


Abstract
A wide range of nuclear proteins are involved in the spatio-temporal organization of the genome through diverse biological processes such as gene transcription and DNA replication. Upon stimulation by testosterone and translocation to the nucleus, multiple androgen receptors (ARs) accumulate in microscopically discernable foci which are irregularly distributed in the nucleus. Here, we investigated the formation and physical nature of these foci, by combining novel fluorescent labeling techniques to visualize a defined chromatin locus of AR-regulated genes—PTPRN2 or BANP—simultaneously with either AR foci or individual AR molecules. Quantitative colocalization analysis showed evidence of AR foci formation induced by R1881 at both PTPRN2 and BANP loci. Furthermore, single-particle tracking (SPT) revealed three distinct subdiffusive fractional Brownian motion (fBm) states: immobilized ARs were observed near the labeled genes likely as a consequence of DNA-binding, while the intermediate confined state showed a similar spatial behavior but with larger displacements, suggesting compartmentalization by liquid–liquid phase separation (LLPS), while freely mobile ARs were diffusing in the nuclear environment. All together, we show for the first time in living cells the presence of AR-regulated genes in AR foci.
Graphical Abstract
Graphical Abstract.

Introduction
Androgens, such as testosterone, are steroid hormones that play a central role in the development and maintenance of the male phenotype. Androgens bind to the androgen receptor (AR), a member of nuclear steroid hormone receptors (SHRs) such as the estrogen (ER), glucocorticoid (GR) and progesterone receptor (PR). Upon activation, the AR is transported to the nucleus, where it binds as a homodimer to specific short palindromic DNA sequences (Androgen Response Elements — AREs) at mainly promoter and enhancer regions. At these sites, AR interacts with cofactors such as P300 and SRC1, which together form a transcriptional regulatory complex that orchestrates transcription (1,2). ARs, like many other transcription factors (TFs), form irregularly distributed membraneless sub-compartments in the nucleus in which AR molecules are highly concentrated (referred to here as AR foci). These foci are maintained in the same nuclear regions, only interrupted by cell division, while we have previously shown that AR foci are not present in cells expressing AR mutants deficient for DNA-binding (1).
Despite extensive knowledge of the role of AR in gene transcription, the spatio-temporal organization of these factors at chromatin, specifically the formation of nuclear foci, remains poorly understood. Biophysical theoretical models have been previously developed to explain such behavior, including assembly-function-dissociation (3), liquid–liquid phase separation (LLPS) (4) and polymer–polymer phase separation (PPPS) (5) models, of which the LLPS hypothesis is most discussed in recent literature (6,7). It has been proposed that interactions between intrinsically disordered regions (IDRs) of multiple TFs are responsible for the formation of these phase-separated compartments (8), leading to the formation of foci as recently shown for GR (9). Targeting the IDR of AR, located at the N-terminal domain (NTD), by molecules such as EPI001 and ET516 has shown a reduction of the number of AR foci (10), probably by indirect destabilization of AR-DNA interactions. As a consequence, AR-regulated transcription was negatively affected by the effect of these compounds. Several studies have confirmed that other transcription-related co-factors (e.g. RNA Pol II, MED1, BRD4) are able to initiate phase separation at distinct genomic locations such as promoter and enhancer regions (11–13).
Fluorescently labeled proteins in microscopy have been used extensively to investigate the biophysical properties of these TFs. Microscopy techniques such as fluorescence correlation spectroscopy (FCS) or fluorescent recovery after photobleaching (FRAP) were used to investigate LLPS in the nuclear environment (14). FRAP showed binding times of several seconds for chromatin-bound ARs, instead of minutes or even hours as reported for DNA repair and replication factors, underpinning the highly dynamic nature of AR (15–17). Furthermore, FRAP experiments confirmed that cells expressing AR mutants deficient for DNA-binding did not show a long-immobilized AR fraction — which are seemingly essential for AR foci formation (18).
More recently, fluorescent labeling techniques such as HaloTag (19) made it possible to track single molecules for a long period of time with short time intervals, permitting real-time investigation of particle-dynamics through single-particle tracking (SPT) experiments (20). The dynamics of nuclear proteins have been studied by the use of SPT to further estimate fraction sizes, differential behavior and binding times (18,21–25), providing evidence of long immobilization of SHR proteins up to several seconds. Typical motion of nuclear proteins is generally described as undergoing subdiffusion, which was also reported for other intranuclear molecules such as mRNA or chromatin loci (26–28). Analysis of SPT data was initially performed exclusively by computing the mean squared displacement (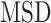 ) analysis which allows the estimation of the diffusion constant. Nevertheless, additional methods are being developed to better characterize the diffusive behavior and to classify/segment trajectories (29). Displacements and angles have also been used for in-depth investigations of particle dynamics (21,27,30).
) analysis which allows the estimation of the diffusion constant. Nevertheless, additional methods are being developed to better characterize the diffusive behavior and to classify/segment trajectories (29). Displacements and angles have also been used for in-depth investigations of particle dynamics (21,27,30).
Visualization of the interaction between TFs and chromatin in live cells remains an elaborate task, particularly due to the challenges of imaging single genomic loci. However, Hager and co-workers recently used a stably integrated MMTV promotor array composed of hundreds of GR-binding sequences (9). Their SPT analysis showed a higher chromatin-bound fraction near the gene array compared to the rest of the nucleus. Moreover, recent technological advancements in the field of genome editing (e.g. CRISPR-Cas9) have opened the doors for stable integrations of novel fluorescent DNA labeling systems at specific chromatin loci, which could facilitate the visualization of biological processes involving DNA, such as the dynamics of TFs at endogenous genes (31).
In this paper, we combined fluorescent labeling techniques to study the dynamics of ARs at AR-regulated genes in prostate cancer cell lines. For this, we used next-generation sequencing (NGS) techniques to determine AR-regulated genes, and selected two non-canonical AR-enriched endogenous genes, PTPRN2 and BANP. We then used the ANCHOR DNA labeling system to visualize these genes, showing the general role of AR as a TF. We used these cell lines to study the interaction between one of the selected genes and either AR foci detected by confocal microscopy, or individual molecules of AR by SPT. We show colocalization of PTPRN2 and BANP loci with AR foci in both fixed and live cells. In addition, the global behavior of AR particles in the nucleus followed a three-state model of diffusion, with the two most confined diffusive states located at the AR-enriched genes. These confined AR molecules are probably the result of DNA-binding at diverse genomic locations which are highly concentrated in TFs, leading to microscopical discernable nuclear foci.
Materials and methods
Cell lines
PC346C cell lines, stably expressing GFP-AR, were cultured, as described earlier (32), in a special medium (Prostate Growth Medium; PGM) based on DMEM/F12 (Lonza: BE12-719F) supplemented with various growth factors, 5% charcoal-stripped FCS (Gibco:26140079) and 100 U/ml penicillin/streptomycin (Lonza: DE17-602E). Cells were passaged every 4 days with 0.05% Tryspin-EDTA solution (Gibco: 25300054).
Chromatin immunoprecipitation assay
Cells were cultured for 2 days in charcoal-stripped medium supplemented with R1881 (100 nM) or vehicle (DMSO). Chromatin immunoprecipitation (ChIP) was performed in three biological replicates per condition. AR ChIPs were performed using 10 μl of AR antibodies (Santa Cruz: SC-816) pre-bound to 50 μl Dynal Protein G magnetic (Thermofischer) beads per sample. Isolated DNA fragments of both samples and inputs were first analyzed by qPCR for AR-target genes KLK2, KLK3 and TMPRSS2. Samples were processed for library generation using the NEXTFlex kit (BioO scientific: NOVA-5143–01) and were sequenced according to the illumina TruSeq v3 protocol using the Illumina HiSeq2000 genome analyzer for 36 bp single reads with 7 bp index read.
The raw and processed data are deposited in the Gene Expression Omnibus database (http://ncbi.nlm.nih.gov/geo/; accession no. GSE229728).
Analysis of next generation sequencing (NGS) data
Expression levels in primary prostate cancer samples (pre- and post-neoadjuvant enzalutamide (ENZ) treatment) for BANP, PTPRN2, FBXO31 and ZFPM1 were extracted from the dataset derived from the DARANA study (GSE197781; EGAS00001006016) (33) and further analyzed. Gene counts were normalized using the DESeq2 Bioconductor package (v1.22.2) (34) and subsequently log-transformed. For visualization purposes, the normalized gene expression data were Z-transformed per gene.
ChIP-seq signals of AR tissues at BANP and PTPRN2 loci in castration resistant metastases (GSE152231) (35) visualized using EaSeq software (v1.101) (36). Results were compared with the AR ChIP-seq data of PC346-GAR cell lines. Publicly available H3K27ac HiChIP data from prostate cancer cell lines were used to generate a gene interaction map for the BANP locus and proximal genes at ±500 kb distance (37).
The CistromeDB Toolkit was used to probe which transcription factors and chromatin regulators had a significant binding overlap with the identified AR binding sites in PC346C cells. For this, the four AR peaks with the highest peak intensity at the BANP or PTPRN2 loci in PC346C cells were integrated with publicly available ChIP-seq data sets using the GIGGLE tool (38).
RNA extraction followed by cDNA synthesis and RT-PCR analysis
PC346C cells were seeded a day prior to hormonal treatment and incubated in regular PGM medium containing 5% dextran-coated charcoal (DCC) treated FCS without R1881 and hydrocortisone. Afterwards, cells were treated for 16 h with either 100 nM R1881, 1 mM Enzalutamide or vehicle (DMSO) to assess gene expression levels of BANP, PTPRN2, FBXO31 and ZFPM1. Subsequently, cells were collected in tubes and afterwards lysed by addition of TRI reagent (Sigma-Aldrich: T9424). RNA was extracted according to the manufacturer's protocol. RNA purity and concentration were measured spectrophotometrically with optical density measurement using the NanoDrop ND-2000 spectrophotometer device (ThermoFisher Scientific) with absorbance values of 260 and 280 nm.
Next, for each sample, 1 μg of RNA was treated by TURBO™ DNase (ThermoFisher: AM1907) according to manufacturer's protocol. Subsequently, complementary DNA (cDNA) were synthesized by using SuperScript IV Reverse Transcriptase (ThermoFisher: 18090010) and Oligo(dT)20 primer (IDT) according to manufacturer's protocol. The RT-PCR reaction mixtures contained 0.1 μl of 5U/μl Platinum Taq polymerase (ThermoFisher: 15966005), 2.5 μl 10× PCR buffer, 0.75 μl 50 mM MgCl2, 0.5 μl 10 mM dNTPs, 0.5 μl 10 μM forward and reverse primer mix, 1 μl cDNA (diluted 1:5), 1 μl SYBR Green (final concentration 0.15× in DMSO – Sigma-Aldrich: S9430), in a final volume of 25 μl. RT-PCRs were performed in a 3 step cycling program consisting of an initial denaturation step at 95 °C for 5 min, followed by 39 cycles of 95 °C – 30 s, 60 °C – 30 s and 72 °C – 30 s. The amplification values of the target genes were normalized to the value of reference gene GAPDH. At least three biological replicates were analyzed by performing two RT-PCR reactions per biological replicate for each condition. Primer sequences for target and reference gene are listed in Supplementary Table S1.
Genome-editing and cell line generation
Genome-editing was performed on PC346C-GFP-AR cell lines using CRISPR/Cas9 technology as previously described with minor modifications (39). The sgRNA sequences for ANCH3 genomic insertions were designed by using the CRISPOR algorithm tool (40) for human BANP (CAAGCCTCCCCAATTGCATA) and PTPRN2 (ATTTGGGGTGCGCCTCGCTG) and cloned into the CRISPR/Cas9-encoded PX459 plasmid (pSpCas9-2A-Puro V2.0; Addgene 62988). The sgRNAs were designed to induce a DSB at approximately 7 kb in front of the transcription start site of either PTPRN2 or BANP. After selecting the gRNAs (and thereby the targeting location for Cas9), 1 kb gene fragments were synthesized (Integrated DNA Technologies) containing 470 bp long homology arms flanked on both sides of the gRNA target site. These gene fragments were cloned into a pCR-Blunt II TOPO backbone vector followed by an insertion of the 1 kB ANCH3 array in between the two homology arms of both PTPRN2 and BANP donor templates using MluI (NEB: R0198S) and BglII (Roche: 10567639001) restriction enzymes. Afterwards, both the PX459-sgRNA plasmid and a repair template were transfected for each gene using Fugene 6 (Promega: E2311) according to the manufacturer's protocol. Cells were pre-seeded on a 6-well plate and transfected the following day using 1 μg of PX459 vector and 1 μg of donor vector (1:1) per well. Cells were transferred to larger surfaces (10 cm plates) 24 h later and cultured in PGM medium supplemented with 2 μg/ml puromycin (Gibco: A1113803). After several days, single colonies were picked using a 200 μl pipette tip and transferred into 96-well plates. DNA extraction was performed after expansion and used for genotyping by applying one primer outside the homology arms and one primer inside the transgene. Selected clones were further expanded and genotypically validated again by PCR for a second time. G4 and G6 clones for PTPRN2 and BANP respectively were selected after validation for further use.
After stably inserting the ANCH3 arrays at a genomic location, the fluorescently tagged OR3 binding proteins were then inserted using the PiggyBac transposition system. The mScarlet-V5-OR3 and IRFP720-OR3 encoding cassettes were cloned into a PiggyBac entry backbone. One of these plasmids was co-transfected to both PTPRN2 and BANP labeled cell lines together with a plasmid encoding the PiggyBac transposase using Fugene 6. Afterwards, cells were sorted by FACS for mScarlet-V5-OR3 or IRFP720-OR3 protein expression.
Confocal microscopy of AR-ANCHOR3 labeled fixed cell lines
PC346C-GFP-AR PTPRN2 (clone G4) and BANP (clone G6) were seeded on 24 mm 1.5H microscope cover glasses (Marienfeld: 0117640) and treated with 100 nM R1881 (Sigma Aldrich: R0908). A day after, cells were fixed at 37 °C for 30 min with 4% paraformaldehyde. Subsequently, cover glasses were mounted on slides with Vectashield antifade mounting medium (vector laboratories: H-1000). Images, with a pixel size of 30 × 30 nm, were made using a Leica TCS SP8 AOBS microscope with a 40 × 1.30 NA HC PL APO CS2 objective and the appropriate laser lines and emission filters (excitation with 488 nm laser and a BP 500–550 nm emission filter for GFP-AR and a 561 nm excitation in combination with BP 575–650 nm emission filter for mScarlet-V5-OR3). Z-stacks were made with a z-step size of 500 nm.
Quantification of the distance between a fluorescently labeled gene (PTPRN2 or BANP) and the nearest AR focus was performed on one single z-plane image from the z-stacks by using an in-house written ImageJ script (https://github.com/ErasmusOIC/NearestNeighbour). In short, all points falling within a certain threshold were selected by the ‘Find Maxima’ function for AR and OR3 channels. The resulting coordinates were later used to measure the distance between the labeled gene and the nearest AR focus. Distances below 300 nm were defined as a colocalizing event.
To assess the randomness of genes colocalizing with AR foci, we simulated 40 000 images of synthetic cell nuclei, each of them containing 300 randomly distributed ‘AR foci’. This number was determined by estimating the average number of AR foci per cell in our confocal images. 30 000 images (75%) were generated with the location of a unique single locus (mimicking a labeled gene), while the remaining 10 000 images were generated with two loci located close to each other, representing two sister chromatids. Distance measurements were therefore conducted between the simulated foci and the single-locus using the method described above.
Time-lapse images of AR-ANCHOR3 labeled cell lines
PTPRN2 and BANP-labeled PC346C-GFP-AR cell lines were pre-treated overnight with 1 μM hydroxyflutamide-containing PGM medium. As a result, AR molecules translocated into the nucleus but without initiating AR foci formation. The following day, cells were transferred to a Leica TCS SP8 AOBS microscope equipped with a temperature-controlled stage and 40× 1.30 NA HC PL APO CS2 objective (37°C and 5% CO2). Cells were then stimulated with 100 nM R1881 (Sigma Aldrich: R0908) to initiate AR-DNA binding, before starting the time-lapse procedure to visualize the AR foci formation in time. Cells were imaged with a pixel size of 30 × 30 nm and by generating Z-stack images with intervals of 500 nm, and with a time interval of one minute between each cycle of Z-stacks, for approximately 20 min. Afterwards, three Z-plane images surrounding the ANCHOR3 spot were used to generate an intensity maximum projection image, later used for the colocalization analysis.
Confocal microscopy of AR foci co-stained with hoechst and propidium iodide
Cells were pre-treated a day before the experiment with 100 nM R1881 (Sigma Aldrich: R0908) and fixed at 37°C for 30 min. Afterwards, cover glasses were mounted on slides with vectashield antifade mounting medium (vector laboratories: H-1000) containing 1:5000 diluted Hoechst 34580 (Thermofisher: H21486) and 1:2000 diluted propidium iodide (Sigma-Aldrich: P4170) to visualize the chromatin and areas containing high concentrations of RNA (e.g. nucleolus), respectively. Cells were imaged by a Zeiss LSM 700 upright confocal microscope equipped with a 63 × 1.40 NA Plan-Apochromat objective and the appropriate laser lines and emission filters (excitation with 405 nm laser and a SP 490 nm emission filter for Hoechst 34580, 488 nm excitation in combination with BP 500–550 nm emission filter for GFP-AR and a 555 nm excitation laser combined with a LP 565 nm emission filter for propidium iodide).
In total 83 nuclei were imaged and used for analysis using ImageJ. For this, we made a mask using the Hoechst channel to exclude the extranuclear region and the propidium iodine channel to exclude the nucleolus regions. Furthermore, AR-GFP foci located within this mask were identified using the ‘Find Maxima’ function. The pixel intensity values of the Hoechst channel were normalized and categorized into five chromatin intensity classes, each class containing a similar area fraction of 20%. The number of AR foci overlapping with a certain class was counted to assess the distribution of AR foci over low-density chromatin (0.0–0.2), euchromatin (0.2–0.8) and heterochromatin (0.8–1.0) regions.
Confocal microscopy of AR foci co-stained with hoechst, h3k27ac and h3k27me3
Cells were pre-treated a day before the experiment with 100 nM R1881 (Sigma Aldrich: R0908) and fixed at 37°C for 30 min. Afterwards, cells were permeabilized for 15 min with 0.05% triton followed by primary antibody staining using rabbit anti-H3K27Me3 (1:1000, Cell signaling, C36B11) and mouse anti-H3K27Ac (1:1000, Actif Motif, 39685). Primary antibodies were visualized by secondary antibodies using goat anti-mouse IgG Alexa 647 (1:1000, ThermoFischer, A-21235) and goat anti-rabbit IgG Alexa 568 (1:1000, ThermoFischer, A-11011) antibodies. Afterwards, cover glasses were mounted on slides with vectashield antifade mounting medium (vector laboratories: H-1000) containing 1:5000 diluted Hoechst 34580 (Thermofisher: H21486). A Leica TCS SP8 AOBS microscope with a 40 × 1.30 NA HC PL APO CS2 objective and the appropriate laser lines and emission filters (excitation with 488 nm laser and a BP 500–550 nm emission filter for GFP-AR, 561 nm excitation in combination with BP 575–650 nm emission filter for H3K27Ac-Alexa 568 and a 633 nm laser in combination with a BP 650–700 for H3K27 Me3-Alexa 647) were used for image acquisition.
Quantification of PTPRN2 and BANP protein expression by confocal microscopy
PC346C-GFP-AR WT cells and both BANP and PTPRN2 labeled PC346C-GFP-AR cell lines were pre-treated a day before the experiment with 100 nM R1881 (Sigma Aldrich: R0908) and fixed at 37°C for 30 min. Afterwards, cells were permeabilized for 15 min with 0.05% triton followed by either primary antibody staining using rabbit anti-PTPRN2 (1:100, ThermoFisher, PA5-99696) or rabbit anti-BANP (1:100, ThermoFisher, PA5-61452). Primary antibodies were visualized by secondary antibodies using goat anti-rabbit IgG Alexa 647 (1:1000, ThermoFischer, A-21235) antibodies. Afterwards, cover glasses were mounted on slides with vectashield antifade mounting medium (vector laboratories: H-1000) containing 1:5000 diluted Hoechst 34580 (Thermofisher: H21486). A Leica TCS SP8 AOBS microscope with a 40 × 1.30 NA HC PL APO CS2 objective and the appropriate laser lines and emission filters (excitation with 488 nm laser and a BP 500–550 nm emission filter for GFP-AR, 561 nm excitation in combination with BP 575–650 nm emission filter for H3K27Ac-Alexa 568 and a 633 nm laser in combination with a BP 650–700 for H3K27Me3-Alexa 647) were used for image acquisition.
Direct stochastic optical reconstruction microscopy (dSTORM)
dSTORM imaging was performed as previously described with minor adaptations (41). Both BANP and PTPRN2 labeled PC346C-GFP-AR cell lines were pre-treated with 100 mM R1881 a day before and fixed with 4% paraformaldehyde at 37°C for 30 min. Cells were permeabilized for 15 min with 0.05% triton followed by primary antibody staining using mouse anti-GFP (1:1000, Abcam: ab13970) and rabbit anti-V5-tag (1:1000, Abcam, ab9116) antibodies. Primary antibodies were visualized by secondary antibodies using goat anti-mouse Alexa 647 (1:1000, Thermofischer, A-21235) and goat anti-rabbit IgG CF568 (1:500, Sigma Aldrich, SAB4600310) antibodies. To perform dSTORM imaging, the following buffer was used: 0.56 mg/ml glucose oxidase (Sigma, G2133), 34 μg/ml catalase (Sigma, C9322), 25 mM MEA (Sigma, m6500) and 20% glucose in Tris–HCl pH8 with 10 mM NaCl. dSTORM imaging data were acquired on a Zeiss Elyra PS1 microscope using a 100×/1.46 NA Alpha Plan-Apochromat DIC (Zeiss) objective. TIRF was used to create a HiLO excitation beam. The data was acquired using a 512 × 512 Andor Ixon DU 897 EMCCD camera. First, a single frame image was taken to visualize the labeled locus as a reference point for the super-resolution images. Next, super-resolution images were generated for both AR and ANCHOR3 labeled cells by acquiring 12000 frames per channel with an acquisition time of 33 ms. High laser power used for dSTORM imaging can bleach fluorophores of higher wavelengths. Consequently, imaging was performed from high to low wavelength, starting with Alexa 647 to detect AR, followed by CF568 to detect OR3 proteins. One cell for PTPRN2 and one cell of BANP were visualized by dSTORM to provide a high-resolution example showing similar distances as found by confocal and SPT.
dSTORM image analysis
Localizations were drift corrected (model-based approach) and grouped (max on time = 50 frames, off gap = 2 frames, capture radius = 4 pixels) using ZEN2012 software (Zeiss). We used the dSTORM channel alignment software of ZEN2012 for alignment of the different channels and selected the Alexa 647 channel as a base. Localization data was analyzed and processed using the R-package SMoLR (42). Detections were clustered using the DBSCAN algorithm (43) with the radius set to 50 nm and the minimal points to 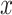 . Distance between the center of mass of these clusters were measured. Images with a 5 nm pixel size of the whole cell were constructed using a Gaussian distribution based on the precise localizations.
. Distance between the center of mass of these clusters were measured. Images with a 5 nm pixel size of the whole cell were constructed using a Gaussian distribution based on the precise localizations.
Single-particle tracking and trajectory extraction
ARWT-HaloTag, ARΔNTD-HaloTag and ARR585K-HaloTag were transfected and stably integrated in both fluorescently labeled (OR3-IRFP720) PTPRN2 and BANP PC346C cell lines. AR-HaloTag proteins were labeled 2 hours prior imaging for 20 min with PGM medium containing 250 pM JF549 dye. These cells were imaged using the Zeiss Elyra PS1 super-resolution microscope, consisting of a Tokai Hit temperature-controlled stage and objective (37°C and 5% CO2). Images were acquired using a TIRF lens in combination with a 100x/1.46 NA Alpha Plan-Apochromat DIC (Zeiss) objective to create a highly inclined and laminated optical sheet (HiLo). First, a snapshot (300 by 50 pixels – 100 nm pixel size) was taken of the OR3-IRFP720 spot(s) utilizing 0.4% of the 642 nm laser (100 mW). Afterwards, in the same field of view, most AR-HaloTag-JF549 molecules were bleached with a laser power intensity of 30% (100 mW, 561 nm laser) for 3 s. Subsequently, 15 000 frames were captured per cell by using a lowered laser intensity of 10%. Emitted photons were filtered by a 570–650 nm bandpass (for AR-HaloTag-JF549) or 655 longpass (OR3-IRFP720) filters before being collected by an EM-CCD camera (Andor DU 897) at 7 ms intervals with a gain of 300. Even though most of the fluorescent AR particles were bleached before imaging acquisition, a high population of fluorescent ARs remained at the beginning of the time-lapse images which could constrain the extraction of trajectories, therefore the first 1000 frames were discarded from each image.
The tracking of the SPT data was performed by using the ImageJ SOS plugin for spot detection and linking (http://smal.ws/wp/software/sosplugin/) (44). The position of diffracted-limited fluorescent spots of AR particles in each image frame were estimated by fitting a Gaussian point spread function. The positions were then linked using the nearest-neighbor criterion to construct trajectories.
Analysis of single-particle tracking data
For each time-lapse SPT image, the particle tracking resulted in a set 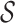 of trajectories
of trajectories 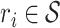 , i = {1, ... , N} where N is the total number of trajectories in the whole image and r(t) = (x(t), y(t)) are the coordinates of a trajectory in 2D at time
, i = {1, ... , N} where N is the total number of trajectories in the whole image and r(t) = (x(t), y(t)) are the coordinates of a trajectory in 2D at time 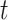 .
.
Segmentation of trajectories into diffusive states using deep learning
The trajectory displacements were classified into one of three diffusive states using a deep learning framework. We developed a refined version of the method of Arts et al. (45) which major changes include the computation of angles as an additional feature for better distinction of the trajectory confinement, the choice of using the 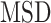 analysis instead of the moment scaling spectrum (MSS) analysis, and the development of a user-friendly python software for replicability on other datasets, available at: https://zenodo.org/record/7767750.
analysis instead of the moment scaling spectrum (MSS) analysis, and the development of a user-friendly python software for replicability on other datasets, available at: https://zenodo.org/record/7767750.
The network was trained using synthetic trajectories of mixed states. For this purpose, we simulated 10000 fBm trajectories with a switching mode between states and a total length of 27 frames. The fBm process is characterized using the fBm kernel (46) defined as:
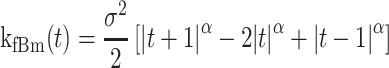 |
(1) |
with 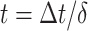 (
(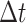 the time measured between two frames and
the time measured between two frames and 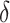 the discrete time interval),
the discrete time interval), 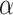 the anomalous exponent and
the anomalous exponent and 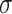 the scaling factor. The latter can be translated to the diffusion constant
the scaling factor. The latter can be translated to the diffusion constant 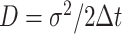 . The motion parameters
. The motion parameters 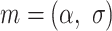 used in the simulation for each state were:
used in the simulation for each state were: 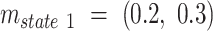 ,
, 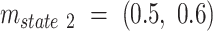 and
and  . The probabilities of transitioning between states were defined by the following probability transition matrix (similar probability values were used in Arts et al. (2019) (45)):
. The probabilities of transitioning between states were defined by the following probability transition matrix (similar probability values were used in Arts et al. (2019) (45)):
 |
(2) |
We estimated the states of the AR trajectory dataset (selecting the ones having a length larger than 10). Beforehand, 1-length frame gaps were filled with a randomly generated point, and gaps having a length of two or more were split in two separate trajectories. Based on the state classification, the trajectories were finally segmented for further computations within each diffusive state.
Transition probabilities between states
The probability 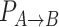 of transitioning from a state
of transitioning from a state 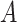 to a state
to a state 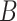 was estimated by dividing the number of observed transitions from
was estimated by dividing the number of observed transitions from 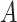 to
to 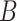 with the total number of track points classified as
with the total number of track points classified as 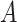 .
.
Estimation of the motion parameters (D and α)
The diffusion constant 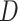 and anomalous exponent
and anomalous exponent 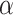 for each individual trajectory or tracklet were estimated using the mean squared displacement (
for each individual trajectory or tracklet were estimated using the mean squared displacement (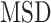 ) analysis. It consists of computing the (time-averaged)
) analysis. It consists of computing the (time-averaged) 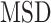 at different time lags
at different time lags 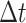 for a single particle imaged at
for a single particle imaged at 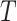 discrete times:
discrete times:
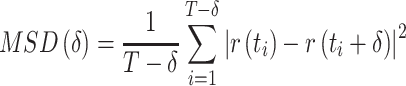 |
(3) |
Secondly, applying a least-square fit from the logarithm form of the  power-law equation (47):
power-law equation (47):
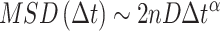 |
(4) |
with  the number of dimensions. This equation being linear for the special case of
the number of dimensions. This equation being linear for the special case of 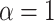 .
.
For the MSD computation we selected track(let)s having a minimum length of 16 frames (or min. 12 frames for our fast control – NLS – since the tracks were shorter) and for the MSD fits we used the first 4 points (or 3 points for trajectories of NLS).
The estimation of  and
and 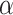 is known to be less accurate on short trajectories (48). However, the performances obtained by the
is known to be less accurate on short trajectories (48). However, the performances obtained by the 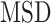 analysis remain the highest on short fBm trajectories compared to other more sophisticated methods (29). Moreover, for trajectories undergoing a similar type of diffusion, the ensemble-averaged
analysis remain the highest on short fBm trajectories compared to other more sophisticated methods (29). Moreover, for trajectories undergoing a similar type of diffusion, the ensemble-averaged 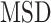 (used to estimate the barycenter) gives more accurate estimates over thousands of tracks (48).
(used to estimate the barycenter) gives more accurate estimates over thousands of tracks (48).
Angles and anisotropy
Angles were computed using each two consecutive displacements at time 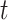 and
and 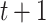 . The fold anisotropy metric
. The fold anisotropy metric 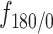 (21) was computed by dividing the number of angles going backwards (
(21) was computed by dividing the number of angles going backwards (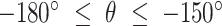 or
or 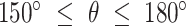 ) by the angles going forwards (
) by the angles going forwards (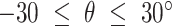 ).
).
Velocity autocorrelation functions (VAC)
The velocity Autocorrelation function is determined as:
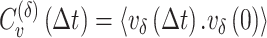 |
(5) |
where the velocity is 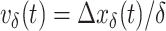 . The theoretical curves of fBm for various
. The theoretical curves of fBm for various 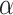 values and
values and 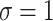 were generated using the fBm kernel (Eq. 1).
were generated using the fBm kernel (Eq. 1).
Ripley's 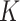 function
function
The 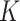 -function measures the expected number of points
-function measures the expected number of points 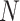 within a distance
within a distance 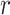 of a given point
of a given point 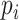 (the sum is taken over
(the sum is taken over 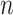 points), and is normalized by the total number of points
points), and is normalized by the total number of points 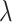 in the entire area (49):
in the entire area (49):
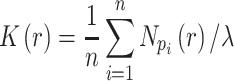 |
(6) |
Ripley's Isotropic Corrector was applied for edge correction when the circles of measurement exceed the limits of the total area. In essence, the correction is applied by multiplying the 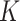 expectation to a weighting term for each search radius
expectation to a weighting term for each search radius 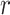 .
.
Results
Selection of AR-bound genes
To identify AR-bound genomic regions, we performed ChIP-seq on R1881-stimulated and unstimulated PC346C cells using AR antibodies. From all regions with highest binding peak intensities, we selected two genes, PTPRN2 (Ch.7q36.3) and BANP (Ch.16q24.2).
PTPRN2 was reported in malignant endocrine tissue such as breast and prostate cancer (50–52). AR ChIP-seq results of primary prostate cancers showed a limited number of peaks present in both PTPRN2 and BANP (Supplementary Figure S1) (33). However, in metastatic tumors numerous AR peaks were observed at both loci (35), partially overlapping with those found in PC346C cells (Figure 1A). To investigate the co-occupancy of AR with other transcription co-regulators, we used a genomics search engine (GIGGLE tool) (38) for the four most AR enriched sites present at the PTPRN2 locus (Supplementary Figure S2a). Using over 14000 individual ChIP-seq databases for TF-binding overlap (results of the search query identified AR-binding sites for all four highest ChIP-seq peaks), we showed a strong overlap with known AR co-regulators (e.g. FOXA1, EP300, GATA2, BRD4). Next to this, RNA-seq results from neoadjuvant enzalutamide treated prostate cancer patients (Figure 1B) (33) and RT-PCR results of PC346C cells (Figure 1C) revealed that PTPRN2 expression was significantly down-regulated after enzalutamide treatment, showing that PTPRN2 is transcriptionally regulated by AR.
Figure 1.

AR regulation of PTPRN2 and BANP gene bodies determined by NGS data. (A) AR ChIP-sequencing results of PC346C cells treated with DMSO or R1881 (top) and AR ChIP-sequencing results of four metastatic castration resistant prostate cancer samples (bottom). Numbering indicating the peaks with the highest intensity, used for a genomics search. (B-D-G-I) Normalized gene expression pre- and post-enzalutamide treatment for PTPRN2 (B), BANP (D), FBXO31 (G) and ZFPM1 (I). Statistical significance was determined using a two-sided Mann–Whitney–Wilcoxon test with Bonferroni correction (****P-value ≤ 10−4; *10−2 < P-value ≤ 0.05; ns: 0.05 < P-value ≤ 1). (F) Gene interaction map of the BANP locus including proximal genes at ±500 kb distance, based on H3K27Ac HiChIP. (C-E-H-J) real-time quantitative PCR analysis of PTPRN2 (C), BANP (E), FBXO31 (H) and ZFPM1 (J) for unstimulated (vehicle), R1881-stimulated (R1881) and enzalutamide-treated (ENZ.) cells.
In contrast to PTPRN2, BANP is not differentially expressed upon enzalutamide treatment (Figure 1D, E), suggesting that BANP is not a direct AR target gene, in spite of a high degree of AR enrichment shown in PC346C cells and AR chromatin binding sites shared between PC346C cells and metastatic castration resistant prostate cancer samples (35) (Figure 1A). However, results derived from the GIGGLE analysis for the four most enriched AR sites showed an overlap with known AR-coregulators including peak no. 4 which was conserved between PC346C cells and metastatic castration resistant prostate cancer samples (Supplementary Figure S2b).
To elucidate the potential function of the BANP locus, we analyzed publicly available H3K27ac HiChIP data to identify all physical DNA interactions around the chromatin regions close to the BANP locus (37) and combined this data with the differential expression profiles derived from the neoadjuvant enzalutamide treated prostate cancer patients for all proximal (±400 kb) located genes. This analysis indicates that promoter/enhancer regions of two genes, FBXO31 and ZFPM1, are present in the BANP coding regions (Figure 1f) and became differentially expressed upon enzalutamide treatment (Figure 1G, I) (53,54).
RT-PCR further confirmed the differential expression of FBXO31 in PC346C cells (Figure 1H). However, in stark contrast to the RNA-seq results of neoadjuvant enzalutamide treated samples, ZFPM1 showed no significant upregulation upon R1881 stimulation (Figure 1J), indicating that the differential expression of this gene might be cell line dependent. We therefore chose the BANP and PTPRN2 loci for further in-depth investigation of AR foci formation.
Colocalization of AR foci with endogenous gene bodies
In order to assess the relationship between AR-DNA binding, shown by ChIP-seq (Figure 1), and AR foci formation, we labeled the PTPRN2 and BANP genes by applying the ANCHOR3 DNA labeling system (55). Briefly, mScarlet tagged OR3 proteins were expressed upon stable genomic integration by the PiggyBac transposon system while the ANCH sequence encoding for the binding regions of the OR3 proteins were knocked-in by CRISPR-Cas9 in PC346C cells (which are aneuploid (56)) expressing AR-GFP (Supplementary Figure S3). To assess the potential influence of the ANCH3 integration on protein expression levels, we applied a quantitative microscopy assay to determine the relative protein expression of either PTPRN2 or BANP for both wild-type and ANCHOR3 labeled PC346C cells. Immunostaining of the two proteins showed no differences in expression levels between cell lines (Supplementary Figure S4). We then used confocal microscopy to visualize 20 cells labeled for the PTPRN2 gene and 17 cells for BANP using the ANCHOR3 system in fixed cells. As a consequence of replication and/or aneuploidy (56) the number of visible labeled loci per cell nucleus varies from one, two or even four, resulting in 37 labeled loci of the PTPRN2 gene and 37 loci of the BANP gene (Figure 2A and Supplementary Figure S5). Approximately 25% of the counted loci were replicated sister chromatids, indicated by the presence of two ANCHOR3 diffraction-limited spots located proximally and at the same focal plane. The presence of more spots at other focal planes within the same cell can be explained by the cancer cell line which is known to contain duplicated chromosomes.
Figure 2.

Colocalization of AR foci and labeled gene loci (PTPRN2 or BANP) in fixed and living cells. (A) Images of R1881-stimulated PC346C cells; PTPRN2 and BANP genes were labeled with the ANCHOR3 system (red), AR foci were labeled with EGFP (green). (B) Pie charts showing percentage of AR foci colocalizing with PTPRN2 loci, BANP loci and in silico simulated loci. (C) Distances between the locus and the closest AR focus. (D) Confocal time-lapse images of PC346C cells. PTPRN2 and BANP genes were labeled with the ANCHOR3 system (red), AR foci were labeled with EGFP (green). The cells were pre-treated overnight with hydroxyflutamide. At 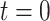 , R1881 was added to induce AR foci formation. (E, F) Measured distance over time between the closest AR focus and either PTPRN2 (E) or BANP (F). The red dashed lines represent the average distance.
, R1881 was added to induce AR foci formation. (E, F) Measured distance over time between the closest AR focus and either PTPRN2 (E) or BANP (F). The red dashed lines represent the average distance.
The distances between the labeled chromatin locus and the nearest AR focus were then measured, where distances lower than 300 nm were defined as colocalizing events (Figure 2B, C). We observed 28 PTPRN2 loci (76%) colocalizing with a nearby AR focus, with an average distance of 174 ± 15nm (SEM). For BANP, 30 labeled loci (81%) colocalized with AR focus with an average distance of 176 ± 12 nm. The average distance for the non-colocalizing events was 400 ± 17 nm and 413 ± 17 nm for PTPRN2 (n = 9) and BANP (n = 7), respectively. To confirm the measured distances between labeled loci and AR foci, we also visualized the labeled loci and AR foci at super resolution by direct stochastic optical reconstruction microscopy (dSTORM) (Supplementary Figure S6). First, a widefield image was acquired to clearly visualize the labeled loci. This image was then used as a reference point in order to correlate the dSTORM clusters to the identified labeled loci. Clustering analysis revealed a measured distance of 179 nm between PTPRN2 and the closest AR cluster, while the distance between BANP and AR was 80 nm, confirming therewith the interparticle distances found by confocal microscopy. Note that, using dSTORM, these particles do not visually overlap. To assess the statistical significance of the observed colocalizations, we generated 40 000 in silico images consisting of a single (or two) ‘gene’ locus and 300 ‘AR foci’ per nucleus (the number of foci and sizes have been estimated based on experimental images). In comparison to the simulated control data, the confocal data had almost two-fold increase of colocalizing events (Figure 2B).
We then acquired time-lapse images to observe the de novo formation of AR foci near the PTPRN2 and BANP labeled loci directly upon R1881 stimulation (Supplementary videos 1, 2). Cells were pre-treated with hydroxyflutamide overnight to facilitate the translocation of AR molecules without inducing AR-DNA binding (57). Image acquisition was started directly after addition of R1881. For both PTPRN2 and BANP, a rapid formation of AR foci was observed after approximately 5 min upon R1881 addition, indicating that foci formation does not require long exposure to R1881 and is fast enough to readily observe AR foci colocalizing with either of the two labeled genes (Figure 2d). After image acquisition, colocalization analysis was performed per time frame to measure the distance between a labeled locus and the nearest AR focus. Distances between the PTPRN2-labeled locus and nearest AR focus ranged between 32 nm and 256 nm over time with an average measured distance of 160 ± 16 nm (SEM) (Figure 2E). For BANP, the distances varied between 50 and 367 nm with an average of 185 ± 20 nm (Figure 2F). These measurements, together with the quantitative colocalization assay of fixed cells, further confirmed a significant spatial and temporal colocalization between the AR foci and the ANCHOR3-labeled PTPRN2 and BANP gene regions.
To further analyze in which chromatin regions the AR foci were localized, we acquired confocal images of cells expressing AR-GFP and stained with Hoechst and propidium iodide (Figure 3A). Propidium iodide was used to segment nucleoli and discard these regions from the analysis, while Hoechst was analyzed in more detail to define five chromatin density classes ranging from low-density DNA regions (blue) to condensed DNA regions, heterochromatin-like areas (yellow), with three classes in-between (green, red, magenta) representing euchromatic regions (Figure 3A, B). Across 83 cell nuclei, quantifications of AR foci in each density class showed that approximately 78% of the AR foci were located at euchromatin regions, 19% at heterochromatin and 3% at low-density chromatin areas (Figure 3c). The large proportion of AR signals in euchromatin further suggests the general role of AR foci in transcriptional processes, since euchromatin is a lightly packed form of the chromatin, facilitating active transcription. These findings were further validated by immunostainings for H3K27Ac and H3K27me3 histone marks, which indicate transcriptionally active and inactive chromatin, respectively. H3K27Ac signal were frequently found near AR foci, whereas H3K27me3 signals were not observed (Supplementary Figure S7).
Figure 3.

AR foci are mostly located in euchromatin. (A) Confocal image of a cell nucleus labeled with AR-GFP to visualize AR foci (green), propidium iodide to segment the nucleolus regions (red), and Hoechst to visualize the chromatin areas (blue). Hoechst normalized intensities were categorized in five classes. (B) Spatial distribution of the detected AR foci at each chromatin intensity class. (C) Bottom: fraction of AR foci in each chromatin intensity class (83 cells). Error bars represent the standard deviation. Top: pie chart showing percentage of foci in low-density chromatin (0.0–0.2), euchromatin (0.2–0.8) and heterochromatin (0.8–1.0) areas, respectively.
Segmentation of SPT data reveals three subdiffusive states
To visualize and quantify the behavior of ARs in foci and the remainder of the nuclear environment, we performed single particle tracking (SPT) of ARs in cells containing either the labeled BANP or PTPRN2 gene bodies. For this purpose, we imaged in two dimensions the genes of interest labeled with the ANCHOR3 system, followed by a time-lapse experiment of 105 seconds with a short time interval of 7 ms (Figure 4A). We recorded 20 time-lapse series in R1881 treated cells expressing ARWT-HaloTag with the ANCHOR3-labeled BANP gene and 20 others in cells expressing ARWT-HaloTag with the ANCHOR3-labeled PTPRN2 gene. The detection and linking of the fluorescent AR spots permitted the construction of trajectories which we then analyzed in-depth to decipher their biological dynamics (Figure 4B). To exclusively analyze the intranuclear region, we generated a mask for each cell nucleus using the ANCHOR3 labeling channel (the fluorescent OR3 proteins accumulate in the cytoplasm allowing visual delineation of the nucleus).
Figure 4.

Single particle tracking data reveals three subdiffusive states. (A) Design of the experiment combining a labeled chromatin locus with single-particle tracking (SPT) of androgen receptors (AR). (B) Analysis pipeline of SPT data: trajectory segmentation using deep learning followed by various geometric computations. (C–G) Results of the AR SPT data analysis per tracklet state combining trajectories from 40 cells. (C) Transition probability diagram of the three states. (D) Anomalous exponent 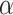 plotted against the diffusion constant
plotted against the diffusion constant 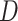 of the tracklets; black circles indicate barycenter of the joint
of the tracklets; black circles indicate barycenter of the joint 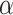 and
and 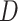 distributions for each state, estimated using the median. (E) Distributions of combined
distributions for each state, estimated using the median. (E) Distributions of combined 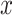 and
and 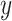 displacements for the time interval
displacements for the time interval 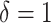 . Inset: Z-score normalization of the distributions, following a Gaussian
. Inset: Z-score normalization of the distributions, following a Gaussian 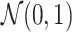 . (F) Angular distributions and fold anisotropy metric
. (F) Angular distributions and fold anisotropy metric 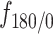 . (G) Velocity Autocorrelation (VAC) functions compared with theoretical fBm curves for various
. (G) Velocity Autocorrelation (VAC) functions compared with theoretical fBm curves for various 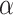 .
.
A particle trajectory can be a mixture of various dynamics which are related to the biological function of the tracked protein. We classified the trajectory displacements in three states by applying a supervised deep-learning method that we have previously developed (45). We segmented the trajectories presenting a mixture of states, and grouped the segmented tracks (further referred to as tracklets) based on their diffusive state (Figure 4B), with the slowest to highest mobility ranging from state 1 to state 3. The state transition probability diagram of AR tracks (Figure 4C) showed that the transitions were occurring more between states 1 and 2, and between 2 and 3 as compared to between states 1 and 3. We estimated that the proportion of tracklets in each group was about the same in the dataset, however this estimation was greatly biased by the 2D imaging and bleaching processes which significantly reduces the number of visible particles and decreased the true underlying number of fast molecules. Transition between states (crossovers) and changes in anomalous exponents with sub- or superdiffusive properties may occur in each independent AR trajectory (58). This may reflect diffusing ARs (state 3) that enter AR foci (state 2) or vice versa, or diffusing ARs (state 3) that collide with binding sequences on the DNA and subsequently are immobilized by DNA binding (state 1), or vice versa. For our real experimental data, without the proposed track segmentation method, such behavioral changes can be barely observed in the separated 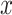 and
and 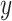 trajectory coordinates and the associated MSD curve (Equation 3 and Supplementary Figure S8a).
trajectory coordinates and the associated MSD curve (Equation 3 and Supplementary Figure S8a).
For each tracklet, we estimated the motion parameters (the diffusion constant 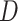 and the anomalous diffusion exponent
and the anomalous diffusion exponent 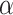 ) by fitting the
) by fitting the 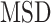 to a power law equation (Equation 4 and Supplementary Figure S8b).
to a power law equation (Equation 4 and Supplementary Figure S8b). 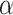 describes the level of confinement of a given trajectory and reveals a nonlinear dependency between the
describes the level of confinement of a given trajectory and reveals a nonlinear dependency between the 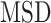 and the time in the case of
and the time in the case of 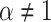 (
(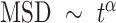 ), with
), with 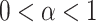 showing specifically subdiffusion,
showing specifically subdiffusion, 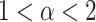 , superdiffusion, and
, superdiffusion, and 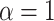 , representing a Brownian motion. Both
, representing a Brownian motion. Both 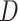 and
and 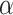 distributions were plotted in a joint scatterplot (Figure 4D) and the barycenter was approximated per state. Each state had a rather characteristic diffusion
distributions were plotted in a joint scatterplot (Figure 4D) and the barycenter was approximated per state. Each state had a rather characteristic diffusion 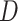 (around
(around 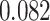 ,
, 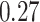 and
and 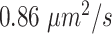 respectively), while
respectively), while 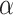 values for states 1 and 2 were similar (
values for states 1 and 2 were similar (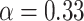 ) compared to the third state (
) compared to the third state (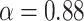 ). Although some fast state tracklets were categorized as superdiffusive, AR tracklets were overall subdiffusive, which is a common behavior of DNA-binding proteins found in the nucleus (22,24) and can be the result of a heterogeneous environment (local variation of AR-cofactor complexes) or obstacles in the path of the particles. Computing the
). Although some fast state tracklets were categorized as superdiffusive, AR tracklets were overall subdiffusive, which is a common behavior of DNA-binding proteins found in the nucleus (22,24) and can be the result of a heterogeneous environment (local variation of AR-cofactor complexes) or obstacles in the path of the particles. Computing the 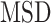 for entire (unsegmented) trajectories produces averaged estimates over the 3 states (barycenter:
for entire (unsegmented) trajectories produces averaged estimates over the 3 states (barycenter: 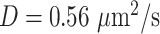 ,
, 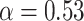 ; Supplementary Figure S8c, d).
; Supplementary Figure S8c, d).
As a control, we tracked histone H2B-HaloTag, largely immobile due to its DNA organizing function (Supplementary Figure S9a), and a HaloTagged peptide containing a nuclear localization signal (NLS — fast diffusion), and found that the motion of H2B closely matches the immobile fraction of ARWT (Supplementary Figure S9b); while NLS particles show similar behavior compared to the mobile fraction of ARWT (Supplementary Figure S9c). The similarities found between the immobile fraction of both H2B and ARwt might also partially represent movement of the chromatin itself, instead of solely AR motion, as proposed by others (59). Furthermore, as controls, we tracked a DNA-binding deficient AR mutant (ARR585K-HaloTag) as well as a mutant AR lacking the first 559 amino acids corresponding to the NTD domain (ARΔNTD-HaloTag). To assess the dynamics of AR regarding either its DBD or NTD. Both ARR585K and ARΔNTD did not form foci, indicating reduced chromatin binding. This was further supported by SPT experiments, showing a much more diffusive behavior (Supplementary Figure S9d, e) for both ARΔNTD and ARR585K (as similarly observed for a fluorescently tagged NLS) in comparison to the wild-type.
Geometric measurements confirmed the existence of three subdiffusive states of fBm
To further investigate each subdiffusive state, we computed additional geometric measures for the tracklets in the different states. 1D displacements between two points at time 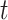 and
and 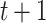 (Figure 4E) showed a different mode of displacement for each state which correlates with the three distinct estimates of diffusion
(Figure 4E) showed a different mode of displacement for each state which correlates with the three distinct estimates of diffusion 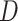 . We then calculated the angles between two consecutive displacements (21,24). In case a trajectory follows a fractional Brownian motion (fBm) (a long-range correlated Gaussian process as described in the Method section), the distribution of angles gives an indication of the nature of the diffusion (29,60,61). A uniform distribution would describe a Brownian motion, while a non-uniform distribution is evidence of anomalous diffusion and can either reflect a preferential directed movement when angles occur more frequently towards
. We then calculated the angles between two consecutive displacements (21,24). In case a trajectory follows a fractional Brownian motion (fBm) (a long-range correlated Gaussian process as described in the Method section), the distribution of angles gives an indication of the nature of the diffusion (29,60,61). A uniform distribution would describe a Brownian motion, while a non-uniform distribution is evidence of anomalous diffusion and can either reflect a preferential directed movement when angles occur more frequently towards 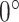 or, on the contrary, a confined diffusion when angles occur towards
or, on the contrary, a confined diffusion when angles occur towards 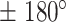 . All states exhibited a non-uniform angular distribution towards
. All states exhibited a non-uniform angular distribution towards 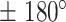 angles (Figure 4F) indicating subdiffusive motion; although, state 3 tracklets explored angles more evenly than angles in states 1 and 2, both of which had a similar distribution. We further computed the fold anisotropy metric
angles (Figure 4F) indicating subdiffusive motion; although, state 3 tracklets explored angles more evenly than angles in states 1 and 2, both of which had a similar distribution. We further computed the fold anisotropy metric 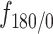 to quantify how likely a step backward is relative to a step forward 21. The fold anisotropy of state 3 tracklets was
to quantify how likely a step backward is relative to a step forward 21. The fold anisotropy of state 3 tracklets was 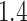 , confirming our observation that they were slightly more diffusive. States 1 and 2 tracklets showed a more pronounced anisotropy with
, confirming our observation that they were slightly more diffusive. States 1 and 2 tracklets showed a more pronounced anisotropy with 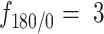 and
and 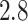 , respectively.
, respectively.
Lastly, we generated the velocity autocorrelation curves (VAC) for each tracklet state and compared them to the theoretical curves of fractional Brownian motion (fBm) with diverse 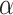 values (Figure 4G). Once more, states 1 and 2 showed a similar trend when compared to state 3. Each curve has a characteristic pattern: including an antipersistence at
values (Figure 4G). Once more, states 1 and 2 showed a similar trend when compared to state 3. Each curve has a characteristic pattern: including an antipersistence at 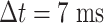 indicating subdiffusion (47), which was also proposed by others (58) and a convergence to zero for longer periods of times, which corresponds to the fBm with an approximate theoretical
indicating subdiffusion (47), which was also proposed by others (58) and a convergence to zero for longer periods of times, which corresponds to the fBm with an approximate theoretical 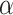 value of
value of 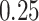 ,
, 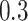 and
and 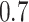 for each diffusive state respectively (relatively close to the estimates shown in Figure 4d). The criterion of Gaussianity is another major characteristic pattern of the fBm model of diffusion, which was verified after normalizing the distributions of 1D displacements with the first discrete time interval
for each diffusive state respectively (relatively close to the estimates shown in Figure 4d). The criterion of Gaussianity is another major characteristic pattern of the fBm model of diffusion, which was verified after normalizing the distributions of 1D displacements with the first discrete time interval 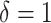 for each state independently (inset of Figure 4E). Although, on entire AR trajectories, the VAC curves seem to follow a fBm at first sight (Supplementary Figure S10a–b), the minimum of the correlator at
for each state independently (inset of Figure 4E). Although, on entire AR trajectories, the VAC curves seem to follow a fBm at first sight (Supplementary Figure S10a–b), the minimum of the correlator at 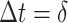 is tending to zero (value which should be constant in the case of pure fBm). In addition, distributions of displacements were non-Gaussian (Laplace) and self-similar for different time intervals (Supplementary Figure S10c, d), as previously reported for RNA (27) and histone H2B (62). All these particularities (including crossovers and non-Gaussianity) are the result of a heterogeneous mixture of diffusive states in the SPT dataset of ARs, therefore reinforcing the use of a three-state segmentation model. These various diffusive states are speculated to be linked with the functions of AR molecules, particularly its property of binding and unbinding to the DNA.
is tending to zero (value which should be constant in the case of pure fBm). In addition, distributions of displacements were non-Gaussian (Laplace) and self-similar for different time intervals (Supplementary Figure S10c, d), as previously reported for RNA (27) and histone H2B (62). All these particularities (including crossovers and non-Gaussianity) are the result of a heterogeneous mixture of diffusive states in the SPT dataset of ARs, therefore reinforcing the use of a three-state segmentation model. These various diffusive states are speculated to be linked with the functions of AR molecules, particularly its property of binding and unbinding to the DNA.
Confined mobility of AR particles close to AR-regulated chromatin loci
We further analyzed the interaction between the PTPRN2 and BANP gene bodies and individual ARWT-HaloTag particles. For that, we overlaid the segmented trajectories from the tracking dataset with the ANCHOR3 channel (Supplementary Figure S11 and Figure 4A). Overlap between AR trajectories and the labeled genes was not always present; we visually estimated 16 cells out of 20 showing PTPRN2-AR colocalization (based on states 1 and 2 tracklets) and 17 cells out of 20 showing a colocalization between ARs and BANP. AR could not be detected on all gene loci due to temporarily unbound states or bleaching of the AR fluorescent label. Additionally, no major differences were observed in the mobility of AR particles between the two gene loci. Note that the BANP region harbors potential promoter/enhancer regions for the FBXO31 and ZFPM1 genes, explaining similar behavior of AR in PTPRN2 and BANP regions, in spite of the fact that BANP is not regulated by AR (see also above). Therefore, we combined the results for both gene regions for further analysis, using 10 labeled loci for each of the two genes.
We cropped the region around the chromatin locus, selected areas of 0.15 μm2 and overlaid the associated AR trajectories on top of each subfigure (Figure 5A, B). We observed several long and confined trajectories residing at the chromatin locus, while several other particles were generally diffusing around or crossing the region. We selected the trajectories observed for at least half a second (representing 70 or more frames), and having at least one third of their track points at 450 nm or less from the center of the labeled chromatin locus (since a circular region of 450 nm radius covers the intensity of a spot and its surrounding). In half of these picture series, we observed two to three selected trajectories close to one another and located near the labeled locus; each of them might represent a unique AR molecule, together with other bleached molecules, forming a nuclear AR focus.
Figure 5.

Behavior of AR trajectories at labeled chromatin loci characterized by a confinement and a spatial organization. (A, B) For each gene (PTPRN2 and BANP), overlays of the labeled locus with AR trajectories (in gray and pink) or tracklets (colored by tracklet state). The trajectories colored in gray are longer than 70 frames and have at least one third of their track points at 4.5 pixels or less from the center of the labeled spot (indicated by a red ‘+’). The points in cyan are the centroid of each gray trajectory (the centroids have been averaged if multiple trajectories spread in the same region). The tracklets in opaque have at least one point within 2 pixels from each trajectory centroid. (C–F) Statistical analysis based on 10 loci per gene. (C) Kernel density plot of the joint 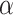 and
and 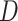 distributions using entire AR trajectories. Estimations were done using the
distributions using entire AR trajectories. Estimations were done using the 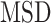 . Points in cyan represent the
. Points in cyan represent the 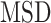 estimations on the gray trajectories (subfigures A and B). (D) Distribution of trajectory length (translated in time) of AR trajectories. Points in cyan are the length of the gray trajectories. (E) Distance of each tracklet point to the trajectory centroid. Statistical significance was determined using a Mann–Whitney–Wilcoxon test two-sided with Bonferroni correction (****P-value ≤ 10−4). (F) Angular distributions and fold anisotropy metric
estimations on the gray trajectories (subfigures A and B). (D) Distribution of trajectory length (translated in time) of AR trajectories. Points in cyan are the length of the gray trajectories. (E) Distance of each tracklet point to the trajectory centroid. Statistical significance was determined using a Mann–Whitney–Wilcoxon test two-sided with Bonferroni correction (****P-value ≤ 10−4). (F) Angular distributions and fold anisotropy metric 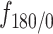 of tracklets in state 3. Tracklet points ‘inside’ are located within the 2-pixel radius threshold.
of tracklets in state 3. Tracklet points ‘inside’ are located within the 2-pixel radius threshold.
The selected tracks were all showing smaller diffusion coefficient and a high degree of confinement after 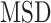 analysis with most of the trajectories having
analysis with most of the trajectories having 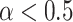 (Figure 5C). In addition, their length ranged from 77 (0.539 s) to 480 (3.36 s) frames, representing a small portion of the longest trajectories in the SPT dataset. The distribution of trajectory lengths was generated using the entire dataset of AR trajectories (Figure 5D, translated in time). This distribution is quite characteristic of SPT microscopy data: the observation of the third dimension being quite limited in 2D images, the fast-diffusing particles tend to promptly move in and out of the focal plane (Supplementary Figure S12a) resulting in short trajectories, while immobile particles remain in-focus until alteration of the dye by photobleaching (or alternatively, by switching to another state and leaving the focus). Moreover, their resemblance to point spread functions facilitates their detection, which contributes to obtaining (extremely) long trajectories (30); our longest trajectory had a length of 951 frames (corresponding to 6.66 s) which might give an idea of the binding-time of AR, while the average length was 32.3 frames (0.226 s) (see Supplementary Figure S12b for tracklet length in each state). The longest trajectories (from 77 to 951 frames) represent in fact only 8.15% of the entire set of trajectories, meaning that these immobile AR particles were most probably located at AR-regulated regions as a consequence of DNA-binding.
(Figure 5C). In addition, their length ranged from 77 (0.539 s) to 480 (3.36 s) frames, representing a small portion of the longest trajectories in the SPT dataset. The distribution of trajectory lengths was generated using the entire dataset of AR trajectories (Figure 5D, translated in time). This distribution is quite characteristic of SPT microscopy data: the observation of the third dimension being quite limited in 2D images, the fast-diffusing particles tend to promptly move in and out of the focal plane (Supplementary Figure S12a) resulting in short trajectories, while immobile particles remain in-focus until alteration of the dye by photobleaching (or alternatively, by switching to another state and leaving the focus). Moreover, their resemblance to point spread functions facilitates their detection, which contributes to obtaining (extremely) long trajectories (30); our longest trajectory had a length of 951 frames (corresponding to 6.66 s) which might give an idea of the binding-time of AR, while the average length was 32.3 frames (0.226 s) (see Supplementary Figure S12b for tracklet length in each state). The longest trajectories (from 77 to 951 frames) represent in fact only 8.15% of the entire set of trajectories, meaning that these immobile AR particles were most probably located at AR-regulated regions as a consequence of DNA-binding.
We then analyzed the spatial organization of the tracklets at these binding regions to identify the different potential areas of AR molecule diffusivity. For this purpose, we estimated the centroid of each selected long trajectories (the centroids have been averaged if multiple selected trajectories overlapped in the same spatial region). We observed that the core of state 1 and 2 tracklets had an average diameter of about 400 nm; therefore, we set a distance threshold around these centroids of 200 nm and kept the whole tracklets having at least one point within this distance (Figure 5A, B). We generally observed tracklets in state 1 being more compact and sitting at the centroid, while tracklets in state 2 were located at the same region, plus in the close neighborhood of tracklets in state 1. Finally, about half of the tracklets in state 3 were located within the 200 nm threshold, in the neighborhood of both state 1 and 2 tracklets, while the other half was diffusing outside of the AR foci region. These observations were confirmed by computing the distance from each tracklet point to the corresponding centroid (Figure 5E). The diameter of the binding regions composed by states 1 and 2 tracklets can be roughly estimated to 400–800 nm from these boxplots. Lastly, we compared the angles of state 3 tracklets in and outside the delimited radius threshold (Figure 5F). The anisotropy value of the fast tracklets located inside was almost 4 times higher than the value obtained for the tracklets outside. This marked a clear change in the behavior of ARs outside and inside of the delimited threshold, probably due to a change of the environment, going from least to most dense in molecules. This anisotropy value is also two times higher than anisotropy values previously calculated for state 1 and 2 tracklets (Figure 4F).
Confined mobility at unlabeled nuclear regions
Next to the distinct observations made (trajectories and state distributions) at the PTPRN2 and BANP labeled gene bodies, we analyzed the SPT data at the unlabeled regions to verify whether we obtained similar patterns. Across the entire imaged region for each cell (Supplementary Figure S13 and Figure 6A, B), we identified similar patterns as previously observed in Figure 5, where tracklets in state 2 were observed close to tracklets in state 1, while tracklets in state 3 were visibly diffusing in the nucleus (examples in Figure 6C). We pursue our statistical investigation across the imaged nuclear region of 6 different cells (Figure 6D–I). The trajectories longer than 70 frames were selected and the few long tracks presenting free diffusion were discarded. The distribution of trajectory lengths (Figure 6D) showed that the selected trajectories among the studied cell had an average length of 179 frames (1.25 s) with the two longest tracks counting 715 and 808 frames (5.01 and 5.66 s). The centroid of each selected trajectories was measured, and their coordinates were used to estimate the Ripley's 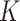 -function to study how the confined AR particles were spatially distributed in the nucleus. For this purpose, we cropped a region of 100 × 40 pixels for each nucleus and averaged the number of centroids within a radius
-function to study how the confined AR particles were spatially distributed in the nucleus. For this purpose, we cropped a region of 100 × 40 pixels for each nucleus and averaged the number of centroids within a radius 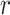 from any centroid. An edge correction was applied to prevent biases, and the obtained curve was compared to a completely spatial random (CSR) pattern (here, we used a Poisson distribution) (Figure 6E). We did not observe an important deviation from the CSR, which would have indicated scales of clustering or dispersion. In addition, we estimated the
from any centroid. An edge correction was applied to prevent biases, and the obtained curve was compared to a completely spatial random (CSR) pattern (here, we used a Poisson distribution) (Figure 6E). We did not observe an important deviation from the CSR, which would have indicated scales of clustering or dispersion. In addition, we estimated the 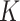 -function from 6 confocal images of AR-GFP expressing cells (by using the location of the maximum pixel intensities, corresponding to AR focus locations) and almost no differences were found in the dispersion of the two types of data, indicating that the immobilized AR molecules have a spatial dispersion similar to AR foci.
-function from 6 confocal images of AR-GFP expressing cells (by using the location of the maximum pixel intensities, corresponding to AR focus locations) and almost no differences were found in the dispersion of the two types of data, indicating that the immobilized AR molecules have a spatial dispersion similar to AR foci.
Figure 6.

Similar diffusion pattern observed over the entire imaged nuclear region. (A–C) Overlay of the ANCHOR3 channel of a single cell with AR trajectory detections (A) or tracklet detections (B). The trajectories colored in gray are longer than 70 frames and having less than 20% of the track points labeled as state 3 (to discard the diffusive tracks); the detections in pink represent all the remaining tracks. The points in cyan are the centroid of each selected gray trajectory. The cropped (1–4) are zoomed-in regions of tracklets illustrated in (C). (D–I) Statistical analysis based on six cells. (D) Distribution of trajectory length (translated in time) of trajectories extracted. (E) Multi-distance spatial pattern analysis (Ripley's 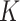 -function) for different radii
-function) for different radii 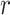 using the SPT data (using centroid locations) and the AR-GFP expressed confocal images from 6 other cells (using the locations of the maximum pixel intensities). The curves were compared to a Poisson distribution as a CSR pattern. (F) Distance of each tracklet point (having at least one point at 2 pixels or less from the closest centroid) to the corresponding centroid. Statistical significance was determined using a Mann–Whitney–Wilcoxon test two-sided with Bonferroni correction (****P-value ≤ 10−4). (G) Fraction of tracklet points for each tracklet state located inside or outside the 2-pixel radius threshold. (H) Angular distributions and fold anisotropy metric
using the SPT data (using centroid locations) and the AR-GFP expressed confocal images from 6 other cells (using the locations of the maximum pixel intensities). The curves were compared to a Poisson distribution as a CSR pattern. (F) Distance of each tracklet point (having at least one point at 2 pixels or less from the closest centroid) to the corresponding centroid. Statistical significance was determined using a Mann–Whitney–Wilcoxon test two-sided with Bonferroni correction (****P-value ≤ 10−4). (G) Fraction of tracklet points for each tracklet state located inside or outside the 2-pixel radius threshold. (H) Angular distributions and fold anisotropy metric 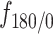 for each tracklet state inside or outside the radius threshold. (I) Distributions of combined
for each tracklet state inside or outside the radius threshold. (I) Distributions of combined 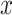 and
and 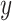 displacements for the time interval
displacements for the time interval 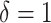 for each tracklet state inside or outside the radius threshold.
for each tracklet state inside or outside the radius threshold.
We selected the entire tracklets having at least one point within a 200 nm distance threshold to each centroid and computed the distance from each tracklet point to the corresponding centroid. The resulting measures for each tracklet group (Figure 6F) were quite close to the distances shown in Figure 5E, confirming the ordered visual aspect of the tracklets around each centroid. Next, we separated the tracklet points in two groups: residing inside or outside of the 200 nm radius. The fraction of tracklets in state 1 were unsurprisingly higher inside, and state 3 tracklets higher outside (Figure 6G). We observed slightly more state 1 tracklets outside than expected which can be explained by our procedure which did not select the trajectories shorter than 70 frame long. The fraction of state 2 tracklets was roughly the same ratio inside and outside. Finally, we measured the distribution of angles and displacements for each tracklet group, inside or outside the delimited regions (Figure 6H, I). We did not observe strong differences for state 1 and 2 tracklets inside versus outside, except the anisotropy values which are a little higher for tracklets inside. However, the fast tracklets (state 3) showed more differences between inside and outside: the displacements were larger for tracklets outside, while tracklets inside showed a higher degree of confinement based on angle distribution, indicating that free molecules become partially obstructed by other ARs in foci. All these observations showed confined diffusion which might result from DNA-binding in AR foci.
Discussion
We investigated the interaction of ARs with chromatin, largely, but not exclusively, focusing on two specific fluorescently labeled endogenous genes. Based on ChIP-seq and previously published RNA-seq we selected the PTPRN2 and BANP genes (Figure 1), and used the ANCHOR DNA labeling system (Supplementary Figure S3) to visualize them in fixed and living cells, and showed by confocal time-lapse imaging that upon activation by R1881, GFP-tagged AR rapidly accumulates in foci at the labeled genes (Figure 2). We also recorded single-particle tracks at the two genes of individual R1881-activated ARs fused to HaloTags, and used a three-state model based on an artificial neural network to classify individual tracks into distinct diffusive states (Figure 4). This classification revealed a spatially organized pattern of ARs at the labeled genes (Figure 5A, B), which consist of a few immobile AR tracklets (state 1) surrounded by an intermediate confined state exhibiting a larger displacement (state 2). Similarly, highly mobile tracklets (state 3) showed sharper angles near state 1 and 2 tracklets, and a free diffusion outside the foci. (Figure 5F).
In addition, we analyzed AR foci in the remainder of the nucleus by confocal microscopy (EGFP-AR) showing that the majority (>75%) of AR foci are located in euchromatin (Figure 3). Single-particle tracking (ARWT-HaloTag) showed that the spatial organization pattern of ARs observed in unlabeled regions of the nucleus were very similar to those studied at labeled genes (Figures 5E and 6F). Note that the spatial distribution and approximate number of foci observed by confocal microscopy were similar to the distribution of colocalizing state 1 and state 2 tracklet clusters (Figure 6E). Intriguingly, a part of the mobile fraction (state 3) has the particularity of being confined to where state 1 and 2 tracklets have been located, in sharp contrast to the outer region (Figure 5F), probably due to the affinity of a local environment which would lead to the recruitment of mobile ARs. This phenomenon could be explained by the property of liquid–liquid phase separated structures, as shown by others (8,9). Alternatively, it is possible that both state 1 and 2 mobilities reflect chromatin movement (59). This explanation is supported by the observation that the AR DNA-binding deficient mutant does not form AR foci. However, from the available data it can not be excluded that the AR DNA-binding deficient mutant is unable to engage in phase separation and therefore does not form foci. Further research is required to test these different models.
Together, confocal (time-lapse) images show that AR foci are formed and maintained at genes, and the SPT data strongly suggest that, after immobilization of a small number of ARs by binding directly to DNA, liquid–liquid phase separation (LLPS) drives further compartmentalization, leading to the formation of foci. Phase separation of ARs was also suggested by Xie et al. (10), but no data was provided that foci are formed near genes. In addition, Zhang et al. (8) showed that in vitro AR droplet formation was enhanced in the presence of MED1, suggesting that the presence of other transcription activators at certain DNA regions contribute to phase separation-based foci formation. Therefore, it might be that not all observed AR foci are initiated by AR itself, but rather AR may reside in foci nucleated by other factors. Then, AR may have a supportive—instead of a regulatory—role, for instance by (helping in) recruiting chromatin remodelers or other co-factors. This could be further investigated for instance by knocking down the transcription factors identified by GIGGLE analysis (Supplementary Figure S2) and performing SPT of AR to determine the impact on the distribution of the three different subdiffusive states.
Our data are compatible with a model where a part of AR molecules is gradually trapped at random after collision with the local environment of a nuclear focus, as a possible consequence of relatively weak protein-protein interactions inside these structures (Figure 7). Fast diffusing ARs (state 3) will thereby get trapped by LLPS as previously suggested for GR (9). This trapping results in a spatial confinement with a significantly reduced diffusion (state 2), likely necessary for probing ARE sequences. Once these molecules encounter an unbound ARE sequence, they will become completely immobilized (bound — state 1) on the DNA due to direct interactions with the DNA-binding domain (DBD). Based on the increasing evidence for LLPS in transcriptional processes (12,13), we propose an essential role for state 1 and 2 AR tracklets together for proper transcriptional activity. In fact, phase-separated droplets effectively facilitate a higher concentration of TFs locally, enhancing the chance of probing unbound ARE sequences at these specific genomic sites. Affecting the condensation of AR by targeting either the DBD, the ligand-binding domain (LBD) or the N-terminal domain (NTD) result almost always to an overall downregulated transcriptional output (1,10). Moreover, targeting experiments (e.g. domain mutations and domain targeting compounds) against these domains showed that they are all required to effectively induce foci condensation (1,8,9). However, further research is required to provide a better understanding of the mechanisms underlying AR function in general and the role of foci formation in particular. Finally, this knowledge may contribute to new therapeutic targeting strategies for novel prostate cancer therapies.
Figure 7.

Model of AR foci dynamics in a 3D-genome space.
Supplementary Material
Acknowledgements
We would like to thank Wytske van Weerden (Department of Urology, Erasmus MC) for providing the PC346C cell line; Guido Jenster (Department of Urology, Erasmus MC) and Andrea Sacchetti (Department of Pathology, Erasmus MC) and Sarra Merzouk (Department of Developmental Biology, Erasmus MC) for insightful discussions and technical support; the Flow Cytometry Facility of the Pathology department of Erasmus MC for technical support; Kerstin S. Wendt and Thomas van Staveren (Department of Cell Biology, Erasmus MC) for assistance with optimizing the ANCHOR system; Maarten W. Paul (Department of Molecular Genetics, Erasmus MC) for support on SPT experiments. Graphical Abstract, Figures 4A–C, 7 and S3 were created using BioRender.com.
Author contributions: S.Y., H.K., M.E.v.R., I.S. and A.B.H. conceived the study; T.A.A., M.E.v.R., M.Q., R.A.P. and A.B.H. performed the ChIP-seq experiments; S.L. and W.Z. analyzed the NGS data; S.Y., T.A.A. and J.v.S. performed the RT-PCR; S.Y. and T.A.A. generated the genetically modified cell lines; J.v.S. and F.G.G. optimized the ANCHOR labeling system; S.Y., W.A.v.C. and A.N. performed the confocal experiments and analyzed the confocal images; J.A.S. implemented the script to generate the simulated confocal data and S.Y. ran the simulations; S.Y, T.A.A. and J.A.S. performed the dSTORM experiments; S.Y. performed the SPT experiments and the tracking of AR particles; H.K. designed the SPT analysis pipeline with the input from I.S., S.Y., A.B.H., M.E.v.R. and J.v.S.; H.K. implemented the trajectory segmentation software and S.Y. ran the software; H.K. performed the SPT analysis; S.Y. and H.K. wrote the manuscript with the input from all co-authors.
Contributor Information
Selçuk Yavuz, Department of Pathology, Erasmus University Medical Center, Rotterdam, The Netherlands.
Hélène Kabbech, Department of Cell Biology, Erasmus University Medical Center, Rotterdam, The Netherlands.
Jente van Staalduinen, Department of Cell Biology, Erasmus University Medical Center, Rotterdam, The Netherlands.
Simon Linder, Division of Oncogenomics, The Netherlands Cancer Institute, Amsterdam, The Netherlands.
Wiggert A van Cappellen, Erasmus Optical Imaging Center, Erasmus University Medical Center, Rotterdam, The Netherlands.
Alex L Nigg, Erasmus Optical Imaging Center, Erasmus University Medical Center, Rotterdam, The Netherlands.
Tsion E Abraham, Erasmus Optical Imaging Center, Erasmus University Medical Center, Rotterdam, The Netherlands.
Johan A Slotman, Erasmus Optical Imaging Center, Erasmus University Medical Center, Rotterdam, The Netherlands.
Marti Quevedo, Department of Cell Biology, Erasmus University Medical Center, Rotterdam, The Netherlands.
Raymond A Poot, Department of Cell Biology, Erasmus University Medical Center, Rotterdam, The Netherlands.
Wilbert Zwart, Division of Oncogenomics, The Netherlands Cancer Institute, Amsterdam, The Netherlands; Laboratory of Chemical Biology and Institute for Complex Molecular Systems, Department of Biomedical Engineering, Eindhoven University of Technology, Eindhoven, The Netherlands.
Martin E van Royen, Department of Pathology, Erasmus University Medical Center, Rotterdam, The Netherlands.
Frank G Grosveld, Department of Cell Biology, Erasmus University Medical Center, Rotterdam, The Netherlands.
Ihor Smal, Department of Cell Biology, Erasmus University Medical Center, Rotterdam, The Netherlands.
Adriaan B Houtsmuller, Department of Pathology, Erasmus University Medical Center, Rotterdam, The Netherlands; Erasmus Optical Imaging Center, Erasmus University Medical Center, Rotterdam, The Netherlands.
Data availability
All PC346C cell line ChIP-seq data generated in this study have been deposited in the Gene Expression Omnibus (GEO) database under the accession code: GSE229728 (PC346C AR ChIPseq). Public ChIP-seq datasets used in this study are available from GEO or European Genome-Phenome Archive (EGA) databases under the following accession codes: GSE152231 (AR ChIPseq) and EGAS00001006017 (AR ChIP-seq). Public RNA-seq datasets used in this study are available from EGA under the following accession code EGAS00001006016. NearestNeighbour ImageJ Plugin is available on Github (https://github.com/ErasmusOIC/NearestNeighbour) and Zenodo (http://doi.org/10.5281/zenodo.8359132). TrackSegNet script is available in Zenodo (http://doi.org/10.5281/zenodo.7767750).
Supplementary data
Supplementary Data are available at NAR Online.
Funding
Dutch Research Council (NWO) through the Building Blocks of Life program (GENOMETRACK project) [737.016.014]. Funding for open access charge: Erasmus MC, Rotterdam, The Netherlands.
Conflict of interest statement. None declared.
References
- 1. van Royen M.E., van Cappellen W.A., de Vos C., Houtsmuller A.B., Trapman J.. Stepwise androgen receptor dimerization. J. Cell Sci. 2012; 125:1970–1979. [DOI] [PubMed] [Google Scholar]
- 2. Nash C., Boufaied N., Badescu D., Wang Y.C., Paliouras M., Trifiro M., Ragoussis I., Thomson A.A.. Genome-wide analysis of androgen receptor binding and transcriptomic analysis in mesenchymal subsets during prostate development. Dis. Model Mech. 2019; 12:dmm039297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Coulon A., Chow C.C., Singer R.H., Larson D.R.. Eukaryotic transcriptional dynamics: from single molecules to cell populations. Nat. Rev. Genet. 2013; 14:572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hyman A.A., Weber C.A., Jülicher F.. Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 2014; 30:39–58. [DOI] [PubMed] [Google Scholar]
- 5. Erdel F., Rippe K.. Formation of chromatin subcompartments by phase separation. Biophys. J. 2018; 114:2262–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Weber S.C., Peng A.. Evidence for and against liquid-liquid phase separation in the nucleus. Noncoding RNA. 2019; 5:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wagh K., Garcia D.A., Upadhyaya A.. Phase separation in transcription factor dynamics and chromatin organization. Curr. Opin. Struct. Biol. 2021; 71:148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang F., Biswas M., Massah S., Lee J., Lingadahalli S., Wong S., Wells C., Foo J., Khan N., Morin H.et al.. Dynamic phase separation of the androgen receptor and its coactivators key to regulate gene expression. Nucleic Acids Res. 2023; 51:99–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Garcia D.A., Johnson T.A., Presman D.M., Fettweis G., Wagh K., Rinaldi L., Stavreva D.A., Paakinaho V., Jensen R.A.M., Mandrup S.et al.. An intrinsically disordered region-mediated confinement state contributes to the dynamics and function of transcription factors. Mol. Cell. 2021; 81:1484–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xie J., He H., Kong W., Li Z., Gao Z., Xie D., Sun L., Fan X., Jiang X., Zheng Q.et al.. Targeting androgen receptor phase separation to overcome antiandrogen resistance. Nat. Chem. Biol. 2022; 18:1341–1350. [DOI] [PubMed] [Google Scholar]
- 11. Boija A., Klein I.A., Sabari B.R., Dall’Agnese A., Coffey E.L., Zamudio A.V., Li C.H., Shrinivas K., Manteiga J.C., Hannett N.M.et al.. Transcription factors activate genes through the phase-separation capacity of their activation domains. Cell. 2018; 175:1842–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chong S., Dugast-Darzacq C., Liu Z., Dong P., Dailey G.M., Cattoglio C., Heckert A., Banala S., Lavis L., Darzacq X.et al.. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science. 2018; 361:eaar2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sabari B.R., Dall’Agnese A., Boija A., Klein I.A., Coffey E.L., Shrinivas K., Abraham B.J., Hannett N.M., Zamudio A.V., Manteiga J.C.et al.. Coactivator condensation at super-enhancers links phase separation and gene control. Science. 2018; 361:eaar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Palikyras S., Papantonis A.. Modes of phase separation affecting chromatin regulation. Open Biol. 2019; 9:190167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Houtsmuller A.B., Rademakers S., Nigg A.L., Hoogstraten D.J., Hoeijmakers J.H., Vermeulen W. Action of DNA repair endonuclease ERCC1/XPF in living cells. Science. 1999; 284:958–961. [DOI] [PubMed] [Google Scholar]
- 16. van Royen M.E., Farla P., Mattern K.A., Geverts B., Trapman J., Houtsmuller A.B.. Fluorescence recovery after photobleaching (FRAP) to study nuclear protein dynamics in living cells. Methods Mol. Biol. 2008; 464:363–385. [DOI] [PubMed] [Google Scholar]
- 17. Weidtkamp-Peters S., Weisshart K., Schmiedeberg L., Hemmerich P.. Fluorescence correlation spectroscopy to assess the mobility of nuclear proteins. Methods Mol. Biol. 2008; 464:321–341. [DOI] [PubMed] [Google Scholar]
- 18. Van Royen M.E., van Cappellen W.A., Geverts B., Schmidt T., Houtsmuller A.B., Schaaf M.J.M.. Androgen receptor complexes probe DNA for recognition sequences by short random interactions. J. Cell Sci. 2014; 127:1406–1416. [DOI] [PubMed] [Google Scholar]
- 19. Los G.V., Encell L.P., McDougall M.G., Hartzell D.D., Karassina N., Zimprich C., Wood M.G., Learish R., Ohana R.F., Urh M.et al.. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 2008; 3:373–382. [DOI] [PubMed] [Google Scholar]
- 20. Manzo C., Garcia-Parajo M.F.. A review of progress in single particle tracking: from methods to biophysical insights. Rep. Prog. Phys. 2015; 78:124601. [DOI] [PubMed] [Google Scholar]
- 21. Hansen A.S., Amitai A., Cattoglio C., Tjian R., Darzacq X.. Guided nuclear exploration increases CTCF target search efficiency. Nat. Chem. Biol. 2020; 16:257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Paul M.W., Sidhu A., Liang Y., van Rossum-Fikkert S.E., Odijk H., Zelensky A.N., Kanaar R., Wyman C.. Role of BRCA2 DNA-binding and C-terminal domain in its mobility and conformation in DNA repair. eLife. 2021; 10:e67926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Miné-Hattab J., Heltberg M., Villemeur M., Guedj C., Mora T., Walczak A.M., Dahan M., Taddei A.. Single molecule microscopy reveals key physical features of repair foci in living cells. eLife. 2021; 10:e60577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Muñoz-Gil G., Romero-Aristizabal C., Mateos N., Campelo F., de Llobet Cucalon L.I., Beato M., Lewenstein M., Garcia-Parajo M.F., Torreno-Pina J.A.. Stochastic particle unbinding modulates growth dynamics and size of transcription factor condensates in living cells. Proc. Natl. Acad. Sci. U.S.A. 2022; 119:e2200667119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Paakinaho V., Presman D.M., Ball D.A., Johnson T.A., Schiltz R.L., Levitt P., Mazza D., Morisaki T., Karpova T.S., Hager G.L.. Single-molecule analysis of steroid receptor and cofactor action in living cells. Nat. Commun. 2017; 8:15896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Arbel-Goren R., McKeithen-Mead S.A., Voglmaier D., Afremov I., Teza G., Grossman A.D., Stavans J.. Target search by an imported conjugative DNA element for a unique integration site along a bacterial chromosome during horizontal gene transfer. Nucleic Acids Res. 2023; 51:3116–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lampo T.J., Stylianidou S., Backlund M.P., Wiggins P.A., Spakowitz A.J.. Cytoplasmic RNA-protein particles exhibit non-Gaussian subdiffusive behavior. Biophys. J. 2017; 112:532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Golding I., Cox E.C.. Physical nature of bacterial cytoplasm. Phys. Rev. Lett. 2006; 96:098102. [DOI] [PubMed] [Google Scholar]
- 29. Muñoz-Gil G., Volpe G., Garcia-March M.A., Aghion E., Argun A., Hong C.B., Bland T., Bo S., Conejero J.A., Firbas N.et al.. Objective comparison of methods to decode anomalous diffusion. Nat. Commun. 2021; 12:6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hansen A.S., Woringer M., Grimm J.B., Lavis L.D., Tjian R., Darzacq X.. Robust model-based analysis of single-particle tracking experiments with Spot-On. eLife. 2018; 7:e33125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. van Staalduinen J., van Staveren T., Grosveld F., Wendt K.S.. Live-cell imaging of chromatin contacts opens a new window into chromatin dynamics. Epigenetics Chromatin. 2023; 16:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van Soest R.J., van Royen M.E., de Morrée E.S., Moll J.M., Teubel W., Wiemer E.A.C., Mathijssen R.H.J., de Wit R., van Weerden W.M.. Cross-resistance between taxanes and new hormonal agents abiraterone and enzalutamide may affect drug sequence choices in metastatic castration-resistant prostate cancer. Eur. J. Cancer. 2013; 49:3821–3830. [DOI] [PubMed] [Google Scholar]
- 33. Linder S., Hoogstraat M., Stelloo S., Eickhoff N., Schuurman K., de Barros H., Alkemade M., Bekers E.M., Severson T.M., Sanders J.et al.. Drug-induced epigenomic plasticity reprograms circadian rhythm regulation to drive prostate cancer toward androgen independence. Cancer Discov. 2022; 12:2074–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Severson T.M., Zhu Y., De Marzo A.M., Jones T., Simons J.W., Nelson W.G., Yegnasubramanian S., Freedman M.L., Wessels L., Bergman A.M.et al.. Epigenetic and transcriptional analysis reveals a core transcriptional program conserved in clonal prostate cancer metastases. Mol Oncol. 2021; 15:1942–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lerdrup M., Johansen J.V., Agrawal-Singh S., Hansen K. An interactive environment for agile analysis and visualization of ChIP-sequencing data. Nat. Struct. Mol. Biol. 2016; 23:349–357. [DOI] [PubMed] [Google Scholar]
- 37. Giambartolomei C., Seo J.-H., Schwarz T., Freund M.K., Johnson R.D., Spisak S., Baca S.C., Gusev A., Mancuso N., Pasaniuc B.et al.. H3K27ac HiChIP in prostate cell lines identifies risk genes for prostate cancer susceptibility. Am. Hum. Genet. 2021; 108:2284–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zheng R., Wan C., Mei S., Qin Q., Wu Q., Sun H., Chen C.-H., Brown M., Zhang X., Meyer C.A.et al.. Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 2019; 47:D729–D735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F.. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013; 8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Concordet J.-P., Haeussler M.. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018; 46:W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Slotman J.A., Paul M.W., Carofiglio F., de Gruiter H.M., Vergroesen T., Koornneef L., van Cappellen W.A., Houtsmuller A.B., Baarends W.M.. Super-resolution imaging of RAD51 and DMC1 in DNA repair foci reveals dynamic distribution patterns in meiotic prophase. PLoS Genet. 2020; 16:e1008595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Paul M.W., de Gruiter H.M., Lin Z., Baarends W.M., van Cappellen W.A., Houtsmuller A.B., Slotman J.A.. SMoLR: visualization and analysis of single-molecule localization microscopy data in R. BMC Bioinf. 2019; 20:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hahsler M., Piekenbrock M., Doran d.. dbscan: fast density-based clustering with R. J. Stat. Softw. 2019; 91:1–30. [Google Scholar]
- 44. Reuter M., Zelensky A., Smal I., Meijering E., van Cappellen W.A., de Gruiter H.M., van Belle G.J., van Royen M.E., Houtsmuller A.B., Essers J.et al.. BRCA2 diffuses as oligomeric clusters with RAD51 and changes mobility after DNA damage in live cells. J. Cell Biol. 2014; 207:599–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Arts M., Smal I., Paul M.W., Wyman C., Meijering E.. Particle mobility analysis using deep learning and the moment scaling spectrum. Sci. Rep. 2019; 9:17160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lundahl T., Ohley W.J., Kay S.M., Siffert R.. Fractional Brownian motion: a maximum likelihood estimator and its application to image texture. IEEE Trans. Med. Imaging. 1986; 5:152–161. [DOI] [PubMed] [Google Scholar]
- 47. Metzler R., Jeon J.-H., Cherstvy A.G., Barkai E.. Anomalous diffusion models and their properties: non-stationarity, non-ergodicity, and ageing at the centenary of single particle tracking. Phys. Chem. Chem. Phys. 2014; 16:24128–24164. [DOI] [PubMed] [Google Scholar]
- 48. Michalet X. Mean square displacement analysis of single-particle trajectories with localization error: brownian motion in an isotropic medium. Phys. Rev. E. 2010; 82:041914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ripley B.D. Modelling spatial patterns. J. R. Stat. Soc. Series B Stat. Methodol. 1977; 39:172–192. [Google Scholar]
- 50. Chen C., Mahalingam D., Osmulski P., Jadhav R.R., Wang C., Leach R.J., Chang T., Weitman S.D., Kumar A.P., Sun L.et al.. Single-cell analysis of circulating tumor cells identifies cumulative expression patterns of EMT-related genes in metastatic prostate cancer. Prostate. 2013; 73:813–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sengelaub C.A., Navrazhina K., Ross J.B., Halberg N., Tavazoie S.F.. PTPRN2 and PLCβ1 promote metastatic breast cancer cell migration through PI(4,5)P2-dependent actin remodeling. EMBO J. 2016; 35:62–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ghashghaei M., Niazi T.M., Aguilar-Mahecha A., Klein K.O., Greenwood C.M.T., Basik M., Muanza T.M.. Identification of a radiosensitivity molecular signature induced by enzalutamide in hormone-sensitive and hormone-resistant prostate cancer cells. Sci. Rep. 2019; 9:8838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Duan S., Moro L., Qu R., Simoneschi D., Cho H., Jiang S., Zhao H., Chang Q., de Stanchina E., Arbini A.A.et al.. Loss of FBXO31-mediated degradation of DUSP6 dysregulates ERK and PI3K-AKT signaling and promotes prostate tumorigenesis. Cell Rep. 2021; 37:109870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dozmorov M.G., Hurst R.E., Culkin D.J., Kropp B.P., Frank M.B., Osban J., Penning T.M., Lin H.-K.. Unique patterns of molecular profiling between human prostate cancer LNCaP and PC-3 cells. Prostate. 2009; 69:1077–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Germier T., Audibert S., Kocanova S., Lane D., Bystricky K.. Real-time imaging of specific genomic loci in eukaryotic cells using the ANCHOR DNA labelling system. Methods. 2018; 142:16–23. [DOI] [PubMed] [Google Scholar]
- 56. van Bokhoven A., Caires A., Maria M.D., Schulte A.P., Lucia M.S., Nordeen S.K., Miller G.J., Varella-Garcia M. Spectral karyotype (SKY) analysis of human prostate carcinoma cell lines. Prostate. 2003; 57:226–244. [DOI] [PubMed] [Google Scholar]
- 57. Farla P., Hersmus R., Trapman J., Houtsmuller A.B.. Antiandrogens prevent stable DNA-binding of the androgen receptor. J. Cell Sci. 2005; 118:4187–4198. [DOI] [PubMed] [Google Scholar]
- 58. Wang W., Balcerek M., Burnecki K., Chechkin A.V., Janušonis S., Ślęzak J., Vojta T., Wyłomańska A., Metzler R.. Memory-multi-fractional Brownian motion with continuous correlations. Phys. Rev. Res. 2023; 5:L032025. [Google Scholar]
- 59. Wagh K., Stavreva D.A., Jensen R.A.M., Paakinaho V., Fettweis G., Schiltz R.L., Wüstner D., Mandrup S., Presman D.M., Upadhyaya A.et al.. Dynamic switching of transcriptional regulators between two distinct low-mobility chromatin states. Sci. Adv. 2023; 9:eade1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Briane V., Vimond M., Kervrann C.. An overview of diffusion models for intracellular dynamics analysis. Brief Bioinform. 2020; 21:1136–1150. [DOI] [PubMed] [Google Scholar]
- 61. Seckler H., Metzler R.. Bayesian deep learning for error estimation in the analysis of anomalous diffusion. Nat. Commun. 2022; 13:6717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ghosh R.P., Franklin J.M., Draper W.E., Shi Q., Beltran B., Spakowitz A.J., Liphardt J.T.. A fluorogenic array for temporally unlimited single-molecule tracking. Nat. Chem. Biol. 2019; 15:401–409. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All PC346C cell line ChIP-seq data generated in this study have been deposited in the Gene Expression Omnibus (GEO) database under the accession code: GSE229728 (PC346C AR ChIPseq). Public ChIP-seq datasets used in this study are available from GEO or European Genome-Phenome Archive (EGA) databases under the following accession codes: GSE152231 (AR ChIPseq) and EGAS00001006017 (AR ChIP-seq). Public RNA-seq datasets used in this study are available from EGA under the following accession code EGAS00001006016. NearestNeighbour ImageJ Plugin is available on Github (https://github.com/ErasmusOIC/NearestNeighbour) and Zenodo (http://doi.org/10.5281/zenodo.8359132). TrackSegNet script is available in Zenodo (http://doi.org/10.5281/zenodo.7767750).