


Abstract
We present the high-resolution structure of stem-loop 4 of the 5′-untranslated region (5_SL4) of the severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2) genome solved by solution state nuclear magnetic resonance spectroscopy. 5_SL4 adopts an extended rod-like structure with a single flexible looped-out nucleotide and two mixed tandem mismatches, each composed of a G•U wobble base pair and a pyrimidine•pyrimidine mismatch, which are incorporated into the stem-loop structure. Both the tandem mismatches and the looped-out residue destabilize the stem-loop structure locally. Their distribution along the 5_SL4 stem-loop suggests a role of these non-canonical elements in retaining functionally important structural plasticity in particular with regard to the accessibility of the start codon of an upstream open reading frame located in the RNA's apical loop. The apical loop—although mostly flexible—harbors residual structural features suggesting an additional role in molecular recognition processes. 5_SL4 is highly conserved among the different variants of SARS-CoV-2 and can be targeted by small molecule ligands, which it binds with intermediate affinity in the vicinity of the non-canonical elements within the stem-loop structure.
Graphical Abstract
Graphical Abstract.

Introduction
Since its outbreak in December 2019, the coronavirus disease 2019 (COVID-19) pandemic has had a tremendous impact on global society. To date, >660 million people worldwide have been infected and >6.7 million people have succumbed to the disease. The causative agent of this respiratory infection is SARS-CoV-2, which belongs to the betacoronaviruses (β-CoVs) within the Coronaviridae family. SARS-CoV-2 is a single-stranded RNA virus with a genome of ∼30 000 nucleotides. The propensity for secondary structure formation of coronavirus RNA genomes in general and SARS-CoV-2 in particular has been predicted to be higher than that of any other previously characterized viral genome (1). Accordingly, chemical probing approaches have demonstrated that >60% of the viral genome is engaged in secondary structure inside virions (2,3). The 5′-genomic end in particular, which includes the 5′-untranslated region (UTR), and the 3′-UTR of the genome contain multiple highly structured stem-loop elements, whose conservation among all β-CoVs is substantially higher than that of the coding part of the genome. These stem-loops have an essential functional role in viral RNA replication, subgenomic mRNA production and viral protein translation (4).
While the three-dimensional structures of virtually all folded viral protein domains and their complexes have been reported, experimental structures of the RNA genome are missing with the exception of stem-loop 2 in the 5′-UTR (5), which is 100% conserved between SARS-CoV and SARS-CoV-2 and the pseudoknot constituting the frameshifting element within the coding region of the RNA genome (6–8). So far, only 3D structure models derived from fragment assembly of RNA with full atom refinement (FARFAR) (9) and molecular dynamics simulations (10) are available for a set of genomic RNA elements.
For proteins, 3D structure determination has had substantial impact on the elucidation of their biomolecular functions within the viral life cycle and also on the development of anti-viral inhibitors, e.g. targeting the two proteases Nsp3d and Nsp5 (11), the RNA-dependent RNA-polymerase (12) as well as the nucleocapsid protein (13).
Within the global consortium Covid19-nmr (14), we have defined and investigated 15 individually folded RNA elements of the SARS-CoV-2 genome—located in the 5′-genomic end, the 3′-UTR and the frameshifting region—by nuclear magnetic resonance (NMR) spectroscopic studies and reported their secondary structures (15). These RNA elements have also been screened against small molecule libraries (16). For the further development of screening hits toward viral inhibitors with high affinity and specificity, precise information on the 3D structure and dynamics of the target RNAs is indispensable. Here, we report the solution NMR structure of the fourth stem-loop within the 5′-UTR of the genome (5_SL4) comprising nucleotides 86 to 125. 5_SL4 was initially predicted based on its high conservation within the Coronaviridae family (4,17). In all β-CoVs, it contains an upstream ORF (uORF), which appears to be under positive evolutionary selection pressure (17) and is suggested to weakly reduce translation of the viral proteins Nsp1-16 (18). The putative function of 5_SL4 is to enhance fidelity of discontinuous transcription by presenting a structural roadblock by guiding the replication–transcription complex to the proper location (19). Accordingly, the location and stability of this RNA stem-loop are more conserved than its sequence within the Coronaviridae family (20). Furthermore, only recently, recognition of the GUGUG motif in the lower helix of 5_SL4 (residues 86 to 90) by the host RNA-binding protein 24 (RBM24) could be demonstrated (21). An interaction between a similar motif and RBM24 has been shown to inhibit viral transcript translation for Hepatitis B and C viruses previously (22,23). Interestingly, the consensus GUGUG motif exists twice in the SARS-CoV-2 5′-UTR, the second one located in 5_SL5b, 18 nts upstream of the start codon. However, although both sequences are located in very similar secondary structure elements, no RBM24 binding to the 5b RNA element could be detected, whereas RBM24 unambiguously bound to SL4 (21). This observation underscores the need for experimental structural and dynamic data as a basis for the predictive study of RNA and its interactions.
From the secondary structure analysis, it is apparent that 5_SL4 contains non-canonical structural elements: three bulge motifs as well as a five-nucleotide loop, which are conserved in all SARS-CoV-2 variants. Knowledge on the precise structure of these non-canonical elements is indispensable as they significantly contribute to stem-loop stability and, hence, to the folding landscape of the RNA element in general. Moreover, non-canonical nucleotide interactions interrupt the formation of the classical A-form RNA helix and thereby provide specific binding sites for cellular interaction partners as well as for potential anti-viral inhibitors of low molecular weight. 5_SL4 has been shown to be druggable by the class of amiloride compounds, which selectively bind to the non-canonical parts of RNA stem-loop structures (24,25). The 3D structure of 5_SL4 should both aid in gaining further insight into the function of this stem-loop within the SARS-CoV-2 genome such as, for example, interactions with host proteins, and in facilitating the structure-guided development of compounds targeting this RNA element.
Materials and methods
Two different RNA sequences were used for the structural investigation of 5_SL4, a longer 44 nt sequence comprising the complete 5_SL4 (residues 86–125 of the SARS-CoV-2 genome) elongated by two G–C base pairs (5_SL4, 5′-GGGUGUGGCUGUCACUCGGCUGCAUGCUUAGUGCACUCACGCCC-3′) and a shorter 25 nt sequence containing only the upper stem-loop region (residues 96–116) also elongated by two G–C base pairs (5_SL4sh, 5′-GGCACUCGGCUGCAUGCUUAGUGCC-3′). RNA synthesis, NMR sample preparation and NMR resonance assignment have been described in detail previously (26). NMR assignments were deposited in the BMRB (entries 50347 and 50760 for 5_SL4 and the shorter apical stem-loop sequence 5_SL4sh, respectively) (26).
NMR spectroscopy
NMR spectra were collected on 600, 800, 900 and 950 MHz Bruker Avance NMR-spectrometers equipped with 5-mm cryogenic triple resonance TCI-N probes, a 700 MHz spectrometer equipped with a quadruple resonance QCI-P cryogenic probe and an 800 MHz spectrometer equipped with a 13C-optimized TXO cryogenic probe. 19F measurements were carried out on a 600 MHz Bruker Neo NMR-spectrometer equipped with a 5-mm cryogenic quadruple resonance QCI probe. Measurements were performed in 25 mM KPi, pH 6.2, 50 mM KCl at 10°C in 5% D2O/95% H2O for the exchangeable protons and at 25°C in 100% D2O for the non-exchangeable protons. NMR spectra were recorded and processed using TOPSPIN (Bruker). For spectra analysis, CARA was used (27).
In addition to the previously described NOESY experiments (26), the following NMR experiments were recorded: For the determination of the sugar pucker for 5_SL4sh, a forward directed HCC-TOCSY-CCH-E.COSY spectrum was obtained and 3J(H1’,H2’) and 3J(H3’,H4’) coupling constants were extracted (28,29). Structural dynamics were analyzed by recording {1H},13C heteronuclear NOE (hetNOE) experiments optimized for aromatic H6C6- and H8C8-groups for 5_SL4 and 5_SL4sh and aliphatic H1’C1’-groups for 5_SL4sh with selective decoupling of C5 or C2’, respectively, during 13C chemical shift evolution. Experiments were acquired as duplicates. Residual dipolar couplings (RDCs) were recorded using liquid crystalline Pf1 phage as alignment medium (ASLA biotech). Next, 11.6 mg/ml Pf1 phage was included in 550 μl samples of A,C-13C,15N- or G,U-13C,15N-labeled 5_SL4 RNA with a final concentration of 200 μM. An excellent agreement of the degree of alignment could be achieved for the two selectively 13C,15N-labeled samples. Isotropic reference samples with the same RNA concentrations were prepared by adding appropriate amounts of the corresponding buffer solution (10 mM potassium phosphate buffer pH 7.6) instead of the phage solution. 1DHN and 1DHC RDCs were obtained using IPAP separation schemes of the doublet components (30). 1DHN of the U and G imino groups were extracted from 1H,15N-IPAP-sofast-HMQC spectra (31). The spectra were recorded with a 15N resolution of 21 Hz (21) and a recycle delay of 0.3 s. 1DH1’C1’, 1DH5C5, 1DH6C6 and 1DH8C8 were extracted by recording gradient coherence selected 1H,13C- IPAP-HSQC spectra (30). 1DH6C6 and 1DH8C8 were obtained within the same spectrum, which was selectively C5 decoupled in the 13C dimension. 1DH1’C1’ and 1DH5C5 were obtained in separate spectra with selective C2’ or C6/C4 decoupling, respectively. Due to a larger line width, spectra of the aligned samples were typically recorded with ∼10 times the number of transients and half the resolution in the indirect dimension compared to the isotropic samples resulting in experimental times of ∼12 hours compared to the ∼1.5 hours for the isotropic sample. All RDC measurements were carried out at 800 MHz at a temperature of 25°C.
To probe the protonation state of C100, 5_SL4sh was titrated with a 1 M HCl solution in steps of 1 μl. After each addition of HCl, the pH was measured using a pH electrode dedicated to NMR tubes. pH-induced chemical shift perturbances (CSPs) were followed by recording 2D-H5(C5)C4 spectra at pH values of 6.6, 6.2, 5.9, 5.5, 4.8, 4.5 and 4.3, all at 25°C. In addition, a 2D-H5(C5C4)N3 spectrum at 25°C and a 1H,15N-HSQC spectrum for the imino group region at 10°C was recorded at pH 4.3. The C100 C4 chemical shift (ΩC4) was plotted against the pH of the NMR sample. The pKa for C100 protonation was derived from a curve fit in Origin using Equation (1):
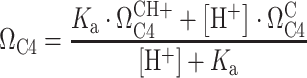 |
(1) |
In Equation (1), Ka is the association constant of the protonation reaction and 10−pKa, 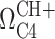 is the C4 chemical shift of the protonated state,
is the C4 chemical shift of the protonated state, 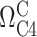 is the C4 chemical shift of the non-protonated state and [H+] is 10−pH.
is the C4 chemical shift of the non-protonated state and [H+] is 10−pH.
Titration of the ligand DMA0043 with 5_SL4 was carried out at 25°C with a sample volume was 170 μl in 3-mm NMR tubes. The screening buffer was 25 mM KPi, pH 6.2, 50 mM KCl in 95% H2O/5% [D6]DMSO. 19F 1D spectra were recorded with 256 scans without 5_SL4 and in the presence of 125 μM of 5_SL4. Binding site mapping on the RNA side was carried out using 50 μM samples of a selectively A,C- and a selectively G,U-13C,15N labeled 5_SL4 RNA titrated in a sample volume of 500 μl with increasing amounts of DMA0043 up to a final concentration of 1 mM. The titration was followed by recording 1H,13C-sofast-HMQC spectra of the aromatic CH-moieties. Combined 1H and 13C CSPs were extracted from comparison of the 1H and 13C chemical shifts of the spectrum of free 5_SL4 to the end-point of the titration (ΔΩH and ΔΩC, respectively) using Equation (2).
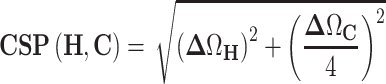 |
(2) |
Input restraints and structure calculation
1H-1H distance restraints were obtained from NOE cross peak intensities extracted from 2D 1H,1H-NOESY, 2D 15N-CPMG-NOESY and 3D aromatic and aliphatic 1H,13C-NOESY-HSQC spectra. NOE intensities were referenced to the average of H5/H6 cross peak intensities of pyrimidine residues that were set to 2.4 Å and then classified into five classes of upper limit distance restraints (3.5, 4.3, 5.4, 6.5 and 7.5 Å). Nucleobase and ribose moieties with {1H},13C-hetNOE values <1.2 were considered rigid. For residue U95 with a considerably higher hetNOE value of the nucleobase moiety, only intraresidual distance restraints were incorporated to account for the conformationally averaged NOEs of this residue. For nucleobase and ribose moieties of residues in the apical loop, which displayed elevated hetNOE values >1.2, distance restraints were loosened to the next longest upper limit class.
Confirmed hydrogen bonds of canonical Watson–Crick and wobble G•U base pairs were incorporated using two upper limit and two lower limit restraints between the donor hydrogen and the acceptor heteronucleus (2.0 and 1.8 Å) and between the two heteronuclei (3.0 and 2.8 Å). The backbone torsion angles α, β, γ, δ, ε and ζ as well as the glycosidic torsion angle χ of all canonical A-form residues were amply set to typical A-form helical values (±20°).
Residues with 3J(H1’,H2’) coupling constants <2 Hz and 3J(H3’,H4’) >8 Hz were restrained to the C3’-endo conformation, residues with 3J(H1’,H2’) >8 Hz and 3J(H3’,H4’) <2 Hz were restrained to the C2’-endo conformation. In addition and for residues, for which no 3J(H,H) coupling constants could be determined due to spectral overlap, canonical coordinates were used to delineate the ribose pucker (32,33).
Structure calculation was carried out using CYANA v.3.98.13 (34). A total of 100 structures were calculated using 32 000 refinement steps per conformer with 12 000 high temperature steps of torsion angle dynamics followed by 20 000 steps of slow cooling and 8000 steps of conjugate gradient minimization. The 10 structures with the lowest target function were refined in explicit water using ARIA protocols (35). MOLMOL (36) and PyMOL (Schrödinger, Inc.) were used for structure visualization.
SAXS
SAXS data on 5_SL4 were collected at 20°C in phosphate buffer (25 mM KPi pH 6.5, 150 mM KCl, supplemented with 2 mM TCEP to reduce radiation damage) with an RNA concentration of 4.5 mg/ml. SAXS measurements were carried out remotely at beamline P12 of the DESY synchrotron Hamburg with the PETRA III source (37). Measurements were performed under continuous flow with a total exposure time of 3.8 s (40 × 95 ms frames). Referencing was carried out with BSA. Data were processed and analyzed using the ATSAS v.3.1.3 software suite (38) by averaging of frames followed by subtraction of buffer scattering taken from the flow-through of RNA sample concentration. The final curve was further processed, i.e. trimmed by noise in the high-q area and initial points removed to enable shape fitting. Clean curves were used to derive Dmax from the pair-wise distribution function P(r) and Rg values. Subsequently, an estimate of the molecular weight was obtained using Bayesian inference and an arbitrary globular shape as part of the ATSAS Primus tool. Finally, we used the SAXS data to create an ab initio dummy shape model for 5_SL4 with the DAMMIF program and for the evaluation of the NMR structure via comparison to a back-calculated, theoretical scattering curve using the CRYSOL procedure (39).
Results and discussion
SARS-CoV-2 5_SL4 is comprised of nucleotide (nt) positions 86 to 125 of the viral 5′-UTR (Figure 1a). It is predicted to form a hairpin structure with a 5 nt apical loop, a single unpaired residue (U95) and two tandem mismatches (TM1 and TM2), each consisting of a pyrimidine•pyrimidine mismatch and a G•U base pair (Figure 1b). For our structural investigation, we elongated the 5_SL4 stem-loop sequence by two G–C base pairs resulting in a 44 nt long stabilized stem-loop optimized for in vitro transcription (5_SL4; Figure 1b, left). To allow for a more detailed structural analysis of the apical loop and TM2, we also investigated a shortened 25 nt construct of 5_SL4 comprising only the apical stem-loop (nt 96–116) closed by two additional G–C base pairs (5_SL4sh; Figure 1b, right). In an 1H,15N-HSQC, the NMR signature of the imino group signals of 5_SL4 coincides with the one of a larger construct comprising 5_SL1 to 5_SL4 (Figure 1c), strongly suggesting that the 5_SL4 stem-loop is present in the larger context of the SARS-CoV-2 5′-UTR and folds context-independently. 5_SL4sh displays imino group signals that are a subset of the 5_SL4 spectrum indicating that the smaller RNA construct also folds as expected (Figure 1c).
Figure 1.

(A) Genomic context of 5_SL4: Schematic representation of the SARS-CoV-2 genome (top) and overview of the stem-loop elements of the 5′-genomic end (bottom). 5_SL1-4ext and 5_SL4 are highlighted with a gray and blue box, respectively. (B) Sequence and secondary structure of 5_SL4 (left) and 5_SL4sh (right). The uORF start codon is highlighted by open letters. The two tandem mismatches (TM1 and TM2) are indicated, the respective residues are shown with bold letters. The part of 5_SL4, which constitutes 5_SL4sh, is indicated by a red box. (C) Overlay of the imino 1H,15N-HSQC spectra of 5_SL1-4ext (black), 5_SL4 (blue) and 5_SL4sh (red). Assignments for 5_SL4 and 5_SL4sh are given in blue and red, respectively.
3D structure of 5_SL4
To solve the three-dimensional solution-state structure of 5_SL4, we first performed NMR resonance assignments on both 5_SL4 and 5_SL4sh using standard NMR assignment procedures as described (26). Initial resonance assignment and secondary structure elucidation investigations supported the assumed stem-loop structure of 5_SL4 (15). All predicted Watson–Crick base pairs as well as the three expected G•U base pairs in wobble geometry could be confirmed. At that point, however, the structural arrangement of the two pyrimidine mismatches C92•C119 and C100•U112, the unpaired nucleotide U95 and the structure of the apical loop (U104-U108) had not been addressed. In particular, the structure of the non-canonical intra-helical elements can have a strong effect on overall stem-loop geometry, as they might induce kinks resulting in significant deviations from a predicted rigid rod-like stem-loop structure. In addition, the detailed structure of the non-canonical elements, in particular the loop, together with their conservation pattern allows for insights into their impact on viral function.
In determining the structure of 5_SL4, we relied on NOE-derived 1H-1H distance restraints from a fully 15N-labeled and two selectively 13C, 15N-labeled samples (A, C and G, U) of 5_SL4 to decrease signal overlap as well as a fully 13C, 15N -labeled sample of 5_SL4sh. For the lower part of the stem-loop (residues G-2 to 96 and G116 to C+2), the upper limit distance restraints derived from NOE cross peaks extracted from the 5_SL4 samples were used, while for the apical stem loop (residues C96 to G116) the distance restraints obtained from the better resolved NOEs of the smaller 5_SL4sh were incorporated into the structure calculation. We could obtain on average 12 interresidual NOEs per residue. However, in the proximity of the two tandem mismatches and the single unpaired U95, this number was significantly reduced, mainly due to the absence of observable imino proton resonances. This local lack of NOE data resulted in a considerable variability in the calculated global structures, ranging from a completely rod-like overall fold to L- and U-shaped structures (Supplementary Figure S1a, left). To test whether this local scarcity in NOE data reflects real conformational flexibility, we exploited the {1H},13C-hetNOE of the nucleobase H6C6 and H8C8 moieties along the sequence of 5_SL4 as probes for the NOE-relevant sub-τc dynamics of the individual nucleobases (Figure 2a). Distinctly increased hetNOE values (>1.2) indicate that the unpaired residue U95 and G105 to U108 of the apical loop are flexible, while all other residues including the four nucleotides of the two tandem mismatches exhibit lower hetNOE values suggesting stable structural arrangements. Analysis of the NOEs in the vicinity of the flexible unpaired U95 revealed a number of stacking NOEs between the adjacent G94-C117 and C96-G116 base pairs, demonstrating that U95 is looped-out of an otherwise continuously stacking helix (Figure 2b). Of note, U95 is completely conserved among the different SARS-CoV-2 variants, suggesting a functional role, e.g. serving as a recognition hub for host or viral protein binding partners.
Figure 2.

NMR structure and structural details of 5_SL4. (A) {1H},13C-hetNOE of the aromatic pyrimidine H6C6 and purine H8C8 moieties of 5_SL4. Residues deviating from canonical A-form conformation are assigned and highlighted in different colors according to the color scheme in (C). Residues of the two pyrimidine•pyrimidine mismatches are connected by dotted lines. (B) NOE derived upper limit distance restraints of residue U95 (orange) and between its two adjacent base pairs (gray). The RNA is shown as a stick representation. The residues are assigned. Distance restraints are depicted as gray lines. (C) NMR structure of 5_SL4. Overlay of the 10 structures with the lowest target function (middle). The heavy atoms of all residues with H6/8C6/8 {1H},13C-hetNOE values <1.2 were used for the overlay. The RNA is shown as stick representation. Residues deviating from canonical A-form structure are highlighted in different colors according to the secondary structure (left). The structure with the lowest target function is shown to the right in cartoon representation displaying the same color scheme. (D) SAXS de novo envelope of 5_SL4 generated with the program DAMMIF superimposed with the best fitting NMR-structure (ninth lowest target function) using the program CIFSUB of the ATSAS v.3.1.3 software suite (38). The RNA is shown as a cartoon representation, the SAXS envelope as mesh.
To analyze the global structure of the 5_SL4 stem-loop, we measured NH and CH RDCs. Incorporation of these RDC values into the structure calculation resulted in a global rod-like structure (Supplementary Figure S1a, right) as well as an improvement in the average pairwise heavy atom RMSD of all non-flexible residues of the ensemble of the 10 lowest target function structures from 10.3 ± 5.8 Å to a final value of 2.1 ± 0.5 Å. The combined NMR data show that 5_SL4 in fact forms a continuous straight stem with the unpaired U95 looped out, a rather unstructured apical loop and the two pyrimidine•pyrimidine mismatches consistently located within the helical structure (Figure 2c). The NMR statistics of the structure of 5_SL4 are summarized in Table 1.
Table 1.
Details of the NMR-structure determination of 5_SL4
| Distance restraints | 1126 |
| Intra-residue | 510 |
| Sequential | 368 |
| Long-range | 156 |
| Hydrogen bond | 92 |
| Dihedral angle restraints | 275 |
| Ribose pucker | 82 |
| Backbone | 155 |
| Glycosidic torsion | 38 |
| RDCs | 64 |
| 1DHN | 11 |
| 1DHC | 53 |
| Structural statistics | |
| Average pairwise heavy atom RMSD (Å) | 2.1 ± 0.5 |
| for G-1-G94,C96-C103,G109-C+1 |
To independently cross-validate the global NMR structure of 5_SL4 by a complementary method, we subjected the RNA to SAXS measurements. The ab initio-modeled SAXS envelope of 5_SL4 can be described as a roughly axially symmetric elongated stick with a radius of gyration (Rg) of 21.3 Å and a maximal distance distribution (Dmax) of 69.5 Å. These dimensions agree well with the 10 structures of the NMR bundle (Rg = 20.3 ± 0.7 Å and Dmax = 71 ± 4 Å; Figure 2d). The agreement between the back-calculated scattering of the structures of the NMR ensemble and the SAXS data is very good (χ2 = 13.4 ± 12.3; Supplementary Figure S1b). In summary, SAXS data support that 5_SL4 adopts a rod-like global structure.
The apical loop is flexible with residual structural features
The apical loop of 5_SL4 consists of five nucleotides with the highly conserved sequence 5′-UGCAU-3′. The two 3′-residues of the loop are part of the start codon of the uORF. High {1H},13C-hetNOE values (>1.2) indicate that most of the loop residue nucleobase and ribose moieties are more dynamic than the stem (Figure 3a). In line with this observed flexibility, the apical loop is not converged in the NMR-bundle but largely disordered (Figure 3b,c). While {1H},13C-hetNOE values report on the presence of sub-nanosecond flexibility of individual C-H moieties, they do not allow to draw conclusions on whether the observed dynamics originate from a correlated motion of all the affected residues, independent motions of the individual residues or a mixture thereof. Therefore, we have focused on residual structural features of the loop, which can be delineated from chemical shifts, NOE patterns and scalar coupling constants. U104—the first nucleotide in the loop—is the only structurally stable loop residue with low {1H},13C-hetNOE for both its ribose and nucleobase moiety (Figure 3a), canonical chemical shifts (Figure 3d) and a sequential NOE pattern to the preceding C103 (Supplementary Figure S2a). These data demonstrate a continuation of the canonical helical structure into the loop by one residue, with U104 stably stacking on top of C103. For G105 as well as C106, both 3J(H,H) coupling constants and canonical coordinates point to a stable C2’-endo ribose conformation (Supplementary Table S1, Supplementary Figure S2b). For C106 unusual upfield chemical shifts can be observed for H2’, H4’, H5’ and H5’’ (Figure 3d), suggesting a proximity of the C106 ribose to a nucleobase moiety. In the NMR structure bundle, the C106 moiety is most often located close to the nucleobase of A107. The proximity of a ribose moiety in C2’-endo conformation to the nucleobase of the following residue is a typical feature of the Z-step structural motif (40). This 2-nucleotide motif is characteristic for Z-DNA, but has also been identified in a large variety of structural contexts in natural RNAs such as riboswitch aptamer domains or ribosomal RNA (41). It involves a lone-pair…π interaction between the ribose O4’ of the 5′-residue and the nucleobase of the 3′-residue (Supplementary Figure S2c). To achieve this interaction, the 5′-ribose moiety is reversed in its orientation. This head-to-head orientation can be observed for the C106 ribose moiety in a number of the structures of the NMR bundle. In a Z-step the 3′-residue is preferably a purine with the nucleobase in syn-orientation. For 5_SL4, the moderate intraresidual NOE between H1’ and H8 for A107, however, suggests that this residue adopts predominantly an anti-conformation. Taken together, the NMR data indicate that C106 and A107 form a subcategory of Z-steps referred to as Zanti-step at least transiently (40).
Figure 3.

Structural details of the apical loop. (A) {1H},13C-hetNOE of the aromatic pyrimidine H6C6 and the purine H8C8 moieties (bottom) and aliphatic H1’C1’ moieties (top) of 5_SL4sh. Residues of the loop region (gray) are highlighted in different colors according to the color scheme in Figure 2. A dashed line indicates the threshold for flexible residues (1.2). (B-C) NMR structural bundle of the apical loop. The loop closing base pair is used for structure alignment. The RNA is shown as a cartoon representation in two different orientations turned by 180° in respect to each other. The loop residues shown in different colors according to the color scheme in Figure 2 and assigned. (D) Aliphatic 1H,13C-HSQC of 5_SL4sh. Resonances of U104 (green), G105 (red), C106 (blue) and A107 (magenta) are assigned in the respective colors. Gray spheres mark the canonical regions of the spectrum and characteristic 13C chemical shift regions are indicated to the right of the graph. (E) Overlay between the apical loop region of the NMR bundle of 5_SL4 and the lowest target function NMR structure of the cUUCGg tetraloop in a 14-nt RNA stem-loop (PDB entry 2koc). The C-G closing base pairs are used for alignment of the loops. The 5_SL4 RNA is shown as a cartoon representation using the color scheme shown in Figure 2. The UUCG tetraloop residues are colored accordingly. (F) Schematic representation of the transient structural features of the 5_SL4 apical loop. Residues are highlighted in different colors according to the color scheme in Figure 2 and assigned. Hydrogen bonds are shown as dashed lines and stacking interactions as gray ellipses. The Z-step with its lone pair...π stacking contact is indicated.
The last residue of the 5_SL4 apical loop, U108, is very dynamic, with a flexible nucleobase moiety and completely averaged ribose pucker conformation. In the NMR structure bundle, U108 is mostly flipped-out of the loop, suggesting that the 5_SL4 pentaloop can be actually considered as a tetraloop with a one-nucleotide 3′-extension. For the first four loop nucleotides, the observed residual structural features, the C2’-endo ribose pucker of loop residues 2 and 3, the Z-step conformation of loop residues 3 and 4 and the stable stacking of the first loop residue are all structural characteristics reminiscent of the YNMG tetraloop motif (Y = C, U; N = any nucleotide; M = A, C) (42). This motif has been shown to tolerate 3′-extensions by one nucleotide without significant changes in structure or stability (described as YNMG(N) consensus motif) (43). Members of the YNMG tetraloop family also share characteristic NMR spectral features. Thus, the upfield ribose proton chemical shifts of the third loop nucleotide found for the 5_SL4 C106 ribose moiety are also generally observed for members of the YNMG family such as the very stable UNCG motif (Supplementary Figure S2d) (44), the CACG tetraloop of the Coxsackievirus D-loop (45), and the UCAG(U) as well as the CUUG(U) pentaloops of a telomerase RNA mutant (43) and mouse hepatitis virus stem-loop 2 (46). Furthermore, downfield chemical shifts of H2’ and H3’ observed for A107 are characteristic for the fourth UNCG loop residue (44).
An additional structural characteristic of the YNMG motif is a hydrogen bond between the amino group of the A or C at position 3 and the phosphate group of the second loop residue (44). Although a similar interaction cannot be observed directly in the apical loop of 5_SL4, the amino group nitrogen resonance of C106 is shifted downfield by ∼1.5 ppm compared to the amino resonance of free monomeric cytidine (Supplementary Figure S2e). In general, the amino nitrogen chemical shift is sensitive to hydrogen bonding interactions with a shift toward larger ppm values signifying a stronger hydrogen bond. In addition, also the temperature dependence of the amino group NMR signals can report on hydrogen bond stability. Two distinct resonances are observed for the two amino hydrogens when the rotation around the exocyclic C–N bond is sufficiently slow, e.g. at low temperatures or in the presence of stabilizing hydrogen bonds. By contrast, at higher temperatures only one degenerate hydrogen resonance is present due to chemical exchange arising from increased rotation around the C–N bond. For C106, the amino group signals can still be observed at temperatures of up to 30°C, whereas the amino resonances of free cytidine vanish already at ∼20°C (Supplementary Figure S2e,f). In contrast, for cytidines in Watson–Crick G–C base pairs, two distinct amino resonances can still be clearly observed at a temperature of 50°C. We therefore conclude that the amino group of C106 forms a weak or transient hydrogen bond. Although the hydrogen bond acceptor group could not be determined experimentally, it is reasonable to assume that in analogy to the YNMG tetraloop motif, the acceptor is one of the non-bridging oxygen atoms of the G105 phosphate group.
In general, all loop sequences that have been included into the extended YNMG consensus motif so far have displayed base pairs between the first and the fourth loop residue. This comprised a trans Sugar•Watson-Crick Y•G base pair (44,45), a sheared G A pseudo base pair without a direct hydrogen bond (47) and a Watson–Crick C–G base pair (46). All of these base pairs are characterized by a syn or a high anti conformation of the fourth loop nucleotide. By contrast, the fourth residue of the 5_SL4 apical loop, A107, adopts an anti-conformation, an averaged ribose conformation and no indication of a stable base-pairing interaction with U104 can be found. The absence of a stable base pair between the first and the fourth loop residue likely rationalizes the decreased stability of the 5_SL4 apical loop compared to the YNMG(N) loop sequences, for which stable structures were reported previously. The transient nature of the structural features in the apical loop of 5_SL4 is also supported by the canonical A-form like chemical shifts of its phosphate resonances likely originating from a less defined or averaged conformation (26). Accordingly, an overlay between the 5_SL4 NMR bundle and the NMR structure of a UUCG tetraloop shows that whereas the overall positioning of the first and fourth loop residue is similar, the distinct backbone conformation of the UUCG tetraloop is not reflected in the various different conformations of the 5_SL4 apical loop (Figure 3e). In conclusion, the apical loop of 5_SL4 can be described as dynamic with transient structural features which share certain similarities with a YNMG(N) motif summarized in Figure 3f.
Interestingly, in agreement with the structural features observed in the NMR-experiments, molecular dynamics simulations of 5_SL4 report a dynamic ensemble for the apical loop structures comprising an YNMG(N)-like fold with a flipped-out 3′-residue. Within the molecular dynamics ensemble, a Z- or Zanti-step between the third and fourth residue is found and a base pair between U104 and A107, which either adopts Watson–Crick or Sugar•Hoogsteen geometry (10).
Given that the observed high sequence conservation of the apical loop is not imposed by requirements to form a stable structure, the question remains as to the underlying reasons for this conservation. It can be assumed that a stable loop structure would interfere with the function of the uORF, whose start codon comprises loop residues A107 and U108. The residual structural features identified could be involved in additional functions of 5_SL4 such as providing an adaptive region for transient interactions with viral or host structures.
Mixed tandem mismatches
5_SL4 contains two mixed tandem mismatches (TMs) both consisting of a pyrimidine•pyrimidine mismatch and a G•U wobble base pair (Figure 4a). In the lower part of 5_SL4, the C92•C119 mismatch is preceded by the G91•U120 wobble base pair (TM1). Further up in the 5_SL4 stem, the U112•C100 mismatch follows the U111•G101 wobble base pair (TM2). Both tandem mismatches are flanked by a Watson–Crick G–C base pair on the G•U side and a Watson–Crick U–A (TM1) or A-U (TM2) base pair on the pyrimidine•pyrimidine mismatch side. For both G•U base pairs the usual wobble-geometry characterized by two hydrogen bonds, one between the U-N3H3 imino group and the G-O6 carbonyl group and one between the G-N1H1 imino group and the U-O2 carbonyl group has been established previously (26). For both pyrimidine•pyrimidine mismatches, {1H},13C-hetNOE values of both the ribose C1’ and the nucleobase C6 spin are <1.2 signifying a stable conformation (see Figures 2a and 3a). The ribose puckers of all four pyrimidine residues determined from 3J(H,H) coupling constants and canonical coordinates of the ribose 13C shifts is C3’-endo (Supplementary Table S1 and Supplementary Figure S3a), and the characteristic aromatic-aliphatic NOE pattern is maintained along the mismatches as expected for an A-form RNA helical arrangement (Supplementary Figure S3b,c). Taken together, these data suggest that the pyrimidine•pyrimidine mismatches in TM1 and TM2 form defined base pairs.
Figure 4.

Tandem mismatches in 5_SL4. (A) Structure of the two tandem mismatches and the two flanking base pairs in the NMR structure with the lowest target function. TM1 is shown in blue, TM2 in red. The RNA is shown as a stick representation, residues are assigned. Left: Top view from the pyrimidine•pyrimidine side of the mismatches. Hydrogen bonds are indicated by dashed lines. Middle: Sequence of the TMs and surrounding base pairs. Right: Side view of the TMs and surrounding base pairs. C1’-C1’ distances are indicated by dashed red lines, distances are given. (B) Relative imino resonance integrals referenced to the stable residue average along the base pairs of 5_SL4. The base pairs of TM1 and TM2 and the flanking base pairs are highlighted in blue and red, respectively. For G•U base pairs, the integral of the G imino resonance was considered in the analysis.
In general, pyrimidine•pyrimidine mismatches are known to be polymorphic (48). In a helical environment, their geometry varies with the identity of the flanking base pairs. Major driving forces seem to be to maintain helical integrity, while at the same time securing optimal base stacking and hydrogen bonding interactions. Considering the type of base pair formed between C92 and C119 in TM1, neither of the two cytidine residues is protonated, as no imino resonance can be detected and nucleobase C4 chemical shifts suggest neutral N3 sites for both C92 and C119 (26). Previous studies of intrahelical C•C mismatches in other RNAs postulated the presence of a dynamical base pair subjected to positional averaging (49–51). In these cases, no amino group signals could be observed due to the presence of chemical exchange. This exchange was attributed to the two C-residues interchanging as donor and acceptor sites in more or less equal proportions with a hydrogen bond between the amino group of one and the N3 of the other cytidine. Also for 5_SL4 the amino resonance of C92 cannot be detected due to exchange broadening. However, two weak, but distinct amino proton signals are found for C119 (Supplementary Figure S4a). Thus, the C92•C119 mismatch in the 5_SL4 sequence context appears slightly asymmetric, with the amino group of C119 serving as the predominant hydrogen bond donor group. The NMR ensemble is also in agreement with conformational averaging with a preference for the conformation with the C119 amino group as the hydrogen bond donor (Supplementary Figure S4b).
For the C100•U112 mismatch in TM2, no imino proton resonance is detectable for U112 in the 1H,15N imino HSQC spectrum (see Figure 1c), suggesting that the U112 imino group is not involved in a stable hydrogen bond. By contrast, two distinct resonances can be observed for the C100 amino group with a nitrogen shift of 96.8 ppm (see Supplementary Figure S2d). Compared to the amino group chemical shifts of free cytidine, the amino group nitrogen of C100 is thus shifted downfield by ∼3 ppm, suggesting that it serves as hydrogen bond donor. Two distinct amino proton resonances can be observed up to a temperature of 35°C, indicating that the hydrogen bonding interaction is weak compared to the one of cytidine amino groups in Watson–Crick base pairs (see Supplementary Figure S2e). In principle, the hydrogen bond acceptor group could be either the U112-O2 or -O4 carbonyl group. Uridine carbonyl chemical shifts are known to be sensitive to their involvement in hydrogen bonds with downfield resonance positions suggesting the presence of a hydrogen bond (43). For U112 both C2 and C4 are found to resonate at an upfield position, thus no clear information on which carbonyl group is hydrogen bonded to the C100 amino group can be obtained from the carbonyl shifts (Supplementary Figure S5). Although both hydrogen bonding arrangements have been reported in earlier structures of C•U mismatches, only the hydrogen bond between the C-amino group and the U-O4 carbonyl group has been found in intrahelical single C•U mismatches (44,52–59). Incorporation of either the hydrogen bond to O2 or O4 of U112 into the structure calculation of 5_SL4 also resulted in a preference for the O4 carbonyl as hydrogen bond acceptor, since in this case the hydrogen bond could be introduced without violation of distance restraints.
A structural comparison of the two tandem mismatches shows that in agreement with the different directionality of the G•U wobble base pairs, their base stacking topology also differs (Figure 4a, left). G•U wobble base pairs are known to be non-isosteric to U•G base pairs. In particular, the twist angle preceding a G•U base pair (as in TM1) is larger than the canonical twist, while the one preceding a U•G base pair (as in TM2) is smaller, implicating sequence dependent effects on base stacking (60). Accordingly, while in TM1 intra-strand stacking mostly occurs between the two mismatch base pairs and stacking to the adjacent Watson–Crick base pairs is limited, the opposite is the case in TM2. In both cases the disruption of the A-form helix introduced by the G•U mismatch in respect of helical twist and stacking interactions, appears to be compensated by the opposite geometry of the pyrimidine•pyrimidine mismatch. Furthermore, C1’-C1’ distances differ only very slightly from canonical A-form geometry across both tandem mismatches (Figure 4a, right) again suggesting that the mismatches adopt a conformation which causes minimal disruption of the A-form helix.
In line with their different structural features, the stability of the two tandem mismatches differs as well. Imino group resonance integrals in 1H,15N-HSQC spectra reflect the stability of the imino proton against exchange with the solvent water, which in turn depends on the stability of the hydrogen bonding interaction in which a given imino group is involved. For both TM regions, significant reductions in imino group resonance integrals compared to the stable stem regions are observed (Figure 4b), indicating that despite the moderate disruption of A-form geometry, both TM regions locally disrupt the stability of the 5_SL4 stem. The extent of this destabilization, however, is different for the two TMs. For TM2, two sharp imino resonances are observed for the 5′-U111•G101-3′ base pair with resonance integrals similar to those of canonical stem residues, indicating that this U•G wobble base pair is stable. By contrast, for 5′-G91•U120-3′ in TM1, the imino group signal integrals are much reduced, indicating unstable base pairing. Furthermore, the imino group signal integral of U99 of the U-A base pair flanking TM2 is significantly larger than the one of U93 flanking TM1. Taken together, these observations suggest that TM1 is less stable than TM2. The local instability of the two TMs as well as their different relative stability observed in our NMR-investigation is in agreement with dimethyl sulfate (DMS) mapping data (61), which reports increased reactivity of all three cytosines in the pyrimidine•pyrimidine mismatches with C92 in TM1 showing the highest reactivity.
C100 in the C100•U112 mismatch is transiently protonated
Interestingly, we could not detect a N3 resonance for C100 under standard slightly acidic NMR-conditions (pH 6.2, (26)). The absence of the N3 resonance suggests that this nitrogen is transiently protonated at pH 6.2, e.g. broadened by chemical exchange between a protonated and a non-protonated state. To test this hypothesis, we performed a step-wise pH titration. As shown by Legault and Pardi, the cytidine C4 chemical shift is extremely sensitive to the protonation state of the neighboring N3 shifting upfield by ∼6 ppm on N3 protonation (62). Indeed, the C4 of residue C100 shifted by exactly 6 ppm from 167.2 ppm at pH 6.2 to 161.2 ppm at pH 4.3 (Figure 5a). The pH dependence of the C4 shift of C100 can be used to fit a pKa of 5.3 ± 0.1 for this residue (Figure 5a, inset). This value is shifted by one pH unit from the pKa of free cytidine mononucleotides (pKa of 4.3, (63)). Thus, at a pH of 6.2, which was used in the structure determination procedure, C100 is only partially protonated with 11% of the protonated species. We therefore report the unprotonated structure in this investigation.
Figure 5.

Structure of the C100•U112 base pair. (A) Overlay of 2D H5(C5)C4 spectra recorded for 5_SL4sh at pH 6.6 (black), pH 6.2 (dark blue), pH 5.9 (light blue), pH 5.5 (green), pH 4.8 (yellow), pH 4.5 (orange) and pH 4.3 (red). Assignments are given. pH-dependent chemical shift changes of resonances of C100 and C106 are indicated by gray dashed lines. The magnetization transfer pathway of the experiment is indicated in red in a schematic representation of an uracil. Inset: pH dependence of the C100 C4 chemical shift, fitted pH dependence and resulting pKa value. (B) 2D H5(C5C4)N3 spectrum recorded for 5_SL4sh at pH 4.3. Assignments are given. The magnetization transfer pathway is highlighted in red in the chemical structure of an uracil. (C) 1H,15N imino HSQC spectrum recorded for 5_SL4sh at pH 4.3. Assignments are given. The chemical structure of an uracil with highlighted imino group is displayed. For the protonated C100 the assignment of the imino resonance at pH 4.3 is followed by dashed lines in the H5(C5)C4, the H5(C5C4)N3 and the 1H,15N-HSQC spectrum. (D) Schematic representation of the C100•U112 mismatch at high pH (top) and low pH (bottom). Hydrogen bonds are shown as red dashed lines.
At a pH of 4.3, an additional imino resonance can be detected in the 1H,15N-HSQC spectrum (Figure 5c), which can be assigned to the C100 H3N3 imino group by connecting the H5 to the N3 via the H5(C5)C4 and the H5(C5C4)N3 spectrum (Figure 5a–c). Both the C100-N3 and its H3 resonance are found in the upfield region of the imino resonances typical for non-canonical interactions with oxygens as hydrogen bond acceptors. Based on the NMR structure, the most likely hydrogen bond acceptor for the C100-N3 imino group is the U112-O4 carbonyl group. The structural context of the C100•U112 mismatch with the hydrogen bond between the C100-amino and the U112-O4 carbonyl group brings C100-N3 in close proximity to the U112-O4. The formation of this second hydrogen bond to the U112-O4 carbonyl group is supported by a slight downfield shift of the U112-C4 resonance at lower pH values (Figure 5a). In this detailed analysis, we observe a pH-dependent polymorphism of the C100•U112 base pair (Figure 5d): At higher pH, only one hydrogen bond is consistently formed between the C100-N4 amino and the U112-O4 carbonyl group. At lower pH, the N3 protonated form of C100 becomes increasingly populated and a bifurcated C+•U base pair with two hydrogen bonds, one between the C+-N4 amino group and the U-O4 carbonyl group and one between the C+-N3 imino group and the U-O4 is predominant. Although the low-pH C+•U base pair is stabilized by two hydrogen bonds, the temperature dependence of its amino proton resonances suggests that it is not significantly more stable than the neutral C•U mismatch (Supplementary Figure S6). Thus, the gain in stability by an additional hydrogen bond is apparently counteracted by a less favorable base pair geometry with regard to base stacking, C1’-C1’ distance or overall hydrogen bonding geometry.
Structure prediction of 5_SL4 using FARFAR2
For proteins, three-dimensional structure prediction from primary sequences provided by programs such as AlphaFold has made considerable progress (64). These deep learning algorithms profit from the vast number of deposited protein structures combined with neural network learning procedures. For RNA, de novo sequence based predictions can be obtained from Rosetta's Fragment Assembly of RNA (FARFAR2 (65)), which uses a helix base pair step in combination with a fragment library. To assess the reliability of the FARFAR2 prediction in the case of 5_SL4 we compared the NMR structural bundle to a bundle of 10 FARFAR conformers predicted based on the 5_SL4 sequence and previously established secondary structure (9). As the experimentally determined structure, FARFAR2 consistently predicts a rod-like stem-loop structure (Supplementary Figure S7a). However, the structural details of the prediction differ significantly from the experimentally determined structure. Thus, U95 is stacked inside the helical stem between the U94-G117 and the C96-G116 base pair in all FARFAR2 structures, whereas the experimental data clearly shows that this residue is excluded from the helix and flexible (Supplementary Figure S7b). Interestingly, the asymmetric insertion of U95 into the helix in the predicted 5_SL4 stem-loops is incorporated without an appreciable kink in the overall structure. The overall straight rod-like shape is similar to the NMR structure, with the difference that the FARFAR2 prediction results in slightly narrower and shorter structures (Supplementary Figure S7c; Rg = 19.0 ± 0.2 Å and Dmax = 66.6 ± 1.3 Å). An evaluation of the solution scattering from the FARFAR2 and the NMR structure with the best agreement to the SAXS data (χ2 = 23 and 2, respectively) by the program CRYSOL (39) illustrates the better fit of the experimental NMR-structure to the SAXS-data (Supplementary Figure S7d).
A general overemphasis of stacking interactions in the prediction is also found for the apical loop, where both the first residue (U104) and the last residue (U108) are found inside the loop stacked on the respective closing base pair residue (Supplementary Figure S7e). In addition, the remaining loop residues are found in different stacked conformations inside the loop. Overall, the FARFAR2 prediction significantly overemphasizes the compactness of the apical loop conformation in contradiction to the high degree of conformational flexibility indicated by the NMR data.
As for the C92•C119 mismatch, two different conformations are predicted with similar frequency, both with one hydrogen bond between the amino group of one of the cytidines to the N3 of the other cytidine, suggesting the presence of a conformational equilibrium between these two locally symmetric states (Supplementary Figure S7f). Apparently, while the hydrogen bonding details of the C•C mismatch are reflected in the FARFAR2 prediction, the asymmetry of the mismatch environment is not sufficiently incorporated into it. For the C100•U112 mismatch, a base pair with two hydrogen bonds is consistently predicted by FARFAR2, one between the C-amino group and U-O4 and one between the U-imino group and C-N3 (Supplementary Figure S7g).
In summary, FARFAR2 predicts an overall rod-like structure for 5_SL4 in agreement with the experimental data. The structural details of the interesting non-canonical elements, however, are characterized by an overestimation of both stacking and hydrogen bonding interactions. Interestingly, despite the availability of the respective structural data, features like the preference for the Zanti-step in the apical loop, which are consistently observed by the NMR investigation and modeled in the molecular dynamics simulations, are not predicted by FARFAR2. Considering that the 5_SL4 stem-loop with its small size and moderate number of non-canonical elements represents a rather simple prediction benchmark, the need for further improvement of the prediction scoring algorithms and cross-validation with experimental data such as accessibility data or hydrogen bonding information from NMR seems still advisable, in particular when the structure constitutes the basis for drug targeting efforts.
5_SL4 chemical shift and structural data as basis for structure-guided drug development
As most of the stem-loop structures in the 5′-UTR of the SARS-CoV-2 genome, also 5_SL4 is highly conserved among the different SARS-CoV-2 variants. In particular, the C92•C119 mismatch is completely conserved, the C100•U112 mismatch is only very rarely substituted by a G•U, the looped out U95 is only very rarely deleted and A107 and U108 of the apical loop are completely conserved as they are part of the conserved uORF. Of the remaining residues of the apical loop, U104 is completely conserved, while G105 and C106 are only very rarely mutated to uridines (66).
5_SL4 has been recently found to be a druggable target for small molecule ligands (16,24). The chemical shift assignment and the information on the three-dimensional structure of the elements of the SARS-CoV-2 genome allow a more detailed characterization of small compound binders identified in these screening efforts. Amiloride has been previously identified as promising scaffold for specific RNA targeting (25). Here, we have selected one of several identified binders from a library of dimethylamiloride (DMA) derivatives carrying fluoride atoms as additional NMR sensitive probes, DMA0043 (Figure 6a). Initial binding to 5_SL4 was verified by 19F compound shifts in 1D NMR spectra on addition of the RNA (Supplementary Figure S8a). To map the binding region of DMA0043 on 5_SL4, we titrated selectively G,U- and A,C-13C,15N-labeled samples with increasing amounts of the compound. The binding event was followed by recording 1H,13C-correlation spectra for the aromatic CH-moieties. Thus, we could monitor site-specific binding for each residue. On titration with DMA0043, several peaks in the 1H,13C-HMQC spectrum of 5_SL4 shift and/or become broader in agreement with binding of the compound to the RNA with an affinity in the μM range (Figure 6b; Supplementary Figure S8b). Regions with large CSPs are located around the two tandem mismatches, the looped-out U95 as well as around the two terminal G-C base pairs (Figure 6c). Given that the mismatches could be regions with the function to locally destabilize the otherwise rigid stem-loop structure, a ligand like DMA0043 could interfere with this function by effectively stabilizing the mismatch region. Considering the small size of the compound, the extended regions on the 5_SL4 RNA, for which the NMR-signals are affected by the presence of DMA0043, suggest multiple interaction regions. Interestingly, also the terminal G–C base pairs show affinity for DMA0043 likely rationalizing the lack in specific binding observed for the compound, which also showed affinity to several other stem-loops in previous screening assays (16). This rationale for the unspecific RNA binding properties of DMA0043 shows the value of our RNA-based site-specific NMR analysis and can serve to direct further combinatorial chemistry efforts to develop tighter and more specific binders. Thus, further diversification of the amiloride scaffold as for example demonstrated for HIV-1 TAR RNA (25), e.g. by introduction of bulky substituents could interfere with unspecific stacking interactions.
Figure 6.

(A) Chemical structure of the screening hit DMA0043. (B) Titration of DMA0043 to 5_SL4 followed by 1H,13C-HMQC spectra of the aromatic pyrimidine H6C6 and purine H8C8 moieties using a selectively G,U-13C,15N labeled sample. The spectra of the different titrations steps are shown in different shades of gray as indicated. For residues displaying significant shifts in the course of the titration, assignments are given and the peak shifts with increasing ligand concentration are indicated by arrows. (C) Combined 1H and 13C chemical shift perturbations of all aromatic H6/8C6/8 moieties of 5_SL4 with resolved resonances derived from the comparison of 1H,13C-HMQC spectra obtained in the absence or in the presence of 1 mM DMA0043. Cut-offs for small (0.05) and large shifts (0.1) are indicated by dashed lines. Non-canonical regions are highlighted. Different binding regions are colored blue (around TM1), red (around TM2) or green (terminal region) with large shifts in dark and small shifts in light colors. (D) CSPs induced by DMA0043 displayed on the secondary structure of 5_SL4. The region comprising TM1 and TM2 are shown in blue and red, respectively. The third binding region at the terminus of 5_SL4 is shown in green. Small CSPs (0.05–0.1) are shown in light red, light blue and light green, large shifts (>0.1) are shown in red, blue and green. (E) CSPs induced by DMA0043 displayed on the NMR structure of 5_SL4. The RNA is shown as a sphere and stick representation. Residues are highlighted according to the color scheme in (D).
Functional implications of the 5_SL4 structure
Our structural investigation of 5_SL4 shows that its non-canonical elements are integrated into a more or less unperturbed A-form stem-loop structure. Given that both the looped-out U95 as well as the two mixed tandem mismatches do not result in pronounced structural deviations from the A-form RNA helix while they are conserved among the SARS-CoV-2 variants, the question remains as to which function these structural elements perform in the viral genome. We observed that the regions around the mismatches and the looped-out residue are less stable than the surrounding stretches of continuous canonical RNA. For G•U mismatches, which are the most frequently occurring mismatches in biological RNAs, their structural deformability is believed to be crucial for molecular recognition (49). The function of the non-canonical elements could therefore be to maintain the conformation and at the same time allow for plasticity of the long stem-loop structure of 5_SL4 in processes such as interactions with viral proteins during the viral life cycle. In particular, effective scanning of the ribosomal 48S pre-initiation complex and translation of the uORF with the start codon located in the apical loop of 5_SL4 likely depend on unstable non-canonical regions in its vicinity.
Supplementary Material
Acknowledgements
We would like to thank Roderick Lambertz and Laura Hahn for the preparation of isotope labeled RNA and Andreas Oxenfarth for helpful discussions on how to conduct RDC measurements. We thank Andreas Schlundt and his group for help with the SAXS measurements and data evaluation. We are grateful to Sridhar Sreeramulu and Andreas Schlundt for carefully reading the manuscript.
Contributor Information
Jennifer Vögele, Institute for Molecular Biosciences, Goethe-University Frankfurt, Max-von-Laue-Strasse 9, 60438 Frankfurt/M., Germany; Center for Biomolecular Magnetic Resonance (BMRZ), Goethe-University Frankfurt, Max-von-Laue-Strasse 7, 60438 Frankfurt/M., Germany.
Daniel Hymon, Center for Biomolecular Magnetic Resonance (BMRZ), Goethe-University Frankfurt, Max-von-Laue-Strasse 7, 60438 Frankfurt/M., Germany; Institute for Organic Chemistry and Chemical Biology, Goethe-University Frankfurt, Max-von-Laue-Strasse 7, 60438 Frankfurt/M., Germany.
Jason Martins, Center for Biomolecular Magnetic Resonance (BMRZ), Goethe-University Frankfurt, Max-von-Laue-Strasse 7, 60438 Frankfurt/M., Germany; Institute for Organic Chemistry and Chemical Biology, Goethe-University Frankfurt, Max-von-Laue-Strasse 7, 60438 Frankfurt/M., Germany.
Jan Ferner, Center for Biomolecular Magnetic Resonance (BMRZ), Goethe-University Frankfurt, Max-von-Laue-Strasse 7, 60438 Frankfurt/M., Germany; Institute for Organic Chemistry and Chemical Biology, Goethe-University Frankfurt, Max-von-Laue-Strasse 7, 60438 Frankfurt/M., Germany.
Hendrik R A Jonker, Center for Biomolecular Magnetic Resonance (BMRZ), Goethe-University Frankfurt, Max-von-Laue-Strasse 7, 60438 Frankfurt/M., Germany; Institute for Organic Chemistry and Chemical Biology, Goethe-University Frankfurt, Max-von-Laue-Strasse 7, 60438 Frankfurt/M., Germany.
Amanda E Hargrove, Department of Chemistry, Duke University, Durham, NC 27708, USA.
Julia E Weigand, Philipps-University Marburg, Department of Pharmacy, Institute of Pharmaceutical Chemistry, Marbacher Weg 6, 35037 Marburg, Germany.
Anna Wacker, Center for Biomolecular Magnetic Resonance (BMRZ), Goethe-University Frankfurt, Max-von-Laue-Strasse 7, 60438 Frankfurt/M., Germany; Institute for Organic Chemistry and Chemical Biology, Goethe-University Frankfurt, Max-von-Laue-Strasse 7, 60438 Frankfurt/M., Germany.
Harald Schwalbe, Center for Biomolecular Magnetic Resonance (BMRZ), Goethe-University Frankfurt, Max-von-Laue-Strasse 7, 60438 Frankfurt/M., Germany; Institute for Organic Chemistry and Chemical Biology, Goethe-University Frankfurt, Max-von-Laue-Strasse 7, 60438 Frankfurt/M., Germany.
Jens Wöhnert, Institute for Molecular Biosciences, Goethe-University Frankfurt, Max-von-Laue-Strasse 9, 60438 Frankfurt/M., Germany; Center for Biomolecular Magnetic Resonance (BMRZ), Goethe-University Frankfurt, Max-von-Laue-Strasse 7, 60438 Frankfurt/M., Germany.
Elke Duchardt-Ferner, Institute for Molecular Biosciences, Goethe-University Frankfurt, Max-von-Laue-Strasse 9, 60438 Frankfurt/M., Germany; Center for Biomolecular Magnetic Resonance (BMRZ), Goethe-University Frankfurt, Max-von-Laue-Strasse 7, 60438 Frankfurt/M., Germany.
Data availability
The coordinates of the 10 5_SL4 structures with the lowest target functions of the NMR-bundle have been submitted to the PDB under the accession code 8CQ1. SAXS data are available in SASBDB under the accession code SASDRV7.
Supplementary data
Supplementary Data are available at NAR Online.
Funding
Work at the Center for Biomolecular Magnetic Resonance (BMRZ) at the Goethe-University Frankfurt is supported by the state of Hesse. Work in COVID-19-NMR was supported by the Goethe Corona Funds, by the IWB-EFRE-program 20007375 of the state of Hesse, the DFG in CRC902: ‘Molecular Principles of RNA-based regulation’ and infrastructure funds (Project Numbers: 277478796, 277479031, 392682309, 452632086, 70653611) as well as the European Union's Horizon 2020 research and innovation program iNEXT-discovery under grant agreement No 871037. H.S. is supported by the DFG in graduate school CLIC (GRK 1986). H.S., J.E.W. and J.W. receive funding by the DFG through grants SCHW 701/27-1, WE 5819/6-1 and WO 901/11-1, respectively. 19F NMR measurements were made possible through grant INST161/981-1 FUGG by the DFG.
Conflict of interest statement. None declared.
References
- 1. Simmonds P. Pervasive RNA secondary structure in the genomes of SARS-CoV-2 and other coronaviruses. mBio. 2020; 11:e01661-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cao C., Cai Z., Xiao X., Rao J., Chen J., Hu N., Yang M., Xing X., Wang Y., Li M.et al.. The architecture of the SARS-CoV-2 RNA genome inside virion. Nat. Commun. 2021; 12:3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tavares R.d.C.A., Mahadeshwar G., Wan H., Huston N.C., Pyle A.M.. The global and local distribution of RNA structure throughout the SARS-CoV-2 genome. J. Virol. 2020; 95:e02190-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yang D., Leibowitz J.L.. The structure and functions of coronavirus genomic 3' and 5' ends. Virus Res. 2015; 206:120–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee C.W., Li L., Giedroc D.P.. The solution structure of coronaviral stem-loop 2 (SL2) reveals a canonical CUYG tetraloop fold. FEBS Lett. 2011; 585:1049–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang K., Zheludev I.N., Hagey R.J., Haslecker R., Hou Y.J., Kretsch R., Pintilie G.D., Rangan R., Kladwang W., Li S.et al.. Cryo-EM and antisense targeting of the 28-kDa frameshift stimulation element from the SARS-CoV-2 RNA genome. Nat. Struct. Mol. Biol. 2021; 28:747–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bhatt P.R., Scaiola A., Loughran G., Leibundgut M., Kratzel A., Meurs R., Dreos R., O’Connor K.M., McMillan A., Bode J.W.et al.. Structural basis of ribosomal frameshifting during translation of the SARS-CoV-2 RNA genome. Science. 2021; 372:1306–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jones C.P., Ferré-D’Amaré A.R.. Crystal structure of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) frameshifting pseudoknot. RNA. 2022; 28:239–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rangan R., Watkins A.M., Chacon J., Kretsch R., Kladwang W., Zheludev I.N., Townley J., Rynge M., Thain G., Das R.. De novo 3D models of SARS-CoV-2 RNA elements from consensus experimental secondary structures. Nucleic Acids Res. 2021; 49:3092–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bottaro S., Bussi G., Lindorff-Larsen K.. Conformational ensembles of noncoding elements in the SARS-CoV-2 genome from molecular dynamics simulations. J. Am. Chem. Soc. 2021; 143:8333–8343. [DOI] [PubMed] [Google Scholar]
- 11. Monica L., G. B., A. L., A. M.. Targeting SARS-CoV-2 main protease for treatment of COVID-19: covalent inhibitors structure-activity relationship insights and evolution perspectives. J. Med. Chem. 2022; 65:12500–12534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu X., Chen Y., Lu X., Zhang W., Fang W., Yuan L., Wang X.. An update on inhibitors targeting RNA-dependent RNA polymerase for COVID-19 treatment: promises and challenges. Biochem. Pharmacol. 2022; 205:115279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pohler A., Abdelfatah S., Riedl M., Meesters C., Hildebrandt A., Efferth T.. Potential coronaviral inhibitors of the nucleocapsid protein identified in silico and in vitro from a large natural product library. Pharmaceuticals (Basel). 2022; 15:1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Duchardt-Ferner E., Ferner J., Fürtig B., Hengesbach M., Richter C., Schlundt A., Sreeramulu S., Wacker A., Weigand J.E., Wirmer-Bartoschek J.et al.. The COVID19-NMR Consortium: a public report on the impact of this new global collaboration. Angew. Chem. Int. Ed. Engl. 2023; 62:e202217171. [DOI] [PubMed] [Google Scholar]
- 15. Wacker A., Weigand J.E., Akabayov S.R., Altincekic N., Bains J.K., Banijamali E., Binas O., Castillo-Martinez J., Cetiner E., Ceylan B.et al.. Secondary structure determination of conserved SARS-CoV-2 RNA elements by NMR spectroscopy. Nucleic Acids Res. 2020; 48:12415–12435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sreeramulu S., Richter C., Berg H., Wirtz Martin M.A., Ceylan B., Matzel T., Adam J., Altincekic N., Azzaoui K., Bains J.K.et al.. Exploring the druggability of conserved RNA Regulatory Elements in the SARS-CoV-2 Genome. Angew. Chem. Int. Ed. Engl. 2021; 60:19191–19200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu H.-Y., Guan B.-J., Su Y.-P., Fan Y.-H., Brian D.A.. Reselection of a genomic upstream open reading frame in mouse hepatitis coronavirus 5'-untranslated-region mutants. J. Virol. 2014; 88:846–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Condé L., Allatif O., Ohlmann T., Breyne S.d.. Translationof SARS-CoV-2 gRNA is extremely efficient and competitive despite a high degree of secondary structures and the presence of an uORF. Viruses. 2022; 14:1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang D., Liu P., Giedroc D.P., Leibowitz J.. Mouse hepatitis virus stem-loop 4 functions as a spacer element required to drive subgenomic RNA synthesis. J. Virol. 2011; 85:9199–9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Raman S., Bouma P., Williams G.D., Brian D.A.. Stem-loop III in the 5' untranslated region is a cis-acting element in bovine coronavirus defective interfering RNA replication. J. Virol. 2003; 77:6720–6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yao Y., Sun H., Chen Y., Tian L., Huang D., Liu C., Zhou Y., Wang Y., Wen Z., Yang B.et al.. RBM24 inhibits the translation of SARS-CoV-2 polyproteins by targeting the 5'-untranslated region. Antivir. Res. 2022; 209:105478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yao Y., Yang B., Cao H., Zhao K., Yuan Y., Chen Y., Zhang Z., Wang Y., Pei R., Chen J.et al.. RBM24 stabilizes hepatitis B virus pregenomic RNA but inhibits core protein translation by targeting the terminal redundancy sequence. Emerg. Microbes & Infect. 2018; 7:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cao H., Zhao K., Yao Y., Guo J., Gao X., Yang Q., Guo M., Zhu W., Wang Y., Wu C.et al.. RNA binding protein 24 regulates the translation and replication of hepatitis C virus. Protein Cell. 2018; 9:930–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zafferani M., Haddad C., Luo L., Davila-Calderon J., Chiu L.-Y., Mugisha C.S., Monaghan A.G., Kennedy A.A., Yesselman J.D., Gifford R.J.et al.. Amilorides inhibit SARS-CoV-2 replication in vitro by targeting RNA structures. Sci. Adv. 2021; 7:eabl6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Patwardhan N.N., Ganser L.R., Kapral G.J., Eubanks C.S., Lee J., Sathyamoorthy B., Al-Hashimi H.M., Hargrove A.E.. Amiloride as a new RNA-binding scaffold with activity against HIV-1 TAR. Medchemcomm. 2017; 8:1022–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vögele J., Ferner J.-P., Altincekic N., Bains J.K., Ceylan B., Fürtig B., Grün J.T., Hengesbach M., Hohmann K.F., Hymon D.et al.. 1H, 13C, 15N and 31P chemical shift assignment for stem-loop 4 from the 5'-UTR of SARS-CoV-2. Biomol. NMR Assign. 2021; 15:335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Keller R. The Computer Aided Resonance Assignment Tutorial. 2004; Goldau, Switzerland: CANTINA Verlag. [Google Scholar]
- 28. Schwalbe H., Marino J.P., Glaser S.J., Griesinger C.. Measurement of H,H-coupling constants associated with.nu.1,.nu. 2, and.nu.3 in uniformly 13C-labeled RNA by HCC-TOCSY-CCH-E.COSY. J. Am. Chem. Soc. 1995; 117:7251–7252. [Google Scholar]
- 29. Glaser S.J., Schwalbe H., Marino J.P., Griesinger C.. Directed TOCSY, a method for selection of directed correlations by optimal combinations of isotropic and longitudinal mixing. J. Magn. Reson., Ser. B. 1996; 112:160–180. [DOI] [PubMed] [Google Scholar]
- 30. Ottiger M., Delaglio F., Bax A.. Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J. Magn. Reson. 1998; 131:373–378. [DOI] [PubMed] [Google Scholar]
- 31. Kern T., Schanda P., Brutscher B.. Sensitivity-enhanced IPAP-SOFAST-HMQC for fast-pulsing 2D NMR with reduced radiofrequency load. J. Magn. Reson. 2008; 190:333–338. [DOI] [PubMed] [Google Scholar]
- 32. Cherepanov A.V., Glaubitz C., Schwalbe H.. High-resolution studies of uniformly 13C,15N-labeled RNA by solid-state NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 2010; 49:4747–4750. [DOI] [PubMed] [Google Scholar]
- 33. Ebrahimi M., Rossi P., Rogers C., Harbison G.S.. Dependence of 13C NMR chemical shifts on conformations of RNA nucleosides and nucleotides. J. Magn. Reson. 2001; 150:1–9. [DOI] [PubMed] [Google Scholar]
- 34. Güntert P., Mumenthaler C., Wüthrich K.. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 1997; 273:283–298. [DOI] [PubMed] [Google Scholar]
- 35. Linge J.P., Williams M.A., Spronk C.A.E.M., Bonvin A.M.J.J., Nilges M.. Refinement of protein structures in explicit solvent. Proteins. 2003; 50:496–506. [DOI] [PubMed] [Google Scholar]
- 36. Koradi R., Billeter M., Wüthrich K.. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graphics. 1996; 14:29–32. [DOI] [PubMed] [Google Scholar]
- 37. Blanchet C.E., Spilotros A., Schwemmer F., Graewert M.A., Kikhney A., Jeffries C.M., Franke D., Mark D., Zengerle R., Cipriani F.et al.. Versatile sample environments and automation for biological solution X-ray scattering experiments at the P12 beamline (PETRA III, DESY). J. Appl. Crystallogr. 2015; 48:431–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Manalastas-Cantos K., Konarev P.V., Hajizadeh N.R., Kikhney A.G., Petoukhov M.V., Molodenskiy D.S., Panjkovich A., Mertens H.D.T., Gruzinov A., Borges C.et al.. ATSAS 3.0: expanded functionality and new tools for small-angle scattering data analysis. J. Appl. Crystallogr. 2021; 54:343–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Franke D., Petoukhov M.V., Konarev P.V., Panjkovich A., Tuukkanen A., Mertens H.D.T., Kikhney A.G., Hajizadeh N.R., Franklin J.M., Jeffries C.M.et al.. ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 2017; 50:1212–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. D’Ascenzo L., Leonarski F., Vicens Q., Auffinger P.. Revisiting GNRA and UNCG folds: U-turns versus Z-turns in RNA hairpin loops. RNA. 2017; 23:259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. D’Ascenzo L., Leonarski F., Vicens Q., Auffinger P.. Z-DNA like’ fragments in RNA: a recurring structural motif with implications for folding, RNA/protein recognition and immune response. Nucleic Acids Res. 2016; 44:5944–5956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Proctor D.J., Schaak J.E., Bevilacqua J.M., Falzone C.J., Bevilacqua P.C.. Isolation and characterization of a family of stable RNA tetraloops with the motif YNMG that participate in tertiary interactions. Biochemistry. 2002; 41:12062–12075. [DOI] [PubMed] [Google Scholar]
- 43. Theimer C.A., Finger L.D., Feigon J.. YNMG tetraloop formation by a dyskeratosis congenita mutation in human telomerase RNA. RNA. 2003; 9:1446–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Allain F.H., Varani G.. Structure of the P1 helix from group I self-splicing introns. J. Mol. Biol. 1995; 250:333–353. [DOI] [PubMed] [Google Scholar]
- 45. Ohlenschläger O., Wöhnert J., Bucci E., Seitz S., Häfner S., Ramachandran R., Zell R., Görlach M.. The structure of the stemloop D subdomain of coxsackievirus B3 cloverleaf RNA and its interaction with the proteinase 3C. Structure. 2004; 12:237–248. [DOI] [PubMed] [Google Scholar]
- 46. Liu P., Li L., Keane S.C., Yang D., Leibowitz J.L., Giedroc D.P.. Mouse hepatitis virus stem-loop 2 adopts a uYNMG(U)a-like tetraloop structure that is highly functionally tolerant of base substitutions. J. Virol. 2009; 83:12084–12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ihle Y., Ohlenschläger O., Häfner S., Duchardt E., Zacharias M., Seitz S., Zell R., Ramachandran R., Görlach M.. A novel cGUUAg tetraloop structure with a conserved yYNMGg-type backbone conformation from cloverleaf 1 of bovine enterovirus 1 RNA. Nucleic Acids Res. 2005; 33:2003–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Leontis N.B., Westhof E.. Conserved geometrical base-pairing patterns in RNA. Q. Rev. Biophys. 1998; 31:399–455. [DOI] [PubMed] [Google Scholar]
- 49. Chang K.-Y., Varani G., Bhattacharya S., Choi H., McClain W.H.. Correlation of deformability at a tRNA recognition site and aminoacylation specificity. Proc. Natl Acad. Sci. USA. 1999; 96:11764–11769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Collier A.J., Gallego J., Klinck R., Cole P.T., Harris S.J., Harrison G.P., Aboul-Ela F., Varani G., Walker S.. A conserved RNA structure within the HCV IRES eIF3-binding site. Nat. Struct. Biol. 2002; 9:375–380. [DOI] [PubMed] [Google Scholar]
- 51. Tavares T.J., Beribisky A.V., Johnson P.E.. Structure of the cytosine-cytosine mismatch in the thymidylate synthase mRNA binding site and analysis of its interaction with the aminoglycoside paromomycin. RNA. 2009; 15:911–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tanaka Y., Kojima C., Yamazaki T., Kodama T.S., Yasuno K., Miyashita S., Ono A., Kainosho M., Kyogoku Y.. Solution structure of an RNA duplex including a C-U base pair. Biochemistry. 2000; 39:7074–7080. [DOI] [PubMed] [Google Scholar]
- 53. Holbrook S.R., Cheong C., Tinoco I., Kim S.H.. Crystal structure of an RNA double helix incorporating a track of non-Watson-Crick base pairs. Nature. 1991; 353:579–581. [DOI] [PubMed] [Google Scholar]
- 54. Harms J., Schluenzen F., Zarivach R., Bashan A., Gat S., Agmon I., Bartels H., Franceschi F., Yonath A.. High resolution structure of the large ribosomal subunit from a mesophilic eubacterium. Cell. 2001; 107:679–688. [DOI] [PubMed] [Google Scholar]
- 55. Lukavsky P.J., Kim I., Otto G.A., Puglisi J.D.. Structure of HCV IRES domain II determined by NMR. Nat. Struct. Biol. 2003; 10:1033–1038. [DOI] [PubMed] [Google Scholar]
- 56. Du Z., Yu J., Ulyanov N.B., Andino R., James T.L.. Solution structure of a consensus stem-loop D RNA domain that plays important roles in regulating translation and replication in enteroviruses and rhinoviruses. Biochemistry. 2004; 43:11959–11972. [DOI] [PubMed] [Google Scholar]
- 57. Theimer C.A., Finger L.D., Trantirek L., Feigon J.. Mutations linked to dyskeratosis congenita cause changes in the structural equilibrium in telomerase RNA. Proc. Natl Acad. Sci. USA. 2003; 100:449–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Berger K.D., Kennedy S.D., Turner D.H.. Nuclear magnetic resonance reveals that GU base pairs flanking internal loops can adopt diverse structures. Biochemistry. 2019; 58:1094–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chen Y., Zubovic L., Yang F., Godin K., Pavelitz T., Castellanos J., Macchi P., Varani G.. Rbfox proteins regulate microRNA biogenesis by sequence-specific binding to their precursors and target downstream Dicer. Nucleic Acids Res. 2016; 44:4381–4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Masquida B., Westhof E.. On the wobble GoU and related pairs. RNA. 2000; 6:9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhu C., Lee J.Y., Woo J.Z., Xu L., Nguyenla X., Yamashiro L.H., Ji F., Biering S.B., van Dis E., Gonzalez F.et al.. An intranasal ASO therapeutic targeting SARS-CoV-2. Nat. Commun. 2022; 13:4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Legault P., Pardi A.. Unusual dynamics and pKa shift at the active site of a lead-dependent ribozyme. J. Am. Chem. Soc. 1997; 119:6621–6628. [Google Scholar]
- 63. Saenger W. Principles of Nucleic Acid Structure. 1984; NY: Springer Verlag. [Google Scholar]
- 64. Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A.et al.. Highly accurate protein structure prediction with AlphaFold. Nature. 2021; 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Watkins A.M., Rangan R., Das R.. FARFAR2: improved de novo Rosetta prediction of complex global RNA folds. Structure. 2020; 28:963–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hadfield J., Megill C., Bell S.M., Huddleston J., Potter B., Callender C., Sagulenko P., Bedford T., Neher R.A.. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. 2018; 34:4121–4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The coordinates of the 10 5_SL4 structures with the lowest target functions of the NMR-bundle have been submitted to the PDB under the accession code 8CQ1. SAXS data are available in SASBDB under the accession code SASDRV7.