

Significance
Many neurological disorders share symptoms, despite disparity among diseases. Treatments are prescribed based on diagnosis rather than individual symptoms. While only treating symptoms may obscure the disease, mechanism-based drug development allows the two approaches to converge. Hub proteins, those that coordinate the expression of proteins that mediate specific cellular functions, may be dysregulated across a broad range of disorders. We show that the RBP DJ-1 controls the activity of L-type voltage-gated calcium channels (L-VGCC), via the expression of its alpha subunit CaV1.2 and the auxiliary subunit alpha2delta2 (α2δ2). Importantly, we demonstrate that this pathway is commonly disrupted among neurological disorders, namely Alzheimer’s disease (AD) and tuberous sclerosis complex (TSC). Collectively, these data rationalize mechanism-based drug therapy to treat disease.
Keywords: voltage-gated calcium channels, RNA-binding protein, tuberous sclerosis complex, Alzheimer’s disease, mammalian target of rapamycin
Abstract
L-type voltage-gated calcium (Ca2+) channels (L-VGCC) dysfunction is implicated in several neurological and psychiatric diseases. While a popular therapeutic target, it is unknown whether molecular mechanisms leading to disrupted L-VGCC across neurodegenerative disorders are conserved. Importantly, L-VGCC integrate synaptic signals to facilitate a plethora of cellular mechanisms; however, mechanisms that regulate L-VGCC channel density and subcellular compartmentalization are understudied. Herein, we report that in disease models with overactive mammalian target of rapamycin complex 1 (mTORC1) signaling (or mTORopathies), deficits in dendritic L-VGCC activity are associated with increased expression of the RNA-binding protein (RBP) Parkinsonism-associated deglycase (DJ-1). DJ-1 binds the mRNA coding for the alpha and auxiliary Ca2+ channel subunits CaV1.2 and α2δ2, and represses their mRNA translation, only in the disease states, specifically preclinical models of tuberous sclerosis complex (TSC) and Alzheimer’s disease (AD). In agreement, DJ-1-mediated repression of CaV1.2/α2δ2 protein synthesis in dendrites is exaggerated in mouse models of AD and TSC, resulting in deficits in dendritic L-VGCC calcium activity. Finding of DJ-1-regulated L-VGCC activity in dendrites in TSC and AD provides a unique signaling pathway that can be targeted in clinical mTORopathies.
Tuberous sclerosis complex (TSC) is considered a linchpin mTORopathy, suggesting that research findings on TSC may be broadly applicable to other neurological disorders with dysregulated mTOR activity, such as Alzheimer’s disease (AD) (1). TSC results from a loss of function of either TSC1 or TSC2, upstream inhibitors of the mTOR signaling (2). Loss of TSC1 repression leads to constitutive activity of mTOR (2). Dysfunction of TSC2 and hyperactive mTOR have been reported in Alzheimer’s disease (AD) (3); however, to date, nothing is known about common downstream signaling mechanisms shared between TSC and AD leading to neuronal dysfunction.
mTOR forms two complexes—mTORC1 and mTORC2. The classic role for mTORC1 is to signal downstream proteins that promote mRNA translation initiation, leading to an increase in protein synthesis (4, 5). In contrast, our laboratory has shown that mTORC1 signaling actively represses protein synthesis of select mRNAs in the brain (6, 7). In agreement with mTOR’s role in repression of mRNA translation, Bear and colleagues found that in a preclinical model of TSC neuronal protein synthesis was reduced (8). Likewise, neuronal protein synthesis deficits have been detected in AD (9). Mechanisms describing how mTORC1 activity represses protein synthesis, which is likely to lead to the pathological state, are beginning to emerge (6, 10).
We previously identified Parkinsonism-associated deglycase (DJ-1), an RNA-binding protein (RBP), as an mTORC1-sensitive protein that is up-regulated with mTORC1 activity and overexpressed in Tsc1-null neuronal dendrites (6). DJ-1 has been reported to repress translation of its target mRNAs (11). Thus, we hypothesize that mTORC1’s upregulation of DJ-1, combined with DJ-1’s repressive RNA-binding properties, makes it a suitable candidate for mediating deficits in protein synthesis in TSC and AD.
Neuronal hyperexcitability is a common symptom in both TSC and AD (12, 13). Loss of ion channel expression/function is a prominent cause of hyperexcitability (14). Indeed, animal models that harbor loss of function mutations in critical ion channels suffer from hyperexcitable circuits (15, 16). Additionally, ion-channel dysfunction has been implicated in neuropsychiatric disorders comorbid with TSC and AD (17–22). To date, ion channels are undercharacterized in TSC and AD. Collectively, these data led us to predict that overactive mTORC1 signaling leads to a loss of critical ion channels due to deficits in protein synthesis in both diseases.
Based on the parallel findings outlined above in TSC and AD, we hypothesize that there will be common mechanisms between these disorders. Herein, we demonstrate in two preclinical models, TSC1 conditional knockout (TSC) and APP/PS1 (AD) mice, that the mTORC1-regulated RNA-binding protein DJ-1 1) represses the protein synthesis of the dendritic L-type voltage-gated calcium channels (CaV1.2) and its auxiliary subunit α2δ2 in TSC and AD mouse models, 2) contributes to deficits in dendritic L-VGCC activity in both disease models, and 3) synaptic expression of DJ-1, CaV1.2, and α2δ2 is disrupted in cortices from human AD patients.
Results
mRNAs Encoding for Calcium Channel Subunits Contain Several Predicted DJ-1 Binding Sites.
How does overactive mTORC1 signaling in TSC lead to deficits in protein synthesis? To begin to address this question, we reasoned that the RNA-binding protein DJ-1 is up-regulated by mTORC1 activity and is predicted to repress mRNA translation and thus, is a promising target. We first identified a putative DJ-1 RNA-binding motif(s), starting with a list of known DJ-1 target mRNAs reported by van der Brug et al. and computationally defined the short, highly represented DJ-1-binding sequence—GNGCNG and CNGCNG (11). To predict which synaptic proteins are putative DJ-1 target mRNAs, we screened a list of 1416 postsynaptic-associated proteins for the DJ-1-binding motifs (6, 23, 24). We found that Grin2a was a predicted DJ-1 mRNA target present in the postsynaptic proteome and common between three overactive mTORC1-related diseases; epilepsy, autism spectrum disorder (ASD), and AD (6). We used Grin2a as a threshold with 81 predicted DJ-1 binding motifs, to identify novel DJ-1 binding mRNA targets. Out of 1,416 postsynaptic-associated proteins, 214 were above the set threshold (Dataset S1). To test the specificity of the predicted DJ-1 binding sites, we compared the frequency/kb of DJ-1 binding sites between putative DJ-1-targets and nontargets. This analysis determined that the difference in binding sites between targets and nontargets are significantly different (P = 1.2 × 10−38, Student’s t test).
Next, we note that several voltage-gated calcium channel subunits are probable DJ-1 target mRNAs (Fig. 1A). Interestingly, Cacna1a and Cacna1c, the pore forming alpha subunits, have the highest numbers of predicted binding sites, 158 and 156 respectively (25), while the second highest number resides in the mRNA coding for the auxiliary subunit, α2δ2 with 98 putative binding sites. Therefore, DJ-1, an RBP, might play a role in differentially regulating de novo protein synthesis of calcium channel subunits in TSC and AD.
Fig. 1.
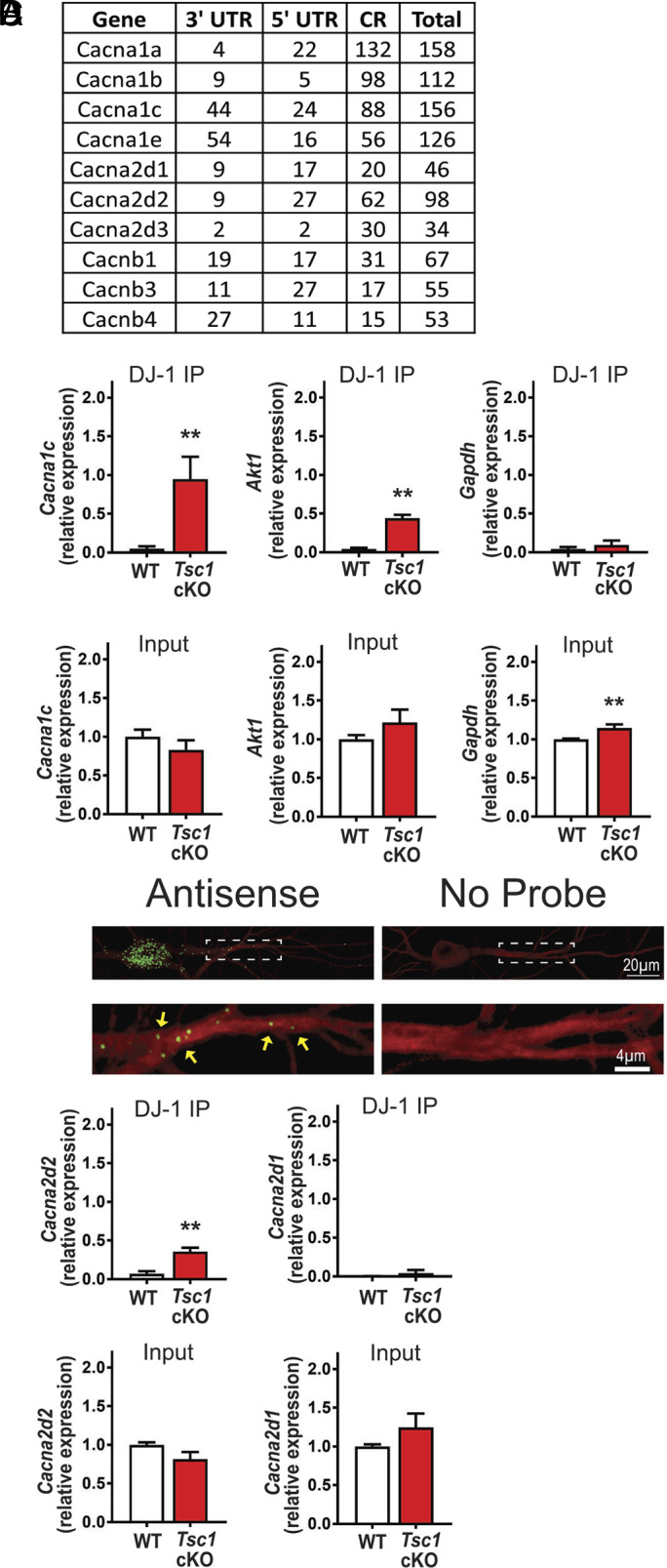
DJ-1 associates with Cacna2d2 and Cacna1c in Tsc1 cKO. (A) List of predicted DJ-1 binding mRNA targets coding for Ca2+ channel subunits (coding region, CR) (B) (Top) qRT-PCR analyses of Tsc1Cre/Cre: KO and Tsc1 fl/fl: WT from DJ-1-RNA-immunoprecipitation. Cacna1c: WT = 0.05 ± 0.03, Tsc1 cKO = 0.95 ± 0.3, P = 0.03. Akt1: WT = 0.04 ± 0.02, Tsc1 cKO = 0.44 ± 0.05, P = 0.001. Gapdh: WT = 0.04 ± 0.03, Tsc1 cKO = 0.09 ± 0.06, P = 0.47. (Bottom) qRT-PCR of 5% of the input. Cacna1c: WT = 1 ± 0.09, Tsc1 cKO = 0.83 ± 0.1251, P = 0.29. Akt1: WT = 1 ± 0.05, Tsc1 cKO = 1.22 ± 0.17, P = 0.24. Gapdh: WT = 1 ± 0.01, Tsc1 cKO = 1.15 ± 0.05, P = 0.006. n = 3 technical replicates, 3 independent mice. Student’s t test, two-tailed. (C) (Top) Representative images of Cacna2d2 mRNA. (Bottom) Magnified dendrites (dashed-lines) Cacna2d2 mRNA, indicated by green puncta/yellow arrowheads. (D) (Top) qRT-PCR of mRNA isolated from DJ-1 RNA-IP. Cacna2d2: WT = 0.07 ± 0.04, Tsc1 cKO = 0.4 ± 0.05, P = 0.01. Cacna2d1: WT = 0.002 ± 0.002, Tsc1 cKO = 0.04 ± 0.04, P = 0.4. (Bottom) qRT-PCR of 5% input RNA from DJ-1 RNA-IP. Cacna2d2: WT = 1 ± 0.03, Tsc1 cKO = 0.8 ± 0.09, P = 0.08. Cacna2d1: WT = 1 ± 0.03, Tsc1 cKO = 1.23 ± 0.21, P = 0.26. N = 3 technical replicates, 3 independent mice, t test, two-tailed.
Cacna1c and Cacna2d2 mRNA Preferentially Associate with DJ-1 in TSC.
To determine the best candidate to pursue, we considered our previous work suggesting that 1) mTORC1 activity promotes DJ-1 de novo protein synthesis in neuronal dendrites, 2) both Cacna1c mRNA and the protein it codes for, CaV1.2, are present in dendrites, and 3) DJ-1 is overexpressed in Tsc1-null dendrites (6, 26). Using a DJ-1 specific antibody, we performed an RNA-immunoprecipitation (RIP) using cortical lysates from Tsc1-null mice and WT littermates [Tsc1fl/flSyn1-Cre+: Tsc1 cKO, and Tsc1fl/fl: WT; (27, 28)]. Western blot analysis shows the specificity of the DJ-1 immunoprecipitation (SI Appendix, Fig. S1). When the DJ-1-bound mRNAs are isolated, Cacna1c mRNA was detected only in Tsc1 cKO lysates via qRT-PCR, but not in WT or IgG, similar to the positive control Akt1 (11). In contrast, the negative control, Gapdh mRNA, does not bind to DJ-1 in either genotype (Fig. 1 B, Top and SI Appendix, Fig. S2). Of note, the input for Cacna1c and Akt1 are similar between genotypes (Fig. 1 B, Bottom). These results suggest that Cacna1c binds to DJ-1 in a disease-specific manner (Fig. 1 B, Top and SI Appendix, Fig. S2).
Next, we hypothesized that mRNAs with coordinated function are repressed together, suggesting that Cacna2d2 may be a target of DJ-1. Importantly, Cacna2d2 has 98 predicted DJ-1 binding sites, more than the other Cacna2d mRNAs (29). Paradoxically, the literature suggests that α2δ2 is expressed at presynaptic terminals (30, 31); however, it has been detected in the postsynaptic proteome (23). To verify the dendritic presence of α2δ2 mRNA, we performed an in situ hybridization in cultured hippocampal neurons. Indeed, Cacna2d2 mRNA was detected in Map2 (microtubule-associated protein 2) positive dendrites using antisense probes [green puncta (RNA) within red dendrite (Map2)], with no signal in the no probe control (Fig. 1C). These data combined with the literature suggest that DJ-1 protein and mRNA coding for CaV1.2 and α2δ2 are present in dendrites (6, 26).
Next, we asked whether Cacna2d2 mRNA binds to DJ-1. Again, DJ-1 RNA-IP revealed that Cacna2d2 mRNA only binds to DJ-1 in Tsc1 cKO cortical lysates (Fig. 1 D, Top and SI Appendix, Fig. S2). As an additional control, we assessed whether Cacna2d1 mRNA binds DJ-1, which bears 46 predicted DJ-1 binding motifs, below our set threshold. Indeed, Cacna2d1 did not associate with DJ-1 in either genotype, as verified by a single t test that was not significant from zero. The input mRNA expression is not significantly different between genotypes (Fig. 1 D, Bottom). These data suggest that Cacna1c and Cacna2d2 selectively bind to DJ-1 in the disease state.
Reduced De Novo Protein Synthesis of CaV1.2 and α2δ2 but Not α2δ1 in the Hippocampus of Tsc1 Conditional Knockout (Tsc1 cKO) Mice.
Based on the DJ-1 RNA-IP experiment, we predict that new protein synthesis of CaV1.2 and α2δ2 is reduced in TSC. Thus, we performed a surface sensing of translation (SUnSET) assay, combined with proximity ligation assay (PLA), to label newly synthesized CaV1.2 and α2δ2 in the hippocampus of Tsc1 cKO mice, that have excess DJ-1 expression (6). As predicted, there was reduced protein synthesis of CaV1.2 in Tsc1 cKO hippocampal slices (Fig. 2 A and B and SI Appendix, Fig. S3). As an additional control, we assessed newly synthesized α2δ1 protein. Interestingly, we saw that there was a significant increase in newly synthesized α2δ1 in Tsc1 cKO hippocampal slices (Fig. 2C). Collectively, the binding of DJ-1 to Cacna1c and Cacna2d2 mRNAs is in line with DJ-1’s function as a translational repressor.
Fig. 2.

Tsc1 cKO mouse model exhibits decreased de novo protein synthesis of CaV1.2 and α2δ2, but not α2δ1. De novo protein synthesis, visualized by SUnSET-PLA (green) in hippocampal dendrites (MAP2, red). WT (W) and TSC (T) dendrites are outlined by broken lines. (A) Basal CaV1.2 protein synthesis is detected in dendrites of WT is markedly reduced in TSC, quantified below (WT = 1.00 ± 0.07, 117 total ROIs, 12 to 18 ROIs/slice, 2 to 3 slices/animal, 3 animals; Tsc1 cKO = 0.01 ± 0.01, 106 total ROIs, 12 to 18 ROIs/slice, 2 to 3 slices/animal, 3 animals). (B) α2δ2 basal new protein synthesis is detected in dendrites of WT but is attenuated in TSC as quantified below (WT = 1.00 ± 0.15, n = 77 total ROIs, 12 to 18 ROIs/slice, 2 to 3 slices/animal, 3 animals; Tsc1 cKO = 0.12 ± 0.02, n = 73 total ROIs, 12 to 18 ROIs/slice, 2 to 3 slices/animal, 3 animals). (C) Basal α2δ1 protein synthesis in dendrites of WT is lower than TSC as quantified below (WT = 1.00 ± 0.17, n = 28 total ROIs, 12 to 18 ROIs/slice, 2 to 3 slices/animal, 3 animals; Tsc1 cKO = 3.97 ± 0.90, n = 17 total ROIs, 12 to 18 ROIs/slice, 2 to 3 slices/animal, 3 animals). For representative images in A through C, CaV1.2, α2δ2, and α2δ1 puncta were dilated once using ImageJ. Bar values represent mean ± SEM. ***P < 0.001, ****P < 0.0001.
Overexpression of DJ-1 Represses CaV1.2 and α2δ2 Expression in Neuronal Dendrites.
To determine whether DJ-1 directly impacts protein expression of these calcium channel subunits, we overexpressed DJ-1 (DJ-1 OE) in the hippocampus of C57Bl6 mice using a recombinant adeno-associated virus (rAAV) coding for DJ-1 and measured CaV1.2, α2δ2, and α2δ1 protein levels by immunocytochemistry (SI Appendix, Fig. S4). Fig. 3A demonstrates that DJ-1 is overexpressed in CA1 by ~threefold. As predicted, overexpression of DJ-1 reduces CaV1.2 and α2δ2 in dendrites by ~34% and ~33%, respectively, consistent with the role for DJ-1 as a translational repressor (Fig. 3B) (11). Of note, DJ-1 overexpression has no effect on α2δ1 (Fig. 3B). As an additional control, CaV1.3, another L-VGCC that is present in the hippocampus (32) yet was not detected in the postsynaptic-proteome (23, 24), shows no difference in protein expression upon DJ-1 OE (SI Appendix, Fig. S5), and its mRNA does not associate with DJ-1 (SI Appendix, Fig. S6). These data suggest that DJ-1 selectively represses the protein expression of CaV1.2 and α2δ2 (Fig. 3B) with no effect on α2δ1 or CaV1.3.
Fig. 3.

Overexpression of DJ-1 represses CaV1.2 and α2δ2 expression in neuronal dendrites and reduces L-VGCC activity in response to BayK. Protein of interest density is denoted by pseudo coloring (fire) and Map2 (grayscale). Representative images of control (CTL, Left) and overexpression (DJ-1 OE, Middle). (A) DJ-1 expression (Left) and quantification (Right); Scale bar, 20 µm, (Right) DJ-1/Map2 intensity ratio (control = 1.00 ± 0.08; OE = 2.99 ± 0.40; n per condition = 5 ROI/slice, 2 slices/animal, 4 animals; P < 0.0001). (B) (Top) CaV1.2. (Top, Right) quantification of CaV1.2/Map2 ratio (control = 1.00 ± 0.09; OE = 0.66 ± 0.06, n per condition = 5 ROI/slice, 2 slices/animal, 4 animals; P < 0.002). (Middle) α2δ2. (Middle, Right) quantification of α2δ2/Map2 ratio (control = 1.00 ± 0.13; OE = 0.67 ± 0.06, n per condition = 5 ROI/slice, 2 slices/animal, 4 animals; P = 0.02). (Bottom) α2δ1. (Bottom, Right) quantification of α2δ1/Map2 ratio (control = 1.00 ± 0.12; OE = 1.06 ± 0.11, n per condition = 5 ROI/slice, 2 slices/animal, 4 animals; P = 0.72). Bar values are shown as mean ± SEM; statistical tests: Student’s t test. (C) L-VGCC activity detected by L-type agonist, BayK-8644 (BayK, 5 μM). (Left) Average traces of Ca2+ fluorescence signal before (baseline; 0 to 60 s) and after (520 to 580 s) the addition of vehicle or BayK at ~90 s. In pcDNA-transfected neurons (control), Ca2+ fluorescence increases with BayK (black) compared to vehicle treatment (gray). In DJ-1 overexpressing neurons, addition of BayK (red) has similar Ca2+ fluorescence as vehicle treatment (pink). (Right) Quantification of change in fluorescence (ΔF) normalized to baseline (F). Significance determined by two-way ANOVA (F1,34 = 12.04, P = 0.0014) and genotype (F1,34 = 7.22, P = 0.0111). pcDNA-Vehicle (gray, 5.80 ± 3.16, n = 4 dendrites, 4 neurons) vs. pcDNA-BayK (black, 57.07 ± 14.81, n = 6 dendrites, 4 neurons), DJ-1-OE-Vehicle (pink, −3.10 ± 6.04, n = 17 dendrites, 11 neurons) vs. DJ-1-OE-BayK (red, 13.44 ± 7.57, n = 11 dendrites, 9 neurons). Statistical tests: A and B by Student’s t test; C by two-way ANOVA. Bar values represent mean ± SEM. *P < 0.05, **P < 0.01.
Overexpressing DJ-1 Attenuates Dendritic L-VGCC Activity.
As our data suggest that DJ-1 represses the expression of CaV1.2 and α2δ2 proteins in dendrites (Figs. 2 A and B and 3B), we asked whether overexpression of DJ-1 reduces L-VGCC activity. Therefore, we assessed L-type channel function by performing live Ca2+ imaging in dissociated rat hippocampal neurons that overexpress DJ-1 filled with the calcium indicator, Oregon Green Bapta (OGB). We note that WT cultured neurons are spontaneously active such that L-VGCC are open, can be further opened with BayK-8644 (BayK), and specifically blocked with the L-VGCC antagonist nimodipine (33) (SI Appendix, Fig. S7). We treated DJ-1 OE neurons with BayK (5 μM) and measured the change in dendritic Ca2+ fluorescence (ΔF/F). Application of BayK on pcDNA-expressing neurons increases dendritic fluorescence by ~50% (Fig. 3C). Interestingly, DJ-1-overexpressing neurons fail to display BayK-induced elevation of Ca2+ signal in dendrites (Fig. 3C). To further verify these findings, we used GCamp6-XC expressing neurons and similarly observed a significant increase in dendritic fluorescence signal upon BayK application in WT, which was absent in DJ-1 OE neurons (SI Appendix, Fig. S8). These data collectively support that DJ-1 regulates the protein expression of the channel subunits and dendritic L-VGCC activity.
Protein Expression of CaV1.2 and α2δ2 Are Reduced and L-VGCC Activity Is Attenuated in Tsc1 cKO.
Since DJ-1 is excessively expressed in dendrites of Tsc1 cKO neurons, we hypothesized that CaV1.2 expression, along with α2δ2, is reduced in hippocampal neurons from Tsc1 cKO mice (11). We first measured CaV1.2 and α2δ2 levels, in vitro, after knocking out the Tsc1 gene by Cre expression (Tsc1 cKO) in dissociated hippocampal Tsc1fl/fl neurons. By immunostaining, Tsc1 cKO dendrites display ~34% less CaV1.2 (Fig. 4A) and ~48% less α2δ2 (Fig. 4B), compared to WT neurons. The control, α2δ1, increases by approximately ~69% (Fig. 4C).
Fig. 4.

Tsc1 cKO neurons have reduced levels of calcium channel-associated proteins and L-VGCC function. (A–C) Map2 staining (red) marks dendrites. (A) CaV1.2 (blue). Compared to wild type (WT, Top), TSC1cKO dendrites (Bottom) have less CaV1.2 as quantified (Right). CaV1.2/Map2: (WT = 1.00 ± 0.10, n = 27 neurons; Tsc1 cKO = 0.66 ± 0.09, n = 23 neurons; P = 0.0129). (B) α2δ2 (blue) expression is greater in WT (Top) than Tsc1 cKO as quantified (Right). α2δ2/Map2 (control = 1.00 ± 0.10, n = 15 neurons; Tsc1 cKO = 0.52 ± 0.08, n = 12 neurons; P = 0.0017). (C) Representative WT (Top) and Tsc1 cKO (Bottom) neurons express α2δ1 (blue) in dendrites. α2δ1/Map2 (WT = 1.00 ± 0.12, n = 30 neurons; Tsc1 cKO = 1.69 ± 0.21, n = 34 neurons; P < 0.0085). (D) (Left) Average traces of calcium fluorescence before (baseline; 0 to 60 s) and after (520 to 580 s) the addition of vehicle or BayK at ~90 s. With BayK, dendritic calcium fluorescence increases in WT (black) but not in Tsc1 cKO (red). (Right) Quantification of change in fluorescence (ΔF) normalized to baseline (F). Two-way ANOVA revealed a significant main effect of treatment (F1,76 = 8.15, P = 0.0055), genotype (F1,76 = 9.64, P = 0.0027), and a significant genotype × treatment interaction (F1,76 = 6.591, P = 0.0122). WT-Vehicle (gray, −0.96 ± 1.12, n = 20 dendrites, 15 neurons) vs. WT-BayK (black, 27.92 ± 6.39, n = 14 dendrites, 11 neurons) Tsc1cKO-Vehicle (pink, −3.82 ± 2.98, n = 28 dendrites, 20 neurons) vs. Tsc1cKO-BayK (red, −2.29 ± 2.44, n = 18 dendrites, 14 neurons). Statistical tests: A through C by t test; D by two-way ANOVA, Tukey post hoc test. Bar values represent mean ± SEM. *P < 0.05, ***P < 0.001, ****P < 0.0001.
Low CaV1.2 and α2δ2 levels predict reduced Ca2+ signaling in Tsc1 cKO dendrites (34). Using hippocampal neurons loaded with OGB, we performed live calcium imaging to assess dendritic L-VGCC activity. WT dendrites show a ~27% increase in ΔF/F with BayK (Fig. 4D) (34–36). As predicted, BayK-induced L-type activity is absent in Tsc1 cKO dendrites, similar to DJ-1 overexpression (Fig. 3C). These data reveal that a preclinical model of TSC has reduced dendritic L-VGCC activity.
DJ-1 Preferentially Associates with Cacna1c and Cacna2d2 mRNAs in APP/PS1 Model of AD.
Having established that DJ-1, CaV1.2, and α2δ2 protein and L-type channel activity are disrupted in TSC, we wanted to examine whether the dysregulation of this pathway is conserved in AD, another mTORopathy (37–39). We used a preclinical mouse model of AD—APP/PS1 transgenic mouse—where we demonstrate that mTORC1 is overactive (SI Appendix, Fig. S9). First, we examined dendritic DJ-1 expression in cultured APP/PS1 hippocampal neurons, and saw that DJ-1 is overexpressed, similar to Tsc1 cKO neurons (6) (Fig. 5A).
Fig. 5.

The APP/PS1 preclinical model of AD exhibits aberrant DJ-1 expression and CaV1.2 and a2d2 de novo translation. (A) In hippocampal neuronal cultures, expression of DJ-1 (blue) in dendrites (Map2, red), yellow arrows indicate puncta, is low in WT (Top) compared to APP/PS1 (Bottom) as quantified (Right). Quantification: WT = 1.00 ± 0.088, n = 133 dendrites, 48 neurons; APP/PS1 = 1.89 ± 0.10, n = 143 dendrites, 50 neurons, ****P < 0.0001. (B) (Top). qRT-PCR from DJ-1-RIP from APP/PS1 and WT animals, showing Cacna1c and Cacna2d2 bind to DJ-1. Cacna1c: WT = 0.06 ± 0.01, APP/PS1 = 0.87 ± 0.2, P = 0.01. Cacna2d2: WT = 0.02 ± 0.01, APP/PS1 = 0.89 ± 0.3, P = 0.03. Cacna2d1: WT = 0.26 ± 0.1, APP/PS1 = 0.27 ± 0.07, P = 0.99. Akt1: WT = 0.01 ± 0.01, APP/PS1 = 0.61 ± 0.05, P = 0.0004. Gapdh: WT = 0.11 ± 0.06, APP/PS1 = 0.15 ± 0.08, P = 0.71. (Bottom) qRT-PCR of 5% of the input from APP/PS1 or WT. Cacna1c: WT = 1 ± 0.06, APP/PS1 = 0.91 ± 0.14, P = 0.61. Cacna2d2: WT = 1 ± 0.08, APP/PS1 = 0.57 ± 0.10, P = 0.0042. Cacna2d1: WT = 1 ± 0.04, APP/PS1 = 1.3 ± 0.22, P = 0.23. Akt1: WT = 1 ± 0.04, APP/PS1 = 1.2 ± 0.11, P = 0.14. Gapdh: WT = 1 ± 0.05, APP/PS1 = 1.28 ± 0.03, P = 0.0001. n = 3 technical replicates, 3 independent mice. t test, two-tailed. (C) De novo protein synthesis, SUnSET-PLA (green) in dendrites (MAP2, red). (Left) CaV1.2 is basally translated in WT but is repressed in APP/PS1 as quantified in rightmost panel. Quantification (Bottom): WT = 1.00 ± 0.09, n = 110 total ROIs, 15 to 21 ROIs/slice, 3 slices/animal, 3 animals; APP/PS1 = 0.01 ± 0.003, n = 114 total ROIs, 15 to 21 ROIs/slice, 3 slices/animal, 3 animal. (Middle) Basal translation of α2δ2 is robust in WT but is attenuated in APP/PS1 as quantified in rightmost panel. Quantification (Bottom): WT = 1.00 ± 0.13, n = 100 total ROIs, 15 to 21 ROIs/slice, 3 slices/animal, 3 animals; APP/PS1 = 0.03 ± 0.005, n = 100 total ROIs, 15 to 21 ROIs/slice, 3 slices/animal, 3 animals. (Right) New synthesis of α2δ1 protein in WT is lower than APP/PS1 as quantified in rightmost panel. Quantification (Bottom): WT = 1.00 ± 0.14, n = 104 total ROIs, 15 to 21 ROIs/slice, 3 slices/animal, 3 animals; APP/PS1 = 2.00 ± 0.14, n = 112 total ROIs, 15 to 21 ROIs/slice, 3 slices/animal, 3 animals. For representative images, CaV1.2, α2δ2, and α2δ1 puncta were dilated once using ImageJ. Statistical tests: A and C by Student’s t test. Bar values represent mean ± SEM. ****P < 0.0001.
Next, we examined whether DJ-1 associates with Cacna1c and Cacna2d2 mRNAs in APP/PS1 mice, as we previously detected in Tsc1 cKO mice. Indeed, we observed that DJ-1 binds to Cacna1c and Cacna2d2 mRNAs exclusively in the disease state, similar to the positive control Akt1 mRNA (Fig. 5 B, Top and SI Appendix, Fig. S10). The negative control Gapdh mRNA did not bind DJ-1 in either genotype. Again, Cacna2d1 does not bind to DJ-1 in either genotype, as determined by a single t test with a value that is not significant from zero (Fig. 5 B, Top and SI Appendix, Fig. S11). While the input mRNAs were more variable in the APP/PS1 lysates, there was no correlation between how much the transcript was bound to DJ-1 (Fig. 5 B, Bottom).
Reduced De Novo Protein Synthesis of CaV1.2 and α2δ2 in the Hippocampus of APP/PS1 Mice.
To determine whether de novo protein synthesis of CaV1.2 and α2δ2 is affected in APP/PS1, we repeated the SUnSET-PLA experiment in hippocampal slices from these mice. We observed the same reduction in new protein synthesis of CaV1.2 and α2δ2 as indicated by the reduced number of puncta in dendritic fields of CA1 relative to WT (Fig. 5C) and similar to Tsc1 cKO mice (Fig. 2). Again, we saw a marked increase in α2δ1 (Fig. 5C). Collectively our data strongly argue that DJ-1 selectively represses CaV1.2 and α2δ2 mRNA translation in two different mTORC1-related diseases.
Aberrant α2δ2 and CaV1.2 Expression and L-type VGCC Activity in APP/PS1.
Consistent with the changes in new protein synthesis, we verified that the total dendritic protein for CaV1.2 and α2δ2 are reduced (Fig. 6 A and B), while α2δ1 is increased in APP/PS1 relative to WT (Fig. 6C). In light of these data, we predict that there will be a parallel decrease in L-type VGCC activity in dendrites of APP/PS1 hippocampal neurons, similar to Tsc1 cKO dendrites. Indeed, live calcium imaging showed that BayK activation of the L-VGCCs is absent in APP/PS1 dendrites, while there was a significant increase in L-type-mediated Ca2+ signal in neurons isolated from WT littermates (Fig. 6D). Evidently, DJ-1 and CaV1.2/α2δ2 pathway is conserved between AD and TSC.
Fig. 6.

APP/PS1 neurons express reduced levels of calcium channel-associated proteins and L-type VGCC function. (A–C) Map2 staining (red) marks dendrites. (A) CaV1.2 proteins (blue) in dendrites, wild type (WT, Top), APP/PS1 dendrites (Bottom) have less CaV1.2. (Right) Quantification of CaV1.2/Map2: WT = 1.00 ± 0.06, n = 114 dendrites, 39 neurons; APP/PS1 = 0.45 ± 0.05, n = 108 dendrites, 38 neurons. (B) α2δ2 (blue) expression is greater in WT (Top) than APP/PS1 (Bottom). (Right) Quantification of α2δ2/Map2: WT = 1.00 ± 0.10, n = 104 dendrites, 38 neurons; APP/PS1 = 0.79 ± 0.08, n = 98 dendrites, 39 neurons. (C) Representative WT (Top) and APP/PS1 (Bottom) neurons express α2δ1 (blue) in dendrites. APP/PS1 dendrites express more α2δ1. (Right) Quantification of α2δ1/Map2 WT = 1.00 ± 0.11, n = 91 dendrites, 35 neurons; APP/PS1 = 1.30 ± 0.10, n = 85 dendrites, 32 neurons. (D) L-VGCC function as measured by BayK-induced calcium fluorescence is absent in APP/PS1. (Left) Average traces of calcium fluorescence before (baseline; 0 to 60 s) and after (520 to 580 s) the addition of vehicle or BayK at ~90 s. In BayK, dendritic calcium fluorescence increases in WT (black) but not in APP/PS1 (red). (Right) Quantification of change in fluorescence (ΔF) normalized to baseline (F). ΔF/F was determined by normalizing ΔF of each condition/genotype to its respective baseline fluorescence (F). Two-way ANOVA revealed a significant main effect of treatment (F1,60 = 9.962, P = 0.0025) but not genotype (F1,60 = 3.512, P = 0.0658). There was a significant genotype × treatment interaction (F1,60 = 16.72, P = 0.0001). Tukey’s multiple comparisons post hoc test revealed a significant difference between WT-Vehicle (gray, −9.86 ± 3.57, n = 14 dendrites, 11 neurons) vs. WT-BayK (black, 26.90 ± 7.27, n = 16 dendrites, 12 neurons), but no significant difference between APP/PS1-Vehicle (pink, 1.3810 ± 3.87, n = 19 dendrites, 15 neurons) vs. APP/PS1-BayK (red, −4.51 ± 4.51, n = 15 dendrites, 12 neurons). There is a significant difference between WT-BayK vs. APP/PS1-BayK. Statistical tests: A through C by Student’s t test; D by two-way ANOVA. Bar values represent mean ± SEM. *P < 0.05, ***P < 0.001, ****P < 0.0001.
AD Patients Have Increased mTORC1 and DJ-1 Expression at Their Synapses, with Reduced CaV1.2 and α2δ2.
As preclinical models of TSC and AD show the same abnormal DJ-1/CaV1.2/α2δ2 signaling, we sought whether this signaling pathway is similarly disrupted in AD patients. Subject gender, age, beta-amyloid load, Braak score, and dementia status are reported in SI Appendix, Fig. S12. In synaptoneurosomes (pre- and postsynaptic nerve endings) isolated from human prefrontal cortices (PFC), we first measured mTORC1 activity in control and AD-diagnosed tissue. Western blot analyses of phospho-S6 are significantly elevated by ~94% in AD samples, consistent with previous reports that mTORC1 is hyperactive in AD (Fig. 7A) (40–42). We assessed DJ-1 expression and observed ~56% more in AD compared to age-matched donors (Fig. 7B). Because DJ-1 interacts with Cacna1c and Cacna2d2 mRNAs (Figs. 1 C and E and 5B) and abates dendritic CaV1.2 and α2δ2 levels (Fig. 3B), we measured CaV1.2 and α2δ2 in human PFC synaptoneurosomes. Recapitulating the observations in AD and TSC mouse models, the human AD samples express ~57% less CaV1.2 (Fig. 7C) and ~54% less α2δ2 (Fig. 7D) relative to control. To demonstrate equal enrichment of synaptic proteins between samples, we measured two additional markers, TTBK1 and PSD-95, with no change between control and AD (SI Appendix, Fig. S13). Altogether, these data suggest that TSC and AD share dysregulated L-VGCC-associated protein expression.
Fig. 7.

Synapses isolated from prefrontal cortex of AD individuals express upregulation of mTORC1 signaling and DJ-1 levels, and downregulation of CaV1.2 and α2δ2 levels. (A) Western blot of phosphorylated S6 ribosomal protein (Ser240/244) (control = 1.00 ± 0.07, n = 3; AD = 1.94 ± 0.34, n = 3) (B) DJ-1 protein (control = 1.00 ± 0.15, n = 5; AD = 1.56 ± 0.18, n = 5;). (C) AD patient synapses have decreased CaV1.2 (control = 1.00 ± 0.20, n = 5; AD = 0.42 ± 0.21, n = 5) and (D) decreased α2δ2 levels (control = 1.00 ± 0.15, n = 4; AD = 0.45 ± 0.20, n = 3). Statistical tests: Student’s t test. Bar values represent mean ± SEM. *P < 0.05. For pathological details, see SI Appendix, Fig. S6.
Discussion
Using an unbiased bioinformatics approach to identify new TSC- and AD-related proteins, we found that the RBP DJ-1, which is elevated in overactive mTOR, preferentially targets Ca2+ channel mRNAs. This approach uncovered a unique L-VGCC pathology shared between TSC and AD. L-VGCC has been suggested to be a homeostat for multiple pathways (36, 37). Our data expand the role of L-VGCC as a central homeostat for mTORopathies. The animal models of TSC and AD that we use here express elevated DJ-1, low CaV1.2, and α2δ2 and repressed L-VGCC activity in dendrites. Interestingly, previous studies have suggested that L-VGCC activity increases in the cell bodies of aged rodents and preclinical models of TSC2, affecting action potential firing (43, 44). The data presented herein demonstrate a signaling pathway that effects the dendritic protein expression.
At the cellular level, dendritic L-VGCC activity triggers a cascade of signaling pathways that mediate gene expression and local protein synthesis (45, 46). Our original question sought to find mTORC1-regulated RBPs that repress mRNA translation as a mechanism for deficits in protein synthesis in overactive mTOR-related diseases. Surprisingly, we identified a potential, additional mechanism that may account for deficits in protein synthesis through reduced dendritic L-VGCC activity.
Interestingly, dendrites isolated from mouse models of TSC and AD show a marked decrease in L-VGCC activity in dendrites with only moderate reduction in total protein levels of CaV1.2 and α2δ2. Since TSC and AD are disorders of dysregulated protein synthesis, it would be expected that other proteins that influence L-VGCC activity have dysregulated protein expression. Potential examples are Ras-related small G-proteins that inhibit L-VGCC plasma membrane expression and activity as well as other dendritic proteins (45–50). While our study focuses on translational regulation, future studies that include methods that detect protein–protein interactions may yield further insight into L-VGCC function in TSC and AD.
The large increase in expression of α2δ1 suggests that α2δ1 should be involved in an upregulation of CaV1.2 and other calcium channels (51). It is unclear why this is not observed here. In addition, α2δ subunits have been implicated in synaptogenesis (31, 52, 53). Future questions exploring the expression of other CaV subunit function and their interaction with α2δ1 are needed to better understand how dendritic calcium channels and/or synaptogenesis contribute to the pathological states of TSC and AD.
α2δ2 is an auxiliary calcium channel subunit that has been best characterized in the cerebellum (16, 35, 54–57). Interestingly, knocking out tsc1 in cerebellar Purkinje cells results in reduced excitability and impaired firing rates (58, 59). Thus, determining whether DJ-1/CaV1.2/α2δ2 signaling is also disrupted in the cerebellum of mouse models of TSC and AD and whether this pathway contributes to the observed changes in firing rates in Purkinje cells is warranted in follow up studies.
It has not escaped our attention that L-VGCC blockers are being considered as potential drug therapies (60–62). Our data, however, suggests that blocking L-VGCC may not be suitable for TSC, AD, and perhaps for other mTORopathies, since L-VGCC function in these neurological disorders is already absent or low at best in dendrites. Moreover, these data collectively may explain why L-VGCC blockade can facilitate subtle, nonconvulsive epilepsy, paralleling what is observed in TSC and dementia patients (63–66). In summary, our work identifies the DJ-1/α2δ2/L-VGCC pathway in AD and TSC, providing new druggable targets for the treatment of these disorders.
Methods
All methods are explained in detail in SI Appendix, Supplemental Methods.
Mice.
Experiments follow the NIH’s Guide for the Care and Use of Laboratory Animals and have been approved and supervised by the Wake Forest Institutional Animal Care and Use Committee.
Stereotaxic Injections.
For DJ-1 overexpression, 3 to 4-mo-old WT male mice were used. Coordinates and detailed methods are outlined in SI Appendix, Fig. S3 and Supplemental Methods.
Surface Sensing of Translation–Proximity Ligation Assay.
SUnSET was performed by combining the methods described by refs. 67 and 68.
Calcium Imaging.
Dissociated hippocampal neurons at DIV 14–21 were used for live calcium imaging. Prior to imaging, cells were incubated in ACSF with Oregon Green 488 BAPTA-1 AM (OGB, 200 μM; 30 min; 37 °C; ThermoFisher) as described by Workman et al. (69) with detailed methods outlined in the SI Appendix, Supplemental Methods. The equation, ΔF/F = ((F – F0)/F0) , was used to measure the change in signal, and data were plotted as a percentage of the baseline (69).
Subjects and Tissues.
Postmortem human prefrontal cortex (PFC) tissue samples were acquired from the University of Washington (UW) BioRepository and Integrated Neuropathology (BRaIN) laboratory and UW ADRC Precision Neuropathology Core in accordance with the UW and Wake Forest University School of Medicine Institutional Review Boards. The donor characteristics are shown in SI Appendix, Fig. S12. Mean age of death is 86.9 y, and the postmortem interval (PMI) ranged between 2 and 5 h with an average of 4.03 h.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Acknowledgments
This study was supported by NIH NINDS R01NS105005 (K.F.R.-G.), R01NS105005-03S1 (K.F.R.-G.); USAMRMC Award W81XWH-14-1-0061 and W81XWH-19-1-0202 (K.F.R.-G.); Alzheimer’s Association AARG-NTF-21-852843 (K.F.R.-G.); NIA Wake Forest School of Medicine Alzheimer’s Disease Research Center Pilot Grant P30AG049638 (K.F.R.-G.), NIH NIAAA R01AA029691 (K.F.R.-G.), NSF Postdoctoral Research Fellowship in Biology DBI-1306528 (F.N.) and DBI-1103738 (L.P.C.), EES 2200474 (F.N.); R16NS134542 (F.N.); Alzheimer’s Association AARF-19-614794 (F.N.), AARF-19-614794-RAPID (F.N.); NIAAA T32AA007565 (C.F.H.), a grant from FRAXA Research (C.F.H. and C.J.M.); NIH F31NS117096 (S.H.B.), T32DA041349 (S.H.B.), R01 AG055581 (T.M.), R01 AG056622 (T.M.), and R01AG073823 (T.M.), R21AG077271 (K.R.F.-G.); R01AA026551 (K.R.F.-G.); NIA Wake Forest School of Medicine Alzheimer’s Disease Core Center P30AG072947 (S.C.), and University of Washington Alzheimer’s Disease Research Center P30 AG066509 (C.D.K.); NIA Adult Changes in Thought (ACT) study (U19 AG066567), and the Nancy and Buster Alvord Endowment (C.D.K.).
Author contributions
F.N., A.U., Z.D., L.P.C., and K.F.R.-G. designed research; F.N., A.U., C.J.M., Z.D., H.X.E.-B., L.P.C., W.C.T., X.W., S.H.B., M.P.S., and C.F.H. performed research; C.R., J.P., S.C., and C.D.K. contributed new reagents/analytic tools; F.N., A.U., C.J.M., Z.D., H.X.E.-B., L.P.C., S.V.N., W.C.T., M.P.S., C.D.K., T.M., and K.F.R.-B. analyzed data; and F.N., A.U., T.M., and K.F.R.-G. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
All study data are included in the article and/or supporting information.
Supporting Information
References
- 1.Modi M. E., Sahin M., Tau: A novel entry point for mTOR-based treatments in autism spectrum disorder? Neuron 106, 359–361 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Davis P. E., Peters J. M., Krueger D. A., Sahin M., Tuberous sclerosis: A new frontier in targeted treatment of autism. Neurotherapeutics 12, 572–583 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Habib S. L., et al. , Role of tuberin in neuronal degeneration. Neurochem. Res. 33, 1113–1116 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saxton R. A., Sabatini D. M., mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsukumo Y., Sonenberg N., Alain T., Transcriptional induction of 4E-BP3 prolongs translation repression. Cell Cycle 15, 3325–3326 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Niere F., et al. , Analysis of proteins that rapidly change upon mechanistic/mammalian target of rapamycin complex 1 (mTORC1) repression identifies Parkinson protein 7 (PARK7) as a novel protein aberrantly expressed in tuberous sclerosis complex (TSC). Mol. Cell. Proteomics 15, 426–444 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raab-Graham K. F., Haddick P. C., Jan Y. N., Jan L. Y., Activity- and mTOR-dependent suppression of Kv1.1 channel mRNA translation in dendrites. Science 314, 144–148 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Auerbach B. D., Osterweil E. K., Bear M. F., Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 480, 63–68 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma T., et al. , Suppression of eIF2alpha kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat. Neurosci. 16, 1299–1305 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sosanya N. M., et al. , Degradation of high affinity HuD targets releases Kv1.1 mRNA from miR-129 repression by mTORC1. J. Cell Biol. 202, 53–69 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van der Brug M. P., et al. , RNA binding activity of the recessive parkinsonism protein DJ-1 supports involvement in multiple cellular pathways. Proc. Natl. Acad. Sci. U.S.A. 105, 10244–10249 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bateup H. S., et al. , Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron 78, 510–522 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ranasinghe K. G., et al. , Neuronal synchrony abnormalities associated with subclinical epileptiform activity in early onset Alzheimer’s disease. Brain 145, 744–753 (2021), 10.1093/brain/awab442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Niere F., Raab-Graham K. F., mTORC1 is a local, postsynaptic voltage sensor regulated by positive and negative feedback pathways. Front Cell Neurosci. 11, 152 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lerche H., et al. , Ion channels in genetic and acquired forms of epilepsy. J. Physiol. 591, 753–764 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barclay J., et al. , Ducky mouse phenotype of epilepsy and ataxia is associated with mutations in the Cacna2d2 gene and decreased calcium channel current in cerebellar Purkinje cells. J. Neurosci. 21, 6095–6104 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu A. J., et al. , Association of cognitive and behavioral features between adults with tuberous sclerosis and frontotemporal dementia. JAMA Neurol. 77, 358–366 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kedra M., et al. , TrkB hyperactivity contributes to brain dysconnectivity, epileptogenesis, and anxiety in zebrafish model of Tuberous Sclerosis Complex. Proc. Natl. Acad. Sci. U.S.A. 117, 2170–2179 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Curatolo P., Moavero R., de Vries P. J., Neurological and neuropsychiatric aspects of tuberous sclerosis complex. Lancet Neurol. 14, 733–745 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Villela D., Suemoto C. K., Pasqualucci C. A., Grinberg L. T., Rosenberg C., Do copy number changes in CACNA2D2, CACNA2D3, and CACNA1D constitute a predisposing risk factor for Alzheimer’s disease? Front. Genet. 7, 107 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nimmrich V., Eckert A., Calcium channel blockers and dementia. Br. J. Pharmacol. 169, 1203–1210 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang L., Wang Z., Wang B., Justice N. J., Zheng H., Amyloid precursor protein regulates Cav1.2 L-type calcium channel levels and function to influence GABAergic short-term plasticity. J. Neurosci. 29, 15660–15668 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Collins M. O., et al. , Molecular characterization and comparison of the components and multiprotein complexes in the postsynaptic proteome. J. Neurochem. 97 (suppl. 1), 16–23 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Bayes A., et al. , Comparative study of human and mouse postsynaptic proteomes finds high compositional conservation and abundance differences for key synaptic proteins. PloS One 7, e46683 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ertel E. A., et al. , Nomenclature of voltage-gated calcium channels. Neuron 25, 533–535 (2000). [DOI] [PubMed] [Google Scholar]
- 26.Cajigas I. J., et al. , The local transcriptome in the synaptic neuropil revealed by deep sequencing and high-resolution imaging. Neuron 74, 453–466 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwiatkowski D. J., et al. , A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Hum. Mol. Genet. 11, 525–534 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Meikle L., et al. , A mouse model of tuberous sclerosis: Neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J. Neurosci. 27, 5546–5558 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keene J. D., Lager P. J., Post-transcriptional operons and regulons co-ordinating gene expression. Chromosome Res. 13, 327–337 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Geisler S., et al. , Presynaptic alpha2delta-2 Calcium channel subunits regulate postsynaptic GABAA receptor abundance and axonal wiring. J. Neurosci. 39, 2581–2605 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schopf C. L., et al. , Presynaptic alpha2delta subunits are key organizers of glutamatergic synapses. Proc. Natl. Acad. Sci. U.S.A. 118, e1920827118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang H., et al. , Association of CaV1.3 L-type calcium channels with Shank. J. Neurosci. 25, 1037–1049 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turrigiano G., LeMasson G., Marder E., Selective regulation of current densities underlies spontaneous changes in the activity of cultured neurons. J. Neurosci. 15, 3640–3652 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dolphin A. C., Voltage-gated calcium channels and their auxiliary subunits: Physiology and pathophysiology and pharmacology. J. Physiol. 594, 5369–5390 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dolphin A. C., Calcium channel auxiliary alpha2delta and beta subunits: Trafficking and one step beyond. Nat. Rev. Neurosci. 13, 542–555 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Dolphin A. C., The alpha2delta subunits of voltage-gated calcium channels. Biochim. Biophys. Acta 1828, 1541–1549 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Pei J. J., Hugon J., mTOR-dependent signalling in Alzheimer’s disease. J. Cell Mol. Med. 12, 2525–2532 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caccamo A., De Pinto V., Messina A., Branca C., Oddo S., Genetic reduction of mammalian target of rapamycin ameliorates Alzheimer’s disease-like cognitive and pathological deficits by restoring hippocampal gene expression signature. J. Neurosci. 34, 7988–7998 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cai Z., Chen G., He W., Xiao M., Yan L. J., Activation of mTOR: A culprit of Alzheimer’s disease? Neuropsychiatr. Dis. Treat. 11, 1015–1030 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.An W. L., et al. , Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer’s disease. A. J. Pathol. 163, 591–607 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li X., Alafuzoff I., Soininen H., Winblad B., Pei J. J., Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer’s disease brain. FEBS J. 272, 4211–4220 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Oddo S., The role of mTOR signaling in Alzheimer disease. Front Biosci. 4, 941–952 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hisatsune C., Shimada T., Miyamoto A., Lee A., Yamagata K., Tuberous Sclerosis Complex (TSC) inactivation increases neuronal network activity by enhancing Ca(2+) influx via L-type Ca(2+) channels. J. Neurosci. 41, 8134–8149 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thibault O., Landfield P. W., Increase in single L-type calcium channels in hippocampal neurons during aging. Science 272, 1017–1020 (1996). [DOI] [PubMed] [Google Scholar]
- 45.Liu G., et al. , Mechanism of adrenergic Ca(V)1.2 stimulation revealed by proximity proteomics. Nature 577, 695–700 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beguin P., et al. , Regulation of Ca2+ channel expression at the cell surface by the small G-protein kir/Gem. Nature 411, 701–706 (2001). [DOI] [PubMed] [Google Scholar]
- 47.Finlin B. S., Crump S. M., Satin J., Andres D. A., Regulation of voltage-gated calcium channel activity by the Rem and Rad GTPases. Proc. Natl. Acad. Sci. U.S.A. 100, 14469–14474 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang S., et al. , Densin-180 controls the trafficking and signaling of L-type voltage-gated Ca(v)1.2 Ca(2+) channels at excitatory synapses. J. Neurosci. 37, 4679–4691 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vierra N. C., Kirmiz M., van der List D., Santana L. F., Trimmer J. S., Kv2.1 mediates spatial and functional coupling of L-type calcium channels and ryanodine receptors in mammalian neurons. Elife 8, e49953 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vierra N. C., O’Dwyer S. C., Matsumoto C., Santana L. F., Trimmer J. S., Regulation of neuronal excitation-transcription coupling by Kv2.1-induced clustering of somatic L-type Ca(2+) channels at ER-PM junctions. Proc. Natl. Acad. Sci. U.S.A. 118, e2110094118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Felix R., Gurnett C. A., De Waard M., Campbell K. P., Dissection of functional domains of the voltage-dependent Ca2+ channel alpha2delta subunit. J. Neurosci. 17, 6884–6891 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eroglu C., et al. , Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139, 380–392 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Risher W. C., et al. , Thrombospondin receptor alpha2delta-1 promotes synaptogenesis and spinogenesis via postsynaptic Rac1. J. Cell Biol. 217, 3747–3765 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cole R. L., et al. , Differential distribution of voltage-gated calcium channel alpha-2 delta (alpha2delta) subunit mRNA-containing cells in the rat central nervous system and the dorsal root ganglia. J. Comp. Neurol. 491, 246–269 (2005). [DOI] [PubMed] [Google Scholar]
- 55.Beeson K. A., Beeson R., Westbrook G. L., Schnell E., alpha2delta-2 protein controls structure and function at the cerebellar climbing fiber synapse. J. Neurosci. 40, 2403–2415 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beeson K. A., Westbrook G. L., Schnell E., alpha2delta-2 is required for depolarization-induced suppression of excitation in Purkinje cells. J. Physiol. 600, 111–122 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lein E. S., et al. , Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176 (2007). [DOI] [PubMed] [Google Scholar]
- 58.Tsai P. T., et al. , Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 488, 647–651 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsai P. T., et al. , Sensitive periods for cerebellar-mediated autistic-like behaviors. Cell Rep. 25, 357–367.e354 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zamponi G. W., Targeting voltage-gated calcium channels in neurological and psychiatric diseases. Nat. Rev. Drug Discov. 15, 19–34 (2016). [DOI] [PubMed] [Google Scholar]
- 61.Mullins C., Fishell G., Tsien R. W., Unifying views of autism spectrum disorders: A consideration of autoregulatory feedback loops. Neuron 89, 1131–1156 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nanou E., Catterall W. A., Calcium channels, synaptic plasticity, and neuropsychiatric disease. Neuron 98, 466–481 (2018). [DOI] [PubMed] [Google Scholar]
- 63.Lin J. W., et al. , Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nat. Neurosci. 3, 1282–1290 (2000). [DOI] [PubMed] [Google Scholar]
- 64.van Luijtelaar E. L., Ates N., Coenen A. M., Role of L-type calcium channel modulation in nonconvulsive epilepsy in rats. Epilepsia 36, 86–92 (1995). [DOI] [PubMed] [Google Scholar]
- 65.Noebels J., A perfect storm: Converging paths of epilepsy and Alzheimer’s dementia intersect in the hippocampal formation. Epilepsia 52 (suppl. 1), 39–46 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shehata H. S., et al. , Clinical patterns and outcomes of status epilepticus in patients with tuberous sclerosis complex. Ther. Clin. Risk Manag. 13, 779–785 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schmidt E. K., Clavarino G., Ceppi M., Pierre P., SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 6, 275–277 (2009). [DOI] [PubMed] [Google Scholar]
- 68.Workman E. R., et al. , Rapid antidepressants stimulate the decoupling of GABA(B) receptors from GIRK/Kir3 channels through increased protein stability of 14-3-3eta. Mol. Psychiatry 20, 298–310 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Workman E. R., Niere F., Raab-Graham K. F., mTORC1-dependent protein synthesis underlying rapid antidepressant effect requires GABABR signaling. Neuropharmacology 73, 192–203 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Data Availability Statement
All study data are included in the article and/or supporting information.