

Abstract
Significance Statement
Several recent studies identified mitochondrial mutations in patients with Gitelman or Fanconi syndrome. Mitochondrial cytopathies are generally not considered in the diagnostic workup of patients with electrolyte disorders. In this systematic review, we investigated the presence of electrolyte disorders in patients with mitochondrial cytopathies to determine the relevance of mitochondrial mutation screening in this population. Our analysis demonstrates that electrolyte disorders are commonly reported in mitochondrial cytopathies, often as presenting symptoms. Consequently, more clinical attention should be raised for mitochondrial disease as cause for disturbances in electrolyte homeostasis. Further prospective cohort studies are required to determine the exact prevalence of electrolyte disorders in mitochondrial cytopathies.
Background
Electrolyte reabsorption in the kidney has a high energy demand. Proximal and distal tubular epithelial cells have a high mitochondrial density for energy release. Recently, electrolyte disorders have been reported as the primary presentation of some mitochondrial cytopathies. However, the prevalence and the pathophysiology of electrolyte disturbances in mitochondrial disease are unknown. Therefore, we systematically investigated electrolyte disorders in patients with mitochondrial cytopathies.
Methods
We searched PubMed, Embase, and Google Scholar for articles on genetically confirmed mitochondrial disease in patients for whom at least one electrolyte is reported. Patients with a known second genetic anomaly were excluded. We evaluated 214 case series and reports (362 patients) as well as nine observational studies. Joanna Briggs Institute criteria were used to evaluate the quality of included studies.
Results
Of 362 reported patients, 289 had an electrolyte disorder, with it being the presenting or main symptom in 38 patients. The average number of different electrolyte abnormalities per patient ranged from 2.4 to 1.0, depending on genotype. Patients with mitochondrial DNA structural variants seemed most affected. Reported pathophysiologic mechanisms included renal tubulopathies and hormonal, gastrointestinal, and iatrogenic causes.
Conclusions
Mitochondrial diseases should be considered in the evaluation of unexplained electrolyte disorders. Furthermore, clinicians should be aware of electrolyte abnormalities in patients with mitochondrial disease.
Keywords: calcium, electrolytes, Gitelman syndrome, hypernatremia, hyperparathyroidism, hypokalemia, hyponatremia, mitochondria
Introduction
Electrolyte disorders are common and can be caused by variability of causes, including hereditary and acquired gastrointestinal disorders, endocrine disturbances, and renal defects. The kidney plays a central role in the homeostasis of the electrolyte and water balance, by tight regulation of these reabsorption processes. As a consequence, the kidneys have a high energy demand to fuel the active reabsorption of electrolytes along the nephron. Defects in energy production, e.g., by primary defects in mitochondrial oxidative phosphorylation, may therefore result in reduced electrolyte reabsorption.
Mitochondrial cytopathies can be caused by single nucleotide variants (SNVs) in the mitochondrial genome (mitochondrial DNA [mtDNA], affecting one of the 37 genes mtDNA genes), structural variants (SVs) in mtDNA (often deletions of 3–9 kilobases), or variants in the nuclear genome (nuclear DNA [nDNA], encoding the other ±1300 genes of the mitoproteome).1 Moreover, in mitochondrial defects affecting mtDNA, the percentage of mtDNA molecules with the pathogenic variant can differ significantly from cell to cell (heteroplasmy).2 Consequently, the presentation of patients with a mitochondrial disease can be highly variable, ranging from single-organ to multiorgan disease, from mild to severe and from prenatal-onset to adult-onset.2
In recent years, electrolyte disturbances have been described as primary presentation of patients with mitochondrial diseases.3,4 For instance, we recently identified specific mtDNA variants in 13 families with a Gitelman-like phenotype comprising hypomagnesemia, hypokalemia, and metabolic alkalosis.4 Similarly, a 7-year-old girl presenting with Bartter-like tubulopathy was diagnosed with a large mtDNA deletion.5 Nevertheless, mitochondrial diseases are generally not considered in the differential diagnosis of Gitelman syndrome, Bartter syndrome, or other electrolyte disorders. However, the relevance mitochondrial mutation testing in hereditary electrolyte disorders has never been systematically investigated.
The aim of this article was to establish which electrolyte disorders occur in primary mitochondrial diseases. To achieve this, we systematically reviewed the literature and performed data synthesis on the 228 articles that met our inclusion criteria. We investigated which electrolyte disorders are most frequently reported, in which combinations they occur and which pathophysiologic mechanisms have been reported.
Methods
The review protocol was submitted to the International prospective register of systematic reviews (PROSPERO, Accession No. CRD42021283468).
Data Sources and Searches
MEDLINE and Embase online were systematically searched for case reports, case series, cross-sectional studies, and cohort studies that studied electrolytes in genetically confirmed mitochondrial disease. We complemented findings by a systematic literature search using Google Scholar because this search engine is the only large database that can perform complementary full-text searches. The exact formulation of the search terms can be found in the Supplemental Methods. None of the searches was restricted by date, language, study design, or otherwise. In addition, we screened all reviews that were identified through our searches to identify additional articles in the reference list. The last search in Google Scholar was performed on July 16, 2021, and the last search in MEDLINE and Embase was performed on November 4, 2022.
Study Selection
The following inclusion criteria were used: (1) the study reports at least one serum electrolyte (potassium, sodium, magnesium, and/or potassium) in at least one patient, regardless of how the electrolyte measurement is reported (i.e., qualitative or qualitative) and regardless of the result (normal or abnormal) and (2) the patient in whom the serum electrolyte is reported has a genetically confirmed mitochondrial disease. Cohort studies and cross-sectional studies were also included, provided that they described data on at least one serum electrolyte for a genetically characterized cohort of patients with primary mitochondrial disease.
Primary mitochondrial disease was defined as a genetic disease that directly perturbs the function of a protein expressed in the mitochondrion or of a protein that is directly involved in mitochondrial function. Both mtDNA-encoded and nDNA-encoded genetic causes were accepted. We re-evaluated the diagnostic accuracy of each variant and each case according to current clinical genetic guidelines (Supplemental Methods).6,7 No restrictions were made on demographic qualities of the studied individuals (e.g., age and sex). Cases that had already been described in previous articles were included, provided that both articles contained unique clinical information on the case. Moreover, if included articles described a patient who was also described in another article, we also included/retrieved the additional article, provided that this article added additional clinical information, even if the article itself did not meet the inclusion criteria (e.g., genetic defect only described in one article, while the other article provides the electrolyte information). Unpublished manuscripts were eligible when identified through our search strategy.
The following exclusion criteria were used: (1) the study only reports on individuals in whom the cause of mitochondrial disease is not genetic or not genetically confirmed; (2) for cohort studies that do not provide individual-level patient data, the study population is not made up of patients with a primary mitochondrial disease; and (3) the cause of the mitochondrial disease is a large nDNA deletion involving multiple genes, or the case has a second genetic diagnosis due to a defect in a gene that is not primarily involved in mitochondrial function. We also excluded reviews, books, theses, and conference abstracts.
For the selection process, all references were uploaded to Sysrev. Two reviewers independently screened the title and abstract of each article to determine eligibility for inclusion. Any disagreement was discussed between the two reviewers, and in case of doubt, articles were included at this stage. In a second round, full texts were studied. Any disagreements in this round were resolved in consultation with a third reviewer.
Data Extraction and Quality Assessment
In short, data were extracted by one reviewer and curated by a second reviewer (software and sources used for genetic analyses are listed in Supplemental Table 1). The reviewers collected data on a prespecified set of parameters. In case of doubt on the accuracy of a genetic diagnosis, the reviewers consulted an independent laboratory geneticist. Cases that did not meet criteria for a genetic diagnosis were excluded. The data represent that the interpretation by authors of whether the serum/urinary concentration of an electrolyte, hormone, or metabolite was too low or too high. We contacted authors to provide missing data when the genetic diagnosis was unclear or when only qualitative information was given on (a) serum electrolyte(s).
To assess the risk of bias, we used the critical appraisal tools developed by the Joanna Briggs Institute (JBI).8 We followed the descriptions provided by JBI to classify the study type. Thus, case series were defined as “studies where only patients with a certain disease or disease-related outcome are sampled.” Consequently, an article was classified as a collection of case reports when sampling was based on an exposure (i.e., genetic variant).
We decided before scoring that all critical appraisal questions would be scored with a focus on the risk of bias regarding the interpretation of the electrolyte disturbances and not the risk of bias of the conclusion of the article (Supplemental Methods). Two reviewers scored the first ten cases independently to standardize scoring. One reviewer evaluated the risk of bias of all subsequent articles.
Data Synthesis and Analysis
Cohort studies and cross-sectional studies were summarized narratively. Patient-level data from case reports and case series were pooled and evaluated together. The number of patients, mean, and proportion of total were calculated for several demographic, phenotypic, and genotypic parameters.
For plotting graphs and other statistical analyses, Prism 9 for Mac was used (Graphpad, version 9.4.1). We calculated correlations between serum electrolyte concentrations (potassium–magnesium, potassium–sodium, magnesium–calcium, calcium–phosphate) using Pearson correlations assuming a Gaussian distribution of electrolyte values. A correlation line was plotted using standard simple linear regression on the basis of the data of all patients combined disregarding genotype class. Statistical significance was defined as P < 0.05 unless specified otherwise.
Role of the Funding Source
External funders were not involved in the conception of the project, data collection, analyses, writing, or the decision to publish.
Results
Two Hundred and Eighty-Eight Studies Report Electrolyte Data in Primary Mitochondrial Disease
The search strategy resulted in 4555 records after deduplication (Figure 1 and Supplemental Figure 1). Screening of title and abstract resulted in exclusion of 3766 articles that did not describe patients with mitochondrial disease. Of the remaining 789 full-text articles, 228 articles met the inclusion criteria. Of these articles, 113 were found with Google Scholar only. Common reasons for exclusion at the last stage were that no serum electrolyte data were reported in the article or that the mitochondrial disease was not genetic or not genetically confirmed. An example of an article that might seem to meet inclusion criteria are the 21 patients described by Martin-Hernandez et al., in all of whom urinary electrolytes were reported.10 However, ten patients did not have a genetic diagnosis, two patients had a chromosomal deletion involving multiple genes (thus more than one genetic defect), and of the remaining patients, serum electrolyte values were only reported in one individual. Thus, only this individual was included in the study.
Figure 1.

PRISMA flowchart. Number of articles identified, screened, assessed for eligibility, and included reported in a flow diagram as directed by the PRISMA guidelines.9 PheWAS, phenome-wide association study.
Cohort and Cross-Sectional Studies: One Study with Low Risk of Bias
Among the included articles are six retrospective cohort studies, two cross-sectional studies, and one phenome-wide association study (Supplemental Table 2). All except one were judged to have a high or very high risk of bias regarding conclusions on electrolyte disorders in mitochondrial disease. The remaining study, written by Wilson et al.,11 had a low risk of bias for establishing a link between one specific mtDNA variant (m.4291T>C) and specific electrolyte disorders. This study describes a large family (142 individuals), among whom 67 carriers of the m.4291T>C variant. Many carriers have hypomagnesemia, hypokalemia, renal magnesium wasting, relative hypocalciuria, hypercholesterolemia, and hypertension. Statistical analyses show significant differences in carriers compared with noncarriers.
Case Reports and Case Series
In addition, two hundred nineteen articles that report on one or more cases were included, together describing 362 patients (Table 1, Supplemental Table 3). The age range of included cases was from birth to 76 years, with 61% of patients being younger than 18 years. In total, 80% of patients had at least one electrolyte abnormality. The electrolyte disorder was the presenting symptom, or main symptom, in 38 patients (15 articles).4,12–25 In some of these cases, the initial evaluation included diagnostic testing for a genetic cause of the electrolyte disorder; a mitochondrial disease was considered only after the development of additional symptoms. Figure 2 shows the reported electrolyte values of all included case reports and case series.
Table 1.
Main demographic, genotypic, and phenotypic characteristics of included patients
| Demographic, genotypic and phenotypic characteristics | All patients (N=362) |
|---|---|
| Age in yra, mean (SD) | 18 (20) |
| Sex (%) | |
| Male | 156 (43) |
| Female | 199 (55) |
| Not mentioned | 7 (2) |
| Children, i.e., <18 yr (%) | 220 (61) |
| Genotypic | |
| No. of patients (%) | |
| mtDNA SVs | 114 (32) |
| mtDNA SNVs | 138 (38) |
| nDNA variants | 110 (31) |
| Phenotypic | |
| No. of patients with and without electrolyte abnormalities (% of total) | |
| None | 73 (20) |
| One or more | 289 (80) |
| No. of patients in which electrolyte was reported (% of total) | |
| Magnesium | 110 (30) |
| Hypomagnesemia | 70 (64) |
| Hypermagnesemia | 4 (4) |
| Calcium | 187 (52) |
| Hypocalcemia | 90 (48) |
| Hypercalcemia | 14 (8) |
| Potassium | 212 (59) |
| Hypokalemia | 87 (41) |
| Hyperkalemia | 57 (27) |
| Sodium | 168 (46) |
| Hyponatremia | 86 (51) |
| Hypernatremia | 10 (6) |
| Phosphate | 134 (37) |
| Hypophosphatemia | 39 (29) |
| Hyperphosphatemia | 33 (25) |
| Bicarbonate | 169 (47) |
| Decreased bicarbonate | 109 (64) |
| Elevated bicarbonate | 14 (8) |
Main characteristics of included patients from case reports and case series. mtDNA, mitochondrial DNA; SVs, structural variants; SNVs, single-nucleotide variants; nDNA, nuclear DNA.
Age at first measurement (if known) or at presentation (if age at measurement not clear).
Figure 2.

Serum electrolyte values per patient. (A–F) Serum electrolyte values as reported in included patients from case reports and case series. If multiple electrolyte values were reported, the most extreme value was plotted. If only qualitative data were available, or when values were unrealistic, no point was plotted. Dotted lines represent the upper and lower limit of normal adults. Of note, phosphate reference ranges (E) are provided for adults but are strongly age-dependent; therefore, the plotted reference range will not apply to all data points. For instance, the normal range for neonates 0–5 days of age is 1.55–2.65 mmol/L,26 and all ages have been included in this graph. mtDNA, mitochondrial DNA; nDNA, nuclear DNA; SNVs, single nucleotide variants; SVs, structural variants.
Calcium
Hypocalcemia (as defined by the reporting authors) was the most commonly reported electrolyte abnormality: 90 patients had hypocalcemia. Figure 2 also shows that calcium levels were lowest in patients with a mtDNA SV (mean: 1.86 mmol/L), which is on average 0.42 mmol/L lower than in patients with a mtDNA SNV and 0.34 mmol/L lower than in patients with a nDNA variant. Episodic calcium disturbances were described in six cases and associated with rhabdomyolysis in two cases.
Potassium
Hypokalemia was detected in 87 patients. In 13 hypokalemic patients, urinary potassium was measured and reported. In all but one case, the values pointed toward renal potassium wasting. Hyperkalemia was commonly reported too (57 patients), in contrast to the other electrolytes. Hyperkalemia was most common among patients with nDNA variants and least common among patients with mtDNA SVs. In two cases, the hyperkalemia was described to be episodic, either spontaneous or in association with heart failure aggravation.27
Sodium
Hyponatremia was reported in 86 cases, evenly distributed among genotype groups. Episodic hyponatremia was described in eight cases, and it occurred spontaneously or in association with stroke-like episodes or heart failure.28
Magnesium
Hypomagnesemia was present in 70 reported cases. In 26 of 31 cases, high urinary magnesium levels pointed toward a renal cause, while in only three cases, the low value pointed toward an extrarenal cause.29 No differences were observed between the different genotype groups. Hypermagnesemia was reported in only four patients.
Combined Electrolyte Disorders
Included patients with mtDNA SV and patients with a nDNA variant had on average a higher number of electrolyte abnormalities compared with patients with mtDNA SNV (more than two abnormalities versus one on average, Figures 3). There was no clear difference in the number of electrolyte abnormalities in male patients versus female patients (Figure 4). mtDNA SVs were relatively prevalent among patients with at least one electrolyte disorder: 35% of all included patients had mtDNA SV versus 12%–20% in the general mitochondrial disease population (Supplemental Table 4).30,31
Figure 3.

Number of electrolyte abnormalities per patient. (A–C) The number of electrolyte abnormalities in included patients from case reports and case series. For classification of an electrolyte value as abnormal, judgment by the reporting author was followed. mtDNA, mitochondrial DNA; nDNA, nuclear DNA; SNVs, single-nucleotide variants; SVs, structural variants.
Figure 4.

Number of electrolyte abnormalities among male and female patients. (A–C) The number of electrolyte abnormalities in included patients from case reports and case series, split by sex. Gray bars represent female patients, and black bars represent male patients. For classification of an electrolyte value as abnormal, judgment by the reporting author was followed. mtDNA, mitochondrial DNA; nDNA, nuclear DNA; SNVs, single-nucleotide variants; SVs, structural variants.
Pearson correlation analysis of the four pairs of electrolytes depicted in Figure 5, A–D reveals that magnesium and potassium values are significantly associated with each other (Pearson r=0.49, P = 0.0002). Phosphate and calcium are significantly associated as well (r=−0.37, P = 0.0010). In line with the negative correlation, low levels of parathyroid hormone (PTH) are observed in a subset of patients despite hypocalcemia (Figure 5E). In line with Figure 5A, a combined deficiency of magnesium and potassium was present in 32 patients (48% of all patients in whom both serum potassium and magnesium were reported, qualitatively or quantitatively). Similarly, hypocalcemia and hyperphosphatemia co-occur in 24 patients (26% of total).
Figure 5.
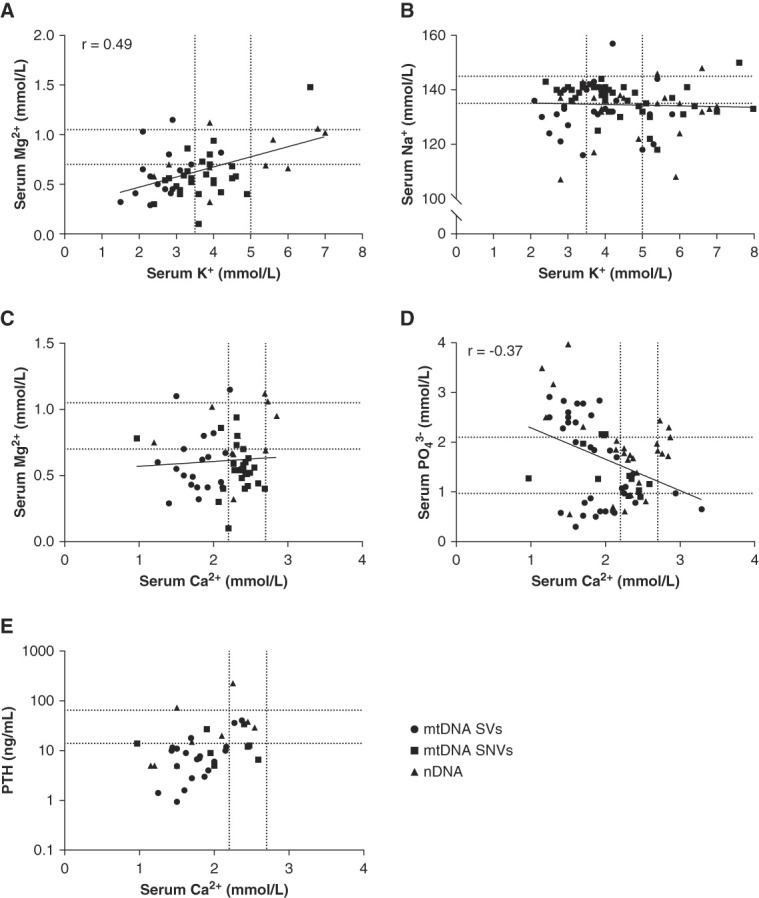
Within-patient correlation between electrolytes. (A–E) Correlation between selected serum electrolytes in the same patient from case reports and case series. If multiple electrolyte values were reported, the most extreme value was plotted. If only qualitative data were available, or when values were unrealistic, no point was plotted. Dotted lines represent the upper and lower limit of normal in adults. Linear correlations were also plotted on the basis of the combined data, and a Pearson correlation coefficient (r) was provided when a statistically significant correlation existed. Phosphate reference ranges (D) are provided for adults but are strongly age-dependent; therefore, the plotted reference range will not apply to all data points. For instance, the normal range for neonates 0–5 days of age is 1.55–2.65 mmol/L,26 and all ages have been included in this graph. PTH values were often reported as below or above a certain measurement limit. In that case, the measurement limit was plotted instead. mtDNA, mitochondrial DNA; nDNA, nuclear DNA; PTH, parathyroid hormone; SNVs, single-nucleotide variants; SVs, structural variants.
Evidence for Specific Pathophysiologic Mechanisms
In addition to investigating the co-occurrence of specific electrolyte abnormalities, we also examined which pathophysiologic mechanisms were reported by the authors to explain their findings (Table 2). Commonly reported mechanisms include hypoparathyroidism causing hypocalcemia with hyperphosphatemia (55 patients),14,24,32–39 proximal tubulopathies causing metabolic acidosis, hyponatremia and/or hyperkalemia (46 patients),10,19,40–45 and tubulopathies affecting distal segments (33 patients and one large family).4,5,11,16,18,20,46–49 In line with the frequent reporting of tubulopathies, 72 patients were identified in whom reported urinary electrolytes provided evidence for renal electrolyte wasting.
Table 2.
Pathophysiology of electrolyte disorders in primary mitochondrial diseases
| Pathophysiologic Mechanism | No. of Patients (% of Total) |
|---|---|
| Hypoparathyroidism | 55 (15) |
| Hyperparathyroidism | 9 (2) |
| Proximal tubulopathy | 46 (13) |
| Bartter/Gitelman-like tubulopathy | 33 (9) |
| Syndrome of inappropriate antidiuretic hormone secretion | 8 (2) |
| Disturbed renin and/or aldosterone | 24 (7) |
| Urinary electrolyte values consistent with renal wasting | 72 (20) |
| Gastrointestinal (e.g., diarrhea or vomiting) | 79 (22) |
Pathophysiologic mechanisms reported in the case reports and case series. Percentages reflect the number of patients in which a mechanism was reported compared with the total number of included patients. Percentages do not take into account in how many patients the item was measured.
Other less frequently reported mechanisms include hyperkalemia and/or hyponatremia due to deficiencies of mineralocorticoids or glucocorticoids or renal resistance to them13,27,28,38,50–62; hyponatremia due to syndrome of inappropriate antidiuretic hormone secretion, cerebral salt wasting, hypovolemia, or heart failure28,63–71; and calcium disturbances due to rhabdomyolysis.24,72,73 Nine studies describe treatment-related adverse events, such as electrolyte disturbances due to L-arginine, succinate, coenzyme Q10 or Calcium beta-hydroxybutyrate, and Sodium beta-hydroxybutyrate.36,39,58,74–79 Finally, several studies produced hypotheses on novel mechanisms, such as hypokalemia due to a nonreabsorbable anion effect of methylmalonate in patients with MMUT variants,80 episodic hypokalemia due to an intracellular potassium shift caused by lactic acid,81 hypocalcemia due to impaired 25-hydroxylation of vitamin D in the liver,82 or hyponatremia caused by dysregulation of sympathetic innervation to the kidney complicated by sodium wasting.71,83
Some symptoms that are common in mitochondrial diseases are also known to associate with electrolyte disorders (Supplemental Table 5).84–90 Gastrointestinal symptoms, such as vomiting and diarrhea, affected at least 79 patients (22% of total). In patients with hearing loss (n=122), potassium levels were slightly higher (4.9 versus 4.1 mmol/L, P = 0.004) and serum calcium levels were slightly lower (2.0 versus 2.2 mmol/L, Supplemental Figure 2). In patients with retinopathy (n=47), calcium levels were lower (1.70 versus 2.16 mmol/L, P < 0.0001), phosphate levels were higher (1.92 versus 1.39 mmol/L, P = 0.014), and bicarbonate levels were lower (20 versus 8 mmol/L, P = 0.002, Supplemental Figure 3). Renal ultrasound was reportedly abnormal in 52 of 77 patients (68%), the most reported abnormality was hyperechogenicity (n=36). Histologic analyses of kidney tissue were reported in 45 patients and showed a large variety in the type, degree, and location of anomalies. Interestingly, electron microscopy of kidney tissue often showed mitochondrial deformities in specific renal cell types.
Quality of Included Studies
Scoring all case reports and case series with the respective JBI checklists revealed that certain aspects are well described, while other features have been understudied. Demographic characteristics are often well described, as well as the age at onset of the electrolyte disorder. However, some other aspects are frequently unrecorded. Examples include the duration of the electrolyte disorder, whether other causes were considered and/or excluded, quantitative values for each electrolyte, relevant reference ranges, and specifics of treatment (Supplemental Table 6).
Discussion
This systematic review provides a comprehensive overview of the literature on electrolyte disorders in primary mitochondrial diseases. We found 219 case reports and case series that provide information on serum electrolytes, together describing 362 patients. Among these, 289 patients had one or more electrolyte disorders. Our analysis demonstrates that the electrolyte disorders are frequently caused by renal tubulopathies. Moreover, the data suggest the presence of genotype–phenotype associations, with a higher number of electrolyte abnormalities in patients with mtDNA SVs.
Deficiencies in calcium, magnesium, potassium, and sodium are not rare in mitochondrial diseases, each was reported in at least 70 patients (>40%). Because reporting and publication bias certainly are a complicating factor in our approach, our dataset does not allow to determine the exact prevalence of these electrolyte disturbances. Nevertheless, this systematic literature review demonstrates that electrolyte disturbances are present in mitochondrial diseases. Interestingly, an electrolyte disorder was the presenting symptom in 38 patients, suggesting that mitochondrial cytopathies should be part of the diagnostic work up of patients presenting with electrolyte disorders.
Several results point toward the presence of genotype–phenotype relationships. Patients with mtDNA SVs had a higher average number of electrolyte abnormalities than patients with mtDNA SNVs (2.4 per patient versus 1.0). Of note, in comparison with the general population of patients with mitochondrial disease, mtDNA SVs were overrepresented in our review (35% versus an expected 12%–20%).30,31 Generally, serum electrolyte levels were lower in mtDNA SV patients, especially calcium and potassium. By contrast, nDNA variants were associated with relatively high potassium and phosphate levels.
We observed a large variation in the group with nDNA variants: Variants in some genes are almost invariably associated with electrolyte disorders, e.g., SARS2 variants,91 whereas electrolyte abnormalities were not reported in patients with pathogenic variants in TK2, PDHA1, or OPA1, despite those genes being a more common cause of mitochondrial disease.30 SARS2 encodes the mitochondrial seryl-tRNA synthetase that loads serine onto its respective tRNA. Interestingly, TARS2 and YARS2 variants have been linked to electrolyte disorders too.92,93 Such findings may generate new hypotheses for molecular mechanisms involved in electrolyte disorders for instance by supporting a recently found link between renal amino acid metabolism and renal electrolyte handling.94,95
Several pathophysiologic mechanisms may explain the development of the electrolyte disorders, comprising renal, gastrointestinal, hormonal, and iatrogenic causes. Primary tubulopathies are rare in the general population96 but were often reported in included studies. Proximal tubulopathies were most common (46 patients), while more distal tubulopathies, including Bartter-like and Gitelman-like tubulopathies, were also reported in 33 patients and one large family. Interestingly, we also found a correlation between serum magnesium and potassium values after data synthesis. This combination is often seen in distal tubulopathies,97 although it might also point to concomitant loss in the gastrointestinal tract97,98 or hypomagnesemia-induced hypokalemia.99
The exact renal cellular mechanisms of how mitochondrial dysfunction can cause electrolyte disorders are currently unknown. An obvious hypothesis is a plain ATP-deficit. The kidney is the most energy-expensive organ in the body together with the heart.100 Most of this energy is used for sodium reabsorption,101 and 95% comes from aerobic ATP production by the mitochondria.102 This ATP is indispensable to power the basolateral Na+/K+-ATPase along the nephron, which is not only necessary for sodium reabsorption itself but also for maintaining an electrochemical gradient to drive reabsorption of other electrolytes.
However, the ATP-deficit hypothesis does not easily explain the observed genotype-phenotype relationships, such as the specific distal tubular defect with variants in MT-TI, MT-TF, or SARS2. It should be realized that genetic heterogeneity and pleiotropy are common phenomena in mitochondrial disorders.4,103–105 The exact mechanisms are incompletely understood across the field, although the exact location of the mtDNA variant and varying heteroplasmy levels in different tissues play a role in many cases.106 However, these cannot explain the genotype–phenotype effect that seems present with certain nDNA or homoplasmic mtDNA variants. A second hypothesis to explain this might be that each genetic mitochondrial defect leads to slightly different metabolic alterations. For example, a deficiency of nitric oxide can lead to either sodium retention or sodium wasting, possibly depending on the specific tubular location and timing (nitric oxide has a short half-life).107–109 Deficiency of nitric oxide is a hallmark of some mitochondrial diseases,110 and it is likely to be present in other mitochondrial diseases as well because of the production of reactive oxygen species.107 Hypothetically, different mitochondrial defects might have different effects on the production of reactive oxygen species and nitric oxide, depending on the cell type and its unique metabolic wiring, and thereby differ in the effect on electrolyte reabsorption.
Given the substantial energy requirement of the kidney, especially for the energy-costly process of sodium reabsorption,101 it seems likely that mitochondrial dysfunction can lead to impaired tubular transport of sodium and other electrolytes.4,111
In addition to renal causes, gastrointestinal symptoms (vomiting, diarrhea, exocrine pancreas insufficiency) were frequently reported. However, their relationship with the electrolyte disturbance was often not described; thus, they might have been a cause, effect, or spurious co-occurrence. Urinary electrolyte values generally pled for a renal cause, but reporting bias might distort conclusions. Hormonal causes encompassed disturbances in aldosterone and PTH signaling. Hypoparathyroidism was commonly reported, which was corroborated by our finding that overall serum calcium concentrations showed an inverse relationship with phosphate. The mechanism underlying hypoparathyroidism remains unclear, although one study reported aplasia of the parathyroid glands on autopsy.112 Furthermore, an in vitro study showed that pharmacologic inhibition of mitochondrial function inhibits PTH production within 10 minutes by 80%–85%.113
The included articles report a large variation in the reporting, type, and efficacy of treatment. A randomized controlled trial to further investigate this will be hardly feasible, given the considerable variation in reported pathophysiologic mechanisms. A mechanism-based choice of treatment, evaluated on a case-by-case basis, would be more reasonable. US Food and Drug Administration–approved pharmacologic therapies for mitochondrial defect are currently not available.114 Interestingly, two articles in our review (three patients) provide evidence for the use of coenzyme Q10 as treatment of hypocalcemia in mitochondrial disease caused by mtDNA deletions.36,39 True n-of-one trials with both blinding and randomization115 or additional mechanistic studies are desirable to confirm this effect.
A strength of our study is the comprehensive search strategy, which allowed for the inclusion of a large number of articles (228) and patients (362, excluding cohort and cross-sectional studies). Among the included patients were a large number of patients with disease-causing nDNA variants, which allows for comparison among mitochondrial diseases caused by different molecular mechanisms. At the same time, inclusion criteria were strict on whether a mitochondrial disease was truly of genetic origin, preventing inclusion of patients with a nonmitochondrial (genetic) disease.
An important limitation of this study is the outcome reporting bias and publication bias of included studies.116,117 Not all electrolytes are routinely measured in patients with mitochondrial disease, and if measured, not all measurements will be reported.118 Moreover, extreme values will be reported more often than normal or subtly abnormal values. Still, the large number of reports, in light of the rarity of mitochondrial diseases, suggests that electrolyte disorders might be a common complication of mitochondrial diseases.2,30,31 As a consequence of this bias, it is impossible to calculate a reliable prevalence or incidence of electrolyte disorders in mitochondrial diseases. Another limitation is that a causal link between the mitochondrial disease and the electrolyte disorder could not be established in all case reports. However, several included studies provide strong clinical and molecular mechanistic evidence for a causal association.4,11,47,48 Especially in older reports, disease-causing nDNA variants that cause multiple DNA deletions might have gone unrecognized, such as variants in POLG1.
In this review, an electrolyte disorder was the presenting symptom in 38 patients, and sometimes, it was also the only symptom. Such findings suggest that a comprehensive analysis for mitochondrial diseases should always be considered when primary genetic testing does not yield a diagnosis. Of note, exome sequencing has an overall molecular diagnostic yield of 35%–70% for mitochondrial diseases, mainly due to incomplete capturing of mtDNA variants.119,120 Therefore, the secondary genetic testing strategy should explicitly aim to capture mtDNA/nDNA variations that could cause a mitochondrial disease.121 A special instance in which first-tier testing for a mitochondrial disease should be considered is in patients with a maternal inheritance pattern or multiorgan involvement. Finally, we propose to incorporate the mtDNA genes MT-TI and MT-TF in the primary genetic testing strategy because patients with variants in these genes usually present without any other organ involvement.4
Our review demonstrates that mitochondrial diseases may contribute to resolving unexplained electrolyte disorders, especially hypocalcemia, hypomagnesemia, hypokalemia, and hyponatremia. Inclusion of electrolyte disorders in the differential diagnosis could reduce the delay in diagnosis that is commonly seen in mitochondrial diseases.122 Our study also shows the urgent need for prospective, mechanistic, and therapeutic studies. Follow-up studies can hopefully address questions on the prevalence of electrolyte disorders in mitochondrial disease and the reasons for the large variation in underlying mechanisms.
Supplementary Material
Acknowledgments
We are very grateful to all authors who provided us with additional clinical and/or genetic information when asked. In this context, we want to express special thanks to Prof. Tomáš Honzík/Dr. Petr Hanák, Dr. Sara Seneca/Dr. Elena Martín Hernández, and Dr. Rebecca Ganetzky as they were especially elaborate in their replies and provided data on multiple patients. Furthermore, we want to express our gratitude to Dr. Richard Rodenburg and especially Dr. Dorien Lugtenberg for their expertise and advice on variant pathogenicity of mitochondrial and nuclear DNA variants.
Footnotes
J.D. and J.H.F.d.B. contributed equally to this work.
Disclosures
R.J.M. Bindels reports Advisory or Leadership Role: REGMED XB Regenerative Medicine Crossing Borders; and Other Interests or Relationships: Scientific Board of the Dutch Kidney Foundation. J.H.F. de Baaij reports Other Interests or Relationships: Research funding from the Dutch Diabetes Research Foundation, the Dutch Kidney Foundation, the Dutch Organization of Scientific Research, and the European Union. J. Deinum reports Speakers Bureau: Health Investment and Prevents. L. Vermeltfoort reports Employer: House of Smart and Jeroen Bosch Ziekenhuis (boyfriend). The remaining author has nothing to disclose.
Funding
J.H.F. de Baaij is financially supported by the Netherlands Organization of Scientific Research (NWO, Vidi 09150172110040) and the European Research Council (ERC STG 101040682). The funding source was not involved in the conception of the project, data collection, analyses, writing, or the decision to publish.
Author Contributions
Conceptualization: Jeroen H.F. de Baaij, Jaap Deinum, Daan H.H.M. Viering.
Data curation: Jaap Deinum, Lars Vermeltfoort, Daan H.H.M. Viering.
Formal analysis: Lars Vermeltfoort, Daan H.H.M. Viering.
Supervision: Jeroen H.F. de Baaij, René J.M. Bindels, Jaap Deinum.
Writing – original draft: Daan H.H.M. Viering.
Writing – review & editing: Jeroen H.F. de Baaij, René J.M. Bindels, Jaap Deinum, Lars Vermeltfoort.
Data Sharing Statement
All data on which the analyses are based are provided in this manuscript or the supplementary. The data collection forms templates were empty versions of the Supplemental Table 2 and Supplemental Table 3. Scripts for statistical analyses and creating figures are available upon request. The protocol for this systematic review was registered at PROSPERO with Accession No. CRD42021283468.
Supplemental Material
This article contains the following supplemental material online at http://links.lww.com/JSN/E524 and http://links.lww.com/JSN/E525.
Supplemental Table 1. Software and websites used for genetic analyses.
Supplemental Table 2. Complete extracted data of cohort and cross-sectional studies.
Supplemental Table 3. Complete extracted data of case reports and case series.
Supplemental Table 4. Genotype distribution in patients with at least one electrolyte abnormality versus general population.
Supplemental Table 5. Secondary outcomes extracted from case reports and case series.
Supplemental Table 6. JBI checklists scores for included case report patients (n=312).
Supplemental Figure 1. PRISMA flowchart.
Supplemental Figure 2. Electrolyte-deafness associations.
Supplemental Figure 3. Electrolyte-retinopathy associations.
References
- 1.Alston CL, Rocha MC, Lax NZ, Turnbull DM, Taylor RW. The genetics and pathology of mitochondrial disease. J Pathol. 2017;241(2):236–250. doi: 10.1002/path.4809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gorman GS Chinnery PF DiMauro S, et al. Mitochondrial diseases. Nat Rev Dis Primers. 2016;2(1):16080. doi: 10.1038/nrdp.2016.80 [DOI] [PubMed] [Google Scholar]
- 3.Pellock J, Behrens M, Lewis L, Holub D, Cartter S, Rowland L. Kearns-Sayre syndrome and hypoparathyroidism. Ann Neurol. 1978;3(5):455–458. doi: 10.1002/ana.410030519 [DOI] [PubMed] [Google Scholar]
- 4.Viering D Schlingmann KP Hureaux M, et al. Gitelman-like syndrome caused by pathogenic variants in mtDNA. J Am Soc Nephrol. 2022;33(2):305–325. doi: 10.1681/ASN.2021050596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choe Y Park E Hyun HS, et al. A 7-year-old girl presenting with a Bartter-like phenotype: answers. Pediatr Nephrol. 2017;32(6):983–985. doi: 10.1007/s00467-016-3480-8 [DOI] [PubMed] [Google Scholar]
- 6.Ellard S Baple E Callaway A, et al. ACGS best practice guidelines for variant classification in rare disease 2020. Assoc Clin Genomic Sci. 2020;1-32. https://www.acgs.uk.com/media/11631/uk-practice-guidelines-for-variant-classification-v4-01-2020.pdf [Google Scholar]
- 7.McCormick EM Lott MT Dulik MC, et al. Specifications of the ACMG/AMP standards and guidelines for mitochondrial DNA variant interpretation. Hum Mutat. 2020;41(12):2028–2057. doi: 10.1002/humu.24107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Munn Z Barker TH Moola S, et al. Methodological quality of case series studies: an introduction to the JBI critical appraisal tool. JBI Evid Synth. 2020;18(10):2127–2133. doi: 10.11124/jbisrir-d-19-00099 [DOI] [PubMed] [Google Scholar]
- 9.Page MJ McKenzie JE Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ 2021;372:n71. doi: 10.1136/bmj.n71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martin-Hernandez E Garcia-Silva MT Vara J, et al. Renal pathology in children with mitochondrial diseases. Pediatr Nephrol. 2005;20(9):1299–1305. doi: 10.1007/s00467-005-1948-z [DOI] [PubMed] [Google Scholar]
- 11.Wilson FH Hariri A Farhi A, et al. A cluster of metabolic defects caused by mutation in a mitochondrial tRNA. Science. 2004;306(5699):1190–1194. doi: 10.1126/science.1102521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choe Y Park E Hyun HS, et al. A 7-year-old girl presenting with a Bartter-like phenotype: questions. Pediatr Nephrol. 2017;32(6):981–982. doi:doi: 10.1007/s00467-016-3473-7 [DOI] [PubMed] [Google Scholar]
- 13.Szabolcs MJ, Seigle R, Shanske S, Bonilla E, DiMauro S, D'Agati V. Mitochondrial DNA deletion: a cause of chronic tubulointerstitial nephropathy. Kidney Int. 1994;45(5):1388–1396. doi: 10.1038/ki.1994.181 [DOI] [PubMed] [Google Scholar]
- 14.Tulinius MH Oldfors A Holme E, et al. Atypical presentation of multisystem disorders in two girls with mitochondrial DNA deletions. Eur J Pediatr. 1994;154(1):35–42. doi: 10.1007/s004310050245 [DOI] [PubMed] [Google Scholar]
- 15.Eviatar L Shanske S Gauthier B, et al. Kearns-Sayre syndrome presenting as renal tubular acidosis. Neurology. 1990;40(11):1761–1763. doi: 10.1212/wnl.40.11.1761 [DOI] [PubMed] [Google Scholar]
- 16.Mihai CM, Catrinoiu D, Toringhibel M, Stoicescu RM, Hancu A. De Toni-Debré-Fanconi syndrome in a patient with Kearns-Sayre syndrome: a case report. J Med Case Reports. 2009;3(1):101. doi: 10.1186/1752-1947-3-101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee YS, Yap HK, Barshop BA, Lee YS, Rajalingam S, Loke KY. Mitochondrial tubulopathy: the many faces of mitochondrial disorders. Pediatr Nephrol. 2001;16(9):710–712. doi: 10.1007/s004670100637 [DOI] [PubMed] [Google Scholar]
- 18.Panetta J, Gibson K, Kirby DM, Thorburn DR, Boneh A. The importance of liver biopsy in the investigation of possible mitochondrial respiratory chain disease. Neuropediatrics. 2005;36(4):256–259. doi: 10.1055/s-2005-865866 [DOI] [PubMed] [Google Scholar]
- 19.Atale A Bonneau-Amati P Rötig A, et al. Tubulopathy and pancytopaenia with normal pancreatic function: a variant of Pearson syndrome. Eur J Med Genet. 2009;52(1):23–26. doi: 10.1016/j.ejmg.2008.10.003 [DOI] [PubMed] [Google Scholar]
- 20.Buglioni A Hasadsri L Nasr SH, et al. Mitochondriopathy manifesting as inherited tubulointerstitial nephropathy without symptomatic other organ involvement. Kidney Int Rep. 2021;6(9):2514–2518. doi: 10.1016/j.ekir.2021.05.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu X-L Yan C-Z Ji K-Q, et al. Clinical, neuroimaging, and pathological analyses of 13 Chinese Leigh syndrome patients with mitochondrial DNA mutations. Chin Med J. 2018;131(22):2705–2712. doi: 10.4103/0366-6999.245265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oláhová M Peter B Szilagyi Z, et al. POLRMT mutations impair mitochondrial transcription causing neurological disease. Nat Commun. 2021;12(1):1135. doi: 10.1038/s41467-021-21279-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oláhová M Berti CC Collier JJ, et al. Molecular genetic investigations identify new clinical phenotypes associated with BCS1L-related mitochondrial disease. Hum Mol Genet. 2019;28(22):3766–3776. doi: 10.1093/hmg/ddz202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Naiki M Ochi N Kato YS, et al. Mutations in HADHB, which encodes the β-subunit of mitochondrial trifunctional protein, cause infantile onset hypoparathyroidism and peripheral polyneuropathy. Am J Med Genet A. 2014;164(5):1180–1187. doi: 10.1002/ajmg.a.36434 [DOI] [PubMed] [Google Scholar]
- 25.Majander A Suomalainen A Vettenranta K, et al. Congenital hypoplastic anemia, diabetes, and severe renal tubular dysfunction associated with a mitochondrial DNA deletion. Pediatr Res. 1991;30(4):327–330. doi: 10.1203/00006450-199110000-00007 [DOI] [PubMed] [Google Scholar]
- 26.Lockitch G, Halstead AC, Albersheim S, MacCallum C, Quigley G. Age- and sex-specific pediatric reference intervals for biochemistry analytes as measured with the Ektachem-700 analyzer. Clin Chem. 1988;34(8):1622–1625. doi: 10.1093/clinchem/34.8.1622 [DOI] [PubMed] [Google Scholar]
- 27.Shimizu J Inatsu A Oshima S, et al. Hyperkalemia in familial mitochondrial cytopathy. Clin Nephrol. 2008;70(10):348–353. doi: 10.5414/cnp70348 [DOI] [PubMed] [Google Scholar]
- 28.Kubota H, Tanabe Y, Takanashi J, Kohno Y. Episodic hyponatremia in mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes (MELAS). J Child Neurol. 2005;20(2):116–119. doi: 10.1177/08830738050200020601 [DOI] [PubMed] [Google Scholar]
- 29.Elisaf M, Panteli K, Theodorou J, Siamopoulos KC. Fractional excretion of magnesium in normal subjects and in patients with hypomagnesemia. Magnes Res. 1997;10(4):315–320. [PubMed] [Google Scholar]
- 30.Bellusci M Paredes-Fuentes AJ Ruiz-Pesini E, et al. The genetic landscape of mitochondrial diseases in Spain: a nationwide call. Genes (Basel). 2021;12(10):1590. doi: 10.3390/genes12101590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gorman GS Schaefer AM Ng Y, et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol. 2015;77(5):753–759. doi: 10.1002/ana.24362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilichowski E Grüters A Kruse K, et al. Hypoparathyroidism and deafness associated with pleioplasmic large scale rearrangements of the mitochondrial DNA: a clinical and molecular genetic study of four children with Kearns-Sayre syndrome. Pediatr Res. 1997;41(2):193–200. doi: 10.1203/00006450-199702000-00007 [DOI] [PubMed] [Google Scholar]
- 33.Guo L, Wang X, Ji H. Clinical phenotype and genetic features of a pair of Chinese twins with Kearns–sayre syndrome. DNA Cell Biol. 2020;39(8):1449–1457. doi: 10.1089/dna.2019.5010 [DOI] [PubMed] [Google Scholar]
- 34.Tengan CH, Kiyomoto BH, Rocha MS, Tavares VL, Gabbai AA, Moraes CT. Mitochondrial encephalomyopathy and hypoparathyrodism associated with a duplication and a deletion of mitochondrial deoxyribonucleic acid. J Clin Endocrinol Metab. 1998;83(1):125–129. doi: 10.1210/jcem.83.1.4497 [DOI] [PubMed] [Google Scholar]
- 35.Maaloul I Aloulou H Kmiha S, et al. Hypoparathyroidism in children: a study of eight cases. Tunis Med. 2018;96(8-9):472–476. [PubMed] [Google Scholar]
- 36.Papadimitriou A, Hadjigeorgiou GM, Divari R, Papagalanis N, Comi G, Bresolin N. The influence of Coenzyme Q10 on total serum calcium concentration in two patients with Kearns-Sayre Syndrome and hypoparathyroidism. Neuromuscul Disord. 1996;6(1):49–53. doi: 10.1016/0960-8966(95)00020-8 [DOI] [PubMed] [Google Scholar]
- 37.Shigemoto M Yoshimasa Y Yamamoto Y, et al. Clinical manifestations due to a point mutation of the mitochondrial tRNAleu(UUR) Ge five families with diabetes mellitus. Intern Med. 1998;37(3):265–272. doi:doi: 10.2169/internalmedicine.37.265 [DOI] [PubMed] [Google Scholar]
- 38.Tzoufi M Makis A Chaliasos N, et al. A rare case report of simultaneous presentation of myopathy, Addison's disease, primary hypoparathyroidism, and Fanconi syndrome in a child diagnosed with Kearns-Sayre syndrome. Eur J Pediatr. 2013;172(4):557–561. doi: 10.1007/s00431-012-1798-1 [DOI] [PubMed] [Google Scholar]
- 39.Jung HH, Hwang HH, Kim DH. A case of the hypercalcemia induced by the coenzyme Q10 and alphacalcidol treatment in a patient with Kearns-Sayre syndrome and hypoparathyroidism. J Korean Soc Pediatr Endocrinol. 2007;12(2):155–158. [Google Scholar]
- 40.Topaloğlu R Lebre AS Demirkaya E, et al. Two new cases with Pearson syndrome and review of Hacettepe experience. Turk J Pediatr. 2008;50(6):572–576. [PubMed] [Google Scholar]
- 41.Krauch G, Wilichowski E, Schmidt KG, Mayatepek E. Pearson marrow-pancreas syndrome with worsening cardiac function caused by pleiotropic rearrangement of mitochondrial DNA. Am J Med Genet. 2002;110(1):57–61. doi: 10.1002/ajmg.10410 [DOI] [PubMed] [Google Scholar]
- 42.Mori K, Narahara K, Ninomiya S, Goto Y, Nonaka I. Renal and skin involvement in a patient with complete Kearns-Sayre syndrome. Am J Med Genet. 1991;38(4):583–587. doi: 10.1002/ajmg.1320380417 [DOI] [PubMed] [Google Scholar]
- 43.Lee JH, Kim MJ, Park SH, Chae JH, Shin K. Case study of an inborn error manifested in the elderly: a woman with adult-onset mitochondrial disease mimicking systemic vasculitis. Int J Rheum Dis. 2019;22(6):1152–1156. doi: 10.1111/1756-185x.13575 [DOI] [PubMed] [Google Scholar]
- 44.Berio A, Piazzi A. Kearns-Sayre syndrome associated with de Toni-Debré-Fanconi syndrome due to cytochrome-c-oxidase (COX) deficiency. Panminerva Med. 2001;43(3):211–214. [PubMed] [Google Scholar]
- 45.Kanako KI Sakakibara N Murayama K, et al. BCS1L mutations produce Fanconi syndrome with developmental disability. J Hum Genet. 2022;67(3):143–148. doi: 10.1038/s10038-021-00984-0 [DOI] [PubMed] [Google Scholar]
- 46.Emma F Pizzini C Tessa A, et al. “Bartter-like” phenotype in Kearns-Sayre syndrome. Pediatr Nephrol. 2006;21(3):355–360. doi: 10.1007/s00467-005-2092-5 [DOI] [PubMed] [Google Scholar]
- 47.Southgate HJ, Penney MD. Severe recurrent renal salt wasting in a boy with a mitochondrial oxidative phosphorylation defect. Ann Clin Biochem. 2000;37(6):805–808. doi: 10.1258/0004563001900002 [DOI] [PubMed] [Google Scholar]
- 48.Goto Y, Itami N, Kajii N, Tochimaru H, Endo M, Horai S. Renal tubular involvement mimicking Bartter syndrome in a patient with Kearns-Sayre syndrome. J Pediatr. 1990;116(6):904–910. doi: 10.1016/s0022-3476(05)80648-1 [DOI] [PubMed] [Google Scholar]
- 49.Belostotsky R Ben-Shalom E Rinat C, et al. Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am J Hum Genet. 2011;88(2):193–200. doi: 10.1016/j.ajhg.2010.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Duran GP, Martinez-Aguayo A, Poggi H, Lagos M, Gutierrez D, Harris PR. Large mitochondrial DNA deletion in an infant with addison disease. JIMD Rep. 2012;3:5–9. doi: 10.1007/8904_2011_33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bruno C Minetti C Tang Y, et al. Primary adrenal insufficiency in a child with a mitochondrial DNA deletion. J Inherit Metab Dis. 1998;21(2):155–161. doi: 10.1023/a:1005347826664 [DOI] [PubMed] [Google Scholar]
- 52.Boles RG, Roe T, Senadheera D, Mahnovski V, Wong LJC. Mitochondrial DNA deletion with Kearns Sayre syndrome in a child with Addison disease. Eur J Pediatr. 1998;157(8):643–647. doi: 10.1007/s004310050902 [DOI] [PubMed] [Google Scholar]
- 53.Listernick R. A 9-year-old boy with labored breathing. Pediatr Ann. 2007;36(5):254–257. doi: 10.3928/0090-4481-20070501-04 [DOI] [PubMed] [Google Scholar]
- 54.Artuch R Pavía C Playán A, et al. Multiple endocrine involvement in two pediatric patients with Kearns-Sayre syndrome. Horm Res. 1998;50(2):99–104. doi: 10.1159/000023243 [DOI] [PubMed] [Google Scholar]
- 55.Sanaker PS, Husebye ES, Fondenes O, Bindoff LA. Clinical evolution of Kearns-Sayre syndrome with polyendocrinopathy and respiratory failure. Acta Neurol Scand Suppl. 2007;115:64–67. doi: 10.1111/j.1600-0404.2007.00850.x [DOI] [PubMed] [Google Scholar]
- 56.Ribes A Riudor E Valcarel R, et al. Pearson syndrome: altered tricarboxylic acid and urea-cycle metabolites, adrenal insufficiency and corneal opacities. J Inherit Metab Dis. 1993;16(3):537–540. doi: 10.1007/bf00711675 [DOI] [PubMed] [Google Scholar]
- 57.D’Aco KE, Manno M, Clarke C, Ganesh J, Meyers KE, Sondheimer N. Mitochondrial tRNA Phe mutation as a cause of end-stage renal disease in childhood. Pediatr Nephrol. 2013;28(3):515–519. doi: 10.1007/s00467-012-2354-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gurrieri C Kivela JE Bojanić K, et al. Anesthetic considerations in mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes syndrome: a case series. Can J Anesth. 2011;58(8):751–763. doi: 10.1007/s12630-011-9528-0 [DOI] [PubMed] [Google Scholar]
- 59.Mordel P Schaeffer S Dupas Q, et al. A 2 bp deletion in the mitochondrial ATP 6 gene responsible for the NARP (neuropathy, ataxia, and retinitis pigmentosa) syndrome. Biochem Biophys Res Commun. 2017;494(1-2):133–137. doi: 10.1016/j.bbrc.2017.10.066 [DOI] [PubMed] [Google Scholar]
- 60.Vinu N, Puri RD, Anand K, Verma IC. Expanding the phenotype of the founder south asian mutation in the nuclear encoding mitochondrial RMND1 gene. Indian J Pediatr. 2018;85(2):87–92. doi: 10.1007/s12098-017-2515-x [DOI] [PubMed] [Google Scholar]
- 61.Ng YS Alston CL Diodato D, et al. The clinical, biochemical and genetic features associated with RMND1-related mitochondrial disease. J Med Genet. 2016;53(11):768–775. doi: 10.1136/jmedgenet-2016-103910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mory PB, Santos MCd, Kater CE, Moisés RS. Maternally-inherited diabetes with deafness (MIDD) and hyporeninemic hypoaldosteronism. Arq Bras Endocrinol Metabol. 2012;56(8):574–577. doi: 10.1590/s0004-27302012000800019 [DOI] [PubMed] [Google Scholar]
- 63.Das BB, Hernandez LE, Jayakar P, Chatfield KC, Chrisant M. Novel loss of function in the AGK gene. JACC Case Rep. 2019;1(1):11–16. doi: 10.1016/j.jaccas.2019.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brecht M, Richardson M, Taranath A, Grist S, Thorburn D, Bratkovic D. Leigh syndrome caused by the MT-ND5 m.13513G>A mutation: a case presenting with WPW-like conduction defect, cardiomyopathy, hypertension and hyponatraemia. JIMD Rep. 2015;19:95–100. doi: 10.1007/8904_2014_375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Swiderska N, Appleton R, Morris A, Isherwood D, Selby A. A novel presentation of inappropriate antidiuretic hormone secretion in Leigh syndrome with the myoclonic epilepsy and ragged red fibers, mitochondrial DNA 8344A> G mutation. J Child Neurol. 2010;25(6):782–785. doi: 10.1177/0883073809347594 [DOI] [PubMed] [Google Scholar]
- 66.Patel IB, Sidani M, Zoorob R. Mitochondrial encephalopathy, lactic acidosis and stroke-like syndrome (MELAS): a case report, presentation, and management. South Med J. 2007;100(1):70–72. doi: 10.1097/01.smj.0000223691.66759.8f [DOI] [PubMed] [Google Scholar]
- 67.Sinnecker T Andelova M Mayr M, et al. Diagnosis of adult-onset MELAS syndrome in a 63-year-old patient with suspected recurrent strokes–a case report. BMC Neurol. 2019;19:91–98. doi: 10.1186/s12883-019-1306-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jameel I, Sreh A, Das P. Recurrent stroke events secondary to a late presentation of mitochondrial encephalomyopathy with lactic acidosis and stroke-like symptoms (MELAS) syndrome. Cureus. 2020;12(12):e11839. doi: 10.7759/cureus.11839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gagliardi D Mauri E Magri F, et al. Can intestinal pseudo-obstruction drive recurrent stroke-like episodes in late-onset MELAS syndrome? A case report and review of the literature. Front Neurol. 2019;10:38. doi: 10.3389/fneur.2019.00038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Inui K Fukushima H Tsukamoto H, et al. Mitochondrial encephalomyopathies with the mutation of the mitochondrial tRNA(Leu(UUR)) gene. J Pediatr. 1992;120(1):62–66. doi: 10.1016/s0022-3476(05)80599-2 [DOI] [PubMed] [Google Scholar]
- 71.Simon MT Eftekharian SS Stover AE, et al. Novel mutations in the mitochondrial complex I assembly gene NDUFAF5 reveal heterogeneous phenotypes. Mol Genet Metab. 2019;126(1):53–63. doi: 10.1016/j.ymgme.2018.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Labarthe F, Benoist JF, Brivet M, Vianey-Saban C, Despert F, Ogier de Baulny H. Partial hypoparathyroidism associated with mitochondrial trifunctional protein deficiency. Eur J Pediatr. 2006;165(6):389–391. doi: 10.1007/s00431-005-0052-5 [DOI] [PubMed] [Google Scholar]
- 73.Chen HZ Jin M Cai NQ, et al. Rhabdomyolysis and respiratory insufficiency due to the common ETFDH mutation of c.250G>A in two patients with late-onset multiple acyl-CoA dehydrogenase deficiency. Chin Med J. 2019;132(13):1615–1618. doi: 10.1097/cm9.0000000000000288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kouga T Takagi M Miyauchi A, et al. Japanese Leigh syndrome case treated with EPI-743. Brain Dev. 2018;40(2):145–149. doi: 10.1016/j.braindev.2017.08.005 [DOI] [PubMed] [Google Scholar]
- 75.Fischer T, Och U, Marquardt T. Long-term ketone body therapy of severe multiple acyl-CoA dehydrogenase deficiency: a case report. Nutrition. 2019;60:122–128. doi: 10.1016/j.nut.2018.10.014 [DOI] [PubMed] [Google Scholar]
- 76.Saitoh S, Momoi MY, Yamagata T, Mori Y, Imai M. Effects of dichloroaeetate in three patients with MELAS. Neurology. 1998;50(2):531–534. doi: 10.1212/wnl.50.2.531 [DOI] [PubMed] [Google Scholar]
- 77.Fang Q, Chen L, Chen Q, Yang F. Clinical features of child mitochondrial encephalopathy with lactic acidosis and stroke with status epileptics. J Clin Pediatr. 2015;33(2):160–163. doi: 10.3969/j.issn.1000-3606.2015.02.015 [DOI] [Google Scholar]
- 78.Motlagh Scholle L, Zierz S, Mawrin C, Wickenhauser C, Lehmann Urban D. Heteroplasmy and copy number in the common m.3243a>G mutation-A post-mortem genotype- phenotype analysis. Genes (Basel). 2020;11(2):212. doi: 10.3390/genes11020212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ni Cathain D, Browne E, Skehan K, Boyle K. MELAS syndrome: an acute stroke-like episode complicated by renal tubular acidosis. BMJ Case Rep. 2021;14(11):e245898. doi: 10.1136/bcr-2021-245898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dao M Arnoux J-B Bienaimé F, et al. Long-term renal outcome in methylmalonic acidemia in adolescents and adults. Orphanet J Rare Dis. 2021;16:220–310. doi: 10.1186/s13023-021-01851-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Miao J, Qian Q, Zand L. Long-standing hypokalemia and lactic acidosis as the primary presentation of mitochondrial myopathy. Kidney Int Rep. 2020;5(5):742–745. doi: 10.1016/j.ekir.2020.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ibdah JA, Dasouki MJ, Strauss AW. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: variable expressivity of maternal illness during pregnancy and unusual presentation with infantile cholestasis and hypocalcaemia. J Inherit Metab Dis. 1999;22(7):811–814. doi: 10.1023/a:1005506024055 [DOI] [PubMed] [Google Scholar]
- 83.Jin S, Long Z, Wang W, Jiang B. Hyponatremia in neuromyelitis optica spectrum disorders: literature review. Acta Neurol Scand. 2018;138(1):4–11. doi: 10.1111/ane.12938 [DOI] [PubMed] [Google Scholar]
- 84.Gennari FJ, Weise WJ. Acid-base disturbances in gastrointestinal disease. Clin J Am Soc Nephrol. 2008;3(6):1861–1868. doi: 10.2215/CJN.02450508 [DOI] [PubMed] [Google Scholar]
- 85.Macafee DA, Allison SP, Lobo DN. Some interactions between gastrointestinal function and fluid and electrolyte homeostasis. Curr Opin Clin Nutr Metab Care. 2005;8(2):197–203. doi: 10.1097/00075197-200503000-00015 [DOI] [PubMed] [Google Scholar]
- 86.Konrad M Schaller A Seelow D, et al. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet. 2006;79(5):949–957. doi: 10.1086/508617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Birkenhager R Otto E Schurmann MJ, et al. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat Genet. 2001;29(3):310–314. doi: 10.1038/ng752 [DOI] [PubMed] [Google Scholar]
- 88.Scholl UI Choi M Liu T, et al. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci U S A. 2009;106(14):5842–5847. doi: 10.1073/pnas.0901749106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bockenhauer D Feather S Stanescu HC, et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. New Engl J Med. 2009;360(19):1960–1970. doi: 10.1056/NEJMoa0810276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zuvarox T, Belletieri C. Malabsorption Syndromes. StatPearls [Internet]. StatPearls Publishing; 2022. [PubMed] [Google Scholar]
- 91.Zhou Y Zhong C Yang Q, et al. Novel SARS2 variants identified in a Chinese girl with HUPRA syndrome. Mol Genet Genomic Med. 2021;9(4):e1650. doi: 10.1002/mgg3.1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gao X Xin G Tu Y, et al. TARS2 variants causes combination oxidative phosphorylation deficiency-21: a case report and literature review [published online ahead of print September 23, 2022]. Neuropediatrics. doi: 10.1055/a-1949-9310 [DOI] [PubMed] [Google Scholar]
- 93.Nakajima J Eminoglu TF Vatansever G, et al. A novel homozygous YARS2 mutation causes severe myopathy, lactic acidosis, and sideroblastic anemia 2. J Hum Genet. 2014;59(4):229–232. doi: 10.1038/jhg.2013.143 [DOI] [PubMed] [Google Scholar]
- 94.Kim SH Choi JH Wang P, et al. Mitochondrial threonyl-tRNA synthetase TARS2 is required for threonine-sensitive mTORC1 activation. Mol Cell. 2021;81(2):398–407.e4. doi: 10.1016/j.molcel.2020.11.036 [DOI] [PubMed] [Google Scholar]
- 95.Schlingmann KP Jouret F Shen K, et al. mTOR-activating mutations in RRAGD are causative for kidney tubulopathy and cardiomyopathy. J Am Soc Nephrol. 2021;32(11):2885–2899. doi: 10.1681/ASN.2021030333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gómez CJB, Gil-Peña H, Álvarez FAO, Rodríguez FS. Outcome of primary tubular tubulopathies diagnosed in pediatric age. Nefrología (Engl Ed). 2021;41(2):182–190. doi: 10.1016/j.nefroe.2020.07.001 [DOI] [PubMed] [Google Scholar]
- 97.Schlingmann KP, de Baaij JHF. The genetic spectrum of Gitelman(-like) syndromes. Curr Opin Nephrol Hypertens. 2022;31(5):508–515. doi: 10.1097/mnh.0000000000000818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bosman W, Hoenderop JGJ, de Baaij JHF. Genetic and drug-induced hypomagnesemia: different cause, same mechanism. Proc Nutr Soc. 2021;80(3):327–338. doi: 10.1017/s0029665121000926 [DOI] [PubMed] [Google Scholar]
- 99.Huang C-L, Kuo E. Mechanism of hypokalemia in magnesium deficiency. J Am Soc Nephrol. 2007;18(10):2649–2652. doi: 10.1681/ASN.2007070792 [DOI] [PubMed] [Google Scholar]
- 100.Wang Z Ying Z Bosy-Westphal A, et al. Specific metabolic rates of major organs and tissues across adulthood: evaluation by mechanistic model of resting energy expenditure. Am J Clin Nutr. 2010;92(6):1369–1377. doi: 10.3945/ajcn.2010.29885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hansell P, Welch WJ, Blantz RC, Palm F. Determinants of kidney oxygen consumption and their relationship to tissue oxygen tension in diabetes and hypertension. Clin Exp Pharmacol Physiol. 2013;40(2):123–137. doi: 10.1111/1440-1681.12034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Soltoff SP. ATP and the regulation of renal cell function. Annu Rev Physiol. 1986;48(1):9–31. doi: 10.1146/annurev.ph.48.030186.000301 [DOI] [PubMed] [Google Scholar]
- 103.Hanna MG, Nelson IP, Morgan-Hughes JA, Wood NW. MELAS: a new disease associated mitochondrial DNA mutation and evidence for further genetic heterogeneity. J Neurol Neurosurg Psychiatry. 1998;65(4):512–517. doi: 10.1136/jnnp.65.4.512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Goto Y, Nonaka I, Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348(6302):651–653. doi: 10.1038/348651a0 [DOI] [PubMed] [Google Scholar]
- 105.Melone MA Tessa A Petrini S, et al. Revelation of a new mitochondrial DNA mutation (G12147A) in a MELAS/MERFF phenotype. Arch Neurol. 2004;61(2):269–272. doi: 10.1001/archneur.61.2.269 [DOI] [PubMed] [Google Scholar]
- 106.Suomalainen A, Battersby BJ. Mitochondrial diseases: the contribution of organelle stress responses to pathology. Nat Rev Mol Cell Biol. 2018;19(2):77–92. doi: 10.1038/nrm.2017.66 [DOI] [PubMed] [Google Scholar]
- 107.Carlström M. Nitric oxide signalling in kidney regulation and cardiometabolic health. Nat Rev Nephrol. 2021;17(9):575–590. doi: 10.1038/s41581-021-00429-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gao Y, Stuart D, Takahishi T, Kohan DE. Nephron-specific disruption of nitric oxide synthase 3 causes hypertension and impaired salt excretion. J Am Heart Assoc. 2018;7(14):e009236. doi: 10.1161/jaha.118.009236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sekii Y Kiuchi H Takezawa K, et al. Dietary salt with nitric oxide deficiency induces nocturnal polyuria in mice via hyperactivation of intrarenal angiotensin II-SPAK-NCC pathway. Commun Biol. 2022;5(1):175. doi: 10.1038/s42003-022-03104-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Almannai M, El-Hattab AW. Nitric oxide deficiency in mitochondrial disorders: the utility of arginine and citrulline. Front Mol Neurosci. 2021;14:682780. doi: 10.3389/fnmol.2021.682780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Xu C Tong L Rao J, et al. Heteroplasmic and homoplasmic m.616T>C in mitochondria tRNAPhe promote isolated chronic kidney disease and hyperuricemia. JCI Insight. 2022;7(11):e157418. doi: 10.1172/jci.insight.157418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tyni T, Rapola J, Palotie A, Pihko H. Hypoparathyroidism in a patient with long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency caused by the G1528C mutation. J Pediatr. 1997;131(5):766–768. doi: 10.1016/s0022-3476(97)70111-2 [DOI] [PubMed] [Google Scholar]
- 113.Wallace J, Scarpa A. Regulation of parathyroid hormone secretion in vitro by divalent cations and cellular metabolism. J Biol Chem. 1982;257(18):10613–10616. doi: 10.1016/s0021-9258(18)33866-3 [DOI] [PubMed] [Google Scholar]
- 114.Weissig V. Drug development for the therapy of mitochondrial diseases. Trends Mol Med. 2020;26(1):40–57. doi: 10.1016/j.molmed.2019.09.002 [DOI] [PubMed] [Google Scholar]
- 115.Lillie EO, Patay B, Diamant J, Issell B, Topol EJ, Schork NJ. The n-of-1 clinical trial: the ultimate strategy for individualizing medicine? Per Med. 2011;8(2):161–173. doi: 10.2217/pme.11.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sterne JA, Egger M, Moher D: Addressing reporting biases. In: Cochrane Handbook for Systematic Reviews of Interventions. Edited by Higgins JPT, Green S, The Cochrane Collaboration, 2008, pp 297–333. [Google Scholar]
- 117.Dickersin K: Publication bias: Recognizing the problem, understanding its origins and scope, and preventing harm. In: Publication Bias in Meta‐Analysis: Prevention, Assessment and Adjustments. Edited by Rothstein HR, Sutton AJ, Borenstein M, John Wiley & Sons, 2005, pp 9–33. [Google Scholar]
- 118.de Baaij JH, Hoenderop JG, Bindels RJ. Magnesium in man: implications for health and disease. Physiol Rev. 2015;95:1–46. doi: 10.1152/physrev.00012.2014 [DOI] [PubMed] [Google Scholar]
- 119.Poole OV Pizzamiglio C Murphy D, et al. Mitochondrial DNA analysis from exome sequencing data improves diagnostic yield in neurological diseases. Ann Neurol. 2021;89(6):1240–1247. doi: 10.1002/ana.26063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rius R Compton AG Baker NL, et al. Application of genome sequencing from blood to diagnose mitochondrial diseases. Genes (Basel). 2021;12(4):607. doi: 10.3390/genes12040607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mavraki E Labrum R Sergeant K, et al. Genetic testing for mitochondrial disease: the United Kingdom best practice guidelines. Eur J Hum Genet. 2022;31(2):148–163. doi: 10.1038/s41431-022-01249-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Grier J, Hirano M, Karaa A, Shepard E, Thompson JL. Diagnostic odyssey of patients with mitochondrial disease: results of a survey. Neurol Genet. 2018;4(2):e230. doi: 10.1212/nxg.0000000000000230 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data on which the analyses are based are provided in this manuscript or the supplementary. The data collection forms templates were empty versions of the Supplemental Table 2 and Supplemental Table 3. Scripts for statistical analyses and creating figures are available upon request. The protocol for this systematic review was registered at PROSPERO with Accession No. CRD42021283468.