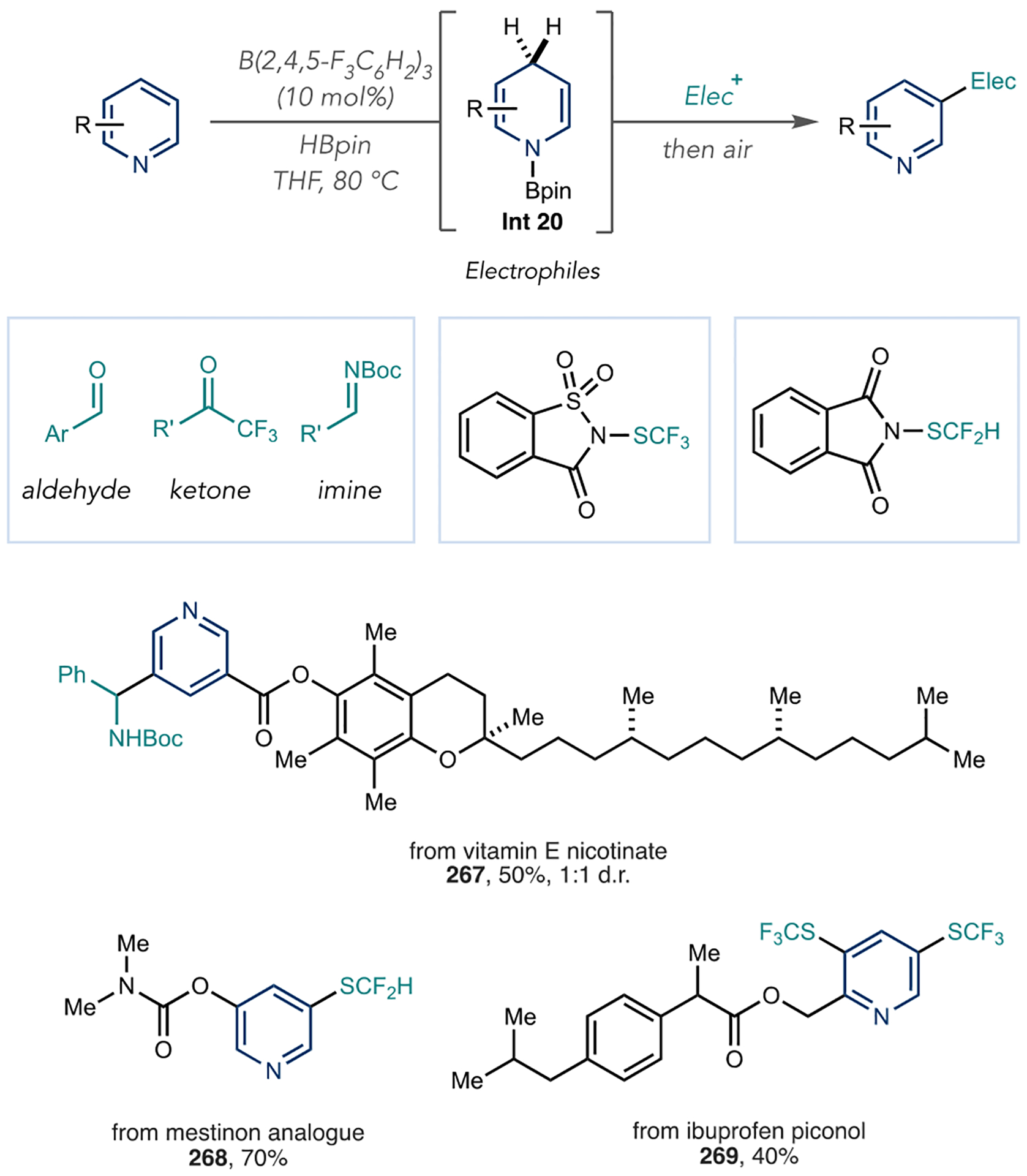
Pyridine Hydroboration and Reaction with Electrophiles for C3-Functionalization
The Kunibobu group developed a 3-selective trifluoromethylation reaction, primarily of quinolines, via a similar mechanistic approach to Scheme 57.146 They exploited an initial borane-catalyzed 1,4-hydrosilylation and reacted the dearomatized intermediates (not shown) with Togni I reagent before reoxidizing with DDQ. While this reaction does not appear to have the same scope as Wang et al.’s system, the authors did report an impressive LSF example in which they transformed quinoxyfen into 3-trifluoromethyl derivative 270 in 60% yield via a one-pot reaction (Scheme 57).
Scheme 57.
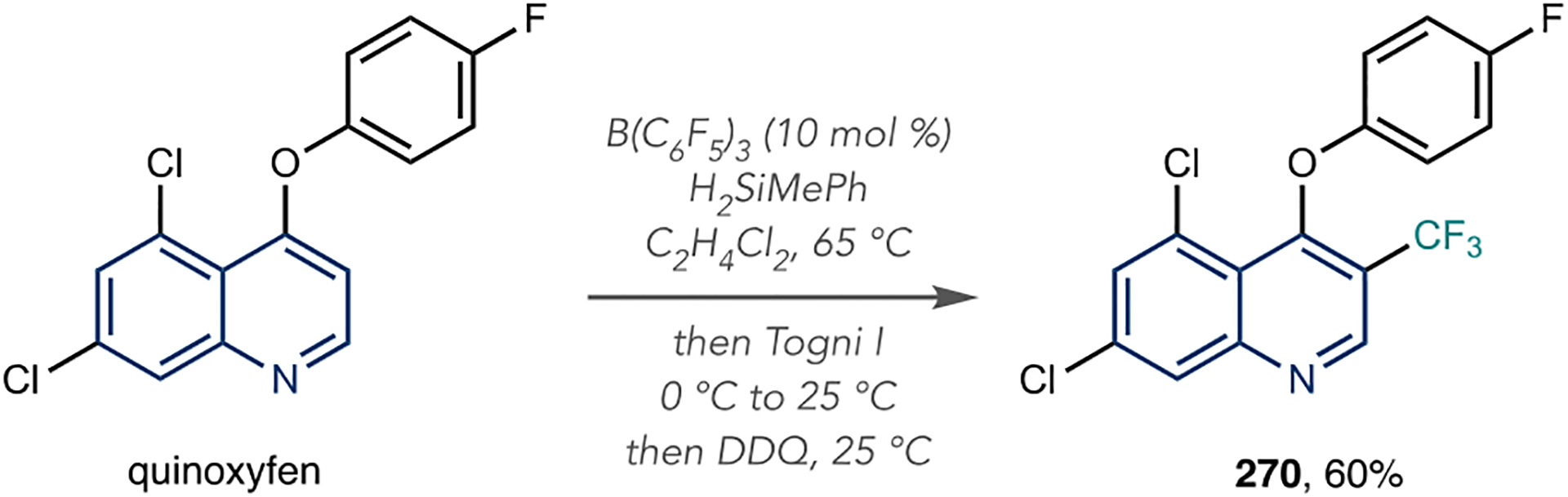
Trifluoromethylation via Hydrosilylated Azine Intermediates
In 2022, McNally’s group reported a one-pot procedure that halogenates the 3-position of pyridines via a ring opening, halogenation, and ring-closing sequence and demonstrated its viability for LSF applications.147 The late-stage halogenation of pyridines is highly enabling because many distinct chemical reactions can transform the C–Halogen bond into a diverse array of derivatives. Halopyridines are also present in pharmaceuticals, such as etoricoxib (Scheme 58), which shows that they can exert favorable properties on the biological profile of these molecules. Therefore, the installation of halides in LSF applications serves as a means for compound diversification and alteration of advanced candidates’ PK/PD properties. Existing methods for pyridine C3-halogenation typically require high temperatures, strong acids, and reactive halogen electrophiles. A longstanding challenge has been to devise methods that operate under mild reaction conditions, tolerate various functional groups, and apply to various pyridine substitution patterns.
Scheme 58.
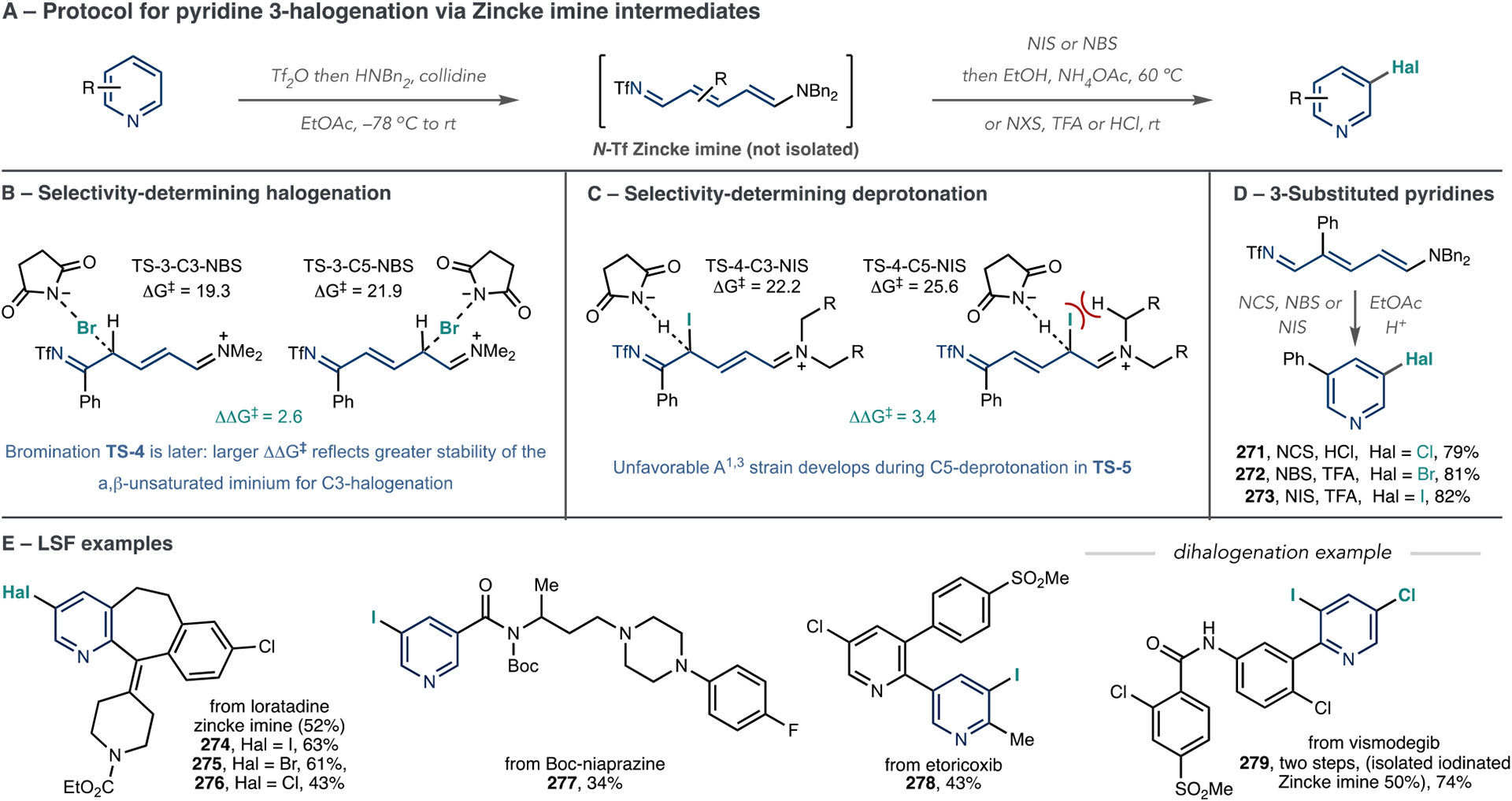
Halogenation of the 3-Position of Pyridines via Ring-Opened Zincke Imine Intermediates
The McNally group’s approach uses a modified version of the classic Zincke ring-opening reaction: dibenzylamine adds to N-Tf pyridinium salts and forms N-Tf Zincke imine intermediates (Scheme 58A). The N-Tf activation step in this process significantly broadens the scope of pyridines amenable to ring-opening compared with the N-dinitrophenylpyridinium salts used in traditional Zincke reactions. The N-Tf Zincke imine intermediates can react with halogen electrophiles selectively, much like electron-rich aromatics (vide infra). Notably, this approach addresses a significant challenge in regiocontrolled synthesis: halogenation is 3-selective for nonsymmetrical pyridines when both the 3- and 5-positions are unsubstituted. After the halogenation step is complete, the addition of ammonium salts to the same reaction vessel facilitates ring-closing to reform the pyridine ring. In most cases, this sequence occurs in the same reaction vessel by adding reagents sequentially to promote each stage.
Scheme 58B shows the rationale for selective bromination and iodination reactions. Bromination is a kinetically controlled process proceeding through a late-transition state with a significant difference in distortion energy when reacting at the C3 and C5 positions. In turn, a more stable α,β-unsaturated iminium ion in TS-3-C3-NBS than TS-3-C5-NBS and a ΔΔG of 2.5 kcal mol–1 results. However, C–I bond formation occurs reversibly, and the selectivity observed results from the rate- and selectivity determining deprotonation from the succinimide anion to reform the conjugated system. In TS-4-C5-NIS, restoration of the C–I bond into planarity results in A1,3 strain with the benzyl substituent and disfavors the formation of the 5-iodo isomer (Scheme 58C). On 2-substituted systems, chlorination shows moderate selectivity and occurs in lower yields. For 3-substituted adducts, Scheme 58D shows halogenation through the corresponding Zincke imine under acidic conditions, which results in sequential C–halogen bond formation and recyclization (271–273).
This reaction has a broad tolerance for various functional groups and substituted pyridines. Other azines, such as quinolines, isoquinolines, and pyrimidines, can be halogenated, and the Zincke imine intermediates can outcompete electronic directing groups, such as alkoxy moieties, and other electron-rich rings. They also showed a large collection of pyridine LSF reactions, and Scheme 58E shows representative examples, including loratadine, N-Boc niaprazine, and etoricoxib (274–278). Halogenation reactions can be sequenced to form dihalogenated products, such as vismodegib derivative 279.
The Studer group developed a redox-neutral dearomatization–rearomatization process to selectively functionalize the 3-position of pyridines via fluoroalkylation, halogenation, nitration, sulfanylation, and selenylation reactions (Scheme 59A).148 They combined pyridines with a dimethyl acetylenedicarboxylate (DMAD) and methyl pyruvate (MP) to result in dearomatized oxazino pyridine Int 21. This dearomatization step has significant scope and is not substantially perturbed by sterics around the N atom in substrates, such as 2-substituted pyridines. These intermediates then partake radical additions (Condition A) or react through two electron pathways with electrophiles, such as N-halosuccinimides (Condition B). After forming a C–C or C–heteroatom bond, the authors added aqueous acid to form the C3-functionalized pyridine.
Scheme 59.

Pyridine C3 Halogenation and Fluoroalkylation via Dearomatized Oxazino Pyridine Intermediates
In the authors’ proposed mechanism for trifluoromethylation, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) forms a complex 280 with CF3–I, and blue light then promotes C–I bond homolysis to form the trifluoromethyl radical and start a radical chain reaction (Scheme 59B). Next, the trifluoromethyl radical adds selectively to the oxazino pyridine Int 22 and generates a radical intermediate (Int 23) that reacts with CF3I in the chain propagation step. DBU then eliminates HI to form C–H trifluoromethylated oxazino pyridine 281 and is the driving force for the endothermic iodine atom transfer step. The addition of aqueous hydrochloric acid and heating of the reaction to 60 °C rearomatizes 281 to give the trifluoromethylated pyridine. For the halogenation reactions, two electron pathways are invoked from the oxazino pyridine intermediates.
Like the McNally group’s approach, this reaction can selectively functionalize the 3-position of pyridines, when both 3- and 5-positions are unsubstituted. Using N-chlorosuccinimide (NCS), 3-chloroinated products are obtained, and the Studer group concluded that chlorination is irreversible and kinetically controlled. Furthermore, they also found that the selectivity switches to the 5-position when NBS and N-iodosuccinimide (NIS) are employed to form 5-brominated and 5-iodinated products, respectively. In these cases, the reactions are reversible, and the 5-position is the less sterically hindered and more thermodynamically stable site. For the fluoroalkylation reactions, they found that mixtures of 3- and 5-position isomers form on 2-aryl pyridines, thereby indicating that regiocontrol is more challenging in open shell pathways. It is important to note that the addition of fluoroalkyl radicals directly to pyridines in Minisci-type reactions often results in regiomeric mixtures. In either case, application of the chemistry in Scheme 59 to 3-substituted pyridines results in 5-functionalized products for both C–heteroatom and C–C bond formations.
This reaction has a broad pyridine scope and shares similar attributes to the McNally group’s approach described above. It also has significant potential for LSF applications, and Scheme 59C shows four examples of functionalized pyridine-containing drugs. The 3-selective chlorination and 5-selective bromination reactions observed in simpler substrates translate to complex pharmaceuticals, such as vismodegib (282 and 283). The authors used their radical fluoroalkylation protocol to obtain a 3-CF3 loratadine analogue 284. Chlorinated fasudil 285 indicates that the protocol also applies to heterocycles beyond pyridines.
5. CONCLUSION AND OUTLOOK
In summary, we have described the current set of chemical reactions viable for azine LSF. We categorized these processes as radical additions, transition-metal-catalyzed C–H activation reactions, and reactions via dearomatized intermediates. There is a catalog of transformations practitioners can peruse and apply to their molecules of interest. The number of azine LSF reactions will continue to grow in concert with factors such as a greater focus on LSF in general or with advances in synthetic method development like photoredox catalysis. This review also highlights that there can be great variety in the approaches taken to functionalize the azine C–H bond, thereby indicating significant depth in how these six-membered heterocycles can react. Thus, further developments in azine LSF will undoubtedly co-opt the traits of synthetic organic chemists to invent new important transformations.
It is helpful to examine several important factors to explain the current state of azine LSF reactions and potentially make predictions about future developments. It is reasonable to argue that the community regards azine chemistry as well developed with an existing toolkit of reactions widely practiced in industry. Thus, new developments may have escaped the attention of researchers in chemical methods development. The reactions in this review show how many important new transformations have emerged by applying modern developments in organic synthesis and reinvigorated interest in the reactivity of these heterocycles. The approaches taken in this review show how another perceptive factor can influence progress in azine LSF. The synthetic community is interested in catalytic processes for self-evident reasons, such as efficiency and sustainability. However, the output of LSF reactions has a different set of criteria; often, only milligram quantities of an analogue compound are required for biological testing, so concerns about waste production, for example, do not carry such significant weight. The development of new catalytic transformations can be particularly difficult because of the need to sequence many individual steps together to turn over the cycle. In this review, there are several examples of new transformations using stoichiometric, reagent-based approaches that may not be possible to exist in a catalytic format. Put another way, being free from the constraints of catalysis can release reaction developers to design powerful transformations. The impact of these methods in the academic community and uptake by industry could spur more developments in this vein in the future.
It is noticeable that there are relatively few industry-lead reports of azine LSF reactions from the collection of papers summarized in this review. This observation is surprising given the direct relevance of these reactions to medicinal and agrochemical programs. There may be more engagement than anticipated in-house. However, the skill set of industrialists, and the technologies available to that cohort, means they have a significant role to play in advancing this field. These scientists apply azine functionalization chemistry regularly and are acutely aware of the deficiencies of the current synthetic toolkit. Many have backgrounds in synthetic method development, and we have seen examples in this review of skillful reaction design for new azine C–H functionalization reactions.54,66,74,140,141 Technologies such as high-throughput experimentation methods are also likely to play an important role.13 Techniques such as informer libraries are an excellent means to evaluate the strengths and weaknesses of a given method, and machine learning will become more common as an assistive tool for reaction development.149 As well as solving major problems in reaction development, industrialists have a unique capacity to describe to the community what the significant challenges are.
Biographies
Andrew McNally grew up in Liverpool in the United Kingdom. He gained his undergraduate degree from the University of Cambridge and completed his final year project in Professor Ian Paterson’s group. Andy stayed in Cambridge for his Ph.D. studies as part of the inaugural class in Professor Matthew Gaunt’s laboratory. He gained a Marie Curie International Outgoing Fellowship and moved to Princeton University to work in Professor David MacMillan’s group as a postdoctoral researcher. For the return phase of the fellowship, he returned to Cambridge to Professor Gaunt’s laboratory and worked on the C–H activation reactions of aliphatic amines. He began his independent career in 2014 at Colorado State University, was promoted to Associate Professor, and awarded the Albert I. Meyers Chair in Organic Chemistry in 2020. His group is developing broadly applicable new transformations of electron-deficient heterocycles using main group elements and dearomatized intermediates.
Celena M. Josephitis is currently a Ph.D. candidate in chemistry under the supervision of Dr. Andrew McNally at Colorado State University. She earned her B.S. in chemistry in 2019 from the University of Wisconsin – Stevens Point while completing undergraduate research in ring-closing reactions to synthesize 5- and 6-membered N-heterocycles. Her current research focuses on transformations of pyridines to saturated and unsaturated derivatives.
Hillary M. H. Nguyen was born and raised in Orange County, California. She obtained her B.S. in chemistry and a minor in biological sciences from the University of California, Irvine (UCI). While at UCI, she worked on Rh-catalyzed hydroacylation reactions under the supervision of Professor Vy Dong. Hillary is currently pursuing her Ph.D. at Colorado State University under the supervision of Professor Andrew McNally. Her research focuses on the development of cross-coupling and functionalization of pyridines.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.chemrev.2c00881
The authors declare no competing financial interest.
Contributor Information
Celena M. Josephitis, Department of Chemistry, Colorado State University, Fort Collins, Colorado 80523, United States.
Hillary M. H. Nguyen, Department of Chemistry, Colorado State University, Fort Collins, Colorado 80523, United States.
Andrew McNally, Department of Chemistry, Colorado State University, Fort Collins, Colorado 80523, United States;.
REFERENCES
- (1).Cernak T; Dykstra KD; Tyagarajan S; Vachal P; Krska SW The Medicinal Chemist’s Toolbox for Late Stage Functionalization of Drug-Like Molecules. Chem. Soc. Rev 2016, 45, 546–76. [DOI] [PubMed] [Google Scholar]
- (2).Borgel J; Ritter T Late-Stage Functionalization. Chem. 2020, 6, 1877–1887. [Google Scholar]
- (3).Jana R; Begam HM; Dinda E The Emergence of the C-H Functionalization Strategy in Medicinal Chemistry and Drug Discovery. Chem. Commun 2021, 57, 10842–10866. [DOI] [PubMed] [Google Scholar]
- (4).Guillemard L; Kaplaneris N; Ackermann L; Johansson MJ Late-Stage C-H Functionalization Offers New Opportunities in Drug Discovery. Nat. Rev. Chem 2021, 5, 522–545. [DOI] [PubMed] [Google Scholar]
- (5).Zhang L; Ritter T A Perspective on Late-Stage Aromatic C-H Bond Functionalization. J. Am. Chem. Soc 2022, 144, 2399–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kelly CB; Padilla-Salinas R Late Stage C-H Functionalization via Chalcogen and Pnictogen Salts. Chem. Sci 2020, 11, 10047–10060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Hota SK; Jinan D; Panda SP; Pan R; Sahoo B; Murarka S Organophotoredox-Catalyzed Late-Stage Functionalization of Heterocycles. Asian J. Org. Chem 2021, 10, 1848–1860. [Google Scholar]
- (8).Cannalire R; Pelliccia S; Sancineto L; Novellino E; Tron GC; Giustiniano M Visible Light Photocatalysis in the Late-Stage Functionalization of Pharmaceutically Relevant Compounds. Chem. Soc. Rev 2021, 50, 766–897. [DOI] [PubMed] [Google Scholar]
- (9).Bellotti P; Huang HM; Faber T; Glorius F Photocatalytic Late-Stage C-H Functionalization. Chem. Rev 2023, DOI: 10.1021/acs.chemrev.2c00478. [DOI] [PubMed] [Google Scholar]
- (10).Parida SK; Hota SK; Kumar R; Murarka S Late-Stage Alkylation of Heterocycles Using N-(Acyloxy)phthalimides. Chem. Asian. J 2021, 16, 879–889. [DOI] [PubMed] [Google Scholar]
- (11).Sap JBI; Meyer CF; Straathof NJW; Iwumene N; Ende CWA; Trabanco AA; Gouverneur V Late-Stage Difluoromethylation: Concepts, Developments and Perspective. Chem. Soc. Rev 2021, 50, 8214–8247. [DOI] [PubMed] [Google Scholar]
- (12).Wencel-Delord J; Glorius F C-H Bond Activation Enables the Rapid Construction and Late-Stage Diversification of Functional Molecules. Nat. Chem 2013, 5, 369–375. [DOI] [PubMed] [Google Scholar]
- (13).Friis SD; Weis E; Johansson MJ, HTE as a Tool in C-H Activation Reaction Discovery and Late-Stage Functionalization of Pharmaceuticals. In The Power of High-Throughput Experimentation: Case Studies from Drug Discovery, Drug Development, and Catalyst Discovery; ACS Symposium Series, Vol. 1420, 2022; pp 161–179. [Google Scholar]
- (14).Zakharychev VV; Kuzenkov AV; Martsynkevich AM Good Pyridine Hunting: A Biomimic Compound, a Modifier and a Unique Pharmacophore in Agrochemicals. Chem. Heterocycl. Compd 2020, 56, 1491–1516. [Google Scholar]
- (15).Baumann M; Baxendale IR An Overview of the Synthetic Routes to the Best Selling Drugs Containing 6-Membered Heterocycles. Beilstein J. Org. Chem 2013, 9, 2265–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles Among U.S. FDA-Approved Pharmaceuticals. J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- (17).Bhutani P; Joshi G; Raja N; Bachhav N; Rajanna PK; Bhutani H; Paul AT; Kumar R US FDA-Approved Drugs from 2015-June 2020: A Perspective. J. Med. Chem 2021, 64, 2339–2381. [DOI] [PubMed] [Google Scholar]
- (18).Joule JA; Mills K Heterocyclic Chemistry, 5th ed.; Wiley: Hoboken, N.J., 2009; pp xxviii, 689. [Google Scholar]
- (19).Kishbaugh TLS Pyridines and Imidazopyridines with Medicinal Significance. Curr. Top. Med. Chem 2016, 16, 3274–3302. [DOI] [PubMed] [Google Scholar]
- (20).Ling Y; Hao ZY; Liang D; Zhang CL; Liu YF; Wang Y The Expanding Role of Pyridine and Dihydropyridine Scaffolds in Drug Design. Drug Des. Devel. Ther 2021, 15, 4289–4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Pennington LD; Moustakas DT The Necessary Nitrogen Atom: A Versatile High-Impact Design Element for Multiparameter Optimization. J. Med. Chem 2017, 60, 3552–3579. [DOI] [PubMed] [Google Scholar]
- (22).Mohamed EA; Ismail NSM; Hagras M; Refaat H Medicinal Attributes of Pyridine Scaffold as Anticancer Targeting Agents. Future J. Pharm. Sci 2021, 7, 24. [Google Scholar]
- (23).Eicher T; Hauptmann S; Speicher A The Chemistry of Heterocycles: Structure, Reactions, Syntheses, and Applications, 3rd, Completely Revised and Enlarged Edition; Wiley-VCH: Weinheim, 2012; pp xiii, 632. [Google Scholar]
- (24).Grimmett MR Halogenation of Heterocycles: II. Six-Membered and Seven-Membered Rings. Adv. Heterocycl. Chem 1993, 58, 271–345. [Google Scholar]
- (25).Murakami K; Yamada S; Kaneda T; Itami K C-H Functionalization of Azines. Chem. Rev 2017, 117, 9302–9332. [DOI] [PubMed] [Google Scholar]
- (26).Sowmiah S; Esperanca JMSS; Rebelo LPN; Afonso CAM Pyridinium Salts: From Synthesis to Reactivity and Applications. Org. Chem. Front 2018, 5, 453–493. [Google Scholar]
- (27).Wang D; Desaubry L; Li GY; Huang MD; Zheng SX Recent Advances in the Synthesis of C2-Functionalized Pyridines and Quinolines Using N-Oxide Chemistry. Adv. Synth. Catal 2021, 363, 2–39. [Google Scholar]
- (28).Dong DQG; Sun YA; Li GHA; Yang H; Wang ZL; Xu XM Recent Progress in the Functionalization of Quinoline N-Oxide. Chin. J. Org. Chem 2020, 40, 4071–4086. [Google Scholar]
- (29).Yan GB; Borah AJ; Yang MH Recent Advances in Catalytic Functionalization of N-Oxide Compounds via C-H Bond Activation. Adv. Synth. Catal 2014, 356, 2375–2394. [Google Scholar]
- (30).Kang QK; Shi H Catalytic Hydrogen Isotope Exchange Reactions in Late-Stage Functionalization. Synlett 2022, 33, 329–338. [Google Scholar]
- (31).Sawada S; Okajima S; Aiyama R; Nokata K; Furuta T; Yokokura T; Sugino E; Yamaguchi K; Miyasaka T Synthesis and Antitumor Activity of 20(S)-Camptothecin Derivatives: Carbamate-Linked, Water-Soluble Derivatives of 7-Ethyl-10-Hydroxycamptothecin. Chem. Pharm. Bull 1991, 39, 1446–1454. [DOI] [PubMed] [Google Scholar]
- (32).Cernak T; Dykstra KD; Tyagarajan S; Vachal P; Krska SW The Medicinal Chemist’s Toolbox for Late-Stage Functionalization of Drug-Like Molecules. Chem. Soc. Rev 2016, 45, 546–576. [DOI] [PubMed] [Google Scholar]
- (33).Duncton MAJ Minisci Reactions: Versatile C-H Functionalizations for Medicinal Chemists. MedChemComm 2011, 2, 1135–1161. [Google Scholar]
- (34).Proctor RSJ; Phipps RJ Recent Advances in Minisci-Type Reactions. Angew. Chem., Int. Ed 2019, 58, 13666–13699. [DOI] [PubMed] [Google Scholar]
- (35).Ma X; Herzon SB Intermolecular Hydropyridylation of Unactivated Alkenes. J. Am. Chem. Soc 2016, 138, 8718–8721. [DOI] [PubMed] [Google Scholar]
- (36).Li M; Jin W; Jiang C; Zheng C; Tang W; You T; Lou L 7-Cycloalkylcamptothecin Derivatives: Preparation and Biological Evaluation. Bioorg. Med. Chem. Lett 2009, 19, 4107–4109. [DOI] [PubMed] [Google Scholar]
- (37).Duncton MAJ; Estiarte MA; Johnson RJ; Cox M; O’Mahony DJR; Edwards WT; Kelly MG Preparation of Heteroaryloxetanes and Heteroarylazetidines by Use of a Minisci Reaction. J. Org. Chem 2009, 74, 6354–6357. [DOI] [PubMed] [Google Scholar]
- (38).Seiple IB; Su S; Rodriguez RA; Gianatassio R; Fujiwara Y; Sobel AL; Baran PS Direct C-H Arylation of Electron-Deficient Heterocycles with Arylboronic Acids. J. Am. Chem. Soc 2010, 132, 13194–13196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Schonherr H; Cernak T Profound Methyl Effects in Drug Discovery and a Call for New C-H Methylation Reactions. Angew. Chem., Int. Ed 2013, 52, 12256–12267. [DOI] [PubMed] [Google Scholar]
- (40).O’Hara F; Blackmond DG; Baran PS Radical-Based Regioselective C-H Functionalization of Electron-Deficient Heteroarenes: Scope, Tunability, and Predictability. J. Am. Chem. Soc 2013, 135, 12122–12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Zhou Q; Gui J; Pan CM; Albone E; Cheng X; Suh EM; Grasso L; Ishihara Y; Baran PS Bioconjugation by Native Chemical Tagging of C-H Bonds. J. Am. Chem. Soc 2013, 135, 12994–12997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Fujiwara Y; Dixon JA; O’Hara F; Funder ED; Dixon DD; Rodriguez RA; Baxter RD; Herle B; Sach N; Collins MR; Ishihara Y; Baran PS Practical and Innate Carbon-Hydrogen Functionalization of Heterocycles. Nature 2012, 492, 95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Ji Y; Brueckl T; Baxter RD; Fujiwara Y; Seiple IB; Su S; Blackmond DG; Baran PS Innate C-H Trifluoromethylation of Heterocycles. Proc. Natl. Acad. Sci. U.S.A 2011, 108, 14411–14415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Fujiwara Y; Dixon JA; Rodriguez RA; Baxter RD; Dixon DD; Collins MR; Blackmond DG; Baran PS A New Reagent for Direct Difluoromethylation. J. Am. Chem. Soc 2012, 134, 1494–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Zhou Q; Ruffoni A; Gianatassio R; Fujiwara Y; Sella E; Shabat D; Baran PS Direct Synthesis of Fluorinated Heteroarylether Bioisosteres. Angew. Chem., Int. Ed 2013, 52, 3949–3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Smith JM; Dixon JA; deGruyter JN; Baran PS Alkyl Sulfinates: Radical Precursors Enabling Drug Discovery. J. Med. Chem 2019, 62, 2256–2264. [DOI] [PubMed] [Google Scholar]
- (47).Gutierrez-Bonet A; Remeur C; Matsui JK; Molander GA Late-Stage C-H Alkylation of Heterocycles and 1,4-Quinones via Oxidative Homolysis of 1,4-Dihydropyridines. J. Am. Chem. Soc 2017, 139, 12251–12258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Revil-Baudard VL; Vors JP; Zard SZ Xanthate-Mediated Incorporation of Quaternary Centers into Heteroarenes. Org. Lett 2018, 20, 3531–3535. [DOI] [PubMed] [Google Scholar]
- (49).Sutherland DR; Veguillas M; Oates CL; Lee AL Metal-, Photocatalyst-, and Light-Free, Late-Stage C-H Alkylation of Heteroarenes and 1,4-Quinones Using Carboxylic Acids. Org. Lett 2018, 20, 6863–6867. [DOI] [PubMed] [Google Scholar]
- (50).Dong J; Wang Z; Wang X; Song H; Liu Y; Wang Q Metal-, Photocatalyst-, and Light-Free Minisci C-H Alkylation of N-Heteroarenes with Oxalates. J. Org. Chem 2019, 84, 7532–7540. [DOI] [PubMed] [Google Scholar]
- (51).Dong J; Wang Z; Wang X; Song H; Liu Y; Wang Q Correction to ″Metal-, Photocatalyst-, and Light-Free Minisci C-H Alkylation of N-Heteroarenes with Oxalates″. J. Org. Chem 2022, 87, 15719. [DOI] [PubMed] [Google Scholar]
- (52).Prier CK; Rankic DA; MacMillan DW Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Chan AY; Perry IB; Bissonnette NB; Buksh BF; Edwards GA; Frye LI; Garry OL; Lavagnino MN; Li BX; Liang Y; Mao E; Millet A; Oakley JV; Reed NL; Sakai HA; Seath CP; MacMillan DWC Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis. Chem. Rev 2022, 122, 1485–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).DiRocco DA; Dykstra K; Krska S; Vachal P; Conway DV; Tudge M Late-Stage Functionalization of Biologically Active Heterocycles Through Photoredox Catalysis. Angew. Chem., Int. Ed 2014, 53, 4802–4806. [DOI] [PubMed] [Google Scholar]
- (55).Wang JH; Li GX; He G; Chen G Photoredox-Mediated Minisci Alkylation of N-Heteroarenes using Carboxylic Acids and Hypervalent Iodine. Asian J. Org. Chem 2018, 7, 1307–1310. [Google Scholar]
- (56).Genovino J; Lian Y; Zhang Y; Hope TO; Juneau A; Gagne Y; Ingle G; Frenette M Metal-Free-Visible Light C-H Alkylation of Heteroaromatics via Hypervalent Iodine-Promoted Decarboxylation. Org. Lett 2018, 20, 3229–3232. [DOI] [PubMed] [Google Scholar]
- (57).Sherwood TC; Li N; Yazdani AN; Dhar TGM Organocatalyzed, Visible-Light Photoredox-Mediated, One-Pot Minisci Reaction Using Carboxylic Acids via N-(Acyloxy)phthalimides. J. Org. Chem 2018, 83, 3000–3012. [DOI] [PubMed] [Google Scholar]
- (58).Garza-Sanchez RA; Tlahuext-Aca A; Tavakoli G; Glorius F Visible Light-Mediated Direct Decarboxylative C-H Functionalization of Heteroarenes. ACS Catal. 2017, 7, 4057–4061. [Google Scholar]
- (59).Wang ZZ; Ji XC; Zhao JW; Huang HW Visible-Light-Mediated Photoredox Decarbonylative Minisci-Type Alkylation with Aldehydes Under Ambient Air Conditions. Green Chem. 2019, 21, 5512–5516. [Google Scholar]
- (60).Matsui JK; Primer DN; Molander GA Metal-Free C-H Alkylation of Heteroarenes with Alkyltrifluoroborates: A General Protocol for 1°, 2° and 3° Alkylation. Chem. Sci 2017, 8, 3512–3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Li GX; Morales-Rivera CA; Wang Y; Gao F; He G; Liu P; Chen G Photoredox-Mediated Minisci C-H Alkylation of N-Heteroarenes Using Boronic Acids and Hypervalent Iodine. Chem. Sci 2016, 7, 6407–6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Yan H; Hou ZW; Xu HC Photoelectrochemical C-H Alkylation of Heteroarenes with Organotrifluoroborates. Angew. Chem., Int. Ed 2019, 58, 4592–4595. [DOI] [PubMed] [Google Scholar]
- (63).Zhang P; Le CC; MacMillan DW Silyl Radical Activation of Alkyl Halides in Metallaphotoredox Catalysis: A Unique Pathway for Cross-Electrophile Coupling. J. Am. Chem. Soc 2016, 138, 8084–8087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Dong J; Lyu X; Wang Z; Wang X; Song H; Liu Y; Wang Q Visible-Light-Mediated Minisci C-H Alkylation of Heteroarenes with Unactivated Alkyl Halides Using O2 as an Oxidant. Chem. Sci 2019, 10, 976–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Perkins JJ; Schubert JW; Streckfuss EC; Balsells J; ElMarrouni A Photoredox Catalysis for Silyl-Mediated C-H Alkylation of Heterocycles with Non-Activated Alkyl Bromides. Eur. J. Org. Chem 2020, 2020, 1515–1522. [Google Scholar]
- (66).Nuhant P; Oderinde MS; Genovino J; Juneau A; Gagne Y; Allais C; Chinigo GM; Choi C; Sach NW; Bernier L; Fobian YM; Bundesmann MW; Khunte B; Frenette M; Fadeyi OO Visible-Light-Initiated Manganese Catalysis for C-H Alkylation of Heteroarenes: Applications and Mechanistic Studies. Angew. Chem., Int. Ed 2017, 56, 15309–15313. [DOI] [PubMed] [Google Scholar]
- (67).Dunet V; Rossier C; Buck A; Stupp R; Prior JO Performance of F18-Fluoro-Ethyl-Tyrosine (F18-FET) PET for the Differential Diagnosis of Primary Brain Tumor: A Systematic Review and Metaanalysis. J. Nucl. Med 2012, 53, 207–214. [DOI] [PubMed] [Google Scholar]
- (68).Trump L; Lemos A; Lallemand B; Pasau P; Mercier J; Lemaire C; Luxen A; Genicot C Late-Stage F18-Difluoromethyl Labeling of N-Heteroaromatics with High Molar Activity for PET Imaging. Angew. Chem., Int. Ed 2019, 58, 13149–13154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Huang Y; Lei YY; Zhao L; Gu J; Yao Q; Wang Z; Li XF; Zhang X; He CY Catalyst-Free and Visible Light Promoted Trifluoromethylation and Perfluoroalkylation of Uracils and Cytosines. Chem. Commun 2018, 54, 13662–13665. [DOI] [PubMed] [Google Scholar]
- (70).Wu X; Zhang H; Tang N; Wu Z; Wang D; Ji M; Xu Y; Wang M; Zhu C Metal-Free Alcohol-Directed Regioselective Heteroarylation of Remote Unactivated C(sp3)-H Bonds. Nat. Commun 2018, 9, 3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Zhao H; Jin J Visible Light-Promoted Aliphatic C-H Arylation Using Selectfluor as a Hydrogen Atom Transfer Reagent. Org. Lett 2019, 21, 6179–6184. [DOI] [PubMed] [Google Scholar]
- (72).Jouffroy M; Kong J Direct C-H Carbamoylation of Nitrogen-Containing Heterocycles. Chem.—Eur. J 2019, 25, 2217–2221. [DOI] [PubMed] [Google Scholar]
- (73).Dong J; Xia Q; Lv X; Yan C; Song H; Liu Y; Wang Q Photoredox-Mediated Direct Cross-Dehydrogenative Coupling of Heteroarenes and Amines. Org. Lett 2018, 20, 5661–5665. [DOI] [PubMed] [Google Scholar]
- (74).Huff CA; Cohen RD; Dykstra KD; Streckfuss E; DiRocco DA; Krska SW Photoredox-Catalyzed Hydroxymethylation of Heteroaromatic Bases. J. Org. Chem 2016, 81, 6980–6987. [DOI] [PubMed] [Google Scholar]
- (75).Jin J; MacMillan DWC Alcohols as Alkylating Agents in Heteroarene C-H Functionalization. Nature 2015, 525, 87–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Bieszczad B; Perego LA; Melchiorre P Photochemical C-H Hydroxyalkylation of Quinolines and Isoquinolines. Angew. Chem., Int. Ed 2019, 58, 16878–16883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Cheng WM; Shang R; Fu Y Photoredox/Bronsted Acid Co-Catalysis Enabling Decarboxylative Coupling of Amino Acid and Peptide Redox-Active Esters with N-Heteroarenes. ACS Catal. 2017, 7, 907–911. [Google Scholar]
- (78).Proctor RSJ; Davis HJ; Phipps RJ Catalytic Enantioselective Minisci-Type Addition to Heteroarenes. Science 2018, 360, 419–422. [DOI] [PubMed] [Google Scholar]
- (79).Proctor RSJ; Chuentragool P; Colgan AC; Phipps RJ Hydrogen Atom Transfer-Driven Enantioselective Minisci Reaction of Amides. J. Am. Chem. Soc 2021, 143, 4928–4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Kim M; Koo Y; Hong S N-Functionalized Pyridinium Salts: A New Chapter for Site-Selective Pyridine C-H Functionalization via Radical-Based Processes under Visible Light Irradiation. Acc. Chem. Res 2022, 55, 3043–3056. [DOI] [PubMed] [Google Scholar]
- (81).Shin S; Lee S; Choi W; Kim N; Hong S Visible-Light-Induced 1,3-Aminopyridylation of [1.1.1]Propellane with N-Aminopyridinium Salts. Angew. Chem., Int. Ed 2021, 60, 7873–7879. [DOI] [PubMed] [Google Scholar]
- (82).Moon Y; Lee W; Hong S Visible-Light-Enabled ortho-Selective Aminopyridylation of Alkenes with N-Aminopyridinium Ylides. J. Am. Chem. Soc 2020, 142, 12420–12429. [DOI] [PubMed] [Google Scholar]
- (83).Vellakkaran M; Kim T; Hong S Visible-Light-Induced C4-Selective Functionalization of Pyridinium Salts with Cyclopropanols. Angew. Chem., Int. Ed 2022, 61, No. e202113658. [DOI] [PubMed] [Google Scholar]
- (84).Jung S; Lee H; Moon Y; Jung HY; Hong S Site-Selective C-H Acylation of Pyridinium Derivatives by Photoredox Catalysis. ACS Catal. 2019, 9, 9891–9896. [Google Scholar]
- (85).Kim I; Park S; Hong S Functionalization of Pyridinium Derivatives with 1,4-Dihydropyridines Enabled by Photoinduced Charge Transfer. Org. Lett 2020, 22, 8730–8734. [DOI] [PubMed] [Google Scholar]
- (86).Jung S; Shin S; Park S; Hong S Visible-Light-Driven C4-Selective Alkylation of Pyridinium Derivatives with Alkyl Bromides. J. Am. Chem. Soc 2020, 142, 11370–11375. [DOI] [PubMed] [Google Scholar]
- (87).Jeon J; He YT; Shin S; Hong S Visible-Light-Induced ortho-Selective Migration on Pyridyl Ring: Trifluoromethylative Pyridylation of Unactivated Alkenes. Angew. Chem., Int. Ed 2020, 59, 281–285. [DOI] [PubMed] [Google Scholar]
- (88).Kim I; Park B; Kang G; Kim J; Jung H; Lee H; Baik MH; Hong S Visible-Light-Induced Pyridylation of Remote C(sp3)-H Bonds by Radical Translocation of N-Alkoxypyridinium Salts. Angew. Chem., Int. Ed 2018, 57, 15517–15522. [DOI] [PubMed] [Google Scholar]
- (89).Kim I; Kang G; Lee K; Park B; Kang D; Jung H; He YT; Baik MH; Hong S Site-Selective Functionalization of Pyridinium Derivatives via Visible-Light-Driven Photocatalysis with Quinolinone. J. Am. Chem. Soc 2019, 141, 9239–9248. [DOI] [PubMed] [Google Scholar]
- (90).Xie DM; Wang YW; Zhang X; Fu ZY; Niu DW Alkyl/Glycosyl Sulfoxides as Radical Precursors and Their Use in the Synthesis of Pyridine Derivatives. Angew. Chem., Int. Ed 2022, 61, No. e202204922. [DOI] [PubMed] [Google Scholar]
- (91).Laha R; Patel TI; Moschitto MJ Desulfinative Alkylation of Heteroarenes via an Electrostatic Electron Donor-Acceptor Complex. Org. Lett 2022, 24, 7394–7399. [DOI] [PubMed] [Google Scholar]
- (92).Kim JH; Constantin T; Simonetti M; Llaveria J; Sheikh NS; Leonori D A Radical Approach for the Selective C-H Borylation of Azines. Nature 2021, 595, 677–683. [DOI] [PubMed] [Google Scholar]
- (93).Marsais F; Queguiner G Review on the Metalation of π-Deficient Heteroaromatic-Compounds: Regioselective ortho-Lithiation of 3-Fluoropyridine: Directing Effects and Application to Synthesis of 2,3-Disubstituted or 3,4-Disubstituted Pyridines. Tetrahedron 1983, 39, 2009–2021. [Google Scholar]
- (94).Queguiner G; Marsais F; Snieckus V; Epsztajn J Directed Metalation of π-Deficient Azaaromatics: Strategies of Functionalization of Pyridines, Quinolines, and Diazines. Adv. Heterocycl. Chem 1991, 52, 187–304. [Google Scholar]
- (95).Schlosser M; Mongin F Pyridine Elaboration Through Organometallic Intermediates: Regiochemical Control and Completeness. Chem. Soc. Rev 2007, 36, 1161–1172. [DOI] [PubMed] [Google Scholar]
- (96).Jaric M; Haag BA; Unsinn A; Karaghiosoff K; Knochel P Highly Selective Metalations of Pyridines and Related Heterocycles Using New Frustrated Lewis Pairs or tmp-Zinc and tmp-Magnesium Bases with BF3.OEt2. Angew. Chem., Int. Ed 2010, 49, 5451–5455. [DOI] [PubMed] [Google Scholar]
- (97).Ni S; Hribersek M; Baddigam SK; Ingner FJL; Orthaber A; Gates PJ; Pilarski LT Mechanochemical Solvent-Free Catalytic C-H Methylation. Angew. Chem., Int. Ed 2021, 60, 6660–6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Li Z; Wang Z; Chekshin N; Qian S; Qiao JX; Cheng PT; Yeung KS; Ewing WR; Yu JQ A Tautomeric Ligand Enables Directed C-H Hydroxylation with Molecular Oxygen. Science 2021, 372, 1452–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Li JF; Pan D; Wang HR; Zhang T; Li Y; Huang G; Ye M Enantioselective C2-H Alkylation of Pyridines with 1,3-Dienes via Ni-Al Bimetallic Catalysis. J. Am. Chem. Soc 2022, 144, 18810–18816. [DOI] [PubMed] [Google Scholar]
- (100).Zhang T; Luan YX; Lam NYS; Li JF; Li Y; Ye M; Yu JQ A Directive Ni Catalyst Overrides Conventional Site Selectivity in Pyridine C-H Alkenylation. Nat. Chem 2021, 13, 1207–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Wang P; Verma P; Xia G; Shi J; Qiao JX; Tao S; Cheng PTW; Poss MA; Farmer ME; Yeung KS; Yu JQ Ligand-Accelerated Non-Directed C-H Functionalization of Arenes. Nature 2017, 551, 489–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Fan Z; Chen X; Tanaka K; Park HS; Lam NYS; Wong JJ; Houk KN; Yu JQ Molecular Editing of Aza-Arene CH Bonds by Distance, Geometry and Chirality. Nature 2022, 610, 87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Shi H; Lu Y; Weng J; Bay KL; Chen X; Tanaka K; Verma P; Houk KN; Yu JQ Differentiation and Functionalization of Remote C-H Bonds in Adjacent Positions. Nat. Chem 2020, 12, 399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Larsen MA; Hartwig JF Iridium-Catalyzed C-H Borylation of Heteroarenes: Scope, Regioselectivity, Application to Late-Stage Functionalization, and Mechanism. J. Am. Chem. Soc 2014, 136, 4287–4299. [DOI] [PubMed] [Google Scholar]
- (105).Murphy JM; Liao X; Hartwig JF Meta Halogenation of 1,3-Disubstituted Arenes via Iridium-Catalyzed Arene Borylation. J. Am. Chem. Soc 2007, 129, 15434–15435. [DOI] [PubMed] [Google Scholar]
- (106).He ZT; Li HQ; Haydl AM; Whiteker GT; Hartwig JF Trimethylphosphate as a Methylating Agent for Cross Coupling: A Slow-Release Mechanism for the Methylation of Arylboronic Esters. J. Am. Chem. Soc 2018, 140, 17197–17202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Fischer DF; Sarpong R Total Synthesis of (+)-Complanadine A Using an Iridium-Catalyzed Pyridine C-H Functionalization. J. Am. Chem. Soc 2010, 132, 5926–5927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Newton JN; Fischer DF; Sarpong R Synthetic Studies on Pseudo-Dimeric Lycopodium Alkaloids: Total Synthesis of Complanadine B. Angew. Chem., Int. Ed 2013, 52, 1726–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Cheng C; Hartwig JF Iridium-Catalyzed Silylation of Aryl C-H Bonds. J. Am. Chem. Soc 2015, 137, 592–595. [DOI] [PubMed] [Google Scholar]
- (110).Campeau LC; Rousseaux S; Fagnou K A Solution to the 2-Pyridyl Organometallic Cross-Coupling Problem: Regioselective Catalytic Direct Arylation of Pyridine N-Oxides. J. Am. Chem. Soc 2005, 127, 18020–18021. [DOI] [PubMed] [Google Scholar]
- (111).Tan Y; Barrios-Landeros F; Hartwig JF Mechanistic Studies on Direct Arylation of Pyridine N-Oxide: Evidence for Cooperative Catalysis Between Two Distinct Palladium Centers. J. Am. Chem. Soc 2012, 134, 3683–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Zhao L; Tsukano C; Kwon E; Takemoto Y; Hirama M Total Syntheses of Complanadines A and B. Angew. Chem., Int. Ed 2013, 52, 1722–1725. [DOI] [PubMed] [Google Scholar]
- (113).Puleo TR; Klaus DR; Bandar JS Nucleophilic C-H Etherification of Heteroarenes Enabled by Base-Catalyzed Halogen Transfer. J. Am. Chem. Soc 2021, 143, 12480–12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Jo W; Baek SY; Hwang C; Heo J; Baik MH; Cho SH ZnMe2-Mediated, Direct Alkylation of Electron-Deficient N-Heteroarenes with 1,1-Diborylalkanes: Scope and Mechanism. J. Am. Chem. Soc 2020, 142, 13235–13245. [DOI] [PubMed] [Google Scholar]
- (115).Nagase M; Kuninobu Y; Kanai M 4-Position-Selective C-H Perfluoroalkylation and Perfluoroarylation of Six-Membered Heteroaromatic Compounds. J. Am. Chem. Soc 2016, 138, 6103–6106. [DOI] [PubMed] [Google Scholar]
- (116).Elbert BL; Farley AJM; Gorman TW; Johnson TC; Genicot C; Lallemand B; Pasau P; Flasz J; Castro JL; MacCoss M; Paton RS; Schofield CJ; Smith MD; Willis MC; Dixon DJ C-H Cyanation of 6-Ring N-Containing Heteroaromatics. Chem.Eur. J 2017, 23, 14733–14737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Friedrich M; Schulz L; Hofman K; Zangl R; Morgner N; Shaaban S; Manolikakes G Direct C-H-Sulfonylation of 6-Membered Nitrogen-Heteroaromatics. Tetrahedron Chem. 2022, 1, 100003. [Google Scholar]
- (118).Ham WS; Choi H; Zhang J; Kim D; Chang S C2-Selective, Functional-Group-Divergent Amination of Pyrimidines by Enthalpy-Controlled Nucleophilic Functionalization. J. Am. Chem. Soc 2022, 144, 2885–2892. [DOI] [PubMed] [Google Scholar]
- (119).Hilton MC; Dolewski RD; McNally A Selective Functionalization of Pyridines via Heterocyclic Phosphonium Salts. J. Am. Chem. Soc 2016, 138, 13806–13809. [DOI] [PubMed] [Google Scholar]
- (120).Dolewski RD; Fricke PJ; McNally A Site-Selective Switching Strategies to Functionalize Polyazines. J. Am. Chem. Soc 2018, 140, 8020–8026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Dolewski RD; Hilton MC; McNally A 4-Selective Pyridine Functionalization Reactions via Heterocyclic Phosphonium Salts. Synlett 2018, 29, 8–14. [Google Scholar]
- (122).Anderson RG; Jett BM; McNally A Selective Formation of Heteroaryl Thioethers via a Phosphonium Ion Coupling Reaction. Tetrahedron 2018, 74, 3129–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Patel C; Mohnike M; Hilton MC; McNally A A Strategy to Aminate Pyridines, Diazines, and Pharmaceuticals via Heterocyclic Phosphonium Salts. Org. Lett 2018, 20, 2607–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Anderson RG; Jett BM; McNally A A Unified Approach to Couple Aromatic Heteronucleophiles to Azines and Pharmaceuticals. Angew. Chem., Int. Ed 2018, 57, 12514–12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Hilton MC; Zhang X; Boyle BT; Alegre-Requena JV; Paton RS; McNally A Heterobiaryl synthesis by Contractive C-C Coupling via P(V) Intermediates. Science 2018, 362, 799–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Zhang X; Nottingham KG; Patel C; Alegre-Requena JV; Levy JN; Paton RS; McNally A Phosphorus-Mediated sp2-sp3 Couplings for C-H Fluoroalkylation of Azines. Nature 2021, 594, 217–222. [DOI] [PubMed] [Google Scholar]
- (127).Anders E; Markus F New Methods for Regiospecific Substitution of Slow-Reacting N-Heteroaromatic Ring-Systems. Tetrahedron Lett. 1987, 28, 2675–2676. [Google Scholar]
- (128).Hilton MC; Dolewski RD; McNally A Selective Functionalization of Pyridines via Heterocyclic Phosphonium Salts. J. Am. Chem. Soc 2016, 138, 13806–13809. [DOI] [PubMed] [Google Scholar]
- (129).Levy JN; Alegre-Requena JV; Liu RR; Paton RS; McNally A Selective Halogenation of Pyridines Using Designed Phosphine Reagents. J. Am. Chem. Soc 2020, 142, 11295–11305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Zhang X; McNally A Phosphonium Salts as Pseudohalides: Regioselective Nickel-Catalyzed Cross-Coupling of Complex Pyridines and Diazines. Angew. Chem., Int. Ed 2017, 56, 9833–9836. [DOI] [PubMed] [Google Scholar]
- (131).Zhang X; McNally A Cobalt-Catalyzed Alkylation of Drug-Like Molecules and Pharmaceuticals Using Heterocyclic Phosphonium Salts. ACS Catal. 2019, 9, 4862–4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Che YY; Yue Y; Lin LZ; Pei B; Deng X; Feng C Palladium-Catalyzed Electrophilic Functionalization of Pyridine Derivatives through Phosphonium Salts. Angew. Chem., Int. Ed 2020, 59, 16414–16419. [DOI] [PubMed] [Google Scholar]
- (133).Koniarczyk JL; Greenwood JW; Alegre-Requena JV; Paton RS; McNally A A Pyridine-Pyridine Cross-Coupling Reaction via Dearomatized Radical Intermediates. Angew. Chem., Int. Ed 2019, 58, 14882–14886. [DOI] [PubMed] [Google Scholar]
- (134).Greenwood JW; Boyle BT; McNally A Pyridylphosphonium Salts as Alternatives to Cyanopyridines in Radical-Radical Coupling Reactions. Chem. Sci 2021, 12, 10538–10543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Fricke PJ; Dolewski RD; McNally A Four-Selective Pyridine Alkylation via Wittig Olefination of Dearomatized Pyridylphosphonium Ylides. Angew. Chem., Int. Ed 2021, 60, 21283–21288. [DOI] [PubMed] [Google Scholar]
- (136).Deguest G; Bischoff L; Fruit C; Marsais F Anionic, in Situ Generation of Formaldehyde: A Very Useful and Versatile Tool in Synthesis. Org. Lett 2007, 9, 1165–1167. [DOI] [PubMed] [Google Scholar]
- (137).Fier PS; Hartwig JF Selective C-H Fluorination of Pyridines and Diazines Inspired by a Classic Amination Reaction. Science 2013, 342, 956–960. [DOI] [PubMed] [Google Scholar]
- (138).Fier PS; Hartwig JF Synthesis and Late-Stage Functionalization of Complex Molecules Through C-H Fluorination and Nucleophilic Aromatic Substitution. J. Am. Chem. Soc 2014, 136, 10139–10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Deng Z; Zhao M; Wang F; Tang P Selective C-H Trifluoromethoxylation of (Hetero)Arenes as Limiting Reagent. Nat. Commun 2020, 11, 2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Fier PS A Bifunctional Reagent Designed for the Mild, Nucleophilic Functionalization of Pyridines. J. Am. Chem. Soc 2017, 139, 9499–9502. [DOI] [PubMed] [Google Scholar]
- (141).Fier PS; Kim S; Cohen RD A Multifunctional Reagent Designed for the Site-Selective Amination of Pyridines. J. Am. Chem. Soc 2020, 142, 8614–8618. [DOI] [PubMed] [Google Scholar]
- (142).Gu YT; Shen YY; Zarate C; Martin R A Mild and Direct Site-Selective sp2 C-H Silylation of (Poly)Azines. J. Am. Chem. Soc 2019, 141, 127–132. [DOI] [PubMed] [Google Scholar]
- (143).Obradors C; List B Azine Activation via Silylium Catalysis. J. Am. Chem. Soc 2021, 143, 6817–6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Liu Z; He JH; Zhang M; Shi ZJ; Tang H; Zhou XY; Tian JJ; Wang XC Borane-Catalyzed C3-Alkylation of Pyridines with Imines, Aldehydes, or Ketones as Electrophiles. J. Am. Chem. Soc 2022, 144, 4810–4818. [DOI] [PubMed] [Google Scholar]
- (145).Zhou XY; Zhang M; Liu Z; He JH; Wang XC C3-Selective Trifluoromethylthiolation and Difluoromethylthiolation of Pyridines and Pyridine Drugs via Dihydropyridine Intermediates. J. Am. Chem. Soc 2022, 144, 14463–14470. [DOI] [PubMed] [Google Scholar]
- (146).Muta R; Torigoe T; Kuninobu Y 3-Position-Selective C-H Trifluoromethylation of Pyridine Rings Based on Nucleophilic Activation. Org. Lett 2022, 24, 8218–8222. [DOI] [PubMed] [Google Scholar]
- (147).Boyle BT; Levy JN; de Lescure L; Paton RS; McNally A Halogenation of the 3-Position of Pyridines Through Zincke Imine Intermediates. Science 2022, 378, 773–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Cao H; Cheng Q; Studer A Radical and ionic meta-C-H Functionalization of Pyridines, Quinolines, and Isoquinolines. Science 2022, 378, 779–785. [DOI] [PubMed] [Google Scholar]
- (149).Kutchukian PS; Dropinski JF; Dykstra KD; Li B; DiRocco DA; Streckfuss EC; Campeau LC; Cernak T; Vachal P; Davies IW; Krska SW; Dreher SD Chemistry Informer Libraries: A Chemoinformatics Enabled Approach to Evaluate and Advance Synthetic Methods. Chem. Sci 2016, 7, 2604–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]