

Abstract
Membrane potential is a property of all living cells1. Nevertheless, its physiological role in non-excitable cells is poorly understood. Resting membrane potential is typically considered fixed and under tight homeostatic control2. Contrary to this paradigm, we find that membrane potential is a dynamic property that directly reflects mechanical forces acting on the cell and that cells use membrane potential to assess their biomechanical state. We show that several important mechano-sensitive signal transduction pathways, like MAPK and Hippo3–9, are directly controlled by membrane potential and this signaling is mediated by upstream membrane-bound receptors, including FAT1. We further show that mechano-transduction via membrane potential plays a critical role in the homeostasis of epithelial tissues, setting cellular biomass density and cell number density by controlling proliferation and cell elimination. In epithelial scratch wound assays, as well as Xenopus tadpole tail regeneration, we observe a wave of depolarization caused by a drop in cellular biomass density due to mechanical stretch and we show that this depolarization wave is critical for wound closure. Together, these data are explained by a first-principles biophysical model, which demonstrates that membrane potential is physically coupled to mechanical pressure and cellular biomass density. Membrane potential thereby provides a quasi-instantaneous, global readout of the biophysical state of the cell and in turn regulates cell growth, resulting in homeostatic feedback control of biomass density and cell number density in tissues. This interplay may be an ancient mechanism for growth control in multi-cellular organisms and its misregulation may play an important role in tumorigenesis.
How the volume, mass and macromolecule density of metazoan cells are regulated is yet to be fully understood, but these variables are critical for biochemical functions at the cellular level, as well as for the growth and homeostasis of tissues and organisms. Cells in confluent tissues need to precisely control biomass synthesis and proliferation to quickly heal injuries10–12, while avoiding excessive growth that is the hallmark of tumorigenesis13,14. Mechanical signaling has been proposed to be involved in this process15–26 and important mechano-sensitive signaling pathways, like the Hippo pathway, appear to play an important role in cell number density homeostasis27–31. However, for mechanical signaling to confer useful information for tissue density homeostasis, it needs to reflect important physiological parameters on the cellular and tissue scale, such as cell volume or total cellular biomass in relation to the available space in the tissue. It is not clear how known mechanisms of mechano-transduction can bridge the gap between local molecular interactions and physiological variables on the cellular scale. For example, tension in the plasma membrane results in calcium influx via stretch-activated channels32–34, but it is not clear how the total amount of plasma membrane would be controlled in relation to total cellular biomass for cells of different size and shape and at different points in the cell cycle. Moreover, it has been experimentally shown that mechanical stretching result in production of additional membrane35 and stretch-mediated calcium flux decays within short distance from the point of stretch by patch pipette36, suggesting that total amount of plasma membrane is not fixed. Similarly, tension of individual cell-cell adherence junctions can mediate mechanical signaling, but how do cells control the amount of cytoskeleton that generates these forces and how do osmotic forces from macromolecular content of the cytoplasm affect these forces? It remains unclear how cells can gain information about global cellular and tissue-level state via localized, short-timescale mechanical signaling.
Cellular biomass density, on the other hand, remains an underexplored physiological variable. It is typically assumed that biomass density is constant for a given cell type, indicating that cell mass and volume are directly proportional, even as cells undergo additional rounds of proliferation in already confluent epithelia37 or when cell volume changes quickly and dramatically during wound healing. However, this assumption has not been thoroughly tested experimentally. Here, by directly measuring cellular biomass density, we find that the assumption of constant biomass density is false in many situations. Instead, we find that biomass density is a central physiological variable that is intimately coupled to cellular growth control and mechano-transduction.
Quantification of tissue biomass density
To measure cellular biomass density, we used normalized Raman imaging (NoRI)38, which enables absolute quantification of cellular macromolecular components and water. Specially, we wanted to quantify intracellular biomass densities at increasing levels of confluence. We therefore grew different cell lines that model epithelial monolayers39 of varying tissue origin, including Madin Darby Canine Kidney (MDCK), MCF10a, and EpH4-Ev. Remarkably, rather than remaining constant, cellular biomass density underwent a striking increase as a function of cell number density in all cases. Fig. 1a shows the nuclei density and biomass density of MDCK cells at 0 hours, 48 hours, and 96 hours after initially reaching confluence, and Fig. S1a–d shows the separate protein and lipid contributions. We observed a similar trend in MCF10a and EpH4-Ev monolayers (Fig. 1c, 1d, Fig. S1e, S1f). We confirmed this observation with a second independent microscopic technique, based on laser interferometry, also referred to as holo-tomographic microscopy (Tomocube) (Fig. 1a, red squares & Fig. S3)40,41.
Figure 1: Membrane potential is a sensor and regulator of tissue density.

a, MDCK cells imaged for 4 consecutive days after confluence with increasing cell number density. Left panel shows number of cell nuclei and cellular biomass density (in units of g/ml) measured via Normalized Stimulated Raman Spectroscopy (NoRI). Plot below shows correlation between number density (mean ± s.d., NoRI: n=8, Tomocube: n=10) and biomass density (mean ± s.d., NoRI: n=15, Tomocube: n=~20) measured using NoRI and Tomocube. Right panel displays snapshots of biomass density in tissue simulation (mean ± s.d., n=4 simulations). b, Left panel shows fluorescence of MDCK-Voltron cells, plot below shows log fold change in fluorescence (mean ± s.d., n=8) against number density. Right panel displays snapshots of membrane potential in tissue simulation (mean ± s.e.m., n=4 simulations). c-d, Top panels: NoRI images and quantification (mean ± s.d., number density: n=~10, NoRI: n=24) of MCF10A/EpH4-Ev cells for 4 consecutive days after confluence with increasing cell number density. Bottom panels: DiOC2(3) imaging from colony edge to colony centre. e. Feedback control loop between biomass growth and membrane potential is crucial for epithelial homeostasis. f-g, Depolarization of membrane potential by ouabain increases the biomass density (box plot, line at mean, error bars represent min to max, n=40) and number density (n = 8) of confluent cells over the homeostatic steady state limit (dotted line; quantified from experiment in panel a).
Tissue density is reflected in membrane potential
Cellular biomass components carry a net negative electric charge42–44. Inspired by the Gibbs-Donnan effect, a phenomenon from equilibrium thermodynamics45, we wanted to know whether changes in biomass density were reflected in changes of membrane potential. Using the hybrid voltage-indicator Voltron46, we quantified membrane potential in MDCK cells as a function of cell number and biomass density. Strikingly, MDCK cells became increasingly hyperpolarized with increasing cell number (Fig. 1b). In MCF10a and EpH4-Ev cells, we also confirmed the hyperpolarization of membrane potential with increasing cell number density (Fig. 1c and Fig. 1d, lower panel) with the membrane potential dye DiOC2(3). These findings are consistent with previous glass-microelectrode-based measurements of resting membrane potential in CHO cells as they reached different levels of confluence over time (Fig. S4a,b) and in different regions of a colony with varying local cell number density47 (Fig. S4d). Similarly, our measurements in different regions of CHO colonies showed depolarization with decreasing cell number density, whereas denser regions of the colony exhibit hyperpolarized membrane potential (Fig. S4e). The mechanistic basis or physiological consequences of these early striking observations has never been explained. We hypothesized that, just like in MDCK cells, changes in CHO cells’ membrane potential might correlate with changes in cellular biomass density. Indeed, using NoRI imaging, we confirmed that biomass density in CHO cells also increases with increasing cell number density (Fig. S4c). Unlike MDCK or MCF10a cells, CHO cells do not form a tight epithelium connected by tight junctions, suggesting the relationship between membrane potential and biomass density is more general.
The correlation of biomass density and membrane potential suggests that these physiological variables are directly coupled. To test the causal nature of this relationship, we applied external osmotic pressure to compress cells, artificially increasing their biomass density. Indeed, we found that the membrane potential of CHO and MDCK cells hyperpolarized with mechanical compression (Fig. S5). Therefore, changes in membrane potential could be a direct result of changes in biomass density, as in the Gibbs-Donnan situation in thermodynamic equilibrium.
Membrane potential regulates biomass density and cell number density
Based on these observations, we hypothesized that rather than being a passive consequence of changes in biomass density, membrane potential could be directly involved in regulating biomass production and proliferation. To test this hypothesis, we first grew MDCK monolayers to their maximum cell number density (Fig. 1e). We confirmed that cells reached homeostatic biomass density threshold by NoRI (Fig. 1e, 1f). After this, we depolarized the cells overnight by inhibiting the sodium-potassium pump (using the pharmacological inhibitor ouabain). Indeed, we found that depolarization pushed cells to a significantly higher biomass density (Fig 1e,f) that was also reflected in a significantly higher cell number density (Fig. 1g). We confirmed that this phenotype was independent of the pharmacological method of depolarization in high-throughput experiments. In these experiments, we grew MDCK cells to maximum density and then applied different depolarizing agents (gramicidin, TCT48,49) or hyperpolarizing drugs (valinomycin) at different concentrations to tissue culture. Cells in depolarizing conditions (Fig. S6a, red) maintained a significantly higher cell number density than cells grown in the absence of drugs (Fig. S6a, cyan). Conversely, the cell number density in hyperpolarizing conditions (Fig. S6a, yellow) dropped substantially and then plateaued at a much lower level. We found that steady-state cell number density could be modulated continuously with the dose of the hyperpolarizing drug (Fig. S6c).
Mechano-electro-osmotic model
To understand how membrane potential might control biomass density and the effect of cell number density on proliferation, we formulated a biophysical mathematical model of ion homeostasis, cellular biomass and mechanical pressure based on first principles. Classical models of membrane potential like the Goldman Hodgkin Katz model do not consider mechanical forces at all, and their systems of equations are only closed by introducing convenient, yet unphysical simplifications2,50. Models correctly rooted in elementary physical principles were developed in the 1960s and showed that osmotic stabilization of cells requires constant ion export that results in a build-up of membrane potential as a by-product51–53. However, it remained unclear how these models should be applied to growing cells, as they change in cell mass, cell membrane and volume, and model parameters like permeability and transport activity are subject to change. More importantly, none of these works uncovered that membrane potential, a by-product of volume stabilization, may directly convey crucial physiological information to cells and direct cellular decision making.
Membrane potential is intimately coupled to osmotic pressure of the cytoplasm and the balance of charged ions and macromolecules inside the cell. As illustrated in Box 1 (top), an imaginary cell resists external forces compressing it, via osmotic pressure resulting from an increased concentration of intracellular macromolecules in the cytoplasm and an increased concentration of counterions. A substantial fraction of these counterions are not condensed on macromolecules and therefore osmotically active even at very high salt concentrations, as demonstrated by classical experimental and theoretical works54–56. However, this results in a steeper ion concentration gradient across the plasma membrane, promoting a larger efflux of ions by diffusion. A buildup of membrane potential due to charged biomolecules inside the cell counteracts this diffusive efflux, retaining an elevated intracellular ion concentration to counteract the external pressure. In equilibrium conditions, this is known as the Gibbs-Donnan effect45, but the same processes are at play far from equilibrium. With a constant expression ratio of ion transporters and channels, membrane potential constitutes a cell size-independent readout of total mechanical pressure on the cell and the corresponding cytoplasmic biomass density (see Box 1 & Supplementary Note 1). In effect, membrane potential physically controls biomass density and mechanical forces, and vice versa, but crucially it provides cells with an accessible readout of biomass density and mechanical forces that can be used for signal transduction. Of note, the polarity of the membrane potential in the model is determined by the net charge on the macromolecules in the cell. Negative charge dominates over positive because the proteome has an average isolectric point above 757 and nucleic acids, especially the significant fraction of the biomass in the cytoplasm comprised of RNA58, are negatively charged. As a result, K+ and Na+ are the abundant cations in the cytoplasm, and the membrane potential polarity is such that there is net negative charge on the inside of the cell.
Box 1: Mechano-electro-osmotic model.
Top: The model combines three elementary conservation laws: Flux balance across the plasma membrane, mechanical force balance, and charge balance. Left-hand side illustrates a cell under low mechanical pressure and the right-hand side a corresponding cell under high mechanical pressure. Cellular biomass like protein and RNA carries a net negative charge. Cells resist compression of their volume via cytoplasmic osmotic pressure that counteracts and balances external forces (force balance). This osmotic pressure originates from an intracellular concentration of counterions that are attracted to balance negatively charged macromolecules (charge balance). As illustrated on the right-hand side, higher mechanical pressure acting in the cell is thus balanced by a higher concentration of counterions. Higher intracellular ion concentrations result in steeper ion concentration gradients across the membrane, as illustrated in the right inset. Thereby, with constant ion channel abundances, a steeper concentration gradient across the membrane results in an augmented diffusive efflux of ions (Fick’s law). With constant active ion transport flux (sodium-potassium-ATPase), this increased diffusive ion efflux results in a buildup of net negative charge in the cytoplasm and a corresponding membrane potential that counteracts the diffusive efflux of positively charged ions until flux balance is achieved (flux balance for each ion species). Bottom: A one-to-one correspondence between mechanical pressure, cytoplasmic biomass density and membrane potential emerges from the mechano-electro-osmotic equilibrium illustrated in the top panel. Crucial physiological properties like total biomass in relation to available space or mechanical and spatial constraints on the entire cell are difficult for cells to assess and integrate from individual molecular players. Membrane potential, on the other hand, provides an instantaneous, global readout of these physiological variables. We hypothesize that membrane potential is therefore the ideal starting point for signal transduction to control cellular decisions like growth, apoptosis, and cell motility to achieve tissue homeostasis.
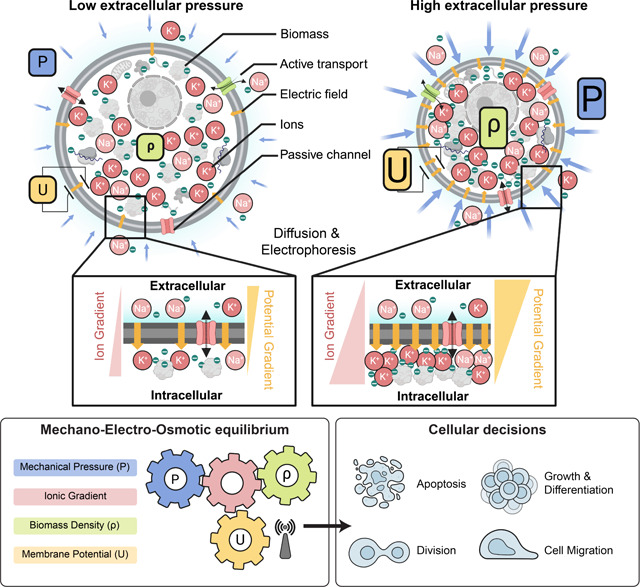
Tissue simulation
We calibrated the mechano-electro-osmotic model with physiological parameters and use it to simulate a mono-layered epithelium (Supplementary Note 2). Membrane potential provides cells in the tissue with a quasi-instantaneous readout of tissue density and the mechanical constraints, which are important for tissue homeostasis (Fig. 1c). Because the coupling between mechanics, density, and membrane potential requires no active regulation and is present even in thermodynamic equilibrium, we hypothesize that regulation via membrane potential could be one of the earliest homeostatic mechanisms employed in multi–cellular organisms. To test the theoretical idea that tissues can achieve homeostasis relying only on the membrane potential as a control variable, we tested a model where the membrane potential is the sole regulator of biosynthesis and apoptosis (see Supplementary Note 2 for details of implementation).
This tissue simulation successfully recapitulated our experimental observations, including the increase in biomass density with cell number density (Fig. 1a, right side) and the corresponding changes in membrane potential (Fig. 1b, right side). Implementing the mechanism of different depolarizing and hyperpolarizing drugs in the simulation, the model recapitulated the observed changes in steady-state tissue density (Fig. S6b). The general effect of these drugs can be understood intuitively: cells perceive mild depolarization to be a state of low confluence. They continue to grow, generating an excessive density (Fig. S6b, red circles). This is remarkable and non-trivial, given that depolarization should normally result in swelling and thereby a decrease in cellular biomass density. Conversely, hyperpolarization by drugs leads to a lower homeostatic tissue density because cells interpret hyperpolarization as excess tissue density and arrest their growth prematurely (Fig. S6b, yellow circles). Thereby, regulation of cell growth by membrane potential correctly predicts changes in steady-state tissue density.
Membrane potential reflects mechanical forces
The mechano-electro-osmotic model (Box 1) predicts that mechanical forces should be directly reflected in the membrane potential. To test this prediction, we imposed external mechanical perturbations on the tissue and measured the response of membrane potential. One method is to culture epithelial cells on an elastic membrane that can be stretched or de-stretched mechanically, thereby expanding or compressing the tissue itself59,60 (Fig. 2a). We cultured MDCK cells on a stretchable PDMS membrane and waited until cells reached maximum cell number density. We employed the voltage sensitive dye FluoVolt, whose fluorescence intensity increases with depolarization61, as it is ideally suited to image changes in membrane potential on short timescales. Indeed, stretching the MDCK monolayer resulted in depolarization (Fig. 2a,b, quantified in panel c), consistent with simulation prediction (Fig. 2d). To check that this change in membrane potential was reversible on short timescales and confirm our findings with another membrane potential indicator, we de-stretched the membrane using DiSBAC2(3)62. As predicted by the model (Fig. 2g), reducing the stretch of the PDMS membrane quickly led to repolarization of membrane potential (Fig. 2e,f). According to the model, depolarization upon stretch is caused by a drop in cellular biomass density due to an increased cell volume, rather than tension in the plasma membrane or changes in cell-cell adhesion. Using NoRI imaging, we quantified biomass density of the confluent monolayer just before and immediately after a 20% uniaxial stretch, and then again after overnight cultivation (Fig. 2h,i). As expected from the tissue simulation (Fig. 2j), we found that stretching resulted in a significant drop in biomass density and induced biomass production and proliferation, as biomass density and cell number recovered to their initial level overnight. Conversely, we also confirmed that de-stretching the PDMS membrane resulted in an increase of cellular biomass density after overnight recovery (Fig. S7).
Figure 2: Cells detect and respond to mechanical forces via membrane potential.

a, Schematic diagram depicting the experimental design with monolayers grown on stretchable membrane. In one case, cells were grown to confluence and then 20% uniaxial stretch was applied (left panel) or cells were grown to confluence on a pre-stretched membrane, and then the stretch was released to induce compression (right panel). b, FluoVolt images of MDCK cells before and after stretching. c, Quantification of panel b (line at mean, n=8, p=0.0006, paired t-test). d, Membrane potential predicted by the tissue simulation upon a 20% uniaxial stretch (line at mean, n=4 simulations, each point is averaged across all cells in simulation). e, DiSBAC2(3) imaging of MDCK cells before stretching, during stretching, and after destretching showing the reversibility of membrane potential change. f, Quantification of panel e (mean ± s.d., n = 8). Increase in fluorescence intensity indicates depolarization and decrease indicates hyperpolarization. Time of stretch and destretch are indicated by cyan and yellow arrows respectively. g, Membrane potential predicted by the tissue simulation upon stretch and subsequent destretch (n=4 simulations, each point is averaged across all cells in simulation). h, NoRI images of cellular biomass in response to tissue stretch. Biomass density recovers to the initial biomass density level overnight. i, Quantification of panel h. 0 hr represents time of stretch. (n=10, p-values: 0.0006 (initial vs 0 hr), 0.0001 (0 hr vs overnight), unpaired t test). j, Tissue simulation recapitulates the drop of biomass density upon stretch and overnight recovery. (mean ± s.d., n=4 simulations) k, Rescue of destretching-induced tissue crowding by ouabain. After 16 hr, a significant number of cells had been eliminated, consistent with previous studies59,60. l, Quantification of panel k (mean ± s.e.m., n = 3 experiments of 20 ROIs each, p-value < 0.0001, unpaired t-test). m, Tissue simulation recapitulates cell elimination after compression and rescue from elimination by depolarizing drugs (mean ± s.e.m., n=4 simulations).
Depolarization rescues cells from elimination after mechanical compression
We wished to directly test the role of the membrane potential in regulating the response of cells to mechanical perturbations. Therefore, we grew MDCK cells on pre-stretched membranes to high cell number density and then released the stretch to induce cell crowding and compression of the tissue (Fig. 2k). Cell extrusion in response to tissue compression is well established59,60. If crowding induced hyperpolarization were to play a central role in determining cell number density, it should be possible to retain a higher cell number density simply by depolarizing the tissue. As we expected, in the presence of depolarizing drugs, cells maintained a significantly higher cell number density after the tissue was compressed (Fig. 2k,l red vs. cyan circles). The observed experimental dynamics are consistent with our model and could be recapitulated with the tissue simulation when we simulated uniaxial compression of the tissue (Fig. 2m). In a separate experiment, we induced crowding while cells were still in growth phase and observed the same increase of steady-state cell number density when cells were treated with a different depolarizing drug, TCT48,49 (Fig. S8).
Mechanical stretch results in a wave of depolarization during wound healing caused by a drop in biomass density
Wound healing is a natural situation where substantial dynamical changes in tissue density and mechanical forces are occurring. All tissues can experience injuries that must be detected and quickly repaired in a controlled fashion. During wound healing, mechanisms of tissue homeostasis are operating in high gear and we hypothesized that mechanical regulation may play an important role. To investigate the role of membrane potential in wound healing, we performed scratch wound assays on confluent MDCK monolayers. Strikingly, we observed a wave of depolarization in the tissue, starting at the wound’s immediate edge and moving deeper into the tissue over several hours (Fig. 3a, quantified in 3b). Depolarization at wound edges has been previously observed, but the mechanistic cause of this depolarization has yet to be explained64,65. According to our model, a drop of biomass density due to mechanical stretching of the tissue could explain depolarization. Indeed, we observed an increase in cell area in the tissue indicative of mechanical stretch (Fig. 3a, middle & 3c), as cells became motile via epithelial mesenchymal transition (Fig. S9) and moved into the wound area, pulling on the cell behind them. Using NoRI, we quantified biomass density as a function of distance from the wound edge (Fig. 3d, e & Fig. S10). Remarkably, we observed a drop in biomass density that closely coincided with the depolarization wave. The tissue simulation with the mechano-electro-osmotic model successfully recapitulates the temporal and spatial dynamics of biomass density and membrane potential during wound healing (Fig. 3a, d, lower panels).
Figure 3: A mechanically induced depolarization wave enhances wound healing.

a, Upper panel: DiSBAC2(3) and fluorescently labeled membrane images of MDCK cells imaged over 3 hours after scratch wound. A depolarization wave was observed from the wound edge up to several layers deep into the tissue. Lower panel: Membrane potential from the tissue simulation over the course of wound healing. b, Quantification of membrane potential as a function of distance from scratch wound at different times (mean ± s.d., n=4 rectangular ROIs of 100 μm width). c, Quantification of cell area shows expansion of cells during wound healing (mean ± s.d., n=9). d, Upper panel: NoRI images over the course of wound healing. Lower panel: Biomass density from the tissue simulation over the course of wound healing. e. Quantification of cellular biomass density as a function of distance from scratch wound at different times after wounding (mean ± s.d., n=4 rectangular ROIs of 50 μm width). f, Representative images of control, depolarizing drug treatment (ouabain) and hyperpolarizing drug treatment (valinomycin) during wound healing. Wound border outlines were generated using Wound healing size tool63. g, Quantification of wound area closure as a function of time (mean ± s.d., n=2 wells). Depolarization resulted in faster wound healing (ouabain, red circles), as compared to the untreated control (cyan circles). Hyperpolarization (valinomycin, yellow circles, 5% PEG, green circles) resulted in slower wound healing.
We hypothesized that the physiological function of this depolarization wave might be to upregulate motility and proliferation in cells that are not immediately adjacent to the wound edge. Based on the previous experiments, this would be expected to speed up the wound healing process. Indeed, we observed both depolarization (Fig. 3a) and a drop in biomass density (Fig. S9) in cells immediately adjacent to the wound, as they underwent the epithelial-mesenchymal transition. It is also known that motility forces of cells many cell rows away from the wound generate most of the tissue tension, resulting in wound closure66,67. Thus, according to this hypothesis, cells at the immediate wound edge become motile first and move into the wound. The migration mechanically stretches cells in rows behind them. As this wave of mechanical stretch propagates deeper into the tissue, it causes a drop in biomass density, resulting in depolarization of membrane potential. Cells can detect tissue stretching via depolarization and may upregulate motility, biomass production and proliferation to enhance wound healing.
Depolarization enhances efficiency of wound healing
By applying depolarizing and hyperpolarizing drugs and quantifying wound healing speed, we tested whether modulation of the membrane potential directly affects efficiency of wound healing We used CHO cells instead of MDCK cells because MDCK tissues tended to detach entirely during scratch wounding when an automated scratch wounding device was used. Indeed, even low concentrations of hyperpolarizing drugs (valinomycin) dramatically slowed down wound healing (Fig. 3f, lower panel & Fig. 3g, yellow circles), compared to the DMSO control. We had previously established that compression of cells by raising the external osmolarity results in hyperpolarization of membrane potential (Fig. S4). Indeed, compression by external osmolarity (Fig. 3g, green circles & Fig. S11) phenocopies the effect of hyperpolarizing drugs. Remarkably, we found that the efficiency of wound healing could be increased, becoming faster than wild-type tissues with the addition of depolarizing drugs (Fig. 3f, middle & Fig. 3g, red circles). This is evident as compared to the untreated control (Fig.3f, upper & Fig. 3g, cyan circles). Depolarization by other drugs showed a similar improvement of wound healing efficiency (Fig. S11). These data suggest that depolarizing cells provides them with a ‘head start’ to upregulate motility and proliferation, as compared to the control tissue, where depolarization is only triggered later, by stretching from other cells moving into the wound area.
Membrane potential modulates MAPK signaling
Membrane potential appears to be a direct readout of mechanical forces and tissue density and causally plays an important role in tissue density homeostasis. But how is membrane potential integrated with signal transduction pathways to mediate these phenotypes? We wondered whether membrane potential might directly affect pathways previously implicated in mechano-sensing such as the MAP Kinase (MAPK) pathway8,67–72. Indeed, in a previous study, it was demonstrated that depolarization of the membrane potential can induce K-Ras clustering, providing a plausible route to MAPK pathway activation, but the physiological role of this signaling and the effect on downstream effectors and pathway intermediates of MAPK was never elucidated. MAPK downstream effectors p38 (p-Tyr 182) and JNK (p-Thr 183, p-Tyr 185) have been previously indicated to be responsive to mechano-transduction, and we wanted to check if membrane potential can alter their cellular signaling response39–41. In sub-confluent cells (hereby called sparse in contrast to fully confluent dense cells), pp38 and pJNK predominantly localize in the cytoplasm, whereas they predominantly localize in the nucleus in dense cells. Since sparse cells are more depolarized, we hypothesized that inducing hyperpolarization would cause pp38 and pJNK to translocate to the nucleus, mimicking a dense, hyperpolarized state (Fig 4c, j). Conversely, inducing depolarization in dense cells should reverse this effect, causing pp38 and pJNK to become cytoplasmic (Fig 4f, m). Indeed, the addition of a hyperpolarizing drug (valinomycin, Fig. 4a–b, h–i) and a depolarizing drug (ouabain, Fig. 4d–e, k-l) caused pp38 and pJNK to exhibit this inversion phenotype in both MDCK and MCF10a cells. Mechanistically, nuclear localization of activated p38 and JNK at high tissue density may contribute to tissue density homeostasis by suppressing proliferation, promoting autophagy, inducing apoptosis, or causing cell extrusion68–70. On the other hand, reduction in the level of nuclear localized pJNK under depolarization may promote proliferation at low tissue density.
Figure 4: MAPK signaling is affected by membrane potential.

a-f, Membrane potential dependence of localization of phosphorylated p38 (pp38) in immunostaining. pp38 localizes predominantly in cytoplasm in sub-confluent (sparse) cells, and hyperpolarization by valinomycin results in augmented nuclear localization of pp38 in MDCK and MCF10 cells (panels a-b). pp38 localizes to cell nuclei in dense tissues, and Depolarization by ouabain results in nuclear exclusion of pp38 (panels c-d). Nuclear to cytoplasmic ratio of pp38 is quantified in accompanying plots (box plot, line at mean, error bars represent min to max, n=24, p-values: <0.0001 (MDCK sparse control vs valinomycin), <0.0001 (MDCK dense control vs ouabain), 0.0001 (MCF10A sparse control vs valinomycin), <0.0001 (MCF10A dense control vs ouabain)). h-m, Membrane potential dependence of localization of phosphorylated JNK in immunostaining. Nuclear to cytoplasmic ratio of pJNK is quantified in accompanying plots (box plot, line at mean, error bars represent min to max, n=24, p-values: <0.0001 (MDCK sparse control vs valinomycin), 0.003 (MDCK dense control vs ouabain), 0.0031 (MCF10A sparse control vs valinomycin), 0.0015 (MCF10A dense control vs ouabain)).
Another important mediator of the MAP kinase signaling cascade is the Src kinase. We found that depolarization stabilized pSrc (Fig. S12a), while hyperpolarization strongly reduced its levels (Fig. S12a). Active Src kinase signaling can activate many downstream targets, including EGFR71–73, PI3K74, and PDK1, which can stimulate proliferation and motility. The downstream cellular responses elicited by membrane-potential-mediated Src signaling are consistent with the role of membrane potential in tissue homeostasis and wound healing, shown in Figs. 1–3.
Membrane potential controls Hippo signaling
Our observations so far indicated that cellular membrane potential is a global readout for mechanical cues and that membrane potential regulates pro and anti-proliferative pathways. The Hippo signaling pathway is another classic mediator of mechano-transduction via the downstream effector YAP, which shuttles between nucleus and cytoplasm and promotes cell proliferation when localized to the nucleus4,9. However, the precise mechanisms that render Hippo signaling mechano-sensitive are incompletely understood and ligands of the upstream membrane-bound mediator of Hippo signaling FAT1 have thus far not been discovered.
Conventionally, the nuclear/cytoplasmic shuttling of YAP is thought to be akin to a step function, as cells become confluent29. Instead, we experimentally observed a continuous modulation of nuclear/cytoplasm ratio of YAP by cell number density (Fig. 5a–c). Next, we wanted to see if Hippo signaling could be modulated and overridden by altering the membrane potential. Strikingly, in both sparse MDCK and MCF10a cells, hyperpolarization causes significant nuclear exclusion and cytoplasmic localization of YAP (Fig. 5e,f upper panels) as compared to control. In Figs. S6&S11, we demonstrate that hyperpolarization causes a lower steady-state cell number density and negatively affects wound healing efficiency, consistent with the anti-proliferative effect of nuclear exclusion of YAP under hyperpolarizing conditions.
Figure 5: Hippo pathway mediates membrane potential signaling.

a, YAP immunostaining and fluorescent nuclei images in low density (top panel) and high density MDCK monolayers (bottom panel). b, Quantification of nuclear/cytoplasmic YAP ratio as a function of cell number density. (mean ± s.d., n = 20337, cells binned together by local number density). c, Schematic representation of YAP localization at different cell number densities. d, Schematic diagram of different signaling intermediates in the Hippo pathway. MAPK activation promotes cytoplasmic pJNK, which activates LIMD1 to inhibit LATS1, causing YAP to translocate to the nucleus. In parallel, MST1 colocalizes with the c-terminus tail of FAT1. Release of MST1 from this complex inhibits LATS1 and causes YAP to translocate to the nucleus. e-f, YAP immunostaining of MDCK/MCF10A cells under sparse (top panels) and dense (bottom panels) conditions with corresponding drug perturbations. g, YAP immunostaining of sparse EpH4-Ev cells under control and JNK inhibitor (SP600125) conditions. h, YAP immunostaining of sparse and dense EpH4–1424 cells with quantification. i-j, YAP immunostaining of sparse MDCK/MCF10A cells with control, valinomycin, and valinomycin with MST inhibitor (XMU-MP-1) conditions. The p-value between sparse and valinomycin+MSTi conditions is 0.0092. k, Sparse MCF10A cells with and without FAT1 knockdown (KD) are stained for FAT1 and YAP under control and valinomycin conditions. The nucleus/cytoplasm ratio for FAT1KD cells under control vs valinomycin has a p-value of 0.558 (ns). l, MST1 immunostaining of MCF10A/MDCK cells under sparse control, sparse valinomycin, dense control, and dense ouabain conditions. White arrows highlight areas of increased MST1 organized membrane clusters. For quantifications in panels e-k, the nucleus/cytoplasm ratio of YAP is measured. In the plots, the line is at mean, n=24, and p-value is measured with an unpaired t-test and is <0.0001 unless otherwise specified,
On the other hand, we found that in dense cells, depolarization was also enough to induce YAP nuclear reentry (Fig. 5e,f lower panels). Consistently, we observed stabilization and increased levels of total YAP under depolarizing conditions, whereas total YAP slightly decreased under hyperpolarization (Fig S12a). The observed regulation of YAP by membrane potential can easily be rationalized in the context of our physiological observations. Low tissue density results in depolarized membrane potential, resulting in localization of YAP to the nucleus, promoting cell growth and cell cycle progression37,38. On the other hand, high tissue density is reflected in hyperpolarized membrane potential, resulting in YAP exiting the nucleus and suppressing proliferation. Another previous observation29 of nuclear reentry of YAP in confluent monolayer upon mechanical stretch can also be mechanistically explained by this model, as stretching causes a drop in biomass density and concomitant depolarization of cellular membrane potential as demonstrated in Fig. 2, which promotes nuclear re-entry of YAP (Fig. 5).
pJNK and MST1 are intermediates in membrane potential-modulated Hippo signaling
Next, we wanted to elucidate the molecular mechanism by which changes in membrane potential lead to changes in YAP localization. Previous work has shown that pJNK-mediated LIMD1 phosphorylation inhibits LATS, which releases YAP from the LATS-YAP complex, promoting its nuclear translocation75. Since we have shown that pJNK is regulated by membrane potential, we wanted to check if pJNK plays a key role in controlling YAP localization under membrane potential perturbations. Experimentally, we took advantage of the twin cell lines EpH4-Ev and EpH4–1424, where the latter expresses constitutively active JNK. We added a JNK inhibitor (SP600125) to sparse EpH4-Ev cells (Fig. 5g) and observed significant nuclear exclusion of YAP. Additionally, JNK inhibition replicated this effect in sparse MDCK cells (Fig. S13a–b) and even caused YAP to become more cytoplasmic in dense MCF10A cells (Fig. S13 c–d).
However, if activated JNK were sufficient to drive YAP localization under depolarization, then the constitutively JNK-active EpH4–1424 cells should not exhibit density and membrane potential-dependent YAP localization. However, we did observe that YAP localization still responded to changes in cell number density in constitutively JNK-active EpH4–1424 cells, which suggests that while activated JNK is required for YAP localization (Fig. 5g, Fig. S13), it is not sufficient for controlling YAP localization.
Therefore, we concluded that there must be parallel routes of signal transduction of membrane potential in Hippo signaling. The canonical Hippo signaling pathway involves the membrane localized receptor FAT1 and the downstream signal transduction mediator MST1 (Fig. 5d). Active MST1 phosphorylates LATS1/2, which in turn phosphorylates YAP to inhibit its nuclear localization. We hypothesized that if the FAT1-MST1 axis is involved in membrane potential signal transduction, then MST1 inhibition should break the transduction. To test this, we added a hyperpolarizing drug (valinomycin) to sparse MDCK and MCF10A cells and observed nuclear exclusion of YAP (Fig. 5i,j). However, adding an MST1 inhibitor (XMU-MP-1) along with the hyperpolarizing drug reversed this effect and even caused YAP to become more nuclear (Fig. 5i, j). Therefore, the canonical Hippo signaling pathway with MST1 is necessary for membrane potential mediated signal transduction. But how exactly does membrane potential mediate the signal from FAT1 to MST1 remains unclear.
Active MST1 is known to associate with the C-terminus tail of FAT1 and initiate a multi-protein signalome assembly76 in which it phosphorylates LATS1 to sequester YAP in the cytoplasm. Therefore, we hypothesized that plasma membrane-bound FAT1 could directly respond to membrane potential and thereby mediate the membrane potential signal in the Hippo pathway. We therefore generated a FAT1 KD MCF10a cell line. Using immunostaining, we observed that FAT1 is indeed localized on the cell membrane in sparse MCF10a cells, and that the FAT1 signal disappeared in the knockdown cell line (Fig. 5k upper panel). Under hyperpolarization, we observed significant nuclear exclusion of YAP in the WT cell line (Fig. 5f), but YAP did not translocate to the nucleus in the knockdown cell line (Fig. 5k lower panel). This means that the FAT1-MST1 axis mediates the membrane potential signal in the Hippo pathway.
But how does FAT1 convert membrane potential changes to changes in signaling activity? Membrane potential can affect clustering of membrane leaflet associated signal transduction proteins like K-Ras and EGFR77,78. We hypothesized that membrane potential can affect the FAT1-MST1 complex assembly in a similar way, which could in turn modulate downstream Hippo signaling. We observed that in sparse MDCK and MCF10a cells, MST1 is diffuse in the cytoplasm, whereas in dense cells, MST1 organizes at the membrane (Fig. 5l), confirming its assembly with the C-terminus tail of FAT1. This inhibitory state is classically described as the ‘Hippo-on’ configuration that sequesters YAP in the cytoplasm. Strikingly, in sparse cells, hyperpolarization by valinomycin causes MST1 to form organized clusters on the membrane, mimicking the dense state (Fig. 5l left panel). Conversely, in dense cells, depolarization by ouabain caused disruption of the membrane-organized distribution of MST1, rendering its localization diffuse in the cytoplasm (Fig. 5l right panel). This should turn off the Hippo inhibitory state and promote YAP nuclear re-entry. Combining these results, we conclude that membrane potential mediated Hippo signaling is mediated via the FAT1-MST1 axis, and the JNK-mediated route can further modulate signaling activity.
Membrane potential depolarization during of Xenopus tail regeneration is caused by a decrease in cellular biomass density
Using in-vitro tissue culture experiments, we showed that during wound healing, a wave of membrane potential depolarization is caused by a decrease of biomass density, initiating at the wound edge and propagating deeper into the tissue over time (Fig. 3a–e). We further showed that this wave of depolarization is essential for efficient wound healing (Fig. 3f–g). Therefore, we wanted to see if we would observe a similar phenomenology during regeneration in vivo. Using the Xenopus embryo (stage 40) tail regeneration as a model system, we first validated that the embryos correctly regenerated their tail after amputation (Fig. S14). Indeed, precisely as we observed in cell culture, using a membrane potential indicator, we observed a gradient of depolarization from the wound edge and spanning over 6 cell layers 10 minutes after tail amputation (Fig. 6b). Moreover, using NoRI, we also observed a reduction in biomass density of cells at the wound edge immediately after tail amputation (Fig. 6c left panels). After 10 minutes, we observed that 5 to 6 layers of cells next to the wound edge had become significantly dilute (Fig. 6c right panels). This drop in biomass density is probably due to stretching from a rapid wound healing response, where cell layers from the two lateral sides rapidly roll over and merge to seal the cut wound edge within a very short period.
Figure 6: Interplay of membrane potential and cytoplasmic dilution occurs at the beginning of Xenopus tail regeneration.

a, Schematic diagram showing the experimental set up for Xenopus embryo tail amputation. b, Brightfield and membrane potential (DiSBAC2(3)) images of Xenopus wound edge, 10 minutes post-amputation. Cell layers get progressively depolarized from deep tissue towards the wound edge. c, NoRI images of Xenopus tail amputation, 0 min and 10 min post-cut. One cell layer becomes dilute in biomass density immediately after the cut, and 5–6 cell layers become dilute in 10 minutes. Images represent different Xenopus embryos amputated and fixed at the specified times.
Finally, as we hypothesized that this wound healing mechanism could be evolutionarily conserved, we conducted a similar experiment in the fresh-water polyp Hydra vulgaris that diverged evolutionarily at an early metazoan stage. Indeed, we observed a similar gradient of biomass density dilution in epithelial and mesogleal cells after amputation of the body trunk (Fig. S15).
Discussion
Membrane potential can convey cell size within a tissue in relation to cellular biomass, as well as the mechanical state of a cell. These cellular and tissue-level variables are of central physiological importance for homeostasis and development. However, these cellular-scale properties are difficult for cells to accurately assess from local protein interactions. Membrane potential on the other hand, provides a quasi-instantaneous, globally integrated readout of cellular state. The coupling between mechanical pressure, density and membrane potential is based only on elementary physical laws, requires no active regulation, and is present even in thermodynamic equilibrium44. Therefore, even primitive cells, lacking active transport and confined by a simple cell wall, could have utilized membrane potential as a readout of their mechanical state and biomass concentration. Membrane-potential-mediated mechano-transduction may therefore have been among the first homeostatic mechanisms to emerge in multicellular organisms.
Note that while we assumed the absence of active regulation or amplification of membrane potential in our model as a proof-of-concept, there are many ways how cells could enhance and amplify changes in membrane potential to reflect even small differences in density and mechanical forces. Fixing membrane potential to a constant homeostatic value (like body temperature in mammals) would result in a loss of valuable information conveyed by membrane potential. Therefore, cells may have evolved to actively amplify changes of membrane potential signaling with mechanical pressure. Indeed, observed changes in membrane potential are sometimes even larger than those expected from our model without signal amplification (Fig. S4).
Note also that while we have shown that several important mechano-transduction pathways are controlled by membrane potential, there are likely other parallel signal transduction routes that are affected by membrane potential. Calcium signaling would be an ideal effector of membranepotential-mediated mechano-transduction. This could occur either via voltage-gated channels in the plasma membrane or the strongly voltage-dependent equilibrium calcium concentration that results from the calcium ion’s two positive charges. Indeed, flux through key calcium channels like Piezo has been shown to strongly depend on membrane potential79 and Piezo has been shown to be voltage-gated80.
The concept of mechano-sensing via membrane potential should not be thought of as limited to epithelia. For instance, stem cell differentiation has been shown to depend on both cell volume and osmotic pressure81,82, as well as membrane potential83, suggesting a role of membrane-potential-mediated mechanical signaling. As another example, the response of membrane potential to cellular compression may play a role in neuronal mechano-transduction if mechanical forces were to affect total neuronal cell volume. The resulting changes in resting membrane potential could trigger voltage-gated channels and calcium siganling79,80, independent of tension in the plasma membrane.
Finally, the importance of membrane potential in tissue growth control that we report, suggests that mis-regulation of membrane potential or its downstream signaling cascade could have dramatic consequences for tissue homeostasis, potentially resulting in tumorigenesis when cells stop responding to the mechanical cues from their environment. In fact, this may explain the frequent occurrence of ectopic expression of neuronal ion channels in tumors84 and the commonly observed phenotype of depolarized resting membrane potential in cancer85.
Supplementary Material
Acknowledgements
We would like to thank Sean Megason, Tim Mitchison, Jan Skotheim, and Xavier Trepat for their comments and feedback on the manuscript. We would like to thank Peter Sorger and Clarance Yapp for access to their high content imaging systems. Thanks to Eugenia Piddini (University of Bristol), J.J. Fredberg and C.Y. Park (Harvard T.H. Chan School of Public Health) for helping us with MDCK cells. We thank Nikon imaging center at Harvard Medical School for help with all fluorescence imaging experiments. Specifically, we thank Jennifer Waters, Tally Lambert, Anna Payne-Tobin Jost for their valuable support with the microscopy. Thanks to Florian Engert for helping us with Hydra experiments, and special thanks to Andrew Murphy of Sean Megason lab for helping us with Hydra husbandry. Thanks to Bill Jia of Megason lab for valuable comments on membrane potential measurement. This project was supported by MIRA grant (5R35GM137895) and an HMS Junior Faculty Armenise grant to M.B. N.C.B was supported by following grants National Science Foundation Graduate Research Fellowship Program (DGE 2140743) and Systems, Synthetic, and Quantitative Biology Training grant award (T32GM135014). YH was supported by HCRP of Harvard College. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation. Leonid Peshkin was supported by R01AG073341 and R24OD031956 and Marc W. Kirschner was awarded R01AG073341 and MIRA R35 GM145248. Christophe Dupre was awarded Swiss National Science Foundation Early Postdoc Mobility Fellowship (P2SKP3-187684).
References
- 1.Molecular Biology of the Cell: Reference Edition, Band 1. (2008).
- 2.Hille B. Ion channels of excitable membranes. in (2001).
- 3.Hino N. et al. ERK-Mediated Mechanochemical Waves Direct Collective Cell Polarization. Dev Cell 53, 646–660.e8 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Panciera T., Azzolin L., Cordenonsi M. & Piccolo S. Mechanobiology of YAP and TAZ in physiology and disease. Nat Rev Mol Cell Biol 18, 758–770 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Niisato N., Post M., Van Driessche W. & Marunaka Y. Cell Swelling Activates Stress-Activated Protein Kinases, p38 MAP Kinase and JNK, in Renal Epithelial A6 Cells. Biochem Biophys Res Commun 266, 547–550 (1999). [DOI] [PubMed] [Google Scholar]
- 6.Kishi H. et al. Osmotic Shock Induces G1 Arrest through p53 Phosphorylation at Ser33 by Activated p38MAPK without Phosphorylation at Ser15 and Ser20. Journal of Biological Chemistry 276, 39115–39122 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Lee S.-H., Park Y., Yoon S. K. & Yoon J.-B. Osmotic Stress Inhibits Proteasome by p38 MAPK-dependent Phosphorylation. Journal of Biological Chemistry 285, 41280–41289 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.HuangFu W.-C., Omori E., Akira S., Matsumoto K. & Ninomiya-Tsuji J. Osmotic Stress Activates the TAK1-JNK Pathway While Blocking TAK1-mediated NF-κB Activation. Journal of Biological Chemistry 281, 28802–28810 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dupont S. et al. Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Zhu J. & Thompson C. B. Metabolic regulation of cell growth and proliferation. Nat Rev Mol Cell Biol 20, 436–450 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hosios A. M. et al. Amino Acids Rather than Glucose Account for the Majority of Cell Mass in Proliferating Mammalian Cells. Dev Cell 36, 540–549 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palm W. & Thompson C. B. Nutrient acquisition strategies of mammalian cells. Nature 546, 234–242 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vander Heiden M. G., Cantley L. C. & Thompson C. B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science (1979) 324, 1029–1033 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pavlova N. N., Zhu J. & Thompson C. B. The hallmarks of cancer metabolism: Still emerging. Cell Metab 34, 355–377 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barbazan J. et al. Cancer-associated fibroblasts actively compress cancer cells and modulate mechanotransduction. Nat Commun 14, 6966 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim S., Uroz M., Bays J. L. & Chen C. S. Harnessing Mechanobiology for Tissue Engineering. Dev Cell 56, 180–191 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Humphrey J. D., Dufresne E. R. & Schwartz M. A. Mechanotransduction and extracellular matrix homeostasis. Nat Rev Mol Cell Biol 15, 802–812 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pensalfini M. & Tepole A. B. Mechano-biological and bio-mechanical pathways in cutaneous wound healing. PLoS Comput Biol 19, e1010902 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosińczuk J., Taradaj J., Dymarek R. & Sopel M. Mechanoregulation of Wound Healing and Skin Homeostasis. Biomed Res Int 2016, 1–13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cambria E. et al. Linking cell mechanical memory and cancer metastasis. Nat Rev Cancer 24, 216–228 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chaudhuri P. K., Low B. C. & Lim C. T. Mechanobiology of Tumor Growth. Chem Rev 118, 6499–6515 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Papavassiliou K. A., Basdra E. K. & Papavassiliou A. G. The emerging promise of tumour mechanobiology in cancer treatment. Eur J Cancer 190, 112938 (2023). [DOI] [PubMed] [Google Scholar]
- 23.Clevenger A. J. et al. Advances in cancer mechanobiology: Metastasis, mechanics, and materials. APL Bioeng 8, (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong S. H. D. et al. Mechanical manipulation of cancer cell tumorigenicity via heat shock protein signaling. Sci Adv 9, (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li C. et al. Extracellular matrix-derived mechanical force governs breast cancer cell stemness and quiescence transition through integrin-DDR signaling. Signal Transduct Target Ther 8, 247 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Labernadie A. et al. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat Cell Biol 19, 224–237 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aragona M. et al. A Mechanical Checkpoint Controls Multicellular Growth through YAP/TAZ Regulation by Actin-Processing Factors. Cell 154, 1047–1059 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Piccolo S., Dupont S. & Cordenonsi M. The Biology of YAP/TAZ: Hippo Signaling and Beyond. Physiol Rev 94, 1287–1312 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Benham-Pyle B. W., Pruitt B. L. & Nelson W. J. Mechanical strain induces E-cadherin–dependent Yap1 and β-catenin activation to drive cell cycle entry. Science (1979) 348, 1024–1027 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo J. et al. The oncogenic roles and clinical implications of YAP/TAZ in breast cancer. Br J Cancer 128, 1611–1624 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H. et al. YAP/TAZ drives cell proliferation and tumour growth via a polyamine–eIF5A hypusination–LSD1 axis. Nat Cell Biol 24, 373–383 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saotome K. et al. Structure of the mechanically activated ion channel Piezo1. Nature 554, 481–486 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ranade S. S. et al. Piezo2 is the major transducer of mechanical forces for touch sensation in mice. Nature 516, 121–125 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coste B. et al. Piezo proteins are pore-forming subunits of mechanically activated channels. Nature 483, 176–181 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kosmalska A. J. et al. Physical principles of membrane remodelling during cell mechanoadaptation. Nat Commun 6, 7292 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shi Z., Graber Z. T., Baumgart T., Stone H. A. & Cohen A. E. Cell Membranes Resist Flow. Cell 175, 1769–1779.e13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Devany J., Falk M. J., Holt L. J., Murugan A. & Gardel M. L. Epithelial tissue confinement inhibits cell growth and leads to volume-reducing divisions. Dev Cell 58, 1462–1476.e8 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oh S. et al. Protein and lipid mass concentration measurement in tissues by stimulated Raman scattering microscopy. Proc Natl Acad Sci U S A 119, e2117938119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buckley C. E. & St Johnston D. Apical–basal polarity and the control of epithelial form and function. Nat Rev Mol Cell Biol 23, 559–577 (2022). [DOI] [PubMed] [Google Scholar]
- 40.Park H. et al. Three-dimensional refractive index tomograms and deformability of individual human red blood cells from cord blood of newborn infants and maternal blood. 10.1117/1.JBO.20.11.111208 20, 111208 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Shin S., Kim K., Yoon J. & Park Y. Active illumination using a digital micromirror device for quantitative phase imaging. Optics Letters, Vol. 40, Issue 22, pp. 5407–5410 40, 5407–5410 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Shaw K. L., Grimsley G. R., Yakovlev G. I., Makarov A. A. & Pace C. N. The effect of net charge on the solubility, activity, and stability of ribonuclease Sa. Protein Science 10, 1206–1215 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanford C. The Interpretation Of Hydrogen Ion Titration Curves Of Proteins. in 69–165 (1963). doi: 10.1016/S0065-3233(08)60052-2. [DOI] [Google Scholar]
- 44.Vallina Estrada E., Zhang N., Wennerström H., Danielsson J. & Oliveberg M. Diffusive intracellular interactions: On the role of protein net charge and functional adaptation. Curr Opin Struct Biol 81, 102625 (2023). [DOI] [PubMed] [Google Scholar]
- 45.Donnan F. G. Theorie der Membrangleichgewichte und Membranpotentiale bei Vorhandensein von nicht dialysierenden Elektrolyten. Ein Beitrag zur physikalisch-chemischen Physiologie. Zeitschrift für Elektrochemie und angewandte physikalische Chemie 17, 572–581 (1911). [Google Scholar]
- 46.Abdelfattah A. S. et al. Bright and photostable chemigenetic indicators for extended in vivo voltage imaging. Science (1979) 365, 699–704 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Cone C. D. & Tongier M. Contact inhibition of division: involvement of the electrical transmembrane potential. J Cell Physiol 82, 373–86 (1973). [DOI] [PubMed] [Google Scholar]
- 48.Barboiu M. et al. Polarized Water Wires under Confinement in Chiral Channels. J Phys Chem B 119, 8707–8717 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Barboiu M. et al. An artificial primitive mimic of the Gramicidin-A channel. Nat Commun 5, 4142 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Goldman D. E. POTENTIAL, IMPEDANCE, AND RECTIFICATION IN MEMBRANES. J Gen Physiol 27, 37 (1943). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.TOSTESON D. C. & HOFFMAN J. F. Regulation of Cell Volume by Active Cation Transport in High and Low Potassium Sheep Red Cells. Journal of General Physiology 44, 169–194 (1960). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kay A. R. & Blaustein M. P. Evolution of our understanding of cell volume regulation by the pump-leak mechanism. Journal of General Physiology 151, 407–416 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Armstrong C. M. The Na/K pump, Cl ion, and osmotic stabilization of cells. Proceedings of the National Academy of Sciences 100, 6257–6262 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Manning G. S. Counterion Binding in Polyelectrolyte Theory. Acc Chem Res 12, 443–449 (1979). [Google Scholar]
- 55.Manning G. S. Limiting Laws and Counterion Condensation in Polyelectrolyte Solutions I. Colligative Properties. J Chem Phys 51, 924–933 (1969). [Google Scholar]
- 56.Manning G. S. Limiting laws and counterion condensation in polyelectrolyte solutions. Biophys Chem 7, 95–102 (1977). [DOI] [PubMed] [Google Scholar]
- 57.Dyer K. F. The Quiet Revolution: A New Synthesis of Biological Knowledge. J Biol Educ 5, 15–24 (1971). [Google Scholar]
- 58.Milo R., Jorgensen P., Moran U., Weber G. & Springer M. BioNumbers—the database of key numbers in molecular and cell biology. Nucleic Acids Res 38, D750 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eisenhoffer G. T. & Rosenblatt J. Bringing balance by force: live cell extrusion controls epithelial cell numbers. Trends Cell Biol 23, 185–192 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eisenhoffer G. T. et al. Crowding induces live cell extrusion to maintain homeostatic cell numbers in epithelia. Nature 484, 546–549 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Park J. et al. Screening fluorescent voltage indicators with spontaneously spiking HEK cells. PLoS One 8, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Burgstahler R. et al. Confocal ratiometric voltage imaging of cultured human keratinocytes reveals layer-specific responses to ATP. Am J Physiol Cell Physiol 284, (2003). [DOI] [PubMed] [Google Scholar]
- 63.Suarez-Arnedo A. et al. An image J plugin for the high throughput image analysis of in vitro scratch wound healing assays. PLoS One 15, e0232565 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chifflet S. et al. Early and late calcium waves during wound healing in corneal endothelial cells. Wound Repair and Regeneration 20, 28–37 (2012). [DOI] [PubMed] [Google Scholar]
- 65.Reid B. & Zhao M. The Electrical Response to Injury: Molecular Mechanisms and Wound Healing. Adv Wound Care (New Rochelle) 3, 184–201 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Trepat X. et al. Physical forces during collective cell migration. Nat Phys 5, 426–430 (2009). [Google Scholar]
- 67.Ladoux B. Cells guided on their journey. Nat Phys 5, 377–378 (2009). [Google Scholar]
- 68.Zeke A., Misheva M., Reményi A. & Bogoyevitch M. A. JNK Signaling: Regulation and Functions Based on Complex Protein-Protein Partnerships. Microbiol Mol Biol Rev 80, 793 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maik-Rachline G., Lifshits L. & Seger R. Nuclear P38: Roles in Physiological and Pathological Processes and Regulation of Nuclear Translocation. International Journal of Molecular Sciences 2020, Vol. 21, Page 6102 21, 6102 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dhanasekaran D. N. & Reddy E. P. JNK signaling in apoptosis. Oncogene 27, 6245–6251 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stover D. R., Becker M., Liebetanz J. & Lydon N. B. Src Phosphorylation of the Epidermal Growth Factor Receptor at Novel Sites Mediates Receptor Interaction with Src and P85α. Journal of Biological Chemistry 270, 15591–15597 (1995). [DOI] [PubMed] [Google Scholar]
- 72.Tice D. A., Biscardi J. S., Nickles A. L. & Parsons S. J. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proceedings of the National Academy of Sciences 96, 1415–1420 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Maa M. C., Leu T. H., McCarley D. J., Schatzman R. C. & Parsons S. J. Potentiation of epidermal growth factor receptor-mediated oncogenesis by c-Src: implications for the etiology of multiple human cancers. Proceedings of the National Academy of Sciences 92, 6981–6985 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Beadnell T. C. et al. Src-mediated regulation of the PI3K pathway in advanced papillary and anaplastic thyroid cancer. Oncogenesis 7, 23 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Codelia V. A., Sun G. & Irvine K. D. Regulation of YAP by Mechanical Strain through Jnk and Hippo Signaling. Current Biology 24, 2012–2017 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Martin D. et al. Assembly and activation of the Hippo signalome by FAT1 tumor suppressor. Nat Commun 9, 2372 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang Y. et al. Regulation of EGFR nanocluster formation by ionic protein-lipid interaction. Cell Res 24, 959–976 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhou Y. et al. Membrane potential modulates plasma membrane phospholipid dynamics and K-Ras signaling. Science (1979) 349, 873–876 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Coste B. et al. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science (1979) 330, 55–60 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moroni M., Servin-Vences M. R., Fleischer R., Sánchez-Carranza O. & Lewin G. R. Voltage gating of mechanosensitive PIEZO channels. Nature Communications 2018 9:1 9, 1–15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Guo M. et al. Cell volume change through water efflux impacts cell stiffness and stem cell fate. Proc Natl Acad Sci U S A 114, E8618–E8627 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fan Y. L., Zhao H. C. & Feng X. Q. Hypertonic pressure affects the pluripotency and self-renewal of mouse embryonic stem cells. Stem Cell Res 56, 102537 (2021). [DOI] [PubMed] [Google Scholar]
- 83.Levin M. Leading Edge Bioelectric signaling: Reprogrammable circuits underlying embryogenesis, regeneration, and cancer. Cell 184, 1971–1989 (2021). [DOI] [PubMed] [Google Scholar]
- 84.Li M. & Xiong Z. G. Ion channels as targets for cancer therapy. Int J Physiol Pathophysiol Pharmacol 3, 156 (2011). [PMC free article] [PubMed] [Google Scholar]
- 85.Yang M. & Brackenbury W. J. Membrane potential and cancer progression. Front Physiol 4, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.